Note: Descriptions are shown in the official language in which they were submitted.
CA 02447703 2003-11-14
DESCRIPTION
METHODS FOR PROLIFERATING TERMINAL DIFFERENTIATED CELLS
AND RECOMBINANT VECTORS THEREFOR
Technical Field
The present invention relates to methods for proliferating terminal
differentiated cells and recombinant vectors therefor. More specifically, the
present invention relates to methods and the like for proliferating terminal
differentiated cells by using cyclins and cyclin dependent kinases.
Background Art
The eucaryotic cell cycle is regulated by cyclin dependent kinases
(CDKs). These are phosphoenzymes (kinases), which are only activated after
binding with a subunit called as cyclin, while they do not have the activity
by
themselves. Various processes in cell cycles, such as replication and
introduction to mitosis, are regulated by different CDKs. However, the
expression levels of CDKs are generally consistent and the activities of CDKs
are dependent on the expression levels of cyclins. Cyclins are transiently
expressed at required stages in the cell cycle and activate CDKs.
Moreover, CDK activity is also regulated by a group of inhibitory
factors, called as CDK inhibitors. These inhibitors are roughly classified
into
two groups based on the feature of their primary structure and the specificity
of the inhibition of CDK, namely, the INK4 (Inhibitor of CDK4) family, and the
CIP/KIP (CDK interacting protein/kinase inhibitory protein) family. Comparing
the progression of cell cycle to the driving an automobile, CDKs play a role
of
accelerator, and CDK inhibitors play a role of break. The progression of cell
cycle is determined by the concerted regulation of both of the accelerator and
the break.
CA 02447703 2003-11-14
When quiescent cells enter cell cycle, D-type cyclins are expressed
during from middle to late G1 phase in response to the mitogenic stimulation.
Ras/Raf-1/MAPK accelerates the transcription of the genes coding for D-type
cyclins, and P13K (phosphatidylinositol 3-kinase)/Akt (protein kinase B)
represses the degradation of the gene products. CDK4 and CDK6 bind to D-
type cyclins, the assembled proteins enter the cell nucleus where they must
be phosphorylated by CAK (CDK -activating kinase) to become active. D-type
cyclins have been reported to be a cytoplasmic sensor for stimulation signals,
i.e., their expression is induced in response to extracellular growth factors,
and they play a role for transferring the signal to CDK2 and CDC2 which
progress the cell cycle. It is also reported that D-type cyclin/CDK4, 6 has
two
roles for cell cycle progression. One is the role of canceling the growth
inhibitory effect of RB (the retinoblastoma protein) through phosphorylation.
Another role is trapping (sequestering) CIP/KIP. When CIP/KIP exists alone in
the cell, the activity of CDK2 appeared in G1 anaphase, has been inhibited. It
is speculated that D-type cyclin/CDK reduces the activity of such inhibitors
as
CIP/KIP to inhibit CDK2 by associating therewith.
In G1 phase of cell cycle, the target of CDK is RB. RB is known as to
associate with many proteins, especially as a key molecule, with a
transcription factor E2F. The E2F regulates the transcription of the genes,
which are necessary for progression of cell cycle and DNA replication, for
example, it activates the transcription of cyclin E. Thus, E2F plays an
important role for initiation of S phase through the function of cyclin
E/CDK2.
The non-phosphorylated RB binds with E2F strongly, and represses the DNA
replication by E2F. When RB is phosphorylated by cell cycle progressive CDK,
the phosphorylated RB loses the function to repress E2F, and becomes
inactive. Recently, it has been reported that the homologous proteins with
each of RB and E2F were existed. They are called RB family and E2F family
respectively. As a result, it has been revealed that the progression of cell
2
CA 02447703 2003-11-14
cycle from G1 to S phase was a highly regulated process by RB and E2F
family proteins. Further, this RB-E2F pathway was revealed to be related to
many biological events such as cell differentiation, malignant transformation
and apoptosis. For example, inactivation of RB and the like causes the
abnormal regulation of cell cycle, and leads to the malignant transformation
of
the cell. A number of these genes, which regulate cell cycle, are reported as
suppressor genes of cancer.
On the other hand, it is known that terminal differentiated cells such
as cardiomyocytes and nerve cells withdraw from the cell cycle and take on
1o the special state called as stationary phase (GO phase). Cardiomyocytes,
one
of these terminal differentiated cells loses proliferative activity soon after
birth.
Thus, the necrosis or loss of cardiomyocytes by infarction or dilated
cardiomyopathy leads to failure of regeneration of cardiomyocytes. Thereafter,
there are a lot of cases of severe heart failure and death and this is a cause
of
high mortality rate of these cardiac diseases. Although it has been thought
that cardiomyocytes are arrested at GO phase and the cell cycle does not
proceed, recent studies suggest that cardiomyocytes have the regulatory
mechanisms of the cell cycles.
For example, adenovirus transforming gene product E1A promotes
transcription of E2F dependent genes by interacting with the RB, and induces
cellular DNA synthesis. As a result of examination whether El A releases E2F
in cardiomyocytes, and induces DNA synthesis and dedifferentiation by using
rat neonatal cultured cardiomyocytes, E1 A alone did not clearly induce the
DNA synthesis but leads to apoptosis. In the presence of E113, El A is
reported to induce DNA synthesis in cardiomyocytes (refer to Kirshenbaum, L.
A. et al., The Journal of Biological Chemistry 270, 7791-7794 (1995)).
Next, as a result of using E2F adenovirus, it has been reported that
E2F represses the expression of myocardial specific genes in cardiomyocytes,
and induces DNA synthesis (refer to Kirshenbaum, L.A. et al., Developmental
3
CA 02447703 2003-11-14
Biology 179, 402-411, (1996)).
On the other hand, the transgenic mouse, in which a wild type cyclin
D1 without nuclear localization signals was overexpressed, showed the
elevation of expression level of CDK4 as well as the DNA synthesis in
cardiomyocytes, however, it was reported that abnormal multinucleated cells
were increased (refer to Soonpaa, M.H et al., Clin. Invest. 99, 2644-2654,
(1997)).
From such a line of research, the DNA synthesis is observed and the
progression of cell cycles is suggested in cardiomyocytes, however,
1o subsequent cell division or increase of cell numbers was not reported.
Thus,
any method for substantially proliferating cardiomyocytes without induction of
apoptosis is not known.
Recently, for the purposes of organ transplantation or treatment of
leukemia and the like, the regeneration medical technology to produce
desired kinds of cells using multipotent cells called as embryonic stem (ES)
cells have been studied. However, because ES cells are produced by
disrupting the embryo that has a potential to grow into fetus, there are many
ethical resistances to the research.
Problem to be solved by the Invention
In view of the above problems, the present invention is directed to
provide a method for proliferating terminal differentiated cells, which method
is applicable for the development of cardiac regenerating therapy and self-
transplantation of well-differentiated organs such as cardiomyocytes.
