Note: Descriptions are shown in the official language in which they were submitted.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
POWDER COMPOSITIONS
Field of the Invention
The invention relates to vaccine compositions. More specifically, the
invention
relates to vaccine compositions suitable for transdermal particle delivery
from a needleless
syringe system.
Back~,TOUnd to the Invention
The ability to deliver pharmaceutical agents into and through skin surfaces
(transdermal delivery) provides many advantages over oral or parenteral
delivery
techniques. In particular, transdermal delivery provides a safe, convenient
and noninvasive
alternative to traditional administration systems, conveniently avoiding the
major problems
associated with oral delivery (e.g. variable rates of absorption and
metabolism,
gastrointestinal irntation and/or bitter or unpleasant drug tastes) or
parenteral delivery (e.g.
needle pain, the risk of introducing infection to treated individuals, the
risk of contamination
or infection of health care workers caused by accidental needle-sticks and the
disposal of
used needles).
However, despite its clear advantages, transdermal delivery presents a number
of
its own inherent logistical problems. Passive delivery through intact skin
necessarily entails
the transport of molecules through a number of structurally different tissues,
including the
stratum corneum, the viable epidermis, the papillary dermis and the capillary
walls in order
for the drug to gain entry into the blood or lymph system. Transdermal
delivery systems
must therefore be able to overcome the various resistances presented by each
type of
tissue.
In light of the above, a number of alternatives to passive transdermal
delivery have
been developed. These alternatives include the use of skin penetration
enhancing agents,
or "permeation enhancers," to increase skin permeability, as well as non-
chemical modes
such as the use of iontophoresis, electroporation or ultrasound. However,
these alternative
techniques often give rise to their own unique side effects such as skin
irntation or
sensitization. Thus, the spectrum of agents that can be safely and effectively
administered
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
2
using traditional transdermal delivery methods has remained limited.
More recently, a novel transdermal drug delivery system that entails the use
of a
needleless syringe to fire powders (i.e., solid drug-containing particles) in
controlled doses
into and through intact skin has been described. In particular, commonly owned
U.S.
Patent No. 5,630,796 to Bellhouse et al. describes a needleless syringe that
delivers
pharmaceutical particles entrained in a supersonic gas flow. The needleless
syringe is used
for transdennal delivery of powdered drug compounds and compositions, for
delivery of
genetic material into living cells (e.g., gene therapy) and for the delivery
of
biopharmaceuticals to skin, muscle, blood or lymph. The needleless syringe can
also be
used in conjunction with surgery to deliver drugs and biologics to organ
surfaces, solid
tumors and/or to surgical cavities (e.g., tumor beds or cavities after tumor
resection). In
theory, practically any pharmaceutical agent that can be prepared in a
substantially solid,
particulate form can be safely and easily delivered using such devices.
One area of the pharmaceuticals field which is of particular interest for
delivery via
this new system is that of vaccine compositions. Suitable vaccines include
those
comprising an antigen adsorbed into a salt adjuvant. Such compositions are
known.in the
art (see for example U.S.Patent No. 5,902,565) and are advantageous since the
adjuvant
enhances the immunogenicity of the vaccine.
However, the storage and transportation of adjuvant vaccines is problematic.
Commercial vaccine compositions containing salt adjuvants cannot be frozen
without
causing damage to the vaccine. Further, one of the common storage techniques
currently
used for vaccines, freeze-drying, is also unavailable for salt adjuvant
containing
compositions. Previous research has demonstrated that freeze-drying causes the
collapse
of the gel structure of the vaccine composition, resulting in aggregation and
precipitation of
the adjuvant salt on resuspension in water (Warren et al, 1986, Annu. Rev.
T_m_m__unol. 4:
pages 369-388; Alving et al , Ann. N. Y. Acad. Sci. 690: pages 265-275). This
is
believed to be due to crystallisation of the water contained in the
composition into large
crystals on freezing and hence the concentration of the solute into specific
regions, known
as freeze concentrate regions. Tn the freeze concentrate regions, adjuvant
salt particles are
brought into close proximity and repulsive forces are overcome, thereby
resulting in
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
3
coagulation. Once the salt has coagulated, the original suspension cannot be
reproduced.
This effect has been found to significantly reduce the immunogenicity of the
vaccine, one
report demonstrating a complete loss in immunogenicity of a freeze-dried alum-
adsorbed
hepatitis B surface antigen (HBsAg) after storage at 4°C for two years
(Diminsky et al,
Vaccine, 18: pages 3-17).
An alternative method for storing adjuvant vaccine compositions is therefore
required, which addresses the problems of aggregation associated with freeze-
drying and
which provides maximum retention of immunogenicity. Prolonged storage of
vaccines is
essential, both for use with the novel transdermal drug delivery systems
mentioned above
and also for use with conventional vaccination techniques. The provision of an
effective
alternative to freeze-drying is therefore of considerable commercial
importance. It is also
desired that the vaccine be produced in a form suitable for needleless
injection. Needleless
injection requires the vaccine composition to be in powder form, each particle
having a
suitable size and strength for transdermal delivery and being capable of
forming a gel on
resuspension.
Alternatives to conventional freeze-drying techniques that have previously
been
reported include the incorporation of additives in the vaccine composition to
improve the
stability of an alum adjuvant. U.S. Patent No. 4,578,270 describes the
addition of large
amounts of both dextran and protein in order to achieve partial retention of
the aluminum
gel structure. This large addition of protein could however act to displace
vaccine antigens
from the aluminum gel and in addition would, in most cases, be immunogenic and
as a
result tend to swamp the immune response to the vaccine antigen.
EP-B-0130619 is also concerned with the addition of stabilisers to
lyophilised, or
freeze-dried, vaccine preparations. Lyophilised preparations of a hepati"tis B
vaccine
comprising an inactivated purified hepatitis B virus surface antigen absorbed
an aluminum
gel and stabiliser are described. The stabiliser is composed of at least one
amino acid or
salt thereof, at least one saccharide and at least one colloidal substance.
Very low
concentrations of aluminum salt adjuvant are used, typically less than 0.1% by
weight.
However, this document relates only to the hepatitis B vaccine and does not
disclose a
generic process, which is non-immunogen-specific.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
4
Spray-dried vaccine preparations comprising an immunogen adsorbed into an
aluminum salt are disclosed in U.S. Patent No. 5,902,565. hnmediate-release
preparations are described which are prepared by spray-drying an aqueous
suspension of
aluminum salt-adsorbed immunogen. In the only Example, Example 1, in which
such
information is given, the resultant microspheres had a size range around 3 ~,m
in diameter.
According to U.S. Patent No. 5,902,565 the gel-forming nature of aluminum gels
is
completely retained during spray-drying even in the absence of any other
materials which
could exert a stabilising effect (apart from minimal quantities of vaccine
antigen, typically 1
to 10 ~g/ml). Addition of water to the spray-dried powder was said to result
in the instant
formation of a typical gel, with sedimentation properties similar to the
starting material.
Summary of the Invention
We investigated whether a gel-forming spray-dried powder of an aluminum salt
could .indeed be formed as described in U.S. Patent No. 5,902,565. We found
that spray
drying a suspension of aluminum hydroxide or aluminum phosphate in water
caused
submicron particles of the aluminum salt to aggregate to larger particles in
the resulting
spray-dried powder. Upon reconstitution of this powder in water, these larger
particles
did not disintegrate into small particles. A geI suspension did not form.
Rather, the
aggregated particles of aluminum hydroxide or aluminum phosphate sedimented
and
precipitated out of the suspension.
Further experiments were carried out. We found that a suitable powder could be
formed by spray-drying when an aluminum salt was utilised with a specific
combination of
other agents. Additionally, the aluminum salt and other agents needed to be
used in
specific proportions. We found too that the particular drying method used has
a significant
effect on the degree of coagulation of the adjuvant salt. These investigations
led to the
finding that a powder suitable for needleless inj ection, and which
substantially retained its
gel structure on reconstitution in water, was obtainable by spray freeze-
drying an alum
adjuvant vaccine composition.
The spray freeze-drying method involves atomizing the suspended vaccine
composition into liquid nitrogen. This process has two important effects:
firstly, the liquid
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
nitrogen acts as a heat transfer agent and provides rapid freezing of the
suspension; and
secondly, the atomisation reduces the volume of each droplet to be frozen,
further
increasing the freezing rate. This combined effect causes extremely rapid
freezing of very
small droplets of suspension and leads to the formation of smaller ice
crystals in the solid.
The freeze concentrate regions which form during a standard freeze-drying
technique are
therefore significantly reduced in size. The rapid freezing of the particles,
and their small
size leads to powders having little or no aggregated adjuvant.
The present invention therefore provides simple, yet effective techniques that
generate salt adjuvant-containing vaccine compositions in a powder form which
is suitable
for long-term storage. The vaccine compositions of the invention show
substantially no
aggregation on reconstitution and therefore immunogenicity is substantially
retained. The
compositions also have well-defined particle size, density and mechanical
properties which
collectively axe suitable for powders for transdermal delivery from a
needleless syringe.
The invention has the further, significant advantage that it is suitable for
use with a
wide range of vaccine compositions and may well also be applicable to other
pharmaceutical compositions, in particular where similar aggregation problems
are
encountered. As yet, the spray freeze-drying technique has been found to be
entirely
formulation independent within the field of adjuvant vaccine compositions.
