Note: Descriptions are shown in the official language in which they were submitted.
n m
CA 02395557 2002-06-21
c
BENZOPHENONE 06-D-GLYCOPYRANOSIDES, PREPARATION AND THERAPEUTIC USE
Field of the invention
The present invention relates, by way of novel industrial products, to
4-cyano-4'-hydroxybenzophenone derivatives of formula I below, which are
benzophenone a-D-glycopyranosides. It further relates to the process for their
preparation and to their use in therapeutics, especially in the form of
compositions
in which they are present as active principles.
Prior art
EP-A-0051023 has disclosed compounds which contain a hydroxybenzo-
phenone residue substituted by a (3-D-xylosyl group and which have valuable
pharmacological activity for the treatment or prevention of venous thrombosis.
Also, EP-A-0133103 has disclosed derivatives of the benzylphenyl
~3-D-xyloside type which possess hypocholesterolemic and hypolipidemic
properties. It is also known that derivatives in which the ~i-D-xylosyl
radical has
been replaced with a (3-D-thioxylosyl radical have been described in EP-A-
0365397
and EP-A-0290321, said compounds being useful on account of their
antithrombotic activity.
Finally, the article by F. BELLAMY et al., J. Med. Chem., 1993, 36 (no. 7),
pages 898-903, has disclosed compounds derived from benzophenone substituted
by glycosyl groups, among which only the derivatives of the (3 configuration
have
antithrombotic activity. A study of these products demonstrated that these
compounds, particularly those containing a [3-D-xylosyl group, were good
substrates for galactosyltransferase I and, consequently, were capable of
initiating
the synthesis of glycosaminoglycans (GAGS). This mode of action, obtained
after
oral administration of the product, is very probably responsible for the
antithrombotic activity, and only those derivatives in which the D-xylose is
of the
(3 configuration exhibit activity in this therapeutic field. There is
therefore a
correlation between the action on GAG synthesis and the antithrombotic
activity
which meant that the compounds other than those derived from (3-D-xylose were
of
no value in this therapeutic field.
Object of the invention
According to the invention, it is proposed to provide a novel technical
solution for obtaining novel products of therapeutic value in respect of
arterial
1H184600/0059 PCT
(PGT/FR 00/03420)
gar
CA 02395557 2002-06-21
2
atheromatous plaque, either for treating said plaque or for preventing its
appearance.
Subject of the invention
According to the novel technical solution of the invention,
[4-(4-cyanobenzoyl)phenyl] a-D-glycopyranoside compounds are used which,
surprisingly, in the light of the publications cited above, exhibit activity
in the
prevention or regression of arterial atheromatous plaque.
The novel products according to the invention are selected from the group
consisting of:
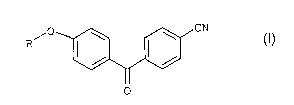
(i) the [4-(4-cyanobenzoyl)phenyl] a-D-glycopyranosides of formula I:
p ~ ~ CN
(z)
0
in which the a-D-glycopyranosyl group R is an a-D-glucopyranosyl,
a-D-galactopyranosyl, a-D-mannopyranosyl, a-D-arabinopyranosyl,
a-D-lyxopyranosyl or a-D-ribopyranosyl group; and
(ii) their esters resulting from the esterification of at least one OH group
on each
glycopyranosyl group by a C2-C4 alkanoic or cycloalkanoic acid.
According to a second feature of the invention, a process is proposed for the
preparation of the compounds of formula I above and their esters.
According to yet a third feature of the invention, a therapeutic composition
is provided which contains, in association with a physiologically acceptable
excipient, a therapeutically effective amount of at least one compound of
formula I
or one of its esters.
According to another feature of the invention, it is also recommended to use
a compound of formula I or one of its esters as an active principle for the
preparation of a drug to be used in therapeutics for combating atheromatous
plaque,
particularly for its prevention or treatment.
Detailed description
The novel compounds according to the invention comprise the products of
formula I and their esters; they are pyranoside derivatives of 4-cyano-4'-
hydroxy
benzophenone [or 4-(4-hydroxybenzoyl)benzonitrile]. The preferred products, in
1H184600/0059 PCT
(PCT/FR 00/03420)
~ar 1 I .