It is a further object of the present invention to provide a recombinant
vector that can be used for above method, or a pharmaceutical composition
comprising thereof.
4
CA 02447703 2010-10-19
Disclosure of the Invention
To solve the above problems, the present inventors have investigated
about the mechanism of cell cycle regulation in the terminal differentiated
cells,
especially, the role of cyclin/CDK in cardiomyocytes induced by mitogenic
stimuli.
As a result, it was found that the D-type cyclin/CDK4 induced by mitogenic
stimuli
remained in cytoplasm of cardiomyocytes, and did not enter the cell nucleus,
whereby neither phosphorylation of RB nor activation of cyclin E/CDK2 took
place.
Therefore, the present inventors constructed adenovirus vectors inserted a
cyclin
D1 gene attached with a nuclear localization signal (NLS), as well as a CDK4
gene respectively, and introduced these adenovirus vectors into cultured
cardiomyocytes. These viruses caused not only expression of cyclin D1/CDK4 in
the nucleus and RB phosphorylation, but also proliferation of cardiomyocytes.
These findings have led to the completion of the present invention.
According to a first aspect of the present invention, there is provided a
method for proliferating terminal differentiated cells, the method comprising
introducing a D-type cyclin and a cyclin dependent kinase that is cyclin
dependent
kinase 4 (CDK4) or cyclin dependent kinase 6 (CDK6) into the nucleus of
terminal
differentiated cells, wherein at least one of said D-type cyclin and said
cyclin
dependent kinase is (are) fused with a nuclear localization signal, and
wherein
said terminal differentiated cells are cardiomyocytes, nerve cells, kidney
cells, or
pancreatic cells. In an embodiment, the method is an in vitro method.
In an embodiment, there is provided a method for proliferating terminal
differentiated cells comprising, introducing a cyclin and a cyclin dependent
kinase
into the nucleus of terminal differentiated cells, and then cultivating or
holding said
cells.
In another embodiment, there is provided a method for proliferating
terminal differentiated cells, the method comprising: fusing a nucleotide
sequence
coding for a nuclear localization signal with at least one of a nucleotide
sequence
coding for a D-type cyclin and a nucleotide sequence coding for a cyclin
CA 02447703 2010-10-19
dependent kinase that is cyclin dependent kinase 4 (CDK4) or cyclin dependent
kinase 6 (CDK6); and introducing each of said nucleotide sequence coding for a
D-type cyclin and said nucleotide sequence coding for a cyclin dependent
kinase to
terminal differentiated cells in vitro, wherein said terminal differentiated
cells are
cardiomyocytes, nerve cells, kidney cells, or pancreatic cells.
In a preferred embodiment for introducing said cyclin and said cyclin
dependent kinase into the nucleus of terminal differentiated cells, there is
provided a
method comprising the steps of adding a nucleotide sequence coding for a
nuclear
localization signal to at lease one of a cyclin gene and a cyclin dependent
kinase
gene; and introducing each of said genes to terminal differentiated cells in
vitro, and
then cultivating said cells, or introducing each of said genes directly to
terminal
differentiated cells in vivo.
In a further preferred embodiment of the present invention, said cyclin is a
cyclin that is capable of activating CDK4 or CDK6, for example, mammalian
cyclin D1,
D2 and D3 are preferable. Said cyclin dependent kinase is a cyclin dependent
kinase
that is activated by D-type cyclin, for example, CDK4 and CDK6 are preferable.
In another preferred embodiment of the present invention, said terminal
differentiated cells are cardiomyocytes, nerve cells, kidney cells, or
pancreatic cells.
In another embodiment, said terminal differentiated cells are cardiomyocytes.
In a still further preferred embodiment of the present invention, said cyclin
gene and said cyclin dependent kinase gene are transferred to the terminal
differentiated cells by using an adenovirus vector.
According to a second aspect of the present invention, there is provided a
recombinant vector comprising a cyclin gene comprising a nucleotide sequence
coding for a nuclear localization signal (NLS; a signal peptide having a
function of
transporting the protein into the nucleus), or a cyclin dependent kinase gene
comprising a nucleotide sequence coding for a NLS. Said recombinant vector may
comprise both a cyclin gene and a cyclin dependent kinase gene, if only at
least
6
CA 02447703 2009-09-14
one of said genes is (are) attached with a nucleotide sequence coding for a
nuclear localization signal.
In another embodiment, there is provided a recombinant vector
comprising: a nucleotide sequence coding for a D-type cyclin fused with a
nucleotide sequence coding for a nuclear localization signal; or a nucleotide
sequence coding for a cyclin dependent kinase fused with a nucleotide sequence
coding for a nuclear localization signal, wherein said cyclin dependent kinase
is
cyclin dependent kinase 4 (CDK4) or cyclin dependent kinase 6 (CDK6).
In a further embodiment, there is provided a recombinant vector
comprising a nucleotide sequence coding for a D-type cyclin and a nucleotide
sequence coding for a cyclin dependent kinase that is cyclin dependent kinase
4
(CDK4) or cyclin dependent kinase 6 (CDK6), wherein at least one of said
nucleotide sequences is (are) fused with a nucleotide sequence coding for a
nuclear localization signal.
In a preferred embodiment of the present invention, said cyclin is a cyclin
that is capable of activating CDK4 or CDK6, and said cyclin dependent kinase
is a
cyclin dependent kinase that is activated by cyclin D1, D2 or D3.
In a further preferred embodiment of the present invention, said
recombinant vector further comprises an adenovirus vector, and the nucleotide
sequence of said nuclear localization signals is that derived from large T
antigen
of SV40. In particular, said sequence is recommended to encode for triplicate
nuclear localization signals derived from large T antigen of SV40.
According to a third aspect of the present invention, there is provided a
mammalian cell or tissue that was proliferated by any one of methods in the
above
first aspect. In another embodiment, there is provided a mammalian cell that
was
proliferated by any one of methods in the above first aspect.
Further, according to a fourth aspect of the present invention, there is
provided a pharmaceutical composition for proliferating terminal
differentiated
cells or tissues, comprising an effective amount of any one of recombinant
vectors
7
CA 02447703 2010-10-19
in the above second aspect of the present invention. In an embodiment, the
pharmaceutical composition may further comprise a pharmaceutically acceptable
carrier. In a further embodiment, the pharmaceutical composition may be for
the
treatment of cardiopathy in a human patient.
In the preferred embodiment to use the pharmaceutical composition of the
aspect, there is provided a method for treating cardiopathy in a human patient
comprising directly introducing into myocardium of the patient the virus
vector
expressing a cyclin gene and a cyclin dependent kinase gene attached with a
nucleotide sequence coding for a nuclear localization signal to at lease one
of said
genes, and proliferating said myocardium of the patient.
