Note: Descriptions are shown in the official language in which they were submitted.
CA 02399634 2009-05-15
30754-31
-1-
Title of the invention
Process for the preparation of 9-deoxo-8a-aza-(8a-alkyl)-8a-
homoerythromycin A derivatives from 9-deoxo-9(Z)-hydroxyiminoerythromycin A.
A subject-matter of the present invention is a process for the preparation of
9-deoxo-8a-aza-8a-homoerythromycin A and of its 8a-alkylated derivatives from
9-deoxo-
9(Z)-hydroxyiminoerythromycin A.
The present invention relates more particularly to the field of macrolide
antibiotics of erythromycin type and more particularly their azamacrolide
derivatives,
which form the subject-matter of Patent EP 508,699 and correspond to the
following
to general formula:
C. H3 NMe2
I HO
Mg ' ' C,H3
r 0 CH3
,,1
Ho
off
CH3
H C C OMe
H3 Ha 4. 3
a
NH2
CH3
in which R is a hydrogen atom or a C1-C1o alkyl, C2-C10 alkenyl or C6-C12
arylsulfonyl
group, if appropriate substituted.
These compounds are obtained from erythromycin A and their synthesis
involves two major stages:
CA 02399634 2009-05-15
30754-31
-2-
- the creation of the 8a-azalide macrocycle from the 9-(E)-oxime of
erythromycin A, isomerized to the 9-(Z)-oxime, which is subjected to a
stereospecific
Beckmann rearrangement, and
- the modification of the cladinose group at the 4"-position, consisting of
the
conversion of the 4"(S)-OH to 4"(R)-NH2.
The present invention relates more particularly to the first stage and is
aimed at providing a novel process which makes it possible to prepare
optionally 8a-
alkylated 9-deoxo-8a-aza-8a-homoerythromycin A directly from 9-(Z)-
oximeerythromycin
A of formula II given hereinbelow.
The conventional synthetic route can be represented diagrammatically in
the following way:
CA 02399634 2009-05-15
30754-31
-3-
T'U
z O U 0
o
be = m U O
F
N O
L(j ff' o =p o
2C) U=
z
v+
r Os o
_ 0 Z p
O =
b ro,, OU = =o
= 2 U p ~
n _.
Z O
Z = a =
0 ~-= O = ea w E
FO z
pry' 2 .....u=
p L 0= O
2 ~ U O =
{ z p p U
=O U
U
V-,,.., O p = U = = Q s 0
z Z: U v
o~ 2
= ~ 2
2 b = = 0 2
U
= b U
2 S
CA 02399634 2009-05-15
30754-31
-4-
In fact, the current reaction conditions cannot be extrapolated to the
industrial scale.
First of all, they require the isolation of the imidate intermediates III and
IV.
This is because the reduction with sodium borohydride directly on the reaction
mixture
comprising said imidate intermediates cannot be linked in because of the
inhibiting nature
of the pyridine. Furthermore, these intermediates are not very stable and
readily
dehydrate on isolation. This therefore results in a significant drop in the
yield.
Furthermore, the imidate of formula IV is sensitive to epimerization at
the 10-position, thus resulting in an additional loss of product.
Finally, the reduction conditions currently used, namely sodium borohydride
in ethylene glycol or methanol, are not sufficiently effective with regard to
the imidate of
formula III.
Consequently, it is difficult under current experimental conditions to obtain
an overall yield for these two stages, namely Beckmann rearrangement and
reduction
with sodium borohydride via the intermediate isolation of the two imidates,
which is
greater than 30%.
A specific object of the present invention is to provide an effective
alternative to the synthetic route discussed hereinabove.
