Note: Descriptions are shown in the official language in which they were submitted.
CA 02449296 2003-12-15
MODULATORS OF AMYLOID AGGREGATION
This is a divisional application of Canadian Patent Application Serial Number
2,214,247, filed March 14, 2003.
S
Baclcaround of the Invention
Alzheimer's disease (AD), first described by the Bavarian psychiatrist Alois
Alzheimer
in 1907, is a progressive neurological disorder that begins with short term
memory loss and
proceeds to disorientation, impairment of judgement and reasoning and,
ultimately, dementia.
The course of the disease usually leads to death in a severely debilitated,
immobile state
between 4 and 12 years after onset. AD has been estimated to afflict 5 to 11
percent of the
population over age 65 and as much as 47 percent of the population over age
85. The societal
cost for managing AD is upwards of 80 billion dollars annually, primarily due
to the extensive
custodial care required for AD patients. Moreover, as adults born during the
population boom
of the 1940's and 1950's approach the age when AD becomes more prevalent, the
control and
treatment of AD will become an even more significant health care problem.
Currently, there
is no treatment that significantly retards the progression of the disease. For
reviews on AD,
see Selkoe, D.J. Sci. Amer., November 1991, pp. 68-78; and Yankner, B.A. et
al. (1991) N.
Eng. J. Med. 325:1849-1857.
It has recently been reported (Games et al. (1995) Nature 373:523-527) that an
Alzheimer-type neuropathology has been created in transgenic mice. The
transgenic mice
express high levels of human mutant amyloid precursor protein and
progressively develop
many of the pathological conditions associated with AD.
Pathologically, AD is characterized by the presence of distinctive lesions in
the
victim's brain. These brain lesions include abnormal intracellular filaments
called
neurofibrillary tangles (NTFs) and extracellular deposits of amyloidogenic
proteins in senile,
or amyloid plaques. Amyloid deposits are also present in the walls of cerebral
blood vessels
of AD patients. The major protein constituent of amyloid plaques has been
identified as a 4
kilodalton peptide called ~i-amyloid peptide (~i-AP)(Glenner, G.G. and Wong,
C.W. (1984)
Biochem. Biophys. Res. Commun. 120:885-890; Masters, C. et al. (1985) Proc.
Natl. Acad.
Sci. USA 82:4245-4249). Diffuse deposits of ~i-AP are frequently observed in
normal adult
brains, whereas AD brain tissue is characterized by more compacted, dense-core
~-amyloid
plaques. (See e.g., Davies, L. et al. (1988) Neurology 38:1688-1693). These
observations
suggest that /3-AP deposition precedes, and contributes to, the destruction of
neurons that
occurs in AD. In further support of a direct pathogenic role for /3-AP, /i-
amyloid has been
CA 02449296 2003-12-15
la
shown to be toxic to mature neurons, both in culture and in vivo. Yankner,
B.A. et al. (1989)
Science 245:417-420; Yankner, B.A. et al. ( 1990) Proc. Natl. Acad. Sci. USA
87:9020-9023;
Roher, A.E. et al. (1991) Biochem. Biophys. Res. Commun. 174:572-579; Kowall,
N.W. et al.
(1991) Proc. Natl. Acad. Sci. USA 88:7247-7251. Furthermore, patients with
hereditary
cerebral hemorrhage with amyloidosis-Dutch-type (HCHWA-D), which is
characterized by
diffuse ~-amyloid deposits within the cerebral cortex and cerebrovasculature,
have been
shown to have a point mutation that leads to an amino acid substitution within
~-AP. Levy,
CA 02449296 2003-12-15
2
E. et o1 (1990) Science x:1124-1126. This observation demonstrates that a
specific
alteration of the (3-AP sequence can cause /3-amyloid to be deposited.
Natural ~i-AP is derived by proteolysis from a much larger protein callod the
amyloid
precursor protein (APP). Kung, J. et al. ( 1987) Nature~:733; Goldgaber, D. et
al. (1987)
Science X5_:877; Robakis, N.K. et al. (1987) Proc. Natl. Acad Sci. USA
84:4190; Tanzi,
RE. et al. (1987) Science~5:880. The APP gene maps to chromosome 21, thereby
providing an explanation for the p-amyloid deposition seen at an early age in
individuals with
Down's syndrome, which is caused by trisomy of chromosome 21. Mann, D:M. et
al. (1989)
Neuropathol. Appl. Neurobiol. 1,x:317; Rumble, B. et al. ( 1989) N. E»g. J.
Med. x:1446.
APP contains a single membrane spanning domain, with a long amino terminal
region (about
two-thirds of the protein) extending into the extracellular environment and a
shorter carboxy-
terlninal region projecting into the cytoplasm. Differential splicing of the
APP messenger
RNA Ieads to at Ieast five forms of APP. composed of either 563 amino acids
(APP-563),
695 amino acids (APP-695), 714 amino acids (APP-714), 7~ 1 amino acids (APP-7~
1 ) or 770
I S amino acids (APP-770).
Within APP. naturally-occurring (3 amyloid peptide begins at an aspattic acid
residue
at amino acid position b72 of APP-770. Naturally-occurring /3-AP derived from
proteolysis
of APP is 39 to 43 amino acid residues in length. depending on the carboxy-
terminal end
point. which exhibits heterogeneity. The predominant circulating form of ~-AP
in the blood
and cerebrospinal fluid of both AD patients and normal adults is ~il-40
("short ~i"). Seubert,.
P, et al. (1992) Nature X59:325; Shoji, M. et al. (1992) Science 258:126.
However. (31-42
and ~il-43 ("long ~") also are forms in ~i-amyloid plaques. Masters. C. et al.
(1985) Proc.
. Natl. Acad Sci. USA x:4245; Miller. D. et al. (1993) Arch. Biochem. Biophys.
301:41; Mori,
H. er al. ( 1992) J. Biol. Chem. X7:17082. Although the precise molecular
mechanism
23 leading to ~i-APP aggregation and deposition is unknown. the process has
been likened to that
of nucleation-dependent polymerizations. such as protein crystallization,
microtubule
formation and actin polymerization. See e.g., Jarrett, J.T. and Lansbury, P.T.
(1993) Cell
7~:105~-1058. In such processes, polymerization of monomer components does not
occur
until nucleus formation. Thus, these processes are characterized by a lag time
before
aggregation occurs. followed by rapid polymerization after nucleation.
Nucleation can be
accelerated by the addition of a "seed" or preformed nucleus, which results in
rapid
polymerization. The long ~ forms of ~i-AP have been shown to act as seeds,
thereby
accelerating polymerization of both long and short ~i-AP forms. Jarrett, J.T.
et al. (1993)
Biochemistry ;x:4693.
In one study. in which amino acid substitutions were made in (3-AP, two mutant
~i
peptides were reported to imerfere with polymerization of non-mutated ~-AP
when the
mutant and non-mutant forms of peptide were mixed. Hilbich, C. et al. (1992)
J. Mol. Biol.
228:460-473. However, equimolar amounts of the mutant and non-mutant (i. e.,
natural) ~3
CA 02449296 2003-12-15
3
amyloid peptides were used to see this effect and the mutant peptides were
reported to be
unsuitable for use in vivo. Hilbich, C. et al. (1992), supra.
Summary of the Invention
It should be understood that the expression "the invention" and the like as
used herein,
encompass the subject matter of both the parent and the divisional
applications.
This invention pertains to compounds, and pharmaceutical compositions thereof,
that
can modulate the aggregation of amyloidogenic proteins and peptides, in
particular
compounds that can modulate the aggregation of natural ~3-amyloid peptides (~-
AP) and
inhibit the neurotoxicity of natural S-APs. In one embodiment, the invention
provides an
amyloid modulator compound comprising an amyloidogenic protein, or peptide
fragment
thereof, coupled directly or indirectly to at least one modifying group such
that the compound
modulates the aggregation of natural amyloid proteins or peptides when
contacted with the
natural amyloidogenic proteins or peptides. Preferably, the compound inhibits
aggregation of
natural amyloidogenic proteins or peptides when contacted with the natural
amyloidogenic
proteins or peptides. The amyloidogenic protein, or peptide fragment thereof,
can be, for
example, selected from the group consisting of transthyretin (TTR), prion
protein (PrP), islet
amyloid polypeptide (IAPP), atrial natriuretic factor (ANF), kappa light
chain, lambda light
chain, amyloid A, procalcitonin, cystatin C, i82 microglobulin, ApoA-I,
gelsolin, procalcitonin,
calcitonin, fibrinogen and lysozyme.
In the most preferred embodiment of the invention, the compound modulates the
aggregation of natural S-AP. The invention provides a ~3-amyloid peptide
compound
comprising a formula:
n
X,aa
wherein Xaa is a (3-amyloid peptide having an amino-terminal amino acid
residue
corresponding to position 668 of ~-amyloid precursor protein-770 (APP-770) or
to a residue
carboxy-terminal to position 668 of APP-770, A is a modifying group attached
directly or
indirectly to the S-amyloid peptide of the compound such that the compound
inhibits
aggregation of natural ~-amyloid peptides when contacted with the natural ~-
amyloid
peptides, and n is an integer selected such that the compound inhibits
aggregation of natural
a-amyloid peptides when contacted with the natural ~-amyloid peptides.
In one embodiment, at least one A group is attached directly or indirectly to
the amino
terminus of the (3-amyloid peptide of the compound. In another embodiment, at
least one A
group is attached directly or indirectly to the carboxy terminus of the ~-
amyloid peptide of the
CA 02449296 2003-12-15
Q
compound. In yet another embodiment, at least one A group is attached directly
or indirectly
to a side chain of at least one amino acid residue of the /i-amyloid peptide
of the compound.
In one aspect, this invention also provides a a-amyloid modulator compound
comprising an A~ aggregation core domain (ACD) coupled directly or indirectly
to at least
one modifying group (MG) such that the compound modulates the aggregation or
inhibits the
neurotoxicity of natural ,~-amyloid peptides when contacted with the natural ~-
amyloid
peptides. Preferably, the A~ aggregation core domain is modeled after a
subregion of natural
(3-amyloid peptide between 3 and 10 amino acids in length.
In one aspect, the invention provides a ~i-amyloid modulator compound
consisting of
an AFB aggregation core domain (ACD) from 4 to 7 amino acids in length and
modeled
after amino acid positions 17 to 20 of natural (3-amyloid peptide coupled
directly or
indirectly to at least one modifying group (MG) that is not naturally coupled
to natural ~i-
amyloid peptides in their native form such that the compound modulates the
aggregation
or inhibits the neurotoxicity of natural /3-amyloid peptides when contacted
with the natural
~-amyloid peptides, wherein said ~3-amyloid modulator compound comprises at
least one
D amino acid residue.
In another aspect, the invention provides a ~3-amyloid modulator compound
consisting
of an AS aggregation core domain (ACD) modeled after amino acid positions 17
to 20 of
natural ~-amyloid peptide coupled directly or indirectly to at least one
modifying group (MG)
that is not naturally coupled to natural /3-amyloid peptides in their native
form such that the
compound modulates the aggregation or inhibits the neurotoxicity of natural (3-
amyloid
peptides when contacted with the natural ~-amyloid peptides, wherein said ~8-
amyloid
modulator compound is from 4 to 7 amino acids in length.
In one aspect, the invention also provides (3-amyloid modulator compound
comprising
a formula:
n
( Y-Xaal-Xaa2-Xaa3-Z
wherein Xaal, Xaa2 and Xaa3 are each amino acid structures and at least two of
Xaa,,
Xaa2 and Xaa3 are, independently, selected from the group consisting of a
leucine structure, a
phenylalanine structure and a valine structure;
Y, which may or may not be present, is a peptidic structure having the formula
(Xaa)a,
wherein Xaa is any amino acid structure and a is an integer from 1 to 15;
CA 02449296 2003-12-15
Z, which may or may not be present, is a peptidic structure having the formula
(Xaa~,
wherein Xaa is any amino acid structure and b is an integer from 1 to 15; and
A is a modifying group attached directly or indirectly to the compound and n
is an
integer;
5 Xaa,, Xaa2, Xaa3, Y, Z, A and n being selected such that the compound
modulates the
aggregation or inhibits the neurotoxicity of natural a-amyloid peptides when
contacted with
the natural ~-amyloid peptides. In a preferred embodiment, Xaal and Xaa2 are
each
phenylalanine structures. In another preferred embodiment, Xaa2 and Xaa3 are
each
phenylalanine structures. In another aspect, at least one of Xaa,, Xaa2, and
Xaa3 is a D-amino
acid.
In one aspect, this invention further provides a ~B-amyloid modulator compound
comprising a formula:
n
Y xaa~_Xaa2_Xaa3_Xaa4_Z
wherein Xaa, and Xaa3 are amino acid structures;
Xaa2 is a valine structure;
Xaa4 is a phenylalanine structure;
Y, which may or may not be present, is a peptidic structure having the formula
(Xaa)a,
wherein Xaa is any amino acid structure and a is an integer from 1 to 15;
Z, which may or may not be present, is a peptidic structure having the formula
(Xaa~,,
wherein Xaa is any amino acid structure and b is an integer from 1 to 15; and
A is a modifying group attached directly or indirectly to the compound and n
is an
integer;
Xaa~, Xaa3, Y, Z, A and n being selected such that the compound modulates the
aggregation or inhibits the neurotoxicity of natural ~-amyloid peptides when
contacted with
the natural (3-amyloid peptides. In a preferred embodiment, Xaal is a leucine
structure and
Xaa3 is phenylalanine structure. In another aspect, at least one of Xaa~,
Xaa2, Xaa3, and Xaa4
is a D-amino acid.
In one aspect, the invention still further provides a compound comprising the
formula:
A-Xaa~-Xaa2-Xaa3-Xaa4-Xaas-Xaab-Xaa~-XaaB-B
wherein Xaa, is a histidine structure;
Xaa2 is a glutamine structure;
Xaa3 is a lysine structure;
Xaa4 is a leucine structure;
CA 02449296 2003-12-15
6
Xaas is a valine structure;
Xaa6 is a phenylalanine structure;
Xaa~ is a phenylalanine structure;
Xaag is an alanine structure;
A and B are modifying groups attached directly or indirectly to the amino
terminus
and carboxy terminus, respectively, of the compound;
and wherein Xaa~-Xaaz-Xaa3, Xaa,-Xaa2 or Xaa, may or may not be present;
Xaag may or may not be present; and
at least one of A and B is present.
In a further aspect, said ~-amyloid modulator compound is from 4 to 7 amino
acids in
length.
In one aspect, the modifying groups A and B are groups that are not naturally
coupled
to natural ~i-amyloid peptides in their native form.
In one aspect, said compound comprises at least one D-amino acid residue.
In one aspect, the invention still further provides a S-amyloid modulator
compound
comprising a modifying group attached directly or indirectly to a peptidic
structure, wherein
the peptidic structure comprises amino acid structures having an amino acid
sequence selected
from the group consisting of His-Gln-Lys-Leu-Val-Phe-Phe-Ala (SEQ ID NO: 5),
His-Gln-
Lys-Leu-Val-Phe-Phe (SEQ ID NO: 6), Gln-Lys-Leu-Val-Phe-Phe-Ala (SEQ ID NO:
7), Gln-
Lys-Leu-Val-Phe-Phe (SEQ ID NO: 8), Lys-Leu-Val-Phe-Phe-Ala (SEQ ID NO: 9),
Lys-Leu-
Val-Phe-Phe (SEQ ID NO: 10), Leu-Val-Phe-Phe-Ala (SEQ ID NO: 11), Leu-Val-Phe-
Phe
(SEQ ID NO: 12), Leu-Ala-Phe-Phe-Ala (SEQ ID NO: 13), Val-Phe-Phe (SEQ ID NO:
19),
Phe-Phe-Ala (SEQ ID NO: 20), Phe-Phe-Val-Leu-Ala (SEQ ID NO: 21), Leu-Val-Phe-
Phe-
Lys (SEQ ID NO: 22), Leu-Val-Iodotyrosine-Phe-Ala (SEQ ID NO: 23), Val-Phe-Phe-
Ala
(SEQ >D NO: 24), Ala-Val-Phe-Phe-Ala (SEQ ID NO: 25), Leu-Val-Phe-Iodotyrosine-
Ala
(SEQ ID NO: 26), Leu-Val-Phe-Phe-Ala-Glu (SEQ ID NO: 27), Phe-Phe-Val-Leu (SEQ
ID
NO: 28), Phe-Lys-Phe-Val-Leu (SEQ ID NO: 29), Lys-Leu-Val-Ala-Phe (SEQ ID NO:
30),
Lys-Leu-Val-Phe-Phe-(3Ala (SEQ ID NO: 31) and Leu-Val-Phe-Phe-DAIa (SEQ ID NO:
32).
In one aspect, said /3-amyloid modulator compound comprises at least one D-
amino acid
residue.
In the compounds of the invention comprising a modifying group, preferably the
modifying group comprises a cyclic, heterocyclic or polycyclic group.
Preferred modifying
groups contain a cis-decalin group, such as a cholanoyl structure. Preferred
modifying groups
include a cholyl group, a biotin-containing group, a diethylene-
triaminepentaacetyl group, a
(-)-menthoxyacetyl group, a fluorescein-containing group or an N-
acetylneuraminyl group.
CA 02449296 2003-12-15
The compounds of the invention can be further modified, for example to alter a
pharmacokinetic property of the compound or to label the compound with a
detectable
substance. Preferred radioactive labels are radioactive iodine or technetium.
In one aspect, the invention also provides a /3-amyloid modulator which
inhibits
S aggregation of natural (3-amyloid peptides when contacted with a molar
excess amount of
natural /3-amyloid peptides.
In one aspect, the invention also provides a S-amyloid peptide compound
comprising
an amino acid sequence having at least one amino acid deletion compared to
~AP,_39, such that
the compound inhibits aggregation of natural /3-amyloid peptides when
contacted with the
natural ~3-amyloid peptides. In one embodiment, the compound has at least one
internal amino
acid deleted compared to /3AP1_39. In another embodiment, the compound has at
least one N-
terminal amino acid deleted compared to aAP,_39. In yet another embodiment,
the compound
has at least one C-terminal amino acid deleted compared to /iAP,_39. Preferred
compounds
include (3AP6_ZO (SEQ ID NO: 13), ~iAPl6-30 (SEQ ID NO: 14), (3AP~_ZO,26~o
(SEQ ID NO: 1 S)
1 S and EEWHHHHQQ-~3AP,6_4o (SEQ ID NO: 16).
The compounds of the invention can be formulated into pharmaceutical
compositions
comprising the compound and a pharmaceutically acceptable carrier. The
compounds can also
be used in the manufacture of a medicament for the diagnosis or treatment of
an
amyloidogenic disease.
Another aspect of the invention pertains to diagnostic and treatment methods
using the
compounds of the invention. The invention provides a method for inhibiting
aggregation of
natural ~-amyloid peptides, comprising contacting the natural (3-amyloid
peptides with a
compound of the invention such that aggregation of the natural ~i-amyloid
peptides is
inhibited. T'he invention also provides a method for inhibiting neurotoxicity
of natural
(3-amyloid peptides, comprising contacting the natural ~i-amyloid peptides
with a compound of
the invention such that neurotoxicity of the natural /3-amyloid peptides is
inhibited.
In another embodiment, the invention provides a method for detecting the
presence or
absence of natural (3-amyloid peptides in a biological sample, comprising
contacting a
biological sample with a compound of the invention and detecting the compound
bound to
natural a-amyloid peptides to thereby detect the presence or absence of
natural ~i-amyloid
peptides in the biological sample. In one embodiment, the /3-amyloid modulator
compound
and the biological sample are contacted in vitro. In another embodiment, the a-
amyloid
modulator compound is contacted with the biological sample by administering
the a-amyloid
CA 02449296 2003-12-15
7a
modulator compound to a subject. For in vivo administration, preferably the
compound is
labeled with radioactive technetium or radioactive iodine.
In another embodiment, the invention provides a method for detecting natural
/3-amyloid peptides to facilitate diagnosis of a ~3-amyloidogenic disease,
comprising contacting
S a biological sample with a compound of the invention and detecting the
compound bound to
natural (3-amyloid peptides to facilitate diagnosis of a ~-amyloidogenic
disease. In one
embodiment, the (3-amyloid modulator compound and the biological sample are
contacted in
vitro. In another embodiment, the (3-amyloid modulator compound is contacted
with the
biological sample by administering the ~-amyloid modulator compound to a
subject. For in
vivo administration, preferably the compound is labeled with radioactive
technetium or
radioactive iodine. Preferably, the method facilitates diagnosis of
Alzheimer's disease.
In one aspect, the invention also provides a method for treating a subject for
a disorder
associated with amyloidosis, comprising administering to the subject a
therapeutically or
prophylactically effective amount of a compound of the invention such that the
subject is
treated for a disorder associated with amyloidosis. The method can be used to
treat disorders
is selected, for example, from the group consisting of familial amyloid
polyneuropathy
(Portuguese, Japanese and Swedish types), familial amyloid cardiomyopathy
(Danish type),
isolated cardiac amyloid, systemic senile amyloidosis, scrapie, bovine
spongiform
encephalopathy, Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker
syndrome, adult
onset diabetes, insulinoma, isolated atrial amyloidosis, idiopathic (primary)
amyloidosis,
myeloma or macroglobulinemia-associated amyloidosis, primary localized
cutaneous nodular
amyloidosis associated with Sjogren's syndrome, reactive (secondary)
amyloidosis, familial
Mediterranean Fever and familial amyloid nephropathy with urticaria and
deafness (Muckle-
Wells syndrome), hereditary cerebral hemorrhage with amyloidosis of Icelandic
type,
amyloidosis associated with long term hemodialysis, hereditary non-neuropathic
systemic
amyloidosis (familial amyloid polyneuropathy III), familial amyloidosis of
Finnish type,
amyloidosis associated with medullary carcinoma of the thyroid, fibrinogen-
associated
hereditary renal amyloidosis and lysozyme-associated hereditary systemic
amyloidosis.
In a preferred embodiment, the invention provides a method for treating a
subject for a
disorder associated with a-amyloidosis, comprising administering to the
subject a
therapeutically or prophylactically effective amount of a compound of the
invention such that
the subject is treated for a disorder associated with (3-amyloidosis.
Preferably the disorder is
Alzheimer's disease.
CA 02449296 2003-12-15
7b
In yet another embodiment, the invention provides a method for treating a
subject for a
disorder associated with (3-amyloidosis, comprising administering to the
subject a recombinant
expression vector encoding a peptide compound of the invention such that the
compound is
synthesized in the subject and the subject is treated for a disorder
associated with
~i-arnyloidosis. Preferably, the disorder is Alzheimer's disease.
In one aspect, the invention provides a commercial package containing a ~i-
amyloid
modulator compound as described herein, together with instructions for its use
for the
treatment of a disorder associated with ~i-amyloidosis. Preferably, said
disorder is
Alzheimer's disease.
In a further aspect, the invention provides a method for inhibiting
aggregation of
natural (3-amyloid peptides, comprising contacting the natural (3-amyloid
peptides with a /3-
amyloid modulator compound as described herein, such that aggregation of the
natural (3-
amyloid peptides is inhibited.
In another aspect, the invention provides use of the S-amyloid modulator
compound as
described herein, for the treatment of a disorder associated with (3-
amyloidosis.
In a further aspect, the invention provides use of the ~3-amyloid modulator
compound
as described herein, for the manufacture of a medicament for the treatment of
a disorder
associated with (3-amyloidosis.
CA 02449296 2003-12-15
Brief Descr~~otion of the Draw~na
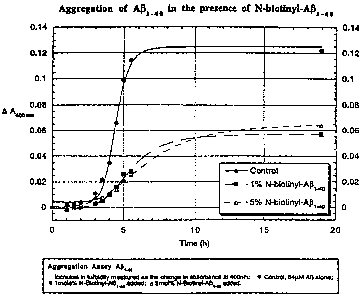
Figure 1 is a graphic representation of the turbidity of a ~-AP ~ ~o solution,
as
measured by optical density at 400 nm, either in the absence of a ~-amyloid
modulator or in
the presence of the (3-amyloid modulator N-biotinyl-~iAP~~o
(1 %, or 5%).
Figure 2 is a schematic representation of compounds which can be used to
modify a
~3-AP or an A~i aggregation core domain to form a ~3-amyloid modulator of the
invention.
Figure 3 is a graphic representation of the toxicity of A~i ~..4o aggregates.
but not
A~i ~ ~o monomers, to cultured neuronal cells.
Figure 4 is a graphic representation of the aggregation of A~ i~o in the
presence of an
equimolar amount of cholyl-A~i6.2o (panel A), a -2-fold molar excess of cholyl-
A(3~2o
(panel B) or a ~6-fold molar excess of cholyl-A(36-20 (panel C) and the
corresponding toxicity
of the aggregates of panels A. B and C to cultured neuronal cells (panels D. E
and F,
respectively).
Detailed Description of the Invention
This invention pertains to compounds. and pharmaceutical compositions thereof,
that
can modulate the aggregation of amyloidogenic proteins and peptides. in
particular
compounds that can modulate the aggregation of natural ~3 amyloid peptides (~i-
AP) and
inhibit the neurotoxicity of natural (3-APs. A compound of the invention that
modulates
aggregation of natural (3-AP, referred to herein interchangeably as a ~i
amyloid modulator
compound. a (3 amyloid modulator or simply a modulator. alters the aggregation
of natural [3-
AP when the modulator is contacted with natural ~i-AP. Thus. a compound of the
invention
acts to alter the natural aggregation process or rate for (3-AP. thereby
disrupting this process.
Preferably. the compounds inhibit ~i-AP aggregation. Furthermore. the
invention provides
subregions of the ~i amyloid peptide that are sufficient. when appropriately
modified as
described herein. to alter (and preferably inhibit) aggregation of natural p
amyloid peptides
when contacted with the natural ~i amyloid peptides. In particular. preferred
modulator
compounds of the invention are comprised of a modified form of an A~i
aggregation core
domain, modeled after the aforementioned A~i subregion (as described further
below). which
is sufficient to alter (and preferably inhibit) the natural aggregation
process or rate for (3-AP.
This A/3 aggregation core domain can comprises as few as three amino acid
residues (or
derivative, analogues or mimetics thereof). Moreover. while the amino acid
sequence of the
A~i aggregation core domain can directly correspond to an amino acid sequence
found in
natural ~i-AP, it is not essential that the amino acid sequence directly
correspond to a (3-AP
sequence. Rather, amino acid residues derived from a preferred subregion of ~-
AP (a
hydrophobic region centered around positions 17-20) can be rearranged in order
and/or
substituted with homologous residues within a modulator compound of the
invention and yet
maintain their inhibitory activity (described further below).
CA 02449296 2003-12-15
w
9
The $ amyloid modulator compounds of the invention can be selected based upon
their ability to inhibit the aggregation of nauual (3-AP in vitro and/or
iahibit the netu~otoxicity
of natural (3-AP fibrils for cultured cells (using assays described herein).
Accordingly, the
preferred modulator compounds inhibit the aggregation of natural (3-AP and/or
inhibit the
neurotoxicity of natural (3-AP. However, modulator compounds selected based on
one or
both of these properties may have additional properties in vivo that may be
beneficial in the
treatment of amyloidosis. For example, the modulator compound may interfere
with
processing of natural (3-AP (either by direct or indirect protease inhibition)
or by modulation
of processes that produce toxic ~i-AP, or other APP firagments, in vivo.
Alternatively,
modulator compounds may be selected based on these latter properties, rather
than inhibition
of A(3 aggregation in vitro. Moreover. modulator compounds of the invention
that are
selected based upon their interaction with natural (3-AP also may interact
with APP or with
other APP fragments.
As used herein. a "modulator" of (3-amyloid aggregation is intended to refer
to an
agent that, when contacted with natural ~3 amyloid peptides. alters the
aggregation of the
natural (3 amyIoid peptides. The term "aggregation of ~i amyloid peptides"
refers to a process
whereby the peptides associate with each other to form a multimeric, largely
insoluble
complex. The term "aggregation" further is intended to encompass ~i amyloid
fibril
formation and also encompasses ~-amyloid plaques.
- The terms "natural p-amyloid peptide", "natural (3-AP" and "natural A(i
peptide", used
interchangeably herein, are intended to encompass naturally occurring
proteolytic cleavage
products of the ~i amyloid precursor protein (APP) which are involved in [3-AP
aggregation
and ~i-amylvidosi$. These natural peptides include ~i-amyloid peptides having
39-43 amino
acids (i.e., A(3~-;g, A(3i~p, A~i~.~~. A~i»~ and A(3~.4;). The amino-terminal
amino acid
residue of natural ~i-AP corresponds to the aspartic acid residue at position
672 of the 770
amino acid residue fotzn of the amyloid precursor protein ("APP-770"). The 43
amino acid
long form of natural ~i-AP has the amino acid sequence
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGWIAT
(also shown in SEQ ID NO: 1 ), whereas the shorter forms have 1-4 amino acid
residues
deleted from the carboxy-tezZninal end. The amino acid sequence of APP-770
from position
672 (l. e., the amino-terminus of natural p-AP) to its C-terminal end ( 103
amino acids) is
shown in SEQ ID NO: 2. The preferred form of natural (3-AP for use in the
aggregation
assays described herein is A~il.~o.
In the presence of a modulator of the invention, aggregation of natural ~
amyloid
peptides is "altered" or "mpdulated". The various forms of the term
"alteration" or
"modulation" are intended to encompass both inhibition of ~3-AP aggregation
and promotion
of ~i-AP aggregation. Aggregation of natural (3-AP is "inhibited" in the
presence of the
modulator when there is a decrease in the amount andlor rate of (3-AP
aggregation as
compared to the amount and/or rate of (3-AP aggregation in the absence of the
modulator.
CA 02449296 2003-12-15
..
I0
The various-forms of the term "inhibition" are intended to include both
complete and partial
inhibition of ~i-AP aggregation. Inhibition of aggregation can be quantitated
as the fold
increase in the lag time for aggregation or as the decrease in the overall
plateau level of
aggregation (t. e., total amount of aggregation), using an aggregation assay
as described in the
Examples. In various embodiments, a modulator of the invention increases the
lag time of
aggregation at least 1.2-fold, I .5-fold, 1.8-fold, 2-fold, 2.5-fold, 3-fold,
4-fold or 5-fold. In
various other embodiments, a modulator of the invention inhibits the plateau
level of
aggregation at least 10%, 20%, 30%, 40 %, SO %, 75 % or 100 %.
