Note: Descriptions are shown in the official language in which they were submitted.
WO 03/012086 CA 02453522 2004-01-091
PCT/1T02/00488
ANTIGEN PRESENTING CELLS, METHOD FOR THEIR
PREPARATION AND THEIR USE FOR CANCER VACCINES
The present invention relates to the medical field, in particular
to products, substances and compositions for use in methods for the
treatment of human or animal subjects, more in particular for the
diagnosis, treatment, and prevention of cancer. The present
invention relates to cancer vaccines and methods for their
preparation.
Background of the invention
Several tumor-associated antigens (TAA) constitutively
expressed by transformed cells of different histotype have been
recently identified (Renkvist N. et al. Cancer Immunol. Immunother.
50:3-15,2001).
A number of these TAA can provide multiple immunodominant
antigenic peptides recognized by CD8+ cytotoxic T lymphocytes (CTL)
in the context of specific HLA class I allospecificities (Renkvist N. et
al. Cancer Immunol. Irnmunother. 50:3-15,2001), furthermore selected
TAA, such as for example MAGE (Jager E. et at., J. Exp. Med., 187:
265-270, 1998), NY-ESO-1 (Jager E. et at., J. Exp. Med., 187: 265-
270, 1998), SSX (Tureci 0, et al. Cancer Res; 56(20):4766-72 1996),
tyrosinase (Topalian S.L. et al., J. Exp. Med., .183: 1965-1971, 1996.),
Melan-A/MART-1 (Zarour H.M. et al., Proc. Natl. Acad. Sci. USA, 97:
400-405, 2000) concomitantly include epitopes recognized by CD4+
T lymphocytes in the context of specific HLA class IT allospecificities,
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
2
thus being able to induce a TAA-directed humoral immune response
(Wang R.F., Trends Immunol., 22: 269-276, 2001).
Different classes of TAA that may play a major role as
therapeutic targets have been identified:
i) cancer-testis antigens (CTA), expressed in tumors of various
histology but not in normal tissues, other than testis and
placenta such as for example MAGE, GAGE, SSX SART-1,
BAGE, NY-ESO-1, XAGE-1, TRAG-3 and SAGE, some of
which represent multiple families (Traversari C., Minerva
Biotech., 11:243-253, 1999);
ii) differentiation-specific antigens, expressed in normal and
neoplastic melanocytes, such as for example tyrosinase,
Me'an-A/MART-1, gp100/ Prnel 1 7, TRP-1/ gp75, TRP-2
(Traversari C., Minerva Biotech., 11:243-253, 1999);
iii) antigens over-expressed in malignant tissues of different
histology but also present in their benign counterpart, for
example PRAME (Ikeda H. et al., Immunity, 6: 199-208,
1997), HER-2/neu (Traversari C., Minerva Biotech., 11: 243-
253, 1999), CEA, MUC-1(Monges G.M. et al., Am. J. Clin.
Pathol., 112: 635-640, /999), alpha-fetoprotein (Meng W.S.
et al., Mol. Immunol., 37: 943-950, 2001);
iv) antigens derived from point mutations of genes encoding
ubiquitously expressed proteins, such as MUM-1, 13-
WO 03/012086 CA 02453522 2004-01-093
PCT/1T02/00488
catenin, HLA-A2, CDK4, and caspase 8 (Traversari C.,
Minerva Biotech., 11: 243-253, 1999);
v) viral antigens (Traversari C., Minerva Biotech., 11: 243-253,
1999).
In addition to TAA, the cellular elements that are crucial for
their effective immunogenicity and efficient recognition by host's T
lymphocytes include HLA class I and HLA class II antigens, and co-
stimulatory/accessory molecules (e.g., CD40, CD54, CD58, CD80,
CD81) (Fleuren G.J. et al., Immunol. Rev., 145: 91-122, 1995).
Among known classes of TAA, CTA are particularly suitable
therapeutic targets for active specific immunotherapy of cancer
patients, because of their limited expression in normal tissues and
their known in vivo immunogenicity in living subjects, in particular
mammals, humans included (Jager E. et al., J. Exp. Med., 187: 265-
270, 1998; Rejnolds S.R. et al., Int. J. Cancer, 72: 972-976, 1997).
However, the heterogeneous expression of specific CTA among
neoplastic lesions of different patients limits their biological
eligibility to CTA-directed therapeutic vaccination. In fact, malignant
lesions of distinct cancer patients can frequently express only
selected CTA (Sahin U. et al., Clin.. Cancer Res., 6: 3916-3922, 2000),
additionally down-regulated (Let.h.2 B. et at., Melanoma Res., 7: S83-
S88, 1997) and/or heterogeneous (dos Santos N.R. et al., Cancer
Res., 60: 1654-1662, 2000) expression of specific CTA within
individual neoplastic lesions has also been reported (Jungbluth A.A.
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
4
et al., Br. J. Cancer, 83: 493-497, 2000). These events, that can occur
in vivo separately or concomitantly, may also contribute to the
constitutively poor immunogenicity of malignant cells favouring
disease progression (Speiser D.E. et al., J. Exp. Med., 186: 645-653,
1997), and may as well lead to in vivo immuno selection of neoplastic
cells with the emergence of CTA-negative clones, in the course of
immunologic treatment against specific CTA. Thus,
immunotherapeutic approaches that focus on the immunologic
targeting of distinct immunogenic epitopes of single CTA cannot be
applied to large numbers of cancer patients, due to the absence or
the possibly down-regulated expression of target CTA in their
neoplastic lesions; furthermore, the immunological targeting of
single CTA in vivo may generate CTA-loss tumor variants that
efficiently escape treatment-induced/amplified CTA-specific immune
response. An additional limit to therapeutic approaches that target
single CTA derive from their heterogeneous intralesional expression (
Schultz-Thater E. et al., Br. J. Cancer, 83: 204-208, 2000), moreover,
the presentation of distinct immunogenic epitopes of single CTA by
specific HLA class I or HLA class II allospecificities allows treatment
only of patients with certain defined HLA phenotypes.
To partially obviate to these limitations, recent therapeutic
strategies are utilizing more than one immunogenic epitope of single
or multiple CTA, or the whole CTA protein as vaccinating agent
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
5
(Conference on Cancer Vaccines, Eds. Ferrantini M. and Belardelli F.,
Rome-Italy, November 15-16, 1999; http://www.cancerresearch.org).
Accordingly, there is a strongly felt need for a cancer vaccine
which can overcome the drawbacks of the state of the art, in
particular poor immunogenicity, in vivo immunoselection, the
possibility to practice a cancer vaccine on a wide population of
cancer patients, not limited to the specific single targeted CTA, or
TAA, and in that the cancer vaccine not be "restricted" to selected
HLA class I and/or HLA class II antigens.
Recent in vitro evidences have demonstrated that the
expression of all CTA genes that have been investigated, among the
so far known, is induced or up-regulated in neoplastic cells of
different histology following their exposure to DNA hypomethylating
agents (dos Santos N.R. et al., Cancer Res., 60: 1654-1662, 2000;
Weber J. et al., Cancer Res., 54: 1766-1771, 1994). CTA induction
was found to be persistent being still detectable several weeks after
the end of treatment . These findings support the notion that CTA
belong to a class of TAA that is comprehensively regulated by DNA
methylation. Furthermore, treatment of neoplastic cells with DNA
hypomethylating agents induced a concomitant and persistent up-
regulation of their expression of HLA class I antigens and of
investigated HLA class I allospecificities, and also up-modulated the
expression of the co-stimulatory/accessory molecules CD54 and
CD58 (Coral S. et al., J. Imm-unother., 22: 16-24, 1999).
WO 03/012086 CA 02453522 2004-01-09 PCT/1T02/00488
6
Notwithstanding their promising therapeutic profile, CTA,
however, show a number of drawbacks, such as that specific CTA so
far investigated show a heterogeneous expression within distinct
neoplastic lesions, with the co-existence of CTA-positive and -
negative malignant cells; that only selected CTA among the ones so
far identified may be expressed on distinct neoplastic lesions,
independently from their hystological origin; that threshold levels of
expression of specific CTA on neoplastic cells are required for their
recognition by CTA-specific CTL and that vaccination against a
specific CTA requires an appropriate HLA class I and, for selected
CTA also HLA class II phenotype of patients.
