Note: Descriptions are shown in the official language in which they were submitted.
CA 02477474 2004-08-25
J .
WO 03/074489 1 PCT/EP03/01596
Cyclic N-substituted alpha-imino carboxylic acids for selective inhibition of
collagenase
The invention relates to the use of cyclic N-substituted alpha-imino
carboxylic acids
of the formula I for selective inhibition of collagenase (MMP 13). The
compounds of
the formula I can therefore be employed for treating degenerative joint
disorders.
io In disorders such as osteoarthritis and rheumatism there is destruction of
the joint
caused in particular by the proteolytic degradation of collagen by
collagenases.
Collagenases belong to the superfamily of metalloproteinases (MP) or matrix
metallproteinases (MMP). The MMPs form a group of Zn-dependent enzymes which
are involved in biological degradation of the extracellular matrix (D. Yip et
al in
Investigational New Drugs 17 (1999), 387-399 and Michaelides et al in Current
Pharmaceutical Design 5 (1999) 787 -819). These MMPs are able in particular to
degrade fibrillary and non-fibrillary collagen, and proteoglycans, both of
which
represent important matrix constituents. MMPs are involved in processes of
wound
healing, of tumor invasion, of metastasis migration and in angiogenesis,
multiple
sclerosis, heart failure and atherosclerosis (Michaelides p. 788; see above).
In
particular, they play an important part in degradation of the joint matrix in
arthrosis
and arthritis, whether osteoarthrosis, osteoarthritis or rheumatoid arthritis.
A selected subgroup within the MMPs is formed for example by collagenases.
Only
collagenases are able to degrade native collagen which, after all, exercises
an
important supporting function in the matrix. This subgroup consists of
interstitial
collagenase MMP-1, of neutrophil collagenase MMP-8, and of MMP-1 3 which was
identified only later. It was virtually impossible to detect MMP-1 3 in the
body of
healthy adult individuals; it is expressed only during the course of diseases,
e.g. in
cancer cells or ulcers. MMP-1 3 has also been detected in the joint matrix of
arthrotic
people and mammals, especially in clinically advanced arthrosis, while it does
not
occur in the joint tissue of healthy adults. Inhibition of MMP-13 by
appropriate
inhibitors is therefore particularly suitable for controlling said diseases.
CA 02477474 2004-08-25
2
A large number of different inhibitors of MMPs and of collagenases are known
(EP 0
606 046; W094/28889; WO 96/27583). It is also known that cyclic and
heterocyclic
N-substituted alpha-imino hydroxamic and carboxylic acids are inhibitors of
metalloproteinases (EP 0 861 236).
After the initial clinical studies on humans, it has now emerged that MMPs
cause
side effects. The side effects which are mainly mentioned are musculoskeletal
pain
or arthralgias. It is unambiguously expected from the prior art that selective
inhibitors
will be able to reduce these side effects mentioned (Yip, page 387, see
above).
A disadvantage of known inhibitors of MMPs is therefore frequently the lack of
specificity of the inhibition for only one class of MMPs. Thus, most MMP
inhibitors
inhibit a plurality of MMPS simultaneously, because the MMPs have a catalytic
domain of similar structure. Accordingly, the inhibitors act in an unwanted
manner on
many enzymes, even those with a vital function (Massova I, et al., The FASEB
Journal (1998) 12, 1075-1095).
The application WO 97/18194 (EP 0 861 236) has already described cyclic and
heterocyclic N-substituted alpha-imino hydroxamic and alpha-imino carboxylic
acids
which have a strong inhibitory effect on MMP-3 and MMP-8. The compounds
disclosed by the description in the examples in WO 97/18194 also show a strong
inhibitory effect on MMP-13, as has been found by our own remeasurements.
In the effort to find effective compounds for the treatment of connective
tissue
disorders, it has now been found that the compounds employed according to the
invention are strong inhibitors of matrix metalloproteinase 13, while the
compounds
employed according to the invention are essentially ineffective for MMPs 3 and
8.
These compounds employed according to the invention are thus more suitable in
a
much more targeted manner for the selective treatment of said diseases than
the
compounds disclosed and described in the examples in WO 97/18194, which
inhibit
other MMPs besides MMP-13. It is therefore to be expected that these selective
inhibitors will show a considerably improved range of side effects on
treatment of
said disorders.
