Note: Descriptions are shown in the official language in which they were submitted.
WO 03/066620 CA 02475093 2004-08-03 PCT/EP03l00653
DIHYDROTHIAPHENANTHRENECARBONYLGUANIDINES, PROCESS FOR THEIR
PREPARATION, THEIR USE AS MEDICAMENT OR DIAGNOSTIC AID
Description
Dihydrothiaphenanthrenecarbonylguanidines, process for their preparation,
their use
as medicament or diagnostic aid, and medicament comprising them
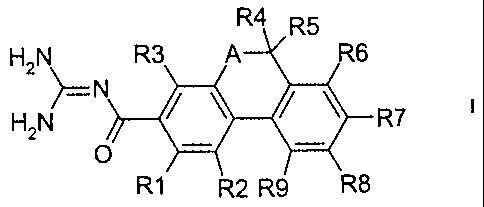
The invention relates to dihydrothiaphenanthrenecarbonylguanidines of the
formula I
R5
H2N R3 A R6
N
H2N ~ ~--R7
O ~ ~
R1~ 2 R9 \R8
in which the meanings are:
R(1 ) and R(3)
independently of one another hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms,
alkoxy having 1, 2, 3 or 4 carbon atoms, F, CI, Br, l, CN, NR(10)R(11 ),
-Op-(CH2)n-(CF2)x-CF3 or -(SOm)p-(CH2)n-(CF2)x-CF3;
R(10) and R(11 )
independently of one another hydrogen, alkyl having 1, 2, 3 or 4
carbon atoms or -(CH2)n-(CF2)x-CF3;
m zero, 1 or 2;
n zero, 1, 2, 3, 4, 5 or 6;
x and p
independently of one another zero or 1;
R(2) hydrogen, F, CI, Br, I, CN, alkyl having 1, 2, 3 or 4 carbon atoms,
methoxy,
cycloaikyl having 3, 4, 5, 6, 7 or 8 carbon atoms,
R(4) and R(5)
independently of one another hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms;
R(6), R(7), R(8) and R(9)
independently of one another hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms;
alkoxy having 1, 2, 3 or4 carbon atoms, F, CI, Br, I, CN, NR(12)R(13),
-Oq-(CH2)r'(CF2)s-CF3 or -(SOw)t-(CH2)u-(CF2)v-CF3;
R(12) and R(13)
CA 02475093 2004-08-03
2
independently of one another hydrogen, alkyl having 1, 2, 3 or 4
carbon atoms;
w zero, 1 or 2;
r and a
zero, 1, 2, 3, 4, 5 or 6;
q, s, t and v
independently of one another zero or 1;
or
R(6) and R(7) or R(7) and R(8) or R(8) and R(9)
together with the phenyl ring carrying them a naphthalene system;
A -S-, -SO- or -S02-
and the pharmaceutically suitable salts thereof.
Preference is given to compounds of the formula I in which the meanings are:
R(1 ) and R(3)
independently of one another hydrogen, methyl, ethyl, methoxy, ethoxy, F, CI,
CN, NR(10)R(11 ), -Op-(CH2)n-CF3 or -(SOm)p-(CH2)n-CF3;
R(10) and R(11 )
independently of one another hydrogen, methyl, ethyl or -CH2-CF3;
m zero, 1 or 2;
n zero, 1, 2 or 3;
p independently of one another zero or 1;
R(2) hydrogen, F, CI, CN, alkyl having 1, 2, 3 or 4 carbon atoms, methoxy,
cycloalkyl having 3, 4, 5 or 6 carbon atoms,
R(4) and R(5)
independently of one another hydrogen, methyl or ethyl;
R(6), R(7), R(8) and R(9)
independently of one another hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms;
methoxy, ethoxy, F, CI, CN, NR(12)R(13), -Oq-(CH2)~CFg or
-(SOw~-(CH2)u-CF3;
R(12) and R(13)
independently of one another hydrogen, methyl or ethyl;
CA 02475093 2004-08-03
3
w zero, 1 or 2;
r and a
zero, 1, 2 or 3;
q and t
independently of one another zero or 1;
or
R(6) and R(7) or R(7) and R(8) or R(8) and R(9)
together with the phenyl ring carrying them a naphthalene system;
A -S-, -SO- or -S02-
and the pharmaceutically suitable salts thereof.
Particular preference is given to compounds of the formula I, in which the
meanings
are:
R(1 )
hydrogen, methyl, ethyl, methoxy, ethoxy, F, CI, NR(10)R(11 ),
-Op-(CH2)n-CF3 or -(SOm)p-(CH2)n-CF3;
R(10) and R(11 )
independently of one another hydrogen, methyl, ethyl or -CH2-CF3;
m zero, 1 or 2;
n, p independently of one another zero or 1;
R(2) hydrogen, F, CI, methyl, cycloalkyl having 3, 4, 5 or 6 carbon atoms,
R(3), R(4) and R(5)
hydrogen;
R(6), R(7), R(8) and R(9)
independently of one another hydrogen, methyl; methoxy, ethoxy, F, CI,
NR(12)R(13), -Oq-(CH2)~CFg or -(SOw)t-(CH2)u-CF3;
R(12) and R(13)
independently of one another hydrogen, methyl or ethyl;
w zero, 1 or 2;
q, r, t and a
independently of one another zero or 1;
or
CA 02475093 2004-08-03
4
R(6) and R(7) or R(7) and R(8) or R(8) and R(9)
together with the phenyl ring carrying them a naphthalene system;
A -S-, -SO- or -S02-
and the pharmaceutically suitable salts thereof.
