Note: Descriptions are shown in the official language in which they were submitted.
CA 02466701 2010-02-01
NEUROPROTECTIVE USE OF CYCLIC PROLYL GLYCINE
BACKGROUND OF THE INVENTION
NMDA AMPA Receptors '
Excessive excitation by neurotransmitters can cause the degeneration and
death of neurons. It is believed that this degeneration is in part mediated by
the
excitotoxic actions of the excitotoxic amino acids (EAA) glutamate and
aspartate at
the N-methyl-D-aspartate (NMDA) receptor, the alpha-amino-3-hydroxy-5-methyl 4-
isoxazole proprionic acid (AMPA) receptor, and the kainate receptor.
AMPAJKainate
receptors may be referred to jointly as non-NMDA receptors.
This excitotoxic action is considered responsible for the loss of neurons in
cerebrovascular disorders such as cerebral ischemia or cerebral infarction
resulting
from a range of conditions, such as thromboembol or hemorrhagic stroke,
cerebral
vascospasm, hypoglycemia, cardiac drowning, pulmonary surgery, and cerebral
trauma, as well as Alzheimer's disease, Parkinson's disease, and Huntington's
disease.
Among excitatory amino acid receptor antagonists recognized for usefulness in
the treatment of neurological disorders are those that block AMPA receptors.
(Bigge
C.F. and Malone T.C., Curr. Opin. Ther. Pat., 1993:951; Rogawski M.A. TiPs,
1993;
14: 325).
AMPA receptor antagonists have prevented neuronal injury in several models
of global cerebral ischernia. (Li H. and Buchan A.M., J. Cerer. Blood Flow
Metab.,1993; 13: 933; Nellga B. and Wielock T. J. Cerer. Blood Flow
Metab.,1992;
12:2) and focal cerebral ischemia (Bullock R., Graham D.I. Swanson S.,
McCullock.,
J. Cerer. Blood Flow Metab. 1994; 14: 466; X ue D. et al J. Cerer. Blood Flow
Metab.,
1994; 14: 251).
CA 02466701 2010-02-01
2
AMPA receptor antagonists have also demonstrated promise in chronic
neurodegenerative disorders such as Parkinson's disease. (Klockgether T. et
al., Ann.
Neurol., 1993; 34 (4): 585-593).
Excitatory amino acid receptor antagonists that block NMDA receptors are
also recognized for usefulness in the treatment of disorders. NMDA receptors
are
intimately involved in the phenomenon of excitotoxicity, which may be a
critical
determinant of outcome of several neurological disorders. Disorders known to
be
responsive to blockade of the NMDA receptor include cerebral ischemia (stroke
or
cerebral trauma, for example), muscular spasm, convulsive disorders,
neuropathic
pain, and anxiety, and may be a significant causal factor in chronic
neurodegenerative
disorders such as Parkinson's disease (Klockgether T., Turski L., Ann. Neurol.
1993;
34: 585-593), human immunodeficiency virus (HIV) related neuronal injury,
amyotrophic lateral sclerosis (ALS); Alzheimer's disease (Francis P.T, el al.
J.
Neurochem. 1993; 60(5): 1589-1604) and Huntington's Disease (Lipton S., TINs,
1993; 16(12): 527-532; Lipton S., Rosenberg P.A. New Eng. J. Med 1 994; 330
(9):
613-622); Bigge C.F. Biochem. Pharmacol. 1993; 45: 1547-156).
NMDA receptor antagonists may also be used to prevent tolerance to opiate
analgesisa or to help control withdrawal symptoms from addictive drugs.
A novel antagonist to NMDA/AMPA
This invention relates to the discovery of the functions of cyclic Prolyl
Glycine (herein referred to as "cyclic PG" or "cPG") as a novel antagonist
that either
blocks the ANIPA and/or the NMDA receptors.
CPG has been found to be an endogenous compound exhibiting anxiolytic
activities in animal studies by Gudasheva TA et al (Biull Eksp Biol
Med 1999 Oct 128:10 411-3) and Seredenin S.B. et al (Butt Exp Biol Med 2002
Apr
133:360-2).
We have surprisingly discovered that cPG is the end product of the
metabolites of glycine-proline-glutamate (GPE), which in turn is one of the
components of the cleavage of insulin-like growth factor 1 (1GF-1).
IGF-1 is a 70 amino acid-long polypeptide with several metabolic actions
known to be expressed in the rat brain during development and after acute
injury.
(D'Ercole, Al et al Molecular Neurobiology 1996;13: 227-255).
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
3
Some of the biological effects of IGF-I are probably facilitated by des (1-3)
IGF-I, an IGF-I derivative lacking the N-terminal tripeptide glycine-proline-
glutamate
(GPE). It was reported that des (1-3) IGF-I is less effective than recombinant
human
IGF-I (nhIGF-I) as a neuronal rescue agent, which suggests that the central
effect of
IGF-I might be partially mediated by the tripeptide GPE. (Guan J, et al
Endocrinology
1996; 137: 893-898).
However, it was Sara et al, who in 1989 showed that GPE is a neuroactive
peptide which facilitates the release of both acetylcholine and dopamine from
cortical
slices in vitro. (Biochem. Biophys. Res. Comn 1989; 165: 766-771).
Sarah's group has Swedish, European, and Japanese patents on GPE as a
neuromodulatory peptide (EP0366638, SE8803847, JP2250895).
The US patent by Bourguignon et al, US 5, 804, 550 or W094/26301 suggests
that GPE is an NMDA antagonist.
The US patent by Gluckman et al, US 6, 187, 906 claims that GPE can be used
to protect dopaminergic neurons of a mammal against death from Parkinson's
disease.
This group reported that the mechanism by which GPE leads to prevention of
cell
death was not known, but was not by modulating neuronal activity.
In their patent claims, the Gluckman group proposed that the "concentration of
GPE and/or analogues thereof in the CNS and in the brain of the patient in
particular
should be increased in order to treat the CNS damage." They also proposed a
suitable
dosage range to be between about 0.1 to 1,000 g of GPE per 100g of body
weight
where the composition is administered centrally.
