Note: Descriptions are shown in the official language in which they were submitted.
CA 02474836 2004-07-16
MASS SPECTROMETER
The present invention relates to an ion source, a
mass spectrometer, an Electrospray Ionisation/
Atmospheric Pressure Chemical Ionisation ("ESI/APCI")
ion source and a method of producing ions. The
preferred embodiment relates to an Atmospheric Pressure
Chemical Ionization ("APCI") ion source.
Chemical ionization involves the transfer of
charged species from reagent ions to analyte molecules
to produce analyte ions that can be subsequently mass
analysed. The charged species most commonly formed in
positive ion mode is the adduct between the analyte
molecule and positive hydrogen ions (H+).
Chemical ionization conducted at atmospheric
pressure is known as Atmospheric Pressure Chemical
Ionization ("APCI"). A sample containing analyte
material is typically delivered to an Atmospheric
Pressure Chemical Ionization ion source as a solution.
The solution containing the analyte is then sprayed into
a heated tube through which a nebulising gas is also
directed. The nebulising gas causes the sprayed
solution to be nebulised into fine droplets which then
impact the inner wall of the heated tube and are
converted into the gas phase. As the solution is
converted into the gas phase the analyte molecules
become desolvated. Hot gas comprising mobile phase
solvents, microdroplets and desolvated analyte molecules
then exit the heated tube and expand towards a corona
needle. The analyte molecules are then ionised by
chemical ionization with reagent ions produced by a
corona discharge in the presence of a reagent gas. In
particular, analyte molecules are ionised by gas phase
CA 02474836 2004-07-16
- 2 -
ion-molecule reactions between reagent ions and analyte
molecules.
In this conventional arrangement, analytes that
exit the heated tube in the form of neutral gaseous
molecules, ions or charged micro-droplets directly pass
the corona needle prior to entering the vacuum section
of a mass spectrometer via an ion sampling orifice.
Only a relatively small proportion of the analyte ions
formed at atmospheric pressure are actually drawn
through a small aperture into the vacuum system of the
mass spectrometer for subsequent mass analysis
Reagent ions which transfer charged species to the
analyte molecules to form analyte ions are produced as a
result of a corona discharge in solvent vapour. The
corona discharge is generated by applying a high voltage
(e.g. 5 kV) to the tip of a sharp corona needle or pin.
Analyte molecules are ionised by gas phase ion-molecule
reactions with reagent ions in the region between the
corona tip and the ion sampling orifice. Analyte ions
are therefore generated in the region of the corona
discharge since this is also where the reagent ions are
formed.
The majority of the gas exits the ion source via an
exhaust port whilst a small proportion of the gas and
analyte ions will be drawn through the ion sampling
orifice into the vacuum system of the mass spectrometer
for subsequent mass analysis.
Analyte samples which are low to moderately polar
when analysed by Atmospheric Pressure Chemical
Ionisation typically exhibit an increase in ion signal
intensity as the voltage or current applied to the
corona needle is increased. In contrast, highly polar
or ionic analytes typically exhibit a decrease in ion
CA 02474836 2004-07-16
- 3 -
signal intensity as the voltage or current applied to
the corona needle is increased. Therefore, in order to
achieve a sufficiently high ion signal intensity for
highly polar or ionic analytes these analytes are
conventionally generated using an ion source other than
an Atmospheric Pressure Chemical Ionisation ion source,
such as, for example, an Electrospray Ionisation ("ESI")
ion source.
It is believed that in Atmospheric Pressure
Chemical Ionisation ion sources highly polar or ionic
analytes emerge from the outlet of the heated tube in
the form of ions or charged micro-droplets before the
analytes have had an opportunity to interact with
reagent ions. As the corona needle is maintained at a
relatively high positive potential (for positive ion
analysis) an electric field is generated in the region
of the corona needle. The electric field generated by
the corona needle will tend to retard and disperse the
already positively charged analyte ions or
micro-droplets which exit the heated tube causing the
analyte ions or charged analyte micro-droplets to become
defocussed in the region of the ion sampling orifice.
Accordingly, if the voltage or current applied to the
corona needle is further increased then the positive
analyte ions or micro-droplets will simply be retarded
and dispersed to an even greater extent and hence even
fewer analyte ions will pass through the ion sampling
orifice into the main body of the mass spectrometer for
subsequent mass analysis and detection. Accordingly,
the ion signal intensity for highly polar or ionic
analytes is significantly decreased as the corona
current is increased.
."
CA 02474836 2004-07-16
- 4 -
It follows that the ion signal intensity for highly
polar or ionic analytes is optimized when a relatively
low current or voltage is applied to the corona needle.
In contrast, the ion signal intensity for low to
moderately polar analytes is optimized when a relatively
high current or voltage is applied to the corona needle.
This is because when a higher current or voltage is
applied to the corona needle a higher number of reagent
ions are generated in the region of the corona needle.
