Note: Descriptions are shown in the official language in which they were submitted.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
1
COMPOUNDS USEFUL FOR TREATMENT OR PREVENTION OF
DISEASE MEDIATED BY ALPHA-2B-ADRENOCEPTOR
The present invention relates to the use of selective alpha-2B-adrenoceptor
antagonists for the manufacture of a pharmaceutical preparation useful for the
treatment or prevention of diseases mediated by the alpha-2B-adrenoceptor in
mammals.
BACKGROUND OF THE INVENTION
The publications and other materials used herein to illuminate the background
of
the invention, and in particular, cases to provide additional details
respecting the
practice, are incorporated by reference.
It is known that alpha-2B-adrenoceptors mediate vascular contractions.
Therefore,
alpha-2B-antagonists are useful in the treatment or prevention of diseases
involving vascular contraction. It has also been found that certain
individuals have
a genetic polymorphism in the alpha-2B-adrenoceptor gene. It has been observed
that the alpha-2B-adrenoceptor protein in some subjects has a deletion of 3
glutamates from the glutamic acid repeat element of 12 glutamates (amino acids
297-309), in an acid stretch of 17 amino acids, located in the third
intracellular
loop of the receptor polypeptide (WO 01/29082; Heinonen et al., 1999).
OBJECTS AND SUMMARY OF THE INVENTION
An object of this invention is to provide compounds useful for the treatment
or
prevention of a disease mediated by the alpha-2B-adrenoceptor in a mammal.
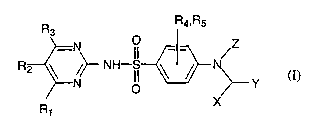
Thus this invention concerns a novel compound of formula (I)
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
2
R R4~Rs
3
-N
R2 ~ ~~ NH II ~ ~ N (I)
N O ~--- Y
R X
1
or a pharmaceutically acceptable salt thereof.
R1 , R2 , R3 , R4 and RS are independently of each other H , a straight or
branched alkyl or alkoxy group with 1 to 4 carbon atoms, or a halogen;
X is H , a straight or branched alkyl chain with 1 to 4 carbon atoms, phenyl
or
-OH;
Z is H , acetyl, -CH2-Ph-O-CF3 or -CH2-Ph-CF3 , where Ph is phenyl;
Y is a ring structure optionally linked to formula (I) with an alkyl chain
having
one or two carbon atoms, wherein the ring structure is
a) phenyl optionally mono- or disubstituted and each substituent is
independently selected from the group consisting of a halogen, a straight or
branched alkyl or alkoxy chain with 1 to 4 carbon atoms, a halogen
substituted methyl or methoxy group, a nitrile, an amide, amino, or a vitro
group;
1 S b) 2-benzimidazolyl, 2-imidazolyl, or 2- or 3-indolyl, wherein one N
optionally has a substituent that is a straight or branched alkyl or alkoxy
chain with 1 to 4 carbon atoms, or benzyl; and wherein the
2-benzimidazolyl, 2-imidazolyl, or 2- or 3-indolyl is optionally mono- or
disubstituted and each substituent can independently be a straight or
branched alkyl or alkoxy group with 1 to 4 carbon atoms, or a halogen;
(c) pyridinyl optionally mono- or disubstituted and each substituent can
independently be a straight or branched alkyl or alkoxy group with 1 to 4
carbon atoms, or a halogen; or
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
3
(d) naphthyl optionally mono- or disubstituted and each substituent can
independently be a straight or branched alkyl or alkoxy group with 1 to 4
carbon atoms, or a halogen.
The following previously known compounds are excluded: 4-[(IH-benzimidazol-
2-ylmethyl)-amino]-N (4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide
(Kumar & Reddy, 1985), N (4,6-dimethylpyrimidin-2-yl)-4-[(1-methyl-1H-
benzimidazol-2-ylmethyl)-amino] -benzenesulfonamide (No 653716, ChemBridge
Corporation, 16981 Via Tazon, Suite G, San Diego CA 92127) and
N-(4,6-dimethylpyrimidin-2-yl)-4-[(1-ethyl-1H-benzimidazol-2-ylmethyl)-amino]-
benzenesulfonamide (No AE-848/34956037, SPECS and BioSPECS B. V.,
Fleminglaan 16, 2289 CP Rijswijk, The Netherlands) and N-(4-methyl-2-
pyrimidinyl)-4-[(1H-benzimidazol-2-ylinethyl)-amino]-benzenesulfonamide
(Farag & El-Mouafi & Khalifa, 1991).
This invention further concerns use of compound of formula (I)
Ra~ Rs
O _~_ /Z
NH-II ~ ~ (I)
-- Y
R X
or a pharmaceutically acceptable salt thereof for the manufacture of a
pharmaceutical preparation useful for the treatment or prevention of a disease
mediated by the alpha-2B-adrenoceptor in a mammal wherein
Rl , R2 , R3 , R4 and RS are independently of each other H , a straight or
branched alkyl or alkoxy group with 1 to 4 carbon atoms, or a halogen;
X is H , a straight or branched alkyl chain with 1 to 4 carbon atoms, phenyl,
-OH or =O ;
Z is H, acetyl, -CH2-Ph-O-CF3 or -CH2-Ph-CF3 , where Ph is phenyl;
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
4
Y is a ring structure optionally linked to formula (I) with an alkyl chain
having
one or two carbon atoms, wherein the ring structure is
a) phenyl optionally mono- or disubstitued and each substituent is
independently selected from the group consisting of a halogen, a straight or
branched alkyl or alkoxy chain with 1 to 4 carbon atoms, a halogen
substituted methyl or methoxy group, an acetyl, a nitrile, an amide, amino,
or a intro group;
b) 2-benzimidazolyl, 2-imidazolyl, or 2- or 3-indolyl, wherein one N
optionally has a substituent that is a straight or branched alkyl or alkoxy
chain with 1 to 4 carbon atoms, or benzyl; and wherein the
2-benzimidazolyl, 2-imidazolyl, or 2- or 3-indolyl is optionally mono- or
disubstituted and each substituent can independently be a straight or
branched alkyl or alkoxy group with 1 to 4 carbon atoms, or a halogen;
(c) pyridinyl optionally mono- or disubstituted and each substituent can
independently be a straight or branched alkyl or alkoxy group with 1 to 4
carbon atoms, or a halogen; or
(d) naphthyl optionally mono- or disubstituted and each substituent can
independently be a straight or branched alkyl or alkoxy group with 1 to 4
carbon atoms, or a halogen.
The following compounds previously known to be selective alpha-2B-
adrenoceptor antagonists are excluded: N (4,6-dimethylpyrimidin-2-yl)-
4-[(1-methyl-1H-benzimidazol-2-ylmethyl)-amino] -benzenesulfonamide
(No 653716, ChemBridge Corporation, 16981 Via Tazon, Suite G, San Diego
CA 92127), N-(4,6-dimethylpyrimidin-2-yl)-4-[(1-ethyl-1H-benzimidazol-2-yl
methyl)-amino] -benzenesulfonamide (No AE-848/34956037, SPECS and
BioSPECS B. V., Fleminglaan 16, 2289 CP Rijswijk, The Netherlands) and N-[4-
(4,6-dimethylpyrimidin-2-ylsulfamoyl)-phenyl]-4-ethoxy-benzamide (No AF-
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
399/36012031, SPECS and BioSPECS B. V., Fleminglaan 16, 2289 CP Rijswijk,
The Netherlands).
DETAILED DESCRIPTION OF THE INVENTION
Preferred compounds of the invention are compounds of formula (I)
R R4~ R5
3
-N
R2 ~ ~~- NH S ~ ~ N (I)~
N ~ ~Y
R X
5
as defined in the preceding summary or pharmaceutically acceptable salts
thereof
wherein Ri and R3 are methyl and R2 , R4 and RS are H.
In some preferable compounds X is H, Y is a phenyl optionally mono- or
disubstituted with a straight or branched alkoxy group and Z is H. Compounds
fulfilling all of the aforementioned characteristics and wherein said phenyl
is
substituted and said alkoxy substituent is methoxy are
4-(2,4-dimethoxybenzylamino)-N-(4,6-dimethylpyrimidin-2-yl)-benzenesulfon-
amide, N (4,6-dimethylpyrimidin-2-yl)-4-(3-methoxybenzylamino)-benzenesulfon-
amide, 4-(3,5-dimethoxybenzylamino)-N (4,6-dimethylpyrimidin-2-yl)-
benzenesulfonamide, 4-(2,5-dimethoxybenzylamino)-N-(4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide and N (4,6-dimethylpyrimidin-2-yl)-4-(2-methoxybenzyl-
amino)-benzenesulfonamide.