However, the Gluckman patent (filed on July 15, 1999 and issued on February
13, 2001) did not cite a publication by Curatolo L. et al (Annals of New York
Academy of Sciences 1995; 765: 145-150 Neuroprotective effect of GPE
Pretreatment on rat Hippocampal Organotypic cultures exposed to NMDA), in
which
the effect of GPE did not seem to be clearly concentration-dependent. The
highest
degree of neuroprotection was obtained with 10 M GPE while lower (1 M) or
higher (50-100 M) concentrations reduced the neuronal damage to a lesser
extent.
The lack of a concentration-dependent effect does not indicate a single
receptor-
mediated effect. In fact, the bell-shaped curve describing the pharmacological
effect
of increasing concentrations of GPE might be the result of multiple mechanisms
of
action.
CA 02466701 2010-02-01
4
Without the clear understanding of the mechanism of action of GPE, it was
very difficult to determine the optimal dosage or human treatment. The
empirical bell-
shaped curve-model for rats may be only applied to rats and might not be
entirely
appropriate for human applications.
Private communication with the Gluckman group showed unpublished results
of recent studies in which high concentrations of GPE administered
intraperitoneally
and intravenously caused severe brain damage to rats. It was questionable
whether
GPE may be a suitable neuroprotective agent based on unreproducible results of
the
bell-shaped curve concentrations of GPE.
The uncertainty of the concentration dependency was not only observed in
GPE, but also with IGF-I. Johnston B. et al (J. Clini. Invest. Volume 97,
Number 2,
January 1996, pp 300-308 and subsequent private communications) reported that
lp.g
IGF-I is more neuroprotective than 10Ong in fetal sheep studies, but when the
dose
increased from lp.g to 1014 all the neurOprotective effects are lost. It was
found that
increasing the dose to 100pg or more usually killed the sheep fetus. It was
not known
why the neuroprotective dose in the fetal sheep (weighs about 3.5kg) was about
500
times less than the effective dose in the rat (weighs about 350g) if the
difference in
weight between the fetus and rat is taken into account (a fetal sheep weighs
about 10
times the rat).
The present invention provides an explanation to the unpredictable bell-shaped
curve effect of the concentrations of IGF-I and GPE. The present invention
provides a
novel mechanism of action of GPE, in which GPE is not a final product of the
cleavage of IGF-I, but GPE is metabolized into cyclic Prolyl Glycine and
glutamic
acid, as illustrated in Figure 1.
Over the past twenty years there have been a large number of publications
reporting the neurological effects of the insulin like growth factors.
(Pimentel E.
(1994) Handbook of Growth Factors, Volume 1-3, CRC Press (Ann Arbor), Now with
the finding that IGF-1 and GPE are the pro-drug of cPG, we now can link the
biological activities of these pro-drugs to cPG, thus not only enabling us to
study the
mechanisms of action of 1GF-1 and GPE, but also to attribute the
neuroprotective
activities of these two compounds to cPG and their neurotoxicity to the
corresponding
glutamic acids.
CA 02466701 2010-02-01
It is noted that the stereoisomers of GPE are important factors and it is
found
that only the cis form of GPE can be metabolized into cyclic Prolyl Glycine
and
glutamic acid. Since glutamic acid or glutamate is a well-known neurotoxic
agent, it
is predicted that cyclic Prolyl Glycine (cPG) must possess very potent
neuroprotective effect to overcome the intrinsic neurotoxicity of glutamate.
As shown in the Figure I and 2, cyclic PG can form chelating complexation
with metal ions such as calcium ions, magnesium ions, and it also can bind to
large
molecules such as 1GF-Binding Proteins, such that cyclic PG can serve as a
neurotransmittance agent or a neurotransportor as well as an energy storage in
the
central nervous system. These attributes can make cyclic PG an anti-necrotic
and anti-
apoptotic agent in the central nervous system.
Our experimental results reported herein, serving as limited examples, showed
that cPG not only acts as a potent neuroprotective agent but also serves as a
neurogenesis agent, which can be considered a novel drug candidate for
treatment of
neurological disorders.
SUMMARY OF THE INVENTION
One aspect the invention provides cyclic Prolyl Glycine compounds suitable
for the treatment or prevention of disease and injury in animals and humans.
The
cyclic PG being selected from the group that includes cPG, cPG analogues, cPG
pepticlomimetrics and relating compounds which promote or cause the formation
of
cPG or cPG analogues in vivo.
Preferably the cPG compounds are administered in a pharmaceutically
acceptable composition.
More preferably the composition additionally includes a therapeutic amount of
a cPG compound in combination with a compound selected from growth factors and
associated derivatives (insulin-like growth factor-I [IGF-1], insulin-like
growth factor-
II GPE, transforming growth factor-ill, activin, growth hormone, nerve
growth factor, growth hormone binding protein, JQF-binding proteins
[especially
JGFBP-3], basic fibroblast growth factor, acidic fibroblast growth factor, the
hst/Kfgk
gene product, FGF-3, FGF-4. FGF-6, keratinocyte growth factor. androgen-
induced
growth factor. Additional members of the FGF family include, for example, int-
2,
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
6
fibroblast growth factor homologous factor-1 (FHF-1) FHF-2 FHF-3 and FHF-4,
karatinocyte growth factor 2, glial-activating factor, FGF- 10 and FGF- 16,
ciliary
neurotrophic factor, brain derived growth factor, neurotrophin 3, neurotrophin
4,
bone morphogenetic protein 2 [BMP-2], glial-cell line derived neurotrophic
factor,
activity-dependant neurotrophic factor, cytokine leukaemia inhibiting factor,
oncostatin M, interleukin), 13,a,x or consensus interferon, TNF-a;
clomethiazole;
kyriurenic acid, Semax, FK506 [tacrolimus], L-threo-l-phey1-2-decanoylamino-3-
morpholino-l-propanol, andrenocorticotropin-(4-9_ analogue [0RG2766] and
dizolcipine [MK-801], selegiline; glutamate antagonists such as, NPS1506,
GV1505260, MK-801, GV150526; AMPA antagonists such as 2,3-dihydroxy-6-
nitro-7- sulfamoylbenzo (Oquinoxaline (NBQX), LY303070 and LY300164; anti-
inflammatory agents directed against the addressin MAdCAM-1 and/or it integrin
a4
receptors (a4131 and a 4p7), such as anti-MAdCAM-11mAb MECA-367 (ATCC
accession no. (HB-9478), interferons including interferon beta lb and
interferon
alfacon-1.