The increased number of reagent ions interact with the
analyte molecules and generate a higher number of
analyte ions. As low to moderately polar analytes do
not generally become charged before they exit the heated
tube and approach the corona needle, the low to
moderately polar analyte molecules are not retarded and
dispersed by the electric field generated by the corona
needle. Accordingly, as the current or voltage applied
to the corona needle is increased a higher number of
analyte ions are generated (due to the increased number
of reagent ions produced) and these analyte ions pass
through the ion sampling orifice for subsequent mass
analysis and hence a greater ion signal intensity is
detected.
It will be appreciated, therefore, that in order to
analyse samples containing a mixture of both low to
moderately polar analytes and also highly polar or ionic
analytes using a conventional Atmospheric Pressure
Chemical Ionisation ion source, that it is necessary to
execute multiple sequential experimental runs in which
different voltages or currents are applied to the corona
needle of the ion source (e.g. a relatively low corona
current is set in a first experimental run so that
ionisation is optimised for highly polar analytes and a
CA 02474836 2004-07-16
- 5 -
relatively high corona current is set in a second
experimental run so that ionisation is optimised for low
to moderately polar analytes). Executing multiple
experimental runs whilst applying different voltages or
currents to the corona needle yields multiple sets of
data which together provide a relatively high ion signal
intensity for each analyte in the sample irrespective of
the polarities or ionic nature of the analytes in the
sample. However, the requirement to repeat the data
acquisition process whilst applying different voltages
or currents to the corona needle increases both the
sample analysis time and the sample consumption volume.
This can be a particular problem especially when only
very small amounts of sample are available for analysis
and also when the sample supplied to the ion source is
dynamically changing in a short period of time, for
example in chromatography applications.
It is therefore desired to provide an improved ion
source.
According to an aspect of the present invention
there is provided an ion source for a mass spectrometer
comprising:
a discharge region with a discharge device arranged
in the discharge region; and
a reaction region;
wherein in use reagent ions created in the
discharge region pass from the discharge region into the
reaction region and analyte molecules and/or analyte
ions pass into the reaction region, wherein ions in the
reaction region are at least partially shielded from an
electric field generated by the discharge device in the
discharge region.
CA 02474836 2004-07-16
- 6 -
The discharge region preferably comprises a
discharge chamber and the discharge device preferably
comprises a corona discharge device such as a corona
needle or pin. In a mode of operation a current of <
0.1 pA, 0.1-0.2 pA, 0.2-0.3 pA, 0.3-0.4 pA, 0.4-0.5 pA,
0.5-0.6 pA, 0.6-0.7 pA, 0.7-0.8 pA, 0.8-0.9 pA, 0.9-1.0
pA or > 1 pA may be applied to the discharge device. In
a mode of operation a voltage of < 1 kV, 1-2 kV, 2-3 kV,
3-4 kV, 4-5 kV, 5-6 kV, 6-7 kV, 7-8 kV, 8-9 kV, 9-10 kV
or > 10 kV may be applied to the discharge device.
According to the preferred embodiment the reaction
region comprises a substantially field free region.
Preferably, the reaction region comprises a reaction
chamber. A passage or orifice preferably connects or
communicates the discharge region to or with the
reaction region, wherein in use reagent ions created in
the discharge region pass from the discharge region to
the reaction region via the passage or orifice. A
housing preferably encloses the discharge region, the
reaction region and the passage or orifice.
According to the preferred embodiment the corona
discharge from the corona discharge device is confined
to the discharge region or the corona discharge chamber.
Accordingly, no discharge occurs within the reaction
region or reaction chamber. As a result analyte
molecules or analyte ions in the reaction region or
reaction chamber are not exposed to a corona discharge.
A gas inlet is preferably arranged upstream of the
discharge region, the gas inlet receiving, in use, a
reagent gas which is supplied to the discharge region.
A gas outlet is preferably arranged downstream of the
reaction region, the gas outlet discharging, in use, gas
and/or analyte ions and/or reagent ions.
CA 02474836 2004-07-16
- 7 -
The ion source preferably comprises an Atmospheric
Pressure Ionisation ion source, further preferably an
Atmospheric Pressure Chemical Ionisation source.
The discharge region and/or the reaction region are
preferably maintained, in use, at a pressure selected
from the group consisting of: (i) < 100 mbar; (ii) 100-
500 mbar; (iii) 500-600 mbar; (iv) 600-700 mbar; (v)
700-800 mbar; (vi) 800-900 mbar; (vii) 900-1000 mbar;
(viii) 1000-1100 mbar; (ix) 1100-1200 mbar; (x) 1200-
1300 mbar; (xi) 1300-1400 mbar; (xii) 1400-1500 mbar;
(xiii) 1500-2000 mbar; and (xiv) > 2000 mbar.