In other preferred compounds X is H, Y is a phenyl optionally mono- or
disubstituted with a straight or branched alkyl and/or a halogen and Z is H.
These comprise compounds such as 4-benzylamino-N-(4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(2-methylbenzyl-
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
6
amino)-benzenesulfonamide, 4-(2,4-dimethylbenzylamino)-N (4,6-dimethyl-
pyrimidin-2-yl)-benzenesulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(3-methyl-
benzylamino)-benzenesulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(4-methyl-
benzylamino)-benzenesulfonamide, 4-(2,5-dimethylbenzylamino)-N-(4,6-
dimethylpyrimidin-2-yl)-benzenesulfonamide, 4-(2,6-dimethylbenzylamino)-N
(4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide, 4-(4-bromobenzylamino)-N
(4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide and 4-(2,6-
dichlorobenzylamino)-N-(4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide.
Further preferred compounds are N (4,6-dimethylpyrimidin-2-yl)-4-[(1-ethyl-1H-
indol-3-ylmethyl)-amino]-benzenesulfonamide, N-(4,6-dimethylpyrimidin-2-yl)-4-
[(1-isobutyl-1H-benzimidazol-2-ylmethyl) -amino] -benzenesulfonamide, N-(4,6-
dimethylpyrimidin-2-yl)-4-(1-phenylethylamino)-benzenesulfonamide, N-(4,6-
dimethylpyrimidin-2-yl)-4-[2-(2-methoxyphenyl)-ethylamino]-benzenesulfon-
amide and N-(4,6-dimethylpyrimidin-2-yl)-4-[(naphthalen-2-ylmethyl)-amino]-
benzenesulfonamide.
According to one embodiment of the invention the compound is N-(4-methyl-2-
pyrimidinyl)-4-[ [( 1-methyl-1H-benzimidazol-2-yl)-methyl]amino]-benzenesulfon-
amide.
The invention also relates to the use of selective alpha-2B-adrenoceptor
antagonists of formula (I)
Ra~ Rs
0 ~Z
NH-II ~ ~ (I)~
--- Y
X
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
7
as defined in the preceding summary, or a pharmaceutically acceptable salt
thereof
for the manufacture of a pharmaceutical preparation.
In many preferable compounds to be used Rl and R3 are typically methyl and
R2, R4 and R5 are typically H.
In some compounds preferably used X is H, Y is a phenyl optionally mono- or
disubstituted with a straight or branched alkoxy group and Z is H. Especially
preferable for use are compounds in which said phenyl is substituted and said
alkoxy substituent is methoxy. Such compound comprise 4-(2,4-
dimethoxybenzylamino)-N-(4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide, N
(4,6-dimethylpyrimidin-2-yl)-4-(3-methoxybenzylamino)-benzenesulfonamide, 4-
(3,5-dimethoxybenzylamino)-N-(4,6-dimethylpyrimidin-2-yl)-
benzenesulfonamide, 4-(2,5-dimethoxybenzylamino)-N-(4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide and N-(4,6-dimethylpyrimidin-2-yl)-4-(2-methoxybenzyl-
amino)-benzenesulfonamide.
In other compounds preferably used X is H, Y is a phenyl optionally mono- or
disubstituted with a straight or branched alkyl and/or a halogen and Z is H.
Such
compound comprise 4-benzylamino-N-(4,6-dimethylpyrimidin-2-yl)-benzene-
sulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(2-methylbenzylamino)-benzene-
sulfonamide, 4-(2,4-dimethylbenzylamino)-N (4,6-dimethylpyrimidin-2-yl)-
benzenesulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(3-methylbenzylamino)-
benzenesulfonamide, N (4,6-dimethylpyrimidin-2-yl)-4-(4-methylbenzylamino)-
benzenesulfonamide, 4-(2,5-dimethylbenzylamino)-N (4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide, 4-(2,6-dimethylbenzylamino)-N (4,6-dimethylpyrimidin-
2-yl)-benzenesulfonamide, 4-(4-bromobenzylamino)-N (4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide and 4-(2,6-dichlorobenzylamino)-N (4,6-
dimethylpyrimidin-2-yl)-benzenesulfonamide.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
8
Further preferred compounds to be used comprise 4-[(1H-benzimidazol-
2-ylmethyl)-amino]-N (4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide, N-(4,6-
dimethylpyrimidin-2-yl)-4- [ ( 1-ethyl-1 H-indol-3-ylmethyl)-amino] -ben-
zenesulfonamide, N-(4,6-dimethylpyrimidin-2-yl)-4-[(1-isobutyl-1H-benz
imidazol-2-ylmethyl)-amino] -benzenesulfonamide, N (4,6-dimethylpyrimidin-2
yl)-4-(1-phenylethylamino)-benzenesulfonamide, N-(4,6-dimethylpyrimidin-2-yl)
4-[2-(2-methoxyphenyl)-ethylamino]-benzenesulfonamide and N (4,6-dimethyl
pyrimidin-2-yl)-4-[(naphthalen-2-ylmethyl)-amino]-benzenesulfonamide.
Alpha-2B-adrenoceptor antagonists are useful in the treatment and/or
prevention
of many diseases.
Individuals having a deletion in the alpha-2B-adrenoceptor protein (WO
01/29082;
Heinonen et al., 1999), particularly the deletion/deletion genotype (D/D
genotype)
is an important target group, which benefits from administration of selective
alpha-2B-adrenoceptor antagonists. These individuals have a deletion of 3
glutamates from the glutamic acid repeat element of 12 glutamates (amino acids
297-309), in an acid stretch of 17 amino acids, located in the third
intracellular
loop of the receptor polypeptide.
It has been found that in a population-based cohort of Finnish middle-aged men
that subjects with a D/D genotype of the alpha-2B-adrenoceptor gene have a
significantly elevated risk for acute myocardial infarction (AMI) in a five-
year
follow-up study. The risk for AMI was increased in subjects who had no
previously diagnosed coronary heart disease (CHD) at the study outset.
Therefore,
it has been postulated that the D/D genotype is related to an impaired
capacity to
down-regulate alpha-2B-adrenoceptor function during sustained receptor
activation. Therefore, alpha-2B-adrenoceptors are believed to be involved in
the
pathogenesis of a significant fraction of all cases of AMI, especially in
subjects
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
9
with the D/D genotype, but also in I/D and I/I subjects (I means "insertion"
and
stands for the "normal" allele).
The alpha-2B-adrenoceptor antagonists as disclosed in this invention would be
particularly useful in the treatment or prevention of coronary heart diseases.
As
examples can be mentioned
a) Acute AMI
If alpha-2B-adrenoceptor dependent vasoconstriction is a causative factor in
some cases of AMI, then antagonism of these receptors should restore
coronary circulation and reduce the ischemic myocardial damage.
b) Unstable angina pectoris
An alpha-2B-adrenoceptor antagonist will relieve the vasoconstrictive
component in the sustained ischemic episode, thus alleviating the symptoms
and preventing AMI.
c) Prinzmetal's variant form of angina pectoris
Vasoconstriction is a key factor in the pathogenesis of Prinzmetal's angina,
and an alpha-2B- adrenoceptor antagonist may resolve and prevent attacks.
d) Other forms of chronic angina pectoris and CHD
An alpha-2B-adrenoceptor antagonist will help to alleviate the
vasoconstrictive component in all types of CHD, providing both symptomatic
relief and protection from AMI. A general reduction in vascular tone will
contribute to this by reducing venous return, cardiac workload and oxygen
consumption (a nitrate-type effect; see below).
e) Prevention of restenosis after coronary angioplasty in cases where
vasoconstriction plays a role in restenosis.
Furthermore, the alpha-2B-adrenoceptor antagonists as disclosed in this
invention
would be useful in the treatment or prevention of essential hypertension,
especially
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
in subjects with increased sympathetic activity and a hyperdynamic circulatory
system.
In the study mentioned above, the D/D variant of the alpha-2B-adrenoceptor
gene
was not clearly associated with blood pressure. The inventors believe that
this was
5 due to two main factors, 1 ) antihypertensive treatment, and 2) complex
regulation
of systemic blood pressure. In another study (Heinonen et al.), it was
observed that
the D/D genotype was associated with reduced basal metabolic rate and reduced
heart rate. These associations probably reflect increased vascular resistance
in
these subjects.