A modulator which inhibits ~i-AP aggregation (an "inhibitory modulator
compound")
can be used to prevent or delay the onset of ~i-amyloid deposition. Moreover,
as
demonstrated in Example 10, inhibitory modulator compounds of the invention
inhibit the
formation andlor activity of neurotoxic aggregates of natural A/3 peptide (t.
e., the inhibitory
compounds can be used to inhibit the neurotoxicity of ~3-AP). Still further,
also as
demonstrated in Example 10. the inhibitory compounds of the invention can be
used to
I 5 reduce the neurotoxicity of preformed ~i-AP aggregates. indicating that
the inhibitory
modulators can either bind to preformed A~i fibrils or soluble aggregate and
modulate their
inherent neurotoxicity or that the modulators can perturb the equilibrium
between monomeric
and aggregated forms of (3-AP in favor of the non-neurotoxic form.
Alternatively, in another embodiment, a modulator compound of the invention
promotes the aggregation of natural A(i peptides. The various forms of the
term "promotion"
refer to an increase in the amount and/or rate of ~i-AP aggregation in the
presence of the
modulator. as compared to the amount and/or rate of (3-AP aggregation in the
absence of the
modulator. Such a compound which promotes Aj3 aggregation is referred to as a
stimulatory
modulator compound. Stimulatory modulator compounds may be useful for
sequestering ~3-
amyloid peptides. for example in a biological compartment where aggregation of
(3 :AP may
not be deleterious to thereby deplete (3-AP from a biological compartment
where aggregation
of ~i-AP is deleterious. Moreover, stimulatory modulator compounds can be used
to promote
A~3 aggregation in in vitro aggregation assays (e.g., assays such as those
described in the
Examples), for example in screening assays for test compounds that can then
inhibit or
reverse this A~ aggregation (i.e., a stimulatory modulator compound can act as
a "seed" to
promote the formation of A(3 aggregates).
In a preferred embodiment, the modulators of the invention are capable of
altering ~i-
AP aggregation when contacted with a molar excess amount of natural ~3-AP. A
"molar
excess amount of natural ~i-AP" refers to a concentration of natural (3-AP, in
moles, that is
greater than the concentration, in moles, of the modulator. For example, if
the modulator and
~i-AP are both present at a concentration of I ~tM, they are said to be
"equimolar", whereas if
the modulator is present at a concentration of 1 ~M and the ~i-AP is present
at a concentration
of 5 ~M. the ~-AP is said to be present at a 5-fold molar excess amount
compared to the
modulator. in preferred embodiments, a modulator of the invention is effective
at altering
CA 02449296 2003-12-15
11
natural ~3-AP aggregation when the natural (3-AP is present at at least a 2-
fold, 3-fold or 5-
fold mole excess compared to the concentration of the modulator. In other
embodiments, the
modulator is effective at altering ~i-AP aggregation when the natural ~i-AP is
present at at
least a 10-fold, 20-fold, 33-fold, 50-fold, 100-fold, 500-fold or 1000-fold
molar excess
compared to the concentration of the modulator.
Various additional aspects of the modulators of the invention. and the uses
thereof,
are described in further detail in the following subsections.
I. Modulator Compounds
In one embodiment, a modulator of the invention comprises a (3-amyloid peptide
compound comprising the formula:
n
( Xaa
wherein Xaa is a (3-amyloid peptide. A is a modulating group attached directly
or
indirectly to the (3-amyloid peptide of the compound such that the compound
inhibits
aggregation of natural (3-amyloid peptides when contacted with the natural (3-
amyloid
peptides, and n is an integer selected such that the compound inhibits
aggregation of natural
(3-amyloid peptides when contacted with the natural (i-amyloid peptides.
Preferably, (3-amyloid peptide of the compound has an amino-terminal amino
acid
residue corresponding to position 668 of (3-amyloid precursor protein-770 (APP-
770) or to a
residue carboxy-terminal to position 668 of APP-770. The amino acid sequence
of APP-770
from position 668 to position 770 (i.e., the carboxy terminus) is shown below
and in SEQ ID
NO: 2:
EVKMDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIATVIVITL
VMLKKKQYTSIHHGVVEVDAAVTPEERHLSKMQQNGYENPTYKFFEQMQN
More preferably, the amino-terminal amino acid residue of the ~3-amyloid
peptide
corresponds to position 672 of APP-770 (position ~ of the amino acid sequence
of SEQ ID
NO: 2) or to a residue carboxy-terminal to position 672 of APP-770. Although
the ~-amyloid
peptide of the compound may encompass the 103 amino acid residues
corresponding to
positions 668-770 of APP-770, preferably the peptide is between 6 and 60 amino
acids in
length, more preferably between 10 and 43 amino acids in length and even more
preferably
between 10 and 2~ amino acid residues in length.
As used herein, the term "~i amyloid peptide", as used in a modulator of the
invention
is intended to encompass peptides having an amino acid sequence identical to
that of the
natural sequence in APP, as well as peptides having acceptable amino acid
substitutions from
the natural sequence. Acceptable amino acid substitutions are those that do
not affect the
CA 02449296 2003-12-15
ability of the peptide to slur natural (3-AP alggregation. Moreover,
particular amino acid
substitutions may further contribute to the ability of the peptide to alter
nanual p-AP
aggregation and/or may confer additional beneficial properties on the peptide
(e.g., increased
solubility, reduced association with other amyloid proteins, etc.). For
example. substitution
of hydrophobic amino acid residues for the two phenylalanine residues at
positions 19 and 20
of natural (3-AP (positions 19 and 20 of the amino acid sequence shown in SEQ
ID NO: 1 )
may further contribute to the ability of the peptide to alter ~i-AP
aggregation (see Hilbich, C.
(1992) J. Mol. Biol. 228:460-473). Thus, in one embodiment, the (i-AP of the
compound
consists of the amino acid sequence shown below and in SEQ ID NO: 3:
DAEFRHDSGYEVHHQKLV(Xaal9)(Xaa2o)AEDVGSNKGAIIGLMVGGWIAT
(or an amino-terminal or carboxy-terminal deletion thereof), wherein Xaa is a
hydrophobic
amino acid. Examples of hydrophobic amino acids are isoleucine. leucine.
threonine. serine.
alanine. valine or glycine. Preferably, F ~ 9F~o is substituted with T ~ gT~o
or G ~ 9I2o~
Other suitable amino acid substitutions include replacement of amino acids in
the
human peptide with the corresponding amino acids of the rodent (3 :AP peptide.
The three
amino acid residues that differ between human and rat (3-AP are at positions
~. 10 and 13 of
the amino acid sequence shown in SEQ ID NOs: 1 and 3. A human ~3-AP having the
human
to rodent substitutions Argg to Gly, Tyro to Phe and Hisl3 to Arg has been
shown to retain
the properties of the human peptide (see Fraser, P.E. et al. (1992)
Biochemistry 31:10716-
10723: and Hilbich. C. et al. ( 1991 ) Eur. J. Biochem. 201:61-69).
Accordingly. a human ~i-
AP having rodent ~i-AP a.a. substitutions is suitable for use in a modulator
of the invention.
Other possible ~i-AP amino acid substitutions are described in Hilbich. C. et
al.
(1991) J. Mol. Biol. 218:149-163: and Hilbich. C. (1992) J. Mol. Biol. ?28:460-
473.
Moreover, amino acid substitutions that affect the ability of ~i-AP to
associate with other
proteins can be introduced. For example, one or more amino acid substitutions
that reduce
the ability of ~-AP to associate with the serpin enzyme complex (SEC)
receptor, a 1-
antichymotrypsin (ACT) and/or apolipoprotein E (ApoE) can be introduced. A
preferred
substitution for reducing binding to the SEC receptor is L34M35 to A;4A35 (at
positions 34
and 35 of the amino acid sequences shown in SEQ ID NOs: 1 and 3). A preferred
substitution for reducing binding to ACT is Sg to Ag (at position 8 of the
amino acid
sequences shown in SEQ ID NOs: 1 and 3).
Alternative to (3- .AP amino acid substitutions described herein or known in
the art. a
modulator composed. at least in part, of an amino acid-substituted (3 amyloid
peptide can be
prepared by standard techniques and tested for the ability to alter (3-AP
aggregation using an
aggregation assay described herein. To retain the properties of the original
modulator,
preferably conservative amino acid substitutions are made at one or more amino
acid
residues. A "conservative amino acid substitution" is one in which the amino
acid residue is
CA 02449296 2003-12-15
13
replaced with an amino acid residue having a similar side chain. Families of
amino acid
residues having similar side chains have been defined in the art, including
basic side chains
(e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid,
glutamic acid),
uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine,
threonine, tyrosine,
cysteine), nonpolar side chains (e.g., alanine, valise, leucine, isoleucine,
proline,
phenylslanine, methionine, tryptophan), ~-branched side chains (e.g.,
threonine, valise,
isoleucine) and aromatic side chains (e.g., tyrosine, phcnylalanine,
tryptophan, histidine).
Accordingly, a modulator composed of a ~i amyloid peptide having an amino acid
sequence
that is mutated from that of the wild-type sequence in APP-770 yet which still
retains the
ability to alter natural ~i-AP aggregation is within the scope of the
invention.
As used herein, the term "~3 amyloid peptide" is further intended to include
peptide
analogues or peptide derivatives or pegtidomimetics that retain the ability to
alter natural ~i-
AP aggregation as described herein. For example, a (3 amyloid peptide of a
modulator of the
invention may be modified to increase its stability. bioavailability,
solubility, etc. The terms
"peptide analogue", "peptide derivative" and "peptidomimetic" as used herein
are intended to
include molecules which mimic the chemical structure of a peptide and retain
the functional
properties of the peptide. Approaches to designing peptide analogs are known
in the art. For
example, see Farmer, P.S. in drug; D~sig~ (E.J. Ariens. ed.) Academic Press,
New York,
1980, vol. 10, pp. 119-143; Ball. J.B. and Alewood, P.F. (1990) J. Mol.
Recognition 3_:55;
Morgan, B.A. and Gainor, J.A. ( 1989) Ann. Rep. Med Chem. 24:243; and
Freidinger, R.M.
(1989) Trendy Pharmacol. Sci. 10:270. Examples of peptide analogues,
derivatives and
peptidomimetics include peptides substituted with one or more benzodiazepine
molecules
(see e.g., James. G.L. et al. (1993) Science 260:1937-1942), peptides with
methylated amide
linkages and "retro-inverso" peptides (see U.S. Patent No. 4.522.752 by
Sisto). Peptide
analogues, peptide derivatives and peptidomimetic are described in further
detail below with
regard to compounds comprising an A~ aggregation core domain.
In a modulator of the invention having the formula shown above, a modulating
group
("A") is attached directly or indirectly to the (3-amyloid peptide of the
modulator (As used
herein, the term "modulating group" and "modifying group" are used
interchangeably to
describe a chemical croup directly or indirectly attached to an A~i derived
peptidic structure).
For example, the modulating group can be directly attached by covalent
coupling to the ~i-
amyloid peptide or the modulating group can be attached indirectly by a stable
non-covalent
association. In one embodiment of the invention, the modulating group is
attached to the
amino-terminus of the ~i-amyloid peptide of the modulator. Accordingly, the
modulator can
comprise a compound having a formula:
A-~-{ Xaa
CA 02449296 2003-12-15
14
Alternatively, in another embodiment of the invention, the modulating group is
attached to
the carboxy-terminus of the (3-amyloid peptide of the modulator. Accordingly,
the modulator
can comprise a compound having a formula:
O
( Xaa ) ~-A
In yet another embodiment, the modulating group is attached to the side chain
of at least one
amino acid residues of the ~i-amyloid peptide of the compound (e.g., through
the epsilon
amino group of a lysyl residue(s), through the carboxyl group of an aspartic
acid residues) or
a glutamic acid residue(s), through a hydroxy group of a tyrosyl residue(s). a
serine residues)
or a threonine residues) or other suitable reactive group on an amino acid
side chain).
The modulating group is selected such that the compound inhibits aggregation
of
natural ~i-amyloid peptides when contacted with the natural (i-amyloid
peptides.
Accordingly. since the ~3-AP peptide of the compound is modified from its
natural state- the
modulating group "A" as used herein is not intended to include hydrogen. In a
preferred
embodiment. the modulating group is a biotin compound of the formula:
W
~X~
~X
O
X;~R~-C-Y
wherein X1-X; are each independently selected from the group consisting of S.
O and NR,.
wherein R~ is hydrogen. or an aryl, lower alkyl, alkenyl or alkynyl moiety; VV
is =O or
NR2; R~ is a lower alkylenyl moiety and Y is a direct bond or a spacer
molecule selected for
its ability to react with a target group on a ~i-AP. At least one of Xl-X3 or
W is an NR~
group.
The term "aryl" is intended to include aromatic moieties containing
substituted or
uasubstituted ring(s), e.g., benzyl, napthyl, etc. Other more complex fused
ring moieties also
are intended to be included.
The term "lower alkyl or alkylenyl moiety" refers to a saturated, straight or
branched
chain (or combination thereof] hydrocarbon containing 1 to about 6 carbon
atoms, more
preferably from 1 to 3 carbon atoms. The terms "lower alkenyl moiety" and
"lower alkynyl
moiety" refer to unsaturated hydrocarbons containing 1 to about 6 carbon
atoms, more
preferably 1 to 3 carbon atoms. Preferably, R2 contains 1 to 3 carbon atoms.
Preferably, R~
contains 4 carbon atoms.
CA 02449296 2003-12-15
The-spacer molecule (~ can be, for lex5ample, a lower alkyl gmup or a linker
peptide.
and is preferably selected for its ability to link with a free amino group
(e.g., the oc-amino
group at the amino-terminus of a ~i-AP). Thus, in a preferred embodiment, the
biotin
compound modifies the amino-terminus of a ~3-amyloid peptide.
Additional suitable modulating groups may include other cyclic and
heterocyclic
compounds and other compounds having similar steric "bulk". Non-limiting
examples of
compounds which can be used to modify a ~3-AP are shown schematically in
Figure 2, and
include N acetylneuraminic acid, cholic acid, traps-4-cotininecarboxylic acid,
2-imino-1-
imidazolidineacetic acid, (S')-(-~indoline-2-carboxylic acid, (-
~menthoxyacetic acid. 2-
norbornaneacetic acid, Y-oxo-5-acenaphthenebutyric acid, (-~2-oxo-4-
thiazolidinecarboxylic
acid, tetrahydro-3-furoic acid, 2-iminobiotin-N hydroxysuccinimide ester,
diethylenetriaminepentaacetic dianhydride, 4-morpholinecarbonyl chloride, 2-
thiopheneacetyl chloride, 2-thiophenesulfonyl chloride, S-(and 6-
~carboxyfluorescein
(succinimidyl ester), fluorescein isothiocyanate. and acetic acid (or
derivatives thereof).
Suitable modulating groups are described further in subsection II below.
In a modulator of the invention. a single modulating group may be attached to
a ~i-
amyloid peptide (e.g., n=1 in the formula shown above) or multiple modulating
groups may
be attached to the peptide. The number of modulating groups is selected such
that the
compound inhibits aggregation of natural ~i-amyloid peptides when contacted
with the natural
(3-amyloid peptides. However, n preferably is an integer between l and 60,
more preferably
between l and 30 and even more preferably between l and 10 or 1 and 5.
In another embodiment, a ~i-amyloid modulator compound of the invention
comprises
an A~3 aggregation core domain (abbreviated as ACD) coupled directly or
indirectly to a
modifying group such that the compound modulates the aggregation or inhibits
the
neurotoxicity of natural ~i-amyloid peptides when contacted with the natural
~3-amyloid
peptides. As used herein, an "A(3 aggregation core domain" is intended to
refer to a structure
that is modeled after a subregion of a natural (3-arnyloid peptide which is
sufficient to
modulate aggregation of natural ~i-APs when this subregion of the natural (3-
AP is
appropriately modified as described herein (e.g., modified at the amino-
terminus). The term
"subregion of a natural (3-amyloid peptide" is intended to include amino-
terminal and/or
carboxy-terminal deletions of natural ~3-AP. The term "subregion of natural (3-
AP" is not
intended to include full-length natural ~i-AP (i.e., "subregion" does not
include Aft-39,
A~1-40~ A~31-41~ Apl-42 ~d A~31-43)~
Although not intending to be limited by mechanism, the ACD of the modulators
of
the invention is thought to confer a specific targeting function on the
compound that allows
the compound to recognize and specifically interact with natural (3-AP.
Preferably, the ACD
is modeled after a subregion of natural ~i-AP that is less than 15 amino acids
in length and
more preferably is between 3-10 amino acids in length. In various embodiments,
the ACD is
CA 02449296 2003-12-15
16
modeled after a subregion of (3-AP that is 10, 9, 8, 7, 6, 5, 4 or 3 amino
acids in length. In
one embodiment, the subregion of ~i-AP upon which the ACD is modeled is an
internal or
carboxy-terminal region of ø-AP (i.e., downstream of the amino-terminus at
amino acid
position 1 ). In another embodiment, the ACD is modeled after a subregion of
(3-AP that is
hydrophobic. In certain specific embodiments, the term Ap aggregation core
domain
specifically excludes ~i-AP subregions corresponding to amino acid positions 1-
15 (A~i1.15)~
6-20 (A~6_20) and 16-40 (A~iI~O).
An A(3 aggregation core domain can be comprised of amino acid residues linked
by
peptide bonds. That is, the ACD can be a peptide corresponding to a subregion
of ~i-AP.
Alternatively, an A~i aggregation core domain can be modeled after the natural
A~i peptide
region but may be comprised of a peptide analogue, peptide derivative or
peptidomimetic
compound. or other similar compounds which mimics the structure and function
of the
natural peptide. Accordingly, as used herein, an "A~3 aggregation core domain"
is intended to
include peptides. peptide analogues, peptide derivatives and peptidomimetic
compounds
which. when appropriately modified. retain the aggregation modulatory activity
of the
modified natural A(3 peptide subregion. Such structures that are designed
based upon the
amino acid sequence are referred to herein as "A~i derived peptidic
structures." Approaches
to designing peptide analogues, derivatives and mimetics are known in the art.
For example,
see Farmer, P.S. in Drub Design (E.J. Ariens, ed.) Academic Press. New York.
1980. vol. 10,
pp. 119-143; Ball. J.B. and Alewood, P.F. (1990) J. Mol. Recognition 3:~5;
Morgan, B.A.
and Gainor. J.A. ( 1989) Ann. Rep. Med Chem. 24:243; and Freidinger, R.M. (
1989) Trends
Pharmacol. Sci. 10:270. See also Sawyer. T.K. ( 1995) "Peptidomimetic Design
and
Chemical Approaches to Peptide Metabolism" in Taylor, M.D. and Amidon. G.L.
(eds.)
Peptide-Based Drug Design: Controlling Transport and Metabolism. Chapter 17:
Smith. A.B.
3rd. et al. (1995) J. Am. Chem. Soc. 117:11113-11123: Smith. A.B. 3rd, et al.
(1994) J. Am.
Chem. Soc. 116:9947-9962; and I~iirschman. R., et al. (1993) J. Am. Chem. Soc.
11~:12550-
12568.
As used herein, a "derivative" of a compound X (e.g., a peptide or amino acid)
refers
to a form of X in which one or more reaction groups on the compound have been
derivatized
with a substituent group. Examples of peptide derivatives include peptides in
which an
amino acid side chain. the peptide backbone, or the amino- or carboxy-terminus
has been
derivatized (e.g., peptidic compounds with methylated amide linkages). As used
herein an
"analogue" of a compound X refers to a compound which retains chemical
structures of X
necessary for functional activity of X yet which also contains certain
chemical structures
which differ from X. An examples of an analogue of a naturally-occurring
peptide is a
peptides which includes one or more non-naturally-occurring amino acids. As
used herein, a
"mimetic" of a compound X refers to a compound in which chemical structures of
X
necessary for functional activity of X have been replaced with other chemical
structures
which mimic the conformation of X. Examples of peptidomimetics include
peptidic
CA 02449296 2003-12-15
compounds in which the peptide backbone is substituted with one or more
benzodiazepine
molecules (see e.g., James, G.L. et al. (1993) Science x:1937-1942), peptides
in which all
L-amino acids arc substituted with the corresponding D-amino acids and "retro-
inverso"
peptides (see U.S. Patent No. 4,522,752 by Sisto), described further below.
The term mimetic, and in particular, peptidomimetic, is intended to include
isosteres.
The term "isostere" as used herein is intended to include a chemical structure
that can be
substituted for a second chemical structure because the steric conformation of
the first
structure fits a binding site specific for the second structure. The term
specifically includes
peptide back-bone modifications (i.e., amide bond mimetics) well known to
those skilled in
the art. Such modifications include modifications of the amide nitrogen, the a-
carbon. amide
carbonyl, complete replacement of the amide bond, extensions, deletions or
backbone
crosslinks. Several peptide backbone modifications are known, including
yr[CH2S], yr
[CH.,NH], yr[CSNH2], yr[NHCO], yr[COCH,], and yr[(E) or (Z) CH=CH]. In the
nomenclature used above, yr indicates the absence of an amide bond. The
structure that
replaces the amide group is specified within the brackets. Other examples of
isosteres
include peptides substituted with one or more benzodiazepine molecules (see
e.g., James,
G.L. et al. (1993) Science x,60:1937-1942)
Other possible modifications include an N-alkyl (or aryl) substitution
(fir[CONR]),
backbone crosslinking to construct lactams and other cyclic structures.
substitution of all D-
amino acids for all L-amino acids within the compound ("inverso" compounds) or
retro-
inverso amino acid incorporation (~y[NHCO]). By "inverso" is meant replacing L-
amino
acids of a sequence with D-amino acids, and by "retro-inverso" or "enantio-
retro" is meant
reversing the sequence of the amino acids ("retro") and replacing the L-amino
acids with D-
amino acids. For example, if the parent peptide is Thr-Ala-Tyr, the retro
modified form is
Tyr-Ala-Thr. the inverso form is thr-ala-tyr, and the retro-inverso form is
tyr-ala-thr (lower
case letters refer to D-amino acids). Compared to the parent peptide. a term-
inverso peptide
has a reversed backbone while retaining substantially the original spatial
conformation of the
side chains, resulting in a retro-inverso isomer with a topology that closely
resembles the
parent peptide. See Goodman et al. "Perspectives in Peptide Chemistry" pp. 283-
294
(1981 ). See also U.S. Patent No. 4,522.752 by Sisto for further description
of "retro-inverso"
peptides.
Other derivatives of the modulator compounds of the invention include C-
terminal
hydroxymethyl derivatives, O-modified derivatives (e.g., C-terminal
hydroxymethyl benzyl
ether), N-terminally modified derivatives including substituted amides such as
alkylamides
and hydrazides and compounds in which a C-terminal phenylalanine residue is
replaced with
a phenethylamide analogue (e.g., Val-Phe-phenethylamide as an analogue of the
tripeptide
Val-Phe-Phe).
In a preferred embodiment, the ACD of the modulator is modeled after tNe
subregion
of (3-AP encompassing amino acid positions 17-20 (i.e., Leu-Val-Phe-Phe; SEQ
ID NO: 12).
CA 02449296 2003-12-15
As described feather in Examples 7, 8 sad 91 peptide subregions of Aø 1.4,0
were per,
amino-terminally modified and evaluated for their ability to modulate
aggregation of natsnal
ø-amyloid peptides. One subregion that was effective at inhibiting aggregation
was Aø~2o
(i.e., amino acid residues 6-20 of the natural Aø~,.4o peptide, the amino acid
sequence of
which is shown in SEQ ID NO: 4). Amino acid residues werc serially deleted
from the
amino-terminus or carboxy terminus of this subregion to further delineate a
minimal
subregion that was su~cient for aggregation inhibitory activity. This process
defined
Aø ~ 7_20 (e. e., amino acid residues 17-20 of the natural Aø 1,.4o peptide)
as a minimal
subregion that, when appropriately modified. is sufficient for aggregation
inhibitory activity.
Accordingly, an "Aø aggregation core domain" within a modulator compound of
the
invention can be modeled after Aø ~ 7_20. In one embodiment. the Aø
aggregation core
domain comprises Aø~7_2o itself (i.e., a peptide comprising the amino acid
sequence leucine-
valine-phenylalanine-phenylalanine; SEQ ID NO: 12). In other embodiments, the
structure
of AøI7_~o is used as a model to design an Aø aggregation core domain having
similar
structure and function to Aøt7.2o. For example, peptidomimetics, derivatives
or analogues of
Aø ~ 7_20 (as described above) can be used as an Aø aggregation core domain.
In addition to
Aa 17-20~ ~e ~~ Aø Peptide is likely to contain other minimal subregions that
arc
suffcient for aggregation inhibitory activity. Such additional minimal
subregions can be
identified by the processes described in Examples 7. 8 and 9, wherein a 1 ~mer
subregion of
Aø l ~o is serially deleted from the amino-terminus or carboxy terminus, the
deleted peptides
are appropriately modified and then evaluated for aggregation inhibitory
activity.
One form of the ø-amyloid modulator compound comprising an Aø aggregation core
domain modeled after Aø ~ 7_2o coupled directly or indirectly to at least one
modifying group
has the formula:
n
( Y-XaamXaa~-Xaa3-Xaa4-Z
wherein Xaal and Xaa3 are amino acid structures;
Xaay is a valine structure;
XaaQ is a phenylalanine structure;
Y, which may or may not be present. is a peptidic structure having the
formula (Xaa)a, wherein Xaa is any amino acid structure and a is an integer
from 1 to I 5;
Z. which may or may not be present. is a peptidic structure having the
formula (Xaa)b, wherein Xaa is any amino acid structure and b is an integer
from 1 to I 5; and
A is a modifying group attached directly or indirectly to the compound and n
is an integer;
Xaa~, Xaa3. Y, Z. A and n being selected such that the compound modulates the
aggregation or inhibits the neurotoxicity of natural ø-amyloid peptides when
contacted with
the natural ø-amyloid peptides.
CA 02449296 2003-12-15
Preferably, a modulator compound of t9he above formula inhibits agon of
natiual (3-amyloid peptides when contacted with the natural ~i-amyloid
peptides and/or
inhibits A~ neurotoxicity. Alternatively, the modulator compound can promote
aggregation
of natural ~-amyloid peptides when contacted with the natural ~3-amyloid
peptides. The type
and number of modifying groups ("A") coupled to the modulator are selected
such that the
compound alters (and preferably inhibits) aggregation of natural ~3-amyloid
peptides when
contacted with the natural ~i-amyloid peptides. A single modifying group can
be coupled to
the modulator (i.e., n=1 in the above formula) or, alternatively, multiple
modifying groups
can be coupled to the modulator. In various embodiments, n is an integer
between 1 and 60,
between I and 30, between I and 10, between 1 and 5 or between I and 3.
Suitable types of
modifying groups are described further in subsection II below.
As demonstrated in Example 9, amino acid positions 18 (Val ~ g) and 20 (Phe2p)
of
A~3~~_2p (corresponding to Xaa, and Xaa4) are particularly important within
the core domain
for inhibitory activity of the modulator compound. Accordingly. these
positions are
conserved within the core domain in the formula shown above. The terms "valise
structure"
and "phenylalanine structure" as used in the above formula are intended to
include the natural
amino acids, as well as non-naturally-occurring analogues, derivatives and
mimetics of valise
and phenylalanine, respectively, (including D-amino acids) which maintain the
functional
activity of the compound. Moreover, although Val ~ g and Phe2a have an
important functional
role, it is possible that Xaa2 and/or Xaa4 can be substituted with other
naturally-occurring
amino acids that are structurally related to valise or phenylalanine,
respectively, while still
maintaining the activity of the compound. Thus, the terms "valise structure"
is intended to
include conservative amino acid substitutions that retain the activity of
valise at Xaa,. and
the term "phenylalanine structure" is intended to include conservative amino
acid
substitutions that retain the activity of phenylalanine at Xaa4. However, the
term "valise
structure" is not intended to include threonine.
In contrast to positions 18 and 20 of A~i ~ ~.2p, a Phe to Ala substitution at
position 19
(corresponding to Xaa3) did not abolish the activity of the modulator,
indicating position I 9
may be more amenable to amino acid substitution. In various embodiments of the
above
formula, positions Xaa~ and Xaa3 are any amino acid structure. The term "amino
acid
structure" is intended to include natural and non-natural amino acids as well
as analogues,
derivatives and mimetics thereof, including D-amino acids. In a preferred
embodiment of the
above formula. Xaal is a leucine structure and Xaa3 is a phenylalanine
structure (i.e.,
modeled after Leu 1 ~ and Phe ~ g, respectively, in the natural A~i peptide
sequence). The term
"leucine structure" is used in the same manner as valise structure and
phenylalanine structure
described above. Alternatively, an another embodiment, Xaa3 is an alanine
structure.
The four amino acid structure ACD of the modulator of the above formula can be
flanked at the amino-terminal side, carboxy-terminal side, or both, by
peptidic structures
derived either from the natural A~i peptide sequence or from non-A~i
sequences. The term
CA 02449296 2003-12-15
"peptidic stin~cnu~e" is intended to include peptide analogues, derivatives
and mimetics
thereof,. as described above. The peptidic structure is composed of one or
more linked amino
acid structiues, the type and number of which in the above formula are
variable. For
example, in one embodiment, no additional amino acid structures flank the Xaat-
Xaa,-Xaa3-
5 Xaa4 core sequence (i.e., Y and Z are absent in the above formula). In
another embodiment,
one or more additional amino acid structures flank only the amino-terminus of
the core
sequences (i. e., Y is present but Z is absent in the above formula). In yet
another
embodiment, one or more additional amino acid structures flank only the
carboxy-terminus of
the core sequences (i.e., Z is present but Y is absent in the above formula).
The length of
10 flanking Z or Y sequences also is variable. For example, in one embodiment,
a and b are
integers from I to 15. More preferably, a and b are integers between I and 10.