According to another aspect, there is provided use of an effective amount of
any one of recombinant vectors in the above second aspect of the present
invention
for the proliferation of terminal differentiated cells that are
cardiomyocytes, nerve cells,
kidney cells, or pancreatic cells. In an embodiment, said terminal
differentiated cells
are cardiomyocytes. In an embodiment, the use is for the treatment of
cardiopathy
in a human patient, wherein the recombinant vector is for directly introducing
into a
myocardium of the patient.
According to a further aspect, there is provided use of: a D-type cyclin; and
a
cyclin dependent kinase that is cyclin dependent kinase 4 (CDK4) or cyclin
dependent
kinase 6 (CDK6), for the proliferation of terminal differentiated cells that
are
cardiomyocytes, nerve cells, kidney cells, or pancreatic cells, wherein at
least one of
said D-type cyclin and said cyclin dependent kinase is (are) fused with a
nuclear
localization signal; and wherein said D-type cyclin and said cyclin dependent
kinase
are for introducing into the nucleus of terminal differentiated cells. In a
further
embodiment, said terminal differentiated cells are cardiomyocytes.
According to another aspect, there is provided use of: a nucleotide sequence
coding for a nuclear localization signal; a nucleotide sequence coding for a D-
type
cyclin; and a nucleotide sequence coding for a cyclin dependent kinase that is
cyclin
dependent kinase 4 (CDK4) or cyclin dependent kinase 6
8
CA 02447703 2010-10-19
(CDK6), for the proliferation of terminal differentiated cells that are
cardiomyocytes,
nerve cells, kidney cells, or pancreatic cells, wherein said nucleotide
sequence coding
for a nuclear localization signal is for fusing with at least one of said
nucleotide
sequence coding for a D-type cyclin and said nucleotide sequence coding for a
cyclin
dependent kinase; and wherein each of said nucleotide sequence coding for a D-
type
cyclin and said nucleotide sequence coding for a cyclin dependent kinase is
for
introducing directly to terminal differentiated cells. In another embodiment,
said
terminal differentiated cells are cardiomyocytes.
Brief Description of the Invention
Fig.1 shows the western blot analysis of the expression of cyclin D1 and the
phosphorylation of RB in cardiomyocytes stimulated by various growth stimuli.
ppRB:
phosphorylated RB protein, pRB: RB protein.
Fig.2 shows the western blot analysis of the expression of D-type cyclin and
CDK4 in cytoplasmic extract or nucleic extract in cardiomyocytes stimulated by
various growth stimuli. RB:RB protein
Fig.3 are the photographs of subcellular localization of cyclin D1 induced by
various stimuli in cardiomyocytes analyzed by immunofluorescence staining with
the
mouse monoclonal anti-sarcomeric actin antibody (Fig. 3a in colour; Fig. 3b in
black
and white).
Fig.4 shows the western blot analysis of the import of cyclin D1 to the
nucleus in cardiomyocytes by the addition of nuclear localization signals.
Fig.5 shows the western blot analysis of subcellular localization of cyclin D1
and CDK4 expressed by various recombinant adenoviruses in cardiomyocytes.
Fig.6 shows the western blot analysis of the phosphorylation of RB in
cardiomyocytes infected by various recombinant adenoviruses. ppRB:
phosphorylated RB protein, pRB: RB protein.
Fig.7 shows the western blot analysis of the expression of cyclin A and cyclin
E in cardiomyocytes infected by recombinant adenoviruses.
8a
CA 02447703 2009-09-14
Fig.8 shows the cell cycle analysis by laser scan cytometer (LSC) of the
cardiomyocytes induced by various stimuli.
Fig.9 shows the analysis of the mitotic cardiomyocytes visualized by LSC
with confocal laser microscope (Fig. 9ai in colour; Fig. 9aii in black and
white).
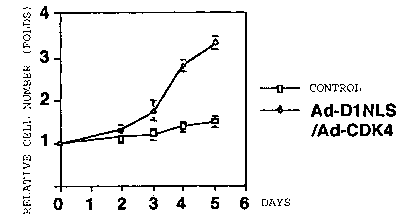
Fig.10 shows the relative cell number counted by cell counter system, at
the indicated days after viruses infection.
Fig.11 shows the , visualized views of expressed proteins by
immunofluorescence staining of heart tissue section after infection of
recombinant
adenovirus comprising D 1 NLS/CDK4 (Fig.11 a and Fig.11 b) or lacZ gene
(Fig.11 c)
in rat cardiomyocytes in vivo. Red, sarcomeric actin; green, Ki-67. Bar, 20pm.
Fig. 11d, Fig. 11e and Fig. 11f are black and white versions of Fig 11a, Fig.
11b
and Fig. 11 c, respectively. Dark grey, sarcomeric actin; light grey, Ki-67.
In each of the above Figs., the symbols mean as follows.
+ : addition of each of the growth factors or infection of each of the
recombinant
adenoviruses.
- : no addition of each of the growth factors or non-infection of each of the
recombinant adenoviruses.
Preferred Embodiments
(Cyclins and cyclin dependent kinases)
Mammalian D-type cyclins consists of 3 kinds of subtypes (D1, D2, D3).
They are expressed during from middle to late G1 phase, and activate CDK4 and
CDK6 by binding therewith. They promote G1-to-S phase progression by
phosphorylating predominantly RB protein. Although cyclin D1 is localized in
cell
nucleus during G1 phase, it is exported to the cytoplasm by the
phosphorylation of
cyclin D1 at threonine 286 by glycogen synthase kinase 3(3 (GSK-3R), which
leads
to degradation of cyclin D1 by proteasome. Various functions of cyclin D1/CDK4
except for cell cycle, such as regulation
8b
CA 02447703 2003-11-14
of differentiation of muscle through the inhibition the transcription factor
MyoD,
and activation of ER (estrogen receptor) by binding therewith in the
epithelial
cell of mammary gland, are reported.
The present invention relates to a method for proliferating terminal
differentiated cells through the progression of cell cycle by introducing such
cyclins and cyclin dependent kinases into the nucleus of terminal
differentiated cells. As for the method of introducing these proteins into
cell
nucleus, there is a physical injection method such as microinjection, however,
it is preferable to use the gene transfer method in view of efficiency of gene
transfer. Any cyclin that can activate CDK4 and CDK6 is preferably introduced,
for example, the above 3 kinds of subtypes (cyclin D1, D2, D3) are
enumerated. Any cyclin dependent kinase which is activated by D-type cyclin
is preferably introduced, for example, CDK4 and CDK6 are enumerated.
(Preparation of terminal differentiated cells)
The terminal differentiated cells used for the methods of the present
invention can be isolated from a living subject and cultured by various
methods. The term "terminal differentiated cells" means the cells staying in
GO phase of cell cycles without a cell division throughout an individual life,
such as cardiomyocytes, nerve cells, kidney cells, pancreatic cells and the
like. For example, when the cultured cardiomyocytes from 1 to 2-day post-
natal rats are analyzed by cell cycle assay such as flow cytometry, it is
confirmed that more than 90% of the cells are GO/G1 phase within 24 hours
after cultivation. In addition, even when the growth stimulation such as serum
is added, the cells do not enter the S phase (refer to Claycomb, W.C., Trends
Cardiovascular Medicine 2, 231-236, (1992)).
