Note: Descriptions are shown in the official language in which they were submitted.
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
POLYCYCLIC DIANTHRAQUINONES AS ANTI-CANCER AND ANTI-
ANGIOGENIC AGENTS
FIELD OF THE INVENTION
The present invention relates to the therapeutic use of polycyclic
dianthraquinones
such as hyperycins and helianthrones as inhibitors of angiogenesis and to the
use of some of
them, particularly of 1,3,4,6-tetrahydroxy-helianthrone and derivatives
thereof, as anti-cancer
agents in the absence of light irradiation.
BACKGROUND OF THE INVENTION
The discovery of the signal transduction pathways that activate cell
proliferation in
response to interactions between growth factors and corresponding cellular
receptors,
triggered an extensive search for inhibitors that can interfere with this
cascade in
malignancies where malignant cells undergo uncontrolled proliferation. The
chemical signals
in this cascade have been identified as phosphorylation of proteins either on
tyrosine residues,
catalyzed by a group of enzymes collectively termed protein tyrosine kinases
(PTK), or on
serine/threonine residues by protein kinases A, B, and C. Protein kinase C
(PKC) is also an
important cellular signal transducer that contains a catalytic domain which
phosphorylates
substrates and a regulatory domain which controls its activity.
Polyhydroxylated flavones
such as genistein and quercetin were identified as inhibitors of the
phosphorylation kinases
(Losiewicz et al., 1994).
Perylene quinones are a unique group of kinase inhibitors (Diwu et al., 1994).
The
first of these compounds to be thoroughly evaluated was hypericin, a potent
photodynamic
agent initially discovered by the present inventors to be virucidal to
retroviruses (Lavie et al.,
1989; Meruelo et al., 1988), and subsequently to all lipid-enveloped viruses
(Tang et al.,
1990). Additional studies identified hypericin as a potent and irreversible
light-dependent
inhibitor of protein kinase C (PKC), particularly when PKC is translocated to
the cell
membrane following cell activation, this PKC inhibitory activity of hypericin
being possibly
related to its antiretroviral activity (Takahashi et al., 1989).
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
Hypericin is able to act within biological systems in the dark, possibly
because of a
low red/ox potential, and this appears to enable electron scavenging from
physiological
transfer reactions (Lavie et al., 1994). The unique combination of properties
of hypericin
prompted its current clinical evaluation in phase II clinical trials as an
anti-tumor agent in the
treatment of malignant glioma (Couldwell et al., 1994). This neoplasia relies
on PKC
signaling for cell proliferation. Hypericin is also a potent photosensitizer
capable of
generating singlet oxygen and free radicals (Hadjur et al., 1994). These
properties also render
it useful in photodynamic therapy (PDT) of superficial tumors accessible to
light irradiation.
Unfortunately, hypericin is active in only half of the cases and, in addition,
may cause
severe side effects, such as prolonged post-treatment sensitivity to light, a
condition medically
known as hypericism. It would be desirable to provide additional
photosensitizing agents and
cell proliferation signal transduction inhibitors which can elicit their
cytotoxic effect with
greater efficiency as compared with existing agents and, potentially, with
lower and less
severe side effects.
The present inventors have disclosed previously that some helianthrone
derivatives
may be useful in photodynamic therapy (PDT) of tumors, to elicit destruction
of tumors in
conjunction with light irradiation (PCT Publication WO 99/06347).
While photodynamic properties have been implicated in the mechanism of the
biological activities of hypericin, many of these activities also occur in the
dark. Effects such
as growth inhibition of malignant glioma cells are independent of light
(Couldwell et al.
1994); the virucidal activity of hypericin, while strongly enhanced by light
has also been
documented in the dark against murine cytomegalovirus (Hudson et al., 1991 )
Nowhere in the background art is it taught or suggested that perihydroxylated
polycyclic dianthraquinones are useful for the inhibition of tumor metastases
and prevention
of angiogenesis. There is thus a widely recognized unmet need for inhibitors
of angiogenesis
which specifically blocks the proliferation of vascular structures,
substantially without affecting
other physiological processes, including inhibition of angiogenesis associated
with tumor
growth or progression, restenosis and ophthalmologic disorders.
2
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
SUMMARY OF THE INVENTION
The present invention is based on the surprising finding that some
helianthrone
deivatives are capable, at micromolar concentrations, of inhibiting
transduction of signalsfor
cell proliferation and cell progression through the cell replication cycle,
indicating that they
can be used as antineoplastic agents for the treatment of cancer in the
absence of light
irradiation.
It is further based on the surprising finding that hypericin and helianthrones
interfere
with the process of angiogenesis (formation of new blood vessels) both in the
eye and in the
formation of primary tumors and particularly metastases, indicating that they
can be used for
treatment of ophthalmologic disorders associated with angiogenesis and for
treatment of
primary tumors and prevention of formation of metastases.
It is thus an object of the present invention to provide pharmaceutical
compositions
comprising helianthrone and hypericin derivatives effective as inhibitors of
angiogenesis and
suitable for the treatment of angiogenesis-associated ophthalmologic disorders
and for
inhibition of formation of metastases and of restenosis. It is a further
object of the present
invention to provide such pharmaceutical compositions comprising such
helianthrone
derivatives effective as anti-cancer agents in the absence of light
irradiation.
It is now disclosed that the compositions of the present invention comprising
the
helianthrone derivatives previously described in WO 99/06347 to act as anti-
cancer agents in
conjunction with light in photodynamic therapy, are unexpectedly effective as
well in the
absence of light irradiation. Furthermore, these compositions and also those
containing
hypericin, known for the treatment of primary tumors, or hypericin
derivatives, are
unexpectedly useful as anti-metastatic agents. The compositions of the present
invention are
now disclosed to possess hitherto unknown anti-angiogenic activity. This
invention thus
further relates to pharmaceutical compositions that are useful for the
treatment of pathological
angiogenesis or in conditions requiring inhibition of angiogenesis.