Even more
preferably, a and b are integers between 1 and 5. Most preferably, a and b are
integers
between 1 and 3
One form of the ~i-amyloid modulator compound comprising an A~3 aggregation
core
15 domain modeled after A~3 ~ ~_~p coupled directly or indirectly to at least
one modifying group
has the formula: -
A-(Y}-XaauXaa~-Xaa;-Xaa4-(Z)-B
wherein Xaai and Xaa3 are amino acids or amino acid mimetics;
20 Xaa~ is valise or a valise mimetic
Xaa4 is phenylalanine or a phenylalanine mimetic;
Y, which may or may not be present. is a peptide or peptidomimetic having
the formula (Xaa)a, wherein Xaa is any amino acid or amino acid mimetic and a
is an integer
from 1 to 15:
~ Z. which may or may not be present, is a peptide or peptidomimetic having
the formula (Xaa)b, wherein Xaa is any amino acid or amino acid mimetic and b
is an integer
from I to I5; and
A and B, at least one of which is present, are modifying groups attached
directly or indirectly to the amino terminus and carboxy terminus,
respectively, of the
compound;
Xaal, Xaa3, Y, Z, A and B being selected such that the compound modulates the
aggregation or inhibits the neurotoxicity of natural ~i-amyloid peptides when
contacted with
the natural ~i-amyloid peptides.
In this embodiment. the modulator compound is specifically modified at either
its
amino-terminus, its carboxy-terminus, or both. The terminology used in this
formula is the
same as described above. Suitable modifying groups are described in subsection
II below. In
one embodiment, the compound is modified only at its amino terminus (i.e., B
is absent aad
the compound comprises the formula: A-(Y)-Xaa ~ -Xaa,-Xaag-Xaa4-(Z)). In
another
embodiment, the compound is modified only at its carboxy-terminus (i.e., A is
absent and the
CA 02449296 2003-12-15
compound comprises the formula: (Y~Xaa~ IXaa,-Xaa3-Xaa4-(Z)-B). In yet another
embodiment, the compound is modified at both its amino- and carboxy termini
(i.e., the
compound comprises the formula: A-(Y~Xaa~-Xaa~-Xaa3-Xaa4-(Z)-B and both A and
B are
present). As described above, the type and number of amino acid structures
which flank the
Xa~aZ-Xaa~-Xaa3-Xaa~ core sequences in the above formula is variable. For
example, in one
embodiment, a and b are integers from 1 to 1 S. More preferably, a and b are
integers between
I and 10. Even more preferably, a and b are integers between 1 and 5. Most
preferably, a
and b are integers between 1 and 3.
As demonstrated in Examples 7, 8 and 9, preferred A~i modulator compounds of
the
invention comprise modified forms of A~ij4.2t (His-GIn-Lys-Leu-Val-Phe-Phe-
Ala; SEQ ID
NO: 5), or amino-terminal or carboxy-terminal deletions thereof, with a
preferred "minimal
core region" comprising A~i 1 ~-2p. Accordingly, in specific embodiments. the
invention
provides compounds comprising the formula:
A-Xaa~-Xaa,-Xaa3-Xaa4-Xaag-Xaa6-Xaa~-Xaa8-B
wherein Xaa I is a histidine structure;
Xaa2 is a glutamine structure;
Xaa3 is a lysine structure;
Xaa4 is a leucine structure;
XaaS is a valine structure;
Xaa6 is a phenylalanine structure;
Xaa7 is a phenylalanine structure;
Xaa8 is an alanine structure;
A and B are niodifying groups attached directly or indirectly to the amino
tenniaus and carboxy terminus. respectively, of the compound;
and wherein Xaa~-Xaa~-Xaa3, Xaa~-Xaa~ or Xaa~ may or may not be present;
Xaag may or may not be present; and
at least one of A and B is present.
In one specific embodiment, the compaund comprises the formula: A-Xaa4-Xaas-
Xaa6-XaaT-B (e.g, a modified form of A~i~~_2p, comprising an amino acid
sequence Leu-Val-
Phe-Phe; SEQ ID NO: 12).
In another specific embodiment, the compound comprises the formula: A-Xaa4-
Xaag-Xaa6-Xaa~-Xaag-B (e.g, a modified form of A~it~_2~, comprising an amino
acid
sequence Leu-Val-Phe-Phe-Ala; SEQ ID NO: I 1).
In another specific embodiment, the compound comprises the formula: A-Xaa3-
Xaad-XaaS-Xaa~-Xaa~-B (e.g., a modified farm of A(3t6.2p, comprising an amino
acid
sequence Lys-Leu-Val-Phe-Phe; SEQ ID NO: 10).
CA 02449296 2003-12-15
22
In apother specific embodimern, the cornpou~nd comprises the forlaula: A-Xaa3-
Xaa4-Xaas~Xaa6-Xaa~-Xaag-B (c g., a modified form of A(3~~.21, comprising an
amino acid
sequence Lys-Leu-Val-Phe-Phe-Ala; SEQ ID NO: 9).
In another specific embodiment, the compound comprises the formula: A-Xaa2-
Xsa3-Xaa4-Xaas-Xaa6-Xaa~-B (e.g., a modified form of A~its-2o~ ~mPnsing an
amino acid
sequence Gln-Lys-Leu-Val-Phe-Phe; SEQ ID NO: 8).
In another specific embodiment, the compound comprises the formula: A-Xaa2-
Xaa3-Xaa4-Xaag-Xaab-Xaa~-Xaag-B (e.g., a modified form of Aøls-21- comprising
an amino
acid sequence GIn-Lys-Leu-Val-Phe-Phe-Ala; SEQ ID NO: 7).
In another specific embodiment, the compound comprises the formula: A-Xaal-
Xaa2-Xaa3-Xaa4-Xaas-Xaa6-Xaa~-B (e.g., a modified form of A~314-2p, comprising
an
amino acid sequence His-Gln-Lys-Leu-Val-Phe-Phe; SEQ ID NO: 6).
In another specific embodiment the compound comprises the formula: A-Xaa~-
Xaa,-Xaa.;-Xaa4-Xaas-Xaa~-Xaa~-Xaag-B (e.g., a modified form of AJ3I4-2 i ~
comprising an
amino acid sequence His-Gln-Lys-Leu-Val-Phe-Phe-Ala; SEQ ID NO: 5).
In preferred embodiments of the aforementioned specif c embodiments, A or B is
a
cholanoyl structure or a biotin-containing structure (described further in
subsection II below).
In further experiments to delineate subregions of A~ upon which an A(3
aggregation
con domain can be modeled (the results of which are described in Example 11 ),
it was
demonstrated that a modulator compound having inhibitory activity can comprise
as few as
three A~i amino acids residues (e.g., VaI-Phe-Phe. which corresponds to A~3I8-
2p or Phe-Phe-
Ala. which corresponds to A[3 ~ q-2 ~ ). The results also demonstrated that a
modulator
compound having a modulating group at its carboxy-terminus is effective at
inhibiting A(3
aggregation. Still further. the results demonstrated that the cholyl group, as
a modulating
group, can be manipulated while maintaining the inhibitory activity of the
compounds and
that an iodotyrosyl can be substituted for phenylalanine (e.g., at position 19
or 20 of the A~
sequence) while maintaining the ability of the compound to inhibit Ap
aggregation.
Still ftuther, the results demonstrated that compounds with inhibitory
activity can be
created using amino acids residues that are derived from the Ap sequence in
the region of
about positions 17-21 but wherein the amino acid sequence is rearranged or has
a substitution
with a non-A~i-derived amino acid. Examples of such compounds include PPI-426,
in which
the sequence of A~i ~ ~-2 t (LVFFA) has been rearranged (FFVLA), PPI-372, in
which the
sequence ofA~it~.2p (KLVFF) has been rearranged (FKFVL), and PPI-388, -389 and
-390, in
which the sequence of A(31~-21 (LVFFA) has been substituted at position 17, 18
or 19,
respectively, with an alanine residue (AVFFA for PPI-388, LAFFA for PPI-389
and LVAFA
for PPI-390). The inhibitory activity of these compounds indicate that the
presence in the
compound of an amino acid sequence directly corresponding to a portion of A~i
is not
essential for inhibitory activity, but rather suggests that maintenance of the
hydrophobic
CA 02449296 2003-12-15
nature of this core region, by inclusion of am~2'3no acid residues such as
phenylalanine, valise,
leucine, regardless of their precise order, can be sufficient for inhibition
of Aø aggregation.
Accordingly, an Aø aggregation core domain can be designed based on the direct
Aø amino
acid sequence or can be designed based on a rearranged Aø sequence which
maintains the
hydrophobicity of the Aø subregion, e.g., the region around positions 17-20.
This region of
Aø contains the amino acid residues Leu, Val and Phe. Accordingly, preferred
Aø
aggregation core domains are composed of at least three amino acid structures
(as that term is
defined hereinbefore, including amino acid derivatives, analogues and
mimeties), wherein at
least two of the amino acid structures are, independently, either a leucine
structure, a valise
structure or a phenylalanine structure (as those terms are defined
hereinbefore, including
derivatives, analogues and mimetics).
Thus, in another embodiment, the invention provides a ø-amyloid modulator
compound comprising a formula:
An
( Y-Xaa ~ -Xaa~-Xaa3-Z
wherein Xaal, Xaa,and Xaa3 are each amino acid structures and at least two of
Xaat,
Xaa., and Xaa3 are. independently, selected from the group consisting of a
leucine structure, a
phenylalanine structure and a valise structure;
ZO Y, which may or may not be present, is a peptidic structure having the
formula (Xaa)a, wherein Xaa is any amino acid structure and a is an integer
from 1 to 15;
Z, which may or may not be present. is a peptidic structure having the
formula (Xaa)b. wherein Xaa is any amino acid structure and b is an integer
from 1 to 1 ~; and
A is a modifying group attached directly or indirectly to the compound and n
is an integer;
Xaal, Xaa~, Xaa3, Y, Z, A and n being selected such that the compound
modulates
the aggregation or inhibits the neurotoxicity of natural ø-amyloid peptides
when contacted
with the natural ø-amyloid peptides.
Preferably, the compound inhibits aggregation of natural ø-arnyloid peptides
when
contacud with the natural ø-amyloid peptides. In preferred embodiments, Xaa~
and Xaa2 are
each phenylalanine structures or Xaa2 and Xaag are each phenylalanine
structures. "n" can
be, for example, an integer between 1 and 5, whereas "a" and "b" can be, for
example,
integers between 1 and 5. The modifying group "A" preferably comprises a
cyclic,
heterocyclic or polycyclic group. More preferably, A contains a cis-decalin
group, such as
cholanoyl structure or a cholyl group In other embodiments, A can comprise a
biotin-
_.
containing group, a diethylene-triaminepentaacetyl group, a (-)-menthoxyacetyl
group, a
fluorescein-containing group or an N-acetylneun3minyl group. In yet other
embodiments. the
compound may promotes aggregation of natural ø-amyloid peptides when contacted
with the
CA 02449296 2003-12-15
24
nataaal ø-amyloid peptides, may be fuztlter modified to alter a
pharmacolcinctic property of
the compound or may be further modified to label the compound with a
detectable substance.
In another embodiment, the invention provides a ø-amyloid modulator compound
comprising a formula:
A-(~-~ 1-~2-~3-~Z~'B
wherein Xaa~, Xaa~and Xaa3 are each amino acid structures and at least two
of Xaal, Xaa~ and Xaa3 are, independently, selected from the group consisting
of a leucine
structure, a phenylalanine structure and a valine structure;
Y, which may or may not be present, is a peptidic structure having the
formula (Xaa)a, wherein Xaa is any amino acid structure and a is an integer
from Z to 15;
Z, which may or may not be present, is a peptidic structure having the
formula (Xaa)b, wherein Xaa is any amino acid structure and b is an integer
from 1 to 1 ~: and
Z S A and B, at least one of which is present are modifying groups attached
directly or indirectly to the amino terminus and carboxy terminus.
respectively. of the
compound;
Xaa~, Xaa,, Xaa3, Y, Z, A and B being selected such that the compound
modulates the aggregation or inhibits the neurotoxicity of natural ø-amyloid
peptides when
contacted with the natural ø-amyloid peptides:
Preferably, the compound inhibits aggregation of natural ø-amyloid peptides
when
contacted with the natural ø-amyloid peptides. In preferred embodiments, Xaa~
and Xaa-, are
each phenylalanine structures or Xaa~ and Xaa3 are each phenylalanine
structures. In one
subembodiment, the compound comprises the formula:
2S A-(y)-~at W?-~3-(Z)
In another subembodiment, the compound comprises the formula:
(Y~-Xaai-Xaa~-Xaa3-(Z}-B
"n" can be, for example, an integer between 1 and 5, whereas "a" and "b" can
be. for example.
integers between 1 and S. The modifying group "A" preferably comprises a
cyclic,
heterocyclic or polycyclic group. More preferably, A contains a cis-decalin
group. such as
cholanoyl structure or a cholyl group In other embodiments. A can comprise a
biatin-
containing group, a diethylene-triaminepentaacetyl group, a (-)-menthoxyacetyl
group, a
fluorescein-containing group or an N-acetylneurarninyl group. In yet other
embodiments, the
compound may promote aggregation of natural ø-amyloid peptides when contacted
with the
3S natural ø-amyloid peptides, may be further modified to alter a
pharmacokinetic property of
the compound or may be further modified to label the compound with a
detectable substance.
In preferred specific embodiments, the invention provides a ø-amyloid
modulator
compound comprising a modifying group attached directly or indirectly to a
peptidic
structure. wherein the peptidic structure comprises amino acid structures
having an amino
CA 02449296 2003-12-15
acid sequence selected From the group consisting of His-Gln-Lys-Leu-Val-
Phe~Phe-Ala (SEQ
ID NO: 5), His-Gln-Lys-Leu-Val-Phe-Phe (SEQ ID NO: 6), GIn-Lys-Leu-Val-Phe-Phe-
Ala
(SEQ ID NO: 7), Gln-Lys-Leu-Val-Phe-Phe (SEQ ID NO: 8), Lys-Leu-VaI-Phe-Phe-
Ala
(SEQ ID N0: 9), Lys-Leu-Val-Phe-Phe (SEQ ID ~NO: 10), Leu-Val-Phe-Phe-Ala (SEQ
ID
5 NO: I l), Leu-Val-Phe-Phe (SEQ ID NO: 12), Leu-AIa-Phe-Phe-Ala (SEQ ID NO:
13), Val-
Phe-Phe (SEQ ID NO: 19), Fhe-Phe-Ala (SEQ ID NO: 20), Phe-Phe-Val-Leu-AIa (SEQ
ID
NO: 21 ), Leu-Val-Phe-Phe-Lys (SEQ ID NO: 22), Leu-Vat-Iodotyrosine-Phe-Ala
(SEQ ID
NO: 23), Val-Phe-Phe-Ala (SEQ ID NO: 24), Ala-Vad-Phe-Phe-Ala (SEQ ID NO: ZS),
Leu-
Val-Phe-Iodotyrosine-Ala (SEQ ID NO: 26), Leu-Val-Phe-Phe-Ala-Glu (SEQ ID NO:
27),
10 Phe-Phe-Val-Leu (SEQ ID NO: 28), Phe-Lys-Phe-VaI-Leu (SEQ ID NO: 29), Lys-
Leu-Val-
Ala-Phe (SEQ ID NO: 30), Lys-Leu-Val-Phe-Phe-~3Ala (SEQ ID NO: 31 ) and Leu-
Val-Phe-
Phe-DAIa (SEQ ID NO: 32).
These specific compounds can be further modified to alter a phannacokinetic
property
of the compound and/or further modified to label the compound with a
detectable substance.
15 The modulator compounds of the invention can be incorporated into
pharmaceutical
compositions (described further in subsection V below) and can be used in
detection and
treatment methods as described further in subsection VI below.
II. Modifvine Grg~, s
20 Within a modulator compound of the invention, a peptidic structure (such as
an A~i
derived peptide. or an A~i aggregation core domain. or an amino acid sequence
corresponding
to a rearranged A~i aggregation core domain) is coupled directly or indirectly
to at least one
modifying gmup (abbreviated as MG). In one embodiment. a modulator compounds
of the
invention comprising an aggregation core domain coupled to a modifying group.
the
25 compound can be illustrated schematically as MG-ACD. The term "modifying
group" is
intended to include structures that are directly attached to the peptidic
structure (e.g., by
coval~t coupling), as well as those that are indirectly attached to the
peptidic structure (e.g.,
by a stable non-covalent association or by covalent coupling to additional
amino acid
residues, or mimetics, analogues or derivatives thereof, which may flank the
A~i-derived
peptidic structure). For example, the modifying group can be coupled to the
amino-terminus
or carboxy-terminus of an A~i-derived peptidic structure, or to a peptidic or
peptidomimetic
region flanking the core domain. Alternatively, the modifying group can be
coupled to a sidc
chain of at least one amino acid residue of an A~i-derived peptidie structure,
or to a peptidic
or peptidomitnetic region flanking the core domain (e.g.. through the epsilon
amino group of
a lysyl residue(s). through the carboxyl group of an aspartic acid residues)
or a glutamic acid
residue(s), through a hydroxy group of a tyrosyl residue(s), a serine
residues) or a threonine
residues) or other suitable reactive group on an amino acid side chain).
Modifying groups
covalently coupled to the pcptidic structure can be attached by means and
using~methods well
CA 02449296 2003-12-15
26
known in the art for linking chemical structures, including, for example,
amide, alkylamino,
carbamate or urea bonds.
The term "modifying group" is intended to include groups that are not
naturally
coupled to natural Aø peptides in their native form. Accordingly, the term
"modifying
S group" is not intended to' include hydrogen. The modifying gmup(s) is
selected such that the
modulator compound alters, and preferably inhibits, aggregation of natural ø-
amyIoid
peptides when contacted with the natural ø-amyloid peptides or inhibits the
neurotoxicity of
natural ø-amyloid peptides when contacted with the natural ø-amyloid peptides.
Although
not intending to be limited by mechanism, the modifying groups) of the
modulator
compounds of the invention is thought to function as a key pharmacophore which
is
important for conferring on the modulator the ability to disrupt Aø
polymerization.
In a preferred embodiment. the modifying groups) comprises a cyclic,
heterocyclic or
palycyclic group. The term "cyclic group", as used herein, is intended to
include cyclic
saturated or unsaturated (i.e., aromatic) group having from about 3 to 10,
preferably about 4
to 8. and more preferably about 5 to 7. carbon atoms. Exemplary cyclic groups
include
cyclopropyl. cyclobutyl, cyclopentyl, cyclohexyl. and cyclooctyl. Cyclic
groups may be
unsubstituted or substituted at one or more ring positions. Thus. a cyclic
group may be
substituted with, e.g.. halogens, alkyls, cycloalkyls, alkenyls, alkynyls,
aryls, heterocycles,
hydroxyls. aminos, nitros, thiols amines, imines. amides, phosphonates.
phosphines,
carbonyls, carboxyls, silyls, ethers, thioethers, sulfonyls. sulfonates.
selenoethers, ketones,
aldehydes, esters, -CF3, -CN, or the Like.
The term "heterocyclic group" is intended to include cyclic saturated or
unsaturated
(i.e.. aromatic) group having from about 3 to 10, preferably about 4 to 8, and
more preferably
about ~ to 7. carbon atoms. wherein the ring structure includes about ane to
four heteroatoms.
Heterocyclic groups include pyrrolidine. oxolane. thiolane. imidazole.
oxazole, piperidine,
piperazine, morpholine. The heterocyclic ring can be substituted at one or
more positions
with such substituents as, for example, halogens, alkyls, cycloalkyls,
alkenyls, alkynyls,
aryls. other heterocycles, hydroxyl, amino. vitro, thiol, amines, imines,
amides,
phosphonates, phosphines, carbonyls, carboxyls, silyls, ethers, thioethers,
sulfonyls,
selcnoethers, ketones, aldehydes, esters. -CF3, -CN, or the Like. Heterocycles
may also be
bridged or fused to other cyclic groups as described below.
The term "polycyclic group" as used herein is intended to refer to two or more
saturated or unsaturated (i.e., aromatic) cyclic rings in which two or more
carbons are
common to two adjoining rings, e.g., the rings are "fused rings". Rings that
are joined
through non-adjacent atoms are termed "bridged" rings. Each of the rings of
the polycyclic
group can be substituted with such substituents as described above. as for
example, halogens,
alkyls, cycloalkyls, alkenyls, alkynyls, hydroxyl, amino. vitro, thiol.
amines, imines, amides,
phosphonates, phosphines. carbonyls, carboxyls, silyls, ethers, thioethers,
sulfonyls,
selenoethers, ketones. aldehydes, esters. -CF;, -CN. or the like.
CA 02449296 2003-12-15
27
A preferred polycyclic group is a group containing a cis-decalin stxuctvre.
Although
not intending to be limited by mechanism, it is thought that the "bent"
conformation
conferred on a modifying group by the presence of a cis-decalin structure
contributes to the
efficacy of the modifying gmup in disrupting A~ polymerization. Accordingly,
other
structures which mimic the "bent" configuration of the cis-decalin structure
can also be used
as modifying groups. An example of a cis-decalin containing structure that can
be used as a
modifying group is a cholanoyl structure, such as a cholyl group. For example,
a modulator
compound can be modified at its amino terminus with a cholyl group by reacting
the
aggregation core domain with cholic acid, a bile acid. as described in Example
4 (the
structure of cho1ie acid is illustrated in Figure 2). Moreover, a modulator
compound can be
modified at its carboxy terminus with a cholyl group according to methods
known in the art
(see e.g., Wess, G. et al. (1993) Tetrahedron Letters, X4_:817-832; Wess, G.
et aL (1992)
Tetrahedron Letters i~:195-198; and Kramer, W. et al. (1992) J. Biol. Chem.
2u7:I8598-
18604). Cholyl derivatives and analogues can also be used as modifying groups.
For
I S example, a preferred cholyl derivative is Aic (3-(O-aminoethyl-iso)-
cholyl). which has a free
amino group that can be used to further modify the modulator compound (e.g., a
chelation
group for 9~Tc can be introduced through the free amino gmup of Aic). As used
herein, the
term "cholanoyl structure" is intended to include the cholyl group and
derivatives and
analogues thereof in particular those which retain a four-ring cis-decalin
configuration.
Examples of cholanoyl structures include groups derived from other bile acids,
such as
deoxycholic acid, lithocholic acid, ursodeoxycholic acid, chenodeoxychoiic
acid and
hyodeoxycholic acid, as well as other related structures such as cholanic
acid. bufalin and
resibufogenin (although the latter two compounds are not preferred for use as
a modifying
group). Another example of a cis-decalin containing compound is 5~3-cholestan-
3a-of (the
cis-decalin isomer of (+~dihydrocholesterol). For further description of bile
acid and steroid
structure and nomenclature, see Nes, W.R. and McKean, M.L. Biochemisrry of
Steroids and
Other Isopentanoids, University Park Press, Baltimore, MD, Chapter 2.
In addition to cis-decalin containing groups, other polycyclic groups may be
used as
modifying groups. For example, modifying groups derived from steroids or (3-
lactams may
be suitable modifying groups. Moreover, non-limiting examples of some
additional cyclic,
heterocyclic or polycyclic compounds which can be used to modify an A~3-
derived peptidic
structure are shown schematically in Figure 2. In one embodiment, the
modifying group is a
"biotinyI structure", which includes biotinyl groups and analogues and
derivatives thereof
(such as a 2-iminobiotinyl group). In another embodiment, the modifying group
can
3S comprise a "fluorescein-containing group", such as a group derived from
reacting an A(3-
derived peptidic structure with 5-(and 6-)-carboxvfluorescein, succinimidyl
ester or
fluorescein isothiocyanate. In various other embodiments, the modifying
groups) can
comprise an N acetylneuraminyl group, a trans-4-cotininecarboxyl group, a 2-
imino-1-
imidazolidineacetyl group, an (S}-(-rindoline-2-carboxyl group, a (-
~menthoxyacetyl group,
CA 02449296 2003-12-15
a 2-norbonoaaeacetyl group, a Y-oxo-S-acen~~thenebutyryl, a (-)-2-oxo-4-
thiazolidinecarboxyl group, a teuahydro-3-furoyl group, a 2-iminobiotinyl
group, a
diethylenetriaminepentaacetyl group, a 4-morpholinecarbonyl group. a 2-
thiopheneacetyl
group or a 2-thiophenesulfonyl group.
Prefeaed modifying groups include groups comprising cholyl structures,
biotinyl
structures, fluorescein-containing groups, a diethylene-triaminepentaacetyl
group, a (-)-
menthoxyacetyl group, and a N-acetylneuraminyl group. More preferred modifying
groups
those comprising a cholyl structure or an iminiobiotinyl group.
In addition to the cyclic. heterocyclic and polycyelic groups discussed above,
other
IO types of modifying groups can be used in a modulator of the invention. For
example, small
hydrophobic groups may be suitable modifying groups. An example of a suitable
non-cyclic
modifying group is an acetyl group.
Yet another type of modifying group is a compound that contains a non-natural
amino
acid that acts as a beta-turn mimetic. such as a dibenzofuran-based amino acid
described in
Tsang. K.Y. et al. ( 1994) J. Am. Chem. Soc. I 16:3988-400; Diaz H and Kelly.
J. W. ( 1991
Tetrahedron Letters 41:5735-5728; and Diaz. H et aI. (1992) J. Am. Chem. Soc.
114:8316-
8318. An example of such a modifying group is a peptide-
aminoethyldibenzofiwanyl-
proprionic acid (Adp) group (e.g., DDIIL-Adp). This type of modifying gmup
further can
comprise one or more N-methyl peptide bonds to introduce additional steric
hindrance to the
aggregation of natural (3-AP when compounds of this type interact with natural
~i-AP.
III. Additional Chemical Modifications of A~i Modulators
A p-amyloid modulator compound of the invention can be further modified to
alter
the specific properties of the compound while retaining the ability of the
compound to alter
A~i aggregation and inhibit A(3 neurotoxicity. For example. in one embodiment,
the
compound is further modified to alter a pharmacokinetic property of the
compound, such as
in vivo stability or half life. In another embodiment. the compound is further
modified to
label the compound with a detectable substance. In yet another embodiment. the
compound
is fiurther modified to couple the compound to an additional therapeutic
moiety.
Schematically, a modulator of the invention comprising an A~i aggregation core
domain
coupled directly or indirectly to at least one modifying group can be
illustrated as MG-ACD,
whereas this compound which has been further modified to alter the properties
of the
modulator can be illustrated as MG-ACD-CM, wherein CM represents an additional
chemical
modification.
To further chemically modify the compound, such as to alter the
pharmacokinetic
properties of the compound, reactive groups can be derivatized. For example,
when the
modifying group is attached to the amino-terminal end of the aggregation core
domain, the
earboxy-terminal end of the compound can be further modified. Preferred C-
terminal
modifications include those which reduce the ability of the compound to act as
a substrate for
CA 02449296 2003-12-15
29
carboxypeptidases. Examples of preferred C-terminal modifiers include an amide
group, an
ethylamide group and various non-natural amino acids, such as D-amino acids
and (3-alanine.
Alternatively, when the modifying group is attached to the carboxy-teTm~inal
end of the
aggregation core domain, the amino-terminal end of the compound can be further
modified,
for example, to reduce the ability of the compound to act as a substrate for
aminopeptidases.
A modulator compound can be fitrther modified to label the compound by
reacting the
compound with a detectable substance. Suitable detectable substances include
various
enzymes, prosthetic groups. fluorescent materials, luminescent materials and
radioactive
materials. Examples of suitable enzymes include horseradish peroxidase,
alkaline
phosphatase, ~i-galactosidase, or acetylcholinesterase; examples of suitable
prosthetic group
complexes include streptavidin/biotin and avidin/biotin; examples of suitable
fluorescent
materials include umbelliferone, fluorescein. fluorescein isothiocyanate,
rhodamine,
dichlorotriazinylamine fluorescein, dansyl chloride or phycoerythrin; an
example of a
luminescent material includes luminol: and examples of suitable radioactive
material include
14C, 1231, 1241. 12~I? 1311, 99mT~~ 35S or'I~i. In a preferred embodiment, a
modulator
compound is radioactively labeled with 14C, either by incorporation of 14C
into the
modifying group or one or more amino acid structures in the modulator
compound. Labeled
modulator compounds can be used to assess the in vivo pharmacokinetics of the
compounds,
as well as to detect A~i aggregation. for example for diagnostic purposes. A~i
aggregation can
be detected using a labeled modulator compound either in vivo or in an in
vitro sample
derived from a subject.
Preferably, for use as an in vivo diagnostic agent. a modulator compound of
the
invention is labeled with radioactive technetium or iodine. Accordingly, in
one embodiment,
the invention provides a modulator compound labeled with technetium.
preferably ~Tc.
2~ Methods for labeling peptide compounds with technetium are known in the art
(see e.g., U.S.
Patent Nos. 5.443,815, 5_5,.180 and 5,405,597, all by Dean et al.: Stepniak-
Biniakiewicz
D., et al. (1992) J. Med Chem. x:274-279; Fritzberg, A.R., et al. (1988) Proc.
Natl. Acad
Sci. USA X5:4025-4029; Baidoo. K.E., et al. (1990) Cancer Res. Suppl. 50:799s-
803s; and
Regan; L, and Smith, C.K. (1995) Science 270:980-982). A modifying group can
be chosen
that provides a site at which a chelation group for ggmTc carr be introduced.
such as the Aic
derivative of cho1ie acid, which has a free amino group (see Example 11 ). In
another
embodiment, the invention provides a modulator compound labeled with
radioactive iodine.
For example. a phenylalanine residue within the A~i sequence (such as Phe 1 g
or Phe2p) can be
substituted with radioactive iodotyrosyl (see Example 11 ). Any of the various
isotopes of
radioactive iodine can be incorporated to create a diagnostic agent.
Preferably, 1231 (half life
= 13.2 hours) is used for whole body scintigraphy, 1241 (half life = 4 days)
is used for
positron emission tomography (PET), 1251 (half life = 60 days) is used for
metabolic turnover
studies and 1311 (half life = 8 days) is used for whole body counting and
delayed low
resolution imaging studies.
CA 02449296 2003-12-15
Fore. an additional modif canon of a modulator compound of the invention
can serve to confer an additional therapeutic property on the compound. That
is, the
additional chemical modification can comprise an additional functional moiety.
For example,
a functional moiety which serves to break down or dissolve amyloid plaques can
be coupled
5 to the modulator compound. In this form, the MG-ACD portion of the modulator
serves to
target the compound to Aø peptides and disrupt the polymerization of the Aø
peptides,
whereas the additional functional moiety serves to break down or dissolve
amyloid plaques
after tt~e compound has been targeted to these sites.
