Note: Descriptions are shown in the official language in which they were submitted.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
3-Desoxy-Vitamin D3 Analog Esters
This invention relates to methods for treating a variety of diseases using
Vitamin D3
analogs and methods for producing these analogs. This invention relates
particularly to 3-
desoxy-20-desmethyl-20-cyclopropyl vitamin D3 analog esters and methods for
producing
and using the same.
Osteoporosis is the most common form of metabolic bone disease and may be
to considered the symptomatic, fracture stage of bone 1055 (osteopenia).
Although
osteoporosis may occur secondary to a number of underlying diseases, 90% of
all cases
appear to be idiopathic. Postmenopausal women are at risk for idiopathic
osteoporosis
(postmenopausal or Type I osteoporosis); another particularly high risk group
for
idiopathic osteoporosis is the elderly of either sex (senile or Type II
osteoporosis).
Osteoporosis has also been related to corticosteroid use, immobilization or
extended bed
rest, alcoholism, diabetes, gonadotoxic chemo-therapy, hyperprolactinemia,
anorexia
nervosa, primary and secondary amenorrhea, transplant immunosuppression, and
oophorectomy. Postmenopausal osteoporosis is characterized by fractures of the
spine,
while femoral neck fractures are the dominant features of senile osteoporosis.
2o The mechanism by which bone is lost in osteoporotics is believed to involve
an
imbalance in the process by which the skeleton renews itself. This process has
been termed
bone remodeling. It occurs in a series of discrete pockets of activity. These
pockets appear
spontaneously within the bone matrix on a given bone surface as a site of bone
resorption.
Osteoclasts (bone dissolving or resorbing cells) are responsible for the
resorption of a
portion of bone of generally constant dimension. This resorption process is
followed by
the appearance of osteoblasts (bone forming cells) which then refill with new
bone the
cavity left by the osteoclasts.
In a healthy adult subject, osteoclasts and osteoblasts function so that bone
formation
and bone resorption are in balance. However, in osteoporotics an imbalance in
the bone
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-2-
remodeling process develops which results in bone being replaced at a slower
rate than it is
being lost. Although this imbalance occurs to some extent in most individuals
as they age,
it is much more severe and occurs at a younger age in postmenopausal
osteoporotics,
following oophorectomy, or in iatrogenic situations such as those resulting
from
corticosteroid therapy or the immunosuppression practiced in organ
transplantation.
Various approaches have been suggested for increasing bone mass in humans
afflicted with osteoporosis, including administration of androgens, fluoride
salts, and
parathyroid hormone and modified versions of parathyroid hormone. It has also
been
suggested that bisphosphonates, calcitonin, calcium, 1,25-dihydroxy vitamin D3
and some
of its analogs, and/or estrogens, alone or in combination, may be useful for
preserving
existing bone mass.
Vitamin D3 is a critical element in the metabolism of calcium, promoting
intestinal
absorption of calcium and phosphorus, maintaining adequate serum levels of
calcium and
phosphorus, and stimulating flux of calcium into and out of bone. Vitamin D3
is
hydroxylated in vivo, with the resulting lcc,25-dihydroxy metabolite being the
active
material. Animal studies with 1,25-(OH)2 vitamin D3 have suggested bone
anabolic
activity. Aerssens et al. in Calcif Tissue Int, 55:443-450 (1994) reported
upon the effect of
Iec-hydroxy Vitamin D3 on bone strength and composition in growing rats with
and
without corticosteroid treatment. However, human usage is restricted to
antiresorption
2o due to the poor therapeutic ratio (hypercalciuria and hypercalcemia as well
as
nephrotoxicity).
Dechant and Goa, in "Calcitriol. A review of its use in the treatment of
postmenopausal osteoporosis and its potential in corticosteroid-induced
osteoporosis,"
Drugs Aging [NEW ZEALAND 5 (4): 300-17 (1994)], reported that 1,25-
dihydroxyvitamin
D3 (calcitriol) has shown efficacy in the treatment of postmenopausal
osteoporosis (and
promise in corticosteroid-induced osteoporosis) based upon a clinical trial in
622 women
with postmenopausal osteoporosis. Patients with mild to moderate disease (but
not those
with more severe disease) who received calcitriol (0.25 microgram twice daily)
had a
significant 3-fold lower rate of new vertebral fractures after 3 years of
treatment compared
3o with patients receiving elemental calcium 1000 mg/day. In patients
commencing long term
treatment with prednisone or prednisolone, calcitriol 0.5 to 1.0
micrograms/day plus
calcium 1000 mg/day, administered with or without intranasal calcitonin 400
IU/day,
prevented steroid-induced bone loss. Overall, calcitriol was well tolerated.
At
recommended dosages hypercalcaemia was infrequent and mild, generally
responding to
reductions in calcium intake and/or calcitriol dosage. However, the narrow
therapeutic
window of calcitriol required that its use be adequately supervised, with
periodic
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-3-
monitoring of serum calcium and creatinine levels. This study clearly
identifies the key
limitation of calcitriol therapy as the close proximity of therapeutic and
tonic doses.
Certain 3-desoxy-20-cyclopropyl vitamin D3 analogs are disclosed as inhibiting
cellular proliferation in vitro in prostate cancer lines ("20-Cyclopropyl-
Cholecalciferol
Vitamin D3 Analogs," M. ICoike et. al., Anticancer Research, 19:1689-1698
(1999))
Hyperpetrathyroidism
Secondary hyperparathyroidism is a common finding in patients with chronic
renal
failure. It is established that the reduction of renal 1,25(0H)2 vitamin D3
(calcitriol)
synthesis is one of the principal mechanisms leading to the secondary
hyperparathyroidism
1o in these patients and it has been shown that calcitriol possesses direct
suppressive action on
PTH synthesis. Therefore, administration of calcitriol has been recommended
for the
treatment of secondary hyperparathyroidism in these patients. However, as
described
above, calcitriol has potent hypercalcemic effects giving it a narrow
therapeutic window
which limits its usage, especially at high doses. It would therefore be
desirable to have an
alternative means of treating hyperparathyroidism without incurring these
undesirable
hypercalcemic effects.
While a variety of compounds are available for treating these and other
diseases,
many of these compounds have undesirable side-effects and/or are relatively
unstable, i.e.,
have short storage period. Therefore, there is a continuing needs for other
compounds
2o which are useful in treating these diseases.
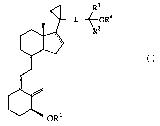
One aspect of the present invention provides a 3-desoxy vitamin D3 analog
ester of
the formula:
vn
or a salt thereof, and methods for using and producing the same, where
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
-CH -CHZ-CHZ-,
so -CH2-CH=CH-,
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-4-
-CH2-C=C-,
-CHZ-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
each of Ra and R3 is independently alkyl or haloalkyl; or RZ and R3 and
together with
the carbon atom to which they are attached to form a cycloalkyl; and
each of Rl and R4 is independently hydrogen, alkyl, acyl group or other
hydroxy
protecting group,
provided at least one of Rl and R4 is an acyl group.
"Acetate" or "Ac" are used interchangeably herein and refer to a moiety of the
formula -C(=O)CH3.
"Acyl" refers to a moiety of the formula -C(O)R', where R' is alkyl,
heteroallcyl,
cycloalkyl, aryl, heteroaryl, aralkyl or heteroaralkyl.
"Alkyl" means a linear fully-saturated hydrocarbon moiety having one to six,
preferably one to four, carbon atoms or a branched fully saturated hydrocarbon
moiety
having three or six carbon atoms.
"Aralkyl" means a moiety of the formula -Ra-Rb, where Ra is alkyl and Rb is
aryl as
defined herein.
"Aryl" means a monocyclic or bicyclic aromatic hydrocarbon moiety. In
addition,
one or more, preferably one, two or three, hydrogen atoms of the aryl moiety
can be
replaced by halo, nitro, cyano, hydroxy, amino, alkyl or alkoxy. Exemplary
aryl groups
include phenyl and naphthalenyl which can be substituted with one or more
substituents
listed above. Preferably, aryl is phenyl.
"Cycloalkyl" means a fully saturated cyclic hydrocarbon moiety of three to six
ring
carbon atoms, e.g., cyclopropyl, cyclopentyl and the like.
"Ester" refers to a compound comprising a moiety of the formula -O-C(=O)-R',
where R' is alkyl, heteroalkyl, cycloalkyl, aryl, heteroaryl, aralkyl or
heteroaralkyl.
"Haloalkyl" refers to an alkyl moiety, as defined above, in which one or more
hydrogen atoms attached to the carbon backbone have been replaced with one or
more
3o halides. Preferred halide is fluoride.
"Heteroalkyl" means an alkyl moiety as defined herein having one or more,
preferably one, two or three, substituents selected from -NRaRb, -OR' wherein
Ra, Rb and R'
are independently of each other hydrogen, alkyl, or the corresponding
protecting group.
"Heteroaralkyl" means a moiety of the formula -Ra-Rb, where Ra is alkyl and Rb
is
heteroaryl as defined herein.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-5-
"Heteroaryl" means a monocyclic or bicyclic radical of 5 to 12 ring atoms
having at
least one aromatic ring containing one, two, or three ring heteroatoms
selected from N, O,
or S, the remaining ring atoms being C, with the understanding that the
attachment point
of the heteroaryl radical will be on an aromatic ring In addition, one or
more, preferably
one, two or three, hydrogen atoms of the heteroaryl moiety can be replaced by
the
substituents described above for the aryl group.
The terms "hydroxy protecting group" and "other hydroxy protecting group" are
used interchangeably herein and refer to hydroxy protecting groups known to
one skilled
in the art excluding alkyl or acyl groups, which are referred herein
specifically.
Representative hydroxy protecting groups include silyl ethers, carbonates,
carbamates,
substituted methyl ethers, substituted ethyl ethers, and the like. A list of
other suitable
hydroxy protecting groups can be found, for example, in Protective Groups in
Organic
Synthesis, 3rd edition, T.W. Greene and P.G.M. Wuts, John Wiley & Sons, New
York, 1999,
which is incorporated herein by reference in its entirety.
"Pharmaceutically acceptable excipient" means an excipient that is useful in
preparing a pharmaceutical composition that is generally safe, non-toxic and
neither
biologically nor otherwise undesirable, and includes an excipient that is
acceptable for
veterinary use as well as human pharmaceutical use. "A pharmaceutically
acceptable
excipient" as used in the specification and claims includes both one and more
than one
2o such excipient.
"Therapeutically effective amount" means the amount of a compound that, when
administered to a mammal for treating or preventing a disease, is sufficient
to effect such
treatment or prevention for the disease. The "therapeutically effective
amount" will vary
depending on the compound, the disease and its severity and the age, weight,
etc., of the
patient to be treated.
"Treating" or "treatment" of a disease includes: ( 1 ) preventing the disease,
i.e.
causing the clinical symptoms of the disease not to develop in a mammal that
may be
exposed to or predisposed to the disease but does not yet experience or
display symptoms
of the disease, (2) inhibiting the disease, i.e., arresting or reducing the
development of the
3o disease or its clinical symptoms, or (3) relieving the disease, i.e.,
causing regression of the
disease or its clinical symptoms.
