Note: Descriptions are shown in the official language in which they were submitted.
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
TITLE OF THE INVENTION
ACTIVE METABOLITE OF ANT1FUNGAL COMPOUND
BACKGROUND OF THE INVENTION
This invention relates to the identification of a metabolite compound of
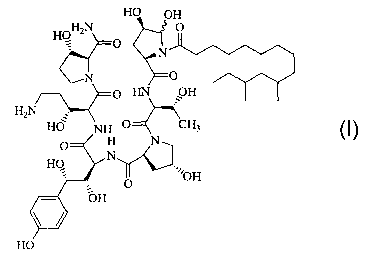
Formula I
HO OH
HO aN
O
1V
O
H2N
HO ~ru
O H N
HO N
I~~~OH
O
OH
Formula I
having antifungal activity. This metabolite has been found to be the major
metabolite
of CANCIDASTM (caspofungin acetate) produced in human plasma. Caspofungin
acetate is a semisynthetic antifungal agent developed for the parenteral
treatment of
Caszdida, Aspergillus, P~zeumocystis carirzii and other mycotic infections.
Its activity
is mediated by inhibition of the synthesis of (3-(1,3)-glucan, an integral
component in
the cell wall of the target organisms. Caspofungin is a macrocyclic peptide
with a
molecular weight of 1094 disclosed in US Patent No. 5,378,804. This compound
possesses a variety of potentially reactive functionalities that include an
aminal
moiety. The metabolite is formed by the hydrolysis of the aminal in the
ornithine
residue, ring opening and recyclization to form two isomeric 5-membered cyclic
hemiaminal products. These two isomers are not easily separable by high
performance liquid chromatography and thus are isolated together as a single
product
and collectively termed "the metabolite".
-1--
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
H2N
NH OH
HO O NH
N
O
H2N
HO Nu O
O N N '2 HOAc
HO ~"J~~'~OH
O
OH
Caspofungin acetate
In prior work, it had been determined that a related ring-opened analog
of pneumocandin Bo was found to possess very poor (3-(1,3)-glucan synthesis
activity.
This hexapeptide metabolite of Formula I, however, has been found to have
surprising
activity against Candida isolates.
A ring-opened analog of pneumocandin Bo was disclosed by Merck
Research Laboratories. See J. M. Balkovec et al., "Synthesis, Stability and
Biological
Evaluation of Water Soluble Prodrugs of a New Echinocandin Lipopeptide.
Discovery of a Potential Clinical Agent for the Treatment of Systemic
Candidiasis and
Pneumocystis cariuii Pneumonia (PCP)" J. Med. Chem. 1992, 35, 194-8. The
synthesis of compound 2 an intermediate in the synthesis of the compound of
Formula
I was first described by F.A. Bouffard et al., "Synthesis and Antifungal
Activity of
Novel Cationic Pneumocandin BO Derivatives" J. Med. Chem. 1994, 37, 222-5. The
preparation of the compound of Formula I begins with pneumocandin Bo that can
be
isolated by following the procedures disclosed by Merck Research Laboratories.
See
R.E. Schwartz, et al., Pneumocandins from Zalerion arboricola I. Discovery and
isolation. J. A~ctibiot. (1992) 45:1853-1866, and P. S. Masurekar et al.,
Pneumocandins from Zalerion arboricola II. Modification of product spectrum by
mutation and medium manipulation. J. Afatibiot. (1992) 45:1867-1874, and US
Patent
-2-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Nos. 5,194,377 and 5,202,309. US Patent Nos. 5,854,212 and 5,939,384 also
disclose
a method for the preparation of Pneumocandin BO See Example 1.
Recent disclosures discuss the formation of a metobolite/degradation
byproduct of caspofungin. See XU, X., DELUNA, F., YUANG, A., CYLC, D.,
DAVIS, M., SAHLY, Y. and LIN, J.H. Slow hepatic uptake and elimination play an
important role in the disposition of L-743,872, a potent antifungal agent, in
rats. For
presentation at: American Association of Pharmaceutical Scientists 10th Annual
Meeting (AAPS), Seattle, Washington, 10/26/1996 - 10/31/1996; KAUFMAN, M.J.
and NERURKAR, M. Degradation of the macrocyclic antifungal agent L-743,872:
Reaction products and kinetics, and stabilization strategies. For presentation
at:
American Chemical Society 31st Middle Atlantic Regional Meeting,
Pleasantville,
New York, 05/27/1997 - 05/30/1997; KAUFMAN, M.J, and NERURKAR, M.
