Note: Descriptions are shown in the official language in which they were submitted.
CA 02406356 2002-10-28
y
~ 1
METHOD AND PRODUCT FOR THE SEQUENCE DETERMINATION
OF PEPTIDES USING A MASS SPECTROMETER
RELATED APPLICATION
This application is a continuation in part of
copending and cosmonly owned application s~trial number
07/891,177 filed May 29, 1992.
FIELD OF THE. INVENTION
This invention relates to rapid and efficient
methods for sequencing formed or forming polypeptides
utilizing a mass spectrometer.
Pailypeptides are a class of compounds composed of
a(-amino acid residues chemically bonded together by amide
linkages with elfaination of water between the c~rboxy
group of one amino acid and the amino group of another.
amino acid. A polypeptide is thus a polymer of ~-amino
acid residues which may contain a large number of such
residues. Peptides are similar to polypeptides, except
that they are comprised of a lesser number of a(-amino
acids. There is no clear-cut distinction between
polypeptides and peptides. For convenience, fn this
disclosure and claims, the term "polypeptide" will be
used to refer generally to peptides and polyp~ptfdes.
CA 02406356 2002-10-28
WO 93/4834 ~ PCT/US93/05070
2
Proteins are polypeptide chains folded into a
defined three dimensional structure. They are c~pl~x
high polymers containing carbon, hydrogen, nitrogen, and
sulfur and are comprised of linear chains of amino acids
connected by peptide links. They are siailar to
polypeptides, but of a much higher molecular weight.
for a complete understanding of physiological
reactions involving proteins it is often nscessary to
understand their structure. There are a number of
facets to the structure of proteins. These are the
primary structure which is concerned with amino acid
sequence in the protein chain and the secondary,
tertiary and quaternary structures which generally
relate to the three dimensional configuration of
~5 proteins. This invention is concerned with s~qusncing
polypeptides to assist in determining the prirxr
structure of proteins. It provides a facile and
accurate procedure for sequencing polypeptides. ~t is
also applicable to sequencing the amino acid residues at
the termini of.proteins.
Many procedures have been used over the years to
deteranine the amino acid sequence, i.e. the primary
structure, of polyp~ptides and proteins. At the present
time, the best method available for such determfnati~ons
CA 02406356 2002-10-28
WO 93I2~34 ~ PCT/LJS93f~0
3
is the Edman degradation. In this procedure, one amino
terminal amino acid residue at a tame is removed f~c~s a
polypeptide to be analyzed. That amino acid is normally
identified by reverse phase high performance liquid
chromatography (I;PLC), but recently mass spectrosetric
procedures have been described for this purpose (1).
The Edman degradation cycle is repeated for each
successive terminal amino acid residue until the
complete polypeptide hes been degraded. The procedure
is tedious and time consuming. Each sequential removal
of a terminal amino acid requires 20 to 30 minutes.
Hence, with a polypeptide of even moderate length, say
for example 5o amino acid residues, a ssque~rce
determination may require many hours. The ~oosdura bas
been automated. The automated machines are available as
sequenators, but it still requires an una~ocepta~le
amount of time to carry out a sequence analysis.
Although the procedure is widely employed, ort~s rich
required less time and which yielded information about a
broader range of modified or unusual amino acid residues
present in a polypeptide would be very useful to the
art. A process which can be used to sequence individual
members of mixtures of polypeptides would be
particularly useful.
CA 02406356 2002-10-28
WO 93/~A$3~ ~ ~'CT/US93111~5070
4
Recent advances in the art of mass sp'ctro~copy
have made it possible to obtain characterizing data Eros
extremely small amounts of polypeptide samples. it is,
for example, presently possible because of the
sensitivity and precision of available instruments to
obtain useful data utilizing from picomole to
subpicomole amounts of products to be analyzed.
Further, the incipient ion-trap technologies pr~eise
even better sensitivities, and have already been
demonstrated to yield useful spectra in the l0-15 to
l0 16 sample range.
In general, both electrospray and matrix-assisted
laser desorption ionizaton methods mainly generate
intact molecular ions. The resolution of the
75 electrospray quadrupole instruments is about 1 in 2,000
and that of the laser desorption time-of-flight
instruments about 1 in 400. Both techniques give sta~ss
accuracies of about 1 in 10-20,000 (i.e. +/- 0.~10 or
better). There are proposed modificdtion~ of tfae-of-
flight analyzer that may improve the resolution by up to
factor of l0-fold, and markedly improve the sensitivity
of that technique.
CA 02406356 2002-10-28
W'U 93/34 ~ ~ ~.ty~93/OSOTO
r
These techniques yield mass measur:menus accurate
to +/- 0.2 atomic mass units, or better. These
capabilities mean that, by employing.the process of this
invention, the polypeptide itself whether already formed
or as it is being formed can be sequenced more readily,
with greater sped, sensitivity,_and precision, than the
amino acid derivative released by stepwise degradation
techniques such as the Edman degradation. ~!s will be
explained in more detail below, the process of this
invention employs a novel technique of sequence
determination in which a mixture containing a family of
~fragments~, each differing by.a single amino acid
residue is produced and thereafter analysed by mass
spectroscopy.
SU~RY OF THE INVENTION
This invention provides a mothoct for the sequential
analysis of polypeptides which may be already formed or
are being formed by producing under controlled
conditions, from the formed polypeptide or from the
segments of the polypeptide as it is being formed, a
mixture containing a series of adjacent polypptides in
which each member of the series differs from the next
adjacent meatier by one amino acid residue. The mixture
is then subjected to mass spectrometric analysis to
generate a spectrum in which the peaks represent the
CA 02406356 2002-10-28
WO 93/14834 ~ PCT/L~593/0!5070
6
ssparate ors of the series. The difference: in
molecular sass between such adjacent ~aeabers fled
with the position of the peaks in the spectrum for such
adjacent members is indicative of the identity of the
said amino acid residue and of its position in the gain
of the formed or forming polypeptide.
The process of this invention which utilizes
controlled cycling of reaction conditions to produce
peptide ladders of predictable structure is to be
contrasted with previous methods employing mass
spectroscopy including exopeptidase digestion on
uncontrolled chemical degradation. See references 2-5.
Because of the uncontrolled nature of these previous
methods, only incomplete sequence information could be
obtained.
BRIEF DESCRIPTION OF THE DRiIWINGS
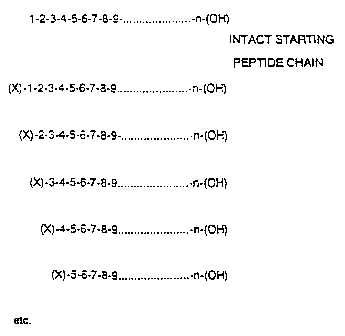
Fig. 1 indicates a family or mixture of
polypeptides (peptide ladder, as defined hereinafter.)
derived from a single formed polypeptide containim~ n
amino acid residues. The mixture is analyzed in
accordance with this invention to determine the amino
acid sequence of the original polypeptide. Each amino
CA 02406356 2002-10-28
WO 9~ ~ PCT/U593/0~78
7
acid in the sequence is denoted by a number with tt~e
numbering starting at the amino terminal of thl peptide.
X denotes a terminating group.
Fig. 2 is an idsalized mass spectrum of the peptide
ladder of a polypeptide similar to the family shown in
Fig. 1.
Fig. 3. shows the reactions involved in generating a
peptide ladder from a formed polypeptide for analysis
utilizing phenyl isothiocyanate (PITC) as the coupling
reagent arid phenyl isocyanate (PIC) as the terminating
reagent.
Fig. 4 is a more precise summary of the process
shown in Fig. 3.
Fig. 5 is an idealised mass spectrum of peptide
t5 ladders obtained from a mi~cture of two formed
polypeptides one of which is identified as A, the other
as B.