In an alternative chemical modification, a ø-amyloid compound of the invention
is
10 prepared in a "prodrug" form, wherein the compound itself does not modulate
Aø
aggregation, but rather is capable of being transformed, upon metabolism in
vivo, into a ø-
amyloid modulator compound as defined herein. For example, in this type of
compound, the
modulating group can be present in a prodrug form that is capable of being
converted upon
metabolism into the form of an active modulating group. Such a prodrug form of
a
15 modifying gmup is referred to herein as a "secondan~ modifying group." A
variety of
strategies are known in the art for preparing pepude prodrugs that limit
metabolism in order
to optimize delivery of the active form of the peptide-based drug (see e.g.,
Moss, J. (1995) in
Peptide-Based Drug Design: Controlling Transport and Metabolism. Taylor, M.D.
and
Amidon. G.L. (eds), Chapter 18. Additionally strategies have been specifically
tailored to
20 achieving CNS delivery based on "sequential metabolism" (see e.g., Bodor,
N., et al. ( 1992)
Science 27:1698-1700; Prokai, L., et al. (1994) J. Am. Chem. Soc. 11~f:2643-
2644: Bodor,
N. and Prokai. L. (1995) in P~tide-Based Drug Des~Qn: Controlling Transport
and
M oli . Taylor, M.D. and Amidon. G.L. (eds). Chapter 14. In one embodiment of
a
prodrug form of a modulator of the invention. the modifying group comprises
~an alkyl ester
25 to facilitate blood-brain barrier permeability.
Modulator compounds of the invention can be prepared by standard techniques
known in the art. The peptide component of a modulator composed, at least 'in
part. of a
peptide, can be synthesized using standard techniques such as those described
in Bodansky,
30 M. Principles ofPeptide Synthesis,, Springer Verlag, Berlin (1993) and
Grant. G.A (ed.).
Synthetic Pegtides: A User's Guide, W.H. Freeman and Company, New York (199?).
Automated peptide synthesizers are commercially available (e.g., Advanced
ChemTech
Model 396: Milligen/ Biosearch 9600). Additionally, one or more modulating
groups can be
attached to the Aø-derived peptidic component (e.g.. an Aø aggregation core
domain) by
standard methods. for example using methods for reaction through an amino
group (e.g., the
alpha-amino group at the amino-terminus of a peptide), a carboxyl group (e.g.,
at the carboxy
terminus of a peptide), a hydroxyl group (e.g., on a tyrosine, serine or
threonine residue) or
other suitable reactive group on an amino acid side chain (see e.g., Greene,
T.W and Wuts,.
P.G.M. Protective Groups in Organic S ,vnthesis. John Wiley and Sons, Inc.,
New York
CA 02449296 2003-12-15
31
( 1991 ). Exemplary syntheses of preferred p amyloid modulators is described
further in
Examples 1, 4 and 11.
IV. Screen,~nQ As~avs
Another aspect of the invention pertains to a method for selecting a modulator
of ~i-
amyloid aggregation. In the method, a test compound is contacted with natural
~ amyloid
peptides, the aggregation of the natural (3-AP is measured and a modulator is
selected based
on the ability of thesest compound to alter the aggregation of the natural p-
AP (e.g., inhibit
or promote aggregation). In a preferred embodiment, the test compound is
contacted with a
molar excess amount of the natural ~-AP. The amount and/or rate of natural ~i-
AP
aggregation in the presence of the test compound can be determined by a
suitable assay
indicative of ~i-AP aggregation. as described herein (see e.g., Examples 2, ~
and 6).
In a preferred assay, the natural (3-AP is dissolved in solution in the
presence of the
test compound and aggregation of the natural ~i-AP is assessed in a nucleation
assay (see
1 ~ Example 6) by assessing the turbidity of the solution over time. as
measured by the apparent
absorbance of the solution at 40~ nm (described further in Example 6; see also
Jarrett et al.
(1993) Biochemistry 32:4693-4697). In the absence of a ~i-amyloid modulator,
the A4og~, of
the solution typically stays relatively constant during a lag time in which
the (3-AP remains in
solution, but then the A4o5nm of the solution rapidly increases as the ~i-AP
aggregates and
comes out of solution, ultimately reaching a plateau level (r.e., the A4o;~ of
the solution
exhibits sigmoidal kinetics over time). In contrast. in the presence of a test
compound that
inhibits j3-AP aggregation. the A4o5nm of the solution is reduced compared to
when the
modulator is absent. Thus, in the presence of the inhibitory modulator. the
solution may
exhibit an increased lag time. a decreased slope of aggregation and/or a lower
plateau level
2~ compared to when the modulator is absent. This method for selecting a
modulator of ~i-
amyloid polymerization can similarly be used to select modulators that promote
(3-AP
aggregation. Thus, in the presence of a modulator that promotes ~i-AP
aggregation, the
A405nm of the solution is increased compared to when the modulator is absent
(e.g., the
solution may exhibit an decreased lag time, increase slope of aggregation
and/or a higher
plateau level compared to when the modulator is absent).
Another assay suitable for use in the screening method of the invention, a
seeded
extension assay, is also described further in Example 6. In this assay, ~i-AP
monomer and an
aggregated j3- .AP "seed" are combined, in the presence and absence of a test
compound. and
the amount of. (i-fibril formation is assayed based on enhanced emission of
the dye
Thioflavine T when contacted with ~i-AP fibrils. Moreover, (3-AP aggregation
can be .
assessed by electron microscopy (EIvi] of the ~i-AP Preparation in the
presence or absence of
the modulator. For example, ~i amyloid fibril formation, which is detectable
by EM, is
reduced in the presence of a modulator that inhibits (i-AP aggregation (i.e.,
there is a reduced
amount or number of ~i-fibrils in the presence of the modulator), whereas ~
fibril formation is
CA 02449296 2003-12-15
32
increased in-the presence of a modulator that promotes ø-AP aggregation (i.e.,
there is as
increased amount or number of ø-fibrils in the presence of the modulator).
An even more prefentd assay for use in the screening method of the invention
to
select suitable modulators is the neurotoxicity assay described in Examples 3
and 10.
Compounds are selected which inhibit the formation of neurotoxic Aø aggregates
and/or
which inhibit the neurotoxicity of preformed Aø fibrils. This neumtoxicity
assay is
considered to be predictive of neurotoxicity in vivo. Accordingly, inhibitory
activity of a
modulator compound in the in vitro neurotoxicity assay is predictive of
similar inhibitory
activity of the compound for neurotoxicity in vivo.
V. ~hg~gubcal Compositions
Another aspect of the invention pertains to pharmaceutical compositions of the
ø-
amyloid modulator compounds of the invention. In one embodiment. the
composition
includes a ø amyloid modulator compound in a therapeutically or
prophylactically effective
amount suff cient to alter. and preferably inhibit, aggregation of natural ø-
amyioid peptides,
and a pharmaceutically acceptable carrier. In another embodiment, the
composition includes
a ø amyloid modulator compound in a therapeutically or prophylactically
effective amount
sufficient to inhibit the neurotoxicity of natural ø-amyloid peptides. and a
pharmaceutically
acceptable carrier. A "therapeutically effective amount" refers to an amount
effective, at
dosages and for periods of time necessary. to achieve the desired therapeutic
result, such as
reduction or reversal or ø-amyloid deposition and/or reduction or reversal of
Aø
neurotoxicity. A therapeutically effective amount of modulator may vary
according to factors
such as the disease state, age, seat. and weight of the individual. and the
ability of the
modulator to elicit a desired response in the individual. Dosage regimens may
be adjusted to
provide the optimum therapeutic response. A therapeutically effective amount
is also one in
which any toxic or deuimental effecu of the modulator are outweighed by the
therapeuticahy
beneficial effects. The potential neurotoxicity of the modulators of the
invention can be
assayed using the cell-based assay described in Examples 3 and 10 and a
therapeutically
effective modulator can be selected which daes not exhibit significant
neurotoxicity. In a
prefetied embodiment, a therapeutically effective amount of a modulator is
sufficient to alter,
and preferably inhibit. aggregation of a molar excess amount of natural ø-
amyloid peptides.
A "prophylactically effective amount" refers to an amount effective, at
dosages and for
periods of time necessary, to achieve the desired prophylactic result, such as
preventing or
inhibiting the rate of ø~amyloid deposition and/or Aø neeuotoxicity in a
subject predisposed
to ø-amyloid deposition. A prophylactically effective amount can be determined
as described
above for the therapeutically effective amount. Typically, since a
prophylactic dose is used
in subjects prior to or at an earlier stage of disease, the prophylactically
effective amount will
be less than the therapeutically effective amount.
CA 02449296 2003-12-15
33
Onrfactor that may be considered when determining a therapeutically or
prophylactically effective amount of a p amyloid modulator is the
concentration of natural ~i-
AP in a biological compartment of a subject, such as in the cerebrospinal
fluid (CSF) of the
subject. The concentration of natural /3-AP in the CSF has been estimated at 3
nM
(Schwartzman, (1994) Proc. Natl. Acaci: Sci. USA X1_:8368-8372). A non-
limiting range for a
therapeutically or prophylactically effective amounts of a ~i amyloid
modulator is 0.01 nM-10
pM. It is to be noted that dosage values may vary with the severity of the
condition to be
alleviated. It is to be further understood that for any particular subject,
specific dosage
regimens should be adjusted over time according to the individual need and the
professional
judgment of the person administering or supervising the administration of the
compositions,
and that dosage ranges set forth herein are exemplary only and are not
intended to limit the
scope or practice of the claimed composition.
The amount of active compound in the composition may vary according to factors
such as the disease state, age. sex. and weight of the individual, each of
which may affect the
amount of natural (3-AP in the individual. Dosage regimens may be adjusted to
provide the
optimum therapeutic response. For example. a single bolus may be administered.
several
divided doses may be administered over time or the dose may be proportionally
reduced or
increased as indicated by the exigencies of the therapeutic situation. It is
especially
advantageous to formulate parenteral compositions in dosage unit form for ease
of
administration and uniformity of dosage. Dosage unit form as used herein
refers to
physically discrete units suited as unitary dosages for the mammalian subjects
to be treated;
each unit containing a predetermined quantity of active compound calculated to
produce the
desired therapeutic effect in association with the required pharmaceutical
carrier. The
specification for the dosage unit forms of the invention are dictated by and
directly dependent
on (a) the unique characteristics of the active compound and the particular
therapeutic effect
to be achieved. and (b) the limitations inherent in the art of compounding
such an active
compound for the treatment of sensitivity in individuals.
As used herein "pharmaceutically acceptable carrier" includes any and all
solvents.
dispersion media, coatings, antibacterial and antifungal agents, isotonic and
absorption
delaying agents, and the like that are physiologically compatible. In one
embodiment. the
carrier is suitable for parenteral administration. Preferably, the carrier is
suitable for
administration into the central nervous system (e.g., intraspinally or
intracerebrally).
Alternatively. the carrier can be suitable for intravenous, intraperitoneal or
intramuscular
administration. In another embodiment, the carrier is suitable for oral
administration.
Pharmaceutically acceptable carriers include sterile aqueous solutions or
dispersions and
sterile powders for the extemporaneous preparation of sterile injectable
solutions or
dispersion. The use of such media and agents for pharmaceutically active
substances is well
known in the art. Except insofar as any conventional media or agent is
incompatible with the
active compound. use thereof in the pharmaceutical compositions of the
invention is
CA 02449296 2003-12-15
contemplated. Supplementary active compounds can also be incorporated into the
compositions.
Therapeutic compositions typically must be sterile and stable under the
conditions of
manufacture and storage. The composition can be formulated as a solution,
microemulsion,
S liposome, or other ordered structure suitable to high drug concentration.
The carrier can be a
solvent or dispersion medium containing, for example, water, ethanol, polyol
(for example,
glycerol, propylene glycol, and liquid polyetheylene glycol, and the like),
and suitable mixtures
thereof., The proper fluidity can be maintained, for example, by the use of a
coating such as
lecithin, by the maintenance of the required particle size in the case of
dispersion and by the
use of surfactants. In many cases, it will be preferable to include isotonic
agents. for example,
sugars, polyalcohols such as manitol, sorbitol, or sodium chloride in the
composition.
Prolonged absorption of the injectable compositions can be brought about by
including in the
composition an agent which delays absorption. for example. monostearate salts
and gelatin.
Moreover. the modulators can be administered in a time release formulation,
for example in a
composition which includes a slow release polymer. The active compounds can be
prepared
with carriers that will protect the compound against rapid release. such as a
controlled release
formulation, including implants and microencapsulated delivery systems.
Biodegradable,
biocompatible polymers can be used. such as ethylene vinyl acetate,
polyanhydrides,
polyglycolic acid. collagen, polyorthoesters, polylactic acid and polylactic,
polyglycolic
copolymers (PLG). Many methods for the preparation of such formulations are
patented or
generally known to those skilled in the art.
Sterile injectable solutions can be prepared by incorporating the active
compound
(e.g., ~3-amyloid modulator) in the required amount in an appropriate solvent
with one or a
combination of ingredients enumerated above. as required. followed by filtered
sterilization.
2~ Generally. dispersions are prepared by incorporating the active compound
into a sterile
vehicle which contains a basic dispersion medium and the required other
ingredients from
those enumerated above. In the case of sterile powders for the preparation of
sterile
injectable solutions. the preferred methods of preparation are vacuum drying
and freeze-
drying which yields a powder of the active ingredient plus any additional
desired ingredient
from a previously sterile-filtered solution thereof.
A modulator compound of the invention can be formulated with one or more
additional compounds that enhance the solubility of the modulator compound.
Preferred
compounds to be added to formulations to enhance the solubility of the
modulators are
cyclodextrin derivatives, preferably hydroxypropyl-y-cyclodextrin. Drug
delivery vehicles
containing a cyclodextrin derivative for delivery of peptides to the central
nervous system are
described in Bodor, N., et al. (1992) Science 257:1698-1700. For the ~3-
amyloid modulators
described herein. inclusion in the formulation of hydroxypropyl-Y-cyclodextrin
at a
concentration 50-200 mM increases the aqueous solubility of the compounds. In
addition to
increased solubility, inclusion of a cyclodextrin derivative in the
formulation may have other
CA 02449296 2003-12-15
beneficial effects, since p-cyclodextrin itself has been reported to interact
with the Ap peptide
and inhibit fibril formation in vitro (Camilleri, P., et al. (1994) FEES
Letters 341:256-2~$.
Accordingly, use of a modulator compound of the invention in combination with
a
cyclodextrin derivative may result in greater inhibition of A~i aggregation
than use of the
5 modulator alone. Chemical modifications of cyclodextrins are known in the
art (Hanessian,
S., et al. (1995) J. Org. Chem. C0_:4786-4797). In addition to use as an
additive in a
pharmaceutical composition containing a modulator of the invention,
cyclodextrin derivatives
may also be useful.as modifying groups and, accordingly, may also be
covalently coupled to
an A(3 peptide compound to form a modulator compound of the invention.
18 In another embodiment. a pharmaceutical composition comprising a modulator
of the
invention is formulated such that the modulator is transported across the
blood-brain barrier
(BBB). Various strategies known in the art for increasing transport across the
BBB can be
adapted to the modulators of the invention to thereby enhance transport of the
riodulators
across the BBB (for reviews of such strategies. see e.g., Pardridge. W.M.
(1994) Trends in
15 Biotechnol. 1?:239-24~: Van Hree, J.B. et al. (1993) Pharm. World Sci. 1~:2-
9: and
Pardridge, W.M. et al. ( 1992) Pharmacol. Toxicol. 7 ~:3-10). In one approach,
the modulator
is chemically modified to form a prodrug with enhanced transmembrane
transport. Suitable
chemical modifications include covalent linking of a fatty acid to the
modulator through an
amide or ester linkage (see e.g., U.S. Patent 4,933,324 and PCT Publication WO
89/07938,
20 both by Shashoua; U.S. Patent 5,284,876 by Hesse et al.; Toth, I. et al.
(1994) J. Drug
Target. 2:217-239; and Shashoua, V.E. et al. (1984) J. Med Chem. 27:659-664)
and
glycating the modulator (see e.g., U.S. Patent 5.260,308 by Poduslo et al.).
Also, N-
acylamino acid derivatives may be used in a modulator to form a "lipidic"
prodrug (see e.g.,
U.S. Patent No. 5,112,863 by Hashimoto et al. issued on May 12'", 1992).
25 In anqther approach for enhancing transport across the BBB, a peptidic or
peptidomimetic modulator is conjugated to a second peptide or protein, thereby
forming a
chimeric protein, wherein the second peptide or protein undergoes absorptive-
mediated or
receptor-mediated transcytosis through the BBB. Accordingly, by coupling the
modulator to
this second peptide or protein, the chimeric protein is uamsported across the
BBB. The
30 second peptide or protein can be a ligand for a brain capillary endothelial
cell receptor ligand.
For example, a preferred ligand is a monoclonal antibody that specifically
binds to the
transferrin .receptor on brain capillary endothelial cells (see e.g., U.S.
Patents 5,182,107 and
5,154.924 and PCT Publications WO 93/10819 and WO 95/02421, all by Friden et
al.).
Other suitable peptides or proteins thai can mediate transport across the BBB
include histot~es
35 (see e.g., U.S. Patent 4,902,505 by Pardridge and Schimmel) and ligands
such as biotin,
folate, niacin, pantothenic acid, riboflavin, thiamin, pryridoxal and ascorbic
acid (see e.g.,
U.S. Patents 5,416,016 and 5,108,921, both by Heinstein). Additionally, the
glucose
transporter GLUT-1 has been reported to transport glycopeptides (L-serinyl-~i-
D-glucoside
analogues of [MetS]enkephalin) across the BBB (Poll, R et al. (1994) Proc.
Natl. Acad Sci.
CA 02449296 2003-12-15
36
USA Q1:7114-I778). Accordingly, a modulator compound can be coupled to such a
glycopeptide to target the modulator to the GLUT-1 glucose transporter. For
example, a
modulator compound which is modified at its amino terminus with the modifying
group Aic
(3-(O-aminoethyl-iso~cholyl, a derivative of cholic acid having a free amino
group) can be
coupled to a glycopeptide through the amino group of Aic by standard methods.
Chimeric
proteins can be formed by recombinant DNA methods (e.g., by formation of a
chimeric gene
encoding a fusion protein) or by chemical crosslinking of the modulator to the
second peptide
or protein to form a chimeric protein. Numerous chemical crosslinking agents
are known in
the (e.g., commercially available from Pierce, Rockford IL). A crosslinking
agent can be
chosen which allows for high yield coupling of the modulator to the second
peptide or protein
and for subsequent cleavage of the linker to release bioactive modulator. For
example, a
biotin-avidin-based linker system may be used.
In yet another approach for enhancing transport across the BBB, the modulator
is
encapsulated in a carrier vector which mediates transport across the BBB. For
example. the
modulator can be encapsulated in a liposome. such as a positively charged
unilamellar
liposome (see e.g., PCT Publications WO 88/07851 and WO 88/07852. both by
Faden) or in
polymeric microspheres (see e.g., U.S. Patent 5,413,797 by Khan et al.. U.S.
Patent
5 71,961 by Mathiowitz et~al. and 5,019,400 by Gombotz et al.). Moreover. the
carrier
vector can be modified to target it for transport across the BBB. For example,
the carrier
vector (e.g., liposome) can be covalently modified with a molecule which is
actively
transported across the BBB or with a ligand for brain endothelial cell
receptors. such as a
monoclonal antibody that specifically binds to transferrin receptors (see
e.g., PCT
Publications WO 91/04014 by Collies et aL and WO 94/02178 by GreiQ et al.).
In still another approach to enhancing transport of the modulator across the
BBB. the
modulator is coadministered with another agent which functions to permeabilize
the BBB.
Examples of such BBB "permeabilizers" include bradykinin and bradykinin
agonists (see
e.g., U.S. Patent 5,112.596 by Malfroy-Canine) and peptidic compounds
disclosed in U.S.
Patent 5,268,164 by Kozarich et al.
A modulator compound of the invention can be formulated into a pharmaceutical
composition wherein the modulator is the only active compound or.
alternatively, the
pharmaceutical composition can contain additional active compounds. For
example, two or
more modulator compounds nay be used in combination. Moreover. a modulator
compound
of the invention can be combined with one or more other agents that have anti-
amyloidogenic
properties. For example. a modulator compound can be combined with the non-
specific
cholinesterase inhibitor tacrine (Cognex~, Parke-Davis).
In another embodiment. a pharmaceutical composition of the invention is
provided as
a packaged formulation. The packaged formulation may include a pharmaceutical
composition of the invention in a container and printed instructions for
administration of the
CA 02449296 2003-12-15
37
composition-for treating a subject having a disorder associated with (3-
amyloidosis, e.g.
Alzheimer's disease.
VI. Methods of Using Aa Modulators
Another aspect of the invention pertains to methods for altering the
aggregation or
inhibiting the neurotoxicity of natural (3-amyloid peptides. In the methods of
the invention,
natural p amyloid peptides are contacted with a ~i amyloid modulator such that
the
aggregation of the aatinal (l amyloid peptides is altered or the neurotoxicity
of the natural ~
amyloid peptides is inhibited. In a preferred embodiment, the modulator
inhibits aggregation
of the natural /3 amyloid peptides. In another embodiment, the modulator
promotes
aggregation of the natural ~i amyloid peptides. Preferably, aggregation of a
molar excess
amount of p-AP. relative to the amount of modulator, is altered upon contact
with the
modulator.
In the method of the invention. natural ~i amyloid peptides can be contacted
with a
1 S modulator either in vitro or in vivo. Thus, the term "contacted with" is
intended to encompass
both incubation of a modulator with a natural ~i-AP preparation in vitro and
delivery of the
modulator to a site in vivo where natural ~i-AP is present. Since the
modulator compound
interacts with natural (3-AP, the modulator compounds can be used to detect
natural (3-AP,
either in vitro or in vivo. Accordingly, one use of the modulator compounds of
the invention
is as diagnostic agents to detect the presence of natural ~3-AP, either in a
biological sample or
in vivo in a subject. Furthermore, detection of natural (3-AP utilizing a
modulator compound
of the invention further can be used to diagnose amyloidosis in a subject.
Additionally, since
the modulator compounds of the invention disrupt (3-AP aggregation and inhibit
(3-AP
neurotoxicity. the modulator compounds also are useful in the treatment of
disorders
2~ associated with (3-amyloidosis, either prophylactically or therapeutically.
Accordingly.
another use of the modulator compounds of the invention is as therapeutic
agents to alter
aggregation and/or neurotoxicity of natural (i-AP.
In one embodiment. a modulator compound of the invention is used in virro, for
example to detect and quantitate natural ~i-AP in sample (e.g., a sample of
biological fluid).
To aid in detection. the modulator compound can be modified with a detectable
substance.
The source of natural ~i-AP used in the method can be, for example, a sample
of
cerebrospinal fluid (e.g., from an AD patient, an adult susceptible to AD due
to family
history, or a normal adult). The natural ~i-AP sample is contacted with a
modulator of the
invention and aggregation of the (3-AP is measured, such as by as assay
described in
Examples 2, ~ and 6. Preferably, the nucleation assay and/or seeded extension
assay
described in Example 6 is used. The degree of aggregation of the ~i-AP sample
can then be
compared to that of a control samples) of a known concentration of (3-AP,
similarly
contacted with the modulator and the results can be used as an indication of
whether a subject
is susceptible to or has a disorder associated with p-amyloidosis. Moreover, ~-
AP can be
CA 02449296 2003-12-15
detected by detecting a modulating gmup 'incorporated into the modulator. For
example,
modulators incorporating a biotin compound as described herein (e.g., an amino-
terminally
biotinylated ~i-AP peptide) can be detected using a streptavidin or avidin
probe which is
labeled with a detectable substance (e.g., an enzyme, such as peroxidase).
Detection of
natural ~i-AP aggregates mixed with a modulator of the invention using a probe
that binds to
the modulating group (e.g., biotinlstreptavidin) is described further in
Example 2.
In another embodiment, a modulator compound of the invention is used in vivo
to
detect, and, if desired, quantitate, natural ~i-AP deposition in a subject,
for example to aid in
the diagnosis of p amyloidosis in the subject. To aid in detection, the
modulator compound
can be modified with a detectable substance, preferably '~"Tc or radioactive
iodine
(described further above), which can be detected in vivo in a subject. The
labeled ~i-amyloid
modulator compound is administered to the subject and, after sufficient time
to allow
accumulation of the modulator at sites of amyloid deposition, the labeled
modulator
compound is detected by standard imaging techniques. The radioactive signal
generated by
the labeled compound can be directly detected (e.g.. whole body counting). or
alternatively,
the radioactive signal can be converted into an image on an autoradiograph or
on a computer
screen to allow for imaging of amyloid deposits in the subject. Methods for
imaging
amyloidosis using radiolabeled proteins are known in the art. For example,
serum amyloid P
component (SAP), radioIabeled with either 1~I or ~Te, has been used to image
systemic
amyloidosis (see e.g., Hawkins, P.N. and Pepys, M.B. (1995) Eur. J. Nucl. Med.
:595-599).
Of the various isotypes of radioactive iodine, preferably 1'-'I (half life =
13.2 hours) is used
for whole body scintigraphy, 1241 (half life = 4 days) is used for positron
emission
tomography (PET). 1'-SI (half life = 60 days) is used for metabolic turnover
studies and I3~I
(half life = 8 days) is used for whole body counting and delayed low
resolution imaging
studies. Analogous to studies using radiolabeled SAP, a labeled modulator
compound of the
invention can be delivered to a subject by an appropriate route (e.g.,
intravenously,
intraspinally, intracerebrally) in a single bolus, for example containing 100
~.g of labeled
compound carrying approximately 180 MBq of radioactivity.
The invention provides a method for detecting the presence or absence of
natural ~i-
arnyloid peptides in a biological sample, comprising contacting a biological
sample with a
compound of the invention and detecting the compound bound to natural /3-
amyloid peptides
to thereby detect the presence or absence of natural ~i-amyloid peptides in
the biological
sample. In one embodiment, the ~i-amyloid modulator compound and the
biological sample
are contacted in vitro. In another embodiment, the ~i-amyloid modulator
compound is
contacted with the biological sample by administering the ~-amyloid modulator
compound to
a subject. For in vivo administration, preferably the compound is labeled with
radioactive
technetium or radioactive iodine.
The invention also provides a method for detecting natural (3-amyloid peptides
to
facilitate diagnosis of a ~i-amyloidogenic disease. comprising contacting a
biological sample
CA 02449296 2003-12-15
with the compound of the invention and det3a~nng the compound bound to natural
~i-amyloid
peptides to facilitate diagnosis of a ~-amyloidogenic disease. In one
embodiment, the ~i-
amyloid modulator compound and the biological sample are contacted in vitro.
In another
embodiment, the p-amyloid modulator compound is contacted with the biological
sample by
administering the (3-amyloid modulator compound to a subject. For in vivo
administration,
preferably the compound is labeled with radioactive technetium or radioactive
iodine.
Preferably, use of the method facilitates diagnosis of Alzheimer's disease.
In another embodiment, the invention provides a method for altering natural ~i-
AP
aggregation or inhibiting ~i-AP neurotoxiciry, which can be used
prophylactically or
therapeutically in the treatment or prevention of disorders associated with (3
amyloidosis, e.g.,
Alzheimer's Disease. As demonstrated in Example 10, modulator compounds of the
invention reduce the toxicity of natural ~3-AP aggregates to cultured neuronal
cells.
Moreover. the modulators not only reduce the formation of neurotoxic
aggregates but also
have the ability to reduce the neurotoxiciry of preformed A~i fibrils.
Accordingly, the
modulator compounds of the invention can be used to inhibit or prevent the
formation of
neurotoxic A(3 fibrils in subjects (e.g., prophylactically in a subject
predisposed to ~i-amyloid
deposition) and can be used to reverse (3-amyloidosis therapeutically in
subjects already
exhibiting ~i-amyloid deposition.
A modulator of the invention is contacted with natural ~i amyloid peptides
present in a
subject (e.g., in the cerebrospinal fluid or cerebrum of the subject) to
thereby alter the
aggregation of the natural (3-AP and/or inhibit the neurotoxicity of the
natural (3-APs. A
modulator compound alone can be administered to the subject. or alternatively,
the modulator
compound can be administered in combination with other therapeutically active
agents (e.g.,
as discussed above in subsection IV). When combination therapy is employed,
the
therapeutic agents can be coadministered in a single pharmaceutical
composition,
coadministered in separate pharmaceutical compositions or administered
sequentially.
The modulator may be administered to a subject by any suitable route effective
for
inhibiting natural ~i-AP aggregation in the subject, although in a
particularly preferred
embodiment, the modulator is administered parenterally, most preferably to the
central
nervous system of the subject. Possible routes of CNS administration include
intraspinal
administration and intracerebral administration (e.g., intracerebrovascular
administration).
Alternatively, the compound can be administered, for example, orally,
intraperitoneally,
intravenously or intramuscularly. For non-CNS administration routes, the
compound can be
administered in a formulation which allows for transport across the BBB.
Certain modulators
may be transported across the BBB without any additional further modification
whereas
others may need further modification as described above in subsection IV.
Suitable modes and devices for delivery of therapeutic compounds to the CNS of
a
subject are known in the art, including cerebrovascular reservoirs (e.g.,
Ommaya or Rikker
reservoirs; see e.g., Raney, J.P. et al. (1988) J. Neurosci. Nurs. ?Q:23-29;
Sundaresan, N. et
CA 02449296 2003-12-15
al. (1989) Oncology x:15-22), catbeters for inzrathxal delivery (e.g., Port-a-
Cath, Y-cathet«a
and the like; see e.g., Plummer, J.L. (1991) Pain 44:215-220; Yaksh, T.L. et
al. (1986)
Pharmacol. Biochem. Behav. ,x:483-485), injectable intrathecal reservoirs
(e.g., Spinalgesic;
see e.g., Brazenor, G.A. (1987) Neurosurgery x:484-491 ), implantable infusion
pump
5 systems (e.g., Infusaid; see e.g:, Zierski, J. et al. ( 1988) Acta
Neurochem. Suppl. 43:94-99;
Kanoff, R.B. (1994) J. Am. Osteopath. Assoc. 94:487-493) and osmotic pumps
(sold by Alza
Corporation). A particularly preferred mode of administration is via an
implantable,
extemal~y programmable infusion pump. Suitable infusion pump systems and
reservoir .
systems are also described in U.S. Patent No. 5, 368,562 by Blomquist and U.S.