Degradation of the macrocyclic antifungal agent L-743,872: Reaction products
and
kinetics, and stabilization strategies. For presentation at: American Chemical
Society
31st Middle Atlantic Regional Meeting, Pleasantville, New York, 05127/1997
05/30/1997; BALANI, S.K., XU, X., ARISON, B.H., S7LVA, M.V., GRIES, A.,
DELUNA, F.A., CUI, D., KARI, P.H., LY, T., HOP, C.E.C.A., SINGH, R.,
WALLACE, M.A., DEAN, D.C., LIN, J.H., PEARSON, P.G. and BAILLIE, T.A.
Metabolites of caspofungin acetate, a potent antifungal agent, in human plasma
and
urine. For submission to: Drug Metabolism and Disposition; and MCQUADE, M.S.,
FORSYTH, R.J., Z>IVVIMERMAN, J. and ROBERTS, J.C. Stability of reconstituted
CancidasTM (caspofungin acetate) in commonly used i.v. solutions and flexible
polyvinyl chloride containers. For submission to: American Journal of Health-
System
Pharmacy.
SUMMARY OF THE INVENTION
A method for treating a fungal infection comprising administering to a
mammalian subject in need of such treatment, an effective amount of a compound
of
Formula I or its pharmaceutically acceptable salt,
-3-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
HO OH
O
O
N ~ ~ a
O
O HN ~OH
Hz~
HU NH O CH3
O H N
N
HO .~''OH
O
OH
Formula I
Additional aspects of the invention include: 1) a method for controlling
mycotic
infections comprising administering to an immune-comprised patient in need of
such
treatment, an effective amount of a compound of Formula I or its
pharmaceutically
acceptable salt; 2) a method for controlling Pheu~raocystis pneumonia
comprising
administering to an immune-comprised patient in need of such treatment, an
effective
amount of a compound of Formula I or its pharmaceutically acceptable salt; 3)
a
pharmaceutical composition comprising a compound of Formula I or its
pharmaceutically acceptable salt and a second antifungal agent selected from
the
group consisting of: azoles or polyenes, purine or pyrimidine nucleotide
inhibitors,
chitin synthesis inhibitors, elongation factor inhibitors, mannan-binding
antifungal
agents, bactericidal/permeability-inducing (BPI) protein products, and complex
carbohydrate antifungal agents; and 4) a method of treating a fungal infection
comprising administering to a mammalian subject in need of such treatment an
effective amount of a combination of a compound of Formula I or a
pharmaceutically
acceptable salt thereof and a second antifungal agent selected from the group
consisting of: azoles or polyenes, purine or pyrimidine nucleotide inhibitors,
chitin
synthesis inhibitors, elongation factor inhibitors, mannan-binding antifungal
agents,
bactericidal/permeability-inducing (BPI) protein products, and complex
carbohydrate
antifungal agents.
-4-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
DETAILED DESCRIPTION OF THE INVENTION
A method for treating fungal infections comprising administering to a
mammalian subject in need of such treatment, an effective amount of a compound
of
Formula I or its pharmaceutically acceptable salt,
HO OH
HOH2N
O
N
O
H2N
HO Nu O
O H N
N
HO I''°OH
O
OH
Formula I
The method as. recited above, wherein the compound of Formula I is
administered as a pharmaceutical composition of said compound with a
pharmaceutically acceptable carrier.
The method as recited above, wherein the pharmaceutical composition
is administered parenterally or orally.
The method as recited above, wherein the pharmaceutical composition
is administered parenterally.
The method as recited above, wherein the pharmaceutical composition
is administered by intravenous infusion.
A method for controlling mycotic infections comprising administering
to a mammalian subject in need of such treatment, an effective amount of a
compound
of Formula I or its pharmaceutically acceptable salt,
-5-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
HO OH
HOH2N O
O I
N
N O
O HN ~OH
HaN
HO ~ O CH3
O H N
N
HO ~''OH
O
OH
Formula I
The method as recited above, wherein the compound of Formula I is
administered as a pharmaceutical composition of said compound with a
pharmaceutically acceptable carrier.
The method as recited above, wherein the pharmaceutical composition
is administered parenterally.
The method as recited above, wherein the pharmaceutical composition
is administered by intravenous injection.
The method as recited above, wherein the pharmaceutical composition
is administered by intravenous infusion.
A method for controlling Pneumocystis pneumonia comprising
administering to an immune-comprised patient in need of such treatment, an
effective
amount of a compound of Formula I or its pharmaceutically acceptable salt,
-6-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
HO OH
HOH2N O
O N
N O
O HN ~OH
H2N
HO ~ O CH3
O H N
N
HO I~''OH
O
OH
Formula I
The method as recited above, wherein the compound of Formula I is
administered as a pharmaceutical composition of said compound with a
pharmaceutically acceptable carrier.