Preferably the cPG compounds may be used in the treatment or prevention of
cell damage or cell death in response to diseases and injury resulting from
septic
shock, ischemia, administration of cytokines, overexpression of cytokines,
ulcers,
gastritis, ulcerative colitis, Crohn's disease, diabetes, rheumatoid
arthritis, asthma,
Alzheimer's disease, Parkinson's disease, multiple sclerosis, stroke,
cirrhosis, allograft
rejection, transplant rejection, encephalomyelitis, meningitis, pancreatitis,
peritonitis,
vasculitis, lymphocytic choriomeningitis glomerulonephritis, uveitis,
glaucoma,
blepharitis, chalazion, allergic eye disease, corneal ulcer, keratitis,
cataract, retinal
disorders, age-related macular degeneration, optic neuritis ileitis,
inflammation
induced by overproduction of inflammatory cytokines, hemorrhagic shock,
anaphylactic shock, burn, infection leading to the overproduction of
inflammatory
cytokines induced by bacteria, virus, fungus, and parasites, hemodialysis,
chronic
fatigue syndrome, stroke, cancers, cardiovascular diseases associated with
overproduction of inflammatory cytokines, heart disease, cardiopulmonary
bypass,
ischemic/reperfusion injury, ischemic/reperfusion associated with
overproduction of
inflammatory cytokines, toxic shock syndrome, adult respiratory distress
syndrome,
cachexia, myocarditis, autoimmune disorders, eczema, psoriasis, heart failure,
dermatitis, urticaria, cerebral ischemia, systemic lupus erythematosis, AIDS,
AIDS
dementia, chronic neurodegenerative disease, chronic pain, priapism, cystic
fibrosis,
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
7
amyotrophic lateral sclerosis, schizophrenia, depression, premenstrual
syndrome,
anxiety, addiction, migraine, Huntington's disease, epilepsy, gastrointestinal
motility
disorders, obesity, hyperphagia, neuroblastoma, malaria, hematologic cancers,
myelofibrosis, lung injury, graft-versus-host disease, head injury, CNS
trauma,
hepatitis, renal failure, chronic hepatitis C, paraquat poisoning, transplant
rejection
and preservation, fertility enhancement, bacterial translocation, circulatory
shock,
traumatic shock, hemodialysis, hangover, and combinations of two or more
thereof.
Preferably the cPG compounds may be used in the restoration of myelination
of axons in mammals where myelin depleted due to neural injury or disease.
Preferably cPG compound may be used in the restoration of myelination
where depletion due to trauma, toxin exposure, asphyxia or hypoxia-ischemia,
perinatal hypoxic-ischemic injury, injury to or disease of the white matter of
the CNS,
acute brain injury, chronic neurodegenerative disease including multiple
sclerosis, and
demyelinating diseases and disorders including acute disseminated
encephalomyelitis,
optic neuritis, transverse myelitis, Devic's disease, the leucodystrophies;
non-
inflammatory involvement; progressive multifocal leukoencephalopathy, and
central
pontine myelinolysis.
Preferably the cPG compound will be administered in combination with IGF-1
or an interferon.
Another related aspect the invention relates to a method of treating or
preventing cell damage or cell death in response to injury and disease by
administering at least one cPG compound.
Preferably the cPG compound will be administered at between about li.tg to
about 150mg per kilogram of bodyweight. A suitable dosage for administration
of
cPG may be, for example, at between 0.1mg to about 100mg per kilogram of body
weight, at between about lmg to about 75 mg per kilogram of body weight, at
between 10mg to about 50mg per kilogram of body weight, or at between about
20mg
to about 40mg per kilogram of bodyvveight.
A further aspect the invention relates to a method of restoring the
myelination
of axons in a mammal in need of restored myelination due to neural injury or
disease,
comprising administering a therapeutic amount of a cPG compound, where a cPG
compound comprises cPG, a biologically active cPG analogue, a biologically
active
cPG peptidomimetic, a compound that increases the concentration of cPG, or a
compound that increases the concentration of cPG analogues, effective to
restore
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
8
myelination of axons in a mammal. In one aspect of the invention, the method
of
restoring myelination of axons comprising administering a therapeutic amount
of a
cPG compound comprises stimulation of astrocytes to promote remyelination. In
another aspect of the invention, the method of restoring myelination of axons
comprising administering a therapeutic amount of a cPG compound comprises
stimulation of oligodendrocytes to produce myelin.
In yet another aspect of the invention, the method of restoring myelination of
axons to a mammal in need of restored myelination further comprises
administering a
therapeutic amount of a cPG compound in combination with a compound selected
from IGF-I or an interferon. In one aspect of the invention, the method of
restoring
myelination of axons comprising administering a therapeutic amount of a cPG
compound in combination with IGF-I or an interferon to stimulate astrocytes to
promote remyelination. In another aspect of the invention, the method of
restoring
myelination of axons comprising administering a therapeutic amount of cPG in
combination with IGF-I or an interferon to stimulate oligodendrocytes to
produce
myelin. In preferred embodiments, the interferon comprises interferon beta lb
(Betaseron). In a further most preferred embodiment, the interferon comprises
consensus interferon (Infergen , interferon alfacon-1).
In still a further aspect of the invention, the methods to treat or prevent
cell
damage and death in response to injury and disease, comprises administration
of a
therapeutic amount of a cPG compound in an amount from about 1 jig to about
150mg
of cPG per kg of body weight of the mammal.