Hypericin and helianthrone and derivatives of both are now disclosed to
interfere with
the process of tumor angiogenesis. This discovery renders these compounds
useful as
treatment modalities in cancer patients undergoing surgical removal of primary
tumors to
prevent the growth of incurable metastases. Surgery has been established to
stimulate the
3
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
growth of micrometastases that were maintained dormant by growth factor
inhibitors secreted
from the primary tumors. These compounds may prevent metastatic growth by
interfering with
the production or activity of vascular endothelial growth factor (VEGF) or
other angiogenic
factors. VEGF, a potent enhancer of vascular permeability, is known to exert
in vivo a key
role in pathological neovascularization associated with many diseases
including tumor
neovascularization, rheumatoid arthritis, and diabetic retinopathy.
The present invention thus provides, in one aspect, the use of a compound
selected
from hypericin, helianthrone or derivatives thereof for the preparation of a
pharmaceutical
composition for inhibition of angiogenesis, said compound having the general
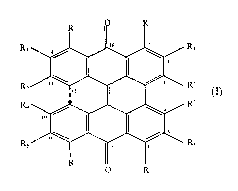
formula (I):
R~ R3
I
R, R'
1 J R' R'
RS R~,
0 R
wherein the dotted line between positions I 1 and 12 represents an optional C1
I-C12 bond; R
is selected from the group consisting of hydroxy, Ci-Cio alkoxy, NH-C,-C,o
alkyl, and NH-
hydroxy(C,-C,o)alkyl but is H at positions 8 and 1~ when there is no C11-C12
bond; R' is
selected from the group consisting of hydroxy and C,-C"~ alkoxy; and R,, R~,
R3, R.~, R; and
Rh are independently selected from the group consistin<~ of hydrogen, hydroxy,
chloro,
bromo, C,-C,o alkyl, Ci-Ci~ alkoxy, and Ci-C,~ alkoxycarbonyl.
In the general formula (I), the helianthrone derivatives are those wherein
there is no
bond between positions 11 and 12 and R is H at positions 8 and 15, and the
hypericin
derivatives are those wherein there is an additional ring formed by the bond
between positions
11 and 12 and R is not H at positions 8 and 15.
Examples of such compounds as the currently most preferred embodiments of the
present invention are hypericin, 10,13-dimethyl-1,3,4,6-
tetrahydroxyhelianthrone and 10,13
dimethyl-1,3,4,6-tetramethoxyhelianthrone of formulas A, B and C as follows:
4
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
OH O OH off o
OCH3 p
i w
-10 ~ I ~ CH3 HO ~ I ~ cry CH30 \ I I ~ CH
3
~O / ~ CH3 HO , \ c~ CH30 / \ CHs
\ ~ ~ i ~ ~ ~ i \ ~ ~ i
OH O OH off o OCFi~ p
A. Hypericin B. Dimethyl tetrahydroxy C. Dimethyl tetramethoxy
helianthrone helianthrone
In one embodiment, the pharmaceutical composition comprising a compound of the
general formula (I) is for use in the treatment of angiogenesis-associated
ophthalmologic
disorders such as, but not being limited to, retinopathies, including but not
limited to diabetic,
retinopathy, macular degeneration and eye, particularly bacterial, infections.
In another embodiment, the pharmaceutical composition comprising a compound of
the general formula (I) is for use in the prevention of metastases.
In a further embodiment, the pharmaceutical composition comprising a compound
of
the general formula (I) is for use in the prevention of restenosis
particularly after percutaneous
I S transluminal coronary angioplasty.
In still a further embodiment, the pharmaceutical composition comprising a
helianthrone compound of the formula (I) is useful for inhibiting transduction
of cell
proliferation signals and is thus suitable for the treatment of cancer in the
absence of light
irradiation.
In another aspect, the present invention provides pharmaceutical compositions
comprising a compound of the general formula (I) as described above for the
uses as
described above.
In a further aspect, the present invention provides a method for the
inhibition of
angiogenesis which comprises administering to a patient in need thereof an
effective amount
of a compound of the general formula (I).
In still a further aspect, the present invention provides a method for
inhibiting
transduction of cell proliferation signals comprising administering to a
patient in need thereof
an effective amount of a helianthrone compound of the formula (I). In a
preferred
5
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
embodiment of this aspect, the helianthrone compound is useful for the
treatment of cancer in
the absence of light irradiation.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 shows the effects of various concentrations of 10,13-dimethyl-1,3,4,6-
tetramethoxy-helianthrone (dimethyl TMH) and hypericin on U251 human
glioblastoma cell
viability in complete darkness.
Fig. 2 shows the effects of various concentrations of dimethyl TMH on LANS
neuroblastoma cell viability in complete darkness.
Fig. 3 shows the effects of various concentrations of dimethyl TMH and
hypericin on
U87MG glioblastoma cell viability in complete darkness.
Fig. 4 shows the effects of various concentrations of dimethyl TMH and TMH on
1 S U87MG glioblastoma cell viability in complete darkness for 48 hours.
Fig. 5 shows the light-dependent photodynamic effects of dimethyl TMH on
primary
post-mitotic human peripheral blood lymphocytes (PBL) viability in the dark
and in
conjunction with light.
Figs. 6A-D show the effects of 10 pM dimethyl TMH on U251 human glioblastoma
cells in culture without (12A) and after treatment for 24 hours (12B), 48
hours (12C), and 72
hours ( 12D).
Figs. 7A-C show the dose response effects of 10 ~M (13B) and 20 ~M (13C)
dimethyl TMH on U251 human glioblastoma cells in culture. Control (untreated,
13A).
Fig. 8 shows percent survival of BALB/c mice inoculated with squamous cell
carcinoma cells after treatment with dimethyl TMH.
Figs. 9A-C show percent survival of BALB/c mice inoculated with DA-3HI -
induced
breast adenocarcinoma cells after treatment with hypericin, at days 89 (9A)
and 100 (9B) after
surgery.
Figs. l0A-B are photographs of eyes of rats after heparanase-induced
angiogenesis
with no treatment (10A, control) and after treatment with hypericin (10B).
6
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
DETAILED DESCRIPTION OF THE INVENTION
The compositions according to the present invention are now disclosed to act
as
effective anti-cancer agents even in the absence of light. Furthermore,
compositions according
to the invention are now disclosed to be especially potent anti-metastatic
agents.
Unexpectedly, the compositions of the invention are now disclosed to be
effective anti
angiogenic agents. Thus these compositions may be used in a variety of
conditions and
diseases involving pathological angiogenesis including but not limited to
restenosis,
angiogenesis-associated ophthalmological diseases and neovascularization
associated with
tumor formation and progression to metastases.