The method as recited above, wherein the pharmaceutical composition
is administered parenterally.
The method as recited above, wherein the pharmaceutical composition
is administered by intravenous injection.
The method as recited above, wherein the pharmaceutical composition
is administered by intravenous infusion.
The present invention also relates to a pharmaceutical composition
comprising a combination of the compound of Formula I and a second antifungal
agent, as well as an antifungal combination therapy comprising the
administration of
the antifungal compound of Formula I and a second antifungal agent.
It is understood that this combination therapy would involve the
sequential, simultaneous or concomitant administration of these two agents.
More
particularly, the invention relates to antifungal combination therapy
comprising the
use of a second antifungal agent such as: azoles or polyenes, purine or
pyrimidine
nucleotide inhibitors, chitin synthesis inhibitors, elongation factor
inhibitors,
mannan-binding antifungal agents, bactericidal/permeability-inducing (BPI)
protein
products, and complex carbohydrate antifungal agents.
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Representative examples of such second antifungal agents are: azoles,
such as fluconazole, voriconazole, itraconazole, ketoconazole, miconazole,
ravuconazole, posaconazole; polyenes such as amphotericin B, nystatin or
liposomal
and lipid forms thereof such as ABELCET, AMBISOME and AMPHOCIL; purine or
pyrimidine nucleotide inhibitors such as flucytosine; or polyoxins such as
nikkomycins, in particular nikkomycin Z or other chitin synthesis inhibitors,
elongation factor inhibitors such as sordarin and analogs thereof, mannan-
binding
antifungal agents such as the pradamicins, bactericidal/permeability-inducing
(BPI)
protein products such as XMP.97 or XMP.127 or complex carbohydrate antifungal
agents such as CAN-296.
In particular, this combination therapy has been shown to be useful
against such opportunistic pathogens as Cryptococcus spp., Candida spp.,
Aspergillus spp., Histoplasma spp., Coccidioides spp., Paracoccidioides spp.
Blastomyces spp., Fusariunz spp., Sporotlzrix spp., Trichospororz spp.,
Rlzizopus spp.,
Pseudallescheria spp., dermatophytes, Paeciliornyces spp., Alternaria spp.,
Curvularia spp., Exophiala spp., Wangiella spp., Penicillium spp.,
Saccharonzyces
spp., Dematiaceous fungi and Ptzeumocystis caritzii.
CANC>DASTM (Caspofungin acetate) is disclosed in U.S. Patent No.
5,378,804 and its preparation is described in that patent, as well as U.S.
Patent No.
5,552,521.
The azole, polyene or other antifungal agent may be administered
orally or parenterally. The compound of Formula I is preferably administered
parenterally, but is not limited to that route and may also be administered by
other
routes such as oral, intramuscular or subcutaneous.
Combination therapy results in enhanced effects using sub-inhibitory
concentrations of all agents. These effects may be demonstrated in vitro and
in vivo
using clinical and environmental strains of C. neoformans, C. albicans and A.
funzigatus.
The compound of Formula I is formed from caspofungin by hydrolysis
of the aminal group, followed by ring opening and recyclization onto N2 of the
ornithine group.
The compound of Formula I may be prepared from pneumocandin Bo
(Compound I) following the procedures outlined in SCHEME 1. Briefly, the 3-
hydroxyglutamine residue of pneumocandin Bo rnay be reduced to give Compound
2.
Borane dimethylsulfide complex is a preferred reducing agent. Compound 2 may
be
_g_
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
treated with a base such as triethylamine, to effect ring opening and
subsequent
recyclization to give the compound of Formula I. The compound of Formula I is
formed as a mixture of two isomers at the newly formed hemiaminal group. The
isomers are not easily separable by standard chromatographic techniques and
thus
may be used as a mixture.
-9-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
SCHEME 1
HOr ,OH
HO O
N
.,.~H H
O N
O
~nH
' BH~SMe2
H2N HO~ NH
O H Hue, N
N THF
HO; ',,H ~~'~~OH
O
OH
Pneumocandin Bo
(Compound I)
H
HO; ,~OH
HO O
r
.,,~H f
N
C
'~-~ nH
H2N HO~ NH Et3N
O H
N CN, H20
HO; .,,~H
OH
2
-10-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
SCHEME 1 (continued)
HO OH
HOH2N O
O N
I O
N
O HN ~OH
HzN
HO ~ O CH3
O H N
N
HO ~~''OH
O
OH
Formula I
The invention also embraces acid addition salts. The compound of
Formula I in the normal course of isolation is obtained as an acid addition
salt. The
salt thus obtained may be dissolved in water and passed through an anion
exchange
column bearing the desired anion. The eluate containing the desired salt may
be
concentrated to recover the salt as a solid product.