Similarly, central nervous cells, especially such as brain cells, have
been completed to divide at the fetal stage, therefore, brain disorder after
birth is difficult to cure. When kidney cells are impaired, treatments such as
9
= CA 02447703 2009-09-14
artificial dialysis are often necessary due to the loss of regeneration.
Pancreatic
cells produce a lot of digestion enzymes and gastrointestinal hormones. When
the
R cell was impaired, insulin secretion was inhibited due to the loss of
regeneration
thereof. Thus, this is known to be a cause of diabetes. In the present
invention,
these terminal differentiated cells can be proliferated by applying the method
of
the present invention in vitro, or in vivo, and then cultivating or holding
said cells.
(Construction of the recombinant vectors)
D-type cyclin genes and cyclin dependent kinase genes have been cloned
from human and various other organisms, and their sequences have been
reported. For example, the nucleotide sequence of mouse cyclin D1 gene is
described in Matsushime, H. et al., Cell, 65, 701-713, (1991), and registered
in
GenBankTM (Accession No. M64403). In addition, human cyclin D1 gene is
described in Lee, D. et al., Cell, 66, 1197-1206, (1991) and the like. Human
CDK4
is a protein consisting of 303 amino acid residues, and cDNA sequence encoding
thereof is registered in GenBankTM (Accession No. M14505). Further, these
genes
can be obtained by polymerase chain reaction (PCR) with specific primers for
desired gene and mammalian genomic DNA as a template.
To introduce proteins encoded in these genes into nucleus of terminal
differentiated cells, it is necessary to import the proteins synthesized in
cytoplasm
into nucleus. For this purpose, nucleotide sequences encoding a nuclear
localization signal is attached to at least one of the above genes, preferably
cyclin
gene. At present, 3 kinds of nuclear localization signals are known. All of
these
sequences have a consensus sequence referred to as a motif.
The first is a type of sequence that hardly has a basic amino acid such as
lysine and arginine and the like. Although the examples of this type of
CA 02447703 2003-11-14
motif is very small in number, there is a nuclear localization signal of the
nucleoprotein of influenza virus (AAFEDLRVLS: SEQ ID No. 1) (refer to Davy,
J. et al., Cell, 40, 667-675, (1985)). The second is a type of sequence that
has
a number of basic amino acids. There are a lot of examples of this type of
motif, for example, a nuclear localization signal of SV40 large T antigen
(PPKKKRKV: SEQ ID No. 2) (refer to Kalderon, D. et al., Nature, 311, 33-38,
(1984)). The third is a type of sequence that basic amino acids form the
cluster approximately 10 amino acid apart, and referred to as Bipartite type
nuclear localization signal. There are also a lot of examples of this type of
motif, for example, a nuclear localization signal of the nucleoplasmin of
Xenopus laevis (KRPAATKKAGQAKKKK: SEQ ID No. 3) (refer to Robbins, J.
et al., Cell, 64, 615-623, (1991)).
The nucleotide sequence coding for the nuclear localization signal is
attached to at least one of a D-type cyclin gene and a cyclin dependent
kinase gene, which is activated by, said cyclin. The two proteins expressed by
these genes forms the complex in the cytoplasm, therefore, if one of these
proteins, preferably cyclin has a nuclear localization signal, said complex is
capable of passing through the nuclear membrane. The recombinant vector
containing the nucleotide sequences coding for these nuclear localization
signals, and that is used for cloning a DNA from which the expressed protein
is imported to nucleus, can be obtained easily. For example, the expression
plasmid pEF/myc/nuc having a nuclear localization signal of SV40 large T
antigen is purchased from Invitrogen Corp.
As for vectors, there is exemplified such vectors derived from virus as
adenovirus, adeno associated virus, retrovirus, vaccinia virus, chick poxvirus
and papovavirus as SV40, and the like.
Adenovirus vectors used in the preferred embodiment of the present
invention can be prepared by homologous recombination method (refer to
Miyake, S. et al., Proc. Natl. Acad. Sci. USA, 93, 1320, (1996)) and in vitro
11
CA 02447703 2003-11-14
ligation method (refer to Mizuguchi, H. et al., Gene Ther. 9, 2577-2583,
(1998)) using human embryonic kidney cell line 293 (HEK293) or E. co/i.
Adenovirus is one of DNA viruses having a linear double stranded DNA
genome, and human adenovirus antigen type 5 (AD5) and human adenovirus
antigen type 2 (AD2) are most well-studied. Non-replicable adenovirus
vectors can be prepared by deleting most of the early gene 1 (El) and the
early gene 3 (E3) of these wild type adenovirus. Several kb exogenous DNA
can be inserted into these viruses without any harmful effect on the virion
morphogenesis. The recombinant adenovirus deletes the transcription factor
El gene, however, the inserted target genes can be expressed predominantly
by the transcription unit specific for the inserted gene, without any
dependent
on the growth of the target cells or presence of other virus genes. For
constructing these expression systems, the Adenovirus Expression Vector Kit
(Takara) is commercially available.
(Gene transfer and cultivation of the cells)
Any conventional method of gene transfer, which is known previously,
can be used for introducing genes. For example, there are tansfection
methods with recombinant DNA using calcium phosphate or liposome, and
transduction methods using various viruses such as retrovirus. The retrovirus
used for the transduction usually infects the specific cell, and integrates
the
genetic information to the cellular DNA itself, however, it may infect more
than
two kinds of cells. The expression of the introduced genes may be transient or
constitutive by integrating to the chromosomal DNA of the recipient cells. It
is
preferable to use the recombinant adenovirus vectors for efficient
transduction and high level expression of the introduced genes. The
introduction of the genes by adenovirus is one of the most powerful methods
for introducing genes into mammalian cells, and can be used for the gene
transfer to actually all kinds of human cells and a number of non-human cells.
12
CA 02447703 2003-11-14
In addition, the infection by adenovirus is not dependent on cell cycles,
therefore, the adenovirus vector can be used for expressing the genes in
various primary culture cells or transformed cell lines. In particular, it can
introduce the genes effectively even in the cells that do not synthesize the
DNA nor take the cell division such as terminal differentiated cells. After
the
infection, a number of cells receives a plural of recombinant DNA copies, thus
the introduced genes are expressed in high level transiently. Further, the
adenovirus DNAs are not integrated to the cellular genome in general, but
remain as episomes. Accordingly, it is an advantage that the gene transfer
to using adenoviruses scarcely causes mutated errors at the random integration
of heterogeneous DNA into the host cell genome.
