Note: Descriptions are shown in the official language in which they were submitted.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
CYCLIC POLYAMINE COMPOUNDS FOR CANCER THERAPY
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to Provisional Patent Application
Number 60/222,522 filed August 2, 2000, the content of which is incorporated
herein by reference in its entirety.
TECHNICAL FIELD
[0002] This invention is directed to compounds and methods useful for
treating cancer and other diseases caused by uncontrolled cell proliferation.
More
specifically, this invention is directed to cyclic polyamine compounds which
display anti-tumor activity in vitro, as well as methods of making and using
those
compounds.
BACKGROUND OF THE INVENTION
[0003] Cancer is one of the leading causes of death in the developed
world. Approximately one-quarter of the deaths in the United States in 1997
were
due to cancer, making it the second most common cause of death after heart
disease. Accordingly, development of new and effective treatments for cancer
is a
high priority for health care researchers.
[0004] Cancer is often treated by using chemotherapy to selectively kill or
hinder the growth of cancer cells, while having a less deleterious effect on
normal
cells. Chemotherapeutic agents often kill rapidly dividing cells, such as
cancer
cells; cells which are dividing less rapidly are affected to a lesser degree.
Other
agents, such as antibodies attached to toxic agents, have been evaluated for
use
against cancers. These agents target the cancer cells by making use of a
characteristic specific to the cancer, for example, higher-than-normal rates
of cell
division, or unique antigens expressed on the cancer cell surface.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0005] One peculiar distinguishing characteristic of malignant cells is their
high rate of glycolysis, even in the presence of oxygen (so-called aerobic
glycolysis, or the Warburg effect). Studies by Otto Warburg over seven decades
ago demonstrated that the vast majority of human and animal tumors display a
high rate of glycolysis. Although Warburg's hypothesis that defective
oxidative
metabolism underlies this high rate of glycolysis is not supported by recent
studies, the original observation has been fully confirmed. See Chesney, J. et
al.,
"An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich
instability element: role in tumor cell glycolysis and the Waxburg effect,"
Proc.
Natl. Acad. Sci. USA (1999) 96(6):3047-52. Human tumors endure profound
hypoxia, and hence adaptation to hypoxic conditions is a crucial step in tumor
progression. The anaerobic use of glucose as an energy source through
glycolysis
is therefore a feature common to most solid tumors. See Dang, C.V. and
Semenza, G.L., "Oncogenic alterations of metabolism," Trends Biochem. Sci.
(1999) 24(2):68-72 and Boros, L.G. et al., "Nonoxidative pentose phosphate
pathways and their direct role in ribose synthesis in tumors: is cancer a
disease of
cellular glucose metabolism?" Med. Hypotheses (1998) 50(1):55-9
[0006] Magnetic resonance spectroscopy and positron-emission
tomography have demonstrated that tumors have an increased uptake of glucose
as compared with normal tissues, and that tumor aggressiveness and prognosis
correlates with glucose uptake. See Imdahl, A. et al., "Evaluation of positron
emission tomography with 2-[18F]fluoro-2-deoxy-D-glucose for the
differentiation
of chronic pancreatitis and pancreatic cancer," Br. J. Surg. (1999) 86(2):194-
9 and
Maublant, J. et al., "Positron emission tomography (PET) and (F-18)-
fluorodeoxyglucose in (FDG) in cancerology," Bull. Cancer (Paris) (1998)
85(11):935-50. The expression of the glucose transporter GLUT1 is also
increased in cancer cells. See Grover-McKay, M. et al., "Role for glucose
transporter 1 protein in human breast cancer," Pathol. Oncol. Res. (1998)
4(2):115-20 and Burstein D.E. et al., "GLUT1 glucose transporter: a highly
sensitive maxker of malignancy in body cavity effusions," Mod. Pathol. (1998)
11 (4):392-6. Glucose utilization through the glycolytic pathway in cancer
cells
2
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
leads to pyruvate formation as is the case in normal cells, but in the absence
of
oxygen, pyruvate is not metabolized through the tricarboxylic cycle. This
deprives the cancer cells of the efficient production of ATP by oxidative
phosphorylation. In cancer cells pyruvate is reduced (by NADPH) to lactate,
leading to the acidic environment of tumors. The cytosolic pH of tumor cells,
however, is maintained as it is in normal cells. See Dang, C.V. and Semenza,
G.L., "Oncogenic alterations of metabolism," Trends Biochem. Sci. (1999)
24(2):68-72.
[0007] Hypoxia is a strong selective force, and it regulates glycolysis by
modulating oncogenes and tumor suppressing genes. Tumor angiogenesis is
stimulated by hypoxia and hypoglycemia, which induce expression of angiogenic
factors that recruit microvessels to allow delivery of nutrients and oxygen,
to
support expansion of the tumor mass. See Moser, T.L. et al., "Angiostatin
binds
ATP synthase on the surface of human endothelial cells," Proc. Natl. Acad.
Sci.
USA (1999) 96(6):2811-6. However, the new microvessels are limited and
disorganized, and the oxygen consumption rate exceeds the supply rate. Glucose
deprivation is a potent inducer of necrosis in transformed cells, and
physiological
and oncogenic transcription factors that stimulate glycolysis by increasing
glucose
transport as well as the activity of key glycolytic enzymes (e.g., hexokinase
II,
lactate dehydrogenase A) play a crucial role in promoting the survival of
cancer
cells in adverse tumor microenvironments. See Blancher C. et al., "The
molecular
basis of the hypoxia response pathway: tumour hypoxia as a therapy target,"
Cancer Metastasis Rev. (1998) 17(2):187-94.
[0008] Cancer cells thus depend mainly on the glycolytic pathway to
generate the necessary ATP to grow, even in the presence of oxygen. It is
known
that the energy provided by one mole of ATP is needed to produce 10 g of
cells.
While the aerobic oxidation of one mole of glucose to carbon dioxide results
in a
net gain of ca. 38 moles of ATP, the anaerobic (glycolytic) transformation of
1
mole of glucose into pyruvate and lactate only results in the gain of 2 moles
of
ATP, 19 times less than in aerobic oxidation. It is clear that ATP is very
much at
3
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
a premium in cancer cells due to the incomplete oxidation of glucose in the
cells,
which in turn requires an elevated rate of glycolysis in tumor cells.
[0009] Because of the limited supply and high demand for ATP in
malignant cells, drugs which can hydrolyze ATP can provide a means to control
cancer growth. Such a drug will disproportionately impact a cancer cell, while
being less deleterious to normal tissue where ATP synthesis is constantly
replenished by oxidative phosphorylation in the mitochondria.
[0010) Early note was taken of the various functions that cyclic
polyamines can perform. These functions include facilitating selective uptake
and
transport of metal ions, metal chelation, and serving as models of catalyst
and
enzyme function. See Kimura, E., "Macrocyclic polyamines with intelligent
functions," Tetrahedron (1992) 48(30):6175.
[0011] Cyclic polyarnines have been observed fo have surprising effects
on ATP hydrolysis. Cyclic polyamines, when protonated, bind ATP, ADP, and
AMP stably and enhance the rate of hydrolysis of ATP by several orders of
magnitude over a wide pH range. Linear polyamines, which do not bind ATP, do
not increase the rate of hydrolysis. Hydrolysis catalyzed by a cyclic compound
yields orthophosphate and ADP as products; the ADP is then hydrolyzed slowly
to AMP. In the cleavage of ATP, the formation of an intermediate
phosphoramidate was detected and the possible form of an initial "perched"
complex and a mechanism of hydrolysis were postulated. See Merthes, M.P. et
al., "Polyammonium macrocycles as catalysts for phosphoryl transfer: the
evolution of an enzyme mimic," Account of Chemical Research (1990) 23:413;
Hosseini, M.W. et al, "Efficient molecular catalysis of ATP hydrolysis by
protonated macrocyclic polyamines," Helv. Chim. Acta (1983) 66:2454; Prakash,
T.P. et al., "Macrocyclic polyamine[16]-N3 and [21]-N4: Synthesis and study of
their ATP complexation by 31P NMR spectroscopy," J. Chem. Soc. Perkin Trans.
(1991) 1:1273; Hosseini, M.W. et al., "Supramolecular catalysis in the
hydrolysis
of ATP facilitated by macrocyclic polyamines: mechanistic studies," J. Am.
Chem. Soc. (1987) 109:537; and Bencini, A. et al., '-'Potential ATPase mimics
by
polyammonium macrocycles: criteria for catalytic activity," Bioorganic Chem.
4
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
(1992) 20:8. These cyclic catalysts were described as "functional mimics" of
ATPases. The hydrolysis of ATP involved an exchange of oxygen at the
beta-phosphate of ATP and occurred in the presence of calcium. Under these
conditions, subsequent hydrolysis of ADP was decreased and the phosphorylated
cyclic compound accumulated. When this reaction mixture was adjusted to pH
4.5, pyrophosphate was formed. The cyclic phosphoramidate was shown to be
capable of phosphorylating ADP to give ATP.
[0012] The phosphatase activity of the cyclic polyamines was also studied
using other biological phosphate esters. It was shown that a cyclic polyamine
catalyst cleaved acetyl phosphate to orthophosphate; the reaction then
proceeded
to the synthesis of pyrophosphate. It has been observed that a cyclic
polyamine
could activate formate in an ATP-dependent reaction in the presence of Ca++ or
Mg-~-~-. The activation appeared to proceed via the hydrolysis of ATP to
generate
the cyclic phosphoramidate, with the latter species forming the proposed
intermediate formyl-phosphate that was then cleaved on the cycle to produce a
cyclic formamide (N-formylation). It has been suggested that this set of
reactions
might mimic the ATP-dependent enzymatic synthesis of Nl°-formyl
tetrahydrofolate and is relevant to the nature of formyl tetrahydrofolate
synthetase. See Jahansouz H. et al., "Formate activation of neutral aqueous
solutions mediated by a polyammonium macrocycle," J. Am. Chem. Soc. (1989)
111:1409.
[0013] Independent of the chemical studies described above with cyclic
polyamines and with cyclic polyethers (Kimura et al., supra) it was known from
the phytochemical literature that cyclic polyamine alkaloids (also called
macrocyclic aminolactams) are an important class of natural products. They
originate mainly from the crossover of the phenylpropanoid biosynthetic
pathway
(the shikimate pathway) and the polyamine (spermine and spermidine) pathway.
Thus, from the plant families Cannabis (indian hemp), Codonocarpus, Equisetum
(horsetail), Lunaria, Maytenus, Oncinotis, Peripterygia, and Pleurostylia the
following cyclic spermidine-derived alkaloids, among others, were isolated:
chaenorhine, aphelandrine, orantin, the ephedradines, and the periphyllines.
See
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Gerardy, R. et al., Phytochemistry (1993) 32:79; Zenk, M.H. et al., J. Chem.
Soc.
Chem. Common. (1989) 1725; Husson, H.-P. et al., Tetrahedron (1973) 29:1405;
Sagner, S. et al., Tetrahedron Letters (1997) 38:2443; Stach, H. et al.,
Tetrahedron
(1988) 44:1573; and Kramer, U. et al., Angew. Chem. (1977) 89:899. Among the
spermine-derived alkaloids are homaline and the mixture of alkaloids called
pithecolobines. The latter were isolated from extracts of Pithecolobium saman
(see Wiesner, K. et al., "Structure of pithecolobine II," Can. J. Chem. (1968)
46:1881 and Wiesner, K. et al., "Structure of pithecolobine III," Can. J.
Chem.
(1968) 46:3617) and their biosynthesis likely derives from the crossover of
the
metabolism of spermine with the metabolic pathways for unsaturated fatty
acids.
From the seeds of the Indian plant Albizia amara, a methanol extract was shown
to contain a mixture of nine alkaloids that were called budmunchiamines A to
I.
See Pezzuto, J.M. et al., "DNA-based isolation and the structure elucidation
of the
budmunchiamines, novel macrocyclic alkaloids from Albizia amara,"
Heterocycles (1991) 32:1961-68; Pezzuto, J.M. et al., Phytochemistry (1992)
31:1795-1800. Their structures were established by physical methods and were
found to be analogous to the pithecolobines. Isolates from the seeds of
Albizia
amara were found to have cytotoxic effects in a general screen for possible
biological effects. Mar, W. et al., "Biological activity of novel macrocyclic
alkaloids (budmunchiamines) from Albizia amara detected on the basis of
interaction with DNA," J. Natural Products (1991) 54:1531. Other studies
regarding budmunchiamine's are described in Rukunga, G.M. et al., J. Nat.
Prod.
59(9):850-3 (1996); Rukunga, G.M. et al., Phytochemistry 42(4):1211-15 (1996);
Misra, L.N. et al., Phytochemistry 39(1):247-249 (1995); Dixit, A.K. et al.,
J. Nat.
Prod. 60(10):1036-1037 (1997); Rukunga, G.M. et al., Bull. Chem. Soc. Ethiopia
10(1):47-51 (1996); Cordell, G.A. et al., Pure Appl. Chem. 66(10-1):2283-2286
(1994); and Onuki, H. et al., Tetr. Lett., 34(35):5609-5612 (1993).
[0014] ~ However, the above-referenced art does not suggest use of isolated
cyclic polyamines for cancer therapy, nor does it provide guidance to use
cyclic
polyamines as ih vivo catalysts of hydrolysis of intracellular ATP in cancer
cells.
Thus, the cyclic polyamines of the present invention represent a new approach
to
6
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
cancery therapy. Additionally, no syntheses of the budmunchiamine compounds
have been reported, and work with budmunchiamines has typically been
performed with plant extracts containing a rriixture of compounds. The present
invention provides methods that allow the synthesis of individual compounds
similar to the structures proposed for the pithecolobines and the
budmunchiamines, in order to design new compounds with ATP-ase-like activity
in vivo and permit study of the isolated compounds. Such new compounds were
created with the methods of the present invention for use in treating cancer
and
other pathological conditions.
DISCLOSURE OF THE INVENTION
[0015] The invention provides compounds and compositions for treating
diseases caused by uncontrolled proliferation of cells, such as cancer,
especially
prostate cancer, and for inducing intracellular ATP hydrolysis for treatment
of
other disorders.
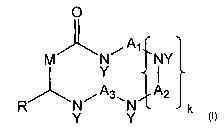
[0016] In one embodiment, the invention provides compounds of the
O
A
M N~ 1 NY
Y
i A~. A2
R Y Y
formula:
where Al, each Aa (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from H or C1-C4 alkyl; where
M
is selected from CI-C4 alkyl; where k is 0, 2, or 3; and where R is selected
from
C1-C32 alkyl; and all stereoisorners and salts thereof. In additional
embodiments,
the Y group is -H or -CH3. In another embodiment, Al, each A2 (if present),
and
A3 are independently selected from C2-C4 alkyl. In yet another embodiment, M
is
-CH2-. The invention also includes compositions of one or more of the
compounds above in combination with a pharmaceutically-acceptable carrier.