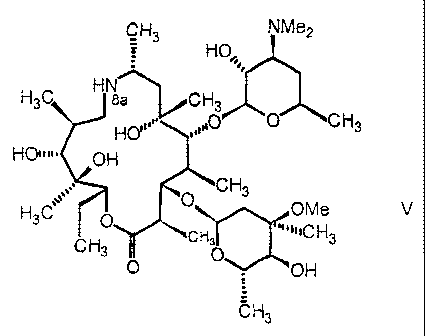
More specifically, a subject-matter of the present invention is a process for
the preparation of a compound of general formula V
CA 02399634 2009-05-15
30754-31
-5-
CH3 NMe2
HO,,, 1
H3C H sa CH3
HO'0 CH3
HO W# OH
CH3
0
3 OMe V
H
C 3 "'"1C H 3
CH3 0 O OH
CH3
said process comprising:
a stereospecific Beckmann rearrangement, in a reaction mixture using organic
solvents
comprising pyridine as main solvent, of a compound of general formula II
CH3 NMe2
HO
HO- - CH
,,,,10 0 CH3
HO'G''HO' 3 '*0
HO
H3C O CH3 OMe II
O ,,,n1CH3
CH3 CH.
0 OH
CH3
into two intermediate imidates III and IV
CA 02399634 2009-05-15
30754-31
-6-
CH3 NMe2
HO.
H3 N CH3
(ya" 0 CH3
HD .... OH
,, 0 CH3 IIl
H3C 0 OMe
CHg H3 ,CH3
o 0 off
CH3
CH3 NMe2
r HO
H3 I CH3
HO' J'=l 141110 0 CH3
H01104 o
1-13C t,.' O CH3 We N ; and
C ,,,111CH3
CH3 :
OH
CH3
a reduction reaction of said compounds 111 and IV, wherein said compounds III
and IV
formed in the reaction mixture of the Beckmann rearrangement are not isolated
from said
mixture and are employed directly in the reduction reaction using a sufficient
amount of
borohydride, after extraction of the pyridine with a hydrocarbon which is
miscible with the
latter and in which said imidates ill and 1V, in the salt form, are insoluble.
CA 02399634 2009-05-15
30754-31
-7-
According to another characteristic, the process according to the invention
is additionally characterized in that the compound of formula V formed on
conclusion of
the reduction reaction is not isolated from the reaction mixture and is
directly N-alkylated
therein by the addition of an alkylating reagent to said mixture in an amount
sufficient to
result in the 9-deoxo-8a-aza-8a-alkyl-8a-homoerythromycin A of formula VI.
The inventors have unexpectedly found that it is possible to reduce the
intermediate imidate compounds III and IV with a highly satisfactory yield
without isolating
them beforehand from said Beckmann rearrangement mixture, with the proviso
that the
pyridine is extracted from this reaction mixture using a hydrocarbon which is
miscible with
lo pyridine and in which the imidates III and IV, protonated at the 3'-
dimethylamino group,
are insoluble.
It is therefore an opposite approach to the conventional route, which
consists in extracting the imidates from the reaction mixture with
dichloromethane, after
partitioning the residue resulting from the rearrangement comprising them in a
water/dichloromethane mixture.
Surprisingly, the use of a hydrocarbon makes it possible to extract and
remove the pyridine from the mixture without significant decomposition of the
imidate IV
and thus makes it possible, on the one hand, to effectively dispense with the
stage of
isolation of the imidate intermediates, which is harmful in terms of yield and
of feasibility
on the industrial scale, and, on the other hand, to reduce them more
efficiently with an
alkali metal borohydride in water and/or an organic solvent. This has the
advantage of
resulting in an overall yield which is markedly improved with respect to the
30% values
mentioned above.
Furthermore, the inventors have also demonstrated that it is possible to N-
alkylate the reduced compound obtained directly in the reaction mixture by
addition of an
aldehyde, without it being necessary to isolate it.
This has the advantages, particularly in the case of N-methylation, of
avoiding the use of chloroform and formic acid, such as in the Eschweiler-
Clarke process
CA 02399634 2009-05-15
30754-31
-8-
used in particular in the abovementioned Application EP 508 699, and of
carrying out the
reaction under conditions which are simplified from the viewpoint of
temperature and
duration.
According to another aspect of the present invention, there is provided a
process as defined above, wherein, after removing the organic solvents,
including
pyridine, from the Beckmann rearrangement mixture, a solvent for the reduction
reaction,
which is an organic solvent selected from C1-C1o alcohol, or which is a
mixture of said
C1-C1o alcohol solvent and water, is added to the residue.