Patent No.
10 4,731.058 by Doan, developed by Pharmacia Deltec Inc.
The method of the invention for altering ~i-AP aggregation in vivo , and in
particular
for inhibiting (3-AP aggregation, can be used therapeutically in diseases
associated with
abnormal ~i amyloid aggregation and deposition to thereby slow the rate of ~i
amyloid
deposition and/or lessen the degree of (3 amyloid deposition. thereby
ameliorating the course
15 of the disease. In a preferred embodiment. the method is used to treat
Alzheimer's disease
(e.g., sporadic or familial AD, including both individuals exhibiting symptoms
of AD and
individuals susceptible to familial AD). The method can also be used
prophylactically or
therapeutically to treat other clinical occurrences of ~i amyloid deposition.
such as in Down's
syndrome individuals and in patients with hereditary cerebral hemorrhage with
amyloidosis-
20 Dutch-type (HCHWA-D). While inhibition of ~3-AP aggregation is a preferred
therapeutic
method. modulators that promote ~i-AP aggregation may also be useful
therapeutically by
allowing for the sequestration of (3-AP at sites that do not lead to
neurological impairment.
Additionally. abnormal accumulation of ~3-amyloid precursor protein in muscle
fibers
has been implicated in the pathology of sporadic inclusion body myositis (IBM)
(Askana. V.
25 et al. (1996) Proc. NatL Acad Sci. USA 93:1314-1319: Askanas. V. et al.
(1995) Current
Opinion in Rhewnatology 7:486-496). Accordingly, the modulators of the
invention can be
used prophylactically or therapeutically in the treatment of disorders in
which ~i-AP. or APP.
is abnormally deposited at non neurological locations, such as treatment of
IBM by delivery
of the modulators to muscle fibers.
VII. Unmodified A~3 Peptides that Iryibit Ag,Qregation of Natural Q-AP
In addition to. the ~i-amyloid modulators described hereinbefore in which an
A(3
peptide is coupled to a modifying group. the invention also provides ~i-
amyloid modulators
comprised of an unmodified A(3 peptide. It has now been discovered that
certain portions of
natural (3-AP can alter aggregation of natural j3-APs when contacted with the
natural ~i-APs
(see Example 12). Accordingly, these unmodified A~i peptides comprise a
portion of the
natural ~3-AP sequence (i.e., a portion of (3AP~_3g, ~3AP1-40~ ~W-4? ~d ~~'~-
43). In
particular these unmodified A(3 peptides have at Ieast one amino acid deletion
compared to
~~1-39~ ~e ~ortest natural (3-AP, such that the compound alters aggregation of
natural.(3-
CA 02449296 2003-12-15
amyloid peptides when contacted with the 4nat ral ~-amyloid peptides. In
various
embodiments, these unmodified peptide compounds can promote aggregation of
natural ~i-
amyloid peptides, or, more preferably, can inhibit aggregation of natural ~i-
amyl'oid peptides
when contacted with the natural (3-amyloid peptides. Even more preferably, the
unmodified
peptide compound inhibits aggregation of natural ~i-amyloid peptides when
contacted with a
molar excess amount of natural ~i-amyloid peptides (e.g., a 10-fold, 33-fold
or 100-fold molar
excess amount of natural ~i-AP).
As discussed above, the unmodified peptide compounds of the invention comprise
an
amino acid sequence having at least one amino acid deletion compared to the
amino acid
sequence of (3AP~.3g. Alternatively, the unmodified peptide compound can have
at least five,
ten, fifteen. twenty, twenty-five, thirty or thirty-five amino acids deleted
compared to
(~AP1-39~ StilI further the unmodified peptide compound can have 1-~. 1-10, 1-
15. I-20, I-
25, 1-30 or 1-3~ amino acids deleted compared to (3AP~_39~ The amino acid
deletions) may
occur at the amino-terminus. the carboxv-terminus. an internal site. or a
combination thereof.
of the (3-AP sequence. Accordingly, in one embodiment. an unmodified peptide
compound
of the invention comprises an amino acid sequence which has at least one
internal amino acid
deleted compared to ~3AP ~ _;g. Alternatively. the unmodified peptide compound
can have at
Least five. ten, fi$een, twenty, twenty-five, thirty or thirty-five internal
amino acids deleted
compared to (3AP~_~g. Still further the unmodified peptide compound can have 1-
5, I-10, 1-
IS, 1-20, 1-25. 1-30 or 1-35 internal amino acids deleted compared to
(3AP~_39~ For peptides
with internal deletions, preferably the peptide has an amino terminus
corresponding to amino
acid residue 1 of natural (3AP and a carboxy terminus corresponding to residue
40 of natural
~iAP and has one or more internal (3-AP amino acid residues deleted (i.e.. a
non-contiguous
A~ peptide).
In another embodiment. the unmodified peptide compound comprises an amino acid
sequence which has at least one N-terminal amino acid deleted compared to
(3AP~_39.
Alternatively, the unmodified peptide compound can have at least five, ten,
fifteen. twenty,
twenty-five. thirty or thirty-five N-terminal amino acids deleted compared to
~3AP~_39. Still
further the unmodified peptide compound can have 1-5, 1-10, 1-I~, 1-20. 1-25.
1-30 or 1-35
N-terminal amino acids deleted compared to ~AP~_39~
In yet another embodiment, the unmodified peptide compound comprises an amino
acid sequence which has at least one C-terminal amino acid deleted compared to
~iAP 1 _39.
Alternatively, the unmodified peptide compound can have at least five, ten,
fifteen. twenty,
twenty-five, thim or thirty-five C-terminal amino acids deleted compared to
~iAP~_;g. Still
further the unmodified peptide compound can have 1-S, 1-10,' I-15. 1-20, I-25,
1-30 or 1-35
C-terminal amino acids deleted compared to ~iAP~_39~
In addition to deletion of amino acids as compared to ~iAP 1 _39, the peptide
compound
can have additional non-~i-AP amino acid residues added to it, for example, at
the amino
terminus, the carboxy-terminus or at an internal site. In one embodiment, the
peptide
CA 02449296 2003-12-15
42
compound has at least one non-/3-amyloid peptide-derived amino acid at iu N-
terminus.
Alternatively, the compound can have, for example, 1-3, 1-5, 1-7, I-10, 1-IS
or I-20 non-~-
amyloid peptide-derived amino acid at its N-terminus. In another embodiment,
the peptide
compound has at least one non-~i-amyloid peptide-derived amino acid at iu C-
terminus.
Alternatively, the compound can have, for example, I-3, 1-5, 1-7, I-10, 1-16
or 1-20 non-U-
amyloid peptide-derived amino acid at its C-terminus.
In specific preferred embodiments,, an unmodified peptide compound of the
invention
comprises A(3~2p (the amino acid sequence of which is shown in SEQ ID NO: 4),
A~i ~ X30
(the amino acid sequence of which is shown in SEQ ID NO: 14), Aril-20, 26-40
(tee ~o
acid sequence of which is shown in SEQ ID NO: 15) or EE~HHHHQQ-~iAP~~o (the
amino acid sequence of which is shown in SEQ ID NO: 16). In the nomenclature
used
herein= ~iAP1-2o, 26-4o represents /3AP~-4o in which the internal amino acid
residues 21-25
have been deleted.
An unmodified peptide compound of the invention can be chemically synthesized
using standard techniques such as those described in Bodansky, M. Principles
of Peptide
Synthesis. Springer Verlag, Berlin (1993) and Grant. G.A (ed.). Synthetic
Peptides: A User's
Guide, W.H. Freeman and Company, New York (1992). Automated peptide
synthesizers are
commercially available (e.g., Advanced ChemTech Model 396; MiIligen/ Biosearch
9600).
Alternatively, unmodified peptide compounds can be prepared according to
standard
recombinant DNA techniques using a nucleic acid molecule encoding the peptide.
A
nucleotide sequence encoding the peptide can be determined using the genetic
code and an
oligonucleotide molecule having this nucleotide sequence can be synthesized by
standard
DNA synthesis methods (e.g., using an automated DNA synthesizer).
Alternatively. a DNA
molecule encoding an unmodified peptide compound can be derived from the
natural (3-
amyloid precursor protein gene or cDNA (e.g.. using the polvmerase chain
reaction and/or
restriction enzyme digestion) according to standard molecular biology
techniques.
Accordingly. the invention further provides an isolated nucleic acid molecule
comprising a nucleotide sequence encoding a /3-amyloid peptide compound, the
~i-amyloid
peptide compound comprising an amino acid sequence having at least one amino
acid
deletion compared to [3AP~-3g such that the p-amyloid peptide compound alters
aggregation
of natural ~i-amyloid peptides when contacted with the natural ~-amyloid
peptides. As used
herein. the term "nucleic acid molecule" is intended to include DNA molecules
and RNA
molecules and may be single-stranded or double-stranded, but preferably is
double-stranded
DNA. The isolated nucleic acid encodes a peptide wherein one or more amino
acids are
deleted from the N-terminus, C-terminus and/or an internal site of ~iAPI_39,
as discussed
above. In yet other embodiments, the isolated nucleic acid encodes a peptide
compound
having one or more amino acids deleted compared to ~iAP~-39 and further having
at least one
non-/3-AP derived amino acid residue added to it, for example, at the amino
terminus, the
carboxy-terminus or at an internal site. In specific preferred embodiments, an
isolated
CA 02449296 2003-12-15
nucleic acid molecule of the invention encodes ~AP6-2o, (3AP 16.3p, SAP ~-20,
26-~0 or
EEWHHHFiQQ-~3AP 16-ao~
To facilitate expression of a peptide compound in a host cell by standard
recombinant DNA techniques, the isolated nucleic acid encoding the peptide is
S incorporated into a recombinant expression vector. Accordingly, the
invention also
provides recombinant expression vectors comprising the nucleic acid molecules
of the
invention. As used herein, the term "vector" refers to a nucleic acid molecule
capable of
transporting anothernucleic acid to which it has been linked. One type of
vector is a
"plasmid", which refers to a circular double stranded DNA loop into which
additional
DNA segments may be Iigated. Another type of vector is a viral vector, wherein
'
additional DNA segments may be Iigated into the viral genome. Certain vectors
are
capable of autonomous replication in a host cell into which they are
introduced (e.g.,
bacterial vectors having a bacterial origin of replication and episomal
mammalian vectors).
Other vectors (e.g., non-episomal mammalian vectors) are inteerated into the
genome of a
host cell upon introduction into the host cell. and thereby are replicated
along with the host
genome. Moreover, certain vectors are capable of directing the expression of
genes to
which they are operatively linked. Such vectors are referred to herein as
"recombinant
expression vectors" or simply "expression vectors". In general. expression
vectors of
utility in recombinant DNA techniques are often in the form of plasmids. In
the present
specification, "plasmid" and "vector" may be used interchangeably as the
plasmid is the
most commonly used form of vector. However, the invention is intended to
include such
other forms of expression vectors. such as viral vectors, which serve
equivalent functions.
In the recombinant expression vectors of the invention. the nucleotide
sequence
encoding the peptide compound are operatively linked to one or more regulatory
sequences.
selected on the basis of the host cells to be used for expression. The term
"operably linked"
is intended to mean that the sequences encoding the peptide compound are
linked to the
regulatory sequences) in a manner that allows for expression of the peptide
compound. The
term "regulatory sequence" is intended to includes promoters, enhancers and
other expression
control elements (e.g., .polyadenylation signals}. Such regulatory sequences
are described, for
example, in Goeddel; Gene Expression Technology: Methods in Enzymology 185,
Academic
Press. San Diego, CA (1990). Regulatory sequences include those that direct
constitutive
expression of a nucleotide sequence in many types of host cell, those that
direct expression of
the nucleotide sequence only in certain host cells (e.g., tissue-specific
regulatory sequences)
and those that direct expression in a regulatable manner (e.g., only in the
presence of an
inducing agent). It will be appreciated by those skilled in the art that the
design of the
expression vector may depend on such factors as the choice of the host cell to
be transformed,
the level of expression of peptide compound desired, etc. The expression
vectors of the
invention can be introduced into host cells thereby to produce peptide
compounds encoded by
nucleic acids as described herein.
CA 02449296 2003-12-15
The recombinant expression vectors of the invention can be designed for
expression
of peptide compounds in prokaryotic or eukaryotic cells. For example, peptide
compounds
can be expressed in bacterial cells such as E. coli, insect cells (using
baculovirus expression
vectors) yeast cells or mammalian cells. Suitable host cells are discussed
further in Goeddel,
Gene Expression Technology: Methods in Enzymology~85, Academic Press, San
Diego, CA
( 1990). Alternatively, the recombinant expression vector may be transcribed
and translated
in vitro, for example using T7 promoter regulatory sequences and T7
polymerase. Examples
of vectors for expression in yeast S. cerivisae include pYepSecl (Baldari et
aL, (1987)
EMBOJ. 6:229-234), pMFa (Kurjan and Herskowitz, (1982) Cell 30:933-943),
pJRY88
(Schultz et al., (1987) Gene 54:113-123), and pYES2 (Invitrogen Corporation,
San Diego,
CA). Baculovirus vectors available for expression of proteins or peptides in
cultured insect
cells (e.g., Sf 9 cells) include the pAc series (Smith et al., (1983) Mol.
Cell. Biol. 3_:2156-
2165) and the pVL series (Lucklow. V.A., and Summers, M.D., (1989) Virology
170:31-39).
Examples of mammalian expression vectors include pCDM8 (Seed. B.. (1987)
Nature
329:840) and pMT2PC (Kaufman et al. (1987). EMBO J. 6_:187-19~). When used in
mammalian cells. the expression vector's control functions are often provided
by viral
regulatory elements. For example. commonly used promoters are derived from
polyoma,
Adenovirus 2, cytomegalovirus and Simian Virus 40.
In addition to the regulatory control sequences discussed above. the
recombinant
expression vector may contain additional nucleotide sequences. For example,
the
recombinant expression vector may encode a selectable marker gene to identify
host cells
that have incorporated the vector. Such selectable marker genes are well known
in the art.
Moreover. the facilitate secretion of the peptide compound from a host cell.
in particular
mammalian host cells. the recombinant expression vector preferably encodes a
signal
sequence operatively linked to sequences encoding the amino-terminus of the
peptide
compound such that upon expression. the peptide compound is synthesized with
the signal
sequence fused to its amino terminus. This signal sequence directs the peptide
compound
into the secretory pathway of the cell and is then cleaved. allowing for
release of the
mature peptide compound (i.e., the peptide compound without the signal
sequence) from
the host cell. Use of a signal sequence to facilitate secretion of proteins or
peptides from
mammalian host cells is well known in the art.
A recombinant expression vector comprising a nucleic acid encoding a peptide
compound that alters aggregation of natural ~i-AP can be introduced into a
host cell to
thereby produce the peptide compound in the host cell. Accordingly, the
invention also
provides host cells containing the recombinant expression vectors of the
invention. The
terms "host cell" and "recombinant host cell" are used interchangeably herein.
It is
understood that such terms refer not only to the particular subject cell but
to the progeny or
potential progeny of such a cell. Because certain modifications may occur in
succeeding
generations due to either mutation or environmental influences, such progeny
may not, in
CA 02449296 2003-12-15
fact, be iden~cal to the parent cell, but are still included within the scope
of the term as used
herein. A host cell may be any prokaryotic or eukaryotic cell. For example, a
peptide
compound may be expressed in bacterial cells such as E. coli, insect cells,
yeast or
mammalian cells. Preferably, the peptide compound is expressed in mammalian
cells. In a
5 preferred embodiment, the peptide compound is expressed in mammalian cells
in vivo in a
mammalian subject to treat amyloidosis in the subject through gene therapy
(discussed
further below). Preferably, the ~i-amyloid peptide compound encoded by the
recombinant
expression vector issecreted from the host cell upon being expressed in the
host cell.
Vector DNA can be introduced into prokaryotic or eukaryotic cells via
conventional
10 transformation or transfection techniques. As used herein, the terms
"transformation" and
"transfection" are intended to refer to a variety of art-recognized techniques
for introducing
foreign nucleic acid (e.g., DNA) into a host cell, including calcium phosphate
or calcium
chloride co-precipitation, DEAE-dextran-mediated transfection, lipofection,
electroporation,
microinjection and viral-mediated transfection. Suitable methods for
transforming or
15 transfecting host cells can be found in Sambrook et al. (Molecular Cloning:
.4 Laboratory
Manual. ?nd Edition, Cold Spring Harbor Laboratory press ( 1989)), and other
laboratory
manuals. Methods for introducing DNA into mammalian cells in vivo are also
known in the
art and can be used to deliver the vector DNA to a. subject for gene therapy
purposes
(discussed further below).
20 For stable transfection of mammalian cells. it is known that, depending
upon the
expression vector and transfection technique used. only a small fraction of
cells may integrate
the foreign DNA into their genome. In order to identify and select these
integrants, a gene
that encodes a selectable marker (e.g., resistance to antibiotics) is
generally introduced into
the host cells along with the gene of interest. Preferred selectable markers
include those that
25 confer resistance to drugs. such as 6418, hygromycin and methotrexate.
Nucleic acid
encoding a selectable marker may be introduced into a host cell on the same
vector as that
encoring the peptide compound or may be introduced on a separate vector. Cells
stably
transfected with the introduced nucleic acid can be identified by drug
selection (e.g., cells
that have incorporated the selectable marker gene will survive, while the
other cells die).
30 A nucleic acid of the invention can be delivered to cells in vivo using
methods known
in the art, such as direct injection of DNA, receptor-mediated DNA uptake or
viral-mediated
transfeetion. Direct injection has been used to introduce naked DNA into cells
in vivo (sec
e.g., Acsadi et al. (1991) Nature 332: 815-818; Wolffet al. (1990) Science
247:1465-1468).
A delivery apparatus (e.g., a "gene gun") for injecting DNA into cells in vivo
can be used.
35 Such an apparatus is commercially available (e.g., from BioRad). Naked DNA
can also be
introduced into cells by complexing the DNA to a canon, such as polylysine,
which is
coupled to a ligand for a cell-surface receptor (see for example Wu, G. and
Wu, C.H. (1988)
J. Biol. Chem. 263:14621: Wilson et al. (1992) J. Biol. Chem. 267:963-967; and
U.S. Patent
No. 5.166320). Binding of the DNA-ligand complex to the receptor facilitates
uptake of the
CA 02449296 2003-12-15
46
DNA by receptor-mediated endocytosis. Additionally, a DNA-ligand complex
linked to
adenovirus capsids which naturally disrupt endosomes, thereby releasing
material into the
cytoplasm can be used to avoid degradation of the complex by intracellular
lysosomes (see
for example Curiel et al. ( 1991 ) Proc. Natl. Acad. Sci. USA 88:8850;
Cristiano et al.
(1993) Proc. Natl. Acad. Sci. USA 90:2122-2126).
Defective retroviruses are well characterized for use in gene transfer for
gene therapy
purposes (for a review see Miller, A.D. (1990) Blood 76:2?1). Protocols for
producing
recombinant retroviruses and for infecting cells in vitro or in vivo with such
viruses can be
found in Current Prst~cols in Molecular BioloQ:v, Ausubel, F.M. et al. (eds.)
Green
Publishing Associates, (1989), Sections 9.10-9.14 and other standard
laboratory manuals.
Examples of suitable retroviruses include pLJ, pZIP, pWE and pEM which are
well known
to those skilled in the art. Examples of suitable packaging virus lines
include yrCrip, yrCre,
yr2 and yrAm. Retroviruses have been used to introduce a variety of genes into
many
different cell types, including epithelial cells, endothelial cells,
lymphocytes, myoblasts,
hepatocytes, bone marrow cells, in vitro and/or in vivo (see for example
Eglitis, et al.
(1985) Science 230:1395-1398; Danos and Mulligan (1988) Proc. Natl. Acad. Sci.
USA
85:6460-6464; Wilson et al. (1988) Proc. Natl. Acad. Sci. USA 85:3014-3018;
Armentano
et al. (1990) Proc. Natl. Acad. Sci. USA 87:6141-6145; Huber et al. (1991)
Proc. Natl.
Acad. Sci. USA 88:8039-8043; Ferry et al. (1991) Proc. Natl. Acad. Sci. USA
88:8377-
8381; Chowdhury et al. (1991) Science 254:1802-1805; van Beusechem et al.
(1992)
Proc. Natl. Acad. Sci. USA 89:7640-7644; Kay et al. (1992) Human Gene Therapy
3:641-
647; Dai et al. (1992) Proc. Natl. Acad Sci. USA 89:10892-10895; Hwu et al.
(1993) ,I.
Immunol. 150:4104-4115; U.S. Patent No. 4,868,116 by Morgan et al., issued on
September 29'", 1989; U.S. Patent No. 4,980,286 by Morgan et al., issued on
December 25",1990; PCT Application WO 89/07136 (PCT/US89/00422); PCT
Application WO 89/02468 (PCT/US88/03089); PCT Application WO 89/05345
(PCT/US88/04383); and PCT Application WO 92107573 (PCT/US91/08127)).
Alternatively, the genome of an adenovirus can be manipulated such that it
encodes .
and expresses a peptide compound but is inactivated in terms of its ability to
replicate in a
normal lytic viral life cycle. See for example Berlrner et al. (1988) Bio
Techniques 6:616;
Rosenfeld et al. (1991) Science 252:431-434; and Rosenfeld et al. (1992) Cell
68:143-155.
CA 02449296 2003-12-15
46a
Suitable adenoviral vectors derived from the adenovirus strain Ad type 5 d1324
or other
strains of adenovirus (e.g., Ad2, Ad3, Ad7 etc.) are well known to those
skilled in the art.
Recombinant adenoviruses are advantageous in that they do not require dividing
cells to
be effective gene delivery vehicles and can be used to infect a wide variety
of cell types,
S including airway epithelium (Rosenfeld et. aL (1992) cited supra),
endothelial cells
(Lemarchand et al. ( 1992) Proc. Natl. Acad Sci. USA 89:6482-6486),
hepatocytes (Herz
and Gerard (1993) Proc. Natl. Acad. Sci. USA 90:2812-2816) and muscle cells
(Quantin et
al. (1992) Proc. Natl. Acad. Sci USA 89:2581-2584). Additionally, introduced
adenoviral
DNA (and foreign DNA contained therein) is not integrated into the genome of a
host cell
but remains episomal, thereby avoiding potential problems that can occur as a
result of
CA 02449296 2003-12-15
47
insertional mutagenesis in situations where introduced DNA becomes integrated
iato the host
genome (e.g., retroviral DNA).
Adeno-associated virus (AAV) can also be used for delivery of DNA for gene
therapy
purposes. AAV is a naturally occurring defective virus that requires another
virus, such as an
adenovirus or a herpes virus, as a helper virus for efficient replication and
a productive life
cycle. (For a review see Muzyczka et al. Curr. Topics in Micro. and Immunol.
(1992)
158:97-129). It is also one of the few viruses that may integrate its DNA into
non-dividing
cells, and exhibits ~ high frequency of stable integration (see for example
Flotte et al. ( 1992}
Am. J. Respir. Cell. Mol. Biol. 7:349-356; Samulski et al. (1989) .l. 1%irol.
63:3822-3828; and
MeLaughlin et al. (1989) J. Yirol. 62:1963-1973). Vectors containing as little
as 300 base
pairs of AAV can be packaged and can integrate. An AAV vector such as that
described in
Tratschin et al. (1985) Mol. Cell. Biol. 5:325I-3260 can be used to introduce
DNA into cells.
A variety of nucleic acids have been introduced into different cell types
using AAV vectors
(see for example Hermonat et al. (1984) Proc. Natl. Acad Sci. USA 81:6466-
6~170; Tratschin
et al. (1985) Mol. Cell. Biol. 4:2072-2081: Wondisford et al. (1988) Mol.
Endocrinol. 2:32
39; Tratschin et al. (1984) J. ~iroL 51:611-619; and Flotte et al. (1993) J.
Biol. Chem.
268:3781-3790}.
The invention provides a method for treating a subject for a disorder
associated with
ø-amyloidosis. comprising administering to the subject a recombinant
expression vector
encoding a ø-amyloid peptide compound, the compound comprising an amino acid
sequence
having at least one amino acid deletion compared to øAP~ ;g, such that the ø-
amyloid
peptide compound is synthesized in the subject and the subject is treated for
a disorder
associated with ø-amyloidosis. Preferably, the disorder is Alzheimer's
disease. In one
embodiment the recombinant expression vector directs expression of the peptide
compound
in neuronal cells. In another embodiment. the recombinant expression vector
directs
expression of the peptide compound in glial cells. In yet another embodiment,
the
recombinant expression vector directs expression of the peptide compound in
fibroblast cells.
General methods for gene therapy. are known in the art. See for example, U.S.
Patent
No. 5,399,346 by Anderson et al. A biocompatible capsule for delivering
genetic material is
described in PCT Publication WO 95/05452 by Baetge et al. Methods for grafting
genetically modified cells to mat central nervous system disorders are
described in U.S.
Patent No. 5,082,670 and in PCT Publications WO 90/06757 and WO 93/10234, all
by Gage
et al. Isolation and/or genetic modification of multipotent neural stem cells
or neuro-derived
fetal cells are described in PCT Publications WO 94/02593 by Anderson et al.,
WO 94/16718
by Weiss et al., and WO 94/23754 by Major et al. Fibroblasts transduced with
genetic
material are described in PCT Publication WO 89/02468 by Mulligan et aL
Adenovirus
vectors for transfering genetic material into cells of the central nervous
system are described
in PCT Publication WO 94/08026 by Kahn et al. Herpes simplex virus vectors
suitable for
treating neural disorders are described in PCT Publications WO 94104695 by
Kaplitt and WO
CA 02449296 2003-12-15
48
90/09441 by-Geller et al. Promoter elements of the filial fibrillary acidic
protein that cxn
confer astrocyte specific expression on a linked gene or gene fragment, and
which thus can be
used for expression of Aø peptides specifically in ast<ocytes, is described in
PCT Publication
WO 93!07280 by Brenner et al. Furthermore, alternative to expression of an Aø
peptide to
modulate amyloidosis, an antisense oligonucleotide that is complementary to a
region of the
ø-amyloid precursor protein mRNA corresponding to the peptides described
herein can be
e~cpressed in a subject to modulate amyloidosis. General methods for
expressing antisense
oligonu~leotides to modulate nervous system disorders are described in PCT
Publication WO
95!09236.
Alternative to delivery by gene therapy, a peptide compound of the invention
comprising an amino acid sequence having at least one amino acid deletion
compared to
øAP~.3g can be delivered to a subject by directly administering the peptide
compound to the
subject as described further herein for the modified peptide compounds of the
invention. The
peptide compound can be formulated into a pharmaceutical composition
comprising a
therapeutically effective amount of the ø-amyloid peptide compound and a
pharmaceutically
acceptable carrier. The peptide compound can be contacted with natural ø-
amyloid peptides
with a ø-amyloid peptide compound such that aggregation of the natural ø-
amyloid peptides
is inhibited. Moreover, the peptide compound can be administered to the
subject in a
therapeutically effective amount such that the subject is treated for a
disorder associated with
ø-amyloidosis, such as Alzheimer's disease.
VIII. Qther Embodiments
Although the invention has been illustrated hereinbefore with regard to Aø
peptide
compounds, the principles described. involving attachment of a modifying
groups) to a
peptide compound. are applicable to any arnyloidogenic protein or peptide as a
means to
create a modulator compound that modulates. and preferably inhibits. amyloid
aggregation.
Accordingly, the invention provides modulator compounds that can be used to
treat
amyloidosis is a variety of forms and clinical settings.
Amyloidosis is a general term used to describe pathological conditions
characterized
by the presence of amyloid. Amyloid is a general term referring to a group of
diverse but
specific extracellular protein deposits which are seen in a number of
different diseases.
Though diverse in their occurrence. all amyloid deposits have common
morphologic
properties, stain with specific dyes (e.g., Congo red), and have a
characteristic red-green
birefringent appearance in polarized light after staining. They also share
common
ultrastructural features and common x-ray diffraction and infrared spectra.
Amyloidosis can
be classified clinically as primary, secondary, familial andlor isolated.
Primary amyloid
appears de »ovo without any preceding disorder. Secondary amyloid is that form
which
appears as a complication of a previously existing disorder. Familial amyloid
is a genetically
CA 02449296 2003-12-15
49
inherited form found in particular geographic populations. Isolated forms of
amyloid are
those that tend to involve a single organ system.
Different amyloids are characterized by the type of pmtein(s) or peptides)
present in
the deposit. For example, as described hereinbefore, amyloid deposits
associated with
Alzheimer's disease comprise the (3-amyloid peptide and thus a modulator
compound of the
invention for detecting and/or treating Alzheimer's disease is designed based
on modification
of the ~i-arnyloid peptide. The identities of the proteins) or peptides)
present in amyloid
deposits associatedwith a number of other amyloidogenic diseases have been
elucidated. '
Accordingly, modulator compounds for use in the detection andlor treatment of
these other
amyloidogenic diseases can be prepared in a similar fashion to that described
herein for ~i-
AP-derived modulators. In vitro assay systems can be established using an
amyloidogenic
protein or peptide which forms fibrils in vitro, analogous to the A~i assays
described herein.
Modulators can be identified using such assay systems, based on the ability of
the modulator
to disrupt the ~i-sheet structure of the fibrils. Initially, an entire
amyloidogenic pmtein can be
modified or. more preferably, a peptide fragment thereof that is known to form
fibrils in vitro
can be modified (e.g., analogous to Aril-40 described herein). Amino acid
deletion and
substitution analyses can then be performed on the modified protein or peptide
(analogous to
the studies described in the Examples) to delineate an aggregation core domain
that is
suffcient, when modified. to disrupt fibril formation.
Non-limiting examples of amyloidogenic proteins or peptides, and their
associated
amyloidogenic disorders. include:
Transthvretin (TTR) - Amyloids containing transthyretin occur in familial
amyloid
polyneuropathy (Portuguese. Japanese and Swedish types), familial amyloid
cardiomyopathy
(Danish type). isolated cardiac amyloid and systennic senile amyloidosis.