10 In transgenic mice with targeted inactivation of the alpha-2B-adrenoceptor
gene,
intravenously administered alpha-2-adrenoceptor agonists fail to induce the
characteristic blood pressure elevation, which is seen in normal animals and
also
in humans after large doses of such drugs (Link et al., 1996). The hypotensive
effect of these drugs was markedly accentuated. This demonstrates that alpha-
2B-
adrenoceptors mediate vascular contraction. Thus, an antagonist should reduce
blood pressure. This effect has not been seen with alpha-2B-non-selective
alpha-2-
adrenoceptor antagonists, because antagonism of alpha-2A-adrenoceptors
increases sympathetic outflow, cardiac output and blood pressure. In mice with
dysfunctional alpha-2A-adrenoceptors, alpha-2-adrenoceptor agonists caused an
accentuated hypertensive response and no hypotension (MacMillan et al., 1996).
An alpha-2B-adrenoceptor antagonist is postulated to have favourable effects
in
hypertensive subjects through their effects on renal function, muscle blood
flow,
and also on vascular resistance in other vascular beds. The anti-AMI effect of
such
a drug will be an additional benefit, as hypertension is a significant risk
factor for
AMI. This protection is due to three factors: 1) a reduction in systemic blood
pressure, 2) decreased risk of coronary vasoconstriction, and 3) a nitrate-
like effect
on venous return, myocardial workload and oxygen consumption.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
11
Moreover, the alpha-2B-adrenoceptor antagonists as disclosed in this invention
would be useful in the treatment or prevention of other vascular diseases.
Specifically, benefits can be expected in the treatment or prevention of
- vasoconstriction and hypoxic brain damage subsequent to subarachnoid
haemorrhage,
- migraine,
- Raynaud's disease and cold intolerance,
- pre-eclampsia,
- male erectile dysfunction, and
- obesity and the metabolic syndrome.
The last mentioned effect is due to the fact that reduced muscle blood flow
and
reduced basal metabolic rate contribute to the development of obesity and
hypertension. An alpha-2B-adrenoceptor antagonist will, by increasing the
muscle
blood flow, increase energy expenditure and shift the caloric balance to a
favourable direction.
The alpha-2B-adrenoceptor antagonists disclosed in this invention are also
useful
in anaesthesia and analgesia to potentiate the clinical efficacy of alpha-2-
adrenoceptor agonists, which are not selective for the alpha-2B-adrenoceptor
subtype. By blocking the vasoconstriction induced by these agonists, a
simultaneously administered alpha-2B-adrenoceptor antagonist will allow the
use
of larger doses of said agonists, up to anaesthetic dose levels which have not
previously been possible in man, only in veterinary anaesthetic practice.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
12
EXPERIMENTAL SECTION
Human alpha-2-adrenoceptor binding affinity
The affinity of test compounds for the three human oc2-adrenoceptor subtypes
(a2A,
oczB and oc2~) was determined in competition binding assays with 3H-
rauwolscine.
The biological material for these experiments consisted of membranes from
Shionogi S 115 cells stably transfected with any of the three human a2
subtypes
(Marjamaki et al. 1992). Membrane (5-10 pg of total protein per sample) and 1-
2
nM 3H-rauwolscine (specific activity 78 Ci/mmol) were incubated in 50 mM
KIi2PO4, pH 7.5 with 6 concentrations of the compounds. Each concentration was
run in duplicate. Non-specific binding was defined by 100 pM oxymetazoline and
corresponded to 5-15% of total binding. After 30 min at room temperature,
incubations were terminated by rapid vacuum filtration through GF/B glass
fiber
filter and three 5 ml washes with ice-cold incubation buffer. The filters were
then
dried, impregnated with scintillate and their radioactivity was measured by
scintillation counting. The analysis of the experiments was carned out by non-
linear least square curve fitting. Experimentally determined IC50 values were
converted to Ki's by making use of the Cheng-Prusoff equation (Cheng and
Prusoff, 1973). Experiments were repeated a minimum of three times.
Table 1: Human oc2-adrenoceptor subtypes binding affinities. Data is presented
as
Ki's in nM (Mean ~ SEM).
Compound alpha-2A alpha-2B alpha-2C
A >13000 160 20 >30000
B >4500 34 2 >10000
C 2000 400 10 2 > 10000
D >10000 440 70 >10000
E >5100 20 4 > 10000
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
13
F >4300 43 7 > 10000
G 2200 600 32 5 >10000
H >30000 8000 500 >30000
Results expressed in the form of " > " means that no numerical values for Ki's
could be established due to lack of displacement or due to incomplete
competition
curves. However, the experimental data indicated that, at a minimum, the Ki's
must be larger than the numbers given.
Antagonist activity on human alpha-2-adrenoceptor subtypes
Antagonist potencies were determined as the ability of test compounds to
competitively inhibit epinephrine-stimulated 35S-GTPyS binding to G proteins
(Tian et al., 1993; Wieland and Jakobs, 1994; Jasper et al., 1998) in
membranes of
CHO cells stably transfected with one of the three human ocz subtypes
(Pohjanoksa
et al., 1997; Marjamaki et al., 1998). Membranes (2-6 pg of protein per
sample)
and 12 concentrations of test compound were preincubated for 30 min with a
fixed
concentration fo epinephrine (5 ~M for oc2A" 15 ~M for a2$, 5 pM for oc2c) in
50
mM Tris, 5 mM MgCl2, 150 mM NaCI, 1 mM DTT, 1 mM EDTA, 10 pM GDP,
30 ~M ascorbic acid, pH 7.4 at room temperature. Binding of radiolabel was
started by the addition of trace amounts of 35S-GTPyS (0.08-0.15 nM, specific
activity 1250 Ci/mmol) to the incubation mixture. After an additional 60 min
at
room temperature, the incubation was terminated by rapid vacuum filtration
through glass fibre filter. Filters were washed three times with 5 ml ice cold
wash
buffer (20 mM Tris, 5 mM MgCl2, 1 mM EDTA pH 7.4 at room temperature),
dried and counted for radioactivity in a scintiallation counter. Analysis of
experiments was carried out by non-linear least square fitting. Results are
based on
a minimum of three experiments.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
14
Table 2: Antagonist effect on human oc2-adrenoceptor subtypes. Data is
presented
as KB's in nM (Mean ~ SEM).
Compound alpha-2A alpha-2B alpha-2C
B 14500 3600 75 9 5700 700
C 5400 1400 17 5 6300 1400
E 7900 3100 29 5 7300 1100
F 8700 1100 240 60 12000 2000
G 3200 500 86 64 4700 1800
For the purpose of the invention, the alpha-2B-adrenoceptor antagonist or its
pharmaceutically acceptable salt can be administered by various routes. The
suitable administration forms include, for example, oral formulations;
parenteral
injections including intravenous, intramuscular, intradermal and subcutanous
injections; transdermal or rectal administration forms. The required dosage of
the
compounds of the alpha-2B-adrenoceptor antagonist will vary with the
particular
condition being treated, the severity of the condition, the duration of the
treatment,
the administration route and the specific compound being employed. The
suitable
dose varies in the range 5 ~,g to 100 mg per kg body weight and day for an
adult
person.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
EXAMPLES
Example 1
N (4 6-Dimethylpyrimidin-2-yl)-4-f(1-ethyl-1H-benzimidazol-2-ylmethyl)-aminol
-benzenesulfonamide
5 Step l: Alkylation of 2-hydroxymethylbenzimidazole
99.2 mg (0.67 mmol) 2-hydroxymethylbenzimidazole was dissolved in 3 ml
methanol. Potassium carbonate (103.1 g, 0.75 mmol) and diethylsulfate (442 ~l
3.38 mmol) were added to the reaction mixture. Solution was stirred and
refluxed
overnight. The reaction mixture was then evaporated to dryness and purified on
10 silica using gradient elution (chloroform to 5% methanol in chloroform) to
obtain
white crystals of 1-ethyl-2-hydroxymethylbenzimidazole, 41 mg (32%).
Step IL Chlorination of 1-ethyl 2-hydroxymethylbenzimidazole
mg (0.11 mmol) 1-ethyl-2-hydroxymethylbenzimidazole was dissolved in 2 ml
dichloromethane. Thionyl chloride (24 ~1, 0.33 mmol) was diluted 20 times with
15 dichloromethane and the solution was added to the reaction mixture.
Reaction
mixture was stirred at room temperature for two hours, evaporated to dryness
and
washed with water to yield 1-ethyl-2-chloromethylbenzimidazole as pale yellow
crystals, 36 mg (95%).
Step III: Coupling reaction between 1-ethyl 2-chloromethylbenzimidazole and
20 sulfamethazine
32.4 mg (0,12 mmol) sulfamethazine and 36 mg (0.18 mmol) 1-ethyl-2-
chloromethylbenzimidazole were dissolved in 4 ml methanol. 64 ~l (0.44 mmol)
triethylamine and catalytic amount of sodium iodide were added to the reaction
CA 02454187 2004-O1-15
~ ~~SG91' , ~~
;' ~?=1'li~t~otl. ~ 4' 0~~2003' , , Y :5
,x.