CA 02395557 2002-06-21
3
which the glycoside radical is in the pyranose form, have the formulae below,
which are given according to the structure of the glycopyranosyl group R of
the
a-D configuration:
(a) a-D-glucose structure (a-D-Glc):
O ,~,v0 ~ ~ ~ ~ CN
R10 ~,,~~ ~~,,~ / /
R10 ORl ~ v C IA)
ORl O
(b) a-D-galactose structure (a-D-Gal):
O ,~,~0 ~ ~ CN
R10
~,,.~ / / I
R10 ORl
ORl O
(c) a-D-mannose structure (a-D-Man):
O ,~,~0 ~ ~ ~ ~ CN
R10
R O~~',~ OR / / ~ Ic)
i i
ORl O
(d) a-D-arabinose structure (a-D-Ara):
O ,~,.0 ~ .~ ~ ,,~ CN
/ / CID)
R10 ~ OR1
ORl O
1H184600/0059 PCT
(PCT/FR 00/03420)
ial / I
CA 02395557 2002-06-21
4
(e) a-D-lyxose structure (a-D-Lyx):
0 ,~,~0 ~ ~ ~ ~ CN
/ '~ (IE)
R10 ORl
ORl O
(f) a-D-ribose structure (a-D-Rib):
0 ,,~~~ O ~ ~ ~ ~ CN
/ /
RiOv..,, ...aORi
ORl 0
In these formulae, R1 is a hydrogen atom or a group COR2, R2 being a
C1-C3 alkyl group selected from methyl, ethyl, propyl, isopropyl and
cyclopropyl
groups.
The process for the preparation of a compound of formula I or one of its
esters according to the invention comprises:
( 1 °) reacting a peracetylated pentose or hexose of the pyranosyl
structure of
formula II:
Z O OAc
(II)
Ac0 OAc
OAc
in which Z is H or CHZOAc,
selected from the group consisting of 1,2,3,4,6-pentaacetyl-D-glucose,
1,2,3,4,6-pentaacetyl-D-galactose, 1,2,3,4,6-pentaacetyl-D-mannose, 1,2,3,4-
tetraacetyl-D-arabinose, 1,2,3,4-tetraacetyl-D-lyxose and 1,2,3,4-tetraacetyl-
D-ribose,
with 4-(4-hydroxybenzoyl)benzonitrile of formula III:
1H184600/0059 PCT
(PCT/FR 00/03420)
1I
CA 02395557 2002-06-21
O
(III)
HO CN
to give, after purification, the corresponding oside compound of formula IV:
Z O O ~ ~ CN
(IV)
Ac0 ~ -OAc
OAc O
5 in which Z is defined as indicated above; and
(2°) if necessary, carrying out a displacement reaction on the acetyl
groups of the
resulting oside compound of formula IV in order to replace them with
hydrogen atoms to give the corresponding compound of formula I in which R1
is H, it being possible for the other esters (in which Rl is other than Ac) to
be
obtained by esterifying the compound of formula I in which R1 is H with a
C3-C4 acid.
Advantageously, the reaction II + III of step ( 1 °) is carried out in
an organic
solvent (especially dichloromethane), in the presence of a Lewis acid (for
example
tin tetrachloride), at a temperature between 25°C and the boiling point
of the
1 S solvent, for 10 to 30 hours.
In step (2°), the replacement of the Ac groups with hydrogen atoms
is
advantageously performed as follows. The compound of formula IV is reacted
with
NH3 in solution in an anhydrous alcohol (especially methanol) in order to
displace
the Ac groups and replace them with H.