7
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0017] The invention also provides compounds of the formula:
O
A
M N~ 1~NY
Y I
R N~A3'NiA2
Y Y
where A1 and A3 are independently selected from C1-C8 alkyl; where Aa is
independently selected from C1-C3 alkyl or CS-C8 alkyl; where each Y is
independently selected from H or Cl-C4 alkyl; where M is selected from Cl-C~
alkyl; and where R is selected from C1-C32 alkyl; and all stereoisomers and
salts
thereof. In additional embodiments, the Y group is -H or -CH3. In another
embodiment, A1 and A3 are independently selected from Ca-C4 alkyl, and Aa is
selected from the group consisting of CZ-C3 alkyl and CS alkyl. In yet another
embodiment, M is -CH2-. The invention also includes compositions of one or
more of the compounds above in combination with a pharmaceutically-acceptable
carrier.
[0018] The invention also provides compounds of the formula:
O
A
M N~ 1~NY
Y I
R N~A3'N~A2
Y Y
where A1 and A3 are independently selected from C1-C8 alkyl; where Aa is
independently selected from C1-C8 alkyl; where each Y is independently
selected
from H or C2-C4 alkyl; where M is selected from C1-C4 alkyl; and where R is
selected from C1-C32 alkyl; and all stereoisomers and salts thereof. In an
additional embodiment, each Y group is -H. In another embodiment, A1 and A3
8
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
are independently selected from Ca-C4 alkyl, and AZ is selected from the group
consisting of C2-CS alkyl. In yet another embodiment, M is -CH2-. The
invention
also includes compositions of one or more of the compounds above in
combination with a pharmaceutically-acceptable carrier.
[0019] The invention also provides a method of synthesizing a compound
of the formula
O
A .(
M~N~ 11~NY
Y
R N~A3~N
Y Y
where Al, each Aa (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from H or C1-C4 alkyl; where
M
is selected from Cl-C4 alkyl; where k is 0, 1, 2, or 3; and where R is
selected from
C1-C32 alkyl; where the method comprises the steps of reacting an w-halo alkyl
alkanoate with an aldehyde or ketone-containing compound to give an alkene-
containing alkanoate compound; reacting the alkene-containing alkanoate
compound with a compound containing two primary amino groups and optionally
containing secondary amino groups to effect addition of one of the amino
groups
across the double bond; cyclizing the other amino group with the alkanoate
group
to form an amide bond; and optionally alkylating the secondary amino groups if
present. In one embodiment, the ~-halo alkyl alkanoate is ethyl bromoacetate.
In
another embodiment, the aldehyde or ketone-containing compound is an
aldehyde-containing compound. In yet another embodiment, the step of reacting
an w-halo alkyl alkanoate with an aldehyde or ketone-containing compound to
give an allcene-containing alkanoate compound is performed by reacting the c~-
halo alkyl alkanoate with triphenylphosphine. In still another embodiment, the
compound containing two primary amino groups is selected from the group
consisting of H2N-Al-(NH-A2)k-NH-A3-NHa where Ai, each A2 (if present), and
9
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
A3 are independently selected from C1-C8 alkyl and k is 0, 1, 2, or 3. The
compound containing two primary amino groups can be selected from the group
consisting of spermine, spermidine, and putrescine in still another
embodiment.
The step of cyclizing the other amino group with the alkyl alkanoate group to
form an amide bond can be performed by reacting the compound with antimony
(III) ethoxide in yet another embodiment. In an additional embodiment, the
step
of optionally alkylating any secondary amino groups, if present, can performed
by
reacting the compound first with an aliphatic aldehyde to result in a Schiff
base,
then reducing the Schiff base, resulting in alkylation of the secondary amino
groups; the step of reducing the Schiff base can be performed by using the
reagent
NaCNBH3.
[0020] The invention also provides a method of synthesizing a compound
of the formula:
O
A
M N~ 1 NY
Y
R N~A3~N
Y Y
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
where A1 is C3 alkyl, and each A2 (if present) and A3 are independently
selected from C1-C8 alkyl; where each Y is independently selected from H or C1-
C4 alkyl; where M is selected from C1-C4 alkyl; where k is 0, I, 2, or 3; and
where
R is selected from C1-C32 alkyl; where the method comprises the steps of
condensing a compound comprising a primary amino group and a
hexahydropyrimidine moiety with an a,(3-unsaturated ester compound, such that
the primary amino group adds at the [3-position of the unsaturated ester
compound, whereby the primary amino group is converted to a secondary amino
group; cleaving the methylene bridge of the hexahydropyrimidine moiety to
generate a secondary amino group and a newly-generated primary amino group;
and condensing the newly-generated primary amino group with the ester group to
form an amide group. The a,[3-unsaturated ester can be of the formula
(C1-C8 alkyl)-O-C(=O)-CH=CH-(C1-C32 alkyl). In another embodiment, the
compound comprising a primary amino group and a hexahydropyrimidine moiety
is of the formula
H2N-A3-(NY-A2)~ N~NH
where each Aa (if present) and A3 are independently selected from C1-C$
alkyl; where each Y is independently selected from H or C1-C4 alkyl; and where
j
is 0, 1, 2, or 3. In a preferred embodiment, j is 0. In another preferred
embodiment, A3 is C4 alkyl. The step of cleaving the methylene bridge of the
hexahydropyrimidine moiety can be performed with anhydrous HCl in an
alcoholic solvent. The step of condensing the newly-generated primary amino
group with the ester group to form an amide group can be performed with the
reagent B(N(CH3)a)3.
[0021] The invention also provides compounds of the formula
11
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
X
A
4
'A
M~N~ 1 NY
R N~A3~N
Y Y
where Al, each A2 (if present), and A3 are independently selected from C1-
C8 alkyl; where A4 is selected from C1-C8 alkyl or a nonentity; where X is
selected from -H, -Z, -CN, -NH2, -C(=O)-C1-C$ alkyl, or -NHZ, with the proviso
that when A4 is a nonentity, X is -H, -C(=O)-C1-C8 alkyl, or -Z; where Z is
selected from the group consisting of an amino protecting group, an amino
capping group, an amino acid, and a peptide; where each Y is independently
selected from H or C1-C4 alkyl; where M is selected from C1-C4 alkyl; where k
is
0, 1, 2, or 3; and where R is selected from C1-C3a alkyl; and all
stereoisomers and
salts thereof. In certain embodiments, A4 is a nonentity. In other
embodiments, X
is -Z, and -Z is -H. In other embodiments, Y is -CH3. In other embodiments, M
is
-CH2-. In still further embodiments, k is 1. In further embodiments, A1 and A3
are -CHZCHZCH2-. In still further embodiments, -CH2CHaCH2CH2-. In still
further embodiments, R is -C13H27. In yet further embodiments, one or more of
the specific limitations on A4, X, Z, Y, M, k, Al, A3, and R are combined.
[0022] In further embodiments of these compounds, A4 is C1-C8 alkyl, X
is -NHZ, and Z is selected from one of the 20 genetically encoded amino acids
(alanine, cysteine, aspartic acid, glutamic acid, phenylalanine, glycine,
histidine,
isoleucine, lysine, methionine, asparagine, proline, glutamine, arginine,
serine,
threonine, valine, tryptophan, tyrosine), a peptide of the formula acetyl-
SKLQL-, a
peptide of the formula acetyl-SKLQ-(3-alanine-, or a peptide of the formula
acetyl-SKLQ-.
[0023] The invention also provides methods of synthesizing compounds of
the formula
12
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
H
A
M~N~ 1 NY
R N~A3~N
Y Y
by reducing the carbonyl of the amide group of a compound of the formula
O
H
A
M N~ 1 NY
R N~A3~N
Y Y
where Al, each A2 (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from H or C1-C4 alkyl; where
M
is selected from C1-C4 alkyl; where k is 0, 1, 2, or 3; and where R is
selected from
Cl-~32 ~kYh ~d all stereoisomers and salts thereof. Lithium aluminum hydride
may be used as the reducing agent. Diborane may also be used as the reducing
agent.
[0024] The invention also provides a method of synthesizing a compound
of the formula
CN
A
M~N~ 1 NY
R N~A3~N
Y Y
13
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
where Al, each AZ (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from C1-C4 alkyl; where M is
selected from C1-C4 alkyl; where k is 0, 1, 2, or 3; and where R is selected
from
Cl-C32 alkyl, comprising reacting a compound of the formula
H
A
M~N~ 1 NY
R N~A3~N
Y Y
where Al, each A2 (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from Cl-C4 alkyl; where M is
selected from C1-C4 alkyl; where k is 0, l, 2, or 3; and where R is selected
from
C1-C32 alkyl, with a compound of the formula HaC=CH-CN.
[0025] The invention also provides a method of synthesizing compounds
of the formula
NH2
A
M~N~ 1 NY
R N~A3~N
Y Y
where Al, each A2 (if present), and A3 are independently selected from C1-
C8 alkyl; where each Y is independently selected from C1-C4 alkyl; where M is
selected from C1-C4 alkyl; where k is 0, l, 2, or 3; and where R is selected
from
C1-C32 alkyl, by reducing the nitrile group of a compound of the formula
14
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
CN
A
M~N~ 1 NY
R N~A3~N
Y Y
(where Al, each A~ (if present), and A3 are independently selected from
C1-Cg alkyl; where each Y is independently selected from Cl-C4 alkyl; where M
is
selected from C1-C4 alkyl; where k is 0, 1, 2, or 3; and where R is selected
from
C1-C32 alkyl) to an amino group. A preferred reducing reagent is gaseous
hydrogen (H2) over Raney nickel.
[0026] The invention also provides methods of treating diseases
characterized by uncontrolled cell proliferation, such as cancer, especially
prostate
cancer, by administration of one or more of the compounds described above. The
invention also provides methods of depleting ATP, particularly in a cancerous
cell, by administration of one or more of the compounds described above. The
invention also includes compositions of one or more of the compounds described
above in combination with a pharmaceutically-acceptable carrier, or with
another
therapeutic agent.
BRIEF DESCRIPTION OF THE DRAWINGS
[0027] Fig. l is a graph showing the in vitro effect of increasing
concentration of SL-11174 on the growth of cultured human prostate cancer cell
DuPro.
[0028] Fig. 2 is a graph showing the in vitro effect of increasing
concentration of SL-11197 on the growth of cultured human prostate cancer cell
DuPro.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0029] Fig. 3 is a graph showing the in vitro effect of increasing
concentration of SL-I 1199 on the growth of cultured human prostrate cancer
cell
DuPro.
[0030] Fig. 4 is a graph showing the in vitro effect of increasing
concentration of SL-I 1200 on the growth of cultured human prostrate cancer
cell
DuPro.
[0031] Fig. 5 is a graph showing the ih vitro effect of increasing
concentration of SL-11208 on the growth of cultured human prostrate cancer
cell
DuPro.
[0032] Fig. 6 is a graph showing the effect of SL-11174 on the survival of
cultured human prostate cancer cell DuPro.
[0033] Fig. 7 is a graph showing the effect of SL-11197 on the survival. of
cultured human prostate cancer cell DuPro.
[0034] Fig. 8 is a graph showing the effect of SL-11199 on the survival of
cultured human prostate cancer cell DuPro.
[0035] Fig. 9 is a graph showing the effect of SL-11200 on the survival of
cultured human prostate cancer cell DuPro.
[0036] Fig.10 is a graph showing the effect of SL-11208 on the survival
of cultured human prostate cancer cell DuPro. ,
[0037] Fig. 1l depicts the effect of SL-11238 on DuPro cell growth.
[0038] Fig.12 depicts the effect of SL-11239 on DuPro cell growth.
[0039] Fig.13 depicts the effect of SL-11238 on survival of DuPro cells.
[0040] Fig.14 depicts the effect of SL-11239 on survival of DuPro cells.
[0041] Fig.15 depicts the in vitro effect of spermine (control) and
SL-11174 on ATP hydrolysis
[0042] Fig.16 depicts the in vitro effect of spermine (control) and
SL-11197 on ATP hydrolysis.
[0043] Fig.17 depicts the ire vitro effect of spermine (control) and
SL-11199 on ATP hydrolysis.
[0044] Fig.18 depicts the i~ vitro effect of spermine (control) and
SL-11200 on ATP hydrolysis.
16
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0045] Fig.19 depicts the in vitro effect of spermine (control) and
SL-11208 on ATP hydrolysis.
[0046] Fig. 20 depicts the in vitro effectlof SL-11238 on ATP hydrolysis.
[0047] Fig. 21 depicts the i~ vitro effect of SL-11239 on ATP hydrolysis.
[0048] Fig. 22 depicts the mean relative changes in luciferin/luciferase
activities and standard deviations in the presence of extracts of 50,000
cultured
human prostate tumor cells (DuPro) treated with varying concentrations of
SL-11174, SL-11197, SL-11199, SL-11200, and SL-11208. Standard deviations,
where not seen, are smaller than the symbol size.
[0049] Fig. 23 depicts the effect of SL-11238 on cellular ATP measured
by the luciferin/luciferase reaction.
[0050] Fig. 24 depicts the effect of SL-11239 on cellular ATP measured
by the luciferin/luciferase reaction.
BEST MODE FOR CARRYING OUT THE INVENTION
[0051] Reference is made throughout the Detailed Description to the
reaction Schedules and Tables included herein. For sake of clarity and
brevity,
reference numerals have been assigned to each chemical structure described.
These reference numerals are used consistently throughout the disclosure to
unambiguously designate the chemical entities discussed.
[0052] The invention includes alI salts of the compounds described herein.
Particularly preferred are pharmaceutically acceptable salts. Pharmaceutically
acceptable salts are those salts which retain the biological activity of the
free
bases and which are not biologically or otherwise undesirable. The desired
salt
may be prepared by methods known to those of skill in the art by treating the
polyamine with an acid. Examples of inorganic acids include, but are not
limited
to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and
phosphoric
acid. Examples of organic acids include, but are not limited to, formic acid,
acetic
acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malefic acid,
malonic
acid, succinic acid, fiunaric acid, tartaric acid, citric acid, benzoic acid,
cinnamic
17
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
acid, mandelic acid, sulfonic acids, and salicylic acid. Salts of the
polyamines
with amino acids, such as aspartate salts and glutamate salts, can also be
prepared.
[0053] The invention also includes all stereoisomers of the compounds,
including diastereomers and enantiomers, as well as mixtures of stereoisomers,
including, but not limited to, racemic mixtures. Unless stereochemistry is
explicitly indicated in a structure, the structure is intended to embrace all
possible
stereoisomers of the compound depicted.
[0054] The term "alkyl" refers to saturated aliphatic groups including
straight-chain, branched-chain, cyclic groups, and combinations thereof,
having
the number of carbon atoms specified, or if no number is specified, having up
to
12 carbon atoms. Examples of alkyl groups include, but are not limited to,
groups
such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-
butyl, n-
pentyl, neopentyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and
adamantyl. Cyclic groups can consist of one ring, including, but not limited
to,
groups such as cycloheptyl, or multiple fused rings, including, but not
limited to,
groups such as adamantyl or norbornyl. Alkyl groups may be unsubstituted, or
may be substituted with one or more substituents including, but not limited
to,
groups such as halogen (fluoro, chloro, bromo, and iodo), alkoxy, acyloxy,
amino,
hydroxyl, mercapto, carboxy, benzyloxy, phenyl, benzyl, cyano, vitro,
thioalkoxy,
carboxaldehyde, carboalkoxy and carboxamide, or a functionality that can be
suitably blocked, if necessary for purposes of the invention, with a
protecting
group. Examples of substituted alkyl groups include, but are not limited to, -
CF3,
-CF2-CF3, and other perfluoro and perhalo groups.