CA 02477474 2004-08-25
3
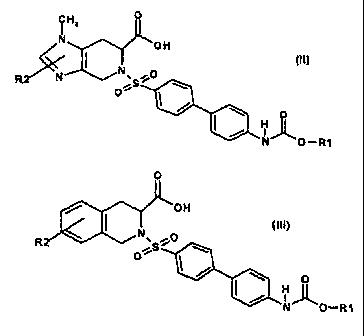
The invention therefore relates to the use of compounds of the formula I
0
A OH
(CH On
I (~~
R .'O
"S
H O
N 'J~
O-R1
and/or all stereoisomeric forms of the compound of the formula I and/or
mixtures of
these forms in any ratio, and/or a physiologically tolerated salt of the
compound of
the formula I
for producing a pharmaceutical for the prophylaxis and therapy of disorders in
the
progression of which an enhanced activity of matrix metalloproteinase 13 is
involved,
where
A is carbon atom or nitrogen atom, in which
A is unsubstituted or substituted by -(C1-C4)-alkyl,
n is the integers 1 or 2,
R1 is 1. -(C1-C1 p)-alkyl in which alkyl is linear or branched,
2. -(C2-C1 p)-alkenyl in which alkenyl is linear or branched,
3. -(C2-C10)-alkynyl in which alkynyl is linear or branched,
4. -(C1-C4)-alkylphenyl,
5. -(C1-C4)-alkyl-(C3-C7)-cycloalkyl,
6. -(C3-C7)-cycloalkyl or
7. -CH2CF3, and
R2 is hydrogen atom or -(C1-C4)-alkyl.
The invention further relates to the use of compounds of the formula I where
A is carbon atom or nitrogen atom, in which
A is unsubstituted or substituted by methyl,
n is the integers 1 or 2,
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
CA 02477474 2004-08-25
4
R2 is hydrogen atom or methyl.
The invention further relates to the use of compounds of the formula 11 where
s
H o
N OH
R N'~O
O S
H O
/ N O- R1
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
R2 is hydrogen atom or methyl.
A further aspect of the invention relates to the use of compounds of the
formula III
where
O
OH
cui)
O
R e
O
:r_ S
LL4AORl
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
R2 is hydrogen atom or methyl.
A further aspect of the invention relates to novel compounds of the formula I,
CA 02477474 2004-08-25
0
A OH
(CHZ)n I (1)
R N,' :o
OAS
H O
N O-R1
and/or all stereoisomeric forms of the compound of the formula I and/or
mixtures of
these forms in any ratio, and/or a physiologically tolerated salt of the
compound of
the formula I, where
5 A is carbon atom or nitrogen atom, in which
A is unsubstituted or substituted by -(C1-C4)-alkyl,
n is the integers 1 or 2,
R1 is 1. -(C1-C10)-alkyl in which alkyl is linear or branched,
2. -(C2-C 1 0)-alkenyl in which alkenyl is linear or branched,
3. -(C2-C10)-alkynyl in which alkynyl is linear or branched,
4. -(C1-C4)-alkylphenyl,
5. -(C1-C4)-alkyl-(C3-C7)-cycloalkyl,
6. -(C3-C7)-cycloalkyl or
7. -CH2CF3, and
R2 is hydrogen atom or -(C1-C4)-alkyl.
The invention further relates to compounds of the formula I where
A is carbon atom or nitrogen atom, in which
A is unsubstituted or substituted by methyl,
n is the integers 1 or 2,
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
R2 is hydrogen atom or methyl.
CA 02477474 2004-08-25
6
A further aspect of the invention relates to compounds of the formula II where
CH3 0
N OH
(II)
R N ' ; 1 1
OAS ` ~
1 ~
H O
N O-RI
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
R2 is hydrogen atom or methyl.
io A further aspect of the invention relates to compounds of the formula III
where
O
OH
` (III)
N ~O
O:S~
H O
N AO-RI
R1 is 1. -(C2-C6)-alkyl in which alkyl is linear or branched,
2. -CH2-phenyl, or
3. -CH2-cyclopropyl, and
R2 is hydrogen atom or methyl.
