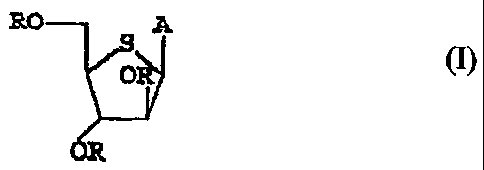Note: Descriptions are shown in the official language in which they were submitted.
WO 00/04866 ~ 02464681 2004-04-07 p~'/Ugg9118630
PREPARATION OF THIOARABrNOFURANOSYL,
CC?1VIPOUNDS AND USE THEREOF
DESCRIPTION
a
Federally Sponsored Research and Development
This invention was supported by Grant CA-34200 from National Institutes of
Dealth_
Technical Field
The present invention is concerned with treating patients suffering horn
cancer by administering to the patients cereaiti thioarabinofuranosyl
compounds.
Compounds employed according to the present invention have exhibited good
anticancer activity. Compounds employed accordix~ to the present invention are
in
the beta configuration as cozitrasted to the alpha configuration, which failed
to
exhibit anticancer activity. The. present invention also relates to inhibiting
DNA
replication in a rnaaunalian cell by contacting with the thioarabinofuranosyl
compounds. The present invention also relates to a nevv process for preparing
the
compounds employed according to the present invention.
Background of Invention
Past amounts of research have accrued over the years related to developing
treatments against cancers to inhibit and kill tumor cells. Some of this
research has
~"0 resulted in achieving some success in finding clinically approved
treauaents:
Nevertheless, efforts continue at an ever-increasing rate in view of the
extreme
difficulty in uncovering promising anticancer treatments. For example, even
when
CA 02464681 2004-04-07
VSO flfl/fl4S66 ' PCTIUS99116630
a compound is found to have cytotaxic activity, there is no predictability of
it being
selective against cancer cells.
One particular compound that has been used rather widespread is cytosine
arablnoside, comfnonly referred to as .lira-C.
S ~ Sununary of Invention
It has been found according to the present invention that certain
thioarabinofuranosyl cytosine compounds are suitable as anticancer agents. The
presence of the thiv sugar moiety surprisingly makes it possible to achieve
good
antitumor activity. More particularly, the present invention relates to
treating a
mammalian host in need of an anticancer treatment by administering to the host
an
effective anticancez amount of at lease one compound represented by the
following
formula 1_
RO ~ A
OR 1
wherein each R individually is H ox an aliphatic or aromatic aryl group;
A is selected from the group consisting of
NHa
N / ~
Z
O
n
CA 02464681 2004-04-07
WC7 00/L148Gt FCTIU$g9116b3a
N N
o~N
> ~
X is selected from the group consisting of hydrogen, flaoto, alkoxy, alkyl,
haloalkyl> alkenyl, haloalkenyl, aIkynyl, amino, monoalkylamina, dialkyiamino,
cyano az~d nitro.
rt has also been found according to the present invention that the above-
disclosed compounds of formula x can be used to inhibit DIVA replication in a
IfJ manunalian cell by contacting the cell with at least one of these
compounds.
The present invention is also concerned with a pzocess for preparing the
above-identified compounds. The compounds employed according eo the present
invention can be prepared by:
3
CA 02464681 2004-04-07
A) reacting a 2,3,5-tri-O-aryl or alkyl-4-xylose diaryl or dialkyl
dithioacetal such as
2,3,5-tri-O-benzyl-L-xylose-dibenzyl dithioacetal in the presence of a leaving
group at the 4
hydroxyl to produce the corresponding 1,4-dithio-D-arabinofuranoside such as
benzyl 2,3,5-
tri-O-benzyl-1,4-dithio-D-arabinofuranoside;
B) subjecting the product from step A) to acidolysis to form the
corresponding. 0-
acetyl-4-thio-D arabinofuranose such as 2,3,5-tri-O-benzyl-1-O-acetyl-4-thio-D-
arabinofuranose;
C) reacting the product of step. B) with a cytosine, a 5- or 6-aza compound or
a
suitably blocked derivative thereof forming a corresponding 4-thin-a,13-D-
arabinofuranosyl
compound such as, in the case of cytosine, 1-(2,3,5-tri-O-benzyl-4-thin-a,13-D-
arabinofuranosyl) cytosine;
D) converting the compound of step C) by hydrolysis to the corresponding thin
sugar
derivative such as 1-(4-thio-a,13-D-arabinofuranosyl) cytosine;
E) separating out the a form of the anomeric mixture of step D) to thereby
obtain the
desired compound of formula l, such as 1-(4-thio-13-D-arabonifuranosyl)
cytosine.
In a broad aspect, then, the present invention relates to the compound
represented by
the formula 1:
RO A
S
RO
RO (1)
wherein each R individually is H, an aliphatic acyl group or an aromatic acyl
group;
4
CA 02464681 2004-04-07
A 1S
NHZ
N
O N
NHz
N~ N
ON
NHZ
N NH
O N
NHZ
X
/N
O N
wherein X is hydrogen, fluorine, alkoxy, alkyl, haloalkyl, alkenyl,
haloalkenyl,
alkynyl, amino, monoalkylamino, dialkylamino, cyano or vitro, for treating a
solid. tumor in a
mammalian host.
4a
CA 02464681 2004-04-07
In another broad aspect; then, the present invention relates to the compound 1-
(4-thio-
13-D-arabinofuranosyl) cytosine for treating a mammalian host suffering from a
cancer
selected from the group consisting of melanoma, prostate cancer, mammary
cancer, renal
cancer, colon cancer and lung cancer.
In yet another broad aspect, then, the present invention relates to use of a
compound represented by the formula 1:
RO A
S
RO
RO
wherein each R individually is H, an aliphatic acyl group or an aromatic acyl
group;
A is
x
NFi
N N
p N
4b
CA 02464681 2004-04-07
NH2
N NH
O N
or
NH2
X
N
~I IN
O N
wherein X is hydrogen, fluorine, alkoxy, alkyl, haloalkyl, alkenyl,
haloalkenyl,
alkynyl, amino, monoalkylamino, dialkylariiino, cyano or nitro, for treating a
solid tumor in a
mammalian host.
In a further broad aspect, then, the present invention relates to use of the
compound represented by the formula 1:
RO A
S
RO
RO'~ ~l~
wherein each R individually is H, an aliphatic acyl group or an aromatic acyl
group;
4c
CA 02464681 2004-04-07
!~ 1S
NHZ
X
N
O N
NHZ
N N
J
O N
NHp .
N ~ NH
O N
NHz
X
N
~N
O N
wherein X is hydrogen, fluorine, alkoxy, alkyl, haloalkyl, alkenyl,
haloalkenyl,
alkynyl, amino, monoalkylamino, dialkylamino, cyano or nitro, for treating a
mammalian
host suffering from a cancer selected from the group of melanoma, prostate
cancer, mammary
cancer, renal cancer, colon cancer or lung cancer.
4d
CA 02464681 2004-04-07
In a still further broad aspect, then, the present invention relates to use of
the
compound represented by the formula 1:
RO A
S
RO
RO (1)
wherein each R individually is H, an aliphatic acyl group or an aromatic acyl
group;
A is
NH2
X
N
i
O N
NHp
N~ N
~J
O N
J
NHZ
N NH
O N
or
4e
CA 02464681 2004-04-07
NHZ
N
~N
O N
wherein ~ is hydrogen, fluorine, alkoxy, alkyl; haloalkyl, alkenyl,
haloalkenyl,
alkynyl, amino, monoalkylamino, dialkylamino, cyano or nitro for inhibiting
DNA
replication in a mammalian cell.
In another broad aspect, then, the present invention relates to use of I-(4-
thin-13-D-
arabinofuranosyl) cytosine for treating a mammalian host suffering from a
cancer selected
from melanoma, prostate cancer, mammary cancer, renal cancer, colon cancer or
lung cancer.
4f
CA 02464681 2004-04-07
WO 00104866 PCTIUS99116630
Summary of Drawings .