[0055] The term "alkenyl" refers to unsaturated aliphatic groups including
straight-chain, branched-chain, cyclic groups, and combinations thereof,
having
the number of carbon atoms specified, or if no number is specified, having up
to
12 carbon atoms, which contain at least one double bond (-C=C-). Examples of
alkenyl groups include, but are not limited to, -CH2-CH=CH-CH3 and
-CH2-CHZ-cyclohexenyl, there the ethyl group can be attached to the
cyclohexenyl moiety at any available carbon valence. The term "alkynyl" refers
to unsaturated aliphatic groups including straight-chain, branched-chain,
cyclic
18
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
groups, and combinations thereof, having the number of carbon atoms specified,
or if no number is specified, having up to 12 carbon atoms, which contain at
least
one triple bond ( -C---C-). "Hydrocarbon chain" or "hydrocarbyl" refers to any
combination of straight-chain, branched-chain, or cyclic alkyl, alkenyl, or
alkynyl
groups, and any combination thereof. "Substituted alkenyl," "substituted
alkynyl," and "substituted hydrocarbon chain" or "substituted hydrocarbyl"
refer
to the respective group substituted with one or more substituents, including,
but
not limited to, groups such as halogen, alkoxy, acyloxy, amino, hydroxyl,
mercapto, carboxy, benzyloxy, phenyl, benzyl, cyano, nitro, thioalkoxy,
carboxaldehyde, carboalkoxy and carboxamide, or a functionality that can be
suitably blocked, if necessary for purposes of the invention, with a
protecting
group.
[0056] "Aryl" or "Ar" refers to an aromatic carbocyclic group having a
single ring (including, but not limited to, groups such as phenyl) or multiple
condensed rings (including, but not limited to, groups such as naphthyl or
anthryl), and includes both unsubstituted and substituted aryl groups.
Substituted
aryls can be substituted with one or more substituents, including, but not
limited
to, groups such as alkyl, alkenyl, alkynyl, hydrocarbon chains, halogen,
alkoxy,
acyloxy, amino, hydroxyl, mercapto, carboxy, benzyloxy, phenyl, benzyl, cyano,
nitro, thioalkoxy, carboxaldehyde, carboalkoxy and carboxamide, or a
functionality that can be suitably blocked, if necessary for purposes of the
invention, with a protecting group.
[0057] "Heteroalkyl," "heteroalkenyl," and "heteroallcynyl" refer to alkyl,
alkenyl, and alkynyl groups, respectively, that contain the number of carbon
atoms specified (or if no number is specified, having up to 12 carbon atoms)
which contain one or more heteroatoms as part of the main, branched, or cyclic
chains in the group. Heteroatoms include, but are not limited to, N, S, O, and
P;
N and O are preferred. Heteroalkyl, heteroalkenyl, and heteroalkynyl groups
may
be attached to the remainder of the molecule either at a heteroatom (if a
valence is
available) or at a carbon atom. Examples of heteroalkyl groups include, but
are
19
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
not limited to, groups such as -O-CH3, -CH2-O-CH3, -CHa-CH2-O-CH3,
-S-CH2-CH2-CH3, -CH2-CH(CH3)-S-CH3, -CH2-CH2-NH-CH2-CH2-,1-ethyl-6-
propylpiperidino, 2-ethylthiophenyl, and morpholino. Examples of heteroalkenyl
groups include, but are not limited to, groups such as
-CH=CH-NH-CH(CH3)-CH2-. "Heteroaryl" or "HetAr" refers to an aromatic
carbocyclic group having a single ring (including, but not limited to,
examples
such as pyridyl, thiophene, or furyl) or multiple condensed rings (including,
but
not limited to, examples such as imidazolyl, indolizinyl or benzothienyl) and
having at least one hetero atom, including, but not limited to, heteroatoms
such as
N, O, P, or S, within the ring. Heteroalkyl, heteroalkenyl, heteroalkynyl and
heteroaryl groups can be unsubstituted or substituted with one or more
substituents, including, but not limited to, groups such as alkyl, alkenyl,
alkynyl,
benzyl, hydrocarbon chains, halogen, alkoxy, acyloxy, amino, hydroxyl,
mercapto, carboxy, benzyloxy, phenyl, benzyl, cyano, nitro, thioalkoxy,
carboxaldehyde, carboalkoxy and carboxamide, or a functionality that can be
suitably blocked, if necessary for purposes of the invention, with a
protecting
group. Examples of such substituted heteroalkyl groups include, but are not
limited to, piperazine, substituted at a nitrogen or carbon by a phenyl or
benzyl
group, and attached to the remainder of the molecule by any available valence
on
a carbon or nitrogen, -NH-S02-phenyl, -NH-(C=O)O-alkyl,
-NH-(C=O)O-alkyl-aryl, and -NH-(C=O)-alkyl. The heteroatom(s) as well as the
carbon atoms of the group can be substituted. The heteroatom(s) can also be in
oxidized form. Unless otherwise specified, heteroalkyl, heteroalkenyl,
heteroalkynyl, and heteroaryl groups have between one and five heteroatoms and
between one and twenty carbon atoms.
[0058] The term "alkylaryl" refers to an alkyl group having the number of
carbon atoms designated, appended to one, two, or three aryl groups.
[0059] The term "allcoxy" as used herein refers to an alkyl, alkenyl,
alkynyl, or hydrocarbon chain linked to an oxygen atom and having the number
of
carbon atoms specified, or if no number is specified, having up to 12 carbon
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
atoms. Examples of alkoxy groups include, but are not limited to, groups such
as
methoxy, ethoxy, and t-butoxy.
[0060] The term "alkanoate" as used herein refers to an ionized carboxylic
acid group, such as acetate (CH3C(=O)-O~-1~), propionate (CH3CH2C(=O)-O~-1~),
and the like. "Alkyl alkanoate" refers to a carboxylic acid esterified with an
alkoxy group, such as ethyl acetate (CH3C(=O)-O-CH2CH3). "w-haloalkyl
alkanoate" refers to an alkyl alkanoate bearing a halogen atom on the
alkanoate
carbon atom furthest from the carboxyl group; thus, ethyl w-bromo propionate
refers to ethyl 3-bromopropionate, methyl w-chloro n-butanoate refers to
methyl
4-chloro n-butanoate, etc.
[0061] The terms "halo" and "halogen" as used herein refer to Cl, Br, F or
I substituents.
[0062] "Protecting group" refers to a chemical group that exhibits the
following characteristics: 1) reacts selectively with the desired
functionality in
good yield to give a protected substrate that is stable to the projected
reactions for
which protection is desired; 2) is selectively removable from the protected
substrate to yield the desired functionality; and 3) is removable in good
yield by
reagents compatible with the other functional groups) present or generated in
such projected reactions. Examples of suitable protecting groups can be found
in
Greene et al. (1991) Protective Groups in Organic Synthesis, 2nd Ed. (John
Wiley
& Sons, Inc., New York). Preferred amino protecting groups include, but are
not
limited to, benzyloxycarbonyl (CBz), t-butyloxycarbonyl (Boc), t-
butyldimethylsilyl (TBDIMS), 9-fluorenylmethyloxycarbonyl (Fmoc), tosyl,
benzenesulfonyl, 2-pyridyl sulfonyl, or suitable photolabile protecting groups
such as 6-nitroveratryloxy carbonyl (Nvoc), nitropiperonyl,
pyrenylmethoxycarbonyl, nitrobenzyl, dimethyl dimethoxybenzil, 5-bromo-7-
nitroindolinyl, and the like. Preferred hydroxyl protecting groups include
Fmoc,
TBDIMS, photolabile protecting groups (such as nitroveratryl oxymethyl ether
(Nvom)), Mom (methoxy methyl ether), and Mem (methoxy ethoxy methyl
ether). Particularly preferred protecting groups include NPEOC (4-
21
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
nitrophenethyloxycarbonyl) and NPEOM
(4-nitrophenethyloxymethyloxycarbonyl).
[0063] The terms "peptide," "polypeptide", "polypeptide moiety",
"protein", and the like are used interchangeably herein to refer to any
polymer of
amino acid residues of any length, i.e., polymers of two or more amino acids.
The
polymer can be linear or non-linear (e.g., branched), it can comprise modified
amino acids or amino acid analogs, and it can be interrupted by chemical
moieties
other than amino acids. The terms also encompass an amino acid polymer that
has been modified naturally or by intervention; for example, by disulfide bond
formation, glycosylation, lipidation, acetylation, phosphorylation, or any
other
manipulation or modification, such as conjugation with a labeling or bioactive
component. Amino acids include the twenty encoded amino acids (including
proline, an imino acid), other alpha-amino acids, and other natural and
artificial
amino acids such as p-iodotyrosine and beta-alanine.
Cyclic polyamine analogs: Synthetic approach.
[0064] Cyclic polyamine derivatives that can affect the hydrolysis of ATP
ih vivo are constructed by condensing spermine (spm), its isomers and its
higher
and lower homologues, as well as spermidine (spd) and its isomers and higher
and
lower homologues, with an a, (3-unsaturated fatty acid chain (Scheme 1).
22
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
H
H2N N
H2N H
spm
O
~N N
H H
N N
H H
Scheme 1
[0065] Although Scheme 1 is very likely the pathway for biogenesis of
cyclic polyamines (e.g., pithecolobines and budmunchiamines), the practical
synthetic approach follows a different route. The latter is depicted in Scheme
2
for several analogs of these series.
[0066] By reaction of ethyl bromoacetate 1 with triphenylphosphine the
Wittig salt 2 was obtained. By condensation of 2 with aliphatic aldehydes 3a-
3c
following procedures of the Wittig reaction, the a, (3-unsaturated esters 4a-
4c
were obtained in ca. 90% yield. By reaction of 4a-4c with spermine (or a
spermine analog), one equivalent of the base adds to the double bond by its
primary amino group and the amino esters Sa-Sc are obtained in ca. 40% yield.
Lactamization of 5a-5c to 6a-6c was achieved using antimony (III) ethoxide in
76% yield. Finally if N-methylation of the secondary amino residues of 6a-6c
is
desired, it can be achieved by a reductive alkylation reaction using
formaldehyde
and sodium cyanoborohydride to give 7a-7c. Yields for this reaction are ca.
80%.
23
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
N-alkylation with homologues of formaldehyde will give the higher homologues
of 7a-7c.
O O O
~OEt a) 'OEt H R
3a R = propyl
Br 1 P2 hs Br 3b R = methyl
3c R = pentyl
b)
i R = propyl
~ R = methyl
R = pentyl
c) R
5a R = propyl
NON N~/~/NH2 5b R = methyl
H H 5c R = pentyl
R
ie
H H e) 'H
HRH ~ N~N~
Me Me
R
6a R = propyl 7a R = propyl (Budmunchiamine A)
6b R = methyl 7b R = methyl (Budmunchiamine B)
6c R = pentyl 7c R = pentyl (Budmunchiamine C)
Scheme 2
[0067] The conditions and reagents used in Scheme 2 are as follows: a)
PPh3, toluene, 2h, 80°C (94% yield); b) NaOEt, 10°C followed
by warming to
room temperature (88% yield); c) spermine, 40°C (43% yield); d)
Sb(OEt)3,
benzene, reflux (76% yield); e) 1. formalin 37%, acetic acid, 0°C
followed by
warming to room temperature; 2. NaCNBH3, room temperature (83% yield).
24
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0068] In general, synthesis of compounds of the invention proceeds by
reacting a haloalkyl alkanoate, preferably an c~-haloalkyl alkanoate, with
triphenylphosphine to give a phosphonium salt. The phosphonium salt is
condensed with an aldehyde or ketone-containing compound, preferably an
aldehyde-containing compound, to give an a,[i-unsaturated alkenyl alkanoate
following the general reaction protocol of the Wittig reaction. Addition of a
polyamine containing at least two primary amino groups across the double bond
yields a (3-aminoalkyl alkanoate, where one of the primary amino groups has
added to the double bond and the other amino group remains free. Condensation
of the free amino group with the ester function gives the cyclic compound.
Derivatization of secondary amino groups, if present in the cycle, can then be
carried out if desired. When amino groups of polyamines are connected by
straight-chain alkyl groups, it can be readily appreciated that by varying the
length
of the alkyl groups and by varying the number of amino groups in the
polyamine,
different ring sizes can be constructed upon condensation of the polyamine to
give
the cyclic compound.
[0069] An alternate method to synthesize compounds of the invention is
depicted in Scheme 3, where reagent 4c is as depicted in Scheme 2.
2,5
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
n
H N N~NH
2
4c
EtOOC
~CHz)~2CHa
COOEt
9
N N~NH
H
MeOH/HCI
Me
H
N NH2
N
H
B(NMe~)3/xylene
O
~ N'
H NH 91
N
H
Scheme 3
[0070] As can be readily appreciated, the synthesis following Scheme 3
utilizes a compound (8) comprising a primary amino group and a
hexahydropyrimidine moiety. The hexahydropyrimidine moiety can be
considered a protected form of 1,3-diaminopropane; the methylene bridge
between the two nitrogens in the hexahydropyrimidine ring is readily cleaved
to
yield the free amino groups. The portion of the molecule containing the free
26
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
primary amino group is attached to one of the hexahydropyrimidine nitrogens;
the
primary amine can be linked to the hexahydropyrimidine nitrogen by any linker
arm. Preferably the linker arm contains at Least one carbon atom. The Linker
arm
can be of the form -A3-(NY-A2)~-, where each A2 (if present) and A3 are
independently selected from Cl-C8 alkyl, where each Y is independently
selected
from H or C1-C4 alkyl, and where j is 0, l, 2, or 3; this compound is
represented
by the structural formula
I
H2N-A3-(NY-A2)~ N~NH
[0071] More preferably, the linker arm is -CH2CH2CH2- as in compound 8
of Scheme 3. Hexahydropyrimidine 8 is readily prepared from spermidine and
formalin (see Chantrapromma, K. et al., "The chemistry of naturally occurring
polyamines. 2. A total synthesis of thermospermine," Tetr. Lett. (1980),
21 (26):2475-6). Addition of the primary amino group across a -C=C- bond
converts the free primary amino group into a secondary amino group. In Scheme
3, the primary amino group adds across the alkene bond of an a,(3-unsaturated
ester compound. The primidine ring can then be opened, releasing a second
primary amino group, which can be condensed with the ester function of the
molecule. Examples 14-16 detail the experimental conditions used in the
synthesis of Scheme 3.
[0072] Using the synthetic methods described herein, the following
compounds listed in Table 1 were synthesized.
27
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Table 1 Synthesized Compounds
Compound Structure MW
No.
SL-11174 0 547
N~N
H H
~3 HCI
NON
H H
SL-11197 0 424
N~NH
H
~3 HCI
NON
H H
SL-11199 0 562
N~N
H H
~3 HCI
NON
H ' H
SL-11200 0 627
H~H
N'~N~NH
H H ~4 HCI
SL-11208 0 454
N~NH
NH ~2 HCI
R
R= n-C~sHz7
SL-11238 ie 612
'N N
H
~4 HCI
C13H27 N N
1e 1e
28
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
SL-11239 I I 706
~5 HCI
C~sH2~ ~ / ' i
Me Me
Reduction of Compounds and Further Derivatization
[0073] The compounds above can be readily reduced with hydride
reagents, such as lithium aluminum hydride and other reducing agents known in
the art, to convert the amide function into a secondary amine. For the cyclic
polyamine compounds containing an amide group where the non-amide nitrogens,
i.e., the amino groups, are alkylated, the amide group will be reduced to a
secondary amino group, while the other nitrogens will be present as tertiary
amino
groups, and this difference can be exploited to perform further chemistry at
the
secondary amino group. The reduction is illustrated in Scheme 4, where Yaik
indicates an alkyl group (e.g., excluding hydrogen). (The reduction can, of
course, be performed where the substituents on the non-amide nitrogens are
hydrogen. In subsequent steps this will lead to derivatization of all
(secondary)
nitrogens in the compound, as opposed to the schemes outlined below, where
only
the nitrogen originally present as an amide was derivatized.)