In yet another aspect of the invention, the method of restoring myelination of
axons to a mammal in need of restored myelination further comprises
administering a
therapeutic amount of a cPG compound in combination with IGF-1 from about 1 to
10mg of IGF-I per 1 Kg body weight of the mammal or an interferon from about
0.1
to 1000p.g of IGF-I per 100g of body weight of the mammal. In a preferred
embodiment, the interferon is interferon beta. In the most preferred
embodiment, the
interferon is interferon beta lb (Betaseron). In a further most preferred
embodiment,
the interferon comprises consensus interferon (Infergen , interferon alfacon-
1).
In a further preferred embodiment of the methods to treat or prevent cell
damage and death in response to injury and disease, comprising administration
of a
cPG compound, the cPG compound is administered to the mammal through a shunt
into a ventricle of the mammal.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
9
In a further preferred embodiment of the methods to treat or prevent cell
damage and death in response to injury and disease, comprising administration
of a
cPG compound, the cPG compound is administered to the mammal by peripheral
administration.
The present invention provides a method of treatment for stimulating mature
astrocytes to promote myelin production after hypoxic-ischemic injury
including the
step of increasing the active concentration of cPG and/or the concentration of
analogues of cPG in the CNS of mammals.
Most preferably, it is the effective amount of IGF-I itself that is increased
within the CNS of the mammal. This can be effected by direct administration of
cPG
and indeed this is preferred. However, the administration of compounds that
indirectly
increase the effective amount of IGF-I (for example a pro-drug which, within
the
patient is cleaved to release cPG) is in no way excluded.
The active compound (IGF-I or its analogue or its mimetic) can be
administered alone, or as is preferred, a part of a pharmaceutical
composition.
The composition can be administered directly to the CNS. The latter route of
administration can involve, for example, lateral cerebro-ventricular
injection, focal
injection or a surgically inserted shunt into the lateral cerebro-ventricle of
the brain of
the patient.
Conveniently, the stimulation and promotion of myelin production in
oligodendrocytes and the support, stimulation and promotion of remyelination
by
mature astrocytes is promoted through the administration of cPG compounds in
the
prophylaxis or therapy of demyelinating diseases such as multiple sclerosis.
BRIEF DESCRIPTION OF DRAWINGS
A better understanding of the invention will be gained from reference to the
following examples and drawings wherein:
Figure 1 illustrates the proposed metabolism pathway of cis-GPE to cyclic
Prolyl
Glycine and glutamic acid.
Figure 2 illustrates the proposed mechanism by which cyclic prolyl glycine may
act
to bind metal ions.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
Figure 3 illustrates in graphic form Glutamate toxicity in cerebellar
microexplants
(P4) and rescue effect by cyclic GP.
Figure 4 illustrates in graphic form prevention of glutamate toxicity by
cyclic GP
monitored within P4-cerebellar microexplants.
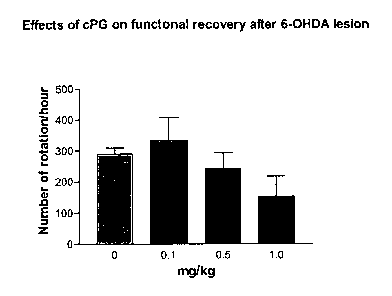
Figure 5 illustrates in graphic form effects of cPG on functional recovery
after 6-
OHDA lesion
DETAILED DESCRIPTION OF THE INVENTION
The following examples are given by way of illustration only and shall not be
taken as limiting the scope of the invention.
It has been surprisingly discovered that the process of the metabolism of IGF1
to the tripeptide GPE and des IGF is only a part of the process.
The cis-isomer of the GPE can further break down to form a cyclic Prolyl
Glycine and glutamic acid. This is shown in Figure 1.
The cyclic PG structure is sufficiently small to allow it to cross the blood-
brain barrier.
In addition, as shown in Figure 2 the structure of the molecule is such that
it is
able to provide ligands for binding metal ions such as Mg2+, Ca2t, Co2+ and
the like
and as such can act as a chelating agent.
The possible role of cPG as an agent is further supported by the companion
break down product, glutamic acid.
Glutamic acid is known to be associated with brain disease. (Johnston, G.A.R.
in Roberts P.J. et al Editors, Glutamate: Transmitter in the Central Nervous
System,
John Wiley & Sons, 1981, pp.77-87).
As used herein, a cPG compound is a compound with biological activity
similar or identical to the biological activity of cPG; cPG compounds comprise
cPG,
biologically active cPG analogues, biologically active cPG mimetics, and
compounds
that increase the concentration of cPG and cPG analogues in a mammal. cPG
compounds include cPG agonist molecules such as truncated portions of IGF-I
compounds as well as other chemical and biological analogues and mimetics.
As used herein, "cPG analogue" is any analogue of cPG, naturally occurring
analogue of cPG, or any variants thereof, which are capable of effectively
binding to
CA 02466701 2004-05-07
WO 03/041655 PC
T/US02/36639
11
mGluR receptors in the CNS and of promoting an equivalent neuroprotective
effect
on CNS nerve cells.