Due to their unique biologic features, selected CTA are being
utilized in different clinical trials that aim to induce or potentiate a
CTA-specific immune response in patients affected by malignant
diseases of different histology. Diverse strategies are currently
utilized for the in vivo administration of therapeutic CTA in the clinic
or for the generation of more powerful vaccinating tools at pre-
clinical level (dos Santos N.R. et al., Cancer Res., 60: 1654-1662,
2000; Weber J. et al., Cancer Res., 54: 1766-1771, 1994) as the
person expert in the art is aware of. Noteworthy, mainly due to a
number of technical and practical limitations, only a limited number
of immunogenic epitopes of specific CTA, or single whole CTA protein
are currently utilized in the clinic for the therapeutic purposes.
Following is a list including the main strategies already utilized, or
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
7
hypothesised so far, to administer CTA to cancer patients; it should
also be emphasised that identical strategies are utilized to
administer to patients TAA that belong to the other classes of so far
known TAA, and that different adjuvants and/or carriers are
sometimes utilized to potentiate the immunogenicity of therapeutic
agents.
= Synthetic peptides representing immunogenic epitope(s) of
single or multiple CTA recognized by CD8+ T cells (Conference
on Cancer Vaccines, Eds. Ferrantini M. and Belardelli F., Rome-
Italy, November 15-16, 1999; http://www.cancerresearch.org).
= Liposome-encapsulated synthetic peptides representing
immunogenic epitope(s) of single or multiple CTA (Steller M.A. et
al., Clin. Cancer Res., 4: 2103-2109, 1998).
= Whole synthetic protein of a single CTA (Conference on Cancer =
Vaccines, Eds. Ferrantini M. and Belardelli F., Rome-Italy,
November 15-16, 1999; http / / www.cancerresearch.org).
= Recombinant viral vectors expressing epitopes of single or
multiple CTA recognized by CD8+ T cells (Jenne L. et at., Trends
Immunol., 22:102-107, 2001) .
= Naked DNA shooting (Park J.H. et a/., Mol. Cells, 9: 384-391,
1999) .
= Autologous PBMC/macrophages loaded ex vivo with synthetic
peptides representing epitopes of single or multiple CTA
recognised by CD8+ T cells (Conference on Cancer Vaccines, Eds.
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
8
Ferrantini M. and Belardelli F., Rome-Italy, November 15-16,
1999; http://www.cancerresearch.org).
= Autologous dendritic cells loaded ex vivo with synthetic peptides
representing epitopes of single or multiple CTA recognised by
CD8+ T cells or loaded with whole synthetic protein of a single
CTA, or loaded with whole tumour cell preparations (Conference
on Cancer Vaccines, Eds. Ferrantini M. and Belardelli F., Rome-
Italy, November 15-16, 1999; http://www.cancerresearch.orq;
Jenne L. et al., Trends Immunol., 22:102-107, 2001).
= Autologous dendritic cells transfected or transduced ex vivo
with DNA/RNA to express full-length CTA or fused with whole
tumor cells (Jenne L. et al., Trends Immunol., 22:102-107, 2001);
= Autologous T lymphocytes transfected or transduced ex vivo
with DNA/RNA to express full-length CTA.
As far as autologous cancer vaccines, which the present
invention refers to as the main object, a number of patent references
may be cited. WO 99/42128 discloses methods for determining the
HLA transcription or expression profile of a solid tumor, for selection
of appropriate treatments and/or for monitoring progress of the
tumor. The purpose of this reference is to inhibit some isoforms of
HLA-G in order to increase the native antitumor response. The
method comprises extracting cells from a tumor sample, lysing them
and reacting the lysate with antibodies directed against HLA Class I
antigens.
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
9
DE 29913522 provides an apparatus for preparing a cancer
vaccine by submitting tumor cells extracted from a patient to
pressures of 200-9000 bar, in order to kill or damage the cells while
leaving their surface intact then reinjecting the cells to the patient.
WO 00/02581 discloses a telomerase protein or peptide,
capable of inducing a T cell response against an oncogene or mutant
tumor suppressor protein or peptide. Said peptides are used for a
cancer vaccine.
WO 00/18933 discloses DNA constructs causing expression of
functionally inactive, altered antigens which are unaltered with
respect to the efficiency of transcription and translation of DNA, RNA
or the generation of antigenic peptides. The patient affected by
cancer is treated by the administration of the RNA or plasmid DNA
encoding an altered human cancer associated antigen, in particular
PSMA antigen. In a different embodiment, autologous dendritic cells
that have been exposed in vitro to the RNA or the plasmid DNA are
used as vaccine.
WO 00/20581 discloses a cancer vaccine comprising a new
isolated MAGE-A3 human leukocyte antigen (HLA) class II-binding
peptide. The peptide can also be used to enrich selectively a
population of T lymphocytes with CD4+ T lymphocytes specific the
said peptide. Said enriched lymphocytes are also used as cancer
vaccine.
WO 03/012086 CA 02453522 2004-01-09 PCT/1T02/00488
10
WO 00/25813 discloses universal Tumor-Associated Antigen
(TAA) binding to a major histocompatibility complex molecule. The
method of treatment comprises administering a nucleic acid
molecule encoding the TAA, which is processed by an antigen-
presenting cell which activates cytotoxic lymphocytes and kills cells
expressing TAA. Other than the specific hTERT peptide, the
identification of different TAAs is enabled by a complex computer-
aided method synthesis of the computer-designed peptide and
biological assays for confirmation of the usefulness of the peptide.
WO 00/26249 discloses fragments of human WT-1 protein or
human gata-1 protein. These peptide fragments are used for cancer
vaccine through activation of cytotoxic T lymphocytes (CTL).
US 6077519 provides a cancer vaccine comprising a
composition of T cell epitopes recovered through acid elution of
epitopes from tumor tissue.
WO 00/46352 provides a cancer vaccine comprising human T
lymphocytes that express a functional CD86 molecule. T
lymphocytes are obtained by subjecting T cells to at least two
sequential stimuli, each involving at least one activator (an antibody
anti CD2, 3 or 28) and a cytokine (interleukine) that stimulates T
cell proliferation.
Coral S. et al. Journal of Immunotherapy 22(1):16-24, 1999,
teach that the immunogenic potential of melanoma cells and their
recognition by the host's cytotoxic cells depend on the presence and
WO 03/012086 CA 02453522 2004-01-09PC T/IT02/00488
11
on the level of expression of Human Leukocytic Antigen (HLA) class I
antigens, costimulatory molecules and melanoma-associated
antigens (MAA) on neoplastic cells. There may be a suggestion that
5-AZA-CdR for use in active and) or passive specific immunotherapy
for human melanoma through its systemic administration might
enhance melanoma cells recognition by cytotoxic cells.
Momparler, Anticancer Drugs Apr; 8(4):358-68, 1997, mentions
5-AZA-CdR as chemotherapic.
Shichijo S. et al Jpn. J. Cancer Res. 87, 751-756, July 1996,
investigated whether the demethylating agent 5-AZA-CDR induces
MAGE 1, 2, 3 and 6 in normal and malignant lymphoid cells in order
to better understand the mechanisms of their expression in the
cells. The authors showed the induction of investigated CTA in
selected samples tested and discussed that demethylation is not a
sufficient stimulus to induce MAGE genes in all cases and that their
results should lead to a better understanding of mechanisms of
MAGE genes expression in cells. No perspective therapeutic
implications were suggested.
Abstract of the Invention
It has now been found a method of generation of antigen
presenting cells, comprising:
a) collecting said cells from a subject,
b) activating said collected cells;
CA 02453522 2012-08-09
31129-22
12
c) culturing and optionally expanding ex vivo said activated cells;
d) treating said cultured and optionally expanded cells with
DNA hypomethylating agents so that said cells concomitantly express multiple
tumor
associated antigens.
In one aspect, the invention provides a method for the generation of
Cancer Testis Antigen (CTA) presenting cells comprising: a) activating PBMC
cells
isolated from a human subject; b) culturing said activated cells; wherein c)
said
cultured cells are treated with 1 pM 5-aza-2'-deoxycytidine once every 12
hours for a
total of four treatments; then replacing half of the culture medium with fresh
medium
and allowing to proceed for additional 48 hours so that said cells
concomitantly
express multiple Cancer Testis Antigens (CTA) of the following families: MAGE-
1,
MAGE-2, MAGE-3, MAGE-4, NY-ESO-1, GAGE-1-6 and SSX-2.