Fig. 6 is a positive ion, matrix assisted laser
desorption mass spectrum of the formed polypeptide
[Glut]fibrinopeptide H.
CA 02406356 2002-10-28
VVO 93J~4834 ~ PCT/L~593/05670
8
Fig. 7 is a positive ion matrix assisted laser
desorption spectrum of [Glut]fibrinopeptide 8 after 7
cycles of sequential reactions in accordance with an
embodiment this invention in which a formed polypeptide
is degraded in a controled manner to produce a mixture
containing a peptide ladder:
Fig. 8 is the spectrum o! the peptide ladder in the
region 87-67 obtained from the mixture 99-67 in Example
2.
Fig. 9 is the spectrum of the mixture 6b-33
obtained in Example 2. v
Fig. 10 is a spectrum of the low mass region
obtained from the mixture 66-33 obtained in Exaa~le 2
showing the side reaction products formed duri~ tht
synthesis of HIV-1 protease.
Fig. 11 is a spectrum of the reaction mixtut~e
obtained in Example 3.
Figs. 127 and 128 show the reaction support systsm
employed in an embodiment of the inventions which
permits multiple simultaneous sequencing of
polypeptfdes.
CA 02406356 2002-10-28
1~HH0 93/4834 ~ PGT/liS93/OS070
9
Figs. 13A and 138 are the mass spectra of the
peptide ladders formed from both phosphorylated (1Z11)
and unphosphorylated (128) 16 residue peptides
containing a serine residue.
Fig. 14 shows the spectrum of a protein ladder
generated by incomplete Edman degradation:
Fig. 15 shows the spectrum of the mixture obtained
in Example 4.
As will be explained in more detail below, Ffgs. 8
through to are spectra obtained in the sequencing of a
forming polypeptida employing the process of this
invention.
The invention will be more easily understood if
certain of the terms used in this specification and
claims are defined.
The term "polypeptids" is used herein in a gens~ric
sense to describe both high and low molecular wsigh!t
products comprising linear covalent polymers of amino
acid residues. As the description of this invention
proceeds, it will be seen that mixtures are produced
which may contain individual components containing 100
CA 02406356 2002-10-28
WO 93/244 . pCf/L9S93/05D70
or more amino acid residues or as few as one or two such
residues. Conventionally, such low molecular weight
products would be referred to a amino acids, dipaptid~s,
tripeptides, etc. liowaver, for convenience hsrsin, all
5 such products will be referred to as polypoptfdas sinv~s
the mixtures which are prepared for mass spactrosetric
analysis contain such components together with products
of sufficiently high molecular weight to be
conventionally identified as polypieptides.
The term ~formed polypeptide~ refers to an existing
polypeptide which is to be sequenced. It refers, for
example to [Glut]fibrinopeptids B which is sequenced for
purposes of illustration in Example 1. The process of
the invention is, of course; most useful for ssquemcing
the primary structure of unknown polypeptides isolated,
for example, by reverse phase I;PLC of an enzyoatic
digest from a protein.
The term forming polypeptide~ refers to such
polypeptides as they are being formed for example by
solid phase synthesis as illustrated in Example 2.
The ttr~ ~paptida laddar~ refers to a mixtures
containing a aeries of polypsptidss produced by the
processes described'herein either from a foraad or a
CA 02406356 2002-10-28
WO 931 ~ ~ ' PC'f/U593/DS070
11
forcing polypeptide. As will ba seen from the various
figures and understood from this description of the
invention, a peptide ladder comprises a mixture of
polypeptides in which the various components of the
mixture differ from the next adjacent member of the
series by the molecular mass of one amino acid residue.
~ coupling reagents is a reactant which forms a
reaction product with a terminal amino acid residue of a
polypeptide to be sequenced and is subsequently removed
together with the residue.
A terminating reagents is a reactant which
similarly forms a reaction product with a terminal amino
acid of polypeptide and is stable to subsequent cycling
procedures.
DETAILED DESCRIPTION OF THE INVENTION
There are several procedures for building peptide
ladders, some applicable to the sequencing of formed
polypeptides, others to sequencing of polypeptidas as
they are being formed.
One such process will be understood from~a study of
Fig. 3 which shows an embodiment of the invention which
is applicable to formed polypsptides. The figure shows
CA 02406356 2002-10-28
VI~O 93/l~ ~ ' hCT/L~S9310~56'10
' . 12
the sequencing of an original formed poiypeptide which
may contain any number of amino acid residues, even as
many as 50 or more. The polypeptide is shown here by
way of illustration as containing thr.e residues, each
residue with a side chain represented by Rl, RZ or R3 in
accordance with conventional practice.
The significant feature of this embodiment of the
invention, as illustrated in the figure, is that the
reaction conditions are cycled to produce a peptide
to ladder in the final mixture. The final mixture is
analyzed by mass spectroscopy to determine the exact
mass of the components of the ladder, thereby to
accumulate the information necessary to sequence the
original polypeptide.
The skilled artisan will recognise that this
procedure of sequencing a formed polypeptide makes use
of degradation chemistry, but is based on a naw
principle, i.e. the original polypeptide is aa~ployed to
generate a family of fragments, each differing by a
2p single amino acid as shown in Fig. 1 wherein X
represents a terminating agent. Typically X will be a
terminating agent that is resistant to all subsequent
reactions or manipulations in tJie cyclic degradation
CA 02406356 2002-10-28
VItO 93/~~8.14 ~ . PCT/US93/05070
13
process of this invention: As will be described below,
in connection with another embodibent of this invention,
X may also ba hydrogen.
In the process illustrated in Fig. 3, PITC is the
coupling reagent and PIC is the t~rsinati~ reagent.
From such a family or peptide ladder of terminatsd
molecular species prepared as outlined in the figure,
the amino acid sequence can be simply read out in a
single mass spectrometry operation, based on the mass
differences between the intact molecular ions.
Fu=thermore, because of the sensitivity of modern mass
spectrometers, the accuracy of the amino acid ssquatice
thus determined is unaffected, over a wide range (5-fold
or more), by the amount of each molecular species
t5 present in the mixture.
Fig. 2 shows an idealized mass spectrum of a
peptide ladder in which each peak is rep~esentatiy. of
one member of a series of terminated polypeptides each
member of which differs from the adjacent member by one
amino acid residue.
Thus, for example, if the peaks of the highest mass
in Fig. 2 represent a polypeptide, the first five
members of which at the amino terminal end may be:
CA 02406356 2002-10-28
VV4 93/1834 ~ PCT/l?S93/O50?0
14
Glyl-Leu-Val-Phe-Alas-,
the next peak of lower mass would represent
Leu2-Val-Phe-Ala5-
Subsequent peaks would represent products with one ia~ss
amino acid residue. The difference in mass between
adjacent members of the series would be indicative o!
the amino acid residue removed. The difference in
aolecular mass between the first product on the right
and the adjacent product would correspond to a glycine
residue. subsequent peaks show the sequential removal
of leucine, valise, phenylalanine and alanine residues
thus establishing the sequence of these amino acid
residues in the original polypeptide.
Fig. 3 illustrates a practical sequence of
reactions by which the idealized procedure of Figs. 1
and 2 can be conducted utilizing PITC and PIC e1 the
reagents for sequencing an original formed polypeptide
by cycling reaction conditions to produce a peptide
ladder for spectrometric analysis.
20' In the first step of the sequwncing procedure the
original polypeptide is reacted with a mixture of PITC
and PIC under basic conditions: A large molar excess of
each reagent is employed. A much,larger amount of PITc
than of PIC is utilized so as to be certain t3~at at each
CA 02406356 2002-10-28
W~ 931~d834 ~ , ~/US93>05~70
cycle of the pra~cedure most of the available polyp~ptids
reacts with the coupling agent but that a small
measurable fraction of the available peptide rea~ets with
the terminating reagent. The fraction rsa~cted with tbs
5 terminating agent will be detorminad by the relative
activities of the coupling agent and th~ terminating
agent, and the molar ratio of the two roegsnts.