Accordingly, the present invention provides a gel-forming free-flowing powder
suitable for use as a vaccine, said powder being obtainable by spray-drying or
spray
freeze-drying an aqueous suspension comprising:
(a) from 0.1 to 0.95% by weight of an aluminum salt or calcium salt adjuvant
having an antigen adsorbed thereon;
(b) from 0.5 to 6% by weight of saccharide;
(c) from 0.1 to 2% by weight of an amino acid or salt thereof; and
(d) from 0.02 to 1% by weight of a colloidal substance.
Free-flowing powder compositions suitable for vaccine use can thus be
produced.
The compositions have well-defined particle size, density and mechanical
properties which
collectively are suitable for powders for transdermal delivery from a
needleless syringe.
The invention further provides:
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
6
- a process for the preparation of a gel-forming free-flowing powder suitable
for use as a vaccine, which process comprises spray-drying or spray ,
freeze-drying an aqueous suspension comprising:
(a) from 0.1 to 0.95% by weight of an aluminum salt or
calcium salt adjuvant having an antigen adsorbed therein;
(b) from 0.5 to 6% by weight of a saccharide;
(c) from 0.1 to 2% by weight of an amino acid or salt thereof;
and
(d) from 0.02 to 1 % by weight of a colloidal substance;
- a dosage receptacle for a needleless syringe, said receptacle containing an
effective
amount of a powder of the invention;
- a needleless syringe which is loaded with a powder of the invention;
- a vaccine composition comprising a pharmaceutically acceptable carrier or
diluent
and a powder of the invention;
- a method of vaccinating a subject, which method comprises administering to
the
said subj ect an effective amount of a powder of the invention; and
- a gel-forming free-flowing powder suitable for use as a vaccine, which
powder
comprises:
(i) from 5 to 60% by weight of an aluminum salt or calcium
salt adjuvant having an antigen adsorbed thereon;
(ii) from 25 to 90% by weight of a saccharide;
(iii) from 4.5 to 40% by weight of an amino acid or salt
thereof; and
(iv) from 0.5 to 10% by weight of a colloidal substance.
Additionally, the present invention provides a powder suitable for use as a
vaccine,
said powder being obtainable by spray freeze-drying an aqueous suspension
comprising an
aluminum salt or calcium salt adjuvant having an antigen adsorbed therein.
The invention further provides:
- a process for the preparation of a powder suitable for use as a vaccine,
wluch
process comprises spray freeze-drying an aqueous suspension comprising an
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
7
aluminum salt or calcium salt adjuvant having an antigen adsorbed therein;
- a dosage receptacle for a needleless syringe, said receptacle containing an
effective
amount of such a spray freeze-dried powder of the invention;
- a needleless syringe which is loaded with this spray freeze-dried powder of
the inventic
- a vaccine composition comprising a pharmaceutically acceptable carrier or
diluent
and the spray freeze-dried powder of the invention; and
- a method of vaccinating a subject, which method comprises administering to
the
said subj ect an effective amount of the spray freeze-dried powder of the
invention.
Brief Description of the Drawings
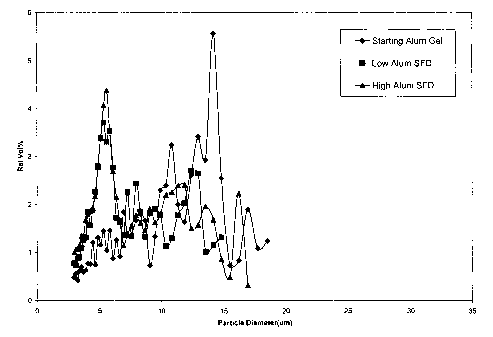
Figure 1 shows the particle size distribution of an HBsAg adsorbed alum gel
(i)
before drying and (ii) after drying using a spray freeze-drying technique
followed by
reconstitution in water.
Figure 2 shows the particle size distribution of a second HBsAg adsorbed alum
gel
before drying and after drying via a conventional freeze drying method.
Figure 3 illustrates the results of an immunogenicity study using mice inj
ected with
HBsAg absorbed alum vaccine which had been dried by either spray freeze-drying
(SFD)
according to present invention, or using freeze-drying (FD). The FD powders
were sieved
into different size fractions and tested for immunogenicity. Two SFD
formulations, varying
in alum contact, were tested.
Figure 4 illustrates the immunogenicity of three different spray freeze-dried
powders in mice irmnunized by either intramuscular injection using a needle or
epidermal
powder immunization using a powder delivery device.
Figure 5 illustrates the immunogenicity of spray freeze-dried diphtheria-
tetanus
toxoid vaccine in guinea pigs. Spray freeze-dried powders of ~0-38 pm and 38-
53 ~,m in
diameter were administered as a powder to the abdominal skin using a powder
delivery
device.
Detailed Description of the Preferred Embodiments
Before describing the present invention in detail, it is to be understood that
this
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
invention is not limited to particularly exemplified compositions or process
parameters as
such may, of course, vary. It is also to be understood that the terminology
used herein is
for the purpose of describing particular embodiments of the invention only,
and is not
intended to be limiting.
All publications, patents and patent applications cited herein, whether supra
or
infra, are hereby incorporated by reference in their entirety.
It must be noted that, as used in this specification and the appended claims,
the
singular forms "a", "an" and "the" include plural referents unless the content
clearly dictates
otherwise. Thus, for example, reference to "a particle" includes a mixture of
two or more
such particles, reference to "an excipient " includes mixtures of two or more
such
excipients, and the like.
A. Definitions
Unless defined otherwise, all technical and scientific terms used herein have
the
same meaning as commonly understood by one of ordinary skill in the art to
which the
invention pertains. Although a number of methods and materials similar or
equivalent to
those described herein can be used in the practice of the present invention,
the preferred
materials and methods are described herein.
In describing the present invention, the following terms will be employed, and
are
intended to be defined as indicated below. By "antigen" is meant a molecule
which
contains one or more epitopes that will stimulate a host's immune system to
make a cellular
antigen-specific immune response or a humoral antibody response. Thus,
antigens include
polypeptides including antigenic protein fragments, oligosaccharides,
polysaccharides and
the like. Furthermore, the antigen can be derived from any known virus,
bacterium,
parasite, plant, protozoan or fungus, and can be a whole organism. The term
also includes
tumor antigens. Similarly, an oligonucleotide or polynucleotide which
expresses an antigen,
such as in DNA immunization applications, is also included in the definition
of an antigen.
Synthetic antigens are also included, for example polyepitopes, flanking
epitopes and other
recombinant or synthetically derived antigens (Bergmann et al (1993) Eur. J.
Immuhol.
23:2777-2781; Bergmann et al. (1996) J. Immuyaol. 157:3242-3249; Suhrbier, A.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
9
(1997) Immunol. and Cell Biol. 75:402-408; Gardner et al. (1998) 12th World
AIDS
Conference, Geneva, Switzerland, June 28-July 3, 1998).
The aduvants having antigen adsorbed thereon of the present invention, alone
or in
combination, are typically combined with one or more added materials such as
carriers,
vehicles, and/or excipients. "Carriers," "vehicles" and "excipients" generally
refer to
substantially inert materials which are nontoxic and do not interact with
other components
of the composition in a deleterious manner. These materials can be used to
increase the
amount of solids in particulate pharmaceutical compositions. Examples of
suitable carriers
include water, silicone, gelatin, waxes, and like materials. Examples of
normally employed
"excipients," include pharmaceutical grades of carbohydrates including
monosaccharides,
disaccharides, cyclodextrans, and polysaccharides (e.g., dextrose, sucrose,
lactose,
trehalose, raffmose, mannitol, sorbitol, inositol, dextrans, and
maltodextrans); starch;
cellulose; salts (e.g. sodium or calcium phosphates, calcium sulfate,
magnesium sulfate);
citric acid; tartaric acid; glycine; high molecular weight polyethylene
glycols (PEG);
Pluronics; surfactants; and combinations thereof. Generally, when Garners
and/or
excipients axe used, they are used in amounts ranging from about 0.1 to 99 wt%
of the
pharmaceutical composition.
The term "powder" as used herein refers to a composition that consists of
substantially solid particles that can be delivered transdermally using a
needleless syringe
device. The particles that make up the powder can be characterized on the
basis of a
number of parameters including, but not limited to, average particle size,
average particle
density, particle morphology (e.g. particle aerodynamic shape and particle
surface
characteristics) and particle penetration energy (P.E.).
The average particle size of the powders according to the present invention
can
vary widely and is generally from 0.1 to 250 Vim, for example from 10 to 100
~m and more
typically from 20 to 70 ~,m. The average particle size of the powder can be
measured as a
mass mean aerodynamic diameter (MMAD) using conventional techniques such as
microscopic techniques (where particles are sized directly and individually
rather than
grouped statistically), absorption of gases, permeability or time of flight.
If desired,
automatic particle-size counters can be used (e.g. Aerosizer Counter, Coulter
Counter,
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
HIAC Counter, or Gelman Automatic Particle Counter) to ascertain the average
particle
size.
Actual particle density or "absolute density" can be readily ascertained using
known quantification techniques such as helium pycnometry and the like.
Alternatively,
5 envelope ("tap") density measurements can be used to assess the density of a
powder
according to the invention. The envelope density of a powder of the invention
is generally
from 0.1 to 25 g/cm3, preferably from 0.8 to 1.5 g/cm3.
Envelope density information is particularly useful in characterizing the
density of
objects of irregular size and shape. Envelope density is the mass of an object
divided by
10 its volume, where the volume includes that of its pores and small cavities
but excludes
interstitial space. A number of methods of determining envelope density are
known in the
art, including wax immersion, mercury displacement, water absorption and
apparent
specific gravity techniques. A number of suitable devices are also available
for determining
envelope density, for example, the GeoPycTM Model 1360, available from the
Micromeritics Instrument Corp. The difference between the absolute density and
envelope
density of a sample pharmaceutical composition provides information about the
sample's
percentage total porosity and specific pore volume.