Figs. la and 1b are graphs showing metabolism of 2-devxycytidiae, 2'-
deoxythiocytidine, ara c and thio ara-c to their respective triphosphates.
Fig. 2 is a graph showing the retention of ara c tiiphosphate and thin ara c
triphosphate in CEM cells.
Best and Various Modes for Carrying ~ut Invention
The present invention is related to treating a mammalian host in need of an
anticancer ra~eacmenc, which comprises administering to the host an effective
anticalzcer amount of at .least compound represented by the formula 1:
RO ~ A .
1.o 0
g
Each R in formula 1 individually is preferably H or an aliphatic or aromatic
acyl Eroup. Typical aliphatic acyl groups contain from 1 to 6 carbon acorns
and
include formyl, acetyl, and propionyl. Typical aromatic acyl groups include
unsubstituted and alkyl substituted aromatic ,groups containdng 7-10 carbon
atoms in.
1$ the arc~mauc group. When substituted, tl3e alkyl group typically contains 1-
6 carbon
atoms. Typical aromat5c acyl groups include benzoyl and para-tol.oyI.
A in formula l is preferably
WD 00104866 ~ 02464681 2004-04-07
PCTlLyS99f 16630
N N
O%'N/
NHa
N! 'NH
and .
o a
o~N~
N
Suitable monoalkylamino groups for X ~contsin 1-6 carbon atoms and intclude
monomethylamino, monoethylamino, mono-isopropylamino, mono-n-propylamino;
mono-isoburyl-amino, mono-n-butylamino and mono-n-hexylamino. The all~yrl
moiety can .be stzaight or branched chain.
Suitable dialkylamino groups for Y and X contain 1-6 carbon atoms in each
Itl alkyl ~roup. The alkyl gzoups can be the same or different and can be
straight or
CA 02464681 2004-04-07
WO 00104866 PCTNS99116G30
branched chain. Examples of some suitable gmups are dimethyIamino,
diethylamino, ethylmethylamino, dipropylamina; dibutylamino, dipentylamino,
dihexylamino, methylpentylamino, ethylpropylamino and ethylhexylamino.
Suitable halogen groups for X include GI, Br and F.
Suitable alkyl groups far X typically contain 1--6 carbon atoms and can be
straight or branched chain. Some examples are methyl, ethyl, i-propyl, n-
propyl, i-
butyl, n-butyl, pentyl and hexyl_
Suitable haloalkyl groups typically contain I-6 carbon atoms and can be
straight Qr branched chain and include Cl, Br ar >substituted alkyl groups
including
LQ the above specifically disclosed alkyl groups.
5uitabie alkoxy groups typically contain 1-6 carbon atoms and include
rnerhaxy, ethoxy, pxopoxy and butoxy,
Suitable alkenyl groups typically contain 2-6 carbon atoms and include
ethenyl and propenyl_
Suitable halaalkenyl groups typically contain 1-6 carbon atoms and include
Cl.. Br or F substituted alltenyl groups including the above specifically
disclosed
alkenyl groups.
Suitable alkynyl groups typically contain 1-6 carbon atoms and include
ethynyl and propynyt. ,
24 The preferred compound employed according to the process of the present
invention is 1-(4-thin-!3-D-arabinofuranosyl} cytosine.
7
CA 02464681 2004-04-07
WO 00104866 pCTlUS991t6630
The present invention is suitable for treating mammalian hosts including
humans suffering from cancer including melanoma, prostate cancer, mammary
cancer, renal cancer, colon cancer, sung cancer, lettkemias and lymphomas.
The compounds employed according to the present invention can be prepared
by reacting a 2,3,5-tri-O-aryl or alkyl-4-xylose diaryl or dialkyl
dithioacetal such as
~.3.~-tri-O-benzyl-I:-xylose-dibenzyl dithioacetal in the presence of a
bearing gmup
at the 4 hydroxyl position to produce the corresponding 1,4-dithio-D-
arabinofuranoside such as Isenzyl 2,3,5-tri-O benzyl-l,4rdithio-D-
arabinofuranoside.
This step can he carried aut using phosphine, iodine and imida2ole. The
product
from the above step is subjected eo acidolysis to form the corresponding 4-
acetyl-4
thin-D arabinofuranose such as 2,3,5-tri-O-benzyl-1-~-acetyl-4-chio-D
arabinofuranose. For instance, acetic acid in the presence of rncrcuric
acetate can
be employed.
The product of the above step is reacted, with a cytosine, a 5- ar 6-aza
1.5 compound or a suitably blocked derivative thereof forming a corresponding
4-thio-
«,13-D-arabinofuranosyl compound such as, in the case of cytosine, 1-(2,3;5-
tri-O-
benzyl-4-thio-cz,t3-D-arabinofuranosyl) cytosine. Suitable blocked derivatives
include acyl and tximethylsilylated derivatives, The compound of the above
step is
converted by hydrolysis to the coaesponding thio sugar derivative such as 1-(4-
thio-
a,li-D-arabinofuranasyl) cytosine.
The cx form of the anomeric mixture of the above step is separated out to
thereby obtain the desired compound of formula l, such as 1-(~i-thio-!3-D-
arabinofuranosyl) cytosine.
Compounds according to the present invention can be prepared by the
2a process sequence shoawn in scheme I and examples 1 and 2 described
hereinbelour,
wlaerein the preferred compound, 1.-(4-thio~B-D-arabinofuranosyI) cytosine is
illustrated for purposes of facilitating an understanding of the process
sequence.
The precursor emgloyed, 2,3,5-thin-O-benzyl-L-xylose dibez~yl dithioacetal,
can be
praduced by the process described by Secrist, 1II et al. "'The Synthesis and
f
CA 02464681 2004-04-07
wo oo~oa8ss PCTlUSS91166~0
Biological Activity of Certain 4'-Thionucleosides, Nucleosides & Nucleotides,
14 (3-
5), 675-686 (1995), disclosure of which is incorporated herein by reference.
The
process of the present invention provides for a relatively efficacious meEhod
far
producing signifzcant quantities of the desired compowtd. Prior arc techniques
are
exuemely complicated and not readily suitable for producing desired amounts of
the
a .
compounds_
9
CA 02464681 2004-04-07
wo oo~o~sss pcr~~99nss~o
c~sHn~ ~
OCFI3 En H ~ S8n
Lxylo.~ --~- RO~ ~~.----,. H Bn -----s
RO HO Ii Hn0
t~OBn
2: R H
1 3: it~Hn 4 s
NF3z
$n g N r
en OAe
O~N
$n0 H
6 XsH ? x~H
X~P 7n X~F
~a
X
... ,~:, '
BQ O g N
so
HO
is X~H
8 X~H 9 10a X=F
8a X-F
1~
6U8STiME SHEET (RULE 26)
CA 02464681 2004-04-07
Wt) QO/U4866 . YCTIUS991i563o
The pharmaceutically acceptable effective dosage of the active compound of
the present invention to be administered is dependent on the species of the
warm-
blooded animal (mammal), the body weight, age and individual condition, and on
the form of administration.
S The pha~cmaeeutical composition may be oral, parenteral, suppository or
other form which delivers the compounds used in the present invention into the
bloodstream of a mammal to be treated.
The compounds of the presern invention can be administered by any
conventional means available for use in conjunction with pharmaceuticals;
either as
30 individual therapeutic agents or m a combination of therapeutic agents.
They can be
administered alone, but generally administered with a pharmaceutical cattier
selected on the basis of the chosen route of administration and standard
pharmaceutical practice.
The dosage administered will, of course; vary depending upon lanovvn
IS factors, such as the pharmacodynamic characteristics of the particular
agent and its
mode and route of administration; the age; health and weight of the recipient;
the
nature and extent of the symptoms; the kind of concurrent treatment; the
frequency
of treatment; and the effect desired. A daily dosage of active ingredient can
be
expected to be about 0:001 to y000 milligram (mg) per kilogram (kg) of body
20 weight, with the preferred dose being 0.1 to about 30 mg/kg.