The compound of Formula I was tested for yeast susceptibility
following protocol M27-A. This protocol is the standard by which all hospital
clinical laboratories test their yeast isolates for susceptibility to
antifungal agents.
See J.N. Galgiani et al. Reference Method for Broth Dilution Antifungal
Susceptibility
Testing: Approved Standard M27-A (Reference F-7), National Committee for
Clinical
Laboratory Standards1997; 17(9):1-29. A description of this method follows:
The
inoculum was standardized with a spectrophotometer (optical density, 550 nm)
and
was diluted to a final concentration of 0.5 x 103 to 2.5 x 103 in RPMI 1640
medium
with L-glutamine, without sodium bicarbonate, buffered with 0.165 M MOPS
(morpholinepropanesulfonic acid) (BioWhittaker, Walkersville, MD).
The compound of Formula I along with appropriate control standards
were prepared as concentrated stock solutions in sterile deionized water and
diluted in
RPMI 1640 medium, and tested at concentrations ranging from 128 (ug/mL down to
-11-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
0.06 ~,g/mL in two-fold serial dilutions. The Minimum Inhibitory Concentration
(MIC) was defined as the lowest concentration of compound which completely
inhibited visible growth after incubation at 35°C for 24 or 48 hr.
The same method as described above was used for Aspergillus except
that the reference standard is M38-P [National Committee for Clinical
Laboratory
Standards. Reference method for broth dilution antifungal susceptibility
testing of
conidium-forming filamentous fungi. Proposed standard M38-P. Wayne, PA:
National Committee for Clinical Laboratory Standards; 1998.] and the inoculum
final
concentration was 1 to 5 x 104 conidia/ml and the MIC is defined as the lowest
concentration of compound which produces prominent reduction in growth as
compared to the growth control (approximately 50% of control). The results are
shown in Table A:
Table A. Minimum Inhibitory Concentrations of Compound I Against Cafadida and
Asper~illus st~ecies
MIC (~. mL)
Or anism Isolate 24 h 48 h
C. albicafZS MY 1055 1-2 2
C. albicans ATCC 24433 1-2 2
~. albicans ATCC 90028 1 2
C. tro icalis ATCC 750 1 1
C. krusei ATCC 6258 16 16-32
A. fumigatus MF 5668 64 >64
A. fur~zigatus MF 5669 64 >64
The compound of Formula I also shows in vivo effectiveness against
fungi which may be demonstrated using the following in vivo assay.
Growth from an overnight SDA culture of Cafzdida albicans MY 1055
was suspended in sterile saline and the cell concentration determined by
hemacytometer count and the cell suspension adjusted to 3.75 x 105 cells/ml.
Then
0.2 milliliter of this suspension was administered LV. in the tail vein of
mice so that
the final inoculum was 7.5 x 104 cells/mouse.
The assay then was carried out by administering aqueous solutions of
compound of Formula I at various concentrations intraperitoneally (LP.),
thrice daily
-12-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
(t.i.d.) for one day to 18 to 20 gram female DBA/2 mice, which previously had
been
infected with Candida albicans in the manner described above. Deionized water
was
administered LP. to C. albicahs challenged mice as controls. After 24 hours,
the mice
were sacrificed by carbon dioxide gas, paired kidneys were removed aseptically
and
placed in sterile polyethylene bags containing 5 milliliters of sterile
saline. The
kidneys were homogenized in the bags, serially diluted in sterile saline and
aliquots
spread on the surface of SDA plates. The plates were incubated at 35°C
for 48 hours
and yeast colonies were enumerated to determine the number of colony forming
units
(CFU) per gram of kidneys. The results are shown in Table B. Compound of
Formula I reduced the tissue burden in the kidneys of infected animals at
doses of
0.31, 1.25, 5 and 10 mg/kg/day.
Table B. In Vivo Anti-Cahdida Activity of Compound of Formula I
Dose Log CFU/gm Reduction
Com ound m da Mortality Kidne from sham
Formula I 10 0% 5.19 -0.41
Formula I 5 0% 5.26 -0.34
Formula I 2.5 0% 5.48 -0.12
Formula I 1.25 0% 5.24 -0.36
Formula I 0.63 0% 5.51) -0.09
Formula I 0.31 0% 5.23 -0.37
Deionized water 0 0% 5.60 ---
The compound of Formula I is also useful for inhibiting or alleviating
Pneumocystis carinii infections in immune-compromised patients. The efficacy
of the
compounds of the present invention for therapeutic or anti-infection purposes
may be
demonstrated in studies on immunosuppressed rats.
