Note: Descriptions are shown in the official language in which they were submitted.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
1
TITLE OF INVENTION
Methods and Kits .for Identifying Scavengers of Reactive
Oxygen Species (ROS)
REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of U.S.
Provisional Application No. 60/266,657, filed February 05,
2001, , the content of which is herein incorporated by
reference.
FIELD OF INVENTION
This invention relates to methods of identifying, and/or
determining the ROS-scavenging ability of, a compound with
ROS-scavenging function.
BACKGROUND OF THE INVENTION
Aerobic metabolism in living organisms can lead to
generation of reactive oxygen species (ROS), which include
hydroxyl radicals, superoxide anion, hydrogen peroxide and
nitric oxide. Production of ROS can be due to various
enzymatic and non-enzymatic processes. In aerobic organisms,
ROS are formed from the partial reduction of molecular oxygen
to water during oxidative metabolism. Bacterial cells produce
endogenous hydrogen peroxide from the dismutation of superoxide~
or hydroxyl radical as a product of the respiratory chain when
oxygen is used as the terminal electron acceptor. Enteric
bacteria (e. g. Salmonella typhimurium and E. coli) encounter
toxic levels of hydrogen peroxide produced by macrophages
during engulfment.
Under normal conditions, ROS may play an important
role in different biological.processes. However, when ROS are
excessively produced under certain unusual conditions, they can
cause oxidative damage to DNA, proteins and lipids.
ROS have been implicated in the pathogenesis of many
different disease situations as well as harmful conditions.
These include aging, AIDS, atherosclerosis, cancer, cataracts,
congestive heart failure, diabetes, inflammatory disorders,
rheumatoid arthritis, and neuro-degenerative diseases such as
Alzheimer's, Parkinson's, multiple sclerosis, and Down's
syndrome, in addition to exposure to pollutants and ionizing
irradiation.
Living organisms have developed different ways of
coping with the R(iS. The capacity of enzymatic or non-
enzymatic antioxidants to quench the ROS can help cells to
defend against th:~ oxidative stress. Therefore, antioxidants
have been linked to and used for disease prevention.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
2
Antioxidants may be proteins, such as ferritin,
lactoferritin and transferritin, or enzymes, such as superoxide
dismutase, catalase and glutathione peroxidase. Nonenzymatic
antioxidants may be macromolecules, such as albumin, copper-
binding ceruloplasmin and hemoglobin, or small molecules which
may be water-soluble antioxidants, such as vitamin C, uric acid
and bilirubin or lipid-soluble antioxidants, such as vitamin E,
carotenoids, retinoids and ubiquinol-10.
However, these natural defenses can be overwhelmed in
many pathological states. More potent antioxidants should be
supplemented to deal with the oxidative stresses. Screening
and assay methods are needed to identify potent antioxidants;
but the current methods are both time-consuming and expensive.
In addition, they cannot measure the intracellular antioxidant
activities, which are more relevant to biological applications.
Therefore, a simple method that can measure the intracellular
antioxidant activities and hence can be used to search for
better oxidant scavenging molecules is of great importance in
the pharmaceutical and nutraceutical fields.
The public has shown an increasing interest in the
natural antioxidants contained in dietary supplements, as
antioxidants can give health benefits by preventing
oxidative damage caused by ROS. Standardized assays to
assess antioxidant activities and distinguish different
antioxidants are useful. Such assay methods are useful to
properly assess and label antioxidant products. Such assays
are also useful for measuring activities of antioxidants for
use as food supplements, natural products and drugs.
Farr (US patents 5,585,252, 5,811,231 and
5,589,337) have described use of stress promoters fused to
reporter genes to determine toxicity.
Catalase is a protein antioxidant. Catalases
catalyze the dismutation of hydrogen peroxide to water and
oxygen. The primary role for catalases is to protect the cells
.,. __. .,,_ ____ , ,___ ____~____ _______. .
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
3
The present invention relates to a method for
determining the ability of a compound to remove an ROS. The
method generally comprises: a) providing a cell containing
S an ROS-inducible promoter (RIP) which drives expression of a
reporter gene. The reporter gene is heterologous to the
promoter to which it is operably linked. b) exposing the
cell to a compound potentially able to remove the ROS. c)
measuring a change in the ROS-inducible expression level of
the reporter gene in the cell when the cell is exposed to
the compound. A reduction in the reporter protein level
would indicate that the compound is able to remove the ROS.
The invention further relates to a method for
selecting a.nucleic acid which encodes a protein~potentially
able to remove an ROS. The method generally comprises: a)
Providing cells containing an ROS-induced promoter which
drives expression of a reporter gene. The reporter gene is
heterologous to the promoter to which it is operably linked.
b) Introducing into the cells expression vectors containing
different nucleic acids, such as those found in a cDNA
library, or in a library where the nucleic acids have been
mutagenized. These nucleic acids encode proteins which are
potentially able to remove the ROS. c) Measuring a change
in the ROS-inducible expression of the reporter gene in the
cells when the nucleic acids are expressed. d) Selecting
for cells with reduced ROS-inducible expression of the
reporter gene. e) Isolating the nucleic acid from the
cells with reduced ROS-inducible expression of the reporter
gene. The nucleic acid isolated by such a procedure likely
encodes a protein able to remove the ROS.
In one embodiment, the different nucleic acids all
encode proteins able to remove ROS. By selecting for cells
with the greatest degree of reduction in the level of
reporter protein, the most efficient ROS remover may be
identified.
The method of the invention does not require that
the cell be exposed to an external source of ROS. Rather,
the ROS which induces the ROS-inducible promoter may be
intracellular. In one embodiment, the intracellular level
of the ROS may be elevated by methods known in the art. The
intracellular level of ROS such as H20z may also be induced
by acid pH, especially in bacteria such as A. tumefaciens.
In another embodiment, the intracellular level of the ROS
may be made constitutively elevated by using a cell which
has been genetically modified.
Cells containing such genetic modifications aze
known in the art and may have, for example, modified genes
of the respiratory chain so that the redox balance of the
cell is disturbed. Other genetic modifications may involve
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
4
knocking out functional enzymes which break down or remove
ROS intracellularly, such as catalase, superoxide dismutase,
alkyl hydroperoxidase, and glutathione reductase.
S As is clear from above, the method of the
invention also does not require that the potential ROS-
removing compound be exposed to the cell extracellularly.
Rather, the compound, in this case a gene product, may be
expressed from a nucleic acid inside the cell.
In a preferred embodiment, the cell expressing the
RIP-reporter does not express the functional native gene
product, i.e. that which is naturally expressed from the
promoter to which the reporter gene is operably linked.
Absence of the native gene function ensures that no
complicating mechanism such as a feedback loop interferes
with the correlation between the ability of a compound to
remove an ROS and the RIP-reporter expression level.
In one embodiment, the ROS-inducible promoter is
from a gene selected from the group consisting of: AhpCF,
Bcp, Dps, gor, KatA, KatB/AnkB, Kate, TrxB, human MAP kinase
phosphatase 1 (MKP-1) genes; mammalian hic-5 genes, the isc
operon; Escherichia coli zwf, fpr, fumC, micF, nfo, and sodA
genes; Azotobacter vinelandii spr gene; Xanthomonas oryzae
pv oryzae katX gene; rat and human haem oxygenase-1 (HO-1);
yeast 2-deoxyglucose-6-phophate phosphatase (DOG2);
catalase; human manganese superoxide dismutase (MnSOD); rat
glutathione S-transferase (GST); human interstitial
collagenase (MMP-1); human glutathione peroxidase ~(GPX2);
fish metallothionein (MT); and rat multidrug resistance type
1 (mdrl ) .
In another embodiment, the ROS is H20z. Where it is
desirable to identify or select for H202-removing compounds,
an~H202-inducible promoter is used. Such a promoter may be
from the following genes: AhpCF, Bcp, Dps, gor, KatA,
KatB/AnkB, Kate, TrxB, human MAP kinase phosphatase 1 (MKP-
1) genes; mammalian hic-5 genes, and the isc operon.
In another embodiment, the cell used in the
methods described above is a bacterial cell. It is
understood that if a bacterial cell is used, the ROS-
responsive promoter must function as such in bacteria and
the reporter gene must encode a protein functional in
bacteria. Likewise, if a plant cell, a yeast cell, or a
mammalian cell is used, the ROS-responsive promoter and the
reporter protein must be functional in the particular chosen
cell type.
The present invention also relates'to diagnostic
kits for determining the ability of a gene product to remove
an ROS. Such a kit generally comprises: a~ a cell which
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
contains an ROS-inducible promoter driving expressing of a
reporter gene. The reporter gene is understood to be
heterologous to the promoter to which it is operably linked.
b) means for introducing in the cell a nucleic acid
5 encoding a gene product; and (c) instructions for
determining a reduction in ROS-inducible expression of the
reporter gene in the cell once the nucleic acid is
expressed. This would indicate whether the gene product is
able to remove the ROS.
The kit of the invention provides components for
carrying out the methods of the invention. Accordingly, in
one embodiment, the kit further contains means for measuring
the level of the product of the reporter gene. The kit may
further comprise means for elevating the intracellular level
of the ROS in the cell. The cell provided in the kit may be
genetically modified to contain an elevated intracellular
level of an ROS, or may lack at least one naturally
occurring ROS-removing activity. The cell of the kit may
also lack a gene encoding an active enzyme such as catalase,
superoxide dismutase, alkyl hydroperoxidase, and glutathione
reductase.
The ROS-inducible promoter contained in the kit
may be from a gene such as AhpCF, Bcp, Dps, gor, KatA,
KatB/AnkB, Kate, TrxB, human MAP kinase phosphatase 1 (MKP-
1) genes; mammalian hic-5 genes, the bacterial isc operon;
Escherichia coli zwf, fpr, fumC, micF, nfo, soi28, and sodA
genes; Azotobacter vinelandii spr gene; Xanthomonas oryzae
pv oryzae katX gene; rat and human haem oxygenase-1 (HO-1);
yeast 2-deoxyglucose-6-phophate phosphatase (DOG2);
catalase; human manganese superoxide dismutase (MnSOD); rat
glutathione S-transferase (GST); human interstitial
collagenase (MMP-1); human glutathione peroxidase (GPX2);
fish metallothionein (MT); and rat multidrug resistance type
1 (mdrl ) .
In one embodiment, the kit is used to determine
the ability of a certain compound to remove H2O2.
Preferably, the kit provides an H2O2-inducible promoter from
a gene such as AhpCF, Bcp, Dps, gar, KatA, KatB/AnkB, Ka tG,
TrxB, human MAP kinase phosphatase 1 (MKP-1) genes;
mammalian hic-5 genes, and the isc operon.
In another embodiment the cell provided by the kit
is a bacterial cell and the reporter gene encodes a protein
functional in bacteria. In a preferred embodiment, the
bacterial cell is Agrobacterium tumefaciens.
In preferred embodiments of the method or kit of
the invention, the ROS-inducible promoter is~from the KatA
gene of Agrobacterium tumefaciens.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
6
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1. Alignment of the amino acid sequence of the
Agrobacterium tumefaciens katA gene product with the
S homologous catalase sequences. Alignment was completed by
using the DNAsis program. Atu KatA represents the A. ,
tumefaciens katA gene product; Lpn KatB represents the
Legionella pneumophila katB gene product; Bst Cat represents
the Bacillus stearothermophilus catalase.
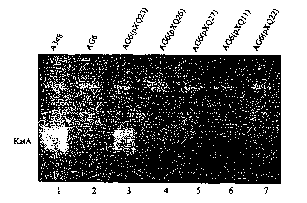
Fig. 2. Catalase isozyme assays. A. Agrobacterium
tumefaciens strains A348 (lane 1) , AG6 (pXQ9) (lane 2) and
AG6 (lane 3) were grown overnight at 28 °C in MG/L liquid
medium. Crude cell extracts were prepared as described in
the Materials and Methods. For each strain, 20 ~1 of crude
cell extract was loaded and electrophoresed on 7.5%
nondenaturing gel. B. 40 ~1 (lane 1) and 20 ~.1 (lane 2) of
A348 cell crude extract (that had been diluted 2x further
after the dilution described in the Materials and Methods)
was loaded and electrophoresed. Catalase isozymes were
visualised by activity staining according to Clare et al.
(1984) .
Fig. 3. The katA-gfp expression in different growth media.
2S Agrobacterium tumefaciens strains A348 and AG6 were grown at
28 °C for 24 hr on agar plates of MG/L, AB, IB (pH S.S), and
IB (pH 7.0) and fresh Kalanchoe leaf tissue and stem tissue
sections. The cells were harvested and then resuspended in
dH20. The fluorescence of each cell suspension was measured
by Luminescence Spectrometer LS50B (Perkin Elmer) as
described in the Materials and Methods using A348 as the
blank.
Fig. 4. Comparison of katA-gfp expression in different
genetic backgrounds. Agrobacterium tumefaciens strains
A348, AG6, AG6(pSW172), AG6(pXQ9), AG613, CGI1 and
CGI1(pXQ9) were grown at 28 °C fox 24 hr on agar plates of IB
(pH 5.5). The cells were harvested and then resuspended in
dHzO. The fluorescence of each cell suspension was measured
by Luminescence Spectrometer LS50B (Perkin Elmer) as
described in the Materials and Methods using A348 as the
blank.
