Note: Descriptions are shown in the official language in which they were submitted.
CA 02469882 2009-02-10
NOVEL STABLE CRYSTALS OF SUBSTITUTED PHENYLPROPIONIC ACID
DERIVATIVE AND PROCESS FOR PRODUCING THE SAME
SPECIFICATION
Novel stable crystals of phenylpropanoic acid derivative and
process for the preparation thereof
Technical field
The present invention relates to a stable crystal form of
(S)-2-[[3-[N-[4-[(4-fluorophenoxy)phenyl]methyl]-carbamoyl]-4-
methoxyphenyl]methyl]butanoic acid (hereinafter referred to as
compound (I)) represented by a formula (I)
O
F ao"'~ N COOH
H
Me0
and process for the preparation thereof.
Background technology
Compound (I) is a useful compound for the therapy of the
abnormality of lipid metabolism as an agonist of human peroxisome
proliferator-activated receptor(PPAR),in particular, as an agonist
of human PPARa isoform, and has been prepared through a process
disclosed in Jpn. Kokai Tokkyo Koho JP 2001-55367.
Preparation of the compound (I) on an industrial scale as a
medicinal drug lies in the finding of homogeneous
crystals with excellent stability and the establishment
of a preparative process thereof.
-1-
CA 02469882 2005-10-11
Disclosure of the invention
When having advanced the research and development on the physical
properties and preparative process of compound (I), the inventors
have found that new type crystals more stable and richer in the
homogeneity being different from the crystals (referred to as old
type crystals) obtainable through the conventional process (Jpn.
Kokai Tokkyo Koho JP 2001-55367) can be obtained on an industrial
scale, leading to the completion of the invention. Namely, it has
been ascertained that, if recrystallizing the crude crystals of
compound (I) prepared according to the conventional process from
a suitable solvent, then stable and homogeneous crystals (referred
to as new type crystals) that exhibit higher melting point than that
of old type crystals can be obtained. Furthermore, it has become
clear that, if purifying these new type crystals by acid-base
treatment, new type crystals in the state of containing no residual
solvent in the crystals can be taken out. Moreover, it has also been
identified that, by further recrystallizing the old type crystals
obtained through the conventional process with a suitable solvent
as described above, it is possible to convert them into the inventive
new type crystals. In this way, the new type crystals of compound
(I) with more preferable properties as original powder of medicinal
drug and the preparative process have been found, leading to the
completion of the invention.
The new type crystals of compound (I) are crystals characterized
by exhibiting the diffraction angles (20) at at least 17.7 , 19.0
-2-
CA 02469882 2005-08-26
and 24 .1 in the X-ray powder diffraction. Moreover, they have higher
melting point than that of conventional crystals.
The new type crystals of compound (I) can usually be obtained
in good reproducibility, if recrystallizing the crude crystals
obtained after completion of the reaction from a suitable solvent
and successively purifying by acid-base treatment.
As the solvents to be used for recrystallization, lower alcohols
such as ethanol and water-containing lower alcohols can be mentioned.
Preferable solvent is water-containing ethanol or water-containing
isopropyl alcohol, and it may also be safe to dissolve previously
into alcohol and then add water. The acid-base treatment is performed
by dissolving the new type crystals obtained by recrystallization
into a common inorganic base, preferably aqueous solution of sodium
hydroxide or potassium hydroxide, neutralizing with a common acid,
preferably hydrochloric acid under warming, preferably at 50 C or
so, and filtering and washing the precipitated crystals.
The new type crystals obtainable according to the invention have
no hygroscopicity, make stable supply possible in terms of
preparation, and leave no residual organic solvent. Such new type
crystals are very advantageous for the industrial production of
compound (I).
According to an aspect of the present invention there
is provided Crystals of (S) -2- [ [3- [N- [4- [ (4-fluorophenoxy)
phenyl ] methyl ] carbamoyl ]-4-methoxyphenyl ] methyl ] butanoic ac.;.d
(compound (I)), exhibiting diffraction angles (20) at
-3-
CA 02469882 2008-02-27
least 17.7 , 19.00 and 24.1 in X-ray powder diffraction.