When referring to a chemical reaction, the terms "treating", "contacting" and
"reacting" are used interchangeably herein and refer to adding or mixing two
or more
reagents under appropriate conditions to produce the indicated and/or the
desired
product. It should be appreciated that the reaction which produces the
indicated and/or
the desired product may not necessarily result directly from the combination
of two
reagents which were initially added, i.e., there may be one or more
intermediates which are
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-6-
produced in the mixture which ultimately leads to the formation of the
indicated and/or
the desired product.
As used herein, the terms "those defined above" and "those defined herein"
when
referring to a variable incorporates by reference the broad definition of the
variable as well
as preferred, more preferred and most preferred definitions, if any.
"Haloalkyl" refers to an alkyl radical, as defined above, in which one or more
hydrogen atoms attached to the carbon backbone have been replaced with one or
more
halides. Preferred halide is fluoride.
1o "Pro-drugs" means any compound which releases an active parent drug
according to Formula (I) in vivo when such prodrug is administered to a
mammalian
subject. Prodrugs of a compound of Formula (I) are prepared by modifying
functional
groups present in the compound of Formula (I) in such a way that the
modifications may
be cleaved in vivo to release the parent compound. Prodrugs include compounds
of
1s Formula (I) wherein a hydroxy group in compound (I) is bonded to any group
that may be
cleaved in vivo to regenerate the free hydroxyl group. Examples of prodrugs
include, but
are not limited to esters (e.g., acetate, formate, and benzoate derivatives),
carbamates (e.g.,
N,N-dimethylaminocarbonyl) and ethers of hydroxy functional groups in
compounds of
Formula (I), and the like. Such compounds are routinely made by one of skill
in the art by
2o acylating or etherifying the hydroxy group in the parent molecule.
"Hydroxy protecting group" refers to a grouping of atoms that when attached
to a hydroxy group in a molecule masks, reduces or prevents the reactivity of
the hydroxy
group. Examples of protecting groups can be found in T.W. Green and P.G. Futs,
Protective Groups in Organic Chemistry, (Wiley, 2nd ed. 1999) and Harrison and
Harrison
25 et al., Compendium of Synthetic Organic Methods, Vols. 1-8 (John Wiley and
Sons, 1971-
1996). Representative hydroxy protecting groups include those where the
hydroxy group is
either acylated or alkylated such as benzyl, and trityl ethers as well as
alkyl ethers,
tetrahydropyranyl ethers, trialkylsilyl ethers and allyl ethers.
It has been surprisingly discovered that certain 3-desoxy-loc-hdroxy-20-
cyclopropyl cholecalciferol vitamin D3 compounds, hitherto known only for
their anti-
proliferative activity are surprisingly efficacious relative to 1,25-dihydroxy
vitamin D3 in
increasing bone formation. Accordingly, one aspect of the present invention
provides a
method for treating osteoporosis or hyperparathyroidism comprising
administering a 3-
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
desoxy-la-hydroxy vitamin D3 analog to the patient, wherein said 3-desoxy
vitamin D3
analog is of the formula:
I
a prodrug or a salt thereof, preferably a compound of formula I or a salt
thereof,
wherein
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
-CHz-CHz-CHz_,
-CHz-CH=CH-,
-CHz-C-C_
-CHz-CHz-C(=O)-, and
-CH=CH-CH=CH-;
each of Rl and R4 is selected from the group consisting of hydrogen and alkyl;
and
each of Rz and R3 is independently selected from the group consisting of alkyl
and
haloalkyl, or Rz and R3 and together with the carbon atom to which they are
attached to
form a cycloalkyl.
Another aspect of the present invention provides a method for producing a
compound of the formula:
R3
L~ORd
Rz
0R'
I
comprising contacting a ketone of the formula:
R3
L-~-OR-0
Rz
O
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
_g_
II
with a phosphine oxide compound of the formula:
Arz
Art-P-O
OR1
III
under conditions sufficient to produce said compound of Formula I,
wherein
each of Arl and Arz is independently optionally substituted aryl;
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
io -CHZ-CHZ-CHZ-,
-CHZ-CH=CH-,
-CHZ-C---C-,
-CHZ-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
15 each of R1 and R4 is selected from the group consisting of hydrogen, alkyl,
and a
hydroxy protecting group; and
each of RZ and R3 is independently selected from the group consisting of
alkyl;
haloalkyl; or RZ and R3 and together with the carbon atom to which they are
attached to
form a cycloalkyl.
2o In one embodiment of the present method for producing a compound of
Formula I, R4 is hydrogen. In a particular embodiment, the method further
comprises the
steps of protecting the hydroxy group of said ketone of Formula II prior to
said step of
contacting with said phosphine oxide compound of Formula III and removing the
hydroxy
protecting group after contacting said ketone of Formula II with said
phosphine oxide
25 compound of Formula III to produce said compound of Formula I.
Preferred is a compound of the formula
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-9-
I
or a salt thereof, preferably a compound of formula I,
wherein
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
-CHZ-CHI-CH2-,
-CHZ-CH=CH-,
-CHZ-C---C-,
-CHZ-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
each of RZ and R3 is independently alkyl or haloalkyl; or R' and R3 and
together with the
carbon atom to which they are attached to form a cycloallcyl; and
each of Rl and R4 is independently hydrogen, alkyl, acyl group or other
hydroxy protecting
group,
provided at least one of Rl and R4 is an acyl group.
Further preferred is a compound of the formula
R3
L-~--OR4
R2
ORI
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-10-
wherein
Rl, Rz, R3, R4 and L are those defined in Claim 1.
Another preferred aspect of the invention is a compound of the formula
R3
L-E-ORd
Rz
OR ~
wherein
L is selected from the group consisting of
_CHz_CHz_CHz_~
-CHz-CH=CH-;
-CHz-C---C-; and
l o -CH=CH-CH=CH-.
Further preferred is a compound of formula I, wherein said linker L is
selected from
the group consisting of
-CHz-CH=CH-; and
-CHz-C---G.
Also preferred is a compound of formula I, wherein Rl is an acyl group.
Another preferred aspect of the invention is a compound according to formula
I,
wherein R4 is an acyl group.
Also preferred is a compound of formula I, wherein each of Rz and R3 is
independently selected from the group consisting of alkyl and haloalkyl .
2o Another preferred aspect of the invention are compounds of formula I,
wherein Rz
and R3 are trifluoromethyl.
Particularly preferred are compounds of formula I selected from
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-11-
[ 1R-( 1 cc(E),3a(3,7acc) ] -Octahydro-7a-methyl-1- [ 5,5,5-trifluoro-4-
hydroxy-4-
(trifluoromethyl)-2-pentynyl] cyclopropyl] -4H-inden-4-ol;
[ 1 R-( 1 oc(E),3a(3,7acc) ] ] Octahydro-7a-methyl-1- [ 1- [ 1- [ 5,5,5-
trifluoro-4-hydroxy-4-
(trifluoromethyl)-2-pentynyl] cyclopropyl]-4H-inden-4-one;
[1R-[lcc(E),3a(3,7aoc)]]-Octahydro-7a-methyl-1-[1-[5,5,5-trifluoro-4-
trifluoromethyl)-4-
[ (trimethylsilyl) oxy] -2-pentynyl] cyclopropyl] -4H-inden-4-one;
3-desoxy-1,25-dihydroxy-20-methyl-23-(E)-ene-26,27-hexafluoro-21,28-cyclochole-
calciferol;
3-desoxy-1,25-dihydroxy-20-methyl-23-(Z)-ene-26,27-hexafluoro-21,28-
to cyclocholecalciferol;
3-desoxy-1,25-dihydroxy-20-methyl-23-yne-21,28-cyclocholecalciferol;
3-desoxy-1,25-dihydroxy-20-methyl-23-yne-26,27-hexafluoro-21,28-
cyclocholecalciferol;
3-desoxy-1,25-dihydroxy-20-methyl-21,28-cyclocholecalciferol;
3-desoxy-1CC-acetoxy-25-hydroxy-20-cyclopropyl-23E-ene-26,27-hexafluoro-
cholecalciferol; and
3-desoxy-lcc,25-diacetoxy-20-cyclopropyl-23E-ene-26,27-hexafluoro-
cholecalciferol.
Further preferred is a method for producing a compound of formula I comprising
(a) contacting a ketone of the formula:
R3
L--~OR4
Rz
O
2o II
with a phosphine oxide compound of the formula:
Arz
Arl-P-O
ORl
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-12-
III
under conditions sufficient to produce said compound of Formula I,
wherein
each of Arl and Arz is independently optionally substituted aryl;
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
-CHz_CHz-CHz-
-CHz-CH=CH-,
-CHz-C---C-,
to -CHz-CHz-C(=O)-, and
-CH=CH-CH=CH-;
each of Rz and R3 is independently alkyl or haloalkyl; or Rz and R3 and
together with the
carbon atom to which they are attached to form a cycloalkyl; and
each of Rl and R4 is independently alkyl, an acyl group or a hydroxy
protecting group, and
(b) when neither Rl nor R4 is an acyl group, acylating the compound of Formula
I with
an acylating agent under conditions sufficient to produce the Compound of
Formula I
where at least one of Rl and R4 is an acyl group.
Particularly preferred is the above method, wherein Rl and R4 are hydroxy
protecting
groups.
Also particularly preferred is the above method, wherein said acylating step
(b)
comprises
(i) removing the hydroxy groups by contacting the resulting compound of said
step (a)
with a hydroxy protecting group removing compound under conditions sufficient
to
produce a 1-hydroxy-3-desoxy vitamin D3 analog of the formula:
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-13-
OH
and
(ii) contacting the 1-hydroxy-3-desoxy vitamin D3 analog with an acylating
agent under
conditions sufficient to produce a 3-desoxy vitamin D3 analog ester of the
Formula
wherein Rla is an acyl group and R4a is hydrogen or an acyl group.
Moreover, particularly preferred is the above method, wherein R4a is an acyl
group.
Further preferred is the above method, wherein Arl and Arz are phenyl.
Another preferred aspect of the invention is a compound according to formula
I,
to when manufactured according to the above method.
Also preferred is a pharmaceutical composition comprising a compound according
to formula I and a pharmaceutically acceptable excipient.
Further preferred are compounds according to formula I for use as
therapeutically
active substance.
15 Another preferred aspect of the present invention are compounds according
to
formula I for the preparation of medicaments for the prophylaxis and therapy
of bone
related diseases.
Preferred are compounds according to formula I for the preparation of
medicaments
for the prophylaxis and therapy of hyperparathyroidism, renal osteodystrophy
or
20 osteoporosis.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
- 14-
Also preferred is a method for treating a bone related disease in a patient
comprising
administering a compound of formula I to the patient.
Further preferred is a method according to formula I, wherein the disease is
selected
from hyperparathyroidism, secondary hyperparathyroidism, renal osteodystrophy
and
osteoporosis.
Another preferred aspect of the invention is a method for treating a bone-
related
disease in a patient comprising administering to the patient a compound
according to
formula
OR4
~o I
or a prodrug or a salt thereof, preferably a compound of formula I or a salt
thereof, most
preferred a compound of formula I,
wherein
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
CHZ-CHZ-CHZ-,
-CHZ-CH=CH-,
-CHz-C---C-,
-CHZ-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
each of Rl and R4 is selected from the group consisting of hydrogen or alkyl;
and
each of RZ and R3 is independently selected from the group consisting of alkyl
or haloallcyl,
or RZ and R3 and together with the carbon atom to which they are attached to
form a
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-15-
cycloalkyl. Particularly preferred is the above method for treating a bone-
related disease in
a patient, comprising administering to the patient a compound according to
formula I.