Fig. 5. Schematic presentation of the wild-type and mutated
katA genes. The lines represent the DNA sequences; the
boxes represent the KatA open reading frames (ORFs). The
vertical lines indicate the restriction sites .or amino acid
positions. The diamond indicates the mini-Tn5 transposon
insertion position. The key restriction endonuclease sites
and primers~used are indicated. The DNA sequences under the
triangles ark the ORF sequences concerned for the site-
directed mutagenesis. ~ represents a deletion of the G of
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
7
the second codon; * represents the stop codon introduced at
the fifth codon. The wild type katA in pXQ9 and the mutant
katA genes encoding KatA050 and KatA486 were driven by the
katA promoter. The wild type katA in pXQ23 and the mutant
genes encoding KatA(98H/D), KatA(94R/Q)(98H/D), OkatA(-.2)
and OkatA(*5) were driven by both the katA and 1ac promoter.
Fig. 6. The effects of katA mutations on the KatA protein
stability. Agrobacterium tumefaciens strains A348 (panel A,
lane 2; panel B, lane 1), AG6 (panel A, lane 3), AG6(pXQ23)
(panel A, lane 4), AG6(pXQ26) (panel A, lane 5), AG6(pXQ27)
(panel A, lane 6), AG6(pXQ30) (panel A, lane 7), AG6(pXQ31)
(panel A, lane 8), AG6(pXQ9) (panel B, lane 2), AG6(pXQll)
(panel B, lane 3), and AG6(pXQ22) (panel B, lane 4) were
.15 grown overnight at 28 °C on'IB plates. The cells were
harvested, washed and diluted to a concentration of
ODsoo=0.3. The cells from 500 ~l of cell suspensions were
harvested by centrifugation and resuspended in the Laemmli
(1970) sample buffer. An aliquot of 2 ~,1 of each sample was
electrophoresed on SDS/10% PAGE gels. The proteins were
transferred onto Immobilon-P membrane and visualized by
(His)6-KatA antibody. The purified (His)6-KatA was used as
the control.
Fig. 7. Detection of the GFP protein expression of the
katA-gfp fusion. Agrobacterium tumefaciens strains A348
(lane 1), AG6 (lane 2), AG6(pSW172) (lane 3), AG6(pXQ23)
(lane 4), AG6(pXQ26) (lane 5), AG6(pXQ27) (lane 6),
AG6 (pXQ30) (lane 7) , AG6 (pXQ31) (lane 8) , AG6 (pXQl1) (lane
9), and AG6(pXQ22) (lane 10) were grown overnight at 28 °C on
IB plates. The cells were harvested and then resuspended
in dH20. One portion of each cell suspension was used to
measure the fluorescence by Luminescence Spectrometer LS50B
(Perkin Elmer) as described in the Materials and Methods
using A348 as the blank (upper panel). Another portion of
each cell suspension was diluted to a concentration of
OD6oo=0.3. The cells from 500 ~l of cell suspensions were
harvested by centrifugation and resuspended in the Laemmli
(1970) sample buffer. An aliquot of 2 ~l of each sample was
electrophoresed on SDS/15% PAGE gels. The proteins were
transferred onto Immobilon-P membrane; the GFP was
visualized by the GFP antibody (lower panel).
Fig. 8. Assays for catalase activity bands. Agrobacterium
tumefaciens strains A348 (lane 1), AG6 (lane 2), AG6(pXQ23)
(lane 3), AG6(pXQ26) (lane 4), AG6(pXQ27) (lane 5),
AG6(pXQll) (lane 6), and AG6(pXQ22) (lane 7) were grown
overnight at 28 °C ixr MG/L liquid medium. Samples of. crude
cell extracts were prepared and electrophore~ed on 7.5%
nondenaturing gel as described previously (Xu and Pan,
2000). Catalase isozymes were visualized by activity
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
8
staining according to Clare et al (1984). Since AG6(pXQ23)
was over-expressing KatA, this sample was diluted 8 fold
before loading.
Fig. 9. Induction of the katA-gfp fusion by HZO2. The cells
of Agrobacterium tumefaciens AG6 grown in MG/L (OD6oo=0.5)
were exposed to 0, 30 ~M, 60 ~M, and 120 ~M Hz02. The cell
suspensions were incubated at 28°C for 2 hours. Aliquots of
1 ml cell cultures were harvested by centrifugation and
resuspended in the Laemmli (1970) sample buffer. An aliquot
of 10 ~,1 of each sample was electrophoresed on SDS/15°s PAGE
gels. The proteins were transferred onto Immobilon-P
membrane; the GFP was visualized by the GFP antibody.
Fig. 10. Repression of katA-gfp expression by surrounding
bacterial cells. The AG6 cells were mixed with at 1:l ratio
with the cells from the bacterial strains A348, Rhizobium
meliloti RCR2011, or E. coli DHSa; the mixtures were spotted
on IB plates. The same amount of bacterial cells from a
single strain A348, AG6(pXQ9), Rhizobium meliloti RCR2011,
or E. coli DHSa was also spotted on IB plates. The plates
were incubated overnight at 28°C. The bacterial fluorescence
under W light was photographed (upper panel). The
fluorescence intensity was measured (lower panel) as
described in the Materials and Methods.
DETAILED DESCRIPTION Of THE EMBODIMENTS
To determine whether a compound is able to remove
an ROS, an ROS-inducible promoter (RIP) is fused to a
reporter gene to drive its expression. The reporter gene is
heterologous to the promoter to which it is operably linked.
The RIP-reporter construct is then stably transformed into
the cell. To test whether a certain compound is an ROS-
remover, the cell is exposed to the test compound.
Preferably, the cell is exposed to the test compound
intracellularly. If necessary, the intracellular level of
the ROS is induced, and the ROS-inducible expression level
of the reporter gene in the cell is measured. A reduction
in the reporter protein level when the cell is exposed to
the compound would indicate that the compound is able to
remove the ROS.
The test compound may also be provided by being
expressed in a neighbouring cell, rather than being
expressed from the cell containing the RIP.
As used herein, the terms "reactive oxygen
species" (ROS) and "oxidants" aye used interchangeably, and
include hydroxyl radicals, superoxide anion,'hydrogen
peroxide and nitric oxide.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
9
ROS-removing compounds are anti-oxidants or
oxidant scavengers. Namely, these compounds have the
ability remove ROS by breaking them down chemically, or by
sequestering them away from solution. Known ROS-removing
compounds include proteins, such as ferritin, lactoferritin
and transferritin, or enzymes, such as superoxide dismutase,
catalase and glutathione peroxidase. Nonenzymatic
antioxidants may be macromolecules, such as albumin, copper-
binding ceruloplasmin and hemoglobin, or small molecules
such as water-soluble antioxidants (e. g. vitamin C, uric
acid and bilirubin) or lipid-soluble antioxidants (e. g.
vitamin E, carotenoids, retinoids and ubiquinol-10).
. In a preferred embodiment, the test compounds with
potential ROS-removing capability-.are proteins expressed
intracellularly. They are often proteins heterologous to the
cells containing the RIP-reporter construct. However, a
polypeptide naturally present in such cells may also be
tested as a ROS-removing compound provided that the gene
encoding the functional polypeptide has been knocked out
from the cell.
The term "heterologous" means, in the context of
the present invention, that the components are not found
naturally together. For example, a reporter gene which is
heterologous to the promoter to which it is linked is not
the natural coding sequence of the gene from which the
promoter is derived.
The term "vector" refers to a nucleic acid
sequence that is capable of propagating in particular host
cells and can accommodate inserts of foreign nucleic acid.
Typically, vectors can be manipulated in vitro to insert
foreign nucleic acids and the vectors can be introduced into
host cells such that the inserted nucleic acid is
transiently or stably present in the host cells.
The term "expression vector" refers to a vector
designed to express inserted nucleic acid sequences. Such
vectors may contain a powerful promoter located upstream of
the insertion site.
The term "expression" in the context of nucleic
acids refers to transcription and/or translation of nucleic
acids into mRNA and/or protein products.
The term "expression library" refers to a library
of nucleic acid fragments contained as inserts in an
expression vector.
The term "stable transformation" refers to the
continued presence of a nucleic acid sentence in a host cell
for a period of time that is at least as long as that
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
required to carry out the methods of the present invention.
Stable transformation can be achieved through integration of
the construct into a host cell chromosome, or engineering
the construct so that it possesses elements that ensure its
5 continued replication and segregation within the host (i.e.,
an artificial chromosome), or alternatively, the construct
may contain a selectable marker (e. g., a drug resistance
gene) so that persistence of the construct in the cell is
ensured by growing the host cells under selective conditions
10 (e. g:, in drug-containing media).
The term "cell" or "host cell" in the_present
invention refers to a cell of prokaryotic or eukaryotic
origin that can serve as a recipient of an introduced
vector. The host cell often allows replication and
segregation of the vector that resides within. In certain
cases, however, replication and/or segregation are
irrelevant; expression of vector or insert DNA is the
objective. Typical bacterial host cells include E. coli, B.
subtilis and A. tumefaciens; fungal host cells include S.
cerevisiae and S. pombe; plant cells include those isolated
from A. thaliana, and Z. maize; insect host cells include
those isolated from D, melanogaster, A. aegypti, and S.
frugiperda; and mammalian cells include those isolated from
human tissues and cancers including melanocyte (melanoma),
colon (carcinoma), prostate (carcinoma), brain (glioma,
neuroblastoma, astrocytoma) and liver (hepatoma).
An ROS-inducible promoter is one which, in
response to the presence of the ROS inside the cell,
expression from the promoter is increased. Numerous ROS
inducible promoters are known in the art. They include:
AhpCF, Bcp, Dps, gor, Ka tA, Ka tB/AnkB, Ka tG, TrxB, human MAP
kinase phosphatase 1 (MKP-1) genes; mammalian hic-5 genes,
the isc operon; Escherichia coli zwf, fpr, fumC, micF, nfo,
and sodA genes; Azotobacter vinelandii spr gene; Xanthomonas
oryzae pv bryzae katX gene; rat and human haem oxygenase-1
(HO-1); yeast 2-deoxyglucose-6-phophate phosphatase (DOG2);
catalase; human manganese superoxide dismutase (MnSOD); rat
glutathione S-transferase (GST); human interstitial
collagenase (MMP-1); human glutathione peroxidase (GPX2);
fish metallothionein (MT); and rat multidrug resistance type
1 (mdrl ) .
It is expected that most of the promoters above
would be functional to some degree in a heterologous cell
type. However, it is preferred that promoters naturally
found in bacteria would be used in bacteria, and yeast
promoter in yeast cells, and mammalian promoters in
mammali;.n cells, according to the methods of the invention.
In a preferred embodiment, the ROS is HzOz, and the
H2O2-inducible promoter is from a gene such 'as AhpCF, Bcp,
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
11
Dps, gor, KatA, KatB/AnkB, Kate, TrxB, human MAP kinase
phosphatase 1 (MKP-1) genes; mammalian hic-5 genes, and the
isc operon.
The RIP-reporter vector is customized so that
reporter expression reflects as closely as possible the ROS
level of the host cell. Thus, the expression vector is
designed so that the reporter gene is placed under control
of ROS-response cis regulatory elements functional in the
host cell. Preferably, the reporter is expressed at a low
level in the absence of the ROS; i.e. the basal activity of
the promoter should be low so that induction by ROS is
readily detectable.
A partial listing of the genes, their organism of
origin, and Genbank accession numbers are provided below. A
brief description of some of these genes and reference
publications are also provided:
Streptococcus mutans ahpC and noxl genes for alkyl
hydroperoxidase and NADH oxidase/alkyl hydroperoxidase
reductase, ACCESSION AB010712.
Mycobacterium marinum alkylhydroperoxide reductase
(ahpC) gene, ACCESSION AF034861.
Bacteroides fragilis alkyl hydroperoxide reductase
subunit C (ahpC) and alkyl hydroperoxide reductase
subunit F (ahpF) genes, ACCESSION AF129406.
Salmonella typhimurium alkyl hydroperoxide reductase
(ahpC) and (ahpF) genes, ACCESSION J05478.
Mycobacterium avium alkyl hydroperoxidase C (ahpC)
gene, ACCESSION U18263.
Mycobacterium tuberculosis alkyl hydroperoxidase C
(ahpC) gene, ACCESSION U18264.
Mycobacterium smegmatis alkyl hydroperoxide reductase C
(ahpC) gene, ACCESSION U43719.
Mycobacterium intracellulare alkyl hydroperoxidase C
(ahpC), ACCESSION U71061.
Staphylococcus aureus alkyl hydroperoxide reductase
subunit C(aphC) and subunit F (aphF) genes, ACCESSION
U92441 X85029.
Escherichia coli,laacterioferritin comigratory protei~l
(bcp), ACCESSION M63654 M37689.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
12
Escherichia coli DNA binding protein Dps (dps) gene,
ACCESSION AF140030.
Bacteroides fragilis non-specific DNA-binding protein
Dps (dps), ACCESSION AF206033.
Synechococcus sp. nutrient-stress induced DNA binding
protein (dpsA) gene, ACCESSION U19762.
Streptococcus thermophilus glutathione reductase (gor)
gene, ACCESSION L27672.
E.coli gor gene encoding glutathione reductase,
ACCESSION M13141.
20
P, aeruginosa gor gene for glutathione reductase (EC
1.6.4.2), ACCESSION X54201.
Agrobacterium tumefaciens catalase (KatA), S$Q ID N0:1.
Vibrio fischeri catalase (katA) gene, ACCESSION
AF011784.
Pseudomonas aeruginosa catalase isozyme A (katA) gene,
ACCESSION AF047025.
Actinobacillus actinomycetemcomitans catalase (katA)
gene, ACCESSION AF162654.