Dosage forms (compositions suitable for administration) typically contain
from about 1 mg to abort 100 mg of active ingredient pet unit. In these
pharmaceutics! compositions, the active ingredient will ordinarily be present
in an
amount of about 0.5-95 % by weight based on the total weight of the
composition.
11
CA 02464681 2004-04-07
s
The active ingredient can be administered orally in solid dosage forms, such
as capsules, tablets, and powders, or in liquid dosage farms, such as elixirs,
syrups,
and suspensions. it can also be administered parenterally, in sterile liquid
dosage
forms. The acEive ingredient can also be administered intranasally (nose
drops) or
by inhalation. Other dosage forms are potentially possible such as
administration
transdermally, via a patch mechanism or ointment:
Gelatin capsules contain the active ingredient grad powdered carriers, such as
lactose, starch, cellulose derivatives, magnesiutn stearate, stearic acid, and
the like.
Similar diluents can be used to make compressed tablets. Both tablets and
capsules
ltl can be manufactured as sustained release products to provide far
continuous release
of medication over a period of hours. CompresseB tablets can be sugar-gated or
film-coated to mask any unpleasant taste and protect the tablet from the
atmosphere,
or enteric coated for selective disintegration in the gastrointestinal tract.
Liquid dosage forms for oral administration can contain coloring and
15 flavoring to increase patient acceptance.
In general, water, a suitable oil, saline, aqueous dextrose (glucose), and
related sugar solutions and glyeols such as propylene glycol or polyethylene
glycols
are suitable carriers for parenteral solutions. Solutions for parenterai
administration
preferably contain a water-soluble salt of the active ingredient; suitable
stabilizing
2~ ~ agents, and, if necessary, buffer substances. Andoxidi2ing agents such as
sodium
bisulfate, sodium sulfite, or ascorbic acid, either alone or combined, are
suitable
stabilizing agents. Also used are citric acid and its salts and sodium EDTA.
In
addition, parenteral solutions can contain preservatives, such as benzalkonium
chloride. methyl- or propylgaraben, and chlombutanol.
25 Suitable pharmaceutical carriers are described in Remington's
Pharmaceutical
Sciences. Mack Publishing Company, a standard reference text in this field.
12
CA 02464681 2004-04-07
wo ov~o4ass FC'rluS99ll~s~o
Useful pharmaceutical dosage forms' for adminisaation of the compounds
according to the present invention can be illustrated as follows:
' Ca es
A large number of unit capsules are prepared by fillitlg standard two-piece
hard gelatin capsules- each with 1~ mg of powdered active ingredient, 150 nitg
of
lactose, 50 mg of cellulose, and 6 mg of magnesium stearate.
Soft GeEati~n Ca~,,~~ules
A mixture of active ingredient in a digestible oil such as soybean oil,
cottonseed oil, or olive oil is prepared and injected by means of a positive
I(3 displacement pump into gelatin to form soft gelatin capsules containing
I00 imu of
the active ingredient., The capsules are washed and dried.
A large number of tablets axe prepared by conventional procedures so that
the dosage unit was 100 mg of active ingredient, 0.2 mg of eolloidal silicon
dioxide, 5 mg of magnesium stearate, 275 mg of microcryscalline cellulose, I 1
mg
of starch, and 98_8 mg of lactose. Apgropriate coatirsgs may be applied to
increase
palatability or delay absorption.
'~Tarious modif:cations of the invention in additaon to those shown and
described herezrr will be apparent to thase skilled in the art from the
foregoing
2U description. Such modifications are alSO intended to fall within the scope
of the
appended claims.
I3
CA 02464681 2004-04-07
WO 0010486d PCTJUS99116630
Tlte foregoing disclosure includes all the information deemed essential to
enable those Skilled in the arc to practice the claimed invention_ Hecause the
cited
applications may provide further useful infarmatian, these cited materials are
hereby
incorporated by reference in their entirery.
The following non-lirtiitiag examples are presented to further illustrate the
present invention.
I4
CA 02464681 2004-04-07
WO ~O1o4866 PCTIUS991I663o
Txample 1
Preparation of 1-(4-Thic~-~-n-Arabinofaranosyi) Cytosine
Z,3,a-Trl-O-benzyl-t~-xylose DibenzYl Dithioaeetal (4). L-Xylose (x; 25g, 16'7
inmol)
was stirred for 5 hours in 0.3°lo hydrogen chloride in methanol (675
mL) at room
temperature and then neutralised with Amberlite IRA-400 OH anion exchange
resin.
Tide filtrate and washings were combined and evaporated to dryness and W a
crude
product was purified by silica gel chromatography (CHC13I MeOH, 92:8) to
afford 26.2
g of methyl L-xylofuranoside (2, 9S% yield) as an a and ~3 ( 1:1 ) mixture. MS
I 64 ,
(M)-. 1 GS (M+H)+, 133 (M-OCH3)".
To an ice-cold solution of 2 (14 g, 60.9 mmol) in dry tetrahydrofuran (350
rnL)
was added sodium hydride (60% dispersion in mineral oil, 14.8 g, 370 mmol) and
the
reaction mixture was stirred for 15 min under N~. To this reaction mixtwre was
added
solid tetrabutylammonium iodide (0.36 g, 0.9& mmol) followed by a dropwise
addition
of bemyl bromide (36.6 g; 214 mrnol). The reaction mixture was stirred for 3
days at
roo~o zernperature. After the addition of methanol (25 mL) the solution was
evaporated.
m3der reduced pressure, and the crude product was purified by silica. gel
chromatography
(cyclolaexanelEtOAc, 9:1 ) to afFord pure methyl 2,3,5-tri-O-benzyl-L-
xylofuxanoside (3,
2. g. 87% yield). MS 435 (1V1+H)'', 433 (M-H)+, 443 (M-OCH;)'; 'H NMIt
(CD~CI;)
b 7.38-7.25 (m, 30H, aromatic H--s), 4.94 (d, JH, H-la, J,~ - 4.3 Hz), 4.87
(d. IH, H-
1 Vii. ,1 ~y = 0.9 Hz), 4.d4-4.45 (m, I2H, PhCH2 s), 4.37 (m. IH, H-4a.),
4..27 (dt. 1 H. H-
4(3, J,,.SO = 3,7 Hz, Ja,~b = 6.5 Hz, J34 = 6.2-Hz), 4.17 (t. 1H~, H-3a, J3.~
= 6:9 Hz. J.,., =
5.6 Hz), 4.a7 (dd, 1 H, H-3~, J3,a
IS
suesrm~rE sHE~r t~uu~ ash
CA 02464681 2004-04-07
= wo aa~aasss PCTNS9911b630
= 6.2 Hz, 3,,3 - ?.5 Hz), 4.00 (dd, 1H, H-2a, J~,3 ~ 5-6 Hz?, 3.95 (t, 1H. H-
2(3, J.,.3
=2.5 Hz), 3.70 (dd, IH, H-5aa, J4,jp ~ 4.5 Hz, Jse,55 = 10.4 Hz), 3.66 (dd.
1H. H-Sa(3,~,
J,~,~~ = 3.7 Hz, Js~~, =10.T Hz), 3.54 {;dd, IH, H-Sba, J~.S~ = 7.5 Hz), 3.49
(dd, 1H. h1-
R
Sb(3_ Ja.sh = 6.5 Hz).
To a solution of 3 (42 g, 97 mrnol) in dichlorometfiane (1000 mL) were added
L~enzyl inercaptan (49.6 g, 400 xnrr3ol) and stannic chloride (4.93 g, 18_9
rnrnol), and the
reaction mixture was stirred at room temperature ouerni~ht. After
neutralization with
~% aqueous NaHCO~ (750mL), the organic layer was separated and the aqueous
layer
was exn~acted with dichloromethane (500 mL). The combined organic layers were
. evaporated, and crude 4 was purified by silica gel chromatography
(cyclohexauelEtpAc,
99:1 ) to afford 4 (8.53 g, 57%) of sufficient purity tv carry forward. MS 657
(M-t-t,i)'';
'H NlvIR (CDC13) 8 7.35-7.29 {m, 19H, aromatic H=s), 7.19-7_13 (m, 4H_
aromatic
H=s). 7.01-6.96 (m, 2H azomatic I~'=s), 4.86.(d, 1H, PhCHX. J = 11.1 Hz),
4.'70 (huo
overlapping d=s, 2H, PhCHH, PhCHH, J = I 1.1 Hz, J = 1 I.2 ITz), 4.43 (d, 1H.