Very particular preference is given to compounds of the formula I in which the
meanings are:
R(1 )
hydrogen, methyl, methoxy, ethoxy, CI, NR(10)R(11 ), -O-CH2-CF3 or
-(SOm)p-(CH2)n-CF3;
R(10) and R(11 )
independently of one another hydrogen, methyl, ethyl or -CH2-CF3;
m zero, 1 or 2;
p zero or 1;
R(2) hydrogen, F, CI or methyl;
R(3), R(4) and R(5)
hydrogen;
R(6), R(7), R(8) and R(9)
independently of one another hydrogen, methyl; methoxy, ethoxy, F, CI, -O-
CH2-CF3 or -(SO~,"~-(CH2)u-CF3;
w zero, 1 or 2;
t and a
independently of one another zero or 1;
or
R(6) and R(7) or R(7) and R(8) or R(8) and R(9) .
together with the phenyl ring carrying them a naphthalene system;
A -S 02-
and the pharmaceutically suitable salts thereof.
The compounds of the formula I may with appropriate substitution exist in
stereoisomeric forms. If the compounds of the formula I contain one or more
centers of
CA 02475093 2004-08-03
asymmetry, these may have, independently of one another, the S configuration
or the
R configuration. All possible stereoisomers, e.g. enantiomers or
diastereomers, and
mixtures of two or more stereoisomeric forms, e.g. enantiomers and/or
diastereomers,
in any ratios, belong to the invention. Thu', enaptiumers fr~r example oelong
to the
5 invention in enantiopure form, both as levorotatory and as dextrorotatory
antipodes,
and in the form of mixtures of the two enantiomers in various ratios or in the
form of
racemates. Individual stereoisomers can be prepared if desired by
fractionation of a
mixture by conventional methods or, for example, by stereoselective synthesis.
If
mobile hydrogen atoms are present, the present invention also encompasses all
tautomeric forms of the compounds of the formula I.
The designated alkyl radicals may be straight-chain or branched. The invention
further
relates to a process for preparing the compound I, which comprises reacting a
compound of the formula ll
R5
~ R7 II
O
R1 ' R2 R9~R8
in which R(1 ) to R(9) and A have the stated meaning, and L is a leaving group
amenable to easy nucleophilic substitution, with guanidine.
The activated acid derivatives of the formula II in which L is an alkoxy,
preferably a
methoxy, group, a phenoxy group, phenylthio, methylthio, 2-pyridylthio group,
a
nitrogen heterocycle, preferably 1-imidazolyl, are advantageously obtained in
a
manner known per se from the underlying carbonyl chlorides (formula II, L =
CI), which
in turn can be prepared in a manner known per se from the underlying
carboxylic acids
(formula II, L = OH), for example with thionyl chloride.
Besides the carbonyl chlorides of the formula II (L = CI), it is also possible
to prepare
other activated acid derivatives of the formula II in a manner known per se
directly from
CA 02475093 2004-08-03
6
the underlying benzoic acid derivatives (formula 11, L = OH), such as the
methyl esters
of the formula II with L = OCH3 by treatment with gaseous HCI in methanol, the
imidazolides of the formula II by treatment with carbonyldiimidazole [L = 1-
imidazolyl,
Staab, Angew. Chem. Int. Ed. Engl. 1, 351 -367 (1962)], the mixed anhydrides
II with
CI-COOC2H5 or tosyl chloride in the presence of triethylamine in an inert
solvent, as
well as activations of benzoic acids with dicyclohexylcarbodiimide (DCC) or
with
O-[(cyano(ethoxycarbonyl)methylene)amino]-1,1,3,3-tetramethyluronium
tetrafluoroborate ("TOTU") [Proceedings of the 21 st European Peptide
Symposium,
Peptides 1990, Editors E. Giralt and D. Andreu, Escom, Leiden, 1991]. A number
of
suitable methods for preparing activated carboxylic acid derivatives of the
formula II
are indicated in J. March, Advanced Organic Chemistry, 3rd Edition (John Wiley
&
Sons, 1985), page 350, indicating the source literature.
Reaction of an activated carboxylic acid derivative of the formula II with
guanidine
takes place in a manner known per se in a protic or aprotic polar but inert
organic
solvent. Those which have proved suitable in the reaction of the methyl
benzoates
(II, L = OMe) with guanidinomethanol, isopropanol or THF from 20°C to
the boiling
point of these solvents. Most of the reactions of compounds II with salt-free
guanidine
have advantageously been carried out in aprotic inert solvents such as THF,
dimethoxyethane, dioxane. However, water can also be used as solvent in the
reaction
of II with guanidine if a base such as, for example, NaOH is employed.
When L is CI, it is advantageous to add an acid scavenger, e.g. in the form of
excess
guanidine to bind the hydrohalic acid.