The term "(C1-C10)-alkyl" means hydrocarbon radicals whose carbon chain is
straight-chain or branched and contains 1 to 10 carbon atoms, for example
methyl,
CA 02477474 2004-08-25
7
ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl, isopentyl,
neopentyl,
hexyl, 2,3-dimethylbutyl, neohexyl, heptyl, octanyl, nonanyl, decanyl.
The term "(C2-C1 o)-alkenyl" means hydrocarbon radicals whose carbon chains is
straight-chain or branched and contains 1 to 10 carbon atoms and, depending on
the chain length, 1, 2 or 3 double bonds. (C3-C7)-cycloalkyl radicals are, for
example, compounds derived from 3- to 7-membered monocycles such as
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
The invention further relates to a process for preparing the compound of the
formula
io I and/or a stereoisomeric form of the compound of the formula I and/or a
physiologically tolerated salt of the compound of the formula I, which
comprises
a) reacting a compound of the formula IV
0
,Re
(CH2)n A N O
R2 ADC
IV
is in which A, R2 and n are as defined in formula I and Re is a hydrogen atom
or
an ester protective group,
with a compound of the formula V,
O
11
O2N S-Rz
V
in which Rz is chlorine atom, imidazolyl or OH,
20 in the presence of a base to give a compound of the formula VI
0
A ORe (VI)
(CH2>
N\
AI
R2 o S NO2
in which A, R2 and n are as defined in formula I, and Re is as defined above,
and
CA 02477474 2004-08-25
8
b) reducing a compound of the formula VI prepared as in a) with hydrogen and a
metal catalyst to an amine of the formula VII
0
A Re (VII)
(CH2) I
N~ O
A
R2 O S NHZ
and subsequently reacting the compound of the formula VII with a compound
of the formula VIII
O
)t...~R1 (VIII)
Cl O
in which R1 is as defined in formula I,
in the presence of a basic compound such as triethylamine, trimethylamine or
pyridine to give a compound of the formula IX
O
(CH 2 )n q N O.Re O (Ix)
I O
R2 C H O
where R1, R2, A and n are as defined in formula I, and Re is as defined
above,
c) fractionating a compound of the formula I which has been prepared by
process a) and which, because of its chemical structure, occurs in
enantiomeric forms into the pure enantiomers by salt formation with
enantiopure acids or bases, chromatography on chiral stationary phases or
derivatization using chiral enantiopure compounds such as amino acids,
separation of the diastereomers obtained in this way, and elimination of the
chiral auxiliary groups, or
CA 02477474 2004-08-25
9
d) either isolating in free form the compound of the formula I prepared by
processes b) or c) or, in the case where acidic or basic groups are present,
converting into physiologically tolerated salts.
It is possible to employ as ester protective group Re the groups as protective
groups
for esters in Protective Groups in Organic Synthesis, T.H. Greene, P. G. M.
Wuts,
Wiley-Interscience, 1991. Preferred ester protective groups are, for example,
methyl,
ethyl, isopropyl, tert-butyl or benzyl.
The starting materials and reagents employed can either be prepared by known
io processes or be purchased. The spinacine ring system can be prepared for
example
as described by S. Klutchko et al. (J. Heterocyclic. Chem., 28, 97, (1991)).
The reactions take place for example as described in WO 97/18194. The reaction
in
process step a) takes place in the presence of a base such as KOH, NaOH, LIOH,
N,O-bis(trimethylsilyl)acetamide (BSA), N-methylmorpholine (NMM),
N-ethylmorpholine (NEM), triethylamine (TEA), diisopropylethylamine (DIPEA),
pyridine, collidine, imidazole or sodium carbonate in solvents such as
tetrahydrofuran (THF), dimethylformamide (DMF), dimethylacetamide, dioxane,
acetonitrile, toluene, chloroform or methylene chloride, or else in the
presence of
water.
The reaction in process step b) takes place for example using the metal
catalysts
Pd/C, SnCI2 or Zn under standard conditions.
