Note: Descriptions are shown in the official language in which they were submitted.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
IMPROVED.SOLID PHARMACEUTICAL DOSAGE
FORMULATION OF HYDROPHOBIC DRUGS
Background of the Invention
The present invention relates to improved solid pharmaceutical dosage
forms. In particular, the present invention is concerned with enhancing the
dissolution of hydrophobic drugs.
H,~phobic Drugs
As is well known, many pharmaceutically active compounds intended
for oral administration are poorly soluble in water. Hydrophobic drugs do not
generally dissolve easily and rapidly in the gastro-intestinal tract. This
hydrophobic
property often makes it difficult to formulate a drug so that it exhibits a
satisfactory
bioavailability profile in vivo. Poor bioavailability may lead to ineffective
therapy,
the need for higher dosing and/or undesirable side effects.
It has been recognized that the addition of a surfactant during the
processing of a hydrophobic drug may improve the dissolution of the dosage
units
within the gastro-intestinal tract. Furthermore, for some hydrophobic drugs,
the
addition of a surfactant during processing may improve the bioavailability of
the
product due to improved wetting of the hydrophobic active ingredient, leading
to
faster dissolution and absorption.
Therefore, in order to compensate for the poor solubility of certain
hydrophobic drugs, various carrier systems have been suggested wherein such
drugs
are co-formulated in intimate admixture with certain surfactants and other
ingredients.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-2-
For example, U.S. Patent No. 4,344,934 discloses a mixture or solution
of a poorly water-soluble drug with a pharmaceutically acceptable water-
soluble
polymer, wherein said mixture or solution has been treated with a minor amount
of a
wetting agent selected from anionic and cationic surfactants. Such
compositions are
formed as follows: First, a mixture or solution of the drug with the water-
soluble
polymer is formed. The mixture can be formed in a solvent or solvent mixture
which
is a mutual solvent for both the drug and the polymer. After the drug-polymer
mixture or solution has been formed in a solvent, it is dried by spray-drying,
flash
evaporation or air drying. The powdered drug-polymer mixture is then treated
with
an amount of a primarily aqueous wetting solution containing a wetting agent
selected
from anionic and cationic surfactants. The treated mixture is then again dried
and, if
necessary, it is milled, screened or ground prior to formulating into suitable
dosage
forms with pharmaceutically acceptable excipients.
U.S. Patent No. 5,827,541 discloses a process for the preparation of an
oral, rapidly disintegrating dosage form of a hydrophobic drug. The process
comprises forming a suspension of the hydrophobic drug in a solvent containing
a
pharmaceutically acceptable surfactant together with a water-soluble or water-
dispersible carrier material; forming discrete units of the suspension; and
removing
solvent from the discrete units under conditions whereby a network of the
carrier
material carrying a dosage of the hydrophobic drug is formed.
Thus, the common approach knovcm in the art tends to focus on the
development of carrier systems wherein the hydrophobic drug must be intimately
admixed with the surfactant and other components. A serious disadvantage of
this
approach is that it has required the development, more or less empirically, of
a
separate carrier system for each hydrophobic drug. Also, admixing of
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-3-
wetting/solubilizing agents with the active ingredient can lead to product
instability
due to interaction between the drug and the wetting/solubilizing agent. The
necessity
to devise a separate carrier system for each drug is, of course, time-
consuming and
expensive. There continues to be a need for a single drug carrier system which
is
suitable for a wide range of different hydrophobic drugs.
Deposition of Drubs
A unique type of solid dosage form may be obtained by deposition of
an active pharmaceutical ingredient on a pharmaceutically acceptable
substrate.
Various means for depositing pure active ingredients, such as weighing,
spraying or
spreading, can be used to generate the dosage form as taught, for example, in
the
following patents and patent publications, the disclosures of which are
incorporated
by reference herein in their entireties: U.S. Patent Nos. 5,845,463,
5,240,049,
5,018,335 and 4,640,322, as well as WO 00/09249, SU 1803328 and GB 2238768.
In a preferred embodiment, electrostatic deposition methodologies can
be used. In the electrostatic deposition process, a cloud or stream of charged
particles
of the active ingredient is exposed to, or directed towards, a substrate, at
the surface of
which substrate a pattern of opposite charges has been established. In this
fashion, a
measured dosage of the active ingredient can be adhered to the substrate.