According to a further aspect of the present invention
there is provided a process for preparing the crystals as
defined above, by recrystallizing the compound (I) from a
suitable solvent.
According to another aspect of the present invention there
is provided a use of the crystals as defined above, for
therapy of abnormality of lipid metabolism as an agonist of
human peroxisome proliferator-activated receptor.
According to a still further aspect of the present
invention there is provided a use of the crystals as defined
above, as an agonist of human peroxisome proliferator-
activated receptor.
According to another aspect of the present invention there
is provided a use of the crystals as defined above, for the
manufacture of a medicament for therapy of abnormality of
lipid metabolism as an agonist of human peroxisome
proliferator-activated receptor.
According to a further aspect of the present invention
there is provided a use of the crystals as defined above,
for the manufacture of a medicament as an agonist of human
peroxisome proliferator-activated receptor.
According to yet another aspect of the present invention
there is provided a pharmaceutical composition for therapy
of abnormality of lipid metabolism as an agonist of human
peroxisome proliferator-activated receptor comprising
crystals of as defined above, in admixture with a
pharmaceutically acceptable diluent or carrier.
According to a still further aspect of the present
invention there is provided a pharmaceutical composition as
an agonist of human peroxisome proliferator-activated
receptor comprising crystals of as defined above, in
- 3a -
CA 02469882 2005-08-26
admixture with a pharmaceutically acceptable diluent or
carrier.
According to a further aspect of the present invention
there is provided a commercial package comprising the
crystal as defined above, together with instructions for its
use for therapy of abnormality of lipid metabolism as an
agonist of human peroxisome proliferator-activated receptor.
According to a still further aspect of the present
invention there is provided a commercial package comprising
the crystal as defined above, together with instructions for
its use as an agonist of human peroxisome proliferator-
activated receptor.
Best embodiment to put the invention into practice.
In following, the invention will be illustrated in more
detail showing examples, but the invention is not confined
to these examples at any rate.
Brief description of the drawings
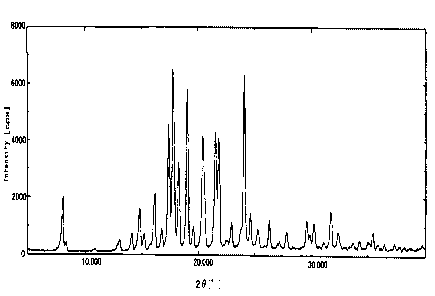
Fig. 1 The X-ray powder diffraction diagram of the inventive new
type crystals.
Fig. 2 The X-ray powder diffraction diagram of the old type
crystals.
Fig. 3 The thermometry diagram of the inventive new type
crystals.
Fig. 4 The thermometry diagram of the old type crystals.
