Note: Descriptions are shown in the official language in which they were submitted.
CA 02478138 2004-08-17
CXCL10-BASED DIAGNOSIS AND TREATMENT OF RESPIRATORY
ILLNESSES
FIELD OF THE INVENTION
The invention relates to the diagnosis and
treatment of patients suffering from respiratory illnesses,
e.g. Severe Acute Respiratory Syndrome (SARS), influenza,
and community-acquired pneumonia. Elevated blood levels of
CXCL10 polypeptide are associated with respiratory illnesses
and are useful in diagnosis and prognosis of patients.
Methods are provided for treating respiratory illnesses by
inhibiting the CXCL10:CXCR3 axis. Methods are also provided
for identifying inhibitors of the CXCL10:CXCR3 axis, for use
in treating patients suffering from respiratory illnesses.
BACKGROUND OF THE INVENTION
Respiratory illnesses (e. g. community-acquired
pneumonia, influenza and SARS) are a major public health
concern.
Severe acute respiratory syndrome (SARS) emerged
in late 2002 from its purported origins in Guangdong
Province, China and infected over 8400 persons worldwide to
date with an accompanying case fatality rate of
approximately 11~ (1-4, World Health Organization,
http://www.who.int/csr/sars/country/en/country2003-OS-l5.pdf
A novel coronavirus (CoV), causing a spectrum of
disease ranging from non-specific flu-like symptoms and lung
inflammation to acute respiratory distress syndrome CARDS)
requiring intensive care, has been identified as the
etiologic agent of SARS (5-8). While the SARS CoV epidemic
of 2003 was largely contained through public health
1
CA 02478138 2004-08-17
measures, it is unknown whether or not human SARS CoV will
cause another global outbreak. With the confirmation of new
unrelated SARS cases in China (9) and the finding that SARS
CoV-related viruses infected persons in Hong Kong at least 2
years prior to the 2003 outbreak (10), it is clear that SARS
will not easily be eradicated and that jumps from animals to
humans will continue.
Lungs from patients with severe SARS show
extensive acute injury with diffuse alveolar damage, acute
vascular and endothelial injury and extensive immune
infiltration (11-13). The molecular, cellular and
pathological determinants that lead to lung injury and poor
outcome in SARS are presently unclear; however the severity
of SARS CoV infection may be partially determined by the
immune system and dysregulated proinflammatory cytokines and
chemokines (14-16).
SUMMARY OF THE INVENTION
In one aspect, the present invention provides a
method for diagnosing respiratory illness in a patient, the
method comprising detecting CXCL10 polypeptide in a
biological sample from a patient, wherein an elevated level
of CXCL10 polypeptide in said sample relative to a healthy
control is diagnostic or prognostic of respiratory illness.
In another aspect, the present invention provides
a commercial package comprising means for detecting CXCL10
polypeptide in a biological sample from a patient, together
with instructions for use for diagnosis and/or prognosis of
respiratory illness in a patient or for monitoring treatment
thereof .
2
CA 02478138 2004-08-17
In another aspect, the present invention provides
the use of CXCL10 polypeptide in the diagnosis of
respiratory illness, wherein said CXCL10 polypeptide is
contained in a biological sample from a patient and wherein
an elevated level of CXCL10 polypeptide in said sample
relative to a healthy control is diagnostic or prognostic of
respiratory illness.
In another aspect, the present invention provides
a method for treating a patient suffering from respiratory
illness, the method comprising administering a compound that
inhibits the CXCL10:CXCR3 axis.
In another aspect, the present invention provides
a method for identifying a therapeutic agent for treating a
respiratory illness, the method comprising:
(a) providing a test compound;
(b) providing a cell that expresses a polypeptide selected
from CXCL10 polypeptide and CXCR3 polypeptide when
cultured under suitable conditions in vitro; and
(c) detecting whether the test compound inhibits expression
of said polypeptide by said cell.
In another aspect, the present invention provides
a method for identifying a therapeutic agent for treating
respiratory illness, the method comprising:
(a) providing a test compound;
(b) providing a polypeptide selected from CXCL10
polypeptide or CXCR3 polypeptide; and
(c) detecting whether the test compound binds to said
polypeptide.
3
CA 02478138 2004-08-17
In another aspect, the present invention provides a
method for identifying a therapeutic agent for treating
respiratory illness, the method comprising:
(a) providing a test compound;
(b) providing a polypeptide selected from CXCL10
polypeptide or CXCR3 polypeptide;
(c) providing a binding partner that binds to said
polypeptide; and
(d) detecting whether the test compound inhibits binding of
said binding partner to said polypeptide.
In another aspect, the present invention provides
a therapeutic agent for treating respiratory illness
identified by the screening method of the invention.
In another aspect, the present invention provides
a pharmaceutical composition for treating respiratory
illness comprising the therapeutic agent a therapeutic agent
identified by the screening method of the invention.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1. High plasma concentrations of CXCL10
polypeptide in SARS patients. CXCL10 polypeptide (Figure
lA) and CXCL9 (Figure 1D) were measured in plasma from
suspect (n = 80) and confirmed (n = 34) SARS patients within
48 hours of onset of symptoms by CBA. CXCL10 polypeptide
and CXCL9 levels were also recorded healthy controls (n =
14) and convalescents (n = 63). Higher levels of CXCL10
polypeptide were measured in SARS patients relative to the
other 3 groups (** P < 0.0001). Other group comparisons
revealed no further statistically significant differences.
4
CA 02478138 2004-08-17
CXCL10 polypeptide (Figure 1B, 1C) and CXCL9 (Figure lE, 1F)
were measured in 25 confirmed SARS patients (Figure 1B, lE)
and 10 confirmed SARS patients in the intensive care unit
(ICU) (Figure 1C, 1F). * significantly lower points of
CXCL10 polypeptide expression at 16-40 days in confirmed
SARS patients compared to those in the ICU (* P < 0.05).
Solid lines represent consecutive measurements in the same
patient and t indicates deceased. Curves produced by
nonlinear regression analysis (hatched lines).
Figure 2. CXCL10 polypeptide and CXCR3
polypeptide are expressed in the lungs of deceased SARS
patients. RNA was isolated from bilateral lower lung
biopsies from deceased confirmed SARS patients (n=3) and
normal cadaveric lungs (n=3). CXCL10 polypeptide (Figure
2A) and CXCR3 polypeptide (Figure 2B) were quantitated by
real time PCR. The relative expression of each gene is
shown normalized to GAPDH.
Figure 3. CXCL10 polypeptide and CXCR3
polypeptide expression by SARS patient PBMCs and CXCL10
polypeptide expression by SARS CoV-infected VERO E6 cells as
determined by real time PCR. CXCL10 polypeptide (Figure 3A)
and CXCR3 polypeptide (Figure 3B) were upregulated in PBMCs
from three random SARS patients within 7 days from onset of
symptoms in comparison to PBMCs from three healthy controls.
CXCL10 polypeptide transcripts were also detected in
triplicate cultures of SARS CoV (Tor2)-infected VERO E6
cells at 12-24 hours (Figure 3C). The relative expression
of each gene is shown normalized to GAPDH.
Figure 4. High proliferative capacity of, and
CXCR3 polypeptide expression by, SARS N protein-specific CD4+
and CD8+ T cells in convalescent SARS patients. 1x106
5
CA 02478138 2004-08-17
cryopreserved PBMCs from convalescent confirmed SARS
patients were thawed, stained with CFSE and cultured in
media alone or in the presence of recombinant SARS CoV N
protein. After 6 d, we measured proliferating (CFSE1°'") CD4+
and CD8+ T cells and CXCR3 polypeptide expression on
proliferating CD4+ and CD8'" T cells from healthy controls
(Figure 4A) and convalescent SARS patients (Figure 4H) by
four FAGS analysis. Results shown are representative of 5
randomly chosen convalescent SARS patients and 5 healthy
controls. The numbers shown in the upper left quadrants
are the percentage of CFSE1°"' CD4'" or CD8+ T cells or those
expressing CXCR3 polypeptide. 50 ng/ml SEA (Toxin
Technology, Sarasota, FL) was used as a positive control.
Average percentages following SEA stimulation were Control
CD4+ CFSE1°"' 74 ~ ( range 6 7 - 8 8 ) , SARS CD4+ CFSEz°W
8 8 ~ ( range
83-92), Control CD4+/CXCR3+ CFSE1°"' 20~ (range 16-23), SARS
CD4+/CXCR3+ CFSE1°'" 22~ (range 15-32) , Control CD8+
CFSE1°"' 86~
(range 81-92) , SARS CD8+ CFSEl°'" 91~ (range 85-96) , Control
CD8+/CXCR3+ CFSE1°W 63~ (range 52-68) , SARS CD4''/CXCR3~ CFSEloW
55~ (range 29-74).
Figure 5. PBMCs from macaques infected by SARS
CoV express CXCL10 polypeptide within 3-5 days post-
infection. Rhesus (n=4) and cynomolgus (n=4) macaques were
inoculated with 1 x 106 p.f.u. SARS-CoV (Tor2)
intratracheally and monitored for 13 days. Blood samples
were collected on the days shown and PBMC RNA was isolated.
Real time PCR was performed using the human CXCL10 primers
listed in the Methods and Materials and the relative
expression of each gene is shown normalized to GAPDH.
Results are shown for one representative cynomolgus and
rhesus monkey. CXCL10 polypeptide/GAPDH ratios in PBMCs
from day 0 monkeys and additional uninfected control monkeys
(n=4) did not exceed 0.03.
6
CA 02478138 2004-08-17
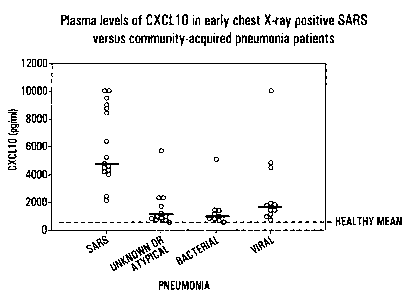
Figure 6. High plasma concentrations of CXCL10
polypeptide in chest X-Ray positive SARS patients and some
severe cases of community acquired pneumonia. CXCL10
polypeptide was measured in plasma from confirmed chest X-
Ray positive SARS patients (n = 16) within 7 d of onset of
symptoms by CBA. CXCL10 polypeptide levels were also
recorded in healthy controls (n = 14, mean shown) and
patients with chest X-Ray positive community acquired
pneumonia (n = 32). Causative agents are listed on the
l0 X axis.
DETAILED DESCRIPTION OF THE INVENTION
Previous findings indicate that CXCL10 polypeptide
(IFN-y inducible protein 10/IP-10) is associated with host
defense in marine models of non-SARS coronavirus infections
(17-20) ,
We profiled the CXCL10:CXCR3 axis in patients of
different SARS diagnosis, status and outcome throughout
their disease and utilized two non-human primate models of
SARS CoV infection to confirm select findings. We found
that the lung tropism of SARS CoV could be linked with early
expression of CXCL10 polypeptide in the plasma and lungs of
SARS patients, which in turn could regulate infiltration of
SARS antigen-specific T cells and potentially result in an
uncontrollable immune responses against SARS CoV. Our data
suggest that lung injury in SARS patients may result from an
unabated proinflammatory immune responses against the SARS
CoV involving CXCL10 polypeptide and its receptor CXCR3
polypeptide.
7
CA 02478138 2004-08-17
Thus, our data shows that the CXCL10:CXCR3 axis
plays a key role in SARS disease course and lung injury and
represents an important therapeutic target.
We obtained specimens within 24-48 hours of onset
of symptoms from both confirmed SARS patients and non-S.AR.S
patients with SARS-like symptoms (suspect SARS in Fig. 1).
Our longitudinal study also delineated patients suffering
from mild to moderate or severe forms of the disease in the
intensive care unit (ICU). Wong et al. described CXCL10
polypeptide increases during the early phase of infection
relative to healthy controls in a recent study of plasma
levels of cytokines and chemokines in a Hong Kong cohort of
SARS patients (15), however our study is the first to
associate the CXCL10:CXCR3 axis with a unique role in the
evolution of SARS illness.
CXCLIO polypeptide was significantly elevated in
all SARS patients and only slightly elevated in a few
suspect cases. In patients with a severe course of disease,
plasma levels of CXCL10 polypeptide remained elevated for
weeks whereas CXCL10 polypeptide levels decreased more
quickly to normal levels in patients with mild disease.
Steroids appeared to have little effect on CXCL10
polypeptide levels since expression of CXCL10 polypeptide
persisted in both SARS patients with mild disease and those
with severe disease after steroid therapy commenced
(typically 3-5 d post-onset of symptoms), at least until the
patient began to recover. Moreover, an ICU patient with the
highest sustained levels of plasma CXCL10 polypeptide died
of SARS while undergoing immunosuppressive therapy to
prevent graft rejection and aggressive steroid therapy for
SARS (Fig. l). Indeed, it has been shown that elevated
levels of CXCL10 polypeptide in multiple sclerosis patients
8
CA 02478138 2004-08-17
are not affected by steroid treatment (23) and CXCL10
polypeptide expression in bronchial epithelial cells is not
affected by steroid treatment (24). We do not yet know what
causes the long-term increased levels of CXCL10 polypeptide
in the plasma of critical care patients, however mechanical
injury due to ventilation culminating in ARDS may be
responsible for continued CXCL10 polypeptide induction (25).
On the other hand, this may only represent a partial
explanation since whether or not a patient had cleared the
virus, not whether or not mechanical ventilation was
required, was more strongly correlated with changes in
plasma CXCL10 polypeptide levels. Indeed, it has been shown
that lung tissues from deceased SARS patients remain
infected with SARS CoV (12,13).
The SARS patients profiled in this study comprised
a highly diverse cohort with many variables in its
composition, such as demographics, symptom presentation,
course of illness and treatment, clinical features and
parameters, microbiology, comorbidities and adverse events.
Non-human primate models for SARS have been developed where
symptoms in macaques are consistent with mild forms of SARS
in humans, such as lung inflammation, alveolar damage and
pneumonia, following experimental inocculation with SARS CoV
(26,27). Using two non-human primate models of SARS
infection we confirmed that CXCL10 polypeptide expression is
indeed upregulated in vivo in SARS-infected rhesus and
cynomolgus macaques in the early stages of infection with
SARS CoV (Tor2).
Antiviral cellular responses are dependent on the
production of cytokines, especially type I and II
interferons and chemokines. Chemokines are known for their
roles in cell recruitment but they can also activate immune
9
CA 02478138 2004-08-17
cells and shape Thl/Th2 responses (28,29). Tn fact,
chemokine expression can be induced upon viral entry into a
cell (30). Based on the fact that SARS CoV induces CXCL10
polypeptide expression directly in infected VERO E6 cells,
we suggest that as SARS CoV establishes infection, CXCL10
polypeptide is induced during primary lung inflammation.
CXCR3 polypeptide, the receptor for CXCL10 polypeptide,
plays a potent role in regulating migration of activated T
cells (especially Thl cells) to sites of inflammation (28).
We expect that the tropism of coronaviruses in
general induces localized expression of CXCL10 polypeptide
and subsequent recruitment of CXCR3 polypeptide-bearing
lymphocytes. In keeping with this idea, we found elevated
RNA levels of CXCR3 polypeptide in the lungs of deceased
SARS-infected individuals, increased RNA levels of CXCR3
polypeptide in PBMCs and expression of cell surface CXCR3
polypeptide on SARS CoV N antigen-specific T cells with high
proliferative capacity.
Others have demonstrated a role for CXCL10
polypeptide in the immunopathology of CoV infections in
murine models of other diseases (17-20). CXCL10 polypeptide
seems to be important for clearance of CoV and may also
regulate autoinflammatory events that lead to severe
pathogenesis in these models, as well as human autoimmune
inflammatory diseases (31-33).
Based on our findings, we propose that CXCL10
polypeptide plays a dual role in SARS CoV infections.
Elevation of CXCL10 polypeptide in 100 of SARS-infected
individuals early in their disease indicates that it is a
necessary host response to clear SARS CoV through the
recruitment of antigen-specific effector CXCR3 polypeptide-
CA 02478138 2004-08-17
bearing T cells, however in a minority of patients CXCL10
polypeptide may participate in SARS pathogenesis through the
continued recruitment of activated T cell and mononuclear
infiltrates resulting in tissue destruction and eventual
organ failure. Once again, lung tissues from deceased SARS
patients express high levels of CXCL10 polypeptide (Fig. 2)
and remain infected with SARS CoV (12,13).
Previous studies have shown that CXCL10
polypeptide can be induced by IFN-y under certain conditions.
We found statistically significant increases (3-5 fold) in
IFN-y levels in the plasma of SARS patients at onset relative
to healthy controls and convalescents (data not shown)
indicating that IFN-y-induced expression of CXCL10
polypeptide may play a role in the SAR.S disease course.