Preferred
electrostatically deposited dosage forms are disclosed in published
international
patent application number WO 99/63972, assigned to the assignee of the present
invention, the disclosure of which is incorporated by reference herein in its
entirety.
Although electrostatic drug deposition generally has certain benefits,
including improved dose uniformity, certain problems still arise when the drug
to be
electrostatically deposited is hydrophobic. Specifically, the final dosage
form may
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-4-
suffer from the same problems of poor dissolution and poor bioavailability
that were
discussed above with respect to conventional solid dosage forms of hydrophobic
drugs. Moreover, the prior art approach, involving the intimate admixture of
the
hydrophobic drug and a surfactant, would be difficult or impossible to
implement in
the context of electrostatic deposition.
For example, if the drug and surfactant powders are to be blended prior
to electrostatic deposition on the substrate, it may be difficult to obtain a
suitably
homogenous blend, or to maintain such homogeneity during the charging and
delivery
to the substrate. Moreover, co-deposition of two different powders would
require that
both powders behave similarly during the deposition, but this is difficult to
achieve
since different powders often have different optimum deposition parameters. In
an
extreme case, the surfactant may deposit only under a charge opposite that
utilized for
the active ingredient.
One possible solution would be to deposit the active ingredient and the
surfactant sequentially. However, there may be difficulty in forming
depositions on
top of pre-existing depositions, due to charge dissipation.
Therefore, it would be desirable to provide a dosage form of a
hydrophobic drug, wherein the problems of poor solubility and poor
bioavailability,
as well as the technical problems identified in the preceding paragraphs, are
overcome.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-5-
Summary of the Invention
In accordance with the teachings of the present invention, improved
solid pharmaceutical dosage formulations are provided, characterized by the
enhanced
dissolution of hydrophobic drugs. The formulations comprise:
a base substrate comprising a first polymer;
a deposit, comprising a therapeutic amount of a hydrophobic drug, deposited
on the base substrate;
a cover substrate comprising a second polymer, the cover substrate covering
the deposit and joined to the base substrate by a bond that surrounds the
deposit; and
a dissolution-enhancing amount of a surfactant, disposed within a carrier that
is segregated from, but in contact with, the deposit.
It is accordingly an object of the present invention to provide solid
pharmaceutical dosage formulations of hydrophobic drugs having enhanced
dissolution and improved bioavailability.
Brief Description of the Drawings
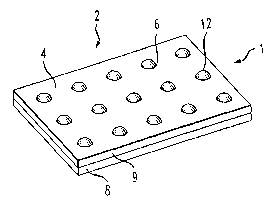
FIG. 1 depicts an isometric view of a product comprising a strip
package containing a plurality of unit forms in accordance with the prior art.
FIG. 2 depicts a cover layer of a prior art strip package partially
separated from a substrate.
FIG. 3 depicts a side view of an illustrative unit form in accordance
with the prior art.
FIG. 4 depicts a top view of the illustrative unit form of FIG. 3.
FIG. 5 depicts components of various embodiments of unit forms of
the present invention.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-6-
FIG. 6 is a graph of dissolution profiles for the drug CCN00401.
FIG. 7 is a graph of dissolution profiles for the drug hydrocortisone.
FIG. 8 is a graph of dissolution profiles for the drug glipizide.
Detailed Description of the Invention
FIGS. 1 through 4 depict the general structure of prior art dosage forms
which are to be improved in accordance with the present invention. In FIG. 1,
product 1 comprises a package 2 that is realized as a strip 4 having an array
of unit
dosage forms 6. Strip 4 comprises a substrate 8 and a cover layer 9.
Substrate 8 and cover layer 9 each comprise a substantially planar,
flexible film or sheet. In some embodiments, one of either substrate 8 or
cover layer 9
includes an array of semi-spherical bubbles, concavities, blisters or
depressions
(hereinafter "bubbles") 12 that are advantageously arranged in columns and
rows. In
the illustrative package depicted in FIG. l, cover layer 9 comprises a three-
by-five
array of such bubbles 12, although more or fewer bubbles may suitably be
provided.
Substrate 8 and cover layer 9 are advantageously formed to have a thickness of
about
0.001 inches (0.0254 mm) and typically comprise a thermoplastic material.