Assembly of the dihydrothiaphenanthrenecarboxylic acid framework
advantageously
starts from appropriately substituted benzylsulfanyls, phenylmethanesulfinyls
or
phenylmethanesulfonyls. These are subjected to an intramolecular aryl-aryl
coupling
as known in principle (see Chem. Rev. 95 (7), 2457 (1995) or "Metal-catalyzed
Cross-
coupling Reactions", Diederich, Francois; Stang, Peter J.; Editors Germany
(1998)
Publisher: (Wiley-VCH, Weinheim, Germany), 517) or Tetrahedron (1998),
54(3/4),
263). Coupling of a boronic acid with a suitable aryl halide such as an aryl
chloride,
aryl bromide, aryl iodide or with a suitable aryl ester such as an aryl
mesylate or aryl
trifluoromethanesulfonate is preferred. In these cases, the boronic acid
function may
CA 02475093 2004-08-03
7
have been introduced both on the benzyl and on the benzoic acid reactant. It
is also
particularly preferred to use bis(pinakolato)diboron as described in
Tetrahedron Lett.
(1997), 38(22), 3841-3844. Formula III describes such a starting material in
which R(1 )
to R(9) and A and L have the stated meaning, and X and Y are each a halogen or
an
-O-S02CH3 or an -O-S02CF3. The preferred catalytic metal is palladium,
particularly
preferably in its complex Pd(dppf)2. The reaction is carried out in a dipolar
aprotic
solvent, preferably DMF or DMA, at a temperature between 0°C and the
boiling point
of the solvent, preferably at temperatures between 40°C and
120°C.
R7
R6 r R8
R5
R4 \ R9 R4 R5
R3 A Y R3 R6
L L
-_'-~.' ~ ~ ~ ~ R7
X O
R ~ R2 R9 R8
R1 R2
III II
The derivatives of the general formula III are preferably prepared from 3-
mercapto-
benzoic acid derivatives or from 3-sulfinobenzoic acid derivatives of the
formula IV with
activated benzyl derivatives of the formula V:
R7
R3 A-H Rfi , R8
L ~ ~ X + R5 ~ ( ~''' III
O/ R4 ~ ~R9
z Y
R1 R2
IV V
In this case, Z is a leaving group amenable to easy nucleophilic substitution,
such as,
for example, chlorine, bromine, iodine, mesylate, tosylate or
trifluoromethanesulfonate.
The derivatives IV and V are reacted in a suitable solvent such as DMF, THF or
CA 02475093 2004-08-03
8
acetonitrile, using a base such as triethylamine or DIPEA, at a temperature
between
-20°C and the boiling point of the solvent, preferably at a temperature
of between 0°C
and 40°C.
Aroylguanidines I are generally weak bases and are able to bind acid to form
salts.
Suitable acid addition salts are salts of all pharmacologically acceptable
acids, for
example halides, especially hydrochlorides, lactates, sulfates, citrates,
tartrates,
acetates, phosphates, methylsulfonates, p-toluenesulfonates.
The compounds I are substituted acylguanidines.
Compared with known compounds, the compounds of the invention are
distinguished
by exceptionally high activity in inhibition of Na+/H+ exchange.
Just like the known compounds, they have no undesired and disadvantageous
saiurific
properties but have very good antiarrhythmic properties as are important, for
example
for the treatment of disorders occurring in association with manifestations of
oxygen
deficiency. As a consequence of their pharmacological properties, the
compounds are
outstandingly suitable as antiarrhythmic medicaments with a cardioprotective
component for prophylaxis of infarction and treatment of infarction, and for
the
treatment of angina pectoris, and they also inhibit or greatly reduce in a
preventive
manner the pathophysiological processes associated with the development of
ischemia-induced damage, especially in the initiation of ischemia-induced
cardiac
arrhythmias. Because of their protective effects against pathological hypoxic
and
ischemic situations, the compounds of the invention, of the formula I, can be
used, as
a consequence of inhibition of the cellular Na+/H+ exchange mechanism, as
medicaments for the treatment of all acute or chronic damage induced by
ischemia or
disorders induced primarily or secondarily thereby. This relates to their use
as
medicaments for surgical operations, e.g. in organ transplants, where the
compounds
can be used both to protect the organs in the donor before and during removal,
to
protect removed organs for example during treatment with or storage thereof in
physiological bath fluids, and on transferring to the recipient organism. The
compounds
are likewise valuable medicaments with a protective effect when angioplastic
surgical
operations are performed for example on the heart and on peripheral vessels.
In
CA 02475093 2004-08-03
9
accordance with their protective effect against ischemia-induced damage, the
compounds are also suitable as medicaments for the treatment of ischemias of
the
nervous system, especially of the CNS, being suitable, for example, for the
treatment
of stroke or cerebral edema. In addition, the compounds of the invention, of
the
formula I, are likewise suitable for the treatment of types of shock such as,
for
example, of allergic, cardiogenic, hypovolemic and bacterial shock.
In addition, the compounds of the invention, of the formula I, are
distinguished by a
strong inhibitory effect on the proliferation of cells, for example fibroblast
cell
proliferation and the proliferation of smooth vascular muscle cells. The
compounds of
the formula I are therefore suitable as valuable therapeutic agents for
disorders in
which cell proliferation represents a primary or secondary cause, and can
therefore be
used as antiatherosclerotics, agents to prevent late complications of
diabetes, cancers,
fibrotic disorders such as pulmonary fibrosis, hepatic fibrosis or renal
fibrosis, organ
hypertrophies and hyperplasias, especially for prostate hyperplasia and
prostate
hypertrophy.