We expect that certain other viral infections can
drive expression of CXCL10 polypeptide early in the course
of infection. Our preliminary data indicates that some
cases of West Nile Virus infection and community-acquired
pneumonia are associated with small short-term increases of
CXCL10 polypeptide. We have found that those infections
also drive the expression of CXCL9, a closely related non-
ELR chemokine that also binds CXCR3 polypeptide (data not
shown). This finding is in contrast to SARS, where levels
of CXCL9 could not be associated with disease onset, course
or severity. This disparate involvement of CXCL10
polypeptide versus CXCL9 has been noted in studies of
autoimmune disease (34).
We have shown that an early pattern of high levels
of circulating CXCL10 polypeptide in the absence of CXCL9 is
associated with a SARS CoV infection. Our data supports a
model in which lung tropism of SARS CoV induces early
11
CA 02478138 2004-08-17
expression of CXCL10 polypeptide in the lungs that in turn
regulates infiltration of SARS antigen-specific T cells,
thereby mediating immune responses against SARS CoV. The
relationship between elevated and sustained levels of CXCL10
polypeptide and poor outcome in SARS patients may be a
result of CXCL10 polypeptide recruitment of autoinflammatory
cells and the failure of the immune system to clear the SARS
CoV. While CXCL10 polypeptide may play a role in the
establishment of other respiratory diseases, including those
induced virally, patients suffering from inflammatory lung
disease may benefit from strategies that neutralize or
inhibit the pivotal function of the CXCL10:CXCR3 axis.
Based on our findings in patients infected with
SARS, we predicted that the CXCL10:CXCR3 axis may play a
role in other respiratory infections (such as influenza,
especially influenza A, and community-acquired pneumonia,
which may be of viral, bacterial, or atypical (unknown)
origin), i.e, that other infectious respiratory agents could
drive the expression of CXCL10 polypeptide during the course
of infection. Our data show that this is indeed the case.
Community-acquired pneumonia refers to a set of potentially
severe lung infections acquired outside of hospitals.
Common pathogens responsible for community-acquired
pneumonia infections are Streptococcus pneumoniae,
Haemophilus influenzae, Mycoplasma pneumoniae, influenza A,
Legionella, Moraxella catarrhalis, Mycobacterium
tuberculosis, and Chlamydophilia pneumoniae. The causative
agent of community acquired pneumonia can remain
unidentified in up to 50~ of cases (atypical pneumonia).
Our data comparing chest X-Ray positive (immune infiltrated)
SARS patients with chest X-Ray positive patients with the
various forms of community acquired pneumonia indicated that
CXCL10 polypeptide plays a role in the establishment of
12
CA 02478138 2004-12-24
severe cases of these other respiratory diseases. Therefore
patients suffering from multiple forms of serious
inflammatory lung disease may benefit from the diagnostic
and prognostic properties of the CXCL10:CXCR3 axis and well
as therapies that neutralize or inhibit the pivotal function
of the CXCL10:CXCR3 axis.
In the present context, patients suffering from
"severe SARS" are those SARS patients requiring increased
levels of supplemental oxygen (i.e. those with Pa02/Fi02
readings <200), supplied by mask or intubation, and/or
admission to the intensive care unit.
The time course of SARS disease highly variable.
Mild cases of SARS typically last 14-20 days. However,
severe cases of SARS can require 2-3 months of
hospitalization.
CXCL10 polypeptides and CXCR3 polypeptides and
polvnucleotides encodina them
"CXCL10" belongs to the a-chemokine (C-X-C) family
and is also referred to as "Interferon-'y inducible protein 10
kD" or "IP-10". Several CXCL10 polypeptide amino acid
sequences and nucleotide sequences encoding them are known,
including for example:
(1) Human CXCL10 polypeptide amino acid sequence: GenBank
accession No. NP 001556.1 (SEQ ID N0: 1).
(2) Human CXCL10 polypeptide mRNA sequence: GenBank
accession No. NM 001565 (SEQ ID N0: 2).
(3) Murine CXCL10 polypeptide amino acid sequence: GenBank
accession No. NP 067249.1 (SEQ ID N0: 3).
13
CA 02478138 2004-12-24
(4) Murine CXCL10 polypeptide mRNA sequence: GenBank
accession No. NM 021274.1 (SEQ ID N0: 4).
"CXCR3" amino acid sequences and nucleotide
sequences encoding them are also known, including for
example:
(1) Human CXCR3 polypeptide mRNA sequence: GenBank
accession No. NM 001504 (SEQ ID N0: 5).
(2) Human CXCR3 polypeptide amino acid sequence: GenBank
accession No. NP 001495 (SEQ ID N0: 6).
(3) Murine CXCR3 polypeptide amino acid sequence: GenBank
accession No. NP 034040.1 (SEQ ID N0: 7).
(4) Murine CXCR3 polypeptide mRNA sequence: GenBank
accession No. NM 009910.1 (SEQ ID N0: 8).
As defined herein, the expressions "CXCL10
polypeptide" and "CXCR3 polypeptide" include variants of
native CXCL10 polypeptide and CXCR3 polypeptide, for
example: deletions, including truncations and fragments;
insertions and additions, including tagged polypeptides and
fusion proteins; substitutions, for example site-directed
mutants and allelic variants; modifications, including for
example peptoids having one or more non-amino aryl groups
(q. v., sugar, lipid, etc.) covalently linked to the peptide
and post-translational modifications; and peptidomimetics.
As used herein, "polypeptide" means any chain of
amino acids, regardless of length or post-translational
modification (e.g., glycosylation or phosphorylation), and
14
CA 02478138 2004-08-17
include: natural proteins; synthetic or recombinant
polypeptides and peptides as well as hybrid molecules (e. g.
a fusion protein or chimera having one portion comprising
all or part of a polypeptide of the invention and a second
portion comprising an amino acid sequence from another
protein or peptide); modified peptides, including for
example peptoids having one or more non-amino acyl groups
(q. v., sugar, lipid, etc.) covalently linked to the peptide;
and peptidomimetics. Typically the protein or polypeptide
may be isolated or substantially pure or recombinant. In
the present context, polypeptides can have a length of for
example at least 6, 8, 10, 12, 14, 16, 18, 20, 50, 100, 200,
300, 400, 500, etc. amino acids.
The term "polynucleotide" refers to a polymeric
form of nucleotides of any length (e.g. at least 9, 12, 15,
18, 20, 50, 100, 200, 500, 1000, 2000, etc. nucleotides) and
may also be referred to in the art as a "nucleic acid" or
"nucleic acid molecule". The nucleotides can be
ribonucleotides, deoxyribonucleotides, or modified forms of
either type of nucleotide (e.g. as described below for
antisense nucleotide molecules). The term includes single
and double stranded forms of DNA or RNA. DNA includes, for
example, cDNA, genomic DNA, chemically synthesized DNA, DNA
amplified by PCR, and combinations thereof. The
polynucleotides of the invention include full-length genes
and cDNA molecules as well as fragments thereof.
Variants can be prepared, for example, by
substituting, deleting or adding one or more amino acid
residues in the amino acid sequence of a native CXCL10
polypeptide or CXCR3 polypeptide or fragment thereof, and
screening for biological activity. Preferably,
substitutions are made with conservative amino acid
CA 02478138 2004-08-17
residues, i.e., residues having similar physical, biological
or chemical properties.
A peptidomimetic is a compound that mimics the
conformation and certain stereochemical features of the
biologically active form of a particular peptide. In
general, peptidomimetics are designed to mimic certain
desirable properties of a compound, but not the undesirable
properties, such as flexibility, that lead to a loss of a
biologically active conformation and bond breakdown.
Peptidomimetics may be prepared from biologically active
compounds by replacing certain groups or bonds that
contribute to the undesirable properties with bioisosteres.
Bioisosteres are known to those of skill in the art. For
example the methylene bioisostere CHZS has been used as an
amide replacement in enkephalin analogs (see, eg., Spatola
(1983) pp. 267-357 in Chemistry and BIochemistry of Amino
Acids, Peptides, and Proteins, Weistein, Ed. volume 7,
Marcel Dekker, New York). By way of illustration, morphine
is a well-known peptidomimetic of the peptide endorphin. For
purposes herein, cyclic peptides are included among
pepidomimetics.
As used herein, "biological activity" includes any
parameter that is indirectly or directly under the influence
of CXCL10 polypeptide or CXCR3 polypeptide expressed either
endogenously or exogenously. It includes binding to a
binding partner and regulating the immune response and local
inflammation e.g. by regulating T-cell recruitment and
adhesion).
A variant of CXCL10 polypeptide may be used, for
example, in a screening assay for identifying compounds that
inhibit interaction between CXCL10 polypeptide and CXCR3
16
CA 02478138 2004-08-17
polypeptide. In that context, the CXCL10 polypeptide
variant must be capable of binding CXCR3 polypeptide.
A variant of CXCR3 polypeptide that binds to
CXCL10 polypeptide can be used, for example, as an inhibitor
of CXCL10 polypeptide or in a screening assay for
identifying compounds that inhibit interaction between CXCR3
polypeptide and CXCL10 polypeptide. For example, a fragment
of CXCR3 polypeptide comprising the extracellular domain of
CXCR3 polypeptide and which can bind CXCL10 polypeptide may
be useful as an inhibitor of CXCL10 polypeptide or as a
binding partner in assays for identifying an inhibitor
binding of CXCL10 polypeptide to CXCR3 polypeptide.
CXCL10 polypeptide can be conveniently detected in
a biological sample (e. g. for practicing the diagnostic
methods of the invention) using standard techniques,
including for example: ELISA; cytometric bead array (CHA);
FAGS (i.e. intracellular fluorescence staining); ELIspot;
immunoblotting (i.e. western blotting); and microarray.
In the present context, the term "biological
sample" denotes any sample obtained from an animal,
including for example, tissue, secretion, hair, blood, blood
cells (e. g, lymphocytes, such as T-cells) and plasma.
There are many ways in which the CXCL10:CXCR3 axis can
be inhibited for treating or ameliorating a respiratory
illness in a patient, including for example administering to
the patient:
(a) a binding partner of CXCL10 polypeptide or CXCR3
polypeptide;
(b) an antibody that binds CXCL10 polypeptide, CXCR3
polypeptide, or IFN-gamma;
17
CA 02478138 2004-08-17
(c) an antisense RNA that that binds to an mRNA encoding
CXCL10 polypeptide, CXCR3 polypeptide, or IFN-gamma;
(d) an RNAi that targets mRNA encoding CXCL10 polypeptide,
CXCR3 polypeptide, or IFN-gamma;
(e) a transcription regulator protein that inhibits
expression of CXCL10 polypeptide, CXCR3 polypeptide or
INF-gamma; and
(f) a small molecule that interacts with CXCL10
polypeptide, CXCR3 polypeptide, or IFN-gamma or
inhibits expression thereof.
Homology/hybridization
"Homology" and "homologous" refers to sequence
similarity between two peptides or two nucleic acid
molecules. Homology can be determined by comparing each
position in the aligned sequences. A degree of homology
between nucleic acid or between amino acid sequences is a
function of the number of identical or matching nucleotides
or amino acids at positions shared by the sequences. As the
term is used herein, a nucleic acid sequence is "homologous"
to another sequence if the two sequences are substantially
identical and the functional activity of the sequences is
conserved (as used herein, the term 'homologous' does not
infer evolutionary relatedness). Two nucleic acid sequences
are considered substantially identical if, when optimally
aligned (with gaps permitted), they share at least about 50%
sequence similarity or identity, or if the sequences share
defined functional motifs. In alternative embodiments,
sequence similarity in optimally aligned substantially
identical sequences may be at least 60%, 70%, 75%, 80%, 85%,
90% or 95%. As used herein, a given percentage of homology
18
CA 02478138 2004-08-17
between sequences denotes the degree of sequence identity in
optimally aligned sequences. An "unrelated" or "non-
homologous" sequence shares less than 40~ identity, though
preferably less than about 25 ~ identity, with any of the
sequences identified herein for CXCL10 polypeptides, CXCR3
polypeptides and the polynucleotides encoding them.
Substantially complementary nucleic acids are nucleic
acids in which the complement of one molecule is
substantially identical to the other molecule. Two nucleic
acid or protein sequences are considered substantially
identical if, when optimally aligned, they share at least
about 70~ sequence identity. In alternative embodiments,
sequence identity may for example be at least 75~, at least
80~, at least 85~, at least 90~, or at least 95~. Optimal
alignment of sequences for comparisons of identity may be
conducted using a variety of algorithms, such as the local
homology algorithm of Smith and Waterman, 1981, Adv. Appl.
Math 2: 482, the homology alignment algorithm of Needleman
and Wunsch, 1970, J. MoI. Biol. 48:443, the search for
similarity method of Pearson and Lipman, 1988, Proc. Natl.
Acad. Sci. USA 85: 2444, and the computerised
implementations of these algorithms (such as GAP, BESTFIT,
FASTA and TFASTA in the Wisconsin Genetics Software Package,
Genetics Computer Group, Madison, WI, U.S.A.). Sequence
identity may also be determined using the BLAST algorithm,
described in Altschul et al., 1990, J. Mol. Biol.°215:403-10
(using the published default settings). Software for
performing BLAST analysis may be available through the
National Center for Biotechnology Information (through the
Internet at http://www.nebi.nlm.nih.gov/). The BLAST
algorithm involves first identifying high scoring sequence
pairs (HSPs) by identifying short words of length W in the
19
CA 02478138 2004-08-17
query sequence that either match or satisfy some positive-
valued threshold score T when aligned with a word of the
same length in a database sequence. T is referred to as the
neighbourhood word score threshold. Initial neighbourhood
word hits act as seeds for initiating searches to find
longer HSPs. The word hits are extended in both directions
along each sequence for as far as the cumulative alignment
score can be increased. Extension of the word hits in each
direction is halted when the following parameters are met:
the cumulative alignment score falls off by the quantity X
from its maximum achieved value; the cumulative score goes
to zero or below, due to the accumulation of one or more
negative-scoring residue alignments; or the end of either
sequence is reached. The BLAST algorithm parameters W, T and
X determine the sensitivity and speed of the alignment. The
BLAST program may use as defaults a word length (W) of 11,
the BLOSUM62 scoring matrix (Henikoff and Henikoff, 1992,
Proc. Natl. Acad. Sci. USA 89: 10915-10919) alignments (B)
of 50, expectation (E) of 10 (or 1 or 0.1 or 0.01 or 0.001
or 0.0001), M=5, N=4, and a comparison of both strands. One
measure of the statistical similarity between two sequences
using the BLAST algorithm is the smallest sum probability
(P(N)), which provides an indication of the probability by
which a match between two nucleotide or amino acid sequences
would occur by chance. In alternative embodiments of the
invention, nucleotide or amino acid sequences are considered
substantially identical if the smallest sum probability in a
comparison of the test sequences is less than about 1,
preferably less than about 0.1, more preferably Less than
about 0.01, and most preferably less than about 0.001.
An alternative indication that two nucleic acid
sequences are substantially complementary is that the two
sequences hybridize to each other under moderately
24
CA 02478138 2004-08-17
stringent, or preferably stringent, conditions.
Hybridisation to filter-bound sequences under moderately
stringent conditions may, for example, be performed in 0.5 M
NaHP04, 7% sodium dodecyl sulfate (SDS), 1 mM EDTA at 65°C,
and washing in 0.2 x SSC/0.1% SDS at 42°C (see Ausubel, et
al. (eds), 1989, Current Protocols in Molecular Biology,
Vol. 1, Green Publishing Associates, Inc., and John Wiley &
Sons, Inc., New York, at p. 2.10.3). Alternatively,
hybridization to filter-bound sequences under stringent
conditions may, for example, be performed in 0.5 M NaHP04, 7%
SDS, 1 mM EDTA at 65°C, and washing in 0.1 x SSC/0.1% SDS at
68°C (see Ausubel, et al. (eds), 1989, supra). Hybridization
conditions may be modified in accordance with known methods
depending on the sequence of interest (see Tijssen, 1993,
Laboratory Techniques in Biochemistry and Molecular Biology
-- Hybridization with Nucleic Acid Probes, Part I, Chapter 2
"Overview of principles of hybridization and the strategy of
nucleic acid probe assays", Elsevier, New York). Generally,
stringent conditions are selected to be about 5°C lower than
the thermal melting point for the specific sequence at a
defined ionic strength and pH.
Antisense nucleic acid molecules
Antisense molecules and ribozymes for exogenous
administration can be used to effect the degradation and/or
inhibition of the translation of a target mRNA involved in
the CXCL10:CXCR3 axis, e.g. a mRNA encoding CXCL10
polypeptide. Examples of therapeutic antisense
oligonucleotide applications, incorporated herein by
reference, include: U.S. Pat. No. 5,135,917, issued Aug. 4,
1992; U.S. Pat. No. 5,098,8'90, issued Mar. 24, 1992; U.S.
Pat. No. 5,087,617, issued Feb. 11, 1992; U.S. Pat. No.
21
CA 02478138 2004-08-17
5,166,195 issued Nov. 24, 1992; U.S. Pat. No. 5,004,810,
issued Apr. 2, 1991; U.S. Pat. No. 5,194,428, issued Mar.