PhC~'Fi,
IS 11.2 Hz), 4.40 (d, 1H, PhCHH, 3 = I1.9 Hz), 4.36 (d, 1H, PhCHH, x = r L.9
Hz), 4.0f
(dd, 1H. H-2, 3~ , = 3.0 Hz, 3~,~ = ?.5 Hz), 3.75-3.67 (m, 4H, two PhC~T==s),
3.68 (d,
1H. H-i. 3,~ ~ 3.0 Hz), 3_36-3.25 (m, 2~i, H-4, H-Sa), 3.15-.3.12 (m, IH, H-
Sb), 2,22
(d. 1 H. 4~OH; ~ = 6.2 Hz).
W
S1188Tfft,JT~ SHEEP (RULE 26)
CA 02464681 2004-04-07
WO 00!0d855 PCT/US99115630
2,3,5-Tri-O-benz~-1-1-~-acetyl-4-thin-n-arabinofuranose (6). To a solution of
4 (13.0
~.. 20 rnmol) in dry 2:1 tolucnelacetonitriie {200 mL) were added
triphenylphosphine
(15.7 g. GO ramoi), iodine (12.? g, 50 mmol) and imidazole (5.44 g, 80 rnmol).
The
' reacuon mixture ~~-as stirred at 90 1C for 24 ti after which time the
solution zvas
S evaporated to di~mess. The crude product was purified by silica geI
chromatography
(cyclohexane/EtOA.c. 4:1) to afford benzyl 2,;,5-tri-O-benzyl-t,4-ditJnio-D-
arabinofuranoside as a syrup (5, 9.0 g, 83°/a). MS 543 (M+Hf; 'H NMR
(CDCI,) ~
7.40-7.~0 (1n, 20H. aromatic H=s), 4.69-4.42 (m, 6H, three PhCH=O=s), 4.37 (m,
1H,
H-1 ): 4.?0 (m, 2H. H-2, H-3), 3.87 (s, 2H, PhCH3S-), 3.80 (dd, 1 H, H-Sa. 3a
5, = 7.4
Hz. JS,~~~, = 9.3 .Hz). 3.55 (dd, IH, H-Sb, Jd.~, = 7.i Hz), 3.47 (m, 1H, H-
4). Anal.
(C~aH=~O,Sz ~ 0.?5 1-1~0) C, H.
Td a suspension of mercuric acetate (x.29 g, 22.9 mmol) in acetic acid (96 g)
was added 5 (5_4? g, 10 mmol), and the resulting mixture w$s stirred at roonri
temperature for 21Z. The reaction mixture Was diluted with dichloromethane
(200 mL)
1$ and washed successively with water, saturated aqueous NaHC03 and 5% aqueous
KGN -
soltttion. The organic layer was dried over Na2SOQ and concentrated.
Chromatography
o f the crude product using cyclohexaue:ethylacetate (98:2) as etuent gave a
mixture of
a and (3 ( 1:1 ) anon~ers of 6 (3.73 g, 78°/a) as a colorless syrup. MS
479 (M+H)''; ~ H
NMR (CDCI;) S 7.35-?.23 (m, 1SH, aromatic H=s), 6.07 (d; 0.25H, H-1(3, 3,, =
4:0
Hz), 5.98 (d, 0.75H. H-1 ce. Js~ --- 2.S Hz), 4.83-4.45 (ra, 6H, PhCH,'s),
4.26 (dd, 0.?5H, .
H-2a. J,., -- 5.4 Hz). 4.17-4.1 I (m, O.SH, H-Z(3, H-3(3), 4.03 {t; 0.75H, H
tea. J;.~ = 6
Hz). 3.80-3.67 (m. 1.25H, H-4a, H-5aa, H-Sa[3), 3.53 x_39 (m, I.75.H, H-Sbcx.
H-4(i,
H-Sb(i), ?.0G (s, 3H. CI33-a and CH3-Vii). Anal. (Cz8H3aO5S ~ Q.75 H=O) C. H.
I7
SUBS'CITUTE_SHEEC (RULE 26)
WU 00/04866 ~ 02464681 2004-04-07 i~C?IUS~9/16630
I-(2,3,5-Tri-O-benzyl-4-thio-~x,~~n-arabinofuranosyl) cytosine ('7). To a
suspension
of 1-n-acetyl 2.3.5-tri-C7-benzyl-4-thio-n-arabinofuranose (478 mg, 1 mmol)
and
cytosine ( I 11.0 m~, 1 nnrial) in anhydrosis acetonitrile (25 rr~mol) were
added
consecutively hexamethyld'iszla2ane (HMDS.162rng,1 tnmoi) and
chlorotrirnethylsilane
a
(TMSCI, 4;4 mg, 4 mmol), and the mixture was stirred at room temperature for
Q.5 ,
hours. This solution was cooled to -78 ° C.
Trimethylsilyltrifluoromethane sulfonate
(267 nag, I .2 mmol) was added and the resulting solution was stirred at -78
° C for
another 2.5 h, after which time the reaction was essentially complete. The
mixture was
warmed to room temperature, concentrated to a small volume (5 mL), diluted
v~rith
methylene chloride (50 mL) and then washed with water (20 rnL) followed by
saturated
sodium bicarbonate and water. The organic layer was dried over MgSO:, and
evaporated to dryness. The residue was purified by chromatography over silica
gel (SO
a. elution with CHCI3/MEOH 98:2) to afford 7 (4I2 mg 77.5%) as a coloriess
syrup
TLC (95:5 CHC13IME(.7lEi) R,t. 0.65; MS zle 536 (M+Li)t.
'H NIV1TZ (CDCI3) ~ $.22 (d, 1, H-dp, 3 ° 7.~6); (d; 1, H-6a; J5,6 =
7.5 I32); x.38-?.09 (m,
30, aromatic H's); 5.65 (d, 1, H-1'~, 3~.,~. = 5.7 I3.z); 6.36 (d, 1, H-I'a,
Jt.,. _ 1_2 Hz);
5.44 (d. 1. H-SQ); 5.26 (d, 1, H-Sp); 4.97-4.33 (averlapgirtg multiplets, 12;
CaH3CH,),
4.26 (dd. I H-2'p, J,..s- = G.S Hz); 4.22 (m, 1, H-3'~, J3~.4. = 1 Hz); 4.1b
(dd; 1, H-3'~,
J;..,. = 6.4 I-iz); 4.13 (m, I. H-Z'a, J2._3. = 1.7 Hz); 3.91 (m, 1_ ~I-4'~);
3.78 (m, 1. H
5~.,); 3.73-3.63 (m. 2. H-S'e); 3.55 (tn, I, H-5'a); 3.46 (in, 1. H-4'~.
I8
SUBS'TIfliT6 SH~61" (RLSt~ 26)
CA 02464681 2004-04-07
WO 00104866 PC'TItJS99116b30
I-(4-'JChio-a, ~-u-arabiu~oturanosyl) cytosine (8). To a solution of boron
irichloride
( 1 M solution) in dry dichloromethane (7mL, 7mmo1) cooled to -78 ~ C, -was
added
dropwise over a period of 30 min, a solution of compound 7 (26S mg, 0.5 mmol)
in clxy
diclUoromethane (10 mL). Stinting was continued overnight at -20 ~C. The
solvent
was removed in vacuo and the residue was coevagorated with dichloromethane (4
x ?0
mL). The residue was neutralized with saturated NaHCOs (25 tnL) and washed
with
chloraforn~ (IS mL). The aqueous iayer'was applied to a canon exchange (H')
column
and eluted with water to remove salts and tlxen eluted with 1N NHdOH to obtain
the
desired compound 8 (11U rng, 85°~°0) MS z!e 260(M+I~i)".