Particle morphology, particularly the aerodynamic shape of a particle, can be
readily assessed using standard light microscopy. It is preferred that the
particles which
make up the instant powders have a substantially spherical or at least
substantially elliptical
aerodynamic shape. It is also preferred that the particles have an axis ratio
of 3 or less to
avoid the presence of rod- or needle-shaped particles. These same microscopic
techniques can also be used to assess the particle surface characteristics,
e.g. the amount
and extent of surface voids or degree of porosity.
Particle penetration energies can be ascertained using a number of
conventional
techniques, for example a metallized film P.E. test. A metallized film
material (e.g. a 125
~,m polyester film having a 350 A layer of aluminum deposited on a single
side) is used as a
substrate into which the powder is fired from a needleless syringe (e.g. the
needleless
syringe described in U.S. Patent No. 5,630,796 to Bellhouse et a~ at an
initial velocity of
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
11
about 100 to 3000 m/sec. The metallized film is placed, with the metal-coated
side facing
upwards, on a suitable surface.
A needleless syringe loaded with a powder is placed with its spacer contacting
the
film, and then fired. Residual powder is removed from the metallized film
surface using a
suitable solvent. Penetration energy is then assessed using a BioRad Model GS-
700
imaging densitometer to scan the metallized film, and a personal computer with
a SCSI
interface and loaded with MultiAnalyst software (BioRad) and Matlab software
(Release
5.1, The MathWorks, Inc.) is used to assess the densitometer reading. A
program is used
to process the densitometer scans made using either the transmittance or
reflectance
method of the densitometer. The penetration energy of the spray-coated powders
should
be equivalent to, or better than that of reprocessed mannitol particles of the
same size
(mannitol particles that are freeze-dried, compressed, ground and sieved
according to the
methods of commonly owned International Publication No. WO 97/48485,
incorporated
herein by reference).
The term "subject" refers to any member of the subphylum cordata including,
without limitation, humans and other primates including non-human primates
such as
chimpanzees and other apes and monkey species; farm animals such as~cattle,
sheep, pigs,
goats and horses; domestic mammals such as dogs and cats; laboratory animals
including
rodents such as mice, rats and guinea pigs; birds, including domestic, wild
and game birds
such as chickens, turkeys and other gallinaceous birds, ducks, geese, and the
like. The
term does not denote a particular age. Thus, both adult and newborn
individuals are
intended to be covered. The methods described herein are intended for use in
any of the
above vertebrate species, since the immune systems of all of these vertebrates
operate
similarly.
The term "transdermal delivery" includes both transdermal ("percutaneous") and
transmucosal routes of administration, i.e. delivery by passage through the
skin or mucosal
tissue. See, e.g., Transdernaal Drug Delivery: Developmental Issues and
Research
Initiatives, Hadgraft and Guy (eds.), Marcel Dekker, Inc., (1989); Controlled
Drug
Delivery: Fundamentals and Applications, Robinson and Lee (eds.), Marcel
Dekker
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
12
Inc., (1987); and Transdermal Delivery ofDrugs, Vols. 1-3, Kydonieus and
Berner
(eds.), CRC Press, (1987).
B. General Methods
The invention is concerned with gel-forming free-flowing powders suitable for
use
as vaccines. The powders are suitable for transdermal administration from a
needleless
syringe delivery system. As such, the particles which make up the powdered
composition
must have sufficient physical strength to withstand sudden acceleration to
several times the
speed of sound and the impact with, and passage through, the skin and tissue.
The
particles are formed by spray-drying or spray freeze-drying an aqueous
suspension
comprising or, in some embodiments, consisting essentially of:
(a) from 0.1 to 0.95% by weight of an aluminum salt or calcium salt adjuvant
having an antigen adsorbed therein;
(b) from 0.5 to 6% by weight of a saccharide;
(c) from 0.1 to 2% by weight of an amino acid or salt thereof; and
(d) from 0.02 to 1 % by weight of a colloidal substance.
The aqueous suspension contains, as component (a), less than 1 % by weight of
the
adjuvant having antigen adsorbed thereon. Preferably, the suspension contains
from 0.2 or
0.3 to 0.6 or 0.75% by weight, preferably from 0.2 to 0.4% by weight, of the
adjuvant
onto which antigen is adsorbed. The aluminum salt adjuvant is generally
aluminum
hydroxide or aluminum phosphate. Alternatively, the adjuvant may be aluminum
sulfate or
calcium phosphate.
Any suitable antigen as defined herein may be employed. The antigen may be a
viral antigen. The antigen may therefore be derived from members of the
families
Picornaviridae (e.g. polioviruses, etc.); Caliciviridae; Togaviridae (e.g.
rubella virus,
dengue virus, etc.); Flaviviridae; Coronaviridae; Reoviridae; Birnaviridae;
Rhabodoviridae
(e.g. rabies virus, etc.); Filovindae; Paramyxoviridae (e.g. mumps virus,
measles virus,
respiratory syncytial virus, etc.); Orthomyxoviridae (e.g. influenza virus
types A, B and C,
etc.); Bunyaviridae; Arenaviridae; Retroviradae (e.g. HTLV-I; HTLV-II; HIV-1
and
HIV-2); and simian immunodeficiency virus (SIV) among others.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
13
Alternatively, viral antigens may be derived from papillomavirus (e.g. HPV); a
herpesvirus; a hepatitis virus, e.g. hepatitis A virus (HAV), hepatitis B
virus (HBV),
hepatitis C (HCV), the delta hepatitis virus (HDV), hepatitis E virus (HEV) or
hepatitis G
virus (HGV); and the tick-borne encephalitis viruses. See, e.g. Vinology, 3rd
Edition
(W.K. Joklik ed. 1988); Fundamental Virology, 2nd Edition (B.N. Fields and
D.M.
Knipe, eds. 1991) for a description ofthese viruses.
Bacterial antigens for use in the invention can be derived from organisms that
cause
diphtheria, cholera, tuberculosis, tetanus, pertussis, meningitis and other
pathogenic states,
including, e.g., Meningococcus A, B and C, Hemophilus influenza type B (HIB),
Helicobacter pyloni, hibnio cholerae, Esche~ichia coli, CampylobacteY,
Shigella,
Salmonella, Streptococcus sp, and Staphylococcus sp. A combination of
bacterial
antigens may be provided, for example diphtheria, pertussis and tetanus
antigens. Suitable
pertussis antigens are pertussis toxin and/or filamentous haemagglutinin
and/or pertactin,
alternatively termed P69. An anti-parasitic antigen may be derived from
organisms causing
malaria and Lyme disease.
Antigens for use in the present invention can be produced using a variety of
methods known to those of skill in the art. In particular, the antigens can be
isolated
directly from native sources, using standard purification techniques.
Alternatively, whole
killed, attenuated or inactivated bacteria, viruses, parasites or other
microbes may be
employed. Yet further, antigens can be produced recombinantly using known
techniques.
See, e.g., Sambrook, Fritsch & Maniatis, Molecular Cloning: A LaboYato~y
Manual,
Vols. I and II (D.N. Glover et. 1985).
Antigens for use herein may also be synthesised, based on described amino acid
sequences, via chemical polymer syntheses such as solid phase peptide
synthesis. Such
methods are known to those of skill in the art. See, e.g. J.M. Stewart and
J.D. Young,
Solid Phase Peptide Synthesis, 2nd Ed., Pierce Chemical Co., Rockford, IL
(1984) and
G. Barany and R. B. Merrifield, The Peptides: Analysis, Synthesis, Biology,
editors E.
Gross and J. Meienhofer, Vol. 2, Academic Press, New York, (1980), pp. 3-254,
for
solid phase peptide synthesis techniques; and M. Bodansky, PYinciples
ofPeptide
Synthesis, Springer-Verlag Berlin (1984) and E. Gross amd J. Meienhofer, Eds.,
The
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
14
Peptides: Ayaalysis, Synthesis, Biology, supra, Vol. 1, for classical solution
synthesis.
One or more saccharides may be present in the aqueous suspension as component
(b). The saccharide content is typically 1.5 to 5% by weight, preferably 2 to
4% by
weight. The saccharide may be a monosaccharide such as glucose, xylose,
galactose,
fructose, D-mannose or sorbose; a disaccharide such as lactose, maltose,
saccharose,
trehalose or sucrose; or a sugar alcohol such as mannitol, sorbitol, xylitol,
glycerol,
erythritol or arabitol.
One or more amino acids or amino acid salts is present in the aqueous
suspension
as component (c). Any physiologically acceptable amino acid salt may be
employed. The
salt may be an alkali or alkaline earth metal salt such as sodium, potassium
or magnesium
salt. The amino acid may be an acidic, neutral or basic amino acid. Suitable
amino acids
are glycine, alanine, glutamine, arginine, lysine and histidine. Monosodium
glutamate is a
suitable amino acid salt. The aqueous suspension generally contains from 0.5
to 1.5% by
weight, more preferably from 0.75 to 1.25% by weight, of the amino acid and/or
amino
acid salt.
The colloidal substance (d) is a divided substance incapable of passing
through a
semi-permeable membrane, comprised of fine particles which, in suspension or
solution,
fail to settle out. Suitable colloidal substances are disclosed in EP-B-
0130619.
Component (d) may be selected from polysaccharides such as dextran or
maltodextran;
hydrogels such as gelatin or agarose; or proteins such as human serum albumin.