In a variant, the reaction II + III ~ IV of step ( 1 °) can be replaced
with the
reaction V + III -~ IV, where V is a corresponding peracetylated
halogenopentose
or halogenohexose. Under these circumstances, step (1°) becomes step
(1') below,
namely:
( 1') reacting a peracetylated halogenopentose or halogenohexose of the
pyranosyl
structure of formula V:
1H184600/0059 PCT
(PCT/FR 00/03420)
i1
CA 02395557 2002-06-21
6
Z o X
(~)
Ac0 OAc
OAc
in which X is a halogen atom (i.e. F, C1, Br or I, the preferred halogen atom
being Br) and Z is H or CHZOAc,
selected from the group consisting of 1-bromo-2,3,4,6-tetraacetyl-D-glucose,
1-bromo-2,3,4,6-tetraacetyl-D-galactose, 1-bromo-2,3,4,6-tetraacetyl-D-
mannose, 1-bromo-2,3,4-triacetyl-D-arabinose, 1-bromo-2,3,4-triacetyl-D-
lyxose and 1-bromo-2,3,4-triacetyl-D-ribose,
with 4-(4-hydroxybenzoyl)benzonitrile of formula III:
O
\ \
~ , (zzz)
HO CN
to give, after purification, the corresponding oside compound of formula IV:
Z O O \ \ CN
(v)
Ac0 ~ OAc
OAc O
in which Z is defined as indicated above.
Advantageously, the reaction V + III -+ N is carried out in an anhydrous
solvent such as dichloromethane, 1,2-dichloroethane or acetonitrile, in the
presence
of a coupling agent such as silver trifluoromethanesulfonate or silver oxide,
at a
temperature of the order of -10 to +10°C, for 5 to 40 hours.
The reactions II + III ~ IV and V + III ~ IV are applicable to the
preparation of all the compounds of formula IV according to the invention.
Other advantages and characteristics of the invention will be understood
more clearly from the following Preparatory Examples and pharmacological
tests.
Of course, these details as a whole do not imply a limitation but are provided
by
way of illustration.
1H184600/0059 PC'f
(PCT/FR 00/03420)
1I
CA 02395557 2002-06-21
7
Example 1 (formula 1,4, RI = COCH3)
j4-(4-Cyanobenzoyl)phenylJ 2,3,4,6-tetra-O-acetyl-~D-glucopyranoside
A suspension of 12.17 g (31.10-3 mol) of 1,2,3,4,6-penta-O-acetyl-(3-D
glucose and 10.36 g (46.10-3 mol) of 4-(4-hydroxybenzoyl)benzonitrile in 500
ml of
dichloromethane is prepared and 8.5 ml (72.6.10-3 mol) of anhydrous tin
tetrachloride are added gradually at 0°C, with stirring. After stirring
for 20 hours at
room temperature, a further 4.25 ml (36.3.10-3 mol) of anhydrous tin
tetrachloride
are added and the reaction mixture is refluxed gently for 24 hours. After
cooling,
the reaction medium is poured onto ice. The organic phase is separated off,
then
washed with water, extracted with 1 N sodium hydroxide solution, then washed
with water until the washings are neutral, and dried over magnesium sulfate.
After
concentration under reduced pressure, the residue is purified by
chromatography on
silica gel using a toluene/ethyl acetate mixture (8/2; v/v) as the eluent to
give 918
mg of the expected product in the form of an amorphous white solid (yield =
5.3%).
M.p. = 63°C
[a]Da9 = +g3.2° (c = 0.25; DMSO)
Example 2 (formula 1,,, Rl = H)
j4-(4-Cyanobenzoyl)phenylJ a-D-glucopyranosirie
A solution of 575 mg ( 1.06.10-3 mol) of the compound obtained according
to Example 1 in 50 ml of methanol is prepared and 5.6 ml of a saturated
solution of
ammonia in methanol are added at 0°C, with stirring. The reaction
mixture is
subsequently stirred for 6 hours at room temperature and the solvent is then
driven
off under reduced pressure. The crude product obtained is purified by chroma
tography on silica gel using a dichloromethane/methanol mixture (9/1; v/v) as
the
eluent to give 137 mg of the expected product in the form of a fine white
solid
(yield = 34%).