~H NMR (Me-,SO-ab) 8 7.9~ (d, .67, H-6-ø, J5.6 = 7.5 Hz); 7.9a (d. 4.33, H-6a,
JS_6 --
7.5 Hz); 7.17-7.03 (overlapping bs, 2, NH's); 6.33 (d, 4.67, H-i' ø, d, :,. =
4.6 Hz); 5.86
(d. 0.33, ~-I-1'oc, J,.,- = 7.3 Hz); 5.77 (d, 0.33, H-Stx): 5.70 (d, 0.67, H-5
j3): 5.61 arid
5.59 (overlapping doubiets, 1, 2'-OHø, J.,.~.,~ = 5.1 Hz, 2'-OHa. J,.,-_oH =
5.9 Hz);
5.47 (d, 0.33, 3'-OHa, J3. ~,~H = 5.1 Hz); 5.38 (d, 0.67. 3'-fJHø, J 3'.3'-(3H
= 4.2 Hz);
5.08 (t. 0.67. 5'-f)H~3; Jg~_S'.OH ~ 5.4 Hz); 4,90 (t, 0.33, S"-UHa, JS~,~,_oH
~ 5:2 Hz); 4.00-
3.93 {m. 1.&7, H-2'Q, H-2's, H-3'~; 3.$6-3.76 (tn, l, IH S'« and H-5'p~; 3.67-
3.55 (xn;
1. H-5' ~, + H-3' «); 3.49-3 .3 3 (m, 0.67; H ~' «, H-5' «); 3 . L7 (m. 0.67,
H-4' ~).
19
SU86'i'ITUi"E SHEET (RULE 26~
WO OO/Od866 ~ 02464681 2004-04-07 pCTNS99/16630 :y
1-(S-D-dimethoxytrityl-4-than-Q-n-strabinofuranosyl) cytosine (9). To a
solution of
compound 8 (I00 mg, 0.38 mmoi) in dry pyridine (10 mL) was added 4,4'-~
Dimeth.oxytritylchloride (135 mg, 0.6 mtnol) and the solution was stirred for
2 It at
room temperatuze_ The reaction mixture was evaporated to dryness and the crude
visas
dissolved in ethyl acetate (Z0 mL) and washed with water and evaporated to
dryness to
afford a solid which was purified on silica geI column (CHCI3/MeOH 98:2) to
obtain
pure compound 9 (96 mg, 90% based on 1:1 mixhire of oc, (3-mixture of 8). MS
~Je
SG8.3 (M+Li)':
'H NMR (Me,SO-db) S 7.77 (d, 1, H-6, Jss ~ 7.5 Hz); 7.42-?.23 (m, 9, aromatic
H's);
?.17 (bs, I, NH); 7,05 (bs, I; NH); 6.91-6.88 (rn, 4, aromatic H's); 6.36 (d,
1, H-1',
.1,. ,. = 4.8 Ha); 5.65 (d, I, H-S); 5.76 (d, 1, 2'-OH, J2.,i..os, = 4.6 Hz);
5.43 (d, 1, 3'-OH,
~Ta-.:--cm ' 3.3 Hz); 3.98-3:9I (m, Z, H-2'; H-3'); 3.75 (s, G. OCIri;); 3.39-
3.25 (m, B, H'
4'. H-~').
~EUBSTtTU'tE SHEEP (RULE 2~
1~V0 ~0/04866 ~ 02464681 2004-04-07
1-(4-Thio-(3-u-arabinofuranospl) cytosine (I4). Compound 9 (90 mg, 0.16 mmol)
was
treated with trifluoroaGetic acid (22 mg) in chlorafaxm (5 mL) at roam
temperature foz
I O min. The reaction mixture was neutralized with aq. NaHC03 and the aqueaits
layer
was applied on a catiou exchange column which was eluted first with water to
remove
salt and finally with IN NHdOH to afford compound 10 (35 mg, 85°/a),
rnp218-Z20 °C
( li t' 3? 1 _322 °C); MS zle 26Q(M+H)+.
'H NMR (Me,SO-d6) 6 7.94 (d, 1, H-6, 3~.6= 7.5 Hz); ?.I2 (bs, 1, NH); ?.U4
(6s, 1.;
NHl'. _6.33 (d, 1. H-1', J' :,' = 4.6 H~); 5.70 (d, 1, H-5); 5.61 (bd, 1, 2'-
OH, .T,.,,.ou =
3.1 Hz); 5.38 (6d. 1, 3'-OH, J3.,3,.cH '- 3:5 Hz); S.Q8 (bt, 1, 5'-OH,
JS.,s..oH ~ 4.9 Hz);
4.U0-3.93 (m, 2, H-2', H-3'); 3.78 (m, I, H-S'a); 3.6I (m, 1, H-5'b); 3.16 (m,
'l, H-4').
Example 2
Preparatioct of 1-(4-Thin-t3-D-Arabinofnranosyl)~.FluoroCytosine
1-(2,3,5-Tri-D-benzyl-4-thio-a,tl-~?-arabiuofnranosyl)~-flunrocytosine ('7a).
To a
suspension of 1-C?-acetyl 2,3,5-tri-O-benzyl-4-thio-D-arabinofmranose (478
m.g,1 ~nmol)
and cytosine ( 129.0 mg, 1 mmol) in anhydrosis acetouitrile (25 mmol) were
added
consecutively I~zexamethyldisilazane (HIVIDS,162 mg, l mmol) and
chlorotrimethylsilane
(TMSCI. 434 mg. 4 mmol), and the mixture was stirred at room temperature for
0.5
llour5. Tlois solution was cooled to -78°C.
Trirnethylsilyltrifluoromethane sulfonate
(2b'l mg, 1.2 mmol) was added and the resulting solution was stirred at -
7&°C for
2fl another 2.5 h, after which time the reaction was essentially. complete.
The mixture was
warnaed to room temperature, concentrated to a small volume (5 mL), diluted
with
ZI
~UBS~IME SHEET (RUCHE 26) .
CA 02464681 2004-04-07
WO 00I0~866 PCTJUS99/16630
tnethyleue clzlor~ide (SO mL) and then washed with water (20 mL) followed by~
saturated
sodium bicarbonate and water. The organic layer was dried over. MgSO., and
evaporated to dryness, The residue was purified by chromatography over silica
gel (SO
g. elution with CHCI~IMEOH 98:2) to afford 7 as 2:1 oe.Ii mixture (425.2 m~
80.0%)
as a colorless syntp TLC (95:5 CHCI3fM,EOH R,. 0.65; MS 7le S53 (IvI+Li)'.
'I-i NMR {CDCI3} 8 8.40 (d, I, H-6a,J=7.6); 8.10 (d, 1, H-6a, Jt,b ~ 7.5 Hz};
7..35-7.09 -
(n3, 30. aromatic H's); 6.55 (d, 1, H-1'a, J, :~- = 5.7 Hz); 6.25 (d, 1, I~-
1'n, 31. ,. = 1.2 '
Hz), 4.90 (d, 1. II-S~); 4.~8 (d, l, H-Sa}; 4.30-4.55 (overlapping multiplets.
12,
C,~H;CHI), 4.15 (dd, 1 H-2'a, Ja~,~~ = 6.8 Hz); 4.i0 (m, 1. H-3'Q, 33,.x. = 'l
Hz): 3.90 (dd,
l, H ~'~,. J3~.,,- = 6.4 Hz); 3.75 (m, 1, H-2'a, Jz.w = 1.7 Hz); 3.70 (m, 1. H-
4'~; 3.65 (m,
I. H-5'~a): 3.50-3.55 (m,2,H-5'a); 3.45 (m, I, H-5'a}; 3.4D (m. 1, H-4'~,
Z-(4-Trio-a-D-arabinofuranosyl)5-ifuorvrytosine (loa). To a solution of boron
trichioride ( 1 M solution} in dry dichlorometbane (7 mL. 7 anmol} cooled to -
78°C. ~~as
added dropwise over a period of 30 min, a solution of corapound 7 (2273 mg.