16
mixture. Solution was stirred and .refluxed overnight. The reaction mixture
was
then evaporated to dryness, and purified on silica using gradient elution
(chloroform to 5 % methanol in chloroform) to provide white crystals of the
title
compound, 10 mg (20%).1H NMR (DMSO-dd, 500 MHz): 7.68 (2H, m), 7.60 (1H,
m), 7.52 (1H, m), 7.22 (1H, m), 7.17 (IH, m), 7.00 (1H, br, s), 6.78 (3H, m),
6.60
(1H, br, s), 4.60 (2H, m), 4.30 (2H, q, 7.2 Hz), 2.18 (6H, s), 1.28 (3H, t,
7.2 Hz);
MS (ESI~): m/z 437 (M + H)~.
Example 2
4-f (1H Benzimidazol-2-ylmethyl)-aminol-1V X4,6-dimeth~pyrimidin-2-
benzenesulfonamide
Following the procedure outlined in Step III of example 1, but without
triethylamine and substituting 1-ethyl-2-chloromethylbenzimidazole by
2-chloromethylbenzimidazole afforded the title compound with the yield of 51
%.
. 1H NMR (DMSO-ds, 500 MHz): 7.70 (2H, m),, 7.48 (2H, br, m), 7.13 (2H, m),
7.I1 (1H, t, 5.8 Hz), 6.70 (3H, m), 4.52 (2H, d, 5.8 Hz), 2.21 (6H, s); MS
(ESI~):
m/z 409 (M + H)+.
Example 3
N (4 6-Dimethylpyrimidin-2-~ -) ~.-f (pyridin-4-ylmethyll-aminol-
benzenesulfonamide . '
Following the procedure outlined in Step III of example 1 without
triethylamine
and substituting 1-ethyl-2-chloxomethylbenzimidazole by 4-picolylchloride
hydrochloride afforded the title compound with the yield of 54%. MS (ESI~):
m/z
392 (M + Na)~, 370 (M + H)~. .
1'~~ RECTIFIED SHEET (RULE 91 ) ~ , t~'~'~4~"Q~,;~Q4~
CA 02454187 2004-O1-15,
~ /'~ ~ ,; ;ig
~~'~"''~~~~~~ ~1t4 n ~ , sz Et~.:2 '~,. 5~. ,f:.~ ' ~, 1.,i " s :"tt,l: ': t
'~.,..:; ~~nt , ..na;w pf yq; ; ~,.6-. ~, . "w, ~..~:.:~ i~ ' ec ~ E ~ it ,~:y-
t r ' ; T'~ !
w,.u.~ ..~.. '0~~48~02; F1~3~~~~~.
_ . .t- ,.».,.,.,..,..: . ' t ;s.~,..,a.: l~ z e~° v ~s
_ . Du.~..a.;~. ~. ~s~,.,u.: s. ,m"
J
17
Example 4
N 4 6-Dimeth I 'din-2- 1 -4- 1-isobut 1-IH benzimidazol-2- lmeth 1 -
am_inol -benzenesulfonamide (Compound A)
Following the procedure outlined in example 1, but substituting in step I
ethyl
5~brom~.de for isobutyl iodide, affoirded the title compound with stepwise
yields of
~15%, 95% and 15%. 1H NMR {DMSO-d6, 500 MHz): '7.76 (2H; m), 7.70 {1H, m),
7.60 (1H, m), 7.27 (1H, m), 7.22 (IH, m), 7.15 (1H, br, t, 5.3 Hz), 6.86 (2H,
m),
6.77 (1H, s), 4.65 (2H, d, 5.3 Hz), 4.13 (2H, d, 7.5 Hz), 2.27 (6H, s), 2.25
(IH, m),
0.91 (6H, d, 6.7 Hz); MS (ESI~): m/z 487 (M + Na)~, 465 (M + H)~.
Example 5
4- I-Benz 1-1H benzimidazol-2- lmeth 1 -amino -N ~ 4 6-diineth' 1 'midin-2-
yl)- benzenesulfonamide
Following the procedure outlined ~in example 1, but substituting in step I
'ethyl
bromide for benzyl bromide, afforded the title compound with stepwise yields
of
I5 23%, 90%,and 18%. rH NMR (DMSO-d6, 500 MHz): 7.81 (2H, m), 7.52 (1H, m),
7.45 -(1H, m), 7.30 (5H, m), 7..16 (2H, m), 6.75 (1H, s), 6.54 (2H, m), 6.02
(1H, br,
s), 5.66 (2H, s), 5.63 (2H, s), 2:21 (6H, s); MS (ESI+): rn/z 521 (M +
1'~ta)~, 499 (M
+ H)~*.
Example 6
4-~(1-Ethy1-1FI benzimidazol-2-ylmethyl) ami,nol N (S methoxynyrinudin 2 v11
benzenesulfonamide ,
Following the procedure outlined in example 1 step III, but substituting
sulfamethazine by 5-methoxysulfadxazine, afforded the title compound with the
,2'rRECTIFIED SHEET (RULE 91 ) ,
~y~ ,0~~ .~0~ ~; 2t:
..:,:L::, ..:...:_ ,.:'.i
r-
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
18
yield of 8%. Dimethylformamide was used as a solvent and additional silica gel
chromatography purification with 2:1 petrol ether:ethylacetate was needed. MS
(ESI+): m/z 461 (M + Na)+, 439 (M + H)+.
Example 7
4-f(1H-Benzimidazol-2- l~yl)-aminol-N-(pyrimidin-2-yl)-benzenesulfonamide
628 mg (3.8 mmol) sulfadiazine and 728 mg (3.0 mmol) 2-chloro-
methylbenzimidazole were dissolved in 10 ml 1 M NaOH. Solution was stirred
and refluxed for four hours. Reaction mixture was neutralised with addition of
1 M
acetic acid until product precipitated. Crystals were filtered and purified on
silica
using gradient elution (chloroform to 5% methanol in chloroform) to obtain the
title compound as white crystals with 38% yield. 1H NMR (DMSO-d6, 500 MHz):
8.46 (2H, m), 7.70 (2H, m) 7.49 (2H, br, m), 7.14 (1H, t, 5.7 Hz) 7.13 (2H,
m),
6.98 (1H, m), 6.72 (2H, m), 4.53 (2H, 5.7 Hz); MS (ESI+): m/z 381 (M + H)+.
Example 8
N-(1H-Benzimidazol-2-~yl)-N f4-(4,6-dimeth~pyrimidin-2-ylsulfamoyl)-
phenyll-acetamide
18 mg (0.044 mmol) 4-[(1H-benzimidazol-2-ylinethyl)-amino]-N (4,6-dimethyl-
pyrimidin-2-yl)-benzenesulfonamide was dissolved in 2 ml of 15% pyridine in
dichloromethane. Acetyl chloride (31 ~ 1, 0.44 mmol) was diluted with 1 ml
dichloromethane and solution was added to the reaction mixture. After three
hours
reaction mixture was washed with acidic water and organic layer was evaporated
to dryness. Crystals were purified on silica using gradient elution
(chloroform to
5% methanol in chloroform) to obtain white crystals with 30% yield. 1H NMR
(DMSO-d6, 500 MHz): 7.97 (2H, m), 7.60 (2H, m), 7.48 (2H, m), 7.14 (2H, m),
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
19
6.68 (1H, s), 5.08 (2H, s), 2.18 (6H, s), 1.93 (3H, s); MS (ESI+): m/z 473 (M
+
Na)+, 451 (M + H)+.
Example 9
N-( 1-Acetyl-1 H-benzimidazol-2-ylmethyl)-N- f 4-(4, 6-dimethylpyrimidin-2-
ylsulfamo.~phenyll-acetamide
Title compound was purified from the reaction mixture produced according to
example 8 with a yield of 14%. 1H NMR (DMSO-d6, 500 MHz): 8.15 (2H, m),
7.80 (2H, m), 7.55 (2H, m), 7.48 (1H, s), 7.21 (3H, m), 5.17 (2H, s), 2.54
(6H, s),
2.03 (3H, s), 1.83 (3H, s); MS (ESI+): m/z 493 (M + H)+.