16, 1993; U.S. Pat. No. 4,806,463, issued Feb. 21, 1989;
U.S. Pat. No. 5,286,717 issued Feb. 15, 1994; U.S. Pat. No.
5,276,019 and U.S. Pat. No. 5,264,423; BioWorld Today, Apr.
29, 1994, p. 3.
Preferably, in antisense molecules, there is a
sufficient degree of complementarity to the target mRNA to
avoid non-specific binding of the antisense molecule to non-
target sequences under conditions in which specific binding
is desired, such as under physiological conditions in the
case of in vivo assays or therapeutic treatment or, in the
case of in vitro assays, under conditions in which the
assays are conducted. The target mRNA for antisense binding
may include not only the information to encode a protein,
but also associated ribonucleotides, which for example form
the 5'-untranslated region, the 3'-untranslated region, the
5' cap region and intron/exon junction ribonucleotides. A
method of screening for antisense and ribozyme
polynucleotides that may be used to provide the antisense
molecules used to practice the invention is disclosed in
U.S. Patent No. 5,932,435 (which is incorporated herein by
reference) .
Antisense molecules (oligonucleotides) used to
practice the invention may include those which contain
intersugar backbone linkages such as phosphotriesters,
methyl phosphonates, short chain alkyl or cycloalkyl
intersugar linkages or short chain heteroatomic or
heterocyclic intersugar linkages, phosphorothioates and
those with CHa--NH--O--CH2, CH2--N(CH3) --O--CH2 (known as
methylene (methylimino) or MMI backbone) , CHZ--O--N(CH3) --CHZ,
CHZ--N(CH3) --N(CH3) --CHz and O--N(CH3) --CH2 --CHZ backbones
22
CA 02478138 2004-08-17
(where phosphodiester is O--P--O--CH2). Oligonucleotides
having morpholino backbone structures may also be used (U. S.
Pat. No. 5,034,506). In alternative embodiments, antisense
oligonucleotides may have a peptide nucleic acid (PNA,
sometimes referred to as "protein nucleic acid") backbone,
in which the phosphodiester backbone of the oligonucleotide
may be replaced with a polyamide backbone wherein
nucleosidic bases are bound directly or indirectly to aza
nitrogen atoms or methylene groups in the polyamide backbone
(Nielsen et al., 1991, Science 254:1497 and U.S. Pat. No.
5,539,082). The phosphodiester bonds may be substituted
with structures which are chiral and enantiomerically
specific. Persons of ordinary skill in the art will be able
to select other linkages for use in practice of the
invention.
Antisense oligonucleotides may also include
species which include at least one modified nucleotide base.
Thus, purines and pyrimidines other than those normally
found in nature may be used. Similarly, modifications on
the pentofuranosyl portion of the nucleotide subunits may
also be effected. Examples of such modifications are 2'-0-
alkyl- and 2'-halogen-substituted nucleotides. Some
specific examples of modifications at the 2' position of
sugar moieties which are useful in the present invention are
OH, SH, SCH3, F, OCN, 0 (CH2) n NH2 or 0 (CHz) n CH3 where n is
from 1 to about 10; C1 to Clo lower alkyl, substituted lower
alkyl, alkaryl or aralkyl; Cl; Br; CN; CF3 ; OCF3 ; O-, S-,
or N-alkyl; 0-, S-, or N-alkenyl; SOCH3 ; SOZ CH3; ONOZ ; NOz
N3; NHZ; heterocycloalkyl; heterocycloalkaryl;
aminoalkylamino; polyalkylamino; substituted silyl; an RNA
cleaving group; a reporter group; an intercalator; a group
for improving the pharmacokinetic properties of an
oligonucleotide; or a group for improving the
23
CA 02478138 2004-08-17
pharmacodynamic properties of an oligonucleotide and other
substituents having similar properties. One or more
pentofuranosyl groups may be replaced by another sugar, by a
sugar mimic such as cyclobutyl or by another moiety which
takes the place of the sugar.
In some embodiments, the antisense
oligonucleotides used to practice the invention may comprise
from about 5 to about 100 nucleotide units. In many cases,
the antisense oligonucleotide used to practice the invention
comprises at least 14, 16 or 30 contiguous nucleotides or
modified nucleotides that are complementary to a contiguous
sequence of nucleotides encoding the target polypeptide
(e.g. CXCL10 polypeptide). As will be appreciated, a
nucleotide unit is a base-sugar combination (or a
combination of analogous structures) suitably bound to an
adjacent nucleotide unit through phosphodiester or other
bonds forming a backbone structure.
RNAi
In a further embodiment, the present invention
relates to the use of RNA interference (RNAi) technology, a
type of post-transcriptional gene silencing, for treating
respiratory illnesses (e.g, community-acquired pneumonia and
SARS). RNAi may be used to create a pseudo "knockout", i.e.
a system in which the expression of the product encoded by a
gene or coding region of interest is reduced, resulting in
an overall reduction of the activity of the encoded product
in a system. As such, RNAi may be used to target a
polynucleotide, to in turn reduce its expression and the
level of activity of the product which it encodes (i.e. a
polypeptide involved in the CXCL10:CXCR3 axis). Such a
system may be used for functional studies of the product, as
24
CA 02478138 2004-08-17
well as to treat disorders related to the activity of such a
product. RNAi is described in for example Hammond et al.
(2001) Nature Rev. Genet. 2: 110-1119; Sharp (2001) Genes
Dev. 15: 485-490; Caplen et al. (2001), Sedlak (2000) and
published US patent applications 20020173478 (Gewirtz;
published November 21, 2002) and 20020132788 (Lewis et al.;
published November 7, 2002), all of which are herein
incorporated by reference. Reagents and kits for performing
RNAi are available commercially from for example Ambion Inc.
(Austin, TX, USA) and New England Biolabs Inc. (Beverly, MA,
USA) .
The initial agent for RNAi in some systems is
thought to be dsRNA molecule corresponding to a target
polynucleotide. The dsRNA is then thought to be cleaved
into short interfering RNAs (siRNAs) which are 21-23
nucleotides in length (19-21 by duplexes, each with 2
nucleotide 3' overhangs). The enzyme thought to effect this
first cleavage step has been referred to as "Dicer" and is
categorized as a member of the RNase III family of dsRNA-
specific ribonucleases. Alternatively, RNAi may be effected
via directly introducing into the cell, or generating within
the cell by introducing into the cell a suitable precursor
(e. g. vector encoding precursor(s), etc.) of such an siRNA
or siRNA-like molecule. An siRNA may then associate with
other intracellular components to form an RNA-induced
silencing complex (RISC). The RISC thus formed may
subsequently target a transcript of interest via base-
pairing interactions between its siRNA component and the
target transcript by virtue of homology, resulting in the
cleavage of the target transcript approximately 12
nucleotides from the 3' end of the siRNA. Thus the target
mRNA is cleaved and the level of protein product it encodes
is reduced.
CA 02478138 2004-08-17
RNAi may be effected by the introduction of
suitable in vitro synthesized siRNA or siRNA-like molecules
into cells. RNAi may for example be performed using
chemically-synthesized RNA (Brown et al., 2002).
Alternatively, suitable expression vectors may be used to
transcribe such RNA either in vitro or in vivo. In vitro
transcription of sense and antisense strands (encoded by
sequences present on the same vector or on separate vectors)
may be effected using for example T7 RNA polymerase, in
which case the vector may comprise a suitable coding
sequence operably-linked to a T7 promoter. The in vitro-
transcribed RNA may in embodiments be processed (e. g. using
E. coli RNase III) in vitro to a size conducive to RNAi.
The sense and antisense transcripts are combined to form an
RNA duplex which is introduced into a target cell of
interest. Other vectors may be used, which express small
hairpin RNAs (shRNAs) which can be processed into siRNA-like
molecules. Various vector-based methods are described in
for example Brummelkamp et al. (2002), Lee et al. (2002),
Miyagashi and Taira (2002), Paddison et al. (2002) Paul et
al. (2002) Sui et al. (2002) and Yu et al. (2002). Various
methods for introducing such vectors into cells, either in
vitro or in vivo (e. g. gene therapy) are known in the art.
Accordingly, expression of CXCL10 polypeptide may
be inhibited by introducing into or generating within a cell
an siRNA or siRNA-Like molecule corresponding to a
polynucleotide encoding either CXCL10 polypeptide or an
inducer of CXCL10 polypeptide expression (such as IFN-y), or
to an polynucleotide homologous thereto. ~~siRNA-like
molecule" refers to a polynucleotide molecule similar to an
siRNA (e. g. in size and structure) and capable of eliciting
siRNA activity, i.e. to effect the RNAi-mediated inhibition
26
CA 02478138 2004-12-24
of expression. In various embodiments such a method may
entail the direct administration of the siRNA or siRNA-like
molecule into a cell, or use of the vector-based methods
described above. In an embodiment, the siRNA or siRNA-like
molecule is less than about 30 nucleotides in length. In a
further embodiment, the siRNA or siRNA-like molecule is
about 21-23 nucleotides in length. In an embodiment, siRNA
or siRNA-like molecule comprises a 19-21 by duplex portion,
each strand having a 2 nucleotide 3' overhang. In
embodiments, the siRNA or siRNA-like molecule is
substantially identical to a polynucleotide encoding either
CXCL10 polypeptide or an inducer of CXCL10 polypeptide
expression (such as IFN-y). In embodiments, the sense strand
of the siRNA or siRNA-like molecule is substantially
identical to the coding sequence for human CXCL10
polypeptide mRNA (as set forth in GenBank accession No.
NM 001565 (SEQ ID N0: 2)).
Polvpeptide production and purification
Recombinant full-length human and murine CXCL10
polypeptide (IP-10) are available commercially for example
from BIODESIGN International.
Polypeptides used to practice the invention can
also be prepared for example by culturing host cells that
have been transformed or injected with polynucleotides
encoding them (or vectors comprising such polynucleotides),
under culture conditions suitable to express the
polypeptide. The polypeptide so expressed may then be
purified from such culture using known purification
processes, such as gel filtration and ion exchange
chromatography. The purification of the polypeptide may
also include an affinity column containing agents which will
27
CA 02478138 2004-08-17
bind to the polypeptide (e.g. an anti-CXCL10 polypeptide or
anti-CXCR3 polypeptide antibody); one or more column steps
over such affinity resins as concanavalin A-agarose,
heparin-toyopearl~ or Cibacrom blue 3GA Sepharose~; one or
more steps involving hydrophobic interaction chromatography
using such resins as phenyl ether, butyl ether, or propyl
ether; or immunoaffinity chromatography. Alternatively, the
polypeptides of interest may also be expressed in a form
that will facilitate purification. For example, it may be
expressed as a fusion polypeptide, such as those of maltose
binding polypeptide (MBP), glutathione-S-transferase (GST)
or thioredoxin (TRX). Kits for expression and purification
of such fusion polypeptides are commercially available from
New England BioLab (Beverly, MA), Pharmacia (Piscataway,
NJ), and InVitrogen, respectively. The polypeptide can also
be tagged with an epitope and subsequently purified by using
a specific antibody directed to such epitope. One such
epitope ("Flag") is commercially available from Kodak (New
Haven, Conn.). Optionally, one or more reverse-phase high
performance liquid chromatography (RP-HPLC) steps employing
hydrophobic RP-HPLC media, e.g., silica gel having pendant
'fiethyl or other aliphatic groups, can be employed to further
purify the polypeptide. Some or all of the foregoing
purification steps, in various combinations, can be employed
to provide a substantially purified recombinant polypeptide,
i.e. a recombinant polypeptide that is substantially free of
other mammalian polypeptides and is defined in accordance
with the invention as a "substantially purified
polypeptide". Polypeptides can also be expressed as a
product of transgenic animals, e.g., as a component of the
milk of transgenic cows, goats, pigs, or sheep which are
characterized by somatic or germ cells containing a
polynucleotide encoding the polypeptide.
28
CA 02478138 2004-08-17
Suitable host cells for expression of the
polypeptide include eukaryotic and prokaryotic cells.
Materials and methods for baculovirus/insect cell expression
systems are commercially available in kit form from, e.g.,
Invitrogen, San Diego, CA, U.S.A. (the MaxBac~ kit), as well
as methods described in Summers and Smith, Texas
Agricultural Experiment Station Bulletin No. 1555 (1987),
and Luckow and Summers, Bio/Technology 6:47 (1988),
incorporated herein by reference. Mammalian host cells
include, for example, the COS-7 line of monkey kidney cells
(ATCC CRL 1651) (Gluzman et al., Cell 23:175, 1981), L
cells, 0127 cells, 3T3 cells (ATCC CCL 163), Chinese hamster
ovary (CHO) cells, HeLa cells, BHK (ATCC CRL 10) cell lines,
the CVI/EBNA cell line derived from the African green monkey
kidney cell line CV1 (ATCC CCL 70) (see, McMahan et al. EMBO
J. 10: 2821, 1991), human kidney 293 cells, human epidermal
A431 cells, human Co1o205 cells, other transformed primate
cell lines, normal diploid cells, cell strains derived from
in vitro culture of primary tissue, primary explants, HL-60,
U937, HaK or Jurkat cells. Alternatively, it may be possible
to produce the polypeptide in lower eukaryotes such as yeast
or in prokaryotes such as bacteria. Potentially suitable
yeast strains include Saccharomyces cerevisiae,
Schizosaccharomyces pombe, Kluyveromyces strains, Candida,
or any yeast strain capable of expressing heterologous
polypeptides. Potentially suitable bacterial strains
include, for example, Escheri.chia coli, Bacillus subtilis,
Salmonella typhimurium, or any bacterial strain capable of
expressing heterologous polypeptides. If the polypeptide is
made in yeast or bacteria, it may be necessary to modify the
polypeptide produced therein, for example by phosphorylation
or glycosylation of the appropriate sites, in order to
obtain the functional polypeptide. Such covalent
29
CA 02478138 2004-08-17
attachments may be accomplished using known chemical or
enzymatic methods. The polypeptide may also be produced by
operably linking a polynucleotide encoding it to suitable
control sequences in one or more insect expression vectors,
and employing an insect expression system.
Cell-free translation systems can also be employed
to produce polypeptides using RNAs derived from nucleic acid
constructs disclosed herein. A host cell that comprises an
isolated polynucleotide used to practice the invention ,
preferably operably linked to at least one expression
control sequence, is a "recombinant host cell".
Established methods for introducing DNA into
mammalian cells have been described (Kaufman, R.J., Large
Scale Mammalian Cell Culture, 1990, pp. 15-69). Additional
protocols using commercially available reagents, such as
Lipofectamine or Lipofectamine-Plus lipid reagent
(Gibco/BRL), can be used to transfect cells (Felgner et al.,
Proc. Natl. Acad. Sci. USA 84:7413, 1987). In addition,
electroporation can be used to transfect mammalian cells
using conventional procedures, such as those in Sambrook et
a1. (Molecular Cloning: A Laboratory Manual, 2 ed. Vol. 1-3,
Cold Spring Harbor Laboratory Press, 1989). Selection of
stable transformants can be performed using methods known in
the art, such as, for example, resistance to cytotoxic
drugs. Kaufman et al., Meth. in Enzymology 185:487, 1990,
describes several selection schemes, such as dihydrofolate
reductase (DHFR) resistance. A suitable strain for DHFR
selection can be CHO strain DX-811, which is deficient in
DHFR (Urlaub et al., Proc. Natl. Acad. Sci. USA 77:4216,
1980). A plasmid expressing the DHFR cDNA can be introduced
into strain DX-B11, and only cells that contain the plasmid
can grow in the appropriate selective media. Other examples
CA 02478138 2004-08-17
of selectable markers that can be incorporated into an
expression vector include cDNAs conferring resistance to
antibiotics, such as 6418 and hygromycin B. Cells harboring
the vector are selected on the basis of resistance to these
compounds.
It is also possible to utilize an affinity column
having an affinity moeity (such as a monoclonal antibody
generated against either CXCL10 polypeptide or CXCR3
polypeptide) to affinity-purity expressed polypeptides.
These polypeptides can be removed from an affinity column
using conventional techniques, e.g., in a high salt elution
buffer and then dialyzed into a lower salt buffer for use or
by changing pH or other components depending on the affinity
matrix utilized, or be competitively removed using the
naturally occurring substrate of the affinity moiety, such
as a polypeptide derived from the invention. In this aspect
of the invention, the affinity moeity (e. g., an antibody
that binds CXCL10 polypeptide or CXCR3 polypeptide) can be
bound to a solid phase support or a similar substrate
suitable for identifying, separating, or purifying cells
that express polypeptides on their surface. Adherence of
the binding protein to the solid phase surface can be
accomplished by any means, for example, magnetic
microspheres can be coated with these binding proteins and
held in the incubation vessel through a magnetic field.
Suspensions of cell mixtures are contacted with the solid
phase that has such binding proteins thereon. Binding
proteins bind those cells having target polypeptides vention
on their surface, and unbound cells (e. g., cells lacking
target polypeptide) are washed away from the bound cells.