4.5
IS mmol) in dry dichloromethane (10 mL).~ Stirring was continued overnight at -
20°C:
tl3e solvent was removed in vacuo and the residue was coevaporated with
dichloramethane (4 x 20 mL). The residue was neutralized with saturated NaHC03
(25
nL) and washed with chlorafomm (15 mL). The aqueous layer was applied to a
cation
exchange (H') column and eluted with ,hater to remove salts and then eluted
with 1N
NH~OH to obtain the desired compound Sa as ?:l a.B mixture which upon
crystallization with water gave pure IOa (32.2 mg, 25%) MS zle 278(M+H)',
'H NMR (Mt~SO-db) 8 8:15 (d, 1, H-6, Js.e = 7.5 Hz); 7.75 (bs. I, NH); 7.50
(bs. 1,
NH); G ~5 (d, I,
22
SUBSTCtUfE SHEEt (RU4E 2.6~
CA 02464681 2004-04-07
wo ooioa866 PCTlU59911~630
H-1', 1~ :,v= 4.t~ Hz); 5.65 (d, 1, H S); 5.60 (bd, 1,2'-OH. J=.,~-~H = 3.1
Hz); 5.X0 (bd.,
1.3'-OH: J~ ,3'-OM = 3.5 Hz); 5.30 (bt, 1, 5'-OH, 3S..S..o~ = 4.9 Hz); 4.00-
3.90 (in. 2. H-?', ~ .
H-3'); 3.75 (m, 1, H-S'n); 3.65 (m, l, H-S'~); 3.1S (m, I, H-4').
' Ototani, H.; Whistler, R.L. Preparation and Antitumo'~r Activity of 4'-Thio
Analogs
0~' ?.?'-Anhydro-1-l3-17-arabinofuranosylcytosme. J. Med. Chem. 1974, 17, 535-
537.
23
SUB~~ SHED' (R!!L~ ~6,
WO 00!04866 ~ 02464681 2004-04-07 PCTlUS99/I6630
Example 3
The following in vitro tests were carried out.
Table 1
iCSfl Molar Values - '
ThioaraC 5-fluorothiaaraC~'ts~e
arabinc~side
LOX IMV 1 Melanoma 7 x 10'6 6 x 10'6 1 x 10-6
PC-3 Prostate $ x 10'6 1 x 10''~ S x IO'~
ZR-75-I Mammary 3 x 10'6 6 x 10'' 4 x 10'
CAKI-I Renal 3 x 10'~ 2 x IO's 6 x 1U-'
DLD-1 Colon 7 x 106 . 8 x 10's 5 x I0'~
ID ~ NC1-H23 Lung 5 x 10'G ' 3 x 10-6 3 x IO'' ,
SK-MEL 28 Melanoma 7 x 10'6 - 2 x IO'6
S1~TB-7 (CNS) 1 x IO'a ~t 2 x 10'
The above tests were carried out with a 72 hour exposure to the compound
using Natural Red assay for all cell lines except SK-MEL 28 which was done
with
SRB assay.
24
CA 02464681 2004-04-07
WU 00!04866 . PC'1'1US99~116630
Example 4
The fallowing in viva tests were carried out to illustrate the effectiveness
of
' the compounds of the present invention.
\7'/~ ~(?~p4~~ CA 02464681 2004-04-07
PCTIUS99/16630
H
~$f.
m
O
m ,n. v
,a
a m
'
G
o i.
.~
m
..
mm
n.
6-
~w
~ O
C'N
~ H ~,
b
H 6t
~ b
n
4 '
O m
D
=
~
V
4
_
~1
Z ! n
G
Ln w
Ij
~t
V
x
4
O Q Q
.
a
y~ u1 m
~
a 0 0
O
V r
r
U a
~
4., ~ a
O r
n n a
x
%' b a a
K N w
A
G
L
t:'__
C
m~
v
F
:e
a
o~ ~m
v
a
n~ g
y
~~
m
D Go
N
=
m V
~' ~1
Mm N V
O a
b ~ I
~'v ~
u
Q
m
0.
T1
CA 02464681 2004-04-07
WD 00!0a865 PCTlUS99/I6630
a. .~e w
n 1't n 71 6 n M _
g n ~ a~ ~~ ,~,a~N
3aa-~
,,.~
m r !. a. m ara ~ .
as wn n n w - ~ .',
n
w
oo' 'o e' ~o n o'a
1~ .a
o,o _
A
~t
~ A
H w a. .
1.
U
a
N .
'~
H~'
// d 0 o 7
0~
~n ~ 0 . a
o ~
H q~
9loa n .w m d o c a
H
Qi~ ~ ' \ \ 1 \ ~
~ D b 41 D 1~N
"~
H '
a
a ~A A
N In H
n . : w
x ~ i r.
a a 4 a~ a ~'
.. w
~
~ ~~ a ~ g n : .
P
m aa a n v o a v
a .tw ~,' H ~ w ..'w
o ~ q
~
ai ~n w ~ H .v r ,~..
Oi
D
~ ~
~ .~ ~ 8 g
g ~
~ ~
~
~ . ~ ~
. R'
~n " ~
a ar w ~
a r ~ a
M
~
o,
ws i
~r a
c pa~ vu a
~
.o a~ ~ , u
P o
. ~ W ,v ~ .
a
~
~ ~ ~u a v v J ~i
~ i
'.e' ~ w'de~ ~ ~ ~ .'..,
-; n
7 O
.~ ~r
Z7
WU 00104$66 ~ 02464681 2004-04-07 ' PCT/US99/16630
b a .r ..
v r
H w ~ i w
p0 n w m
e. ~ .
an
.
,
,. ao a C W
'1
- ?~ T hN .
N
~ An Y A t. H
I
a .,
m w
w a
b p
y
~
l1 ~ H'
ah o n o a
'
~i o
h m
m
~ ~ $~
n
n
~1~~ n N
n
H
0
Y m
4
0 W sa a a r o
W O O0 O O 6
d
. O -'
e-i
C~ ~u
a L' vv, v ~o ~o 0
v m
~cvna 'v o a a o
~
-~
H
.
ri
V vv V b 1v V
r~
~ a a ~ ~
x roa .
~ M
.~ ~ ~~ ~ b 9 ~
a
rn o aa v v o 0
~i.!
DQ w ~N a 1p'11~1 w
A n N
~
A ~ nw w e~ w -1
V
_.
.
eg
9
~ MY~ ~ ~
~
G
y '
a
~
~' ~ ~
aveuu
~ w~
ra
a~ ~ '
w ~
d
~
6
~s ~ o
a
sao N
0 n~
O
s'
a m U 4~ V
~~~
g ~ ~ ~ ~.,~
~~~ u
g.; ~ ~ ~m~
an ~ ~
., ~
e
~n O <n
..r
~uesn~.~~ sN~~r tRU~.E ~~~
CA 02464681 2004-04-07
' WO OO/OA866 PCTJUS99l1663t3
As can be appreciated from the above tests, in most cell lines, A12A-C
exhibits l3igher cytoxicity as comgaaed to that of the present invention which
wauild
tend to discourage use of the compounds of the present invention as a cancer
treaement. However, surprisingly, the J.-(4-thin-i3-D-
arabinofuranosyl)cytosine is
more potent than ARA,-C and more selective in killing cancel cells in vivo.
Compounds employed according to the present invention exhibit better
anticancer
activity along with lower toxicity. This may be due to the fact that 1-(4-thio-
~-Ll-
arabinofuranosyl)cytosine does not get deaminated by cytidine dearninase as
quickly
as AraC does, which is shown below in Table 5 by using radio labeled
substances:
Good substrate activity of cytidine deaminase with AraC has been a major
problem
with this drug because it not only decreases half life of drug but also gets
converted
to Ai'3Uri~lile Wnich is not an active substance.
