Note: Descriptions are shown in the official language in which they were submitted.
f
CA 02475320 2004-08-05 PCT/EP03/00782
L,.~C- I ~~ 3 'J ~ ~' +
-1-
QUINOXALINONES AND THEIR USE ESPECIALLY
IN THE TREATMENT OF CARDIOVASCULAR DISORDERS
The invention relates to quinoxalinones, to a process for their preparation
and to their
use for producing medicaments for the treatment and/or prophylaxis of
diseases,
especially of cardiovascular disorders.
Acetylcholine is the transmitter of the parasympathetic 'nervous system. This
part of
the autonomic nervous system has a crucial influence on fundamental processes
of a
wide variety of organ functions, such as, for example, lung, bladder, stomach
and
intestine, glands, brain, eye, blood vessels and heart.
Acetylcholine itself cannot be used therapeutically because it is very rapidly
inactivated by acetylcholin esterase, but its effect can be imitated by direct
parasympathomimetic agents such as, for example, carbachol. Active substances
which have an agonistic effect like acetylcholine on the muscarinic (M)
acetylcholine
receptors may thus influence and control numerous functions depending on the
organ
or tissue system. For example, activation of muscarinic acetylcholine
receptors in the
brain may influence the memory and learning processes and pain processing.
It is possible, for example, by using receptor subtype-specific agonists to
reduce via
the muscarinic M2 acetylcholine receptor, which is expressed particularly
strongly in
myocardial cells, the heart rate and the contractility after beta-adrenergic
stimulation
(B. Rauch, F. Niroomand, J. Eur. Heart. 1991, 12, 76-82). Both effects reduce
the
myocardial oxygen consumption.
Substances of similar structure to the compounds of the invention are known in
other
indications and for other mechanisms of action. Thus, for example, WO
00/00478,
EP-A 0 728 481 and EP-A 0 509 398 describe quinoxalinone derivatives for the
treatment of HIV infections, DE 4341663 describes quinoxalinone derivatives as
endothelin receptor antagonists, WO 98/09987 describes quinoxalinone
derivatives
as thrombin inhibitors, WO 94/11355 describes 3,4-dihydro-1-phenyl-2(1H)-
quinoxalinone derivatives for the treatment of cardiovascular disorders and
CA 02475320 2004-08-05
_2_
US 3,654,275 describes quinoxaline carboxamides as compounds having anti-
inflammatory activity.
It is an object of the present invention to provide medicaments for the
treatment of
disorders, especially cardiovascular disorders.
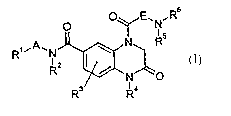
The present invention relates to compounds of the formula
.R6
R3 Ra
in which
A is a C~-C6-alkanediyl chain which is optionally substituted by one or two
hydroxy groups,
E is a C1-C6-alkanediyl chain,
R' is heteroaryl, where heteroaryl is optionally substituted by 1 to 3
substituents
independently of one another selected from the group consisting of halogen,
hydroxy, amino, trifluoromethyl, nitro, cyano, alkyl, alkoxy, alkylamino,
alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
Rz is hydrogen, alkyl or cycloalkyl,
R3 is hydrogen, halogen, alkyl or alkoxy,
R4 is alkyl or cycloalkyl, where alkyl and cycloalkyl are optionally
substituted
by 1 to 3 substituents independently of one another selected from the group
consisting of halogen, hydroxy, amino, alkyl, alkoxy, alkylamino,
CA 02475320 2004-08-05
-3-
alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
RS is hydrogen, alkyl or cycloalkyl,
and
R6 is alkyl, cycloalkyl, heterocyclyl, aryl or heteroaryl, where alkyl,
cycloalkyl,
heterocyclyl, aryl or heteroaryl are optionally substituted by 1 to 3
substituents independently of one another selected from the group consisting
of halogen, hydroxy, amino, trifluoromethyl, nitro, cyano, alkyl which may in
turn be substituted by one or two hydroxy groups, or alkoxy, alkylamino,
alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
or
R5 and R6 form together with the nitrogen atom to which they are bonded a
nitrogen-
containing heterocyclyl ring, where heterocyclyl is optionally substituted by
1
to 3 substituents independently of one another selected from the group
consisting of halogen, hydroxy, amino, trifluoromethyl, nitro, cyano, alkyl,
alkoxy, alkylamino, alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl.
The compounds of the invention may also exist in the form of their salts,
solvates or
solvates of these salts.
The compounds of the invention may exist, depending on their structure, in
stereoisomeric forms (enantiomers, diastereomers). The invention therefore
relates to
the enantiomers or diastereomers and respective mixtures thereof. The
stereoisomerically pure constituents can be isolated in a known manner from
such
mixtures of enantiomers and/or diastereomers.
The invention also relates, depending on the structure of the compounds, to
tautomers
of the compounds.
' CA 02475320 2004-08-05
-4-
Salts which are preferred for the purposes of the invention are
physiologically
acceptable salts of the compounds of the invention.
Physiologically acceptable salts of the compounds (~ of the invention comprise
acid
addition salts of mineral acids, carboxylic acids and sulphonic acids, for
example salts
of hydrochloric acid, hydrobromic acid, sulphuric acid, phosphoric acid,
methanesulphonic acid, ethanesulphonic acid, toluenesulphonic acid,
benzenesulphonic
acid, naphthalenedisulphonic acid, acetic acid, propionic acid, lactic acid,
tartaric acid,
malic acid, citric acid, fiunaric acid, malefic acid and benzoic acid.
Physiologically acceptable salts of the compounds (n of the invention also
comprise
salts of conventional bases such as, for example and advantageously, alkali
metal salts
(for example sodium and potassium salts), alkaline earth metal salts (for
example
calcium and magnesium salts) and ammonium salts derived from ammonia or
organic
amines with 1 to 16 C atoms, such as, for example and preferably, ethylamine,
diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine,
diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol,
procaine,
dibenzylamine, N-methylinorpholine, dihydroabiethylamine, arginine, lysine,
ethylenediamine and methylpiperidine.
Solvates refer for the purposes of the invention to those forms of the
compounds of the
invention which form a complex in the solid or liquid state through
coordination with
solvent molecules. Hydrates are a specific type of solvates in which the
coordination
takes place with water.
For the purposes of the present invention, unless otherwise specified, the
substituents
have the following meaning:
Alkyl per se and 'alk' and 'alkyl' in alkoxy, alkylamino, alkylaminocarbonyl
and
alkoxycarbonyl stand for a linear or branched alkyl radical with, usually, 1
to 6,
preferably 1 to 4, particularly preferably 1 to 3, carbon atoms, by way of
example
and preferably methyl, ethyl, n-propyl, isopropyl, tent-butyl, n-pentyl and n-
hexyl.
' CA 02475320 2004-08-05
-5-
Alkoxy is by way of example and preferably methoxy, ethoxy, n-propoxy,
isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
Alkylamino is an alkylamino radical with one or two alkyl substituents (chosen
independently of one another), by way of example and preferably methylamino,
ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino,
n-hexylamino, N,N dimethylamino, N,N diethylamino, N ethyl-N-methylamino, N
methyl-N n-propylamino, N isopropyl-N n-propylamino, N t-butyl-N methylamino,
N ethyl-N n-pentylamino and N n-hexyl-N-methylamino.
Alkylaminocarbonyl is an alkylaminocarbonyl radical with one or two alkyl
substituents (chosen independently of one another), by way of example and
preferably methylaminocarbonyl, ethylaminocarbonyl, n-propylaminocarbonyl,
isopropylaminocarbonyl, tert-butylaminocarbonyl, n-pentylaminocarbonyl, n-
hexyl-
aminocarbonyl, N,N dimethylaminocarbonyl, N,N diethylaminocarbonyl, N ethyl-
N-methylaminocarbonyl, N methyl-N n-propylaminocarbonyl, N isopropyl-
N n-propylaminocarbonyl, N t-butyl-N methylaminocarbonyl, N ethyl-N n-pentyl-
aminocarbonyl and N n-hexyl-N methylaminocarbonyl.
Alkoxycarbonyl is by way of example and preferably methoxycarbonyl,
ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl, tert-butoxycarbonyl,
n-pentoxycarbonyl and n-hexoxycarbonyl.
Alkanediyl is a straight-chain or branched saturated alkanediyl radical with 1
to 6
carbon atoms. A straight-chain or branched alkanediyl radical with 1 to 4
carbon
atoms is preferred. Mention may be made by way of example and preferably of
methylene, ethane-1,2-diyl, ethane-1,1-diyl, propane-1,3-diyl, propane-1,2-
diyl,
propane-2,2-diyl, butane-1,4-diyl, butane-1,3-diyl, butane-2,4-diyl, pentane-
1,5-diyl,
pentane-2,4-diyl, 2-methylpentane-2,4-diyl.
Cycloalk~~l is a cycloalkyl group with, usually, 3 to 8, preferably 3 to 6,
carbon
atoms, by way of example and preferably cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl and cycloheptyl.
CA 02475320 2004-08-05
A~1 is a mono-, bi- or tricyclic aromatic, carbocyclic radical with, usually,
6 to 14
carbon atoms, by way of example and preferably phenyl, naphthyl and
phenanthrenyl.
Heteroaryl is an aromatic, mono- or bicyclic radical with, usually, 5 to 10,
preferably
5 to 6, ring atoms and up to 5, preferably up to 4, heteroatoms from the
series S, O
and N, by way of example and preferably thienyl, furyl, pyrrolyl, thiazolyl,
oxazolyl,
imidazolyl, pyridyl, pyrimidyl, pyridazinyl, indolyl, indazolyl, benzofuranyl,
benzothiophenyl, quinolinyl, isoquinolinyl.
Heterocycl~ is a mono- or polycyclic, preferably mono- or bicyclic,
nonaromatic
heterocyclic radical with, usually, 4 to 10, preferably 5 to 8, ring atoms and
up to 3,
preferably up to 2, heteroatoms and/or heteroatomic groups from the series N,
O, S,
SO, SO2. The heterocyclyl radicals may be saturated or partially unsaturated.
5- to
8-membered, monocylic saturated heterocyclyl radicals with up to two
heteroatoms
from the series O, N and S are preferred, such as, by way of example and
preferably,
tetrahydrofuran-2-yl, pyrrolidin-2-yl, pyrrolidin-3-yl, pyrrolinyl,
piperidinyl,
morpholinyl, perhydroazepinyl.
Halogen is fluorine, chlorine, bromine and iodine.
A symbol * at a bond means the point of linkage in the molecule.
If radicals in the compounds of the invention are substituted, the radicals
may, unless
otherwise specified, be substituted one or more times identically or
differently.
Substitution with up to three identical or different substituents is
preferred.
Substitution with one substituent is very particularly preferred.
Preference is given to compounds of the formula (I)
in which
. CA 02475320 2004-08-05
A is a C~-C6-alkanediyl chain,
E is a Ci-C6-alkanediyl chain,
Rl is heteroaryl, where heteroaryl is optionally substituted by 1 to 3
substituents
independently of one another selected from the group consisting of halogen,
hydroxy, amino, trifluoromethyl, vitro, cyano, alkyl, alkoxy, alkylamino,
alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
RZ is hydrogen,
R3 is hydrogen,
R4 is alkyl or cycloalkyl, where alkyl and cycloalkyl are optionally
substituted
by 1 to 3 substituents independently of one another selected from the group
consisting of alkyl, alkoxy and alkylamino,
RS is hydrogen,
and
R6 is alkyl, cycloalkyl, heterocyclyl, aryl or heteroaryl, where alkyl,
cycloalkyl,
heterocyclyl, aryl or heteroaryl are optionally substituted by 1 to 3
substituents independently of one another selected from the group consisting
of halogen, hydroxy, amino, trifluoromethyl, vitro, cyano, alkyl, alkoxy,
alkylamino, alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
Particularly preferred compounds of the formula (I) are those
in which
A is methylene or ethane-1,1-diyl,
CA 02475320 2004-08-05
_g_
E is methylene,
Rl is 5- or 6-membered heteroaryl, where heteroaryl is optionally substituted
by
1 to 3 substituents independently of one another selected from the group
consisting of halogen, hydroxy, amino, alkyl, alkoxy, alkylamino,
alkoxycarbonyl, aminocarbonyl and alkylaminocarbonyl,
Rz is hydrogen,
R3 is hydrogen,
R4 is cycloalkyl, where cycloalkyl is optionally substituted by 1 or 2 alkyl
substituents,
RS is hydrogen,
and
R6 is alkyl or cycloalkyl, where cycloalkyl is optionally substituted by 1 to
3
substituents independently of one another selected from the group consisting
of alkyl, alkoxy, alkylamino, alkoxycarbonyl, aminocarbonyl and
alkylaminocarbonyl,
Very particularly preferred compounds of the formula (I) are those
in which
A is ethane-1,1-diyl,
E is methylene,
Rl is 6-membered heteroaryl, where heteroaryl is optionally substituted by 1
or 2
substituents independently of one another selected from the group consisting
CA 02475320 2004-08-05
-9-
of methyl, ethyl, methoxy and ethoxy,
Rz is hydrogen,
R3 is hydrogen,
R4 is cyclopropyl,
RS is hydrogen,
and
R6 is cyclohexyl or cyclopentyl, where cyclohexyl or cyclopentyl are
optionally
substituted by 1 or 2 substituents independently of one another selected from
the group consisting of methyl, ethyl, methoxy and ethoxy.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which A is ethane-1,1-diyl.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which Rl-A- is a radical of the formula
CH3
R1~~''~.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which E is methylene.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which R' is 6-membered heteroaryl, in particular pyridyl.