These hitherto unknown attributes were detected while studying the effects of
hypericin and of helianthrone derivatives on breast adenocarciz~oma tumors
induced in mice
with the DA-3Hl cell line and on murine anaplastic squamous carcinoma tumors
induced with
the SQ-2 cell line. Both are highly metastatic tumors and if surgically
resected after having
reached a diameter larger than 5 mm, the mice will go on to develop metastases
in the lungs
and liver. The metastases cause death of the animals within approximately two
months
following surgery.
Although these two types of tumors are not inhibited by hypericin, it was
unexpectedly
discovered that if the tumors are removed surgically as the tumors reach a
diameter of 8-10 mm,
2-4 injections of hypericin into the peritoneum protect the mice from death
due to metastases.
Hypericin prevents thus the development of metastases.
Furthermore, in trying to understand why primary tumors are less affected by
hypericin
whereas metastases are potently inhibited, it was unexpectedly discovered that
hypericin and also
10,13-dimethyl-1,3,4,6-tetrahydroxyhelianthrone and its derivative 10,13-
dimethyl-1,3,4,6-
tetramethoxyhelianthrone prevented neovascularization (ability of the tumor to
induce formation
of new blood vessels to provide blood and nutrient supply to the growing
tumors). By preventing
development of new blood vessels to supply a growing metastasis, the rapidly
growing metastatic
focus is deprived of nutrients and oxygen for its rapidly multiplying cells
and the lesion
degenerates.
Thus it is now disclosed that hypericin itself and helianthrone as well as
their derivatives
are useful in the inhibition of pathological angiogenesis, including
neovascularization associated
7
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
with tumor progression or ophthalmologic disorders as well as endothelial cell
proliferation
associated with restenosis.
The mechanisms by which cancer cells induce the formation of novel blood
vessels and
direct them towards the tumor has been extensively investigated, and is known
to involve
complex mechanisms. The cancer cells secrete vascular endothelium growth
factor (VEGF) that
directs the growth of newly formed blood vessels in the direction of the VEGF
concentration
gradient towards the higher concentration of VEGF eventually reaching the
tumor (Folkman J.,
1985; Folkman J. et al.; 1989).
In the compounds of formula (I) used in the present invention, R is selected
from the
group consisting of hydroxy, C~-C,o alkoxy, NH-C~-C,o alkyl, and NH-hydroxy(C,-
C,o)alkyl;
R' is selected from the group consisting of hydroxy and C~-Coo alkoxy; and R~,
RZ, R3, R4, RS
and R6 are independently selected from the group consisting of hydrogen,
hydroxy, chloro,
bromo, C~-Coo alkyl, C,-C,o alkoxy, and C~-Coo alkoxycarbonyl.
As used herein, "C,-C,o alkyl, "C~-Coo alkoxy" and "C,-Cio alkoxycarbonyl "
refer to
straight or branched radicals having 1 to 10 carbon atoms. Examples of such
alkyl radicals
are, without being limited to, methyl, ethyl, propyl, isopropyl, butyl, hexyl,
and octyl
Examples of such alkoxy radicals are, without being limited to, methoxy,
ethoxy, propyloxy,
isopropyloxy, butoxy, hexyloxy, and octyloxy. Examples of such alkoxycarbonyl
radicals are,
without being limited to, methoxycarbonyl, ethoxycarbonyl, propyloxycarbonyl.
In one
preferred embodiment, R, R' and R, to R6 are methyl, but longer aliphatic
chains envisaged in
these positions instead of the methyl group may have advantages such as
prolongation of
biological activity due to better retention by cells and requiring less
frequent administration.
Preferred compounds used in the invention are hypericin, helianthrone and
derivatives
thereofof formula (I) wherein the two Rs at positions 1 and 6 are hydroxy,
methoxy,
butylamino or hydroxyethylamino, the two R's at positions 3 and 4 are hydroxy
or
methoxycarbonyl, R~ and RS at positions 14 and 9 are hydrogen, and R3 and R6
at
positions 2 and 5 are hydrogen or bromo. Examples of such preferred compounds
are 1,3,4,6-
tetrahydroxyhelianthrone, 1,3,4,6-tetramethoxyhelianthrone, 10,13-dimethyl-
1,3,4,6-
tetrahydroxyhelianthrone, 10,13-di(methoxycarbonyl)-1,3,4,6-tetramethoxyheli-
anthrone, 1,6-
di-N-butylamino-3,4-dimethoxyhelianthrone, 1,6-di-N-butylamino-3,4- dimethoxy-
10,13-
dimethylhelianthrone, 1,6-di-(N-hydroxyethylamino)-3,4-dimethoxy-
helianthrone, 2,5-
dibromo-1,3,4,6-tetrahydroxyhelianthrone, 2,5-dibromo-10,13- dimethyl-1,3,4,6-
8
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
tetrahydroxyhelianthrone, and, most preferably, 10,13-dimethyl-1,3,4,6-
tetramethoxyhelianthrone.
The compounds of the formula (I) according to the invention in which RZ and R4
are
each lower alkyl can be prepared by the method described in US Patent
5,120,412 using as a
starting material a 1,3-dihydroxy-6-(lower alkyl)-anthraquinone of the formula
(II):
II
R' OH
O
in which R' is lower alkyl. Compound II is reduced to the corresponding
anthrone of the
I S formula (III)
O OH
III
\ /
R' OH
in which R' is as defined above and compound III is condensed to obtain
desired compounds
of formula (I) in which R is lower alkoxy.
Other compounds of formula (I) can be prepared in an analogous manner using
appropriately substituted 1,3-dihydroxy-anthraquinones.
The compounds of formula (I) in which Rz and R4 are each lower alkoxycarbonyl
can be
prepared from the diacetyl derivatives of the compound of formula (II) above
in which R' is
methyl, by oxidation with Cr03 to form the compound of the formula (N):
9
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
IV
O
OA
which is then dimerized by the method of Spitzner (Angew. them. Int. Ed., 16,
46 (1977)) to
form a compound of formula (I) in which R is carboxy which is then esterified
with lower
alkanol to obtain the desired product of formula (I) in which R~ and R4 are
lower
alkoxycarbonyl.