In a representative study, Sprague-Dawley rats (weighing
approximately 250 grams) are immunosuppressed with dexamethasone in the
drinking
water (2.0 mg/L) and maintained on a low protein diet for seven weeks to
induce the
development of Pn.eumocystis pneumonia from a latent infection. Before drug
treatment, two rats are sacrificed to confirm the presence of Pi2eurraocystis
carifaii
pneumonia (PCP). Five rats (weighing approximately 150 grams) are injected
twice
-13-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
daily for four days subcutaneously (sc) with compound of Formula I in 0.25 ml
of
vehicle (distilled water). A vehicle control is also carried out. All animals
continue
to receive dexamethasone in the drinking water and a low protein diet during
the
treatment period. At the completion of the treatment, all animals are
sacrificed, the
lungs are removed and processed, and the extent of disease determined by
microscopic analysis of stained slides.
The outstanding properties are most effectively utilized when the
compound is formulated into novel pharmaceutical compositions with a
pharmaceutically acceptable carrier according to the conventional
pharmaceutical
compounding techniques.
The novel compositions contain at least a therapeutic antifungal or
antipneumocystis amount of the active compound. Generally, the composition
contains at least 1 % by weight of the compound of Formula I. Concentrate
compositions suitable for dilutions prior to use may contain 90% or more by
weight.
The compositions include compositions suitable for oral, topical, parenteral
(including
intraperitoneal, subcutaneous, intramuscular, and intravenous), nasal, and
suppository
administration, or insufflation. The compositions may be prepacked by
intimately
mixing the compound of Formula I with the components suitable for the medium
desired.
Compositions formulated for oral administration may be a liquid
composition or a solid composition. For liquid preparation, the therapeutic
agent may
be formulated with liquid carriers such as water, glycols, oils, alcohols, and
the like,
and for solid preparations such as capsules and tablets, with solid earners
such as
starches, sugars, kaolin, ethyl cellulose, calcium and sodium carbonate,
calcium
phosphate, kaolin, talc, lactose, generally with lubricant such as calcium
stearate,
together with binders disintegrating agents and the like. Because of their
ease in
administration, tablets and capsules represent the most advantageous oral
dosage
form. It is especially advantageous to formulate the compositions in unit
dosage form
(as hereinafter defined) for ease of administration and uniformity of dosage.
Compositions in unit dosage form constitute an aspect of the present
invention.
Compositions may be formulated for injection and may take such
forms as suspensions, solutions or emulsions in oily or aqueous vehicles such
as 0.85
percent sodium chloride or 5 percent dextrose in water and may contain
formulating
agents such as suspending, stabilizing and/or dispersing agents. Buffering
agents as
well as additives such as saline or glucose may be added to make the solutions
-14-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
isotonic. The compound may also be solubilized in alcohol/propylene glycol or
polyethylene glycol for drip intravenous administration. These compositions
also may
be presented in unit dosage form in ampoules or in multidose containers,
preferable
with added preservative. Alternatively, the active ingredients may be in
powder form
for reconstituting with a suitable vehicle prior to administration.
The term "unit dosage form" as used in the specification and claims
refers to physically discrete units, each unit containing a predetermined
quantity of
active ingredient calculated to produce the desired therapeutic effect in
association
with the pharmaceutical carrier. Examples of such unit dosage forms are
tablets,
capsules, pills, powder packets, wafers, measured units in ampoules or in
multidose
containers and the like. A unit dosage of the present invention will generally
contain
from 100 to 200 milligrams of one of the compounds.
When the compound is for antifungal use any method of administration
may be employed. For treating mycotic infections, oral or intravenous
administration
is usually employed.
When the compound is to be employed for control of Pfzeumocystis
infections it is desirable to directly treat lung and bronchi. For this reason
inhalation
methods are preferred. For administration by inhalation, the compounds of the
present inventions are conveniently delivered in the form of an aerosol spray
presentation from pressurized packs or nebulizers. The preferred delivery
system for
inhalation is a metered dose inhalation (MDI) aerosol, which may be formulated
as a
suspension or solution of compound of Formula I in suitable propellants, such
as
fluorocarbons or hydrocarbons.
Although the compounds of the present invention may be employed as
~ tablets, capsules, topical compositions, insufflation powders, suppositories
and the
like, the solubility of the compounds of the present invention in water and
aqueous
media render them adaptable for use in injectable formulations and also in
liquid
compositions suitable for aerosol sprays.
The following examples illustrate the invention but are not to be
construed as limiting.