Compounds of the formula (I) which are likewise very particularly preferred
are
CA 02475320 2004-08-05
- 10-
those in which RZ is hydrogen.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which R3 is hydrogen.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which R4 is cycloalkyl, in particular cyclopropyl.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which RS is hydrogen.
Compounds of the formula (I) which are likewise very particularly preferred
are
those in which R6 is cyclohexyl or cyclopentyl, where cyclohexyl and
cyclopentyl
are optionally substituted by 1 or 2 substituents independently of one another
selected from the group consisting of methyl and ethyl.
The definitions of radicals indicated specifically in the respective
combinations or
preferred combinations of radicals are replaced irrespective of the particular
combinations indicated for the radicals as desired also by definitions of
radicals of
another combination.
Combinations of two or more of the abovementioned preferred ranges are very
particularly preferred.
The invention further relates to a process for preparing the compounds of the
formula (I), characterized in that compounds of the formula
O_ l E.N.Rs
N R$
N O
R4
- CA 02475320 2004-08-05
-11-
in which
E, R3, R4, RS and R6 have the meaning indicated above, are reacted
with compounds of the formula
R'~A~NH
R2 W
in which
A, R' and RZ have the meaning indicated above,
in the presence of conventional condensing agents, where appropriate in the
presence
of a base.
The reaction takes place where appropriate in inert solvents, preferably in a
temperature range from room temperature to 50°C under atmospheric
pressure.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, ethers such as diethyl ether, methyl tent-
butyl
ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
dimethyl
ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or
petroleum fractions, or other solvents such as nitromethane, ethyl acetate,
acetone,
dimethylformamide, dimethylacetamide, 1,2-dimethoxyethane, dimethyl
sulphoxide,
acetonitrile or pyridine, with preference for tetrahydrofuran,
dimethylformamide or
methylene chloride.
Examples of conventional condensing agents are carbodiimides such as, for
example,
N,N'-diethyl-, N,N'-dipropyl-, N,N'-diisopropyl-, N,N'-
dicyclohexylcarbodiimide,
N-(3-dimethylaminoisopropyl)-N'-ethylcarbodiimide hydrochloride (EDC), N-cyclo-
CA 02475320 2004-08-05
-12-
hexylcarbodiimide-N'-propyloxymethyl-polystyrene (PS-carbodiimide) or carbonyl
compounds such as carbonyldiimidazole, or 1,2-oxazolium compounds such as
2-ethyl-5-phenyl-1,2-oxazolium 3-sulphate or 2-tert-butyl-5-methylisoxazolium
perchlorate, or acylamino compounds such as 2-ethoxy-1-ethoxycarbonyl-
1,2-dihydroquinoline, or propanephosphonic anhydride, or isobutyl
chloroformate, or
bis(2-oxo-3-oxazolidinyl)phosphoryl chloride or benzotriazolyloxytri(dimethyl-
amino)phosphonium hexafluorophosphate, or O-(benzotriazol-1-yl)-N,N,N',N'-
tetra-
methyluronium hexafluorophosphate (HBTLI), 2-(2-oxo-1-(2H)-pyridyl)-1,1,3,3-
tetramethyluronium tetrafluoroborate (TPTLT) or O-(7-azabenzotriazol-1-yl)-
N,N,N',N'-tetramethyluronium hexafluorophosphate (HATLI), or 1-hydroxy-
benzotriazole (HOBt), or benzotriazol-1-yloxytris(dimethylamino)phosphonium
hexafluorophosphate (BOP), or mixtures thereof.
Examples of bases are alkali metal carbonates such as, for example, sodium or
potassium carbonate, or bicarbonate, or organic bases such as trialkylamines,
e.g.
triethylamine, N-methylmorpholine, N-methylpiperidine, 4-dimethylaminopyridine
or diisopropylethylamine.
The combination of N-(3-dimethylaminoisopropyl)-N'-ethylcarbodiimide hydro-
chloride (EDC), 1-hydroxybenzotriazole (HOBt) and diisopropylethylamine in
methylene chloride is preferred.
Compounds of the formula (III) are known or can be prepared in analogy to
known
processes.
The compounds of the formula (II) are prepared by reacting compounds of the
formula
Xt
H3
H (~~
O
CA 02475320 2004-08-05
-13-
in which
E, R3 and R4 have the meaning indicated above, and
X1 is halogen, preferably bromine or chlorine,
initially with compounds of the formula
s
HN~R
I s (~
R
in which
RS and R6 have the meaning indicated above,
and subsequently with trifluoroacetic acid to cleave the tert-butyl ester.
The reaction in the first stage takes place where appropriate in inert
solvents, where
appropriate in the presence of a base, preferably in a temperature range from
0°C to
SO°C under atmospheric pressure.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, ethers such as diethyl ether, methyl tent-
butyl
ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
dimethyl
ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or
petroleum fractions, or other solvents such as nitromethane, ethyl acetate,
acetone,
dimethylformamide, dimethylacetamide, 1,2-dimethoxyethane, 2-butanone,
dimethyl
sulphoxide, acetonitrile or pyridine, with preference for tetrahydrofuran or
methylene
chloride.
- CA 02475320 2004-08-05
' - 14-
Examples of bases are alkali metal hydroxides such as sodium or potassium
hydroxide, or alkali metal carbonates such as caesium carbonate, sodium or
potassium carbonate, or amides such as lithium diisopropylamide, or other
bases
such as DBU, triethylamine or diisopropylethylamine, preferably diisopropyl-
ethylamine or triethylamine.
The reaction in the second stage takes place in inert solvents, preferably in
a
temperature range from 0°C to 50°C under atmospheric pressure.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, or other solvents dimethylformamide or
tetrahydrofuran, with preference for methylene chloride.
Compounds of the formula (V) are known or can be prepared in analogy to known
processes.
Compounds of the formula
H C CH3 ~ ~~XZ
N
H O ~
3
N p
R3 Ra
which are compounds of the formula (IV) in which
E is methylene, and
R3, R6 and X1 have the meaning indicated above,
are prepared by reacting compounds of the formula
CA 02475320 2004-08-05
-15-
H C CH3 O
~ NH
' 'O
HsC
'NH
R3 R4
in which
R3 and R4 have the meaning indicated above,
with compounds of the formula
O
X'
X
in which
X1 has the meaning indicated above, and
XZ is halogen, preferably bromine or chlorine.
The reaction takes place in two stages. The reaction in the first stage takes
place in
inert solvents with 2 equivalents of the compounds of the formula (VII) based
on the
compounds of the formula (VI), in the presence of 2 equivalents of a base,
preferably
in a temperature range from 0°C to 50°C under atmospheric
pressure. The second
stage follows without working up the reaction mixture and takes place by
adding
another base.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, ethers such as diethyl ether, methyl tert-
butyl
ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
dimethyl
CA 02475320 2004-08-05
-16-
ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or
petroleum fractions, or other solvents such as nitromethane, ethyl acetate,
acetone,
dimethylformamide, dimethylacetamide, 1,2-dimethoxyethane, 2-butanone,
dimethyl
sulphoxide, acetonitrile or pyridine, with preference for methylene chloride.
Examples of bases are alkali metal hydroxides such as sodium or potassium
hydroxide, or alkali metal carbonates such as caesium carbonate, sodium or
potassium carbonate, or amides such as lithium diisopropylamide, or other
bases
such as DBU, triethylamine or diisopropylethylamine, preferably diisopropyl-
ethylamine or triethylamine for the first stage, and preferably DBU for the
second
stage.
Compounds of the formula (VII) are known or can be prepared in analogy to
known
processes.
Compounds of the formula (VI) are prepared by reacting compounds of the
formula
H C~ 3 O O+
N.p_
HaC
~NH
R3 Ra
in which
R3 and R4 have the meaning indicated above,
with reducing agents in the presence of a catalyst.
The reaction takes place in inert solvents, preferably in a temperature range
from
room temperature to 50°C under atmospheric pressure.
Examples of inert solvents are alcohols such as methanol, ethanol, propanol,
CA 02475320 2004-08-05
-17-
isopropanol or butanol or ethyl acetate or diethyl ether, with preference for
methanol
or ethanol.
The reducing agent is, for example, hydrogen; examples of catalysts are tin
dichloride, titanium trichloride or palladium on activated carbon. The
combination of
palladium on activated carbon and hydrogen is preferred.
Compounds of the formula (VIII) are prepared by reacting the compound of the
formula
H C C
3 ~ i
'o-
c C~
in which
R3 has the meaning indicated above,
with compounds of the formula
R4-NHz fix)
in which
R4 has the meaning indicated above,
where appropriate in the presence of a base.
The reaction takes place in inert solvents, preferably in a temperature range
from
room temperature to 50°C under atmospheric pressure.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
CA 02475320 2004-08-05
-18-
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, ethers such as diethyl ether, methyl tent-
butyl
ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
dimethyl
ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or
petroleum fractions, or other solvents such as nitromethane, ethyl acetate,
acetone,
dimethylformamide, dimethylacetamide, 1,2-dimethoxyethane, 2-butanone,
dimethyl
sulphoxide, acetonitrile or pyridine, with preference for tetrahydrofuran or
methylene
chloride.
Examples of bases are alkali metal hydroxides such as sodium or potassium
hydroxide, or alkali metal carbonates such as caesium carbonate, sodium or
potassium carbonate, or amides such as lithium diisopropylamide, or other
bases
such as DBU, triethylamine or diisopropylethylamine, preferably diisopropyl-
ethylamine or triethylamine.
Compounds of the formula (IX) and (X) are known or can be prepared in analogy
to
known processes.
In an alternative method for preparing the compounds of the formula (IV),
compounds of the formula
H C~ 3 C H
s ~ ~ N
C
'N O
R4
in which
R3 and R4 have the meaning indicated above, are reacted with compounds of the
formula
CA 02475320 2004-08-05
-19-
O
x~E~CI
in which
XI and E have the meaning indicated above,
where appropriate in the presence of a base.
The reaction takes place in inert solvents, preferably in a temperature range
from
room temperature to 50°C under atmospheric pressure.
Examples of inert solvents are halohydrocarbons such as methylene chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
di-
chloroethane or trichloroethylene, ethers such as diethyl ether, methyl tent-
butyl
ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
dimethyl
ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or
petroleum fractions, or other solvents such as nitromethane, ethyl acetate,
acetone,
dimethylformamide, dimethylacetamide, 1,2-dimethoxyethane, 2-butanone,
dimethyl
sulphoxide, acetonitrile or pyridine, with preference for tetrahydrofuran or
methylene
chloride.
Examples of bases are alkali metal hydroxides such as sodium or potassium
hydroxide, or alkali metal carbonates such as caesium carbonate, sodium or
potassium carbonate, or amides such as lithium diisopropylamide, or other
bases
such as DBU, triethylamine or diisopropylethylamine, preferably triethylamine
or
diisopropylethylamine.
Compounds of the formula (XI) are known or can be prepared in analogy to known
processes (for example as disclosed in EP-A 0 509 398).
Compounds of the formula (XII) are known or can be prepared in analogy to
known
CA 02475320 2004-08-05
-20-
processes.
The preparation of the compounds of the invention can be illustrated by the
following synthesis scheme.
Synthesis scheme:
CH3
H3C~CHs
O O THF H3C~ 3 O NO
\ I --~ H3C ~ ~ / 2
N02
F
Palladium on
activated carbon
(10°~), hydrogen,
methanol
or ethanol
1) 2 eq. of chloroacetyl chloride,
H C O O~CI 2 eq. of triethylamine, CH O
N dichloromethane, RT H3C
H3C'~"O ~ '~ H C~O ~ NH2
~ 3
CH3 / N_ 'O 2) DBU, reflux ~ /
N H.