The compounds of formula (I) in which each R at positions I and 6 is
alkylamino or
hydroxy alkylamino may be obtained by amination of the corresponding compound
of
Formula I, in which each R is alkoxy, with an alkyl amine such as butyl amine,
or a
hydroxyalkyl amine such as ethanolamine.
According to the present invention, compounds are provided which inhibit cell
proliferation through the mitotic cycle. It was surprising to discover that
these compounds,
and particularly, 10,13-dimethyl-1,3,4,6-tetramethoxy-helianthrone (herein
designated
"dimethyl TMH"), are highly potent in deregulating several cell-cycle related
checkpoints,
which coordinate the orderly passage of cells through the different phases of
the mitotic cycle.
In this cycle, cells in GO resting phase move into G 1 protein and RNA
accumulation phase.
The cells then enter the S phase in which the genomic DNA is duplicated. As
DNA
duplication is completed, the cells are in the G2 phase with double the amount
of DNA, ready
for division, and progress into cell division M phase (mitosis), in which the
cell divides into
two daughter cells. Thus, dimethyl TMH was found to possess basic inhibitor
activity of
transduction of cell proliferation signals and to arrest malignant cells,
including glioblastoma
and neuroblastoma cells at mid S and G2 phases of the cell replication cycle.
In mice bearing
squamous cell carcinoma tumors, dimethyl TMH completely inhibited the spread
of the tumor
into multiple foci and the tumors hardened, became necrotic, and fell off
after prolonged
treatment.
In human malignant glioblastoma cell lines, the blockage of orderly advance of
the
cells through the different cycle phases culminated in cell death (Fig. I),
with dimethyl-TMH
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
identified to be more potent than hypericin in killing the tumor cells in
culture in complete
darkness. Cell killing by dimethyl-TMH occurred at doses in which hypericin
had no effect
on the cultures. Surprisingly, dimethyl-TMH was equally more potent than
hypericin in the
photodynamic induction of cell death when treatments were performed in
conjunction with
light. The mechanisms that operate in the dark were very different from those
that mediate
light-induced photosensitization. In the dark, cell death occurs approximately
four days after
the compound is administered, whereas the cells died within 2-3 hours with
light.
On normal human peripheral blood mononuclear cells, dimethyl-TMH had no effect
on cell viability. Furthermore, intraperitoneal administration of the compound
to BALB/c
mice on a daily basis for one week had no adverse effect on the animals. In
BALB/c mice
bearing anaplastic squamous cell carcinoma tumors, treatments with 200
~g/mouse every
other day resulted in significant inhibition of tumor growth compared to tumor
bearing
untreated control mice.
The pharmaceutical compositions of the invention will be administered to the
patient by
standard procedures. The amount of compound to be administered and the route
of
administration will be determined according to the kind of tumour, stage of
the disease, age and
health conditions of the patient. The preferable routes of administration are
intravenous or direct
injection into the solid tumor of the aqueous solution of the active compound
comprising
conventional pharmaceutically acceptable carriers and additives, and topical
treatment of the
skin tumors with suitable topical compositions. In disseminated tumors with
metastases or
systemic cancers such as leukemias and lymphomas, the preferential routes are
systemic
routes, the intravenous or the oral routes being preferred.
The compounds of the present invention can be used to treat various types of
cancers
and their metastases, including, but without being limited to, squamous cell
carcinoma, basal
cell carcinoma, melanoma, Kaposi sarcoma, breast carcinoma, prostate
carcinoma,
hemangioma, meningioma, astrocytoma, neuroblastoma, carcinoma of the pancreas,
gastric
carcinoma, colorectal carcinoma, colon carcinoma, transitional cell carcinoma
of the bladder,
and carcinoma of the larynx, chronic myeloid leukemia, acute lymphocytic
leukemia, acute
promyelocytic leukemia, multiple myeloma, T-cell lymphoma and B-cell
lymphomas.
The compound used according to the invention can be formulated by any required
method to provide pharmaceutical compositions suitable for administration to a
patient.
11
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
The novel compositions contain, in addition to the active ingredient,
conventional
pharmaceutically acceptable carriers, diluents and the like. Solid
compositions for oral
administration, such as tablets, pills, capsules or the like, may be prepared
by mixing the active
ingredient with conventional, pharmaceutically acceptable ingredients such as
corn starch,
lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium
phosphate and gums,
with pharmaceutically acceptable diluents. The tablets or pills can be coated
or otherwise
compounded with pharmaceutically acceptable materials known in the art to
provide a dosage
form affording prolonged action or sustained release. Other solid compositions
can be prepared
as microscapsules for parenteral administration. Liquid forms may be prepared
for oral
administration or for injection, the term including subcutaneous,
intramuscular, intravenous, and
other parenteral routes of administration. The liquid compositions include
aqueous solutions,
with or without organic cosolvents, aqueous or oil suspensions, emulsions with
edible oils, as
well as similar pharmaceutical vehicles. In addition, the compositions of the
present invention
may be formed as encapsulated pellets or other depots, for sustained delivery.
The active dose for humans is generally in the range of from 0.1 micrograms to
about 1
mg per kg body weight, in a regimen of one or more times a day. However,
administration at
longer intervals may also be possible, for compounds or formulations having
prolonged action.
In general, the preferred range of dosage is from 1 to 200 micrograms per kg
body
weight. It is evident to one skilled in the art that dosage form and regimen
would be determined
by the attending physician, according to the disease to be treated, method of
administration, and
the patient's general condition. It will be appreciated that the most
appropriate administration of
the pharmaceutical compositions of the present invention will depend first and
foremost on the
clinical indication being treated. The prophylactic treatment of a healthy
individual at high risk
for pathological angiogenesis will necessitate a sustained maintenance dosage
regimen
sufficient to inhibit angiogenesis. This type of treatment might be applied to
individuals at risk
for diabetic retinopathy, retinopathy of prematurity, macular degeneration and
other conditions
that are known to afflict particular sets of patients. In contradistinction,
the treatment of existing
disease might require higher doses at more frequent intervals. It is further
anticipated that the
treatment of certain conditions known to involve abnormal vascular smooth
muscle cell
proliferation, including restenosis, will be treated beneficially with
compositions according to
the present invention in an amount sufficient to inhibit vascular smooth
muscle cell
proliferation.