In another aspect, the invention provides cells obtained by the method
as described above concomitantly expressing multiple Cancer Testis Antigens of
the
MAGE-1, MAGE-2, MAGE-3, MAGE-4, NY-ESO-1, GAGE 1-6, and SSX-2 families.
In the foregoing, the present invention shall be disclosed in detail also
by means of examples and figures, wherein:
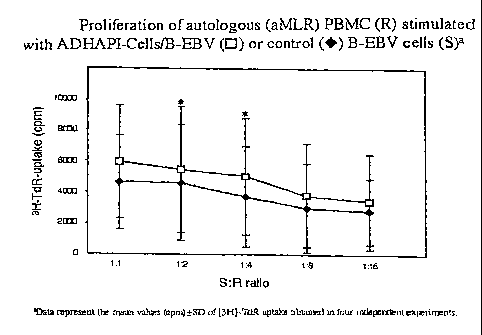
Figure 1 shows the proliferation of autologous (aMLR) PBMC (R)
stimulated with ADHAPI-Cells/B-EBV or control B-EBV cells (S);
Figure 2 shows the proliferation of autologous (aMLR) PBMC (R)
stimulated with ADHAPI-Cells/PWM-B or control PWM-B cells (S);
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
13
Figure 3 shows the proliferation of autologous (aMLR)PBMC (R)
stimulated with ADHAPI-Cells/CD4OL-B or control CD4OL-B cells
(S);
Figure 4 shows the proliferation of autologous (aMLR)PBMC (R)
stimulated with ADHAPI-Cells/PWM-PBMC or control PWM-PBMC
cells (S);
Figure 5 shows the proliferation of autologous (aMLR)PBMC (R)
stimulated with ADHAPI-Cells/PHA-PBMC and control PHA-PBMC;
Figure 6 shows the proliferation of autologous (aMLR)PBMC (R)
stimulated with ADHAPI-Cells/PHA-+PWM-PBMC or control PHA-
+PWM-PBMC (S);
Detailed Disclosure of the Invention
According to the present invention, there is virtually no limit as
to the type of cells that can be treated in order to generate the
antigen-presenting cells, provided that they are suitably activated
and treated with a hypomethylating agent.
According to the present invention, the cells are collected from
a subject, in particular a mammal, more in particular a human. In a
possible embodiment of the present invention, said human is a
cancer patient.
In a first preferred embodiment of the present invention,
antigen-presenting cells obtainable by the method above described
are immune cells.
WO 03/012086 CA 02453522 2004-01-09 PCT/1T02/00488
14
In a second preferred embodiment of the present invention,
antigen-presenting cells obtainable by the method above described
are non-immune cells.
The cells obtainable according to present invention can express
shared immunodominant cancer antigens or can express shared not
immunodominant cancer antigens.
In certain specific embodiments of the present invention, cells
suitable for the method herein disclosed are:
= Epstein-Barr virus-immortalized, DNA hypomethylating
agent-treated B-lymphoblastoid cell lines, generated from
peripheral blood mononuclear cells (PBMC) of cancer
patients in advanced stage of disease or healthy subjects
(ADHAPI-Cells/B-EBV).
= Pokeweed mitogen (PWM)-activated, DNA hypomethylating
agent-treated B-lymphocytes, generated from B-
lymphocytes purified from PBMC of cancer patients in
advanced stage of disease or healthy subjects (ADHAPI-
Cells/PWM-B).
= CD40 activated, DNA hypomethylating agent-treated B-
lymphocytes, generated from B-lymphocytes purified from
PBMC of cancer patients in advanced stage of disease or
healthy subjects (ADHAPI-Cells/CD4O-B).
= Pokeweed mitogen (PW114)-activated, DNA hypomethylating
agent-treated PBMC, generated from purified PBMC of
WO 03/012086 CA 02453522 2004-01-0915
PCT/1T02/00488
cancer patients in advanced stage of disease or healthy
subjects (ADHAPI-Cells/PWM-PBMC)
= Phytohemagglutinin (PHA) + recombinant human
interleukin-2 (rhIL-2)-activated, DNA hypomethylating
agent-treated PBMC, generated from purified PBMC of
cancer patients in advanced stage of disease or healthy
subjects (ADHAPI-Cells/PHA-rhIL2-PBMC)
= Phytohemagglutinin (PHA) + recombinant human
interle-ukin-2 (rhIL-2) + pokeweed mitogen (PWM)-activated,
DNA hypomethylating agent-treated PBMC, generated from
purified PBMC of cancer patients in advanced stage of
disease or healthy subjects (ADHAPI-Cells/PHA-rhIL2-
PWM-PBMC)
= Dendritic cells, monocytes, macrophages.
= CD34+ cells, fibroblasts, stem cells, fibroblasts and
cheratinocytes.
The cells obtainable by the method according to the present
invention are suitable for use as agents for the prevention and
treatment of malignancies of different histotype that constitutively
express one or more of cancer antigens, whether immunodominant
or not immunodominant.
Another possible embodiment of the present invention is
applicable to those cases wherein it is not wished or necessary to
utilize the direct antigen presenting ability of vaccinating cells. In
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
16
this case, vaccinating cells or their cellular components obtainable
by the method of the present invention can be used as "reservoir" of
pooled cancer antigens to vaccinate patients.
In a preferred embodiment of the present invention, the
selected TAA are CTA.
This embodiment of the present invention offers to the skilled
person the following advantages:
CTA are immunogenic since they include epitopes recognized
by HLA class I-restricted CTA-specific CD8+ CTL.
CTA are immunogenic since they include epitopes recognized
by HLA class II-restricted CTA-specific CD4+ T lymphocytes.
Selected CTA simultaneously include epitopes presented by
HLA class I and by HLA class II antigens; thus, selected CTA can
concomitantly induce CD8+ CTL and CD4+ T lymphocytes reactions.
CTA are not expressed in benign tissues with the exception of
testis and placenta.
Different CTA can be concomitantly expressed in neoplastic
cells of solid and hemopoietic malignancies, providing multiple
therapeutic targets that are co-expressed on transformed cells.
Distinct CTA are homogeneously expressed among
concomitant and sequential metastatic lesions of given patients.
Distinct CTA can be expressed in malignant tissues of different
hystological origin providing common therapeutic targets shared by
human neoplasia regardless of their specific hystotype.
WO 03/012086 CA 02453522 2004-01-0917
PCT/1T02/00488
Distinct CTA may encode for multiple immunogenic peptides
presented in the context of different HLA class I and HLA class II
allospecificities.
In a further embodiment of the present invention, histone
deacetylase inhibitors can sinergize with DNA hypomethylating
agents in inducing/up-regulating the expression of CTA, of HLA
antigens and of co-stimulatory/accessory molecules on neoplastic
cells of different histology. In fact, DNA methylation and histone
deacetylation act as synergistic layers for the epigenetic gene
silencing in cancer (Fuks F. et al., Nat. Genet., 24: 88-91, 2000), and
a strong reactivation of selected hypermethylated genes, with tumor
suppressor function, has been observed in colorectal carcinoma cells
after treatment with histone deacetylase inhibitors, following an
initial minimal DNA dernethylation (Cameron E.E. et al., Nat. Genet.,
21: 103-107, 1999).
=
The activation step in the method according to the present
invention is carried out following the general common knowledge, in
any case reference can be made to Current Protocols in Immunology,
Coligan J.E. et al. Eds, Wiley.
The demethylation treatment in the method according to the
present invention is generally well-known and the literature
generally reports the procedure, for further information see also
Santini V. et al., Ann. Intern. Med., 134: 573-586, 2001.
WO 03/012086 CA 02453522 2004-01-0918
PCT/1T02/00488
Hypomethylating agents, also known in the art as
demethylating agents, useful for the purposes of the present
invention are well known in the art. DNA demethylating agents are
widely disclosed in the literature, see for example WO 01/29235, US
5,851,773. A preferred DNA demethylating agent is 5-aza-cytidine
or, more preferred, 5-aza-2'-deoxycytidine (5-AZA-CdR).