THF
O N.~ O
O ~ Trifluoroacetic acid O '~N
H3C N H Dichloromethane, RT~ N H
H3C~O I ~ HO ~ \
CH3 / N O ~ N O
CA 02475320 2004-08-05
-21-
' 1-Hyd roxy-1 H-be nzotriazo le
1-(3-Dimethylaminopropyl}3-ethyl- ~C NHz
carbodiimide -hydrochloride
(EDGHCI) W
N, N-Diisopropylethylamine,
Dichlormethane~ N
~ F. Zaragoza, H. Stephensen, J. Org. Chem., 1999, 64, 2555-2557
describe the solid-phase synthesis of analogous carboxylic acid derivatives
@ Review of coupling methods: Y.S. Klausner, M. Bodansky, Synthesis 1972, 453-
463.
The compounds of the invention show a valuable range of pharmacological and
pharmacokinetic effects which could not have been predicted.
They are therefore suitable for use as medicaments for the treatment and/or
prophylaxis of diseases in humans and animals.
They are distinguished as agonists of the muscarinic M2 acetycholine receptor.
The compounds of the invention can by reason of their pharmacological
properties
by employed alone or in combination with other active ingredients for the
treatment
and/or prophylaxis of cardiovascular diseases, especially of coronary heart
disease,
angina pectoris, myocardial infarction, stroke, ateriosclerosis, essential,
pulmonary
and malignant hypertension, heart failure, heart failure, cardiac arrythmias
or
thromboembolic disorders.
They are additionally suitable for the treatment and/or prophylaxis of
disorders of the
eye (glaucoma), stomach and intestines (atonias), of the brain (e.g.
Parkinson's
disease, Alzheimer's disease, chronic sensation of pain), kidney failure or
erectile or
renal dysfunctions.
CA 02475320 2004-08-05
-22-
The present invention further relates to medicaments which comprise at least
one
compound of the invention, preferably together with one or more
pharmacologically
acceptable excipients or carriers, and to the use thereof for the
aforementioned
purposes.
The active ingredient may have systemic and/or local effects. It can for this
purpose
be administered in a suitable way such as, for example, by the oral,
parenteral,
pulmonary, nasal, sublingual, lingual, buccal, rectal, transdermal,
conjunctival or otic
route or as implant.
The active ingredient can be administered for these routes of administration
in
suitable administration forms.
Administration forms suitable for oral administration are known ones which
deliver
the active ingredient rapidly and/or in a modified manner, such as, for
example,
tablets (uncoated and coated tablets, e.g. with tablets provided with enteric
coatings
or film-coated tablets), capsules, sugar-coated tablets, granules, pellets,
powders,
emulsions, suspensions, solutions and aerosols.
Parenteral administration can take place with avoidance of an absorption step
(intravenous, intraarterial, intracardiac, intraspinal or intralumbar) or with
inclusion
of absorption (intramuscular, subcutaneous, intracutaneous, percutaneous, or
intraperitoneal). Administration forms suitable for parenteral administration
are, inter
alia, preparations for injection and infusion in the form of solutions,
suspensions,
emulsions, lyophilisates and sterile powders.
Examples suitable for the other routes of administration are pharmaceutical
forms for
inhalation (inter alia powder inhalers, nebulizers), nasal drops/solutions,
sprays;
tablets or capsules to be administered lingually, sublingually or buccally,
suppositories, preparations for the ears and eyes, vaginal capsules, aqueous
suspensions (lotions, shaking mixtures), lipophilic suspensions, ointments,
creams,
milk, pastes, dusting powders or implants, for example stems.
CA 02475320 2004-08-05
- 23 -
The active ingredients can be converted in a manner known per se into the
stated
administration forms. This takes place with use of inert, non-toxic,
pharmaceutically
suitable excipients. These include, inter alia, carriers (for example
microcrystalline
cellulose), solvents (for example liquid polyethylene glycols), emulsifiers
(for
example sodium dodecyl sulphate), dispersants (for example
polyvinylpyrrolidone),
synthetic and natural biopolymers (for example albumin), stabilizers (for
example
antioxidants such as ascorbic acid), colours (for example inorganic pigments
such as
iron oxides) or masking flavours andlor odours.
It has generally proved advantageous to administer on parenteral
administration
amounts of about 0.0001 to 10 mg/kg, preferably about 0.001 to 1 mg/kg, of
body
weight to achieve effective results. The amount on oral administration is
about 0.1 to
10 mg/kg, preferably about 0.5 to S mg/kg, of body weight.
It may nevertheless be necessary where appropriate to deviate from the amount
mentioned, in particular as a function of the body weight, route of
administration,
individual response to the active ingredient, nature of the preparation and
time or
interval level in which administration takes place. Thus, it may be sufficient
in some
cases to make do with less than the aforementioned minimum amount, whereas in
other cases the stated upper limit must be exceeded. It may in the event of
administration of larger amounts be advisable to divide these into a plurality
of
individual doses over the day.
The percentage data in the following tests and examples are, unless otherwise
indicated, percentages by weight; parts are parts by weight. Solvent ratios,
dilution
ratios and concentration data on liquid/liquid solutions are in each case
based on
volume.
CA 02475320 2004-08-05
-24-
A. Examples
Abbreviations:
aq. aqueous
Boc tent-butoxycarbonyl
CDCl3 deuterochloroform
DMSO dimethyl sulphoxide
DMF dimethylformamide
EDC N'-(3-dimethylaminopropyl)-N ethylcarbodiimide
x HCl
eq. equivalent
ESI electrospray ionization (in MS)
HATU O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate
HBTU O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate
HOBt 1-hydroxy-1H-benzotriazole x H20
h hour
HPLC high pressure, high performance liquid chromatography
LC-MS coupled liquid chromatography and mass spectroscopy
MS mass spectroscopy
MeOH methanol
M.p. melting point
NMR nuclear magnetic resonance spectroscopy
Pd/C palladium/carbon
Rf retention index (in TLC)
RT room temperature
R, retention time (in HPLC)
sat. saturated
CA 02475320 2004-08-05
- 25 -
HPLC and LC-MS methods:
Method 1:
Instrument: HP 1100 with DAD detection; column: Kromasil RP-18,
600 mm x 2 mm, 3.5 l,un; eluent A = 5 ml HC104/1 HZO, B = ACN; gradient: 0 min
2%B, 0.5 min 2%B, 4.5 min 90%B, 6.5 min 90%B; flow rate: 0.75 ml/min, temp.:
30 degrees C, UV detection: 210 nm.
Method 2:
Instrument: Micromass Platform LCZ, HP 1100; column: Symmetry C 18,
50 mm x 2.1 mm, 3.5 ~.m; eluent B: water + 0.1 % formic acid, eluent A:
acetonitrile
+ 0.1 % formic acid; gradient: 0.0 min 10%A ~ 4.0 min 90%A ~ 6.0 min 90%A;
oven: 40°C, flow rate: 0.5 ml/min, UV detection: 208-400 nm.
Method 3:
Instrument: Finnigan MAT 900S, TSP: P4000,AS3000,LTV30000HR; column:
Symmetry C 18, 150 mm x 2.1 mm, 5.0 ~.m; eluent C: water, eluent B: water +
0.6 g
35% HCI, eluent A: acetonitrile; gradient: 0.0 min 2%A, 49%B, 49%C --~ 2.5 min
95%A, 2.5%B, 2.5%C -~ 5.5 min 2%A, 49%B, 49%C; oven: 70°C, flow rate:
1.2 ml/min, UV detection: 210 nm.
Method 4:
Instrument: Micromass Quattro LCZ, HP 1100; column: Symmetry C 18,
50 mm x 2.1 mm, 3.5 Vim; eluent A: acetonitrile + 0.1% formic acid, eluent B:
water
+ 0.1 % formic acid; gradient: 0.0 min 10%A ~ 4.0 min 90%A ~ 6.0 min 90%A;
oven: 40°C, flow rate: 0.5 ml/min, UV detection: 208-400 nm.
Method 5:
Prep. HPLC enantiomer separation; Packing material: Daicel Chiralpak
AD 250*20 mm ID: ADOOCJ-GL001; used in g: 0.13; eluent A: iso-hexane,
eluent B: ethanol + 0.2% DEA; gradient: O.Omin 30%A-~ 21 min 30%A; injected
volume: 1000 ~1 temperature: 25°C, flow rate: 10 ml/min, wavelength:
250 nm,
range: 1.
CA 02475320 2004-08-05
-26-
Method 6:
Instrument: Micromass Platform LCZ, HP1100; column: Symmetry C18,
50 mm x 2.1 mm, 3.5 Vim; eluent A: water + 0.05% formic acid, eluent B:
acetonitrile + 0.05% formic acid; gradient: 0.0 min 90%A ~ 4.0 min 10%A ~
6.0 min 10%A; oven: 40°C, flow rate: 0.5 ml/min, W detection: 208-400
nm.
Method 7:
Instrument: Micromass Quattro LCZ. HP1100; column: Symmetry C18,
50 mm x 2.1 mm, 3.5 Vim; eluent A: water + 0.05% formic acid, eluent B:
acetonitrile + 0.05% formic acid; gradient: 0.0 min 90%A -~ 4.0 min 10%A ~
6.0 min 10% A; oven: 40°C, flow rate 0.5 ml/min, UV detection: 208-400
nm.
Method 8:
Instrument: MS Micromass ZQ; HPLC: Waters Alliance 2790; column: Symmetry C
18, 50 mm x 2.1 mm, 3.5 ~.m; eluent B: acetonitrile + 0.05% formic acid,
eluent A:
water + 0.05% formic acid; gradient: 0.0 min 10%B -~ 3.5 min 90%B --~ 5.5 min
90%B; oven: 50°C, flow rate: 0.8 ml/min, LTV detection: 210 nm.
CA 02475320 2004-08-05
-27-
Starting compounds:
Example I
tert-Butyl 4-fluoro-3-nitrobenzoate
H3C CH3 O
~ N ~Z
H C _O
3
/ F
21.5 g (116.15 mmol) of 4-fluoro-3-nitrobenzoic acid and 30.5 g (139.4 mmol)
of
t-butyl trichloroacetimidate are introduced under an argon atmosphere into 250
ml of
diethyl ether. 0.64 g (4.52 mmol) of boron trifluoride-diethyl ether complex
is added
dropwise, and the mixture is stirred at room temperature for 16 hours.
6 g of solid sodium bicarbonate are added to the reaction mixture, which is
concentrated in vacuo. The resulting residue is purified by chromatography on
silica
gel (mobile phase gradient cyclohexane -~ cyclohexane/ethyl acetate 1:1).
17.8 g (64% of theory) of product are obtained.
'H NMR (300 MHz, DMSO-d6): 8 = 1.57 (s, 9H), 7.2 (dd, 1H), 8.25-8.3 (m, 1H),
8.52 (dd, 1H)
MS (ESIpos): m/z = 242 (M+H)+
HPLC (method 1): Rt = 5.07 min
Example II
tert-Butyl4-(cyclopropylamine)-3-nitrobenzoate
CH3 O
~ NOz
H3C' 1 _O
CH3
/ N--
H
CA 02475320 2004-08-05
-28-
7.8 g (32.3 mmol) of the compound from Example I are introduced into 150 ml of
tetrahydrofuran. At 0°C, a solution of 3.88 g (67.9 mmol) of
cyclopropylamine in
50 ml of tetrahydrofuran is added. The mixture is stirred at 0°C for 30
minutes and
S then at room temperature for 16 hours.
The reaction mixture is concentrated in vacuo. The residue is taken up in 500
ml of
ethyl acetate and washed three times with 100 ml of water and once with 100 ml
of
saturated sodium chloride solution. The solution is dried over sodium sulphate
and
concentrated in vacuo.
8.95 g (99% of theory) of product are obtained.
1H NMR (300 MHz, DMSO-db): 8 = 0.64-0.69 (m, 2H), 0.87-0.93 (m, 2H), 1.54 (s,
9H), 2.67-2.71 (m, 1H), 7.44 (d, 1H), 8.01 (dd, 1H), 8.33 (br. s, 1H), 8.54
(d, 1H).
MS (DCI): m/z = 296 (M+NH4)+
HPLC (method 1): Rt = 5.47 min
Alternative synthesis to Example II:
1 g (3.88 mmol) of the compound from Example XXXIII is introduced into
tetrahydrofuran (15 ml). 0.44 g (7.76 mmol) of cyclopropylamine is added at
room
temperature. The solution is stirred at 55°C for 2 hours. The mixture
is then poured
into ice-water (50 ml). The precipitated solid is filtered off with suction
and dried.
0.65 g (58% of theory) of product is obtained.