Table 5
Cytidine deaminase activity
I5 determined with radioactive substrates
Compound 1~", V""x '~,n:~/~",
AraC I47 9 0.06
l.-(4-thio-13-D-arabinofuranosyl2571 17 0.007
cytosine
The following tests ware carried out which illustrate the abiliey of the
thioarabinofuranosyl compounds according to tkte present invention to inhibit
DhTA
replication in tttarumalian cells. In the following tests, dCyd refers to 2'-
deoxycytidine, TCyd refers to thiodeoxycytidine, T-araC refers to 1-(4-thio(3-
D-
axabinofuranosyl) cytosine and Ara-C refers to ''cytosine arabinoside" _ The
terms
°'d CTP", "T-d CTP", "ara CTP" and "T ara CTP°' refer to the
corresponding
.. 2S u-iphosphates. The team "dLIItD" refers to 2-deoxyuridine. The term "F-
d.Urd"
refers to fluoxinated deoxyuridine and "F-dUMP" refers to fluorinated
deoxyuridine
monophosphate.
29
CA 02464681 2004-04-07
WO OOIp4866 , PCTlLTS94/16534
Reference is made to Figs. la and 1b which illustrate the metabolism of d
Cyd, T-d Cyd, ara C and T-era C to their respective triphasphates. Ln
particular, ..
CEM cells were incubated with 2040 nM of [5-3H)T-dCyd, 5 nM [5-3H]araC, or 2S
nm [5 3H]T-araC for the times indicated. The radioactivity in the ~'-
triphosphate ,
peak was deterrnin:ed using S,A~ T-1PLC. Por eack~ compound. the amount of
radioactivity in the 5'-triphosphate peak was more than 903 of the total -
radioactivity eluting from the S~ HPLC colunm..
Test results related to the inhibition of CEM cell growth are shown below in
Table 6.
CEM cells were incubated with various concentrations of eaclx compound
and the effect on cell IIUIrib~rS Wa5 determined using a Goulter Counter. The
IC~o
was detexmined from a plot of the perceat of control growth versus drug
concentration. The data presented are the mean and standard deviation from 3
separate experiments. The araC-resistant cells were obtained by incubating CEM
IS cells in she presence of 150 nM araC for approximately 1 month at which
rime the
CEM cells were growing at control rakes.
Table 6
Inhibition of CEM CeII Growth by dCyd Analogs
Compound 'Wild-type ara.C-resistant Resistam/VPT
ICso ~~
T-dCyd ~ 2200 ~ I400 ?240 t 7600 3
T-araC 24 ~ 9 125 t 84 5
araC 6 ~ 3 800 ~ 400 145
. WU Oul04866 CA 02464681 2004-04-07 ~'CTIU$gg~I6630
Test results related to metaboiism in wild type and araC resistant CEM cells
are shown below in Table 7.
CEM cells (wild-type or araC-resistant cells) were incubated with 100 nM of
[5-3H)dCyd, [,5-3H~T-dCyd, [5 3I3]araC, or [5 3H]T-araC for the times
indicated.
' S The radioactivity in the 5'-triphasphate peak and the incurporauon of
radioactivity
is the acid-insoluble fraction (DNA) was determined. Fox each compound, the
amouni of radioactivity in the 5'-triphosphate peak was more than 90 ~ of the
total
radioactivity eluting fmm the SA~~ HPLC column.
Table 7
IO Metabolism of dCyd and Its Anaiags in
Wild-Tppe and araC Resistant CEM Cells
Compound IncubationCeil Type Tri- DNA Total
Time
phosphate
Hours gmoles11 06cells
(gercent
of WT)
dCyd ~ a.25 wT 2.27 I.I9 3.~.~
Resistant 0.021 0.0I1(0.89)0.032
(0.93) (0.91)
T-dCyd 0.25 WT 0. S77 1.29 Z. I7
Resistant 0.036 (4. 0.051 (4. 0.487 (4.0)
I ) 0) .
I~ araC 1 WT I4.7 0.28 14.98
Resistant 0.19 ( 0.0I 8 0.208 ( I .4)
I .2) {b.3)
T-araC I WT 0.202 0.0056 0.2076
Resistant 0:051 {25)O.OD25 0.053$ (26)
(45)
31
W~ Q~~~866 CA 02464681 2004-04-07 ~ p~~g99116fi30
Results illustrating phtnphorylation in cell-free CEM extracts are shown below
in Table 8.
A crude cell extract was prepared from wild-typo and araC~resistant CEM cells
and the
ability to phosphorylate dCyd, araC; and T-araC was determined. The number in
parentheses is the ,
number of experiments performed. Reactions were performed in soiutioas
containing 50 mM Tris
(pN 8.Q); 5 tnN! ATP. 9.5 tnM MgCI:, 20 taM NaF, extract> and 1 ~m IS-'H]dCyd>
(5-~H)araC or .
[5'H]T-araC. At various times after the i~itiadon of the experiment,.an
aliquot of each reaction
volume was removed and placed on DE~81 filters. The filters were washed with 1
mM ammonium
formate attd ethanol, and the amount of radioactivity on each filter was
deterrained. The
phosphorylation of each eompouad increased in a linear manner with respect to
time. This assag was
verified by HPLC.
Tal'le 8
Phosphorylation of dCyd, araC, attd T-at~C
in CeII-Free CEM E~ttraets
Compound Wild Type araC-resistaatt CE.M Percern of wild-type
cells
pmoleslmg pmtefrirrtinnte
7.5 dCyd 240 ~ 33 (3) 3_5 t 0.2 (3) 1.~
araC 94 (1) L2 (I) 1.3
T-trraC U,83 (2) O.OIi (1) L3
Tabte 9 below t'llustrates deoxycydidine deaminase activity, In particular,
deo~cycytidinc
deamittase acciviry was pariftcd from Mvtt-d cells as described (Shewach et
al, Mol. Pharmacot. 42:
SI8-524-; 1992). Each assay was done in dupl3catz and the kinetic constants
were deternuned from
tinear double-reciprocal plots of livelot:iry versus Ilconcentration of the
substrate. The best line was
32
SUBSTnt1'tE SHEEP (RULE 26~
WO alllti4866 ~ 02464681 2004-04-07 p~.~SggJ16534
detetmineti by linear regression from a! least S dacuttt points, and the IC",
and ~t""~ were determined
from the x and y intereepta.
Table 9
Substrate Characteristics of dCyd, araC, and T-araC
with dCyd Kinase Activity lsoiated from Mott-4 Celts
Cottipound K," (uM) Relative V,~", v,nu~m
dCyd 1.~ 1 ~:8 '
araC 15 t3.1 O.Ot?6
W-thin-araC 93 o_a6 0.005
Tabte IO below also illustrates deoxyeycidine deamiaase activity.
Deoxycyddine deaminase activiey was p~srified froth human placenta and the Km
and V~x of
dCyd, T-dCyd, araC, and T-araC were determined. Reaeuans were carried oat is
salucians
containing 20 raM potassium phosphate (pFl ?.4), 100 ntM KCI,~ various
concentrations of
radiolabeted nucleoside, and enzyme: The reacxions were stopped with acid, the
substrate was
separated from the product by paper chromatography, and the radiaacdvity in
each vreze detezrnined.
The kinetic constants were determined from linear doublereciproeal plots of
IJvetocity versus
l~concentration of the substrate. The best line was determined by linear
regression from at least 5
datum points, and the K~, and V~ were detettuiited froth the x and y
intercepts. The data presented
are the mean and standard deviation from 3 separate experiments.