29
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
alk
~N N
H
R N N
Yalk Yalk
LAH
alk
~N N
H
R N N
Yalk Yalk
Scheme 4
[0074] The resulting secondary amine can then be reacted with a
compound such as acrylonitrile to derivatize the secondary amine, as outlined
in
Scheme 5. Alternatively, the resulting secondary amine can then be reacted
with
a compound such as an cu-haloalkyl nitrile, for example, but not limited to,
where
the alkyl group is a Cl-C8 alkyl group and the halogen is iodo or bromo.
Alternatively, the secondary amine can be acylated with an acyl group
(-C(=O)-C1-C8 alkyl), or an amino acid or peptide can be coupled directly to
the
secondary amine. In the event of acylation with a group of the formula
(-C(=O)-C1-C8 alkyl), the acyl group can be reduced with lithium aluminum
hydride or other organometallic agents to form an alkyl group. An omega-cyano
acyl group can also be introduced (i.e., -C(=O)-C1-C8 alkyl-CN), which, upon
reduction by lithium aluminum hydride, will yield a group of the form -CH2-C1-
C8 alkyl-CH2NH2. Alternatively, the secondary amine can be alkylated by alkyl
halides in the presence of bases.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
alk
~N N
H
R N N
Yalk Yalk
~CN
CN
alk
~N N
R N N
Yalk Yalk
Scheme 5
[0075] Reduction of the cyano group to a primary amino group is then
conveniently performed by using Raney-Ni reagent under hydrogen.
[0076] The free primary amino group thus produced can be derivatized in
various manners. One such manner is to use it as the starting point for
peptide
synthesis, by coupling the free acid group of an N-protected amino acid to the
primary amino group of the aminoalkylcyclopropylamine. Various methods of
coupling amino acids or peptides are known in the art. The polypeptides can be
produced by recombinant methods (i.e., single or fusion polypeptides) or by
chemical synthesis. Polypeptides, especially shorter polypeptides up to about
50
amino acids, are conveniently made by chemical synthesis, such as the Fmoc or
Boc synthesis methods. See, for example, Atherton and Sheppard, Solid Phase
31
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Peptide Synthesis: A Practical Approach, New York: IRL Press, 1989; Stewart
and Young: Solid-Phase Peptide Synthesis 2nd Ed., Rockford, Illinois: Pierce
Chemical Co., 1984; and Jones, The Chemical Synthesis of Peptides, Oxford:
Clarendon Press, 1994. The polypeptides can be produced by an automated
polypeptide synthesizer employing the solid phase method, such as those sold
by
Perkin Elmer-Applied Biosystems, Foster City, California, or can be made in
solution by methods known in the art.
[0077] Synthesis of peptides by repetitive coupling of amino acids to the
primary amine of the aminoalkylated cyclic polyamine is readily performed by
solution-phase peptide synthesis techniques, such as those extensively
discussed
in Bodanszky, M., Principles of Peptide Synthesis, 2nd Edition, Springer-
Verlag:
Berlin, 1993; Bodanszky, M., Peptide Chemistry: A Practical Textbook, 2nd
edition, Springer-Verlag: Berlin, 1993, and Bodanszky, M., Bodanszky, A., The
Practice of Peptide Synthesis, Springer-Verlag: Berlin, 1984, and other
techniques well-known in the art. Peptides can also be attached to the cyclic
polyamines by coupling of small protected peptide fragments, using the widely-
known techniques for fragment condensation methods in peptide synthesis.
Individual amino acids, such as leucine, can also be coupled to the cyclic
polyamimes simply by stopping the peptide synthesis procedure after attachment
of the first amino acid. Longer peptides, such as acetyl-Ser-Lys-Leu-Gln-Leu-,
can be attached to the cyclic polyamines by either stepwise synthesis or
fragment
coupling methods.
[0078] By such peptide synthetic methods, the following compound:
N H-Le u-G I n-Leu-Lys-Se r-acetyl
Me
'N N
C13H27
Me Me
32
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
(SL-11243) was synthesized. The peptide sequence, from N-terminus to
C-terminus of the peptide in SL-11243, is acetyl-Ser-Lys-Leu-Gln-Leu-, where
the C-terminal leucine is coupled to the (formerly) primary amino group of the
cyclopolyamine compound. Peptides of interest for use in the peptide-
derivatized
compounds described above include peptides which are substrates of prostate
specific antigen (PSA) or cathepsin B. Peptides of length 25 amino acids or
less,
or 10 amino acids or less, can be used. Examples of such sequences cleaved by
PSA are HSSKLQ, SKL,Q-(3-alanine,SI~LQL, or SKLQ, with or without N-
terminal protecting or capping groups such as Boc, Fmoc, acetyl, or other acyl
capping groups, and with or without side-chain protecting groups (such as
carbobenzyloxycarbonyl, Boc or Fmoc on the s-amino group of lysine).
[0079] Examples of polypeptides recognized and cleaved by cathepsin B
include the peptide sequence Zl-Pa-P1-, where Zl is hydrogen, an amino-
protecting group, or an amino-capping group attached to the N-terminus of Pa;
where P2 is the N-terminal amino acid and P1 is the C-terminal amino acid; and
where P2 is a hydrophobic amino acid and P~ is a basic or polax amino acid. In
another embodiment, the peptide sequence is Zl-P2-Pi-Y-, where Zl is hydrogen,
an amino-protecting group, or an amino-capping group attached to the N-
terminus
of P~; P2 is a hydrophobic amino acid; P1 is a basic or polar amino acid; and
where
Y is leucine, (3-alanine, or a nonentity. In a further embodiment, Zl is a 4-
morpholinocarbonyl group. In yet another embodiment, PZ is selected from the
group consisting of leucine, isoleucine, valine, methionine, and
phenylalanine;
and P1 is selected from the group consisting of lysine, arginine, glutamine,
asparagine, histidine and citrulline.
Utility of Cyclic Polyamihes as Ahti-neoplastic Agev~ts.
[0080] To assess the utility of the subject compounds in the treatment of
neoplastic cell growth, the ability of the compounds to inhibit the ire vitro
growth
characteristics of several commonly used cancer models were studied. For
instance, the subject polyamines induce cell growth inhibitions in several
cultured
33
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
human prostate tumor cell lines such as LnCap, DuPro, and PC-3 as determined
by an accepted MTT assay (Table 2). All three cell lines are sensitive to the
cyclic polyamines, with IDSO values ranging from 500 nM to 2,600 nM. The
results with the DuPro cell line are given; these results are representative
of the
results with human prostate cell lines. The cyclic polyamines of the present
invention have been shown to inhibit cell growth and to cause cell death in
accepted in vitro test cultures of human prostate cancer cell lines as shown
in
Figs. 1-14. The figures are described in detail in the Example section below.
The
uptake of the cyclic polyamines by the DuPro cells and the resultant changes
in
the cellular polyamine levels are shown in Tables 3a and 3b.
[0081] The hydrolysis of ATP is believed to be one of the probable causes
of cell kill. In a standardized method for measuring acid hydrolysis of ATP, a
marked increase was observed in ATP hydrolysis in the presence of the cyclic
polyamines (see Figs. 15-21) as compared to the lack of ATP hydrolysis in the
presence of the naturally occurring linear polyamine spermine. The cyclic
polyamines were also found to hydrolyze ATP in vivo in the cancer cells (Fig.
22-
24). The concentration of cyclic polyamines required to hydrolyze
intracellular
ATP in the cancer cells parallels the concentrations at which they produce
cell kill
(Figs. 6-14), lending support to the hypothesis that cell kill is due to
intracellular
ATP depletion. However, the invention is not to be construed as limited by any
particular theory of biological or therapeutic activity.
Therapeutic use of polyamihe analogs
[0082] Polyamine analogs of the present invention are likely to be useful
for treatment of a variety of diseases caused by uncontrolled proliferation of
cells,
including cancer, particularly prostate cancer and other cancer cell lines.
The
analogs are used to treat mammals, preferably humans. "Treating" a disease
using
a cyclic polyamine of the invention is defined as administering one or more
cyclic
polyamines of the invention, with or without additional therapeutic agents, in
order to prevent, reduce, or eliminate either the disease or the symptoms of
the
disease, or to retard the progression of the disease or of symptoms of the
disease.
34
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
"Therapeutic use" of the cyclic polyamines of the invention is defined as
using
one or more cyclic polyamines of the invention to treat a disease, as defined
above.
[0083] In order to evaluate the efficacy of a particular novel cyclic
polyamine for a particular medicinal application, the compounds can be first
tested against appropriately chosen test cells in vitro. In a non-limiting
example,
polyamine analogs can be tested against tumor cells, for example, prostate
tumor
cells. Exemplary experiments can utilize cell lines capable of growing in
culture
as well as in vivo in athymic nude mice, such as LNCaP. Horoszewicz et al.
(1983) Cancer Res. 43:1809-1818. Culturing and treatment of carcinoma cell
lines, cell cycle and cell death determinations based on flow cytometry;
enzyme
assays including ODC, SAMDC and SSAT activities; and high pressure liquid
chromatography detection and quantitation of natural polyamines and polyamine
analogs are described in the art, for example, Mi et al. (1998) Prostate 34:51-
60;
Kramer et al. (1997) Cancer Res. 57:5521-27; and Kramer et al. (1995) J. Biol.
Chem. 270:2124-2132. Evaluations can also be made of the effects of the novel
cyclic polyamine analog on cell growth and metabolism.
[0084] Analysis begins with ICSO determinations based on dose-response
curves ranging from 0.1 to 1000 ~,M performed at 72 hr. From these studies,
conditions can be defined which produce about 50% growth inhibition and used
to: (a) follow time-dependence of growth inhibition for up to 6 days, with
particular attention to decreases in cell number, which may indicate drug-
induced
cell death; (b) characterize analog effects on cell cycle progression and cell
death
using flow cytometry (analysis to be performed on attached and detached
cells);
(c) examine analog effects on cellular metabolic parameters. Analog effects
can
be normalized to intracellular concentrations (by HPLC analysis), which also
provide an indication of their relative ability to penetrate cells. Marked
differences in analog uptake can be further characterized by studying analog
ability to utilize and regulate the polyamine transporter, as assessed by
competition studies using radiolabeled spermidine, as previously described in
Mi
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
et al. (1998). Cyclic polyamines could also enter the cells by a diffusion
mechanism.
In vivo testing of cyclic polyamine analogs
[0085] Analogs found to have potent anti-proliferative activity i~c vitro
towards cultured carcinoma cells can be evaluated in in vivo model systems.
The
first goal is to determine the relative toxicity of the analogs in non-tumor-
bearing
animals, such as DBA/2 mice. Groups of three animals each can be injected
intraperitoneally with increasing concentrations of an analog, beginning at,
for
example, 10 mg/kg. Toxicity as indicated by morbidity is closely monitored
over
the first 24 hr. A well-characterized polyamine analog, such as BE-333, can be
used as an internal standard in these studies, since a data base has already
been
established regarding acute toxicity via a single dose treatment relative to
chronic
toxicity via a daily x 5 d schedule. Thus, in the case of new analogs, single
dose
toxicity relative to BE-333 is used to project the range of doses to be used
on a
daily x 5 d schedule.
[0086] After the highest tolerated dosage on a daily x 5 d schedule is
deduced, antitumor activity is determined. Typically, tumors can be
subcutaneously implanted into nude athymic mice by trocar and allowed to reach
100-200 mm3 before initiating treatment by intraperitoneal injection daily x 5
d.
Most analogs can be given in a range between 10 and 200 mg/kg. Analogs can be
evaluated at three treatment dosages with 10-15 animals per group (a minimum
of
three from each can be used for pharmacodynamic studies, described below). .
Mice can be monitored and weighed twice weekly to determine tumor size and
toxicity. Tumor size is determined by multi-directional measurement from which
volume in mm3 is calculated. Tumors can be followed until median tumor volume
of each group reaches 1500 mm3 (i.e., 20% of body weight), at which time the
animals can be sacrificed. Although the initial anti-tumor studies focuses on
a
daily x 5 d schedule, constant infusion can be performed via Alzet pump
delivery
for 5 days since this schedule dramatically improves the anti-tumor activity
of
BE-333 against A549 human large cell hung carcinoma. Sharma et al. (1997)
36
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Clih. Cancer Res. 3:1239-1244. In addition to assessing anti-tumor activity,
free
analog levels in tumor and normal tissues can be determined in test animals.
Methods of administration of cyclic polyamihe analogs
[0087] The polyamine analogs of the present invention can be
administered to a mammalian, preferably human, subject via any route known in
the art, including, but not limited to, those disclosed herein. Preferably
administration of the novel polyamine analogs is intravenous. Other methods of
administration include but are not limited to, oral, intrarterial,
intratumoral,
intramuscular, topical, inhalation, subcutaneous, intraperitoneal,
gastrointestinal,
and directly to a specific or affected organ. The novel polyamine analogs
described herein are administratable in the form of tablets, pills, powder
mixtures,
capsules, granules, injectables, creams, solutions, suppositories, emulsions,
dispersions, food premixes, and in other suitable forms. The compounds can
also
be administered in liposome formulations. The compounds can also be
administered as prodrugs, where the prodrug undergoes transformation in the
treated subject to a form which is therapeutically effective. Additional
methods of
administration are known in the art.
[0088] The pharmaceutical dosage form which contains the compounds
described herein is conveniently admixed with a non-toxic pharmaceutical
organic
carrier or a non-toxic pharmaceutical inorganic carrier. Typical
pharmaceutically-
acceptable carriers include, for example, mannitol, urea, dextrans, lactose,
potato
and maize starches, magnesium stearate, talc, vegetable oils, polyalkylene
glycols,
ethyl cellulose, poly(vinylpyrrolidone), calcium carbonate, ethyl oleate,
isopropyl
myristate, benzyl benzoate, sodium carbonate, gelatin, potassium carbonate,
silicic acid, and other conventionally employed acceptable carriers. The
pharmaceutical dosage form can also contain non-toxic auxiliary substances
such
as emulsifying, preserving, or wetting agents, and the like. A suitable
carrier is
one which does not cause an intolerable side effect, but which allows the
novel
cyclic polyamine analogs) to retain its pharmacological activity in the body.