The compounds listed in Table 1 are prepared in analogy to the compound from
Example II.
CA 02475320 2004-08-05
-29-
Table 1
Example Structure Analytical data
L1T IH-NMR (300MHz, DMSO-d6):
8 =
H C CH3 ~ 1.28 (d, 6H), 1.53 (s,
N0 9I~, 3.94-4.01
H3C~O I ~ (m, 1H), 7.16 {d, 1H),
2 7.93 {dd, 1H~,
NH 8.15 (d, 1H), 8.56 (d,
1H)
C"CH MS (DCI]: mlz = 298 (M+NH4)+
H
9 HPLC ethod l~ = 5.59 min
3
rV iH-NMR {300MHz, DMSO-ds):
8 =
H C CH3 O 1.53 (s, 9H), 1.73-1.87
NO {m, 2H), 1.98-
H3C~.D l . ~ 2.12 (m, 2I~, 2.4-2.51
Z (m, 2H), 4.13-
~ NH 4.25 (m, 1I~, 6.98 (d,
1H), 7.92 (dd,
1H), 8.30 (d, 1H), 8.55
(d, 1H)
MS (ESIpos): m/z = 293
(M+I~+
HPLC CMethod 1~: Rr =
5.48 min
V 1H-NMR (300MHz, DMSO-ds):
8 =
H C i H3 O 1.53 (s, 9H), 1.58-I.72
NO (m, 6H), 2.08-
3
C~O [ ~ 2.12 (m, 2I~, 4.07-4.12
Z (m, 1H), 7.16
H
3
N H (d~ 1 H), 7.94 (dd, 1
H), 8 .24 {d, 1 H),
8.55 (d, 1H)
MS {ESIpos): m/z = 307
(M+H)+
HPLC (Method 17: Rr =
5.65 min
CA 02475320 2004-08-05
-30-
Example VI
tert-Butyl 3-amino-4-(cyclopropylamine)benzoate
CH3 O
~ NHZ
H3C_ ' _O
CH3
N--
H
8.85 g (31.8 mmol) of the compound from Example II are introduced into 400 ml
of
methanol under an argon atmosphere, and 0.30 g (1.33 mmol) of palladium on
activated carbon (10% Pd) is added. The mixture is stirred under a hydrogen
atmosphere at atmospheric pressure overnight. The mixture is filtered through
Celite,
and the filtrate is concentrated in vacuo. The product resulting after drying
under
high vacuum for 2 hours (8.20 g, 94% of theory) is reacted further without
delay.
1H NMR (200 MHz, DMSO-d6): 8 = 0.37-0.47 (m, 2H), 0.68-0.80 (m, 2H), 1.48 (s,
9H), 2.35-2.45 (m, 1H), 4.67 (br. s, 2H), 5.64 (br. s, 1H), 6.75 (d, 1H), 7.08
(d, 1H),
7.15 (dd, 1H)
MS (ESIpos): m/z = 249 (M+H)+
HPLC (method 1): Rt = 4.18 min
The compounds listed in Table 2 are prepared in analogy to the compound from
Example 1V. Ethanol is used as solvent. The resulting products are reacted
further
without delay.
CA 02475320 2004-08-05
-31-
Table 2
Example Structure Analytical data
VII IH-NMR (300MHz, DMSO-db): S =
1.I7 (d, 6H), 1.48 (s, 9H), 3.58-3.69
NHz
(m, 1H), 4.67 (s, 2H), 4.80 (d, lI-I),
6.42 (d, 1H), 7.09 (d, 1H), 7.13 (dd,
~c C~ 1~
MS (ESIpos): mlz = 251 (M+I-~+
HPLC (Method 1 }: R; = 4.02 min
VIB 1H-NMR (300MHz, DMSO-d6): b =
H3C_ I s O NH 1.48 (s, 9H), 1.68-1.95 (m, 4H),
H3C~O I ~ 2 2.30-2.42 (m, 2H), 3.82-3.94 (m,
NH 1~~ 4.70 (s, 2H), 5.30 (d, 1H), 6.29
(d, 1H), 7.08-7.12 (m, 2H)
MS (ESIpos): m/z = 263 (M+H)+
HPLC (Method 1): Rt = 4.08 min
TX MS {ESIpos): m/z = 277 (M+H)+
H C i H' O HPLC (Method 1 ): Rt = 4? 6 ~nin
H3C~O I ~ NH2
NH
CA 02475320 2004-08-05
-32-
Example X
tert-Butyl 4-(chloroacetyl)-1-cyclopropyl-2-oxo-1,2,3,4-tetrahydro-6-
quinoxaline-
carboxylate
0s n
CH3
H3C- 1 _O
CH3
7.90 g (31.8 mmol) of the compound from Example VI are introduced into 200 ml
of
dichloromethane and, at 0°C, 8.98 g (79.5 mmol) of chloroacetyl
chloride are added.
The mixture is stirred at room temperature for 30 minutes. At 0°C,
11.2 ml
(79.5 mmol) of triethylamine are added. The mixture is stirred at room
temperature
for 4 hours. Then 7.12 ml (7.26 g, 47.7 mmol) of 1,8-diazabicyclo(5.4.0)undec-
7-ene
are added, and the mixture is heated to reflux. After 16 hours, the reaction
mixture is
cooled and concentrated in vacuo.
The residue is purified by chromatography on silica gel (mobile phase gradient
cyclohexane -~ cyclohexane/ethyl acetate l:l).
4.69 g (62% of theory) of product are obtained as an amorphous solid.
1H NMR (400 MHz, CDCl3): 8 = 0.68-0.72 (m, 2H), 1.16-1.21 (m, 2H), 1.60 (s,
9H),
2.78-2.82 (m, 1H), 4.21 (s, 2H), 4.51 (br. s, 2H), 7.46 (d, 1H), 7.98 (m, 2H)
MS (DCI): m/z = 382 (M+NH4)+
HPLC (method 1 ): Rt = 4.72 min
CA 02475320 2004-08-05
-33-
Example XI
tent-Butyl 4-(chloroacetyl)-1-isopropyl-2-oxo-1,2,3,4-tetrahydro-6-quinoxaline-
carboxylate
O
CH3 O ~CI
~ N
HCI1'O
CH3 ~ /~
_N O
H C- 'CH
3 3
Preparation takes place in analogy to Example X from the appropriate
precursors.
The mixture is worked up by washing three times with 100 ml of water and once
with 100 ml of saturated sodium chloride solution. The organic phase is dried
over
sodium sulphate and concentrated in vacuo.
The residue is purified by chromatography on silica gel (mobile phase gradient
cyclohexane ~ cyclohexane/ethyl acetate 2:1 ).
1H NMR (300 MHz, DMSO-d6): 8 = 1.45 (d, 6H), 1.54 (s, 9H), 4.37 (s, 2H), 4.51
(br. s, 2H), 4.61 (quintet, 1H), 7.49 (d, 1H), 7.81 (dd, 1H), 8.12 (d, 1H)
MS (DCI]: m/z = 384 (M+NH4)+
HPLC (method 1): Rt = 4.71 min
CA 02475320 2004-08-05
-34-
Example XII
tent-Butyl 4-(chloroacetyl)-1-cyclopentyl-2-oxo-1,2,3,4-tetrahydro-6-
quinoxaline-
carboxylate
4s n _.
H3C
Preparation takes place in analogy to Example X from the appropriate
precursors.
The mixture is worked up by washing three times with 100 ml of water and once
with 100 ml of saturated sodium chloride solution. The organic phase is dried
over
sodium sulphate and concentrated in vacuo.
5.63 g (61 % of theory) of a dark brown foam are obtained. The product is
reacted
further without further purification.
DC: Rf= 0.24 (cyclohexane/ethyl acetate S:1)
Example XIII
tent-Butyl 4-(chloro acetyl)-1-c yclobutyl-2-oxo-1,2, 3,4-tetrahydro-6-
quinoxaline-
carboxylate
O
CH3 O ~Cf
~ N
H3C_ 1 _O
CH3
_N O
CA 02475320 2004-08-05
-35-
Preparation takes place in analogy to Example X from the appropriate
precursors.
The mixture is worked up by washing three times with 100 ml of water and once
with 100 ml of saturated sodium chloride solution. The organic phase is dried
over
sodium sulphate and concentrated in vacuo.
5.42 g (82% of theory) of dark brown foam are obtained. The product is reacted
further without further purification.
Rf= 0.53 (cyclohexane/ethyl acetate 2:1)
Example XIV
tent-Butyl 4-(N-cyclopentylglycyl)-1-cyclopropyl-2-oxo-1,2,3,4-tetrahydro-6-
quin-
oxalinecarboxylate
O
cH3 0
~ N
N3CI 1 _O
CH3
_N O
0.10 g (0.27 mmol) of the compound from Example X and 0.07 g (0.82 mmol) of
cyclopentylamine are dissolved in 10 ml of tetrahydrofuran, and the mixture is
stirred
at 40°C for 6 hours. It is left to stand at room temperature for 12
hours.
The reaction mixture is concentrated in vacuo. The residue is taken up in 20
ml of
ethyl acetate and washed twice with 10 ml of saturated sodium chloride
solution. The
organic phase is dried over sodium sulphate and concentrated in vacuo. The
resulting
product is reacted further immediately without further purification.
LCMS (method 2): Rt = 2.85 min
MS (ESIpos): m/z = 414 (M+H)+
CA 02475320 2004-08-05
-36-
The compounds listed in Table 3 are prepared in analogy to the compound from
Example XN from the appropriate precursors and reacted further without
purification.
Table 3
Example Structure Analytical data
XY LCMS (Method 6): R~ = 3.29
~~ O O H Ells
H c c~ I ~ N~ ca' MS (ESIpos): m/z = 442
N O ~~"~+
XVI LCMS (Method 2): R= = 3.06
min
MS (ESIpos): mlz = 442
~,~~+
c o ° N
~H
~c
N O
A'VII ~ LCMS (Method 7): Rt = 2.98
cIHa °~ min
MS (ESIpos): m/z = 400
CM+~+
o
XVIII ~ LCMS (Method 'n: R~= 332
~, O O~H CH, nun
N
"'c cry, ~ ~ ~ MS (ESIpos): m/z = 442
o ~+~+
CA 02475320 2004-08-05
-37-
Example XIX
tert-Butyl 4-(N-cyclohexylglycyl)-1-cyclopropyl-2-oxo-1,2,3,4-tetrahydro-6-
quin-
oxalinecarboxylate
Ow n N
H
H
O
4.5 g (12.33 mmol) of the compound from Example X and 4.24 ml (3.67 g,
37.0 mmol) of cyclohexylamine are dissolved in 175 ml of tetrahydrofuran, and
the
mixture is left to stand at room temperature for 72 h.
The reaction mixture is concentrated in vacuo. The residue is taken up in 400
ml of
ethyl acetate and washed with water (3 x 100 ml) and once with saturated
sodium
chloride solution (100 ml). The solution is dried over sodium sulphate and
concentrated in vacuo.
It is purified by chromatography on silica gel [mobile phase gradient
dichloromethane -~ dichloromethane/methanol 7.5% (v/v)J.
5.30 g (92% of theory) of product are obtained.
1H NMR (300 MHz, DMSO-db): b =0.50-0.57 (m, 2H), 0.85-1.90 (m, 12H, 1.54 (s,
9H), 2.22-2.37 (m, 1H), 2.78-2.85 (m, 1H), 3.49 (br. s, 2H), 4.42 (s, 2H),
7.52 (d,
1H), 7.83 (dd, 1H), 8.06 (s, 1H)
MS (ESIpos): m/z = 428 (M+H)+
HPLC (method 1): Rt = 4.42 min
CA 02475320 2004-08-05
-38-
Example XX
tent-Butyl 4-(N-cyclohexylglycyl)-1-isopropyl-2-oxo-1,2,3,4-tetrahydro-6-quin-
oxalinecarboxylate
O\~
CH3 O
IN
H3C~0
CH3
'N O
H3C- 'CH3
300 mg (0.82 mmol) of the compound from Example XI and 243 mg (2.45 mmol) of
cyclohexylamine are stirred in 10 ml of tetrahydrofuran at room temperature
for 16
hours.
The reaction mixture is worked up by concentration in vacuo. The residue is
purified
by chromatography on silica gel [mobile phase gradient: dichloromethane
dichloromethane/methanol 7.5% (v/v)).
LCMS (method 2): Rt = 3.27 min
MS (ESIpos): m/z = 430 (M+H)+
Example XXI
tent-Butyl 4-(N-cyclopentylglycyl)-1-isopropyl-2-oxo-1,2,3,4-tetrahydro-6-quin-
oxalinecarboxylate
O
CH3 O ~H
~ N
H3C- 1 l0
CH3
'N O
H3C_ _CH3
The compound is prepared in analogy to Example XX from the appropriate
CA 02475320 2004-08-05
-39-
precursors.