-3b-
CA 02469882 2005-10-11
<Example 1>
Preparation of new type crystals of (S)-2-[[3-[N-[4-[(4-
fluorophenoxy)phenyl]methyl]carbamoyl]-4-methoxyphenyl]methyl]
butanoic acid (compound (I))
To a solution of 104g (0.207mo1) of benzyl 5-[3-[4-(R)-
benzyl-2-oxo-1,3-oxazolidine-3-yl]-2-(S)-ethyl-3-oxopropyl] -2-
methoxybenzoate (Jpn. Kokai Tokkyo Koho JP 2001-55367 (Example
165)) in 728m1 of ethyl acetate (AcOEt) were added 6.93g of 10%
palladium on carbon ( Pd-C )( dry article), and the mixture was stirred
for 5 hours at inner temperature of 35 C under an atmosphere of
hydrogen. After the catalyst was filtered off, the reaction mixture
was concentrated under reduced pressure to obtain colorless oily
5-[3-[4-(R)-benzyl-2-oxo-1,3-oxazolidine-3-yl]-2-(S)-ethyl-3-
oxopropyl]-2-methoxybenzoic acid. This oily product was dissolved
into 413mL of dehydrated dimethylformamide (DMF) and, after 62.9g
(0.622mo1) of triethylamine (Et3N) were added, the mixture was
stirred under cooling with ice water. At inner temperature of 5 C,
22.5g (0.207mo1) of ethyl chloroformate were added dropwise (inner
temperature increased to 8 C) and thereafter the mixture was stirred
for 1 hour. Then, 55.2g (0.218mo1) of [4-(4-
fluorophenoxy)phenyl]methylamine hydrochloride were put in at inner
temperature of 6 C and the mixture was stirred for 1 hour. The reaction
mixture was poured into 826mL of water, which was extracted twice
(516mL, 309mL) with ethyl acetate. The ethyl acetate layers were
combined, washed with 207mL of lmol/L aqueous solution of sodium
hydroxide (NaOH aq.), 405mL of water and 36mL of saturated brine
-4-
CA 02469882 2004-06-10
in sequence, then dried over anhydrous sodium sulfate, and solvent
was distilled off under reduced pressure. The product was dried
(vacuum pump) for 2 hours at room temperature under reduced pressure
to obtain 126g of pale yellow oily N-[4-(4-
fluorophenoxy)phenyl]methyl-5-[3-[4-(R)-benzyl-2-oxo-1,3-
oxazolidine-3-yl]-2-(S)-ethyl-3-oxopropyl]-2-methoxybenzamide.
This oily product was dissolved into 806mL of tetrahydrofuran (THF)
and, after 202mL of purified water were added, the mixture was stirred
under cooling with ice water. To this solution were added dropwise
9lmL (0.829mo1) of 31% aqueous hydrogen peroxide (H202) over 10
minutes at inner temperature of 8 C. Following this, a solution of
13.9g(0.332mo1)of lithium hydroxide monohydrate(LiOH=H20)in556mL
of purified water were added dropwise over 80 minutes at inner
temperature of 8 to 10 C and the mixture was stirred further for 1
hour. After a solution of 86.2g of sodium hydrogensulfite (NaHS03 )
in 432mL of purified water were added dropwise over 20 minutes, the
mixture was stirred for 1 hour. After allowed to stand statically,
the aqueous layer was separated, which was extracted with 259mL of
ethyl acetate (pH value of aqueous layer 1. 33 : pH meter). The organic
layers were combined and solvent was distilled off under reduced
pressure. The residue was dissolved into 518mL of ethyl acetate and,
after washing with 415mL of cold water, the solution was extracted
twice with cold aqueous solution of alkali prepared with 570mL of
ice water and 570mL of lmol/L NaOH aq. The aqueous layers were combined
and 273mL of 6mol/L hydrochloric acid (HC1 aq.) were added, which
was then extracted with 829mL and 311mL of ethyl acetate. The ethyl
-5-
CA 02469882 2004-06-10
acetate layers were combined, washed with270mL of saturated aqueous
solution of sodium hydrogencarbonate (pH value of washed liquid:
7.38, pH meter) and 4lmL of saturated brine in sequence, then dried
over anhydrous sodium sulfate and concentrated under reduced
pressure. To the residue were added 207mL of ethyl acetate and 2lmL
of isopropyl alcohol (IPA). After dissolved by heating, 311mL of
hexane were added. The mixture was stirred at room temperature under
cooling by allowing to stand and stirred at inner temperature of
30 C under cooling with ice water. After stirring for 1 hour at inner
temperature of below 10 C, the precipitated crystals were collected
by filtration at inner temperature of 6 C. After washing with 173mL
of mixed solution of hexane/ethyl acetate (3:1), the crystals were
dried for 3 hours at 50 C by blower to obtain 75.Og of crude crystals
of compound (I). After dissolved 75. Og of crude crystals into 483mL
of ethanol (EtOH) by heating, 257mL of purified water were added.