Legionella pneumophila catalase-peroxidase (katA) gene,
ACCESSION AF276752.
Staphylococcus aureus catalase gene, strain ATCC12600.
ACCESSION AJ000472.
Lactobacillus sake catalase (katA) gene, ACCESSION
M84015.
Rhizobium meliloti catalase (katA) gene, ACCESSION
U59271.
Pseudomonas fluorescens plasmid pAM10.6 catalase
isozyme (katA) ACCESSION U72068:
H.pylori katA gene, ACCESSION 270679.
B.subtilis 25 kb genomic DNA segment (from sspE to
katA), ACCESSION 282044.
Pseudomonas aeruginosa paraquat inducible catalase
isozyme B (katB), ankyrin (ankB), ACCESSION U89384.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
13
Caulobacter crescentus catalase-peroxidase (katG) gene,
ACCESSION AF027168.
Mycobacterium smegmatis catalase-peroxidase (katG)
S gene, ACCESSION AF196484.
Synechococcus PCC6301 catalase-peroxidase gene,
ACCESSION AF197161.
Mycobacterium leprae DNA for catalase-peroxidase,
ACCESSION D89336.
E.coli katG gene encoding catalase HP1, ACCESSION
M21516.
Salmonella typhimurium Kat G gene for hydroperoxidase
I. ACCESSION X53001.
M.tuberculosis katG gene for catalase-peroxidase.
ACCESSION X68081 S42739.
M.bovis katG gene. ACCESSION X83277.
M.smegmatis katG gene. ACCESSION X98718,
M.fortuitum katGI gene. ACCESSION Y07865.
M.fortuitum katGII gene. ACCESSION Y07866.
Mycobacterium smegmatis thioredoxin reductase (trxB)
and thioredoxin (trxA) genes, ACCESSION AF023161.
Streptomyces coelicolor sigT, trxB and trxA genes,
ACCESSION AJ007313.
Clostridium litorale thioredoxin reductase (trxB), and
thioredoxin (trxA) genes, ACCESSION U24268.
Mycoplasma pneumoniae thioredoxin reductase K04 orf315
(trxB) gene, ACCESSION U51988.
The sodA gene encodes superoxide dismutase and is
strongly induced when cells are exposed to chemicals
that produce superoxide radicals in the cell, such as
paraquat, plumbagin, menadione, streptonigrin,
methylene blue and phenazine methyl sulfate. SodA gene
induction depends upon an increase in steady state
superoxide concentration, not necessarily upon cellular
damage caused by superoxides.
The soi28 gene encodes a pyruvate:flavo~loxin
oxidoreductase. This gene is induced by superoxide-
producing reagents only. Specifically,' the soi2ii gene
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
14
is induced when two small, thiol-containing proteins,
flavodoxin and ferredoxin, become oxidized.
The ahp gene is induced by hydrogen peroxide and
organic hydroperoxides, both exogenous and those formed
upon peroxidation of proteins and fatty acids.
soil? and soil9 respond to superoxides [T. Kogoma et
al . , (1988) ] .
zwf encodes glucose-6-hydrogenase and is induced by
superoxide-producing compounds and nitric oxide
[Greenberg et a1.(1990)].
micF encodes antisense RNA that shuts off translation
of the porin gene, ompF and is induced by superoxides
[Greenberg et a1.(1990)].
The nfo gene encodes a DNA repair enzyme and is
specifically induced by redox active agents, such as
paraquat and menadione [Farr et a1.(1991)].
If the nucleotide sequence of the ROS-inducible
gene is known, polymerase chain reaction may be used to
produce fusions with the promoter. Specifically, primers
are synthesized which are complementary to the 5' and 3'
ends of the ROS- inducible promoter portion of the gene,
hybridizes those primers to denatured, total DNA~under
appropriate conditions and performs PCR. In this manner,
clonable quantities of any sequenced promoter may be
obtained. Once the promoter DNA has been obtained, it is
ligated to a DNA encoding the reporter gene in an
appropriate vector, such as pRS415 for E. coli, which
contains a multiple cloning site just upstream from the lacZ
gene. Numerous vectors for expressing reporter genes are
known in the art or are commercially available. The methods
are well-known in the art.
A reporter gene as used in the present invention
essentially encodes any gene product that can be expressed
in the cell of interest and is assayable and detectable.
The reporter gene must be sufficiently characterized such
that it can be operably linked to the promoter. Reporter
genes used in the art include the LacZ gene from E. coli
(Shapiro S. K., Chou J., et al., Gene Nov.; 25: 71-82
(1983)), the CAT gene from bacteria (Thiel G., Petersohn D.,
and Schoch S., Gene Feb. 12; 168: 173-176 (1996)), the
luciferase gene from firefly (could S. J., and Subramani S.,
1988), the GFP gene from jellyfish (Chalfie M. and Prashner
D. C., U.S. Pat. No. 5,491,084), galactose kinase (encoded
by the galK gene), and beta-glucosidase (encbded by the gus
gene). These have been primarily used to monitor expression
of genes in the cytoplasm. To monitor expr8ssion at the
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
cell surface, a labeled antibody that binds to the cell
surface marker (e.g., CD20) may be used to quantify the
level of reporter (Koh J., Enders G. H., et al, 1995).
5 Of these reporters, autofluorescent proteins
(e.g., GFP) and the cell surface reporters are preferred for
use in monitoring living cells, because they act as "vital
dyes". Their expression can be evaluated in living cells,
and the cells can be recovered intact for subsequent
10 analysis. Vital dyes, however, are not specifically
required by the methods of the present invention. It is also
very useful to employ reporters whose expression can be
quantified rapidly and with high sensitivity. Thus,
fluorescent reporters (or reporters that can be labeled
15 directly or indirectly with a fluorophore) are especially
preferred. This trait permits high throughput screening on a
flow sorter machine such as a fluorescence activated cell
sorter (FACS).
GFP is a member of a family of naturally occurring
fluorescent proteins, whose fluorescence is primarily in the
green region of the spectrum. GFP has been developed
extensively for use as a reporter and several mutant forms
of the protein have been characterized that have altered
spectral properties. High levels of GFP expression have
been obtained in cells ranging from yeast to human cells. It
is a robust, all-purpose reporter, whose expression in the
cytoplasm can be measured quantitatively using a flow sorter
instrument such as a FACS.
The diagnostic kits and methods of this invention
rely on the induction of specific ROS-inducible promoters to
alter expression of the reporter gene. This change in
expression level is measured both qualitatively and
quantitatively. In order to be useful in those kits and
methods, the particular stress promoter must be operably
linked to the gene which encodes the reporter product.
The term "operable linkage" refers to the
positioning of the promoter relative to the gene encoding
the reporter product such that transcription of the gene is
regulated by the promoter. Such positioning is well known
in the art and involves positioning the promoter upstream
(5') of the gene so that transcription is not impeded by
extraneous termination signals and where the spacing between
the promoter initiation site and the regulatory sequences of
the promoter are optimal for transcription.
Also within the scope of this invention are
constructs wherein the reporter product is in fusion with
the N-terminal portion of the native gene prbduct, i.e. the
gene product of the pr,~moter to which the reporter is fused.
It is important that the portion of native gene product
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
16
fused to the reporter does not retain the function of the
full length native gene product.
The choice of bacterial strain to express the
particular RIP-reporter construct and thus useful in the
methods and kits of this invention is only limited by the
strain's ability to produce the functional reporter and its
inability to synthesize the reporter in its untransformed
state. Most preferably, the strain used should be defective
in genes which endogenously remove ROS intracellularly.
Such genes include those encoding catalase, superoxide
dismutase, alkyl hydroperoxidase, and glutathione reductase.
For example, where an H202-inducible promoter is used, it is
preferred that the endogenous catalase genes be knocked out
or mutated in the cells so that the cells lose or have
decreased capacity to break down H202 endogenously.
Eukaryotic cells useful in the methods and kits of
the invention include cell lines established from primary
tissue, as well as those cell lines and cultures available
from the American Type Culture Collection (ATCC, Rockville,
Md . ) .
The method and kits of the invention rely on a
detectable reduction in ROS-inducible reporter expression to
test whether a compound is capable of removing ROS. This
requires that the level of reporter expression be
sufficiently high in the absence of an ROS-removing
compound, so that a reduction is detectable. In some
embodiments, the method involves elevating the intracellular
level of the ROS. Methods used to elevate the concentration
of various intracellular ROS are known.
In bacteria, intracellular levels of H202 may be
elevated by using glucose/glucose oxidase (GOX) or reduced
glutathione (GSH) as Hz02-generating systems (Saliim et al.
2001). In Agrobacterium, intracellular levels of H202 may be
elevated by acid conditions. In mammalian cells and in
yeast, depletion of intracellular glutathione raises
intracellular ROS. In at least mammalian cells, glutathione
may be depleted by application of buthionine sulfoximine.
Insuline stimulation also generates a burst of intracellular
H202 in insulin-sensitive hepatoma and adipose cells (Mahadev
et al. 2001). In Arabidopsis, application of dexamethasone
activates MAP kinases and results in the generation of Hz02
{Ren et al. 2002).
In other embodiments, the method involves using
cells where the cells have been genetically modified so that
there is an elevated intracellular level of an ROS. In
bacteria, E. coli strains where modulation oi~ expression of
superoxide dismutase results in modulation of intracellular
superoxide (Gort and Imlay, 1998). In yeast, expression of
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
17
cytochrome peroxidase, superoxide dismutase or the GSHl gene
may be modulated. In fibroblasts, cells that stably express
Noxl produces a marked increase in intracellular H202, as
well as some increase in superoxide level (Arnold et al.
2001) .
In an exemplary embodiment, an assay testing for a
compound for its ability remove H20z would proceed along the
following line. An expression construct which expresses a
potential H202-remover is introduced into a cell line which
contains a reporter gene under control of an H2O2-inducible
promoter, such as the A. tumefaciens strain AG6. Production
of intracellular H202 may be induced, for example by exposing
the cells to low pH medium. The level of reporter protein,
as indicated by the level of fluorescence if the reporter is
GFP, would be reduced in the cells expressing an H2O2-
removing compound, compared to the control cells in which
the compound is absent.
An aspect of the invention relates to a method for
selecting a nucleic acid which encodes a protein potentially
able to remove an ROS. In this method, cells are provided
which contain the RIP-reporter gene construct as described
above. Expression vectors containing different nucleic
acids, such as those found in a cDNA library, or in a
library where the nucleic acids have been mutagenized, are
used to transform the cells. These nucleic acids encode
proteins which are potentially able to remove the ROS. Any
reduction in the ROS-inducible expression of the reporter
gene is measured, as described above, when the nucleic acids
are expressed. The cells with reduced ROS-inducible
expression of the reporter gene are then selected and the
nucleic acid used to transform the cell is isolated. This
nucleic acid would likely encode an ROS-removing protein.
The term "library" refers to a collection of
nucleic acid fragments that may individually range in size
from about a few base pairs to about a million base pairs.
These fragments are contained as inserts in vectors capable
of propagating in certain host cells such as bacterial,
fungal, plant, insect, or mammalian cells.
The term "plurality of nucleic acids" refers to a
set of nucleic acid molecules from any source. For example,
a plurality of nucleic acids may comprise total genomic DNA,
genomic DNA from one or more chromosomes, cDNA that has been
reverse-transcribed from total cellular RNA or from
messenger RNA (mRNA), total cellular RNA, mRNA, or a set of
nucleic acid molecules synthesized in vitro either
individually, or using combinatorial methoas. Plurality of
nucleic acids is understood to include, e.g.'an expression
library.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
18
The terms "bright" and "dim" in the context of a
cell sorter refer to the intensity levels of fluorescence
(or other modes of light emission) exhibited by particular
cells: Bright cells have high intensity emission relative to
the bulk population of cells, and by inference, high levels
of reporter gene expression; dim cells have low intensity
emission relative to the bulk population.
The term "flow sorter" refers to a machine that
analyzes light emission intensity from cells or other
objects and separates these cells or objects according to
parameters such as light emission intensity.
In one embodiment, the method using GFP as a
reporter protein, to select for ROS-removing proteins is as
follows. The ROS-inducible promoter-GFP expression
construct is introduced into the chosen host cells and a
stable expresser is selected. This GFP-expressing line is
clonally expanded to generate a population that is bright
green. A library encoding potential ROS-removers is
introduced into the host cells to generate a population of
GFP-containing cells, some of which also express ROS-
removers. This population is examined using a flow sorter
device and cells are sorted into two populations: cells that
continue to express GFP at levels similar to the cells
before introduction of the library inserts.; and, cells that
express reduced levels of GFP. The inserts encoding ROS-
removers from such "dim" cells are isolated and either used
to determine their DNA sequences, or reintroduced into the
GFP-containing host cells for another cycle of selection and
enrichment.
One can envision a flow sorter profile diagram of
the selection procedure described above. The fluorescence
intensity of a population of host cells containing the
library inserts prior to selection would have a normal
distribution. This presorted population is used to select
cells on the left tail of the distribution. The dim cells
on the left of the distribution are selected and inserts
from these cells are reintroduced into the original host
cells. The fluorescence intensity distribution that ensues
from cells transformed with such a sub-library of sequences
would become skewed to the left (i.e., the mean fluorescence
intensity decreases).