The ion source preferably comprises a spray device
for spraying a sample and for causing the sample to form
droplets. A nebulising gas is preferably supplied to
further nebulise the droplets formed by the spray
device. A heated tube is preferably provided upon
which, in use, at least some of the droplets formed by
the spray device impinge. The heated tube preferably
discharges or supplies, in use, analyte molecules and/or
analyte ions into the reaction region.
The ion source may preferably comprise a pneumatic
nebuliser or a pneumatically assisted electrospray
nebuliser.
According to another aspect of the present
invention there is provided a mass spectrometer
comprising an ion source as described above.
The mass spectrometer preferably further comprises
an ion sampling orifice. At least one electrode may be
arranged opposite or adjacent to the ion sampling
orifice so as to deflect, attract, direct or repel at
least some ions towards the ion sampling orifice.
The ion source may be connected, in use, to a gas
or liquid chromatograph.
CA 02474836 2004-07-16
- 8 -
The mass spectrometer preferably further comprises
a mass analyser such as a Time of Flight mass analyser,
a quadrupole mass analyser, a Penning mass analyser, a
Fourier Transform Ion Cyclotron Resonance ("FTICR") mass
analyser, a 2D or linear quadrupole ion trap, a Paul or
3D quadrupole ion trap or a magnetic sector mass
analyser.
According to another aspect of the present
invention there is provided an Electrospray
Ionisation/Atmospheric Pressure Chemical Ionisation
("ESI/APCI") ion source comprising:
a corona discharge device arranged in a corona
discharge chamber;
wherein, in use, analyte molecules having a
relatively low polarity are ionised by gas phase ion-
molecule reactions with reagent ions; and
wherein, in use, analyte molecules having a
relatively high polarity are ionised by electrospray
ionisation to form analyte ions, at least x% of the
analyte ions being arranged to bypass, in use, the
corona discharge chamber.
Preferably, x is selected from the group consisting
of: (i) < 1; (ii) 5; (iii) 10; (iv) 15; (v) 20; (vi) 25;
(vii) 30; (viii) 35; (ix) 40; (x) 45; (xi) 50; (xii) 55;
(xiii) 60; (xiv) 65; (xv) 70; (xvi) 75; (xvii) 80;
(xviii) 85; (xix) 90; and (xx) 95.
The analyte ions which bypass, in use, the corona
discharge chamber preferably at least partially avoid
the effect of an electric field generated by the corona
discharge device in the corona discharge chamber.
According to another aspect of the present
invention there is provided an ion source comprising:
CA 02474836 2004-07-16
- 9 -
a reaction chamber for receiving analyte molecules
and/or analyte ions; and
a corona discharge chamber;
wherein, in use, reagent ions formed in the corona
discharge chamber exit the corona discharge chamber and
enter the reaction chamber and wherein analyte molecules
and/or analyte ions do not substantially enter the
corona discharge chamber.
According to another aspect of the present
invention there is provided a method of producing ions
comprising:
providing a discharge region with a discharge
device arranged in the discharge region, and a reaction
region;
creating reagent ions in the discharge region and
passing the reagent ions from the discharge region into
the reaction region; and
passing analyte molecules and/or analyte ions into
the reaction region, wherein ions in the reaction region
are at least partially shielded from an electric field
generated by the discharge device in the discharge
region.
According to another aspect of the present
invention there is provided a method of producing ions
using an Electrospray Ionisation/Atmospheric Pressure
Chemical Ionisation ("ESI/APCI") ion source comprising:
providing a corona discharge device arranged in a
corona discharge chamber;
ionising analyte molecules having a relatively low
polarity by gas phase ion-molecule reactions with
reagent ions; and
ionising analyte molecules having a relatively high
polarity by electrospray ionisation to form analyte
-
CA 02474836 2004-07-16
- 10 -
ions, at least x% of the analyte ions being arranged to
bypass the corona discharge chamber.
Preferably, x is selected from the group consisting
of: (i) < 1; (ii) 5; (iii) 10; (iv) 15; (v) 20; (vi) 25;
(vii) 30; (viii) 35; (ix) 40; (x) 45; (xi) 50; (xii) 55;
(xiii) 60; (xiv) 65; (xv) 70; (xvi) 75; (xvii) 80;
(xviii) 85; (xix) 90; and (xx) 95.
According to another aspect of the present
invention there is provided a method of producing ions
comprising:
providing a reaction chamber for receiving analyte
molecules and/or analyte ions, and a corona discharge
chamber; and
causing reagent ions formed in the corona discharge
chamber to exit the corona discharge chamber and enter
the reaction chamber, wherein analyte molecules and/or
analyte ions do not substantially enter the corona
discharge chamber.