In the present invention, to express both genes of D-type cyclins and
cyclin dependent kinases that are activated by said cyclins, these genes can
be infected by the same recombinant virus or separate recombinant viruses.
When more than two kinds of viruses are used for co-infection, these viruses
can be infected simultaneously or separately after a set period of time. In
the
present invention, the amount of infected virus is adjusted preferably
approximately 100/cell (MOI=100) using for example 109 to 1011/ml virus stock
solution. The efficiency of gene transfer by co-infection of two different
types
of adenovirus is also high, and the survival rate of the cell after co-
infection
can be high. The virus amount (titer) can be easily measured by plaque
assay.
The terminal differentiated cells of which a D-type cyclin and a cyclin
dependent kinase which can be activated by said cyclin, are expressed in the
nuclei, are proliferated by cultivating according to the conventional methods
previously known. The preferable method of cultivating said cells can be
selected from the group consisting of microtiter plate, static culture in
petri-
dish or flask, rolling bottle culture, micro carrier culture and the like,
depending on the kind of said cells. For example, cardiomyocytes can be
13
CA 02447703 2003-11-14
cultivated in minimum eagles medium (MEM) and the like, supplemented with
growth factors such as 5 to 20% fetal calf serum and the like, in the presence
of 5% carbon dioxide gas, at 37 C. Nerve cells, kidney cells and pancreatic
cells can be also cultivated by the same method. Various culture media for the
growth of each cell in the most preferable condition are developed and
purchased, for example, RPMI1640 , CMRL1066 and the like. These various
culture media are available from the market such as Flow Laboratories, Gibco
and the like.
In the present invention, the genes are also introduced in vivo, for
example, they are directly introduced into mammalian terminal differentiated
cells or tissues and held said cells or said tissues in vivo for
proliferation. Here,
the term "hold" means to maintain said cells or said tissues in the
physiological conditions such as body temperature and bloodstream without
loss of physiological function thereof.
(Proliferated cells and tissues)
The present invention also relates to cells and tissues that were
proliferated by the method of the present invention. For example, a D-type
cyclin gene attached with nuclear localization signals and a cyclin dependent
kinase gene which can be activated by said cyclin are introduced to the
terminal differentiated cells extirpated from patients by the aforementioned
method, and cultured to proliferate. These proliferated cells and tissues can
be used for regenerating the necrotic cells or tissues by injecting into said
patients. Specifically, cardiomyocytes, nerve cells, kidney cells and
pancreatic cells can be transplanted to the patients.
(Pharmaceutical compositions to regenerate the terminal differentiated cells
and/or tissues)
The recombinant vectors of the present invention can be used for the
14
CA 02447703 2003-11-14
prevention or the treatment of diseases. When the specific cells or tissues
are
injured, the necrotic tissues or organs can be repaired or recruited by
proliferating such terminal differentiated cells or tissues to which the
pharmaceutical compositions comprising an effective amount of the
recombinant vector of the present invention were injected. For example, the
recombinant vectors of the present invention can be used for the treatment of
cardiac infarction or dilated cardiomyopathy. In this case, the pharmaceutical
compositions comprising the above recombinant vectors can be directory
injected to the heart of the patient to proliferate the patient's cardiac
muscle
(cardiomyocytes).
Examples
The present invention is explained in more detail by reference to the
following examples which uses cardiomyocytes as a terminal differentiated
cell. Although these examples show embodiments in which rat
cardiomyocytes are proliferated in vitro, they do not restrict the scope of
the
present invention.
[Example 1 ] Preparation of recombinant adenoviruses
1) Preparation of Ad-CDK4
The plasmid pCMV-CDK4 were provided by Dr. Sander van den
Heuvel (Massachusetts General Hospital Cancer Center, refer to van den
Heuvel et al., Science, 262, 2050-2054 (1993)). The plasmid pCMV-CDK4
contained a mouse CDK4 gene. The recombinant Ad-CDK4 was constructed
from pCMV-CDK4 by using Adenovirus Expression Vector Kit (Takara, Tokyo,
Japan, Code No. 6150) as follows.
pCMV-CDK4 was digested by BamHl to isolate an approximately 920
bp DNA fragment. Both terminals of the isolated DNA fragment was blunted
by using DNA Blunting Kit (Takara, Code No. 6025 ) and the DNA fragment
CA 02447703 2009-09-14
was inserted into the Swal site of cosmid pAxcw. The obtained cosmid pAd-CDK4
and DNA-TPC (terminal peptide complex) derived from human adenovirus type 5
were co-tranfected to HEK293 cells. The recombinant adenovirus Ad-CDK4 was
obtained by in vitro recombination in 293 cells.
2) Preparation of Ad-D1 NLS
The cyclin D1/NLS plasmid comprising a cyclin D1 gene attached with
nucleotide sequences encoding nuclear localization signals (NLS) was
constructed by ligation of the mouse cyclin D1 fragment from pRSV-cyclin D1
(refer to aforementioned Matsushime, H. et al.) and NLS from pEF1 myc/nuc
plasmid (Invitrogen Corp.). Namely, plasmid pEF/myc/nuc was digested with
restriction enzymes Ncol and Xhol, and the first DNA fragment of approximately
5.5kb in length was prepared using 1.0% agarose gel electrophoresis and
QlAquickTM Gel Extraction Kit (QIAGEN Cat. No. 28704). Next, plasmid
pRSV-cyclinDl was digested with restriction enzyme Ncol, and the second 603bp
DNA fragment was prepared by the same method. Further, the third DNA fragment
encoding C-terminal region of mouse cyclin D1 was prepared by polymerase
chain reaction (PCR) using the following two primers and plasmid pRSV-cyclinDI
as a template.
Ncol primer: 5' ACCCTCCATGGTAGCTGCTGGGA 3' (SEQ ID No.4), and
Xhol primer: 5' TGATCTCGAGGTCGATGTCCACATCTCGCACGT 3' (SEQ ID
No.5).
These three DNA fragments were ligated by using DNA Ligation Kit
(Takara Code No. 6022), and constructed a plasmid comprising nucleotide
sequences encoding nuclear localization signals (NLS) derived from SV40 large
T
antigen in triplicate at the C-terminal region of mouse cyclin D1 gene. From
this
plasmid, a DNA fragment was excised by restriction enzymes PmaCl and Smal,
and inserted into the Swal site of cosmid pAxcw as described above to prepare
the recombinant adenovirus (Ad-D1 NLS) using Adenovirus Expression Vector Kit
(Takara Code No. 6150). This recombinant
16
CA 02447703 2003-11-14
adenovirus Ad-D1 NLS comprises mouse cyclin D1 gene attached with
nucleotide sequences encoding said NLS between the CAG promoter (chick
(3-actin promoter + cytomegalovirus enhancer) and the polyadenylation signal
of rabbit R-globin gene in DNA sequence of adenovirus deleted El and E3
genes.