This affinity-binding method is useful for purifying,
screening, or separating such polypeptide-expressing cells
from solution. Methods of releasing positively selected
31
CA 02478138 2004-08-17
cells from the solid phase are known in the art and
encompass, for example, the use of enzymes. Such enzymes are
preferably non-toxic and non-injurious to the cells and are
preferably directed to cleaving the cell-surface binding
partner. Alternatively, mixtures of cells suspected of
containing cells expressing polypeptides of interest are
first incubated with a biotinylated binding partner.
Incubation periods are typically at least one hour in
duration to ensure sufficient binding to polypeptides. The
resulting mixture then is passed through a column packed
with avidin-coated beads, whereby the high affinity of
biotin for avidin provides the binding of the cells to the
beads. Use of avidin-coated beads is known in the art (see,
Berenson, et al. J. Cell. &iochem., lOD:239, 1986). Wash of
unbound material and the release of the bound cells is
performed using conventional methods.
Polypeptides used to practice the invention may
also be produced by known conventional chemical synthesis.
Methods for constructing the polypeptides by synthetic means
are known to those skilled in the art. The synthetically-
constructed polypeptide sequences, by virtue of sharing
primary, secondary or tertiary structural and/or
conformational characteristics with a native polypeptides
may possess biological properties in common therewith,
including biological activity. Thus, the synthesized
polypeptides may be employed as biologically active or
immunological substitutes for natural, purified polypeptides
in screening of therapeutic compounds and in immunological
processes for the development of antibodies.
The desired degree of purity depends on the
intended use of the polypeptide. A relatively high degree
of purity is desired when the polypeptide is to be
32
CA 02478138 2004-08-17
administered in vivo, for example. In such a case, the
polypeptides are purified such that no polypeptide bands
corresponding to other polypeptides are detectable upon
analysis by SDS-polyacrylamide gel electrophoresis (SDS-
PAGE). It will be recognized by one skilled in the
pertinent field that multiple bands corresponding to the
polypeptide can be visualized by SDS-PAGE, due to
differential glycosylation, differential post-translational
processing, and the like. Most preferably, the polypeptide
used to practice the invention is purified to substantial
homogeneity, as indicated by a single polypeptide band upon
analysis by SDS-PAGE. The polypeptide band can be visualized
by silver staining, Coomassie blue staining, or (if the
polypeptide is radiolabeled) by autoradiography.
Antibodies
Antibodies that bind CXCL10 polypeptide or the
extracellular domain of CXCR3 polypeptide can be used to
practice the methods of the invention. Specifically, such
antibodies can be used in methods for diagnosing respiratory
illnesses (e.g. community-acquired pneumonia and SARS) and
for monitoring the course of treatment and determining the
prognosis of a patient suffering from respiratory illnesses.
Antibodies that bind CXCL10 polypeptide or the
extracellular domain of CXCR3 polypeptide are also useful as
binding partners for CXCL10 polypeptide in screening methods
of the invention.
Antibodies that bind CXCL10 polypeptide or the
extracellular domain of CXCR3 polypeptide are also useful
for treating respiratory illnesses and in the preparation of
medicaments and pharmaceutical compositions therefor.
33
CA 02478138 2004-08-17
Anti-CXCR3 polypeptide antibodies can be used for
example for screening for host cells that express CXCR3
polypeptides or for affinity purification of CXCR3
polypeptides.
R & D Systems (http://www.rndsystems.com/)
supplies anti-murine and anti-human CXCL10 polypeptide and
CXCR3 polypeptide antibodies and related reagents useful for
laboratory ELISAs.
Antibodies used to practice the invention may be
either polyclonal or monoclonal. Antibodies may be
recombinant, e.g., chimeric (e. g., constituted by a variable
region of murine origin associated with a human constant
region), humanized (a human immunoglobulin constant backbone
together with hypervariable region of animal, e.g., murine,
origin), and/or single chain. Both polyclonal and
monoclonal antibodies may also be in the form of
immunoglobulin fragments, e.g., F(ab)'2, Fab or Fab'
fragments. The antibodies used to practice the invention
are of any isotype, e,g., IgG or IgA, and polyclonal
antibodies are of a single isotype or a mixture of isotypes.
Detection of an antibody of the present invention
can be facilitated by coupling (i.e., physically linking)
the antibody to a detectable substance. Examples of
detectable substances include various enzymes, prosthetic
groups, fluorescent materials, luminescent materials,
bioluminescent materials, and radioactive materials.
Examples of suitable enzymes include horseradish peroxidase,
alkaline phosphatase, beta-galactosidase, or
acetylcholinesterase; examples of suitable prosthetic group
complexes include streptavidin/biotin and avidin/biotin;
examples of suitable fluorescent materials include
34
CA 02478138 2004-08-17
umbelliferone, fluorescein, fluorescein isothiocyanate,
rhodamine, dichlorotriazinylamine fluorescein, dansyl
chloride or phycoerythrin; an example of a luminescent
material includes luminol; examples of bioluminescent
materials include luciferase, luciferin, and aequorin, and
examples of suitable radioactive material include l2sl, 13 I,
asS or 3H .
Accordingly, a further aspect of the invention
provides a diagnostic method for detecting the presence of a
CXCL10 polypeptide and/or activity in a tissue or body
fluid, by contacting the tissue or body fluid with an anti-
CXCL10 polypeptide antibody such that an immune complex is
formed, and by detecting such complex to indicate the
presence of the CXCL10 polypeptide and/or activity in the
sample or the organism from which the sample is derived.
The tissue or body fluid sample can be obtained from a
mammal suspected of being infected with respiratory
illnesses.
Those skilled in the art will readily understand
that the immune complex is formed between a component of the
sample and the antibody, and that any unbound material can
be removed prior to detecting the complex. It is understood
that diagnostic method of the invention may be used for
screening a sample, such as, for example, blood, plasma,
lymphocytes, cerebrospinal fluid, urine, saliva, epithelia,
fibroblasts, or a host cell, for the presence of a CXCL10
polypeptide.
For diagnostic applications, the antibody may be
either in a free state or immobilized on a solid support,
such as a tube, a bead, or any other conventional support
used in the field. Immobilization may be achieved using
CA 02478138 2004-08-17
direct or indirect means. Direct means include passive
adsorption (non-covalent binding) or covalent binding
between the support and the reagent. By "indirect means" is
meant that an anti-reagent compound that interacts with a
reagent is first attached to the solid support. Indirect
means may also employ a ligand-receptor system, for example,
where a molecule such as a vitamin is grafted onto the
reagent and the corresponding receptor immobilized on the
solid phase. This is illustrated by the biotin-streptavidin
system. Alternatively, a peptide tail is added chemically
or by genetic engineering to the reagent and the grafted or
fused product immobilized by passive adsorption or covalent
linkage of the peptide tail.
Such diagnostic agents may be included in a kit
which also comprises instructions for use. The reagent can
be labeled with a detection means which allows for the
detection of the reagent when it is bound to its target.
Suitable detection means include for example fluorescent
agents such as fluorescein isocyanate or fluorescein
isothiocyanate, enzymes such as horse radish peroxidase or
luciferase or alkaline phosphatase, and radioactive elements
such as 125I or SlCr.
In a preferred embodiment, the CXCL10 polypeptide
assay is a cytometric bead array (CHA) assay. CBA assays
are commercially available, e.g. from Becton Dickinson.
For use in a purification, the antibody is either
polyclonal or monoclonal, and preferably is of the IgG type.
Purified IgGs are prepared from an antiserum using standard
methods (see, e.g., Coligan et al. (1994) supra).
Conventional chromatography supports, as well as standard
methods for grafting antibodies, are described in, e.g.,
3F
CA 02478138 2004-08-17
Antibodies: A Laboratory Manual, Harlow and Lane (eds.) Cold
Spring Harbour Laboratory Press (1988), herein incorporated
by reference, and outlined below.
Briefly, the sample, preferably in a buffer
solution, is applied to a chromatography material,
preferably equilibrated with the buffer used to dilute the
sample so that the polypeptide used to practice the
invention (i.e., the antigen) is allowed to adsorb onto the
material. The chromatography material, such as a gel or a
resin coupled to an antibody of the invention, is in either
a batch form or a column. The unbound components are washed
off and the antigen is then eluted with an appropriate
elution buffer, such as a glycine buffer or a buffer
containing a chaotropic agent, e.g., guanidine HC1, or high
salt concentration (e.g., 3 M MgCl2). Eluted fractions are
recovered and the presence of the antigen is detected, e.g.,
by measuring the absorbance at 280 nm.
A further aspect of the present invention is a
diagnostic imaging method for diagnosing respiratory
illness, which comprises introducing into a biological
system an anti-CXCL10 polypeptide antibody which is used in
conjunction with an appropriate detection system to identify
areas where CXCL10 polypeptide or its activity is present or
absent.
A further aspect of the present invention provides
therapeutic applications of antibodies, whereby antibodies
that bind CXCL10 polypeptide, CXCR3 polypeptide, or TFN-y are
administered to a patient to treat or ameliorate respiratory
illnesses by downregulating or inhibiting the CXCL10:CXCR3
axis.
Screening assays
37
CA 02478138 2004-08-17
Another aspect of the invention relates to the use
of CXCL10 polypeptide or CXCR3 polypeptide as a target in
screening assays that may be used to identify a therapeutic
agent for treating respiratory illnesses.
In some embodiments, such a screening assay may
comprise the steps of:
(a) providing a test compound;
(b) providing a polypeptide selected from CXCL10
polypeptide and CXCR3 polypeptide; and
(c) detecting whether the test compound binds to said
polypeptide.
Another type of assay for identifying a inhibitor
of interaction between CXCL10 polypeptide and CXCR3
polypeptide is a competitive binding assay, utilizing a
"binding partner" of between CXCL10 polypeptide or CXCR3
polypeptide. Such a screening assay may comprise the steps
of
(a) providing a test compound;
(b) providing a polypeptide selected from between CXCL10
polypeptide and CXCR3 polypeptide;
(c) providing a source of a binding partner that binds to
said polypeptide; and
(d) detecting whether the test compound inhibits
interaction between said binding partner and said
polypeptide.
Binding partners that bind to between CXCL10 polypeptide and
CXCR3 polypeptide include for example antibodies and
38
CA 02478138 2004-08-17
polypeptides. For example, suitable binding partners for
CXCL10 polypeptide include a polypeptide comprising the
extracellular domain of CXCR3 polypeptide that is capable of
binding CXCL10 polypeptide.
Binding of the binding partner to CXCL10
polypeptide can be determined by conventional methods, for
example by measuring release of a radiolabelled binding
partner or by measuring CXCL10 polypeptide activity (e. g.
binding to anti-CXCL10 polypeptide antibodies or CXCR3
polypeptide) .
Test compounds include, for example, 1) peptides
such as soluble peptides, including Ig-tailed fusion
peptides and members of random peptide libraries (see, e.g.,
Lam et al., Nature 354:82-84 (1991); Houghten et al., Nature
354:84-86 (1991)) and combinatorial chemistry-derived
molecular libraries made of D- and/or L-configuration amino
acids; 2) phosphopeptides (e.g., members of random and
partially degenerate, directed phosphopeptide libraries,
see, e.g., Songyang et al., Cell 72:767-778 (1993)); 3)
antibodies (e. g., polyclonal, monoclonal, humanized, anti-
idiotypic, chimeric, and single chain antibodies as well as
Fab, F(ab~)2, Fab expression library fragments, and epitope-
binding fragments of antibodies); and 4) small organic and
inorganic molecules (e. g., molecules obtained from
combinatorial and natural product libraries), including
peptidomimetics.
Compounds that may be useful for inhibiting CXCL10
polypeptide activity, in vivo or in vitro, by binding
thereto include for example: polypeptides comprising the
extracellular domain of CXCR3 polypeptide; and anti-CXCL10
polypeptide antibodies.
39
CA 02478138 2004-08-17
In another aspect, the invention provides a
reporter assay-based method of selecting agents which
inhibit expression of CXCL10 polypeptide or CXCR3
polypeptide, i.e. for use in identifying a compound for the
treatment of respiratory illnesses. Such a method may
comprise assaying expression of CXCL10 polypeptide or CXCR3
polypeptide in the presence versus the absence of a test
compound. Such gene expression may be measured by detection
of the corresponding RNA or protein, or via the use of a
suitable reporter construct comprising a transcriptional
regulatory elements) normally associated with the gene
encoding CXCL10 polypeptide or CXCR3 polypeptide, operably-
linked to a reporter gene. The expression of such a
reporter gene may be measured on the transcriptional or
translational level, e.g. by the amount of RNA or protein
produced. RNA may be detected by for example Northern
analysis or by the reverse transcriptase-polymerase chain
reaction (RT-PCR) method (see for example Sambrook et al
(1989) Molecular Cloning: A Laboratory Manual (second
edition), Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, New York, USA). Protein levels may be detected
either directly using affinity reagents (e.g. an antibody or
fragment thereof [for methods, see for example Harlow, E.
and Lane, D (1988) Antibodies: A Laboratory Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY); a
ligand which binds the protein) or by other properties (e. g.
fluorescence in the case of green fluorescent protein) or by
measurement of the protein's activity, which may entail
enzymatic activity to produce a detectable product (e. g.
with altered spectroscopic properties) or a detectable
phenotype (e. g. alterations in cell growth). Suitable
reporter genes include but are not limited to
CA 02478138 2004-08-17
chloramphenicol acetyltransferase, beta-D galactosidase,
luciferase, or green fluorescent protein.
Mention is made of VERO 6 cells, which express
CXCL10 polypeptide when infected with SARS CoV, and U937
cells, which express CXCL10 polypeptide when induced with
IFN-y, as examples of cells that can be used in the present
methods for identifying compounds that inhibit expression of
CXCL10 polypeptide.
Inhibitors of CXCL10 polypeptide expression
include for example antibodies that bind to IFN-y, which is
known to induce CXCL10 polypeptide expression in certain
cell types.
These screening methods can be used to identify
compounds that inhibit the biological activity of CXCL10
polypeptide and/or CXCR3 polypeptide. These assays may
further comprise a step of assaying the compounds so
identified for their ability to inhibit the CXCL10:CXCR3
axis in a biological system, e.g. by assaying for inhibition
of inflammation and injury in the lungs of a SARS model
animal such as a mouse or a monkey.
The above-noted methods and assays may be employed
either with a single test compound or a plurality or library
(e.g. a combinatorial library) of test compounds. In the
latter case, synergistic effects provided by combinations of
compounds may also be identified and characterized. The
above-mentioned compounds may be used for inhibiting the
activity of the CXCL10:CXCR3 axis, i.e. for treatment of
respiratory illness in a patient, or may be used as lead
compounds for the development and testing of additional
compounds having improved specificity, efficacy and/or
pharmacological (e. g. pharmacokinetic) properties.
41
CA 02478138 2004-08-17
In certain embodiments, one or a plurality of the
steps of the screening/testing methods of the invention may
be automated. Such assay systems may comprise a variety of
means to enable and optimize useful assay conditions. Such
means may include but are not limited to: suitable buffer
solutions, for example, for the control of pH and ionic
strength and to provide any necessary components for optimal
CXCL10 polypeptide or CXCR3 polypeptide activity and
stability (e. g. protease inhibitors), temperature control
means for optimal a CXCL10 polypeptide or CXCR3 polypeptide
activity and or stability, and detection means to enable the
detection of CXCL10 polypeptide activity. A variety of such
detection means may be used, including but not limited to
one or a combination of the following: radiolabelling (e. g.
32P), antibody-based detection, fluorescence,
chemiluminescence, spectroscopic methods (e.g. generation of
a product with altered spectroscopic properties), various
reporter enzymes or proteins (e. g. horseradish peroxidase,
green fluorescent protein), specific binding reagents (e. g.
biotin/streptavidin), and others. Binding may also be
analysed using generally known methods in this area, such as
electrophoresis on native polyacrylamide gels, as well as
fusion protein-based assays such as the yeast 2-hybrid
system or in vitro association assays, or proteomics-based
approachs to identify CXCL10 polypeptide binding proteins.
The assay may be performed in vitro using cell-
based or cell-free systems as a source of CXCL10 polypeptide
or CXCR3 polypeptide. Such assays may be performed in an
array format. One or more assay steps can be automated.
To perform cell free drug screening assays, it is
sometimes desirable to immobilize either the CXCL10
polypeptide or CXCR3 polypeptide or the binding partner, to
42
CA 02478138 2004-08-17
facilitate separation of complexes from uncomplexed forms of
one or both components, as well as to accommodate automation
of the assay.
Techniques for immobilizing proteins on matrices
can be used in the drug screening assays. In one embodiment,
a fusion protein can be provided which adds a domain that
allows the protein to be bound to a matrix. For example,
glutathione-S-transferase (GST) fusion proteins can be
adsorbed onto glutathione sepharose beads (Sigma Chemical,
St. Louis, Mo.) or glutathione derivatized microtitre
plates, which are then combined with the cell lysates (e. g.,
35 S-labeled) and the candidate compound, and the mixture
incubated under conditions conducive to complex formation
(e. g., at physiological conditions for salt and pH).