The preferred embodiment relates to an Atmospheric
Pressure Chemical Ionization ion source wherein reagent
ions are formed in an ancillary or discharge chamber
separate from the region or reaction chamber through
which the sample to be analysed flows. The reagent ions
are carried by gas flow from the ancillary or discharge
chamber to the reaction chamber whereupon the reagent
ions can then interact with the desolvated analyte
molecules and ionise the analyte molecules by chemical
ionization. However, highly polar analytes which are
already ionised by the time that they enter the reaction
chamber are at least partially shielded from the effects
of the electric field generated in the ancillary or
discharge chamber. Accordingly, the corona current can
CA 02474836 2004-07-16
- 11 -
be set high without affecting the signal intensity when
highly polar analytes are ionised by the ion source.
Various embodiments of the present invention will
now be described, by way of example only, and with
reference to the accompanying drawings in which:
Fig. 1 illustrates how the ion signal intensity for
a highly polar sample (Reserpine) and a low to
moderately polar sample (Corticosterone) vary as a
function of the current applied to a corona needle of a
conventional APCI ion source;
Fig. 2 shows two superimposed ion signal
intensities obtained from two separate LC/MS MRM
analyses of a sample comprising four different analytes
wherein during the first acquisition the corona current
was maintained at 0.2 pA (i.e. relatively low) and
wherein during the second acquisition the corona current
was maintained at 5 pA (i.e. relatively high);
Fig. 3 shows a dual chamber APCI ion source
according to a first embodiment of the present invention
wherein a pneumatic nebuliser is used;
Fig. 4 illustrates how the ion signal intensity for
a highly polar sample (Reserpine) and a low to
moderately polar sample (Corticosterone) vary as an
function of the current applied to a corona needle of an
ion source according to an embodiment of the present
invention;
Fig. 5 shows two superimposed ion signal
intensities obtained from two separate LC/MS MRM
analyses of a sample comprising four different analytes
wherein during the first acquisition the corona current
was maintained at 0.2 pA (i.e. relatively low) and
wherein during the second acquisition the corona current
was maintained at 5 pA (i.e. relatively high); and
CA 02474836 2004-07-16
- 12 -
Fig. 6 shows a dual chamber APCI ion source
according to a second embodiment of the present
invention wherein an electrospray nebuliser is used.
Referring to Fig. 1, this figure shows how the ion
signal intensity varies as a function of the current
applied to a corona needle of a conventional Atmospheric
Pressure Chemical Ionisation ("APCI") ion source for two
different types of analytes. As can be seen from
Fig. 1, the ion signal intensity for a low to moderately
polar sample (e.g. Corticosterone) increases relatively
rapidly and then plateaus at a certain point as the
current applied to the corona needle is further
increased. The initial increase in ion signal intensity
is believed to be due to the ion source producing more
reagent ions as the current applied to the corona needle
is increased. The increased number of reagent ions
interact with the analyte molecules emitted from the
nebuliser tube and hence more analyte ions are produced.
Accordingly, an increased number of analyte ions are
then subsequently mass analysed and hence an increase in
the ion signal intensity is observed.
It can also be seen from Fig. 1 that increasing the
current applied to the corona needle of the ion source
has the opposite affect for a highly polar sample (e.g.
Reserpine). As the current applied to the corona needle
is increased, the ion signal intensity for Reserpine
decreases relatively rapidly and then remains at a
substantially constant low level. In contrast to low to
moderately polar samples it is believed that relatively
highly polar analytes such as Reserpine exit the
nebuliser tube in an already charged state most likely
due to thermal ionisation effects. The already charged
analyte ions are therefore then effectively retarded by
CA 02474836 2004-07-16
- 13 -
the electric field resulting from the voltage applied to
the corona needle. The highly polar analyte ions are
therefore deflected and dispersed by the electric field
generated by the corona needle. Increasing the
potential of the corona needle (which may as a
consequence increase the current drawn from the corona
needle) merely increases the strength of the electric
field in the region of the corona needle and hence in
the region adjacent to the exit of the nebuliser tube.
Therefore, increasing the current applied to the corona
needle merely increases the level of retardation,
deflection and dispersal of the charged analyte ions
which exit the nebuliser tube. As a result, as the
corona current is increased, fewer analyte ions will
ultimately pass through the ion sampling orifice and
into the main body of the mass spectrometer for
subsequent mass analysis.
In view of the different responses of low to
moderately polar analytes and highly polar analytes to
the current applied to the corona needle as shown in
Fig. 1, the conventional approach when seeking to ionise
a mixture containing both low to moderately polar
analytes and also highly polar or ionic analytes is
either to set the current applied to the corona needle
at some compromise level (e.g. 0.25 pA for the example
shown in Fig. 1) which results in sub-optimal ionisation
for both types of analytes, or alternatively to perform
two separate acquisitions in which a first acquisition
is performed at a first corona current setting followed
by a second acquisition performed at a second different
corona current setting. The conventional approaches
therefore either result in ion signals which are not
maximised (if a single acquisition at a compromise
CA 02474836 2004-07-16
- 14 -
corona current is performed) or alternatively the total
analysis time and sample consumption is effectively
doubled (if two separate acquisitions at two different
corona currents are performed).