Antigen presenting cells according to the present invention are
suitable for the preparation of cancer vaccines. In a preferred
embodiment of the present invention, said vaccines are autologo-us
vaccines.
In another preferred embodiment of the present invention, said
vaccines are allogeneic vaccines. In this embodiment, the cells
obtainable according to the method above disclosed may be used
both as antigen presenting cells and as in the form of "reservoir" of
pooled cancer antigens, whether as cells or cellular components
thereof. In a still further another embodiment of the present invention,
the cells and/or the cellular components can be used in a method
for generating effector immune cells, said effector immune cells
being used for the preparation of a product useful in the well-known
adoptive immunotherapy. In another embodiment of the present
invention, the vaccine herein disclosed can be used in combination
with a systemic pre-treatment of the cancer patient with a
hypomethylating agent, for example decitabine. This embodiment
CA 02453522 2010-03-29
29072-42
19
may be performed with an article of manufacture, for example a kit,
comprising a vaccine according to the present invention and a
pharmaceutical composition suitable for systemic administration of
a hypomethylating agent, for example decitabine.
Vaccines can be prepared according to techniques well-know to
the person skilled in this art, just resorting to the general common
knowledge. For example, the patent references mentioned in the
present description are a sufficient disclosure for the preparation of
cancer vaccines, see for example WO 00/25813 or WO 00/46352.
The skilled person will have no difficulty in establishing the
proper manner for using the vaccines according to the present
invention, in particular as to the administration protocol.
The following examples further illustrate the present invention.
EXAMPLE 1
ADHAPI-Cells/B-EBV
PBMC purification
PBMC were purified by standard Ficoll-Hypaque* density
gradient centrifugation from heparinized peripheral blood of cancer
patients in advanced stage of disease or healthy subjects.
Generation of autologous B-lymphoblastoid cell lines by the
immortalization of PBMC with Epstein-Barr Virus (EBV)
B-EBV+ lymphoblastoid cell lines were generated by incubating
PBMC with supernatant from 1395.8 marmoset cell line at 37 C in a
5% CO2 humidified atmosphere, in RPMI 1640 medium
*Trade-mark
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
20
supplemented with 10% heat-inactivated foetal calf serum (or
human AB serum) and 2 mM L-glutamine.
Generation of ADHAPI-Cells/B-EBV and control B-EBV cells
B-EBV+ lymphoblastoid cell lines (7.5x105 cells/ml) were
cultured in RPMI 1640 medium supplemented with 10% heat-
inactivated foetal calf serum (or 10% heat-inactivated human AB
serum) and 2 mM L-glutamine at 37 C in a 5% CO2 humidified
atmosphere, and pulsed four times with 1 jiM 5-aza-2'-deoxycytidine
(5-AZA-CdR) every 12 h; then, half of the culture medium was
replaced with fresh medium and cultures were allowed to proceed
for additional 48 h. Then cells were utilized for experimental
procedures and/or frozen under viable conditions. Control cells
(B-EBV cells) were cultured under similar experimental conditions
but without pulses of 5-AZA-CdR.
Final recovery of ADHAPI-Cells/B-EBV and control B-EBV= cells
For the results, see Table I.
Autologous Mixed Lymphocyte Reaction (aMLR) and MLR
ADHAPI-Cells/B-EBV and control B-EBV cells (stimulators=S)
were collected, washed twice with Hanks' balanced salt solution
(HBSS) and x-ray treated (75 Gy). For aMLR and MLR scalar
concentrations (from 1x106 cells/ml to 6x104 cells/ml) of ADHAPI
Cells/B-EBV or control B-EBV cells were added to autologous or
allogeneic PBMC (1x106 cells/ml) (responder=R) in Basal Iscove's
medium supplemented with 10% heat-inactivated human AB serum,
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
21
2 mM L-glutarnine, 100 Wm]. penicillin, 100 jig/m1 streptomycin
sulphate, and seeded in 96 well U-bottom plates to a final volume of
200 pl/well. After a 24 h incubation at 37 C in a 5%CO2 humidified
atmosphere, 100 1 of culture supernatant were collected and
immediately stored at -80 C until use for cytokine assay. Then, 100
ill of fresh medium were added to each well and cultures were
allowed to proceed for additional five days, when cultures were
pulsed 0/N with 3H-TdR (1 pLCi/well); then plates were harvested
and 3H-TdR incorporation by R cells was measured by a 13-counter.
Proliferation of autologous PBMC (R) stimulated with
ADHAPI-Cells/B-EBV or control B-EBV cells (S) in aMLR
See Figure 1.
Phenotypic profile of ADHAPI-Cells/B-EBV and control B-EBV
cells
See Table II for results."
RT-PCR analysis of CTA expressed by ADHAPI-Cells/B-EBV
and control B-EBV cells
Experimental conditions and primers utilized to assess CTA
expression on investigated cells were as follows:
for MAGE-1, -2, -3, -4 Brasseur, F., et al. Int. J. Cancer 63: 375-
380, 1995; for GAGE 1-6 Van den Eynde, B., et al. J. Exp. Med. 182:
689-698, /995; for NY-ESO-1 Stockert, E. et al. J. Exp. Med. 187:265-
270, 1998; for SSX-2 ; Sahin, U., et al. Clin. Cancer Res. 6: 3916-
3922, 2000.
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
22
5-AZA-CdR
MAGE-1 0/4a 4/4
MAGE-2 NT NT
MAGE-3 0/4 4/4
MAGE-4 NT NT
NY-ESO-1 0/4 4/4
GAGE-1-6 0/4 4/4
SSX-2 2/4 4/4
a positive/tested; NT, not tested;
ELISA evaluation of IFN-y released by PBMC (R) stimulated in
aMLR by ADHAPI-Cells/B-EBV or control B-EBV cells (S)
See Table III for results.
EXAMPLE 2
ADHAPI-Cells/PWM-B
B-Lymphocyte purification
PBMC were purified by standard Ficoll-Hypaque density
gradient centrifugation from heparinized peripheral blood of cancer
patients in advanced stage of disease or healthy subjects, and
purified B lymphocytes were obtained by conventional E rosetting
technique utilizing neuraminidase-treated sheep red blood cells.
Generation of PWM-activated B cells
Purified B-Lymphocytes (1.5x106 cells/ml) were added with
PWM (3 Ag/m1) and cultured for 48 h at 37 C in a 5% CO2
humidified atmosphere in Basal Iscove's medium supplemented with
10% heat-inactivated human AB serum, 2 mM L-glutamine, 100
U/ml penicillin, 100 g/ml streptomycin sulphate.
Generation of ADHAPI-Cells/PWM-B and control PWM-B cells
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
23
PWM-activated B-Lymphocytes were pulsed four times with 1
1.1M 5-aza-2'-deoxycytidine (5-AZA-CdR) every 12 h; then, half of the
culture medium was replaced with fresh medium and cultures were
allowed to proceed for additional 48 h. Then cells were utilized for
experimental procedures and/or frozen under viable conditions.
Control cells (PWM-B) were cultured under similar experimental
conditions but without pulses of 5-AZA-CdR.
Final recovery of ADHAPI-Cells/PWM-B and control PWM-B
cells
See Table I for results
Autologous Mixed Lymphocyte Reaction (aMLRI and MLR
ADHAPI-Cells/PWM-B and control PWM-B cells (stimulators-S)
were collected, washed three times with Hanks' balanced salt
solution supplemented with 0.5% a-methylmannopyranoside, and
x-ray treated (30 Gy). For aMLR and MLR scalar concentrations
(from 1x106 cells/m1 to 6x104 cells/ml) of ADHAPI-Cells/PWM-B or
control PWM-B cells were added to autologous or allogeneic PBMC
(1x106 cells/ ml) (responder= R) in Basal I scove' s medium
supplemented with 10% heat-inactivated human AB serum, 2 mM
L-glutamine, 100 U/ml penicillin, 100 pg/m1 streptomycin sulphate,
and seeded in 96 well U-bottom plates to a final volume of 200
1.1.1/well. After a 6 day incubation at 37 C in a 5% CO2 humidified
atmosphere, 100 i_t1 of culture supernatant were collected from each
well and immediately stored at -80 C until use for cytokine assay.
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
24
Then, 100 1 of fresh medium were added to each well and cultures
were pulsed 0/N with 3H-TdR (1 Ci/well); then, plates were
harvested and 3H-TdR incorporation by R cells was measured by a
13-counter.