Also preferred is the use of compounds according to formula
R3
L-~OR4
R2
OR ~
or a prodrug or a salt thereof, preferably compounds of formula I or a salt
thereof, most
preferred compound of formula I, for the preparation of medicaments for the
treatment
and prophylaxis of bone related diseases,
wherein
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
CHZ-CHz-CHZ-,
-CHZ-CH=CH-,
-CHZ-C=C-,
1s -CHZ-CHz-C(=O)-, and
-CH=CH-CH=CH-;
each of Rl and R4 is selected from the group consisting of hydrogen or alkyl;
and
each of RZ and R3 is independently selected from the group consisting of alkyl
or haloalkyl,
or RZ and R3 and together with the carbon atom to which they are attached to
form a
2o cycloalkyl.
Particularly preferred is the above use, wherein the bone related disease is
hyperparathyroidism, renal osteodystrophy or osteoporosis.
Preferred is the above method or use, wherein the compound is of the formula
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
- 16-
wherein
Rl, Rz, R3, R4 and L are defined as before.
Furthermore, preferred is the above method or use, wherein said linker L is
selected
from the group consisting of
-CHz_CHz-CHz-
-CHz-CH=CH-;
-CHz-C=C-; and
-CH=CH-CH=CH-.
1o Also preferred is the above method or use as mentioned before, wherein said
linker L
is selected from the group consisting of
-CHz-CH=CH-; and
-CHz-C---C-.
Further preferred is the above method or use, wherein Ri is hydrogen.
Also preferred is the above method or use, wherein Rl and R4 are hydrogen.
Particularly preferred is the above method or use, wherein each of Rz and R3
is
independently selected from the group consisting of alkyl and haloalkyl.
Further particularly preferred is the method or use as mentioned above,
wherein Rz
and R3 are both trifluoromethyl.
2o Moreover, particularly preferred is the above method or use, wherein the
disease is
osteoporosis.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
- 17-
Additionally, preferred is the above method or use, wherein the disease is
healing or
reducing the incidence of a fracture.
Further particularly preferred is the above method or use, wherein the patient
is at
increased risk of fracture due to decline in estrogen levels.
Also preferred is the above method or use, wherein the patient is a female.
Further preferred is the method or use as mentioned above, wherein the bone
mineral density of the patient increases.
Also particularly preferred is the method or use as mentioned above, wherein
the
patient is also treated with a bisphosphonate, estrogen, selective estrogen
receptor
l0 modulator or anabolic agent.
Further preferred is a method for producing a compound of the formula:
R3
L~OR4
Rz
OR ~
comprising contacting a ketone of the formula:
R3
L~OR4
Rz
15 0
II
with a phosphine oxide compound of the formula:
Arz
ArWP-O
ORl
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
- l~ -
III
under conditions sufficient to produce said compound of Formula I,
wherein
each of Arl and Arz is independently optionally substituted aryl;
s dotted line is optionally a double bond;
L is a linker selected from the group consisting of
_CHa-CHa-CHa-
-CHz-CH=CH-,
-CHZ-C---C-,
to -CHZ-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
each of R1 and R4 is selected from the group consisting of hydrogen and alkyl;
and
each of RZ and R3 is independently selected from the group consisting of alkyl
and
haloalkyl, or RZ and R3 and together with the carbon atom to which they are
attached form
1s a cycloalkyl.
Particularly preferred is the above method for producing, wherein Rø is
hydrogen.
Moreover, particularly preferred is the above method for producing, further
comprising the steps of protecting the hydroxy group of said ketone of Formula
II prior to
said step of contacting with said phosphine oxide compound of Formula III and
removing
2o the hydroxy protecting group after contacting said ketone of Formula II
with said
phosphine oxide compound of Formula III to produce said compound of Formula I.
One aspect of the present invention provides a 3-desoxy-20-desmethyl-20-
cyclopropyl vitamin D3 analog ester of the formula:
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-19-
Vn
or a salt thereof,
wherein
dotted line is optionally a double bond;
I
L is a linker selected from the group consisting of
-CHz-CHz_CHz-
-CHz-CH=CH-,
_CHz-C-C-
to -CHz-CHz-C(=O)-, and
-CH=CH-CH=CH-;
each of Rz and R3 is independently alkyl or haloalkyl; or Rz and R3 and
together with
the carbon atom to which they are attached to form a cycloalkyl; and
each of Rl and R4 is independently hydrogen, alkyl, acyl group or other
hydroxy
protecting group, provided at least one of Rl and R4 is an acyl group.
It has been surprisingly discovered that compounds of Formula I, where at
least one
of Rl and R4 is an acyl group are unexpectedly stable and crystalline relative
to the
compound where both of Rl and R4 are hydrogen, i.e., the parent diol.
When the cyclopentane ring moiety of Formula I does not contain a double bond,
2o i.e., when the dotted line is absent, the stereochemistry of the side chain
on the
cyclopentane ring system can be alpha or beta. Preferably, the stereochemistry
of the side
chain on the cyclopentane ring system is beta, i.e., of the formula:
In one particular embodiment of the present invention, the 3-desoxy vitamin D3
analog ester is of the formula:
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-20-
where Rl, R2, R3, R4 and L are those defined herein.
In still another embodiment, the linker L is selected from the group
consisting of-
CH2-CHZ-CHI-; -CHZ-CH=CH-; -CHz-C=C-; and -CH=CH-CH=CH-. Preferably, L
s is -CHZ-CH=CH- or -CHI-C---G. More preferably L is -CHZ-CH=CH- where the
double bond is traps.
Yet in another embodiment, Rl is preferably an acyl group, more preferably
acetyl.
Still in another embodiment, Rl is an acyl group and R4 is hydrogen or an acyl
group.
In another embodiment, Rl is an acyl group and each of RZ and R3 is
independently
selected from the group consisting of methyl, ethyl and trifluoromethyl.
In yet another embodiment, RZ and R3 are alkyl or haloalkyl, preferably methyl
or
trifluoromethyl, most preferably trifluoromethyl.
A number of different substituent preferences have been given above and
following
any of these substituent preferences results in a compound of the invention
that is more
preferred than one in which the particular substituent preference is not
followed.
However, these substituent preferences are generally independent, although
some
preferences are mutually exclusive, and following more than one of these
preferences may
result in a more preferred compound than one in which fewer of the substituent
preferences are followed.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-21-
Selected Compounds of the Invention
Ex. No. Structure Mass
M+
1 ' ' 535(M+H)
'OH
CFA
I
I
OH
H
FCC CFA 518
I
I
OH
3 408.3
'OH
I
OH
4 F' 516
'OH
CFA
I
I
OH
-OH
I
I
OH
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
Ex. No. Structure Mass
M+
6 _
~CF~
FCC OH
O~O
7 _
~CFa
F~
O
O"O
In a preferred aspect, the present invention is directed to a method for
treating
osteoporosis, hyperparathyroidism or an autoimmune disease in a patient by
administering
a 3-desoxy vitamin D3 analog of Formula I, where Rl, RZ, R3, R4 and L are as
defined in the
Summary of the Invention.
R3
L--E-ORa
Rz
OR ~
When the cyclopentane ring moiety of Formula I does not contain a double
bond, i.e., when the dotted line is absent or is not a double bond, the
stereochemistry of the
l0 side chain on the cyclopentane ring system can be alpha or beta.
Preferably, the
stereochemistry of the side chain on the cyclopentane ring system is beta,
i.e., of the
formula:
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-23-
In one embodiment, the 3-desoxy vitamin D3 analog is the formula:
~n
where R1, R2, R3, R4 and L are as defined in the Summary of the Invention.
In yet another embodiment, the linker L is selected from the group consisting
of -CHI-CHZ-CHz-; -CHZ-CH=CH-; -CHZ-C=C-; and -CH=CH-CH=CH-.
Preferably, L is -CHZ-CH=CH-; -CHZ-C---C-. More preferably L is -CHz-CH=CH-
where the double is traps.
to In another embodiment, preferably Rl is hydrogen.
In another embodiment, preferably R4 is hydrogen.
In another embodiment, both Rl and R4 are hydrogen.
Still in another particular embodiment, each of RZ and R3 is independently
alkyl
or haloalkyl, preferably methyl or trifluoromethyl.
15 A number of different substituent preferences have been given above and
following any of these substituent preferences results in a compound of the
invention that
is more preferred than one in which the particular substituent preference is
not followed.
However, these substituent preferences are generally independent, although
some
preferences are mutually exclusive, and following more than one of these
preferences may
2o result in a more preferred compound than one in which fewer of the
substituent
preferences are followed.
General Synthetic Scheme
25 While a variety of synthetic methodologies are available for preparation of
compounds of Formula I, one particular embodiment for preparing compounds of
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-24-
Formula I includes coupling a ketone with a phosphine oxide. Specifically,
compounds of
Formula I can be prepared by contacting a compound of Formula II,
R3
L--~-ORd
R2
O
II
with a phosphine oxide compound of the formula:
Ar2
Art-P=O
OR ~
III
under conditions sufficient to produce the compound of Formula I,
wherein
each of Arl and Ar2 is independently optionally substituted aryl;
dotted line is optionally a double bond;
L is a linker selected from the group consisting of
-CH2-CHa-CH2-,
-CHZ-CH=CH-,
-CHZ-C---C-,
-CHz-CHZ-C(=O)-, and
-CH=CH-CH=CH-;
each of RZ and R3 is independently alkyl or haloalkyl; or R' and R3 and
together with
the carbon atom to which they are attached to form a cycloalkyl; and
2o each of Rl and R4 is independently alkyl, an acyl group or a hydroxy
protecting
group, and
(b) when neither Rl nor R4 is an acyl group, acylating the compound of Formula
I
with an acylating agent under conditions sufficient to produce a Compound of
Formula I
where at least one of Rl and R4 is an acyl group.
25 In one embodiment, Rl and R4 are hydroxy protecting groups. In this
particular
embodiment, the acylating step (b) comprises:
(i) removing the hydroxy groups by contacting the resulting compound of said
step (a) with a hydroxy protecting group removing compound under conditions
sufficient
to produce a 1-hydroxy-3-desoxy vitamin D3 analog of the formula:
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-25-
and
UH
(ii) contacting the 1-hydroxy-3-desoxy vitamin D3 analog with an acylating
agent
under conditions sufficient to produce a 3-desoxy vitamin D3 analog ester of
the Formula:
V i\
wherein Rla is an acyl group and R4a is hydrogen or an acyl group.
In another embodiment, R4a is an acyl group.
In yet another embodiment, Arl and Ar2 are phenyl.
Suitably hydroxy protecting groups are well known to one of ordinary skill in
the art
to and examples of such hydroxy protecting groups are disclosed in Protective
Groups in
Organic Synthesis, 3rd edition, T.W. Greene and P.G.M. Wuts, John Wiley &
Sons, New
York, 1999, and Harrison and Harrison et al., Compendium of Synthetic Organic
Methods,
Vols. 1-8 (John Wiley and Sons, 1971-1996), which are incorporated herein by
reference in
their entirety. Representative hydroxy protecting groups include benzyl and
trityl ethers,
15 tetrahydropyranyl ethers, trialkylsilyl ethers, carbamates and allyl
ethers.