Example 10
4-f (1-Acetyl-1H-benzimidazol-2-ylmethXl)-aminol-N-(4,6-dimethylpyrimidin-2-
yl)-benzenesulfonamide
Following the procedure of example 8, but instead of 15 % pyridine in
dichloromethane only few drops of pyridine in dichloromethane were used as a
solvent. Method afforded the title compound with a yield similar to that for N-
(1H-
benzimidazol-2-ylmethyl)-N-[4-(4,6-dimethylpyrimidin-2-ylsulfamoyl)-phenyl]-
acetamide. MS (ESI+): m/z 451 (M + H)+.
Example 11
4-BenzKlamino-N (4,6-dimethylpyrimidin-2-yl)-benzenesulfonamide
100 mg (0.36 mmol) sulfamethazine and 70.8 p1 (0.60 mmol) benzyl bromide were
dissolved in 4 ml methanol. Caesium carbonate (113.4 mg, 0.35 mmol) was added
and solution was refluxed overnight with stirring. The reaction mixture was
then
CA 02454187 2004-O1-15
?. y~~~~ fj R r
~4f~t~~~~~~;~. ,7 4 ~~ u.k~-4 f h,~t t A '.l C rv . . R '~ F : Y
~,F~ .~J1 E f ~~~,c~~ Et ~S 3~ ~, ~ ~y ~ I'y~y 2f-'- f~J~A~~~9..[t
,. a, r~,. ,A. ~,::,," .. r. ,.... .. ax,.:".. .. a ~ w E i
,m:v~ ~ ~r.N : ~ 0~~7~.8~a~:~F~~a.~0~4~-
T . "s,~. ;. _ , . ~ F r
. m.'r°u..,~.~,ri>':.e., t ~e~m 1 .dk~,.....ue..~,y ~..xF,.hf~saa
evaporated to dryness, -and purified on silica using gradient elution
(chloroform to
2% methanol in chloroform) to obtain white crystals in a yield similar to that
described in example 1 for step III. 1H NMR (CDCl3, 500 MHz): 7.93 (2H, m),
7.31 (5H, m), 6.58 (3H, m), 4.36 (2H, s), 2.34 (6H, s); MS (ESh'): m/z 369 (M
+
H)+. , ' ,
Example I2
4-(4-Bromobenzylamino) N (4 6-dimethylpyrimidin 2 y1) benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 4-bromobenzyl bromide, afforded .the title compound with a yield of 8%. 1H
NMR (CDC13, 500 MHz): 7.91 (2H, m}, 7.45 (2H, m), 7.19 (2H~ m), 6.59 (1H,..s)
6.55 (2H, m), 4.32 (2H, s), 2.34 (6H, s); MS (ESI'~): mlz 469 (M + Na)+, 447
(M +
H)~.
Example 13
N (4,6-Dimethyltwrimidin-2-yl)-4-(2-meth lbenzylamino) benzenesulfonamide
(Compound B)
Following the procedure outlined in example 11, but substituting benzyl
bxomide
by 2-methylbenzyl bromide, afforded the title compound in a yield similar to
that
described,in example 1 for step III. 1H NMR (CDCl3, 500 MHz): 7.95 (2H, m),
7.22 (4H, m), 6.58 (3H, m), 4.30 (2H, s), 2.35 (3H, s), 2.34 (6H, s); MS
(ESI~):
m/z 405 (M + Na)+.
3~ RECTIFIED SHEET (RULE 91 )
. , ~4 ,09, X002;
r
CA 02454187 2004-O1-15 -
r sYf r 7 i:.4'.~ ;7 , A,
,~~~r~~~tec~l~~i4 (~4~2OO~'~5 ~ ~? ~ >: ~ ~f~'~* ,.:f ~: r:..~~ ~~,~_,~
~ ~.ar:,~ .,.:: ?;~:: " ~ c a ~, ~,., 4 ~..,~ , >na:,i;:f ' x ~ ~ ..x k ~~.
CT~~~8~Q2, FI~~006~.3
r ''C~ .~.~..F.,.,a:d~~x xn.,.a.NA . ~f a.."..-ash; ~ ~ "
_ Ai..~ av.-,
~21
Example 14
N(4,6-Dimethylpyrun,_'din-2-yI)-4-(4 methylbenz lamino) benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 4-methylbenzyl bromide, afforded the title compound in a yield similar to
that
described: in example I for step III. iH NMR (CDC13, 500 MHz): 7.92 (2H, m),
7.20 (2H, m), 7.15 (2H, m), 6.58. (3H, m), 4.31 (2H, s)~ 2.34 (3H, s}, 2.33
(6H, s};
MS (ESI~): m/z 405 (M + Na)*, 383 (M + H)~.
Example 15
N (4,6-Dimethylpyrimidin-2-yl)-4-(1 phen 1y ethylamino) benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by (1-bromoethyl)-benzene, afforded the title compound with a yield of 12%. 1H
NMR (CDC13, 500 MHz): 7.84 (2H, m), 7.31 (5H, m), 6.57 (1H, s), 6.46 (2H, m),
4.53 (1H, q,..6.7 Hz), 2.30 (6H, s), 1.54 (3H, d, 6.7 Hz); MS (ESI~'): rn/z
405 (M +
Na}'~, 383 (M + H)+.
Example 16
N (4,6-Dimethyloyrimidin-2-yl)-4-(2-methoxybenzvlamino) benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 2-methoxybenzyl bromide, afforded the title compound in a yield similar to
that
described in example 1 for step In. 1H NMR (CDC13, 500 MHz): 7.92 (2H, m),
7.25 (21~, m), 6.90 (2H, m), 6.59 (3H, m}, 4.36 (2H, s), 3.86 (3H, s), 2.32
(6H, s);
MS (ESh''): m/z 421 (M + Na)+, 399 (M + H)~.
"1P
",
L4~~ RECTIFIED SHEET (RULE 91)
~'a'".E:~..~,'1. . ~~~'~,~f
,._
CA 02454187 2004-O1-15
tx
~ a It'~6 ~ ~ ~'~~' ~ ~ ;-~s3 x. r ~~ ., s.:: t~~4 -x
~ ~A rir~ted ~h~ '= ' ~ 5 h,n~F , , ~ F 6 ~ 4 h~
4 04~ X005:, t ~~ ,.,;; ~. E~: ~ ~ ,.~ ,s..~ ~,
,I~ESC9~ yf s0~748~ . r~ , , , ~ . ..
~..,.ct..,:l: t,1" ".u:r. .,Mk..:~.~~
~2 FIil2~l
~'~ ~.,.:i~ z.w..~..v:. x5 ,ix..roas Y.~ a~.=,r~~ .. e.f.,.z ~~~~ <u..~'r. ~~~
p
22
Example 17
4-(2.4-Dimethylbenzylamino)-N (4 6-dimeth~pyrimidin 2
benzenesulfonamide
Following the procedure outlined in example 11, Gut substituting benzyl
bromide
5~by 2;4=dlmet~ylbenzy~~romi ed , a~'fordecT~t a t a m oun ith a yield of 23%.
1H NMR (CDC13, 500 MHz): 7.94 (2H, m), 7.14 (1H, m), 7.03 (IH, s), 6.98 (1H,
m), 6.58 (3H, m), 4.26 ~(2H, s), 2.34 (6H, s), 2.31 {3H, s), 2.30 (3H, s); MS
(ESI~):
m/z 419 (M + Na)+, 397 (M + H)+.
Example 18
N (4 6-Dimethyl~yrimidin-2-yl)-4-(3-methylbenz~amino) benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 3-methylbenzyl bromide, afforded the title compound with a yield of 10%. 1H
NMR (CDC13, 500 MHz): 7.93 (2H, m), 7.23 (1H, m), 7.11 (3H, m), 6:62 (1H, s),
6.58 (2H, m), 4.31 (2H, s), 2.36 (6H, s), 2.34 (3H, s); MS (ESI+): m/z 405 {M
+
Na)+, 383 {M + H)~.
Example 19
4-(2,6-Dichlorobenzylamino)-N (4 6-dimethy~~rimidin 2 y1) benzenesulfonarnide
(Compound C)
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 2,6-dichlorobenzyl bromide, afforded the title compound with the yield of
10%.
1H NMR (CDCl3, 500 MHz): 7.96 (2H, m), 7.34 (2H, m), 7.20 (1H, m), 6.71 (2H,
m), 6.59 (1H, s), 4.63 (2H, s), 2.34 (6H, s); MS (ESI+): 437 (M + H)~.
.,
5,~ RECTIFIED SHEET (RULE 91 )
~,~~~~09 ~ 2{102;
r ,;4. . "_.;...,..,. ,:....r
CA 02454187 2004-O1-15
~i~~r~~~ ~~~'''~ 4 ~4. 4~0 ~- J's . ' i :i: ~ i ~ .;, r t ~ ~- e::z
~dyt.- ~~ FTS:..~..xw ~I ~~..,. , f 03, ~t ,SC9fli x ~",rxv. t ' X902
FIIO~Q'0~~~~
~4.rM,r~ , a~ t "f., ~.~.~_ar . -..~i~s~~..~: ~n~S. _.,r.u.. : ~.~~, ~,. m
'1~;~'.