Formulations for parenteral and nonparenteral drug delivery are known in the
art
37
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
and are set forth in Remihgtoh's Pharmaceutical Sciences, 18th Edition, Mack
Publishing (1990). Solid forms, such as tablets, capsules and powders, can be
fabricated using conventional tableting and capsule-filling machinery, which
is
well known in the art. Solid dosage forms, including tablets and capsules for
oral
administration in unit dose presentation form, can contain any number of
additional non-active ingredients known to the art, including such
conventional
additives as excipients; dessicants; colorants; binding agents, for
example.syrup,
acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrollidone; fillers, for
example
lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine;
tabletting
lubricants, for example magnesium stearate, talc, polyethylene glycol or
silica;
disintegrants, for example potato starch; or acceptable wetting agents such as
sodium lauryl sulphate. The tablets can be coated according to methods well
known in standard pharmaceutical practice. Liquid forms for ingestion can be
formulated using known liquid carriers, including aqueous and non-aqueous
carriers, suspensions, oil-in-water and/or water-in-oil emulsions, and the
like.
Liquid formulations can also contain any number of additional non-active
ingredients, including colorants, fragrance, flavorings, viscosity modifiers,
preservatives, stabilizers, and the like. For parenteral administration, novel
cyclic
polyamine analogs can be administered as injectable dosages of a solution or
suspension of the compound in a physiologically acceptable diluent or sterile
liquid carrier such as water or oil, with or without additional surfactants or
adjuvants. An illustrative list of carrier oils would include animal and
vegetable
oils (peanut oil, soy bean oil), petroleum-derived oils (mineral oil), and
synthetic
oils. In general, for injectable unit doses, water, saline, aqueous dextrose
and
related sugar solutions, and ethanol and glycol solutions such as propylene
glycol
or polyethylene glycol are preferred liquid carriers. The pharmaceutical unit
dosage chosen is preferably fabricated and administered to provide a final
concentration of drug at the point of contact with the cancer cell of from 1
~M to
mM. More preferred is a concentration of from 1 to 100 pM. The optimal
effective concentration of novel cyclic polyamine analogs can be determined
empirically and will depend on the type and severity of the disease, route of
38
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
administration, disease progression and health and mass or body area of the
patient. Such determinations are within the skill of one in the art. Cyclic
polyamine analogs can be administered as the sole active ingredient, or can be
administered in combination with another active ingredient, including, but not
limited to, cytotoxic agents, antibiotics, antimetabolites, nitrosourea, vinca
allcaloids, polypeptides, antibodies, cytokines, etc.
EXAMPLES
Chemical Synthesis Examples
(0089] The following examples are illustrative of the manufacture of
several compounds according to the present invention, and axe not intended to
limit the invention disclosed and claimed herein in any fashion. The Examples
are included herein solely to aid in a more complete understanding of the
present
invention. Reference numerals 1-11 refer to compounds in Reaction Schemes 2
and 3 described above. Reference numbers 12-14 refer to compounds shown in
the Examples and so labeled.
[0090] All commercially available reagents were used without further
purification. All reactions were followed by TLC (silica gel F264 precoated,
Merck); column chromatography was carried out with silica gel (Merck 60,
0.040-0.063 mesh). The detection was performed either with UV light or the
following reagents: I~Mn04 soln. (1:l mixture of 1% aq. KMnO4 soln. and 5% aq.
Na2C03 soln.); Schlittler reagent (iodine platinate) (1 g H2PtCI6 in 6 ml HaO,
20
ml 1N HCI and 25.5 g KI in 225 ml HZO diluted to 1 L) for amides and amines.
IR measurements are presented in units of [cm 1] and were recorded on a
Perkin-Elmer 781 instrument. NMR spectra were recorded on Broker-300 or
Broker AMX-600 instruments with 8 in ppm and using the appropriate solvent as
internal standaxd. MS spectra were generated on Finnigan MAT SSO 700 or
Finnigan MAT 90 instruments using chemical ionization (CI) with NH3 and
39
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
electron impact (EI; 70 eV), and on a Finnigan TSQ 700 instrument using
electrospray ionisation (ESI).
[009I] Numerals included in the structure drawings denote atom numbers
for spectroscopic data which are not otherwise identified; e.g., the numbers l
and
2 for the compound 2 in Example 1 identify carbons 1 and 2 for the carbon
nuclear magnetic resonance assignments.
Example 1
(Ethoxycarbonylmethyl)triphenylphosphonium bromide (2).
O
1 2 ~OEt
PPh3+ Br
[0092] To a suspension of 22 g (84 mmol) triphenylphosphine in 200 ml
toluene were added 14 g (84 mmol) ethyl bromoacetate. The mixture was heated
2 h at 80.°C and stirred overnight at room temp. It was filtered,
washed with
toluene and the precipitated phosphonium bromide was dried 15 h at 10-s mbar
to
give 34 g (94%) 2 as colorless crystals. For analytical purposes, 300 mg were
recrystallized from (CHCl3/hexane l: 2) to give 292 mg.
Rf(CHC13/MeOH 9:1, UV2s4): 0.16.
mp.: 150-155° (CHC13/hexane 1 :2).
IR (CHCl3): 3360w, 2915s. 2705w, 2400w, 1725s, 1585w, 1335m, 1370w,
1305m, 1210m, 1110s, 1020w, 995w, 845w, 660m, 620w.
1H-NMR (CDCl3): 7.93-7.33 (m, 15 arom. H); 5.47 (d, J= 13.8, H2C(1));
4.02 (q, J= 7.1, OCH2CH3), 1.05 (t, J= 7.2, OCH2CH3).
i3C-NMR (CDC13):164.2 (s, C(2)); 135.1, 133.9, 133.7, 130.2, 130.1 (5d,
15 arom. C); 118.4, 117.2, (2s, 3 arom. C); 62.7 (t, OCH2CH3); 33.4, 32.7 (2t,
C(1)); 13.6 (q, OCH2CH3).
ESI-MS: 349 ([M - Br]+).
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Example 2
Ethyl 2-Tetradecenoate (4a).
[0093] To a soln. of NaOEt.prepared from 575 mg (25 mmol) of Na and
100 ml EtOH were added portionwise at 10° 10.7 g (25 mmol) 2 and
stirred 1 h at
room temp. After the addition of 4.37 g (23.75 mmol) laurinaldehyde 3 in 20 ml
CH2C12 and overnight stirring at room temp., the mixture was evaporated and
the'
crude product filtered through 50 g Si02 (Et20/hexane 1:2) to afford 5.1 g
(88%)
4a as an (E/~ mixture (~ 2:1). For analytical purpose, 450 mg of 4a were
purified by chromatography (Et20/hexane 2:98) to give 140 g (31 %) of the (~
isomer and 285 mg (64%) of the (E) isomer as a colorless oils.
Rf (Et2O/hexane 3:97, KPM): 0.67 (~ isomer, 0.47 (E) isomer.
IR (CHC13): 2920vs, 2850s, 1720s, 1640m, 1455m, 1415m, 1360m,
1295w, 1235m, I175s, I I 15s, 1030m, 925w, 820m, 660m, 620w.
1H-NMR (CDC13): (~ 6.21 (dt, J= 11.5, 5.7, HC(3)); 5.75 (d, J=11.5,
HC(2)); 4.16 (q, J= 7.2, OCH2CH3): 2.63 (q, J= 7.3, H2C(4)); 1.41 (t, J= 6.3,
H2C(5)); 1.30-1.25 (m, 8 CH2, OCHaCH3), 0.88 (t, 6.9, H3C(14)).
13C-NMR (CDC13): (~ 166.6 (s, C(1)); 150.5 (d, C(2)); 119.5 (d. C(3));
101.6 59.6 (t, OCH2CH3); 34.3 (t, C(4)); 31.8 (t, C(12)); 29.6 (t, C(5));
29.5, 29.4,
29.3, 29.2, 28.9, 23.4 (6t, 6 C); 22.5 (t, C(13)); 14.1 (q, OCH2CH3); 13.9 (q,
C(14)).
1H-NMR (CDCl3): (E) 6.94 (dt, J=15.6, 7.0, HC(3)); 5.80 (d, J=15.6,
HC(2)); 4.17 (q, J= 7.1, OCH2CH3); 2.19 (q, J, 7.0, H~C(4)), 1.44 (t, J= 7.2,
H2C(5)); 1.32-1.26 (m, 8 CH2, OCH2CH3); 0.87 (t, 6.9, H3C(14)).
41
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
13C-NMR (CDC13): (E) 166.8 (s, C(1)), 149.9 (d, C(2)); 121.2 (d, C(3));
60.1 (t, OCH2CH3); 32.2 (t, C(4)); 31.9 (t, C(12)); 29.6 (t, C(5)); 29.5,
29.4, 29.3,
29.1, 28.1. 23.5 (6t, 6C); 22.7 (t. C(13)); 14.3 (q, C(14)); 14.1 (q,
OCHaC'H3).
EI-MS: 254 (5, [M+ ~ ]), 209 (9, [M- OEt]+), 157 (18), 127 (46), 113 (27),
99 (47), 8I (37), 67 (24), 55 (58), 43 (100).
Example 3
Ethyl 16 Amino-3-undecyl-4, 8,13-triazahexadecanoate (5a).
7 H
N~NHZ
N 12 16
H lo'
[0094] A soln. of 3.49 g (13.7 mmol) 4a in 20 ml EtOH was added over a
period of 30 min to a stirred soln. of 2.77 g (13.7 mmol) spermine in 150 ml
EtOH
and the mixture heated for 3 d at 40°. Evaporation of the solvent and
chromatography of the residue over 100 g SiO2 (CH2C12/EtOH/NH40H 6:3:1)
gave 2.5 g (43%) of Sa as a colorless oil.
Rf (CHC13/MeOH/25% aq. NH4OH 7:4:1, Schlittler): 0.26.
IR (CHC13): 2920vs, 2850vs, 1720vs, 1580w, 1460s, 1370s, 1300m,
1180m, 1115s, 1025m, 920m, 885m, 845m, 655s, 620w.
iH-NMR (CDCl3): 4.12 (q, J= 7.1, OCHZCH3); 2.92 (quiht, J= 6.4,
HC(3)); 2.78 (t, J= 6.8, H2C(16)); 2.69 (t, J= 7.0, H2C(9), H2C(12)); 2.45 (t,
J=
6.8, H2C(5), H2C(7), H2C(14)); 2.38 (d, J= 6.3, H2C(2)); 2.19 (b~. s, NH,
NHa);
1.66 (quint, J= 6.9, HaC(6), H2C(15)); 1.54 (quint, J= 7.1, H2C(10), HaC(11));
1.28-1.23 (m, 10 CH2, OCH2CH3); 0.88 (t, 7.0, H3C(11')).
i3C-NMR (CDCI3): 172.5 (s, C(1)); 60.0 (t, OCHaCH3); 54.7 (d, C(3));
49.7 (t, C(12)); 48.2 (t, C(14)); 47.6 (t, C(9)); 45.2 (t, C(7)); 40.3 (t,
C(5)); 39.1 (t,
C(16)); 34.3 (t, C(2)); 33.5 (t, C(1')); 31.7 (t, C(9)); 30.3, 29.5, 29.4 (3t,
6 C); 29.1
(t, C(6), C(15)); 27.7 (t, C(2'), C(10)); 25.6 (t, C(11)); 22.4 (t, C(10'));
14.0 (q,
OCH2CH3); 13.9 (q, C(11')).
42
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
ESI-MS: 457 (28, [M+ 1]+), 229 (100, [M+ 2]2+).
Example 4
4-Uudecyl- l, 5, 9,14-tetr°aazacycloheptadecan-2-ohe (6a).
O
1~ 13
2~N N
H H
4
1. ~N
H H
lo'
[0095] A solution of 1.3 g (2.85 mmol) Sa in 180 ml dry benzene was
heated over molecular sieves for 2 h under reflux. After cooling to room
temp.,
950 mg (3.7 mmol) antimony(III) ethoxide in 10 ml benzene was added under an
argon atmosphere and the mixture was stirred for 16 h under reflux. The
mixture
was cooled at 10°, quenched with EtOH and evaporated. The residue was
purified
by chromatography (50 g SiOa, CH2C1~/EtOH/NH40H 15:4:1) to give 915 mg
(78%) of 6a as a colorless oil.
Rf(CHC13/MeOH/25% aq. NH4OH 7:4:1, Schlittler): 0.47.
IR (CHCl3): 3240w, 2920vs, 2850vs, 1640s, 1520m, 1460m, 1370m,
1220m, 1120m, 1045w, 925w, 805m, 660m, 620w.
1H-NMR (CDC13): 8.44 (b~. s, NH), 3.37 (t, J= 7.7, H2C(17)); 2.83 (quint,
J= 7.0, HC(4)); 2.76-2.72 (m, H2C(6), HaC(8)); 2.68 (t, J= 5.4, H2C(10),
H2C(13), H2C(15)); 2.37 (dd, J=15.2. 3.3, HaC(3)); 2.25 (br. s, NH); 2.14 (dd,
J
=15.3, 7.2, HbC(3)); 1.67 (quint, J= 6.1, H2C(7), H2C(16)); 1.59 (quint, J=
8.0,
HaC(11), H2C(12)); 1.41-1.25 (m, 10 CH2); 0.88 (t, J= 7.0, H3C(11)).
i3C-NMR (CDC13): 172.2 (s, C(1)); 55.4 (d, C(4)); 48.4 (t, C(13)); 48.2 (t,
C(15)); 48.0 (t, C(8)); 47.3 (t, C(10)); 45.7 (1, C(6)); 40.3 (t, C(3)); 37.6
(t,
C(17)); 34.0 (t, C(1')); 31.7 (t, C(9')); 29.6, 29.4 (2t, 6 C); 29.2 (t,
C(7)); 28.8 (t,
C(16)); 26.7 (t, C(2')); 26.6 (t, C(11)); 25.7 (t, C(12)): 22.5 (t, C(10'));
13.9 (q,
C(11')).
43
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
ESI-MS: 411 (44, [M+ 1]+), 206 (100, [M+ 2]2+).
Example 5
5, 9,14-Trimethyl-4-undecyl-l, 5, 9,14-tetraazacycloheptadecan-2-one
(Budmunchiamine A) (7a).
O
17 ~ a 13
2 ~N N
H
4 9
1, \N~ N\
Me Me lo'
[0096) A soln. of 90. mg (0.21 mmol) 6a and 3 ml formalin (37%) in 10 mI
AcOH was stirred at 0°. After 7 min, 250 mg (4 mmol) of NaCNBH3 in
1 ml
MeOH were added and the mixture was stirred overnight at room temp. After
cooling to 5°, the mixture was quenched with 2N HCl and the org.
solvent
evaporated. The residue was dissolved in 5 ml sat. aq. I~aC03 soln., extracted
with CH2Cl2 and dried over Na2SO4. After evaporation of the solvent and
chromatography of the residue (10 g Si02, CHC13/MeOH/25% aq. NH40H
90:10:0.7) 78 mg (83%) of 7a was obtained as a colorless oil.
Rf (CHC13/MeOH/25% aq. NH40H 85:14:1, Schlittler): 0.41.
IR (CHCl3): 3420m, 2920vs, 2850vs, 2800s, 1640vs, 1520s, 1460s, 1370m,
1230m, 1135m, 1050m, 920w, 845w, 680w, 655m.