The
substance may have a molecular weight of 500 to 80,000 or higher, for example
from
1000 or 2000 to 30,000 or from 5,000 to 25,000. Component (d) is generally
present in
the aqueous suspension in an amount of from 0.05 to 0.5% by weight, preferably
from
0.07 to 0.3% by weight.
The adjuvant having antigen adsorbed thereon and the saccharide, amino acid or
salt thereof and colloidal substance are suspended in water. The aqueous
suspension is
spray dried or spray freeze-dried. The spray-drying or spray freeze-drying
conditions are
selected to enable the desired particles to be produced. The air inlet
temperature, air
outlet temperature, feed rate of the aqueous suspension, air flow rate, etc.
can thus be
varied as desired. Any suitable spray drier may be used. The nozzle size may
vary as
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
necessary. Particular spray freeze-drying conditions are described in more
detail below.
A gel-forming free-flowing powder can thus be provided which is suitable for
use
as a vaccine. The proportions of the various components of the powder can be
adjusting
by adjusting the composition of the suspension that is spray-dried or spray
freeze-dried.
5 However, the powder typically comprises or, in some embodiments, consists
essentially of:
(i) from 5 to 60%, for example from 7 to 50% such as from 10 to 30%, by
weight of an aluminum salt or calcium salt adjuvant having an antigen
adsorbed thereon; .
(ii) from 25 to 90%, for example from 30 to 80% such as from 40 to 70%, by
10 weight of a saccharride;
(iii) from 4.5 to 40%, for example from 7 to 30% such as from-10 to 20%, by
weight of an amino acid or salt thereof; and
(iv) from 0.5 to 10%, for example from 0.8 to 6% such as from 1 to 3%, by
weight of a colloidal substance.
15 The invention is concerned generally with powders suitable for use as
vaccines that
are formed by spray freeze-drying an aqueous suspension comprising an aluminum
salt or
calcium salt adjuvant having an antigen adsorbed therein. Such powders are
suitable for
transdennal achninistration from a needleless syringe delivery system. As
such, the
particles which make up the powdered composition must have sufficient physical
strength
to withstand sudden acceleration of up to several times the speed of sound and
the impact
with, and passage through, the skin and tissue.
Preferably, the aqueous suspension, prior to spray freeze-drying, contains
less than
10% by weight, for instance less than 5% weight and preferably less than 3% by
weight, of
the salt adjuvant having antigen adsorbed thereon. The aqueous suspension
typically
contains at least 0.05% by weight, for instance at least 0.1% by.weight or at
least 0.6% by
weight, of the adjuvant having antigen adsorbed thereon. More preferably, the
suspension
contains from 0.2 or 0.3 to 0.6%, 0.75% or 1% by weight, preferably from 0.2
to 0.4%
by weight, of adjuvant onto which antigen is adsorbed. At concentrations above
about
10% by weight of adjuvant salt, the aqueous suspension becomes highly viscous.
This
limits the ability to atomize the suspension.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
16
It should be understood that the preferred upper limit of adjuvant
concentration
applies to the aqueous suspension prior to spray freeze-drying. The content of
adjuvant
salt having antigen adsorbed thereon may be as high as 50% by weight or more
in the
spray freeze-dried powders of the invention.
The adjuvant is generally an aluminum salt, for example aluminum hydroxide or
aluminum phosphate. Alternatively, the adjuvant salt may be aluminum sulfate
or calcium
phosphate.
Again, any suitable antigen as defined herein may be employed. The antigen may
be a viral antigen. The antigen may therefore be derived from members of the
families
Picomaviridae (e.g. polioviruses, etc.); Caliciviridae; Togaviridae (e.g.
rubella virus,
dengue virus, etc.); Flaviviridae; Coronaviridae; Reoviridae; Birnaviridae;
Rhabodoviridae
(e.g. rabies virus, etc.); Filoviridae; Paramyxoviridae (e.g. mumps virus,
measles virus,
respiratory syncytial virus, etc.); Orthomyxoviridae (e.g. influenza virus
types A, B and C,
etc.); Bunyaviridae; Arenaviridae; Retroviradae (e.g. HTLV-I; HTLV-lI; HIV-1
and
HIV-2); and simian immunodeficiency virus (SIV) among others.
Alternatively, viral antigens may be derived from papillomavirus (e.g. HPV); a
herpesvirus; a hepatitis virus, e.g. hepatitis A virus (HAV), hepatitis B
virus (HBV),
hepatitis C (HCV), the delta hepatitis virus (HDV), hepatitis E virus (HEV) or
hepatitis G
virus (HGV); and the tick-borne encephalitis viruses. See, e.g. Virology, 3rd
Edition
(W.I~. Joklik ed. 1988); Fundarneratal Virology, 2nd Edition (B.N. Fields and
D.M.
6
Knipe, eds. 1991) for a description of these viruses.
Bacterial antigens for use in the invention can be derived from organisms that
cause
diphtheria, cholera, tuberculosis, tetanus, pertussis, meningitis and other
pathogenic states,
including, e.g., Meningococcus A, B and C, Hemophilzss influenza type B (HIB),
Helicobacter pylori, Vibrio cholerae, Esclaericlaia coli, Campylobacter,
Shigella,
Salmonella, Streptococcus sp, and Staphylococcus sp. A combination of
bacterial
antigens may be provided, for example diphtheria, pertussis and tetanus
antigens. Suitable
pertussis antigens are pertussis toxin and/or filamentous haemagglutinin
and/or pertactin,
alternatively termed P69. An anti-parasitic antigen may be derived from
organisms causing
malaria and Lyme disease.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
17
Antigens for use in the present invention can be produced using a variety of
methods known to those of skill in the art. In particular, the antigens can be
isolated
directly from native sources, using standard purification technques.
Alternatively, whole
killed, attenuated or inactivated bacteria, viruses, parasites or other
microbes may be
employed. Yet further, antigens can be produced recombinantly using known
techniques.
See, e.g., Sambrook, Fritsch & Maniatis, Molecular Cloning: A Labot~atory
Manual,
Vols. I and II (D.N. Glover et. 1985).
A~ztigens for use herein may also be synthesised, based on described amino
acid
sequences, via chemical polymer syntheses such as solid phase peptide
synthesis. Such
methods are known to those of skill in the art. See, e.g. J.M. Stewart and
J.D. Young,
Solid Phase Peptide Synthesis, 2nd Ed., Pierce Chemical Co., Rockford, IL
(1984) and
G. Barany and R. B. Mernfield, The Peptides: Analysis, Synthesis, Biology,
editors E.
Gross and J. Meienhofer, Vol. 2, Academic Press, New York, (.1980), pp. 3-254,
for
solid phase peptide synthesis techniques; and M. Bodansky, Principles of
Peptide
Synthesis, Springer-Verlag Berlin (1984) and E. Gross and J. Meienhofer, Eds.,
The
Peptides: Analysis, Synthesis, Biology, supra, Vol. 1, for classical solution
synthesis.
The aqueous suspension may consist essentially of water and adjuvant having an
antigen adsorbed thereon, or further additives may be included in the
suspension. Any
additives may be employed provided that they are substantially non-toxic and
pharmacologically inert. The spray freeze-drying process has been found to be
effective
when applied to suspensions comprising a wide range of different additives
and, as yet, the
process of the invention, and therefore the powders of the invention, have
been found to be
entirely formulation independent.
Typically, the aqueous suspension comprises suitable excipients, along with
protectants, solvents, salts, surfactants, buffering agents and the like.
Suitable excipients
can include free-flowing particulate solids that do not thicken or polymerize
upon contact
with water, which are iimocuous when administered to an individual, and do not
. signif candy interact with the pharmaceutical agent in a manner that alters
its pharmaceutical
activity. Examples of normally employed excipients include, but are not
limited to,
monosaccharides such as glucose, xylose, galactose, fructose, D-mannose or
sorbose,
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
18
disaccharides such as lactose, maltose, saccharose, trehalose or sucrose,
sugar alcohols
such as mantutol, sorbitol, xylitol, glycerol, erythritol or arabitol,
polymers such as dextran,
starch, cellulose or high molecular weight polyethylene glycols (PEG), amino
acids or their
salts, such as glycine, alanine, glutamine, arginine, lysine or histidine or
their salts with alkali
or alkaline earth metals such as a sodium, potassium or magnesium salts, or
sodium or
calcium phosphates, calcimn carbonate, calcium sulfate, sodium citrate, citric
acid, tartaric
acid, and combinations thereof. Suitable solvents include, but are not limited
to, methylene
chloride, acetone, methanol, ethanol, isopropanol and water. Typically, Water
is used as
the solvent. Generally pharmaceutically acceptable salts having molarities
ranging from
about 1 mM to 2M can be used. Pharmaceutically acceptable salts include, for
example,
mineral acid salts such as hydrochlorides, hydrobromides, phosphates,
sulfates, and the
Like; and the salts of organic acids such as acetates, propionates, malonates,
benzoates,
and the like. A thorough discussion of pharmaceutically acceptable excipients,
vehicles
and auxiliary substances is available in REMINGTON'S PHARMACEUTICAL
SCIENCES (Mack Pub. Co., N.J. 1991), incorporated herein by reference.
Preferred excipients for use in the aqueous suspension include saccharides,
amino
acids or salts thereof and polymers. Typically, the suspension contains one or
more
saccharides, such as a combination of rnannitol and trehalose. Saccharides are
typically
present in an amount of from 0.5 to 30% by weight. An amino salt, such as
arginine
glutamate or aspartate in an amount of from 0.1 to 30% by weight, and/or a
polymer, such
as dextran, in an amount of from 0 to 30% may also be included, typically in
an amount of
from 0 to 30 % by weight. Typical excipient combinations include one or more
saccharides and a polymer and include substantially no amino salt. The total
amount of
excipients present in the aqueous suspension is typically from 0 to 50%, more
preferably
from 10 to 30%.