23
Example 20
.~V (4,6-Dimethyltwrimidin-2-yl)-4 f(naphthalen 2 ylmethyl) aminol
benzenesulfonamide (Compound Dl
Following the procedure outlined in example I1, but substituting benzyl
bromide
~5 bye-b~omo~methytnaph~alene, affo~ded~.e tli~.e compound wit~the yield of
20%.
1H NMR (DMSO-d6, 500 MHz): 7.85 (4H, m), 7.68 (2H, m), 7.47 (3H, m), 7.19
(1H, t, 5.8 Hz), 6.70 (IH, s), 6.66 (2H, m), 4.49 (2H, d; 5.8 Hz), 2.21 (6H,
s); MS
(EST): m/z 44I (M + H)~.
Example 21
N (4.6_Dimethylpyrimi.din-2-vl)-4-(3-methoxvhenzylarnino) benzenesulfonamide
(Compound E)
Following the procedure outlined in example I I, but substituting benzyl
bromide
by 3-methoxybenzyl bromide, afforded the.title compound with the yield of 28%.
1H NMR (CDCl3, 500 MHz): 7.93 (2H, m), 6.86 (3H, m),, 6.58 (3H, m), 4.33 (2H,
s), 3.78 (3H, s), 2.33 (6H, s); MS (ESI~: m/z 42I (M + Na)~, 399 (M + H)f.
Example 22
N (4,6-Dimethylpyrimidin-2-yl)-4 (4 nitrobenz~ no) benzenesulfonamlde . . .
Following the procedure outlined in example II, but substituting benzyl
bromide
by 4-nitrobenzyl bromide, afforded the title compound with a yield lower than
I %.
MS (ESI+): m/z 414 (M + H)'~.
" ..,,,,}
RECTIFIED SHEET (RULE 91 )
. I ~4,' ~~Q~9 ~0(72~'=
. .,. :.,......, ,..,....,..:~
,_
CA 02454187 2004-O1-15
a E . ~ ~ t
~ a . v a E
~ F';rl~~~d ~s4 04 20~~ ~ ' E ~ . , ~ ~ b ~ ~:~<n ,
.ad....a t :(..,~~.- F ~ p y d o
,. .~1.2.,.... . ,.v~.':5~,.i' ~.~.i.'.~x ., S , 4
t DESC~~ , r E F (3~7~!':80 ~ ~' FI'0~~b~, '~~
~- , a,ky .:~.~an..,v
t ...!..",e t't ,.....'~. ~ F ~ .,.N~~m..
_ .~ ~ax,.:.,.. ~...
24
Example 23
N (4,6-Dimethylpyrimidin-2- 1y )~4-(4-trifluoromethylbenz~oL
benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
_.._...._..._._~.. ~~y 4-~tluoromeThy~benzy ~bromid:e; affo~.e~tbe t~f a
mpound in y ~~1~: _-
to that described in example 1 for step III. MS (ESI~): m/z 459 (M + Na)~, 437
(M
+ H)+.
Example 24
4-fBis-(4-trzfluoromethylbenzyl)-aminol N (4 6 dimeth l~yrimidin 2 y1)
benzenesulfonamide
Title compound was purified from the reaction mixture produced according to
example 23 with the yield of 20%. MS (ESI~): 617 m/z (M + Na)~, 595 (M + H)~".
Example 25
N (4,6-Dimethylpyrimidin-2-y1)~4-(4-trifluoromethoxybenzylamino)
benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by 4-trifluoromethoxybenzyl bromide, afforded the title compound with a yield
of
25%. MS (ESI+): zn/z 475 (M + Na)+, 453 (M + H)*.
r. a ....
RECTIFIED SHEET (RULE 91 )
j ~~,~0'9 ~ao~~
a ,: .... ,.._..,.._. A..
CA 02454187 2004-O1-15
[..3,y'yy~~ Y 5 'n <~ t
il~~~~~~~"I. ~ ~F4~! va .i a~ r f Fet u,Y.: ~ i. r t S ><.a: ,1 't t ', x~
1,~'~'7 5 , a
i.~c;JFS~ 7 ~... F t3~ ~ .. ~~~~ '~~.rz.i ~"f . (~ :;.
....a..a~.,s- ..t s . ,..r .adz . .. r~ "r.' f ' f V
~t lm,.!xo,xM~ s~L~,uHs;D.~ t a wi;~F
-='.~y".,. ~,~ ':rx
Example 26
4-fBis-(4-trifluoromethoxybenzyl)-aminol N (4,6-dimeth~p~irimidin 2 ~)
benzenesulfonanude
Title compound was purified from the reaction mixture produced according to
- ' example 2~ wnh a yell of 1~%: lVfS~E~~ ~ : n z 64-9~1V.~+~a)T; 627 (.M +
I~)':~~__....'
Example 27
4-(2,5-Dimethylbenzylatnino)-N (4,6-dimeth~p rimidin
benzenesulfonamide.
Following the procedure outlined in example 11 without caesium carbonate and
IO substituting benzyl bromide by 2,5-dimethylbenzyl bromide afforded the
title
compound with. a yield of 35%. 1H NMR (CDCl3, 500 MHz): 7.93 (2H, m), 7.07
(3H, m), 6.59 (3H, m), 4.25 (2H, s), 2.33 (6H, s), 2.30 {3H, s), 2.28 (3H, s);
MS
(ESI~): m/z 419 (M + Na)~.
Example 28
15 4-(2,6-Dimethylbenzylamino) N (4 6-dimeth~pyrimidin-2-yl)
benzenesulfonamide
Following the procedure, outlined in example 1I ~vvithout caesium carbonate
and
substituting benzyl bromide by 2,6-dimethylbenzyl bromide afforded the title
compound with a yield of 25%. 1H NMR (CDC13, 500 MHz): 7.99 (2H, m), 7.0~
20 (3H, m), 6.62 (3H, rn), 4.26 (2H, s), 2.37 (12H, m); MS (EST: m/z 419 (M +
Na)+.
~O~y RECTIFIED SHEET (RULE 91) ~ ~~~~~~~j~~~~~0~
a..,..F.~M~_",..:~f..~.u,. ..:,..._...
,.
CA 02454187 2004-O1-15
~ ~~~~ , ~ ~~,~~ off. ~00~,a~sC9~ ~~~~~ .~F K o~~4 .90 _y , ~ F my
..r..: ~.u... ~, ~ Fo~o~~4~
~~..a . ~.~.~~.r.~Y
~....~.~.~~
26
Example 29
4-(3,5-Dirnethoxybenzylamino)-N (4 6-dimethvl
p~2~)
benzenesulfonamide
Following the procedure outlined in example 11 without caesium carbonate and
substituting benzy~bromide by 3;5-dimethoxybenzyl chloride afforded the title
compound with a yield of 15%. 1H NMR (CDC13, 500 MHz): 7.92 (2H, m), 6.60
(IH, s), 6.58 (2H, m), 6.46 (2H, m), 6.38 (1H, m), 4.30 (2H, s), 3.76 (6H, s),
2.33
(6H, s); MS (ESI*): mlz 429 {M + H)~.
Example 30
4-(2,5-Dimethoxybenzylarnino)-N~4 6-dimethylpyrLmsdin-2-yI)
benzenesulfonamide
Following the procedure outlined in example 1I without caesium carbonate and
substituting benzyl bromide by 2,5-methoxybenzyl chloride afforded the title
compound with the yield of I3%. Reaction time was three days.1H NMR (CDCl3,
IS 500 MHz): 7.91 (2H, m), 6.79 (3H, m), 6.58 {3H, m), 4.33 {2H, s), 3.8I (3H,
s),
3.7I (3H, s), 2.33 (6H, s); MS (EST+): m/z 429 (M + H)~.