Materials
suitable for use as substrate 8 and/or cover layer 9 include, without
limitation,
polymers and copolymers of polyvinyl alcohol, polyvinyl pyrrolidinone,
polysaccharide polymers, acrylate polymers, methacrylate polymers, phthalate
polymers, polyvinyl acetate, methyl cellulose, carboxymethylcellulose,
hydroxyethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose,
ethyl
cellulose, polyethylene oxide, polypropylene, polyester and polyamide films,
Eudragits (that is, polymers and copolymers containing methacrylic acid),
starch-
based polymers, gelatin and the like. Polyvinyl alcohol films suitable for use
as the
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
_7_
substrate andlor cover layer are commercially available from Polymer Films,
Inc. of
West Haven, CT; Chris Craft of Gary, IN; Aquaflm of Winston-Salem, NC;
Idroplast
S.p.A. of Montecatini Terme (PT), Italy; Aicello Chemical Co., Ltd. of
Toyohashi;
Japan; and Soltec of Paris, France.
S As depicted in FIG. 2 (showing cover layer 9 partially "peeled" back
from substrate 8) and FIG. 3, a deposit of a dry active ingredient 14, in the
form of
powder(s)/grains (hereinafter, "powder") is disposed between substrate 8 and
cover
layer 9 within a bubble 12. Active ingredient 14 is deposited on substrate 8.
As
depicted via a cross-sectional view in FIG. 3 and plan view in FIG. 4 (each
showing
only a single bubble 12), substrate 8 and cover layer 9 are attached to one
another via
bonds or welds 7 that are near to and encircle bubble 12. Bonding can be
effected, for
example, via heat or ultrasonic welding or via suitable adhesives. Unit form 6
comprises a deposit of active ingredient 14, bubble 12, and a region of
substrate 8
within bonds 7. Unit form 6 is a stable "core" (hereinafter, an "AccudepTM
Core"),
which may be further processed into a dosage form resembling a conventional
tablet,
capsule, caplet and the like or processed into a non-conventional wafer or
stamp-like
presentation. The preferred dosage forms may be suitable for oral, transdermal
or
buccal dosing of appropriate drugs.
Suitable means of electrostatic deposition of active ingredient 14 are
described in, for example, U.S. Patent Nos. 5,714,007, 5,846,595 and
6,074,688, the
disclosures of which are incorporated by reference herein in their entireties.
In
addition to the electrostatic powder cloud deposition method, active
ingredient may be
coated onto the substrate in the form of a solution or a suspension of finely
divided
medicament; e.g., a collodial suspension. The liquid utilized for these
operations can
be water, an organic solvent, e.g., ethanol, or a hydroalcoholic solvent. One
method
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
_g_
of loading active ingredient in a liquid form onto a substrate is by
electrostatic jet
spray deposition. In this method, the active ingredient containing solution or
suspension is metered into an apparatus which projects a spray of
microdroplets
which are concentrated on a particular area of the substrate through the use
of a
defined area electrostatic field.
In addition to electrostatic jet spray deposition, certain other coating
techniques recognized in other arts as being amenable to the coating of a
substrate
with a liquid may be utilized in loading a pharmaceutically acceptable
substrate with
active ingredient. For example, the substrate may be passed under a roll which
is
immersed in a bath of saturating fluid. As the substrate passes the roller,
the excess
fluid is "wiped" from the substrate by another roller, a jet of air, a rubber
wiping bar, a
wire-wound rod, e.g., a Meier rod, or the like.
The present invention improves upon the prior art dosage forms
depicted in FIGS. 1 through 4 by providing a dissolution-enhancing amount of a
surfactant, disposed within a carrier that is segregated from, but in contact
with, the
active ingredient. The invention is based on the surprising fording that,
contrary to
the teachings in the prior art, a surfactant can improve the dissolution (and,
consequently, the bioavailability) of a hydrophobic drug even though the drug
and the
surfactant are not co-formulated in intimate admixture with one another.
Certain embodiments of the present invention are depicted in FIG. 5.
In the drawing with the legend "Deposition," active ingredient ("drug") 14 is
shown
after being deposited on substrate 8, prior to sealing with cover layer 9. In
the first
drawing with the legend "Cover Film" ("Surfactant in Pouch"), the surfactant
is
incorporated on the cover layer ("cover film") 9 in a pouch I6, and cover
Iayer 9 is
aligned to place the pouch 16 in contact with active ingredient 14. The pouch
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-9-
material may be any polymer, and preferably the same material as substrate 8
or cover
layer 9. Upon administration of the dosage form, during dissolution of cover
layer 9
and/or substrate 8, pouch 16 similarly dissolves and releases the surfactant
in the
immediate vicinity of the drug, thereby improving drug dissolution.