1H-NMR (CDCl3): 8.55 (br. s, NH); 3.32 (dt, J= 6.6, 6.8, HZC(17)); 2.84
(quint, J= 4.7, HC(4)); 2.62 (dt, J= 12.3, 7.0, HaC(6)); 2.49-2.41 (m,
H2C(13));
2.41 (m, HbC(6)); 2.39 (m, H2C(8)); 2.41-2.32 (m, H2C(10)); 2.44-2.34 (m,
H2C(15)); 2.37 (m, HaC(3)); 2.24 (dd, J= 6.2, 1.6, HbC(3)); 2.27 (s, H3CN(9));
2.19 (s, H3CN(14), H3CN(5)); 1.67 (quint, J= 6.5, H2C(16)); 1.64 (quint, J=
6.4,
HaC(7)); 1.52 (quint, J= 6.7, H2C(11), H2C(12)); 1.30-1.17 (m, 10 CH2); 0.88
(t, J
= 6.9, H3C(11')).
44
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
13C-NMR (CDCl3): 172.5 (s, C(I)); 6I.I (d, C(4)); 56.3 (t, C(10)); 56.2 (t,
C(13)); 55.8 (t, C(15)); 54.5 (t C(8)); 51.5 (t, C(6)); 42.8 (q, H3CN(9));
42.3 (q,
H3CN(14)); 37.6 (t, C(17)); 37.0 (t, C(3)); 35.1 (q, H3CN(5)); 31.8 (t,
C(9')): 29.7,
29.6, 29.5, 29.4, 29.2 (St, 6 C); 27.5 (t C(16)); 27.3 (t, C(2')); 27.1 (t,
C(1')); 26.0
(t, C(7)); 24.4 (t, C(11)); 23.3 (t C(12)); 22.5 (t, C(10')); 13.9 (q,
C(11')).
ESI-MS: 453 (100, [M+ 1]+), 227 (80, [M+ 2]2+).
EI-MS: 452 (40, [M+ ~ ]), 437 (28, [M- CH3]+), 380 (16), 366 (31), 295
(25), 273 (19), 243 (31), 238 (20), 226 (20), 212 (19), 200 (28), 186 (16),
169
(15), 149 (33), 127 (18), 112 (21), 100 (29), 98 (35), 86 (76), 84 (100), 70
(39), 58
(57), 49 (95), 43 (69).
Example 6
Ethyl 2-Dodecenoate (4b).
[0097] Analogous to Example 2: From 1.7 g (74 mmol) of Na, 32 g (74
mmol) of 2 and 10.82 g (69.37 mmol) of caprinaldehyde 3b in 200 mI EtOH, 14.1
g (90%) 4b as an (E/~ mixture (= 2:1) was obtained after workup as a colorless
oil. For analytical purpose, 320 mg of 4b were purified by chromatography
(Et20/hexane 2:98) to give 104 mg (32%) of (~ isomer and 205 mg (64%) of (E)
isomer as a colorless oils.
Rf (Et20/hexane 3:97, KPlI~: 0.68 (~ isomer, 0.47 (E) isomer.
IR (CHC13): 2920vs, 2850vs, 1710vs, 1650s, 1460m, 1370s, 1310s, 1275s,
1170m, 1130m, 1035m, 980m, 825w, 660w, 620w.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
1H-NMR (CDCl3): (~ 6.22 (dt, J=11.5, 7.5, HC(3)); 5.74 (dt, J=11.5,
1.7, HC(2)); 4.16 (q, J= 7.1, OCH2CH3); 2.63 (q, J= 7.3, HZC(4)); 1.43 (t, J=
6.3, H2C(5)); 1.30-1.25 (m, 6 CHa, OCH2CH3); 0.87 (t, J= 7.0, H3C(12)).
13C-NMR (CDCl3): (~ 166.4 (s, C(1)); 150.4 (d, C(2)); 119.5 (d, C(3));
59.6 (t, OCH2CH3); 31.7 (t, C(4)); 29.4 (t, C(10)); 29.3, (t, C(5)); 29.1,
29.0, C(8));
28.9, (3t, 4 C); 22.5 (t, C(11)); 14.1 (q, C(12)); 13.9 (q, oCH2cH3).
1H-NMR (CDC13): (E~ 6.95 (dt, J=15.6, 7.0, HC(3)); 5.80 (dt, J= 15.6,
1.6, HC(2)); 4.16 (q, J= 7.2, OCH2CH3); 2.18 (q, J= 7.1, H2C(4)); 1.45 (t, J =
7.2, H2C(5)); 1.32-1.26 (m, 6 CH2, OCH2CH3); 0.88 (t, 6.9, H3C(12)).
I3C-NMR (CDC13): (E) 166.6 (s, C(1)); 149.3 (d, C(2)); 121.1 (d, C(3));
59.9 (t, OCH2CH3), 32.0 (t, C(4)); 31.7 (t, C(10)), 29.3, (t, C(5)); 29.2,
29.1, 29.0,
27.9 (4t, 4 C); 22.5 (t, C(11)), 14.1 (q, C(12)); 13.9 (q, OCH2CH3).
CI-MS: 227 (76, [M+ 1]+), 226 (52, [M+ NH4 - H20]+), 181 (52, [M-
OEt]+), 138 (16),127 (100), 144 (22), 99 (80), 88 (28), 81 (30), 55 (39), 43
(41).
Example 7
Ethyl 16 Amiho-3-~conyl-4-8,13-triazahexadecanoate (5b)
H
wN~NH2
12 16
9'
[0098] Analogous to Example 3: From 6.46 g (32 mmol) of spermine and
7.23 g (32 mmol) of 4b in 500 ml EtOH, 5.2 g (40%) of Sb were obtained after
worlcup as a colorless oil.
Rf (CHC13/MeOH/25% aq. NH40H 7:4:1, Schlittler: 0.25.
IR (CHCl3):2920vs, 2850s, 1720m, 1600w, 1510m, 1370w, 1300w, 1220w,
1115m, 1025w, 925w, 840w, 660m, 620w.
1H-NMR (CDC13): 4.15 (q, J= 7.2, OCH2CH3), 2.92 (quint, J= 6.2,
HC(3)), 2.77 (t, J= 6.8, H2C(16)); 2.75-2.70 (m, H2C(9), H2C(12)); 2.69-2.67
(m,
H2C(5)); 2.62 (m, H2C(7), H2C(14)); 2.38 (d, J= 6.2, H2C(2)); 1.98 (br. s, NH,
46
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
NHZ), 1.64 (quint, J= 6.9, H2C(6), H2C(1S)); 1.52 (quint, J= 6.4, H2C(10),
HaC(11)); 1.28-1.23 (m, 8 CH2, OCH2CH3):0.88 (t, 6.5, H3C(9')).
13C-NMR (CDCI3): 172.5 (s, C(1)); 60.0 (t, OCH2CH3); 54.7 (d, C(3));
49.6 (t, C(12)); 48.2 (t, C(14)); 47.6 (t, C(9)); 45.2 (t, C(7)); 40.3 (t,
C(S)); 39.1 (t,
C(16)); 34.2 (t, C(2)); 33.5 (t, C(1')); 31.7 (t, C(T)); 30.2, 29.6, 29.5,
29.4 (4t, 4C);
29.1 (t, C(6), C(1S)); 27.6 (t, C(2'), C(10)); 25.6 (t, C(11)), 22.5 (t,
C(8')); 14.1 (q,
OCH2CH3): 13.9 (q, C(9')).
ESI-MS: 429 (43, [M+ 1]+), 21S (100, [M+ 2]2+).
Example 8
4-Nonyl-l, 5, 9,14-tetraazacycloheptadecan-2-one (6b).
1~ 13
~~N N
H H
9
1~ ~N N
H H
9'
[0099] Analogous to Example 4: From 4.2 g (9.8 mmol) Sb and 3 g (11.7
mmol) antimony(III) ethoxide in 190 ml benzene, 2.85 g (76%) of 6b were
obtained after workup as a colorless oil.
Rf(CHC13/MeOH/2S% aq. NH40H 7:4:1, Schlittle~: 0.46.
IR (CHCl3): 2920vs, 28SOvs, 1640vs, 1 S20m, 1460m, 1 S70w, 1220m,
1120m, lOSOw, 92Sw, 80Sm, 660m, 620w.
1H-NMR (CDC13): 8.47 (b~. s, NH); 3.36 (dt, J-- 7.1, 6.1, H2C(17)); 2.83
(quint, J= 6.6, HC(4)); 2.78-2.76 (m, H2C(8), H2C(10)); 2.74-2.72 (m, HaC(6));
2.69-2.66 (m, H2C(13), H2C(1S)); 2.37 (dd, J=15.2, 3.4, HaC(3)); 2.14 (dd, J=
15.2, 7.8, HbC(3)); 2.06 (br. s, NH); 1.67 (quint, J= 6.2, H2C(7), H2C(16));
1.59
(quint, J= 5.6, H2C(11), H2C(12)); 1.48-1.32 (m, H2C(1')); 1.25 (m, 8 CH2);
0.88
(t, J= 6.4, H3C(11')).
47
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
13C_NMR (CDC13);172.3 (s, C(1)); 55.7 (d, C(4)); 48.6 (t, C(13)); 48.4 (t,
C(15)); 48.2 (t, C(8)); 47.6 (t, C(10)); 45.9 (t, C(6)); 40.4 (t, C(3)); 37.8
(t, C(17));
34.1 (t, C(1')); 31.8 (t, C(T)); 29.8, 29.7, 29.6, 29.3 (4t, 4 C); 29.3 (t,
C(7)); 29.0
(t, C(16)); 26.8 (t, C(11)); 25.9 (t, C(2'), C(12)); 22.6 (t, C(8')); 14.1 (q,
C(11')).
ESI-MS: 383 ([M+ 1 ]+).
Example 9
5, 9,1 ~-Trimethyl-4-nonyl-1, 5, 9,14-tet~'aazacycloheptadecah-~-one
(Budmu~cchiamine B) (7b).
17 ~ a 13
2 ~N N
H
4 9
1' \N~ N\
Me Me
9'
[0100] Analogous to Example 5: From 70 mg (0.18 mmol) of 6b, 3 ml of
formalin (37%) and 200 mg (3.2 mmol) NaCNBH3 in 8 ml AcOH, 62 mg (80%)
of 7b were obtained after workup as a colorless oil.
Rf(CHCl3/MeOH/25% aq. NH40H 85:14:1, Schlittler: 0.40.
IR (CHCl3): 2920vs, 2850s, 2800m, 1640s, 1520m, 1455m, 1370w, 1235m,
1130w, 1050w, 1005w, 925w, 800w, 660w, 620w.
1H-NMR (CDC13): 8.52 (b~. s, NH); 3.31 (dt, J= 6.9, 6.4, H2C(17)); 2.84
(quint, J= 4.6, HC(4)); 2.62 (dt, J=12.4, 7.0, HaC(6)); 2.50-2.42 (m,
H2C(13));
2.41 (m, HbC(6)); 2.37 (m, HaC(8)); 2.42-2.33 (m, H2C(10)); 2.44-2.35 (m,
H2C(15)); 2.37 (m, HaC(3)); 2.24 (d, J= 4.8, HbC(3)); 2.27 (s, H3CN(9)), 2.20
(s,
H3CN(14), H3CN(5)); 1.65 (quit, J= 6.9, HaC(16)); 1.54 (quint, J= 6.8,
H2C(11), HaC(12)); 1.30-1.17 (m, $ CH2); 0.88 (t, J= 7.0, H3C(9')).
13C-NMR (CDC13): 172.5 (s, C(1)); 61.1 (d, C(4)); 56.3 (t, C(10)); 56.1 (t,
C(13)); 55.6 (t, C(15)); 54.4 (t, C(8)); 51.3 (t, C(6)); 42.5 (q, H3CN(9));
42.2 (q,
H3CN(14)); 37.5 t, C(17)); 37.1 (t, C(3)); 35.3 (q, H3CN(5)); 31.7 (t, C(T));
29.6,
48
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
29.5, 29.4, 29.1 (4t, 4 C); 27.4 (t, C(16)); 27.3 (t, C(2')); 27.1 (t, C(1'));
2S.S (t,
C(7)); 24.2 (t, C(11)); 23.2 (t, C(12)), 22.5 (t, C(8')); 13.9 (q, C(9')).
ESI-MS: 425 (S2, [M+ I]+), 213 (100, [M+ 2]2+).
EI-MS: 424 (S1, [M+ ~ ]), 409 (28, [M- CH3]+), 3S2 (19), 338 (S1), 297
(41), 281 (1S), 224 (14), 212 (2S), 210 (38), 198 (23), 184 (36), 169 (27),
1SS
(17), 112 (2S), 100 (32), 98 (S9), 86 (62), 84 (100), 72 (36), 70 (43), S8
(7S), S7
(29), 43 (29).
Example 10
Ethyl ~-Hexadecenoate (4c).
O
[0101] Analogous to Example 2: From 1.17 g (S 1 mmol) of Na, 21.9 g (S 1
mmol) of 2 and 10 g (47.1 mmol) of myristinaldehyde 3c in 200 ml EtOH, 11.6 g
(86%) 4c as an (El.~ mixture (~ 2:1) were obtained after workup as a colorless
oil. For analytical purpose, 3S0 mg of 4c were purified by chromatography.
(Et20/hexane 2:98) to give 109 mg (3I%) of (~ isomer and 230 mg (6S%) of (E)
isomer as a colorless oils.
Rf(Et20/hexane 3:97, KPH: 0.67 (~ isomer, 0.47 (E) isomer.
IR (CHC13): 2920vs, 28SOvs, 1710vs, l6SOs, 1460s, 136Sm, 127Ss, 1180s,
112Sm, 109Sw, 103Sm, 980m, 92Sw, 610w, 660w, 620w.
1H-NMR (CDC13): (~ 6.22 (dt, J= 11.4, 7.5, HC(3)); 5.73 (dt, J=11.5,
1.6, HC(2)); 4.18 (q, J= 7.1, OCH2CH3); 2.62 (q, J= 7.3, H2C(4)); 1.43 (quint,
J
= 6.5, HaC(S)); 1.32-1.25 (m, 10 CH2, OCH2CH3); 0.88 (t, J= 7.0, H3C(16)).
i3C-NMR (CDCl3): (~ 169.3 (s, C(1)); 150.4 (d, C(2)); 119.5 (d, C(3));.
59.6 (t, OCH2CH3); 32.0 (t, C(4)); 31.8 (t, C(14)); 29.5, (t, C(S)); 29.4,
29.3, 29.2,
49
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
29.1, 29.0, 28.9, 27.9 (7t, 8 C); 22.5 (t, C(15)); 14.1 (q, C(16)); 13.9 (q,
OCH2CH3).
1H-NMR (CDCl3): (E) 6.95 (dt, J=15.6, 7.0, HC(3)); 5.80 (dt, J= 15.6,
1.6, HC(2)); 4.18 (q, J= 7.1, OCH2CH3); 2.18 (q, J= 7.5, H2C(4)); 1.43 (quint,
J
= 6.5, H2C(5)); 1.32-1.25 (m, 10 CH2, OCH2CH3); 0.88 (t, J-- 7.0, H3C(16)).
13C-NMR (CDC13): (E) 166.6 (s, C(1)); 149.3 (d, C(2)); 121.1 (d, C(3));
59.9 (t, OCH2CH3); 32.0 (t, C(4)); 31.8 (t, C(14)); 29.5, (t, C(5)); 29.4,
29.3, 29.2,
29.1, 29.0, 28.9, 27.9 (7t, 8 C); 22.5 (t, C(15)); 14.1 (q, C(16)); 13.9 (q,
OCH2CH3).