LCMS (method 7): Rt = 3.16 min
MS (ESIpos): m/z = 416 (M+H)+
Example XXII
tert-Butyl 4-(N-cyclohexylglycyl)-1-cyclopentyl-2-oxo-1,2,3,4-tetrahydro-6-
quin-
oxalinecarboxylate
CH ~~ ~N
H
HC p .
CH3
0
to
The compound is prepared as described from Example XIX from the appropriate
precursors. The resulting crude product is purified by chromatography on
silica gel
[mobile phase dichloromethane -~ dichloromethane/methanol 5% (v/v)]. A pale
brown oil (410 mg, 21 % of theory) is obtained as product.
'H NMR (400 MHz, DMSO-db): 8 =0.90-2.10 (m, 27H), 2.31 (m, 1H), 2.42 (m, 1H),
3.53 (br. s, 2H), 4.39 (br., s, 2H), 4.36 (quintet, 2H), 7.43 (d, 1H), 7.79
(dd, 1H), 8.08
(br., s, 1 H)
MS (ESIpos): m/z = 456 (M+H)+
CA 02475320 2004-08-05
-40-
Example XXIII
tent-Butyl 4-(N-cyclohexylglycyl)-1-cyclobutyl-2-oxo-1,2,3,4-tetrahydro-6-quin-
oxalinecarboxylate
CH3 O
H3C~0
CH3
O
The synthesis takes place as described for the compound in Example XIX.
The resulting crude product is purified by chromatography on silica gel mobile
phase
gradient [dichloromethane ~ dichloromethane/methanol 5% (v/v)].
A brown oil (701 mg (21 % of theory) is obtained as product.
1H NMR (400 MHz, DMSO-db): 8 = 0.90-1.24 (m, 6H), 1.54 (s, 9H), 1.45-1.82 (m,
8H), 2.00-2.18 (mn, 2H), 2.22-2.40 (m, 2H), 3.56 (br. s, 2H), 4.37 (br. s,
2H), 4.45
(quintet, 1 H), 7.14 (d, 1 H), 7.76 (d, 1 H), 8.07 (br. s, 1 H).
MS (ESIpos): m/z = 442 (M+H)+
CA 02475320 2004-08-05
-41 -
Example XXIV
4-(N-Cyclop entylglycyl)-1-cyclopropyl-2-oxo-1,2, 3,4-tetrahydro-6-quinox
aline-
carboxylic acid
O
O ~~ H
N
Ho
N O
0.11 g (0.27 mmol) of the compound from Example XIX is mixed with 2 ml of a
mixture of trifluoroacetic acid and dichloromethane in the ratio 1:1. The
mixture is
stirred at room temperature for 30 minutes.
The solution is concentrated in vacuo and dried under high vacuum. The residue
is
taken up in 10 ml of a 1:1 dichloromethane/methanol mixture and stirred with 1
g of
solid sodium bicarbonate for 60 minutes. The mixture is diluted with 20 ml of
dichloromethane and filtered with suction, and the filtrate is concentrated.
The
resulting product is reacted further without further purification.
LCMS (method 3): Rt = 0.84 min
MS (ESIpos): m/z = 358 (M+H)+
The compounds listed in Table 4 are prepared analogously from the appropriate
precursors and either immediately reacted further or purified by preparative
HPLC
(column material: GROM-SIL 120 OSD4 HE, 10 Vim; mobile phase gradient
acetonitrile:water 10:90 ~ 95:5).
CA 02475320 2004-08-05
- 42 -
Table 4
Example Structure Analytical data
XXV LCMS (Method 3): Rt
= 0.83
0 o~N min
H
HO \ N MS (ESIpos): mlz =
I / N~O 386
CM+~+
XXVI 'H-NMR (300MHz, DMSO-d6):
O O~N b = 0.50-0.63 (m,
2I~, 0.95-
HO ~ N H 1.30 (m, 7H), 1.45-1.90
~ (m,
/ N~0 5H), 2.40-2.50 (m,
lid, 2.78-
2.85 (m, 1H), 3.67
(br. s, 2H),
4.43 (s, 2H), 7.52
(d, 1H), 7.88
(d, 1H), 8.12 (s,
1H)
MS (ESIpos): m/z=372
(M+~+
HPLC (Method 1): Ri
= 3.64
X~.'VII ~ LCMS (Method 4): Ri=
2.64
O O~H min
HO ~ ~ N~ MS (ESIpos): m/z =
374
N~.O (M+H)+
H3C~CH3
X~'VIL1 LCMS (Method: 6):
~ Rt= 3.17
O min
O ~N
H
N MS (ESIpos): m/z =
HO 400
/ N~O
~+~+
CA 02475320 2004-08-05
- 43 -
Example Structure Analytical data
XXIX LCMS (Method 6): Rt= 3.03
0 o~N min
~ N N MS (ESIpos): m/z = 3 86
(M+~+
N
XXX ~ LCMS (Method 6): R~= 2.7s
O °~~ CH3
HO ~ ~ N1 MS (ESIpos): m/z=386
N o (M+H~T
LCMS (Method $) 1~= l.so
° °~~~ »in
HO ~ N
MS (ESIpos): m/z= 360
N ° (M+I~+
H3C~CH3
XXXII LCMS (Method 6): R~ = 3.48
O O~ N min
N
N~ CH3 MS (ESIpos): m/z = 386
N o (M+~+
Example XXXIII
tert-Butyl 4-chloro-3-nitrobezoate
O
H3C
NOZ
H3C~--O
H3C
CI
10 g (4s.4s mmol) of 4-chloro-3-nitrobenzoyl chloride are dissolved in DMF
CA 02475320 2004-08-05
-44-
(100 ml). 5.10 g of potassium tent-butoxide are added in portions at room
temperature. The solution is stirred at room temperature for one hour. The
mixture is
then poured in portions into ice-water (S00 ml). The precipitated solid is
filtered off
with suction and dried.
7.1 g (60% of theory) of product are obtained.
1H NMR (300 MHz, DMSO-d6): 8 = 1.59 (s, 9H), 7.9 (dd, 1H), 8.18 (m, 1H), 8.49
(dd, 1 H).
MS (ESIpos): m/z = 258 (M+H)+
HPLC (method 1): Rt = 5.10 min
CA 02475320 2004-08-05
- 45 -
R~ Method Mass 'H-NMR
xampl Structure
1H-NMR (400MHz,
off DMSO-d6): d =
0.48-0.60 {m, 2H),
0.83-1.22 {m, 5H),
~s 0 H .57-
H,C~ ~H 1,54 {s, 9I~, I
C p w H 1.70 {m, SIB, 2.18
XJOQV ~ j 2.81 8 444 230 (m, lI~, 2.80
0 2.88 {m, 2H), 3.50
(hr. s, 2I~, 3.55 (m,
1H], 4.42 (s, 2H),
737 (d, 1H), 7S2
(dd, IH), 7.88 (dd,
1
1~ ~ (400
OH DMSO-d6): d =
0.48-0.60 (m, 2H),
O O 0.83-1 ~ (m, 5H),
1.57-1.70 (m, SH),
HO ~ ~~ 2.18-2.30 (m, 1H),
X3C~CV ~ / 195 8 387 2.80-2.88 (m, 2H),
O . . 3.50 (br. s, 2H),
3.55 (m, III, 4.42
(s, 2H), 7.37 (d,
1H), 7.52 (dd, 1H),
7.88 (dd, 1H~, 9.88
(s, 1H).
0~ ~'CHy
H3C~0 0 rv H LhCH3
3
XXXYI 3 ~ ~ ~ 0 293 1 416
3
HO
1.93 1 360
N O
H3C'CI~ 0 O~H~CH3
H3C~O \ N CH3
I I / N~O 3.01 8 434
CA 02475320 2004-08-05
-46-
xample Structure Re Method Mass 'H-NMR
' [min] LCMS (M+H)
O O~H~CH3
\ N CN3
HO
1.99 1 378
N 0
~C CHI O~H
pH 3.15 1 457
~, HaC~O ~ N
_N O
O O~H
XLI HO I ~ N~ OH 226 1 401
N O
O~ ~CH3
HaC~~ O \ 'N( H C~Ha H3
~~ HOC O I ~ ~ 2.98 1 430
N O
O Cti3
O ~H CHCH3
XLIB ~ I ~ ~ 3 1.95 1 374
N O
CA 02475320 2004-08-05
-47-
xample Structure Re Method Mass 1H-NMit
[minj LCMS {M+H)
O O~tNi~C~
H30~
XI,N N3C"O \ N CH3 2.93 1 415
0
O O~~~CH3
N 01-!3
XI,V ~O ~ ~ L94 1 359
N O
(300 MHz, CDC13):
d=7.87(d,1=10
H~ CIH3 O . O~~CH' Hz, 113], 72s (s,
H3~o \ N O~ ~ 1H), 6.92 (d, J = 10
( ~ Hz, IH) 4.48-4,39
\~N~O 2.9s 8 430 (m, 3H), 3.64 (s,
2H), 2.60 (sb, ZH),
236 (s, 2H), 22s
2.21 (m, 2H), 1.88
1.81 (m, 2H), 1.80
(s, 9I~, Ø94 (s, 9H)
O CH3
O yN-lj~OH3
HO
XLVII
NI 'O C~ 2.45 g 373
(M-HCI)
vn
O O N.,,
~H
~~ H,c o ~ % ~ 2.s3 s 4ss
N o
CA 02475320 2004-08-05
-48-
xampl Structure ~ Method Mass 1H-NMR
LcMS ~IVt+g)
OH
o ~..~
o
~ HO I ~ ~ Cl 1.97 g 401
(M-HCn
N O
O
Hs~Ha O ~H
H3C O f / OH 2.85 8 472
N O
O O~N
N Hz
LI HO ~ i ~ CI OH Z42 8 (Iyj HC1)
N O
CA 02475320 2004-08-05
-49-
Exemplary embodiments:
Example 1
4-(N-Cyclohexylglycyl)-1-cyclopropyl-N-(3-furylmethyl)-2-ox o-1, 2,3,4-
tetrahydro-
6-quinoxalinecarboxamide
° °~H
N
,J H ~~.
° N o
200 mg (0.27 mmol) of the compound of Example XXV are introduced into 10 ml of
dichloromethane and, at room temperature, 36.4 mg (0.27 mmol) of 1-hydroxy-
1H-benzotriazole and 51.4 mg (0.27 mmol) of 1-(3-dimethylaminopropyl)-3-ethyl-
carbodiimide hydrochloride are added. After stirring for 20 minutes, 34.8 mg
(0.27 mmol) of N,N-diisopropylethylamine and 26.2 mg (0.27 mmol) of
3-aminomethylfuran are added. The mixture is stirred at room temperature for
16 hours.
The reaction mixture is mixed with 20 ml of dichloromethane and washed once
with
ml of water and once with 20 ml of saturated sodium chloride solution. It is
dried
over sodium sulphate and concentrated in vacuo.
20 The residue is purified by chromatography on silica gel [mobile phase
gradient
dichloromethane -~ dichloromethane/methanol 10% (v/v)].
51 mg (42% of theory) of product are obtained.
1H NMR (300 MHz, DMSO-d6): 8 = 0.48-0.57 (m, 2H), 0.79-1.70 (m, 12H),
2.19-2.30 (m, 1H), 2.79-2.87 (m, 1H), 3.51 (s, 2H), 4.31 (d, 2H), 4.41 (s,
2H), 6.45
(s, 1H), 7.50 (d, 1H), 7.58 (s, 2H), 7.83 (d, 1H), 8.02 (s, 1H), 8.80 (t, 1H).
CA 02475320 2004-08-05
-50-
MS (ESIpos): m/z = 451 (M+H)+
HPLC (method 1): Rt = 3.92 min
Example 2
4-(N-Cyclobutylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
O.
H
Preparation takes place from the appropriate precursors as described for
Example 1.
The dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (300 MHz, DMSO-d6): b = 0.48-0.57 (m, 2H), 1.02-1.12 (m, 2H), 1.23 (s,
1H), 1.45-1.65 (m, 7H), 1.9-2.0 (m, 2H), 2.79-2.87 (m, 1H), 3.08-3.15 (m, 1H),
3.45
(br. s, 2H), 4.4 (s, 2H), 5.2 (quintet, 1H), 7.35 (dd, 1H), 7.51 (d, 1H), 7.78
(d, 1H),
7.87 (d, 1 H), 8.04 (br. s, 1 H), 8.44 (dd, 1 H), 8.6 (d, 1 H), 8.81 (d, 1 H).