The compounds of the invention are effective inhibitors of the cellular sodium-
proton
antiporter (Na+/H+ exchanger) which in numerous disorders (essential
hypertension,
atherosclerosis, diabetes etc.) is also increased in cells which are readily
amenable to
measurements, such as, for example, in erythrocytes, platelets or leukocytes.
The
compounds of the invention are therefore suitable as excellent and simple
scientific
tools, for example in their use as diagnostic aids for the determination and
differentiation of particular forms of hypertension, but also of
atherosclerosis, of
diabetes, proliferative disorders etc. In addition, the compounds of the
formula I are
suitable for preventive therapy to prevent the development of high blood
pressure, for
example of essential hypertension.
It has additionally been found that compounds of the formula I show a
beneficial effect
on serum lipoproteins. It is generally acknowledged that blood lipid levels
which are too
high, so called hyperlipoproteinemias, represent a considerable risk factor
for the
development of arteriosclerotic vascular lesions, especially coronary heart
disease.
The lowering of elevated serum lipoproteins therefore has exceptional
importance for
the prophylaxis and regression of atherosclerotic lesions. Besides a reduction
in total
CA 02475093 2004-08-03
serum cholesterol, it is particularly important to reduce the proportion of
specific
atherogenic lipid fractions of this total cholesterol, especially the low
density
lipoproteins (LDL) and the very low density lipoproteins (VLDL), because these
lipid
fractions represent an atherogenie risk factoi. By e;ontrast, a protective
function against
5 coronary heart disease is ascribed to the high density lipoproteins.
Accordingly,
hypolipidemics should be able to reduce not only total cholesterol but, in
particular, the
VLDL and LDL serum cholesterol fractions. It has now been found that compounds
of
the formula I show valuable therapeutically utilizable properties in. relation
to the effect
on the serum lipid levels. Thus, they significantly reduce the elevated serum
LDL and
10 VLDL concentration which are to be observed for example due to increased
dietary
intake of a cholesterol- and lipid-rich diet or in association with
pathological metabolic
changes, for example, geneticaNy related hyperlipidemias. They can therefore
be used
for the prophylaxis and regression of atherosclerotic lesions through the
elimination of
a causal risk factor. These include not only primary hyperlipidemias but also
certain
secondary hyperlipidemias as occur, for example, in association with diabetes.
In
addition, the compounds of the formula I lead to a marked reduction in the
infarctions
induced by metabolic abnormalities and, in particular, a significant reduction
in the
induced infarct size and its severity. Compounds of the formula I further lead
to
effective protection against endothelial damage induced by metabolic
abnormalities.
This protection of vessels against the syndrome of endothelial dysfunction
makes
compounds of the formula I valuable medicaments for the prevention and
treatment of
coronary vasospasms, of atherogenesis and of atherosclerosis, of left-
ventricular
hypertrophy and of dilated cardiomyopathy, and thrombotic disorders.
Said compounds are therefore advantageously used for producing a medicament
for
the treatment of hypercholesterolemia; for producing a medicament for the
prevention
of atherogenesis; for producing a medicament for the prevention and treatment
of
atherosclerosis, for producing a medicament for the prevention and treatment
of
disorders induced by elevated cholesterol levels, for producing a medicament
for the
prevention and treatment of disorders induced by endothelial dysfunction, for
producing a medicament for the prevention and treatment of atherosclerosis-
induced
hypertension, for producing a medicament for the prevention and treatment of
atherosclerosis-induced thromboses, for producing a medicament for the
prevention
CA 02475093 2004-08-03
11
and treatment of hypercholesterolemia - induced and endothelial dysfunction -
induced
ischemic damage and postischemic reperfusion damage, for producing a
medicament
for the prevention and treatment of hypercholesterolemia - induced and
endothelial
dysfunction - induced cardiac hypertrophies. cardiorr~yopaties and congestive
heart
failure (CHF), for producing a medicament for the prevention and treatment of
hypercholesterolemia - induced and endothelial dysfunction - induced coronary
vasospasms and myocardial infarctions, for producing a medicament for the
treatment
of said disorders in combinations with hypotensive substances, preferably with
angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor
antagonists,
a combination of an NHE inhibitor of the formula ! with a hypolipidemic active
ingredient, preferably with an HMG-CoA reductase inhibitor (e.g. lovastatin or
pravastatin), the latter having a hypolipidemic effect and thus increasing the
hypolipidemic properties of the NHE inhibitor of the formula I proves to be a
favorable
combination with enhanced effect and reduced active ingredient usage.
The administration of sodiumlproton exchange inhibitors of the formula I as
novel
medicaments for reducing elevated blood lipid levels, and the combination of
sodium/proton exchange inhibitors with hypotensive medicaments and/or
medicaments
having hypolipidemic activity are claimed.
Also claimed are the administration of sodium/proton exchange inhibitors of
the
formula I, and the combination of sodium/proton exchange inhibitors with
hypotensive
medicaments, especially with ACE inhibitors (for example ramipril) and with
angiotensin receptor antagonists (for example losartan) as novel medicaments
for the
treatment of CHF.
Medicaments comprising a compound I can moreover be administered orally,
parenterally, intravenously, rectally or by inhalation, with the preferred
administration
being dependent on the particular appearance of the disorder. The compounds I
can
moreover be used alone or together with pharmaceutical excipients, both in
veterinary
medicine and in human medicine.