3) Preparation of virus stock solution
The 293 cells were previously cultured in each of two 6cm dishes
coated with collagen. Each 4pg of Ad-CDK4 and Ad-D1 NLS prepared above
1) and 2) was mixed with 2.5p1 of the restriction enzyme treated DNA-TPC
(component of Takara Adenovirus Expression Vector Kit), and transfected to
the 293 cells cultured in 6cm dish by lipofection method respectively
(FuGENETM6 Transfection Reagent, Roche cat#1814443 was used). On the
following day, the cells were detached, and the recovered cell suspensions
were re-inoculated on the 96 well plates coated with collagen. After 7 to 15
days, the virus grew and the cells were killed in several wells. From each of
the wells in which the cells completely died, the culture medium was collected
into a sterilized tube, and freeze thawed 6 times repeatedly. The supernatants
after the centrifugal separation at 5000rpm for 5 minutes, were stored at -
80 C as a first virus stock solution. 10 l of the first virus stock solution
were
infected to the 293 cell cultivated in a 24 well plate coated with collagen,
and
collected the killed cell/media after 3 to 4 days in a sterilized tube.
Freezing
and thawing was repeated 6 times, and centrifuged at 5000 rpm for 5 minutes
to recover the supernatant and store at -80 C as a second virus stock
solution.
15 I of the second virus stock solution were infected to the 293 cell
cultivated
in a 25 cm3 bottle coated with collagen, and collected the killed cell/media
after 3 to 4 days in a sterilized tube. The virus was released from the cell
by
freezing and thawing, or homogenizing the cell by sealed sonicator, and
centrifuged at 3000 rpm for 10 minutes at 4 C to recover the supernatant and
17
CA 02447703 2003-11-14
store at -80 C as a third virus stock solution. 50 I of the third virus stock
solution were infected to the 293 cell cultivated in a 75 cm3 bottle coated
with
collagen, and collected the killed cell/media after 3 to 4 days in a
sterilized
tube. The virus was released from the cell by freezing and thawing, or
homogenizing the cell by sealed sonicator, and centrifuged at 3000 rpm for 10
minutes at 4 C to recover the supernatant and store at -80 C as a fourth virus
stock solution. The titer of the fourth virus stock solution was determined by
plaque assay with the 293 cell, and found that it was in the range from 109 to
1011 pfu/ml constantly.
[Example 2] Preparation of cardiomyocytes and infection of the recombinant
adenovirus
Cardlomyocytes from 1- or 2-day post-natal Sprague-Dawley (SD)
rats were isolated and subjected to Percoll gradient centrifugation for
recovering the layer (fraction) of cardiomyocytes to suspend in MEM
(minimum essential medium; Sigma, Cat. M-4655) containing 5% calf serum
(refer to Tamamori, M. et al., Am.J.Physiol. 275, (Hert Cirs. Physiol.44),
H2036-H2040 (1988)). We routinely obtained cultures in which more than
95% of the cells are cardiomyocytes as assessed by immunostaining with the
mouse monoclonal anti-sarcomeric actin antibody (Dakopatts, Denmark).
Neonatal rat cardiomyocytes in culture were incubated in Eagle's minimum
essential medium (MEM) (Flow Laboratories) with 5% FCS (Flow
Laboratories) for 24 hours. The next day, the culture medium replaced with
serum-free MEM and cells were further incubated for 24 hours before virus
infection. The cells were incubated with one or more than two of the
recombinant adenoviruses prepared in Example 1. Cells were infected with
each recombinant adenovirus (100 pfu/cell), and cultured for various periods.
A fibroblast REF52 cells were prepared as controls.
18
CA 02447703 2009-09-14
[Example 3] Analysis of the gene expression in cardiomyocytes by Western,
Kinase Assays and Immunofluorescence Staining
Various growth stimulators and/or recombinant adenoviruses prepared in
Example 1 were used to stimulate rat cardiomyocytes prepared in the method of
Example 2. Whole cell extract, cytoplasmic extract and nuclear extract from
the
cell cultivated in various periods after virus infection, were purified as
follows.
Cells on the culture dishes were collected by cell scraper after washing with
ice-cold (4 C) PBS (Phosphate buffered saline), and then centrifuged to
discard
the supernatant. The obtained pellets were washed again with small amount of
PBS, and then transferred to an eppendorf tube of 1.5ml volume. To the cell
pellets, ten volumes of ice-cold (4 C) Lysis buffer (50mM HEPES (pH7.9), 150mM
NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NONIDETTM P-40, 0.4mM NaF, 0.4mM
Na2VO4, 10% glycerol) was added, followed by pipetting on ice and mixing for
15
seconds by vortex mixer, and then stood on ice for 30 minutes. After that, the
mixture was centrifuged at 15,000 rpm for 10 minutes at 4 C, and the
supernatant
was stored as the whole cell extract (rapidly frozen by liquid nitrogen and
stored at
-80 C).
The cytoplasmic extract and the nuclear extract were fractionated as
follows. Cells on the culture dishes were collected by cell scraper after
washing
with ice-cold (4 C) PBS, and then centrifuged to discard the supernatant as in
the
above case. The obtained pellets were washed again with small amount of PBS,
and then transferred to an eppendorf tube of 1.5m1 volume. To the cell
pellets, five
volumes of ice-cold Buffer A (10mM HEPES (pH7.9), 1.5mM MgCl2, 10mM KCI,
0.5mM DTT) was added, followed by pipeting on ice and mixing for 15 seconds by
vortex mixer, and then stood on ice for 10 minutes. NONIDETTM P-40 was further
added to 0.2% in final concentration, mixed by vortex mixer and stood on ice
for 5
minutes. Finally, the mixture was centrifuged at 5,000 rpm for 5 minutes, and
the
supernatant was stored as the cytoplasmic extract (rapidly frozen by liquid
nitrogen and
19
CA 02447703 2009-09-14
stored at -80 C). The pellets were suspended in equal volume of Buffer C (20mM
HEPES (pH7.9), 25% glycerol, 0.42M NaCl, 1.5mM MgCl2, 0.2mM EDTA), and
pipetted. After mixing by vortex mixer for 15 seconds, the mixture was stood
on
ice for 30 minutes. Finally, the mixture was centrifuged at 15,000 rpm for 10
minutes, and the supernatant was stored as the nuclear extract (rapidly frozen
by
liquid nitrogen and stored at -80 C). Incidentally, each of the above Lysis
buffer,
Buffer A and Buffer C was supplemented with 1mM DTT, 1mM PMSF, lpg/ml
aprotinin, 1 pg/ml leupeptin, lpg/ml pepstatin (each was purchased from Sigma)
just before use.
The protein concentration of each sample extracted by the above method
was corrected as each sample was extracted from the same number of cells.