After stirring under cooling with water, the solution was cooled
with ice water at inner temperature of 25 C and stirred for 1 hour
at inner temperature of below 10 C. The precipitated crystals were
collected by filtration at inner temperature of 4 C and, after washing
with 357mL of 25% EtOH/water, the crystals were dried for 1 hour
by blower. These were dried at 50 C until they became constant weight
to obtain 73.3g (yield 78%) of the compound of the present
application.
Furthermore, 73.3g of these crystals were suspended into 344mL
of purified water and dissolved by adding 390mL of 0.5mol/L NaOH
aq., followed by filtration. The filtrate was heated to inner
-6-
CA 02469882 2004-06-10
temperature of 45 to 50 C and, after 89mL of 0.5mol/L HC1 aq. were
added, the solution was stirred for 5 minutes. Following this, 444mL
of 0.5mol/L HC1 aq. were added dropwise and then the solution was
cooled with water to make inner temperature 25 C. The precipitated
crystals were filtered and, after washing with 733mL of purified
water, they were dried for 8 hours at 65 C by blower to obtain 72.6g
(yield 78%) of the new type crystals of compound (I) as white
crystalline powder.
Melting point: 132-133 C (hot plate method).
Mass analysis: EI-MS(m/z): 235, 451(M+, base peak).
Angle of rotation: [a]D25 +29 (C=1.0, MeCN).
Elemental analysis: Anal. Calcd. for C26H26FN05(MW 451.49):
C, 69.17; H, 5.80; N, 3.10.
Found: C, 69.03; H, 5.84; N, 3.11.
NMR spectra: 1H-NMR(DMSO-d6, ppm, 400MHz)6: 0.85(3H, t, J=7.8Hz),
1.41-1.53(2H, m), 2.38---2.45(1H, m), 2.65(1H, dd, J=6.4, 13.7Hz),
2.76(1H, dd, J=8.3, 13.7Hz), 3.85(3H, s), 4.45(2H, d, J=6.4Hz),
6. 93-6 . 97 ( 2H, m), 7. 01-7 . 05 ( 3H, m), 7.16-7 .22 ( 2H, m), 7. 27 (1H,
dd,
J=2.4, 8.8Hz), 7.33(2H, d, J=8.3Hz), 7.57(1H, d, J=2.4Hz), 8.66(1H,
t, J=6.4Hz), 12.09(1H, s).
Purity test: HPLC relative purity ; 99.9% [HPLC conditions
measuring wavelength ; 210nm, column ; Inertsil ODS-3 (4.6mmID x
250mm), precolumn ; Inertsil ODS-3 ( 4. OmmID x 10mm), mobile phase ;
MeCN/diluted phosphoric acid (1-1000)=60:40, column temperature ;
30 C, flow rate ; 1.OmL/min, injection level(solvent)
41Ag/2 L(MeCN)].
-7-
CA 02469882 2005-10-11
Optical purity test : HPLC relative purity ; 100%ee [HPLC conditions :
measuring wavelength ; 210nm, column; CHIRALPAK AD-RH (4.6mmID x
150mm), precolumn ; Inertsil ODS-3 ( 4. OmmID x 10mm), mobile phase;
diluted phosphoric acid (1-1000) : acetonitrile (MeCN)=55:45,
column temperature ; 40 C, flow rate ; 1.OmL/min, injection
level(solvent); 4Etg/2EtL(MeCN)].