Typically, hydroxy groups are protected as silyl ethers; however, the scope of
the
invention includes the use of alternative hydroxyl protecting groups known in
the art as
described in the above disclosed Protective Groups in Organic Synthesis, 3rd
edition, and
Compendium of Synthetic Organic Methods, Vols. 1-8.
2o In general, a phosphine oxide of Formula III in tetrahydrofuran is reacted
with n-
butyllithium typically at about -78 °C. To this mixture is then added
solution of a ketone
of Formula II in tetrahydrofuran to provide a compound of Formula I. As stated
above,
when Ri and/or R4 are hydrogen, they are protected with hydroxy protecting
groups prior
to the coupling reaction. In such a case, the hydroxy protecting groups are
then removed
25 to provide a compound of Formula I.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-26-
Synthesis and purification of compounds of Formula III are known and
conventional
in this art. See, for example, M.R. Uskokovic et al. "Vitamin D Gene
Regulation, Structure
Function Analysis and Clinical Application," Paris, France, pp 139-145 (1991),
U.S. Patent
Nos., 5,086,191 and 5,616,759 to DeLuca et al., 5,087,619 to Baggiolini et
al., 5,384,314 to
Doran et al., 5,428,029 to Doran et al., 5,451,574 to Baggiolini et al.;
European patent
publication EP 0 808,832 A2, patent publication WO 96/31216 to Brasitus et
al.; Shiuey et
al., J. Org. Chem., 55:243-247 (1990), Kiegel, J. et al. and Tetr. Lett.,
32:6057-6060 (1991),
Perlman, K. L., et al., Tetr. Lett., 32:7663-7666 (1991).
Reaction Scheme 1 illustrates a synthetic method for preparing a compound of
to Formula IA.
/ CF3
Example 1 I OOH
CF3 CF3
H Example 2
FsC V
IV HO
HO
/ CF3 / CF3
-OSi(CH3)3 Example 3 I OOH
CF3 CF3
VII ~ VI
O O
Ph2P=p
~OH ORa
IIIA CFs CF3
Example 4
OSi(CH3)Zt-Bu
Example 9 or 10 ~ where
R~ is H or-C(=0)CH3
IA~
Scheme 1
IA
In Reaction Scheme l, the compound of Formula IV is a known compound prepared
by the method described in WO99/12894, published March 18, 1999 (Preparation
of 1,3-
dihydroxy-20,20-dialkyl vitamin D3 analogs). The compound of Formula IV is
converted
to the compound of Formula V by selective partial reduction of the triple bond
to anb E-
double bond using lithium aluminum hydride in inert organic acid such as
2o tetrahydrofuran. The reaction is typically conducted by adding the compound
of Formula
IV to a suspension of LiAlH4 in THF at 0 ~C or 5 ~C. The reaction mixture is
heated under
refluxing condition to provide the compound of Formula V. The compound of
Formula V
is converted to the ketone of Formula VI by oxidation using an oxidizing agent
such as
pyridinium dichromate. The reaction is generally conducted in a halogenated
solvent such
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
_ 27 -
as methylene chloride at room temperature. The hydroxy group of compound of
Formula
VI is then protected as a silyl ether of Formula VII using a silylating agent,
such as 1-
(trimethylsilyl)imidazole, trimethylsilyl chloride or trimethylsilyl triflate,
in an inert
solvent, such as a halogenated solvent (e.g., methylene chloride), at room
temperature.
The compound of Formula IIIA is reacted with n-butyllithium and the resulting
compound
is reacted with the compound of Formula VII in tetrahydrofuran at temperature
of
generally about -78 ° C, and the silyl protecting groups are then
removed, for example, with
tetrabutylammonium fluoride in tetrahydrofuran solvent to give the compound of
Formula
IA'. The free hydroxyl groups are then acetylated to provide the compound of
Formula IA,
1o for example, with acetic anhydride in pyridine. Because the secondary
hydroxyl group is
generally more reactive, it can be selectively acetylated depending on the
amount of
acetylating agent used andlor the reaction conditions used, e.g., the reaction
temperature
and/or the reaction time. Alternatively, both the secondary and the tertiary
hydroxyl
groups can be acteylated by using an excess amount of the acetylating agent
and longer
reaction time.
Similarly, a Z-stereoisomer analog or a saturated carbon chain analog of
compound
of Formula IA can be prepared by reduction of the compound of Formula IV with
hydrogen in the presence of an appropriate hydrogenation catalyst, such as Pd-
S or Pd,
respectively. The resulting compounds can be subjected to similar reaction
conditions as
2o shown in Scheme I to produce the corresponding a Z-isomer analog and a
saturated carbon
chain analog of the compound of Formula IA.
As shown in Reaction Scheme II, a compound of Formula II comprising an
acetylenic
alcohol and varying alkyl, haloalkyl and cycloalkyl groups of RZ and R3 can be
prepared by
condensing an acetylide anion derived from a compound of Formula VIII (where
Pg is a
hydroxy protecting group) with an appropriate ketone, haloketone (e.g.
hexafluoroacetone) and cycloketone, and removing the protecting group.
R2 2
1. nBuLi ~
~OH ~ ~OH
2. R4-C(=O)-Rs R R
OPg VIII OPg IX OH x
Scheme II
The compound of Formula X is then subjected to a similar reaction conditions
3o shown above in Reaction Scheme I (i.e., oxidation, protection and coupling)
to produce a
compound of Formula I having an acetylenic linker moiety.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
_~g_
In certain preferred embodiments, the dotted line is a double bond, i.e., a
compound
of the formula:
IB
In yet another preferred embodiment, the linker L is selected from the group
consisting of
_CHz-CHz-CHz_
-CHz-CH=CH-;
-CHz-C---C-; and
-CH=CH-CH=CH-.
to Preferably, Rl is an acyl group.
Preferably, R4 is hydrogen or an acyl group.
Preferably, each of Rz and R3 is independently selected from the group
consisting of
methyl, ethyl and trifluoromethyl or Rz and R3 together with the carbon atom
to which
they are attached to form a cyclopentyl ring.
1s A number of different substituent preferences have been given above and
following
any of these substituent preferences results in a compound of the invention
that is more
preferred than one in which the particular substituent preference is not
followed.
However, these substituent preferences are generally independent, although
some
preferences are mutually exclusive, and following more than one of these
preferences may
2o result in a more preferred compound than one in which fewer of the
substituent
preferences are followed.
Another aspect of the invention provides salts of a compound of Formula I.
25 While a variety of synthetic methodologies are available for preparation of
compounds of Formula I, one particular embodiment for preparing compounds of
Formula I is illustrated below.
Compounds of Formula I can be prepared from compounds of Formula II,
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-29-
R3
L-~ORd
Rz
c II
by reaction with a phosphine oxide compound of the formula:
Arz
Art-P=O
oR' III
where each of Arl and Arz is independently optionally substituted aryl, and
the dotted
line, Rl, R2, R3, R4 and L are as defined in the Summary of the Invention.
Preferably, Arl
and Ar2 are phenyl. When Rl and/or R4 are hydrogen, the corresponding hydroxy
groups
are preferably protected with hydroxy protecting groups that are compatible
with the
coupling reaction conditions prior to the coupling reaction between the ketone
of Formula
II and the phosphine oxide compound of Formula III. Suitable hydroxy
protecting groups
1o are well known to one of ordinary skill in the art and examples of such
hydroxy protecting
groups are disclosed in Protective Groups in Organic Synthesis, 3rd edition,
T.W. Greene and
P.G.M. Wuts, John Wiley & Sons, New York, 1999, and Harrison and Harrison et
al.,
CompendiT.cm of Synthetic Organic Methods, Vols. 1-8 (John Wiley and Sons,
1971-1996),
which are incorporated herein by reference in their entirety. Representative
hydroxy
protecting groups include benzyl and trityl ethers, tetrahydropyranyl ethers,
trialkylsilyl
ethers and allyl ethers.
Typically, hydroxy groups are protected as silylethers; however, the scope of
the
invention includes the use of alternative hydroxyl protecting groups known in
the art as
described in the above disclosed Protective Groups in Organic Synthesis, 3rd
edition, and
Compendium of Synthetic Organic Methods, Vols. 1-8.
In general, a phosphine oxide compound of Formula III in tetrahydrofuran is
reacted with n-butyllithium typically at about -78 ~C. To this mixture is then
added
solution of a ketone of Formula II in tetrahydrofuran to provide a compound of
Formula I.
As stated above, when Rl and/or R4 are hydrogen, they are protected with
hydroxy
protecting groups prior to the coupling reaction. In such a case, the hydroxy
protecting
groups are then removed to provide a compound of Formula I.
Synthesis and purification of compounds of Formula III are known and
conventional in this art. See, for example, M.R. Uskokovic et al. "Vitamin D
Gene
Regulation, Structure Function Analysis and Clinical Application," Paris,
France, pp 139-
145 (1991), U.S. Patent Nos., 5,086,191 and 5,616,759 to DeLuca etal.,
5,087,619 to
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-30-
Baggiolini et al., 5,384,314 to Doran et al., 5,428,029 to Doran et al.,
5,451,574 to Baggiolini
et al.; European patent publication EP 0 808,832 A2, patent publication WO
96/31216 to
Brasitus et al.; Shiuey et al., J. Org. Chem., 55:243-247 ( 1990), Kiegel, J.
et al. and Tetr. Lett.,
32:6057-6060 ( 1991 ), Penman, K. L., et al., Tetr. Lett., 32:7663-7666 ( 1991
).
Reaction Scheme Ia illustrates a synthetic method for preparing a compound of
Formula IA.
/ CF3
Example I I ~OH
CF3 CF3
Example 2
OH
FsC V
N HO
HO CF
CF3
/ _Example 3 OH
SOS i(CH 3)3 CF3
CF3
VI
O
VII
O
Ph2P=O
IIIA Example 4
OS i(CH 3)Zt-Bu
Scheme la
to
In Reaction Scheme la, the compound of Formula IV is a known compound
prepared by the method described in W099/12894, published March 18, 1999
(Preparation
of 1,3-dihydroxy-20,20-dialkyl vitamin D3 analogs). The compound of Formula IV
is
converted to the compound of Formula V by selective partial reduction of the
triple bond
15 to an E-double bond using lithium aluminum hydride in inert organic acid
such as
tetrahydrofuran. The reaction is typically conducted by adding the compound of
Formula
IV to a suspension of LiAlH4 in THF at 0 °C or 5 °C. The
reaction mixture is heated under
refluxing condition to provide the compound of Formula V. The compound of
Formula V
is converted to the ketone compound of Formula VI by oxidation using an
oxidizing agent
2o such as pyridinium dichromate. The reaction is generally conducted in a
halogenated
solvent such as methylene chloride at room temperature. The hydroxy group of
compound
of Formula VI is then protected as a silyl ether of Formula VII using a
silylating agent, such
as 1-(trimethylsilyl)imidazole, trimethylsilyl chloride or trimethylsilyl
triflate, in an inert
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-31-
solvent, such as a halogenated solvent (e.g., methylene chloride), at room
temperature.
The compound of Formula IIIa is reacted with n-butyllithium and the resulting
compound
is reacted with the compound of Formula VII in tetrahydrofuran at a
temperature of -78° C
to give the compound of Formula IA after removal of silyl protecting groups,
for example,
with tetrabutylammonium fluoride in tetrahydrofuran solvent.