12
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
It will be appreciated by the skilled artisan that in some instances
treatments may
benefically include the administration of the compostions according to the
presnt invention in
conjunction with a depot or medical device. Thus by way of example the
treatment of
angiogenesis in the eye may necessitate an intraocular implant. Similarly the
treatment of
restenosis associated with endothelial cell proliferation may necessitate
application of the
compositon in conjunction with angioplasty , e.g., as a coating on a stmt or
similar device.
The invention will now be illustrated by the following non-limiting Examples.
EXAMPLES
All the compounds used in the present invention are known compounds and
methods
for their preparation are disclosed in the literature and in detail in PCT
Publication 99/06347
of the present applicants, the teachings of which are incorporated herein in
their entirety by
reference as if set forth herein.
Experimental Procedures
A. Cell lines:
Human HL-60 leukemic cells were grown in RPMI-1640 supplemented with 15% fetal
calf serum, 100 mM glutamine and 100 units/ml penicillin-streptomycin. Human
erythroleukemia K-562 cells (derived from a chronic myeloid leukemia patient)
were grown in
the same medium supplemented with 10% fetal calf serum. These cells and the
human U251
glioblastoma, U87MG glioblastoma and LANS neuroblastoma cells used in the
experiments are
available from the ATCC. All cell lines were cultured in a humidified 5%
COZ/95% air
atmosphere at 37°C.
B. Cell viability:
Cell viability was monitored by the MTT assay which measures reduction of 3-
(4,5-
dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide to fonnasan by
mitochondria of viable
cells as described in Mossman, T., J. Irnmunogen., 21, 235-248 (1983). The
cells are incubated
with MTT for four hours at 37°C and analyzed in an ELISA reader at 560
nm. The optical
density of formasan generated by untreated cell cultures (0.D. control) is
defined as one MTT
unit. The number of MTT units in culture samples undergoing treatments is
calculated as the
ratio (O.D.sample - O~D~blank)/O~D~control)~
13
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
C. Photodynamic stress:
Photodynamic (PD) stress is the level of phototoxicity inflicted upon target
cells by
photodynamic compounds and exposure to light. Light irradiation was performed
from a
fluorescent source of two parallel 40 Watts tubes placed at a fixed distance
of 16 cm and
measured to emit an incidence of 4 mWatt/cmz. Light intensities were
quantitated using the IL
1350 Radiometer/Photometer, from International Light Inc., U.S.A.
D. Determination of percentage of apoptotic cells:
Percentage of apoptotic cells was determined by light microscopy on cytospin
cell
preparations stained with May-Grunwald-Giemsa. A total of 400 cells were
counted by two
individuals, independently, and the data are given as the average of the
counts. Apoptotic cells
were recognized by their smaller size and nuclei that were fragmented into
condensed chromatin
bodies.
E. Flow cytometry analysis:
Cells harvested 5 hours after application of photodynamic stress were rinsed
with
phosphate buffered saline (PBS) and fixed with 70% aqueous ethanol. The cells
were then
resuspended in phosphate-citrate buffer (PC buffer) pH 7.8 (192 parts of 0.2 M
NaZPH04 and 8
parts of 0.1 M citric acid) at room temperature for 30 minutes and stained
with propidium
iodide in PC buffer containing 10 pg/ml RNase A. The cells were then analyzed
in a Coulter
EPICS XL-MCL flow cytometer with the entire field gated to include the various
changes that
affected the cells.
14
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
F. DNA Fragmmentation Assay:
DNA fragmentation in cells undergoing apoptosis was assayed as described
previously
(Lotem, J. and Sachs, L., Cell Growth and Dyer., 6, 647-653 (1995). 2x106
cells pelleted in
Eppendorf tubes were lysed in 0.5 ml lysis buffer containing 10 mM Tris-HCI,
pH 7.5, 0.6%
SDS, 10 mM EDTA and 15 ~g/ml RNA mixture (Ambion Corp., Austin TX). After
incubation
at 37°C for 10 minutes, NaCI was added to 1 M and the mixture was kept
overnight at 4°C. The
preparation was spun at 14,000 g for 30 minutes at 4°C, the supernatant
collected, phenol
extracted and DNA precipitated overnight at -20°C by adding 1 ml
ethanol. The DNA pellet
was air-dried, dissolved in 20 p1 TE buffer ( 10 mM Tris, 10 mM EDTA, pH 7.5)
at 4°C for 24
hours, electrophoresed for 4 hours at 2V/cm in 1.5% agarose gel containing 0.5
pg/ml ethidium
bromide and photographed under U.V. light.
Example 1. Killing of Malignant Tumor Cells in Culture by Dimethvl-TMH and
TMH in the Dark
Three human malignant cell lines were evaluated to sensitivity to dimethyl TMH
in
vitro. Human U251 glioblastoma, U87MG glioblastoma and LANS neuroblastoma
cells were
plated (2x 105 per well) in 96-well flat bottom microculture plates, treated
with dimethyl-
TMH and hypericin at dose ranges of 0 (control), 0.1 - 20 pM in complete
darkness for a
period of 72 hours. The medium was aspirated, the adherent monolayer was
washed with
phosphate-buffered saline, and cell viability was monitored by the MTT assay.
Figs. 1, 2 and 3 show the results for the U251 glioblastoma, LANS
neuroblastoma
and U87MG glioblastoma cells, respectively, comparison of the cytotoxic
activity with
hypericin being shown in Figs. 1 and 3. Cell viability was lost in all three
after exposure to
dimethyl-TMH for at least 72 hours, as measured in MTT viability assays. Loss
of cell
viability following treatment with dimethyl-TMH in the dark of the two
glioblastoma cells
was more effective than the treatment with hypericin.
The experiment was then repeated with U251 glioblastoma cells treated with
dimethyl-TMH or tetramethoxy-helianthrone (TMH) at dose ranges of 0.1-12 pM in
complete
darkness. Cell viability was monitored by the MTT assay. The results, in Fig.
4, show that
both dimethyl-TMH and TMH exhibited comparable cytotoxic activities to U251
cells.