In process step c), the compound of the formula I is, if it occurs in
diastereoisomeric
or enantiomeric form and results as mixtures thereof in the chosen synthesis,
separated into the pure stereoisomers, either by chromatography on an
optionally
chiral support material or, if the racemic compound of the formula I is able
to form
salts, by fractional crystallization of the diastereomeric salts formed with
an optically
active base or acid as auxiliary. Examples of suitable chiral stationary
phases for
separation of enantiomers by thin-layer or column chromatography are modified
silica gel supports (called Pirkle phases) and high molecular weight
carbohydrates
such as triacetyl cellulose. For analytical purposes, methods of gas
chromatography
on chiral stationary phases can also be used after appropriate derivatization
known
CA 02477474 2004-08-25
to the skilled worker. To separate enantiomers of the racemic carboxylic
acids, an
optically active, usually commercially available, base such as (-)-nicotine,
(+)-and (-)-
phenylethylamine, quinine bases, L-lysine or L- and D-arginine are used to
form the
diastereomeric salts which differ in solubility, the less soluble component is
isolated
5 as solid, the more soluble diastereomer is deposited from the mother liquor,
and the
pure enantiomers are obtained from the diastereomeric salts obtained in this
way. It
is possible in the same way in principle to convert the racemic compounds of
the
formula I which contain a basic group such as an amino group with optically
active
acids such as (+)-camphor-10-sulfonic acid, D- and L-tartaric acid, D- and L-
lactic
1o acid and (+)- and (-)-mandelic acid into the pure enantiomers. Chiral
compounds
which contain alcohol or amine functions can also be converted with
appropriately
activated or optionally N-protected enantiopure amino acids into the
corresponding
esters or amides, or conversely chiral carboxylic acids can be converted with
carboxyl-protected enantiopure amino acids into the amides or with enantiopure
hydroxy carboxylic acids such as lactic acid into the corresponding chiral
esters. It is
then possible to make use of the chirality of the amino acid or alcohol
residue
introduced in enantiopure form for separating the isomers by carrying out a
separation of the diastereomers which are now available by crystallization or
chromatography on suitable stationary phases and then eliminating the included
chiral moiety using suitable methods.
Acidic or basic products of the compound of the formula I may be in the form
of their
salts or in free form. Preference is given to pharmacologically acceptable
salts, e.g.
alkali metal or alkaline earth metal salts, and hydrochlorides, hydrobromides,
sulfates, hemisulfates, all possible phosphates, and salts of amino acids,
natural
bases or carboxylic acids.
Preparation of physiologically tolerated salts from compounds of the formula I
which
are able to form salts, including the stereoisomeric forms thereof, in process
step d)
takes place in a manner known per se. The compounds of the formula I form
stable
alkali metal, alkaline earth metal or optionally substituted ammonium salts
with basic
reagents such as hydroxides, carbonates, bicarbonates, alcoholates, and
ammonia
or organic bases, for example trimethylamine or triethylamine, ethanolamine or
triethanolamine or else basic amino acids, for example lysine, ornithine or
arginine. If
CA 02477474 2004-08-25
11
the compounds of the formula I have basic groups, it is also possible to
prepare
stable acid addition salts with strong acids. Suitable for this purpose are
both
inorganic and organic acids such as hydrochloric, hydrobromic, sulfuric,
phosphoric,
methanesulfonic, benzenesulfonic, p-toluenesulfonic, 4-bromobenzenesulfonic,
cyclohexylamidosulfonic, trifluoromethylsulfonic, acetic, oxalic, tartaric,
succinic or
trifluoroacetic acid.
The invention also relates to pharmaceuticals which have an effective content
of at
least one compound of the formula I and/or of a physiologically tolerated salt
of the
io compound of the formula I and/or an optionally stereoisomeric form of the
compound of the formula I, together with a pharmaceutically suitable and
physiologically tolerated carrier, additive and/or other active ingredients
and
excipients.
Because of the pharmacological properties, the compounds of the invention are
suitable for the selective prophylaxis and therapy of all disorders in the
progress of
which an enhanced activity of metalloproteinase 13 is involved. These include
degenerative joint disorders such as osteoarthroses, spondyloses, chondrolysis
after
joint trauma or prolonged joint immobilization after meniscus or patellar
injuries or
ligament tears. They also include connective tissue disorders such as
collagenoses,
periodontal disorders, wound-healing disturbances and chronic disorders of the
locomotor system such as inflammatory, immunologically or metabolism-related
acute and chronic arthritides, arthropathies, myalgias and disturbances of
bone
metabolism. The compounds of the formula I are also suitable for the treatment
of
ulceration, atherosclerosis and stenoses. The compounds of the formula I are
furthermore suitable for the treatment of inflammations, cancers, tumor
metastasis,
cachexia, anorexia and septic shock. Said disorders can be employed with the
compounds employed according to the invention considerably more specifically
and
with a smaller range of side effects because essentially only MMP-13 is
inhibited.