MS (ESIpos): m/z = 448 (M+H)+
HPLC (method 1 ): Rt = 3.19 min
CA 02475320 2004-08-05
-51-
Example 3
4-(N-Cyclopentylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
O~ n
N
H
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (300 MHz, DMSO-d6): b = 0.48-0.58 (m, 2H), 1.05-1.3 (m, 4H), 1.33-1.7
(m, 9H), 2.78-3.0 (m, 2H), 3.5 (s, 2H), 4.42 (s, 2H), 5.14 (quintet, 1H), 7.37
(d, 2H),
7.52 (d, 1H); 7.89 (d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.83 (d, 1H).
MS (ESIpos): m/z = 462 (M+H)+
HPLC (method 1 ): Rt = 3.28 min
Example 4
4-(N-Cyclopentylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
O
CH3 O ~H
\ H I \ N
y /
N ~N O
CA 02475320 2004-08-05
-52-
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
'H NMR (300 MHz, DMSO-d6): 8 = 0.48-0.58 (m, 2H), 1.05-1.25 (m, 5H),
1.33-1.65 (m, 8H), 2.78-3.0 (m, 2H), 3.48 (br. s, 2H), 4.42 (s, 2H), 5.20
(quintet,
1 H), 7.3 5 (dd, 1 H), 7.51 (d, 1 H), 7.78 (dt, 1 H), 7. 87 (d, 1 H), 8.05
(br. s, 1 H), 8.44
(dd, 1H), 8.60 (d, 1H), 8.82 (d, 1H).
MS (ESIpos): m/z = 462 (M+H)+
HPLC (method 1): Rt = 3.28 min
Example 5
4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
O~ nN
H
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (200 MHz, DMSO-db): 8 = 0.48-0.60 (m, 2H), 0.8-1.25 (m, 8H), 1.40-1.75
(m, 8H), 2.13-2.36 (m, 1H), 2.78-2.9 (m, 1H), 3.51 (br. s, 2H), 4.43 (s, 2H),
5.13
(quintet, 1H), 7.37 (d, 2H), 7.52 (d, 1H), 7.88 (d, 1H), 8.07 (br. s, 1H),
8.50 (d, 2H),
CA 02475320 2004-08-05
-53-
8.89 (d, 1H).
MS (ESIpos): m/z = 476 (M+H)+
HPLC (method 1): Rt = 3.57 min
Example 6
4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CH3 O
I \ H I \ N
N / ~ N O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (400 MHz, DMSO-db): $ = 0.48-0.60 (m, 2H), 0.83-1.22 (m, 7H), 1.48
(d, 3H), 1.52-1.70 (m, 5H), 2.18-2.30 (m, 1H), 2.80-2.88 (m, 1H), 3.50 (br. s,
2H),
4.42 (s, 2H), 5.13 (quintet, 1H), 7.37 (d, 2H), 7.52 (d, 1H), 7.88 (d, 1H),
8.07 (br. s,
1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 476 (M+H)+
HPLC (method 1): Rt = 3.56 min
CA 02475320 2004-08-05
-54-
Example 7
(+)-4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
O, n
CH3
N / 'H
The product is obtained as fraction 2 by chromatographic separation (method 5)
of
the enantiomers of the compound from Example 6.
1H NMR (400 MHz, DMSO-db): 8 = 0.52 (s, 2H), 0.80-0.98 (m, 2H), 1.02-1.18 (m,
6H), 1.48 (d, 3H), 1.53-1.70 (m, 4H), 1.78-1.90 (m, 1H), 2.18-2.30 (m, 1H),
2.78-2.88 (m, 1H), 3.51 (br. s, 2H), 4.42 (s, 2H), 5.14 (quintet, 1H), 7.36
(d, 2H),
7.51 (d, 1H), 7.87 (d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 476 (M+H)+
HPLC (method 1): Rt = 3.41 min
Specific rotation: +29.7° (ethanol, T = 20.7°C)
The second product as fraction 1 is: (-)-4-(N-cyclohexylglycyl)-1-cyclopropyl-
2-oxo-
N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
'H NMR (400 MHz, DMSO-db): 8 = 0.52 (s, 2H), 0.80-0.98 (m, 2H), 1.02-1.18 (m,
6H), 1.48 (d, 3H), 1.53-1.70 (m, 4H), 1.78-1.90 (m, 1H), 2.18-2.30 (m, 1H),
2.78-2.88 (m, 1H), 3.51 (br. s, 2H), 4.42 (s, 2H), 5.14 (quintet, 1H), 7.36
(d, 2H),
7.51 (d, 1H), 7.87 (d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 476 (M+H)+
CA 02475320 2004-08-05
-55-
HPLC (method 1): Rt = 3.41 min
Specific rotation: -36.2° (ethanol, T = 20.9°C)
Example 8
1-Cyclopropyl-4-[N-(2-methylcyclohexyl)glycyl]-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
O_,
C~;
CH3
N /
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (300 MHz, DMSO-d6): 8 = 0.48-0.60 (m, 2H), 0.76-1.40 (m, 11H), 1.48
(d, 3H), 1.52-1.65 (m, 4H), 1.82-1.95 (m, 1H), 2.80-2.88 (m, 1H), 3.42-3.60
(m, 2H),
4.43 (s, 2H), 5.14 (quintet, 1H), 7.36 (d, 2H), 7.52 (d, 1H), 7.88 (d, 1H),
8.07 (br. s,
1H), 8.49 (d, 2H), 8.83 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): R, = 3.67 min
CA 02475320 2004-08-05
- -56-
Example 9
1-Cyclopropyl-4-[N-(2-methylcyclohexyl)glycyl]-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH3 4 C~H
C \ H I \ N CHa
NJ ~N O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (300 MHz, DMSO-d6): 8 = 0.58-0.68 (m, 2H), 0.72-1.35 (m, 13H), 1.51
(d, 3H), 1.52-1.65 (m, 1H), 1.82-1.95 (m, 1H), 2.76-2.88 (m, 1H), 3.42-3.60
(m, 2H),
4.43 (s, 2H), 5.20 (quintet, 1H), 7.35 (dd, 2H), 7.51 (d, 1H), 7.78 (d, 1H),
7.85 (d,
1 H), 8.06 (br. s, 1 H), 8.44 (dd, 1 H), 8.60 (d, 1 H), 8. 81 (d, 1 H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.68 min
Example 10
1-Cyclopropyl-4-[N-(3-methylcyclohexyl)glycyl]-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CA 02475320 2004-08-05
-57-
O, n
H CHa
O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
'H NMR (300 MHz, DMSO-d6): 8 = 0.48-0.58 (m, 2H), 0.68-0.95 (m, SH),
1.02-1.38 (m, 6H), 1.48 (d, 3H), 1.45-1.72 (m, 4H), 2.20-2.32 (m, 1H), 2.79-
2.88 (m,
1H), 3.42-3.58 (m, 2H), 4.42 (s, 2H), 5.14 (quintet, 1H), 7.36 (d, 2H), 7.51
(d, 1H),
7.88 (d, 1H), 8.08 (br. s, 1H), 8.50 (dd, 2H), 8.83 (d, 1H):
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.71 min
Example 11
1-Cyclopropyl-4-[N-(3-methylcyclohexyl)glycyl]-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH ~ ~CH3
N
Preparation takes place in analogy to the preparation of the compound from
CA 02475320 2004-08-05
-58-
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
'H NMR (300 MHz, DMSO-d6): 8 = 0.48-0.58 (m, 2H), 0.68-0.95 (m, 5H),
1.02-1.38 (m, 6H), 1.51 (d, 3H), 1.53-1.75 (m, 4H), 2.20-2.32 (m, 1H), 2.79-
2.88 (m,
1H), 3.42-3.58 (m, 2H), 4.41 (s, 2H), 5.20 (quintet, 1H), 7.34 (dd, 1H), 7.51
(d, 1H),
7.78 (d, 1H), 7.86 (d, 1H), 8.06 (br. s, 1H), 8.44 (dd, 1H), 8.60 (d, 1H),
8.81 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.72 min
Example 12
4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-(3-thienylmethyl)-1,2,3,4-tetra-
hydro-6-quinoxalinecarboxamide
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (300 MHz, DMSO-db): 8 = 0.48-0.58 (m, 2H), 0.82-0.95 (m, 2H),
1.05-1.18 (m, 5H), 1.23 (s, 1H), 1.45-1.72 (m, 5H), 2.20-2.32 (m, 1H), 2.79-
2.88 (m,
1H), 3.51 (s, 2H), 4.42 (s, 2H), 4.47 (d, 2H), 7.08 (dd, 1H), 7.29-7.33 (m,
1H),
7.45-7.50 (m, 1H), 7.52 (s, 1H), 7.85 (d, 1H), 8.04 (br. s, 1H), 8.93 (t, 1H).
CA 02475320 2004-08-05
-59-
MS (ESIpos): m/z = 467 (M+H)+
HPLC (method 1 ): Rt = 4.05 min
Example 13
4-(N-Cyclohexylglycyl)-1-isopropyl-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CHa O O~H
w
I \ H I \ N
'~ N O
Ha
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 5:1 is used as
solvent.
1H NMR (200 MHz, DMSO-db): 8 = 0.70-1.85 (m, 12H), 1.17 (d, 3H), 1.47 (t, 6H),
3.54 (br. s, 2H), 4.37 (s, 2H), 4.60 (quintet, 1H), 5.14 (quintet, 1H), 7.36
(d, 2H),
7.50 (d, 1H), 7.85 (d, 1H), 8.10 (br. s, 1H), 8.50 (d, 2H), 8.92 (d, 1H).
MS (ESIpos): m/z = 478 (M+H)+
HPLC (method 1): R, = 3.63 min
Example 14
4-(N-Cycloheptylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CA 02475320 2004-08-05
-60-
CHa O D~H
N
i~ ~H ~ /
N 'N ~O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
1H NMR (400 MHz, DMSO-d6): 8 = 0.53 (s, 2H), 1.03-1.65 (m, 16H), 1.48 (d, 3H),
2.79-2.87 (m, 1H), 3.49 (br. s, 2H), 4.42 (s, 2H), 5.14 (quintet, 1H), 7.36
(d, 2H),
7.52 (d, 1H), 7.88 (d, 1H), 8.08 (br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.58 min
Example 15
4-(N-Cycloheptylglycyl)-1-cyclopropyl-2-oxo-N-[1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CH3 O O~H
N
i~ ~H ~ /
N 'N ~O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
CA 02475320 2004-08-05
-61 -
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (400 MHz, DMSO-d6): 8 = 0.52 (s, 2H), 1.03-1.88 (m, 16H), 1.51 (d, 3H),
2.80-2.85 (m, 1H), 3.47 (br. s, 2H), 4.41 (s, 2H), 5.19 (quintet, 1H), 7.34
(dd, 1H),
7.65 (dd, 2H), 7.85 (d, 1H), 8.06 (br. s, 1H), 8.63 (dd, 2H), 8.60 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.58 min
Example 16
4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CH3
I ~~ \H
N
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (400 MHz, DMSO-d6): 8 = 0.82-1.20 (m, 6H), 1.51 (d, 3H), 1.53-2.05
(m, 13H), 2.25-2.38 (m, 1H), 3.58 (s, 2H), 4.38 (br. s, 2H), 4.63 (quintet,
1H), 5.20
(quintet, 1H), 7.34 (dd, 1H), 7.60 (dd, 2H), 7.78 (d, 1H}, 8.09 (br. s, 1H),
8.44 (d,
1H}, 8.60 (d, 1H), 8.86 (d, 1H}.
MS (ESIpos): m/z = 504 (M+H)+
- CA 02475320 2004-08-05
-62-
HPLC (method 1): Rt = 3.79 min
Example 17
1-Cyclobutyl-4-(N-cyclohexylglycyl)-2-oxo-N-[ 1-(4-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CH3 O O~H
I \ H I \ N
N~ ''~'N O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
1H NMR (400 MHz, DMSO-d6): 8 = 0.80-1.13 (m, 8H), 1.48 (d, 3H), 1.49-2.12 (m,
9H), 2.25-2.33 (m, 1H), 3.55 (br. s, 2H), 4.30-4.50 (m, 3H), 5.13 (quintet,
1H), 7.13
(d, 1H), 7.81 (d, 1H), 7.90 (dd, 4H), 8.08 (br. s, 1H), 8.87 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.64 min
Example 18
1-Cyclobutyl-4-(N-cyclohexylglycyl)-2-oxo-N-[ 1-(3-pyridinyl)ethyl]-1,2,3,4-
tetra-
hydro-6-quinoxalinecarboxamide
CA 02475320 2004-08-05
- 63 -
CH3 O O~H
H I ~ N
N ~ N O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
A dichloromethane/dimethylformamide mixture in the ratio 2:1 is used as
solvent.