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
Example 2. Light-dependent, Photodvnamic Effects of Dimethyl-TMH on Normal
Primary Human Peripheral Blood Lymphocvtes
Human peripheral blood lymphocytes (PBL) are non-proliferating cells in the
absence
of mitogenic stimuli. The effects of different doses of dimethyl-TMH on PBL
were examined
in the presence or absence of irradiation with polychromatic white light. PBL
(post-mitotic)
were plated (2 x 105 cells/well) in two separate round bottom 96-well plates
(in triplicates).
Dimethyl-TMH was added to the cultures. One plate was kept in the dark, and
the other was
exposed to polychromatic white light at a fluence rate of 8 mW/cm2 for 30 min
(a total of
14.4 J/cm'). Both plates were then cultured at 37°C, 5% COZ for 72
hours and cell viability
was assayed by the MTT assay. The results, in Fig. 5, show that dimethyl-TMH
had no effect
on PBL viability in the absence of light irradiation, however,
photosensitization with light
caused cell death with an LDSp of approximately 0.65 pM dimethyl TMH,
indicating that
dimethyl-TMH is a potent photodynamic reagent but does not act on non-
proliferating cells in
the absence of light irradiation.
Example 3: Determination of the Cell Cycle Phases in which Dimethvl-TMH
Arrests Vlali~nant Tumor Cells Growth and Proliferation in the Dark
Cell cycle and DNA content analyses were conducted in U251 human glioblastoma
cells after treatment with ~ pg/ml (10 uM) dimethyl-TMH for 24, 48 and 72
hours, and on
LANS neuroblastoma cells after 48 hours. The cells were then stained with
propidium iodide,
washed with PBS and analyzed in a fluorescence activated cell sorter (FACS). A
computer
program arranged the DNA-related fluorescence as follows: the minimal amount
of
fluorescence is considered to be one whole set of cellular DNA related to the
resting G,
phase. A double amount of fluorescence is considered to be Gz phase, in which
the whole
genome is duplicated following complete DNA synthesis, and the in-between
amounts are
considered to be the DNA synthetic S-phase, in which the total DNA synthesis
is not yet
completed.
The results, shown in Figs. 6-7, reveal that administration of 10 pM dimethyl-
TMH to
U251 human glioblastoma cells produced cell proliferation arrest at mid-S
phase ( 12 B). The
proportion of cells found in the S-phase increased steadily with the duration
of exposure to
dimethyl-TMH (Figs. 6A, 6B, 6C). When the dose of dimethyl-TMH was increased
from 10
16
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
~M to 20 ~M (Figs. 7B, 7C), an exclusive arrest at the S phase occurred.
Fluorescence in situ
hybridization (FISH) studies confirmed this imbalance in DNA replication at
the gene level.
This cell cycle arrest causes the toxic effects which elicits cell death.
Example 4: Prevention of formation of metastases in BALB/c mice bearing
highly invasive sguamous cell carcinoma with dimethvl
tetramethoxvhelianthrone.
The effective cytocidal activity of dimethyl-TMH in vitro encouraged the
evaluation
of its safety and anti-tumoral efficacy profile in tumor-bearing mice.
Experiments were
carried out in mice bearing tumors derived from the SQ2 highly metastatic
anaplastic
squamous cell carcinoma (SCC) line. This tumor develops as multifocal centers
that spread at
the vicinity of the primary tumor and metastases develop approximately two
months after cell
inoculation. Treatments with 300 - 600 ~M dimethyl-TMH/mouse, administered
twice or
three-times a week were initiated when the tumors reached 5-7 mm in diameter.
Table 1 shows the results of one of the experiments, in which BALB/c mice were
inoculated with 5 x 105 cells of the SQ2 anaplastic squamous cell carcinoma
line,
intradermally in shaved backs, 8 mice per group. When the primary tumors
reached a
diameter of 5 mm, therapy with 300 pM dimethyl-TMH/mouse, administered
intraperitoneally twice per week, was initiated. Three weeks after the
initiation of therapy, the
number of tumor foci, which have developed at the primary tumor site, was
recorded. The
number of foci, which developed 21 days after start of therapy, was
considerably reduced by
dimethyl-TMH administered at therapeutic doses that were non-toxic to the
animals. In
addition to preventing the multifocal spread of this tumor, the primary tumors
hardened and
fell off in 5 of the treated mice, indicating that complete cure of this tumor
may be achieved
once treatment regimens are optimized.
17
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
Table 1
The Number of Tumor Foci observed 21 days after the Start of Therapy with
Dimethyl
TMH
Mice 1 focus 2 foci 3 foci 4 foci
Control 3 0 2 5
Dimethyl.-TMH 8 1 0 0
Example 5: Survival of Sauamous Cell Carcinoma-Bearing Mice treated
with Dimethvl-TMH
In another experiment, BALB/c mice were inoculated with 5 x 105 cells of the
SQ2
highly metastatic anaplastic squamous cell carcinoma line, intradermally in
shaved backs.
When the primary tumors reached a diameter of 3-4 mm, i.p. administration of
dimethyl
TMH 200 ~g/mouse (400 ~M/mouse) was initiated on day 7 after tumor cell
inoculation and
then administered twice weekly to the tumor-bearing mice 2x per week for six
weeks (total of
12 doses). Animal survival was then followed. The results in Fig. 8 show that
animal survival
was prolonged by approximately 40.3% compared to untreated controls. It is
noteworthy that
the primary tumors continued to grow during the treatment and nevertheless
animal survival
was prolonged. This appears to be the result of reduced metastatic growth as
evident from
Table 1 in Example 4 above.