According to still another aspect of the present invention, there is provided
a
process as defined above, wherein, after removing the organic solvents,
including
pyridine, from the Beckmann rearrangement mixture, a solvent for the reduction
reaction,
which is of amide type or which is a mixture of said amide type solvent and
water, is
added to the residue.
According to yet another aspect of the present invention, there is provided a
process as defined above, wherein, after removing the organic solvents,
including
pyridine, from the Beckmann rearrangement mixture, a solvent for the reduction
reaction,
which is of cyclic urea type, or which is a mixture of a said cyclic urea type
solvent and
water, is added to the residue.
The process according to the invention will be described in more detail
herein below with reference to the following reaction schemes:
CA 02399634 2009-05-15
30754-31
-9-
U N S
m Ov 0
Z ~ ~ 2
o Mp U ~p
~ = y o ~
t
o = _
= O O
U =
= a 2
U
N
~ C
q}
N
O
+ V
~C
N C)
W O
L
O
p ca
~ O
m E
N
g O z O c
_ ~ Lr1
z o z 20 Z
ca
, % _ ,u,,HUU C
O c=7 Q n~ C C p
Z = S ~ O N
U 0 c~ o
_ 1 U Z7
U w Q
N
t p Q % z
U i~
N Z = M
CA 02399634 2009-05-15
30754-31
-10-
N U U =
p _ 0
O Q p
U O %
Ouui~, Q
U - U
Z O 2
7
a)
c
L N
!~ >
CIS
I
0
N
Q)
4
"O O
=- 7
O 0
E
is
m rn
cl)
U
r~i X CO
U 2 v
O i O z
0
2 Q =0
z I z
2 +~+U 0 c
m
0 Mp 10 c a
T
~' - 0
CL
2 v
N `' '= IY
E _ ~p U
a) _ F.. L Q~7 O
N Q
o
~ O O O fU
CA 02399634 2009-05-15
30754-31
-11-
With reference to Reaction Scheme 1, the 9-(Z)-oxime of formula II is
first of all subjected to a Beckmann rearrangement in a pyridine-based
mixture,
resulting in the formation of the imidate intermediates III and IV, which are
not
isolated.
This rearrangement is carried out with sulfonyl chloride preferably
selected from tosyl chloride, benzenesulfonyl chloride and mesyl chloride.
According to a preferred embodiment of the invention, the 9-(Z)-oxime of
formula li is treated in anhydrous pyridine with tosyl chloride, in the solid
form or in
solution in an organic solvent, for example in toluene. The amount of tosyl
chloride is
generally between 1 and 10 equivalents with respect to the 9-(Z)-oxime,
preferably
between 1.5 and 4 equivalents. The organic solvent is generally present in an
amount
sufficient to dissolve the tosyl chloride. The reaction is carried out at a
temperature
preferably of between 0 and 5 C.
After reaction, a hydrocarbon is added to the mixture, which hydrocarbon
is miscible with pyridine and the other organic solvents present in said
reaction
mixture and in which hydrocarbon the imidate intermediates, protonated at the
dimethylamino group at the 3'-position, are insoluble. The hydrocarbon is
preferably
selected from linear-, branched- or cyclic-chain hydrocarbons comprising 5 to
15
carbon atoms. Mention may be made, as representative examples, of pentane,
cyclohexane, methylcyclohexane and heptane. It is more preferably heptane.
The mixture is subsequently allowed to separate by settling and the
upper phase, comprising the organic solvents, including pyridine, is removed.
The solvent for the reduction reaction, which can be water and/or an
organic solvent preferably selected from C1-C1o alcohols, preferably methanol
or
isopropanol, solvents of the amide type, preferably N,N-dimethylformamide or
N,N-
dimethylacetamide, or solvents of cyclic urea type, preferably 1,3-dimethyl-
3,4,5,6-
tetrahydro-2(1 H)-pyrimidone (DMPU) or 1,3-dimethyl-2-imidazolinone (DMEU), is
then
added to the residue.