The first reaction products which fo~~m during the
basic step of the cycle comprise a mixture of original
polypeptide terminated with PIC (PC-polypeptide) and an
original polypeptide terminated with PITC (P'fC-
polypeptide). The PIC terminated polypeptide (FaC-
polypeptide) is stable or essentially stable under ail
subsequent reaction conditions with the rmsult thtt it
15 will be present in a measureable amount in the final
mixture when that mixture is ready for analysis.
The next step in the procedure is to subj~act the
PTC-polypeptide/PC-polypeptide mixture to acid
conditions whereupon a reaction product separates from
the PTC-polypeptide. This reaction product contains the
terminal amino acid residue of the original peptide.
The separation of this product results in the~.foruetion
CA 02406356 2002-10-28
W4 93/24 ~ . ~ PCT/US93/05070
16
of a new polypeptide which, because the terminal amino
acid has bean cleaved contains one less wino acid than
the original polypeptide.
The reaction mixture formed at the end of this
cycle contains as the principal products:
1. unreacted coupling and terminating
reagents,
2. a first reaction product which is the
reaction product between the original
polypeptide and the terminnting reagent. it
is a PC terminated polypeptids (PC-
polypeptide).
3. a new polypeptide from which the amino
terminal amino acid residue hae been ~~1.
t5 The skilled artisan will readily understand that
sequential repeats of the cycle just described will
result in the formation of a mixture which contains as
the principal msasureable components a series of PC-
polypsptides each member of which contains one less
aaino acid residue than the next higher aasber of the
series. The member of the series with the highest
molecular mass will be the first reaction product
between the original polypsptide and the terminating
CA 02406356 2002-10-28
~p 93/?A~4 ~ . P'G'f/US93/05070
.
17
reagent. The molecular mass of ench subsequent rea~c~tion
product in tt~e series will ba the molecular mass of the
next higher adjacent member of the series minus the
molecular mass of the terminal amino acid residue
removed by reaction with the PITC. The molecular mass
of the PIC, blocking group or any other blocking group
selected is irrelevant to the spectrometric analysis
since the identity of each amino acid residue removed
from the next adjacent peptide is determined by
differences in molecular mass. These differences
identify the amino acid residue, and the position of
that mesas difference in the spectrum data set deffines
the position of the identified residue in the original
polypeptide.
A constant 5~ termination of the available
polypeptide at each cycle for ten cycles of the
described chemistry would yield a peptide ladder in
which the mole fraction of the original polypeptide
after each cycle would be approximately
MOhE
FRACTION
(X)-i-2-3-4-5-6-7-8-9-10-li-12-.........-n-(OH) .050
(X)-2-3-4-5-6-7-8-9-10-il-12-..........-n-(OH-) .048
(X)-3-4-5-6-7-8-9-10-11-12-..........-n-(OH) .045
(X)-4-5-6-7-8-9-l0-li-12-..........-n-(OH) .043
(X)-5-6-7-8-9-10-li-12-..........-n-(OH) .041
(X)-6-7-8-9-10-11-12-..........-n-(OH) .039
(X)-7-8-9-10-11-12-..........-n-(OH) .037
(X)-8-9-10-il-12-..........-n-(OH) .035
CA 02406356 2002-10-28
WO 93/34834 ~ PGT/13593/05070
18
(X)-9-i0-ii-12-..........-n-(OH) .033
(X)-10-ii-12-..........-n-(O~i) .031
(X)-ii-12-............-n-(Oii) .60
remain
The differences in molecular mass between each
successive member of the series in the peptide ladder
can be readily determined with high precision by mass
spectroscopy.
With relatively low molecular weight polypeptides,
it is possible to repeat each cycle without removal of
unreacted PITC or PIC. However, as illustrated in
Example 1, it is generally preferred to remove unreacted
coupling and terminating reagents at the ,completion of
each cycle. Such removal may also include romoval of
the cleavage reaction product between the coupling
reagent and the terminal amino acid.
Fig. 4 is a more precise summary of the procedure
illustrated in Fig. 3 and described in detail above. It
specifically illustrates the process utilizing a ~one
pots technique. In the figure ~AA~ stands for amino
acid and ATZ represents 5-anilinothiazolinons. The
other symbols have the same meaning as above.
CA 02406356 2002-10-28
W4 93/Z4834 ~ , ~ PCT/US93/03070
19
The figure illustrates the preparation of a peptide
ladder from a fors~d polypeptide using controlled
ladder-generating chemistry. The stepwise degradation
is conducted with a saall asount of PIC and a aajor
proportion of PITC. Successive cycles of peptide ladder
generating chemistry are performed as described above
without intermediate isolation or analysis of released
amino acid derivatives. Finally the mixture containing
the peptide ladder is read out in one step by laser
desorption time-of-flight mass spectrometry (LDHS).
The coupling and terminating reagents are not
liaited to the pair described above. Those skilled in
the art can readily select other equfvaient reagents.
of course, the procedure can be adapted to either the
wino terminal or the carboxy terminal o~ the
polypeptide under analysis.
Another procedure for constructing a peptide ladder
from a formed polypeptide is to conduct each cycle in a
manner to insure incomplete termination. The process is
similar to the above described procedure except that
only a coupling reagent is employed and the peptide
ladder comprises a series of polypeptides none of which
is terminated with a terminating reagent but each of
which differs from the adjacent member of the aeri,eis by
CA 02406356 2002-10-28
WO 93/t483d ~ ~ PCT/U593/050 70
, ~ 20
one amino acid residue. In this procedure, X of Fig. 1
is hydrogen. The principle of this embodiment of the
invention is that only the coupling reagent is amploy~d
in the cycle, and the extant of reaction is limited for
example by limiting reaction times so that all of the
original forced polypeptide does not rsect. ~s a
result, after the cycle has been moved to the acid step,
the reaction mixture produced will contain:
1. Unreacted PITC,
2. The reaction product of PITC and the terminal
amino acid residue with which it has reacted (PTC-
polypeptide),
3. Unreacted original formed polypeptide,
4. A polypeptide with one less amino acid residue
~5 than the original polypeptide.
It will be apparent that by suitable adjusnt of
reaction conditions; continued repetition of the cycle
any selected number of times will produce a desired
peptide ladder similar to the ladder produced in the
procedure which eaploys both coupling and terminating
reagents except that the polypeptide members of the
ladder are not end blocked with a terminating reagent.
This process is similarly applicable to a mixture of
polypeptiaes.
CA 02406356 2002-10-28
WO 9314 ~ PCT/US93/05074
21
J~nother procedure for generating a peptide ladder
with only one reagent involves termination by side
reaction. In one such process, PITC is employed as a
coupling reagent; and, under controlled conditions o#'
oxidation, a small amount of PITC terminated polyQeptide
is converted to stable PIC terminnted peptide to form a
peptide ladder after a selected number of cycles. The
key to this aspect o! the invention is the controlled
oxidation of a small amount of the PITC terminated
polypeptide to form PIC terminated polypeptide which i=
stable, or essentially stable, under subsequent
reactions conditions.
To describe the process with more specificity, the
reaction steps are as follows:
1. React the polypeptide to be ssqwnced
under basic conditions with an excess of PITiC
to convert substantially all of the
polypeptide to PITC terminated polypeptide
(PTC-polypeptide).
2. React the PTC-polypeptide with a
controlled amount of oxygen to convert a
small portion of the PTC-polypeptide, say. St,
to PC-polypeptide while leaving the balance
unchanged.