Peptide fragments
2~ of transthvretin have been shown to form amyloid fibrils in vitro. For
example. TTR 10-20
and TTR 105-11 ~ form amyloid-like fibrils in 20-30% acetonitrile/water at
room temperature
(Jarvis, J.A., et al.(I994) Int. J. Pept. Protein Res. 44:388-398). Moreover,
familial
cardiomyopathy !Danish type) is associated with mutation of Leu at position
111 to Met. and
an analogue of TTR 105-l la in which the wildtype Leu at position 111 has been
substituted
with Met (TTR 10~-115Met111 ) also forms amyloid-like fibrils in vitro (see
e.g.,
Hermansen, L.F., et al. (1995) Eur. J. Biochem. 227:772-779; Jarvis et al.
supra). Peptide
firagrnents .of TTR that form amyloid fibrils in vitro are also described in
Jarvis, J.A., et al.
( 1993 ) Biochem. Biophys. Res. Commun. 192:991-998 and Gustavsson, A., et al.
( 1991 )
Biochem. Biophys. Res. Commu». 17 :1159-1104. A peptide fragment of wildtype
or .
mutated transthyretin that forms amyloid fibrils can be modified as described
herein to create
a modulator of amvloidosis that can be used in the detection or treatment of
familial amyloid
polyneuropathy (Portuguese. Japanese and Swedish types), familial amyloid
cardiomyopathy
(Danish type). isolated cardiac amyloid or systemic senile amyloidosis.
CA 02449296 2003-12-15
S
dip (PrP) - Amyloids in a number of spongiform encxphslopathies,
including scrapie in sheep, bovine spongiform encephalopathy in cows and
Creutzfeldt-Jakob
disease (CJ) and Gerstrnann-Straussler-Scheinker syndrome (GSS) in himzans,
contain PrP.
Limited proteolysis of PrPSc (the prion protein associated with scrapie) leads
to a 27-30 kDa
fragment (PrP27-30) that polymerizes into rod-shaped amyloids (see e.g., Pan,
K.M., et al.
(1993) Proc. Natl. Acad Sci. USA 9:10962-10966; Gusset, M., et al. (1993)
Proc. Natl.
Acad Sci. USA 90:1-5). Peptide fragments of PrP from humans and other mammals
have
been shown to form amyloid fibrils in vitro. For example, polypeptides
corresponding to
sequences encoded by normal and mutant alleles of the PRNP gene (encoding the
precursor
of the prion protein involved in CJ), in the regions of codon 178 and codon
200,
spontaneously form amyloid fibrils in vitro (see e.g., Goldfarb, L.G., et al.
(1993) Proc. Natl.
Acad Sci. USA X0_:4451-4454). A peptide encompassing residues 106-126 of human
PrP has
been reported to form straight fibrils similar to those extracted from GSS
brains, whereas a
peptide encompassing residues 127-147 of human PrP has been reported to form
twisted
fibrils resembling scrapie-associated fibrils (Tagliavini. F.. et al. ( 1993)
Proc. Natl. Acad
Sci. USA 90:9678-9682). Peptides of Syrian hamster PrP encompassing residues
109-122,
113-127,113-120, 178-191 or 202-218 have been reported to form amyloid
fibrils, with the
most amyloidogenic peptide being Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala (SEQ ID NO:
17),
which corresponds to residues 113-120 of Syrian hamster PrP but which is also
conserved in
PrP from other species (Gusset, M., et al. (1992) Proc. Natl. Acad Sci. USA
89:10940-
10944). A peptide fragment of PrP that forms amyloid fibrils can be modified
as described
herein to create a modulator of amyloidosis that can be used in the detection
or treatment of
scrapie. bovine spongiform encephalopathy, Creutzfeldt-Jakob disease or
Gerstmann-
Straussler-Scheinker syndrome.
I,~let Amvioid Polvge tp ide (IAPP. also known as amylin) - Amyloids
containing IAPP
occur in adult onset diabetes and insuIinoma. IAPP is a 37 amino acid
polypeptide formed
from an 89 amino acid precursor protein (see e.g., Betsholtz. C., et al.
(1989) Exp. Cell. Res.
,3:484-493; V~%estermark, P., et al. (1987) Proc. Natl. Acad. Sci. USA ~4-
:3881-3885). A
peptide corresponding to IAPP residues 20-29 has been reported to form amyloid-
Iike fibrils
in vitro, with residues 25-29, having the sequence Ala-Ile-Leu-Ser-Ser (SEQ ID
N0: 18),
being strongly amyloidogenic (Westetmark, P., et al. (1990) Proe. Natl. Acad
Sci. USA
X7:5036-5040; Glenner, G.G., et al. (1988) Biochem. Biophys. Res. Common.
X55:608-614).
A peptide fragment of IAPP that forms amyloid fibrils can be modified as
described herein to
create a modulator of amyloidosis that can be used in the detection or
treatment of adult onset
diabetes or insulinoma.
Atrial Natriuretic Factor (ANF) - Amyloids containing ANF are associated with
isolated atrial amyloid (see e.g., Johansson, B., et al. (198'7) Biochem.
Biophys. Res.
Com~nun. x:1087-1092). ANF corresponds to amino acid residues 99-126 (proANF99-
126) ofthe ANF prohormone (proANPl-126) (Pucci, A., et al. (1991) J. Pathol.
165:235-
CA 02449296 2003-12-15
241 ). ANF, er a fiagment thereof,, that forms ~yloid fibrils can be modified
as described
herein to create a modulator of amyloidosis that can be used in the detection
or treatment of
isolated atrial amyloid.
KaBoa ~r Lambda Light - Amyloids containing kappa or lambda light chains
are associated idiopathic (primary) amyloidosis, myeloma or macroglobulinemia-
associated
amyloidosis, and primary localized cutaneous nodular amyloidosis associated
with Sjogren's
syndrome. The structure of amyloidogenic kappa and lambda light chains,
including amino
acid sequence analygis, has been characterized (see e.g., Buxbaum, J.N., et
al. (1990) Ann.
Intern. Med _1:455-464; Sehormann, N., et al. (1995) Proc. Natl. Aead Sei. USA
92:9490-
9494; Hurle, M.R., et al. (1994) Proc. Natl. Acad Sci. USA X1_:5446-5450;
Liepnieks, J.J., et
al. (1990) Mol. Immunol. 27:481-485; Gertz, M.A., et al. (1985) Scand J.
Immunol. 2:245-
250; Inazumi, .T., et al. (1994) Dermatology 18Q:125-128). Kappa or lambda
light chains, or
a peptide fragment thereof that forms amyloid fibrils, can be modified as
descriued herein to
create a modulator of amyloidosis that can be used in the detection or
treatmem of idiopathic
(primary) amyloidosis, myeloma or macroglobulinemia-associated amyloidosis or
primary
localized cutaneous nodular amyloidosis associated with Sjostren's syndrome.
Amvloid A - Amyloids containing the amyloid A protein (AA protein), derived
from
serum amyloid A, are associated with reactive (secondary) amyloidosis (see
e.g., Liepnieks,
J.J., et al. (1995) Biochim. Biophys. Acta x:81-86), familial Mediterranean
Fever and
familial amyloid nephropathy with urticaria and deafness (Muckle-Wells
syndrome) (see e.g.,
Linke. R.P., et al. (1983) Lab. Invest. 48:698-704). Recombinant human serum
amylvid A
forms amyloid-like fibrils in vitro (Yamada, T., et al. (1994) Biochim.
Biophys. Acta
1 6:323-329) and circular dichroism studies revealed a predominant ~i
sheet/turn structure
(McCubbin, W.D., et al. (1988) Biochem J. x,56:775-783). Serum amyloid A,
amyloid A
protein or a fraatnent thereof that forms amyloid fibrils can be modified as
described herein
to create a modulator of amyloidosis that can be used in the detection or
treatment of reactive
(secondary) amyloidosis, familial Mediterranean Fever and familial amyloid
nephropathy
with urticaria and deafness (Muckle-Wells syndrome).
sta ' C - Amyloids containing a variant of cystatin C are associated with
hereditary cerebral hemorrhage with amyIoidosis of Icelandic type. The disease
is associated
with a leucine to glycine mutation at position 68 and cystatin C containing
this mutation
aggregates in vitro (Abrahatrtson, M. and Grubb, A. (1994) Proc. Natl. Acad
Sci. USA
Ql_:1416-1420). Cystatin C or a peptide fragment thereof that forms amyloid
fibrils can be
modified as described herein to create a modulator of amyloidosis that can be
used in the
detection or treatment of hereditary cerebral hemorrhage with amyloidosis of
Icelandic type.
13 micrqglobulin - Amyloids containing X32 microglobulin (~i2M) are a major
complication of long term hemodialysis (see e.g., Stein, G., et al. (1994)
Nephrol. Dial.
Transplant. 9:48-50; Floege, J., et al. (1992) Kidney Int. Suppl. 38:S78-S85;
Maury, C.P.
(1990) Rheumatol. Int. 10:1-8). The native ~32M protein has been shown to fomn
amyloid
CA 02449296 2003-12-15
52
fibrils in vitae (Connors, L.H., et al. (1985) Biochem. Biophys. Res. Common.
131:1063-
1068; Ono, K., et al. (1994) Nephron 66:404-407). ~i2~I, or a peptide fragment
thereof that
forms amyloid fibrils, can be modified as described herein to create a
modulator of
amyloidosis that can be used in the detection or treatment of amyloidosis
associated with
long term hemodialysis.
Ap~lip~»rotein A-I (ApoA-I) - Amyloids containing variant forms of ApoA-I have
been found in hereditary non-neuropathic systemic amyloidosis (familial
amyloid
polyneutopathy III). For example. N-tttminal fragments (residues 1-86,1-92 and
1-93) of an
ApoA-I variant having a Trp to Arg mutation at position SD have been detected
in amyloids
(Booth, D.R., et al. ( I 995) QJM $$:695-702). In another family, a Ieucine to
arginine
mutation at position 60 was found (Soutar, A.K., et at. ( 1992) Proc. Natl.
Acad Sci. USA
89:7389-7393). ApoA-I or a peptide fragment thereof that fomns amyloid fibrils
can be
modified as described herein to create a modulator of amyloidosis that can be
used in the
detection or treatment of hereditary non-neumpathic systemic amyloidosis.
1 ~ Ge oli - Amyloids containing variants of gelsolin are associated with
familial
amyloidosis of Finnish type. Synthetic gelsoIin peptides that have sequence
homology to
wildtvpe or mutant geIsolins and that form amyloid fibrils in vitro are
reported in Maury,
C.P. et al. (1994) Lab. Invest. 70:558-564. A nine residue segment surrounding
residue 187
(which is mutated in familial gelsolin amyloidosis) was defined as an
amyloidogenic region
(Maury, et al., supra; see also Maury, C.P., et al. (1992) Biochem. Biophys.
Res. Common.
11:227-231: Maury, C.P. (1991) J. Clin. Invest. 87:I I95-1 I99). Gelsolin or a
peptide
fragment thereof that forms amyloid fibrils can be modified as described
herein to create a
modulator of amvloidosis that can be used in the detection or treatment of
familial
amyloidosis of Finnish type.
Procalcitonin or calcnto~,n - Amyloids containing procalcitonin. calcitonin or
caleitonin-like immunoreactivity have been detected in amyloid fibrils
associated with
medullary carcinoma of the thymid (see e.g., Butler, M. and Khan, S. ( 1986)
Arch. Pathol.
Lab. Med. 110:647-649; Sletten, K., et al. ( 1976) J. Exp. Med. 143:993-998).
Calcitonin has
been shown to form a nonbranching fibrillar structure in vitro (Kedar, L, et
al. ( 1976) Isr. J.
Med. Sei. 12:1137-1140). Procalcitonin. calcitonin or a fragment thereof that
forms amyloid
fibrils can be modified as described herein to create a modulator of
amyloidosis that can be
used in the detection or treatment of amyloidosis associated with medullary
carcinoma of the
thyroid.
Fibrinogen - Amyloids containing a variant form of fibrinogen alpha-chain have
been
found in hereditary renal amyloidosis. An arginine to Ieucine mutation at
position 534 has
been reported in amyloid fibril protein isolated from postmortem kidney of an
affected
individual (Benson. M.D., et al. (1993) Nature Genetics x:252-255). Fibrinogen
alpha-chain
or a peptide fragment thereof that forms amyloid fibrils can be modified as
described herein
CA 02449296 2003-12-15
to create a modulator of amyloidosis that cur be used in the detection or
treatment of
fibrinogen-associated hereditary renal amyloidosis.
vso a - Amyloids containing a variant form of lysozyme have been found in
hereditary systemic amyloidosis. In one family the disease was associated with
a threonine to
isoleucine mutation at position ~6, whereas in another family the disease was
associated with
a histidine to aspartic acid mutation at position 67 (Pepys, M.B., et al.
(1993) Nature
362:53-5~7). Lysozyme or a peptide fragment thereof that forms amyloid fibrils
can be
modified as describFd herein to create a modulator of amyloidosis that can be
used in the
detection or treatment of lysozyme-associated hereditary systemic amyloidosis.
I0
This invention is further illustrated by the following examples which should
not be
construed as limiting. A modulator's ability to alter the aggregation of (3-
amyloid peptide in
the assays described below are predictive of the modulator's ability to
perform the same
function in vivo.
1~
EXAMPLE 1: Construction of [3-Amyloid Modulators
A ~i-amyloid modulator composed of an amino-terminally biotinylated (3-amyloid
20 peptide of the amino acid sequence:
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGW
(positions 1 to 40 of SEQ ID NO: 1 ) was prepared by solid-phase peptide
synthesis using an
Na-9-fluorenylinethyloxycarbonyl (FMOC)-based protection strategy as follows.
Starting
with 2.~ mmoles of FMOC-Val-Vliang resin. sequential additions of each amino
acid wen
25 performed using a four-fold excess of protected amino acids.. l-
hydroxybenzotriazole (HOBt)
and diisopmpyl carbodiimide (DIC). Recouplings were performed when necessary
as
determined by ninhydrin testing of the resin after coupling. Each synthesis
cycle was
minimally described by a three minute deprotection (25 % piperidine/N-methyl-
pyrrolidone
(NMP)), a 1 ~ minute deprotection, five one minute NMP washes, a 60 minute
coupling cycle,
30 five NMP washes and a ninhydrin test. To a 700 mg portion of the fully
assembled peptidc-
resin, biotin (obtained commercially from Molecular Probes, Ine.) was
substituted. for an
FMOC-amino acid was coupled by the above protocol. The peptide was removed
from the
resin by treatment with trifluoroacetic acid (TFA) (82.~ %), water (5 %),
thioanisole (5 %),
phenol (5 %). ethanedithiol (2.5 %) for two hours followed by precipitation of
the peptide in
35 cold ether. The solid was pelleted by centrifugation (Z400 rpm x 10 min.),
and the ether
decanted. It was resuspended in ether, pelIeted and decanted a second time.
The solid. was
dissolved in 10 % acetic acid and lyophilized to dryness to yield 230 mg of
crude biotinylated
peptide. 60 mg of the solid was dissolved in 25 % acetonitrile (ACN) /0.1 %
TFA and
applied to a C 18 reversed phase high performance liquid chromatography (HPLC)
column.
CA 02449296 2003-12-15
Biotinyl ~A~~.4p was eluted usiag a linar Sg adient of 30-45 %
acetonitrilel0.1 % TFA over
40 miautes. One primary fraction (4 mg) and several side fractions were
isolated. The main
fraction yielded a mass spectrum of 4556 (matrix-assisted laser desorption
ionization -time of
flight) which matches the theoretical (4555) for this peptide.
A p-amyloid modulator composed of an amino-terminally biotinylated [3-amyloid
peptide of the amino acid sequence:
DAEFRHDSGYEVHHQ
(positiops 1 to 15 of SEQ ID NO: 1) was prepared on an Advanced ChemTech Model
396
multiple peptide synthesizer using an automated protocol established by the
manufacturer for
0.025 mrnole scale synthesis. Double couplings were performed on all cycles
using
2-(1H-benzotriazol-1-y1r1,1,3,3-tetramethyluronium hexafluorophosphate
(HBTU)/N,N-
diisopropylethylamine (DIEA)/HOBt/FMOC-AA in four-fold excess for 30 minutes
followed
by DIC/HOBtlFMOC-AA in four-fold excess for 4~ minutes. The peptide was
deprotected
and removed from the resin by treatment with TFA/water (95 %/5 %) for three
hours and
precipitated with ether as described above. The pellet was resuspended in 10 %
acetic acid
and lyophilized. The material was purified by a preparative HPLC using 15 %-40
acetonitrile over 80 minutes on a Vydac C18 column (21 x 250 mm). The main
isolate eluted
as a single symmetrical peak when analyzed by analytical HPLC and yielded the
expected
molecular weight when analyzed by electrospray mass spectrometry. Result =
2052.6 (2052
theoretical).
~-amyloid modulator compounds comprising other regions of the ~i-AP amino acid
sequence (e.g.. an A~i aggregation core domain) were similarly prepared using
the synthesis
methods described above. Moreover, modulators comprising other amyloidogenic
peptides
can be similarly prepared.
EXAMPLE 2: Inhibition of p-Amyloid Aggregation by Modulators
The ability of ~3-amyloid modulators to inhibit the aggregation of natural ~i-
AP when
combined with the natural (3-AP was examined in a series of aggregation
assays. Natural (3-
AP (~i-APl,.4p) was obtained commercially from Bachem (Torrance, CA). Amino-
terminally
biotinylated ~i-AP modulators were prepared as described in Example 1.
A. (optical Density Assay
In one assay, (3-AP aggregation was measured by determining the increase in
turbidity
of a solution of natural /3-AP over time in the absence or presence of various
concentrations
of the modulator. Turbidity of the solution was quantitated by determining the
optical
density at 400 nm (AQpp ~,) of the solution over time.
The aggregation of natural (3-AP in the absence of modulator was determined as
follows. (3-AP~-4o was dissolved in hexafluoro isopropanol (HFIP; Aldrieh
Chemical Co.,
CA 02449296 2003-12-15
SS
Inc.) at 2 mglml. Aliquou of the HFIP solution (87 ~1) were transferred to
individual 10 mm
x 75 mm test tubes. A stream of argon gas was passed through each tube to
evaporate the
HFIP. To the resulting thin film of peptide, dimethylsulfoxide (DMSO; Aldrich
Chemical
Co., Inc.) (25 ~1) was added to dissolve the peptide. A 2 mm x 7 mmTeflon-
coated magnetic
stir bar was added to each tube. Buffer (475 pL of 100 mM NaCI, 10 mM sodium
phosphate,
pH 7.4) was added to the DMSO solution with stirring. The resulting mixture
was stirred
continuously and the optical density was monitored at 400 nm to observe the
formation of
insoluble peptide aggregates.
Alternatively, (3-AP~.,40 was dissolved in DMSO as described above at
1.6 mM (6.9 mg/ml) and aliquots (25 p1) were added to stirred buffer (475 ~1),
followed by
monitoring of absorbance at 400 nm.
For inhibition studies in which a ~3-amyloid modulator was dissolved in
solution
together with the natural (3-AP, the modulators were dissolved in DMSO either
with or
without prior dissolution in HFIP. These compounds were then added to buffer
with stirring,
followed by addition of ~i-AP ~ ~0 in DMSO. Alternatively. HFIP solutions of
modulators
were combined with ~i-AP ~ ~0 in HFIP followed by evaporation and
redissolution of the
mixture in DMSO. Buffer was then added to the DMSO solution to initiate the
assay. The
amino-terminally biotinylated ~3-amyloid peptide modulators N-biotinyl-~iAP
1~0, and N-
biotinyl-(iAP 1 _ 15 were tested at concentrations of 1 % and 5 % in the
natural ~i-AP ~ ~0
solution.
A representative example of the results is shown graphically in Figure 1,
which
depicts the inhibition of aggregation of natural ~i-AP ~ ~0 by N-biotinyl-~iAP
~ ~0. In the
absence of the modulator. the optical density of the natural p-AP solution
showed a
characteristic sigmoidal curve. with a lag time prior to aggregation
(approximately 3 hours in
Figure 1 ) in which the A400 nm w~ low . followed by rapid increase in the
A4o0 nm~ which
quickly reached a plateau level. representing aggregation of the natural ~i
amyloid peptides.
In contrast. in the presence of as little as 1 % of the N-biotinyl-(3AP1~0
modulator,
aggregation of the natural [3 amyloid peptides was markedly inhibited,
indicated by an
increase in the lag time, a decrease in the slope of aggregation and a
decrease in the plateau
level reached for the turbidity of the solution (see Figure 1 ). N-biotinyl-
~iAP t.40 at a
concentration of 5 % similarly inhibited aggregation of the natural ~i amyloid
peptide.
Furthermore, similar results were observed when N-biotinyl-~iAP 1 _ 15 ''Was
used as the
modulator. These results demonstrate that an N-terminally biotinylated [3-AP
modulator can
effectively inhibit the aggregation of natural ~i amyloid peptides, even when
the natural ~i
amyloid peptides are present at as much as a 100-fold molar excess
concentration.
B. Fluorescence Assav
In a second assay, (3-AP aggregation was measured using a fluorometric assay
essentially as described in Levine, H. (1993) Protein Science 2_:404-410. In
this assay, the
CA 02449296 2003-12-15
56
dye thioflaviae T (ThT) is contacted with the ~i-AP solution. Association of
ThT with
aggregated ~i-AP, but not monomeric or loosely associated ~-AP, gives rise to
a new
excitation (ex) maximum at 450 nm and an enhanced emission (em) at 482 nm,
compared to
the 385 nm (ex) and 445 nm (em) for the free dye. ~i-AP aggregation was
assayed by this
method as follows. Aliquots (2.9 ~1) of the solutions used in the aggregation
assays as
described above in section A were removed from the samples and diluted in 200
p1 of
potassium phosphate buffer (50 mM, pH 7.0) containing thiotlavin T ( 10 ~uM;
obtained
commerFially from Aldrich Chemical Co., Inc.). Excitation was set at 450 nm
and emission
was measured at 482 nm. Similar to the results observed with the optical
density assay
described above in section A, as little as 1 % of the N-biotinylated ~-AP
modulators was
effective at inhibiting the aggregation of natural (3 amyloid peptides using
this fluorometric
assay.
C. Static gre~ation A~av
In a third assay, [3-AP aggregation was measured by visualization of the
peptide
aggregates using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). In this
assay, ~i-AP
solutions were allowed to aggregate over a period of time and then aliquots of
the reaction
were run on a standard SDS-PAGE gel. Typical solution conditions were 200 pM
of J3-APB.
4p in PBS at 37 °C for 8 days or 200 ~M ~i-AP~~p in 0.1 M sodium
acetate at 37 °C for 3
days. The peptide aggregates were visualized by Coomassie blue staining of the
gel or, for ~3-
AP solutions that included a biotinylated [3-AP modulator, by western blotting
of a filter
prepared from the gel with a streptavidin-peroxidase probe, followed by a
standard
peroxidase assay. The ~i-AP aggregates are identifiable as high molecular
weight, low
mobility bands on the gel, which are readily distinguishable from the low
molecular weight,
high mobility (3-AP monomer or dimer bands.
When natural (3-AP~.Qp aggregation was assayed by this method in the absence
of any
~i amyloid modulators, high molecular weight aggregates were readily
detectable on the gel.
In contrast, when N-biotinyl-~i-AP~.,4p modulator self aggregation was assayed
(i.e.,
aggregation of the N-biotinyl peptide alone, in the absence of any natural (3-
AP), few if any
high molecular weight aggregates were observed, indicating that the ability of
the modulator
to self aggregate is significantly reduced compared to natural ~i-AP. Finally,
when
aggregation of a mixture of natural ~3-AP ~ ~p and N.biotinylated ~3-AP ~ ~p
was assayed by
this method, reduced amounts of the peptide mixture associated into high
molecular weight
aggregates, thus demonstrating that the ~i amyloid modulator is effective at
inhibiting the
aggregation of the natural ~i amyloid peptides.
CA 02449296 2003-12-15
Nenroto:ieity AnsiyaT~ of (3-Amyioid Modulators
The neumtoxicity of the p-amyloid modulators is tested in a cell-based assay
using
the neuronal precursor cell line PC-12, or primary neuronal cells, and the
viability indicator
3,(4,4-dimethylthiazol-2-yl)2,5-Biphenyl-tetrazolium bromide (MT'I~. (See
Shearman. M.S.
et al. (I994) Proc. Natl. Acad Sci. USA x:1470-1474; Hansen, M.B. et al.
(1989) J. Immun.
Methods x_9:203-210). PC-12 is a rat adrenal pheochromocytoma cell line and is
available
from the American,Type Culture Collection, Rockville, MD (ATCC CRL 1721 ). MTT
(commercially available from Sigma Chemical Co. ) is a chromogenic substrate
that is
converted from yellow to blue in viable cells, which can be detected
spectrophotometrically.
To test the neurotoxicity of a p-amyloid modulator (either alone or combined
with
natural p-AP). cells first are plated in 96-well plates at 7,000-10,000
cells/well and allowed to
adhere by overnight culture at 37 °C. Serial dilutions of freshly
dissolved or "aged"
modulators (either alone or combined with natural (3-AP) in phosphate buffered
saline (PBS)
I S are added to the wells in triplicate and incubation is continued for two
or more days. Aged
modulators are prepared by incubating an aqueous solution of the modulator at
37 °C
undisturbed for a prolonged period (e.g., five days or more). For the final
two hours of
exposure of the cells to the modulator preparation, MTT is added to the media
to a final
concentration of 1 mg/ml and incubation is continued at 37 °C.
Following the two hour
incubation with MTT, the media is removed and the cells are lysed in
isopropanoU0.4N HCl
with agitation. An equal volume of PBS is added to each well and the
absorbance of each
well at 570 nm is measured to quantitate viable cells. Alternatively, MTT is
solubilized by
addition of 50 % N.N-dimethyl formamide/20 % sodium dodecyl sulfate added
directly to the
. media in the v~°ells and viable cells are likewise quantitated by
measuring absorbance at 570
nm. The relative neurotoxicity of a ~i-amyloid modulator (either alone or in
combination
with mtural ~i-AP) is determined by comparison to natural ~i-AP alone (e.g.,
(31-40, p1-42),
whica exhibits neurotoxicity in this assay and thus can serve as a positive
control.
EXAMPLE 4: Syntheses of Additional Modified p-Amyloid Peptide Compounds
In this example. a series of modified p-APs. having a variety of N terminal or
random
side chain modifications were synthesized.
A series ofN terminally modified p-amyloid peptides was synthesized using
standard
methods. Fully-protected resin-bound peptides corresponding to Ap(I-15) and
Ap(I-40)
were prepared as described in Example 1 on Wang resin to eventually afford
carboxyl
terminal peptide acids. Small portions of each peptide resin (13 and 20
~cmoles, respectively)
were aliquoted into the wells of the reaction block of an Advanced ChemTech
Model 396
Multiple Peptide Synthesizer. The N-terminal FMOC protecting group of each
sample was
removed in the standard manner with 25% piperidine in NMP followed by
extensive washing
CA 02449296 2003-12-15
SO
with NMP.-The unprotected N terminal a-amino group of each peptide-resin
sample was
modified using one of the following methods:
Metho~g, coupling of modifying reagents containing free carboxylic acid
groups:
The modifying reagent (five equivalents) was predissolved in NMP, DMSO or a
mixture of
those two solvents. HOBT and DIC (five equivalents of each reagent) were added
to the
dissolved modifier and the resulting solution was added to one equivalent of
free-amino
peptide-resin. Coupling was allowed to proceed overnight, followed by washing.
If a
ninhydFin test on a small sample of peptide-resin showed that coupling was not
complete, the
coupling was repeated using I-hydroxy-7-azabenzotriazole (HOAt) in place of
HOBt.
N~ethod B. coupling of modifying reagents obtained in preactivated forms: The
modifying reagent (five equivalents) was predissolved in NMP, DMSO or a
mixture of these
two solvenu and added to one equivalent of peptide-resin.
Diisopropylethylamine (DIEA;
six equivalents) was added to the suspension of activated modifier and peptide-
resin.
Coupling was allowed to proceed overnight, followed by washing. If a ninhydrin
test on a
I 5 small sample of peptide-resin showed that coupling was not complete. the
coupling was
repeated.
After the second coupling (if required) the N terminally modified peptide-
resins were
dried at reduced pressure and cleaved from the resin with removal of side-
chain protecting
groups as described in Example 1. Analytical reversed-phase HPLC was used to
confirm that
a major product was present in the resulting crude peptides which were
purified using
Millipore Sep-Pak cartridges or preparative reverse-phase HPLC. Mass
spectrometry was
used to confirm the presence of the desired compound in the product.
Method A was used to couple N aeetylneuraminic acid, cholic acid. trans-4-
cotininecarboxylic acid. 2-imino-I-imidazolidineacetic acid, (S')-(-)-indoline-
2-carboxylic
acid, (-)-menthoxyacetic acid. 2-norbornaneacetic acid. r-oxo-5-
acenaphthenebutyric acid.
(-~2-oxo-4-thiazolidinecarboxylic acid, and tetrahydro-3-furoic acid. Method B
was.used to
couple 2-iminobiotin N hydroxysuccinimide ester. diethylenetriaminepentaacetic
dianhydride, 4-morpholinecarbonyl chloride, 2-thiopheneacetyl chloride, and 2-
thiophenesulfonyl chloride.
In a manner similar to the construction of N terminally modified A~(1-IS) and
Ap(1-
40) peptides described above, N fluoresceinyl Ap(I-IS) and Aa(I-40) were
prepared in two
alternative manners using the preactivated reagents ~-(and 6)-
carboxyfluorescein
succinimidyl ester and fluorescein-5-isothiocyanate (FITC Isomer I). Both
reagents were
obtained from Molecular Probes Ine. Couplings were performed using four
equivalents of
reagent per equivalent of peptide-resin with DIEA added to make the reaction
solution basic
to wet pH paper. Couplings of each reagent to A~i(1-IS)-resin appeared to be
complete aRer a
single overnight coupling. Coupling to Aa( 1-40)-resin was slower as indicated
by a positive
ninhydrin test and both reagents were recoupled to this peptide-resin
overnight in
CA 02449296 2003-12-15
59
te~hydrofiaan-NMP ( 1 ~ v/v). The resulting N urminally modified peptide-
resins were
cleaved, deprotected and purified as described in Example A.