The present invention may use a flow sorter such
as a FACS or equivalent device to screen through large
numbers of host cells containing expression library inserts
encoding potential ROS-removing proteins, to identify those
that can remove ROS; namely, cells that have reduced levels
of reporter molecule expression. Host cells'which have an
elevated level of ROS and which have the reporter (e. g.,
GFP) present under control of the ROS-inducible promoter
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
19
will have a constitutively high level of reporter
expression. When the expression library inserts are
expressed in these cells, the large majority of cells that
are analyzed by FRCS are expected to have retained this high
level. However, a small number may exhibit reduced
expression, detected on the FACS as cells that fall on the
dimmer side of the cell fluorescence distribution. These dim
cells can be collected and grown in isolation of the others.
Such a procedure results in enrichment from the starting
population of cells for those that contain ROS-removers,
which effectively reduce the level of inducer ROS, thereby
reducing the level of reporter expression., These selected,
dim cells can be used to reisolate the perturbagen fragments
by, e.g., PCR using primer sites that flank the library
inserts, so as to build a sub-library of library inserts
enriched for those that cause reduced reporter expression.
The sub-library of fragments can be recloned (using e.g.,
the same expression vector) and reintroduced into the host
cells, and the screening/selection process can be repeated
as many times as necessary.
After a sufficient number of cycles, a substantial
difference should be observed in the fluorescence intensity
distribution of the original reporter-containing host cells
as compared to the host cells harboring the enriched ROS-
removing sub-library inserts. Preferably, the procedure
should be repeated until a minimal overlap is observed
between these two fluorescence intensity distributions.
Ultimately, the process of FACS sorting and cycling should
result in a population of nucleic acids encoding ROS-_
removers that inhibit expression of the reporter. These can
be isolated and studied individually by molecular cloning
and DNA sequence analysis.
In order that the invention described herein may
be more fully understood, the following examples are set
forth. It should be understood that these examples are for
illustrative purposes only and are not to be construed as
limiting this invention in any manner.
EXAMPLE 1: Materials, Techniques and Assay Conditions
Strains, plasmids, and growth conditions.: The strains and
plasmids used in this study are listed and described in Table
1. Agrobacterium tumefaciens strains were grown in MG/L, IB or
AB medium (Cangelosi et al., 1991) at 28°C, supplemented with
100 ~g/ml kanamycin, 5 ~g/ml tetracycline, or 100 ~,g/ml
carbenicillin as required. Escherichia coli strains were grown
on Luria-Bertani (LB) medium (Sambrook et al,,, 1989) at 37 °C,
supplemented with 50 ~g/ml kanamycin, 10 ~.g/ml tetracycline, or
50 ~g/ml ampicillin as required. Mini-Tn5 transposon
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
mutagenesis of A. tumefaciens strain A348 was carried out by
pAG408 as described (Suarez et al., 1997).
Southern analysis: Total DNA was extracted as described
5 previously (Charles and Nester, 1993) from the A.
tumefaciens mutant AG6. Approximately 1 Pg of total DNA was
digested with ClaIor NruI and then electrophoresed on a 0.9%
agarose gel. The DNA fragments were then transferred onto
nylon membrane Zeta-Probe GT (Bio-Rad) using a transfer
10 apparatus, PosiBlot (Stratagene). The plasmid pAG408 was
labelled as the probe by random priming with the enhanced
chemiluminescence kit (Amersham). The labelling;
hybridisation and signal detection were conducted according
to the manufacturer.
Catalase isozyme assay: A. tumefaciens strains A348, AG6,
AG6(pXQ23), AG6(pXQ26), AG6(pXQ27), AG6(pXQll), and
AG6(pXQ22) were grown overnight at 28 °C in MG/L liquid
medium to 1.4 OD6oo. The cells were harvested by
centrifugation at 4000 rpm for 10 min at 4 °C. The cell
pellets were washed and resuspended in 5 ml extraction
buffer containing 0.05 mM phosphate and 0.4 mM EDTA (pH
7.8). The cells were sonicated for 30 sec for 6 times on
ice with a 2-min cooling on ice between sonications.- The
cell debris was removed by centrifugation at 1100 rpm for 10
min at 4°C. The cell-free supernatant was diluted 2x with
the extraction buffer, and 20 ~1 of each diluted extract was
electrophoresed on 7.5% native polyacrylamide gels. The
resolving gel buffer was prepared at pH 8.1 instead of pH
8.9. Electrophoresis was performed at 150 V for 3 hours.
Catalase isozymes were visualised by an activity staining .
procedure according to Clare et al. (1984).
Protein analysis: SDS/PAGE was conducted in 10% or 15%
polyacrylamide gels to analyze the KatA or GFP expression,
respectively. The proteins were transferred onto Immobilon-
P membranes (Millipore). The KatA or GFP proteins were
visualized with the enhanced chemiluminescence (ECL) western
blot detection system according to the recommendations of
the manufacturer (Amersham).
Table 1. Bacterial strains and plasmids used in this study
Strain/plasmid Relevant characteristics* Source/
Reference
Strains
Agrobacterium
tumefaciens
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
21
A348 Wild type, A136(pTiA6NC) (octopine-type) Laboratory
collection
AG6 Derivative of A348 in which katA was disrupted by the
GFf'-tagged mini-Tn5 transposon at 995 by This study
downstream from the start condon of katA; KmR, GmR
AG613 AG6 containing pXQl3 integrated into the
chromosome (containing a single copy of the wild type This study
katA and katA-g fp fusion); KmR, GmR, CbR
CGI1 Derivative of C58 in which aopB was disrupted by the
GFP-tagged mini-Tn5 transposon; KmR, GmR This study
Escherichia
coli _
DHSa supE dlac(d80ZdM1 S) hsdR recA endA gyrA thi relA Bethesda
Research
Laboratories
MT607 Pro-82 thi-1 hsdR 17 supE44 endAl recA56 Finan, et al.,
1986
Plasmids
pTZl9R Cloning vector, ColEl oriYbla, AmpR ' US
Biochemical
pSW172 Broad-host-range IncP plasmid containingChen and
P,Q~ and
downstream polylinker sequence, TcR Winans,
1991
pXQ6 pBluescript II KS(-) containing a
6-kb CIaI DNA
fragment containing the sequences This study
downstream of
the mini-Tn5 insertion at the katA
gene.
pXQ7 pTZl9R containing a 5-kb NruI DNA
fragment
containing the sequences upstream This study
of the mini-Tn5
insertion at the katA gene.
pXQ9 pSW 172 carrying a 2.8 kb XbaI-NheI
fragment
containing the wild type katA, TcR This study
pXQ pSW 172 carrying a 2.3 kb XbaI-NheI
11 fragment
containing a KatA with a 86 amino This study
acid deletion at the
C-terminus, TcR
pXQ 13 pTZ 19R carrying a 2.8 kb EcoRI fragment from pXQ9
containing the wild type katA, AmpR This study
pXQ 1 S pRSETA carrying a 2.17 kb XhoI-KpnI fragment
containing the full length KatA ORF fused in-frame This study
Wlth (H1S)6, AmpR
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
22
pXQ22 pSW 172 carrying a 2.4 kb XbaI-NheI fragment
containing a KatA with a 50 amino acid deletion at the This study
C-terminus, TcR
pXQ23 pSW 172 ligated with pXQl3 at CIaI,
containing the
wild type katA, AmpR This study
pXQ24 pTZl9R carrying a 2.8 kb EcoRI fragment
from pXQ9
containing a katA with His 98 replacedThis study
by Asp, AmpR
pXQ25 pTZl9R carrying a 2.8 kb EcoRI fragment
from pXQ9
containing a katA with Arg 94 replacedThis study
by Gln and His
98 replaced by Asp, AmpR
pXQ26 pSW 172 ligated with pXQ24 at CIaI,
containing a katA
with His 98 replaced by Asp, AmpR, This study
TcR
pXQ27 pSW 172 ligated with pXQ25 at CIaI,
containing a katA
with Arg 94 replaced by Gln and His This study
98 replaced by
Asp, AmpR , TcR
pXQ28 pTZl9R carrying a 2.8 kb EcoRI fragment
from pXQ9
containing the a katA with SerS replacedThis study
by a stop
codon (TGA), AmpR
pXQ29 pTZI9R carrying a 2.8 kb EeoRI fragment from pXQ9
containing the a katA with a G base pair deletion in the This study
second codon of katA ORF, AmpR
pXQ30 pSW172 ligated with pXQ29 at CIaI, containing a katA
with a G base pair deletion in the second codon of katA This study
ORF, AmpR, TcR
pXQ31 pSW 172 ligated with pXQ28 at CIaI, containing a katA
with SerS replaced by a stop codon (TGA), AmpR, TcR This study
Km, kanamycin; Tc, tetracycline; Amp, ampicillin; Gm, gentamycin.
Measurement of intracellular HZOZ concentrations: The
intracellular concentrations of H202 were measured by the
procedures previously described (Gonzalez-Flecha and Demple,
1995; 1997) with modifications.
Briefly, A. tumefaciens strains A348, AG6 and
AG6(pXQ9) were grown at 28 °C for 24 hr on agar plates of AB
or IB. The cells were harvested, washed and resuspended at
ODsoa = 1 . 0 in 50 mM phosphate-buf fer (pH 7 . 4 ) . H202
generated within the cells passed through membranes and
equilibrated with the buffer. Complete equilibration of the
intracellular and ~~xtracellulare Hz02 levels occurred within
10 min in the assay (Gonzalez-Flecha and Demple, 1997).
After 20 min of equilibration, the cell suspensions were
centrifuged for 1 min at 6,000 rpm at 4°C.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
23
H202 concentrations in the supernatants were then
measured by the Amplex Red Hydrogen Peroxide Assay Kit
(Molecular Probes Inc., USA), which contains a highly
sensitive and specific fluorogenic probe (N-acetyl-3,7-
dihydroxyphenoxazine) for H202 and horse radish peroxidase
(HRP) (Mohanty, et al, 1997). Briefly, 100 ~l supernatant
was mixed with 100 ~1 of the probe at 100 ~M and 1 U/ml HRP.
The fluorometric assay was conducted in a 96-well microplate
and measured by Luminescence Spectrometer LS50B (Perkin
Elmer). The excitation wavelength was 540 nm; the emission
wavelength was 590 nm. The assays were run in four
replicates; the concentrations were then calculated based on
the H202 standard curves generated simutaneously.
Catalase activity assay: The catalase activity in whole
bacterial cells was determined as previously described
(Maciver and Hansen, 1996) except using the Amplex Red
Catalase Assay Kit (Molecular Probes Inc., USA). Briefly,
A. tumefaciens strains were grown at 28 °C for 24 hr on IB
plates. The cells were harvested, washed and resuspended at
OD6oo = 1.0 in 50 mM phosphate-buffer (pH 7.4). For the
strains with the wild type katA [A348, AG6(pXQ9), and
AG6(pXQ23)], 100 ~.1 cell suspension of each sample was
incubated with 50 ~1 40 ~M HZOZ at time intervals of 0, 30
sec, 1 min and 2 min.
For the strains with the katA mutants [AG6,
AG6 (pXQl1) , AG6 (pXQ22) , AG6 (pXQ26) and AG6 (pXQ27) ] , 100 ~,l
cell suspension of each sample was incubated with 50 ~,1 5 ~M
H202 at time intervals of 0, 1, 2, and up to 6 min. The
amount of H202 left after degradation by the bacterial
catalase was then determined by adding 50 ~1 Amplex Red
reagent N-acetyl-3, 7-dihydroxyphenoxazine at 25 N.M and 0.4
U/ml horse radish peroxidase provided by the kit. The
fluorometric assay was conducted by the same procedure as
described above for the measurement of intracellular H202.
The cell suspensions without the added H20z were used as the
blank. Solutions of crystalline bovine catalase (Sigma)
were used to standardize this assay.
EXAMPLE 2: Cloning, sequencing and characterization of the
katA gene encoding catalase from Agrobac-terium
The total DNA was extracted from AG6 which
contains a mini-Tn5 insertion at the katA gene. Southern
analysis revealed a 6-kb ClaI DNA fragment containing the
sequences downstream of the m:.ni-Tn5 insertion at the katA
gene and a 5-kb NruI DNA fragment containing~the sequences
upstream of the insertion. Those DNA fragments were
extracted from the agarose gems and were cleaned by using
GENECLEAN II Kit (BIO 101). The ClaI DNA fragment was
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
24
cloned into pBluescript II KS(-) at the ClaI site, and the
NruI DNA fragment was cloned into pTZl9R at the SmaI site.
The resulting plasmids were designated as pXQ6 and
pXQ7, respectively. Sequencing of pXQ6 and pXQ7 was carried
out using a mini-Tn5 specific primer and the M13 reverse and
-40 universal primers. The resulting sequence data were
then used to generate primers for further sequencing. DNA
sequencing was carried out using the ABI PRISM 377 DNA
Sequencer.
In _order to clone the full length katA gene,
primers p83 (5'-GGTGCGCTAGCCAAATTCGTCACCAAGC-3') and n84
(5'-CAATCGCTAGCGTTCGGCCCTCTG-3') were designed that can
respectively reanneal to the upstream and downstream
sequences of the katA gene. Both primers had a NheI site to
facilitate subsequent cloning. The total DNA from A.
tumefaciens strain A348 was used as the template for PCR to
amplify a 2.9 kb DNA fragment. The PCR product was digested
with NheI and ligated into pSW172 (Chen and Winans, 1991)
that had been digested with XbaI. The resulting plasmid
pXQ9 was sequenced in both directions independently to
obtain unambiguous sequence data. Plasmid pXQ9 was
introduced into the mini-Tn5 mutant AG6 to create AG6(pXQ9)
by triparental mating (Ditta et al., 1980) based on
selection on MG/L medium supplemented with 100 ~g/ml of
kanamycin and 5 ~g/ml of tetracycline.