The particles of the invention are formed by first suspending the adjuvant
having an
antigen adsorbed therein, and any required additives, in water. The aqueous
suspension is
then spray freeze-dried. Any known technique in the art (for example the
methods
described by Mumenthaler et al, Int. J. Pharmaceutics (1991) 72, pages 97-110
and Maa
et al, Phar. Res. (1999) Vol. 16, page 249) may be used to carry out the spray
freeze-
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
19
drying step. A typical spray freeze-drying technique involves atomising the
aqueous
suspension into stirred liquid nitrogen. The liquid nitrogen containing frozen
particles is then
held at reduced temperature, for example from -60°C to -20°C,
followed by vacuum
drying preferably under a pressure of from 20 to 500 mT (2.666 to 66.65 Pa),
and at
reduced temperature such as from -50°C to 0°C. Drying is
typically carried out in two
stages, primary drying and secondary drying. Primary drying time typically
ranges from 4
to 24 hours and secondary drying time typically ranges from 6 to 24 hours. The
temperature may be gradually increased, whilst still under reduced pressure
until room
temperature is reached.
This technique involves the rapid freezing of the aqueous suspension into
droplets.
The drying step then removes the ice by sublimation without the need for high
air
temperatures. The powder may be collected by any known technique. The precise
spray
freeze-drying conditions used may be selected according to the desired
properties of the
particles to be produced. Thus, the temperatures, pressures and other
conditions may be
varied as desired.
The powders of the invention are generally free-flowing. The powders contain
very little or no agglomerated adjuvant salt and are therefore capable of
forming a gel on
resuspension in water. Typically, substantially no precipitate forms upon
resuspension.
After a powder has been added to distilled water (1:500 by weight) and shaken
for three
minutes, a gel-like suspension without any precipitate is typically obtained.
No precipitates
settling out are observed after 3 hours. No precipitates may form after
standing overnight,
for example for 12 hours.
The presence of a precipitate, and the degree of agglomeration of the
reconstituted
gel formulation, is typically assessed by the ability of the reconstituted
formulation to
diffract a beam of light. The degree of agglomeration can also be
quantitatively assessed
by standard light microscopy and/or sedimentation. Another suitable test for
particle
agglomeration can be to determine particle size before and after
reconstitution using any of
a number of standard particle size determination techniques, e.g. laser-based
or light
obscuration.
The particles of the invention have a size appropriate for high-velocity
transdenmal
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
delivery to a subject, typically across the stratum co>"rzeum or a
transmucosal membrane.
The mass mean aerodynamic diameter (MMAD) of the particles is from about 0.1
to 250
~,rn. The MMAD may be from 5 to 100 ~m or from 10 to 100 Vim, preferably from
10 to
70 ~.m or from 20 to 70 Vim. Generally, less than 10% by weight of the
particles have a
5 diameter which is at least 5 ~m more than the MMAD or at least 5 ~,m less
than the
MMAD. Preferably, no more than 5% by weight of the particles have a diameter
which is
greater than the MMAD by 5 ~.m or more. Also preferably, no more than 5% by
weight
of the particles have a diameter which is smaller than the MMAD by 5 ~m or
more.
The particles have an envelope density of from 0.1 to 25 g/cm3, preferably
from
10 0. ~ to 1.5 g/cm3. While the shape of the individual particles may vary
when viewed under
a microscope, the particles are preferably substantially spherical. The
average ratio of the
major axis:minor axis is typically from 3:1 to 1:1, for example from 2:1 to
1:1.
The individual particles of a powder have a substantially spherical
aerodynamic
shape with a substantially uniform, nonporous surface. The particles will also
have a
15 particle penetration energy suitable for transdermal delivery from a
needleless syringe
device.
A detailed description of needleless syringe devices useful in this invention
is found
in the prior art, as discussed herein. These devices are referred to as
needleless syringe
devices and representative of these devices are the dermal PowderJect~
needleless syringe
20 device and the oral PowderJect~ needleless syringe device (PowderJect
Technologies
Limited, Oxford, UI~). By using these devices, an effective amount of the
powder of the
invention is delivered to the subject. An effective amount is that amount
needed to deliver
sufficient of the desired antigen to achieve vaccination. This amount will
vary with the
nature of the antigen and can be readily determined through clinical testing
based on known
activities of the antigen being delivered. The "Physicians Desk Refez~ence"
and
"Good~yzan and Gilman's Tlae Phamacological Basis of Thefapeutics" are useful
for
the purpose of determined the amount needed.
Needleless syringe devices for delivering particles were first described in
commonly owned U.S. Patent No. 5,630,796 to Bellhouse et al, incorporated
herein by
reference. Although a number of specific device configurations are now
available, such
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
21
devices are typically provided as a pen-shaped instrument containing, in
linear order
moving from top to bottom, a gas cylinder, a particle cassette or package, and
a
supersonic nozzle with an associated silencer medium. An appropriate powder
(in the
present case, a spray dried or spray freeze-dried powder of the invention) is
provided
within a suitable container, e.g., a cassette formed by two rupturable polymer
membranes
that are heat-sealed to a washer-shaped spacer to form a self contained sealed
unit.
Membrane materials can be selected to achieve a specific mode of opening and
burst
pressure that dictate the conditions at which the supersonic flow is
initiated. In operation,
the device is actuated to release the compressed gas from the cylinder into an
expansion
chamber within. the device. The released gas contacts the particle cassette
and, when
sufficient pressure is built up, suddenly breaches the cassette membranes
sweeping the
particles into the supersonic nozzle for subsequent delivery. The nozzle is
designed to
achieve a specific gas velocity and flow pattern to deliver a quantity of
particles to a target
surface of predefined area. The silencer is used to attenuate the noise
produced by the
membrane rupture.
A second needleless syringe device for delivering particles is described in
commonly owned International Publication No. WO 96/20022. This delivery system
also
uses the energy of a compressed gas source to accelerate and deliver powdered
compositions; however, it is distinguished from the system of US Patent No.
5,630,796 in
its use of a shock wave instead of gas flow to accelerate the particles. More
particularly,
an instantaneous pressure rise provided by a shock wave generated behind a
flexible dome
strikes the back of the dome, causing a sudden eversion of the flexible dome
in the
direction of a target surface. This sudden eversion catapults a powdered
composition
(which is located on the outside of the dome) at a sufficient velocity, thus
momentum, to
penetrate target tissue, e.g., oral mucosal tissue. The powdered composition
is released at
the point of full dome eversion. The dome also serves to completely contain
the high-
pressure gas flow, which therefore does not come into contact with the tissue.
Because the
gas is not released during this delivery operation, the system is inherently
quiet. This design
can be used in other enclosed or otherwise sensitive applications for example,
to deliver
particles to minimally invasive surgical sites.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
22
In yet a further aspect of the invention, single unit dosages or multidose
containers,
in which a powder of the invention may be packaged prior to use, can comprise
a
hermetically sealed container enclosing a suitable amount of the powder that
makes up a
suitable dose. The powder can be packaged as a sterile formulation, and the
hermetically
sealed container can thus be designed to preserve sterility of the formulation
until use. If
desired, the containers can be adapted for direct use in the above-referenced
needleless
syringe systems.
Powders of the present invention can thus be packaged in individual unit
dosages
for delivery via a needleless syringe. As used herein, a "unit dosage" intends
a dosage
receptacle containing a therapeutically effective amount of a powder of the
invention. The
dosage receptacle typically fits within a needleless syringe device to allow
for transdermal
delivery from the device. Such receptacles can be capsules, foil pouches,
sachets,
cassettes or the like.
The container in which the powder is packaged can further be labeled to
identify
the composition and provide relevant dosage information. In addition, the
container can be
labeled with a notice in the form prescribed by a governmental agency, for
example the
Food and Drug Administration, wherein the notice indicates approval by the
agency under
Federal law of the manufacture, use or sale of the powder contained therein
for human
administration.
The actual distance which the delivered particles will penetrate a target
surface
depends upon particle size (e.g., the nominal particle diameter assuming a
roughly spherical
particle geometry), particle density, the initial velocity at which the
particle impacts the
surface, and the density and kinematic viscosity of the targeted skin tissue.
In this regard,
optimal particle densities for use in needleless injection generally range
between about 0.1
and 25 g/cm3 such as between about 0.8 and 1.7 g/cm3, preferably between about
0.9 and
1.5 g/cm3. Inj ection velocities generally range between about 100 and 3,000
xn/sec. With
appropriate gas pressure, particles having an average diameter of 10-70 ~,m
can be
accelerated through the nozzle at velocities approaching the supersonic speeds
of a driving
gas flow.
If desired, the needleless syringe systems can be provided in a preloaded
condition
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
23
containing a suitable dosage of the powder of the invention. The loaded
syringe can be
packaged in a hermetically sealed container, which may fuxther be labeled as
described
above.
A nuanbex of novel test methods have been developed, or established test
methods
modified, in order to characterize performance of a needleless syringe device.
These tests
range from characterization of the powdered composition, assessment of the gas
flow and
particle acceleration, impact on artificial or biological targets, and
measures of complete
system performance. One, several or all of the following tests can thus be
employed to
assess the physical and functional suitability of the powder of the invention
for use in a
needleless syringe system.