Following incubation, the beads are washed to remove any
unbound label, and the matrix immobilized and radiolabel
determined directly, or in the supernatant after the
complexes are dissociated. Alternatively, the complexes can
be dissociated from the matrix, separated by SDS-PAGE, and
the level of CXCL10 polypeptide or CXCR3 polypeptide found
in the bead fraction quantitated from the gel using standard
electrophoretic techniques. For example, either the CXCL10
polypeptide or its target molecule can be immobilized
utilizing conjugation of biotin and streptavidin using
techniques well known in the art (biotinylation kits and
streptavidin-coated 96-well plates are commercially
available e.g. from Pierce Chemicals, Rockford IL).
Alternatively, antibodies reactive with the CXCL10
polypeptide or CXCR3 polypeptide but which do not interfere
with binding of the polypeptide to its target molecule can
be derivatized to the wells of the plate, and the protein
trapped in the wells by antibody conjugation. Preparations
of a binding partner and a test compound are incubated in
43
CA 02478138 2004-08-17
the wells presenting CXCL10 polypeptide or CXCR3 polypeptide
and the amount of complex trapped in the well can be
quantitated. Methods for detecting such complexes, in
addition to those described above for the GST-immobilized
complexes, include immunodetection of complexes using
antibodies, as well as enzyme-linked assays which rely on
detecting an enzymatic activity associated with the complex
(e.g, horseradish peroxidase, alkaline phosphatase, or
luciferase).
l0 In one embodiment, the test compound is labeled.
Either the test compound, or the binding partner, or both,
is added first to a CXCL10 polypeptide or CXCR3 polypeptide
for a time sufficient to allow binding. Incubations may be
performed at any temperature which facilitates optimal
activity, typically between 4 and 40 degrees C. Incubation
periods are selected for optimum activity, but may also be
optimized to facilitate rapid high throughput screening.
Typically between 0.1 and 1 hour will be sufficient. Excess
reagent is generally removed or washed away. The second
component is then added, and the presence or absence of the
labeled component is followed, to indicate binding.
The interaction between two molecules can also be
detected, e.g., using a fluorescence assay in which at least
one molecule is fluorescently labeled. One example of such
an assay includes fluorescence energy transfer (FET or FRET
for fluorescence resonance energy transfer) (see, for
example, Lakowicz et a2., U.S. Patent No. 5,631,169;
Stavrianopoulos, et al., U.S. Patent No. 4,868,103).
Another example of a fluorescence assay is fluorescence
polarization (FP) (see, e.g., Nasir et a1. (1999) Comb C~em
XTS 2:177-190; Jameson et al. (1995) Methods Enzymol
246:283; Seethala et al., (I998) Anal Hiochem. 255:257),
44
CA 02478138 2004-08-17
which can be monitored in multiwell plates, e.g., using the
Tecan Polarion~" reader (see, e.g., Parker et al. (2000)
Journal of Biomolecular Screening 5 :77 - 88; and Shoeman,
et al. (1999) 38, 16802-16809) .
The ability of a binding partner to bind to a
CXCL10 polypeptide or CXCR3 polypeptide can also be
accomplished using real-time Biomolecular Interaction
Analysis (BIA) (see, e.g., Sjolander, S. and Urbaniczky, C.
(1991) Anal. Chem. 63:2338-2345 and Szabo et al. (1995)
Curr. Opin. Struct. Biol. 5:699-705). Agents that inhibit
CXCL10 polypeptide or CXCR3 polypeptide can be identified
using one or more of the above assays, alone or in
combination. It is generally preferable to use a cell-based
or cell free system first and then confirm activity in a
model system such as an animal, e.g. using a mammal (such as
a mouse, rat, primate or other non-human). Thus, the above-
described assay methods may further comprise determining
whether any compounds so identified can be used for treating
SARS, such as examining their effects) on disease symptoms
in suitable disease animal model systems. The above-
mentioned methods may similarly be used to identify and
characterize compounds for the inhibition of CXCL10
polypeptide or CXCR3 polypeptide in a biological system,
e.g. a whole-animal system such as mouse or monkey.
In another embodiment, polynucleotides encoding
CXCL10 polypeptides or CXCR3 polypeptide can be used in a
yeast two-hybrid system to identify other protein partner
pairs of CXCL10 polypeptide or CXCR3 polypeptide.
Therapeutic formulations
In various embodiments, inhibitors of CXCL10
polypeptide or CXCR3 polypeptide activity may be used
CA 02478138 2004-08-17
therapeutically in formulations or medicaments to treat SARS
in a subject such as mammalian subject. The terms "treat",
"treating" and "treatment" used herein include curative,
preventative (e.g. prophylactic) and palliative or
ameliorative treatment. The invention provides
corresponding methods of medical treatment, in which a
therapeutic dose of an inhibitor of CXCL10 polypeptide or
CXCR3 polypeptide is administered in a pharmacologically
acceptable formulation, e.g. to a patient or subject in need
thereof.
Accordingly, the invention also provides
therapeutic compositions comprising a compound capable of
inhibiting CXCL10 polypeptide or CXCR3 polypeptide activity
and a pharmacologically acceptable excipient or carrier. In
one embodiment, such compositions include an inhibitor of
CXCL10 polypeptide or CXCR3 polypeptide in a therapeutically
or prophylactically effective amount sufficient to treat
respiratory illnesses. The therapeutic composition may be
soluble in an aqueous solution at a physiologically
acceptable pH.
Inhibitors of the CXCL10:CXCR3 axis that may be
used as therapeutic agents include for example:
(a) a binding partner of CXCL10 polypeptide or CXCR3
polypeptide;
(b) an antibody that binds CXCLIOpolypeptide, CXCR3
polypeptide, or IFN-gamma;
(g) an antisense RNA that that binds to an mRNA encoding
CXCL10 polypeptide, CXCR3 polypeptide, or IFN-gamma;
(h) an RNAi that targets mRNA encoding CXCL10 polypeptide,
CXCR3 polypeptide, or IFN-gamma;
4b
CA 02478138 2004-08-17
(i) a transcription regulator protein that inhibits
expression of CXCL10 polypeptide, CXCR3 polypeptide or
INF-gamma; and
(j) a small molecule that interacts with CXCL10
polypeptide, CXCR3 polypeptide, or IFN-gamma or
inhibits expression thereof, as can be identified using
the methods described herein.
A "therapeutically,effective amount" refers to an
amount effective, at dosages and for periods of time
necessary, to achieve the desired therapeutic result, such
as a inhibition of CXCL10 polypeptide or CXCR3 polypeptide
activity and in turn a reduction in SARS disease
progression. The therapeutically effective amount of
inhibitor of CXCL10 polypeptide or CXCR3 polypeptide may
vary according to factors such as the disease state, age,
sex, and weight of the individual, and the ability of the
compound to elicit a desired response in the individual.
Dosage regimens may be adjusted to provide the optimum
therapeutic response. A therapeutically effective amount is
also one in which any toxic or detrimental effects of the
compound are outweighed by the therapeutically beneficial
effects. A "prophylactically effective amount" refers to an
amount effective, at dosages and for periods of time
necessary, to achieve the desired prophylactic result, such
as inhibiting the rate of SARS disease progression. A
prophylactically effective amount can be determined as
described above for the therapeutically effective amount.
For any particular subject, specific dosage regimens may be
adjusted over time according to the individual need and the
professional judgement of the person administering or
supervising the administration of the compositions.
47
CA 02478138 2004-08-17
As used herein "pharmaceutically acceptable
carrier" or "excipient" includes any and all solvents,
dispersion media, coatings, antibacterial and antifungal
agents, isotonic and absorption delaying agents, and the
like that are physiologically compatible. In one
embodiment, the carrier is suitable for parenteral
administration. Alternatively, the carrier can be suitable
for intravenous, intraperitoneal, intramuscular, sublingual
or oral administration. Pharmaceutically acceptable
carriers include sterile aqueous solutions or dispersions
and sterile powders for the extemporaneous preparation of
sterile injectable solutions or dispersion. The use of such
media and agents for pharmaceutically active substances is
well known in the art. Except insofar as any conventional
media or agent is incompatible with the active compound, use
thereof in the pharmaceutical compositions of the invention
is contemplated. Supplementary active compounds can also be
incorporated into the compositions.
Therapeutic compositions typically must be sterile
and stable under the conditions of manufacture and storage.
The composition can be formulated as a solution,
microemulsion, liposome, or other ordered structure suitable
to high drug concentration. The carrier can be a solvent or
dispersion medium containing, for example, water, ethanol,
polyol (for example, glycerol, propylene glycol, and liquid
polyethylene glycol, and the like), and suitable mixtures
thereof. The proper fluidity can be maintained, for
example, by the use of a coating such as lecithin, by the
maintenance of the required particle size in the case of
dispersion and by the use of surfactants. In many cases, it
will be preferable to include isotonic agents, for example,
sugars, polyalcohohs such as mannitol, sorbitol, or sodium
chloride in the composition. Prolonged absorption of the
48
CA 02478138 2004-08-17
injectable compositions can be brought about by including in
the composition an agent which delays absorption, for
example, monostearate salts and gelatin. Moreover, a
therapeutic agent can be administered in a time release
formulation, for example in a composition which includes a
slow release polymer. The active compounds can be prepared
with carriers that will protect the compound against rapid
release, such as a controlled release formulation, including
implants and microencapsulated delivery systems.
Biodegradable, biocompatible polymers can be used, such as
ethylene vinyl acetate, polyanhydrides, polyglycolic acid,
collagen, polyorthoesters, polylactic acid and polylactic,
polyglycolic copolymers (PLG). Many methods for the
preparation of such formulations are patented or generally
known to those skilled in the art.
Sterile injectable solutions can be prepared by
incorporating the active compound in the required amount in
an appropriate solvent with one or a combination of
ingredients enumerated above, as required, followed by
filtered sterilization. Generally, dispersions are prepared
by incorporating the active compound into a sterile vehicle
which contains a basic dispersion medium and the required
other ingredients from those enumerated above. In the case
of sterile powders for the preparation of sterile ~injectable
solutions, the preferred methods of preparation are vacuum
drying and freeze-drying which yields a powder of the active
ingredient plus any additional desired ingredient from a
previously sterile-filtered solution thereof. In accordance
with an alternative aspect of the invention, a therapeutice
agent may be formulated with one or more additional
compounds that enhance the solubility of the agent.
49
CA 02478138 2004-08-17
In accordance with another aspect of the
invention, therapeutic compositions of the present
invention, comprising an inhibitor of CXCL10 polypeptide
CXCL10 polypeptide or CXCR3 polypeptide, may be provided in
containers or commercial packages which further comprise
instructions for use of the pharmaceutical composition for
treatment of respiratory illnesses.
Accordingly, the invention further provides a
commercial package comprising an inhibitor of CXCL10
polypeptide or CXCR3 polypeptide or the above-mentioned
composition together with instructions for the treatment of
respiratory illnesses. The invention further provides a use
of an inhibitor of CXCL10 polypeptide CXCL10 polypeptide or
CXCR3 polypeptide for treatment of respiratory illnesses.
The invention further provides a use of an inhibitor of
CXCL10 polypeptide CXCL10 polypeptide or CXCR3 polypeptide
in the preparation of a medicament for treatment of
respiratory illnesses.
All publications, patent applications, patents and
other references mentioned herein are hereby incorporated by
reference in their entirety.
Although various embodiments of the invention are
disclosed herein, many adaptations and modifications may be
made within the scope of the invention in accordance with
the common general knowledge of those skilled in this art.
Such modifications include the substitution of known
equivalents for any aspect of the invention in order to
achieve the same result in substantially the same way.
Numeric ranges are inclusive of the numbers defining the
range. In the claims, the word "comprising" is used as an
CA 02478138 2004-08-17
open-ended term, substantially equivalent to the phrase
~~including, but not limited to~~.
The following examples are illustrative of various
aspects of the invention, and do not limit the broad aspects
of the invention as disclosed herein.
EXAMPLES
Materials and Methods
Patients
Study subjects were enrolled without bias to age,
sex or previous medical history Informed consent was
,obtained from all subjects under the approval of the
Research Ethics Boards of the University Health Network and
participating hospitals. Clinical details were provided to
us following completion of our analysis and can be found in
other studies describing this overlapping patient cohort
(3,5,12,13). Our cohort consisted of 63 confirmed SARK
patients (median age 40 years, 23 males and 40 females) as
defined by Health Canada case definitions (Health Canada:
Severe acute respiratory syndrome (SARS),
http://www.sars.gc.ca). These patients were identified with
early clinical presentation of SARS symptoms, radiographic
evidence consistent with SARS and laboratory evidence of
SARS-associated CoV infection (positive PCR, seroconversion
and/or virus isolation results). SARS patients were treated
similarly with steroids (approx. 20 to 50 mg/d
hydrocortisone for 10-14 d, not beginning within 48 h from
onset of symptoms) during their disease course, however 10
critical SARS patients required intubation, admission to the
intensive care unit (ICU) and longer periods of steroid
51
CA 02478138 2004-08-17
treatment. Three of these SARS patients died in the ICU
during our study and right and left lower lung samples were
obtained during autopsy. Recovered SARS patients, i.e.
those taken off supplemental oxygen or steroid therapy or
those discharged from the hospital, were termed as
convalescents.
Suspect SARS patients (n=80) were also included in
our analysis having concurrently presented to emergency
rooms or been admitted to hospital with SARS-like
symptoms/respiratory ailments during the Toronto outbreaks.
Patients in this group (39 males and 41 females with a
median age of 42 years) were excluded as SARS when an
alternative medical or microbiological diagnosis was found.
Lastly, 14 healthy normal volunteers (7 males and 7 females
with a median age of 36 years) were sampled.
Ribavirin (5-7 d course at 3-5 d post onset of
symptoms) was administered to approx. 20~ of SARS patients
enrolled in the longitudinal experiments. No significant
effects of ribavirin treatment were noted in our analyses.
Patient Sample Collection and Preparation
Peripheral blood was collected in heparinized
blood collection tubes from study participants upon onset of
SARS-like symptoms, after every five days during
hospitalization and at discharge from the hospital. Whole
blood RNA was isolated using Qiagen Paxgene RNA kits
(Mississauga, ON, Canada). Plasma was obtained by
centrifugation. Peripheral blood mononuclear cells (P$MCs)
were isolated by density centrifugation. Some PBMCs were
cryopreserved using patients' plasma supplemented with 10~
DMSO and maintained in liquid nitrogen for use in the
proliferation assays.
52
CA 02478138 2004-08-17
Cytometric Bead Array (CBA)
Cytokine and chemokine levels were assayed by
human CBA kits according to the manufacturer's protocols (BD
Biosciences, San Jose, CA). Briefly, specific capture beads
for cytokines and chemokines were mixed with 50 ~1 of
patient plasma or standards and multiple phycoerythrin-
conjugated detection antibodies were added. Following a 3h
incubation period with recombinant protein standards or test
samples, samples were fixed with 2% paraformaldehyde and the
acquisition of sample data was performed using a two-color
flow cytometer. Results were generated in graphical and
tabular format using BD CBA Analysis Software.
Real-time PCR
Total RNA was purified from lung tissues and cells
using TriPure Reagent (Roche, Basel, Switzerland)
(Mississauga, ON, Canada) according to the manufacturers'
specifications. RNA was cleaned up using an RNeasy
Purification Kit (Qiagen) and was treated with RNAse-free
DNAse on-column treatment to remove genomic DNA (Qiagen).
250 ug mRNA was reverse transcribed into cDNA using
Superscript II (Invitrogen, Burlington, ON, Canada). 0.25u1
cDNA was amplified with the SYBR Green Master Mix (Applied
Biosystems, Warrington, UK): 15 min at 95°C for initial
denaturing, followed by 40 cycles of 95°C for 15 s
denaturation step and 60°C for 1 min annealing/extension
step using the ABI 7900 Sequence Detection System (Applied
Biosystems). Real-time PCR Primers: CXCL10 F 5' - TCC ACG
TGT TGA GAT CAT TGA - 3', CXCL10 R 5' - TCT TGA TGG CCT TCG
ATT CTG - 3', CXCR3 F 5' - GGT GCC CTC TTC AAC ATC AAC - 3',
CXCR3 R 5' - GGT GGC ATG AAC TAT GTT CAG GTA - 3', GAPDH F
5' - GCA CCA CCA ACT GCT TAG CAC - 3', GAPDH R 5' - TCT TCT
53
CA 02478138 2004-08-17
GGG TGG CAG TGA TG - 3'. The relative expression of each
gene of interest was normalized to GAPDH.