Similarly, a Z-stereoisomer analog or a saturated carbon chain analog of
compound of Formula IA can be prepared by reduction of the compound of Formula
IV
with hydrogen in the presence of an appropriate hydrogenation catalyst, such
as Pd-S or
Pd, respectively. The resulting compounds can be subjected to similar reaction
conditions
as shown in Scheme Ia to produce the corresponding a Z-isomer analog and a
saturated
carbon chain analog of the compound of Formula IA.
As shown in Reaction Scheme 2a, a compound of Formula II comprising an
acetylenic alcohol and varying alkyl, haloalkyl and cycloalkyl groups of Rz
and R3 can be
prepared by condensing an acetylide anion derived from a compound of Formula
VIII
(where Pg is a hydroxy protecting group) with an appropriate ketone,
haloketone (e.g.
hexafluoroacetone) and cycloketone, and removing the protecting group.
Rz z
1. nBuLi ~
I 3 -OH ~ ~OH
2. Rd-C(=O)-RS R R
OPg VIII OPg IX OH ?C
Scheme 2a
2o The compound of Formula X is then subjected to a similar reaction
conditions
shown above in Reaction Scheme Ia (i.e., oxidation, protection and coupling)
to produce a
compound of Formula I having an acetylenic linker moiety.
Ut111
The compounds of the present invention are useful for the prevention and
treatment
of a variety of mammalian conditions manifested by loss of bone mass. All such
conditions
are referred to as "bone-related diseases" and are described in more detail
hereunder. In
particular, the compounds of this invention are indicated for the prophylaxis
and
therapeutic treatment of osteoporosis and osteopenia in mammals without
inducing
hypercalciuria, hypercalcemia, or nephrotoxicity. "Hypercalcemia" is an
excessive
concentration of calcium in the serum; in humans (and rats) this corresponds
to greater
than about 10.5 mg/dl. "Intolerable hypercalcemia", usually occurring at serum
calcium
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-32-
concentrations greater than about 12 mg/dl, is associated with emotional
lability,
confusion, delirium, psychosis, stupor, and coma.
The compounds of the present invention are useful in the treatment of Type I
(postmenopausal), Type II (iatrogenic), and Type III (senile) osteoporosis,
including that
associated with corticosteroid treatment (e.g. for asthma), as well in the
treatment of
osteodystrophy due to renal dialysis and hyperparathyroidism. Treatment with
the vitamin
D3 analogs as described herein results in increased bone mineral density and
unlike
conventional treatments provides bone of good quality. Therefore, the
treatments
described herein may reduce the incidence of fracture and result in faster
healing of pre-
to existing fractures. Such treatments are particularly useful for patients
suffering from
estrogen withdrawal (e.g. elderly females) who would otherwise be at risk for
an increased
fracture rate. Types of fractures treatable include both traumatic and
osteoporotic
fractures, e.g., fractures of the hip, neck of the femur, wrist, vertebrae,
spine, ribs, sternum,
larynx and trachea, radius/ulna, tibia, patella, clavicle, pelvis, humerus,
lower leg, fingers
and toes, face and ankle.
The compounds of the present invention are also useful in treating diseases
caused by
elevated levels of parathyroid hormone. In one aspect, compounds of the
invention are
used in treating secondary hyperparathyroidism associated with renal failure
and in
particular with reversing or reducing the bone loss associated with renal
insufficiency.
2o Other aspects include the treatment of renal osteodystrophy associated with
late stage
secondary hyperparathyroidism. Other aspects include the treatment of primary
hyperparathyroidism.
Compounds of Formula I are also useful in treating neoplastic diseases such as
leukemia, colon cancer, breast cancer and prostate cancer.
Generally, compounds of the present invention do not cause the elevated
calcium
levels observed with other vitamin D3 analogs such as 1,25 (OH)Z vitamin D3,
thus
providing an improved therapeutic ratio and better treatment of the above
diseases.
Administration & Pharmaceutical compositions
In general, the compound of this invention may be administered in amounts
between
about 0.0002 and 0.5 mg compound/kg body weight per day, preferably from about
0.001
to about 0.1 mg/kg body weight per day, more preferably from about 0.002 to
about 0.02
mg/kg body weight per day, most preferably from about 0.005 to about 0.010
mg/kg body
weight per day. For a 50 kg human subject, the daily dose of active ingredient
may be from
about 0.01 to about 25 figs, preferably from about 0.05 to about 10 ~.gs, most
preferably
from about 1.0 ~.g to about 10 ~,g per day. This dosage can be delivered in a
conventional
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-33-
pharmaceutical composition by a single administration, by multiple
applications, or via
controlled release, as needed to achieve the most effective results,
preferably once or twice
daily by mouth. In certain situations, alternate day dosing can prove adequate
to achieve
the desired therapeutic response.
The selection of the exact dose and composition and the most appropriate
delivery
regimen are influenced by, inter alia, the pharmacological properties of the
formulation,
the nature and severity of the condition being treated, and the physical
condition and
mental acuity of the recipient. In general, the requisite dose is greater for
higher doses of
corticosteroids in the treatment of corticosteroid induced osteopenia.
1o Representative delivery regimens include oral, parenteral (including
subcutaneous,
intramuscular and intravenous), rectal, buccal (including sublingual),
pulmonary,
transdermal, and intranasal, most preferably oral. Administration can be
continuous or
intermittent (e.g., by bolus injection).
A related aspect of this invention relates to combination therapies of
compounds of
Formula I with other active agents such as bisphosphonates, estrogen, SERMS
(selective
estrogen receptor modulators), calcitonins or anabolic therapies. Examples of
bisphosphonates include alendronate, ibandronate, pamidronate, etidronate and
risedronate. Examples of SERMS include raloxifene, dihydroraloxifene and
lasofoxifene.
Calcitonins include human and salmon calcitonin. Anabolic agents include
parathyroid
2o hormones (PTH) e.g. hPTH(1-34), PTH(1-84), and parathyroid hormone-related
protein
(PTHrP) and analogs thereof. Particular analogs of PTHrP are described in
"Mono- and
Bicyclic Analogs of Parathyroid Hormone-Related Protein. 1. Synthesis and
Biological
Studies," Michael Chorev et al. Biochemistry, 36:3293-3299 ( 1997) and "Cyclic
analogs of
PTH and PTHrP," WO 96/40193 and U.S. Patent No. 5,589,452 and WO 97/07815. The
other active agent may be administered concurrently, prior to or after the
compound of
Formula I and may be administered by a different delivery method.
A further aspect of the present invention relates to pharmaceutical
compositions
comprising a compound of the present invention as an active ingredient in
admixture with
a pharmaceutically acceptable non-toxic carrier. As mentioned above, such
compositions
3o can be prepared for parenteral (subcutaneous, intramuscular or intravenous)
administration, particularly in the form of liquid solutions or suspensions;
for oral or
buccal administration, particularly in the form of tablets or capsules; for
pulmonary or
intranasal administration, particularly in the form of powders, nasal drops or
aerosols; and
for rectal or transdermal administration.
The compositions of the present invention can conveniently be administered in
unit
dosage form and can be prepared by any of the methods well-known in the
pharmaceutical
art, for example, as described in Remington's Pharmaceutical Sciences, 17th
ed., Mack
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-34-
Publishing Company, Easton, PA ( 1985). Formulations for parenteral
administration can
contain as excipients sterile water or saline, alkylene glycols such as
propylene glycol,
polyalkylene glycols such as polyethylene glycol, oils of vegetable origin,
hydrogenated
naphthalenes and the like. Formulations for nasal administration can be solid
and can
contain excipients, for example, lactose or dextran, or can be aqueous or oily
solutions for
use in the form of nasal drops or metered spray. For buccal administration
typical
excipients include sugars, calcium stearate, magnesium stearate,
pregelatinated starch, and
the like.
Orally administrable compositions can comprise one or more physiologically
1o compatible carriers and/or excipients and can be in solid or liquid form.
Tablets and
capsules can be prepared with binding agents, for example, syrup, acacia,
gelatin, sorbitol,
tragacanth, or poly-vinylpyrollidone; fillers, such as lactose, sucrose, corn
starch, calcium
phosphate, sorbitol, or glycine; lubricants, such as magnesium stearate, talc,
polyethylene
glycol, or silica; and surfactants, such as sodium lauryl sulfate. Liquid
compositions can
contain conventional additives such as suspending agents, for example sorbitol
syrup,
methyl cellulose, sugar syrup, gelatin, carboxymethylcellulose, or edible
fats; emulsifying
agents such as lecithin, or acacia; vegetable oils such as almond oil, coconut
oil, cod liver
oil, or peanut oil; preservatives such as butylated hydroxyanisole (BHA) and
butylated
hydroxytoluene (BHT). Liquid compositions can be encapsulated in, for example,
gelatin
2o to provide a unit dosage form.
Preferred solid oral dosage forms include tablets, two-piece hard shell
capsules and
soft elastic gelatin (SEG) capsules. SEG capsules are of particular interest
because they
provide distinct advantages over the other two forms (see Seager, H., "Soft
gelatin capsules:
a solution to many tableting problems"; Pharmaceutical Technology, 9, ( 1985).
Some of the
advantages of using SEG~ capsules are: a) dose-content uniformity is optimized
in SEG
capsules because the drug is dissolved or dispersed in a liquid that can be
dosed into the
capsules accurately, b) drugs formulated as SEG capsules show good
bioavailability because
the drug is dissolved, solubilized or dispersed in an aqueous-miscible or oily
liquid and
therefore when released in the body the solutions dissolve or are emulsified
to produce
3o drug dispersions of high surface area, and c) degradation of drugs that are
sensitive to
oxidation during long-term storage is prevented because the dry shell of soft
gelatin
provides a barrier against the diffusion of oxygen.
The dry shell formulation typically comprises of about 40% to 60%
concentration of
gelatin, about a 20% to 30% concentration of plasticizer (such as glycerin,
sorbitol or
propylene glycol) and about a 30 to 40% concentration of water. Other
materials such as
preservatives, dyes, opacifiers and flavours also can be present. The liquid
fill material
comprises a solid drug that has been dissolved, solubilized or dispersed (with
suspending
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-35-
agents such as beeswax, hydrogenated castor oil or polyethylene glycol 4000)
or a liquid
drug in vehicles or combinations of vehicles such as mineral oil, vegetable
oils, triglycerides,
glycols, polyols and surface-active agents.
The following examples are given to enable those skilled in the art to more
clearly
understand and to practice the present invention. They should not be
considered as
limiting the scope of the invention, but merely as being illustrative and
representative
thereof.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-36-
EXAMPLES
The following examples are offered to illustrate, but not to limit the claimed
invention.
Example 1
This example illustrate a method for producing [1R-(lcc(E),3a(3,7acc)]-
Octahydro-
7a-methyl-1- [ 5,5,5-trifluoro-4-hydroxy-4-(trifluoromethyl)-2-pentynyl]
cyclopropyl] -4H-
inden-4-ol.
/ CF3
CF3 v I 'OH
OH CF3
F3C
HO HO
To a stirred, chilled (5 °C) suspension of LiAlH4 (237.2 mg; 6.25 mmol)
in anhyd.