Assessment of Effect on Artificial Film Targets
A functional test that measures many aspects of powder injection systems
simultaneously has been designated as the "metallized film" or "penetxation
energy" (PE)
test. It is based upon the quantitative assessment of the damage that
particles can do to a
precision thin metal layer supported by a plastic film substrate. Damage
correlates to the
kinetic energy and certain other characteristics of the particles. The higher
the xesponse
from the test (i.e., the higher the film damage/disruption) the more energy
the device has
imparted to the particles. Either electrical resistance change measurement or
imaging
densitometry, in reflectance or transmission mode, provide a reliable method
to assess
device or formulation performance in a controllable and reproducible test.
The film test-bed has been shown to be sensitive to particle delivery
variations of
all major device parameters including pressure, dose, particle size
distribution and material,
etc. and to be insensitive to the gas. Aluminum of about 350 Angstrom
thickness on a
125 ~m polyester support is currently used to test devices operated at up to
60 bar.
Assessment of Impact Effect on EngineeYirag Foam Targets
Another means of assessing particle performance when delivered via a
needleless
syringe device is to gauge the effect of impact on a rigid polymethylimide
foam (Rohacell 5
IIG, density 52 kg/m3, Rohm Tech Inc., Maiden, MA). The experimental set-up
for this
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
24
test is similar to that used in the metallized film test. The depth of
penetration is measured
using precision calipers. For each experiment a processed mannitol standard is
nuz as
comparison and all other parameters such as device pressure, particle size
range, etc., are
held constant. Data also show this method to be sensitive to differences in
particle size and
pressure. Processed mannitol standard as an excipient for drugs has been
proven to
deliver systemic concentrations in preclinical experiments, so the relative
performance
measure in the foam penetration test has a practical in viv~ foundation.
Promising
powders can be expected to show equivalent or better penetration to mannitol
for
anticipation of adequate performance in preclinical or clinical studies. This
simple, rapid
test has value as a relative method of evaluation of powders and is not
intended to be
considered in isolation.
Particle Attrition Test
A further indicator of particle performance is to test the ability of various
candidate
compositions to withstand the forces associated with high-velocity particle
injection
techniques, that is, the forces from contacting particles at rest with a
sudden, high velocity
gas flow, the forces resulting from particle-to-particle impact as the powder
travels through
the needleless syringe, and the forces resulting from particle-to-device
collisions also as the
powder travels through the device. Accordingly, a simple particle attrition
test has been
devised which measures the change in particle size distribution between the
initial
composition, and the composition after having been delivered from a needleless
syringe
device.
The test is conducted by loading a particle composition into a needleless
syringe as
described above, and then discharging the device into a flask containing a
carrier fluid in
Which the particular composition is not soluble (e.g., mineral oil, silicone
oil, etc.). The
carrier fluid is then collected, and particle size distribution in both the
initial composition
and the discharged composition is calculated using a suitable particle sizing
apparatus, e.g.,
an AccuSizer~ model 780 Optical Particle Sizer. Compositions that demonstrate
less than
about 50%, more preferably less than about 20% reduction in mass mean diameter
(as
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
determined by the AccuSizer apparatus) after device actuation are deemed
suitable for use
in the needleless syringe systems described herein.
Delivery to Human Skin in vitro and Transepidermal Water Loss
5 For a powder performance test that more closely parallels eventual practical
use,
candidate powder compositions can be injected into dermatomed, full thickness
human
abdomen skin samples. Replicate skin samples after injection can be placed on
modified
Franz diffusion cells containing 32°C water, physiologic saline or
buffer. Additives such as
surfactants may be used to prevent binding to diffusion cell components. Two
kinds of
10 measurements can be made to assess performance of the formulation in the
skin.
To measure physical effects, i.e. the effect of particle injection on the
barrier
function of skin, the transepidermal water loss (TEWL) can be measured.
Measurement is
performed at equilibrium (about 1 hour) using a Tewameter TM 210~ (Courage &
I~hazaka, Koln, Ger) placed on the top of the diffusion cell cap that acts
like a ~12 mrn
15 chimney. Larger particles and higher injection pressures generate
proportionally higher
TEWL values in vitro and this has been shown to correlate with results in
vivo. Upon
particle injection in vitro TEWL values increased from about 7 to about 27
(g/mzh)
depending on particle size and helium gas pressure. Helium injection without
powder has
no effect. In vivo, the skin barner properties return rapidly to normal as
indicated by the
20 TEWL returning to pretreatment values in about 1 hour for most powder
sizes. For the
largest particles, 53-75 Vim, skin samples show 50% recovery in an hour and
full recovery
by 24 hours.
Delivery to Human Skin in vitro and Drug Diffusion Rate
25 To measure the formulation performance ira vitro, the antigen components)
of
candidate powders can be collected by complete or aliquot replacement of the
Franz cell
receiver solution at predetermined time intervals for chemical assay using
HPLC or other
suitable analytical technique. Concentration data can be used to generate a
delivery profile
and calculate a steady state permeation rate. This technique can be used to
screen
formulations for early indication of antigen binding to skin, antigen
dissolution, efficiency of
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
26
particle penetration of stratum corneum, etc., prior to in. vivo studies.
These and other qualitative and quantitative tests can be used to assess the
physical
and functional suitability of the present powders for use in a high-velocity
particle inj ection
device. It is preferred, though not required, that the particles of a powder
have the
following characteristics: a substantially spherical shape (e.g. an aspect
ratio as close as
possible to 1); a smooth surface; a suitable active loading content; less than
20% reduction
in particle size using the particle attrition test; an envelope density as
close as possible to
the true density of the constituents (e.g. greater than about 0.8 g/ml); and a
MMAD of
about 20 to 70 ~,m with a narrow particle size distribution. The compositions
are typically
free -flowing (e.g. free-flowing after 8 hours storage at 50% relative
humidity and after 24
hours storage at 40% relative humidity). All of these criteria can be assessed
using the
above-described methods, and are further detailed in the following
publications,
incorporated herein by reference. Etzler et al (1995) Part. Part. Syst.
Charact.12:217;
Ghadiri, et al (1992) IFPRI Final Report, FRR 16-03 University of Surrey, UK;
Bellhouse et al (1997) "Needleless delivery of drugs in dry powder form, using
shock
waves and supersonic gas flow," Plenary Lecture 6, 21 St International
Symposium on
Sl2ock YYaves, Australia; and Kwon et al (1998) Pharm. Sci. suppl.l (1), 103.
A powder of the invention may alternatively be used to vaccinate a subj ect
via
other routes. For this purpose, the powder may be combined with a suitable
earner or
diluent such as Water for Injections or physiologically saline. The resulting
vaccine
composition is typically administered by injection, for example subcutaneously
or
intramuscularly.
Whichever route of administration is selected, an effective amount of antigen
is
delivered to the subject being vaccinated. Generally from 50 ng to 1 mg and
more
preferably from 1 ~.g to about 50 ~,g of antigen will be useful in generating
an immune
response. The exact amount necessary will vary depending on the age and
general
condition of the subject to be treated, the particular antigen or antigens
selected, the site of
administration and other factors. An appropriate effective amount can be
readily
determined by one of skill in the art.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
27
Dosage treatment may be a single dose schedule or a multiple dose schedule. A
multiple dose schedule is one in which a primary course of vaccination may be
with 1-10
separate doses, followed by other doses given at subsequent time intervals,
chosen to
maintain and/or reinforce the immune response, for example at I-4 months for
second
dose and, if needed, a subsequent doses) after several months. The dosage
regimen will
also, at least in part, be determined by the need of the subject and be
dependent on the
judgement of the practitioner. Vaccination will of course generally be
effected prior to
primary infection with the pathogen against which protection is desired.
C. Experimental
Below axe examples of specific embodiments for carrying out the present
invention.
The examples axe offered for illustrative purposes only, and are not intended
to limit the
scope of the present invention in any way.
Efforts have been made to ensure accuracy with respect to numbers used (e.g.,
I S amounts, temperatures, etc.), but some experimental error and deviation
should, of course,
be allowed for.
Reference Example I
A spray-dried immediate-release vaccine preparation was obtained according to
the procedure described in US Patent No. 5,902,565. A formulation containing
5% by
weight mannitol and 5% by weight aluminum phosphate (Adju-Phos) was spray
dried using
a bench-top spray dryer (Buchi 190). The spray-drying conditions were: inlet
temperature
=130°C; outlet temperature = 70°C, liquid feed rate = 3 ml/min;
atomizing airflow rate =
5001/hr; and a full scale of drying air. The free-flowing powder that was
obtained had a
particle size of about 10 ~,m. The powder was reconstituted in distilled water
(1:500 by
weight). The solution failed to form a gel with the suspended particles
setting in I S
minutes. By optical microscopy, the particles after reconstitution maintained
their shape
and size, suggesting that the alum remained coagulated and did not
disintegrate.