The following is a characterization of katA
encoding catalase from Agrobacterium. A. tumefaciens A348
was mutagenized with a mini-Tn5 transposon containing a
promoter-less gene encoding a green fluorescent protein
(GFP) variant, which produces bright green fluorescence
under W light. The mini-Tn5 transposon was carried on a
plasmid pAG408 (Suarez et al., 1997). One of the mutants
AG6 contained the transposon insertion at a gene that was
differentially induced by pH on a minimal medium.
The leaves of Kalanchoe plants were inoculated
with this mutant strain AG6 and compared with the parent
strain A348. AG6 was highly attenuated in the ability to
cause tumors on plants as compared with A348. In order to
isolate the mini-Tn5 containing DNA fragments, Southern
analysis was conducted to estimate their- sizes. A 6-kb ClaI
DNA fragment containing the sequences downstream of the
transposon insertion site and a 5-kb NruI DNA fragment
containing the sequences upstream of the insertion site were
cloned into the vectors, resulting in plasmids pXQ6 and
pXQ7, respectively. Sequence analysis of pXQ6 and pXQ7 .
revealed that the transposon was inserted at~a gene that is
homologous ~to bacterial genes encoding catalases. This gene
is designated as katA.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
In order to determine the complete sequence of the
gene, the DNA fragment was amplified from A348 by polymerase
chain reaction (PCR). A fragment of 2.9 kb was obtained
that contained both the upstream and downstream sequences of
5 katA. The resulting fragment was cloned into pSW172 (Chen
and Winans, 1991) to generate plasmid pXQ9. When pXQ9 was
introduced into AG6, it could fully restore the ability of
the mutant to cause tumors, suggesting that pXQ9 carried a
full length katA gene. Sequence analysis indicated that the
10 katA locus carried a single open reading frame (ORF) which
encodes a putative protein of 723 amino acids with a
molecular weight of 78.7 kDa. This putative protein was
highly homologous to other bacterial catalases (Fig. 1).
15 To determine whether the katA gene encodes a
functional catalase, the catalase isozyme patterns were
analyzed for the mutant, parent strain and complemented
strain by using a catalase activity staining procedure. As
shown in Fig. 2A, both the parent strain A348 and the
20 complemented strain AG6(pXQ9) had three distinct catalase
activity bands (I, II and III), whereas the mutant AG6 had
only one band (I). This demonstrated that the transposon
insertion at the katA gene in AG6 knocked out two catalase
activity bands.
To investigate whether these two catalase activity
bands originated from the same katA gene, different amounts
of the bacterial cell extracts were loaded on the
polyacrylamide gels for the catalase activity staining. It
was found that the catalase activity band III disappeared
even in A348 when less amount of the cell extract was loaded
(Fig. 2B). This suggests that the catalase activity bands
II and III originated from the same katA gene product. The
catalase activity band III appeared only when a sufficient
amount of cell extract was loaded, suggesting that the band
III was an aggregated form of catalase activity band II.
It was important to determine whether this katA
gene encoded a protein that possessed both catalase and
peroxidase activities like some of the catalase genes
(Loewen, 1997). When the peroxidase activities with the
same cell extracts were stained (Gregory and Fridovich,
1974), no peroxidase activity was found to be associated
with the catalase activity bands. Taken together, these
suggest that the katA gene encodes a catalase isozyme in the
A. tumefaciens cells.
EXAMPLE 3: Determination of katA-gfp expression based on GFP
To stud~° the katA gene expression in different growth
conditions,' A. tumefaciens strains A348 and ~G6 were grown at
28°C for 24 hr on agar plates of MG/L, ALA; I_B (Cangelosi et
al., 1991), and fresh Kalanchoe leaf tissue and stem tissue
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
26
sections which were. sterile and placed on MS medium (Murashige
and Skoog, 1962). The cells were harvested, washed and diluted
to a concentration of approximately OD6oo=~.5. The fluorescence
of each cell suspension was measured by Luminescence
Spectrometer LS50B (Perkin Elmer) using A348 as the blank. The
excitation wavelength was 423 nm; the emission wavelength was
509 nm. The fluorescence levels were expressed as the
fluorescence values divided by the corresponding OD6oo.
To study the katA gene expression in different
genetic backgrounds, A. tumefaciens strains A348, AG6,
AG6(pSW172), AG6(pXQ9), A613, CGI1 and CGI1(pXQ9) were grown at
28 °C for 24 hours on IB (pH 5.5) agar plates. The
fluorescence of each strain was determined as described above.
To determine if H20z can induce the katA expression,
AG6 was grown in MG/L liquid medium overnight at 28°C. The
cells were harvested and resuspended in fresh MG/L liquid
medium to a final concentration of ODsoo=0.5. Ahiquots (2 ml)
of the cultures were transferred into sterile tubes, and H20z
was added to the tubes to final concentrations of 30 ~M, 60 ~,M
and 120 ~M. The cell suspensions were incubated at 28°C for 2
hours. Then lml of each cell suspension was centrifuged,
washed and resuspended in the Laemmli (1970) sample buffer, and
subjected to Western blot as described later.
EXAMPLE 4: Mutagenesis
The C-terminus of KatA was deleted by PCR to generate
pXQll and pXQ22. Site-directed mutagenesis of katA was
performed by overlap extension PCR (Ho et al, 1989). Four
oligonucleotides were designed to mutate a single one amino
acid of the KatA protein. Two residues Arg 94 and His 98 in
the putative catalase motif are highly conserved. His 98 was
changed to Asp; alternatively, Arg 94 was changed to Gln and
His 98 was changed to Asp. A 450 by NsiI -AatII PCR fragment
containing a single mutation at His 98 or double mutations at
both His 98 and Arg 94 was used to replace the corresponding
NsiI -AatII fragment of pXQl3 containing the wild-type katA, in
order to generate pXQ24 or pXQ25, respectively. The presence
of the expected point mutations in NsiI-AatII fragment was
confirmed by DNA sequencing using the ABI PRISM 377 DNA
Sequencer. Similarly, A 670 by MfeI-AatII PCR fragment .
containing an introduced stop codon at Ser 5 or a frameshift
deletion at the second codon of the katA ORF was used to
replace the corresponding Mfel-AatII fragment of pXQl3
containing the wild-type katA, to generate pXQ28 and pXQ29,
respectively.
EXAMPLE 5: Purification of His-KatA fusion protein and
generation of~antibody to KatA
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
27
To generate a (His)6-KatA fusion construct, an
oligomer (containing XhoI site) complementary to the start of
the KatA ORF and an oligomer (containing KphI site) was used to
amplify the KatA ORF fragment. The 2.17 kb XhoI-KpnI fragment
was inserted in-frame downstream of (His)6 harbored on pRSETA.
The resulting pXQl5 was introduced into BL21 by tranformation.
BL21(pXQlS) was grown overnight in LB medium in the presence of
100 ~g/ml of carbenicillin at 37°C. The cell culture was
harvested. Purification of (His)6-KatA was conducted with
TALON metal affinity resin according to the manufacturer
(Clontech). The purified protein was injected into rabbits to
generate the primary antibody. Protein analysis of KatA or GFP
was carried out as described above.
EXAMPLE 6: Complementation
Plasmids pXQl3, pXQ24, pXQ25,.pXQ28 and pXQ29 were
digested by ClaI and ligated with Clal digested pSW172 to
generate pXQ23, pXQ26, pXQ27 pXQ30 and pXQ3l: Plasmids
pSW172, pXQll, pXQ22, pXQ23, pXQ26, pXQ27, pXQ30 and pXQ31
were introduced into the mini-Tn5 mutant AG6 by triparental
mating (Ditta et al., 1980) or electroporation (Cangelosi et
al., 1991). Plasmid pXQl3 was introduced into AG6 by
electroporation, followed by selection in the presence of
carbenicillin. The resulting strain AG613 was obtained that
underwent a single crossover homologous recombination at the
katA locus; it was confirmed by Southern analysis. These
transformant strains were analyzed for the GFP and KatA
expression levels.
The intracellular concentrations of H20z were
measured according to the technique described above in
Example 1 ("Measurement of intracellular H202
concentrations").
The catalase activity in whole bacterial cells was
determined as described above in Example 1 ("Catalase
activity assay").
EXAMPLE 7: Intercellular repression of katA-gfp expression
To determine whether catalase activity of one
bacterial cell could affect the katA-gfp gene expression of
another cell, AG6 was co-cultured with other bacteria which
contained catalase activity. The bacterial cells grown
overnight were suspended in IB liquid and adjusted to ODsoo =
1Ø The AG6 cell suspension was mixed with another
bacterial suspension at 1:1 ratio. An aliquot of 12 ~l
bacterial suspension of a single strain or a mixture of two
strains was, spotted onto IB plates. The plates ~~~ere
incubated overnight at 28°C.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
28
The bacterial fluorescence under UV light was
photographed and the intensity for GFP expression was
measured by the procedure described earlier. To check the
growth viability of each strain in the co-culture mixture, a
portion of each co-culture mixture was harvested to test the
viable cell count on MG/L (for total viable cell count) and
MG/L supplemented with 100 ~tg/ml kanamycin (for AG6 viable
cell count).
EXAMPLE 8: katA is inducible by acidic pH
A. tumefaciens mutant AG6 contained a mini-Tn5
transposon containing a promoter-less green fluorescent
protein (GFP) variant; the mini-Tn5 transposon was inserted
at 995 by downstream from the start codon of katA. Since
the gfp gene was under the control of the katA promoter
(designated as katA-gfp), the katA expression would lead to
the accumulation of GFP, which could be visualized as bright
green fluorescence under UV light. Therefore, the
differential expression of katA in A. tumefaciens could be
determined by measuring the GFP expression of the mutant AG6
in different conditions.
The katA-gfp expression was examined by growing
AG6 on different growth media, MG/L (a rich medium; pH 7.0),
AB (a minimal medium; pH 7.0), and IB (a minimal medium; pH
5.5), as well as on fresh Kalanchoe leaf tissue and stem
tissue sections. As shown in Fig. 3, the fluorescence level
of the bacteria grown on IB was about 10-20 fold higher than
that on neutral pH media including AB and MG/L. The
fluorescence levels on Kalanchoe leaf tissue and stem tissue
sections were about 5-10 fold higher than those in the
neutral pH media. This indicates that katA might be induced
by acidic pH, as the plant tissues also have acidic pH and
minimal nutrition (Li et al., 1999).
Previous experiments have demonstrated that IB
medium is representative of the growth conditions the
bacteria encounter inside plant tissues during the infection
process (Li et al., 1999). To confirm that acidic pH can
induce the katA expression, the fluorescence level on IB (pH
5.5) was compared with that on the medium having the same IB
ingredients but with the pH adjusted to pH 7.0 (IB pH 7.0)'.
As shown in Fig. 3, the fluorescence level on IB (pH 7.0)
was reduced to a level that was similar to other neutral
media, including AB and MG/L. This demonstrates that acidic
pH can induce the katA expression.
EXAMPLE 9: Repression of katA-gfp expression by katA
As shown earlier, the plasmid pXQ9 which carried a
full-length katA gene could fully complement the katA
mutation. When the fluorescence level in the complemented
strain AG6(pXQ9) was analyzed, it was surprisingly found
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
29
that this strain had a highly reduced fluorescence (60-70
fold reduction, as shown in Fig. 4). It appeared that the
wild type katA could repress the katA-gfp expression.
To determine if katA could specifically repress
katA-gfp, pXQ9 was introduced into a different mini-Tn5
transposon mutant strain CGI1, which contained the
transposon insertion at a chromosomal gene'aopB and could
produce bright green fluorescence under W light. As shown
in Fig. 4, the katA gene did not repress the aopB-gfp
expression. This suggests that katA can specifically
repress katA-gfp expression.
It was of interest to determine whether the copy
number and the location of katA could affect the ability to
repress. katA was integrated into the chromosome of AG6
through single-crossover homologous recombination. The
resulting strain AG613 contained a single copy of the wild-
type katA and a single copy of the katA-gfp fusion as
verified by Southern analysis. AG613 had a very low level
of katA-gfp expression, just like AG6(pXQ9) harboring the
katA gene on a plasmid (Fig. 4). This suggests that only
one copy of katA was sufficient to repress the katA-gfp
expression, no matter whether katA is located on plasmid or
chromosome.
EXAMPLE 10: Requirements of the katA-gfp repression
It was of interest to determine if the repression
by katA occurred at the mRNA level or protein level. Site-
directed mutagenesis was conducted to generate mutants that
produced no or truncated KatA protein. The C at the
nucleotide position 14 within the katA open reading frame
(ORF) was changed to G. This created a stop codon at Ser 5
[designated as OkatA(*5)J; the resulting plasmid was named
(pXQ31) (Fig. 5; Table. l). A frameshift deletion at katA
was then created by deleting the G of the second codon in
the katA ORF [designated as DkatA(--2)J; the resulting plasmid
was named pXQ30 (Fig. 5).
As shown in Fig. 6, the pXQ31 construct did not
generate any KatA protein. pXQ30 generated a trace amount
of KatA-like protein, presumably produced from an
alternative translation site downstream from the start codon
or due to infrequent frame shifting of the ribosome, which
could restore the translation of the protein. These two
constructs did not repress the katA-gfp expression as
determined by both the GFP fluorescence and western analysis
using the antibody to GFP (Fig. 7). This suggests that
proauction.of the KatA protein is required for the
repression.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
It was then important to determine whether a full-
length KatA polypeptide was required for the repression.