Phenotypic profile of ADHAPI-CellsJPWM-B and control PWM-B
cells.
See Table II for results
RT-PCR analysis of CTA expressed by ADHAPI-Cells/PWM-B
and control PWM-B cells
5-AZA-CdR - +
MAGE-1 0/4a 4/4
MAGE-2 0/4 4/4
MAGE-3 0/4 4/4
MAGE-4 0/4 4/4
NY-ESO-1 0/4 4/4
GAGE- 1-6 0/4 4/4
SSX-2 1/4 4/4
a positive/tested; NT, not tested.
Proliferation of autologous PBMC (R) stimulated with
ADHAPI-Cells/PWM-B or Control PWM-B cells (S) in aMLR
See Figure 2 for results.
ELISA evaluation of IFN-y released by allogeneic (MLR) and
autologous (aMLR) PBMC (R) stimulated with ADHAPI-Cells/PWM-B
or control PWM-B cells (Si
See Table III for results.
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
25
EXAMPLE 3
ADHAPI-Cells/CD4OL-B
PBMC purification
PBMC were purified by standard Ficoll-Hypaque density
gradient centrifugation from heparinized or acid citrate dextrose
(ACD)-anticoagulated peripheral blood of cancer patients in
advanced stage of disease or healthy subjects.
Generation of NIH3T3-CD4OL-activated PBMC
PBMC (2x106 cells/ml) were co-cultured with semiconfluent,
x-ray treated (75 Gy) NIH3T3-CD4OL at 37 C in a 5% CO2 humidified
atmosphere, in Basal Iscove's medium supplemented with 10%
heat-inactivated human AB serum, 2 mM L-glutamine, 2 ng/ml
recombinant human (rh) interleukin 4 (rhIL-4), 50 ig/m1 human
transferrin, 5 ii.g/m1 rh insulin, 5.5x10-7 M cyclosporin A (CsA), 100
U/m1 penicillin, and 100 jig/m1 streptomycin sulphate (complete
medium). After six days of incubation, PBMC were collected, washed
twice with HBSS, resuspended at 1X106 cells/ml in complete
medium and co-cultured for additional 3 days at 37 C in a 5% CO2
humidified atmosphere with NIH3T3-CD4OL freshly prepared as
described above. This procedure was repeated every 2-3 days to a
maximum culture time of 16-18 days.
Generation of ADHAPI-Cells/CD4OL-B and control CD4OL-B
cells
WO 03/012086 CA 02453522 2004-01-09 PCT/1T02/00488
26
After 16-18 days of culture, activated PBMC were harvested
and restimulated with NIH3T3-CD4OL as described above; after an
0/N incubation at 37 C in a 5% CO2 humidified atmosphere,
cultures were pulsed four times with 1 piM 5-aza-2'-deoxycytidine
(5-AZA-CdR) every 12 h; then, cells were harvested and
restimulated with NIH3T3-CD4OL as described above and cultures
were allowed to proceed for additional 48 h. Then cells were utilized
for experimental procedures and/or frozen under viable conditions.
Control cells (CD4OL-B cells) were cultured under similar
experimental conditions but without pulses of 5-AZA-CdR.
Final recovery of ADHAPI-Cells/CD4OL-B and control CD4OL-B
cells
See Table I for results.
Autologous Mixed Lymphocyte Reaction (aMLR) and MLR
ADHAPI-Cells/CD4OL-B and control CD4OL-B cells
(stimulators=S) were collected, washed three times with Hanks'
balanced salt solution and x-ray treated (50 Gy). For aMLR and MLR
scalar concentrations (from 1x106 cells/ml to 6x104 cells/ml) of
ADHAPI-Cells/CD4OL-B or control CD4OL-B cells were added to
autologous or allogeneic PBMC (1x106 cells/ml) (responder=R) in
Basal Iscove's medium supplemented with 10% heat-inactivated
human AB serum, 2 mM L-glutamine, 100 U/ml penicillin, 100
jig/m1 streptomycin sulphate, and seeded in 96 well U-bottom plates
to a final volume of 200 1/well. After a 24 h incubation at 37 C in a
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
27
CO2 humidified atmosphere, 100 pi of culture supernatant were
collected and immediately stored at -80 C until use for cytokine
assay. Then, 100 .1 of fresh medium were added to each well and
cultures were allowed to proceed for additional 5 days when cultures
were pulsed 0/N with 3H-TdR (1 pei/well), then plates were
harvested and 3H-TdR incorporation by R cells was measured by a
13-counter.
Phenotypic profile of ADHAPI-Cells/CD4OL-B and control
CD4OL-B cells
See Table II for results.
RT-PCR analysis of CTA expressed by ADHAPI-Cells/CD4OL-B
and control CD4OL-B cells
5-AZA-CdR - +
MAGE-1 0/10a 10/10
MAGE-2 0/10 9/10
MAGE-3 0/11 10/11
MAGE-4 0/11 11/11
NY-ESO-1 0/14 14/14
GAGE- 1-6 0/14 14/14
SSX-2 0/14 13/14
a positive/tested.
Proliferation of autologous (aMLR) PBMC (R) stimulated with
ADHAPI-Cells/CD4OL-B or control CD4OL-B cells (S) in aMLR
See Figure 3 for results.
ELISA evaluation of IFN-y released by PBMC (R1 stimulated in
aMLR by ADHAPI-Cells/CD4OL-B or control CD4OL-B cells (S)
WO 03/012086 CA 02453522 2004-01-0928
PCT/1T02/00488
See Table III for results.
ADHAPI-Cells/PWM-PBMC EXAMPLE 4
PBMC purification
PBMC were purified by standard Ficoll-Hypaque density
gradient centrifugation from heparinized peripheral blood of cancer
patients in advanced stage of disease or healthy subjects.
Generation of PWM-activated PBMC
PBMC (1.5x106 cells/ml) were added with PWM (3 ,g/m1) and
cultured for 48 h at 37 C in a 5% CO2 humidified atmosphere in
Basal Iscove's medium supplemented with 10% heat-inactivated
human AB serum, 2 mM L-glutarnine, 100 U/ml penicillin, 100
jig/m1 streptomycin sulphate.
Generation of ADHAPI-Cells/PWM-PBMC and control
PWM-PBMC cells
PWM-activated PBMC were pulsed four times with 1 pM
5-aza-2'-deoxycytidine (5-AZA-CdR) every 12 h; then, half of the
culture medium was replaced with fresh medium and cultures were
allowed to proceed for additional 48 h. Then cells were utilized for
experimental procedures and/or frozen under viable conditions.
Control cells (PWM-PBMC) were cultured under similar experimental
conditions but without pulses of 5-AZA-CdR.
Final recovery of ADHAPI-Cells/PWM-PBMC and control
PWM-PBMC cells
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
29
See Table I for results.
Autologous Mixed Lymphocyte Reaction (aMLR) and MLR
ADHAPI-Cells/PWM-PBMC and control PWM-PBMC cells
(stimulators=S) were collected, washed three times with Hanks'
balanced salt solution supplemented with 0.5%
a-methylmannopyranoside, and x-ray treated (30 Gy). For aMLR and
MLR scalar concentrations (from 1X106 cells/ml to 6x104 cells/ml) of
ADHAPI-Cells/PWM-PBMC or control PWM-PBMC cells were added
to autologous or allogeneic PBMC (1X106 cells/m1) (responder=R) in
Basal Iscove's medium supplemented with 10% heat-inactivated
human AB serum, 2 mM L-glutamine, 100 U/m1 penicillin, 100
jig/m1 streptomycin sulphate, and seeded in 96 well U-bottom plates
to a final volume of 200 111/well. After a 6 day incubation at 37 C in
a 5% CO2 humidified atmosphere, 100 jil of culture supernatant
were collected from each well and immediately stored at -80 C until
use for cytokine 100 jii of fresh medium were added to each well
were pulsed 0/N with 3H-TdR (1 viCi/well), then harvested and
3H-TdR incorporation by R cells was measured by a 13-counter.
Phenotypic profile of ADHAPI-Cells/PWM-PBMC and control
PWM-PBMC cells
See Table II for results.