Example 6: The utilization of dimethvl TMH in antineoplastic therapy of
malignant tumors in mice
The antineoplastic effects of dimethyl TMH in vivo can be examined in a number
of
murine experimental tumors. These include Esb murine lymphoma, MCA-105 sarcoma
and
B16 melanoma which are evaluated in C57BL/6J mice. DA3h1 murine breast
carcinoma cells,
a highly metastatic variant of DA3, which generates metastatic breast
adenocarcinoma in
BALB/c mice, and A431 cells which generate epidermoid tumors in NIH Swiss
mice, are
18
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
evaluated for sensitivities to treatment with dimethyl TMH or with TMH. Tumors
are
propagated in mice, 8-10 animals per group, by intradermal inoculations of
tumor generating
cells. Dimethyl TMH dose escalations ranging between 20-1000 ~M (10-S00
pg/mouse) are
examined. Frequencies of administrations are varied from daily
administrations, 3x weekly to
lx weekly, administered for periods ranging from 2 -12 weeks. Animals are
monitored for
differences in primary tumor size compared to untreated tumor bearing control
mice. To
analyze for spread of metastases all mice are sacrificed at the death of the
first control group
mouse or at times designated for termination of the experiment. Endpoints used
in previous
examples are applied. Spleen, liver and lung weights are parameters which we
use for
determination of metastatic load. Total number of metastatic foci in each of
these organs is a
second parameter determined after fixation in Bouins solution. Animal survival
is another
endpoint that is examined. The mean and median survival times, after tumor
cell inoculation,
is determined. The significance of prolongation of survival is calculated by
comparison to
controls of untreated tumor bearing animals without exposure to light
(compound's dark
effects), in the Paired Student's t-test.
In one experiment, the anti tumoral activity of dimethyl TMH to human tumors
in an
in vivo model is evaluated in the C.B-17 SCID mouse strain (Fox Chase). Human
epidermoid
and glioblastoma tumors have been induced in the skin of these mice by
inoculation with the
corresponding human cell lines. The animals are then subjected to various
dimethyl TMH
treatment protocols, the compound administered intraperitoneally. The animals
are monitored
for tumor size and for survival.
Example 7: Prevention of formation of metastases in DA-3HI induced
breast adenocarcinoma tumors in BALB/c mice with hvpericin.
The primary tumor size at which metastases occur in DA-3HI - derived breast
adenocarcinoma tumors was initially calibrated in BALB/c mice inoculated with
Sx 1 O5 DA-
3HI tumor cells intradermally. It was found that if surgical removal of the
primary tumors was
performed when tumors reached a diameter of Smm or less, the resection of the
primary tumor
cured the mice. If the resection was performed on tumors with larger
diameters, the mice died
of metastases. A diameter of approximately 5 mm appears to be the cutoff at
which metastases
begin to spread.
19
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
DA-3HI tumors were induced in 12-week old female BALB/c mice as described
above. When the tumors reached diameters of 8-10 mm the mice were divided into
four
groups. One group of 19 mice was left untreated and in the three other groups
the tumors were
surgically removed. One of the resected groups received two intraperitoneal
(i.p.) injections
with 200 ~g hypericin (HY) each 5 days apart beginning two days prior to
surgery (16 mice).
Another resected group received five i.p. injections with 200 ~g hypericin
each, 5 days apart
beginning two days prior to surgery ( 17 mice). One resected group was not
treated with
hypericin ( 16 mice). The mice were then followed for survival. Fig. 9A shows
that none of the
tumor-bearing untreated mice survived and 20% of the mice that underwent
surgery also
survived at day 89. However, administration of 2 i.p. injections of hypericin
increased the
survival rate to 35% and administration of 5 hypericin doses increased the
survival rate to
60%.
Fig. 9B shows the cumulative survival of mice which received 2 and 5 doses of
hypericin (200 ~g/mouse each) through 100 days following surgery (these values
persisted for
164 days after tumor inoculation). They suggest complete prevention of
metastases in the
surviving group of mice, particularly in the group which received five doses
of hypericin.
These results indicate that hypericin protects the mice from developing
metastases and thus,
prevents animal death form the outcome of systemic dissemination of cells from
the primary
tumor.
EXAMPLE 8. Prevention of formation of metastases in BALB/c mice bearin
hi~hlv invasive squamous cell carcinoma with hvpericin.
In another set of experiments squamous cell carcinoma tumors were generated in
BALB/c mice by inoculating Sx 105 SQ2 cells per mouse. When the tumors reached
a
diameter of 1.0-1.2 cm in diameter they were removed by surgery (resected).
One group of 5
mice also received three i.p. injections of hypericin of 100 ~g/mouse prior to
surgery and two
regimens of 50 ~g/mouse post surgery at intervals of 5 days between each
dosing. Another
group of 8 mice received six hypericin i.p. injections of 100 ~g/mouse prior
to surgery and
five regimens of 50 ~g/mouse post surgery at intervals 5 days apart. One
control group of 17
mice underwent surgery only without treatments with hypericin and another
control group
remained untreated (22 mice). Animal survival was then followed. It was then
found that
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
60% of the mice which received three hypericin injections remained alive
beyond 240 days
following tumor cell inoculation; of the mice receiving 6 hypericin injections
40% remained
alive, whereas of the mice which underwent surgery only, 20% remained alive.
These results
also show protection rates of 20-40% due to hypericin administration.
In an effort to understand how hypericin prevents the growth of metastases the
experiment was repeated and the morphology of the metastatic lesions then
followed. Primary
DA-3HI tumors were induced in BALB/c mice. Surgery was conducted on the tumors
when
diameters reached 8-10 mm. These mice (17 animals) were divided into two
groups, one
treated with 5 doses of hypericin 200 ~g/mouse at 5 day intervals as
previously described (9
animals) and another group served as untreated control (8 mice). The animals
were grown for
two more months. The mice were then sacrificed and the internal organs
examined for
metastatic lesions. The physical examination revealed numerous well developed
metastatic
lesions in untreated mice that were supplied with large visible blood vessels.
The few lesions
that did develop in some of the mice treated with hypericin were much smaller,
somewhat
more necrotic and devoid of such vasculature (supplying blood vessels). This
was evident only
when hypericin injections were initiated very early prior to the resection of
the primary tumor.
These observations indicate that hypericin inhibits angiogenesis (growth of
new vasculature).
It is likely that this lack of blood supply prevented the development of
metastases and not any
direct anti-cancer effects. Since angiogenesis is primarily mediated by
vascular endothelium
growth factor (VEGF), hypericin may interfere with either the formation or
secretion of
VEGF from tumor cells or with its targeting of growth inducing receptors on
vascular
endothelial cells. Without wishing to be bound to any proposed mechanism,
hypericin has
been shown to inhibit protein kinase C and the latter is essential in VEGF
production.
Interference with the signal transduction pathway that culminates in the
production of VEGF
might be the mechanism for hypericin or helianthrone derivative-mediated
inhibition of the
VEGF effect that results in inhibition of metastatic lesion growth.