A borohydride, preferably sodium borohydride or potassium borohydride,
is subsequently added, the reaction temperature preferably being maintained
CA 02399634 2009-05-15
30754-31
-12-
between 0 and 5 C. The amount of borohydride is generally between 3 and 15
equivalents with respect to the compound to be reduced, preferably between 4
and 5 equivalents.
After reaction, the amine of formula V is either isolated according to
Reaction Scheme 1 or directly N-alkylated at the 8a-position according to
Reaction
Scheme 2.
The amine V can thus be isolated from the reaction mixture according to
a conventional procedure which generally involves extraction, washing and then
drying operations.
According to another embodiment of the invention, the amine of formula
V can be converted directly in the reduction reaction mixture by adding
thereto a
sufficient amount of an aldehyde of formula R'CHO, with R' being a hydrogen
atom or
a C1-C9 alkyl or C2-C9 alkenyl group, so as to obtain a compound of general
formula
VII
CH3 NMe2
R'CH2 = HO,
H3C N8a CH3
Hole ..,..0 O CH3
HO~II,, OH
CH3
% H3C'l 0 CH3 OMe 0 41,11,
H3 3
O OH
VII
CH3
with R' as defined hereinabove.
CA 02399634 2009-05-15
30754-31
-13-
The aldehyde used is preferably a C, to C4 aldehyde and more
preferably formaldehyde, ethanal or propanal.
In a specific alternative form of this embodiment of the claimed process,
formaldehyde is added directly to the reduction reaction mixture comprising
the amine
V, without isolating it. The reaction temperature is between -10 and +30 C.
The
reaction duration is generally 2 hours.
The 8a-N-methylated amine of formula VI thus formed
CH3 NMe2
Me = HO,
H3C N8a CH3
HO~$ ,,`O O CH3
HOB41.. OH
CH
H3C`~ 3 OMe
"CH
CH3 CH O 3
O OH
CH3 (VI)
can be isolated according to a conventional procedure which generally
involves extraction, washing and then drying operations.
The process according to the invention therefore has the advantage of
making it possible to link together the Beckmann rearrangement reaction on 9-
(Z)-
oximeerythromycin A, carried out in a pyridine-based mixture, and the reaction
for the
reduction of the imidate intermediates thus formed by a borohydride, without
resorting
to their isolation.
CA 02399634 2009-05-15
30754-31
-14-
According to another embodiment, the process according to the
invention makes it possible to link in an alkylation reaction, such as a
methylation, on
the reduced compound, directly in the reduction reaction mixture, under
simplified
conditions.
The examples which appear hereinbelow are given by way of illustration
and without limitation of the present invention.
All the tests are carried out under an inert atmosphere.
EXAMPLE 1:
Preparation of 9-deoxo-8a-aza-8a-homoerythromycin A (V) by addition
of solid tosyl chloride and reduction in water, according to Reaction Scheme
1:
2 g of (9Z)-9-deoxo-9-hydroxyiminoerythromycin A (2.67 mmol) and
then 16 ml of pyridine are introduced into a dry round-bottomed flask rendered
inert
with argon. The solution is cooled to 0 C and then 1.32 g of tosyl chloride
(6.9 mmol, 2.6 eq.) are introduced portionwise. The reaction mixture is
stirred at 0 C
for 1 h 30 and then 20 ml of heptane are added at this temperature. Stirring
is halted
and the reaction mixture is then allowed to separate by settling. The upper
phase is
removed by suction, 20 ml of heptane are again added without stirring and then
the
upper phase is removed by suction.
ml of distilled water are added, with stirring and still at 0 C, followed
by 0.4 g of sodium borohydride (10.5 mmol, 4 eq.) portionwise. The reaction
mixture is
allowed to return to room temperature and is stirred for one hour at this
temperature 10 ml of methanol are then added and, after 1/2 hour, the reaction
mixture is acidified to pH = 3 using a 2N aqueous hydrochloric acid solution.
The
aqueous phase is extracted with two times 10 ml of dichloromethane, the
organic
phases are removed and then the aqueous phase is basified to pH = 11 using a
2N
aqueous sodium hydroxide solution. After extracting twice with 20 ml of
dichloromethane, the organic phases are combined, dried over magnesium sulfate
and concentrated under vacuum.