CI-MS: 283 (<5, [M+ 1]+), 282 (16, [M+NH4 - H20]+), 237 (37, [M-
OEt]+), 194 (16), 127 (61), 114 (21), 101 (59), 99 (51), 88 (43), 81 (33), 69
(35),
57 (49), 43 (89), 41 (100).
Example 11
Ethyl 16 Amino-3-t~idecyl-4, 8,13-t~iazahexadecanoate (5c).
1
'~H H 16
[0102] Analogous to Example 3: From 4.3 g (21.28 mmol) of spermine
and 6 g (21.28 mmol) of 4c in 900 ml EtOH, 4.25 g (41%) of 5c were obtained
after workup as a colorless oil.
Rf(CHCl3/MeOH/25% aq. NH40H 7:4:1, Schlittle~): 0.27.
IR (CHC13): 2920vs, 2850s, 1720s, 1600w, 1460m, 1370m, 1220m, 1180m,
1115m, 1025w, 925w, 805m, 660m, 620w.
1H-NMR (CDCl3): 4.13 (q, J= 7.2, OCH2CH3); 2.92 (quint, J= 6.2,
HC(3)); 2.76 (t, J= 6.8, H2C(16)); 2.69-2.68 (m, H2C(9), H2C(12)); 2.64 (m,
H2C(5)); 2.62-2.60 (m, H2C(7), HaC(14)); 2.38 (d, J= 6.3, HZC(2)); 1.63
(quint, J
= 7.0, H2C(6), H2C(15)); 1.51-1.45 (m, HaC(10), H2C(11), NH, NH2); 1.32-1.25
(m, 12 CH2, OCH2CH3); 0.88 (t, J= 7.0, H3C(13')).
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
13C-NMR (CDC13): 172.7 (s, C(1)); 60.2 (t, OCHaCH3); 54.9 (d, C(3));
49.9 (t, C(12)); 48.5 (t, C(14)); 47.8 (t, C(9)); 45.4 (t, C(7)); 40.6 (t,
C(5)); 39.3 (t,
C(16)); 34.2 (t, C(2)); 33.5 (t, C(1')); 31.9 (t, C(11')); 30.6, 29.8, 29.7,
29.6, 29.3,
29.2 (6t, 8 C); 27.9 (t, C(2'), C(10)); 25.8 (t, C(11)); 22.6 (t, C(12'));
14.2 (q,
OCH2CH3); 14.1 (q, C( 3')).
ESI-MS: 485 (20, [M+ 1]+), 243 (100, [M+ 2]2+).
Example 12
4-Tridecyl-l, 5, 9,14-tetraazacycloheptadecan-2-one (6c).
1~ 13
2~N N
H H
4
1~ ~N N
H H 12'
[0103] Analogous to Example 4: From 1.7 g (3.5 mmol) of 5c and 1.16 g
(4.55 mmol) of antimony(III) ethoxide in 180 ml benzene, 1.2 g (78%) of 6c
were
obtained after workup as a colorless oil.
Rf(CHC13/MeOH/25% aq. NH40H 7:4:1, Schlittler): 0.49.
IR (CHC13): 3300m, 2920vs, 2850vs, 1640s, 1520m, 1460s, 1370m,
1220m, 1115m, 1045w, 925w, 805w, 660m, 620w.
1H-NMR (CDC13): 8.43 (br. s, NH); 3.37 (t, J= 5.6, HaC(17)); 2.86-2.83
(m, HC(4)); 2.77-2.74 (m, H2C(8), H2C(10)); 2.72-2.78 (m, H2C(6)); 2.68-2.66
(M, H2C(13), H2C(15)); 2.37 (dd, J=15.2, 2.4, HaC(3)); 2.32 (br. s, NH)); 2.14
(dd, J= 15.3, 7.8, HbC(3)); 1.67 (quint, J= 6.0, H2C(7), H2C(16)); 1.59-1.50
(m,
H2C(11), H2C(12)); 1.25 (m, 12 CHa); 0.88 (t, J= 6.6, H3C(13')).
13C-NMR (CDC13): 172.2 (s, C(2)); 55.6 (d, C(4)); 48.6 (t, C(13)); 48.2 (t,
C(15)); 48.3 (t, C(8)); 47.5 (t, C(10)); 45.9 (t, C(6)); 40.3 (t, C(3)); 37.8
(t, C(17));
34.0 (t, C(1')); 31.8 (t, C(7')); 29.7, 29.6, 29.3 (4t, 4C); 29.3 (t, C(7));
29.1 (t,
C(16)); 26.7 (t, C(11)); 25.9 (t, C(2'), C(12)); 22.6 (t, C(12')); 14.0
(C(13')).
51
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
ESI-MS: 439 ([M+1]~.
Example 13
5, 9, 14-Trimethyl-4-tridecyl-l, 5, 9, 14-tet~~aazacycloheptadecah-2-one
(Budmunchiamine C) (7c).
m i a is
a ~N N
H
4 9
\N~ N~
Me Me
9'
[0104] Analogous to Example 5: From 77 mg (0.2 mmol) of 6c, 3 ml of
formalin (37%) and 200 mg (3.2 mmol) of NaCNBH3 in 8 ml AcOH, 65 mg
(81 %) of 7c were obtained after workup as a colorless oil.
Rf(CHC13/MeOH/25% aq. NH40H 85:14:1, Schlittler): 0.42.
IR (CHC13): 3420s, 2920vs, 2850s, 2800s, 1640s, 1520m, 1460s, 1370s,
1230m, 1135m, 1050m, 920s, 840w, 670w, 650m.
1H-NMR (CDC13): 8.50 (br. s, NH); 3.30 (dt, J=6.6, 6.8 CH2(17)); 2.80
(quint, J--4.5, CH(4)); 2.60 (dt, J--12.3, 7.0, HaC(6)); 2.50-2.40 (m, CH2
(13));
2.38 (m, HbC(6)); 2.37 (m, CH2 (8)); 2.40-2.30 (m, CH2 (10)); 2.44-2.34 (m,
CHa
(15)); 2.35 (m, HaC(3)); 2.21 (dd, J--6.2, 1.6 HbC(3)); 2.20 (s, H3CN(9));
2.15 (s,
H3CN(14), H3CN(S)); 1.67 (quint, J=6.5, H2C(16)); 1.64 (quint, J=6.4, CH2(7));
1.50 (quint, J--6.7, CH2(11), CH2(12)); 1.30-1.17 (m, CHZ(12)); 0.9 (t, J--
6.9,
CH3(13')).
isC_NMR (CDC13):172.5 (s, C(2)); 61.0 (d, C(4)); 56.3 (t, C(10)); 56.1 (t,
C(13)); 55.5 (t, C(15)); 54.5 (t, C(8)); 51.0 (t, C(6)); 43.0 (q, CH3N(9));
42.5 (q,
CH3N(14)); 37.6 (t, C(17)); 37.1 (t, C(3)); 35.0 (q, CH3N(5)); 32.0 (t,
C(9')); 29.8,
29.6, 29.5, 29.4, 29.3 (St, 8 C); 27.5 (t, C(16)); 27.3 (t, C(2')); 27.1 (t,
C(1')): 25.9
(t, C(7)); 24.3 (t, C(11)); 23.2 (t, C(12)); 22.5 (t, C(12')); 13.9 (q,
C(13')).
52
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
ESI-MS: 503 (<5, [M+Na]+), 481 (100, [M+ 1]+).
EI-MS: 481 (18 [M+ 1]+), 480 (68 [M+ ~ ]), 465 (37 [M- CH3]+), 408
(21), 394 (56), 339 (16), 297 (58), 266 (42), 254 (38), 240 (50), 238 (30),
226
(36), 169 (32), 155 (22), 112 (27), 100 (35), 98 (69), 86 (69), 84 (100), 72
(32), 70
(41), 58 (62), 43 (25).
Example 14
N (4)-((3-Ethoxyca~bonyl-1-tridecyl)ethyl)ami~obutyl)hexahydropyrimidi
he (9).
COOEt
I
N N~NH
H
H3
[0105] A solution of ethyl 2-hexadecenoate'(4c, 2.82 g, 10 mmol) and
N-(4-aminobutyl)hexahydropyrimidine (8, 1.57 g, 10 mmol; see McManis, J.S.,
Ganem, B., J. Org. Chem. (1980), 45: 2042 and U.S. Pat. No. 5,869,734) in 400
ml of abs. EtOH was stirred for 4 days at 40°. After evaporation the
residue was
purified by column chromatography (Si02, CH2Cla/MeOH/25% aq. NH40H
100:10:1) to give 1.21 g (27%) of 9 as a colorless oil.
Rf= 0.44 (CH2C12/MeOH/25% aq. NH40H 40:6:1).
1H-NMR: 4.13 (q, OCH2); 3.48 (s, N-CHZ-N); 2.92 (m, CH); 2.80 (t, CHa);
2.59 (m, 2 CHa); 2.41 (d, CH2C0); 2.25 (m, CH2); 1.45-1.70 (m, 10 H, 4 CH2+2
NH); 1.15-1.35 (m, 11 CH2); 0.98 (m, 2 CH3).
CI-MS: 440 [M+1]+.
53
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Example 15
Methyl 12 Amino-3-tt~idecyl-4, 9-diazadodecanoate (10).
Me
H
N NH2
N
H
H3
[0106] A solution of 9 (663 mg, 1.51 mmol) in 50 ml of MeOH saturated
with dry HCl gas was refluxed for 10 h. After evaporation the residue was
dried in
vacuum and converted to free base by column chromatography (Si02,
CH2C12/EtOH/25% aq. NH40H 70:30:5) to give 517 mg (83%) of 10 as a
colorless oil.
Rf= 0.60 (CHCl3/MeOH/25% aq. NH40H 7:3:1).
1H-NMR: 3.64 (s, OCH3); 3.38 (br~. s, NH); 2.91 (m, CH); 2.74 (m, CH2);
2.50-2.70 (m, 4 CH2); 2.47 (d, CH2C0); 1.75-1.90 (m, 2 CH2+NHa); 1.63 (m,
CH2); 1.45-1.55 (m, 2 CH2); 1. 15-1.35 (m, 11 CH2); 0.88 (t, CH3).
CI-MS: 414. [M+1]+.
Example 16
2-Tridecyl-1,5,9-triaza cyclotridecan-4 -one (11).
54
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0107] To a solution of 10 (190 mg, 0.46 mmol) in anhydrous xylene was
added B(NMe2)3 (0.09 ml, 75 mg, 0.5 mmol) and NH4Cl (5 mg). The mixture
was refluxed in Na atm. for 15 h; after cooling to room temp., 5 ml of EtOH
was
added. After evaporation the residue was purified by column chromatography
(Si02, CH2C12/MeOH/25% aq. NH40H 70:30:3) to give 88 mg (50%) of 11 as a
white solid, m.p. 72-73 °.
Rf= 0.28 (CHC13/MeOH/25% aq. NH40H 70:3:5).
1H-NMR: 8.56 (br. s, CONH); 3.60-3.43 (m, 1H); 3.30-3 . 10 (m, 1 H);
2.90-2.42 (m, 3 CH2); 2.41 (dd, J~ = 15.1, J2 = 2.9, 1 H); 2.14 (dd, JI =
15.1, J2 =
9.2, 1 H); 1.8-1.1 (m, 15 CH2); 0.87 (t, CH3).
13C-NMR: 172.11 (s, CO); 55.72 (d, CH); 49.42, 48.74, 45.06, 40.96,
39.35, 33.80, 31.94 (7t, 7 CHa); 29.66-29.76 (7 CH2); 29.61, 29.36, 28.05,
27.66,
26.93, 25.65 (6t, 6 CH2); 14.11 (q, CH3).
ESI-MS: 382 [M+1]+.
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Example 17
Reduction and Alkylation of Cyclic Polyamines
O
~N
H
C13H27 N N
(7c)
LAH, THF
70°C, 4h
81%
Me
~N N
H
C13H27
Me Me
(12)
(0108] A solution of 7c (2.0 g) in dry THF (32 mL) was added carefully to
a cooled (0 °C) solution of LAH (95%, 4 eq) in dry THF (11 mL). The
grey
suspension was stirred at 0 °C for 10 min, and then heated to reflux
(oil bath 85
°C) for 4 h. The reaction was cooled to 0 °C, diluted with ether
(60 mL),
quenched with water (4 mL), dried (Na2S04), filtered through a Celite pad, and
concentrated under reduced pressure to give a thick oil, which was subjected
to a
flash column using CHC13-EtOH-28% NH40H (70: 27: 3) as the eluant to afford
12 (81%) as a clear thick oil. 1H NMR (250 MHz, CDC13) 8 2.71-2.56 (m, SH);
2.43-2.25 (m, 10H); 2.21 (s, 3H); 2.17 (s, 3H); 2.16 (s, 3H); 1.66-1.48 (m,
12H);
1.25 (m, 22H), 0.88 (t, J= 6.9, 3H). 13C NMR (62.5 MHz, CDC13) 8 62.39,
57.67, 57.16, 56.28, 54.85, 51.51, 49.37, 48.84, 43.04, 42.74, 36.79, 31.88,
31.46,
29.97, 29.62, 29.31, 28.32, 27.51, 27.40, 26.50, 25.12, 24.65, 22.64, 14.05.
MS-EI m/z 467.8(M+1)+.
56
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0109] (Tetraamine 12 was converted to its tetra-HCl salt, SL-11238, by
dissolving it in MeOH, adding equal volume of concentrated HCI, stirring for 5
min, and evaporating to dryness. The melting point of its crystals from EtOH
was
241.5-244.5 °C. Anal. Calcd for C29H7oN4O2C14 - formula for 12 plus
four
molecules of HCl and two molecules of H20: C, 53.69; H, 10.88; N, 8.64. Found:
C, 53.29; H, 11.08; N, 8.32.)
Me
~N N
H
C13H27
Me Me
(12)
~CN
MeOH, O/N
100%
C13H27
Me Me
(13)
[0110] Acrylonitrile (1.4 mL) was added to a solution of 12 (1.0 g) in
MeOH (5.5 mL), and the reaction was stirred at room temperature overnight (18
h). The solvent and excess acrylonitrile were evaporated under reduced
pressure
at 35 °C, and the residue was purified by a flash column using CHC13-
EtOH-28%
NH40H (70: 27: 3) as the eluant to give 13 (100%) as a clear thick oil. 1H NMR
(250 MHz, CDC13) 8 2.79 (t, J-- 6.7, 2H); 2.66-2.28 (m, 17H); 2.20 (s, 3H);
2.19
(s, 3H); 2.14 (s, 3H); 1.64-1.39 (m, 10H); 1.26 (m, 24H); 0.88 (t, J-- 6.5,
3H). 13C
NMR (62.5 MHz, CDC13) 8 119.14; 61.38; 56.95; 56.54; 54.86; 54.73; 52.12;
57
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
51.70; 49.56; 43.12; 42.94; 36.16; 31.92; 30.05; 29.68; 29.35; 29.08; 28.33;
27.55; 26.23; 25.69; 24.65; 24.49; 22.67; 15.99; 14.08. MS-EI m/z 520.8
(M+1)+.