Example 31
2,6-Dichloro-N f4-(4,6-dimethylnyrimidin-2- lsulfamo 1)- henyll benzamide
Following the procedure outlined in example 1I, but substituting benzyl
bromide
by 2,6-dichlorobenzoyl chloride, afforded the title compound with almost
quantitative yield. 1H NMR (CDC13, 500 MHz): 8.I I (2H, m), 7.80 (2H, m), 7.33
(3H, m), 6.59 (1H, s), 2.34 (6H, s); MS (ESh'): m/z 473 (M + H)~, 451 (M +
H)'''.
i19~ RECTIFIED SHEET (RULE 91 ) ~ ~4 ~'0'~ ~p,~ ~~,
,., ,
,._
CA 02454187 2004-O1-15 -.._
14 ,~~'w n ~~,~~~,~~y(1~:':7~t i ,.~ y'!~~ t':~(si ' t ;~:" y ..-"i_ ~ t .y:,
r ,t. ~~ .~t:itt~, .. 5~::,... t t F 4 e,A,
77~.~i~~~~~~9~t~f4 ~:~~~i - f 1 d kx.k . ~ j'~ k 'l.rt ~ ,.. ..;
-t
_ N. . ". ~u ~ - k~. ~> . ,.r,>,~~. rra ,k~~.,: t .~,.. t,~~a:'
27
Example 32
4-(2-Cyariobenzylamino) N (4 6-dimeth~pyrimidin-2-yl)-benzenesulfonamide
Following the procedure outlined in example 11, but substituting benzyl
bromide
by cc-bromo-o-tolunitrile, afforded the title compound with a yield of 21 %.
1H
___~._ 5 NMR (DMSO-d6, 500 MHz): 7.83 (1H, m), 7.71 (2H, m), 7.65 (1H, rn),
7.47 (2H,
m), 7.15 (1H, br, t, 5.8 Hz), 6.73 (1H, m), 6.63 (2H, m), 4.50 {2H, d, 5.8
Hz), 2.24
{6H, s); MS (ESI+): mlz 416 (M + Na)+, 394 (M + H)~".
Example 33
4-(2,4-Dimethox benz l~~no)~4 6-dimethylpyrimidin-2-,~,
benzenesulfonamide
62 mg (0.37 mmol) 2,4-dimethoxybenzylaldehyde and 100.9 mg . (0.36 mmol)
sulfamethazine were dissolved in 4 ml of 1,2-dichloroethane. Acetic acid (168
~.I,
2.8 mmol) was added to the reaction mixture and solution was stirred with
reflex
overnight. Sodium triacetoxyborohydride {162.9 mg, 0.77 mmol) was dissolved to
the reaction solution and refluxing was continued for three hours. The
reaction
mixture was then evaporated to dryness, and purified on silica using gradient
elution {chloroform to 2% methanol in chloroform) and 2:1 petrol
ether:ethylacetate to obtain the title compound as white crystals with a yield
of
10%. . 1H NMR (CDC13, 500 MHz): 7.91 (2H, m), 7.13 (1H, m), 6.58 (3H, m), .
6.47 (1H, m), 6.42 (1H, m), 4.53 (1H, t, 5.0 Hz), 4.30 (2H, d, 5.0 Hz), 3.82
(3H, s),
3.79 (3H, s), 2.33 (6H, s); MS (ESIk): m/z 429 (M + H)~.
r 1~0",~ RECTIFIED SHEET (RULE 91 )
'~4"~09"200~~i
a ,,.
CA 02454187 2004-O1-15
r~~''~' j ' ' t ' - ' ~:P ~.; ~-"i r y: ' ' ~ sr.
;.. _;~ t vs t -. r .. r . ';. ~~ ! r
E"Pt .,~ , s: .t- r , " ~:; . t' a t .~. i t : n ~, $~ r.:'.'' ~Ek
~t~~;'>.'~..
,~ r''~ef1~14 Q~, ~~(}~r 7;17E~C91 ' y~~
k~a;: , "",.,_....1.. ' ~ i:......> .x.'.E,x;:,:-:.:'a ~i;.S a ~x c ...:,.::f
. '9.u.»~''.4,: F ~ ~~~~p9
~.ust.e ..: -,st ,oxt_F.,i.»ai_._...w:-..M~a..,w;.Y7.
Example 34
N:(4,6-Dimethylpyrimidin-2-yl)-4-((1-ethyl-lHindol-3- l~yl)-aminol-
benzenesulfonamide (Compound F~
1-ethylindole-3-carboxaldehyde was prepared from alkylation reaction of indole-
3-
carboxaldehyde. Indole-3-carbaxaldehyde (900 ing; 6.2 mmol~as clissolved in 5
m1 dimethylformamide, ethyl bromide (918 p1, 12 mmol) and sodium hydride
(282.8 mg, 12 mmol) were added to the reaction miXture. Solution was stirred
and
refluxed for three hours. The reaction mixture was then evaporated to dryness
and
washed with water. Pale brown crystals were obtained with 80% yield.
Following , the procedure outlined in example 33, but substituting
2,4-dimethoxybenzylaldehyde by 1-ethylindole-3-carboxaldehyde, afforded the
title compound with a yield similar to that of 4-(2,4-dimethoxybenzylamino) N
(4,6-dimethylpyrimidin-2-yl}-benzenesulfonamide. Instead of sodium
triacetoxyborohydride sodium borohydride was used to .reduce the imine
intermediate. 1H NMR (CDCl3, 500 MHz): 7.88 (2H, m), 7.52 (1H, m), 7.29 (1H,
m), 7.18 (1H, m), 7.05 (1H, m}, 7.03 (1H, s}, 6.56 (2H, m), 6.52 (1H, s), 4.41
(2H,
d, 4.7 Hz), 4.33 (1H, t, 4.7 Hz), 4.08.(2H, q, 7.3 Hz), 2.27 (6H, s), 1.38
(3H, t, 7.3
Hz); MS (ESI+): mlz 458 (M + Na)+.
Example 35
N (4-(4,6-Dimeth~pyrimidin-2-ylsulfamo 1)-phenyll-2-(2-methoxXphenyl)-
acetamide
2-Methoxyphenylacetic acid (33 mg, 0.20 mmol) and sulfamethazine (55 mg, 0.20
lnlriol) were dissolved in 4 ml of chloroform. Triethylarnine (70 p.1, 0.55
rnmol)
and diisopropylcarbodiimide (50 ~1, 0.20 mmol) were added to the reaction
mixture. Solution was stirred and refluxed overnight. The reaction mixture was
t ll,;~ai''~ RECTIFIED SHEET (RULE 91 ) ' ~~q, pg' ~ptj~;~
E ~__.. ".~~ ' ~ .,._~f
~.~~'~~a~~~3~~~~ ~ f p
~e~.~:,z a .'.~." sd.,:.~.,..'' ., t -~ ...,~,.-~"
v
CA 02454187 2004-O1-15
29
then evaporated to dryness, washed with water and purified on silica using
gradient elution (chloroform to 2% methanol in chloroform) to obtain white
crystals with the yield of 40%. 1H NMR (CDCl3, 500 MHz): 8.04 (2H, m), 7.54
(2H, m), 7.30 (2H, m), 6.98 {2H, m), 6.60 {1H, s), 3.95 (3H, s), 3.72 (2H, s),
2.33
(6H, s); MS (ESI+): m/z 449 {M + Na)~'', 427 (M + H)~.
Example 36
2-Acetyl N f4-(4,6-dimeth~rlpyrimidin-2-ylsulfamo~~phenyll-benzamide
Following the procedure outlined in example 35, but substituting 2-meth-
oxyphenylacetic. acid by 2-acetylbenzoic acid, afforded the title compound
with a
yield of 11 %. MS (ESI~): m/z 447 (M + Na)*, 425 (M + H)+.
Example 37
1-Methyl-1H indole-2-carboxylic acid [4-(4,6-dimethylpyrimidin-2-vlsulfamoyl)-
phenyll-amide
Following the procedure outlined in example 35, but substituting
2-methoxyphenylacetic acid by 1-niethylindole-2-carboxylic acid and using
dimethylformamide as a solvent afforded the title compound with a yield
similar to
that of N [4-(4,6-dimethylpyrimidin-2-ylsulfarnoyl)-phenyl]-2-(2-
methoxyphenyl)-
acetamide. MS (ESI'"): m/z 458 (M + Na)+.
°i ;i, , -, ~ . ~: r ~
I
RECTIFIED SHEET (RULE 91) j,~~ 09' 200~i
c i k
;ni;.a, , . a .... ~... r ~ "..,.
CA 02454187 2004-O1-15
fi) ~ e~ 't7. ~ p 4 ' tk -° ~i d,~'3~ ~-', ~ ~~ 1..t ~- -t," a ~ ~. h
'S~~ ~ f,'4
0
~pI~~~ted 14 04 20t~~'~ . ~C)ESC9'L ~ =fl~~489fl~=F(. '
fl~ fl843
sM..,d~..,vYac 5~w~r._: 3 ..:...n... u's ....,t'..z.n_w~r 4 r. ~~. ~ ~., Ue.'