3o Administration of the pharmaceuticals of the invention can take place by
oral,
inhalational, rectal or transdermal administration or by subcutaneous,
intraarticular,
intraperitoneal or intravenous injection. Oral administration is preferred.
CA 02477474 2004-08-25
12
The invention also relates to a process for producing a pharmaceutical which
comprises converting at least one compound of the formula I into a suitable
dosage
form with a pharmaceutically suitable and physiologically tolerated carrier
and,
where appropriate, further suitable active ingredients, additives or
excipients.
Examples of suitable solid or pharmaceutical preparations are granules,
powders,
coated tablets, tablets, (micro)capsules, suppositories, syrups, oral
solutions,
suspensions, emulsions, drops, injectable solutions, and products with
protracted
release of active ingredient, in the production of which conventional aids
such as
io carriers, disintegrants, binders, coating agents, swelling agents, glidants
or
lubricants, flavorings, thickeners and solubilizers are used. Excipients which
are
often used and which may be mentioned are magnesium carbonate, titanium
dioxide, lactose, mannitol and other sugars, talc, milk protein, gelatin,
starch,
cellulose and its derivatives, animal and vegetable oils such as fish liver
oil,
sunflower, peanut or sesame oil, polyethylene glycol and solvents such as, for
example, sterile water and monohydric or polyhydric alcohols such as glycerol.
The pharmaceutical products are preferably produced and administered in dosage
units, each unit containing as active ingredient a particular dose of the
compound of
the invention, of the formula I. In the case of solid dosage units such as
tablets,
capsules, coated tablets or suppositories, this dose can be up to about 1 000
mg,
but preferably about 50 to 300 mg, and in the case of solutions for injection
in
ampoule form up to about 300 mg, but preferably about 10 to 100 mg.
The daily doses indicated for the treatment of an adult patient weighing about
70 kg
are from about 20 mg to 1 000 mg of active ingredient, preferably about 100 mg
to
500 mg, depending on the activity of the compound of the formula I. However,
in
some circumstances, higher or lower daily doses may also be appropriate. The
daily
dose may be administered both by administration once a day in the form of a
single
3o dosage unit or else a plurality of smaller dosage units, and by
administration more
than once a day of divided doses at defined intervals.
Final products are usually determined by mass spectroscopic methods (FAB-, ESI-
MS), with the main peak being indicated in each case. Temperatures are stated
in
CA 02477474 2004-08-25
13
degrees Celsius, RT means room temperature, (22 C to 26 C). Abbreviations used
are either explained or correspond to the usual conventions.
The invention is explained in detail below by means of examples.
Preparation examples
Compounds 1, 3, 4 and 5 in table 1 were prepared in analogy to the procedures
described in example 2.
io Example 2
Stage 1: Preparation of 2-(4"-nitrobiphenyl-4-sulfonyl)-1,2,3,4-tetrahydro-
isoquinoline-3-(R)-carboxylic acid
44.2 g (250 mmol) of 1,2,3,4-tetrahydroisoquinolyl-3-(R)-carboxylic acid were
dissolved in 500 ml of IN aqueous sodium hydroxide solution and, while
stirring, 150
g (300 mmol) of 4,4'-nitrobiphenylsulfonyl chloride dissolved in 500 ml of
tetrahydrofuran were added. During this, the reaction temperature was kept
below
C, and the pH of the solution was adjusted to pH 10 by adding 1 N NaOH. The
reaction mixture was stirred at room temperature for 12 hours (h).
For workup, the reaction solution was filtered through a clarifying layer of
Celite
and then the filtrate was concentrated to one half the original volume in a
rotary
evaporator under reduced pressure. The resulting solution was acidified to pH
2 to 3
by adding 20% strength aqueous citric acid solution, whereupon the reaction
product
precipitated as a colorless solid. The resulting solid was filtered off and
dried in a
vacuum oven at 40 C.