-15-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
EXAMPLE 1
Preparation of Pneumocandin Bo
R~ R2
Rs O NH O
N
R5 N H
O
H2NOCH2C O ~ ,OH
HO~' ~ O R3
O H N
N
HO .,~~OH
O
R4
Com ound R1 RZ R3 R4 RS RG
I OH OH CH3 OH H OH
II OH H CH3 OH H OH
III OH =O CH3 OH H OH
IV OH OH H OH H OH
V OH OH CH3 H H OH
VI OH OH CH3 OH H H
VII H H CH3 OH OH OH
VIII H OH CH3 OH H OH
IX H H CH3 OH H OH
X OH OH CH3 OH OH H
XI OH OH CH3 OH OH OH
XII OH OH CH3 OH CH3 OH
Part A. Pneumocandin Bo (also referred to as compound I) is disclosed in U.S.
Patent No. 5,202,309 which issued April 13, 1993 and is produced by
cultivating the
fungus Glarea lozoye~csis (formerly identified as Zalerio~c arboricola) under
aerobic
-16-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
conditions. A process for the production of Pneumocandin Bo is disclosed in
U.S.
Patent 5,194,377 which issued March 16, 1993. Pneumocandin Bo is produced by
cultivating Glarea lozoyensis, ATCC No. 20868, deposited under the Budapest
Treaty
in the Culture Collection of American Type Culture Collection at 12301
Parklawn
Drive, Rockville, Md. 20852. Pneumocandin Bo can also be prepared following
the
procedures described in US Patent No. 5,939,384. Alternatively, Pneumocandin
Bo
can be isolated following the procedure described below.
Part B. Alternate Synthesis for Pneumocandin Bo
Culture
The fungus Glarea lozoyerZSis (ATCC 74030) is used to produce
Compound I and the structurally related analogues. This improved production
strain
was derived ultimately from the wild-type organism, ATCC 20868, (isolated from
a
sample of fresh water) by sequential steps of N-methyl-N'-nitro-N-
nitrosoguanidine
mutagenesis. The culture was maintained as aliquots of a mycelial suspension
in 5%
(v/v) glycerol stored at -70°C.
Shake-Flask Scale Fefmefitations-Control Process
A 250 ml Erlenmeyer flask containing 50 ml of LYCP-5 medium
was inoculated aseptically with 1 ml of a thawed culture stock. This first
stage seed
culture was incubated at 25°C with 220 rpm agitation for 3-5 days. A 1
ml aliquot of
the first stage seed was transferred to a second 250 ml Erlenmeyer flask
containing 50
ml of LYCP-5 medium. This second stage seed culture was incubated as above for
3
days.
_I7_
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Table 1: LYCP-5 seed medium
Concentration
Com onent ( er liter)
~2P04 9
Lactic acid (80-90%)2 mL
Glucose 25
Yeast Extract 5
Pharmamedia 10 g
FeS04 7H20 10 m
MnS04 H20 IO m
CuCl2 2H20 0.25 mg
CaCl2 2H20 0.1 mg
H3B~3 0.56 mg
(NH4)6Mo7024 4Ha0 0.19 m
ZnS04 7H20 2 mg
For each variable tested (i.e., treatment group), several 250 ml
Erlenmeyer flasks each containing 25 ml FGY medium (Table 2) or a variation
thereof (described below) were inoculated at 5% (v/v) with second stage seed.
The
flasks were incubated at 25°C with 220 rpm agitation for 14 days. The
pH for each
treatment group was adjusted as required by removing one flask from the group,
adding acid or base to return the pH to 5.0-5.5, and then adding this same
volume of
sterile titrant to the remaining flasks in the group. Where required, a volume
of a
sterile fructose solution was added during the fermentation to maintain the
residual
concentration within a specific range.
_18_
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Table 2: FGY production medium
Concentration
Com onent ( er liter)
Monosodium lutamate 10 g
Fructose 125
Yeast Extract 8.3
KHZPO4 1.5
MgS04 7H20 0.4
Proline 15
MES Buffer 15 g
FeS047H20 8.3 mg
MnS04 7H20 8.3 mg
CuC127H20 0.21 mg
CaC127H20 0.83 mg
H3B03 0.47 mg
(~4)GM07O24 4H20 0.16 m
ZnSO 7H20 1.7 mg
Analysis of the pneumocandins produced was carried out by extracting
the whole broth with organic solvent followed by chromatographic analysis
using
standard reverse phase and normal phase procedures. The titer of Pneumocandin
Bo is
expressed as arbitrary "units". The levels of the structural analogues is
expressed as a
ratio percent of the amount of Pneumocandin Bo produced.
Amino Acid Supplementation
On or about day 6 (i.e., mid-cycle) of the fermentation, sterile solutions
of L-proline, traps-3-hydroxy-L-proline, traps-4-hydroxy-L-proline, threonine,
serine,
arginine, ornithine or glutamine were added to the fermentation to give
appropriate
final concentrations. Pneumocandin extraction and analysis was carried out
after 14
days of fermentation.