CA 02406356 2002-10-28
WO 93/~a4834 ~ ~ PCT/U593/05070
22
3. Cycle the mixture to the acid step to
cleave the PITC bound terminal amino acid
from the PTC-polypeptide and leave a
polypeptide with one less amino acid residue
than the original polypeptide.
4. Repeat tile cycle any selected number of
times to generate a peptide ladder for mass
spectrometric analysis.
A very significant practical advantage of the
process of this invention is that it is possible to
sequence a plurality of peptides in one reaction system.
This advantage arises principally from the high degree
of accuracy that is possible because of the recent
advances in mass spectroscopy.
This aspect of the invention will be understood by
reference to Figs. 12A and 12H which show a suitable
device for producing a plurality of peptide ladders. In
the figure, 1 is a reaction support member shown in the
form of a cylinder with a holding basin 2 and a through
bore 3 permitting the passage of chemicals. A series of
absorbent members or discs 4, for axample absorbent
CA 02406356 2002-10-28
WO 93/?A$34 ~ PCT/US93/05070
' 23
meabranes era supported by a thin filter member 5 which
may be simply a glass fiber or other suitable filttr
material.
In practice, the support member would be in a
closed system adapted to permit the appropriate
reactants for the preparation of a peptide ladder on
each disc to contact each polypeptide to be eequa~ed.
After each step of the cycle, the reactants exit the
support member through the bore 3. The reactants are
i0 delivered to the reaction zone by any conventional
pumping system of the type employed to collect reactant;
from a series of reservoirs., mix them and pass the
mixture through a delivery nozzle.
Sequencing of forced polyp~ptides on sappimrs
i~nobilized on a solid support, ns in the this
embodiment of the invention is especially advantageous
because it is applicable to very small amounts of total
sample and because there are reduced handling losses and
increased recoveries.
As applied to the system illustrated in the
figures, any convenient number of polypeptides tro be
sequenced are separately absorbed on separate discs 4
which may be, for example, an absorbent membrane such as
CA 02406356 2002-10-28
WO 93/-f~183~1 ~ PCT/L~S93/05070
' 2q
tha cati~ni~c, hydrophilic, charge modified
polyvinylidene flupride membrane available from
Millipore Corp. as Imobilon CD.
The discs are spaced apart on the filter paler 5
which is supported over the through bore 3 on support
aember 1 which is then placed in a closed system to
conduct the controlled cyclic reactions appropriate to
the production of a peptide ladder in accordance with
this invention.
The amount of polypeptide absorbed on each segment
may be as small as one picomole or even less.
Generally, it is from about.i to about 10 pi~comoles.
In a typical operation, 1 to 10 picoaoles of each
polypepti~de are separately absorbed on the sela~ted
membrane discs and placed separately on the filter deer
which is then placed on the support member a: shown. The
peptides are subjected to the PITC/PIC/base/acid cycle
described above to generate a peptide ladder on each
disc. Each separate peptide ladder containing mixture
to be analyzed may be extracted from each separate
membrane with an organic solvent containing a small
CA 02406356 2002-10-28
VI~O 93/?,~3d ~ . ~.;./~93/05070
amount of surfactant. one useful extraction solvent is
2.5~ trifluoroacetic acid in a 1:i mixture of
acetonitrile and 1-O-n-octyl-~ -glucopyranoside.
Fig. 14 shows the spectrum obtained using the
5 absorbent membrane technology coupled with incomplete
termination described above. To generate the peptide
ladder which was analyzed, 50 picomoles of [Glu-1]
fibrinopeptide 8 on Immobilon-CD membrane was applied to
ABI-471A protein sequences (Applied Biosystem). The
10 sequences was programmed using 5.5 minute cycle time
with a cartridge temperature of 56°C so as to insure
incomplete reaction at each cycle. Six cycles were
performed. Under these conditions, a reaction yield of
about 56~ was estimated. The resulting peptide ladder
15 is comprised of free N-terminal amines.
This example illustrates the speed with which the
sequencing can be performed. Similar spectra were
obtained with a total loading of only 1 picomole of
polypeptide on the membrane.
20 Although this multiple, simultaneous, sequence
analysis of separate formed polypeptides utilizing the
same chemical reagents for separate reactions with the
said polypeptides has been specifically described by
CA 02406356 2002-10-28
WO 93/24834 ~ ~ PCT/U593/OSfTO
' 26.
reference to the use of a mixture of specitic.aoupling
and terminating reagents in the same reaction zone, it
will be apparent that the process is equally applicable
to the other processes described above.
The system is; of course, applicable to the use of
only one disc for the sequencing of a polypeptide or
polypeptide mixture.
Although the discs are shown separately on the
support, they may also be stacked ox replaced with a
column of suitably absorbent packing materials.
Further, there may be a number of support members
in one device and the chemicals fed to the separate
support members through a manifold system so that
instead of only one reaction zone, there may be a
plurality of reaction zones to still further increase
the number of polypeptidas which can be simultaneously
sequenced.
An especially important embodiment of this
invention is that it provides a method of locating
covalent modifications on a polypeptide chain
particularly post translational modifications of
biologically important products which on chemical or
CA 02406356 2002-10-28
WO 93/1834 ~ ~ PCT/US93/05090
' 27
enzymatic hydrolysis produce polypeptidss which are
phosphorylated, aceylated, glycosylatad, cross-linked by
disulfide bonds or otherwise modified. Such
polypeptides are referred to in this specification and
claims as modified polypaptides~.
The inability to directly identify, locate, and
quantify modified amino acid residues such as
phosphorylated residues in a modified polypeptide is a
major shortcoming of standard sequencing methods, and
has imposed major limitations on currently important
areas of biological research, such as mechanisms of
signal transduction. The process of this invention has
general application to the direct identification of
post-translation modifications present in a peptide
chain being sequenced. A modified amino acid residue
that is stable to the conditions used in generating the
peptide ladder from a formed peptide reveals itself as
an additional mass difference at the site of the
covalent modification. As described above, from the
2p mass difference, both the position in the amino acid
sequence and the mass of the modified amino acid can be
determined. The data generated can provide unambiguous
identification of the chemical nature of the post
translational modification.
CA 02406356 2002-10-28
WO 93/Z4$34 . PCT/US93/05070
' ~ 28
A typical example of #:his aspect of the intention
is the analysis of both phosphorylated and
unphosphorylated forms of the 15 residue peptide
LRRASGLIYNNTLMAR amide prepared by the method of
Schnolzer et al (9) containing a phosphorylatsd satins
residue prepared by enzymatic reaction using 3~, 5~-
cyclic AMP-dependent kinase. After ten cycles of
PITC/PIC chemistry on each form of the peptide using the
procedures described above and illustrated in Example 1,
the two separate sequence-defining fragment mixtures
(peptide ladders) were each read out by laser desorption
mass spectrometry. The resulting protein ladder data
sets are shown in Figs. 13A and 13B. Again, the sass
differences define the identity and order of the amino
acids. For the phosphopeptide (Fig. 13A); a mass
difference of 166.7 daltons was observed for the fifth
amino acid from the N-terminal, compared with the mass
difference of 87.0 for the same residue in the
unphosphorylated peptide (Fig: 138). This measured mass
difference corresponds to a phosphyorylated satins
residue, calculated mass 167.1 daltons. Thus, the
protein ladder sequencing method has directly identified
and located a Ser(Pi) at position five in the peptide.
There was no detectable loss of phosphate from tie
CA 02406356 2002-10-28
WO 93/~ ~ PCT/~93/05070
phosphoserine residue, which has besn regard in the
art as the most sensitive and unstable of the
phosphorylated amino acids.
Altough only ten cycles of ladder generating
chemistry were performed, sequence-defining fragments
corresponding to eleven residues ware observed,
apparently arising from a small amount of premature
cleavage (l0). This side reaction which can have
serious consequences for standard Edman methods, has no
effect on the ladder sequencing approach.