33
suss-rrru~ shot t~utE Zs~
WO 00!048b6 ~ 02464681 2004-04-07 . ' pCT/US99/16630
Table IO
Suliactrmte Characterist3rs of dCyd, T-dCyd, araC, and T~raC
vrith dCyd Deaatlnase Activity Isolated fotri I3uman Placenta
Compound FCC, (u.M) V,~ (pmoles/mg/tttin)V,~IF~,
dCyd z3 t 2.7 . 13 t i.5 O.SS
T-dCyd 111 d: 7? 3? t 12 0.33
araC 238 t 13 t 1~ -~- 7 0.056
T-araC 2944 f 1023 21 t 6 0.0092
Results of half life tests in CEM cells are reported below in Table 11
After incubation of CEM celia for 1 hear with 100 nM of jS-~H]dCyd, [5-'HST
dG~d; j5-
'H]araC, or [5 3H]T-anC, the cells were collected, washed with fresh tnedinm.
and resuspended in
fresh mcdinm that did not contain radiolabeled nudeosides. Samples were
calleceed at variaus times
after the oells were resuspended in fresh medium, and the amount of
radioactivity itt the 5'-
criphosphate peak was determined using SAX HPLC. The data presented are the
mean and standard
deviation from 3 separate experiments.
Tsble II
xnitisl Half Life of dCTP, T-dCTP, araCTP,
and T ar~CTP its CEM Cells
Nucleotide Hours (SI?)
acTP 0.9a . 0.16
T-dCTP 1.10 0.35
araCTP i.31 ~ 0.31
T-araCTP 10.8 t 1.80
34
SUB6TITUTE SHEEt (RULE 26)
CA 02464681 2004-04-07
WO 00!0486b PC?fUS99/166~.i0
Results of tests related to retention of araC'TP and T-araCTP in CEM chlls
are shown in Fig. 2. The tests were carried out as follows:
' After incubation of CEM cells for 1 hour with either 5 nM ~5-3H]araC yr
200 nm [S 3H]T-araC; the cells were collected, washed with fresh medium, and
resuspended in fresh medium that did not contain radialabeled nucleosides.
Samples
were collected at various times after the cells were resuspended in fresh
medium,
and the amount of radioactivity in the 5'-triphosphate peak was determined
using
SAX HPLC. Tn this experiment there was 0.639 pmoles araCTPllOb cells and
0.246 pmoles of T-araC'TP after the 1 hour incubation with radiolabeIed
compound.
Results related to metabolism in CEM cells are reported below in Table 12.
In particular, CEM cells were incubated with 100 nM of [5 3HjdCyd, [5-3H]T-
dCyd, [5 3H]araC, or [5-3H]T-araC for the times indicated. The complete
metabolism of each compound was determined: The medium was analyzed for the
original compound, its deaxninated. form using reverse phase HPLC, and H~~C7;
the
IS acid-soluble extract was analyzed by SAX HPLC fox phosphorylated
metabolites;
and the incorporation of radioactivity into acid-precipitable material (1?NA)
was
determined. All of the original radioactivity was accounted for in these
fractions.
Far each compound, the amount of radioactivity in the 5'-trighosphate peak was
more than 9C1 % ' of the total radioaccavity eluting from the SAX HPLC column.
WC? OOlOd866 ~ 02464681 2004-04-07 . ' p~~s9911663U
Table Ix
RQetabolism of dCyd, T-dCyd, scraC, and T-araC in CEM Cells
Nucleoside Time OriginalDeazxtirtaiedH=O nucleoside- DNA
of
IncubationCompoundCompottttd, TP
(hours) ~ (percent of mtal)
dCyd 1 38 0 55 4 3
T-dCyd 1 90 0 Q 2 8
araC 1 g'7 0 4 13 0.4
T-aaaC 24 96 0 0 4 0.3
Resuhs from tests related to the effect of F-dUrd on metabolism are shown
below in Tahle
13.
CEM cells were incubated with I00 nM of [S-'HJdCyd, [3-'H]T-dCyd, [~',li]araC,
dr (~-
'H]T-araC for the times indicated in the presence ox absence of 10 ~.m F-dUrd,
which is metabolized
to F-iIUMP (a potent iahibitor of thynudylate syuthetase). Tho complete
meuaholism of each
compound was deterntlned as described in the legend to Table 12, and the
percent of deami>lated
metabolites for each compound was determined_ 1a the absence of F..dUrd, the
destitinated product
1~ for dCyd was [3H]13,0 due to the removal of ['H] at the 5 position of F-
dUMP by chymidylate
synchetase. Isi the presence of F-dUrd, the deaminsted product of dCyd, T-
dCyd, and araC was in
the medium.as dUrd. T-dUrd, or araU, respectively.
36
_ . CA 02464681 2004-04-07
W O Ii0/04866 PC?11J599116630
Table 13
Effect of F-dUrd on the M~abolism of
dCyd, T dCyd, araC, or T-araC
Compound F-DUrd (~M) Intrubation Percent Deaminacion
Time ,
~~)
S acya o ~ 32
ac~ya la 1 zz
r-acya o s
T-dCyd ~ 10 g ~
araC 0 g Q
exec to s to
T-araC U 24 p
T-araC IO 24 Q
Table I4 below reports results of tests concerning effects of DNA, 1t1~1A and
protein
syndtesis.
I5 CEM cells vrreze incubated with 60 EsM T-dCyd, 15Q nM azaC, or 7S0 uM T-
araC.
Radiolabeled precursors of DNA [SHadThd, RNA (yi]Urd), or protein
(['HJlenoine) were added to
each treatment 3Q~ minutes after, the addidan of each compound. Samples were
taken I , 2, 3 and 4
hours after the additlan of radialabeted precursors, and the incorpo~acion of
radioactivity into RNA,
DNA, or protein were determined as desezibed (Parker et al, Biocltent.
pharmncol. S5: I6~3-168I,
t998). Ench number represents the average of two experiments.
37
SUBSTfNTE SHEET (MULE 26)
WO Ot1/0486b CA 02464681 2004-04-07 ~ pCTlUS99I16G30
Table 14
Effect of T~dCyd, araC, and T-araC on
DNA, RNA, and Protein Syntheses
Compound DNA RNA ~ protein
Percent of control
60 ~cM T-dCyd 3B 95 78
150 nM araC g 9b 96
750 nRri T ataC 16 8? 72
As can be appreciated ftoTn the above, thioarobitzofuranosyl compounds
according to the
present invention can be used to inhibit DNA replication in mammalian cells.
These results tend to
suggest their ability as itttmunomodtiiatars, which would render them suitable
for seating . .
autolmmune diseases. This is further supported by the fact that several
guanosine analogs have bees
shown tv stimulate the immune system (VW'eigic, W.O., CRD Crit.Rev.lmmunal.,
1987, 7, 285; Lin
ec al, .l.Med.Chetrl., 1985, 28, 1194-1198: Reitzet al, J.Med.Chem., 1994, 3?,
3561-35?8; Michael
et al., l.Med.Chent., 1993, 3G. 3431-3436). Certain 3-B-
ribofuranosylthiazalo(4.,5-d]pyrimidines
hnve also been shown to have significant immunoactivity, including marine
spleen cell proliferation
and in vivo activity against Semliki Forest virus (Nagahara et aL, J
Med.Chem., 1990, 33, 40?-4iS).
The foregoing description of the invention illustrates and describes the
present invention.
Additionally. the disclosure shows attd describes only the preferred
embodiments of the invenuott
but, as mantioned above, it is to be understood that errs irlvencion is
capable of use in various other
24 combinations, modifications, and environments and is capable of chaztges or
modifications within the
scope of the inventive concept as expressed herein, comtttensurate with the
above teachiFtgs andlor
the skill or knowledge of the relevant axt. The embodiments described
hezcinabove are further
intended to explain best modes known of
38
.SUBSTlf UTE SHEET ~RULF 2~
CA 02464681 2004-04-07 F(~T~f~,~9~'~~~3~
practicing the invention and to enable others skilled in the art to utilise
the invernion
in such, or ocher, embodiments and with the various modifications required by
the
gartieular applications or uses of the invention. Accordingly, the description
as not
intended to limit the invention to the form disclosed herein. Also, it is
intended that
the appended claims be construed to include alternative embodiments.
39