RT-PCR analysis of CTA expressed by ADHAPI-
Cells/PWM-PBMC and control PWM-PBMC cells
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
30
5-AZA-CdR - +
MAGE-1 0/4a 4/4
MAGE-2 0/4 3/4
MAGE-3 0/4 4/4
MAGE-4 1/4 3/4
NY-ESO-1 0/4 4/4
GAGE-1-6 0/4 3/4
SSX-2 0/4 3/4
apositive/tested; NT, not tested.
Proliferation of autologous (aMLR) PBMC (R) stimulated with
ADHAPI-Cells/PWM-PBMC or control PWM-PBMC cells (S) in
aMLR
See Figure 4 for results.
ELISA evaluation of IFN-7 released by autologous PBMC -(R)
stimulated in aMLR by ADHAPI-Cells/PWM-PBMC or control
PWM-PBMC cells (S)
Sec Table III for results.
EXAMPLE 5
ADHAPI-Cells/PHA-PBMC
PBMC purification
PBMC were purified by standard Ficoll-Hypaque density
gradient centrifugation from heparinized peripheral blood of cancer
patients in advanced stage of disease or healthy subjects.
Generation of PHA-activated PBMC
PBMC (1.5x106 cells/ml) were added with PHA-M (10 p.g/m1)
and 100 UI/ml rhIL-2, and cultured for 48 h at 37 C in a 5% CO2
CA 02453522 2004-01-09
WO 03/012086 31 PCT/1T02/00488
humidified atmosphere in RPMI 1640 medium supplemented with
10% heat-inactivated foetal calf serum (or in Basal Iscove's medium
supplemented with 10% heat-inactivated human AB serum), 2 mM
L-glutamine, 100 U/ml penicillin, 100 1.1g/m1 streptomycin sulphate
(complete medium).
Generation of ADHAPI-Cells/PHA-PBMC and control
PHA-PBMC
PHA-activated PBMC were pulsed four times with 1 1.i.M
5-aza-2'-deoxycytidine (5-AZA-CdR) every 12 h; then, half of the
culture medium was replaced with fresh complete medium without
PHA-M and cultures were allowed to proceed for additional 48 h.
Then cells were utilized for experimental procedures and/or frozen
under viable conditions. Control cells (PHA-PBMC) were cultured
under similar experimental conditions but without pulses of
5-AZA-CdR.
Final recovery of ADHAPI-Cells/PHA-PBMC and control
PHA-PBMC
See Table I for results.
Autologous Mixed Lymphocyte Reaction (aMLR) and MLR
ADHAPI-Cells/PHA-PBMC and control PHA-PBMC
(stimulators=S) were collected, washed three times with Hanks'
balanced salt solution supplemented with 0.5% a-
methylmannopyranoside, and x-ray treated (50 Gy). For aMLR and
MLR scalar concentrations (from lx106cells/m1 to 6x104cells/m1) of
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
32
ADHAPI-Cells/PHA-PBMC or control PHA-PBMC were added to
autologous or allogeneic PBMC (1x106 cells/ml) (responder-R) in
Basal Iscove's medium supplemented with 10% heat-inactivated
human AB serum, 2 mM L-glutamine, 100 U/ml penicillin, 100
p.g/m1 streptomycin sulphate and seeded in 96 well U-bottom plates
to a final volume of 200 I/well. After a 24 h incubation at 37 C in a
5% CO2 humidified atmosphere, 100 [11 of culture supernatant were
collected from each well and immediately stored at -80 C until use
for cytokine assay. Then, 100 1 of fresh medium were added to each
well and cultures were allowed to proceed for additional 5 days when
cultures were pulsed 0/N with 3H-TdR (1 pei/well); then, plates
were harvested and all-TdR incorporation by R cells was measured
by a p-counter.
Phenotypic profile of ADHAPI-Cells/PHA-PBMC and control
PHA-PBMC.
See Table II for results.
RT-PCR analysis of CTA expressed by
ADHAPI-Cells/PHA-PBMC and control PHA-PBMC
5-AZA-CdR - +
MAGE-1 0/12a 12/12
MAGE-2 0/3 3/3
MAGE-3 0/12 12/12
MAGE-4 0/4 4/4
NY-ESO-1 0/6 6/6
GAGE-1-6 0/4 4/4
SSX-2 0/6 6/6
WO 03/012086 CA 02453522 2004-01-0933
PCT/1T02/00488
apositive/ tested; NT, not tested.
Proliferation of autologous PBMC (R) stimulated with ADHAPI-
Cells/PHA-PBMC and control PHA-PBMC in aMLR
See Figure 5 for results.
ELISA evaluation of IFN-y (released by allogeneic (MLR) and
autologous (aMLR) PBMC (R) stimulated with ADHAPI-Cells/PHA-
PBMC or control PHA-PBMC(S)
See Table III for results.
ADHAPI-Cells/PHA+PVC/M-PBMC EXAMPLE 6
PBMC purification
PBMC were purified by standard Ficoll-Hypaque density
gradient centrifugation from heparinized or ACD-anticoagulated
peripheral blood of cancer patients in advanced stage of disease or
healthy subjects.
Generation of PHA+PWM-activated PBMC
PBMC (1.5x106 cells/ml) were added with PHA-M (10 tig/m1),
PWM (3 lig/m1), 100 UI/ml rhIL-2 and cultured for 48 h at 37 C in a
5% CO2 humidified atmosphere in Basal Iscove's medium
supplemented with 10% heat-inactivated human AB serum (or with
10% heat-inactivated autologous serum), 2 mM L-glutarnine, 100
/ ml penicillin, 100 p.g/m1 streptomycin sulphate (complete
medium).
Generation of ADHAPI-Cells/PHA+PWM-PBMC and control
WO 03/012086 CA 02453522 2004-01-0934
PCT/1T02/00488
PHA+PWM-PBMC
PHA+PWM-activated PBMC were pulsed four times with 1 tilµn
5-aza-2'-deoxycytidine (5-AZA-CdR) every 12 h; then, half of the
culture medium was replaced with fresh complete medium without
PHA or PWM and cultures were allowed to proceed for additional 48
h. Then cells were utilized for experimental procedures and/or
frozen under viable conditions. Control cells (PHA+PWM-PBMC) were
cultured under similar experimental conditions but without pulses
of 5-AZA-CdR.
Final recovery of ADHAPI-Cells/PHA+PWM-PBMC and control
PHA+PWM-PBMC
See Table I for results.
Autologous Mixed Lymphocyte Reaction (aMLR) and MLR
ADHAPI-Cells/PHA+PWM-PBMC and control PHA+PWM-PBMC
(stimulators-S) were collected, washed three times with Hanks'
balanced salt solution supplemented with 0.5% a-
methylmannopyranoside, and x-ray treated (50 Gy). For aMLR and
MLR scalar concentrations (from lx106cells/m1 to 6x104cells/m1) of
ADHAPI-Cells/PHA-rhIL2-+PWM-PBMC or control PHA+PWM-PBMC
were added to autologous or allogeneic PBMC (1x106cells/m1)
(respon.der=R) in Basal Iscove's medium supplemented with 10%
heat-inactivated human AB serum, 2 mM L-glutamine, 100 U/ml
penicillin, 100 vt.g/m1 streptomycin sulphate, and seeded in 96 well
U-bottom plates to a final volume of 200 Ill/well. After a 6 day
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
35
incubation at 37 C in a 5% CO2 humidified atmosphere, 100 l of
culture supernatant were collected from each well and immediately
stored at -80 C until use for cytokine assay. Then, 100 pl of fresh
medium were added to each well and cultures were pulsed 0/N with
3H-TdR (1 pCi/well); then, plates were harvested and 3H-TdR
incorporation by R cells was measured by a (3-counter.
Phenotypic profile of ADHAPI-Cells/PHA+PWM-PBMC and
control
PHA+PWM-PBMC
See Table II for results.
RT-PCR analysis of CTA expressed by
ADHAPI-Cells/PHA+PWM-PBMC and control PHA+PWM-PBMC
5-AZA-CciR - +
MAGE-1 0/7a 7/7
MAGE-2 0/7 7/7
MAGE-3 0/7 7/7
MAGE-4 0/7 7/7
NY-ESO-1 0/7 7/7
GAGE-1-6 0/7 7/7
SSX-2 0/7 7/7
apositive/ tested.