The term "cPG agonist molecules" includes peptide fragments and truncated
portions of longer IGF-I compounds as well as other chemical and biological
analogues and mimetics. cPG compounds can be used in the treatment of mammals,
suffering from neutral injury or disease. In particular the cPG compounds can
be used
to treat human patients, suffering from neural injury or disease. Still more
generally,
the compositions and methods of the invention find use in the treatment of
mammals,
such as human patients, suffering from nerve damage or potential apoptotic
and/or
necrotic cell death, due to injuries and diseases such as septic shock,
ischemia,
administration of cytokines, overexpression of cytokines, ulcers, gastritis,
ulcerative
colitis, Crohn's disease, diabetes, rheumatoid arthritis, asthma, Alzheimer's
disease,
Parkinson's disease, multiple sclerosis, stroke, cirrhosis, allograft
rejection, transplant
rejection, encephalomyelitis, meningitis, pancreatitis, peritonitis,
vasculitis,
lymphocytic choriomeningitis, glomerulonephritis, uveitis, glaucoma,
blepharitis,
chalazion, allergic eye disease, corneal ulcer, keratitis, cataract, retinal
disorders, age-
related macular degeneration, optic neuritis ileitis, inflammation induced by
overproduction of inflammatory cytokines, hemorrhagic shock, anaphylactic
shock,
bum, infection leading to the overproduction of inflammatory cytokines induced
by
bacteria, virus, fungus, and parasites, hemodialysis, chronic fatigue
syndrome, stroke,
cancers, cardiovascular diseases associated with overproduction of
inflammatory
cytokines, heart disease, cardiopulmonary bypass, ischemic/reperfusion injury,
ischemic/reperfusion associated with overproduction of inflammatory cytokines,
toxic
shock syndrome, adult respiratory distress syndrome, cachexia, myocarditis,
autoimmune disorders, eczema, psoriasis, heart failure, dermatitis, urticaria,
cerebral
ischemia, systemic lupus erythematosis, AIDS, AIDS dementia, chronic
neurodegenerative disease, chronic pain, priapism, cystic fibrosis,
amyotrophic lateral
sclerosis, schizophrenia, depression, premenstrual syndrome, anxiety,
addiction,
migraine, Huntington's disease, epilepsy, gastrointestinal motility disorders,
obesity,
hyperphagia, neuroblastoma, malaria, hematologic cancers, myelofibrosis, lung
injury, graft-versus-host disease, head injury, CNS trauma, hepatitis, renal
failure,
chronic hepatitis C, paraquat poisoning, transplant rejection and
preservation,
fertility enhancement, bacterial translocation, circulatory shock, traumatic
shock,
hemodialysis, hangover, and combinations of two or more thereof.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
12
In addition, cPG may be used to treat mammals suffering from white matter
insult as the result of acute brain injury, such as perinatal hypoxic-ischemic
injury; or
from chronic neural injury or neurodegenerative disease, such as multiple
sclerosis, or
from other demyelinating diseases and disorders including inflammatory
involvement,
such as acute disseminated encephalomyelitis, optic neuritis, transverse
myelitis,
Devic's disease, the leucodystrophies; non-inflammatory involvement;
progressive
multifocal leukoencephalopathy, central pontine myelinolysis. Patients
suffering
from such diseases or injuries will benefit greatly by a treatment protocol
able to
initiate re-myelination.
The present invention has application in the induction of myelin production
following insult in the form of trauma, toxin exposure, asphyxia or hypoxia-
ischemia,
and has application in the treatment or prevention of apoptosis in response to
injury or
disease in the form of cancers, viral infections, autoimmune diseases,
neurological
diseases and injuries and cardiovascular diseases.
cPG treatment may be given before (as well as alter) an injury - as for
example
before elective surgery. Examples of relevant elective procedures include
neural
surgery, in which retraction of lobes of the brain may lead to cerebral
oedema, or
heart operations, such as valve replacement, in which inevitable small emboli
are said
to lead to detectable impairment of brain function in some 75% of cases.
Pharmacology and Utility
cPG can act as an anti-necrotic and anti-apoptotic in a process of cell death.
Its
anti-apoptotic and anti-necrotic activity in vivo can be measured by cell
counts. cPG
can also be measured in vitro. (Gudasheva T.A. et al. FEBS Letters, Vol. 391,
Issues
1-2, 5 August 1996, pp. 149-152). CNS damage may for example be measured
clinically by the degree of permanent neurological deficit cognitive function,
and/or
propensity to seizure disorders. (Rakic L.J et al, in Ralcic L.J et al Peptide
and Amino
Acid Transport Mechanisms in The Central Nervous System, 1988, The MacMillan
Press Ltd. (London) pp.167-181).
Pharmaceutical compositions and administration
CPG itself is used to prevent or treat cell damage and death and the induction
of myelin production. Usually this is effected through the direct
administration of
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
13
cGP to the patient. If desired, a combination of the cPG compounds and its
analogues
can be administered in a pharmaceutically acceptable composition.
Those skilled in the art will appreciate there is no intention on the part of
the
applicants to exclude administration of other forms of cPG. By way of example,
the
effective amount of cPG in the CNS= can be increased by administration of a
pro-drug
from of cPG, which comprises cPG and a carrier, cPG and the carrier being
joined by
a linkage which is susceptible to cleavage or digested within the patient. Any
suitable
linkage can be employed which will be cleaved or digested to release cPG
following
administration.
In addition, it is envisaged cPG levels may be increased through an implant
that includes a cell line capable of expressing cPG in an active from within
the CNS
of the patient.
cPG can be administered as part of a medicament or pharmaceutical
preparation. This can involve combining cPG with any pharmaceutically
appropriate
carrier, adjuvant or excipient. The selection of the carrier, adjuvant or
excipient will
of course usually be dependent upon the route of administration to be
employed.
The administration route can vary widely. An advantage of cPG is that it can
be administered peripherally. This means it need not be administered directly
to the
CNS of the patient in order to have effect in the CNS.
Any peripheral route known in the art can be employed. These can include
parenteral routes for example injection into the peripheral circulation,
subcutaneous,
intraorbital, ophthalmic, intraspinal, intracisternal, topical, infusion
(using e.g.,
controlled release devices or minipumps such as osmotic pumps or skin
patches),
implant, aerosol, inhalation, scarification, intraperitoneal, intracapsular,
intramuscular, intranasal, oral, buccal, pulmonary, rectal or vaginal. The
compositions can be formulated for parenteral administration to humans or
other
mammals in therapeutically effective amounts (e.g., amounts which eliminate or
reduce the patient's pathological condition) to provide therapy for the
neurological
diseases described above.
Two of the preferred administration routes will be by subcutaneous injection
(e.g., dissolved in 0.9% sodium chloride) or orally (in a capsule).