An alternative embodiment of the present invention is depicted in the
second drawing in FIG. 5 with the legend "Cover Film" ("Surfactant in
Adhesive").
In this embodiment, the surfactant is incorporated in an ingestible adhesive
10 that is
applied to cover layer 9. After sealing cover layer 9 to substrate 8, the
surfactant is in
contact with, but segregated from, active ingredient 14. Upon administration
of the
dosage form and dissolution of cover layer 9 andlor substrate 8, the adhesive
dissolves
and releases the surfactant in the immediate vicinity of the drug, again
improving drug
dissolution.
In a preferred embodiment (not specifically shown in FIG. 5), neither a
pouch 16 nor a special adhesive 10 is required. Rather, the surfactant is
incorporated
directly in cover layer 9, so that the dissolving cover layer 9 releases the
surfactant in
the immediate vicinity of the encapsulated hydrophobic drug, allowing the
surfactant
to interact with the drug to help with dissolution.
In the context of the present invention, "hydrophobic drug" means a
drug that ranges from "sparingly soluble" to "practically insoluble or
insoluble," as
shown in the following table:
Parts of Solvent Required
Descriptive Term For 1 Part of Solute
Sparingly soluble From 30 to 100
Slightly soluble From 100 to 1000
Very slightly soluble From 1000 to 10,000
Practically insoluble, or Insoluble 10,000 and over
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
- 10-
The hydrophobic drugs, and their pharmaceutically acceptable salts,
which may be formulated in accordance with the present invention include,
without
limitation, the following:
Analgesics and anti-inflammatory agents: acetaminophen, aloxiprin, auranofm,
azapropazone, benorylate, celecoxib, diflunisal, etodolac, fenbufen,
fenoprofen,
flurbiprofen, ibuprofen, indomethacin, ketoprofen, meclofenarizic acid,
mefenamic
acid, nabumetone, naproxen, oxyphenbutazone, phenylbutazone, piroxicam,
rofecoxib, salicylamide, salicylic acid, sulindac.
Anthelmintics: albendazole, bephenium hydroxynaphthoate, cambendazole,
dichlorophen, ivermectin, mebendazole, oxamniquine, oxantel embonate,
oxfendazole, praziquantel, pyrantel embonate, thiabendazole.
Anti-arrhythmic agents: amiodarone, disopyramide, flecainide, quinidine.
Anti-bacterial agents: benethamine, cefaclor, cinoxacin, ciprofloxacin,
clarithromycin,
clofazimine, cloxacillin, demeclocycline, doxycycline, erythromycin,
ethionamide,
imipenem, nalidixic acid, nitrofurantoin, penicillin, rifampicin, spiramycin,
sulphabenzamide, sulphacetamide, sulphadiazine, sulphadoxine, sulphafurazole,
sulphamerazine, sulphamethoxazole, sulphapyridine, tetracycline, trimethoprim.
Anti-coagulants: dicoumarol, dipyridamole, nicoumalone, phenindione.
Anti-depressants: amoxapine, maprotiline, mianserin, nortriptyline,
oxypertine,
trazodone, trimipramine.
Anti-diabetics: acetohexamide, chlorpropamide, glibenclamide, gliclazide,
glipizide,
tolazamide, tolbutamide.
Anti-epileptics: beclamide, carbamazepine, clonazepam, ethotoin, metharbital,
methoin, methsuximide, methylphenobarbitone, oxcarbazepine, paramethadione,
phenacemide, phenobarbitone, phensuximide, phenytoin, primidone, sulthiame,
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-11-
valproic acid.
Anti-fungal agents: amphotericin, butoconazole, clotrimazole, econazole,
fluconazole,
flucytosine, griseofulvin, itraconazole, ketoconazole, miconazole, natamycin,
nystatin, sulconazole, terbinafine, terconazole, tioconazole, undecenoic acid.
Anti-gout agents: allopurinol, probenecid, sulphinpyrazone.
Anti-hypertensive agents: amlodipine, benidipine, darodipine, diazoxide,
dilitazem,
felodipine, guanabenz, isradipine, methyldopa, minoxidil, nicardipine,
nifedipine,
nimodipine, phenoxybenzamine, prazosin, reserpine, terazosin.
Anti-malarials: amodiaquine, chloroquine, chlorproguanil, halofantrine,
mefloquine,
IO proguanil, pyrimethamine, quinine.