<Example 2>
Preparation of new type crystals of (S)-2-[[3-[N-[4-[(4-
fluorophenoxy)phenyl]methyl]carbamoyl]-4-methoxyphenyl]methyl]
butanoic acid (compound (I))
To a solution of 50.2g (100mmo1) of benzyl 5-[3-[4-(R)-
benzyl-2-oxo-1,3-oxazolidine-3-yl]-2-(S)-ethyl-3-oxopropyl] -2-
methoxybenzoate (Jpn. Kokai Tokkyo Koho JP 2001-55367, Example
165)in 351mL of ethyl acetate (AcOEt) were added 3.34g of 10%
palladium on carbon ( Pd-C )( dry article), and the mixture was stirred
for 2 hours at 35 C under an atmosphere of hydrogen. After the catalyst
was filtered off,the reaction mixture was concentrated under reduced
pressure to obtain colorless oily product (41.2g). Triethylamine
( 30 . 4g, 300mmol) was added to a solution of the oily product (41.2g)
obtained in 200mL of dehydrated N,N-dimethylformamide and, after
ethyl chloroformate (10. OmL, 105mmo1) was added at inner temperature
of 5 C under cooling with ice water, the mixture was stirred for 1
hour.[4-(4-Fluorophenoxy)phenyl]methylamine hydrochloride (26.6g,
105mmo1) was put in the mixture, which was stirred for 1 hour. To
the reaction mixture were added 400mL of water, and the mixture was
-8-
CA 02469882 2004-06-10
extracted with ethyl acetate (200mL x twice ). The ethyl acetate layer
was washed with lmol/L aqueous solution of sodium hydroxide(100mL),
water (200mL) and saturated brine (100mL) in sequence, then dried
over anhydrous sodium sulfate and concentrated under reduced
pressure to obtain yellow oily product (61.1g).
After 31% aqueous hydrogen peroxide ( 43 . 9mL, 400mmol) was added
to a solution of the oily product (61. lg ) obtained in 488mL of mixed
solution of tetrahydrofuran-water (4:1) at inner temperature of 8 C
under cooling with ice water, a solution of lithium hydroxide
monohydrate (6.71g, 160mmo1) in 268mL of water was added dropwise
over 20 minutes at inner temperature of 8 C, and thereafter the
mixture was stirred for 2 hours. A solution of sodium hydrogensulfite
(41.6g) in water ( 208mL ) was added dropwise to the reaction mixture
and the mixture was stirred for 1 hour, which was then allowed to
stand statically. After the organic layer was separated and
concentrated under reduced pressure, the residue was dissolved into
ethyl acetate (500mL), which was extracted with 0.5mol/L aqueous
solution of sodium hydroxide (550mL x twice). The aqueous layers
were combined and, after 6mol/L hydrochloric acid (125mL) was added
(pH value1.91),thesolution was extracted with ethyl acetate (400mL,
150mL).The ethyl acetate layers were combined, washed with saturated
aqueous solution of sodium hydrogencarbonate (127mL, pH value of
washed liquid 6.95) and saturated brine (200mL) in sequence, then
dried over anhydrous sodium sulfate and concentrated under reduced
pressure. Ethyl acetate ( 50mL ) and IPA ( 5mL ) were added to the solids
(50.2g) obtained and, after dissolved by refluxing, hexane (76mL)
-9-
CA 02469882 2004-06-10
was added and the inner temperature was made to be 65 C. The solution
was stirred at room temperature under cooling by allowing to stand
(crystal precipitation started at inner temperature of 59 C).
Thereafter, the solution was cooled with ice water at inner
temperature of 30 C and stirred for 1 hour at inner temperature of
below 10 C. The precipitated crystals were collected by filtration
(inner temperature 6 C) and, after washing with a mixed solution
(126mL) of hexane/ethyl acetate (3:1), the crystals were dried for
hours at 50 C by blower to obtain 35.Og of crude crystals of compound
(I). Ethanol ( 228mL ) was added to 35 . Og of crude crystals obtained
and, after dissolved by heating (inner temperature 50 C), the
solution was filtered and purified water (121mL) was added, followed
by stirring under cooling with water (crystal precipitation started
at inner temperature of: 38 C). After crystal precipitation, the
suspension was stirred for 1 hour, cooled with ice water at inner
temperature of 25 C, and stirred for 1 hour at inner temperature of
below 10 C. The precipit:ated crystals were collected by filtration
(inner temperature 6 C) and, after washing with 25% ethanol/water
(126mL), the crystals were dried for 13.5 hours at 50 C by blower
to obtain 34.7g (77%) of the new type crystals of compound (I) as
white needle-like crystals.