77.9 g (71% of theory, colorless solid) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 439.4
Stage 2: Preparation of methyl 2-(4"-nitrobiphenyl-4-sulfonyl)-1,2,3,4-
tetrahydro-
isoqu inoline-3-(R)-carboxylate
- - ------ -------
CA 02477474 2004-08-25
14
' 77.5 g (177 mmol) of the carboxylic acid prepared in stage 1 were added in
portions
to a solution of 65 ml of thionyl chloride (885 mmol) in 800 ml of methanol at
a
temperature of -15 C to -10 C. The mixture was then stirred at 25 C for 1 hour
and
at 50 C for 1 hour.
For workup, the reaction solution was concentrated under reduced pressure and
stirred with 300 ml of dichloromethane and 300 ml of saturated aqueous NaHCO3
solution. The organic phase was then washed with water until neutral and dried
over
Na2SO4, and the solvent was removed under reduced pressure. The crude product
(80 g, brown oil) obtained in this way was purified by chromatography on
silica gel
(40p - 63p) with n-heptane:ethyl acetate = 2:1 as mobile phase.
27 g (34% of theory, colorless oil) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 453.4
Stage 3: Preparation of methyl 2-(4"-aminobiphenyl-4-sulfonyl)-1,2,3,4-
tetrahydro-
isoquinolinyl-3-(R)-carboxylate
53 g (117 mmol) of the nitro compound prepared in stage 2 were dissolved in
1.5 I of
a 1:1 mixture of tetrahydrofuran (THF) and methanol and, after addition of 1 g
of Pd
(10% on activated carbon), hydrogenated with H2 in a hydrogenation apparatus
until
H2 uptake ceased (hydrogen consumption 7.2 1).
For workup, the catalyst was filtered through a clarifying layer of Celite ,
and the
filtrate was concentrated in a rotary evaporator under reduced pressure. The
oily
residue was taken up in dichloromethane and dried over Na2SO4, and the solvent
was removed under reduced pressure. The crude product obtained in this way was
recrystallized from diethyl ether.
45.7 g (93% of theory, colorless solid) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 423.4
CA 02477474 2004-08-25
" Stage 4: Preparation of methyl 2-(4'-isopropoxycarbonylaminobiphenyl-4-
sulfonyl)-
1,2,3,4-tetrahydroisoquinoline-3-(R)-carboxylate
16.9 g (40 mmol) of the compound of stage 3 were dissolved in 200 ml of
absolute
5 dichloromethane and, while stirring at -70 C to -60 C, successively 4.9 ml
of
pyridine (60 mmol) and 44 ml (44 mmol) of isopropyl chloroformate were added.
The
mixture was then stirred at this temperature for 3 h. For workup, the reaction
mixture
was hydrolyzed at 0 C by adding 10 ml of water. After removal of the organic
phase
it was washed until neutral and dried with Na2SO4, and the solvent was removed
1o under reduced pressure in a rotary evaporator. The residue obtained in this
way
crystallized after addition of 100 ml of n-pentane in the form of salmon-
colored
crystals.
20.1 g (98%, salmon-colored crystals) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 509.5
Stage 5: Preparation of 2-(4'-isopropoxycarbonylaminobiphenyl-4-sulfonyl)-
1,2,3,4-
tetrahydroisocquinoline-3-(R)-carboxylic acid
20 g of the carboxylic ester (39.4 mmol) prepared in stage 4 were dissolved in
200
ml of THE and, at 25 C, 48 ml 1 M aqueous LiOH solution were added. The
reaction
solution was stirred at 25 C for 3 h.
The reaction mixture was then concentrated under reduced pressure, the residue
was taken up in 750 ml of water and filtered through a clarifying layer of
Celite with
addition of activated carbon, and the mother liquor was acidified with aqueous
citric
acid. The precipitated reaction product was filtered and purified by
chromatography
on silica gel (40 p - 63 p) using dichlormethane:methanol in the ratio 9:1 as
mobile
phase.
14.1 g (72% of theory, colorless solid) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 495.2
CA 02477474 2004-08-25
16
Example 6
Stage 1: 5-(4'-Nitrobiphenyl-4-sulfonyl)-1-methyl-4,5,6,7-tetrahydro-1 H-
imidazo[4,5-c]pyridine-6-carboxylic acid
2 g of 1-methylspinacine (9.19 mmol) were dissolved in 15 ml of water and, at
0 C, 3
equivalents of a 3N NaOH solution (9.19 ml) were added. After 10 minutes
(min), a
solution of 4'-nitrobiphenyl-4-sulfonyl chloride (2.74g, 9.19 mmol) in
acetonitrile was
slowly added dropwise and, after room temperature was reached, the mixture is
stirred for a further 12 h. The crude product was purified by chromatography.
io 1.9 g (47% of theory, colorless solid) were obtained.