Anti-migraine agents: dihydroergotamine, ergotamine, methysergide, pizotifen,
sumatriptan.
Anti-muscarinic a ents: atropine, benzhexol, biperiden, ethopropazine,
hyoscyamine,
mepenzolate, oxyphencylcimine, tropicamide.
Anti-neoplastic agents and immunosuppressants: aminoglutethimide, amsacrine,
azathioprine, busulphan, chlorambucil, cyclosporin, dacarbazine, estramustine,
etoposide, fmasteride, lomustine, melphalan, mercaptopurine, methotrexate,
mitomycin, mitotane, mitozantrone, procarbazine, raloxifene, tamoxifen,
testolactone.
Anti-Parkinsonian aged: bromocriptine, lysuride.
Anti-protazoal agents: benznidazole, clioquinol, decoquinate,
diiodohydroxyquinoline, diloxanide, dinitolmide, furzolidone, metronidazole,
nimorazole, nitrofurazone, ornidazole, tinidazole.
Anti-thyroid agents: carbimazole, propylthiouracil.
Anxiolytics, sedatives, hypnotics and neuroleptics: allobarbitone,
allylbarbituric acid,
alprazolam, amylobarbitone, barbitone, bentazepam, bromazepam, bromperidol,
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
- 12-
brotizolam, butobarbitone, carbromal, carphenazine, chlordiazepoxide,
chlormethiazole, chlorpromazine, clobazam, clotiazepam, clozapine,
cyclobarbitone,
diazepam, droperidol, ethinamate, flunanisone, flunitrazepam, fluopromazine,
flupenthixol, fluphenazine, flurazepam, haloperidol, lorazepam, lormetazepam,
medazepam, meprobamate, methaqualone, midazolam, nitrazepam, oxazepam,
pentobarbitone, perphenazine, pimozide, prochlorperazine, sulpiride,
temazepam,
thioridazine, triazolam, zopiclone.
-Blockers: acebutolol, alprenolol, atenolol, labetalol, metoprolol, nadolol,
oxprenolol, pindolol, propranolol.
Cardiac Inotropic agents: amrinone, digitoxin, digoxin, enoximone, lanatoside
C,
medigoxin.
Corticosteroids: beclomethasone, betamethasone, budesonide, cortisone,
desoxymethasone, dexamethasone, flucortolone, fludrocortisone, flunisolide,
fluticasone, hydrocortisone, methylprednisolone, prednisolone, prednisone,
triamcinolone.
Diuretics: acetazolamide, amiloride, amisometradine, bendroflumethiazide,
bumetanide, chlorothiazide, chlorthalidone, ethacrynic acid, furosemide,
hydrochlorothiazide, metolazone, spironolactone, triamterene.
Gastro-intestinal a ents: aminosalicylic acid, bisacodyl, cimetidine,
cisapride,
diphenoxylate, domperidone, famotidine, loperamide, mesalazine, nizatidine,
omeprazole, ondansetron, ranitidine, sulphasalazine.
Histamine HI-Receptor Antagonists: acrivastine, astemizole, cinnarizine,
cyclizine,
cyproheptadine, dimenhydrinate, fexofenadine, flunarizine, loratadine,
meclozine,
oxatomide.
Lipid-r~yulatin~~ents: atorvastatin, bezafibrate, clofibrate, dextrothyroxine,
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-13-
fenofibrate, gemfibrozil, lovastatin, probucol, simvastatin.
Nitrates and other anti-anginal agents: amyl nitrate, glyceryl trinitrate,
isosorbide
dinitrate, isosorbide mononitrate, pentaerythritol tetranitrate.
Nutritional agents: betacarotene, vitamin A, vitamin B, vitamin D, vitamin E,
vitamin
K.
Opioid anal eg sics: codeine, dextropropyoxyphene, diamorphine,
dihydrocodeine,
meptazinol, methadone, morphine, nalbuphine, pentazocine.
Respirator a ents: montelukast, pranlukast (CCN00401), zafirlukast, zileuton.
Sex hormones: clomiphexie, conjugated estrogens, danazol, estradiol,
ethinyloestradiol, medrogestone, medroxyprogesterone acetate, mestranol,
methyltestosterone, norethisterone, norgestimate, norgestrel, progesterone,
stanozolol,
stiboestrol, testosterone, tibolone.