Proliferation of autologous (aMLR) PBMC (R) stimulated with
ADHAPI-Cells/PHA+PWM-PBMC or control PHA+PWM-PBMC in
aMLR
See Figure 6 for results.
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
36
ELISA evaluation of IFN-y released by allo_eneic (MLR) and
autologous (aNILR) PBMC (RI stimulated with ADHAPI-
Cells/PHA+PWM-PBMC or control PHA+PWM-PBMC (S).
See Table III for results.
In Vivo Tumorigenicity of ADHAPI-Cells
Single subcutaneous xenografts of viable ADHAPI
Cells/PHA-rhIL2-PWM-PBMC (12x106) and their control cells
(14x106), ADHAPI-Cells /CD4OL-B (8x106) and their control cells
(8x106) or x-ray-treated (30 Gy) ADHAPI-Cells/PHA-rhIL2
PWM-PBMC (12x106) and their control cells (14x106), x-ray treated
(50 Gy) ADHAPI-Cells /CD4OL-B (15x106) and their control cells
(18x106), neither induced tumor formation at injection or distant
(clinically explorable) sites, nor affected general health and weight of
BALB/c nu/nu mice 180 days after ADHAPI-Cells administration.
Repeated subcutaneous xenografts of viable ADHAPI-Cells/B-EBV
(5x106/ 1st injection; 1x107/ 2nd and subsequent injections) and
control B-EBV cells (5x106/1st injection; 1X107/2nd and subsequent
injections) or x-ray-treated ADHAPI-Cells/B-EBV (75 Gy) (5x106/1st
injection; lx 1 07 / 2nd and subsequent injections) and x-ray-treated
(75 Gy) control B-EBV cells (5x106/ 1st injection; 1x107/ 2nd and
subsequent injections), at day 0, 33, 63 and 96, neither induced
tumor formation at injection or distant (clinically explorable) sites,
nor affected general health and weight of BALB/c nu/nu mice 180
days after the first administration. General health and weight of
WO 03/012086 CA 02453522 2004-01-0937
PCT/1T02/00488
ADHAPI-Cells-treated animals was comparable to control animals,
untreated or grafted with B-EBV cells.
Advantages of ADHAPI- Cells as Polyvalent Cellular CTA
Vaccines
As compared to the main strategies already utilized, or so far
hypothesised, to most effectively administer known CTA to cancer
patients, ADHAPI-Cells represent a totally new and innovative
approach, and comprise a number of prominent/remarkable
advantages. Among these:
ADHAPI-Cells vs not genetically-modified cellular CTA vaccines
ADHAPI-Cells are new and unique APC vaccines as they
concomitantly express multiple/all methylation-regulated CTA;
being endogenously synthesised, CTA
can directly and
simultaneously access both HLA class I and HLA class II antigen
processing pathways within ADHAPI-Cells (Jenne L. et al., Trends
Immunol., 22:102-107, 2001).
Thus, due to their constitutive cell membrane expression of
both HLA class I and HLA class II antigens, ADHAPI-Cells can
concomitantly present immunogenic epitopes of endogenously
synthesised CTA both to CD8+ and to CD4+ T autologous
lymphocytes; therefore, ADHAPI- Cells can simultaneously
induce/amplify a CTA-directed CTL and humoral immune
responses. Additionally, ADHAPI-Cells may express and present to
host's T cells methylation-regulated CTA that have not been
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
38
identified and characterized yet (as well as not immunodominant
epitopes of known and still unknown CTA).
Opposite to ADHAPI-Cells, synthetic CTA peptide(s)-pulsed,
synthetic CTA whole protein-pulsed, or whole tumor cell
preparations-pulsed autologous APC vaccines (e.g., dendritic cells,
PBMC), as well as electrofusion-generated tumor cell dendritic cell
hybrids (Kugler A. et at., Nat. Med., 6: 332-336, 2000. Tureci 0. et at.,
Cancer Res., 56: 4766-4772, 1996. Eds), share major limitations
including: i) the unknown fate in vivo of the ex vivo-loaded synthetic
CTA peptide(s), of whole synthetic CTA protein or of tumor-derived
CTA, which may significantly affect the longevity of antigen
presentation to host's immune system; ii) limited amounts of
synthetic CTA peptide(s), of whole synthetic CTA protein or of
tumor-derived CTA that can be loaded ex vivo onto HLA class I
and/or HLA class II antigens of cellular vaccines, which may
significantly hamper the immunogenicity of administered CTA;
the restriction by the patient's HLA phenotype, and the still
relatively limited number of known HLA class I antigens- and even
more HLA class II antigens restricted immunogenic epitopes of so far
identified CTA; iv) availability of adequate amounts of fresh tumor
tissue, that should also be sufficiently representative of the diverse
CTA expressed in neoplastic lesions (Jenne L. et al., Trends Irnmunol.,
22:102-107, 2001).
WO 03/012086 CA 02453522 2004-01-0939
PCT/1T02/00488
The expression of endogenously synthesised CTA by ADHAPI-
Cells is long lasting; thus, at variance with ex vivo synthetic CTA
peptide(s)-pulsed or synthetic CTA whole protein-pulsed or whole
tumor cell preparations-pulsed autologous APC vaccines, ADHAPI-
Cells can provide a prolonged stimulation in vivo of hosts immune
response and with a lower number of administrations to patients.
This hypothesis is reinforced by the foreseen possibility to
administer ADHAPI-Cells as a viable, not x-ray-treated, cellular
vaccines due to their absence of long term tumorigenicity in vivo.
Furthermore, once ADHAPI-Cells would undergo physiological death
in vivo, they could still act as a "reservoir" of endogenously
synthesised CTA peptides and proteins, that could further and
efficiently boost the presentation of HLA class I-restricted epitopes of
CTA to CD8+ T cells by patient's dendritic cells, through the
immunologic mechanism of cross-priming, as well as the
presentation of HLA class II-restricted epitopes of CTA to CD4+ T
cells, through the well-defined exogenous pathway of antigen
processing.
ADHAPI-Cells retain their APC function; in fact, they efficiently
stimulate the proliferation and IFN-y release of autologous and
allogeneic PBMC; furthermore, ADHAPI-Cells are in most instances
more potent stimulators as compared to their respective control
cells. In this respect, it is relevant that in addition to CTA, ADHAPI-
Cells may concomitantly express higher levels of HLA class I
WO 03/012086 CA 02453522 2004-01-0940
PCT/1T02/00488
antigens and/or of different co-stimulatory/accessory molecules as
compared to their respective control cells. These evidences clearly
represent a great advantage of ADHAPI-Cells as autologous cellular
vaccines, compared to autologous tumor cells that are poorly
immunogenic, and do not constitutively express several
co-stimulatory/accessory molecules. Furthermore, as compared to
ex vivo-generated and expanded autologous dendritic cells, ADHAPI-
Cells vaccines are generated by fully mature and immunocompetent
APC; this aspect overcomes the potential limitation represented by
the maturation stage of dendritic cells utilized for the generation of
cellular vaccines, which may influence their tolerogenic rather than
immunogenic potential.
As compared to other cellular vaccines, the ex vivo generation
of ADHAPI-Cells vaccines, that concomitantly express multiple/all
methylation-regulated CTA, is simple, in most cases rapid, does not
=
require cumbersome in vitro cellular manipulations, does not involve
genetic manipulations, does not require autologous tumor tissue,
and it is highly reproducible both from PBMC of healthy individuals
and cancer patients.
Furthermore, close to 100% of ADHAPI-Cells preparations
express all investigated CTA that are demethylation-inducible in
APC. Due to these characteristics, the generation of ADHAPI-Cells
vaccines is easier to standardize and to control for quality (for
example by flow cytometry for selected cell surface molecules and
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
41
RT-PCR for selected CTA) and potency (for example by quantitative
RT-PCR for selected CTA). Additionally, compared to other cellular
vaccines that to date must be freshly prepared each time they must
be administered to patients, thus generating obvious
inter-preparations variability (e. g., cellular viability, phenotypic
profile of vaccinating cells, amount of loaded synthetic CTA
peptide(s) or synthetic CTA whole protein or of whole tumor cell
preparations, efficiency of generation of tumor cell-dendritic cell
hybrids by electrofusion), ADHAPI-Cells vaccines, once prepared and
checked for viability, quality and potency, can be aliquoted,
appropriately frozen, and stored under viable conditions until use for
therapeutic purposes. Furthermore, since they do not require the
availability of autologous tumor tissue to pulse autologous cellular
vaccines or to generate tumor cell-dendritic cell hybrids ex vivo, and
since they can be rapidly prepared in large number from repeated
leukaphereses, ADHAPI-Cells vaccines represent a practically
unlimited source of therapeutic agent for each patient.