Namely, the proteins extracted from 1 x 106 cells/each sample were
electrophoresed on 6% or 11 % SDS-PAGE Gel, and then the gel was transferred
to nitrocellulose membrane which was further soaked in PBS containing 5% skim
milk, 0.2% TweenTM20 and shaken for one hour (blocking). To this solution, the
mouse monoclonal anti-cyclin D1 antibody (Ab-3; Oncogene science), the mouse
monoclonal anti-RB antibody (14001A Pharmingen), the rabbit polyclonal
anti-cyclin A antibody (SC-751; Santa Cruz Biotechnology), the rabbit
polyclonal
anti-cyclin E antibody (SC-481; Santa Cruz Biotechnology) and the rabbit
polyclonal anti-CDK4 antibody (SC-260; Santa Cruz Biotechnology) were added,
and the solutions were shaken for further one hour. Then, after the membrane
was washed with PBS containing 5% skim milk, 0.2% TweenTM20 to remove these
antibodies, it was reacted with the sheep anti-mouse Ig, horseradish
peroxidase
linked whole antibody (Amersham LifeScience; NA931) or the sheep anti-rabbit
Ig,
horseradish peroxidase linked whole antibody (Amersham LifeScience; NA934).
The membrane was washed with PBS containing 5% skim milk, 0.2% TweenTM20
again, and then stained immunofluorescently using ECL kit (Western blotting
detection reagents; Amersham LifeScience; RPN 2109) to
CA 02447703 2003-11-14
analyze Western blot analysis.
At first, the cultured neonatal cardiomyocytes were stimulated with
serum as a growth stimulator, phenylephrine that is an cx -adrenergic
receptor agonist, and a recombinant adenovirus Ad-ras6l L provided by Dr.
Nevins,J.R. (Leone,G et al., Nature, 387, 422-426, (1997))that expresses a
Ras constitutively active mutant protein as a hypertrophic stimulator. The
induction of cyclin D1 expression and phosphorylation of RB in response to
these stimuli were analyzed by western blot analysis. Fig.1 showed the
results of electrophoresis of cell extracts stained by each antibody. Each
cell
was firstly infected with Ad-Ras6l L or Ad-Con which is an adenovirus without
Ras gene as a control, and then the culture medium was exchanged with that
containing 10% serum and 10-6 M Phenylephrine (PE) or serum-free medium
after 18 hours, followed by further 18-hour cultivation. As shown in Fig.1,
these stimuli induced cyclin D1 expression and CDK4 expression (data not
shown). In spite of the accumulation of cyclin D1/CDK4, cyclin D1 associated
kinase was not activated (data not shown), and also RB protein (pRB) was
not phosphorylated, whereas RB was hyperphosphorylated state in
stimulated REF52 cells, because the higher molecular weight band (ppRB) by
phosphorylation was detected. In proliferating cells, RB is phosphorylated,
followed by the expression of D-type cyclins. However, contrary to
expectation, RB was not phosphorylated in stimulated cardiomyocytes. These
data indicate that the reaction of cardiomyocytes to the growth stimuli is
different from that of the growing cells in the point of the lack of RB
phosphorylation.
To determine the reason of the lack of RB phosphorylation in spite of
the accumulation of cyclin D1, we investigated the subcellular localization of
D-type cyclins and CDK4. Fig.2 shows the results of western blot analysis of
the nuclear extracts and cytoplasmic extracts from cardiomyocytes and
REF52 cells. D-type cyclins and CDK4 were not expressed in nucleus and
21
CA 02447703 2003-11-14
expressed only in the cytoplasmic fraction in stimulated cardiomyocytes,
while cyclin D1, D3 and D4 were imported to the nucleus in stimulated REF52
cells. RB protein is in the nucleus in cardiomyocytes as well as REF52 cells.
Fig.3 are the microphotographs of cardoimyocytes stimulated by fetal
calf serum or recombinant adenovirus Ad-RAS61 L analyzed by
immunofluorescence staining with the mouse monoclonal anti-sarcomeric
actin antibody. It is observed that cyclin D1 was accumulated in cytoplasm in
each cell.
Moreover, Fig.4 shows that despite overexpression of cyclin D1 and
CDK4 by using adenovirus Ad-D1 and Ad-CDK4, cyclin D1 did not
accumulate in the nucleus. Therefore, we constructed adenovirus vectors
coding for cyclin D1 with nuclear localization signals (Ad-D1 NLS). As shown
in Fig.4 and Fig.5, co-infection of Ad-D1 NLS and Ad-CDK4 induced
significant accumulation of cyclin D1 and CDK4 in the nucleus of
cardiomyocytes. Incidentally, the adenovirus Ad-D1 which expresses human
cyclin D1 was provided by Dr. Albrecht, J. of Minneapolis Medical Research
Foundation (refer to Albrecht, J.H. et al., Cell Growth & Differentiation, 10,
397-404, (1999)).
To progress from G1 to S phase, assembled cyclin D/CDK4 enter the
cell nucleus where they must be phosphorylated by CDK-activating kinase
(CAK) to be able to phosphorylate RB (refer to Sherr,C.J. et al., 1999, Genes
and Dev). We next investigated whether the cyclin D1 and CDK4 entered into
the nucleus cause RB phosphorylation and cell cycle progression. Fig.6
shows that co-infection of Ad-D1 NLS and Ad-CDK4 causes RB
phosphorylation in the nucleus of cardiomyocytes. In Fig. 6, adenovirus
vectors Ad-p16 and Ad-p21 that express CDK inhibitors p16 and p21 were
co-infected with the above Ad-D1 NLS/Ad-CDK4, and RB phosphorylation
was inhibited. Therefore, it was confirmed that the RB phosphorylation by
Ad-D1 NLD/Ad-CDK4 infection is dependent on the activity of the cyclin
22
CA 02447703 2003-11-14
dependent kinase. Fig.7 shows the expression of cyclin A and cyclin E that
are the targets of RB/E2F in the nucleus of cardiomyocytes. These results
suggest that the activity of cyclin D1/CDK4 in the nucleus possibly leads to
RB phosphorylation and progression of cell cycle.
[Example 4] Cell cycle Analysis
Next, to analyze cell cycles, the recombinant adenoviruses prepared
in Example 1 were infected to rat cardiomyocytes prepared in Example 2.
After each of several hour incubations, cells plated on 25-mm glass coverslips
were fixed with 70% ethanol and stained with propidium iodide for
measurement of DNA content. Namely, cells fixed with ethanol were mixed
with anti-sarcomeric actin antibody labeled by fluorescein isothiocyanate
(FITC) (1:1000), and after washing with PBS, 50 g/ml of PI, and 500 g/ml of
RNase A were added, and further incubated at room temperature for 15
minutes. The cells were detected the cell cycle position by laser scanning
cytometry (LSC) (Olympus, Japan). Cardiomyocytes were identified by
double staining of mouse monoclonal sarcomeric actin antibody and PI.