Mass spectrum: ESI: [M + H+]: m/z= 443
Stage 2: 5-(4'-Aminobiphenyl-4-sulfonyl)-1-methyl-4,5,6,7-tetrahydro-1 H-
imidazo[4,5-c]pyridine-6-carboxylic acid
1 g (2.3 mmol) of 5-(4'-nitrobiphenyl-4-sulfonyl)-1 -methyl-4,5,6,7-tetrahydro-
1 H-
imidazo[4,5-c]pyridine-6-carboxylic acid was dissolved in 15 ml of
dimethylformamide (DMF) and, after addition of 0.1 g of hydrogenation catalyst
(10% Pd on activated carbon), quantitatively hydrogenated within 2 h. After
removal
of the solvent, the crude product was purified by chromatography.
0.74 g (78% of theory, colorless solid) were obtained.
Mass spectrum: ESI: [M + H+j: m/z= 413
Stage 3: 5-(4'-Alkoxycarbonylaminobiphenyl-4-sulfonyl)-1-methyl-4,5,6,7-
tetrahydro-
1 H-imidazo[4,5-c]pyridine-6-carboxylic acids
500 mg (1.2 mmol) of 5-(4'-aminobiphenyl-4-sulfonyl)-1-methyl-4,5,6,7-
tetrahydro-
1 H-imidazo[4,5-c]pyridine-6-carboxylic acid were dissolved in 3 ml of DMF
and, after
cooling to 0 C in an ice bath, 2.4 mmol of pyridine were added. After stirring
at 0 C
for 15 min, 1.8 mmol of isopropyl chloroformate were added. The reaction
solution
was then stirred at room temperature for 2 h. The crude product was purified
by
chromatographic methods.
CA 02477474 2004-08-25
17
0.38 g (64% of theory, colorless solid) was obtained.
Mass spectrum: ESI: [M + H+]: m/z= 499
s Compounds 7 to 10 in table 1 were prepared in analogy to the procedures
described
in example 6.
CA 02477474 2004-08-25
18
Table 1
Example Structure MS (ESI+)
1
C) Chiral 481
O
N,/
4S S
O
N
O O
2 0 495
\ o
Chiral
otN,O
cO
O
N0
3 537
O
S;O
O
O
4 O 509
NHS
O/
O
0
CA 02477474 2004-08-25
19
(D 507
O0
N O~
6
499
\~
N
N
N 0
7
547
CO2H
N\ ~0
N OS
O
N-~
O
8
527
~N CO2H
N~N-S=0
~ N -- O
O
9
~N I CO2H 513
N~N. 'O
piS
N--<C
CA 02477474 2004-08-25
10
511
N CO ZH
< N O
0 r~
CA 02477474 2009-12-11
21
Pharmacological examples
Preparation and determination of the enzymatic activity of the catalytic
domain of
human stromelysin (MMP 3) and neutrophil collagenase (MMP8).
The two enzymes - stromelysin (MMP-3) and neutrophil collagenase (MMP-8) -
were prepared as described by Ye et al. (Biochemistry; 31 (1992) pages
11231-11235). To measure the enzymic activity or the enzyme inhibitory effect,
70 pi
of buffer solution and 10 N1 of enzyme solution are incubated at physiological
pH
1o with 10pI of a 10% strength (v/v) aqueous dimethyl sulfoxide solution which
optionally contains the enzyme inhibitor for 15 minutes. After addition of 10
,u1 of a
10% strength (v/v) aqueous dimethyl sulfoxide solution which contains 1 mmol/I
of
the substrate, the enzymic reaction is followed by fluorescence spectroscopy
(328
nm (ex)/393 nm(em)).
The enzymic activity is represented as increase in extinction/minute. The IC50
values
listed in table 2 are measured as the inhibitor concentrations leading in each
case to
50% inhibition of the enzyme.