'H NMR (400 MHz, DMSO-d6): 8 = 0.80-1.30 (m, 6H), 1.51 (d, 3H), 1.52-1.92 (m,
9H), 2.00-2.18 (m, 2H), 2.22-2.36 (m, 1H), 3.55 (br. s, 2H), 4.37 (br. s, 2H),
4.45
(quintet, 1 H), 5 .19 (quintet, 1 H), 7.13 (d, 1 H), 7.3 2-7.3 7 (m, 1 H), 7.
77-7. 81 (m, 2H),
8.07 (br. s, 1 H), 8.84 (d, 1 H), 8.60 (d, 1 H), 8.85 (d, 1 H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.63 min
Example 19
4-(N-Cyclohexylglycyl)-1-cyclopropyl-N-[ 1-(6-methyl-3-pyridinyl)ethyl]-2-oxo-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH3 O O~N
H ~ ~ N
H3C N ~N O
Preparation takes place in analogy to the preparation of the compound from
CA 02475320 2004-08-05
-64-
Example 1 from the appropriate precursors.
'H NMR (400 MHz, DMSO-d6): S = 0.52 (s, 2H), 0.80-1.20 (m, 8H), 1.49 (d, 3H),
1.52-1.92 (m, 5H), 2.15-2.30 (m, 1H), 2.43 (s, 3H), 2.82 (s, 1H), 3.49 (br. s,
2H),
4.42 (s, 2H), 5.15 (quintet, 1 H), 7.19 (d, 1 H), 7.5 0 (d, 1 H), 7.66 (d, 1
H), 7. 8 5 (d, 1 H),
8.04 (br. s, 1H), 8.45 (s, 1H), 8.78 (d, 1H).
MS (ESIpos): m/z = 490 (M+H)+
HPLC (method 1): Rt = 3.50 min
Example 20
4-(N-Cyclohexylglycyl)-1-cyclopropyl-N-[ 1-(6-methoxy-3-pyridinyl)ethyl]-2-oxo-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH3
~N
H
H3C~~ N
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
1H NMR (400 MHz, DMSO-db): 8 = 0.51 (s, 2H), 0.80-0.98 (m, 2H), 1.00-1.20 (m,
6H), 1.48 (d, 3H), 1.52-1.89 (m, 5H), 2.15-2.30 (m, 1H), 2.82 (s, 1H), 3.49
(br. s,
2H), 3.81 (s, 3H), 4.41 (s, 2H), 5.14 (quintet, 1H), 6.78 (d, 1H), 7.49 (d,
1H), 7.73
(dd, 1H), 7.83 (d, 1H), 8.04 (br. s, 1H), 8.16 (d, 1H), 8.75 (d, 1H).
MS (ESIpos): m/z = 506 (M+H)+
HPLC (method 1): Rt = 3.58 min
CA 02475320 2004-08-05
-65-
Example 21
4-(N-Cyclohexylglycyl)-1-cyclopropyl-2-oxo-N-(3-pyridinylmethyl]-1,2,3,4-tetra-
hydro-6-quinoxalinecarboxamide
I ~ H
N
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors.
The crude product is purified by preparative HPLC chromatography [column
material: GROM-SIL 120 OSD4 HE, 10 pm, mobile phase gradient
acetonitrile/water 10:90 ~ 95:5 (v/v)].
1H NMR (300 MHz, DMSO-d6): 8 = 0.53 (m, 2H), 0.79-1.28 (m, 6H), 1.40-1.76
(m, 6H), 2.26 (m, 1H), 2.82 (m, 1H), 3.51 (s, 2H), 4.42 (s, 2H), 4.51 (d, 2H),
7.35
(dd, 1H), 7.52 (d, 1H), 7.72 (dt, 1H), 7.86 (d, 1H), 8.05 (br. s, 1H), 8.45
(dd, 1H),
8.56 (d, 1H), 9.05 (dd, 1H).
MS (ESIpos): m/z = 462 (M+H)+
CA 02475320 2004-08-05
-66-
Example 22
4-N-[(trans-4-Cyclohexanoyl)glycyl]-1-cyclopropyl-2-oxo-N-[ 1-S-(4-pyridinyl)-
ethyl]-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
,,.~H
N
H
O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
1H NMR (400 MHz, DMSO-d6): 8 = 0.48-0.60 (m, 2H), 0.83-1.22 (m, SH), 1.48
(d, 3H), 1.52-1.70 (m, 5H), 2.18-2.30 (m, 1H), 2.80-2,88 (m, 2H), 3.50 (br. s,
2H),
3.55 (m, 1H), 4.42 (s, 2H), 5.13 (quintet, 1H), 7.37 (d, 2H), 7.52 (d, 1H),
7.88
(d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 492 (M+H)+
HPLC (method 1): Rt = 3.43 min
Example 23
4-N-[(2,2-Dimethylbutyl)glycyl]-1-cyclopropyl-2-oxo-N-[ 1-S-(4-
pyridinyl)ethyl]-
1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CA 02475320 2004-08-05
-67-
CH3 O O~H~CH3
~ ~,,~,~N .,~ N CH3 a
N / H ~ /
'N O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
1H NMR (400 MHz, DMSO-d6): 8 = 0.56 (m, 2H), 0.82 (s, 9H), 1.11 (m, 2H), 1.48
(d, 3H), 2.25 (br. s, 1H), 2.85 (m, 2H), 3.50 (s, 2H), 4.42 (s, 2H), 5.13
(quintet, 1H),
7.37 (d, 2H), 7.51 (d, 1H), 7.88 (d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.86
(d, 1H).
MS (ESIpos): m/z = 464 (M+H)+
HPLC (method 1): Rt = 3.42 min
Example 24
4-N-[( 1-S-Methyl-2,2-dimethylbutyl)glycyl]-1-cyclopropyl-2-oxo-
N-[ 1-S-(4-pyridinyl)ethyl]-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH3
CH3
CH Hs
3
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
CA 02475320 2004-08-05
-68-
1H NMR (400 MHz, DMSO-d6): 8 = 0.56 (m, 2H), 0.78 (s, 9H), 1.11 (m, 2H), 1.49
(d, 3H), 2.12 (d, 3H), 2.25 (br. s, 1H), 2.85 (m, 1H), 2.90 (m, 1H), 3.50 (s,
2H), 4.42
(s, 2H), 5.13 (quintet, 1H), 7.37 (d, 2H), 7.51 (d, 1H), 7.88 (d, 1H), 8.07
(br. s, 1H),
8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 478 (M+H)+
HPLC (method 1): Rt = 3.53 min
Example 25
4-N-[(1-S-Methyl-(2-methylbutyl)glycyl]-1-cyclopropyl-2-oxo-N-[ 1-S-(4-
pyridinyl)-
ethyl]-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
H3
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
1H NMR (400 MHz, DMSO-db): 8 = 0.56 (m, 2H), 0.74 (d, 6H), 1.1 l (m, 2H), 1.25
(m, 1H), 1.49 (d, 3H), 2.12 (d, 3H), 2.22 (br. s, 1H), 2.85 (m, 1H), 2.90 (m,
IH), 3.50
(s, 2H), 4.42 (s, 2H), 5.13 (quintet, 1H), 7.37 (d, 2H), 7.51 (d, 1H), 7.88
(d, 1H), 8.07
(br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 464 (M+H)+
HPLC (method 1 ): Rt = 3.40 min
CA 02475320 2004-08-05
-69-
Example 26
4-N-[(1-S-Methyl-(2-methylbutyl)glycyl)-1-cyclopropyl-2-oxo-N-[(3-pyridinyl-
4-methoxy)ethyl]-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
CH3
O.. ~-~,~ CH3
H
CH3
O
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
IH NMR (400 MHz, DMSO-d6): b = 0.57 (m, 2H), 0.73 (d, 6H), 1.12 (m, 2H), 1.25
(m, 1H), 1.49 (d, 3H), 2.15 (d, 3H), 2.22 (br. s, 1H), 2.85 (m, 1H), 2.90 (m,
1H), 3.23
(s, 3H), 3.50 (s, 2H), 4.42 (s, 2H), 5.06 (quintet, 1H), 7.49 (d, 2H), 7.52
(m, 1H),
7.83 (d, 1H), 8.20 (d, 1H), 8.85 (d, 1H).
MS (ESIpos): m/z = 464 (M+H)+
HPLC (method 1 ): Rf = 3.40 min
Example 27
4-N- { [2,2-Dimethyl-(3-fluoro)butyl] glycyl} -1-cyclopropyl-2-oxo-
N-[ 1-S-(4-pyridinyl)ethyl)-1,2,3,4-tetrahydro-6-quinoxalinecarboxamide
F
O
CH Hs
3
O
CA 02475320 2004-08-05
-70-
Preparation takes place in analogy to the preparation of the compound from
Example 1 from the appropriate precursors. The residue is purified by
preparative
HPLC (acetonitrile/water).
1H NMR (400 MHz, DMSO-d6): 8 = 0.56 (m, 2H), 1.10 (m, 8H), 1.61 (d, 3H), 2.90
(m, 4H), 4.15 (s, 1H), 4.42 (s, 2H), 5.13 (m, 1H), 7.37 (d, 2H), 7.51 (d, 1H),
7.88
(d, 1H), 8.07 (br. s, 1H), 8.50 (d, 2H), 8.86 (d, 1H).