M.p. = 130°C
[a]D23 = +145° (c = 0.33; DMSO)
Example 3 (formula 1~, RI = COCH3)
j4-(4-Cyanobenzoyl)phenylJ 2,3,4,6-tetra-O-acetyl-~D-mannopyranoside
A solution of 9.9 g (144.10-3 mol) of 4-(4-hydroxybenzoyl)benzonitrile and
26 g (63.10-3 mol) of 2,3,4,6-tetra-O-acetyl-a-D-mannopyranosyl bromide in
300 ml of 1,2-dichloroethane is prepared in the presence of about 4 g of a
molecular sieve. The mixture is cooled to -20°C and 34 g ( 132.10-3
mol) of silver
trifluoromethanesulfonate are added at this temperature. The reaction mixture
is
1H184600/0059 PCT
(PCT/FR 00/03420)
YI
CA 02395557 2002-06-21
8
stirred for 24 hours at 0°C and then filtered to remove the solid
particles. The
organic phase is washed with dilute hydrochloric acid solution, then with
water,
then with dilute sodium hydroxide solution and finally with water. After
drying
over magnesium sulfate, the solution is concentrated under reduced pressure
and
the crude product obtained is purified by chromatography on silica gel using a
toluene/ethyl acetate mixture as the eluent to give 19 g of the expected
product in
the form of a pale yellow solid (yield = 77%).
M.p. = 60°C
[a]D2' _ +64° (c = 0.62; DMSO)
Example 4 (formula 1~, RI = H)
~4-(4-Cyanobenzoyl)phenylJ o~D-mannopyranoside
16.7 g (30.10-3 mol) of the product obtained according to Example 3 are
dissolved in 50 ml of methanol, and 100 ml of a saturated solution of ammonia
in
methanol are added at 0°C. The reaction mixture is stirred for 6 hours
at 0-10°C
and then concentrated under reduced pressure. The crude product is purified by
chromatography on silica gel using a dichloromethane/methanol mixture (1511;
v/v)
as the eluent. The pure product fraction is crystallized from acetone to give
7.9 g of
the expected product in the form of fine light beige crystals (yield = 68%).
M.p. = 145°C
[a]D2'= +102° (c = 0.17; DMSO)
Example 5 (formula IB, R1 = COCH3)
f4-(4-Cyanobenzoyl)phenylJ 2,3,4,6-tetra-O-acetyl-~,~.D-galactopyranoside
The expected product is obtained in the form of an amorphous solid with a
yield of 4% by following a procedure analogous to Example 1 and starting from
1,2,3,4,6-penta-O-acetyl-D-galactose.
M.p. = 64-66°C
[a]D29 = +156° (c = 0.26; DMSO)
Example 6 (formula IB, Rl = H)
~4-(4-Cyanobenzoyl)phenylJ ~z-D-galactopyranoside
The expected product is obtained in the form of a white solid with a yield of
90% by following a procedure analogous to Example 2 and starting from the
compound obtained according to Example 5.
M.p. = 240°C
[a]D29 = +173° (c = 0.25; DMSO)
1H184600/0059 PCT
(PCT'/FR 00/03420)
uo 1 I
CA 02395557 2002-06-21
9
Example 7 (formula ID, R1= COCH3)
~4-(4-Cyanobenzoyl)phenylJ 2,3,4-tri-O-acetyl-a-D-arabinopyranoside
The expected product is obtained in the form of a yellow oil with a yield of
15% by following a procedure analogous to Example 3 and starting from 2,3,4-
tri-
O-acetyl-D-arabinopyranosyl bromide.
[a]D24 - -10.7° (c = 0.38; CH2C12)
Example 8 (formula IA Rl = H)
~4-(4-Cyanobenzoyl)phenylJ a.D-arabinopyranoside
The expected product is obtained in the form of a fine beige solid with a
yield of 75% by following a procedure analogous to Example 4 and starting from
the compound obtained according to Example 7.
M.p. = 160°C
[a]p26 - -66° (c = 0.32; DMSO)
Example 9 (formula IE, RI = COCH3)
[4-(4-Cyanobenzoyl)phenylJ 2,3,4-tri-O-acetyl-c~-D-lyxopyranoside
The expected product is obtained in the form of an oil with a yield of 60%
by following a procedure analogous to Example 1 and starting from 1,2,3,4-
tetra-O-
acetyl-D-lyxopyranose.