Virus tissue culture
Vero E6 cells (American Type Culture Collection,
Manassas, VA, USA) were cultured in Dulbecco's Modification
of Eagle's Medium supplemented with 1~
penicillin/streptomycin and 1% glutamine (Sigma, St. Louis,
MO, USA) and 10% fetal calf serum (Cambrex Corporation, East
Rutherford, NJ, USA). Upon confluence (approximately 1-2 x
10' cells per 162 cm2 flask), cells were infected with 100
p.f.u. of the Tor2 strain of the SARS CoV. At the
appropriate time points, media was removed and the cells
were washed twice with PBS and removed. Total RNA was
purified from the mechanically homogenized cells using
TriPure Reagent (Roche).
Proliferation assay and FAGS analysis
Recombinant SARS CoV N protein was kindly provided
by J. Mahony (Hamilton, Canada). 5 x 106 cryopreserved PBMCs
were thawed and labeled with a predetermined concentration
of CFSE (Molecular Probes, Eugene, OR). The final
concentration of CFSE used for PBMC labeling was 1.5 ACM.
Cells were washed twice in PBS and resuspended in RPMI media
supplemented with 10% human serum. 106/ml CFSE-labeled PBMCs
were cultured either in the presence of 2 ug/ml SARS CoV N
protein or in media alone. 50 ng/ml SEA (Toxin Technology,
Sarasota, FL) was used as a positive control (data not
shown) . After 6 d of in vitro incubation at 37°C 5% C02,
cells were collected and stained with anti-CD4-APC, anti-
CD8-PerCP and anti-CXCR3-PE (HD Biosciences). A minimum of
2-5 x 104 events, gated on viable CD4+ or CD8+ T cells, were
54
CA 02478138 2004-08-17
collected on a four colour FACSCaliburTM cytometer and
analyzed using FlowJo~ software (TreeStar).
SARS Infection of Rhesus and Cynomolgus Macaques
Macaques were housed in an animal BSL-3 facility
at the Southern Research Institute in Birmingham, AL.
Rhesus (n=4) and cynomolgus (n=4) macaques were infected
with 1 x 106 p.f.u. SARS-CoV (Tor2) intratracheally and
monitored for 13 days. Uninfected macaques were used as
controls (n=4). Blood samples were collected at day 0, 3,
5, 7 and 13 or whenever containment procedures permitted.
PBMCs were isolated by density centrifugation after which
RNA was purified using TriPure Reagent as above.
Statistical Methods
Our data was found to be suitable for parametric
statistical tests (SigmaStat). Results were compared using
Student's t test for unpaired samples and P < 0.05 was
chosen as the level of significance).
Results
In a strategy adopted as a first step in examining
dysregulated proinflammatory immune responses as reflected
peripherally in SARS-infected patients, we measured plasma
levels of cytokines and chemokines by CHA in approximately
700 blood samples taken from consenting confirmed SARS
patients, suspect SARS patients (non-SARS, but exhibiting
SARS-like symptoms) and healthy controls during the 2003
SARS outbreaks in Toronto, Canada. In general, and as
reported in previous studies (14-16), we observed examples
of increased expression of proinflammatory cytokine, such as
IFN-y, TNF-a, IL-la and IL-6, in SARS patients at onset of
CA 02478138 2004-08-17
symptoms relative to healthy controls (data not shown).
However, we noted a striking pattern of CXCL10 polypeptide
expression in SARS patients of different retrospective
severity and outcome of disease, so we focused herein on
elucidating the role of the CXCL10:CXCL3 axis in SARS
immunopathogenesis.
Firstly, we observed significantly increased
levels of CXCL10 polypeptide in the plasma of confirmed SARS
patients (mean=4,564 pg/ml) within 48 h of onset of symptoms
and under no treatment compared to healthy controls
(mean=528 pg/ml) (Fig. lA). This was the only cytokine or
chemokine to show elevated levels in 100% of SARS cases at
onset. CXCL10 polypeptide levels were also significantly
increased when compared to suspect SARS patients (mean=807
pg/ml) and convalescents (mean=596 pg/ml). Indeed, all
patients in the suspect SARS group were excluded as SARS
patients when an alternative medical or microbiological
diagnosis was found that explained their SARS-like
presentation. No confirmed SARS patients had a CXCL10
polypeptide level in plasma of less than 1000 pg/ml, a
plateau below which nearly all non-SARS patients and all
healthy control levels fell.
To understand the longitudinal relationship
between CXCL10 polypeptide and SARS pathogenesis or disease
course we performed a timeline comparison of CXCL10
polypeptide levels in 25 SARS patients with mild to moderate
symptoms (Fig. 1B) and 10 severe SARS patients admitted to
the ICU and intubated (Fig. 1C). CXCL10 polypeptide levels
in non-ICU patients decreased to convalescent levels within
10-12 d (Fig. 1B), whereas the levels in ICU patients (Fig.
1C) remained significantly elevated versus non-ICU SARS
patients until at least day 40 (P < 0.05). The return of
56
CA 02478138 2004-08-17
CXCL10 polypeptide expression to convalescent levels in mild
SARS patients (approx. 10-12 d since onset) and the return
of CXCL10 polypeptide expression to convalescent levels in
severe SARS patients (approx. 40-50 d since onset) generally
correlated with the median duration of SARS illness in mild
and severe SARS patients (14 d and 52 d respectively). As
described in the Materials and Methods, all recovered SARS
patients, i.e. those that no longer required supplemental
oxygen or steroid therapy before discharge or those
discharged from the hospital, were classified as
convalescents. Some convalescent patients required lengthy
hospital stays (z 90 d) following their SARS illness due to
age or comorbidities.
The highest levels of CXCL10 polypeptide beyond
10 d were recorded in five ICU patients (Fig. 1C, solid
lines). Three of these individuals died during the study of
complications due to SARS, including an organ transplant
recipient on immunosuppression. A fourth critical patient
required an unusually lengthy course of steroids before
quickly regaining health after day 30. A fifth patient
remained in critical condition with protracted SARS
infection and ARDS and began to recover at around day 80 in
conjunction with CXCL10 polypeptide levels returning to
normal. This data indicates that CXCL10 polypeptide is
induced early in the course of SARS and continues to be
elevated in patients with a poor prognosis.
CXCL9 (monokine induced by IFN-Y/MIG) and CXCL10
are non-ELR, CXC chemokines that bind CXCR3 polypeptide and
were originally described as being induced by IFN-y. In
contrast to CXCL10 polypeptide, CXCL9 was not significantly
increased in the plasma of SARS patients, suspect SARS cases
or convalescents (Fig. 1D-F). Also, CXCLB, an ELR CXC
57
CA 02478138 2004-08-17
chemokine that binds to CXCR1 and CXCR2 mainly expressed on
neutrophils, was not significantly expressed in confirmed
SARS patients at onset. However, CXCLB was highly elevated
in the plasma of a small subset of SARS patients at 2 to 5
wk from onset of symptoms (data not shown). Interestingly,
all of the samples from SARS patients showing highly
elevated levels of CXCL8 were obtained from non-ICU SARS
patients treated with ribavirin indicating an additional
effect of ribavirin treatment (21).
The persistence of elevated levels of CXCL10
polypeptide in patients with severe illness lead us to
postulate that infected tissues may be the main source of
CXCL10 polypeptide. Using real time PCR, we found that lung
tissues from deceased individuals had elevated levels of
CXCL10 polypeptide transcripts compared to tissue from
normal lungs (Fig. 2A). We also determined whether SARS-
infected lungs had increased levels of CXCR3 polypeptide.
All lung samples with elevated levels of CXCL10 polypeptide
also had elevated levels of CXCR3 polypeptide (Fig. 2B).
Increased transcript levels of CXCL10 polypeptide and CXCR3
polypeptide could also be noted in PBMCs from confirmed SARS
patients at 7 days (Fig. 3A, B). These data suggest that
SARS CoV infection induces CXCL10 polypeptide expression
that in turn recruits activated T cells into infected
tissues.
To test whether the SARS CoV itself may induce
CXCL10 polypeptide expression, we conducted two experiments.
Firstly, we found that SARS CoV infection of VERO E6 cells
induced CXCL10 polypeptide expression in vitro within 12
hours (Fig. 3C). Secondly, we screened PBMCs of
convalescent SARS patients for the presence of SARS antigen-
specific, CXCR3 polypeptide-expressing T cell responses.
58
CA 02478138 2004-08-17
We used a highly sensitive proliferation assay based on the
labeling of T cells with the cell tracking dye CFSE (22).
Cryopreserved PBMCs from convalescent confirmed SARS
patients were thawed, stained with CFSE and cultured for 6 d
in the presence of recombinant SARS CoV N protein. Using
FRCS, we quantitated proliferating (CFSElow) CD4+ or CD8+ T
cells from convalescent SARS patients and healthy controls
and their expression of CXCR3 polypeptide. We found that a
high proportion of CD4+ and CD8+ T cells from recovered SARS
patients proliferated vigorously in response to SARS CoV N
protein and that a high proportion of those expressed CXCR3
polypeptide (Fig. 4). This finding shows that proliferating
SARS antigen-specific T cells express CXCR3 polypeptide and
thus would have the capacity to infiltrate tissues
expressing elevated levels of CXCL10 polypeptide.
Lastly, we confirmed that CXCL10 polypeptide
expression could be induced in non-human primates following
infection with SARS CoV. We examined longitudinal CXCL10
polypeptide expression by real time PCR in two macaque
models of experimental SARS infection. We found that PBMCs
isolated from cynomolgus and rhesus macaques inoculated with
SARS CoV (Tor2) expressed increased CXCL10 polypeptide
transcripts within 3-5 d post-infection (Fig. 5). During
the 2 wk duration of this preliminary experiment, cynomolgus
macaques (n=4) expressed CXCL10 polypeptide in increasing
intensities (up to approx. 40-fold increase at day 13
relative to day 0), whereas CXCL10 polypeptide expression
peaked in rhesus macaques (n=4) at day 3 (approx. 15-fold
increase relative to day 0). CXCL10 polypeptide/GAPDH
ratios in PBMCs from day 0 monkeys and additional uninfected
control monkeys (n=4) were very small or negligible.
Overall, these data indicate a role for the CXCL10:CXCR3
axis in SARS immunopathogenesis and in mediating host immune
59
CA 02478138 2004-12-24
responses against SARS CoV infection during the course of
disease.
Our CBA study of patients with community-acquired
pneumonia whose presentations were initially confused with
SARS indicated that some severe cases were associated with
increases of CXCL10 polypeptide (Figure 6).
Exemplary CXCL10 polypeptide and CXCR3 polypeptide and amino
acid and nucleotide seauences:
(1) Human CXCL10 polypeptide amino acid sequence: GenBank
accession No. NP 001556.1 (SEQ ID N0: 1).
1 MNQTAILICC LIFLTLSGIQ GVPLSRTVRC TCISISNQPV NPRSLEKLEI IPASQFCPRV
61 EIIATMKKKG EKRCLNPESK AIKNLLKAVS KEMSKRSP
(2) Human CXCL10 polypeptide mRNA sequence: GenBank
accession No. NM 001565 (SEQ ID N0: 2).
1 gagacattcctcaattgcttagacatattctgagcctacagcagaggaacctccagtctc
61 agcaccatgaatcaaactgcgattctgatttgctgccttatctttctgactctaagtggc
121 attcaaggagtacctctctctagaaccgtacgctgtacctgcatcagcattagtaatcaa
2 181 cctgttaatccaaggtctttagaaaaacttgaaattattcctgcaagccaattttgtcca
0
241 cgtgttgagatcattgctacaatgaaaaagaagggtgagaagagatgtctgaatccagaa
301 tcgaaggccatcaagaatttactgaaagcagttagcaaggaaatgtctaaaagatctcct
361 taaaaccagaggggagcaaaatcgatgcagtgcttccaaggatggaccacacagaggctg
421 cctctcccatcacttccctacatggagtatatgtcaagccataattgttcttagtttgca
2 481 gttacactaaaaggtgaccaatgatggtcaccaaatcagctgctactactcctgtaggaa
5
541 ggttaatgttcatcatcctaagctattcagtaataactctaccctggcactataatgtaa
601 gctctactgaggtgctatgttcttagtggatgttctgaccctgcttcaaatatttccctc
661 acctttcccatcttccaagggtactaaggaatctttctgctttggggtttatcagaattc
721 tcagaatctcaaataactaaaaggtatgcaatcaaatctgctttttaaagaatgctcttt
30 781 acttcatggacttccactgccatcctcccaaggggcccaaattctttcagtggctaccta
841 catacaattccaaacacatacaggaaggtagaaatatctgaaaatgtatgtgtaagtatt
901 cttatttaatgaaagactgtacaaagtataagtcttagatgtatatatttcctatattgt
961 tttcagtgtacatggaataacatgtaattaagtactatgtatcaatgagtaacaggaaaa
1021 ttttaaaaatacagatagatatatgctctgcatgttacataagataaatgtgctgaatgg
35 1081 ttttcaaataaaaatgaggtactctcctggaaatattaagaaagactatctaaatgttga
1141 aagatcaaaaggttaataaagtaattataact
(3) Murine CXCL10 polypeptide amino acid sequence: GenBank
accession No. NP 067249.1 (SEQ ID N0: 3).
CA 02478138 2004-12-24
1 MNPSAAVIFC LILLGLSGTQ GIPLARTVRC NCIHIDDGPV RMRAIGKLEI IPASLSCPRV
61 EIIATMKKND EQRCLNPESK TIKNLMKAFS QKRSKRAP
(4) Murine CXCL10 polypeptide mRNA sequence: GenBank
accession No. NM 021274.1 (SEQ ID N0: 4).
1 catcccgagc caaccttccg gaagcctccc catcagcacc atgaacccaa gtgctgccgt
61 cattttctgc ctcatcctgc tgggtctgag tgggactcaa gggatccctc tcgcaaggac
121 ggtccgctgc aactgcatcc atatcgatga cgggccagtg agaatgaggg ccatagggaa
181 gcttgaaatc atccctgcga gcctatcctg cccacgtgtt gagatcattg ccacgatgaa
241 aaagaatgat gagcagagat gtctgaatcc ggaatctaag accatcaaga atttaatgaa
301 agcgtttagc caaaaaaggt ctaaaagggc tccttaactg gagtgaagcc acgcacacac
361 cccggtgctg cgatggatgg acagcagaga gcctctctcc atcactcccc tttacccagt
421 ggatggctag tcctaattgc ccttggtctt ctgaaaggtg accagccgtg gtcacatcag
481 ctgctactcc tcctgcagga tgatggtcaa gccatggtcc tgagacaaaa gtaactgccg
541 aagcaagaat tctttaaggg ctggtctgag tcctcgctca agtggctggg atggctgtcc
601 tagctctgta ctgtaagcta tgtggaggtg cgacgccctt caccatgtgc catgcccagg
661 ctgctcccca caccctcctt gtcctcccta gctcaggctc gtcagttcta agtttacctg
721 agctctttta tttcagatgt aagactacaa atttaagttt gtaagcacga acttaaccac
781 catcttccca aggggttatc aagatactca gaggaacctg aaaatgtatg tgtaaatact
841 atttaatgaa cgactgtaca aagtagaatt cctaatgtat tttttgtatg ctttgcattg
901 tatatggaag aacttgtgtc atcaagtatg tatcaatggg tagttaaagt ttatttttaa
961 aaccgtccaa taccttttgt attatgtaac attcaaaaga caatgtactg tattgaaagt
1021 agtaagagac ccaaaatgta ataaagtaat aataactgac atg
(5) Human CXCR3 polypeptide mRNA sequence: GenBank accession
No. NM 001504 (SEQ ID NO: 5).
1 ccaaccacaagcaccaaagcagaggggcaggcagcacaccacccagcagccagagcacca
61 gcccagccatggtccttgaggtgagtgaccaccaagtgctaaatgacgccgaggttgccg
121 ccctcctggagaacttcagctcttcctatgactatggagaaaacgagagtgactcgtgct
181 gtacctccccgccctgcccacaggacttcagcctgaacttcgaccgggccttcctgccag
241 ccctctacagcctcctctttctgctggggctgctgggcaacggcgcggtggcagccgtgc
301 tgctgagccggcggacagccctgagcagcaccgacaccttcctgctccacctagctgtag
361 cagacacgctgctggtgctgacactgccgctctgggcagtggacgctgccgtccagtggg
421 tctttggctctggcctctgcaaagtggcaggtgccctcttcaacatcaacttctacgcag
481 gagccctcctgctggcctgcatcagctttgaccgctacctgaacatagttcatgccaccc
541 agctctaccgccgggggcccccggcccgcgtgaccctcacctgcctggctgtctgggggc
601 tctgcctgcttttcgccctcccagacttcatcttcctgtcggcccaccacgacgagcgcc
661 tcaacgccacccactgccaatacaacttcccacaggtgggccgcacggctctgcgggtgc
721 tgcagctggtggctggctttctgctgcccctgctggtcatggcctactgctatgcccaca
781 tcctggccgtgctgctggtttccaggggccagcggcgcctgcgggccatgcggctggtgg
841 tggtggtcgtggtggcctttgccctctgctggaccccctatcacctggtggtgctggtgg
901 acatcctcatggacctgggcgctttggcccgcaactgtggccgagaaagcagggtagacg
961 tggccaagtcggtcacctcaggcctgggctacatgcactgctgcctcaacccgctgctct
4 1021 atgcctttgtaggggtcaagttccgggagcggatgtggatgctgctcttgcgcctgggct
5
1081 gccccaaccagagagggctccagaggcagccatcgtcttcccgccgggattcatcctggt
1141 ctgagacctcagaggcctcctactcgggcttgtgaggccggaatccgggctcccctttcg
1201 cccacagtctgacttccccgcattccaggctcctccctccctctgccggctctggctctc
1261 cccaatatcctcgctcccgggactcactggcagccccagcaccaccaggtctcccgggaa
1321 gccaccctcccagctctgaggactgcaccattgctgctccttagctgccaagccccatcc
1381 tgccgcccgaggtggctgcctggagccccactgcccttctcatttggaaactaaaacttc
61
CA 02478138 2004-12-24
1441 atcttcccca agtgcgggga gtacaaggca tggcgtagag ggtgctgccc catgaagcca
1501 cagcccaggc ctccagctca gcagtgactg tggccatggt ccccaagacc tctatatttg
1561 ctcttttatt tttatgtcta aaatcctgct taaaactttt caataaacaa gatcgtcagg
1621 accaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
(6) Human CXCR3 polypeptide amino acid sequence: GenBank
accession No. NP 001495 (SEQ ID N0: 6).