The buffer solution contains 0.05% Brij (Signma, Deisenhofen, Germany) and
0.1 mol/I Tris/HCI, 0.1 mol/I NaCI, 0.01 mol/I CaCl2 and 0.1 mol/I piperazine-
N,N'-
bis[2-ethanesulfonic acid] (pH=7.5).
The enzyme solution contains 5 pg/ml of one of the enzyme domains prepared as
described by Ye et al. The substrate solution contains 1 mmol/I of the
fluorogenic
substrate (7-methoxycoumarin-4-yl)acetyl-Pro-Leu-Gly-Leu-3-(2',4'-
dinitrophenyl)-L-
2,3-diaminopropionyl-Ala-Arg-NH2 (Bachem, Heidelberg, Germany).
Determination of the enzymatic activity of the catalytic domain of human
collagenase
3 (MMP-13).
This protein is obtained as inactive proenzyme from INVITEK, Berlin (catalog
No. 30
100 803). Activation of the proenzyme:
2 parts by volume of proenzyme are incubated with 1 part by volume of APMA
solution at 37 C for 1.5 hours. The APMA solution is prepared from a 10 mmol/L
CA 02477474 2004-08-25
22
p-aminophenylmercuric acetate solution in 0.1 mmol/L NaOH by dilution with 3
parts
by volume of Tris/HCI buffer pH7.5 (see below). The pH is adjusted to between
7.0
and 7.5 by adding 1 mmol/L HCI. After activation of the enzyme it is diluted
with the
Tris/HCI buffer to a concentration of 1.67 pg/mL.
To measure the enzymic activity, 10 pL of enzyme solution are incubated with
10 PL
of a 3% strength (v/v) buffered dimethyl sulfoxide solution (reaction 1) for
minutes. To measure the enzyme inhibitory activity, 10 pL of enzyme solution
are
incubated with 101.& of a 3% strength (v/v) buffered dimethyl sulfoxide
solution
io which contains the enzyme inhibitor (reaction 2).
The enzymic reaction both in reaction 1 and in reaction 2 is followed, after
addition
of 10 pL of a 3% strength (v/v) aqueous dimethyl sulfoxide solution which
contains
0.75 mmol/L of the substrate, by fluorescence spectroscopy (328 nm
(extinction) /
15 393 nm(emission)).
The enzymic activity is represented as increase in extinction/minute.
The inhibitory effect is calculated as percentage inhibition by the following
formula:
% inhibition = 100 - [(increase in extinction/minute in reaction 2) /
(increase in
extinction/minute in reaction 1) x 100].
The IC50, i.e. the inhibitor concentration necessary for 50% inhibition of
enzymic
activity, is found by plotting a graph of the percentage inhibitions at
various inhibitor
concentrations.
The buffer solution contains 0.05% Brij (Sigma, Deisenhofen, Germany) and
0.1 mol/L Tris/HCI, 0.1 mol/L NaCl, 0.01 mol/L CaCI2 (pH=7.5).
The enzyme solution contains 1.67 pg/mL of the enzyme domain.
The substrate solution contains 0.75 mmol/L of the fluorogenic substrate
(7-methoxycoumarin-4-yl)acetyl-Pro-Leu-Gly-Leu-3-(2',4'-dinitrophenyl)-L-2,3-
diaminopropionyl-Ala-Arg-NH2 (Bachem, Heidelberg, Germany).
CA 02477474 2004-08-25
23
" Table 2 below shows the results.
Table 2
Example MMP 3 MMP 8 MMP 13
No. IC50 (nM) IC50 (nM) IC50 (nM)
1 3000 2300 40
2 3000 6000 30
3 >10000 2000 70
4 2000 3000 20
3000 4000 20
6 10000 4000 80
7 >10000 1000 60
8 >10000 5000 70
9 10000 200 60
>10000 700 20
Comparative examples
5 The following compounds specified in table 3 were prepared as described in
WO
97/18194.
Table 3:
Example Structure MMP MMP 3 MMP 8
No. 131) (nM) (nM)
(nM)
34 0 50 500 10
NS~
I
CA 02477474 2004-08-25
24
= 35 0 30 100 5
OH
"IS
O
39 O 2.9 100 1
O
1) Determination took place as described above
2) and 3) The data were taken from WO 97/18194
Table 3 shows that structurally similar compounds from the prior art show no
selectivity in inhibition only of MMP 13.