THF (6.0 ml) was added powdered NaOMe (338 mg, 6.25 mmol). The mixture was
stirred
under Ar at 5 °C for 15 min, treated with a solution of [1R-
(lcc,3a(3,4oc,7acc)]-octahydro-
7a-methyl-1- [ 1- [5,5,5-trifluoro-4-hydroxy-4-(trifluoromethyl)-2-pentynyl]
cyclopropyl] -
4H-inden-4-of (500 mg, 1.25 mmol) in anhyd THF (6.0 ml), and then boiled under
reflux
for 2.5 h. After cooling, the mixture was diluted with EtzO (25 ml), quenched
by the drop-
wise addition of water (2.0 ml) and 2 M NaOH (2.0 ml), and stirred at room
temperature
for 30 min. MgS04 (5 g) was added, and after an additional 30 min of stirring,
the mixture
was diluted with EtzO (25 ml) and filtered over Celite ( 15 g), which was
washed with
2o EtOAc (3 x 20 ml). Evaporation gave a gum (508 mg), which was purified by
flash
chromatography (50 g of silica gel, 3.5 cm diameter column, 30% EtOAc in
hexanes),
taking 20-ml fractions. Evaporation of fractions 7-12 gave colorless crystals
(486 mg),
which were triturated with hexane and collected by filtration to give the
title compound
(442 mg, 88%): mp 122-123 °C; [cc]D + 42.1° (EtOH, c = 0.80); IR
3540, 1602, 965 cm 1; 1H
NMR S 0.05 ( 1 H, m), 0.27 ( 1 H, m), 0.34 ( 1 H, m), 0.74 ( 1 H, m), 0.98 ( 1
H, m), 1.00 ( 3
H, s), 1.17-1.25 (2 H, m), 1.35-1.60 (6 H, m), 1.65 (1 H, dd, J= 12, 5), 1.75-
1.87 (3 H, m),
2.02(lH,d,J=11),2.78(lH,dd,J=14,8.5),2.92 (l H, s,OH),4.05(lH,brs),5.59(1
H, d, J= 16), 6.30 (1 H, ddd, J= 16, 8.5, 6); MS rnlz 400 (M+, 10). Anal.
Calcd for
Ci9HzsF60z: C, 56.99; H, 6.55; F, 28.47. Found: C, 56.87; H, 6.33; F, 28.69.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-37-
Examyle 2
This example illustrate a method for producing [1R-
(lcc(E),3a(3,7acc)]]Octahydro-
7a-methyl-1- [ 1- [ 1- [ 5,5,5-trifluoro-4-hydroxy-4-(trifluoromethyl)-2-
pentynyl] cyclopropyl] -4H-inden-4-one.
/ CF3 / CF3
OH ~ ~OH
Fa CF3
- v
HO O
To a stirred solution of [1R-(1CC(E),3a~3,4oC,7aCC)]]octahydro-7a-methyl-1-
[5,5,5-
trifluoro-4-hydroxy-4-(trifluoromethyl)-2-pentynyl]cyclopropyl]-4H-inden-4-of
(400 mg,
1.00 mmol) in CHZC12 (8.0 ml) was added pulverized pyridinium dichromate (1.25
g, 3.3
to mmol). The mixture was stirred at room temperature for 4.5 h, diluted with
diisopropyl
ether ( 15 ml), and worked up to give 395 mg of a colorless gum. Flash
chromatography of
which (50 g of silica gel, 3.5 cm diameter column, 30% EtOAc in hexanes),
collecting 20-ml
fractions, gave after evaporation of fractions 7-12, the title compound (376
mg, 94%) as
colorless crystals: mp 79-80 °C; [oc]D + 6.9° (EtOH, c= 1.00);
IR 3334, 1704, 964 cm 1; 1H
15 NMR 8 0.14 (1 H, m), 0.33 (1 H, m), 0.69 (1 H, m), 0.70 (3 H, s), 1.46-1.80
(5 H, m), 1.91-
2.30(6H,m),2.44(lH,dd,J=11,6),2.74(lH,dd,J=15,8.5),2.98 (l H, s,OH),5.62(1
H, d, J= 15), 6.33 (1 H, ddd, J= 15, 8.5, 6); MS mlz 398 (M+, 20) Anal. Calcd
for
C19H24F6~2~ C~ 57.28. H, 6.07; F, 28.61. Found: C, 57.19; H, 6.25; F, 28.71.
2o Example 3
This example illustrates a method for producing [1R-[la(E),3a(3,7acc)]]-
Octahydro-
7a-methyl-1-[1-[5,5,5-trifluoro-4- trifluoromethyl) -4-[(trimethylsilyl)oxy]-2-
pentynyl] cyclopropyl] -4H-inden-4-one.
CFa ~ CFa
'OH ~ ~OSi(CH3)a
CF3 '~ CF3
O O
25 A stirred solution of [1R-(lcc)(E),3a(3,7aa)]]-octahydro-7a-methyl-1-[1-
[5,5,5-
trifluoro-4- hydroxy-4-trifluoromethyl)- 2-pentynyl] cyclopropyl]-4H-inden-4-
one ( 165
mg, 0.41 mmol) in anhyd CHZCIz (5 ml) was reacted with 1-
(trimethylsilyl)imidazole (0.5
ml, 3.4 mmol) during 5 h to give after work up crude title compound ( 193
mg,). Flash
chromatography (25 g of silica gel, 20 % EtOAc in hexanes) gave the title
compound (161
3o mg, 83%) as an oil: [oc]D + 3.4 ° CHCl3, c = 1.0); IR 1706 cm 1; 1H
NMR b 0.13 (1 H, m),
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-38-
0.22 (9 H, s), 0.32 ( 1 H, m), 0.67 ( 1 H, m), 0.70 (3 H, s), 1.10 ( 1 H, m),
1.50-2.40 ( 11 H,
m),2.44(lH,dd,J=11,6),2.68(lH,dd,J=16,8.5),5.57 (l H,d,J=16),6.16 (1 H,
ddd, J= 16, 8.5, 6); MS mlz 470 (M+, 33). Anal. Calcd for C22H3aFsOzSI: C,
56.15; H, 6.85;
F, 24.22. Found: C, 56.42; H, 6.63; F, 24.37.
Example 4
This example illustrates a method for producing 3-desoxy-1,25-dihydroxy-20-
methyl-23-(E)-ene-26,27-hexafluoro-21,28-cyclochole-calciferol.
Ph~P=O
/ CF3
OSI(CH3)3 OSI(CH3)Zt-BU
CF3
O
1~
Homer reagent [R-(Z)]-[2-[3-[[(l,l-dimethylethyl)-dimethyl-silyl]oxy]-2-
methylenecyclohexylidene] ethyl] diphenylphosphine oxide (395 mg, 0.872 mmol)
in
anhydrous THF (5.0 ml) was deprotonated with a 1.6 M solution of n-BuLi in
hexanes
(0.55 ml, 0.88 mmol) at-78 °C and after 8 min reacted with ketone [1S-
15 [lct(E),3a(3,7aCC]]octahydro-7a-methyl-1-[1-[5,5,5-trifluoro-4-
(trifluoromethyl)-4-
[trimethylsilyl)oxy-2-pentenyl]cyclopropyl]-4H-inden-4-one (170 mg, 0.361
mmol) in
anhyd THF (2.0 ml) during 3 h and worked up. Flash chromatography (45 g of
silica gel,
20% EtOAc in hexanes) gave a gum (215 mg). This was dissolved in THF (3 ml),
treated
with 1.0 M solution of n-Bu4N+F- in THF (2.8 ml) and stirred for 19 h. Work up
followed
2o by flash chromatography (40 g silica gel, 40% EtOAc in hexanes)gave a gum,
which was
dissolved in HCOZMe (5.0 ml), filtered through a 0.45 [um filter and
evaporated. High
vacuum drying (3 h) gave title compound (144 mg) as a colorless foam: (oc]D
4.0° (MeOH,
c = 0.35); UV 7~m~ 265 (~ = 15,837), 211 (~ =14,458) nm; IR 3598, 1651 cm 1;
1H NMR 8
0.11 (1 H, m), 0.29 (2 H, m), 0.60 (3 H, s), 0.61 (1 H, m), 1.10 (1 H, m),
1.21-1.35 (1 H,
25 m), 1.50 (6 H, m), 1.70 (2 H, m), 1.90 (2 H, m), 2.00 (3 H, m), 2.30 (2 H,
m), 2.60 (1 H, d,
J = 12), 2.85 (2 H, m), 2.90 ( 1 H, s), 4.22 ( 1 H, s), 4.42 ( 1 H, s), 4.99 (
1 H, s), 5.32 ( 1 H, s),
5.42(lH,d,J=12),5.99(lH,d,J=11),6.10(lH,ddd,J=12,7,6),6.36 (l H,d,J=11);
MS (FAB) m/z 535 (M+ + H).
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-39-
Example S
This example illustrates a method for producing 3-desoxy-1,25-dihydroxy-20-
methyl-23-(Z)-ene-26,27-hexafluoro-21,28-cyclocholecalciferol.
PhZP=O
~OSi(CH3)Zt-Bu
Hornerreagent [R-(Z)]-2-[3-[[(1,1-dimethylethyl)-dimethylsilyl]oxy]-2-
methylenecyclohexylidene] ethyl] diphenylphosphineoxide (236 mg, 0.5 mmol) in
THF (3.0
ml) was treated with a 1.6 M solution of n-BuLi in hexanes(0.32 ml, 0.512
mmol). After 8
min, ketone [1S-[1CC(Z), 3a[3, 7aCC]]octahydro-7a-methyl-1-[1-[5,5,5-trifluoro-
4-
(trifluoromethyl)-4-[(trimethylsilyl)oxy]-2-pentenyl]cyclopropyl-4H-inden-4-
one (117.5
mg, 0.25 mmol) in THF (2.0 ml) was added and stirring continued for 2.5 h.
Work up gave
a gum, which was purified by flash chromatography (40 g of silica gel, 20%
EtOAc in
hexanes) to give a gum ( 120 mg). This was dissolved in THF (2.0 ml), treated
with a 1 M
solution of n-Bu4N+F- in THF (2.0 ml) and stirred at room temperature for 20
h, and
worked up. Flash chromatography (40 g of silica gel, 40% EtOAc in hexanes)
gave title
compound (29 mg) as a colorless foam: [cc]D - 41°(MeOH, c = 0.14); IR
3569 cm 1; UV ~,m~
214 ( 10,968), 219 ( 12,931), 259 ( 12,818) nm; 1H NMR 8 0.02 ( 1 H, m), 0.32
(2 H, m), 0.60
(lH,m),0.61(3H,s),1.1-1.7(llH,m),1.85(2H,m),2.0(3H,m),2.2(3H,m),2.85
(3H,m),4.12(lH,brs),4.90(lH,s),5.30(lH,s),5.40(lH,d,J=12.8),6.0 (l H,d,J=
11),6.12(lH,dd,J=12.8,6.8),6.29(lH,d,J=11),6.'12 (l H,dd,J=12.8,6.8),6.29(1
H, d, J= 11); MS mlz 518 (M+, 22).
Example 6
This example illustrates a method for producing 3-desoxy-1,25-dihydroxy-20-
methyl-23-yne-21,28-cyclocholecalciferol.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-40-
PhzP-O
-OH
~OSi(CH3)3
OSi(CH3)2t-Bu
O
OH
Homer reagent [R-(Z)]-[2-[3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-
methylenecyclo-hexylidene]ethyl]diphenylphosphine oxide (375 mg, 0.828) in THF
(5.0
ml) was treated with 1.6 M solution of n-BuLi in hexanes (0.51 ml, 0.81 mmol).