Fig. 2 shows the results of a four channel Multiple
Reaction Monitoring ("MRM") experiment performed using a
conventional APCI ion source in conjunction with a
triple quadrupole mass spectrometer. In particular Fig.
2 shows an overlay of the ion signals resulting from two
separate acquisitions in which a mixture comprising
Verapamil, Corticosterone, Hydroxyprogesterone and
Reserpine was analysed using Liquid Chromatography Mass
Spectrometry ("LCMS").
As will be understood by those skilled in the art,
in a MRM experiment a first mass filter (e.g. quadrupole
rod set mass filter) is set to transmit parent ions
having a certain (specific) mass to charge ratio. The
selected parent ions having a particular mass to charge
ratio are then introduced into a collision or
fragmentation cell wherein the parent ions are
fragmented into daughter or fragment ions. A second
mass filter (e.g. quadrupole rod set mass filter)
provided downstream of the collision or fragmentation
cell is then arranged to transmit daughter or fragment
ions having a certain (specific) mass to charge ratio.
In this and the subsequent described MRM
experiment, Verapamil parent ions having a mass to
charge ratio of 455.1 were transmitted by the first mass
filter and were fragmented in a collision or
fragmentation cell. Characteristic daughter or fragment
ions having a mass to charge ratio of 165.1 were
arranged to be transmitted by the second mass filter.
Corticosterone parent ions having a mass to charge ratio
CA 02474836 2004-07-16
- 15 -
of 347.1 were transmitted by the first mass filter and
were fragmented in the collision or fragmentation cell.
Characteristic daughter or fragment ions having a mass
to charge ratio of 329.1 were arranged to be transmitted
by the second mass filter. Hydroxyprogesterone parent
ions having a mass to charge ratio of 331.1 were
transmitted by the first mass filter and were fragmented
in the collision or fragmentation cell. Characteristic
daughter or fragment ions having a mass to charge ratio
of 109.1 were arranged to be transmitted by the second
mass filter. Finally, Reserpine parent ions having a
mass to charge ratio of 609.1 were transmitted by the
first mass filter and were fragmented in the collision
or fragmentation cell. Characteristic daughter or
fragment ions having a mass to charge ratio of 195.1
were arranged to be transmitted by the second mass
filter.
A first experimental run or acquisition was
performed over a period of 20 minutes (including column
equilibrium) during which time the four analytes eluted
within a time of 7 minutes and wherein a current of
0.2 pA was applied to the corona needle. A second
experimental run or acquisition was then subsequently
performed over another period of 20 minutes (including
column equilibrium), again wherein the four analytes
eluted within a time of 7 minutes but wherein a current
of 5 pA was applied to the corona needle. The analytes
in order of elution were Verapamil, Corticosterone,
Hydroxyprogesterone followed lastly by Reserpine.
Verapamil and Reserpine are highly polar
analytes/molecules whereas Corticosterone and
Hydroxyprogesterone are moderately polar
analytes/molecules.
CA 02474836 2004-07-16
- 16 -
It can be seen from Fig. 2 that the difference in
the resulting ion signal intensities detected for the
two separate experimental runs or acquisitions is
relatively large, especially for the relatively highly
polar analyte Verapamil. As can also be seen from Fig.
2, as the current applied to the corona needle was
increased in the second experimental run or acquisition
from 0.2 pA to 5 pA, the ion signal intensity for the
relatively highly polar analytes Verapamil and Reserpine
significantly decreased whereas the ion signal intensity
for the low to moderately polar analytes Corticosterone
and Hydroxyprogesterone increased.
In this conventional technique utilising two
separate experimental runs in which different currents
are applied to the corona needle a sufficiently high ion
signal intensity is obtainable for each of the two
different types (i.e. polarities) of analytes in the
sample during one or other of the experimental runs.
However, as the analysis is effectively repeated whilst
applying the different currents to the corona needle,
the time required to analyse a sample using such a
conventional technique is relatively long. For example,
the total analysis time for each chromatogram can be 20
minutes including column equilibration. Furthermore,
repeating the experimental run whilst applying a
different current to the corona needle increases the
sample consumption volume.
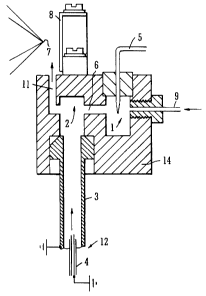
Fig. 3 shows an Atmospheric Pressure Ionisation ion
source according to a first embodiment of the present
invention. The ion source comprises a corona discharge
chamber 1 which houses the tip of a corona needle 5. A
reaction chamber 2 is provided downstream of the corona
discharge chamber 1 and is in communication with the
CA 02474836 2004-07-16
- 17 -
corona discharge chamber 1 via a passage or orifice 6.