1 MVLEVSDHQV LNDAEVAALL ENFSSSYDYG ENESDSCCTS PPCPQDFSLN FDRAFLPALY
61 SLLFLLGLLG NGAVAAVLLS RRTALSSTDT FLLHLAVADT LLVLTLPLWA VDAAVQWVFG
lO 121 SGLCKVAGAL FNINFYAGAL LLACISFDRY LNIVHATQLY RRGPPARVTL TCLAVWGLCL
181 LFALPDFIFL SAHHDERLNA THCQYNFPQV GRTALRVLQL VAGFLLPLLV MAYCYAHILA
291 VLLVSRGQRR LRAMRLVVVV VVAFALCWTP YHLVVLVDIL MDLGALARNC GRESRVDVAK
301 SVTSGLGYMH CCLNPLLYAF VGVKFRERMW MLLLRLGCPN QRGLQRQPSS SRRDSSWSET
361 SEASYSGL
(7) Murine CXCR3 polypeptide amino acid sequence: GenBank
accession No. NP 034040.1 (SEQ ID N0: 7).
1 MYLEVSERQV LDASDFAFLL ENSTSPYDYG ENESDFSDSP PCPQDFSLNF DRTFLPALYS
61 LLFLLGLLGN GAVAAVLLSQ RTALSSTDTF LLHLAVADVL LVLTLPLWAV DAAVQWVFGP
2 O 121 GLCKVAGALF NINFYAGAFL LACISFDRYL SIVHATQIYR RDPRVRVALT CIVVWGLCLL
181 FALPDFIYLS ANYDQRLNAT HCQYNFPQVG RTALRVLQLV AGFLLPLLVM AYCYAHILAV
241 LLVSRGQRRF RAMRLVVVVV AAFAVCWTPY HLVVLVDILM DVGVLARNCG RESHVDVAKS
301 VTSGMGYMHC CLNPLLYAFV GVKFREQMWM LFTRLGRSDQ RGPQRQPSSS RRESSWSETT
361 EASYLGL
(8) Murine CXCR3 polypeptide mRNA sequence: GenBank
accession No. NM 009910.1 (SEQ ID N0: 8).
1 gcaagttccc aaccacaagt gccaaaggca gagaagcagg cagcacgaga cctgacccca
61 gcagccacag ccggagcacc agccaagcca tgtaccttga ggttagtgaa cgtcaagtgc
121 tagatgcctc ggactttgcc tttcttctgg aaaacagcac ctctccctac gattatgggg
181 aaaacgagag cgacttctct gactccccgc cctgcccaca ggatttcagc ctgaactttg
241 acagaacctt cctgccagcc ctctacagcc tcctcttctt gctggggctg ctaggcaatg
301 gggcggtggc tgctgtgcta ctgagtcagc gcactgccct gagcagcacg gacaccttcc
361 tgctccacct ggctgtagcc gatgttctgc tggtgttaac tcttccattg tgggcagtgg
421 atgctgctgt ccagtgggtt ttcggccctg gcctctgcaa agtggcaggc gccttgttca
481 acatcaactt ctatgcaggg gccttcctgc tggcttgtat aagcttcgac agatatctga
541 gcatagtgca cgccacccag atctaccgca gggacccccg ggtacgtgta gccctcacct
601 gcatagttgt atggggtctc tgtctgctct ttgccctccc agatttcatc tacctatcag
661 ccaactacga tcagcgcctc aatgccaccc attgccagta caacttccca caggtgggtc
721 gcactgctct gcgtgtactg cagctagtgg ctggtttcct gctgcccctt ctggtcatgg
62
CA 02478138 2004-08-17
781 cctactgcta tgcccatatc ctagctgttc tgctggtctc cagaggccag aggcgttttc
841 gagctatgag gctagtggta gtggtggtgg cagcctttgc tgtctgctgg accccctatc
901 acctggtggt gctagtggat atcctcatgg atgtgggagt tttggcccgc aactgtggtc
961 gagaaagcca cgtggatgtg gccaagtcag tcacctcggg catggggtac atgcactgct
1021 gcctcaatcc gctgctctat gcctttgtgg gagtgaagtt cagagagcaa atgtggatgt
1081 tgttcacgcg cctgggccgc tctgaccaga gagggcccca gcggcagccg tcatcttcac
1141 ggagagaatc atcctggtct gagacaactg aggcctccta cctgggcttg taattctgga
1201 ctggaactgt agcctgcgca gcccaagtcc taacacactc caagtgcttg tcctcctggt
1261 agttgggcta gctcgaactt acccgtaact ttgctgccag gatgcactga cagctcagca
1321 tatatccagc tctcctgaga atcaatctca gcaacaagga caacaccatt actgtgcctt
1381 agctgccatg ccctatcttg ctgttttaga actagctgcc tggagcccca ccgccctact
1441 aaattagcaa gtagaactca gccatccctg tgtgagaaga gggagaggca aatagcacag
1501 agggccaggc gttgtcagca ctgaatgtgc ccatctcagt atctcaatat ttgcccaatt
1561 ttatttctag aaacctcact taaactttca ataaacaagg taatgagg
63
CA 02478138 2004-08-17
References
1. Tsang, K. W., P. L. Ho, G. C. Ooi, W. K. Yee, T.
Wang, M. Chan-Yeung, W. K. Lam, W. H. Seto, L. Y.
Yam, T. M. Cheung, P. C. along, B. Lam, M. S. Ip,
J. Chan, K. Y. Yuen, and K. N. Lai. 2003. A
cluster of cases of severe acute respiratory
syndrome in Hong Kong. N. Engl. J. Med. 348:1977.
2. Lee, N., D. Hui, A. Wu, P. Chan, P. Cameron, G. M.
Joynt, A. Abuja, M. Y. Yung, C. B. Leung, K. F.
To, S. F. Lui, C. C. Szeto, S. Chung, and J. J.
Sung. 2003. A major outbreak of severe acute
respiratory syndrome in Hong Kong. N. Engl. J.
Med. 348:1986.
3. Poutanen, S. M., D. E. Low, B. Henry, S.
Finkelstein, D. Rose, K. Green, R. Tellier, R.
Draker, D. Adachi, M. Ayers, A. K. Chan, D. M.
Skowronski, I. Salit, A. E. Simor, A. S. Slutsky,
P. W. Doyle, M. Krajden, M. Petric, R. C. Brunham,
and A. J. McGeer. 2003. Identification of severe
acute respiratory syndrome in Canada. N. Engl. J.
Med. 348:1995.
4. Centers for Disease Control and Prevention. 2003.
Update: severe acute respiratory syndrome--
worldwide and United States, 2003. MMWR Morb.
Mortal. Wkly. Rep. 52:664.
5. Booth, C. M., L. M. Matukas, G. A. Tomlinson, A.
R. Rachlis, D. B. Rose, H. A. Dwosh, S. L.
walmsley, T. Mazzulli, M. Avendano, P. Derkach, I.
E. Ephtimios, I. Kitai, B. D. Mederski, S. B.
Shadowitz, W. L. Gold, L. A. Hawryluck, E. Rea, J.
64
CA 02478138 2004-08-17
S. Chenkin, D. W. Cescon, S. M. Poutanen, and A.
S. Detsky. 2003. Clinical features and short-term
outcomes of 144 patients with SARS in the greater
Toronto area. JAMA 289:2801.
6. Ksiazek, T. G., D. Erdman, C. S. Goldsmith, S. R.
Zaki, T. Peret, S. Emery, S. Tong, C. Urbani, J.
A. Comer, W. Lim, P. E. Rollin, S. F. Dowell, A.
E. Ling, C. D. Humphrey, W. J. Shieh, J. Guarner,
C. D. Paddock, P. Rota, B. Fields, J. DeRisi, J.
Y. Yang, N. Cox, J. M. Hughes, J. W. LeDuc, W. J.
Bellini, and L. J. Anderson. 2003. A novel
coronavirus associated with severe acute
respiratory syndrome. N. Engl. J. Med. 348:1953.
7. Peiris, J. S., S. T. Lai, L. L. Poon, Y. Guan, L.
Y. Yam, W. Lim, J. Nicholls, W. K. Yee, W. W. Yan,
M. T. Cheung, V. C. Cheng, K. H. Chan, D. N.
Tsang, R. W. Yung, T. K. Ng, and K. Y. Yuen. 2003.
Coronavirus as a possible cause of severe acute
respiratory syndrome. Lancet 361:1319.
8. Drosten, C., S. Gunther, W. Preiser, W. S. van
der, H. R. Brodt, S. Becker, H. Rabenau, M.
Panning, L. Kolesnikova, R. A. Fouchier, A.
Berger, A. M. Burguiere, J. Cinatl, M. Eickmann,
N. Escriou, K. Grywna, S. Kramme, J. C.
Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S.
Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz,
and H. W. Doerr. 2003. Identification of a novel
coronavirus in patients with severe acute
respiratory syndrome. N. Engl. J. Med. 348:1967.
9. Cyranoski, D. 2004.. Swift response greets return
of SARS in China. Nature 427:89.
CA 02478138 2004-08-17
10. Zheng, B. J., K. H. along, J. Zhou, K. L. along, B.
W. Young, L. W. Lu, and S. S. Lee. 2004. SARS-
related virus predating SARS outbreak, Hong Kong.
Emerg. Infect. Dis. 10:176.
11. Nicholls, J. M., L. L. Poon, K. C. Lee, W. F. Ng,
S. T. Lai, C. Y. Leung, C. M. Chu, P. K. Hui, K.
L. Mak, W. Lim, K. W. Yan, K. H. Chan, N. C.
Tsang, Y. Guan, K. Y. Yuen, and J. S. Peiris.
2003. Lung pathology of fatal severe acute
respiratory syndrome. Lancet 361:1773.
12. Mazzulli, T., G. A. Farcas, S. M. Poutanen, B. M.
Willey, D. Low, J. Butany, S. L. Asa, and K. C.
Kain. 2004. Severe acute respiratory syndrome-
associated Coronavirus in lung tissue. Emerg.
Infect. Dis. 10:20.
13. Hwang, D.M., Chamberlain, D.W., Poutanen, S.M.,
Low, D.E., Asa, S.A., and Butany, J. 2004.
Pulmonary pathology of severe acute respiratory
syndrome in Toronto. Mod. Pathol. online
publication, 23 July 2004;
doi:10.1038/modpatho1.3800247
14. Ng, P. C., C. W. Lam, A. M. Li, C. K. along, F. W.
Cheng, T. F. Leung, E. K. Hon, I. H. Chan, C. K.
Li, K. S. Fung, and T. F. Fok. 2004. Inflammatory
cytokine profile in children with severe acute
respiratory syndrome. Pediatrics 113:e7.
15. along, C.K., Lam, C.W.K., Wu, A.K.L., Ip, W.K.,
Lee, N.L.S., Chan, I.H.S., Lit, L.C.W., Hui,
D.S.C., Chan, M.H.M., Chung, S.S.C., and Sung,
J.J.Y. 2004. Plasma inflammatory cytokines and
66
CA 02478138 2004-08-17
chemokines in severe acute respiratory syndrome.
Clin. Exp. Immunol. 136:95.
16. Lee, C.H., Chen, R.F., Liu, J.W., Yeh, W.T.,
Chang, J.C., Liu, P.M., Eng, H.L., Lin, M.C., and
Yang, K.D. 2004. Altered p38 mitogen-activated
protein kinase expression in different leukocytes
with increment of immunosuppressive mediators in
patients with severe acute respiratory syndrome.
J. Immunol. 172:7841
17. Dufour, J.H., Dziejman, M., Liu, M.T., Leung,
J.H., Lane, T.E., and Luster, A.D. 2002. IFN-y-
inducible protein 10 (IP-10; CXCL10)-deficient
mice reveal a role for IP-10 in effector T cell
generation and trafficking. J. Immunol. 168:3195.
18. Liu, M.T., Keirstead, H.S., and Lane, T.E. 2001.
Neutralization of the chemokine CXCL10 reduces
inflammatory cell invasion and demyelination and
improves neurological function in a viral model of
multiple sclerosis. J. Immunol. 167:4091.
19. Trifilo, M.J., Montalto-Morrison, C., Stiles,
L.N., Hurst, K.R., Hardison, J.L., Manning, J.E.,
Masters, P.S., and Lane, T.E. 2004. CXC
chemokine ligand 10 controls viral infection in
the central nervous system: evidence for a role in
innate immune response through recruitment and
activation of natural killer cells. J. Virol.
78:585
20. Liu, M. T., B. P. Chen, P. Oertel, M. J.
Buchmeier, T. A. Hamilton, D. A. Armstrong, and T.
E. Lane. 2001. The CXC chemokines IP-10 and Mig
67
CA 02478138 2004-08-17
are essential in host defense following infection
with a neurotropic coronavirus. Adv. Exp. Med.
Biol. 494:323.
21. Knowles, S.R., Phillips, E.J., Dresser, L., and
Matukas, L. 2003. Common adverse events
associated with the use of ribavirin for severe
acute respiratory syndrome in Canada. Clin.
Infect. Dis. 37:1139.
22. Younes, S. A., B. Yassine-Diab, A. R. Dumont, M. ,
R. Boulassel, Z. Grossman, J. P. Routy, and R. P.
Sekaly. 2003. HIV-1 viremia prevents the
establishment of interleukin 2-producing HIV-
specific memory CD4+ T cells endowed with
proliferative capacity. J. Exp. Med. 198:1909.
23. Sorensen, T.L., Sellebjerg, F., Jensen, C.V.,
Strieter, R.M., and Ransohoff, R.M. 2001.
Chemokines CXCL10 and CCL2: differential
involvement in intrathecal inflammation in
multiple sclerosis. Eur. J. Neurol. 8:665.
24. Sauty, A., M. Dziejman, R. A. Taha, A. S. Iarossi,
K. Neote, E. A. Garcia-Zepeda, Q. Hamid, and A. D.
Luster. 1999. The T cell-specific CXC chemokines
IP-10, Mig, and I-TAC are expressed by activated
human bronchial epithelial cells. J. Immunol.
162:3549.
25. Ranieri, V. M., P. M. Suter, C. Tortorella, R. De
Tullio, J. M. Dayer, A. Brienza, F. Bruno, and A.
S. Slutsky. 1999. Effect of mechanical ventilation
on inflammatory mediators in patients with acute
68
CA 02478138 2004-08-17
respiratory distress syndrome: a randomized
controlled trial. JAMA 282:54.
26. Enserink, M. 2003. SARS researchers report new
animal models. Science 302:213
27. Kuiken, T., Fouchier, R.A., Scutten, M.,
Rimmelzwaan, G.F., van Amerongen, G., van Riel,
D., Laman, J.D., de Jong, T., van Doornum, G.,
Lim, W., Ling, A.E., Chan, P.K., Tam, J.S.,
Zambon, M.C., Gopal, R., Drosten, C., van der
Werf, S., Escriou, N., Manuguerra, J.C., Stohr,
K., Peiris, J.S., and Osterhaus, A.D. 2003.
Newly discovered coronavirus as the primary cause
of severe acute respiratory syndrome. Lancet
362:263.
28. Luster, A.D. 1998. Chemokines--chemotactic
cytokines that mediate inflammation. N. Engl. J.
Med. 338:436.
29. Rossi, D., and Zlotnik, A. 2000. The biology of
chemokines and their receptors. Annu. Rev.
Immunol. 18:217.
30. Melchjorsen, J., Sorensen, L.N., and Paludan, S.R.
2003, Expression and function of chemokines during
viral infections: from molecular mechanisms to in
vivo function. J. Leukoc. Biol. 74:331.