A specific and very important advantage of this
invention is that it is not limited to analysis of one
polypeptide. Mixtures of polypeptides can be analyzed
simultaneously in one reaction vessel. Each polypeptide
will give a separate spectrum as shown in idealized form
in Fig. 4. In this figure, the molecular masses of the
original components of the mixture differ by any
arbitrary mass difference. Each of the separate spectra
can be analyzed as described above even though there aay
be appreciable overlapping in molecular mass among the
polypeptides to be sequenced. This will be clenr from
the figure. As a result, it is possible to sequence
CA 02406356 2002-10-28
wo 93r.. ~ pcrit~93ioso~o
proteins by analyzing mixtures of palypeptides obtained
by chemical or enzymatic hydrolysis of the protein. The
process can be outlined as follows:
Protein sample in quantities of nanomoles or less
5 enzymatic or
chemical
hydrolysis
fragments
separate - s.~.
10 by HPLC or gel
electrophormais
collection of separated peptides
parallel cyclic
l adder
15 generating
chemistry
mixture of peptide ladders
mass
spectrometry
20 readout
analysis of data
In most cases, gel electrophoresis will be employed
to separate proteins and HPLC to separate polypeptides.
Thus, for example, a protein mixture can be separated
25 into its protein components by electrophoresis and each
separate component sequenced by digestion into
polypeptides, separation and ladder sequencing in
CA 02406356 2002-10-28
WO 9314834 ~ ~ PCT/US93/OS070
31
accordance with the process of this invention to yield
data from which the sequence of the entire protein can
be deduced. The process of the invention may also be
employed to obtain extensive data relating to the
S primary structure of intact proteins at their amino or
carboxy terminals.
There follows a description of the application of
this invention to a forming peptide.
Stepwise solid phase peptide synthesis involves the
assembly of a protected peptide chain by repetition of a
series of chemical steps (the synthetic cycle) which
results in the addition of one amino acid residue to as
amino acid or peptide chain bound to a support, usually
a rsin such as methylbenzhydrylamine. The final
polypeptide chain is built up one residue at a time,
usually from the C-terminal, by repetition of the
synthetic cycle. As is well known to peptide chemists,
the solid phase synthetic method does not always proceed
according to plan. For any of a number of reasons, some
of the polypeptfde formed may terminate before the finnl
product is produced. For example, a synthesis designed
to produce a polypeptide containing twenty amino acid
residues may produce as side products a variety of
CA 02406356 2002-10-28
wo 93~aa83a . ~ PCT/Li593l05070
32
polypeptides containing lesser numbers of amino acid
residues, e.g. tripeptides, octapeptides and
dodecapeptides.
To utilize the advantages of this invention in
solid phase synthesis, polypeptide-resin samples are
collected after each cycle of amino acid addition.
Mixing approximately equal amounts of all samples
obtained in the course of a synthesis yields a peptide
ladder containing all possible lengths of resin bound
polypeptide. Cleavage of the resin from such a mixture
produces a mixture of free polypeptide chains of all
possible lengths containing a common carboxy or amino
terminal. Usually, stepwise solid phase synthesis
proceeds starting from the carboxy terminal. in these
cases, the resulting peptide ladder will contain
polypeptides all having a common carboxy terminal.
Consideration of the steps involved in the
production of a heptapeptide will explain the procedure.
It the heptapeptide to be produced is of the structure:
Alal-Val-Gly-Leu-Phe-Ala-Gly~,
the first synthetic step is the attachment of Gly to tie
resin, usually with a spacer molecule betw~ean the resin
and the Gly. The next step is the attachment of Nd -
blocked Ala to the Gly following well kno~m, coupling
CA 02406356 2002-10-28
WO 93/24534 ~ ~ PiCf/U593/45070
33
and deblocking procedures so that the synths:is i~
controlled. The cycle is repeated to form the
heptapeptide on the resin from which it may be isolated
by standard methods.
In accordance with the procedure of this invention,
a small sample of polypeptide attached to resin is
removed after each cycle. After completion of the
synthesis, the seven samples are added together to
produce a peptide ladder which contains the following
~0 components.
Gly-Resin
Ala-Gly-Resin
Phe-Ala-Gly-Resin
Leu-Phe-Ala-Gly-Resin
Gly-Leu-Phe-Ala-Gly-Resin
Val-Gly-Leu-Phe-Ala-Gly-Resin
Ala-Val-Gly-Leu-Phe-Ala~Gly-Resin
The mixture is then treated, for example with
hydrogen fluoride to generate a resin-free peptide
ladder which is analyzed mass spectrometrically to
assure that the final heptapeptide is of the desired
amino acid structure.
CA 02406356 2002-10-28
WO 93!24834 ~ ~ PC'T/L)S93/05070
' 34
one possible type of side reaction in stepwise
solid phase synthesis is low level blocking at a
particular residue (step) in the synthesis.
It will be apparent that each has occurred and
mixed separate sample collected subsequent to the step
at which a side reaction such as low level blocking has
occurred above during the assembly of the final
polypeptide will contain a portion of such terminated
side product with the result that the amount of such
terminated peptide is amplified in the final mixture as
prepared for mass spectrometric analysis. Thus, for
example, if for some reason such as low level blocking
there was a termination of some polypeptide at the
decapeptide stage in a synthesis designed to produce a
20-residue polypeptide, the simple from each subsequent
synthetic cycle would contain terminated decapeptide and
the final analytical sample would contain a 10-fold
amplification of this side product. The inforaation
obtained by this method of analysis is very useful in
designing optimum procedures for synthesizing
polypeptides, especially those of high molecular weight.
One adaptation of this invention to solid phase
synthesis is illustrated in Example 2.
CA 02406356 2002-10-28
WO 931~B3d ~ ~ PCT/LiS93/05070
optionally, the peptide train samples collected as
described above may be assayed colorimetrically, for
example by a ninhydrin procedure to determine reaction
yields prior to mixing to form a peptide ladder. This
5 procedure provides a complimentary method of controlling
and assessing the process.
In the foregoing process, a sample of polypeptide
attached to the resin is collected at each step of the
synthetic cycle far the preparation of the #inal
10 analytical mixture. An alternative procedure for
preparing the final sample is deliberate termination of
a small portion of the forming peptide at each step of
the synthetic cycle followed by removal of all of the
peptides from the resin to form the analytical mixture
15 directly.
This can be.accomplished by utilizing, instead of
one reversibly blocked amino acid residue at each step
in the cycle, a mixture of the selected amino acid
residue one portion of which is stable under the
20 reaction conditions, another portion of which is
susceptible to removal of the blocking group under
controlled conditions.
CA 02406356 2002-10-28
WO 93/24834 l ~ PCT/tiS93/05070
36
If, for example, the amino acid residue to be added
to the forming polypeptide is alanine, tha peptide i~ond
could be formed utilizing a mixture of Boc-alanine and
Fmoc-alanine in which the carboxyl group is in the
appropriate form for reaction, for example in the dorm
of an hydroxybenzotriazole ester. After the peptide
bond has been formed, one of the blocking groups, the
removable group, can ba removed under conditions such
that the other blocking group remains intact.
Repetition of this cycle will result in the formation of
the desired polypeptide on the resin together with a
peptide ladder comprising a series of polypeptides each
member of which is joined to the resin and is t~etminated
by the selected blocking group.
The procedure will be more readily understood by
reference to the prepazation of a specific polypeptide
such as:
Glyl-Phe-Ala-Lau-IleS.
The chemistry involved in the preparation of such
pentapeptide is standard solid phase polypsptide
synthesis applied in such a manner as to produce a
peptide ladder. As applied to this invention, by way of
example, the C-terminal amino acid residue would be
CA 02406356 2002-10-28
WO 93/t4~63d ~ PGT/1i593/05070
' 37
joined to the resin, typically through a linker, as a
mixture containing a major proportion of t-Boc-
isoleucine and a minor proportion of Fmoc-i~soleucine,
e.g. in a 19:1 ratio.