The reaction chamber 2 is preferably arranged adjacent
to the corona chamber 1 within a housing 14. The
reaction chamber 2 is also preferably in communication
with a source of a sample to be analysed. The ion
source preferably comprises a nebuliser probe 12. The
nebuliser probe 12 preferably comprises a pneumatic
nebuliser 4 and a heated tube 3 for heating a liquid
sample sprayed from the nebuliser 4 to convert the
sample into a gaseous state for subsequent ionisation
and mass analysis. The reaction chamber 2 is preferably
arranged in the region of the exit of the heated tube 3
of the nebuliser probe 12.
During operation of the preferred ion source a
sample is preferably delivered to the ion source by, for
example, a chromatography system. The sample is
preferably delivered to the pneumatic nebuliser 4 of the
nebuliser probe 12 in a liquid state and is then sprayed
from the nebuliser 4 and nebulised by a relatively high
velocity stream of gas, preferably nitrogen gas. The
sample droplets which result from the nebulisation
comprise mobile phase solvents and analytes. These
preferably enter and pass through the heated tube 3.
The nebulised droplets of sample solution are preferably
heated in the heated tube 3 such that the sample is
converted from a liquid state into the gaseous phase.
After the sample has been converted into the gaseous
phase it preferably passes into the reaction chamber 2.
Reagent ions are generated in the ion source in a
discharge region 1 which preferably comprises the corona
chamber 1 housing the corona needle or pin 5. In order
to generate the reagent ions a reagent gas such as, for
example, nitrogen and a solvent such as, for example,
CA 02474836 2004-07-16
- 18 -
methanol are arranged to flow into the corona chamber 1
via a gas inlet 9. The voltage applied to the corona
needle 5 (e.g. -3 kV) preferably generates a corona
discharge in the corona chamber 1 which serves to ionise
molecules in the reagent gas. As a result, a population
of stable reagent ions are formed within the vicinity of
the tip of the corona needle 5. The polarity of the
voltage applied to the corona needle 5 is preferably
positive for positive ion analysis and is preferably
negative for negative ion analysis. The reagent ions
generated in the corona chamber 1 are then preferably
transmitted from the corona chamber 1 to the reaction
chamber 2 through the passage or orifice 6 which links
the two chambers 1,2 preferably by the flow of reagent
gas through the corona chamber 1.
The reagent ions passing from the corona chamber 1
into the reaction chamber 2 preferably mix and interact
with the gaseous sample exiting from the heated tube 3.
The reagent ions preferably undergo gas phase ion-
molecule interactions with any analyte molecules in the
gaseous sample within the reaction chamber 2. These
ion-molecule interactions result in at least some of the
reagent ions transferring a charged species to the
analyte molecules such that the analyte molecules
preferably become ionised and the reagent ions
preferably become neutralised.
In the preferred embodiment, any low to moderately
polar analytes present in the sample to be analysed pass
through the heated tube 3 and into the reaction chamber
2 predominantly as neutral analyte molecules. In
contrast, relatively highly polar or ionic analytes
which may be present in the sample preferably exit the
heated tube 3 and enter the reaction chamber 2 already
CA 02474836 2004-07-16
- 19 -
as ions i.e. the highly polar or ionic analytes are
already ionised (most likely by thermal ionization)
prior to encountering reagent ions in the reaction
chamber 2.
Any neutral analyte molecules which exit the heated
tube 3 and which enter the reaction chamber 2 preferably
undergo interactions with the reagent ions and become
ionised such that at least some, preferably
substantially all of the analytes in the sample are
ionised. The resulting analyte ions, other particles
and gas in the reaction chamber 2 then preferably exits
the reaction chamber 2 via an outlet passage or port 11
preferably under the influence of both the flow of gas
exiting the heated tube 3 and also the flow of gas
through the corona chamber 1 which also passes into the
reaction chamber 2.