Increasing the proline concentration in the base medium (0-15 gm/1)
resulted in a dose-dependent reduction in the levels of Compounds X and XI
while the
level of Compound VI increased as a function of proline concentration (Table
3). A
-19-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
15 gm/1 addition of proline to each of these treatments on or around day 6
resulted in
comparable titers for each treatment but was unable to off-set the effects of
the initial
level of proline in the medium.
Table 3: Effect of varying the initial proline
concentration in the base medium
0 gm/L 5 gm/L 10 gm/L 15 gm/L
Com ound Proline Proline Proline Proline
I 100 units 112 105 106
X 7.0 % 5.8 5.1 4.4
VI 2.1 % 2.5 2.6 2.7
XI 0.9 % 0.5 0.4 0.3
IX 2.6 % 3.6 3.4 3.7
The mid-cycle addition of hydroxyprolines impacted the fermentation
as well (Table 4). A 5 gm/L addition of traps-3-hydroxy-L-proline resulted in
a
50% improvement in titer with the levels of Compounds X, VI and XI reduced
dramatically. Conversely, a 5 gm/L addition of traps-4-hydroxy-L-proline
resulted in
a doubling of the level of Compound X with minimum impact on the other
analogues
or the titer of Pneumocandin Bo.
Table 4: Effect of 15 gm/L traps-3 and
traps-4-hydroxyproline added on or about day 6
Control traps-3- traps-4-
Com ound Process hydroxyproline hydroxyproline
I I00 units140 110
X ' 4.8 O.I 9.4
%
VI 2.7 % 0.7 2.4
XI 0.5 % 0.2 0.7
IX 2.7 % 4.3 2.5
-20-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Amino acids such as glutamine, arginine, and ornithine which can be
metabolized to ~l-pyrroline-5-carboxylate (P5C) also appear to have an impact
on the
analogues which are defined by the specific amino acid incorporated at the
position
"occupied" by 3-hydroxyproline in pneumocandin Bo (Table 5).
Table 5: Effect of proline "related" amino
acids added (5 gm/L) on or about day 6.
Control
Com ound Process Arginine Ornithine Glutamine
I 100 units 105 97 107
X 6.5 % 9.8 6.6 8.1
VI 2.9 % 2.2 1.8 4.0
XI 0.9 % 1.2 0.9 0.5
Supplementation of the medium with 5 gm/L threonine or serine
resulted in a complete elimination ox Iarge increase in the Ievel of Compound
IV
respectively (Table 6). In both cases, the titer of Compound I was reduced by
30%.
Additional work has shown that 1 gm/L threonine is sufficient to maintain
Compound
IV at acceptable levels while having no impact on the titer of Compound I.
Table 6: Effect of adding 5 gm/L serine or threonine on or about day 6
Com ound Control ProcessThreonine Serine
I 100 units 70 70
IV 2.2 % 0 17.4
Effect of TYace Elements
Several trace elements were examined for their impact on the titer
of Compound I and the spectrum of structural analogues produced. When added at
concentrations equal to the ferrous salt, zinc, cobalt, and nickel salts had
the most
-21-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
pronounced effects (Table 7). Zinc reduced the titer of Compound I by 50% and
doubled the level of Compound VI. Cobalt affected a 25% reduction in the titer
of Compound I while increasing the levels of Compounds VI, VIII, IX and V.
The addition of nickel had no impact on the titer of Compound I but increased
the
level of Compound V.
Table 7: Effects of trace elements
Com ound Control ProcessZinc Cobalt Nickel
I 100 Units SO 75 100
II 4.8 % 3.9 7.2 4.4
VIII 0.5 % 0.8 2.5 0.7
IV 2.8 % 1.9 6.0 3.1
V 1.6 % 2.8 14.1 7.5
VI 2.2 % 4.3 1.8 1.7
Osmolarity
In this fermentation, osmolarity can be controlled by maintaining the
residual fructose concentration at high (>75 gm/L) or low (<30 gm/L). The
initial
fructose concentration in the control process is 125 gm/L and is kept high by
making
two 50 gm/L additions during the 14 cycle. Alternatively, the initial fructose
concentration can be lowered to 40 gm/L and several 25 gm/L additions made
during
the course of the fermentation to maintain a low residual sugar level. When
the "low"
fructose process is run, there is a increase in the titer of Compound I along
with an
increase in the level of Compound X (Table 8). This increase in the level of
Compound X can be offset by adding an inorganic salt such as sodium chloride
or
sodium sulfate. The addition of inorganic reduces the effects of running at a
reduced
concentration of fructose. These results suggest that osmolarity plays a role
in
pneumocandin synthesis.