Me
~N
C13H27 ~~
Me Me
(13)
Raney-Ni, NaOH
95% aq. EtOH
H2 (50psi), OIN
100%
NH2
Me
'~N
C13H27
Me Me (14)
(0111] Raney nickel (0.8 g suspension in water) was added to a solution of
13 (1.0 g) and NaOH (5.0 eq) in 95% aq. EtOH (80 mL) in a Parr-shaker. The
suspension was purged 5 times with hydrogen, and then shaken under hydrogen
(50 psi) overnight. The catalyst was filtered off through a Celite pad and
destroyed with 2N HCI, and the filtrate was concentrated. The residue was
dissolved in water (10 mL), extracted with CHCl3 (4 x 30 mL), separated, and
the
organic layers were combined, dried (NaaS04), and concentrated under reduced
pressure to afford 14 (quantitative, NMR & TLC pure) as a clear thick oil.
[0112] 1H NMR (250 MHz, CDCl3) 8 2.73 (t, J= 6.8, 2H); 2.58-2.29 (m,
17H); 2.20 (s, 6H); 2.14 (s, 3H); 1.64-1.33 (m, 14H); 1.26 (m, 24H); 0.88 (t,
J=
58
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
6.9, 3H). 13C NMR (62.5 MHz, CDCl3) 8 61.50; 56.81; 56.18; 55.14; 54.60;
52.36; 52.11; 51.96; 43.14; 42.77; 40.79; 36.09; 31.77; 31.09; 29.89; 29.53;
29.20; 29.09; 27.90; 27.36; 26.05; 25.33; 24.38; 24.30; 22.52; 13.95. MS-EI
mlz
524.6 (M+1)+.
[0113] (Pentaamine 14 was converted to its penta-HCl salt, SL-11239, by
dissolving it in MeOH, adding equal volume of concentrated HCI, stirring for 5
min, and evaporating to dryness. The melting point of its crystals from EtOH
was
242.3-245.2 °C. Anal. Calcd for C32H78NSOZC15 - formula for 14 plus
five
molecules of HCl and two molecules of H2O: C, 51.78; H, 10.59; N, 9.44. Found:
C, 51.68; H, 10.91; N, 9.14.)
Example 18
Cyclic Polyamines as Anti-heoplastic Agents.
[0114] To assess the utility of the subject compounds in the treatment of
neoplastic cell growth, the ability of the compounds to inhibit the ih vitro
growth
characteristics of several commonly used cancer models were studied. The
subject polyamines induce cell growth inhibitions in several cultured human
prostate tumor cell lines such as LnCap, DuPro, and PC-3 as determined by the
accepted MTT assay (Table 2) (Hansen, M.B. et al., "Re-examination and fuxther
development of a precise and rapid dye method for measuring cell growth/cell
kill," J. Immunol. Methods (1989)1I9(2):203-IO). All three cell lines are
sensitive to the cyclic polyamines with IDso values ranging between 500 nM to
2,600 nM. The results with the DuPro cell line are representative of the
results
with human prostate cell lines.
[0115] As shown in Figs. 1-14, the cyclic polyamines of the present
invention have been shown to inhibit cell growth and even to cause cell death
in
accepted in vitro test cultures of human prostate cell cancer. The Figures are
described in more detail below. The uptake of the cyclic polyamines by the
DuPro cells and the resultant changes in the cellular polyamine levels axe
shown
in Tables 3a and 3b.
59
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[OII6j As the hydrolysis of ATP is proposed as a possible mechanism for
the antitumor activity of the cyclic polyamine analogs, assays to measure ATP
hydrolysis were carried out. In a standardized method for measuring hydrolysis
of ATP, a marked increase in ATP hydrolysis was observed in the presence of
the
cyclic polyamines (Figs. 15-21) as compared to the naturally occurring linear
polyamine spermine.
[0117] The effects of the cyclic polyamines on the intracellular ATP
content were measured using the Enliten ATP Assay test kit (Promega Corp.,
Madison, WI). The results of the cellular ATP measurements after a 72 hour
incubation with varying concentrations of cyclic polyamines are shown in Figs.
22-24. All cyclic polyamines depleted the intracellular ATP pool. The relative
abilities of the cyclic polyamines in depleting the intracellular ATP pool
correspond to their relative cytotoxicities.
[0118] In Figs. 1-5 and Figs. 11-22, the X-axes depict the number of days
after seeding DuPro cells and the Y-axes depict the number of cells harvested
under control (no drug) conditions (Figs. 1-5) and in the presence of 10 ~u,M
of the
drug SL-11174 (Fig. 1), 5 p,M SL-11197 (Fig. 2), 5 ~uM SL-11199 (Fig. 3), 10
l.iM
SL-11200 (Fig. 4) and 5 p,M SL-11208 (Fig. 5), 2 ~m of SL-11238 (Fig. 11) and
5
p,m of SL-11239 (Fig. 12).
[0119] The X-axes of Figs. 6-10 and Figs. 13-14 depict the concentrations
of the cyclic polyamines and the Y-axes depict the fraction of surviving cells
after
days treatment with the drug SL-11174 (Fig. 6), SL-11197 (Fig. 7), SL-11199
(Fig. 8), SL-11200 (Fig. 9), SL-11208 (Fig. 10), SL-11238 (Fig. 13), and SL-
11239 (Fig. 14) as determined by the colony forming efficiency (CFE) assay
(Wilson A.P., "Cytotoxicity and viablity assays." See Freshney, R.I. (ed)
Animal
Cell Culture: A Practical Approach. Oxford: IRL Press, 1992, p. 183.)
[0120] The X-axes of Figs. 15-21 depict the concentrations of the
polyamines and the Y-axes depict the relative increase in inorganic phosphate
(PP;) released from 100 p,M ATP in 24 hour in the presence of spermine and
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
SL-11174 (Fig. 15), SL-11197 (Fig. 16), SL-11199 (Fig. 17), SL-11200 (Fig.
18),
SL-11208 (Fig. 19), SL-11238 (Fig. 20), and SL-11239 (Fig. 21) as compared to
the inorganic phosphate released from the same amount of ATP. under identical
conditions in the absence of any polyamines.
[0121] Marked growth inhibition and cell kill were observed for all cyclic
polyamines at concentrations as low as 5 l.~M. Such high intracellular levels
and
such strong growth inhibitory and cytotoxic effects have not been reported for
any
other polyamine analogs tested so far. All cyclic polyamines were observed to
be
more efficient than the linear naturally occurring polyamine spermine in
catalyzing hydrolysis of ATP.
[0122] The following standardized protocol was used to evaluate the test
cultures and to generate data shown in Figs. 1-14. For Figs. 1-5 and Figs. 11-
12,
cells were seeded into 75 cm2 culture flasks with 15 ml of Eagle's minimal
essential medium supplemented with 10% fetal calf serum and nonessential amino
acids. The flasks were then incubated in a humidified 95% air/5% C02
atmosphere. The cells were grown for at least 24 h to ensure that they were in
the
log phase of growth, then treated with the polyamine analogs. Cells were
harvested by treatment for 5 min with STV (saline A, 0.05% trypsin, 0.02%
EDTA) at 37 °C. The flasks were rapped on the lab bench, pipetted
several times
and aliquots of cell suspension were withdrawn and counted using a Coulter
particle counter that had been standardized for counting each cell line using
a
hemacytometer.
[0123] For Figs. 6-10 and Figs. 13-14, cells were washed, harvested, and
replated in quadruplicate at appropriate dilution into 60 mm plastic Petri
dishes.
The Petri dishes were prepared not more than 24 hr in advance with 4 ml of
supplemented Eagle's minimum essential medium containing 5-10% fetal bovine
serum (standardized for each cell line). Cells were incubated for the
previously
standardized number of days in a 95% airl5% C02 atmosphere. The plates were
stained with 0.125% crystal violet in methanol and counted.
j0124] ~ For Figs. 15-21, an ATP hydrolysis assay was standardized. In a
96 well microtiter plate, the first two columns were routinely used for
standard
61
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
curve generation. For the standard curve, 40 ~,l of 0-70 ~uM phosphate buffer
was
used by serially diluting 1 mM NaH2P04 solution in 1 N HCI. For ATP
hydrolysis, the rest of the microtiter plate was equally divided into two
sections to
run two analogs at a time. Each section was divided into the appropriate
number
of columns to serially dilute each analog between 0 - 10 mM in 29 ~,l 2N HCI
for
the final concentration of 2 N HCI. Each drug concentration was run in
quadruplicate. Equal volumes of 500 ~,M ATP solution (pH 7.5) were added to
each well and the plates were incubated at 37° C for appropriate
lengths of time
between 2-24 h. At the end of the incubation period, 160 ~1 of coloring agent
(0.045% Malachite Green in water and 4.2% ammonium molybdate in 4 N HC1
(3:1 v/v)) was added and the plates were incubated at 37° C for another
30 wins.
All plates were read at 595 nm using a Emax precision microplate reader
(Molecular Device, San Jose, CA). A control plate containing analog solutions
without ATP was created for each experiment. Control plate reading was
subtracted from each experimental plate and all data was normalized to ATP
hydrolysis at zero concentration of polyamine analog. The average and standard
deviation for quadruplicate runs were plotted.
[0125] For the data shown in Table 2, an accepted MTT assay protocol
was used. A trypsinized cell suspension was diluted to seed 80 p1 suspension
containing 500 cells in each well of a 96 well microtiter plates and incubated
overnight at 37°C in a humidified incubator in 5% C02. Twenty ~1 of
appropriately diluted stock solution of each drug was added to the middle 8
columns of cell suspension in the microtiter plates. Each drug concentration
was
run in quadruplicate. Outer columns of the plates were used for buffer
controls.
Cells were incubated with the drug for 6 days at 37°C in 5%
C02/H20
atmosphere. Twenty five ~.l of 5 mg/ml solution of 3-(4,5-dimethythiazol-2-yl)-
2,
5-diphenyl tetrazolium bromide (MTT) was added to each well and incubated for
4 hours at 37° C in 5% CO2/H2O incubator. Cells were lysed by
incubating
overnight with 100 ~,1 lysis buffer (500 ml of the lysis buffer containing:
100 g
lauryl sulfate (SDS), 250 ml of N,N-dimethyl formamide in 2 ml of glacial
acetic
acid, pH 4.8). The color was monitored at room temperature at 570 nm in a
62
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
E-max Precision Microplate Reader (Molecular Devices Corporation, Sunnyvale,
CA) and data were analyzed using cell survival software supplied by Molecular
Devices Corporation.
[0126] For data shown in Tables 3a and 3b, intracellular polyamine levels
were determined using a standard protocol. About 0.5-1 x 106 cells were taken
from harvested samples and centrifuged at 1000 rpm at 4°C for 5 min.
The cells
were washed twice with chilled Dulbecco's isotonic phosphate buffer (pH 7.4)
by
centrifugation at 1000 rpm at 4°C and resuspended in the same buffer.
After the
final centrifugation, the supernatant was decanted, and 250 ~,1 of 8%
sulfosalycilic
acid was added to the cell pellet. The cells were then sonicated, and the
mixture
was kept at 4° C for at least 1 h. After centrifugation at 8,000 g for
5 min, the
supernatant was removed for analysis. An appropriate volume (50-100 ~1) was
fluorescence-labeled by derivatizing with dansyl chloride. Labeled polyamines
were loaded onto a C-18 high-performance liquid chromatography column and
separated by gradient elution with acetonitrilelwater at 50°C. Peaks
were detected
and quantitated using a Shimadzu HPLC fluorescence monitor coupled with a
Spectra-Physics peak integrator. Because polyamine levels vary with
environmental conditions, control cultures were sampled for each experiment.
[0127] For the data shown in Figs. 22-24, a protocol was standardized
using the Enliten ATP Assay System (Promega Corp., Madison, WI).
Approximately 1x106 cells from each treatment flask were harvested, counted,
washed twice with chilled PBS and the cell pellets were stored at 4°C
overnight.
On the following day the pellets were resuspended in calculated volumes of
treatment buffer (Enliten ATP Assay System, Promega Corp.) to remove the
extracellular ATP. An aliquot of 140 ~,1 of the cell suspension containing
50,000
cells was plated in each well of a 96 well luminometer plate and was allowed
to
equilibrate to room temperature. Each compound concentration was plated in
quadruplicate. To each well, 40 ~.1 Extraction Buffer (Promega Enliten ATP
Assay System) was added and the plates were placed~in an EG&G luminometer
(Berthold Inc., Bundoora, Victoria, Australia). Then 40 p,1 L/L Reagent
(Promega
Enliten ATP Assay System) containing luciferase/luciferin mixture in assay
buffer
63
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
was injected and each well was read for 5 seconds after a one-second delay
time.
The relative changes in cellular ATP content were measured as relative light
units
(RLL~ generated by the luciferase/luciferin reaction.
64
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Table 2
Effects of Cyclic Polyamines on Prostate Tumor Cell Growth
(IDso values)
Analog IDSO (~M)values
DuPro PC-3 LnCap
SL-11174 0.83 0.60 2.20
SL-11197 0.58 0.5 2.60
SL-11199 1.20 1.4 1.50
SL-11200 1.40 1.30 Nd
SL-11208 1.80 1.70 Nd
Nd = Not determined
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Table 3a
Polyamine Levels in DuPro Cells Treated with Low Conc. of Cyclic
Polyamine Analogs.
Poly-
amine Polyamines Polyamines
(nmoles/106 (nmolesl106
cells) cells)
on on
AnalogsDay Day
4 6
of of
Treatment Treatment
used
Put Spd Spm AnalogPut Spd Spm Analog
Control0.83 1.58 2.48 0.31 0.39 1.07 -
SL- ND ND 0.002 2.788 ND ND 0.001 17.206
11174
(1 pM)
SL- ND ND 0.004 47.566ND ND 0.002 35.913
11197
(0.5
~uM)
SL- ND ND 0.002 26.959ND 0.005 0.003 26.841
11199
(0.5
pM)
SL- 0.004 0.029 0.089 10.089ND 0.014 0.043 46.776
11200
(1 p,M)
ND = Not Detectable. Put = putrescine; Spd = spermidine; Spm =
spermine; Analog as indicated in leftmost column.
66
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
Table 3b
PoIyamine Levels in DuPro Cells Treated with 2 ~M Cyclic Polyamine
Analogs.
Poly-
amine Polyamines Polyamines
(nmolesl106 (nmoles/106
cells) cells)
on on
AnalogsDay Day
4 6
of of
Treatment Treatment
used
Put Spd Spm AnalogPut Spd Spm Analog
Control0.832 1.579 2.484 - 0.31 0.39 1.07 -
SL- ND ND 0.001 25.074ND ND 0.002121.472
11174
SL- ND ND ND 49.072ND ND ND 42.341
11197
SL- ND ND ND 17.198ND ND ND 10.241
11199
SL- 0.020 0.027 0.095 5.989 0.011 0.018 0.053 49.309
11200
ND = Not Detectable. Put = putrescine; Spd = spermidine; Spm = spermine;
Analog as indicated in leftmost column.
67
CA 02417064 2003-O1-22
WO 02/10142 PCT/USO1/24282
[0128] All references, publications, patents and patent applications
mentioned herein are hereby incorporated by reference herein in their
entirety.
[0129] Although the foregoing invention has been described in some
detail by way of illustration and example for purposes of clarity and
understanding, it will be apparent to those skilled in the art that certain
changes
and modifications may be practical. Therefore, the description and examples
should not be construed as limiting the scope of the invention, which is
delineated
by the appended claims.
68