The t-Hoc blocking group is next removed with an
acid such as trifluoroacetic acid. Since the ~'moc group
is stable under acid conditions the Fmoc-isoleucine
attached to the resin will retain its blocking group and
will be stable to all subsequent reactions.
0 In the next step of this synthesis, a 19:1 mixture
of Boc- leucine and P'moc-leucine will be joined to the
Ile-Resin, and the Hoc blocking group selectively
removed under acid conditions. As a result of this step
in the synthetic cycle, the state of the resin may be
~5 indicated by:
Fmoc-ile-Resin
Fmoc-Leu-Ile-Resin
Leu-Ile-Rasin
Repetition of these reactions will result in a
20 final resin mixture comprising a peptide ladder which
may be represented by:
CA 02406356 2002-10-28
WO 93/~d834 , PCT/(~93/05fl7~
' 38
Fmoc-Ile-Resin
Fmoc-Leu-Ile-Resin
Fmoc-Ala-Leu-Ile-Resin
Fmoc-Phe-Ala-Leu-Ile-Resin
Fmoc-Gly-Phe-Ala-Lsu-Ile-Resin
Gly-Phe-Ala-Leu-Iie-Resin
This peptide mixture is removed from the resin by
standard solid phase procedures which, optionally, will
also remove the Fmoc group to produce an analytical
sa~sple ready for analysis by mass spectroscopy as
described above.
The peptide ladder can also be formed by the
reverse procedure of employing Fmoc as the removable
group and t-Boc as the terminating group.
The adaptation of this invention to solid phase
synthesis techniques is illustrated in Example 3 and
~'ig: Z1
Any blocking group stable to the conditiona of
chain assembly synthesis can be used in this application
of the invention. For example, acetic acid could be
added to each reversibly N-protected amino acid in a
CA 02406356 2002-10-28
WO 93/1834 ~ ~ PCT/tlS93/f5070
' 39
stepwise solid phase synthesis in an aaount suitable to
cause a few percent permanent blocking of the growing
peptide chain at each step of the synthesis. The mass
of the blocking group is without effect on the ability
to read out the sequence of the peptide synthesized
since the readout relies on mass differences between
adjacent members of the polypeptide series as described
above.
Using the procedures described, each i~ividual
resin bead carries the mixture of target full-length
peptide arid the peptide ladder. Typically each bead
carries from 1 to 1o or more picomoles of polypeptides.
Thus, cleavage of the products from a single bead
permits the direct determination of the sequence of the
~5 polypeptide on that bead.
It is recognized that the foregoing procedures are
described in an idealized form which does not include
possible interference by other functional groups such as
the hydroxyl group in tyrosine and-serine, the dextral
carboxyl groups in dicarboxylic amino acids or the
~extra~ amino groups in dibasic amino acids. This
method of description has been adopted to avoid
unnecessarily lengthening the specification. The
artisan will recognize the problems which will be
CA 02406356 2002-10-28
WO 93/4834 ~ ~ PGT/ZIS93/f50'~0
. . 40
introduced by the other functional group: and will kTiow
how to deal with them utilizing techniques well known to
peptide chemists.
It will also be recognized that the procedures
described have been applied to relatively small
polypeptides. They are equally applicable to large
polypeptides. For example, if the forming polypeptide
is one which contains twenty or more amino acid
residues, it may be expedient to sequence the
pentapeptide, the decapeptide and the pentadecapeptide
to bs certain that the synthesis is going aocordfng to
plan.
A variety of other chemical reaction syst~ems~can be
employed to generate peptide ladders for analysis in
~5 accordance with this invention.
It will be recognized that there are a number of
significant advantages to the processes of this
invention. For example, the demands on yield of the
chemical degradation reactions are much less stringent
and more readily achieved than by wet chemical stepwise
degradation techniques such as the Edman degradation is
which low molecular weight derivatives are recover8d and
analyzed at each chemical step. Other advantages
CA 02406356 2002-10-28
WO 93/~483d ~ PCT/1iS93/tf~070
' 41
include accuracy, speed, convenience, sample reco~rery,
and the ability to recognize modifications in the
peptide such as phosphorylation. Relatively
unsophisticated and inexpensive mass spectrometric
equipment, a.g. time of flight; single quadrupole; etc.
can be used.
By employing the process of this invention, it is
routinely possible to sequence polypeptides containing
or more amino acid residues from one picomole, yr
10 even a smaller amount of a polypeptide in one hour or
less including cyclic degradation, mass spectrometry,
and interpretation.
The processes described may be readily automated
i.e., carried out for example in microtiter plates,
using an x, y, z chemical robot. Furthermore, the
determination of amino acid sequence from mass
spectrometric data obtained from the protein sequencing
ladders is readily carried out by simple computer
algorithms. The process of the invention therefore
includes coaputer read-out of the spectra of the peptide
ladders prcducad.
CA 02406356 2002-10-28
WO 93/24834 ~ PCT/US93/05070
42
The skilled artisan will recognize that t~rete are
some limitations to the procea of the invention as
described above.
For example, some pairs of amino acids such as
laucine and isoleucine have the same molecular weights.
Therefore, they can not be distinguish~d by mass
differences of terminated polypeptides in a series:
There are several procedures for avoiding this
difficulty. One is to differentiate them by CDNA
sequencing. They are highly degenerate codons, so they
can be accommodated by inosine substitution in DNA
probes/primers for isolation/identification of the
corresponding gene. This limitation will have little
impact on practical application of the invention.
Further, several amino acids differ by only 1 emu.
This places stringent requirements on accuracy of mass
determination. However, this invention utilizes a
determination of mass differences between adjacent
peaks, not a determination of absolute masses. Since
mass differences can be determined with great accuracy
by mass spectroscopy, the limitation will also be of
little practical significance.
CA 02406356 2002-10-28
Wfl 93/24834 ~ ~ PCT/C~S93/05070
43
Finally, samples which are blocked at the amino or
carboxy terminal may not be susceptible to the
generation of peptide ladders. This problem can be
circumvented by chemical or enzymatic fragmentation of
the blocked polypeptide chain to yield unblocked
segments which can be separately analyzed.
The following non-limiting examples are given by
way of illustration only and are not to be considered a~
limitations of the invention many apparent variations of
which may be made without departing from the spf~cit or
scope thereof.
Exa~tple 1
Sectuencing of [Glul~Fibrino~tide B
[Glut]Fibrinopeptide B was purchased from Sigma
Chemical Co. (St. Louis, Mo.). The reported sequence
was: Glul-Gly-Val-Asn-Asp-Asn-aGlu-Glu fly-Phel~-Ph~e-
Ser-Ala-Argl4. Matrix assisted laser dssorption mass
spectrometry gave MW 1570.6 dalton (Calculated: 1570.8
dalton) and showed high purity of the starting peptiale.