CA 02399634 2009-05-15
30754-31
-15-
1.73 g of a white solid are obtained, which solid has a purity,
quantitatively determined by HPLC, of 69% with regard to 9-deoxo-8a-aza-8a-
homoerythromycin A, i.e. a quantitatively determined yield of 60%.
EXAMPLE 2:
Preparation of 9-deoxo-8a-aza-8a-homoerythromycin A (V) by addition
of a solution of tosyl chloride in toluene and reduction in a
water/isopropanol mixture,
according to Reaction Scheme 1:
g of (9Z)-9-deoxo-9-hydroxyiminoerythromycin A (13.35 mmol) and
then 70 ml of pyridine are introduced into a dry round-bottomed flask rendered
inert
with argon. The solution is cooled to 0 C and then 6.6 g of tosyl chloride
(34.5 mmol,
2.6 eq.), in a solution of 40 ml of toluene, are introduced. The reaction
mixture is
stirred at 0 C for 1 h 30 and then 80 ml of heptane are added at this
temperature.
Stirring is halted and the reaction mixture is then allowed to separate by
settling. The
upper phase is removed by suction.
70 ml of distilled water and 30 ml of isopropanol are added, with stirring
and still at 0 C, followed by 2 g of sodium borohydride (53 mmol, 4 eq.)
portionwise.
The reaction mixture is allowed to return to room temperature and is stirred
for one
hour at this temperature. 10 ml of methanol are then added and, after 1/2
hour, the
reaction mixture is acidified to pH = 3 using a 2N aqueous hydrochloric acid
solution.
The aqueous phase is extracted with two times 40 ml of dichioromethane, the
organic
phases are removed and then the aqueous phase is basified to pH = 11 using a
2N
aqueous sodium hydroxide solution. After extracting twice with 40 ml of
dichioromethane, the organic phases are combined, dried over magnesium sulfate
and then concentrated under vacuum.
8.6 g of a white solid are obtained, which solid has a purity, quantitatively
determined by HPLC, of 75.5% with regard to 9-deoxo-8a-aza-8a-homoerythromycin
A, i.e. a quantitatively determined yield of 65%.
CA 02399634 2009-05-15
30754-31
-16-
EXAMPLE 3:
Preparation of 9-deoxo-8a-aza-8a-homoerythromycin A (V) by addition
of tosyl chloride in solution in toluene and reduction in N,N-
dimethylformamide,
according to Reaction Scheme 1:
20 g of (9Z)-9-deoxo-9-hydroxyiminoerythromycin A (26.7 mmol) and
then 160 ml of pyridine are introduced into a dry round-bottomed flask
rendered inert
with argon. The solution is cooled to 0 C and then 10.5 g of tosyl chloride
(53.4 mmol, 2 eq.), in solution in 60 ml of toluene, are introduced. The
reaction mixture
is stirred at 0 C for 1 h 30 and then 260 ml of heptane are added at this
temperature.
Stirring is halted and the reaction mixture is then allowed to separate by
settling. The
upper phase is removed by suction.
200 ml of DMF are added, with stirring and still at 0 C, followed by 4 g of
sodium borohydride (106 mmol, 4 eq.) portionwise. The reaction mixture is
allowed to
return to room temperature and is stirred for one hour at this temperature. 20
ml of
methanol are then added and, after 1/2 hour, the reaction mixture is acidified
to pH = 3
using a 2N aqueous hydrochloric acid solution. The aqueous phase is extracted
with
two times 50 ml of dichloromethane, the organic phases are removed and then
the
aqueous phase is basified to pH = 11 using a 2N aqueous sodium hydroxide
solution.
After extracting twice with 100 ml of dichloromethane, the organic phases are
combined, dried over magnesium sulfate and then concentrated under vacuum.
18.25 g of a white solid are obtained, which solid has a purity,
quantitatively determined by HPLC, of 64.6% with regard to 9-deoxo-8a-aza-8a-
homoerythromycin A, i.e. a quantitatively determined yield of 59%.