In light of their concomitant expression of multiple/all
methylation-regulated CTA that are endogenously synthesised, and
that can directly and simultaneously access the HLA class I and
HLA class II antigen-processing pathway, owing to their possibility to
express and present to host's T cells methylation regulated CTA that
have not been identified and characterized yet (as well as not
immunodominant epitopes of known and still unknown CTA), and
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
42
due to the still limited number of known HLA class I antigens- and
HLA class II antigens-restricted immunogenic epitopes of so far
identified CTA that can thus be utilized for therapeutic applications
according to patient's HLA phenotype, an additional advantage of
ADHAPI-Cells is that they are most likely able to concomitantly
present known and still unknown immunogenic epitopes of different
CTA in the context of any and multiple HLA class I and HLA class II
allospecificities. Thus, as compared to synthetic CTA
peptide(s)-pulsed or synthetic CTA whole protein-pulsed cellular
vaccines, treatment with ADHAPI-Cells vaccines is not limited to
patients with defined HLA phenotypes; hence, all cancer patients
whose neoplastic lesions express one or more CTA can be candidate
to treatment with ADHAPI-Cells vaccines, regardless of their HLA
phenotype. In this respect, among the so far known CTA, one or
more of them is generally expressed in most investigated
malignancies of different histotype; therefore, vaccination with
ADHAPI-Cells is suitable in the large majority of cancer patients. A
significant information is that MAGE, GAGE or NY-ESO-1 are
expressed in 96% of human tumors (Cancer Immunol. Immunother.
50:3-15, 2001).
Compared to synthetic CTA peptide(s)-pulsed and synthetic
CTA whole protein-pulsed cellular vaccines, in which limited
amounts of protein(s) can be loaded ex vivo onto HLA class I and/or
HLA class II antigens of cellular vaccines, significantly hampering
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
43
the immunogenicity of administered CTA, and due to their
concomitant expression of multiple all methylation-regulated CTA,
ADHAPI-Cells vaccines can overcome the immunoselection of
CTA-negative tumor variants occurring in the course of treatment
against single or few CTA, and overcome the constitutively
heterogeneous and sometimes down-regulated expression of distinct
CTA occurring in specific neoplastic lesions.
ADHAPI-Cells vaccines are constituted by autologous
functional APC that concomitantly express multiple! all known
methylation-regulated CTA, and that most likely express still
unidentified CTA whose expression is regulated by DNA methylation;
furthermore, ADHAPI-Cells vaccines can be utilized in patients
affected by CTA-positive tumors of different histotype. These
functional and phenotypic features represent a clear advantage over
currently utilized allogeneic tumor cell vaccines (e.g., lysates of
whole pooled neoplastic cell lines or their non-purified extracts, shed
antigens from pooled neoplastic cell lines). In fact, -these tumor cell
vaccines may not contain or may contain insufficient amounts of
known and of still unknown immunologically-relevant CTA, contain
irrelevant cellular components that may compete with CTA for
immunological responses, may have increased toxicity being
allogeneic, require efficient processing by patients' immune system,
and can be utilized exclusively in patients affected by malignancies
of the same histologic type.
WO 03/012086 CA 02453522 2004-01-0944
PCT/1T02/00488
ADHAPI-Cells vs genetically modified cellular CTA vaccines
The generation of ADHAPI-Cells does not involve the ex vivo
genetic manipulations of autologous dendritic cells or of other
autologous APC, that are required to produce genetically-modified
cellular vaccines expressing selected CTA following transfection or
transduction. Furthermore, as compared to ADHAPI/Cells, a
number of limitations affect genetically-modified cellular vaccines;
among these are: i) the relative low
efficiency of available
transfection methodologies; ii) the induction of cellular immune
responses against antigens of the viral vectors utilized for cellular
transduction, which leads to the destruction of genetically-modified
vaccinating cells; the presence of
pre-existing or
vaccination-induced neutralizing antibodies that interfere with
vaccine administration(s); iv) direct effects of viral vectors on the
viability, maturation and antigen-presentation ability of transduced
cells (Jenne L. et al., Trends Imm-unol., 22:102-107, 2001).
WO 03/012086 CA 02453522 2004-01-0945
PCT/1T02/00488
Table I
Recovery of ADHAPI-Cells and control cells
Cell Type ADHAPI-Cells
Control cells
B-EBVa 114 25
175 51
.PWM-Bb 16 5
38 17
CD4OL-Be 75 27
96 5
PWM-PBMCd 26 11
45 16
PHA-PBMCe 23 10
63 25
PHAA-PWM-PBMCf 35 28
63 36
a Data represent the mean % SD of recovered cells as compared to
the number of cells (100%) utilized for their generation in 3 (a), 4 (b),
4 (c), 4 (d), 7 (e) and 5 (f) independent experiments.
=
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
46
Table II
Phenotypic profile of ADHAPI-Cells compared to
autologous control cells*
Antigen ADHAPI Cells
CD4OL- B-EBITh Vw-m- PWM-Bd PHA- PHA+P
Bat PBMCc PBMCe WM-PB
MCf
HLA ns f ns ns ns ns 0.02g '
Class I _
HLA-A ns ns nth nt 0.004 nt
Locus _ _
HLA-B ns ns nt nt 0.05 nt
Locus
HLA-A 0.01 ns ns ns 0.008 0.006
Alleles
HLA-B ns ns nt nt ns nt
Alleles _
CD40 ns 0.01 ns 0.005 ns ns
CD54 0.03 0.01 ns ns 0.003 ns '
HLA ns ns ' ns 0.03 ns 0.05
Class II 1
CD56 nt nt nt ns nt nt '
CD58 ns ns nt ' nt ns ns
-1
CD59 nt nt ns ' ns nt 0.04
CD80 0.05 ns ns ns ns ns
CD81 nt 0.002 nt nt ns nt
_
CD86 0.008 nt ns ns nt ns
* Data were obtained comparing by Student's paired t-test the
mean values of mean fluorescence intensity obtained by flow
cytometry in 6 (a), 6 (b), 4 (c), 4 (d), 6 (e), and 2 (f) independent
CA 02453522 2004-01-09
WO 03/012086 PCT/1T02/00488
47
experiments. Statistically significant differences were invariably
representative of an up-regulated expression of investigated antigen
on ADHAPI-Cells compared to autologous control cells.
f not significant;
g p value;
h not tested;
tADHAPI-Cells/CD4OL-B= 82-100% CD20+; Control CD4OL-B
cells= 87-99% CD20+.
Table III.
Enzyme-linked immunosorbent assay (ELISA) evaluation of
IFN-y released by autologous (R) (aMLR) and allogeneic (MLR) PBMC
(R) stimulated by ADHAPI-Cells (S) or by control cells (S).*
Cell type AMLR _ MLR
Control ADHAPI- Control cells ADHAPI-Cells
cells Cells
B-EBVa 1770 919 2360 85 nth nt
0.5g
PWM-Bh 4330 629 5530 804 4040 721 4950 476
0.06 0.08
CD40L-Bc 330 197 429 153 nt nt
0.1
PWM-PBMCd 1500 135 1520 175 nt nt
0.6
PHA-pBMCe 140 70 956 436 267 119 1040 545
0.1 0.07
PHA+PWM+ 790 236 831 244 819 184 830 169
PBMCf 0.09 0.7
*Data represent the mean values SD of IFN-y (pg/m 1)
released in two (a), four (b), four (c), four (d), three (e) and four (f)
WO 03/012086 CA 02453522 2004-01-09PCT/1T02/00488
48
independent experiments. S/R ratios were: 3:1(a), 1: 1 (b), 1:2 (e), 1:
1 (d), 1: 1 (e), 1: 2 (f). IFN-y release was assayed 24 h (a), six days (b),
24 h (c), six days (d), 24 h (e) and 6 days (f) after the beginning of
culture; g p value vs control cells obtained by Student's paired t-test;
h not tested.