Fig.8 shows the results of the cell cycle analysis by using laser scan
cytometer (LSC). Fig.8a shows the distribution (correlation between DNA
content and cell numbers) of the cardiomyocytes induced by several viral
infection or growth factors after each of incubation times. Serum starved
cardiomyocytes infected with Ad-D1 NLS/Ad-CDK4 were clearly induced cell
cycle progression, as revealed by the increase in cells with S phase and G2
DNA content, in time dependent manner. At the presence of 5% CS,
D1 NLS/CDK4 induced cell cycle faster than serum starved condition. In
contrast, serum stimulation and Ad-D1/Ad-CDK4 (cyclin D1 without NLS) had
no or little effect on cell cycle. Almost all of the cells cultured in 5% CS
throughout after plating arrested in G1. These results suggest that
D1NLS/CDK4 enable the induction of cell cycle of post-mitotic
23
CA 02447703 2009-09-14
cardiomyocytes.
Moreover, to determine the population of cells that enter the cell cycle, we
blocked the cells in G2/M phase with nocodazole (Sigma). As shown in Fig.8b,
LSC analysis revealed that approximately 95% of the cells
(DI NLS/CDK4+Nocodazole) were in the G2/M fraction. Thus virtually all of the
cells co-infected with AdD1 NLS/AdCDK4 were entered the cell cycle. In
contrast,
the cardiomyocytes infected with Ad-D1/Ad-CDK4 or stimulated with serum were
remained in G1 phase. After washing out nocodazole from the synchronized
cardiomyocytes in G2/M, these cells re-entered G1 phase. These results show
that cyclin D1/CDK4 activity in the nucleus lead cardiomyocytes to
proliferation
once at least.
Fig.9 shows the cardiomyocytes co-infected with Ad-D1 NLS/Ad-CDK4 for
48 hours, and visualized by laser scan cytometer (LSC) with confocal laser
microscope (Olympus Japan). As shown in Fig.9ai and Fig. 9aii, we found many
mitotic and immediately after mitotic cells. Cell cycle position of each cell
was
decided by DNA content measured by PI staining (PI fluorescence value) and PI
fluorescence peak (maximum value). The mitotic cells increase in their DNA
content (PI fluorescence value), whereas the PI fluorescence peak of the cells
immediately before or after mitosis is high (bright) due to their DNA
aggregation.
Based on these indications, each cell (added No.1 to 27) in the
microphotograph
was classified into any one of the groups (a. to f.) correlated with cell
cycle
positions (G1 to M phase) (Fig.9b). As a result, cells of respective positions
of G1
to M phase existed together, many mitotic and immediately after mitotic cells
were
observed.
Fig. 10 shows the relative cell number measured by corter counter (cell
number counter machine). Actually, cell number of cardiomyocytes co-infected
with Ad-D1 NLS/Ad-CDK4 increased in 3-times after 5 days. On the contrary,
cell
number of cardiomyocytes infected with only adenovirus vector as a control,
did
not increase.
24
CA 02447703 2009-09-14
[Example 5] Study of the possibility for proliferating cardiomyocytes in vivo
To test the effects of nuclear import of cyclin D1 and CDK4 on adult
cardiomyocyte proliferation in vivo, two kinds of adenovirus such as Ad-D1 NLS
and Ad-CDK4 prepared in Example 1 and Example 2, were injected into apical
region of rat hearts. Apical injections of viruses into the myocardium were
performed under direct visualization after thoracotomy of Wister rats (250-
300g).
As a control, an adenovirus comprising LacZ gene was also injected as
described
above. Four days after injections, hearts were fixed with 4% paraformaldehyde
by
perfusion. The sections of tissues were stained with anti-Ki-67 and
anti-sarcomeric actin antibodies. Antibodies were visualized with anti-rabbit
AlexaTM 488 or anti-mouse AlexaTM 568 antibodies (Molecular Probes). Images
were obtained with the laser-scanning confocal image system (ZEISS LSM510)
and shown in Fig. 11. In Fig. 11a, Fig. 11b and Fig. 11c, red color indicates
sarcomeric actin and green color indicates Ki-67. In Fig. 11d, Fig. 11e and
Fig.
11f, dark grey indicates sarcomeric actin and light grey indicates Ki-67.
Since the Ki-67 nuclear protein is expressed in proliferating cells in all
phases of the cell cycle (Scholzen, T. et al., J. Cell. Physiol. 182, 311-22
(2000)),
we stained sections of the infected hearts with a Ki-67 antibody to detect
cells
entered into the cell cycle. In the images of heart sections co-infected with
the two
kinds of adenovirus Ad-D1 NLS and Ad-CDK4 (Fig 11 a and 11 b; Fig. 11 d and
Fig.
11 e), the Ki-67 nuclear protein was expressed in a number of cardiomyocytes
and
non-cardiomyocytes. On the other hand, the expression of the Ki-67 nuclear
protein was not observed in the cardiomyocytes injected with the adenovirus
containing IacZ gene (Fig.11c; Fig. 11f). These results strongly suggest that
nuclear import of cyclin D1 and CDK4 could promote cell cycle entry of
cardiomyocytes in adult hearts
On the basis of these data, it was concluded that cardiomyocytes
obtained the ability of proliferation by the expression of cyclin D1/CDK4 in
the
nucleus.
CA 02447703 2003-11-14
Industrial Applicability
According to the present invention, the cell division of terminal
differentiated cells is induced, and cells and tissues for transplantation can
be
prepared by proliferating said terminal differentiated cells. The terminal
differentiated cells, in particular, such as cardiomyocytes, nerve cells,
kidney
cells and pancreatic cells are proliferated by themselves. The proliferated
cells and tissues are expected to be used for regeneration medical care more
safely and certainly compared with the case of differentiation of
undifferentiated cells such as ES cells.
26
CA 02447703 2003-11-14
SEQUENCE LISTING
<110> Ikeda, Masaaki
Adachi, Mimi
<120> Methods for proliferating terminal differentiated cells
and recombinant vectors therefor
<130> 82599-5
<140> PCT/JP01/08208
<141> 2001-09-21
<150> JP P2001-148266
<151> 2001-05-17
<160> 5
<170> Patentln Ver. 2.1
<210> 1
<211> 10
<212> PRT
<213> Influenza virus
<400> 1
Ala Ala Phe Glu Asp Leu Arg Val Leu Ser
1 5 10
<210> 2
<211> 8
<212> PRT
<213> Simian virus 40
<400> 2
Pro Pro Lys Lys Lys Arg Lys Val
1 5
<210> 3
<211> 16
<212> PRT
<213> Xenopus laevis
<400> 3
Lys Arg Pro Ala Ala Thr Lys Lys Ala Gly Gln Ala Lys Lys Lys Lys
1 5 10 15
<210> 4
<211> 23
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: NcoI primer
27
CA 02447703 2003-11-14
<400> 4
accctccatg gtagctgctg gga 23
<210> 5
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: XhoI primer
<400> 5
tgatctcgag gtcgatgtcc acatctcgca cgt 33
28