The 86 amino acids (designated as KatA086) and 50 amino
acids (KatA~50) at the C-terminus of KatA were deleted to
5 generate pXQll and pXQ22, respectively (Fig. 5). These
constructs produced smaller sizes of KatA proteins as
expected (Fig. 6B, lanes 3 and 4). However, the amounts of
these truncated KatA proteins were much less than the wild
type KatA (harbored on pXQ9) (lane 2), indicating that these.
10 truncated KatA proteins were unstable. These truncated
proteins did not repress the kat:A-gfp expression at a
significant level (Fig. 7, lanes 9 and 10), presumably
because they did not exhibit any significant catalase
activity (Fig. 8; Table 2).
Table 2. Catalase activity in whole bacterial cells containing the wild type
or mutant katA genesa
Strain ~ Protein expressed ~ Catalase g ctivity
(unit/10 cells)
AG6(pXQ23) KatA 77145.5 440.3
AG6(pXQ9) KatA 3997.3 459.1
A348 KatA 1486.2 149.5
AG6(pXQ26) KatA(98H/D) 219.3 19.5b
AG6(pXQ 11 KatA~86 193.6 25.2
)
AG6(pXQ27) KatA(94R/Q)(98H/D) 184.0 25.1'
AG6(pXQ22) KatA050 182.7 21.5
AG6 KatA' 175.2 20.2'
a Agrobacterium tumefaciens cells were grown on IB plates and then harvested.
The
2 0 catalase activities in whole bacterial cells were measured as described in
the
Materials and Methods.
b The catalase activity of AG6(pXQ26) was significantly different from that of
AG6
(P < 0.05)~based on Student's t-test.
The catalase activity of AG6(pXQI 1), AG6/(pXQ27) or AG6/(pXQ22) was not
2 5 significantly different from that of AG6 based on Student's t-test.
Since the truncated KatA proteins did not possess
any catalase activity detected by an isozyme staining
procedure (Fig. 8)~, it was important to know whether a
30 functional catalase activity was required for this feedback
repression. The amino acid sequence of A. t:umefaciens
catalase KatA was analyzed by a motif search program
(http://www.motif.ge;~ome.ad.jp). It revealed that a motif
of 12 amino acids (VGMMARVTWHAA) located from amino acid 89
35 to X00 from the start codon was qualified fox a peroxidases
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
31
active site signature. This motif might be involved in the
catalase activity.
Site-directed mutagenesis was conducted to
inactivate the catalase activity. Computer analysis
revealed that Arg 94 and His 98 in the conservative motif of
KatA might be crucial for the catalase activity. Previous
studies have indicated that the corresponding residues Arg
102 and His 106 of the E. coli homolog HPI are important for
the catalase activity (Hillar et al., 2000). His 98 was
changed to Asp (designated as KatA(98H/D)); Arg 94 was
changed to Gln and His 98 was changed to Asp (designated as
KatA(94R/Q)(98H/D)~. The resulting plasmids were named pXQ26
and pXQ27, respectively.
As shown in Fig. 8, both the wild-type strain A348
and the complemented strain AG6(pXQ23) had the KatA catalase
activity bands, whereas they were missing in the mutant AG6,
AG6(pXQ26) and AG6(pXQ27). This suggests that alteration of
His 98 or both Arg 94 and His 98 in the conservative motif
of KatA abolished the KatA catalase activity detected by the
staining procedure. Western blot analysis was performed to
check the stability of the mutant proteins. As shown in
Fig. 6A, AG6(pXQ26) and AG6(pXQ27) produced the same size of
KatA proteins as the wild type, and a high level of the
point mutant proteins accumulated in the cells. As shown in
Fig. 7, these two point mutant proteins could slightly
repress the katA-gfp expression. The GFP expression was
virtually undetectable in AG6(pXQ23) (lane 4), but it was
reduced in AG6(pXQ26) and AG6(pXQ27) (lanes 5 and 6), as
compared with the strains expressing no or truncated protein
KatA (lanes 7, 8, 9 and 10).
It was important to determine if the mutant KatA
proteins possessed any trace amount of catalase activity.
The genes encoding those mutant proteins were introduced
into AG6 that lacks katA. Then the catalase activity in
whole bacterial cells was measured, because this could
presumably avoid any inactivation of catalase activity due
to the cell break-up process.
As shown in Table 2, the catalase activity in the
bacteria containing the mutant protein, KatA086,
KatA(94R/Q)(98H/D) or KatA~50 was slightly higher than that
in AG6, but not at a statistically significant level. This
suggests that these mutant KatA proteins did not possess any
significant catalase activity. The activities in whole
cells for those bacteria were apparently due to the catalase
other than Kai.~., since AG6 (which lacks katA) possessed
catalase activity (Fig. 8 and Table 2). The~catalase
activity in AQ6(pXQ26) was statistically higher than that of
AG6, suggesting that KatA(98H/D) possessed a low level of
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
32
catalase activity. This low activity presumably has
contributed to the low repression of katA-gfp (Fig. 7).
It is interesting to note that KatA(94R/Q)(98H/D)
[in the strain AG6(pXQ27)] repressed the expression of katA-
gfp at a low level (Fig. 7), since the activity for this
protein was not statistically significant and was lower (if
any) than that of KatA~86 (Table 2), which did not repress
the katA-gfp expression at a significant level (Fig. 7).
This suggests that both the KatA catalase activity and the
protein itself were involved in the repression of katA-gfp.
In summary, the repression of katA-gfp expression
required the production of a functional KatA protein that
possessed the catalase activity. In addition, among the
mutant KatA proteins only KatA(98H/D) and KatA(94R/Q)(98H/D)
could significantly repress the katA-gfp expression at a
level much lower than the wild-type KatA (Fig. 7).
Incidentally, only these two mutant KatA proteins
accumulated at a very high level in the bacteria (Fig. 6).
This suggests that the sheer amount of the KatA proteins
present in the cells might contribute to the repression,
presumably because the mutant KatA proteins could still bind
to HzOz to reduce the availability of intracellular HZO2 to
induce katA-gfp expression. KatA(98H/D) repressed the katA-
gfp expression at a level slightly higher than
KatA(94R/Q)(98H/D) (Fig. 7), presumably due to its low
catalase activity while KatA(94R/Q)(98H/D) did not possess
any significant catalase activity (Table' 2) because both two
important residues at the putative active site have been
altered (Fig. 5) .
EXAMPLE 11: katA can be induced by hydrogen peroxide
It was then important to determine how the
catalase activity repressed the katA-gfp expression. One
possibility is that catalase may reduce the intracellular
HZO2 levels to repress the katA-gfp expression. To determine
whether the expression of katA can be induced by H202, like
some of other bacterial catalases (Loewen, 1997), AG6 was
treated with low concentrations of H2O2 (30-120 ~M) in liquid
MG/L medium and the katA-gfp expression levels were then
determined.
As shown in Fig. 9, the katA-gfp expression levels
increased significantly in the presence of H2O2, suggesting
that the katA expression could be induced by HZOz. This
indicates that the reason that the catalase activity could
repress the katA-gfp expression is that catalase could
deplete the katA inducer level. This implies that the
endogenous Hz02 acts as the. intracellular inducer for the
katA expression insi~e A. tumefaciens cells and that
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
33
induction of katA by acidic pH involves the increase of
intracellular H202 levels .
EXAMPLE 12: Acidic pH could enhance the accumulation of
S intracellular H202 in katA- cells
Since katA could be induced by H202, it was
important to determine whether the induction of katA by
acidic pH was indeed due to the enhanced levels of
endogenous H202 in the bacterial cells. The intracellular
H202 concentrations were measured for the bacteria grown on
the acidic IB plates (pH 5.5) or AB plates of neutral pH (pH
7.0), according to the previously described procedures
(Gonzalez-Flecha and Demple, 1995; 1997) that were based on
a complete equilibration of the intracellular and ..
extracellular H2O2 levels of the bacterial cells that were
grown on IB or AB and then resuspended in a phosphate buffer
(pH 7.4) (see the Materials and Methods).
As shown in Table 3, the intracellular H2O2
concentration for the AG6 cells grown on IB was about 10
times higher than those for the bacterial cells containing
the wild-type katA [A348 and AG6(pXQ9)]. The level for the
AG6 cells grown on AB was about 4 times higher than those
for the bacteria having the wild type katA. The level of
intracellular H202 increased about 3 times when the AG6 cells
were switched from pH 7.0 to pH 5.5. The intracellular HZOZ
concentrations were virtually constant for the bacteria
having the wild type katA, no mater whether they were grown
on AB or IB plates. These indicate that mutation at katA
enhanced the accumulation of intracellular HZOzfor the
bacteria grown at acidic pH. The elevated level of
intracellular Hz02 caused by acidic pH in the absence of
functional katA induced the expression of katA-gfp.
Table 3. Intracellular H202 concentrations of bacteria grown on IB and AB
platesa
H2~2~wM)
Strain I AB (pH 7.0) ~ IB (pH 5.5)
A348 ~ 0.12 ~ 0.03 ~ 0.13 ~ 0.02
AG6 ~ 0.41 ~0.08 I 1.16~0.14
AG6(pXQ9) ~ 0.10 ~ 0.02 ~ 0.09 ~ 0.01
Agrobacterium tumefaciens cells wee grown on AB or IB plates. The
intracellular
4 0 H202 concentrations were measured as described in the Mata~ials and
Methods,
based on a complete equilibration of the intracellular and extracellular H202
levels of
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
34
the bacterial cells that were grown on IB or AB and then resuspended in a
phosphate
buffer (pH 7.4).
EXAMPLE 13: Intracellular H202 scavenging assay technology
The studies indicate that mutation at katA can
enhance the accumulation of intracellular HzOz in the
bacterial cells grown at acidic pH. The elevated level of
intracellular H202 caused by acidic pH in the absence of
functional katA can induce the expression of katA-gfp. When
a katA gene encoding a functional catalase is introduced
into the cells lacking the katA gene, katA-gfp expression is
dramatically repressed (Figs. 4, 7, 8 and 10).
Two mutant KatA proteins truncated at the C-
terminus exhibited a very low level of accumulation in the
cells and no significant catalase activity; neither mutant
repressed katA-gfp expression at any significant level
(Figs. 7 and 8; Table 2). These indicate that the katA-gfp
expression levels can reflect the intracellular H2O2
scavenging capacity of the cells. If the cells are
introduced with molecules, large or small, that can scavenge
the intracellular HzOz levels, the katA-gfp expression will
be repressed. Therefore, measuring the katA-gfp expression
can monitor the intracellular H202 scavenging capacity of a
molecule that is introduced into the cells.
In the present study, the fully functional
catalase gene katA repressed the katA-gfp expression at the
highest level; thus it has the highest capacity to scavenge
intracellular Hz02 (Figs. 4, 7 and 10). Two mutant KatA
proteins truncated at the C-terminus exhibited a very low
level of accumulation in the cells and no significant
catalase activity; neither repressed the katA-gfp expression
at any significant level (Figs. 7 and 8; Table 2). Thus,
they have no significant capacity to scavenge intracellular
H20z. Two other mutant KatA proteins, KatA(98H/D) and
KatA(94R/Q)(98H/D), could significantly repress the katA-gfp
expression at a level much lower than the wild-type KatA
(Fig. 7) .
Incidentally, only these two mutant KatA proteins
accumulated at a very high level in the bacteria (Fig. 6).
This suggests that the sheer amount of the KatA proteins
present in the cells might contribute to the repression,
presumably because the mutant KatA proteins could still bind
to H20z to reduce the availability of intracellular H2O2.
KatA(98H/D) repressed the katA-gfp expression at a level
slightly higher than KatA(94R/Q)(98H/D) (Fig. 7), presumably
due to its low catalase activity while KatA(94R/Q)(98H/D)
did not possess any significant catalase activity (Table 2).
All of these indicate that this assay technology is
extremely sensitive in determining the intracellular Hz02
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
scavenging capacities of different molecules and that this
assay can analyze molecules that can scavenge intracellular
HZOZ directly or indirectly.
5 EXAMPLE 14: Repression of katA-gfp expression by
surrounding bacterial cells
To investigate whether the repression of katA-gfp
expression could occur intercellularly, AG6 was co-cultured
10 with the wild type stain A348 (see Examphe 7). As shown in
Fig. 10, A348 indeed could repress the katA-gfp expression
in the surrounding AG6 cells, when A348 was mixed with AG6
(upper panel). The fluorescence level of the A348 + AG6
mixture was reduced about 20 fold as compared to that of AG6
15 alone (lower panel). Evaluation of the viable cell count of
the AG6 + A348 mixture showed that AG6 was 50% of the total
cells as expected, suggesting that the repression in the
mixture was caused intercellularly and not simply due to the
dilution effect from the non-fluorescent A348 cells.
This intercellular repression phenomenon was also
observed when AG6 was co-cultured with R. meliloti and E. coli
which contained the catalase genes (Herouart et al, 1996;
Loewen, 1997) and possessed the catalase activity as measured
by the catalase activity assay. The repression of katA-gfp
expression by R. meliloti and E. coli was weaker than that of
A348 (Fig. 10), presumably because they grew slower than AG6 on
IB plate as demonstrated by the viable cell count experiment.
These suggest that H202 generated in AG6 could pass through the
bacterial cell .membranes and enter into the surrounding cells
that possessed the catalase activity equivalent to KatA. The
repression of katA-gfp expression could be achieved by
catalases from other bacterial species. This indicates that
intracellular H202 scavengers can be applied, external and
adjacent to the cells under oxidative stress, in the form of
live cells or non-living systems that can receive H20z from the
adjacent cells under oxidative stress.