-22-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
Table 8: Effect of osmolarity
Low Low
Low Fructose, Fructose,
Control Fructose, 150 mM 116 mM
Com ound Process No Salt NaCI Na2S04
I 100 units134 111 110
X 4.3 % 8.2 5.5 6.1
In summary, hydroxylation patterns of amino acids of Pneumocandin
Bo are sensitive to zinc, cobalt and nickel. Additionally, amino acid
additions to the
production medium have a direct effect on the pneumocandins produced by the
fermentation. Supplementation of the production medium with proline, traps-3-
hydroxyproline and traps-4-hydroxyproline effects the incorporation of traps-3
or traps-4-hydroxyproline residues in Pneumocandin Bo. The addition of
threonine
to the fermentation controls the level of the serzne analogue, Compound IV.
Thus, the impact of amino acids and trace elements on the fermentation
provides insights into factors affecting the biosynthesis of Pneumocandin Bo
(Compound I) and has provided for an improved fermentation process by
decreasing
the levels of structural analogue and increasing the titer of Pneumocandin Bo.
EXAMPLE 2
Preparation of the Compound of Formula I
Part A. Pneumocandin Bo (15.9 g, 89% area % pure, 3.4 wt % water, 0.0128
mol) was added to dry THF (0.64 L) and the suspension was dried to <I0 mol%
water
0
by refluxing through a bed of 3 A molecular sieves. Additional dry THF was
added to
reconstitute the mixture to the original volume and the suspension was cooled
to <4°
C. with an icelwater/methanol bath. Neat BH3'SMe2 (10.91 g, 0.144 mol) was
added
over ten minutes and the reaction mixture was monitored by HPLC until the
ratio of
starting material to product was 1:l indicating the end of the reaction (3.5
h). At 4
hours, the mixture was cooled to -12° C. and slowly quenched with 2N
HCl (0.036 L.
This solution was diluted to 1.14 L with water. The assay yield of Compound II
was
6.60 g (47%). The quenched solution was diluted to 4 L with water and loaded
onto a
-23-
CA 02433652 2003-07-03
WO 02/055022 PCT/US02/00160
medium-pressure column of LiChroprep RP-C18 adsorbent (158 g). After loading,
the column was washed with I.2 L of water and the amine was eluted with 1.9 L
of
1:4 vlv acetonitrile/water, and then 0.38 L of 1:3 v/v acetonitrile/water. The
rich cuts
(>80 area %) were combined and diluted with water to a 1:7.3 v/v
acetonitrile/water
(1.70 L total). This mixture was loaded to the same column described above,
and the
column was washed with 0.57 L of water. The desired compound was eluted with
0.57 L methanol. The rich cut fractions (>85 area %) were combined and
concentrated by rotary evaporation and static high vacuum to give 6.81 g (87
wt %
pure, 6.8 wt % water) containing 5.92 g of Compound 2 hydrochloride salt for
an
isolated yield of 43%. Partial 1H NMR (400 MHz, CD30D): 8 7.I2 (d, ZH), 6.75
(d,
2H), 5.18 (d, 1H), 4.97 (d, 1H), 1.19 (d, 3H), 0.89 (t, 3H), 0.86 (d, 6H).
Mass
spectrum (FAB) m/z (M+Li)+: 1058.
Part B. Compound 2 (0.220 g, 0.202 mmol), as recited in Scheme 1, was
dissolved in 1:l v/v acetonitrile/water (8 mL). Triethylamine (0.060 mL, 0.43
mmol)
was added and the clouded mixture was stirred at room temperature overnight.
In the
morning, an additional 0.060 mL of triethylamine was added, and the mixture
was
stirred an additional 2 hours. HPLC analysis (45:55 v/v acetonitrile/water/0.1
% TFA,
Zorbax C18, 1.5 mL/min) showed a preponderance of the desired product (RRT
5.68
min) over the starting material (RRT 4.83 min). The reaction mixture was
acidified
with 0.5 mL of glacial acetic acid and concentrated in vacuo. Purification by
preparative HPLC (42:58 v/v acetonitrile/water/0.1% TFA, Waters DELTAPAK C-18
19X300 mm, 12 mL/min) gave the compound of Formula I as a mixture of two
isomers at the hemiaminal center (98% pure by analytical HPLC). Partial 1H NMR
(400 MHz, CD30D): b 7.20 and 7.18 (d, 2H), 6.74 (d, 2H), 5.32 and 5.27 (d,
1H).
Mass spectrum (ESI) m/z (M+H)+: 1051.7.
-24-