It will also be appreciated that on occasion it may desirable to directly
administer IGF-I compounds to the CNS of the patient. Again, this can be
achieved
by any appropriate direct administration route. Examples include
administration by
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
14
lateral cerebroventricular injection or through a surgically inserted shunt
into the
lateral cerebroventricle of the brain of the patient.
The calculation of the effective amount of cPG compounds to be administered
is within the skill of one of ordinary skill in the art, and will be routine
to those
persons skilled in the art. Needless to say, the final amount to be
administered will be
dependent upon the route of administration and upon the nature of the
neurological
disorder or condition that is to be treated. Preferably the cPG compound will
be
administered at between about 11.tg to 100mg of cPG compound per 100g of body
weight where the dose is administered centrally. A suitable dosage for
administration
of cPG may be, for example, at between 0.1mg to about 10mg per 100g of body
weight, or at between about lmg to about 5 mg per 100g body weight.
For inclusion in a medicament, cPG compounds can be obtained from a
suitable commercial source such as Bachem AG of Bubendorf, Switzerland.
Alternatively, cPG, cPG analogues and cPG mimetics can be directly synthesized
by
conventional methods such as the stepwise solid phase synthesis method of
Merryfield etal., 1963. Alternatively synthesis can involve in the use of
commercially available peptide synthesizers such as the Applied Biosystems
model
430A.
As a general proposition, the total pharmaceutically effective amount of the
cPG agonist compound administered parenterally per dose will be in a range
that can
be measured by a dose response curve. One can administer increasing amounts of
the
cPG agonist compound to the patient and check the serum levels of the patient
for
cPG. The amount of cPG agonist to be employed can be calculated on a molar
basis
based on these serum levels of cPG.
Specifically, one method for determining appropriate dosing of the compound
entails measuring cPG levels in a biological fluid such as a body or blood
fluid.
Measuring such levels can be done by any means, including RIA and ELISA. After
measuring cPG levels, the fluid is contacted with the compound using single or
multiple doses. After this contacting step, the cPG levels are re-measured in
the fluid.
If the fluid cPG levels have fallen by an amount sufficient to produce the
desired
efficacy for which the molecule is to be administered, then the dose of the
molecule
can be adjusted to produce maximal efficacy. This method can be carried out in
vitro
or in vivo. Preferably, this method is carried out in vivo, i.e., after the
fluid is
extracted from a mammal and the cPG levels measured, the compound herein is
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
administered to the mammal using single or multiple doses (that is, the
contacting step
is achieved by administration to a mammal) and then the cPG levels are
remeasured
from fluid extracted from the mammal.
The compound may also be suitably administered by a sustained-release
system. Suitable examples of sustained-release compositions include semi-
permeable
polymer matrices in the form of shaped articles, e.g., films, or
microcapsules.
Sustained-release matrices include polylactides (U.S. Pat. No. 3,773,919; EP
58,481),
copolymers of L-glutamic acid and gamma-ethyl-L-glutamate (Sidman et al.,
1983),
poly(2-hydroxyethyl methacrylate) (Langer et al, 1981), ethylene vinyl acetate
(Langer et al., supra), or poly-D-(-)-3-hydroxybutyric acid (EP 133,988).
Sustained-
release compositions also include a liposomally entrapped compound. Liposomes
containing the compound are prepared by methods known per se: DE Patent
3,218,121; Epstein et al., 1985; Hwang et al., 1980; EP Patent 52,322; EP
Patent
36,676; EP Patent 88,046; EP Patent 143,949; EP Patent 142,641; Japanese Pat.
Appin. 83-118008; U.S. Pat. Nos. 4,485,045 and 4,544,545; and EP 102,324.
Ordinarily, the liposomes are of the small (from or about 200 to 800
Angstroms)
unilamellar type in which the lipid content is greater than about 30 mol.
percent cholesterol, the selected proportion being adjusted for the most
efficacious
therapy.
PEGylated peptides having a longer life can also be employed, based on, e.g.,
the conjugate technology described in WO 95/32003 published November 30,
1995.
If parenteral administration is preferred, the compound is formulated
generally
by mixing each at the desired concentration, in a unit dosage injectable form
(solution, suspension, or emulsion), with a pharmaceutically, or parenterally,
acceptable carrier, i.e., one that is non-toxic to recipients at the dosages
and
concentrations employed and is compatible with other ingredients of the
formulation.
Generally, the formulations are prepared by contacting the compound with
liquid carriers or finely divided solid carriers or both. Then, if necessary,
the product
is shaped into the desired formulation. Preferably the carrier is a parenteral
carrier,
more preferably a solution that is isotonic with the blood of the recipient.
Examples
of such carrier vehicles include water, saline, Ringer's solution, a buffered
solution,
and dextrose solution. Non-aqueous vehicles such as fixed oils and ethyl
oleate may
also be used.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
16
The carrier may additionally contain additives such as substances that enhance
isotonicity and chemical stability. Such materials are non-toxic to recipients
at the
dosages and concentrations employed, and include buffers such as phosphate,
citrate,
succinate, acetic acid, and other organic acids or their salts; antioxidants
such as
ascorbic acid; low molecular weight (less than about ten residues)
polypeptides, e.g.,
polyarginine or tripeptides; proteins, such as serum albumin, gelatin, or
immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; glycine;
amino
acids such as glutamic acid, aspartic acid, histidine, or arginine;
monosaccharides,
disaccharides, and other carbohydrates including cellulose or its derivatives,
glucose,
mannose, trehalose, or dextrins; chelating agents such as EDTA; sugar alcohols
such
as mannitol or sorbitol; counter-ions such as sodium; non-ionic surfactants
such as
polysorbates, poloxamers, or polyethylene glycol (PEG); and/or neutral salts,
e.g.,
NaC1, KC1, MgC1<sub>2</sub>, CaC1<sub>2</sub>, etc.