[a]D24 = +4Ø30 (c = 0.67; CH2C12)
Example 10 (formula IE, RI = H)
(4-(4-Cyanobenzoyl)phenylJ o~D-lyxopyranoside
The expected product is obtained in the form of a white solid with a yield of
90% by following a procedure analogous to Example 2 and starting from the
compound obtained according to Example 9.
M.p. = 173°C
[a]DZ8 = +125° (c = 0.175; DMSO)
Example 11 (formula IF, Rl =H)
(4-(4-Cyanobenzoyl)phenylJ ~D-ribopyranoside
[4-(4-Cyanobenzoyl)phenyl] 2,3,4-tri-O-acetyl-a-D-ribopyranoside is
obtained in the form of a yellow solid by following a procedure analogous to
Example 1 and starting from 1,2,3,4-tetra-O-acetylribopyranose. It is treated
with a
solution of ammonia in methanol according to the protocol described in Example
2
to give the expected product in the form of a white powder with an overall
yield of
6%.
M.p. = 164°C
1H184600/0059 PCT
(PCT/FR 00/03420)
n ~I
CA 02395557 2002-06-21
[oc]D26 = +77.7 (c = 0.21; DMSO)
The antiatheromatous activity of the compounds according to the invention
was evaluated as a function of their ability to lower the serum cholesterol
level in
mice subjected to a fatty diet. Several publications have in fact demonstrated
a
5 close correlation between an excess of lipids and a marked increase in the
risk of
atheroma (cf. Lancet 1996, 348, pages 1339-1342; Lancet 1990, 335, pages 1233-
1235). This correlation affords a test which is more rapid than direct
experiments
on the atheromatous plaque, which require a lengthy treatment of the animals
and
an expensive histological study of the walls of the aortic arch.
10 The test used consists in administering a single dose of the compound to
female mice of the C57BLJ6J strain. The protocol is as follows: On the first
day
(DO), the mice are fasted from 9 am to 5 pm, a blood sample being taken at 2
pm.
At 5 pm, a given amount of food (a fatty diet comprising 1.25% of cholesterol
and
0.5% of cholic acid) is distributed. On the second day (Dl), the food
leftovers are
weighed at 9 am and the mice are fasted from 9 am to 2 pm. A blood sample is
taken at 2 pm. For the treated groups of mice, the compound is administered at
9 am on the second day (D 1 ) by tubage in the form of a suspension in a 3 %
aqueous solution of gum. The control groups receive only the aqueous gum.
The compounds were tested at a dose of 100 mg/kg. The total serum
cholesterol is assayed and the results are expressed as the percentage
inhibition of
the increase in cholesterolemia compared with the control group. The results
obtained are given in the "Activity" column of Table I. It may furthermore be
noted that analysis of the cholesterol content of the different classes of
serum
lipoproteins shows a favorable effect of the product on the ratio HDL
cholesterol/total cholesterol.
It was also demonstrated that the compounds of formula I according to the
invention do not induce GAG synthesis.
The products of formula I and their esters according to the invention can
preferably be administered orally in the form of tablets or gelatin capsules
each
containing 20 to 500 mg of a compound of formula I or one of its esters as the
active principle, in association with excipients. The dosage will be about 1
to 4
units per day. The products according to the invention are advantageously
prescribed for atheromatous plaque and particularly for preventing or treating
the
risk of atheroma.
1 H 184600/0059 PC'f
(PC'f/FR 00/03420)
CA 02395557 2002-06-21
11
Table 1
CN
O
Ex. R R1 Activity ( %
)
1 a-D-Glc COCH3 - 27
2 a-D-Glc H - 23
3 a-D-Man COCH3 - 10
4 a-D-Man H - 38
5 a-D-Gala COCH3 - 32
6 a-D-Gala H - 35
7 a-D-Ara COCH3 - 25
8 a-D-Ara H - 29
9 a-D-Lyx COCH3 -
10 a-D-Lyx H -48
11 a-D-Rib H - 26
1H184600/0059 PCT
(PCT/FR 00/03420)