EXAMPLE 4:
Preparation of 9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin A (VI)
by addition of tosyl chloride in solution in toluene and reduction in a
water/isopropanol
mixture, according to Reaction Scheme 2:
CA 02399634 2009-05-15
30754-31
-17-
20 g of (9Z)-9-deoxo-9-hydroxyiminoerythromycin A (26.7 mmol) and
then 160 ml of pyridine are introduced into a dry round-bottomed flask
rendered inert
with argon. The solution is cooled to 0 C and then 10.5 g of tosyl chloride
(53.4 mmol, 2 eq.), in solution of 60 ml of toluene, are introduced. The
reaction
mixture is stirred at 0 C for 1 h 30 and then 240 ml of heptane are added at
this
temperature. Stirring is halted and the reaction mixture is then allowed to
separate by
settling. The upper phase is removed by suction.
140 ml of water and 60 ml of isopropanol are added, with stirring and still
at 0 C, followed by 5 g of sodium borohydride (132.5 mmol, 5 eq.) portionwise.
The
reaction mixture is allowed to return to room temperature and is stirred for
one hour at
this temperature.
22.8 g of 35% aqueous formaldehyde (267 mmol, 10 eq.) are
subsequently added and the reaction mixture is left for 1 h 30 at room
temperature. 20
ml of methanol are then added and, after 1/2 hour, the reaction mixture is
acidified to
pH = 3 using a 2N aqueous hydrochloric acid solution. The aqueous phase is
extracted with two times 50 ml of dichloromethane, the organic phases are
removed
and then the aqueous phase is basified to pH = 11 using a 2N aqueous sodium
hydroxide solution. After extracting twice with 100 ml of ethyl acetate, the
organic
phases are combined, dried over magnesium sulfate and then concentrated under
vacuum.
18.4 g of a white solid are obtained, which solid has a purity,
quantitatively determined by HPLC, of 69% with regard to
9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin A, i.e. a quantitatively
determined
yield of 68%.
EXAMPLE 5:
Preparation of 9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin A (VI)
by addition of a solution of tosyl chloride in toluene and reduction in a
water/1,3-
dimethyl-2-imidazolinone (DMEU) mixture, according to Reaction Scheme 2:
(9Z)-9-Deoxo-9-hydroxyiminoerythromycin A (150 g, 0.19 mol;
purity 94% w/w) and then pyridine (1089 g) are introduced into a dry round-
bottomed
CA 02399634 2009-05-15
30754-31
-18-
flask rendered inert with argon. The solution is cooled to -10 C and then
tosyl chloride
(76.7 g, 0.4 mol, 2.1 equiv.), in solution in toluene (281 g), is run in over
30 minutes.
The reaction mixture is subsequently stirred between -8 and -4 C for 1 h 30
and then
heptane (1011 g) is added at this temperature. Stirring is halted and the
reaction
mixture is then allowed to separate by settling. The upper phase is removed by
suction. The lower viscous phase is diluted a second time with heptane (393 g)
without stirring. The upper phase is subsequently removed by suction. The
reaction
mixture is then diluted with DMEU (140.4 g) and then this solution is added to
a
solution of sodium borohydride (49.1 g, 1.28 mol, 6.85 equiv.) in water (1227
g). After
returning to room temperature, the reaction mixture is stirred for 2 hours at
this
temperature. The mixture is subsequently treated with methanol (351.1 g) and
then
with 37% aqueous formaldehyde (140 g, 1.73 mol, 9.2 equiv.). The mixture is
maintained at room temperature for 2 h. The reaction mixture is subsequently
acidified
to pH = 4 using a 36% aqueous hydrochloric acid solution. The aqueous phase is
extracted with toluene (355 g). The aqueous phase is subsequently basified to
pH = 10 using a 30% w/w aqueous sodium hydroxide solution. After extracting
twice
with toluene (2 x 355 g) at 50 C, the organic phases are combined, washed at
50 C
with water (322 g) and concentrated under vacuum. The organic phase thus
obtained
is taken up several times in heptane in order to distil off the
heptane/pyridine
azeotrope under vacuum.
151 g of a white solid are obtained, which solid has a purity,
quantitatively determined by HPLC, of 65% with regard
to 9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin A, i.e. a quantitatively
determined
yield of 60%.