Interestingly, active catalase added to the medium
of AG6 cells did not repress katA-gfp expression. Catalase
purchased from Sigma was added into IB plates at a final
concentration of 1 or 5 microgram per ml. AG6 cells
(carrying the katA-gfp fusion) were then grown on
the IB plate. The bacterial cells exhibited the same level
of GFP expression as the AG6 cells grown in the absence of
catalase. To ensure that the catalase in the IB medium was
still active, hydrogen peroxide was dropped onto the
catalase-containing plate. Air bubbles were instantly
visible in the catalase-containing plate; no air bubble was
found in the control cat:~lase-free IB plate. This
demonstrated that the catalase in the medium~was
active. Therefore, it can be concluded that extracellular
catalase could not remove the intracellular'hydrogen
<IMG>
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
37
REFERENCES
Arnold et al. (2001) Proc. Natl. Acad. Sci. USA, 98(10):
15550-55.
Cangelosi, G.A., Best, E.A., Martinetti, G. and Nester, E.W.
(1991) Genetic analysis of Agrobacterium. Methods Enzymol
204: 384-397.
Chalfie M. and Prashner D. C., U.S. Pat. No. 5,491,084.
Charles, T.C., and Nester, E.W. (1993) A chromosomally
encoded two-component sensory transduction system is
required for virulence of Agrobacterium tumefaciens. J
Bacteriol 175: 6614-6625.
Chen, C.Y., and Winans, S.C. (1991) Controlled expression of
the transcriptional activator gene virG in Agrobacterium
tumefaciens by using the Escherichia coli lac promoter. J
Bacteriol 173: 1139-1144.
Clare, D.A., Duong, M.N., Darr, D., Archibald, F. and
Fridovich, I. (1984) Effects of molecular oxygen on
detection of superoxide radical with nitroblue tetrazolium
and on activity stains for catalase. Anal Biochem 140:
532-537.
Ditta, G., Stanfield, S., Corbin, D. and Helinski, D.R.
(1980) Broad host range DNA cloning system for gram-
negative bacteria: construction of a gene bank of
Rhizobium melioti. Proc Natl Acad Sci 77: 7347-7351.
Farr et al., Microbiol. Rev., 55, pp. 561-85 (1991).
Farr US patent 5,585,252.
Farr US patent 5,811,231.
Farr US patent 5,589,337.
Gonzalez-Flecha, B. and Demple, B. (1995) Metabolic sources
of hydrogen peroxide in aerobically growing Escherichia
coli. J Biol Chem 270: 13681-13687.
Gonzalez-Flecha, B. and Demple, B. (1997) Homeostatic
regulation of intracellular hydrogen peroxide
concentration in aerobically growing Escherichia coli. J
Bacteriol 179:382-388.
Gort and Imlay, (1998) J Bacteriol. 1998 Mar 180(6):1402-10.
Gould S. J., and Subramani S., Anal Biochem. 1988 Nov
15; 175 (1) :5-13 .
Greenberg et al., Proc. Natl. Acad. Sci. USA, 87, pp. 6181-
85 (1990) .
Gregory, E.M., and Fridovich, I. (1974) Visualization of
catalase on acrylamide gels. Anal Biochem 58: 57-62
Herouart, D., Sigaud, S., Moreau, S., Frendo, P., Touati,
D., and Puppo, A.( 1996) Cloning and characterization of
the katA gene of Rhizobium meliloti encoding a hydrogen
peroxide-inducible catalase. J Bacteriol 178: 6802-6809.
Hillar, A., Peters, B., Pauls, R., Loboda, A., Zhang, H.
Mauk, A.G., and Loewen, P.C. (2000) Modulation,of the
activities of catalase-peroxidase HPI of Escherich~a coli
by site-directed mutagenesis. Biochemistry 39: 5868-5875.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
38
Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease,
L.R. (1989) Site-directed mutagenesis by overlap extension
using the polymerase chain reaction. Gene 77: 51-59.
Kogoma et al., Proc. Natl. Acad. Sci USA, 85, pp. 4799-803
(1988) .
Koh J., Enders G. H., et al, Nature. Jun 8; 375(6531):506-10
(1995) .
Laemmli, U.K. (1970). Cleavage of structural proteins during
the assembly of the head of bacteriophage T4. Nature 227:
680-685.
Li, L.P., Li, Y., Lim, T.M., and Pan, S.Q. (1999) GFP-aided
confocal laser scanning microscopy can monitor
Agrobacterium tumefaciens cell morphology and gene
expression associated with infection. FEMS Microbiol Lett
179: 141-146.
Loewen, P.C. (1997) Bacterial catalases. In Oxidative stress
and the molecular biology of antioxidant defenses.
Scandalios, J.G. (eds). Cold Spring Harbor Laboratory
Press, pp. 273-308.
Maciver, I., and Hansen, E.J. (1996) Lack of expression of
the global regulator OxyR in Haemophilus influenzae has a
profound effect on growth phenotype. Infect Immun 64:
4618-4629.
Mahadev et al. (2001) J. Biol. Chem. 15;276(24):21938-42.
Mohanty, J. Jonathan, G., Jaffe, S., Schulman E. S., and
Raible, D.G. (1997) A highly sensitive fluorescent micro-
assay of Hz02 release from activated human leukocytes
using a dihydroxyphenoxazine derivative. J Immunol Methods
202: 133-141.
Murashige, T., and Skoog, F. (1962) A revised medium for
rapid growth and bioassays with tobacco tissue cultures.
Physiol Plant 15: 473-497.
Ren et al. (2002) J. Biol. Chem. 277(1):559-65.
Saliim and Abu-Shakra (2001) Teratog Carcinog Mutagen
21 (5) :349-59.
Sambrook, J.F., Fritsch, E.F., and Maniatis, T. (1989)
Molecular Cloning: a laboratory manual, 2nd ed. Cold
Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Shapiro S. K., Chou J., et al., Gene Nov:; 25: 71-82 (1983).
Suarez, A., Guttler, A., Stratz, M., Staendner, L.H.,
Timmis, K.N., and Guzman, C.A. (1997) Green fluorescent
protein-based reporter systems for genetic analysis of
bacteria including monocopy applications. Gene 196: 69-74.
Thiel G., Petersohn D., and Schoch S., Gene Feb. 12; 168:
173-176 (1996).
Xu, X.Q. and Pan, S.Q. (2000) An Agrobacterium catalase as a
virulence factor involved in tumorigenesis. Mol Microbiol
2:407-14.
0 While a number of embodiments have been presented,
it is apparent that the basic construction can be altered to
provide other embodiments which utilize the methods and kits
of the invention. The scope of the invention is to be
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
39
defined by the claims appended hereto rather than the
specific embodiments presented herein by way of example.
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
SEQUENCE LISTING
<110> Pan, Shen Quan
<120> Methods and Kits for Identifying Scavengers of Reactive Oxy
gen Species (ROS)
<130> 79612-19
<150> US60/266,657
<151> 2001-02-05
<160> 5
<170> PatentIn version 3.0
<210> 1
<211> 522
<212> DNA
<213> Agrobacterium tumefaciens
<400> 1
ggcacgatcg cctatgacgt cgcgggtctg aagaccttcg gcttcgcctt cggccgcgaa
gacatctggg cgccggaaaa ggacacctat tggggtgacg aaaaggaatg gctggcgccg
120
agcgacggcc gttatggcga cgtgagcaag cccgagacgc tggaaaaccc gcttgccgcc
180
gtgcagatgg gcctgatcta cgtcaacccg gaaggtgtca acggcaagtc cgatccgctg
240
gcgacggcgg cgcagatgcg cgaaaccttt gcccgcatgg ggatggatga cgaggaaacc
300
gttgccctga cggccggcgg ccacaccatc ggcaagtccc atggcaatgg cagtgctgcc
360
aatctcagcc ccgatccgga agctgctggc ccggaatatc agggtctcgg ctggatcaat
420
accaagggcc gcggcattgg ccgtgacacc gtggtgtcgg gtatcgaagg cgcatggaca
980
agcgaaccaa ccaagtggga caacggcttc ttcgacatgc tg
522
1
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
<210> 2
<211> 2839
<212> DNA
<213> Agrobacterium tumefaciens
<220>
<221> CDS
<222> (308)..(2476)
<400> 2
aggctgggag atggcgcagg cttccgccgc gcgcccgaaa tggccgattt tggccagcgc
atcgaaataa cggagatgtt tcatggagag ggcaatcata agctcagcat atcgcagcct
120
ttagaatata caattggaaa ttatggaacc cggctgttag tcatcttcat gacagaaaga
180
agctgtagct gtggatgatc atcggcatcg cagcaggtcg tttgagacct gccgctgcct
240
ctggctcgga atgctgcaca tgtcgaactg ataatttgtt taattgctaa tcccatcgga
300
gggcgaa atg gac gca act tca aaa ccg get ggc aag tgt ccc gtc atg
349
Met Asp Ala Thr Ser Lys Pro Ala Gly Lys Cys Pro Val Met
I 5 10
cat gga ggc aat acg gcc tcc ggc aaa tcg gtg acc gaa tgg tgg ccg
397
His Gly Gly Asn Thr Ala Ser Gly Lys Ser Val Thr Glu Trp Trp Pro
15 20 25 30
aac gcg cta aac ctc gac atc ctg cat cag cac gac acc aag acc aat
445
Asn Ala Leu Asn Leu Asp Ile Leu His Gln His Asp Thr Lys Thr Asn
35 40 45
2
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
ccg ctc ggc acc tcc ttc aac tac cgc gaa gcg ctg aag acg ctt gat
493
Pro Leu Gly Thr Ser Phe Asn Tyr Arg Glu Ala Leu Lys Thr Leu Asp
50 55 60
gtc gaa gcc ctc aag gcc gat ctg cgc gcg ctt atg acc gac agc cag
541
Val Glu Ala Leu Lys Ala Asp Leu Arg Ala Leu Met Thr Asp Ser Gln
65 70 75
gaa tgg tgg ccg gcc gac tgg ggc agt tat gtc ggc atg atg gcc cgt
589
Glu Trp Trp Pro Ala Asp Trp Gly Ser Tyr Val Gly Met Met Ala Arg
80 85 90
gtt acc tgg cat gcc gcc ggt tcc tat cgt gtc aca gac ggt cgc ggc
637
Val Thr Trp His Ala Ala Gly Ser Tyr Arg Val Thr Asp Gly Arg Gly
95 100 105 110
ggc gcc aat acc ggc aac cag cgt ttt gca ccg ctc aat tcc tgg ccg
685
Gly Ala Asn Thr Gly Asn Gln Arg Phe Ala Pro Leu Asn Ser Trp Pro
115 120 125
gac aac gtc aac acc gac aag ggc cgc cgc ctg ctg tgg ccg atc aag
733
Asp Asn Val Asn Thr Asp Lys Gly Arg Arg Leu Leu Trp Pro Ile Lys
130 135 140
aag aaa tac ggc aac aag att tcc tgg gcc gac ctt atc gcg ctc gcc
781
Lys Lys Tyr Gly Asn Lys Ile Ser Trp Ala Asp Leu Ile Ala Leu Ala
145 150 155
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
ggc acg atc gcc tat gac gtc gcg ggt ctg aag acc ttc ggc ttc gcc
829
Gly Thr Ile Ala Tyr Asp Val Ala Gly Leu Lys Thr Phe Gly Phe Ala
160 165 170
ttc ggc cgc gaa gac atc tgg gcg ccg gaa aag gac acc tat tgg ggt
877
Phe Gly Arg Glu Asp Ile Trp Ala Pro Glu Lys Asp Thr .Tyr Trp Gly
175 180 185 190
gac gaa aag gaa tgg ctg gcg ccg agc gac ggc cgt tat ggc gac gtg
925
Asp Glu Lys Glu Trp Leu Ala Pro Ser Asp Gly Arg Tyr Gly Asp Val
195 200 205