A mixture of PITC plus 5% v/v phenylisocyanate PhC was
us~d in the coupling step. PIC reacts with the NH~-
of a polypeptide chain to yield an N~ -phenylcarba~nyl-
peptide which is stable to tha conditions of the ~dman
CA 02406356 2002-10-28
WO 93/~~B34 ~ F'(,'f/I~S93/0"5070
' 4~
degradation. A modification of a standard manual ~dman
degradation procedure (6) was used. All reactions wars
carried out in the sameØ5mL polypropylene microfugs
tube under a blanket of dry nitrogen. Peptide
(200pmoles to 10 nmole) was dissolved in 20u1 of
pyridine/water (i:iv/v; pH10.1); 20uL of coupling
reagent containing
PITC:PIC:pyridine:hexafluoroisopropanol (20:1:76:4 v/v)
was added to the reaction vial. The coupling reaction
was allowed to proceed at 50°C for 3 minutes. The
coupling reagents and non-peptide coproducts ware
extracted by addition of 300uL of heptane:ethyl acetate
(l0:iv/v), gentle vortexing, followed by centrifugation
to separate the phases. The upper phase was aspirated
and discarded. This washing procedure was repeated
once, followed by washing twice with hsptane:ethyl
acetate (2:iv/v). The remaining solution containing the
peptide products was dried on a vacuum centrifuge. The
cleavage step was carried out by addition of 20uL of
anhydrous trifluoroacstic acid to the dry residue in the
reaction vial and reaction at 5o°C for 2 minutes,
followed by drying on a vacuum centrifuge. Coupling-
wash-cleavage steps were regeated for a predetermined
number of cycles. The low MW ATZ/PT~i derivatives
released at each cycle were not separated/analysed.
Finally, the total product mixture was subjset~ed to an
CA 02406356 2002-10-28
WO 93f24834 ~ PCTfIdS93fDS9'f0
additional treatment with PIC to convert any remaining
unblocked peptides to their phenyicarbamyl derivatives.
In this final step, the sample was dissolved in 20uL of
trimsthylamine/water (25~wt/wt) in pyridine (i:lv/v);
5 20uL of PIC/pyridine/IiFIP (1:76:4v/v) was added to the
reaction vial. The coupling reaction was carried out at
50°C for 5 min. The reagents were extracted as
described above. After the last cycle of ladder
generating chemistry, the product mixture was dissolved
in O.i~ aqueous trifluoroacetic acid: acetonitrile (2:1,
v/v). A 1uL aliquot ( 250pmol total peptide, assuming
no losses) was mixed with 9uL of ~-cyano-4-hydroxy-
cinnammic acid (5g/L in 0.1~ trifluoroacetic acid:
acetonitrile, 2:i v/v), and l.OuL of this mixture of
~5 total peptide products (25pmo1) and aatrix was applied
to the probe tip and dried in a stream of air at room
temperature. Mass spectra ware acquired in positive ion
mode using a laser desorption time-of-flight instrument
constructed at The Rockefellsr University (7). Ths
20 spectra resulting from 200 pulses at a wavelength of
355nm, 15 mJ per pulse, were acquired over 80 seconds
and added to give a mass spectrum of the protein
sequencing ladder shown in Fig. 7. Masses were
calculated using matrix peaks of known mass as.
25 calibrants.
CA 02406356 2002-10-28
WO 93/t4834 , ~ PCT/U593/05090
fb
Peptide seuuence read-out. Positive ion (MAhDMS)
spectra of [Glut]Fibrinopeptide B is shown in Fig. 6. A
protonated molecular ion [M+H] was observed at m/z
1572.5 (calculated value is 1571.8).
Its positive ion MAhDMS spectrum of the reaction
mixture obtained after seven cycles is shown in Fig. 6.
Each of the peaks in the spectrum represents a related
phenylcarbamoylpeptide derivative in the peptide ladder
(except a few peaks which will discussed later). The
~0 amino acid sequence can be easily read-out from the mass
difference of adjacent two peaks. for instance, the
mass difference are 129.1, 56.9, and 99.2 between peak$
at m/z 1690.9 and 1561.8, peaks at m/z 1561.8 and 1'504.9
and peaks at m/z 1504.9 and 1405.7. Which correspond to
glutamic acid (ca. 129.12), glycine (ca. 57.05) and
vaiine (ca. 99.13) residues, respectively. One set of
paired peaks gives mass difference 119.0 (1062.1-943.1)
which corresponds to the phenylcarbamoyl group. ~n
other words, these two peaks represent one piece of
Zp peptide with or without phenylcarbamoyl group. Peak at
m/z 1553.8 corresponds partially blocked peptide with
pyroglutamic acid at the N-terminus. This results from
cyclization of the N-terminal Glu under the reaction
CA 02406356 2002-10-28
WO 93124834 ~ ~ PCTfidS93f05070
~7
conditions used, such products are readily identified
from the accurately measured mass and know chemical
reaction tendencies.
Example 2
Stepwise solid phase synthesis of the 99 amino acid
residue polypeptide chain corresponding to the monomer
of the HIV-1 protease (SF2 isolate):
PQITLWQRPLVTIRIGGQLKEALLDTGADDTVLHEMNLPGKWKPKMI~GGI"GGFIX9~t
QYDQIPVEI(Aba)GHKAIGTVLVGPTPVNIIGRNLLTQIG(Aba)TLNF99
[where Aba = °(-amino-n-butyric acid] was undertaken.
Highly optimised Boc-chemistry instrument-assisted
stepwise assembly of the protected peptide chain ~raa
carried out on a resin support, according to the metfiod
described by S.B.H. Kent (8). Samples (3-8mg, about
i5 lumole each) were taken after each cycle of amino acid
addition. The protected peptide-resin samples were
mixed in three batches of consecutive samples: (number
corresponds to the amino acid after which sample was
taken, i.e. residue number in the target sequence.) 99-
67; 66-33; 32-1: The first such mixture contained the
peptises:
CA 02406356 2002-10-28
WO 93/24834 ~ ~ PCTIUS93/~'5070
~l 8
99-Resin
98-99-Resin
97-98-99-Resin
96-97-98-99-Resin
. . . . (etc. ) .
70: . . 96-97-98-99-Resin
69-70. . . . 96-97-98-99-Resin
68-69-70. . . . 96-97-98-99-Resin
67-68-69-70. . . . 96-97-98-99-Resin
Similarly for the other two mixtures. The mixed batches
of peptide-resin were deprotected and cleaved with IiF (i
hours, at OoC, plus 5% cresol/5%/thiacresol). The
products were precipitated with diethyl ether, dissolved
in acetic acid-water 950/50%, v/v) and then lyophilized.
Each peptide mixture was dissolved in 0.1% TFA, 1
uL of the peptide mixture (10 uM per p8pt~d.ie component)
was added to 9uL of 4-hydroxy- -cyanoci~nnamic acid in a
1:2 (v/v) ratio of 30% acetonitrile/0.1% aqueous
trifluoroacetic acid. 0.5uL of the resulting mixture
was applied to the mass spectrometer probe and inserted
into the instrument (7). The spectra shown in Figs. 8
and 9 are the result of adding the data of each of 100
CA 02406356 2002-10-28
wo 9~m ~ ~ Pcriu5g3ro5o~o
49
laser shots performed at a rats of 2.5 laser
shots/second. Figure 8 shows the mass spectrum obtained
from the mixture resulting from cleaving mixed sampl,as
from residues 99-67 of the synthesis: Fig. 9 shows the
mass spectrum obtained from the mixture resulting from
cleaving mixed samples from residues 66-33 of the
synthesis: Table 1 shows the measured mass differences
between consecutive peaks of a selection of these peaks
and compares them with the mass differences calculated
from known sequences of the target peptides. The
agreements are sufficiently close to allow confirmation
of the correctness of the synthesis:
Figure 11 shows mass spectra of the mixture
obtained from mixed samples from residues (66-33) of the
synthesis.
The sequence of the assembled polypeptide chain can
be read out in a straightforward fashion from the mass
differences between consecutive peaks in the mass
spectra of the peptide mixture. This confirmed the
sequence of amino acids in the peptide chain actually
synthesized. The identity of the amino acids as
determined by such mass differences is shown in Table 1
CA 02406356 2002-10-28
WO 9314834 ~ ~ PCT/US93/050T0
Tabte 1. the identify of amino acid by the mass differences in protein ladder
seqvwrx~cltg
using matrix-assisted laser desotption mass spectrometry.