After 8
min, [1R-(loc,3a[3,7acc)]-Octahydro-7a-methyl-1-[4-methyl-4-
[(trimethylsilyl)oxy]-2-
pentynyl] cyclopropyl]-4H-inden-4-one (180 mg 0.50 mmol) in THF (4.0 ml) was
added
and the mixture worked up after 3.5 h. Flash chromatography (45 g of silica
gel, 7
EtOAc in hexanes) gave a syrup (273 mg), which was dissolved in THF (3.3 ml)
and stirred
to with a 1.0 M solution of n-Bu4N+F- in THF (3.3 ml) for 28 h, and worked up.
Flash
chromato-graphy (45 g silica gel, 40% EtOAc in hexanes) gave title compound
(114 mg) as
a colorless foam: [cc]D - 70.32° (EtOH, c = 0.539); LJV ~,m~ 215 (~ =
13,326), 262 (~ _
17,661) nm; IR 3601 cm 1; 1NMR 8 0.28 (2 H, m), 0.41 (1 H, m), 0.59 (1 H, m),
0.60 (3 H,
s), 1.10 ( 1 H, m), 1.50 (6 H, s), 1.55-2.0 ( 18 H, m), 2.09 ( 1 H, d, J =
17), 2.22 (2 H, m), 2.60
(lH,d,J=17),2.80(lH,d,J=11),4.11(lH,brs),4.91(lH,s),5.30 (l H,s),5.99(1H,
d, J= 11), 6.27 (1 H, d, J= 11); MS m/z 408.3 (M+, 60).
Example 7
This example illustrates a method for producing 3-desoxy-1,25-dihydroxy-20-
2o methyl-23-yne-26,27-hexafluoro-21,28-cyclocholecalciferol.
PhZP=p CF3
-OH
CFA
OF3
'OH
OSi(CH3)Zt-Bu
CF3
O
OH
Homer reagent [R-(Z)]-[2-[3-[[(1,1-dimethylethyl)-dimethylsilyl]oxy]-2-
methylenecyclohexylidene] ethyl] diphenylphosphineoxide (350 mg, 0.773 mmol)
in THF
(5.0 ml) was deprotonated with 1.6 M n-BuLi in hexanes (0.49 ml, 0.784 mmol)
at -78 °C
and after 8 min reacted withketone [1S-loc,3a(3,7acc)]octahydro-7a-methyl-1-[1-
[5,5,5-
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-41-
trifluoro-4-hydroxy-4-(trifluoromethyl)-2-pentynyl]cyclopropyl]-4H-inden-4-one
(158
mg, 0.40 mmol) during 3.0 h. Flash chromatography of the crude product (45 g
silica gel,
20% EtOAc in hexanes) followed by desilylation during 18 h at room temperature
with a 1
M solution of n-Bu4N+F- in THF ( 1.8 ml) and flash chromato-graphic
purification (45 g
silica gel, 40% EtOAc in hexanes) gave title compound ( 117 mg) as a colorless
foam: [a] o -
58.3° (EtOH, c = 0.456); UV ~,m~ 214 (~ = 12,900), 260 (E =15,701) nm;
1H NMR ~ 0.29 ( 1
H,m),0.35(lH,m),0.37(lH,m)0.59(3H,s),0.64(l H,m),1.4-1.90 (l2 H,m)2.00(4
H,m),2.18(lH,d,J=17),2.25 (2 H,m),2.72(lH,d,J=17),2.81 (l H,m),3.34(lH,s,
OH),4.12(lH,brs),4.92(lH,s),5.28(lH,s),5.98(lH,d,J=11),6.27 (l H,d,J=11);
l0 MS rnlz 516.2 (M+, 90).
Example 8
This example illustrates a method for producing 3-desoxy-1,25-dihydroxy-20-
methyl-21,28-cyclocholecalciferol.
PhzP=O
'OH
\O$I(CH3)3 ~ ~
~OSi(ON3)zt-Bu
O
OH
A magnetically stirred 25 ml, 3-neck round bottom flask equipped with a
thermometer, a rubber septum on the side and a Claisen adapter containing a
nitrogen
sweep and rubber septum at the center, was charged with 0.564 gr ( 1,246 mmol)
of [R-
(Z)]-2-[3-[[(1,1-dimethylethyl)-dimethylsilyl]oxy]-2-
methylenecyclohexylidene] ethyl] diphenyl-phosphine oxide. This material was
dried under
high vacuum, and was added 5 ml tetrahydrofuran; the solution was stirred and
cooled to -
70 °C, and 0.78 ml ( 1.246 mmol) of 1.6 M BuLi in hexane was added.
(The red color
persisted after the initial 0.16 ml was added). The solution was stirred at -
70 °C for 10 min,
and then a solution of 0.2904 gr (0.796 mmol) of [1S-(loc, 3a~3, 7aoc)]
octahydro-7a-
methyl-1-[1--[4-methyl-4-oxy]-2-pentanyl-cyclo-propyl]-4H-inden-4-one
dissolved in 8
ml tetrahydrofuran was added dropwise. When the reaction was virtually
complete (TLC,
1:9 ethyl acetate-hexane), the mixture was allowed to warm to -30 °C,
then 12 ml of pH7
phosphate buffer ( 139.4 gr of dipotassium phosphate in 400 ml of water plus
10 ml of 2M
3o phosphoric acid) was added dropwise through the center port. The mixture
was stirred for
5 min, then transferred to a separatory funnel with the aid of 35 ml of
hexane. The
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-42-
aqueous phase was re-extracted with 30 ml of hexane. The two hexane layer were
combined, washed with 20 ml of brine, dried by passage through a plug of
sodium sulfate,
then evaporated to a colorless syrup. This material was taken in hexane. White
solids were
present so that hexane suspension had to be filtered through a flash column 25
x 120 mm.
After fraction #2 (20 ml each) the mobile phase was changed to 1:79
ethylacetate-hexane.
Fractions 11-18 (according to TLC in 1:19 ethyl acetate-hexane) were pooled
and
evaporated. It gave 0.4202 gr (88.1%) of silylated title compound.
A 100 ml brown round-bottom flask was charged with 0.4202 gr of silylated
title
compound. To this material was added 5 ml tetrahydrofuran and 3.5 ml of a 1 M
solution
to of tetrabutylammonium fluoride in tetrahydrofuran and stirred at room
temperature for 17
hrs. TLC ( 1:1 ethyl acetate-hexane and ethyl acetate) showed one major spot.
The reaction
mixture was then diluted with 13 ml of brine, stirred for 15 min, then
transferred to a
separatory funnel with aid of 40 ml ethyl acetate. The aqueous layer was re-
extracted with
20 ml of ethyl acetate. Both organic layers were combined and washed with 5 x
35 ml water
and once with brine, then passed through a plug of sodium sulfate and
evaporated to a
crystalline, white residue, 0.3523 gr. This material was chromatographed on a
25 x 110 mm
column using 1:1 ethyl acetate-hexane as mobile phase. Fractions 3-4, already
crystallizing
in the tubes. The suspension so obtained was concentrated to a volume of ca 5
ml, diluted
with hexane and concentrated, and filtered to give crystalline title compound
0.2567 gr.
[cc~D2s - 59.1° (c 0.325, EtOH). UV~,maX (MeOH): 214, 262; 7~Sh 222 nm.
Anal. Calcd for
CZ$H44O: C, 81.50; H, 10.75; 0, 7.75. Found: C, 81.43; H, 10.69; 0, 7.48.
Table 1: Representative compounds of Formula I.
C d. # Structure
24 Chiral
I
'O
26
F
~~ F
I H v F
F F
I
'O
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-43-
C d. # Structure
27 F F
F
0
.~H F
H
O
32 F
F O
'--r ~F
F F
O
34
sb
v
Fi
O
In the above Table, unshared valences on oxygen are intended to be occupied
by hydrogen.
Example 9
This example illustrate a method for synthesizing 3-desoxy-lcc-acetoxy-25-
hydroxy-
20-cyclopropyl-23E-ene-26,27-hexafluoro-cholecalciferol.
A 25-mL round bottom flask was charged with 0.1702 g of 3-desoxy-lcc,25
dihydroxy-20-cycl~propyl-23E-ene-26,27-hexafluoro-cholecalciferol and 2.85 g
of
pyridine. The solution was stirred and cooled in an ice bath, then 0.5 mL of
acetic
anhydride was added and stirring in the ice bath continued for one hour. At
that time the
solution was placed in the refrigerator overnight. The flask was returned to
the ice-bath
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-44-
and 0.2 mL of acetic anhydride was added. After 6 hours the solution was
diluted with 10
mL of ethyl acetate and, while still immersed in the ice bath, 2 mL of water
was added. The
mixture was stirred for 5 min, then transferred to a separatory funnel with
the aid of 20 mL
of water and 20 mL of 1:4 ethyl acetate - hexane. The aqueous phase was re-
extracted with
10 mL of 1:4 ethyl acetate-hexane. TLC in 1:2 ethyl acetate-hexane showed no
product in
this second extract. Thus, the original extract was washed with 4 x 20 mL of
water, 10 mL
of brine and then passed through a plug of sodium sulfate and evaporated. The
residue was
taken up in 1:4 ethyl acetate-hexane and flash chromatographed on a 15 x 150
mm column
using 1:4 ethyl acetate-hexane as mobile phase and taking 10 mL fractions. The
bulk of the
1o major product (0.1050 g) was contained in fraction 3. Fraction 3 still
contained a trace of
the faster running material observed in fraction 2. As an attempt was made to
dissolve the
residue in 1:6 ethyl acetate-hexane crystallization commenced. Thus, a small
quantity of
pentane was added and the mixture allowed to crystallize in the refrigerator.
The mother
liquor was withdrawn, and evaporated, 0.0068 g. The crystals were rinsed once
with
pentane, dried to give 0.0899 g of the title compound.
[cc]D -30.8° (0.54%, methanol),
NMR (CDCl3): 2.07 (OAc),
Anal. Calcd C 64.27, H 6.83 Found: C 64.63, H 6.93; C 64.52, H6.94
LJV max, nm (absorbance) 121 (0.3639), 250 (0.4591), 262 (4486), 244 sh
(0.4482).
Example 10
This example illustrate a method for synthesizing 3-desoxy-1x,25-diacetoxy-20-
cyclopropyl-23E-ene-26,27-hexafluoro-cholecalciferol.
A 25-mL round bottom flask, equipped with magnetic stirred and Claisen adapter
containing a nitrogen sweep and stopper, was charged with 0.315 g (0.6 mmol)
of 3-
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-45-
desoxy-lcc, 25-dihydroxy-20-cyclopropyl-26,27-hexafluoro-cholecalciferol, 60
mg of
dimethylaminopyridine, 5 mL of pyridine. The pyridine was added while the
flask was
immersed in an ice bath. After 10 min, 1.0 mL of acetic anhydride was added
dropwise and
stirring in the ice bath continued. After 1 hour starting material was
virtually undetectable.