The cPG compound is typically formulated in such vehicles at a pH of
between about 5.5 to 8. Typical adjuvants which may be incorporated into
tablets,
capsules, and the like are a binder such as acacia, corn starch, or gelatin;
an excipient
such as microcrystalline cellulose; a disintegrating agent like corn starch or
alginic
acid; a lubricant such as magnesium stearate; a sweetening agent such as
sucrose or
lactose; a flavoring agent such as peppermint, wintergreen, or cherry. When
the
dosage form is a capsule, in addition to the above materials, it may also
contain a
liquid carrier such as a fatty oil. Other materials of various types may be
used as
coatings or as modifiers of the physical form of the dosage unit. A syrup or
elixir
may contain the active compound, a sweetener such as sucrose, preservatives
like
propyl paraben, a coloring agent, and a flavoring agent such as cherry.
Sterile
compositions for injection can be formulated according to conventional
pharmaceutical practice. For example, dissolution or suspension of the active
compound in a vehicle such as water or naturally occurring vegetable oil like
sesame,
peanut, or cottonseed oil or a synthetic fatty vehicle like ethyl oleate or
the like may
be desired. Buffers, preservatives, antioxidants, and the like can be
incorporated
according to accepted pharmaceutical practice.
The compound to be used for therapeutic administration must be sterile.
Sterility is readily accomplished by filtration through sterile filtration
membranes
(e.g., 0.2 micron membranes). Therapeutic compositions generally are placed
into a
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
17
container having a sterile access port, for example, an intravenous solution
bag or vial
having a stopper pierceable by a hypodermic injection needle.
The compound ordinarily will be stored in unit or multi-dose containers, for
example, sealed glass ampules or vials, as an aqueous solution or as a
lyophilized
formulation for reconstitution. As an example of a lyophilized formulation, 10-
mL
vials are filled with 5 ml of sterile-filtered 1% (w/v) aqueous solution of
compound,
and the resulting mixture is lyophilized. The infusion solution is prepared by
reconstituting the lyophilized compound using bacteriostatic Water-for-
Injection.
Combination therapy with the cPG agonist compound herein and one or more other
appropriate reagents that increase total cPG in the blood or enhance the
effect of the
cPG agonist is also contemplated. These reagents generally allow the cPG
agonist
compound herein to release the generated cPG.
In addition, it is envisaged using gene therapy for treating a mammal, using
nucleic acid encoding the cPG agonist compound, if it is a peptide. Generally,
gene
therapy is used to increase (or overexpress) cPG levels in the mammal. Nucleic
acids,
which encode the cPG agonist peptide can be used for this purpose. Once the
amino
acid sequence is known, one can generate several nucleic acid molecules using
the
degeneracy of the genetic code, and select which to use for gene therapy.
There are two major approaches to getting the nucleic acid (optionally
contained in a vector) into the patient's cells for purposes of gene therapy:
in vivo and
ex vivo. For in vivo delivery, the nucleic acid is injected directly into the
patient,
usually at the site where the cPG agonist compound is required. For ex vivo
treatment, the patient's cells are removed, the nucleic acid is introduced
into these
isolated cells, and the modified cells are administered to the patient either
directly or,
for example, encapsulated within porous membranes which are implanted into the
patient. See, e.g., U.S. Pat. Nos. 4,892,538 and 5,283,187.
There are a variety of techniques available for introducing nucleic acids into
viable cells. The techniques vary depending upon whether the nucleic acid is
transferred into cultured cells in vitro, or in vivo in the cells of the
intended host.
Techniques suitable for the transfer of nucleic acid into mammalian cells in
vitro
include the use of liposomes, electroporation, microinjection, cell fusion,
DEAE-
dextran, the calcium phosphate precipitation method, etc. A commonly used
vector
for ex vivo delivery of the gene is a retrovirus.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
18
The currently preferred in vivo nucleic acid transfer techniques include
transfection with viral vectors (such as adenovirus, Herpes simplex I virus,
or adeno-
associated virus) and lipid-based systems (useful lipids for lipid-mediated
transfer of
the gene are DOTMA, DOPE and DC-Chol, for example). In some situations it is
desirable to provide the nucleic acid source with an agent that targets the
target cells,
such as an antibody specific for a cell-surface membrane protein or the target
cell, a
ligand for a receptor on the target cell, etc. Where liposomes are employed,
proteins
which bind to a cell-surface membrane protein associated with endocytosis may
be
used for targeting and/or to facilitate uptake, e.g., capsid proteins or
fragments thereof
tropic for a particular cell type, antibodies for proteins which undergo
internalization
in cycling, and proteins that target intracellular localization and enhance
intracellular
half-life. The technique of receptor-mediated endocytosis is described, for
example,
by Wu eta!, 1987; Wagner et al., 1990). For review of the currently known gene
marking and gene therapy protocols, see Anderson 1992. See also WO 93/25673
and
the references cited therein.
Kits are also contemplated for this invention. A typical kit would comprise a
container, preferably a vial, for the cPG agonist compound formulation
comprising
cPG agonist compound in a pharmaceutically acceptable buffer and instructions,
such
as a product insert or label, directing the user to utilize the pharmaceutical
formulation.
EXAMPLES
Experiment 1
Cyclic PG prevents glutamate induced neuronal death in vitro in a dose related
manner.
Materials and Methods:
Cerebellar Cell Culture Preparing and coating of cover slips
Ten coverslips were placed into a large petri dish and washed in 70% alcohol
for 5 minutes, then washed with Millipore H20. The coverslips were air dried,
then
coated with Poly-D-Lysine (1 mg/ml stock solution in PBS, 90-100 I) and
incubated
for 2 hours at 34 C.
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
19
Extraction
Postnatal day 4 Wistar rats were used for the study. Rats were placed in ice
for 1 minute, the heads were decapitated and the cerebellum removed on ice.