Amino Mass DifferenceDeviation Amino Mass Differencedeviation
Aad Measured, Da) Acid (Meastued,
( Da)
5 Leu~ 113.3 . ~ 114.8 ~ ,0.3
~ 0.1 Aso
~ ~
Glu" _ 129.7 ~ GIn6' t 28.7 ~ 0 6
~ 0.6 ~
Glu' 129.5 ~ ~ 113.2 ~ 0.0
~ - llea
~.4 ~
Mete 130.8 -0:4 ~ 97.0 ~ -0.1
~ Prom
~
Asn~ t 15.0 ~ Val~' 99.4 ~ 0,3
~ 0.9
Leu' 1124 ~ ~ 128:G ~ -0.5
~ -0.8 Glum
~
Prop 97.9 ( ~~ 113.3 ~ 0.1
~ 0.8 Item
~
Gly' 56.1 ~ j~ 84.9 ~ -0.2
~ -0.9 Aba~'
~
~ys" - 1 ~.1 ~0.0 j~ 57.0 . ~ 0.0
~ Gty
~
TrQ'~ 186.4 ~0.2 ~~ 137.3 ~ ~ 02
~ His'
,
15 ~Ys" ~ 128.2 ~ ~~ 127.8 ( X0.4'
0.0 Lys'
~
Pro" ' 97.1 ' ~~Ala" ~ 71.4 ~ 0.3
0.0 ~
I_ys' ~ 128.0 ( ~ Ilen ~ 113.4 , 0.2
-0.2
Met" ~ 131.9 ~0.7 G1y" Sfi.B ( -0.2
Ile" ' 112.6 -0.6 Thr" ; 101.1 ( 0.0
2 ' Gly'~ ( 57.9 ~ ~~Val's~ 99.2 ' ' 0.1
0 0.9
G1y' ~ 56.3 ~ -fl.7 ~ ~eu~ ~ 113.1 ~ -0.1
Ile' f 112.4 ~ -0.8 ~ Val'~~ 99.1 ~ 0 0
GIv3' ~ 57.6 ~ 0.6 ~ Gly' ~ 57.1 ~ 0.1
Glv~ ~ 57.5 ~ ~~Pro' 97.2 ~ d.1
0.5
2 Phe~ ~ 147.3 ~ 0.1 ~ Thr ~ 101.1 0.0 '
5
Ile' ' 112.5 ~ ' Pro' ~ 97.1 ~ 0:0
-fl.7
Lys~ ~ 128.9 ~ ~ ~ f 99.2 ~ 0.1
0.8 Val~ y
Val~ ; 99.0 ( f ~ 113.8 ~ 0.3
-0.1 Asn" j
Arg~' ! 156.2 ~ ~ ~ 113.4 ~ 0.2
0.0 tte~'
Gin~ ~ ~ 28.4 - ' ~~ ~ 113.1 ~ 0:0
0.3 ilea
T yrs' ; 1626 ~ f) ~ 57.1 ~ 0 O
-0.6 Glv
CA 02406356 2002-10-28
VVO 93/2 ~ ~ PCT/tlS93/05070
S1
In addition, terminated by-products (where the
peptide chain has become blocked and doss not grow
anymore) are present in every peptide-resin sample taken
after the step in which the block occurred. Thus, there
is an amplification factor equal to the number of resin
samples in the batch after the point of termination.
This can be seen in Fig. l0 (samples #66-33) which
contains a peak at 3339Ø This corresponds to the
peptide 71-99, 3242.9 (N~terminal His71) plus 96.1
0 dalton. The characteristics mass, together with
knowledge of the chemistry used in the synthesis
identifies the blocking group as CF3C0-(97.1-H = 96.1
dalton). The observed by product is the
trifluoroacetyl-peptide, N'°~-Tfa-(71-99). The ratio of
the amount of this component to the average amount of
the other components is about 2:1. There wets 34
samples combined in this sample. Thus, the terminated
byproduct N°~-Tfa-(71-99) had occurred at a level of
about 5mol~. This side reaction, specific to the N-
terminal His-peptide chain, has not previously been
reported. This illustrates the important sensitivity
advantage provided by this amplification effect in
detecting terminated pegtidss. Such byproducts are not
readily detected by any other means.
CA 02406356 2002-10-28
WO 93/?4834 ~ ~ PCT/tiS93/05070
52
Exa~ple 3
Boc/Fmoc Terminations
Synthesis of the peptide LRRAFGLIGNNPLMAR-amide was
performed manually on a 0.2 mmol scale using p-
methylbenzhydrylamine resin and 0.8 mmoles amino acid
(95 mol%
N-°< -Boc, 5 mol% N-"< -FSnoc) according to the ink
neutralization methods of Schnolzer et al (9). The
following side chain protecting groups were used: 8oc-
Arg, tosyl; Fmoc-Arg, 2,3,6-trimethyl-9-
methoxybenzenesulfonyl (Mtr). Fmoc-Arg(Mtr) was used
for its greater stability in trifluoroacetic acid (TFA).
After completion of the chain assembly, Fmoc groups were
removed using 50% piperidine/DMF, followed by 8oc grog
removal in TFA. The peptide fragments were then cleaved
from the resin by treatment with HF-10% p-cresol (OoC, 1
hour). The resulting crude peptide products were
precipitated and washed with ether, dissolved in 50%
acetic acid, diluted with water and lyophilized. The
mass spectra of the reaction mixture thus produced ie
shown in Fig. 11.
CA 02406356 2002-10-28
W4 93/24834 ~ ~ PCTlUS93/g5070
' 53
Exal4ple 4
Post-ninhYdrin Experiment The machine-assisted
assembly of the peptide LRRASGLIYNNPI~tAR-amide was
performed according to the in situ neutralization
methods of Schnolzer and Kent (9) on a 0.25 mmol scale
using I~HA resin and 2.2 mmol N-~ -Boc amino acids . The
following side chain protecting groups were used: Arg,
tosyl; Asn, xanthyl; Ser, benzyl(Bzl); Tyr,
bromobenzyloxycarbonyl(BrZ). Resin samples were
collected at each step in the synthesis and each sample
was individually subjected to the quantitative ninhydrin
reaction. These samples were then pooled and the Boc
groups removed in neat TFA. Cleavage of the peptide
fragments from the resin was performed by treatment with
HF-10~ p-cresol (0C, 1 hour). The resulting crudre
peptide products were precipitated and Washed with
ether, dissolved in 50~ acetic acid, diluted with water
and lyophillized. The mass spectrum of the mixture is
shown in Fig. 15:
CITATIONS
The following publications are referred to in this
specifications. The complete disclosure of each of them
is hereby incorporated by references.
CA 02406356 2002-10-28
WO 93/24x34 ~ ~ PCT/ C~S9310'5070
54
1. Aebersold et al, Protein Science 1, X94 (1992)
2. R: Self, A. Parents, Biomed. Mass_Sp~ectrom. 10, 78
(1983)
3. L.A. Smith, R.M. Caprioli, Biomed. Mass ~pectroW.
10, 98 (1983)
4. B.T. Chait, T. Chaudhary, F.H. Field, ~Methods in
Protain sequence Analysis 1986, R.A. Walsh, ed.,
8umana Press 1987, pp. 483-493, and uncontrolled
chemical degradation
5. A. Tsugita, K. Takamoto, M. Kamo; H. Iwadate, Eur.
J. Biochem. 206, 691 (1992)
6. G.E. Tarr (1977), in Methods En2Ymoloqy 47, 355.
7. R.C. Beavis and B.T: Chait (1989), Rapid Commun.
Mass Spectrom. 3, 233.
8. S.B.H. Rent, Annual Rev. Hiochem. 5?, 957-984
(1988)
9. Schnolzer et al, Int. J. Peptide Protein Res. 40,
1992, 180-193
10. W.A. Schroeder, Meth. Enzymol. 25, 298 (1972)