In addition to the N fluoresceinyl A(i peptides described above, a p-amyloid
modulator comprised of random modification of Ap(1-40) with fluorescein was
prepared.
Ap(1-40) purchased from Bachem was dissolved in DMSO at approximately 2 mg/mL.
5-
(and-6)-Carboxyfluorescein purchased from Molecular Probes was added in a 1.5
molar
excess and DIEA was added to make the solution basic to wet pH paper. The
reaction was
allowed to proceedfor 1 hour at mom temperature and was then quenched with
triethanolamine. The product was added to assays as this crude mixture.
(3-amyloid modulator compounds comprising other regions of the p-AP amino acid
sequence (e.g., an Aj3 aggregation core domain) were similarly prepared using
the synthesis
methods described above. Moreover, modulators comprising other amyloidogenic
peptides
can be similarly prepared.
1 S E~AMP~ S: Identification of Additional [i-Amyloid Modulators
In this Example, two assays of A~i aggregation were used to identify ~i-
amyloid
modulators which can inhibit this process.
The first assay is referred to as a seeded static assay (SSA) and was
performed as
follows:
To prepare a soitition of A(3 monomer. the appropriate quantity of A(3(1-40)
peptide
(Bachem) was weighed out on a micro-balance (the amount was corrected for the
amount of
water in the preparation, which, depending on lot number,.was 20-30% w/w). The
peptide
was dissolved in 1125 volume of dimethysulfoxide (DMSO), followed by water to
1/2
volume and 1l2 volume 2x PBS (10x PBS: NaCI 137 mM. KCl 2.7 mM Na~HP04 ~ 7H~0
4.3 mM. KH2P04 1.4 mM pH 7.2) to a final concentration of 200 pM.
To prepare a stock seed, l ml of the above A(3 monomer preparation. was
incubated
for 8 days at 37 °C and sheared sequentially through an 18, 23, 26 and
30 gauge needle 25,
25, 50, and 100 times respectively. 2 ~1 samples of the sheared material was
taken for
fluorescence measurements after every 50 passes through the 30 gauge needle
until the
fluorescence units (FU) had plateaued (approx. 100-150x).
To prepare a candidate inhibitor, the required amount of candidate inhibitor
was
weighed out and the stock dissolved in lx PBS to a final concentration of 1 mM
(10x stock).
If insoluble. it was dissolved in 1/10 volume of DMSO and diluted in lx PBS to
1 mM. A
further 1/10 dilution was also prepared to test each candidate at both 100 pM
and 10 ~eM.
For the aggregation assay, each sample was set up in tirplicate [50 ~1 of 200
~M
monomer,125 FU sheared seed (variable quantity dependent on the batch of seed.
routinely
3-6 ~1), 10 ~1 of lOx inhibitor solution, final volume made up to 100 ~1 with
lx PBS]. Two
concentrations of each inhibitor were tested 100 ~M and 10 ~M, equivalent to a
1:1 and a
CA 02449296 2003-12-15
1:10 molar ratio of monomer to inhibitor. The controls included an unneeded
reaction to
confirm that the fresh monomer contained no seed, and a seeded reaction in the
absence of
inhibitor, as a reference to compare against putative inhibitors. The assay
was incubated at
37 °C for 6 h, taking 2 ~1 samples hourly for fluorescence
measuremenTM. To measure
5 fluorescence, a 2 g1 sample of A/3 was added to 400 p1 of Thioflavin-T
solution (50 mM
Potassium Phosphate 10 mM Thioflavin-T pH 7.5). The samples were vortexed and
the
fluorescence was read in a 0.~ ml micro quartz cuvette at EX 450 nm and EM 482
nm
(Hitach14~00 ~luorlmeter). ~i-aggregation results in enhanced emission of
Thioflavin-TT'"'
Accordingly, samples including an effective inhibitor compound exhibit reduced
emission as
10 compared to control samples without the inhibitor compound.
The second assay is referred to as a shaken plate aggregation assay and was
performed
as follows:
A~3(.1-40) peptide from Bachem (Torrance. CA) was dissolved in HFIP
(1,I,I,3,3,3-
1 ~ Hexafluoro-?-proganol: Aldrich I0.5?2-8) at a concentration of 2 mg
peptide/ml and incubated
at room temperature for 30 min. HFIP solubilized peptide was sonicated in a
waterbath
sonicator for ~ min at highest setting. then evaporated to dryness under a
stream of argon. The
peptide film was resuspended in anhydrous dimethylsulfoxide (DMSO) at a
concentration of
6:9 mglml, sonicated for ~ min as before, then filtered through a 0.2 micron
nylon syringe filter
20 (VWR cat. No. 28196-050). Candidate inhibitors were dissolved directly in
DMSO, generally
at a molar concentration 4 times that of the A~3(1-40) peptide.
Candidates were assayed in triplicate. For each candidate to be tested, 4
parts A~i{1-40)
peptide in DMSO were combined with 1 part candidate inhibitor in DMSO in a
glass vial, and
mixed to produce a 1:1 molar ratio of A(i peptide to candidate. For different
molar ratios,
25 candidates were diluted with DMSO prior to addition to A~i( 1-40). in order
to keep the final
DMSO and A~i(1-40) concentrations constant. Into an ultra low binding 96 well
plate (Corning
Costar cat. No. 2500, Cambridge MA) 100 ~1 PTL buffer ( 150 mM NaCI,10 mM
NaH?P04;
pH 7.4) was aliquotted per well. For each candidate, 10 ~1 of peptide mixture
in DMSO was
aliquotted into each of three wells containing bt~'er. The covered plate was
vigorously
30 vortexed on a plate shaker at high speed for 30 seconds. An additional 100
~1 of PTL buffer
was added to each well and again the plate was vortexed vigorously for 30 sec.
Absorbance at
405 nnZ was immediately read in a plate reader for a baseline reading. The
plate was returned
to the plate shaker and vortexed at moderate speed for 5 hours at room
temperature, with
absorbance readings taken at 15-20 min intervals. Increased absorbance
indicated aggregation.
35 Accordingly, effective inhibitor compounds cause a decrease in absorbance
in the test sample
as compared to a control sample without the inhibitor compound.
Representative results of the static seeded assay and shaken plate assay with
prefentd
~-arnyloid modulators are shown below in Table I.
CA 02449296 2003-12-15
TABLE I
61
' CandidsteA~ Amino Modifying Ene~ m i Errec><
~ m
Inhibitor ~ Reagent shaken plate. Seeded Static
Acids
I, ~ ,sss I Assa ' i
~
Cholic Complete ,~",~,
~ acid '
~
174 A 1-15 ~ inhibition
at
100% conc
Diethylene- Decreased ,~"~, I
~
176 ' ' A~i1-15 ~ Plateau
. triamine
yenta
.
. ;
f acetic
acid
i ; None "~,~,
(-)-Menthoxy
~
180 ~ A~1-15 ~ . .
i acetic
acid
I
i
Fluorescein Decreased ~ ,~"~, ;
;
190 A~i1-15 . Plateau
carboxylic
acid
;
(FICO)
h-EVHHHHQ~K- Complete
++
220 ~ A~i16-40 ' inhibition
IAIi at
(16-40)]-OH
100%, increased
mutant ' i
i to at 10
%
Increased ~ ~",~,
~ I tag
224 A(31-40 F~sFzo-'T~sT2o
mutant ~
'
. I. accelerated i ~,~,
L33 H17~5-lU ~VC«v awu , oyy cyouv~ ~ w
~ 10% ConC
* '~"~' = A strong inhibitor of aggregation. The rate of aggregation in the
presence of the inhibitor was decreased compared to the control by
at feast 30-50%
These results indicate that (3-APs modified by a wide variety of N-terminal
modifying
groups are effective at modulating (3-amyloid aggregation.
EXAMPLE 6: Additional [3-Amyloid Aggregation Assays
Most preferably, the ability of (3-amyloid modulator compounds to modulate
(e.g.,
inhibit or promote) the aggregation of natural ~-AP when combined with the
natural (3-AP is
examined in one or both of the aggregation assays described below. Natural ~i-
AP (~i-AP t gyp)
for use in the aggregation assays is commercially available from Bachem
(Torrance, CA).
A. Nucleation Assay,
The nucleation assay is employed to determine the ability of test compounds to
alter
(e.g. inhibit) the early events in formation of p-AP fibers from monomeric (3-
AP.
Characteristic of a nucleated polymerization mechanism, a lag time is observed
prior to
CA 02449296 2003-12-15
62
nucleation, after which the peptide rapidly forms fibers as reflected in a
linear rise in
turbidity. The time delay before polymerization of (i-AP monomer can be
quantified as well
as the extent of formation of insoluble fiber by light scattering (turbidity).
Polymerization
reaches equilibrium when the maximum turbidity reaches a plateau. The
turbidity of a
solution of natural ~i-AP in the absence or presence of various concentrations
of a ~i-amyloid
modulator compound is determined by measuring the apparent absorbance of the
solution at
405nm (A4o5 "",) over time. The threshold of sensitivity for the measurement
of turbidity is
in the rapge of 15-20 pM (3-AP. A decrease in turbidity over time in the
presence of the
modulator. as compared to the turbidity in the absence of the modulator,
indicates that the
modulator inhibits fornzation of ~i-AP fibers from monomeric (3-AP. This assay
can be
performed using stirring or shaking to accelerate polymerization. thereby
increasing the speed
of the assay. Moreover the assay can be adapted to a 96-well plate format to
screen multiple
compounds.
To perform the nucleation assay, first A~i~.~p peptide is dissolved in HFIP
(1,1,1,3,3.3-Hexafluoro-2-propanoh Aldrich 10.522-8) at a concentration of 2
mg peptide/ml
and incubated at room temperature for 30 min. HFIP-solubilized peptide is
sonicated in a
waterbath sonicator for ~ min at highest setting, then evaporated to dryness
under a stream of
argon. The peptide film is resuspended in anhydrous dimethylsulfoxide (DMSO)
at a
concentration of 6.9 mg/ml (25x concentration), sonicated for 5 min as before.
then filtered
through a 0.2 micron nylon syringe filter (VWR cat. No. 28196-050). Test
compounds are
dissolved in DMSO at a 100x concentration. Four volumes of 25x A(3 tip peptide
in DMSO
are combined with one volume of test compound in DMSO in a glass vial. and
mixed to
produce a 1:1 molar ratio of A(3 peptide to test compound. For different molar
ratios, test
compounds are diluted with DMSO prior to addition to A(3 ~-4p, in order to
keep the final
DMSO and A~t.~p concentrations constant. Control samples do not contain the
test
compound. Ten microliters of the mixture is then added to the bottom of a well
of a Corning
Costar ultra low binding 96-well plate (Corning Costar, Cambridge MA; cat. No.
2500).
Ninety microliters of water is added to the well, the plate is shaken on a
rotary shaken at a
constant speed at room temperature for 30 seconds, an additional 100 ~I of 2x
PTL buffer (20
mM NaH2P04, 300 mM NaCI, pH 7.4) is added to the well, the plate is reshaken
for 30
seconds and a baseline (t=0) turbidity reading is taken by measuring the
apparent absorbance
at 405 nm using a Hio-Rad Model 450 Microplate Reader. The plate is then
returned to the
shaker and shaken continuously for 5 hours. Turbidity readings are taken at 1
~ minute
intervals.
~i-amyloid aggregation in the absence of any modulators results in enhanced
turbidity
of the natural ~-AP solution (i.e., an increase in the apparent absorbance at
405 nm over
time). Accordingly, a solution including an effective inhibitory modulator
compound
exhibits reduced turbidity as compared to the control sample without the
modulator
CA 02449296 2003-12-15
63
compound (f. e., less apparent absorbance at 405 nm over time as compared to
the control
sample).
B. Seeded Extension Assay
The seeded extension assay can be employed to measure the rate of A~3 fiber
formed
in a solution of Ap monomer following addition of polymeric A~ fiber "seed".
The ability of
test compounds to prevent further deposition of monomeric A~i to previously
deposited
amyloid is determined using a direct indicator of ~i-sheet formation using
fluorescence. In
contrast with the nucleation assay, the addition of seed provides immediate
nucleation and
continued growth of preformed fibrils without the need for continuous mixing,
and thus
results in the absence of a lag time before polymerization starts. Since this
assay uses static
polymerization conditions, the activity of positive compounds in the
nucleation assay can be
confirmed in this second assay under different conditions and with an
addition2i probe of
amvloid structure.
In the seeded extension assay. monomeric A~il~p is incubated in the presence
of a
"seed" nucleus (approximately ten mole percent of Al3 that has been previously
allowed to
polymerize under controlled static conditions). Samples of the solution are
then diluted in
thioflavin T (Th-T). The polymer-specific association of Th-T with A~i
produces a
fluorescent complex that allows the measurement of the extent of fibril
fornnation (Levine, H.
(1993) Protein Science 2_:404-410). In particular. association of Th-T with
aggregated (3-AP,
but not monomeric or loosely associated ~3-AP, gives rise to a new excitation
(ex) maximum
at 450 nm and an enhanced emission (em) at 482 nm, compared to the 38~ nm (ex)
and 445
nm (em) for the free dye. Small aliquots of the polymerization mixture contain
sufficient
fibril to be mixed with Th-T to allow the monitoring of the reaction mixture
by repeated
2~ sampling. A linear growth curve is observed in the presence of excess
monomer. The
formation of thioflavin T responsive (3-sheet fibrils parallels the increase
in turbidity observed
using the nucleation assay.
A solution of A(3 monomer for use in the seeded extension assay is prepared by
dissolving an appropriate quantity of A~i~"4p peptide in 1/25 volume of
dimethysulfoxide
(DMSO), followed by water to 1/2 volume and 1/2 volume 2x PBS (10x PBS: NaCI
137 mM,
KCI 2.7 mM Na2HP04 ~ 7H20 4.3 mM, KH2P041.4 mM pH 7.2) to a final
concentration
of 200 ~M. To prepare the stock seed, 1 ml of the A~i monomer preparation, is
incubated for
approximately 8 days at 37 °C and sheared sequentially through an 18,
23, 26 and 30 gauge
needle 25, 25, 50, and 100 times respectively. 2 p1 samples of the sheared
material is taken
for fluorescence measurements after every 50 passes through the 30 gauge
needle until the
fluorescence units (F~ plateau (approx.100-150x). Test compounds are prepared
by
dissolving ar. :.ppropriate amount of test compound in lx PBS to a final
concentration of 1
mM (1 Ox stock). If insoluble. the compound is dissolved in 1/10 volume of
DMSO and
CA 02449296 2003-12-15
64
diluted in la PBS to 1 mM. A further 1/10 dilution is also prepared to test
each candidate at
both 100 pM and 10 ~M.
To perform the seeded extension assay, each sample is set up with 50 ~:1 of
200 pM
monomer, 125 FU sheared seed (a variable quantity dependent on the batch of
seed. routinely
3-6 p1) and 10 ~1 of l Ox modulator solution. The sample volume is then
adjusted to a final
volume of 100 p1 with lx PBS. Two concentrations of each modulator typically
are tested:
100 ~M and 10 ~M, equivalent to a 1:1 and a 1: I 0 molar ratio of monomer to
modulator.
The controls include an unneeded reaction to confirm that the fresh monomer
contains no
seed, and a seeded reaction in the absence of any modulators, as a reference
to compare
IO against candidate modulators. The assay is incubated at 37 °C for 6
h. taking 2 ~1 samples
hourly for fluorescence measurements. To measure fluorescence, a 2 ~I sample
of A/3 is
added to 400 gel of Thioflavin-T solution (50 mM Potassium Phosphate 10 mM
Thioflavin-T~
pH 7.5). The samples are vortexed and the fluorescence is read in a 0.~ ml
micro Quartz
TM
cuvette at EX 450 nm and EM 482 nm (Hitachi 4500 Fluorimeter).
15 p-amyloid aggregation results in enhanced emission of Thioflavin-T.
Accordingly,
samples including an effective inhibitory modulator compound exhibit reduced
emission as
compared to control samples without the modulator compound.
~XAMPI~ 7: Ef~'ect of Different Amino Acid Subregions of A[3 Peptide on the
20 Inhibitory Activity of ~i~Amyloid Modulator Compounds
To determine the effect of various subregions of A~i »p on the inhibitory
activity of a
a ~i-amyloid modulator. overlapping A/3 peptide 1 Smers were constructed. For
each 1 Smer,
four different amino-terminal modif crs were tested: a cholyl group, an
iminobiotinyl group.
25 an N-acetyl neuraminyl group (NANA) and a 5-(and 6-~carboxyfluoresceinyl
group (FICO).
The modulators were evaluated in the nucleation and seeded extension assays
described in
Example 6.
The results of the nucleation assays are summarized below in Table II. The
concentration of Aj3l.dp used in the assays was 50 pM. The "mole %" value
listed in Table II
30 refers to the % concentration of the test compound relative to A(3~-4p.
Accordingly, 100%
indicates that A~i ~,.4p and the test compound were equimolar. Mole % values
less than 100%
indicate that A~i ~~p was in molar excess relative to the test compound
(e.g.,10% indicates
that A~i l.~p was in 10-fold molar excess relative to the test compound). The
results of the
nucleation assays for each test compound are presented in Table II in two
ways. The "fold
35 increase in lag time". which is a measure of the ability of the compound to
delay the onset of
aggregation, refers to the ratio of the observed lag time in the presence of
the test compound
to the observed lag time in the control without the test compound. Accordingly
a fold
increase in lag time of 1.0 indicates no change in lag time. whereas numbers >
1.0 indicate an
increase in lag time. The "% inhibition of plateau", which is a measure of the
ability of the
CA 02449296 2003-12-15
compound to dect~se the total amount of aggregation, refers to the reduction
of the final
turbidity in the presence of the test compound expressed as a percent of the
control without
the test compound. Accordingly, an inhibitor that abolishes aggregation during
the course of
the assay will have a % inhibition of 100. N-terminally modified A~i
subregions which
5 exhibited inhibitory activity are indicated in bold in Table II.
Table II
N-terminal Fotd iacresse% Inhibition
Reference lylodificationg,,d Peptide1e "/ a Lay Tjtnef atea
#
PPI-174 cholyl A~ - 10 0 >4.5 100
PPI-264 c6olyl A~ p 100 >4.5 100
PPI-269 cholyl A(3 _~ 100 1.5 ~-,0
PPI-274 choiyl A~i ~ p 100 >4.5 100
PPI-279 cholyl A l_ 100 1.6 51
PPI-284 cholyl A(3 ~ 100 >4.5 87
PPI-173 NANA A~i ~ _ 100 -1 ~0
1
PPI-266 NANA A~i6.2 100 1.3 64
PPI-271 NANA A(3 _Z 100 13 77
PPI-276 ~1ANA A(31 p 100 ~1 ~-~0
'
PPI-281 NANA A(32 _3 100 1 53
PPI-286 NANA A ~p 100 1.3 -~0
PPI-172 IminobiotinylA/3 _~ 100 1.2 -r0
PPI267 IminobiotinylA~i Ip 100 1.6 44
~
PPI-272 IminobiotinylA~ _2 100 1.2 40
PPI-277 IminobiotinylA~t~3e 100 1.2 55
PPI-282 IminobiotinylA 100 -1 66
PPI-287 IminobiotinylA~i ~ ' 100 2.3 -~0
PPI-190 FICO A~ _ 100 -1 30
PPI-268 FICO A~i _2 100 1.9 .r0
PPI-2?3 FICO A~i -Z 100 1.7 34
PPI-278 FICO A~i ~. 100 1.6 59
PPI-283 FICO A~ 100 L2 25
PPI-288 FICO A~i2~p 100 2 75
These results indicate that certain subregions of A~3l~p, when modified with
an
10 appropriate modifying group, are effective at inhibiting the aggregation of
A[3~.~p. A cholyl
group was an effective modifying group for several subregions. Cholic acid
alone was tested
for inhibitory activity but had no effect on A(3 aggregation. The A(3~2o
subregion exhibited
high levels of inhibitory activity when modified with several different
modifying gmups
(cholyl, NANA. in~inobiotinyl), with choIyl-A~36.2p (PPI-264) being the most
active fonm.
CA 02449296 2003-12-15
66
Accordingly, this modulator compound was chosen for fiuther analysis.
described in
Example 8.
EXAMPLE 8: Identification of a Five Amino Acid Subregion of A~ Peptide
Sufficient for Inhibitory Activity of a ~i-Amyloid Modulator Compound
To further delineate a minimal subregion of cholyl-A(3~2o su~cient for
inhibitory
activity,, a series of amino terminal and carboxy terminal amino acid
deletions of cholyl-
A~3~2o were constructed. The modulators all had the same cholyl amino-terminal
modification. Additionally, for the peptide series having carboxy terminal
deletions, the
carboxy terminus was further modified to an amide. The modulators were
evaluated as
described in Example 7 and the results are summarized below in Table III.
wherein the data is
presented as described in Example 7.
1 S Table III
N-Terns. C-Term. Fold increase~0 Inhibition
~
Ref # ~d. A Pe 'de Mod. Mole in LaQ of Plateau
% Time
PPI-264 cholyl A~~~o - 100 >4.5 100
10 2 43
PPI-341 cholyl A~-2o - 100 >4.5 100
33 2 .-0
PPI-342 cholyl A(3g_ - 100 1.~ --0
33 2.1 -r0
PPI-343 cholyl A/39_ o - 33 2.0 --0
344 cholyl = -.i j. 2.1 -r0
PPI -A(It -20
PPI-345 cholyl A~3 _~o - 33 1.5 ~.-0
PPI-346 cholvl A~3 _~o - 33 2.1 --U
PPI-347 cholyl A~3~3- - 33 2.6 ~-0
o
PPI-348 cholyl A~itZp - 33 2.0 49
PPI-349 cholyl A[3 Z - 33 2.3 50
PPI-350 cholyl A(31~20 - 38 3.4 23
PPI-296 cholyl A~i p amide 33 1.8 ~0
PPI-321 cholyl A(3~~9 amide 33 1.4 -~-0
PPI-325 cholyl A/3 ~ ~ amide 33 1.8 ~.0
PPi-331 cholyl A~i~~4 amide 33 1.0 29
PPI-339 cholyl A[3~1o amide 33 1.1 13
These results indicate that activity of the modulator is maintained when amino
acid
residue 6 is removed from the amino terminal end of the modulator (i.e.,
cholyl-A~i~-2o
retained activity) but activity is lost as the peptide is deleted further at
the amino-terminal end
by removal of amino acid position 7 through to amino acid position 12 (s. e..
cholyl-A(3g.2o
CA 02449296 2003-12-15
67
through cholyl-A~ilg-20 did inhibit the plateau level of A~ agony. However,
fi>rtber
deletion of amino acid position 13 resulted in a compound (i.e., cholyl-A~314-
20) in which
inhibitory activity is restored. Furthermore, additional deletion of amino
acid position 14
(i.e., cholyl-A(ils_2o) or Positions 14 and 15 (i.e:, cholyl-A~i16.2o) still
maintained inhibitory
activity. Thus, amino terminal deletions of A~i~zo identified A~i 120 ~ a
subregion
which is sufficient for inhibitory activity when appropriately modified. In
contrast, carboxy
terminal deletion of amino acid position 20 resulted in loss of activity which
was not fully
restored as the peptide was deleted further at the carboxy-terminal end. Thus,
maintenance of
position 20 within the modulator may be important for inhibitory activity.
~.XAMPLE 9: Identification of a Four Amino Acid Subregion of A~i Peptide
Sufficient for Inhibitory Activity of a (3-Amyloid Modulator Compound
In this example, the smallest effective modulator identified in the studies
described in
Example 8. cholyl-A~i 1 X20 (PPI-3 SO), was analyzed further. Additional amino-
and carboxy-
terminal deletions were made with eholyl-A~i 16.20, as well as an amino acid
substitution
(Val1 g->Thr), to identify the smallest region sufficient for the inhibitory
activity of the
modulaxor. A peptide comprised of five alanine residues, (AIa)5, modified at
its amino-
terminus with cholic acid, was used as a specificity control. The modulators
were evaluated
as described in Example 7 and the results are summarized below in Table IV,
wherein the
data is presented as described in Example 7.
Table IV
N-Term. C-Term. Fold Increase% lnhib'rcion
ef. ~ ~ A~i Mod. le % !~ Time of Plateau
Pe
ric
a
PPI-264 cbolyl A~6. - 10 2.0 43
~ o
PPI-347 cbolyl A~t~zo - 10 ZZ 57
PPI-349 cbotyl A~i1 - 100 >5.0 100
20
33 2.6 35
10 2.1 --~0
PPI-350 cboiyl A lit - 100 >5.0 100
o
10 2.4 40
PPI368 cbolyl A~l~-Zt - 100 >5.0 100
PPI-374 imino- Apl6.2o - 100 13 86
~ biotinyl
PPI-366 cholyl A(31 - 100 3.1 --0
-19
10 1.6 --0
PPI-369 cholyl A~i - 100 ~ 1 --0
1
x.20
(Val
8->Thr)
PPI-370 cholyl A(3t6-2p - 100 2.6 73
~ (Phe
Ala)
PPI-365 cholyl (Ala) - 100 --1 -0
(
CA 02449296 2003-12-15
68
PPI-319 _ cholylA[3 smide 33 S.6 ~-4
~
10 2.7 ~0
PPI-321 cholyl A~ amide 100 1.2 0
PPI-377 - A~ - 100 ~l --0
As shown in Table IV, cholyl-A~il~2o (PPI-350) and cholyl-A~i~~-z~ (PPI-368)
both
exhibited inhibitory activity, indicating that the four-amino acid minimal
subregion of
positions 17-20 is sufficient for inhibitory activity. Loss of.position 20
(e.g., in PPI-366 and
PPI-321) resulted in loss of inhibitory activity, demonstrating the importance
of position 20.
Moreover, mutation of valine at position 18 to threonine (in PPI-369) also
resulted in loss of
activity, demonstrating the importance of position 18. In contrast, mutation
of phenylalanine
at position 19 to alanine (cholyl-A~316-2o Phel9->Ala; PPI-370) resulted in a
compound
which still retained detectable inhibitory activity. Accordingly, the
phenylalanine at position
19 is more amenable to substitution, preferably with another hydrophobic amino
acid residue.
Cholyl-penta-alanine (PPI-365) showed no inhibitory activity. demonstrating
the specificity
of the A~i peptide portion of the modulator. Moreover, unmodified A~i~6-2o
(PPI-377) was
not inhibitory, dern~onstrating the functional importance of the amino-
terminal modifying
group. The specific functional group influenced the activity of the modulator.
For example,
iminobiotinyl-A~it~2o (PPI-374) exhibited inhibitory activity similar to
cholyl-A(3i~2o,
whereas an N-acetyl neuraminic acid (NANA)-modified A~i 1 X20 was not an
effective
inhibitory modulator (not listed in Table IV). A C-terminal amide derivative
of cholyl-A~i~6.
20'(PPI-319) retained high activity is delaying the lag time of aggregation,
indicating that the
carboxy-terminus of the modulator can be derivatized without loss of
inhibitory activity.
Although this amide-derivatized compound did not inhibit the overall plateau
level of
aggregation over time. the compound was not tested at concentrations higher
than mole 33 %.
Higher concentrations of the amide-derivatized compound are predicted to
inhibit the overall
plateau level of aggregation, similar to cholyl-A(31~20 (PPI-350).
EXAMPLE 10: Effect of ~-Amyloid Modulators on the Neurotoxicity
of Natural (3-Amyloid Peptide Aggregates
The neurotoxicity of natural ~i-amyloid peptide aggregates, in either the
presence or.
absence of a (3-amyloid modulator, is tested in a cell-based assay using
either a rat or human
neuronally-derived cell line (PC-12 cells or NT-2 cells, respectively) and the
viability
indicator 3,(4,4-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MT'17.
(See e.g.,
Shearman, M.S. et al. (1994) Proc. Natl. Acad Sci. USA 9_x:1470-1474; Hansen,
M.B. et al.
(1989) J. Immun. Methods 11 :203-210 for a description of similar cell-based
viability
assays). PC-12 is a rat adrenal pheochromocytoma cell line and is available
from the
American Type Culture Collection, Roclcville, MD (ATCC CRL 1721 ). MTT
(commercially
CA 02449296 2003-12-15
69
available from Sigma Chemical Co.) is a chromogeaic substrate that is
converted from yellow
to blue in viable cells, which can be detected specuophotometrically.
To test the neurotoxicity of natural ~3-amyloid peptides, stock solutions of
fresh A(3
monomers and aged A~ aggregates were first prepared. A~i~~p in I00% DMSO was
prepared from lyophilized powder and immediately diluted in one half the final
volume in
H20 and then one half the final volume in 2X PBS so that a final concentration
of 200 ~M
peptide, 4% DMSO is achieved. Peptide prepared in this way and tested
immediately on
cells is referred to ~s "fresh" A~ monomer. To prepare "aged" A~i aggregates,
peptide
TM
solution was placed in a 1.~ ml Eppendorf tube and incubated at 37 °C
for eight days to allow
fibrils to form. Such "aged" A~ peptide can be tested directly on cells or
frozen at
-80°C. The neurotoxicity of fresh monomers and aged aggregates were
tested using PC12
and NT2 cells. PC12 cells were routinely cultured in Dulbeco's modif:ed
Eagle's medium
(DNIEM) containing 10% hone serum, 5% fetal calf serum, 4mM glutamine, and I%
genumycin. NT2 cells were routinely cultured in OPTI-MEM medium (GIBCUBRL CAT.
16 31985) supplemented with 10% fetal calf serum, 2 mM glutamine and 1 %
gentamycin.
Cells were plated at 10-15,000 cells per well in 90 p1 of fresh medium in a 96
-well tissue
culture plate 3-4 hours prior to treaunent. The fresh or aged A~ peptide
solutions ( I 0 pL)
were then diluted 1:10 directly into tissue culture medium so that the final
concentration was
in the range of I-I O pM peptide. Cells are incubated in the presence of
peptide without a
change in media for 48 hours at 37°C. For the final three hours of
exposure of the cells to the
(3-AP preparation. MTI' was added to the media to a final concentration of 1
mglml and
incubation was continued at 37 °C. Following the two hour incubation
with MTT, the media
was removed and the cells were Iysed in 100 pL isopropanoU0.4N HCI with
agitation. An
equal volume of PBS was added to each well and the plates were agitated for an
additional 10
minutes. Absorbance of each well at 570 nm was measured using a microtiter
plate reader to
quantitate viable cells.