Cerebellum tissue was placed in 1 ml of 0.65% glucose supplemented PBS (10 1
65% stock D (+)glucose/lml PBS) in a large petri dish, chopped up into smaller
sections and triturate with a lml insulin syringe via a 23 G (0.4mm) needle,
and then
squirted back into the glucose solution on the large petri dish. The tissue
was sieved
through (125 m pore size gaze) and centrifuged (2minutes at 60g) two times for
a
medium exchange into serum-free BSA-supplemented START V medium
(Biochrom). The second centrifugation step was done with lml of START V
medium. The microexplants were reconstituted into 500 1 of START V medium and
put on ice
Cultivation and fixation of cerebellar cells
Two hours after PDL-coating, the slides were washed with Millipore H20 and
air dried. Each slide was placed into a small 35 mm petri dish and 40111 of
START
V/cell suspension added. The tissue was incubated for 2 hours at 34 C
(settlement
period). START V-medium (1m1) was then added to the petri dish and cultivated
at
34 C/ 5% CO2/100% humidity for 48 hours. Cells were rinsed in PBS and then
fixed
for 2-3 minutes in increasing concentrations of paraformaldehyde (500 I of
0.4%
PFA was applied; then 1.2% PFA; then 3% PFA and finally 4% PFA - all fixation
solutions contain 0.2% glutardialdehyde). Finally, the microexplants were
rinsed in
PBS.
Drug Application
I of toxin (L-glutamate-100 mM in Millipore water) was applied
simultaneously with cPG (from bachem, 10 mM stock prepared in PBS and diluted
to
final concentrations between 1-100 nM) for Study 1. A delay in administration
of
cPG at 6 hours after glutamate treatment was performed for Study 2.
Result:
Study 1: Glutamate treatment resulted in 85% loss of cerebellum neurons.
Cyclic PG significantly reduced the glutamate induced neuronal death in a dose
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
response manner when administered simultaneously with glutamate (Figure 3).
The
treatments with lower doses of cPG (10-100nM) showed significant recovery from
glutamate-induced neurotoxicity.
Study 2: Cyclic PG showed a significantly recovery from glutamate induced
neurotoxicity in a dose range of 1-100nM when given 6 hours after the
glutamate
treatment compared to the vehicle treated group (Figure 4).
A further lower dose of cPG also showed a significant increase in neuron
number compared to the normal control group, suggest a role for cPG in
neuronal
proliferation and differentiation.
Conclusions
Excessive glutamate can cause neuronal excitotoxicity by active NMDA
receptors. Cyclic PG completely prevented the glutamate-induced neurotoxicity,
when given either immediately or 6 hours after the glutamate treatment by
acting as a
direct or indirect NMDA antagonist. Given that cPG can agonise mG1u2/3
receptor,
which can inhibit NMDA activity. GPE, the pre- hormone for cPG has been shown
to
be partial NMDA receptor agonist in promoting pCREB, probably due to its
antagonistic effect on mG1u2/3 receptors. CPG may be involved in preventing
neurons undergoing apoptosis because cPG appeared to be still effective as a
delayed
treatment, and promoted the neuronal proliferation.
Experiment 2
Effects of cPG after 6-0HDA induced nigral-striatal lesion.
Materials and Methods
Twenty male Wistar rats (280-310g) were used. After exposing the skull, 6-
OHDA (8pig in a base of 21.1.10.9% saline containing 1% ascorbic acid) was
administered into the right medial forebrain bundle (MFB) using co-ordinates
AP
+4.7mm, R 1.6inmv ¨8inm under 3% halothane anaesthesia. 6-0HDA was injected
through a 25G needle connected via a polyethylene catheter to a 100 1 Hamilton
syringe. The 6-0HDA was infused by a microdialysis infusion pump at a rate of
0.5 1/min. The needle was left in the brain for a further 3 minutes before
being
CA 02466701 2004-05-07
WO 03/041655
PCT/US02/36639
21
slowly withdrawn. The skin was sutured with 2.0 silk and the rats were allowed
to
recover from anaesthesia. The rats were housed in a holding room with free
access to
food and water at all times except during behavioural testing.
Cyclic PG was dissolved in saline. Four different doses of cPG (0, 0.1 0.5
lmg/kg, Bachem) were administered intraperitoneally 2h post lesion.
At 7 days post-lesion, rats were injected with 0.1mg/kg apomorphine and the
number of contralateral rotations/hour was recorded and calculated using a
computerised Rotameter (St Diego Instruments). Experimenter was blinded from
the
treatment groups.
Result:
The group treated with lmg cPG (n=5, 154 64.1) showed a trend toward a
reduction in the number of rotations compared to the vehicle treated group
(n=5,
290.08 18.9) indicating a role for cPG in improving functional recovery in 6-
0HDA
induced nigrostriatal injury. (Figure 5)
Conclusions
Cyclic PG improved the functional recovery after 6-0HDA induced nigral-
striatal lesions in a dose related manner.
The highest dose tested (lmg/kg) reduced functional deficit (apomorphine
induced rotations) by 47% (ns). This data suggested cPG has potential as a
treatment
for Parkinson's disease.
ADVANTAGES
Some advantages offered by the present invention, especially over IGF-I and
the GPE include:
The active ingredients are easy to synthesise either in vitro or by other
means
such as recombinant techniques.
The dipeptide as a small molecule can diffuse readily through the body and
between compartments (e.g. the blood-brain barrier, and mucous membranes),
aiding
in the choice of methods for its administration and its ability to reach sites
where
injury has occurred.
CA 02466701 2011-12-12
?2
cPG is a very stable molecule and is unlikely to present a challenge to the
immune system, so it may be administered over extended periods and it may be
administered prophylactically.
With their antagonistic and agonistic effects, GPE/cPG, the present invention
provides a novel therapeutic method for preventing brain injury and
degenerative
diseases by regulating mGluRs particularly 2/3 leading to long-term benefits
of brain
recovery.
With a role in regulating IGF-1 induction, cPG will provide further
neuroprotection with less potential for growth side-effects.
While various embodiments of the present invention have been described
above, it should be understood that they have been presented by way of
examples
only, and not limitation. It will be understood by those skilled in the art
that various
changes in form and detail may be made therein without departing from the
spirit and
scope of the present invention as defined in the appended claims. Thus, the
breadth
and scope of the present invention should not be limited by any of the above-
described exemplary embodiments, but should be defined in accordance with the
following claims and their equivalents.
=