CA 02475093 2004-08-03
12
Excipients suitable for the desired pharmaceutical formulation are familiar to
the skilled
worker on the basis of his expert knowledge. Besides solvents, gel formers,
suppository bases, tablet excipients and other active ingredient carriers, it
is possible
to use, for example, antioxidants, dispersants, emulsifiers, antifoams,
masking flavors,
preservatives, solubilizers or colors.
For a form for oral use, the active compounds are mixed with the additives
suitable for
this purpose, such as carriers, stabilizers or inert diluents, and converted
by
convenional methods into suitable dosage forms such as tablets, coated
tablets, two-
piece capsules, aqueous, alcoholic or oily solutions. Examples of inert
carriers which
can be used are gum arabic, magnesia, magnesium carbonate, potassium
phosphate,
lactose, glucose or starch, especially corn starch. Preparation can moreover
take place
both as dry and as wet granules. Examples of suitable oily carriers or
solvents are
vegetable or animal oils such as sunflower oil or fish liver oil.
For subcutaneous or intravenous administration, the active compounds are
converted
into a solution, suspension or emulsion, if desired with the substances
customary for
this purpose, such as solubilizers, emulsifiers or other excipients. Examples
of suitable
solvents are: water, physiological saline or alcohol, e.g. ethanol, propanol,
glycerol,
either as sugar solutions such as glucos or mannitol solutions, or else a
mixture of the
various solvents mentioned.
Suitable as pharmaceutical formulation for administration in the form of
aerosols or
sprays are, for example, solutions, suspensions or emulsions of the active
ingredient of
the formula i in a pharmaceutically acceptable solvent such as, in particular,
ethanol or
water, or a mixture of such solvents. The formulation may if required also
comprise
other pharmaceutical excipients such as surfactants, emulsifiers and
stabilizers, and a
propellant gas. Such a preparation normally contains the active ingredient in
a
concentration of about 0.1 to 10, in particular of about 0.3 to 3 % by weight.
The dosage of the active ingredient of the formula I to be administered and
the
frequency of administration depend on the potency and duration of action of
the
compounds used; also on the nature and severity of the disorder to be treated,
and on
the sex, age, weight and individual response of the mammal to be treated.
CA 02475093 2004-08-03
13
On average, the daily dose of a compound of the formula I for a patient
weighing about
75 kg is at least 0.001 mg/kg, preferably 0.01 mg/kg, up to a maximum of 10
mglkg,
preferably up to a maximum of 1 mglkg, of bodyweight. For acute episodes of
the
disorder, for example immediately after suffering a myocardial infarction, it
may be
necessary for the dosages also to be higher and, in particular, more frequent,
e.g. up
to 4 single doses per day. Especially on i.v. use, for example for an infarct
patient in an
intensive care unit, up to 200 mg per day and kg of bodyweight may be
necessary.
List of abbreviations:
DIPEA diisopropylethylamine
DMA N,N-dimethylacetamide
DME 1,2-dimethoxyethane
DMF N,N-dimethylformamide
EA ethyl acetate (EtOAc)
eq. equivalent
MeOH methanol
Pd(dppf)2 [1,1'-bis(diphenylphosphino)ferrocene]palladium(II) chloridel-
methylene chloride complex (1:1 )
RT room temperature
m.p. melting point
THF tetrahydrofuran
Experimental part
General method for the synthesis of dihydrothiaphenanthrenecarbonylguanidines
Stage 1 ) Methyl 4-bromo-5-chlorosulfonyl-2-methylbenzoate
12 g of 4-bromo-5-chlorosulfonyl-2-methylbenzoic acid (J.Med.Chem. 1997, 40,
2017)
and 20 ml of thionyl chloride were boiled under refiux with exclusion of
moisture for
8 hours. The excess thionyl chloride was removed in vacuo in a rotary
evaporator, and
CA 02475093 2004-08-03
14
the residue was taken up in about 50 ml of dry toluene and again evaporated.
The
crude acid chloride was dissolved in 25 ml of anhydrous toluene and, after
addition of
1.7 ml of MeOH, stirred at 50°C for 2 h. A further 1.7 ml of MeOH were
then added,
followed by stirring at 50°C for 4 h. The reaction mixture was diluted
with 200 ml of EA
and washed with 100 ml of a saturated aqueous NaHC03 solution. After drying
over
Na2S04, the solvents were removed in vacuo. 11.0 g of a pale yellow oil were
obtained and were used without further purification.
Stage 2) 2-Bromo-5-methoxycarbonyl-4-methylbenzenesulfinic acid
550 mg of Na2S03 were dissolved in 2 ml of water and, at 70°C, a
solution of 337 mg
of methyl 4-bromo-5-chlorosulfonyl-2-methylbenzoate in 2 ml of DME was added
dropwise. During the dropwise addition, the solution became weakly acidic (pH
= 5).
The mixture was then stirred at 70°C for 2.5 h, allowed to cool and
adjusted to
pH = 1-2 with aqueous HCI solution. It was diluted with 50 ml of EA and washed
with
50 ml of a saturated aqueous NaCI solution. After drying over Na2S04, the
solvents
were removed in vacuo. 228 mg of a pale yellow oil were obtained and were used
without further purification.