The neurotoxicity of aged (S day or 8 day) A~ Lip aggregates alone, but not
fresh
A~3 ~.~p monomers alone, was confirmed in an experiment the results of which
are shown in
Figure 3, which demonstrates that incubating the neuronal cells with
increasing amounts of
fresh A~i ~.~p monomers was not significantly toxic to the cells whereas
incubating the cells
with increasing amounts of 5 day or 8 day A(31~p aggregates led to increasing
atriount of
neurotoxicity. The ECSO for toxicity of aged A~i~.~p aggregates was I-2 pM for
both the
PC12 cells and the NT2 cells.
To determine the effect of a ~i-amyloid modulator compound on the
neurotoxicity of
A~~.4p aggregates. a modulator compound, cholyl-A~iøZp (PPI-264), was
preincubated with
A~il~p monomers under standard nucleation assay conditions as described in
Example 6 and
at particular time intervals post-incubation, aliquots of the ~-AP/modulator
solution were
removed and 1 ) the turbidity of the solution was assessed as a measure of
aggregation and 2)
the solution was applied to cultured neuronal cells for 48 hours at which time
cell viability
CA 02449296 2003-12-15
was assased~usiag IvtTT to determine the n'eurotoxicity of the solution. The
results of the
turbidity analysis are shown in Figure 4, panels A, B and C. In panel A, Aø
1.4p and cholyl-
Aø6.zp were both present at 64 pM. In panel B, Aøt.~p was print at 30 ~NI and
cholyl-
Aø~.2p was present at 64 E,eM. In panel C, Aøl.~p was present at 10 ~M and
cholyl-Aø6.2o
was present at 64 pM. These data show that an equimolar amount of cholyl-
Aø~.2p is
effective at inhibiting aggregation of Aø1"4p (see Figure 4, panel A) and that
as the
concentration of Aø lip is reduced, the amount of detectable aggregation of
the Aø 1-4p
monomer is cowespondingly reduced (compare Figure 4, panels B and C with panel
A). The
corresponding results of the neurotoxicity analysis are shown in Figure 4,
panels D, E, and F.
These insults demonstrate that the ø-amyloid modulator compound not only
inhibits
aggregation of Aø~.4p monomers but also inhibits the neurotoxicity of the
Aø~~p solution,
illustrated by the reduced percent toxicity of the cells when incubated with
the'
Aø~.4plmodulator solution as compared to Aø~.~p alone (see e.g., Figure 4,
panel D).
Moreover, even when Aø l.dp aggregation was not detectable as measured by
light scattering,
the modulator compound inhibited the neurotoxicity of the Aø ~.4p solution
(see Figure 4,
panels E and F). Thus, the formation of neumtoxic Aø t.~p aggregates precedes
the fonaation
of insoluble aggregates detectable by light scattering and the modulator
compound is
effective at inhibiting the inhibiting the formation and/or activity of these
neurotoxic
aggregates. Similar results were seen with other modulator compounds, such as
iminobiotinyl-Aø6_2p (PPI-267), cholyl-Aø»2p (PPI-350) and cholyl-Aø1~2o-amide
(PPI-
319).
Additionally, the ø-amyloid modulator compounds have been demonstrated to
reduce
the neurotoxicity of preformed Aø ~.4p aggregates. In these experiments, Aø
~.~p aggregates
were preformed by incubation of the monomers in the absence of any modulators.
The
modulator compound was then incubated with the preformed A(3~.4p aggregates
for 24 hours
ai 37 °C, after which time the ø-AP/modulator solution was collected
and its neurotoxicity
evaluated as described above. Incubation of preformed Aøl.~p aggregates with
the modulator
compound prior to applying the solution to neuronal cells resulted in a
decrease in the
neurotoxicity of the Aø~-4p solution. These results suggest that the modulator
can either bind
to Aø fibrils or soluble aggregate and modulate their inherent neurotoxicity
or that the
modulator can perivrb the equilibrium between monomeric and aggregated forms
of Aø1.4p
in favor of the non-neurotoxic form.
EXAMPLE 11: Characterization of Additional ø-Amyioid Modulator Compounds
In this example, additional modulator compounds designed based upon amino
acids
17-20 of Aø, LVFF (identified in Example 9), were prepared and analyzed to
further
delineau the structural features necessary for inhibition of ø-amyloid
aggregation. Types of
compounds analyzed included ones having only three amino acid residues of an
Aø
CA 02449296 2003-12-15
71
aggregation-core domain. compounds in which the amino acid residues of an A~
aggregation
core domain were rearranged or in which amino acid substitutions had been
made,
compounds modified with a carboxy-terminal modifying group and compounds in
which the
modifying group had been derivaxized. Abbreviations used in this example are:
h- (free
amino terminus), -oh (free carboxylic acid terminus), -nh2 (amide terminus),
CA (cholyl, the
aryl portion of cholic acid), NANA (N acetyl neuraminyl), IB (iminobiotinyl),
~iA (~i-alanyl),
DA (D-alanyl), Adp (aminoethyldibenzofiuanylpropanoic acid), Aic (3-(O~-
aminoethyl-iso~
cholyI, a derivative,of cholic acid), IY (iodotyrosyl), o-methyl (carboxy-
terminal methyl
ester), N me (N methyl peptide bond), DeoxyCA (deoxycholyl) and LithoCA
(lithocholyl).
Modulator compounds having am Aic modifying group at either the amino- or
carboxy-terminus (e.g., PPI-408 and PPI-418) were synthesized using known
methods (see
e.g., Wess, G, et al. (1993) Tetrahedron Letters, x:817-822; Wess, G. et al.
(1992)
Tetrahedron Letters x:195-198). Briefly, 3-iso-O-(2-aminoethyl)-cholic acid
(3(3-(2-
aminoethoxy)-7a.12a-dihydroxy-S~i-cholanoic acid) was converted to the FMOC-
protected
derivative using FMOC-OSu (the hydroxysuccinimide ester of the FMOC group,
which is
commercially available) to obtain a reagent that was used to introduce the
cholic acid
derivative into the compound. For N-terminal introduction of the cholic acid
moiety, the
FMOC-protected reagent was coupled to the N-terminal amino acid of a solid-
phase peptide
in the standard manner, followed by standard FMOC-deprotection conditions and
subsequent
cleavage from the resin, followed by HPLC purification. For C-terminal
introduction of the
cholic acid moiety, the FMOC-protected reagent was attached to 2-chlorotrityl
chloride resin
in the standard manner. This amino aryl derivatized resin was then used in the
standard
manner to synthesize the complete modified peptide.
The modulators were evaluated in the nucleation and seeded extension assays
described in Example 6 and the results are summarized below in Table V. The
change in lag
time (~L,ag) is presented as the ratio of the lag time observed in the
presence of the test
compound to the lag time of the control. Data are reported for assays in the
presence of 100
mole % inhibitor relative to the concentration of A~ii".4p, except for PPI-
315, PPI-348,
PPI-380, PPI-407 and PPI-418, for which the data is reported in the presence
of 33 mole
inhibitor. Inhibition (% Inuclv) is listed as the percent reduction in the
maximum observed
turbidity in the control at the end of the assay time .~iod: .. inhibition in
the extension assay
('~o Ice") is listed as the percent reduction of thioflavin-T $tmraceace of ~i-
structure in the
presence of 25 mole % inhibitor. Compounds with a % Inucl'n of at least 30%
are highlighted
in bold.
CA 02449296 2003-12-15
T~~,1~ V 72
N-Tens. C-Tam.
PPI-293 CA - -oh 1.0 0 ND*
PPI-315 CA HQKLVFF 1.1 5 ND
PPI-316 NANA HQKLYFF -ah 1.5 -15 ND
PPI-319 CA KLVFF -nh 5.4 70 52
PPI-339 CA HDSGY 1.1- -18 ND
PPI-348 CA HQKLVFF -06 2.0 70 ND
PPI-349 CA Q KLVFF -06 >5 100 56
PPI-350 CA KLVFF -06 1.8 72 11
PPI-365 CA AAAAA -oh 0.8 -7 0
PPI366 CA QKLVF -oh 3.1 -23 ND
PPI-368 CA LVFFA -06 >5 100 91
PPI-369 CA KLTFF oh 1.1 -16 44
PPI-370 CA KLVAF -oh 2.6 73 31
PPI-371 CA KLVFF(A) -oh 2.5 76 80
PPI-372 CA FKFVL -06 0.8 45 37
PFI-373 NANA KLVFF -oh 0.9 16 8
PPI-374 IB KLVFF -oh 13 86 0
PPI-375 CA KTVFF -oh 1.2 18 21
PPI-377 h- KLVFF -oh 1.1 0 8
~PPI-379 CA LVFFAE -oh 1.4 55 16
PPI-380 CA LVFF -06 1.8 72* 51
PPI-381 CA LVFF(DA) -oh 23 56 11
PPI-382 CA LVFFA -nh~ 1.0 -200 91
PPI-383 h-asLa~o~VFF -oh 0.4 14 0
PPI-386 h- LVFFA -oh 1.0 15 11
PPI-387 h- KLVFF -nh~ 1.3 ~ -9 39
PPI-388 CA AVFFA -06 1.4 68 44
PPI-389 CA LAFFA -oh 1.5 47 b6
PPI390 CA LVAFA -oh 2.7 25 0
PPI-392 CA VFFA -06 2.0 76 10
PPI393 CA LVF -oh 1.3 I 0
PPI-394 CA VFF -oh 1.8 55 0
PPI-395 CA FFA -oh 1.0 51 6
PPI-39b CA LV(IY)FA -oh >5 100 71
PPI-401 CA LVFFA -o-methylND . ND 0
PPI-405 h- LVFFA -ah~ 1.3 11 70
PPI-407 CA LVFFK -oh >5 100** 85
-.
PPI-408 6- LVFFA (Aic)-oh3.5 46 3
PPI-418 h-(Aic) LVFFA -oh >5 100** 87
PPI-426 CA FFVLA -oh >5 100 89
PPI-391 CA LVFAA -oh 1.6 40 ND
PPI-397 CA LVF(IY)A -06 >5 95 ND
PPI-400 CA AVAFA -oh 1.0 -I S ND
CA 02449296 2003-12-15
73
PPI-403 * * * HQKLVFF -oh 1.4 -75 0
PPI-404 * * * LKLVFF -oh 1.8 -29 7
*
PPI-424 DeoxyCA LVFFA -oh 3.0 -lI4 82
PPI-425 LithoCA LVFFA -oh 2.8 -229 0
PPI-428 CA FF -oh 1.7 -78 15
PPI-429 CA FFV -oh Z.2 -33 7
PPI-430 CA FFYL -oh 4.1 33 75
PPI-433 CA LVFFA -oh 2.8 27 ND
(all D amino
acids)
PPI-435 t-Boc LVFFA -oh 3.0 -5 ND
PPI-438 CA GFF -oh 1.0 0 ND
' ND = not done
" = 33 mol
"" = h-DDIII(N Me-Val)DLL(Adp)
""= h-DDII(N Me-Leu)VEH(Adp)
Certain compounds shown in Table V (PPI-319, PPI-349, PPI-350, PPI-368 and PPI-
426) also were tested in neurotoxicity assays such as those described in
Example Z 0. For
each compound. the delay of the appearance of neurotoxicity relative to
control coincided
with the delay in the time at which polymerization of A~i began in the
nucleation assays.
This correlation between the prevention of formation of neurotoxic A~i species
and the
prevention of polymerization of A~i was consistently observed for all
compounds tested.
The results shown in Table V demonstrate that at an effective modulator
compound
can comprise as few as three A~i amino acids residues (see PPI-394, comprising
the amino
acid sequence VFF, which corresponds to A~3I$_2o, and PPI-395, comprising the
amino acid
sequence FFA. which corresponds to A(3I9-21 )- The results also demonstrate
that a modulator
compound having a modulating group at its carboxy-terminus is effective at
inhibiting A~i
aggregation (see PPI-408, modified at its C-terminus with Aic). Still further.
the results
demonstrate that the cholyl gmup, as a modulating group, can be manipulated
while
maintaining.the inhibitory activity of the compounds (see PPI-408 and PPI-418,
both of
which comprise the cholyI derivative Aic). The free amino group of the Aic
derivative of
cholic acid represents a position at which a chelation group for ~nTc can be
introduced, e.g.,
to create a diagnostic agent. Additionally, the ability to substitute
iodotyrosyl for
phenylalanine at position 19 or 20 of the A~i sequence (see PPI-396 and PPI-
397) while
maintaining the ability of the compound to inhibit A~i aggregation indicates
that the
compound could be labeled with radioactive iodine, e.g., to create a
diagnostic agent, without
loss of the inhibitory activity of the compound.
Finally, compounds with inhibitory activity were created using A~i derived
amino
acids but wherein the amino acid sequence was rearranged or had a substitution
with a non-
A~-derived amino acid. Exarnples of such compounds include PPI-426, in which
the
seQuence of A~i 1 ~-21 (LVFFA) has been reawanged (FFVLA), PPI-372, in which
tire
CA 02449296 2003-12-15
74
sequence of~Pr~t~.2o (KLVFF~ has been reaeraanged (FKF'VL), and PPI-388, -389
and -390, in
which the sequence of A~i t 7_21 (LVFFA) has been substituted at position
17,18 or 19,
respectively, with an alanine residue (AVFFA for PPI-388, LAFFA for PPI-389
and LVAFA
for PPI-390). The inhibitory activity of these compounds indicate that the
presence in the
compound of an amino acid sequence directly corresponding to a portion of A~i
is not
essential for inhibitory activity, but rather suggests that maintenance of the
hydrophobic
nature of this core region, by inclusion of amino acid residues such as
phenylalaaine, valine,
leucine"regardless of their precise order, can be sufficient for inhibition of
A~i aggregation.
EXAMPLE IZ: Characterization of ~i-Amyloid Modulator Compounds
Comprising an Unmodified ~i-Amyloid Peptide
To examine the ability of unmodified A~ peptides to modulate aggregation of
natural
j3-AP, a series of A~ peptides having amino- and/or carboxy terminal deletions
as compared
to A~it~o, or having intenzal amino acids deleted (i.e.. noncontiguous
peptides), were
prepared. One peptide (PPI-220) had additional, non-A/3-derived amino acid
rasidues at its
amino-terminus. These peptides all had a free amino group at the amino-
terminus and a free
carboxylic acid at the carboxy-terminus. These unmodified peptides were
evaluated in assays
as described in Example 7. The results are summarized below in Table VI,
wherein the data
is presented as described in Example 7. Compounds exhibiting at least a 1.5
fold increase in
lag time are highlighted in bold.
Table VI
Fold Increase% Inhibition
lteferenc~#~ o % _ in g Timeo f plateau
AIi
Peo
'~,ie
PPI ZZ6 ~ ~ 100 i.66 76
A
PPI-227 A~ i 100 -1 47
PPI-Z28 A ~ 100 >4.5 100
PPI-229 A 1 ~ N
PPI-230 A 100 0.8 -.0
o
PPI-231 A~ l~- - ~1 18
-
PPI-247 A~i I00 ~-1 -.~0
.
(Q3I-35)
PPI-?.48 A ~i I00 1.58 -0
_2
(d26-30)
PPI-249 A ~ 100 237 -~0
_
o
(A21-25)
PPI-I50 A~3 100 1.55 --0
,1
(A1620)
PPI-251 A~ 100 ~1.2 ~-0
.
o
t
o
(A11-15)
PPI 152 A~i 100 1.9 33
-
(A6-10)
PPI-253 A~ 100 1.9 -~0
PPI?SO EEVVHHHHQQ-A/3 100 >4 100
CA 02449296 2003-12-15
The results shown in ?able VI demonstrate that limited portions of the A~
sequence can have
a significant inhibitory effect on natural ~-AP aggregation even when the
peptide is not
modified by a modifying group. Preferred unmodified peptides are Aj3g,2o (PPI-
226),
A~1~-30 (PPl-228), A(31-20, 26.40 (fPI-249) and EEwHHHHQQ-A~mo (PPI-Z20), the
amino
5 acid sequences of which are shown in SEQ ID NOs: 4,14,15, and 16,
respectively.
Foaming part of this disclosure is th,e appended Sequence Listing, the
contents of
Which STC summarize in Tabla VII below.
Table VII
SEO I_D NO: gmino A_~ ~gtide Sequence
1 43 amino acids Aft
2 143 amino acids APP C-terminus
3 43 amino acids A(3t~ (19. 20 muted)
4 HDSGYEVHHQKLVFF A~i~
5 HQKLVFFA A(3 4_ t
6 HQKLVFF A~t~. o
7 QKLVFFA A~ t 5-
8 Q~CLVFF A~i
9 KLVFFA A~t6_2t
10 KLVFF A~3t
11 LVFFA A
12 LVFF A(it~_
13 LAFFA A~ ~. t ~ t ~A)
14 KLVFFAEDVGSNKGA Aft o
15 3 5 amino acids ~ A~i t _
16 35 amuio acids EEWHHHHQQ-~iAP t ~o
17 AGAAAAGA FrP peptide
18 AILSS amylin peptide
19 VFF A~it~
20 FFA A
21 FFVLA A(3 ~_ t (scrambled)
22 LVFFK A(31~- t (AZ -~K)
23 ~ LV(I~FA A~ t ~- t ~ t 9-~1~
24 VFFA A~i t g_ t
25 AVFFA A~i ~_ (Lt~-~A)
26 LVF(I~A A~t~- (F2o~I~
27 LVFFAE A(3 ~ ~. ~
28 FFVL A~it~_ (scrambled)
CA 02449296 2003-12-15
76
29 - FKFVL A ~ (scrambled)
30 KLVAF A~i
~
(F
~
->A)
31 KLVFF(~A) A ~ (A2 ->~A)
32 LVFF(DA) A ~i~~. (A2~-FDA)
Those skilled in the art will recognize, or be able to ascertain using no more
than
mutine experimentation, many equivalents to the specific embodiments of the
invention
dcsaibed herein. Such equivalents are intended to be encompassed by the
following claims.
CA 02449296 2003-12-15
77
SEQUSZ1C8 LISTING
(1) GgNBRAL INFORMATION:
S (i) APPLICANT:
(A) NAME: PHARMACBUTICAL PEPTIDES INCORPORATED
(B) STREET: ONE HAMPSHIRE STREET
(C) CITY: CAMBRIDGB
(D) STATE: MASSACHUSETTS
IO (E) COUNTRY: USA
(F) POSTAL CODE (ZIP): 02139-1572
(G) TELEPHONE: (617) 494-8400
(H) TELEFAX: (617) 494-8414
1S (ii) TITLE OF INVENTION: Modulators of Amyloid Aggregation
(iii) NUMBER OF SEQUENCES: 32
(iv) CORRESPONDENCE ADDRESS:
ZO (A) ADDRESSEE: LAIiIVE & COCRFIELD
(B) STREET: 60 State Street, Suite 510
(C) CITY: Boston
(D) STATE: Massachusetts
(E) COUNTRY: USA
ZS (F) ZIP: 02109-1875
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
3O (C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.25
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: 000000
3S (B) FILING DATE: Herewith
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NU1'~ER: USSN 08/404,831
40 (B) FILING DATE: 14-MAR-1995
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: USSN 08/475,579
(B) FILING DATE: 07-JLJN-1995
4S
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NL1MBER: USSN 08/548, 998
(B) FILING DATE: 27-OCT-1995
SO (viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: DeConti, Giulio A.
(B) REGISTRATION NON~ER: 31,503
(C) REFERENCE/DOCKET NDf~ER: PPI-002C2PC
SS (ixf TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (617) 227-7400
(B) TELEFAX: (617)227-5941
CA 02449296 2003-12-15
(2) INP~RMATION FOR SFQ ID NO:1:
(i) SEQUENCE CHAR.P~CTERISTICS:
(A) LENGTH: 43 amino acids
(B) TYPE; amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
IO (v) FRAGMENT TYPE: internal
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: l:
Asp Ala Glu Phe Arg His Asp Ser Gly Tyr Glu Val His His Gln Lys
I5 1 5 10 15
Leu Val Phe Phe Ala Glu Asp Val Gly Ser Asn Lys Gly Ala Ile Ile
20 25 30
20 Gly Leu Met Val Gly Gly Val Val Ile Ala Thr
35 40
(2) INFORMATION FOR SEQ ID N0:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 103 amigo acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(v) FRAGMENT TYPE: internal
3S (xi) SEQUENCE DESCRIPTION: SEQ ID N0:2:
Glu Val Lys Met Asp Ala Glu Phe Arg His Asp Ser Gly Tyr Glu Val
1 5 10 15
His His Gln Lys Leu Val Phe Phe Ala Glu Asp Val Gly Ser Asn Lys
20 25 30
Gly Ala Ile Ile Gly Leu Met Val Gly Gly Val Val Ile Ala Thr Val
35 40 45
Ile Val Ile Thr Leu Val Met Leu Lys Lys Lys Gln Tyr Thr Ser Ile
55 60
His His Gly Val Val Glu Val Asp Ala Ala Val Thr Pro Glu Glu Arg
50 ss ~o ~s so
His Leu Ser Lys Met Gln Gln Asn Gly Tyr Glu Asn Pro Thr Tyr Lys
85 90 95
SS Phe Phe Glu Gln Met Gln Asn
100
(2) INFORMATION FOR SEQ ID N0:3:
CA 02449296 2003-12-15
° i ,
79
(i) SEQU8NCE CiiARACTERISTICS:
(A) LENGTH: 43 amino :cads
(B) TYPE: amino acid
S (D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(v) FRAGMENT TYPE: internal
(ix) FEATURE:
(A) NAME/I~Y: Modified site
(B) LOCATION: 19
(D) OTHER INFORMATION: /note= Xaa is a hydrophobic amino
acid
(ix) FEATURE:
(A) NAME/I~Y: Modified site
(B) IACATION: 20
(D) OTHER INFORMATION: /note= Xaa is a hydrophobic amino
acid
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:3:
2S Asp Ala Glu Phe Arg His Asp Ser Gly Tyr Glu Val His His Gln Lys
1 5 10 15
Leu Val Xaa Xaa Ala Glu Asp Val Gly Ser Asn Lys Gly Ala Ile Ile
20 25 30
Gly Leu Met Val Gly Gly Val Val Ile Ala Thr
40
35 (2) INFORMATION FOR SEQ ID N0:4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 15 amino acids
(8) TYPE: amino acid
(D) TOPOLOGY: linear
4S
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:4:
His Asp Ser Gly Tyr Glu Val His His Gln Lys Leu Val Phe Phe
5 10 15
SO (2) INFORMATION FOR SEQ ID N0:5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 8 amino acids
(B) TYPE: amino acid
SS (D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:5:
CA 02449296 2003-12-15
80
His Gln Lys Leu Val Pht phe Ala
5
(2) INFORMATION
FOR
SEQ
ID N0:6:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 7 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
1$ (xi) SEQUENCE DESCRIPTION: SEQ ID
N0:6:
His Gln Lys Leu Val Phe Phe
5
(2) INFORMATION FOR SEQ ID N0:7:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 7 amino acids
2$ (B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
3O (xi) SEQUENCE DESCRIPTION: SEQ ID N0:7:
Gln Lys Leu Val Phe Phe Ala
5
(2) INFORMATION FOR SEQ ID N0:8:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 6 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
4$ (xi) SEQUENCE DESCRIPTION: SEQ ID NO: B:
Gln Lys Leu Val Phe Phe
5
$0
(2) INFORMATION FOR SEQ ID N0:9:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 6 amino acids
$$ (B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
CA 02449296 2003-12-15
~1
(xi_) SEQ~1C8 DBSCRIPTION: SBQ ID
N0:9:
Lys Leu Val Phe Phe Ala
5
(2) INFORMATION
FOR
SEQ
ID NO:10:
(i) SEQUENCE CHARrACTERISTICS:
(A) LBNGTH: 5 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID
NO:10:
Lys Leu Val Phe Phe
S
(2) INFORMATION
FOR
SEQ
ID NO:11:
(i) SEQUENCE C$AR.ACTERISTICS
(A) LENGTH: 5 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID
NO:11:
Leu Val Phe Phe Ala
5
(2) INFORMATION FOR SEQ ID N0:12:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 4 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:12:
Leu Val Phe Phe
(2) INFORMATION FOR SEQ ID N0:13:
(i) SEQUENCE CHARACTERISTICS:
SS (A) LENGTH: 5 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
CA 02449296 2003-12-15
82
(v) FRAGMENT TYPE: internal
S (xi) SEQUENCE DESCRIPTION: SEQ ID N0:13:
Leu Ala Phe Phe Ala
1 5
(2) INFORMATION FOR SEQ ID N0:14:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 15 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(v) FRAGMENT TYPE: internal
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:14:
2S Lys Leu Val Phe Phe Ala Glu Asp Val Gly Ser Asn Lys Gly Ala
1 5 10 15
(2) INFORMATION FOR SEQ ID N0:15:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 35 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
3S
(ii) MOLECULE TYPE: peptide
(v) FRAGMENT TYPE: internal
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:15:
Asp Ala Glu Phe Arg His Asp Ser Gly Tyr Glu Val His His Gln Lys
1 . 5 IO 15
4S
Leu Val Phe Phe Ser Asn Lys Gly Ala Ile Ile Gly Leu Met Val Gly
20 25 30
Gly Val Val
SO 3s
(2) INFORMATION FOR SEQ ID N0:16:
SS (i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 35 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
CA 02449296 2003-12-15
(i~,) MOLECULE TYPE: peptide 83
(v) FRAGMENT TYPE: internal
$
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:16:
Glu Glu Val Val His His His His Gln Gln Lys Leu Val Phe Phe Ala
1 s 10 is
Glu Asp Val Gly Ser Asn Lys Gly Ala Ile Ile Gly Leu Met Val Gly
2s 30
Gly Val Val
1$ 3s
(2) INFORMATION FOR SEQ ID N0:17:
ZO (i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 8 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
2$ (ii) MOLECULE TYPE: peptide
(v) FRAGMENT TYPE: internal
3O (xi) SEQUENCE DESCRIPTION: SEQ ID N0:17:
Ala Gly Ala Ala Ala Ala Gly Ala
1 5
3$
(2) INFORMATION FOR SEQ ID N0:18:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: s amino acids
40 (B) TYPE: amino acid
(D) TOPOLOGY: linear
iii) MOLECULE TYPE: peptide
4$ (v) FRAGMENT TYPE: internal
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:18:
$0 Ala Ile Leu Ser Ser
1 5
(2) INFORMATION FOR SEQ ID N0:19:
$$
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 3 amino'acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
CA 02449296 2003-12-15
(ii) MOLECULE TYPE: peptide 84
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:19:
S
Val Phe Phe
1
(2) INFORMATION FOR SEQ ID N0:20:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 3 amino acids
(8) TYPE: amino acid
1S (D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:20:
Phe Phe Ala
1
2S (2) INFORMATION
FOR SEQ
ID N0:21:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: S amino acids
(8) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID
N0:21:
3S
Phe Phe Val Leu Ala
1 5
(2) INFORMATION
FOR SEQ
ID N0:22:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 5 amino acids
(B) TYPE: amino acid
4S (D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID
N0:22:
Leu Val Phe Phe Lys
1 5
SS (2) INFORMATION FOR SEQ ID N0:23:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 5 amino acids
(B) TYPE: amino acid
CA 02449296 2003-12-15
r
(D) TOPOLOGY: linear 85
(ii) MOLECULE TYKE: peptide
(ix) FEATURE:
(A) NAME/I~Y: Modified site
(B) LOCATION: 3
(D) OTBER INFORMATION: /note= Xaa is iodotyrosyl
1O (xi) SEQUENCE DESCRIPTION: SEQ ID N0:23:
Leu Val Xaa Phe Ala
1 5
is
(2) INFORMATION FOR SEQ ID N0:24:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 4 amino acids
20 (B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
25 (xi) SEQUENCE DESCRIPTION: SEQ ID N0:24:
Val Phe Phe Ala _
1
(2) INFORMATION
FOR SEQ
ID N0:25:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 5 amino acids
(B) TYPE: amino acid
(D) TOFOLDaY: linear
(ii) MOLECULE TYPE: peptide
4O (xi) SEQUENCE DESCRIPTION: SEQ ID
N0:25:
Ala Val Phe Fhe Ala
1 5
(2) INFORMATION FOR SEQ ID N0:26:
(i) SEQUENCE CHARACITsRISTICS:
(A) LENGTH: 5 amino acids
SO (B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
SS (ix) FEATURE:
(A) NAME/REY: Modified site
(B) LOCATION: 4
(D) OTHER INFORMATION: /note= Xaa is iodotyrosyl
CA 02449296 2003-12-15
r
86
(x~ SBQUErtCE DESCRIPTION: SEQ ID N0:26:
Leu Val Phe Xaa Ala
1
(2) INFORMATION FOR SEQ ID N0:27:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 6 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:27:
Leu Val Phe Phe Ala Glu
1 5
(2) INFORMATION FOR SEQ ID N0:28:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 4 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:28:
Phe Phe Val Leu
1 s
(2) INFORMATION FOR SEQ ID N0:29:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: s amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: peptide
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:29:
Phe Lys Phe Val Leu
1 5
(2) INFORMATION FOR SEQ ID N0:30:
(i) SEQUENCE CHARACTERISTICS:
$$ (A) LENGTH: 5 amino acids
(B) TYPE: amino acid
(D ) TOPOLOGY : l inear
(ii) MOLECULE TYPE: peptide
CA 02449296 2003-12-15
07
(xi) SEQDENCB DBSCRIPTI~1: SEQ ID N0:30:
Lys Leu Val Ala Phe
1 5
(2) INFORMATION FOR SEQ ID N0:31:
1U (i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 6 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
15 (ii) I~LECVLE TYPE: peptide
(ix) FEATURE:
(A) NAME/KEY: Modified site
(B) LOCATION: 6
2~ (D1 OTHER INFORMATION: /note= Xaa is beta-alanyl
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:31:
Lys Leu Val Phe Phe Xaa
25 i
(2) INFORMATION FOR SEQ ID N0:32:
3U (i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 5 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: liaear
35 (ii) MOLECULE TYPE: peptide
(ix) FEATURE:
(A) NAME/KEY: Modified site
(B) LOCATION: 5
4() (D) OTHER INFORMATION: /note= Xaa is D-alanyl
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:32:
Leu Val Phe Phe Xaa
45 i