Mp 131 C (hot plate method).
[cx]D24 = +30 (c=1.0, acetonitrile).
Anal. Calcd. for C26H26FN05(MW 451.49): C, 69.17; H, 5.80; N, 3.10.
Found: C, 69.19; H, 5.64; N, 3.34.
EI-MS m/z: 235, 451[M+].
- 10-
CA 02469882 2005-10-11
1H-NMR(DMSO-d6, 400MHz)S: 0.86(3H, t, J= 7.3Hz), 1.42-1.54(2H, m),
2.39-2.46(1H, m), 2.65(1H, dd, J=5.9, 13.7Hz), 2.76(1H, dd, J=8.3,
13.7Hz), 3.85(3H, s), 4.45(2H, d, J=6.4Hz), 6.95(2H, d, J=8.8Hz),
7.00-7.05(3H, m), 7.17-7.23(2H, m), 7.27(1H, dd, J=2.4, 8.3Hz),
7.33(2H, d, J=8.3Hz), 7.58(1H, d, J=2.OHz), 8.66(1H, t, J=5.9Hz),
12.08(1H, s).
<Example 3> Conversion of crystal form
Into 9.9mL of ethanol were dissolved by heating (45 C) 1.53g of
the old type crystals (melting point 95-V96 C) of compound (I) obtained
through the conventional process (Example 174 of Jpn. Kokai
Tokkyo Koho JP 2001-55367). To this were added 5.4mL of water, the
solution was cooled by allowing to stand, and then cooled (50C) with
ice water. The precipitated crystals were filtered and washed with
7. 5mL of 25% ethanol/water. These were dried for 2 hours at 50 C under
reduced pressure to obtain 1.48g (96.7%) of the new type crystals
of compound (I).
Melting point: 132--133 C (hot plate method)
<Testing example 1> Measurement of X-ray powder diffraction
The X-ray powder diffraction was measured by CuKa line, employing
wide-angle goniometer of X-ray diffraction apparatus RINT2200 from
Rigaku Co. The diffraction angle (20) and the relative intensity
(cps) for the crystals of the compound in said examples are shown
in Fig. 1. The X-ray powder diffraction pattern for the crystals
(old type crystals) obtained through the conventional process is
- 11 -
CA 02469882 2004-06-10
shown in Fig. 2. As a result, the crystals obtained in the examples
of the invention exhibit a characteristic diffraction pattern at
at least 20=17.7 , 19.0 and 24.1 , which differs from that of the
conventional crystals.
<Testing example 2> Differential thermal analysis (DTA) and
thermogravimetry (TG)
The thermal stability of crystals was compared by using apparatus
for thermal analysis (SII: TG/DTA6200). Fig. 3 shows a chart of
thermal analysis and thermogravimetry of the new type crystals of
compound (I) and Fig. 4 those of the old type crystals. With the
new type crystals, the endothermic phenomenon due to transition was
seen from 131.4 C and the endothermic peak due to melting was shown
at 132.0 C. On the other hand, the old type crystals showed the
endothermic peak at 109.7 C. Moreover, in all cases, no weight change
was recognized. From this fact, it has become clear that the new
type crystals are thermally stable crystals.
Utilizability in the industry
It is possible to obtain stable and homogeneous new type crystals
with no residual organic solvent by recrystallizing the crystals
of compound (I) obtainable through the conventional process from
a lower alcoholic suitable solvent, thus obtaining new type crystals,
and further by performing acid-base treatment. The homogeneous and
stable new type crystals to be provided according to the invention
have no hygroscopicity and make a stable supply possible in terms
- 12-
CA 02469882 2005-08-26
of preparation, which is very advantageous in the industrial
production of compound (I).
-13-