Stage 3) Methyl 4-bromo-5-(2-bromophenylmethanesulfonyl)-2-methylbenzoate
150 mg (0.51 mmol) of 2-bromo-5-methoxycarbonyl-4-methylbenzenesulfinic acid
(stage 2) were dissolved in 1.5 ml of DMF: To this were added 128 mg (0.51
mmol) of
2-bromobenzyl bromide dissolved in 0.5 ml of DMF, and 0.1 ml (0.56 mMol) of
DIPEA,
and the mixture was stirred at room temperature with exclusion of moisture for
16 hours. The reaction solution was filtered, diluted with 20 ml of EA and
washed with
20 ml of 1 N hydrochloric acid and then 20 mi of 5% strength brine. The
organic phase
was forced through a drying cartridge (anhydrous sodium sulfate), and the
cartridge
was washed with 5 ml of EA. The filtrate was evaporated. The crude product was
purified by preparative HPLC.
CA 02475093 2004-08-03
In accordance with the general reaction equation
R7 0
Br ~ , o
Br R8 O
O~H ~ / O .~. ~ ~ II
S=O R6
O ,O Br R9 ~ R7
Br
Br / R8
R9
Product stage 2 Product stage 3
5
the following products were prepared analogously:
No.Benzyl bromide Product
2 1-Bromo-2- Methyl 4-bromo-5-(1-bromonaphthalen-2-
(bromomethyl)naphthaleneylmethanesulfonyl)-2-methylbenzoate
3 2-Bromo-4-methylbenzylMethyl4-bromo-5-(2-bromo-4-methyl-
bromide phenylmethanesulfonyl)-2-methylbenzoate
4 1-Bromo-2-bromomethyl-Methyl 4-bromo-5-(2-bromo-5-chloro-
4-chlorobenzene phenylmethanesulfonyl)-2-methylbenzoate
5 2-Bromo-1-bromomethyl-Methyl4-bromo-5-(2-bromo-4-fluoro-
4-fluorobenzene phenylmethanesulfonyl)-2-methylbenzoate
6 1-Bromo-2-bromomethyl-Methyl4-bromo-5-(2-bromo-6-fluoro-
3-fluorobenzene phenylmethanesulfonyl)-2-methylbenzoate
7 2-Bromo-1-bromomethyl-Methyl4-bromo-5-(2-bromo-4-chloro-
4-chlorobenzene phenylmethanesulfonyl)-2-methylbenzoate
8 1-Bromo-2-bromomethyl-Methyl4-bromo-5-(2-bromo-5-methoxy-
4-methoxybenzene phenylmethanesulfonyl)-2-methylbenzoate
CA 02475093 2004-08-03
16
9 1-Bromo-2-bromomethyi-Methyl4-bromo-5-(2-bromo-6-chloro-
3-chlorobenzene phenylmethanesulfonyl)-2-methylbenzoate
2-Bromo-1-bromomethyl-~Methyl4-bromo-5-(2-bromo-3-methyl-
3-methylbenzene phenylmethanesulfonyl)-2-methylbenzoate
11 1-Bromo-2-bromomethyl-Methyl4-bromo-5-(2-bromo-5-methyl-
4-methylbenzene phenylmethanesulfony()-2-methylbenzoate
Those benzyl bromides employed which could not be purchased were prepared
either
from the corresponding methyl aromatic compounds by free-radical bromination
with
N-bromosuccinimide or from the benzyl alcohols by reaction with aqueous HBr or
5 methanesulfonyl chloride/triethylamine followed by tetrabutylammonium
bromide.
The crude products were stirred with acetonitrilelwater = 9:1 (1 ml), possibly
with the
addition of 0.2 ml of DMF, filtered with suction through cartridges and washed
with
acetonitrile/water = 9:1 (0.5 ml). The precipitated solids were dried in a
vacuum oven at
50°C, and the purities were >80% according to HPLC/MS. The mother
liquors were
10 purified by preparative HPLC because they still contained a large
proportion of
product.
Stage 4) Methyl 6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
68 mg (0.266 mmoi) of bis(pinacolato)diboron, 71 mg (0.725 mmol) of potassium
acetate and 9 mg (0.012 mmol) of Pd(dppf)2 were introduced into 2 ml of DMA.
To this
were added 112 mg (0.242 mmol) of methyl 4-bromo-5-(2-bromophenylmethane-
sulfonyl)-2-methylbenzoate (stage 3) dissolved in 4 ml of DMA, and the mixture
was
stirred at 80°C under protective gas overnight. The reaction solution
was filtered
through silica gel and washed with 20 ml of EA. The organic phase was washed
with
water and 5% strength brine, dried over anhydrous sodium sulfate and
evaporated in
vacuo. The crude product was purified by preparative HPLC.