agc aag ccc gag acg ctg gaa aac ccg ctt gcc gcc gtg cag atg ggc
973
Ser Lys Pro Glu Thr Leu Glu Asn Pro Leu Ala Ala Val Gln Met Gly
210 215 220
ctg atc tac gtc aac ccg gaa ggt gtc aac ggc aag tcc gat ccg ctg
1021
Leu Ile Tyr Val Asn Pro Glu Gly Val Asn Gly Lys Ser Asp Pro Leu
225 230 235
gcg acg gcg gcg cag atg cgc gaa acc ttt gcc cgc atg ggg atg gat
1069
Ala Thr Ala Ala Gln Met Arg Glu Thr Phe Ala Arg Met Gly Met Asp
240 - 245 250
gac gag gaa acc gtt gcc ctg acg gcc ggc ggc cac acc atc ggc aag
1117
Asp Glu Glu Thr Val Ala Leu Thr Ala Gly Gly His Thr Ile Gly Lys
255 260 265 270
4
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
tcc cat ggc aat ggc agt get gcc aat ctc agc ccc gat ccg gaa get
1165
Ser His GIy Asn Gly Ser Ala Ala Asn Leu Ser Pro Asp Pro Glu Ala
275 280 285
get ggc ccg gaa tat cag ggt ctc ggc tgg atc aat acc aag ggc cgc
1213
Ala Gly Pro Glu Tyr Gln Gly Leu Gly Trp Ile Asn Thr Lys Gly Arg
290 295 300
ggc att ggc cgt gac acc gtg gtg tcg ggt atc gaa ggc gca tgg aca
1261
Gly Ile Gly Arg Asp Thr Val Val Ser Gly Ile Glu Gly Ala Trp Thr
305 310 315
agc gaa cca acc aag tgg gac aac ggc ttc ttc gac atg ctg ttc aag
1309
Ser Glu Pro Thr Lys Trp Asp Asn Gly Phe Phe Asp Met Leu Phe Lys
320 325 330
cac gag tgg acc ctg acg cac agc ccc gcg ggt gca tcg caa tgg gcg
1357
His Glu Trp Thr Leu Thr His Ser Pro Ala Gly Ala Ser Gln Trp Ala
335 340 345 350
ccg att acc atc gcc gaa gaa gac aag cct gtt gat gtc gag gat gcg
1405
Pro Ile Thr Ile Ala Glu Glu Asp Lys Pro Val Asp Val Glu Asp Ala
355 360 365
tcg atc cgc acc atc ccg atg atg acc gac gcc gac atg gcc ctg aag
1453
Ser Ile Arg Thr Ile Pro Met Met Thr Asp Ala Asp Met Ala Leu Lys
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
370 375 380
gtc gat ccg atc tac cgc gag att tcg ctg aag ttc aag gac gat cag
1501
Val Asp Pro Ile Tyr Arg Glu Ile Ser Leu Lys Phe Lys Asp Asp Gln
385 390 395
gac cat ttc tct gat gtc ttc gcc cgc gcc tgg ttc aag.ctg acg cat
1549
Asp His Phe Ser Asp Val Phe Ala Arg Ala Trp Phe Lys Leu Thr His
400 405 410
cgc gac atg ggg ccg aag tcc cgt tac gtc ggc ccg gat gtt ccg get
1597
Arg Asp Met Gly Pro Lys Ser Arg Tyr Val Gly Pro Asp Val Pro Ala
415 420 425 430
gaa gac ctg atc tgg cag gat ccg atc ccg gca ggc tcc acg agc tac
1645
Glu Asp Leu Ile Trp Gln Asp Pro Ile Pro Ala Gly Ser Thr Ser Tyr
435 440 445
gat gtc get gcc gtc aag get aag atc get gcc tcc ggc ctt tct gtc
1693
Asp Val Ala Ala Val Lys Ala Lys Ile Ala Ala Ser Gly Leu Ser Val
450 455 460
gcc gat ctg gtt tca acc gca tgg gac agt gcc cgc acc ttc cgt ggt
1741
Ala Asp Leu Val Ser Thr Ala Trp Asp Ser Ala Arg Thr Phe Arg Gly
465 470 475
tcg gac aag cgc ggc ggc gcc aat ggc gcg cgt att cgt ctc gca ccg
1789
Ser Asp Lys Arg Gly Gly Ala Asn Gly Ala Arg Ile Arg Leu Ala Pro
6
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
480 485 490
cag aag gat tgg gaa ggc aat gag ccc gcc cgt ctt tcc cgc gtg ctt
1837
Gln Lys Asp Trp Glu Gly Asn GIu Pro Ala Arg Leu Ser Arg Val Leu
495 500 505 510
tcg gtt ctg gag ccg att gcc cgc gaa acc ggt gca agc atc gcc gat
1885
Ser Val Leu Glu Pro Ile Ala Arg Glu Thr Gly Ala Ser Ile Ala Asp
515 520 525
gtg atc gtt ctg get ggc aat tac ggc gtg gag cag gcg gcg aaa gcg
1933
Val Ile Val Leu Ala Gly Asn Tyr Gly Val Glu Gln Ala Ala Lys Ala
530 535 540
get ggt ttc gat atc gcc gtg ccc ttc gcg gcc ggt cgt ggt gac get
1981
Ala Gly Phe Asp Ile Ala Val Pro Phe Ala Ala Gly Arg Gly Asp Ala
545 550 555
tcc gcc gag cag acg gat gcc gac agc ttt gcg ccg ctt gag ccg ctg
2029
Ser Ala Glu Gln Thr Asp Ala Asp Ser Phe Ala Pro Leu Glu Pro Leu
560 565 570
gcg gat ggt ttc cgc aac tgg gtg aag aag gac tat gtc gtc agc ccc
2077
Ala Asp Gly Phe Arg Asn Trp Val Lys Lys Asp Tyr Val Val Ser Pro
575 580 585 590
gaa gag ctg ctg ctc gat cgg gca cag ctt ctt ggc ctc acc gcg ccg
2125
7
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
Glu Glu Leu Leu Leu Asp Arg Ala Gln Leu Leu Gly Leu Thr Ala Pro
595 600 605
gaa ctc acc gtc ctc atc ggc ggc ctg cgc gtc atc ggc gcc aat tac
2273
Glu Leu Thr Val Leu Ile Gly Gly Leu Arg Val Ile Gly Ala Asn Tyr
610 615 620
ggc ggt gcg gcg cat ggc gtc ttc acc gat aag ccg ggg gcg ctt aca
2221
Gly Gly Ala Ala His Gly Val Phe Thr Asp Lys Pro Gly Ala Leu Thr
625 630 635
acg gac ttc ttc acg acg ttg acg gac atg gcc tat tcc tgg gtc ccg
2269
Thr Asp Phe Phe Thr Thr Leu Thr Asp Met Ala Tyr Ser Trp Val Pro
640 645 650
acc ggc aac aat ctc tat gag atc cgt gat cgc aag acc ggc gca gcc
2317
Thr Gly Asn Asn Leu Tyr Glu Ile Arg Asp Arg Lys Thr Gly Ala Ala
655 660 665 670
aga tat tcg gca acc cgc gtc gat ctc gtg atc ggc tcc aac tcc atc
2365
Arg Tyr Ser Ala Thr Arg~Val Asp Leu Val Ile Gly Ser Asn Ser Ile
675 680 685
ctg cgc get tat gcg gaa gtt tat gcg cag gac gac aac agg gaa aaa
2413
Leu Arg Ala Tyr Ala Glu Val Tyr Ala Gln Asp Asp Asn Arg Glu Lys
690 695 700
ttc gcc cgc gac ttc att gcc gcc tgg acg aag gtg atg aac gcc gac
8
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
2461
Phe Ala Arg Asp Phe Ile Ala Ala Trp Thr Lys Val Met Asn Ala Asp
705 710 715
cgt ttc gat ctg atc tgagcggaag cgattagccg aaaagacaac acctccccga
2516
Arg Phe Asp Leu Ile
720
gcgatcgggg aggtgttttt gtggcggctt cctctcgatg acggaggccc catattcatt
2576
caacggcggt cggaagacgg aacctgccgc tcgggtgacg acttttcagt tggtccagca
2636
ggcgtcttgc cactcttctc gttttcatgc gcatccgggc cagcgacaat cacgccacgg
2696
ccctgttctt cacgcttctt gtcggcgaaa tcgcccgctt cctttttctc gttcatcgtt
2756
ttctcccttc gcgttgtttc gtccttcatt caacgaatga cgaagaggtg ggttccagat
2816
agacaattcc gcagagggcc gaa
2839
<210> 3
<211> 723
<212> PRT
<213> Agrobacterium tumefaciens
<400> 3
Met Asp Ala Thr Ser Lys Pro Ala Gly Lys Cys Pro Val Met His Gly
1 5 10 15
Gly Asn Thr Ala Ser Gly Lys Ser Val Thr Glu Trp Trp Pro Asn Ala
20 25 30
Leu Asn Leu Asp Ile Leu His Gln His Asp Thr Lys Thr Asn Pro Leu
9
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
35 40 45
Gly Thr Ser Phe Asn Tyr Arg Glu Ala Leu Lys Thr Leu Asp Val Glu
50 55 60
Ala Leu Lys Ala Asp Leu Arg Ala Leu Met Thr Asp Ser Gln Glu Trp
65 70 75 80
Trp Pro Ala Asp Trp Gly Ser Tyr Val Gly Met Met Ala Arg Val Thr
85 90 95
Trp His Ala Ala Gly Ser Tyr Arg Val Thr Asp Gly Arg Gly Gly Ala
100 105 110
Asn Thr Gly Asn Gln Arg Phe Ala Pro Leu Asn Ser Trp Pro Asp Asn
115 120 125
Val Asn Thr Asp Lys Gly Arg Arg Leu Leu Trp Pro Ile Lys Lys Lys
130 135 140
Tyr Gly Asn Lys Ile Ser Trp Ala Asp Leu Ile Ala Leu Ala Gly Thr
145 150 155 160
Ile Ala Tyr Asp Val Ala Gly Leu Lys Thr Phe Gly Phe Ala Phe Gly
165 170 175
Arg Glu Asp Ile Trp Ala Pro Glu Lys Asp Thr Tyr Trp Gly Asp Glu
180 185 190
Lys Glu Trp Leu Ala Pro Ser Asp Gly Arg Tyr Gly Asp Val Ser Lys
195 200 205
Pro Glu Thr Leu Glu Asn Pro Leu Ala Ala Val Gln Met Gly Leu Ile
210 215 220
Tyr Val Asn Pro Glu Gly Val Asn Gly Lys Ser Asp Pro Leu Ala Thr
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
225 230 235 240
Ala Ala Gln Met Arg Glu Thr Phe Ala Arg Met Gly Met Asp Asp Glu
245 250 255
Glu Thr Val Ala Leu Thr Ala Gly Gly His Thr Ile Gly Lys Ser His
260 265 270
Gly Asn Gly Ser Ala Ala Asn Leu Ser Pro Asp Pro Glu Ala Ala Gly
275 280 285
Pro Glu Tyr Gln Gly Leu Gly Trp Ile Asn Thr Lys Gly Arg Gly Ile
290 '295 300
Gly Arg Asp Thr Val Val Ser Gly Ile Glu Gly Ala Trp Thr Ser Glu
305 310 315 320
Pro Thr Lys Trp Asp Asn Gly Phe Phe Asp Met Leu Phe Lys His Glu
325 330 335
Trp Thr Leu Thr His Ser Pro Ala Gly Ala Ser Gln Trp Ala Pro Ile
340 345 350
Thr Ile Ala Glu Glu Asp Lys Pro VaI Asp Val Glu Asp Ala Ser Ile
355 360 365
Arg Thr Ile Pro Met Met Thr Asp Ala Asp Met Ala Leu Lys Val Asp
370 375 380
Pro Ile Tyr Arg Glu Ile Ser Leu Lys Phe Lys Asp Asp Gln Asp His
385 390 395 400
Phe Ser Asp Val Phe Ala Arg Ala Trp Phe Lys Leu Thr His Arg Asp
405 410 415
Met Gly Pro Lys Ser Arg Tyr Val Gly Pro Asp Val Pro Ala Glu Asp
11
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
420 425 430
Leu Ile Trp Gln Asp Pro Ile Pro Ala Gly Ser Thr Ser Tyr Asp Val
435 440 445
Ala Ala Val Lys Ala Lys Ile Ala Ala Ser Gly Leu Ser Val Ala Asp
450 455 460
Leu Val Ser Thr Ala Trp Asp Ser Ala Arg Thr Phe Arg Gly Ser Asp
465 470 475 480
Lys Arg Gly Gly Ala Asn Gly Ala Arg Ile Arg Leu Ala .Pro Gln Lys
485 490 495
Asp Trp Glu Gly Asn Glu Pro Ala Arg Leu Ser Arg Val Leu Ser Val
500 505 510
Leu Glu Pro Ile Ala Arg Glu Thr Gly Ala Ser Ile Ala Asp Val Ile
515 520 525
Val Leu Ala Gly Asn Tyr Gly Val Glu Gln Ala Ala Lys Ala Ala Gly
530 535 540
Phe Asp Ile Ala Val Pro Phe Ala Ala Gly Arg Gly Asp Ala Ser Ala
545 550 555 560
Glu Gln Thr Asp Ala Asp Ser Phe Ala Pro Leu Glu Pro Leu Ala Asp
565 570 575
Gly Phe Arg Asn Trp Val Lys Lys Asp Tyr Val Val Ser Pro Glu Glu
580 585 590
Leu Leu Leu Asp Arg Ala Gln Leu Leu Gly Leu Thr Ala Pro Glu Leu
595 600 605
Thr Val Leu Ile Gly Gly Leu Arg Val Ile Gly Ala Asn Tyr Gly Gly
12
CA 02437498 2003-08-O1
WO 02/063032 PCT/SG02/00018
610 615 620
Ala Ala His Gly Val Phe Thr Asp Lys Pro Gly Ala Leu Thr Thr Asp
625 630 635 640
Phe Phe Thr Thr Leu Thr Asp Met Ala Tyr Ser Trp Val Pro Thr Gly
645 650 655
Asn Asn Leu Tyr Glu Ile Arg Asp Arg Lys Thr Gly Ala Ala Arg Tyr
660 665 670
Ser Ala Thr Arg Val Asp Leu Val Ile Gly Ser Asn Ser Ile Leu Arg
675 680 685
Ala Tyr Ala Glu Val Tyr Ala Gln Asp Asp Asn Arg Glu Lys Phe Ala
690 695 700
Arg Asp Phe Ile Ala Ala Trp Thr Lys Val Met Asn Ala Asp Arg Phe
705 710 715 720
Asp Leu Ile
<210> 4
<211> 28
<212> DNA
<213> Agrobacterium tumefaciens
<400> 4
ggtgcgctag ccaaattcgt caccaagc
28
<210> 5
<211> 24
<212> DNA
<213> Agrobacterium tumefaciens
<400> 5
caatcgctag cgttcggccc tctg
13
<IMG>