In a preferred embodiment the gas and ions which
exit the reaction chamber 2 via the passage or orifice
11 flow into a region adjacent an ion sampling cone
having an ion sampling orifice 7. The ion sampling
orifice 7 is preferably arranged off-axis with respect
to the axis of the passage or orifice 11 such that the
gas and ions exiting the passage or orifice 11
preferably do not flow directly through the ion sampling
orifice 7. At least one electrode is preferably
arranged in the region of the ion sampling orifice 7 in
order to provide an electric field which deflects (or
less preferably attracts) at least some of the analyte
ions through the ion sampling orifice 7 and into the
main body of the mass spectrometer. A pusher electrode
8 may, for example, be arranged substantially opposite
to the ion sampling orifice 7 such that the gas and ions
exiting the passage or orifice 11 flows between the
CA 02474836 2004-07-16
- 20 -
pusher electrode 8 and the ion sampling orifice 7. In
the preferred embodiment, the pusher electrode 8 causes
at least some of the ions exiting the passage or orifice
11 to be deflected into and through the ion sampling
orifice 7. Preferably, the pusher electrode 8 deflects
at least some of the ions exiting the passage or orifice
11 substantially at right angles to the axis of the
passage or orifice 11. The arrangement of the ion
sampling orifice 7 and the provision of the pusher
electrode 8 therefore enables at least some ions to be
directed into and through the ion sampling orifice 7 for
subsequent mass analysis whilst not assisting neutral
molecules and gas to pass through the ion sampling
orifice 7. In a preferred embodiment, the voltage
applied to the pusher electrode 8 is in the range of 0-
300 V.
In a less preferred embodiment the pusher electrode
8 may be omitted and the ion sampling orifice 7 and the
passage or orifice 11 may be arranged such that the axis
of the passage or orifice 11 is substantially coaxial
with the axis of the ion sampling orifice 7. In this
embodiment at least one additional electrode (not shown)
may be provided to focus or direct at least some of the
ions into and through the ion sampling orifice 7.
Gas passing through the ion sampling orifice 7 is
preferably allowed to expand into the volume of a first
vacuum chamber which preferably includes an exhaust port
to exhaust the gas. Ions preferably then pass from the
first vacuum chamber into a mass analyser for mass
analysis. The entire process of generating analyte ions
described above preferably occurs at or close to
atmospheric pressure.
CA 02474836 2004-07-16
- 21 -
Fig. 4 shows how the ion signal intensity varies
with the current applied to the corona needle 5 of a
preferred dual chamber ion source for the moderately
polar analyte Corticosterone and for the relatively
highly polar or ionic analyte Reserpine. It can be seen
that the ion signal intensity observed for
Corticosterone using the preferred ion source increases
at a relatively high rate and then saturates at a
relatively constant ion signal intensity as the current
applied to the corona needle is increased. The
variation of the ion signal intensity with current
applied to the corona needle 5 for Corticosterone has
some similarities to the response obtained using a
conventional ion source as shown in Fig. 1. With
regards Reserpine, it can be seen that as the current
applied to the corona needle of the preferred ion source
is increased, the ion signal intensity for Reserpine
remains substantially constant (within experimental
error) and certainly shows no significant fall off as
the corona current is increased. This improved response
is in direct contrast to the response obtained using a
conventional ion source as shown in Fig. 1. The ion
signal intensity for Reserpine does not show an increase
with an increase in current applied to the corona needle
due to the fact that Reserpine is already seemingly
highly ionised by the time that it enters the reaction
chamber 2. Increasing the current applied to the corona
needle to increase the number of reagent ions produced
does not therefore generate a significantly higher
number of analyte ions in the case of a highly polar
analyte.
The ion signal intensity obtained for Reserpine
using the preferred ion source shows that the
CA 02474836 2004-07-16
- 22 -
detrimental effects observed with conventional APCI ion
sources when attempting to ionize highly polar or ionic
analytes caused by the electric field generated by the
corona needle are substantially eliminated when using an
ion source according to the preferred embodiment of the
present invention. Accordingly, the preferred ion
source does not suffer from the problem of analyte ions
being defocused or dispersed due to the effects of the
corona discharge process.
According to the preferred embodiment the gaseous
sample comprising the analytes passes through into the
reaction chamber 2 without being significantly
influenced by the electric field generated by the
relatively high potential which is preferably applied to
the corona needle 5 located in the adjacent corona
chamber 1. The relatively highly polar analytes which
typically enter the reaction chamber 2 as ions are
therefore preferably not significantly retarded or
dispersed in the ion source due to the electric field
generated by the corona needle 5. As ions from
relatively highly polar analytes are not dispersed in
the preferred ion source they are able to be transmitted
to the ion sampling orifice 7 preferably arranged
downstream of the reaction chamber 2 for subsequent mass
analysis with an increased efficiency. The ion signal
intensity for relatively highly polar analytes is
therefore increased compared with the ion signal
intensity obtained when using a conventional ion source.
This is particularly advantageous since it follows that
relatively high ion signal intensities can be obtained
for both highly polar or ionic analytes and also low to
moderately polar analytes whilst supplying a constant
current to the corona needle 5 (e.g. 5 pA). Therefore,
CA 02474836 2004-07-16
- 23 -
a single experimental run can be conducted in which
sufficiently high ion signal intensities can be obtained
for all analytes in the sample irrespective of their
polarity. Accordingly, the time required to analyse the
sample and the volume of the sample required to conduct
the analysis are significantly reduced compared with
conventional APCI ion sources.