After two hours, the solution was diluted with 10 mL of ethyl acetate and,
while still
immersed in the ice bath, 10 min later, 2 mL of water was added dropwise. The
mixture
was stirred for 10 min, then transferred to a separatory funnel with the aid
of 20 mL of
water and 20 mL of 1:4 ethyl acetate - hexane. The aqueous phase was re-
extracted with 10
mL of 1.4 ethyl acetate - hexane. TLC in 1:2 ethyl acetate - hexane showed no
product in
to this second extract. Thus, the original extract was washed with 4 x 20 mL
of water, 20 mL
of brine and then passed trough a plug of sodium sulfate and evaporated. The
residue was
taken up in 1:6 ethyl acetate - hexane and flash chromatographed on a 25 x 150
mm
column using 1:6 ethyl acetate - hexane as mobile phase and taking 20-ml
fractions. Pure
material was contained in fraction 3, fraction 4-6 contained the product
contaminated with
slower-moving materials (TLC 1:2). Thus, fractions 4-6 were combined and the
residue re-
chromatographed on a 15 x 150 mm column using 1:9 ethyl acetate - hexane as
mobile
phase and taking 20 mL fractions. Fractions 2 and 3 contained the bulk of the
product in
pure form. These fractions were added to fraction 3 of the first chromatogram,
then
evaporated. The residue was taken up in 1:9, filtered and concentrated,
diluted with
2o pentane and refrigerated. The mother liquor was withdrawn and the crystals
rinsed with
pentane, then dried at hivac for 2 h, to give 0.190 g of the title compound.
NMR (CDC13): 8 2.08 and 2.20 (2 OAc),
[alpha~D - 110.3, 0.59 % (methanol),
MA 192020: found C, 63.99; H, 6.62; calcd C, 63.78; H, 6.69,
UV max (Absorbance) 212 (0.3573), 251 (0.4392), 261 (0.4276).
Example 11
This example illustrates a method of determining effectiveness of the
compounds of
the present invention for bone anabolism in the rat.
Three month old rats are ovariectomized (Ovx) and administered either 1,25-
dihydroxy vitamin D3 or one of the compounds of the present invention once a
day by
mouth starting at 3 weeks post-ovariectomy and continuing until final
sacrifice at 6 weeks
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-46-
post-ovariectomy. Control groups, both sham (rats that were not
ovariectomized) and
Ovx, receive vehicle only. Blood and urine samples are collected twice, at 4
weeks post-
ovariectomy and again at the 6 week mark and the amount of serum and urine
calcium is
determined. The final femoral calcium is determined upon sacrifice 6 weeks
post-
ovariectomy.
The bone mineral density of the right femur is determined by using a High
Resolution Software Package on a QDR-1000W Bone Densitometer~ (Hologic,
Walthan,
MA). The animals are scanned by placing them on a scanning block in a supine
position
such that the right leg was perpendicular to the main body and the tibia was
perpendicular
1o to the femur.
Example 12
This example illustrates a method for determining comparative in vivo efficacy
of
Compounds of the present invention and 1,25-(OH)ZVitamin D3.
Comparison of the efficacy of compounds of the present invention to that of
1,25-
dihydroxy vitamin D3, can be made using the standard animal model for post
menopausal
osteopenia, the rat ovariectomy model. Three month old rats were
ovariectomized, and
then treated for 8 weeks beginning 1 week post OVX. Drugs were administered
once/day
orally in miglyol (medium chain triglyceride) vehicle. Blood and urine samples
were
2o collected at the 3wk and 6wk time point, and bone mineral density (BMD) was
determined
in vivo at 6wk using Dual Energy X-ray Absorptiometry (Hologic QDR-4500T"'
Bone
Scanner). At 8 weeks, the animals were sacrificed, and the lumbar vertebrae
and femur
bones removed for ex vivo BMD determination (Lunar PixiMusT"" Bone Scanner)
and
biomechanical testing for strength. Data for each compound are then determined
for the
highest safe doses. The highest safe dose is defined as that which does not
produce
hypercalcemia as defined by serum calcium levels greater than 10.0 mg/dl.
Example 13
This example illustrates comparative in vivo efficacy of Compound 27 and 1,25-
(OH)ZVitamin D3.
Comparison of the efficacy of Compound 27 to that of 1,25-dihydroxy vitamin
D3 (i.e., VD3), was made using the standard animal model for post menopausal
osteopenia,
the rat ovariectomy model. Three month old rats weighing 285gm were
ovariectomized
(OVX), and then treated for 8 weeks beginning 1 week post OVX. Drugs were
administered once/day orally in miglyol (medium chain triglyceride) vehicle.
Blood and
urine samples were collected at the 3wk and 6wk timepoint, and bone mineral
density
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-47-
(BMD) was determined in vivo at 6wk using Dual Energy X-ray Absorptiometry
(Hologic
QDR-4500T"' Bone Scanner). At 8 weeks, the animals were sacrificed, and the
lumbar
vertebrae and femur bones removed for ex vivo BMD determination (Lunar
PixiMusT""
Bone Scanner) and biomechanical testing for strength. Data for each compound
are
reported in the accompanying tables for the highest safe doses. The highest
safe dose is
defined as that which does not produce hypercalcemia as defined by serum
calcium levels
greater than 10.0 mg/dl.
Table 3 shows the safety (serum calcium, urinary calcium) results. The doses
being compared all gave 3wk serum calcium levels of 9.3mg/dl, and 6wk levels
of 9.8 ~
l0 O.lmg/dl. At these doses, the urinary calcium output per 24hr sample was
the same for the
0.4nmo1/kg dose of VD3 and the 0.5nmo1/kg dose of Compound 27. Table 4 shows
the
efficacy parameters for the 2 compounds (BMD, biomechanics, and urinary
deoxypyridinolines). BMD at all of the bone sites listed is significantly
higher in the
animals dosed with Compound 27 than those dosed with VD3 at doses which give
equivalent serum calcium levels. These doses represent those expected to give
maximal
efficacy achievable safely, as higher doses of each compound are known to be
hypercalcemic. Urinary deoxy-pyridinolines, a marker of bone resorption were
significantly less for Compound 27 than VD3 at 6 weeks showing greater ability
of
Compound 27 to inhibit bone resorption. Compound 27 treatment resulted in
stronger
2o vertebral bone. Failure Load, the amount of force needed to fracture the L5
vertebra, was
determined in axial compression testing. Significantly more force was needed
to fracture
the vertebrae of animals treated with Compound 27 compared to those treated
with VD3 at
the highest safe doses.
30
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-48-
Table 3. Safety Comparison of Compound 27 to 1,25-dihydroxy-Vitamin D3 (VitD)
PARAMETER SHAMOVX VitD V'rtD Compound
27
ControlContrd0.012 0.04 0.5
nmoUkgnmd/tegnmollteg
P'~ P'~ P'~ P~0.012pvs0.04
contrdscantrdscontrolsVD3 VD3
SERUM & URINE
CALCIUM
SERUM CALCIUMWeek 9.4 9.2 9.3 9.3 9.3 ns ns
3 Mean ns ns ns
(mg/dl) S.EM. 0.1 0.1 0.1 0.1 0.1
Ngroup 10 10 10 10 10
Week 9.5 9.3 9.8 9.7 9.9 ns ns
6 Mean "+ *' "+
S.EM. 0.1 0.1 0.1 0.1 0.2
Ngroup 10 10 10 9 10
URINARY CALCIUMWeek 0.310.220.31 0.51 0.51 $ ns
3 Mean " "+ '*+
(MGlNNAOLCreatlnindday)S.EM. 0.020.050.04 0.05 0.06
Ngroup 10 10 10 10 10
Week 0.340.260.43 0.71 0.72 $ ns
6 Mean " "++ *'++
S.EM. 0.030.050.04 0.05 O.D7
Ngroup 10 10 10 9 10
ns _-not significantly different
pc0.05,'* pN.01 vs OVX Control
+ p~0.05, ++ p<0.01 vs OVX Control
S p<0.05, $S p<0.01 vs V'rt-D
1~
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-49-
Table 4. Efficacy Comparison of Compound 27 to 1,25-dihydroxy-Vitamin D3
(VitD)
SHAMOVX VitD VitD Compound p
27 vs
p
vs
0.0120.04
PARAMETER VD3VD3
ControlControl0.012 0.04 0.5
nmoUk nmoUkg nmoUkg
PERCENT BMD
> OVX CONTROL
in vivo
L2-L4 VertebraeWeek 10.00.0 3.3 5.4 10.3 $$ $
6 Mean ++ * *"
S.E.M. 1.9 1.6 1.6 1.6 2.1
Ngroup 9 9 10 9 10
L5 vertebra Week 11.60.0 2.7 4.8 8.7 $ ns
6 Mean ++ + **
S.E.M. 1.2 1.2 2.3 1.4 2.0
Ngroup 9 9 10 9 10
Whole Femur Week 7.3 0.0 3.3 1.9 6.3 ns $
6 Mean + ++ **
S.E.M. 1.1 0.8 1.7 1.7 1.1
Ngroup 9 9 10 9 10
Prox Femur Week 6.5 0.0 3.5 0.7 5.7 ns $$
6 Mean ns ++ **
S.E.M. 1.5 1.4 1.7 1.5 1.2
Ngroup 9 9 10 9 10
Distal Femur Week 11.70.0 2.8 2.8 8.5 $$ $$
6 Mean ++ ++ **
S.E.M. 1.2 1.1 1.8 2.0 1.3
Ngroup 9 9 10 9 10
L5 Bone Density
Ex Vivo
BMD (g/cm~2) Week 0.1130.0980.103 ND 0.112 $$ ND
8 Mean ++ **
S.E.M. 0.0020.0020.003 0.002
Ngroup 10 7 9 10
BMC (g) Week 0.0380.0330.035 ND 0.039 $$ ND
8 Mean ns **
S.E.M. 0.0010.0010.002 0.001
Ngroup 10 7 9 10
URINARY D-PYD
Week 0.380.90 0.81 0.75 0.65 ns ns
3 Mean ++ ++ *+
(nNUmM Creatinine)S.E.M. 0.040.06 0.09 0.07 0.09
Ngroup 10 10 9 9 10
Week 0.310.84 0.78 0.76 0.57 $ $
6 Mean ++ ++ **+
S.E.M. 0.030.07 0.09 0.09 0.06
Ngroup 10 10 10 8 10
L5 Vertebral
Body BIOMECHANICS
Failure Load Week 386 261 342 ND 419 $ ND
(N) 8 Mean * **
S.E.M. 32 20 26 23
Ngroup 7 6 9 10
ND = no data available
ns = not sign'rficantly different
* p<0.05, ** p<0.01 vs OVX Control
+ p<0.05, ++~ p<0.01 vs OVX Control
$ p<0.05, $$ p<0.01 vs Vit-D
The compounds of the invention possess biological activity comparable to that
of
compound 27.
The foregoing invention has been described in some detail by way of
illustration and example, for the purposes of clarity and understanding. It
will be obvious
to one of ordinary skill in the art that changes and modifications may be
practiced within
the scope of the appended claims. Therefore, it is to be understood that the
above
to description is intended to be illustrative and not restrictive. The scope
of the invention
should, therefore, be determined with reference to the following appended
claims, along
with the full scope of equivalents to which such claims are entitled.
CA 02459789 2004-03-04
WO 03/027065 PCT/EP02/10233
-50-
The patents, patent applications and publications cited in this application
are
hereby incorporated by reference in their entirety for all purposes to the
same extent as if
each individual patent, patent application or publication were so individually
denoted.