In accordance with the general reaction equation
CA 02475093 2004-08-03
17
\ \
O
O
O
I I
S=O R6
R7
Br
Br ~ R8 R6
R9 R8 R7
Product stage 3) Product stage 4)
the following products were prepared analogously:
No.Product
2 Methyl2-methyl-5,5-dioxo-5,6-dihydro-5-thiabenzo[c]phenanthrene-3-
carboxylate
3 Methyl3,6-dimethyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
4 Methyl2-chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-
7-carboxylate
Methyl3-fluoro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
CA 02475093 2004-08-03
18
6 Methyl1-fluoro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
7 Methyl3-chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-
7-carboxylate
8 Methyl2-methoxy-6-methyl-9,9-dioxo-9,10-dihydro-9-thia-
phenanthrene-7-carboxylate
9 Methyl1-chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-
7-carboxylate
Methyl4,6-dimethyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
11 Methy! 2,6-dimethyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carboxylate
Stage 5) Dihydrothiaphenanthrenecarbonylguanidines, general method
\ NH2
N
O NH2
H2N NH ~0
-' ~ ~ S' O
NH2 HCI
R6 R9-y i~--R6
5 R8 R7 R8
Product stage 4) Product stage 5)
53 mg (0.5 mmol) of potassium tert.-butoxide were suspended in 2 ml of dry
DMF. To
this were added 50 mg (0.55 mmol) of guanidine hydrochloride, and the
suspension
10 was stirred at RT with exclusion of moisture for 30 min. Then 0.1 mmol of
the methyl
ester from stage 4 dissolved in 1 ml of DMF was added, and the mixture was
stirred at
CA 02475093 2004-08-03
19
RT overnight. The precipitated salts were filtered off, and the filtrate was
immediately
purified by preparative HPLC (column material Merck Supersphere RP18e,
acetonitrile/water gradient with 0.1 % formic acid as buffer). The resulting
products
were characterized by analytical HRLCIMS urn ar~ hgiient sei iej 1100
systerr~.
Mass detection was carried out with positive ionization.
Method A:
Column: MERCK LiChroCart 55-2
Packing: PuroSpher STAR RP18
Flow rate: 0.75 ml/min
Temperature: 40 °C
Gradient:
Solvent A acetonitrile/water (90:10) + 0.5% formic acid
Solvent B acetonitrile/water (10:90) + 0.5% formic acid
Time Solv.
[min] B
[%]
0.00 95.0
0.50 95.0
1.75 5.0
4.25 5.0
4.50 95.0
5.00 95.0
6.20 STOP
Method B:
Column: YMC J'Sphere ODS H80
Packing: 4 ~.
Flow rate: 1.0 ml/min
CA 02475093 2004-08-03
Temperature: 30 °C
Gradient:
Solvent A /water + 0.05% trifluoroacetic acid
Solvent B acetoni.riie
5
Time Solv.
B
[min][%]
0.00 10.0
2.50 95.0
3.30 95.0
3.35 10.0
3.60 STOP
The title compounds of examples 1-11 were synthesized by the general method of
synthesizing dihydrothiaphenanthrenecarbonylguanidines:
Example 1: N-(6-Methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-carbonyl}-
guanidinium formate
H HN H O,S O
HO O H2N
O '_'
MS (ES) : 330 (M+1 )+ retention time 2.384 min (220 nm, method A)
Example 2: N-(2-Methyl-5,5-dioxo-5,6-dihydro-5-thiabenzo[c]phenanthrene-3-
carbonyl)guanidinium formate
CA 02475093 2004-08-03
21
O, , O
H HN
HO O H2N
MS (ES) : 380 (M+1 )+ retention time 1.793 min (220 nm, method B)
Example 3: N-(3,6-Dimethyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)-
guanidinium formate
O, ,,O
H HN H S
N
HO O H2N
O
MS ( ) : 344 (M+1 )+ retention time 2.411 min (220 nm, method A)
Example 4: N-(2-Chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)guanidinium formate
HN ~~S O
H ~-N
HOI '0 HN ~ ~ ~ ~ CI
/ ~ ~/
O
MS (ES) : 364 (M+1 )+ Retention time 2.390 min (220 nm, method A)
Example 5: N-(3-Fluoro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)guanidinium formate
CA 02475093 2004-08-03
22
H HN H ~~S O
N
HO O H2N
O
F
MS ES) : 348 (M+1 )+ retention time 2.215 min (220 nm, method A)
Example fi: N-(1-Fluoro-6-methyl-9,9-dioxo-9,10-dihydrd-9-thiaphenanthrene-7-
carbonyl)guanidinium formats
O
H HN H O S~ F
N
HO O H2N
O ~_'
MS (ES) : 348 (M+1 )+ retention time 2.218 min (220 nm, method A)
Example 7: N-(3-Chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)guanidinium formats
H HN H ~,S O
N
HO O H2N
O
CI
MS (ES) : 364 (M+1 )+ retention time 2.394 min (220 nm, method A)
Beispiel 8: N-(2-Methoxy-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)guanidinium formats
CA 02475093 2004-08-03
23
H HN H ~ S O
N ~ 1
HO O HzN -,- l
O '--
MS (ES) : 360 (M+1 )+ retention time 2.271 min (220 nm, method A)
Example 9: N-(1-Chloro-6-methyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)guanidinium formate
O
H HN H O S~ CI
1
HO O H2N
O
MS (ES) : 364 (M+1 )+ retention time 2.358 min (220 nm, method A)
Example 10: N-(4,6-Dimethyl-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)-
guanidinium formate
H H H ~,S,O
N
HO O H2N
O
MS (ES) : 344 (M+1 )+ retention time 2.302 min (220 nm, method A)
Example 11: N-(2,6-Dimethyi-9,9-dioxo-9,10-dihydro-9-thiaphenanthrene-7-
carbonyl)-
guanidinium formate
CA 02475093 2004-08-03
24
O, , O
H HN H S,
N
HO O H2N
O '='
MS (ES) : 344 (M+1 )+ retention time 2.671 min (220 nm, method A)
CA 02475093 2004-08-03
Jansen NHE inhibition method
The NHE-1 inhibition IC50 [nM] was determined as follows:
FLIPR assay for determining NHE-1 inhibitors by measurement of the recovery in
pHi
5 in transfected cell lines which express human NHE-1
The assay is carried out in an FLIPR (fluorescent imaging plate reader) with
black-
walled 96-well microtiter plates with clear bases. The transfected cell lines
expressing
the various NHE subtypes (the parental cell line LAP-1 [obtained from Prof.