Fig. 5 shows an overlay of the ion signal
intensities as a function of time for two separate
Liquid Chromatography Mass Spectral ("LC/MS") MRM
analyses of a sample comprising four different analytes
using an ion source according to the preferred
embodiment. The four different analytes and the four
channel MRM experiment is essentially the same as
described above in relation to Fig. 2. It can be seen
from comparing Figs. 2 and 5 that as with the ion signal
intensities obtained when generating ions using a
conventional ion source, when using the preferred ion
source significantly different ion signal intensities
are obtained for low to moderately polar analytes (e.g.
Corticosterone and Hydroxyprogesterone) when relatively
low and relatively high currents were applied to the
corona needle (e.g. 0.2 pA and 5 pA respectively). The
increase in ion signal intensity for low to moderately
polar analytes in response to the increase in the
current applied to the corona needle corresponds to an
increase in the number of reagent ions generated in the
corona chamber 1. Accordingly, there are an increased
number of analyte molecule-reagent ion interactions in
the reaction chamber 2 resulting in a higher number of
analyte ions being produced which are then subsequently
mass analysed.
CA 02474836 2004-07-16
- 24 -
In contrast to the ion signal intensities obtained
using a conventional ion source, the ion signal
intensities detected for the relatively highly polar
analytes Verapamil and Reserpine using the preferred ion
source varied relatively little when the current applied
to the corona needle 5 was increased from 0.2 pA to 5
pA. Indeed there was hardly any discernible reduction
in intensity for Resperine when the corona current was
increased from 0.2 pA to 5 pA. Advantageously, when the
current applied to the corona needle is maintained
relatively high (i.e. 5 pA) the ion signal intensities
detected for Verapamil and Reserpine are significantly
higher when using the preferred ion source as compared
to a conventional ion source.
It will be appreciated, therefore, that when the
ion source according to the preferred embodiment is
employed and a relatively high corona current (e.g. 5
pA) is applied to the corona needle a relatively high
ion signal intensity can be obtained both for relatively
highly polar and also for low to moderately polar
analytes. This avoids the need to operate the corona
needle of the ion source at different currents during
two separate acquisitions. Accordingly, samples
comprising analytes having both low to moderately polar
analytes and also highly polar analytes can be analysed
in a single experimental run wherein a moderate to high
current (e.g. 3-10 pA) is applied to the corona needle
5. This single acquisition is advantageous in that both
the total analysis time and the sample consumption
volume are significantly reduced compared with
conventional techniques.
The preferred Atmospheric Pressure Ionization ion
source is further advantageous over conventional ion
CA 02474836 2004-07-16
- 25 -
sources in that the sample gas flow is arranged such
that analytes, involatiles and other contaminants in the
sample gas do not flow past the corona needle 5.
Material present in the sample gas flow is therefore not
deposited on the tip of the corona needle 5 and hence
the operation of the corona needle 5 is not degraded
during use. The preferred ion source therefore also has
a significantly improved long-term stability compared
with conventional arrangements. The preferred ion
source also reduces the carry-over of tuning compounds
and enables reagent ions to be formed which are
independent of the mobile phase, provided that the
reaction thermodynamics are permitted.
Fig. 6 shows an ion source according to another
preferred embodiment. This embodiment is substantially
similar to the embodiment shown and described in
relation to Fig. 3 except that the nebuliser probe 13
comprises a pneumatically assisted electrospray
nebuliser 10 and a heated tube 3. According to this
embodiment, the heated tube 3 is preferably grounded and
the electrospray nebuliser 10 is preferably maintained
at a relatively high voltage (e.g. 3 kV) with respect to
the heated tube 3. Advantageously, the pneumatically
assisted electrospray nebuliser probe ionises relatively
highly polar analytes present in the sample with an
increased efficiency compared with the pneumatic
nebuliser 12 as shown in Fig. 3. Preferably
substantially all relatively highly polar analytes are
likely to be ionised by the pneumatically assisted
electrospray nebuliser 13 before they pass into and
through the reaction chamber 2.
Low to moderately polar analytes, which may not be
efficiently ionised by the pneumatically assisted
CA 02474836 2012-02-21
- 26 -
electrospray nebuliser 13 are preferably converted from
the liquid to gas phase by the electrospray nebuliser 10
in combination with the heated tube 3. The low to
moderately polar analytes then exit the heated tube 3
and are ionised in the reaction chamber 2 by molecule-
ion reactions with reagent ions generated in the corona
chamber 1 and passed into the reaction chamber 2. This
embodiment forms the basis of an Electrospray
Ionisation/Atmospheric Pressure Chemical Ionisation
("ESI/APCI") ion source that can ionise a wide range of
compound classes and is particularly suited for use over
a wide range of Liquid Chromatograph ("LC") flow rates
with a high efficiency.
The scope of the claims should not be limited by
the preferred embodiments described herein, but should
be given the broadest interpretation consistent with the
description as a whole.