Stimulants: amphetamine, cocaine, dexamphetamine, dexfenfluramine,
fenfluramine,
mazindol.
Thyroid agents: levothyroxine.
By "surfactant" is meant, for purposes of the present invention, that the
material is a surface active agent which displays wetting, detergent or soap-
like
qualities as those agents are understood by those of ordinary skill in the
art. Thus, the
term "surfactant," as used herein, represents ionic and nonionic surfactants
or wetting
agents commonly used in the formulation of pharmaceuticals, such as
ethoxylated
castor oil, benzalkonium chloride, polyglycolyzed glycerides, acetylated
monoglycerides, sorbitan fatty acid esters, poloxamers, polyoxyethylene fatty
acid
esters, polyoxyethylene derivatives, monoglycerides or ethoxylated derivatives
thereof, diglycerides or polyoxyethylene derivatives thereof, sodium docusate,
sodium
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-14-
lauryl sulfate, magnesium lauryl sulfate, triethanolamine, cetrimide, sucrose
laurate
and other sucrose esters, glucose (dextrose) esters, simethicone, ocoxynol,
dioctyl
sodium sulfosuccinate, polyglycolyzed glycerides, sodium dodecylbenzene
sulfonate,
dialkyl sodium sulfosuccinate, fatty alcohols such as lauryl, cetyl, and
steryl,
glycerylesters, cholic acid or derivatives thereof, lecithins, and
phospholipids.
The surfactants of the invention may be classified by an "HLB
number." The HLB number provides a means for ranking surfactants based on the
balance between the hydrophilic and lipophilic portions of the surfactant.
That is, the
higher the HLB number, the more hydrophilic the surfactant.
In a broader implementation of the present invention, many other types
of pharmaceutical additives (instead off, or in addition to, the surfactant)
may be
included in the dosage form disposed within a carrier that is segregated from,
but in
contact with, the deposited active ingredient. Such pharmaceutically
acceptable
additives include, but are not limited to, antioxidants, antimicrobial agents,
complexing agents, acidity boosting agents, alkalinity boosting agents,
buffering
agents, carrier molecules, chelating compounds, preservatives and the like.
"Pharmaceutically acceptable" here means that the additive may be introduced
safely
into the human or animal body, for example, taken orally and digested.
Examples of
such additives include, but are not limited to, the following:
Acidifying agents: citric acid, malefic acid, lactic acid, malic acid,
succinic acid,
tartaric acid.
Alkalinity buffering agents: calcium carbonate, monoethanolamine, potassium
citrate, sodium bicarbonate, sodium citrate, triethanolamine.
Anti-microbial agents: benzethonium chloride, benzoic acid, bronopol,
butylparaben,
cetrimide, chlorhexidine, chlorobutanol, chlorocresol, cresol, editic acid,
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-15-
ethylparaben, glycerol, imidurea, methylparaben, phenol, phenolic acid,
phenoxyethanol, phenyl ethyl alcohol, phenylmercuric salts (acetate, borate
and
nitrate), potassium sorbate, propylene glycol, propylparaben, sodium benzoate,
sodium propionate, sorbic acid, thimerosol.
Anti-oxidants: alpha tocopherol, ascorbic acid, ascorbic acid palmitate,
butylated
hydroxyanisole, fumaric acid, malic acid propyl gallate, sodium ascorbate,
sodium
metabisulfate.
Complexing agents: EDTA, potassium citrate, sodium citrate.
EXAMPLES
The following materials were used in the Examples:
Hydroxypropylmethylcellulose E50 ("HPMC"), available from Dow Chemical
Company, Midland, Michigan.
Hydroxypropylcellulose JFP ("HPC"), available from Hercules Inc., Wilmington,
Delaware.
Polyethylene Glycol 400 ("PEG"), available from Union Carbide Corporation,
Danbury, Connecticut
Sodium lauryl sulfate ("SLS"), HLB = 40, available from Spectrum Quality
Products,
New Brunswick, New Jersey
Polysorbate 80 (Tween 80), HLB = 15, available from Uniqema, a division of
ICI,
Wilmington, Delaware
Polysorbate 20 (Tween 20), HLB= 16.7, available from Uniqema
Hydrocortisone, available from Spectrum Quality Products
Glipizide, available from Fine Chemicals Corporation, Capetown, South Africa.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
- 16-
Example 1
A model compound, CCN00401, was used to test the effect sodium
lauryl sulfate (SLS) has on dissolution of an AccudepTM Core. Several types of
AccudepT"~ Cores were made as follows:
Control AccudepTM Core: Sealed a 1 mg deposition of CCN00401
between two polymer sheets with the following composition: 45% HPMC, 45%
HPC, 10% PEG.