10 Pouyssegur, Nice] shows no endogenous NHE activity as a result of
mutagenesis and
subsequent selection) are the preceding day at a density of --25 000
cellslwell.
[The growth medium for the transfected cells (Iscove +10% fetal calf serum)
additionally contains 6418 as selection antibiotic in order to ensure the
presence of the
transfected sequences.]
The actual assay starts with the removal of the growth medium and addition of
100 ~.I
of loading buffer per well (5 NM BCECF-AM [2',7'-bis(carboxyethyl)-5-(and-6)-
carboxyfluorescein, acetoxymethyl ester] in 20 mM NH4CI, 115 mM choline
chlorides,
1 mM MgCl2, 1 mM CaCl2, 5 mM KCI, 20 mM HEPES, 5 mM glucose; pH 7.4
[adjusted with KOH]). The cells are then incubated at 37°C for 20
minutes. This
incubation leads to loading of the cells with the fluorescent dye whose
fluorescence
intensity depends on pHi, and with NH4CI which makes the cells slightly
alkaline.
[The nonfluorescent dye precursor BCECF-AM is, as ester, membrane-permeable.
The actual dye BCECF is not membrane-permeable but is liberated inside cells
by
esterases.J
After this incubation for 20 minutes, the loading buffer which contains NH4C1
and free
BCECF-AM is removed by washing three times in a cell washer (Tecan Columbus)
with in each case 400 NI of washing buffer (133.8 mM choline chlorides, 4.7 mM
KCI,
1.25 mM MgCl2, 1.25 mM CaCl2, 0.97 mM K2HP04, 0.23 mM KH2P04, 5 mM
HEPES, 5 mM glucose; pH 7.4 [adjusted with KOH]). The residual volume
remaining in
CA 02475093 2004-08-03
26
the wells is 90 NI (50-125 NI possible). This washing step removes the free
BCECF-AM
and results, as a consequence of the removal of the external NH4+ ions, in
intracellular
acidification (-- pHi 6.3 - 6.4).
Since the equilibrium of intracellular NH4+ with NH3 and H+ is disturbed by
the
removal of the extracellular NH4* and by the subsequent instantaneous passage
of
the NH3 through the cell membrane, the washing process results in H+ remaining
inside the cells, which is the cause of the intracellular acidification. This
may eventually
Lead to cell death if it persists long enough.
It is important at this point that the washing buffer is sodium-free (<1 mM)
because
extracellular sodium ions would lead to an instantaneous recovery of the pHi
through
the activity of the cloned NHE isoforms.
It is likewise important for all the buffers used (loading buffer, washing
buffer, recovery
buffer) not to contain any HC03- ions, because the presence of bicarbonate
would
lead to activation of interfering bicarbonate-dependent pHi regulatory systems
present
in the parental LAP-1 cell line.
The microtiter plates with the acidified cells are then (up to 20 minutes
after the
acidification) transferred to the FLIPR. In the FLIPR, the intracellular
fluorescent dye is
excited by light with a wavelength of 488 nm generated by an argon laser, and
the
measured parameters (laser power, illumination time and aperture of the CCD
camera
incorporated in the FLIPR) are chosen so that the average fluorescence signal
per well
is between 30 000 and 35 000 relative fluorescence units.
The actual measurement in the FLIPR starts with a photograph being taken by
the
CCD camera every two under software control. After ten seconds, the recovery
of the
intracellular pH is initiated by adding 90 NI of recovery buffer (133.8 mM
NaCI, 4.7 mM
KCI, 1.25 mM MgCl2, 1.25 mM CaCl2, 0.97 mM K2HP04, 0.23 mM KH2P04, 10 mM
CA 02475093 2004-08-03
27
HEPES, 5 mM glucose; pH 7.4 [adjusted with NaOH]) by means of the 96-well
pipettor
incorporated in the FLIPR.
Positive control wells (100% NHE activity) are those to which pure recovery
buffer is
added, white negative controls (0% NHE activity) receive washing buffer.
Recovery
buffer with twice the concentration of test substance is added to all the
other wells.
Measurement in the FLIPR terminates after 60 measurements (two minutes).
The raw data are exported into the Activity Base program. This program firstly
calculates the NHE activities for each tested substance concentration and,
from these,
the IC50 values for the substances. Since the progress of pHi recovery is not
linear
throughout the experiment, but falls at the end owing to decreasing NHE
activity at
higher pHi values, it is important to select for evaluation of the measurement
the part in
which the increase in fluorescence of the positive controls is linear.
Example NHE-1 inhibition IC50
[nM]
- _._- X5.6__ _ _
2 24.2
3 8.6
4 10.8
5 15.6
6 16.5
7 11.7
8 10.2
_. 13.g
10 67.7
11 19.5