Irrespective of the proposed
mechanism of action it is now demonstrated that these compositions are highly
potent
inhibitors of angiogenesis.
21
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
Example 9. Prevention of vascularization (an~io~enesis ) of the anterior
chamber of
the eve by systemic administration of hvpericin.
Four rats (250 g each) were given three intraperitoneal injections of
hypericin (750 pg
per dose in 5 ml water containing 3.5% ethanol) at four day intervals. The
following day the
animals were anesthetized with xylazine-ketamine and angiogenesis (formation
of new blood
vessels) was induced by inoculating 2 p1 heparanase (30 pg/ml) into the
frontal compartment
of the eye in the cornea of one of the two eyes in each rat. A fourth
intraperitoneal injections
of 750 pg hypericin was applied the next day. Two positive control animals
received only 2 p1
heparanase (30 pg/ml) into the frontal compartment of the eye. Angiogenesis
was then
allowed to develop for 5 days at which time animals were anesthetized with
xylazine-
ketamine and examined and photographed under a binocular microscope for
development of
blood vessels in the anterior chamber of the eye. The photograph in Fig. 10A
shows the blood
vessels in a control eye of a rat after heparanase-induced angiogenesis and no
treatment with
hypericin while the photograph in Fig. lOB shows the absence of blood vessels
in the eye of a
hypericin-treated rat. Similar protection was obtained when angiogenesis was
induced in rat
eyes with bFGF (basic fibroblast growth factor) (not shown).
Example 10. Hvpericin interferes with an~io~enesis.
Rat aorta is carved into rings which are embedded in fibrin gels and cultured
in MCDB
131 medium. Endothelial cells that detach from the aorta rings generate
branching
microvessels according to a method previously described (Nicosia R.F. and
Ottinetti A,.
Growth of microvessels in serum-free matrix culture of rat aorta. Laboratory
Investigation 63:
11 S, 1990). Addition of hypericin at a dose range of between 0.1-10 ug/ml
(0.2 - 20 N.M), or
dimethyl tetrahydroxyhelianthrone at a dose range of between 0.1-10 pg/ml (0.2
- 20 NM)
results in the inhibition of formation of the organized microvessels.
The foregoing description of the specific embodiments will so fully reveal the
general nature
of the invention that others can, by applying current knowledge, readily
modify and/or adapt
for various applications such specific embodiments without undue
experimentation and
without departing from the generic concept, and, therefore, such adaptations
and
22
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
modifications should and are intended to be comprehended within the meaning
and range of
equivalents of the disclosed embodiments. It is to be understood that the
phraseology or
terminology employed herein is for the purpose of description and not of
limitation. The
means, materials, and steps for carrying out various disclosed functions may
take a variety of
alternative forms without departing from the invention.
23
CA 02396704 2002-07-08
WO 01/56558 PCT/ILO1/00091
REFERENCES
Couldwell W.T., R. Gopalakrishna, D.R. Hinton, S. He, M.H. Weiss, R.E. Law,
M.L. Apuzzo
and R.E. Law. Hypericin: a potential antiglioma therapy. Neurosurgery 35:705-
710, 1994.
Diwu Z., Zimmermann J., Meyer Th., & Lawn J. W. Design, synthesis, and
investigation of
mechanisms of action of novel protein kinase C inhibitors: perylene quinonoid
pigments.
Biochem. Pharmacol. 47; 373-385, 1994.
Folkman J.: Angiogenesis and its inhibitors. In: Important Advances in
Oncology. De Vita
VT, Hellman S, Rosenberg SA - Editors. p 42. Philadelphia, Lippincott Co.,
1985.
Folkman J. Watson K., Ingber D., Hanahan D. Induction of angiogenesis during
the transition
from hyperplasia to neoplasia. Nature 339: 58, 1989.
Hadjur C., Jeunet A. and Jardon P. Photosensitization by hypericin: ESR
evidence for singlet
oxygen and superoxide anion radicals formation in an in vitro model. J.
Photochem. &
Photobiol. B. Biol 26:67-74, 1994.
Hudson J.B., Lopez-Bazzocchi I. and Towers G.H. Antiviral activities of
hypericin. Antiviral
Res. 15:101, 1991.
Lavie G., F. Valentine, B. Levin, Y. Mazur, G. Gallo, D. Lavie, D. Weiner and
D. Meruelo.
Studies of the mechanisms of action of the antiretroviral agents hypericin and
pseudohypericin. Proc. Nat. Acad. Sci.(USA) 86:5963, 1989.
Lavie G., Y. Mazur, D. Lavie and D. Meruelo. The chemical and biological
properties of
hypericin - A compound with a broad spectrum of biological activities.
Medicinal Res. Rev.
15: 111-119, 1994.
Lavie G., Mazur Y., Lavie D., Prince A. M., Pascual D., Liebes L., Levin B.
and Meruelo D.
Hypericin as an inactivator of infectious viruses in blood products.
Transfusion 35: 392-400,
1995.
24
CA 02396704 2002-07-08
WO 01/56558 PCT/ILOi/00091
Losiewicz M.D., Bradley A.C., Kaur G., Sausville E.A. and Worland P.J. Potent
inhibition of
CDC2 kinase activity by the flavonoid L86-8275. Biochem. Biophys. Res. Commun.
201:589-
595, 1994.
Meruelo D., G. Lavie, D. Lavie. Therapeutic agents with dramatic
antiretroviral activity and
little toxicity at effective doses: aromatic polycyclic diones hypericin and
pseudohypericin.
Proc. Nat. Acad. Sci.(USA) 85: 5230-5324, 1988.
Mossman T. Rapid colorimetric assay for cellular growth and survival:
application to
proliferation and cytotoxicity assays. J. Immunogen. 21:235, 1983.
Takahashi I, S. Nakanishi, E. Kobayashi, H. Nakano, K. Suzuki and T. Tamaoki.
1989.
Hypericin and pseudohypericin specifically inhibit protein kinase C: possible
relation to their
antiretroviral activity. Biochem. Biophys. Res. Commun 165:1207.
Tang J., J.M. Colacino, S.H. Larsen and W. Spitzer. Virucidal activity of
hypericin against
enveloped and non-enveloped DNA and RNA viruses. Antiviral Res.13:313-326,
1990.
25