SLS incorporated in film AccudepTM Core: Sealed a 1 mg deposition of
CCN00401 between two polymer sheets with the following composition: .33.75%
HPMC, 33.75% HPC, 7.5% PEG, 25% SLS (equivalent of about 1.2 mg SLS
incorporated in each AccudepT"~ Core).
SLS mixed directly with CCN00401 AccudepT"" Core: Sealed mixture
of 2 mg of CCN00401/SLS mixture (50!50) between two polymer sheets with the
following composition: 45% HPMC, 45% HPC, 10% PEG.
I S Dissolution profiles for the AccudepT~~ Cores listed above were
generated under the following conditions: SO rpm, paddles, pH 8.0 TRIS buffer.
In
addition, a set of Control AccudepTM Cores were tested in dissolution media
that also
contained Polysorbate 20.
Fig. 6 shows the average dissolution profiles fox all the CCN00401
dissolution runs (n=3). As seen in Fig. 6, the addition of SLS in the polymer
film or
mixed directly with the drug led to significantly faster dissolution at 15
minutes (even
faster than the case where Polysorbate 20 is present in the media). At 30
minutes all
experimental sets had average dissolution in the mid-to-high 90% range except
for the
case with no surfactant in AccudepT~~ Core or dissolution media.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-17-
Example 2
Hydrocortisone, CCN90306A, was used to test the effect SLS and
Polysorbate 80 have on dissolution of an AccudepT~~ Core. AccudepT"" Cores
were
made as follows:
Control AccudepT"~ Core: Sealed a 1 mg deposition of CCN90306A
between two polymer sheets with the following composition: 45% HPMC, 45%
HPC, 10% PEG.
SLS incorporated in film AccudepTM Core: Sealed a 1 mg deposition of
CCN90306A between two polymer sheets with the following composition: 36%
HPMC, 36% HPC, 8% PEG, 20% SLS (equivalent of about 5 mg SLS incorporated in
each AccudepT"~ Core).
Polysorbate 80 incorporated in film AccudepT"~ Core: Sealed a 1 mg
deposition of CCN90306A between two polymer sheets with the following
composition: 36% HPMC, 36% HPC, 8% PEG, 20% Polysorbate 80 (equivalent of
about 5 mg Polysorbate 80 incorporated in each AccudepT"" Core).
Dissolution profiles for the AccudepT~" Cores listed above were
generated under the following conditions: 75 rpm, paddles, distilled water.
Fig. 7 shows the average dissolution profiles for all the CCN90306A
dissolution runs (n=3). As seen in Fig. 7, the addition of SLS and Polysorbate
in the
polymer film led to faster dissolution at 20- and 30-minute sample points.
CA 02417813 2003-O1-29
WO 02/13762 PCT/USO1/24949
-18-
Example 3
Glipizide, CCN90906A, was used to test the effect SLS and
Polysorbate 80 have on dissolution of an AccudepT~~ Core. AccudepT"~ Cores
were
made as follows:
SLS incorporated in film AccudepT"" Core: Sealed a 1 mg deposition of
CCN90906A between two polymer sheets with the following composition: 36%
HPMC, 36% HPC, 8% PEG, 20% SLS (equivalent of about 5 mg SLS incorporated in
each AccudepT"" Core).
Polysorbate 80 incorporated in film AccudepT"" Core: Sealed a 1 mg
deposition of CCN90906A between two polymer sheets with the following
composition: 36% HPMC, 36% HPC, 8% PEG, 20% Polysorbate 80 (equivalent of
about 5 mg Polysorbate 80 incorporated in each AccudepT"~ Core).
Dissolution profiles for the AccudepT"~ Cores listed above were
generated under the following conditions: 50 rpm, paddles, simulated
intestinal fluid.
Fig. 8 shows the average dissolution profiles for all the CCN90906A
dissolution runs (n=6). As seen in Fig. 8, the addition of SLS and Polysorbate
in the
polymer film led to faster dissolution, especially during the first 60
minutes.
Although the present invention has been described with particular
reference to certain preferred embodiments thereof, variations and
modifications of
the present invention can be effected within the spirit and scope of the
following
claims.