Example 1
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
28
The following formulations were prepared by mixing the components listed in
the
Table below in 15 ml of distilled water:
Formulation Aluminum Salt Mannitol GlycineDextran
1 (comparison)14.5g of Alhydrogell>322mg 131mg l7.Smg
2 (invention)2.5g of Alhydrogeh~693mg 130mg l8mg
3 (comparison)15g of Adju-PhosZ)438mg 173mg 16.9mg
4 (comparison)7.7g of Adju-Phos2~882mg 172mg 16.2mg
1~ Alhydrogel: 3% by weight aluminum hydroxide
Adju-Phos: 2% by weight aluminum phosphate
These formulations were spray dried using a Buchi 190 Mini-Spin Drier
operating
under the following conditions: air inlet temperature =130°C; air
outlet temperature =
70°C; Q liquid feed: setting 5; and Q atomising air: 5001/hr. Drying
air was set at the full
scale. Free-flowing powders were obtained. Yields were as follows:
FormulationPowder yield % Yield MMAD
(g)
1 0.52 68.4 8-10 ~,m
2 0.48 53.9 8-10 ~m
3 0.91 74.1 8-10 ~m
4 0.38 31.0 8-10 ~m
The composition of the-powders obtained in relation to the solids content of
the
suspension subjected to spray drying was as follows:
AI(OH)3MannitolGlycineDextranTotal
Solid
Formulation 1
Solid content in suspension2.9 2.1 0.9 0.1 6
for
spray drying (%)
Powder content 48.3% 35.0% 15.0% 1.7%
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
29
A1(OH)3MannitolGlycineDextranTotal
Solid
Formulation 2
Solid content in suspension0.5 4.6 0.9 0.1 6.1
for
spray drying (%)
Powder content 8.2% 75.4% 14.8% 1.6%
Formulation 3
Solid content in suspension4 2.9 1.2 0.1 8.2
for
spray drying (%)
Powder content 48.8% 35.4% 14.6% 1.2%
Formulation 4
Solid content in suspension1 5.9 1.1 0.1 8.1
for
spray drying (%)
Powder content 12.3% 72.8% 13.6% 1.2%
The spray dried powders were resuspended in distilled water. Specifically,
each
powder was added to distilled water (1:500 by weight) and shaken for 3
minutes. The
resulting suspensions were examined for aggregation. Only Formulation 2
according to the
invention formed a gel-Iike suspension without precipitate. The results are
shown below:
- Formulation 1: 32.59 mg of spray-dried powder was added to 1 ml of distilled
water. A white precipitate formed after the resulting suspension
has been allowed to stand overnight.
- Formulation 2: 37.1 mg of spray-dried powder was added to 1 ml of distilled
water. An off white, grey, gel-like suspension formed. No
precipitate was observed after the suspension had been allowed to
stand oversight.
- Fornulation 3: 44.34 mg of spray-dried powder was added to 1 ml of distilled
water. A white precipitate formed after the resulting suspension
had been allowed to stand overnight.
- Formulation 4: 29.4 mg of spray-dried powder was added to 1 ml of distilled
water. A white precipitate formed after the resulting suspension
had been allowed to stand overnight.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
Example 2
Two vaccine formulations were prepared as follows:
5 Formulation A:
A concentrated alum-HBsAg suspension was prepared by first washing an alum-
adsorbed HBsAg vaccine obtained from Rhein Americana S.A. contaiung 20~g of
HBsAg (approximately 1 human dose) adsorbed on SOO~,g of alum (approximately
1500~g of aluminum hydroxide) with distilled, deionised water to remove buffer
salt. Alum
10 gel was allowed to settle overnight in a 250-mL Nalgene narrow-mouth square
polycarbonate bottle at 2-8°C. The supernatant (150mL) was removed and
the same
volume of water was added to the precipitates and mixed. This procedure was
repeated
for a second time.
1008 of the washed alum-HBsAg formulation~was weighed in a Nalgene square
15 bottle and allowed to settle overnight at 2-8°C. After 90mL of
supernatant was removed,
the remaining suspension was transferred to a SOmL polypropylene centrifuge
tube and
centrifuged at 200 rpm for 4 minutes using a bench-top centrifuge (Allegra 6R,
Beckman).
The supernatant was further removed to obtain 3.369g of concentrated alum-
HBsAg
suspension. This suspension was then mixed with 315.24mg mannitol, 81.73mg
glycine,
20 101.91mg dextran and placebo alum gel (A1203 at 2%) to achieve a liquid
alum-HBsAg
formulation having an alum concentration of 3%.
Formulation B:
An alum-HBsAg suspension was washed in accordance with the method
25 described for formulation A. 20.79g of the suspension was weighed in a SOmL
centrifuge
tube and allowed to settle overnight at 2-8°C. After l7mL of
supernatant was removed,
the remaining concentrated suspension (3.572g) was mixed with 113.06mg
mannitol,
47.31mg glycine and 23.22mg dextran to produce a liquid formulation having an
alum
concentration of 0.6%.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
31
The two formulations were dried using the techniques set out in Table 1 below:
Table 1: Dryin techniques
Powder Formulation Drying technique
1 (comparison) A Freeze-drying
2 (invention) A Spray freeze-drying
3 (invention) B Spray freeze-drying
4 (comparison) A Freeze-drying followed
by C/G/S
(using <20~,m fraction)
5 (comparison) A Freeze-drying followed
by C/G/S
(using 38-45~.m fraction)
6 (comparison) A Freeze-drying followed
by C/G/S
(using 53-75~,m fraction)
Freeze Drying:
A Dura-Stop freeze dryer (FTS System, Stone Ridge, NY) was used to freeze dry
the alum-adsorbed HBsAg formulation based on the freeze-drying cycle in Table
2.
Table 2: Freeze-dryi~cycle
Stage/Cycle Conditions
Freezing pre-cool shelf temperature (ST) = 0C
ramp at 1. 0 C/min to ST = -5 5 C,
hold for 15 min
wait for product temp (PT) _ -48 C,
hold for 120 min
Primary Dryingcondenser/vacuum (C/V) switched "on"
when condenser temp. reaches -40C, vacuum
pump
turned on
wait for chamber vacuum to reach 150
mT (20.0 Pa)
wait for foreline vacuum to reach 100
mT (13.3 Pa)
ramp at 1.0 C/min to ST = -25 C, hold
for 18 hours
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
32
Secondary ~ ramp at 1. 0 ° C/min to ST =10 ° C, hold for 4
hours
Drying ramp at 1.0 ° C/min to ST = 20 ° C, hold for 11
hours
A vacuum of 100 mT (I3.3 Pa) was maintained throughout primary and secondary
S drying.
Spray-freeze-drying:
Each suspension solution was sprayed into liquid nitrogen stirred in a
stainless steel
pain using an ultrasonic atomizer (Sono Tek Corporation, Milton, NY) with a
nozzle
frequency of 60 kHz. Sonic energy for atomization was set at S.0 watts. Liquid
feed was
delivered by a MasterFlex C/L peristaltic pump at 1.S mL/min. The pan
containing frozen
particles in liquid nitrogen was loaded into the Dura-lyophilizer pre-cooled
to -SO°C and
freeze-dried based on the condition of Table 3.
1 S Table 3: Freeze-dryin~ c.
Stage/Cycle Conditions
Freezing pre-cool shelf temperature (ST) _ -SOC
ramp at 1.0 C/min to ST = -SS C, hold
for 1 S min
wait for product temp (PT) _ -48 C, hold
for 120 min
Primary Dryingcondenser/vacuum (C/V) switched "on"
when condenser temp. reaches -40C, vacuum
pump turne
on
wait for chamber vacuum to reach 1S0 mT
(20.0 Pa)
wait for foreline vacuum to reach 100
mT (13.3 Pa)
ramp at 1.0C/min to ST =-2SC, hold for
18 hours
Secondary ramp at 1.0 C/min to ST = 20 C, hold for
9 hours
Drying
A vacuum of 200 mT (16.6 Pa) was maintained throughout primary and secondary
2S drying.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
33
Com~ress/Grind/Sieve:
The lyophilized material was rendered into particulate form using a compress,
grind
and sieve ("C/G/S") techtuque. More particularly, the lyophilized material was
compressed in a stainless steel dye of 13-mm in diameter (Carver Press,
Wabash, INS at a
pressure of 12,000 psi for 5-10 minutes. The compressed discs were ground
manually
using a mortar and pestle. The ground powder was manually sieved through a
stack of
sieves (3-in diameter) into three size fractions, 53-75 Vim, 38-53 ~,m, and 20-
38 ~,m.
Experiment 1: Effect of drying_process on the extent of coagulation
Powders 1 to 3 were reconstituted in water at a ratio of 1:SOOw/w and examined
using optical microscopy in accordance with standard techniques. Visual
analysis of the
particles was performed using an optical microscope (Model DMR, Leica,
Germany) with
l Ox-eyepeice lens and Sx-objective lens. The system was equipped with a
Polaroid
camera system for image output. Optical microscopy provides a qualitative
analysis of the
IS degree of alum coagulation. In this experiment, powder I produced very
large aggregates
on reconstitution, whereas powder 2 coagulated only slightly. Powder 3
produced almost
no aggregates at all.
The particle size of the reconstituted powders was also measured
quantitatively.
The reconstituted powder sample was vortexed/sonicated to make a homogeneous
suspension. The suspension was then added to the glass container of a particle
size
analyzer (AccuSizer 780, Particle Sizing Systems, Santa Barbara, CA) for
particle size
distribution measurement. The results of the measurements carried out on
powders 2 and
3 both before and after spray freeze-drying are shown in Figure 1. Similar
comparative
results for powder 1 showing particle size before and after freeze-drying are
shown in
Figure 2. These results illustrate the similar particle size distribution of
powders 2 and 3
before and after drying, demonstrating that little or no alum coagulation
occurred during
freeze-drying. In contrast, the particle size of powder 1 increases
significantly after freeze-
drying, indicating that significant alum coagulation has occurred.