... rMw ~ ~~ ~ 1. b"~;.s~~ f ~ '. ~ y ~5E F
. ,crs.:cii-.w"w,.r" ~""":::. z ~~ ,D.xw..»::. ~r ~.",~.f
Example 38
2-(3,5-Dimethoxyphenyl) N f4-(4 6-dimethyl~yrimidin-2-ylsulfamoyl)-phenyll-
acetarnide
Following the procedure outlined in example 35, but substituting 2-meth-
5 oXyphen~iacetic acia by 3;5=dimethoxyphenyiacetic acid; affo~deci the title
compound with a yield of 54%. MS (ESI+}: m/z 457 (M + H)~.
Example 39
N (4,6-Dimethylpyrimidin-2-yl)-4-(2-(2-methoxyphenyl)-eth~aminol-
benzenesulfonamide (Compound G)
10 33 mg (0.08 mmol) N [4-(4,6-dimethylpyrimidin-2-ylsulfomoyl)-phenyl]-2-(2-
methoxyphenyl)-acetamide was dissolved in 2 ml tetrahydrofurane. 340 ~.l of 1
M
borane tetrahydrofurane complex was added to the reaction mixture under
nihogen
atmosphere and solution was stirred overnight with reflex. Reaction~was
quenched
with 6 M HCl and the reaction mixture was neutralised with 1 M NaOH. Product
15. was extracted with chloroform' and purified on silica using gradient
elution
(chloroform to 2% methanol in chloroform) to obtain white crystals with a
yield of
20%. iH NMR. (CDCl3, 500 MHz): 7.92 (2H, m}, 7.22 (1H, m}, 7.12 (1H, m), 6.89
(2H, m}, 6.60 (1H, br, s), 6.54 (2H, m), 3.85 (3H, s), 3.38 (2H, t, 6.9 Hz),
2.93
(2H, ~t, 6.9 Hz), 2.33 (6H, s); MS (ESI+): mlz 435 (M + Na)~, .413 (M + H}+. .
~,~1,3~;~ RECTIFIED SHEET (RULE 91) ;~'~q, p~ ~p,~~~'~
,.,..... . , i . ""rt.
CA 02454187 2004-O1-15
31
Example 40
t~ rt ( - '~ ~~. ~ i ;.~ ~-s yw~~~' jP~
f 0~~~aso~, ~~0~~~~4~t
t
~..r~.M~.~ s~ x~+r.., "~ 3~f ~_,h ,~. ~x~,.~.:~»
4-f2-(3,5-Dimethoxyphen l~ylaminolN (4 6-dimeth~p ry i~din-2-Xl)-
benzenesulfonamide
Following the procedure outlined in example 39, but substituting N [4_(4,6-di-
~~°-~ a iiieihylpyrimzdin=2=yi5uiiamoyi j plienyi j=2=~~-
meihoxypiienyi)-acetaniiae icy 2-~--
(3,5-dimethoxyphenyl)-N-[4-(4,6-dimethylpyrimidin-2-ylsulfamoyl)-phenyl]-
acetamide, afforded the title compound with a yield of 11%. IH NMR (CDCl3, 500
MHz): 7.92 (2H, m), 6.58 (1H, s), 6.54 (2H, m), 6.34 (3H, s), 4.20 (1H, t, 5.8
Hz),
3.76 (6H, s), 3.42 (2H, m), 2.85 (2H, t, 6.9 Hz), 2.34 (6H, s); MS (ESI~: m/z
465
~ (M + Na)~, 443 (M + H)+.
Example 41
4-(Benzhy~lamino)-N X4,6-dimethylpyrimidin-2-y1)-
benzenesulfonamide (Compound H)
Sulfamethazine (105 mg, 0.38 mmol) and diphenylchloromethane (77 ~.1, 0.38
mmol) were dissolved in 3 ml pyridine. Solution was stirred and refluxed
overnight. The reaction mixture was evaporated to dryness and dissolved to 1 M
NaOH. Product was precipitated with 1 M acetic acid. Precipitation cycle was
repeated for three times to give white crystals of the title compound with a
yield of
8%. MS {ESI+): mlz 467(M + Na)+, 445 (M + H)''~. , . , ,
Example 42
4-(4-Aminobenzylamino)-N (4,6-dimethylp~din-2-yl~benzenesulfonamide
18 mg (0.04 mmol) of N (4,6-dimethylpyrimidin-2-y1)-4-(4-nitrobenzylamino)-
benzenesulfonamide was dissolved in 1 ml of tetrahydrofurane. Hydrazinium
(~ 14~ RECTIFIED SHEET (RULE 91 ) ~'2;~~Qg,;2p~~
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
32
hydrate (50 p l, 1.5 mmol) and catalytic amount of palladium on charcoal were
added to the reaction mixture. Solution was stirred overnight. The reaction
mixture
was evaporated to dryness and purified on silica using gradient elution
(chloroform to 4% methanol in chloroform) to obtain with a yield of 59%. MS
(ESI+): m/z 406 (M + H)+, 384 (M + H)+.
Example 43
4-( f4-(4,6-Dimeth~pyrimidin-2-ylsulfamoyl~-phenylaminol-methyl}-benzamide
4-{ [4-(4,6-Dimethylpyrimidin-2-ylsulfamoyl)-phenylamino]-methyl}-benzoic acid
methyl ester was prepared like described on example 11 and was treated with 25
%
ammonia to obtain the title compound with a yield lower than 1 %. 1H NMR
(CDCl3, 500 MHz): 7.81 (2H, m), 7.67 (2H, m), 7.37 (2H, m), 7.12 (1H, br, t,
6.0
Hz), 6.72 (1H, s), 6.60 (2H, m), 4.37 (2H, d, 6.0 Hz), 2.33 (6H, s); MS
(ESI+): m/z
412 (M + H)+.
Example 44
4-( f (2,6-Dichloro-phenyl)-hydroxy-methyll-amino 1-N-(4,6-dimeth ly~~rrimidin-
2-
xl)-benzenesulfonamide
11 mg (0.02 mmol) of 2,6-dichloro-N [4-(4,6-dimethylpyrimidin-2-ylsulfamoyl)-
phenyl]-benzamide was dissolved in 2 ml tetrahydrofurane. Lithium aluminium
hydride (6 mg, 0.16 mmol) was added to the reaction mixture under nitrogen
atmosphere and solution was stirred overnight. The reaction mixture was
filtered,
evaporated to dryness and purified by on silica using gradient elution
(chloroform
to 4% methanol in chloroform) to obtain the title compound with a yield of
18%.
MS (ESI+): m/z 475 (M + Na)+.
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
33
It will be appreciated that the methods of the present invention can be
incorporated
in the form of a variety of embodiments, only a few of which are disclosed
herein.
It will be apparent for the specialist in the field that other embodiments
exist and
do not depart from the spirit of the invention. Thus, the described
embodiments are
illustrative and should not be construed as restrictive.
REFERENCES
Cheng, Y., and Prusoff, W.H., 1973. Biochem. Pharmacol. 22: 3099
Jasper, J.R., Lesnick, J.D., Chang, L.K., Yamanashi, S.S., Chang, T.C., Hsu,
S.A.O., Daunt, D.A., Bonhaus, D.W., and Egen, R.M., 1998. Biochem. Pharmacol.
55: 1035
Marjamaki, A., Ala-Uotila, S., Luomala, K., Perala, M., Jansson, C., Jalkanen,
M.,
Regan, J.W., and Scheinin, M., 1992. Biochem. Biophys. Acta 1134: 169
Marjamaki, A., Pihlavisto, M., Cockcroft, V., Heinonen, P., Savola, J.-M., and
Scheinin, M., 1998. Mol. Pharmacol. 53: 370
Pohjanoksa, K., Jansson, C.C., Luomala, K., Marjamaki, A., Savola, J.-M., and
Scheinin, M., 1997. Eur. J. Pharmacol. 35: 53
Tian, W.-N., Duzic, E., Zanier, S.M., and Deth, R.C., 1993. Mol. Pharmacol.
45:
524
Kumar, V.B., Reddy, M.V., Indian J. Chem., Sect. B (1985), 24B(12), 1298-1301
Farag, A.M., El-Mouafi, H.M., Khalifa, M., 1991. Egypt. J. Pharm. Sci. 32 (3-
4),
951-9
Wieland, T., and Jakobs, K.H., 1994. Meth. Enzymol. 237: 3
CA 02454187 2004-O1-15
WO 03/008387 PCT/FI02/00643
34
Heinonen et a1.1999, The Journal of Clinical Endocrinology & Metabolism,
84:2429
Link R E et al., 1996, Science 273:803
MacMillan L B et al., 1996, Science 273:801