MS (ESIpos): m/z = 482 (M+H)+
HPLC (method 1): Rt = 3.35 min
CA 02475320 2004-08-05
-71-
R~ Method Mass 1H-NMR
Exa- Structure h°~1 ~M~ (M~'~
mple
0
~H
N
28 t ~ I~~H ~ ~ 3.7 1 490
0
(300 MHz, CDC13):
d=8,58(d,J=5.1
Hz, 2H), 7.96-7.95
(m, 1H), 7.68 (d, 7.4
Hz, 1H), 7.28 (d, J
t~ o °~~oH' S.I Hz, 2H), 6.9~ (d,
\ \ ca-t,~' J = 8.5 Hz, lI~,
6.41 (d; 7.4 Hz, 1H),
29 ~0 3.6 1 478
5.34-5.24 (m, lI~,
4.46-4.37 (m, 3H),
3.66 (s, ?.~, 2.59
(sb, 2H), 2.38 (s,
2H), 2.29-2?2 (m,
ZH), 1.88-1.81 (m,
2H), 1.79 (s, 9H),
1.59 d, 7.2 Hz, 3
OH
o~~..~'
30 t ~ .I~H ~ 2 8 506
N 0
OH
0 N.~
' H
\ \
31 ~ ~ ~ ,I'H ~ 0 2.6 8 540
CA 02475320 2004-08-05
-72-
Exa- ~ Method Mass lg_ivT'YIR
mple Structure [mm] LG'MS (M+Fn
o~
' H
OH
32 N ~ ~,H I ~ ~0 2.13 8 520
cy ~
33 ~ ~ ~ ° I_97 8 478
O~H~CHj
34 ~ ~ H ~ ~ ~ 3.5 1 478
N ~ O
H3
O CNy
C O ~~ ~ H'
35 I N H I ~ 3.3 I 506
0
o~
H
36 ~ i H ~ ~ ~ flH 2.07 8 492
N ~ ~ 0
CA 02475320 2004-08-05
-73-
Exa- Structure & Method Mass 'g-NMR
mple [mint LCMS (M-~1~
37 H ~ 3.8 1 520
H,co ~~ o
~ o o i H l' ~
H
38 ' ~~0 3.7 1 47s
o~
39 H,~ 0 3.8 1 508
~ o o~~~
w w
40 ~ ' ~ ~ 0 3.6 1 494
H,co
Ho 0 o~ ~cH,
H I "CH,
C
41 ~ ~ H ~ ~ ~ ~ 3.3 1 480
N ~ O
HO O
42 I ~ H I w IN~~H~ 3,3 1 492
N ~ ~ O
CA 02475320 2004-08-05
-74-
Exa- Rc Method Mass 'H-NMR
mple Structure (mm] LCMS (M+H)
.Q
43 ~ p .~ H 2.75 8 507
H,CO~~ H ~o
O O'~~,~~N~
' H
44 I ~ ~ ~' 3.51 I 478
i
0
° °~r~''~-~1~
H
45 1 ~ a ~ ~ 3.6 1 508
H,co ~ o
°~
46 ~ y~~~ 3.8 1 520
H,co ~ ~ ~ o
1H-NMR (400MHz,
DM50-d6): d =
0.48-0.60 {m, 2~,
0.83-1.22 (m, SIB,
°H 1.48 (d, 3I~, 1.52
0 0~~~ 1.70 (m, 5I-1], 2.18
47 N ~ H ~ ~ 3.4 1 492 2.88 (m, 2F~~, 38 0
/~~' -'~o . s
3.55
(~
iT~, 4.42 {s, 2~,
5.I3 (quintet, 1~,
7.37 (d, 2I~, 7.52 (d,
1~, 7.88 (d, 11~,
8.07 (s, lI~, 8.48 (d,
1 , 8.86 d, 1
CA 02475320 2004-08-05
-75-
Method Mass Ig_~t
Exa- Structure tm ] L~ (M+g)
mple
1H-NMR (400MHz,
DMSO-d6):
0.48-0.6g (m, 2F~,
0.80-1.2i (m, SH),
L48 (d, 3H)> 1.52-
1.70 (m, SH), 2.18-
0 230 (m, lI-~, 2.33 (s,
3I~, 2.80-2.88 (m,
w w 2H), 3.50 (br. s, 2I~,
48 ~c~~~ 3.6 1 506 3,55 {m, Il~, 4.42 {s,
2H), 5.13 (quintet,
IH), 737 (d, 2I~,
7.s2 (a, IH), 7.$s (d,
1H), 8.07 (br. s, 1F~,
8.50 (d, 2I~, 8.86 (d,
1H)
1H-NMR (400MFiz,
DMSO-d6):
0.48-0.60 (m, 2H),
0.80-1.21 (m, SH),
1.48 (d, 3H), 1.52-
1.70 (m, 51~, 2.18-
2.30 (m, 1H), 2.55 (s,
o °~~~, 3H), 2.80-2.88 {m,
49 ~ 3.8 1 522 2H), 3.50 (br. s, 2H),
rycro ~ i o 3.55 (m, 1~, 4.42 (s,
2H), 5.13 (quintet,
1H), 7.37 (d, 2H),
7.52 {d, 1I~, 7.88 (d,
1H), 8.07 {br. s, 1i-n,
8.50 (d, 2i~, 8.86 (d,
1Hj
o °~~
~~~'v'~o
1 N
50 N~H ~ i 3.5 1 490
CA 02475320 2004-08-05
-76-
Exa- Structure & Method Mass 'H-htMR
[min] LG'MS (M+~
m ple
~ o o~~~
51 N ~ ~ I ~ 3.6 1 464
0
~H
52 N ~ ~ I ~ ~ off 3.3 1 492
0
H,c o~~ct~,
1~ ~
5; ~ j ~ I i a cH~ 3.6 1 492
cH,
o~rNt~c~
1 w N w c~
54 ~ H I i ~ 3.5 1 492
N ~ O
O~N
H
55 ~ w c"~ 3.b 1 492
~ ~ H ~o
CA 02475320 2004-08-05
-77_
Exa- Structure Re Method Mass 'H-NMR
mple (min)LCMS (M+H)
cH,
~o 0
56 ~ % H ~ i ~ 3.4 1 492
0
B. Assessment of the uhysiological activity
Abbreviations:
DMEM Dulbecco's modified Eagle medium
FCS fetal calf serum
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
1. in vitro Tests to determine the M2 activity and selectivity
a) Cellular functional in vitro test
A recombinant cell line was used to identify agonists of the human M2
acetylcholine
receptor (M2AChR) and to quantify the activity of the substances described
herein.
The cell is originally derived from a hamster ovary epithelial cell (Chinese
Hamster
Ovary, CHO Kl, ATCC: American Type Culture Collection, Manassas, VA 20108,
USA). The test cell line constitutively expresses a modified form of the
calcium-
sensitive photoprotein aequorin which after reconstitution with the cofactor
coelenterazine emits light when the free calcium concentration in the inner
mitochondrial compartment is increased (Rizzuto R, Simpson AW, Brini M,
Pozzan T.; Nature 358 (1992) 325-327). The cell is additionally stably
transfected
with the human M2AChR (Peralta EG, Ashkenazi A, Window JW, Smith DH,
Ramachandran J, Capon, DJ, EMBO Journal 6 (1987) 3923-3929) and with the gene
which codes for the promiscuous Gal6 protein (Amatruda TT, Steele DA, Slepak
VZ,
Simon MI, Proceedings in the National Academy of Science USA 88 (1991), 5587-
CA 02475320 2004-08-05
_7g_
5591). The resulting M2AChR test cell responds to stimulation of the
recombinant
M2ACh receptor with an intracellular release of calcium ions, which can be
quantified through the resulting aequorin luminescence with a suitable
luminometer
(Milligan G, Marshall F, Rees S, Trends in Pharmacological Sciences 17 (1996)
235-237).
The in vitro selectivity for the muscarinergic acetylcholine receptor subtypes
M 1 to
MS is determined by using appropriate CHO Kl cells which are stably
transfected
likewise with the gene of the calcium-sensitive photoprotein aequorin and the
gene
of the M1, M3 or MS receptor subtypes or, in the case of the M4 receptor
subtypes,
additionally with the gene of the promiscuous G°16 protein.
Test procedure: The cells are plated out on the day before the test in culture
medium
(DMEM, 10% FCS, 2 mM glutamine, 10 mM HEPES; Gibco Cat.# 21331-020; now
belongs to: Invitrogen GmbH, 76131 Karlsruhe) in 384 (or 1536) well microtiter
plates and kept in a cell incubator (96% humidity, 5% v/v COZ, 37°C).
On the day of
the test, the culture medium is replaced by a Tyrode solution (in mM: 140
NaCI,
5 KCI, 1 MgClz, 2 CaClz, 20 glucose, 20 HEPES) which additionally contains the
cofactor coelenterazine (SO p.M), and the microtiter plate is then incubated
for a
further 3-4 hours. Immediately after the test substances have been transferred
into the
wells of the microtiter plate, the resulting light signal is measured in the
luminometer. The results are shown in Table A:
CA 02475320 2004-08-05
-79-
Table A
Example No. ECSO (nlVn
2 1000
12 1800
_
.
7 5
11 320
_ _..
37
22 150
23 18
24 32
25 6
26 16
b) Binding studies on human muscarinergic acetylcholine receptors
5 Stably transfected CHO K1 cells which express the human muscarinergic M2
receptor are, after 80% confluence is reached, suspended in 10 ml of binding
buffer
(20 mM 40(2-hydroxyethyl)-1-piperazineethanesulphonic acid, 5 mM in magnesium
chloride, pH 7.4) per 175 cmz cell culture bottle and homogenised using an
Ultra-
Turrax apparatus. The homogenates are centrifuged at 1000 g and 4°C
for 10
minutes. The supernatant is removed and centrifuged at 20 000 g and 4°C
for 30 min.
The membrane sediment with the M2 receptors is taken up in 10 ml of binding
buffer
and stored at -70°C.
For the binding test, 2 nM 3H-oxotremorine M (3200 GBq/mmol, Perkin Elmer) are
incubated with 100-1000 pg/ml M2 membranes per mixture (0.2 ml) in the
presence
of the test substances at room temperature for 60 minutes. The incubation is
stopped
by centrifugation at 10 000 g for 10 minutes and subsequent washing wit 0.1%
bovine serum albumin in binding buffer at 4°C. Centrifugation is again
carried out at
10 000 x g and 4°C for 10 minutes. The sediment is resuspended in 0.1
ml of 1 N
sodium hydroxide solution and transferred into scintillation vials. After
addition of
4 ml of Ultima Gold scintillator, the radioactivity bound to the membranes is
CA 02475320 2004-08-05
-80-
quantified using a BeckmanCoulter LS6000 IC scintillation counter. The
nonspecific
binding is defined as radioactivity in the presence of 10 NM oxotremorine M
and is
usually less than 5% of the bound total radioactivity. The binding data (ICso
and
dissociation constant Ki) are determined using the graph pad prism version
3.02
programme.
Z. in vivo Test to detect the cardiovascular effect
a) Langendorff guinea pig heart
The heart is removed from anaesthetised guinea pigs after opening the thoracic
cavity and introduced into a conventional Langendorff apparatus. The coronary
arteries are perfused at constant volume (10 ml/min) and the profusion
pressure
arising during this is recorded via an appropriate pressure transducer. A
decrease in
the profusion pressure in this arrangement corresponds to a relaxation of the
coronary arteries. At the same time, the pressure developed by the heart
during each
contraction is measured via a balloon inserted into the left ventricle and a
further
pressure transducer. The rate of the heart beating in isolation is found by
calculation
from the number of contractions per unit time.
b) Blood pressure measurements on anaesthetised rats
Male Wistar rats with a body weight of 300-350 g are anaesthetised with
thiopental
(100 mg/kg i.p.). After tracheotomy, a catheter is introduced into the femoral
artery
to measure the blood pressure. The substances to be tested are administered
orally in
Transcutol, Cremophor EL, HZO ( 10%/20%/70%) in a volume of 1 ml/kg.
c) Effect on the mean blood pressure of conscious spontaneously
hypertensive rats
Continuous blood pressure measurements over 24 hours are carried out on
spontaneously hypertensive female rats (MOL:SPRD) weighing 200-250 g and
moving freely. For this purpose, pressure transducers (Data Sciences Inc., St.
Paul,
CA 02475320 2004-08-05
-81 -
MN, USA) are implanted chronically in the descending abdominal aorta below the
renal artery of the animals, and the transmitter connected thereto is fixed in
the
abdominal cavity. The animals are kept singly in type III cages which are
positioned
on the individual receiving stations and are adapted to a 12-hour lightldark
rhythm.
Water and feed are freely available. For data acquisition, the blood pressure
of each
rat is recorded for 10 seconds every S minutes. The measurements are combined
in
each case for a period of 15 minutes and the mean is calculated from these
values.
The test compounds are dissolved in Transcutol (10%), Cremophor (20%), Hz0
(70%) mixture and administered orally by gavage in a volume of 2 mllkg of body
weight. The test doses are between 0.3-30 mg/kg of body weight.
d) Blood pressure and heart rate measurements on anaesthetised dogs
The experiments are carried out on dogs (mongrel) of both sexes with a body
weight
between 20 and 30 kg. Anaesthesia is induced by a slow i.v. injection of 25
mg/kg
thiopental (Trapanal~) and continued during the experiment by continuous
infusion
of 0.08 mglkg/h fentanyl (Fentanyl~) and 0.25 mg/kg/h droperidol
(Dehydrobenzperidol~). Alloferin (0.02 mg/kg/h) is added as muscle relaxant.
The
dogs are artificially ventillated with 1 part of nitrous oxide and 3 parts of
oxygen.
The test substances are administered intravenously via the femoral vein.
A MillarTip catheter is passed via the carotid artery into the left ventricle
to pick up
the left ventricular pressure and calculate the contractility. A hollow
catheter is
introduced via the femoral artery into the aorta and connected to a pressure
transducer to measure the peripheral blood pressure. After a left-sided
thoracotomy,
the left circumflex (LCX) or the left anterior descending (LAD) coronary
artery is
exposed and an electromagnetic flow head is sited to measure the coronary
flow. The
ECG is recorded via an extremity lead and an ECG amplifier, and the heart rate
and
ECG parameters are found from the recorded ECG. The oxygen saturation at the
coronary sinus is determined via a Swan-Gantz oximetry TD catheter.
CA 02475320 2004-08-05
-82-
B. Exemplary embodiments of pharmaceutical compositions
The compounds of the invention can be converted into pharmaceutical
preparations
in the following ways:
Tablet:
Composition:
100 mg of the compound of Example 1, 50 mg of lactose (monohydrate), 50 mg of
maize starch (native), 10 mg of polyvinylpyrolidone (PVP 25) (from BASF,
Ludwigshafen, Germany) and 2 mg of magnesium stearate.
Tablet weight 212 mg. Diameter 8 mm, radius of curvature 12 mm.
Production:
The mixture of active ingredient, lactose and starch is granulated with a 5%
strength
solution (m!m) of the PVP in water. The granules are dried and then mixed with
the
magnesium stearate for 5 min. This mixture is compressed using a conventional
tablet press (see above for format of the tablet). A compressive force of 15
kN is
used as guideline for the compression.
Suspension which can be administered orally:
Composition:
1000 mg of the compound of Example 1, 1000 mg of ethanol (96%), 400 mg of
Rhodigel (xanthan gum from FMC, Pennsylvania, USA) and 99 g of water.
10 ml of oral suspension correspond to a single dose of 100 mg of the compound
of
the invention.
Production:
The Rhodigel is suspended in ethanol, and the active ingredient is added to
the
suspension. The water is added while stirring. The mixture is stirred for
about 6 h
until the swelling of the Rhodigel is complete.