Experiment 2: Effect of coagulation on the stability of alum
containin~hepatitis B vaccine
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
34
A study was carried out to assess the effect of alum coagulation on the
immunogenicity of alum-absorbed hepatitis B vaccine. As stated earlier, severe
coagulation occurred when hepatitis B vaccine (containing alum) was dried by
the freeze-
drying process, whereas spxay-freeze-drying of hepatitis B vaccine did not
cause
coagulation. In this mouse experiment, the immunogenicity of freeze-dried and
spray-
freeze-dried hepatitis B vaccines were compared. Further, the immunogenicity
of unsieved
freee-dried vaccine and various sieved fractions (<20, 38-45, 53-75 ~,m in
diameter) were
compared to determine which size fraction was more immunogenic. The
experimental
design is shown in Table 4.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
Table 4: Experimental design of the mouse immunogenicit~tudy
Group Formulation Drying Technique Particle Injection
* size route
(reconstituted)
1 A freeze-drying unsieved intraperitoneal
5 2 A . freeze-drying <20 ~.m intraperitoneal
3 A freeze-drying 38-45 intraperitoneal
~,m
4 A freeze-drying 53-75 intraperitoneal
~rn
5 A Spray-freeze-drying10-75 intraperitoneal
~,m
6 B Spray freeze-drying10-75 intraperitoneal
~.m
10 7 Not treated Liquid alumn vaccine- intraperitoneal
used
* Details of the formulation A and B are described above
Powders were reconstituted with distilled water and used to immunize Balb/C
mice
15 (female, 8 .per group, 5-7 weeks old at the beginning of the study).
Reconstituted vaccines
were administered by intraperitoneal injection using a 23 1/5 needle. Each
injection
administered 200 ~,1 of solution containing 2 ~,g of hepatitis B surface
antigen absorbed on
alum. Control mice were immunized with untreated liquid hepatitis B vaccine.
Following a
prime (day 0) and a boost immunisation (day 28), immune responses to the
hepatitis B
20 vaccine were determined with serum collected on day 42 in an ELISA. The
antibody titers
were determined by comparing to reference a serum.
The results of these trials, as set out in Figure 3, clearly indicated that
the alum
coagulation caused by freeze-drying resulted in a decrease and even loss of
immunogencity
of the hepatitis B vaccine. Compared to the untreated liquid vaccine, freeze-
dried hepatitis
25 B vaccine (group 1) had dimiiushing immunogenicity. The immunogenicity of
the freeze-
dried particle had an adverse correlation with the size of the particles
(groups 2, 3 and 4).
The larger particle fractions were less immunogenic than the smaller particle
size fraction.
This clearly indicated that large size particles associated with coagulation
had lost its
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
36
vaccine potency. The spray-freeze dried hepatitis B vaccine maintained its
immunogenicity
(groups 5 and 6) when compared with the untreated vaccine. The amount of alum
in the
total dry mass (50% or 12%) did not affect the potency.of the dry powder.
Neither of the
spray freeze-dried powders had a coagulation problem. This is significant that
the spray-
s freeze-drying formulation preserves the potency of alum salt adjuvant at a
very high
concentrations (3% by weight).
Taken together all these data, it can be concluded that alum coagulation is
associated with the potency loss of alum vaccine when freeze-dried. It is
believed that the
laxge sizes of coagulated particles, which may fail to solubilize ih vivo, can
not be
processed by the cells of the immune system and, thus, have no potency. More
importantly, the process of the invention can prepare stable dry powders with
alum
containing vaccine without causing coagulation. It is believed that the quick
freezing in the
liquid nitrogen employed in the spray-freeze-drying process is critical for
preventing the
coagulation, thus preserving the vaccine potency.
Experiment 3:Effect of excipient and drvin~ processes on the stabilitv of
snrav-freeze-
dried hepatitis B vaccine
In this study, the effect of excipients and a variant spray-freeze-drying
process on
the stability of alum vaccines was evaluated. Hepatitis B surface antigen
(HBsAg)
absorbed on alum hydroxide was used as a model antigen. In addition, the
immunogenicity
of spray-freeze-dried powders was evaluated in mice following two different
routes of
immunisation, intramuscular injection using a needle and epidermal powder
immunisation
using a needleless powder delivery device. The excipients for the spray-freeze-
dried
formulations are shown in Table 5. In this case, the spray-freeze-dried
formulations used
the combination of two sugaxs and one polymer. There was no amino acid/salt
involved.
The conditions for spray-freeze-drying are the same as that shown in Table 3.
However,
compress/grind/sieve step was not used. The particle size distribution of the
spray-freeze-
dried powders is also indicated in Table 5.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
37
Table S: Composition of spray-freeze-drying formulations
FormulationVaccine Excipient Process Particle
size,
p.m
(Aerosizer)
Dvl Dv5 Dv9
0 0 5
SFD-C 2 ~.g Trehalose/Spray-freeze-dry23 38 57
HBsAg/50 mannitol/
~.g Alum PEG (3:4:3)
SFD-D 2 ~,g Trehalose/Spray-freeze-dry26 39 S9
HBsAg/SO mannitol/
~g Alum 37 kD dextran
(3:4:3)
SFD-E 2 ~,g Trehalose/Spray-freeze-dry24 36 S6
HBsAg/SO mannitol/
~ g Alum 10 kD dextran
(3:4:3)
The immunogenicity of spray-freeze-dried formulations was evaluated in a mouse
study. Balb/C mice (female, 8 per group, S-7 weeks old at the beginning of the
study)
were used. The study design is shown in Table 6. For intramuscular (llVI)
injection,
powders were reconstituted with distilled water and administered by injection
200 ~,l of
solution containing 2 ~g of hepatitis B surface antigen absorbed on alum into
the quadriceps
muscle using a 23 1/S needle. For epidermal (EPA powder immunisation, powders
were
1 S administered to the shaved abdominal skin of mice using a re-chargeable
powder delivery
device. Control mice were immunised with untreated liquid hepatitis B vaccine
by
intramuscular inj ection. Following a prime (day 0) and a boost immunisation
(day 28),
immune responses to the hepatitis B vaccine were determined with serum
collected on day
42 in an ELISA. The antibody titers were determined by comparing to reference
a serum.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
38
Table 6: Experimental design of the mouse immunogenicity study
Group FormulationReconstitution Route
1 SFD-C yes IM
2 SFD-D yes IM
3 SFD-E yes IM
4 SFD-C no EPI
5 SFD-D no ' EPI
6 SFD-E no EPI
7 untreated Not applicable IM
The results of this study, as shown in Figure 4, clearly indicate that all
three spray-
freeze-dried hepatitis B vaccines are immunogenic in mice whether it is'
administered by the
intramuscular route after reconstitution or by the epidermal route as powders.
Different
excipients were used in these formulations and there were no significant
differences in the
immmlogenicity among these formulations. All three formulations had no
coagulation
problem when reconstituted in water (data not shown). This provides further
evidence that
the quick-freezing step in the spray-freeze-drying process is a critical step
to stabilize the
alum. Excipients may play a less important role. This study also demonstrated
that spray-
freeze-dried vaccines absorbed on alum can be useful for immunisation via
different routes,
e.g. intramuscularly injection when reconstituted or epidermal powder
immunisation in a
powder form.
Experiment 4: Tmmuno e~ nicit~of sprayfreeze-dried diphtheria-tetanus toxoid
vaccine
To determine of spray freeze-drying process can be used prepare stable powders
with other alum-containing vaccine, spray-freeze dried powders using
diphtheria-toxoid
vaccine obtained from CSL Limited (Australia) were prepared. This bulk
contained 5%
w/v aluminium phosphate adsorbed with both diphtheria toxoid and tetanus
toxoid at a
concentration of 563 Lf/mL each. The spray freeze-dried diphtheria-tetanus-
toxoid
vaccine was prepared under the conditions as described in Table 3 and followed
by
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
39
compress/grind/sieve to generate particles with mean size of 20-38 ~m and 38-
53 ~,m in
diameter. The formulation information is summarised in Table 7. These
particles do not
have coagulation problems when reconstituted in water and examined under
optical
microscopy (data not shown).
Table 7
HBsAg-Alum Trehalose GlycineDextranTotal DT dose
solid
phosphate dihydrate (mg) (mg) content
(mg) (mg)
(%)
250 292.9 66.1 86.6 4 3 Lf/1-mg
powder
The immunogenicity of spray-freeze-dried diphtheria-tetanus-toxoid vaccine was
determined in guinea pigs (Charles River). Guinea pigs (4/group) were
vaccinated on days
0 and 28 by administering powders to the abdominal skin using a powder
delivery device.
Each animal received O.S mg powders containing 1.5 Lf diphtheria toxoid and
1.5 Lf
tetanus toxoid absorbed on 250 wg of aluminum phosphate. Control animals were
vaccinated with untreated vaccine by intramuscular injection using a 23 %2
needle. Serum
antibody responses to diphtheria toxoid and tetanus toxoid were measured in an
ELISA
using sera collected on days 42.
The results of the immunogenicity study are shown in Figure S. Epidermal
powder
immunisation with spray-freeze-dried diphtheria toxoid absorbed on alum
elicited antibody
responses to each of the vaccine components and the tiers are comparable to
that elicited
by intramuscular injection of untreated vaccine. The size of the spray-freeze-
dried
powders did not appear to affect the immunogenicity significantly since these
powders did
not have coagulation problem ira vivo. The smaller particle fraction of the
spray-freeze
dried formulation appears to have elicited slightly lower antibody titers to
the diphtheria
toxoid than the larger size fraction. This may reflect the relatively lower
delivery efficiency
for the smaller size fraction. Tlus study again demonstrated that spray freeze-
drying
process preserves the potency of alum-containing vaccine the dry solid dosage
form.
CA 02412197 2002-12-09
WO 01/93829 PCT/USO1/18494
Accordingly, novel freeze spray-dried powder compositions and methods for
producing these compositions have been described. Although preferred
embodiments of
the subject invention have been described in some detail, it is understood
that obvious
variations can be made without departing from the spirit and the scope of the
invention as
5 defined by the appended claims.