31. Sorensen, T. L., C. Trebst, P. Kivisakk, K. L.
Klaege, A. Majmudar, R. Ravid, H. Lassmann, D. B.
Olsen, R. M. Strieter, R. M. Ransohoff, and F.
Sellebjerg. 2002. Multiple sclerosis: a study of
CXCL10 and CXCR3 co-localization in the inflamed
central nervous system. J. Neuroimmunol. 127:59.
69
CA 02478138 2004-08-17
32. Nicoletti, F., I. Conget, M. Di Mauro, R. Di
Marco, M. C. Mazzarino, K. Bendtzen, A. Messina,
and R. Gomis. 2002. Serum concentrations of the
interferon-gamma-inducible chemokine IP-10/CXCL10
are augmented in both newly diagnosed Type I
diabetes mellitus patients and subjects at risk of
developing the disease. Diabetologia 45:1107.
33. Patel, D. D., J. P. Zachariah, and L. P. Whichard.
2001. CXCR3 and CCRS ligands in rheumatoid
arthritis synovium. Clin. Immunol. 98:39.
34. Christen, U., McGavern, D.B., Luster, A.D., von
Herrath, M.G., and Oldstone, M.B. 2003. Among
CXCR3 chemokines, IFN-y-inducible protein of 10 kDa
(CXC chemokine ligand (CXCL) 10) but not monokine
induced by IFN-y (CXCL9) imprints a pattern for the
subsequent development of autoimmune disease. J.
Imrrrunol . 171 : 6 8 3
CA 02478138 2004-12-24
SEQUENCE LISTING
(1) GENERAL INFORMATION:
86363-8
(i) APPLICANT: UNIVERSITY HEALTH NETWORK
(ii) TITLE OF INVENTION: CXCL10 - BASED DIAGNOSIS AND TREATMENT OF
RESPIRATORY ILLNESSES
(iii) NUMBER OF SEQUENCES: 8
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: SMART & BIGGAR
(B) STREET: P.O. BOX 2999, STATION D
(C) CITY: OTTAWA
(D) STATE: ONT
(E) COUNTRY: CANADA
(F) ZIP: K1P 5Y6
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: ASCII (text)
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE: 17-AUG-2004
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE:
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: SMART & BIGGAR
(B) REGISTRATION NUMBER: 86363-8
(C) REFERENCE/DOCKET NUMBER:
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (613j-232-2486
(B) TELEFAX: (613)-232-8440
(2) INFORMATION FOR SEQ ID NO.: 1:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 98
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 1:
71
CA 02478138 2004-12-24
Met Asn Gln Thr Ala Ile Leu Ile Cys Cys Leu Ile Phe Leu Thr Leu
1 5 10 15
Ser Gly Ile Gln Gly Val Pro Leu Ser Arg Thr Val Arg Cys Thr Cys
20 25 30
Ile Ser Ile Ser Asn Gln Pro Val Asn Pro Arg Ser Leu Glu Lys Leu
35 40 45
Glu Ile Ile Pro Ala Ser Gln Phe Cys Pro Arg Val Glu Ile Ile Ala
50 55 60
Thr Met Lys Lys Lys Gly Glu Lys Arg Cys Leu Asn Pro Glu Ser Lys
65 70 75 80
Ala Ile Lys Asn Leu Leu Lys Ala Val Ser Lys Glu Met Ser Lys Arg
85 90 95
Ser Pro
(2) INFORMATION FOR SEQ ID NO.: 2:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 1172
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 2:
GAGACATTCC TCAATTGCTT AGACATATTC TGAGCCTACA GCAGAGGAAC CTCCAGTCTC 60
AGCACCATGA ATCAAACTGC GATTCTGATT TGCTGCCTTA TCTTTCTGAC TCTAAGTGGC 120
ATTCAAGGAG TACCTCTCTC TAGAACCGTA CGCTGTACCT GCATCAGCAT TAGTAATCAA 180
CCTGTTAATC CAAGGTCTTT AGAAAAACTT GAAATTATTC CTGCAAGCCA ATTTTGTCCA 240
CGTGTTGAGA TCATTGCTAC AATGAAAAAG AAGGGTGAGA AGAGATGTCT GAATCCAGAA 300
TCGAAGGCCA TCAAGAATTT ACTGAAAGCA GTTAGCAAGG AAATGTCTAA AAGATCTCCT 360
TAAAACCAGA GGGGAGCAAA ATCGATGCAG TGCTTCCAAG GATGGACCAC ACAGAGGCTG 420
CCTCTCCCAT CACTTCCCTA CATGGAGTAT ATGTCAAGCC ATAATTGTTC TTAGTTTGCA 480
72
i i i ii i. ~ i i I
CA 02478138 2004-12-24
GTTACACTAAAAGGTGACCAATGATGGTCACCAAATCAGCTGCTACTACTCCTGTAGGAA 540
GGTTAATGTTCATCATCCTAAGCTATTCAGTAATAACTCTACCCTGGCACTATAATGTAA 600
GCTCTACTGAGGTGCTATGTTCTTAGTGGATGTTCTGACCCTGCTTCAAATATTTCCCTC 660
ACCTTTCCCATCTTCCAAGGGTACTAAGGAATCTTTCTGCTTTGGGGTTTATCAGAATTC 720
TCAGAATCTCAAATAACTAAAAGGTATGCAATCAAATCTGCTTTTTAAAGAATGCTCTTT 780
ACTTCATGGACTTCCACTGCCATCCTCCCAAGGGGCCCAAATTCTTTCAGTGGCTACCTA 840
CATACAATTCCAAACACATACAGGAAGGTAGAAATATCTGAAAATGTATGTGTAAGTATT 900
CTTATTTAATGAAAGACTGTACAAAGTATAAGTCTTAGATGTATATATTTCCTATATTGT 960
TTTCAGTGTACATGGAATAACATGTAATTAAGTACTATGTATCAATGAGTAACAGGAAAA 1020
TTTTAAAAATACAGATAGATATATGCTCTGCATGTTACATAAGATAAATGTGCTGAATGG 1080
TTTTCAAATAAAAATGAGGTACTCTCCTGGAAATATTAAGAAAGACTATCTAAATGTTGA 1140
AAGATCAAAAGGTTAATAAAGTAATTATAACT 1172
(2) INFORMATION FOR SEQ ID NO.: 3:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 98
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Murinae gen. sp.
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 3:
Met Asn Pro Ser Ala Ala Val Ile Phe Cys Leu Ile Leu Leu Gly Leu
1 5 10 15
Ser Gly Thr Gln Gly Ile Pro Leu Ala Arg Thr Val Arg Cys Asn Cys
20 25 30
Ile His Ile Asp Asp Gly Pro Val Arg Met Arg Ala Ile Gly Lys Leu
35 40 45
Glu Ile Ile Pro Ala Ser Leu Ser Cys Pro Arg Val Glu Ile Ile Ala
50 55 60
Thr Met Lys Lys Asn Asp Glu Gln Arg Cys Leu Asn Pro Glu Ser Lys
65 70 75 80
73
CA 02478138 2004-12-24
Thr Ile Lys Asn Leu Met Lys Ala Phe Ser Gln Lys Arg Ser Lys Arg
85 90 95
Ala Pro
(2) INFORMATION 4:
FOR SEQ ID NO.:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 1063
(B) TYPE: nucleic id
ac
(C) STRANDEDNESS:
(D) TOPOLOGY:
( i. i ) MOLECULE DNA
TYPE :
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Murinae gen.
sp.
(xi) SEQUENCE DESCRIPTION:
SEQ ID
NO.:
4:
CATCCCGAGC CAACCTTCCGGAAGCCTCCCCATCAGCACCATGAACCCAA GTGCTGCCGT60
CATTTTCTGC CTCATCCTGCTGGGTCTGAGTGGGACTCAAGGGATCCCTC TCGCAAGGAC120
GGTCCGCTGC AACTGCATCCATATCGATGACGGGCCAGTGAGAATGAGGG CCATAGGGAA180
GCTTGAAATC ATCCCTGCGAGCCTATCCTGCCCACGTGTTGAGATCATTG CCACGATGAA240
AAAGAATGAT GAGCAGAGATGTCTGAATCCGGAATCTAAGACCATCAAGA ATTTAATGAA300
AGCGTTTAGC CAAAAAAGGTCTAAAAGGGCTCCTTAACTGGAGTGAAGCC ACGCACACAC360
CCCGGTGCTG CGATGGATGGACAGCAGAGAGCCTCTCTCCATCACTCCCC TTTACCCAGT420
GGATGGCTAG TCCTAATTGCCCTTGGTCTTCTGAAAGGTGACCAGCCGTG GTCACATCAG480
CTGCTACTCC TCCTGCAGGATGATGGTCAAGCCATGGTCCTGAGACAAAA GTAACTGCCG540
AAGCAAGAAT TCTTTAAGGGCTGGTCTGAGTCCTCGCTCAAGTGGCTGGG ATGGCTGTCC600
TAGCTCTGTA CTGTAAGCTATGTGGAGGTGCGACGCCCTTCACCATGTGC CATGCCCAGG660
CTGCTCCCCA CACCCTCCTTGTCCTCCCTAGCTCAGGCTCGTCAGTTCTA AGTTTACCTG720
AGCTCTTTTA TTTCAGATGTAAGACTACAAATTTAAGTTTGTAAGCACGA ACTTAACCAC780
CATCTTCCCA AGGGGTTATCAAGATACTCAGAGGAACCTGAAAATGTATG TGTAAATACT840
ATTTAATGAA CGACTGTACAAAGTAGAATTCCTAATGTATTTTTTGTATG CTTTGCATTG900
TATATGGAAG AACTTGTGTCATCAAGTATGTATCAATGGGTAGTTAAAGT TTATTTTTAA960
AACCGTCCAA TACCTTTTGTATTATGTAACATTCAAAAGACAATGTACTG TATTGAAAGT1020
AGTAAGAGAC CCAAAATGTAATAAAGTAATAATAACTGACATG 1063
74
CA 02478138 2004-12-24
(2) INFORMATION 5:
FOR SEQ
ID NO.:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH:
1670
(B) TYPE:nucleic id
ac
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE DNA
TYPE:
(vi) ORIGINAL SOURCE:
(A) ORGANISM: sapiens
Homo
(xi) SEQUENCE TION:
DESCRIP SEQ ID
NO.:
5:
CCAACCACAAGCACCAAAGCAGAGGGGCAGGCAGCACACCACCCAGCAGC CAGAGCACCA60
GCCCAGCCATGGTCCTTGAGGTGAGTGACCACCAAGTGCTAAATGACGCC GAGGTTGCCG120
CCCTCCTGGAGAACTTCAGCTCTTCCTATGACTATGGAGAAAACGAGAGT GACTCGTGCT180
GTACCTCCCCGCCCTGCCCACAGGACTTCAGCCTGAACTTCGACCGGGCC TTCCTGCCAG240
CCCTCTACAGCCTCCTCTTTCTGCTGGGGCTGCTGGGCAACGGCGCGGTG GCAGCCGTGC300
TGCTGAGCCGGCGGACAGCCCTGAGCAGCACCGACACCTTCCTGCTCCAC CTAGCTGTAG360
CAGACACGCTGCTGGTGCTGACACTGCCGCTCTGGGCAGTGGACGCTGCC GTCCAGTGGG420
TCTTTGGCTCTGGCCTCTGCAAAGTGGCAGGTGCCCTCTTCAACATCAAC TTCTACGCAG480
GAGCCCTCCTGCTGGCCTGCATCAGCTTTGACCGCTACCTGAACATAGTT CATGCCACCC540
AGCTCTACCGCCGGGGGCCCCCGGCCCGCGTGACCCTCACCTGCCTGGCT GTCTGGGGGC600
TCTGCCTGCTTTTCGCCCTCCCAGACTTCATCTTCCTGTCGGCCCACCAC GACGAGCGCC660
TCAACGCCACCCACTGCCAATACAACTTCCCACAGGTGGGCCGCACGGCT CTGCGGGTGC720
TGCAGCTGGTGGCTGGCTTTCTGCTGCCCCTGCTGGTCATGGCCTACTGC TATGCCCACA780
TCCTGGCCGTGCTGCTGGTTTCCAGGGGCCAGCGGCGCCTGCGGGCCATG CGGCTGGTGG840
TGGTGGTCGTGGTGGCCTTTGCCCTCTGCTGGACCCCCTATCACCTGGTG GTGCTGGTGG900
ACATCCTCATGGACCTGGGCGCTTTGGCCCGCAACTGTGGCCGAGAAAGC AGGGTAGACG960
TGGCCAAGTCGGTCACCTCAGGCCTGGGCTACATGCACTGCTGCCTCAAC CCGCTGCTCT1020
ATGCCTTTGTAGGGGTCAAGTTCCGGGAGCGGATGTGGATGCTGCTCTTG CGCCTGGGCT1080
GCCCCAACCAGAGAGGGCTCCAGAGGCAGCCATCGTCTTCCCGCCGGGAT TCATCCTGGT1140
CTGAGACCTCAGAGGCCTCCTACTCGGGCTTGTGAGGCCGGAATCCGGGC TCCCCTTTCG1200
CCCACAGTCTGACTTCCCCGCATTCCAGGCTCCTCCCTCCCTCTGCCGGC TCTGGCTCTC1260
CCCAATATCCTCGCTCCCGGGACTCACTGGCAGCCCCAGCACCACCAGGT CTCCCGGGAA1320
CA 02478138 2004-12-24
GCCACCCTCC CAGCTCTGAG GACTGCACCA TTGCTGCTCC TTAGCTGCCA AGCCCCATCC 1380
TGCCGCCCGA GGTGGCTGCC TGGAGCCCCA CTGCCCTTCT CATTTGGAAA CTAAAACTTC 1440
ATCTTCCCCA AGTGCGGGGA GTACAAGGCA TGGCGTAGAG GGTGCTGCCC CATGAAGCCA 1500
CAGCCCAGGC CTCCAGCTCA GCAGTGACTG TGGCCATGGT CCCCAAGACC TCTATATTTG 1560
CTCTTTTATT TTTATGTCTA AAATCCTGCT TAAAACTTTT CAATAAACAA GATCGTCAGG 1620
ACCAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA 1670
(2) INFORMATION FOR SEQ ID NO.: 6:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 368
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 6:
Met Val Leu Glu Val Ser Asp His Gln Val Leu Asn Asp Ala Glu Val
1 5 10 15
Ala Ala Leu Leu Glu Asn Phe Ser Ser Ser Tyr Asp Tyr Gly Glu Asn
20 25 30
Glu Ser Asp Ser Cys Cys Thr Ser Pro Pro Cys Pro Gln Asp Phe Ser
35 40 45
Leu Asn Phe Asp Arg Ala Phe Leu Pro Ala Leu Tyr Ser Leu Leu Phe
50 55 60
Leu Leu Gly Leu Leu Gly Asn Gly Ala Val Ala Ala Val Leu Leu Ser
65 70 75 80
Arg Arg Thr Ala Leu Ser Ser Thr Asp Thr Phe Leu Leu His Leu Ala
85 90 95
Val Ala Asp Thr Leu Leu Val Leu Thr Leu Pro Leu Trp Ala Val Asp
100 105 110
Ala Ala Val Gln Trp Val Phe Gly Ser Gly Leu Cys Lys Val Ala Gly
115 120 125
76
CA 02478138 2004-12-24
Ala Leu Phe Asn Ile Asn Phe Tyr Ala Gly Ala Leu Leu Leu Ala Cys
130 135 140
Ile Ser Phe Asp Arg Tyr Leu Asn Ile Val His Ala Thr Gln Leu Tyr
145 150 155 160
Arg Arg Gly Pro Pro Ala Arg Val Thr Leu Thr Cys Leu Ala Val Trp
165 170 175
Gly Leu Cys Leu Leu Phe Ala Leu Pro Asp Phe Ile Phe Leu Ser Ala
180 185 190
His His Asp Glu Arg Leu Asn Ala Thr His Cys Gln Tyr Asn Phe Pro
195 200 205
Gln Val Gly Arg Thr Ala Leu Arg Val Leu Gln Leu Val Ala Gly Phe
210 215 220
Leu Leu Pro Leu Leu Val Met Ala Tyr Cys Tyr Ala His Ile Leu Ala
225 230 235 240
Val Leu Leu Val Ser Arg Gly Gln Arg Arg Leu Arg Ala Met Arg Leu
245 250 255
Val Val Val Val Val Val Ala Phe Ala Leu Cys Trp Thr Pro Tyr His
260 265 270
Leu Val Val Leu Val Asp Ile Leu Met Asp Leu Gly Ala Leu~Ala Arg
275 280 285
Asn Cys Gly Arg Glu Ser Arg Val Asp Val Ala Lys Ser Val Thr Ser
290 295 300
Gly Leu Gly Tyr Met His Cys Cys Leu Asn Pro Leu Leu Tyr Ala Phe
305 310 315 320
Val Gly Val Lys Phe Arg Glu Arg Met Trp Met Leu Leu Leu Arg Leu
325 330 335
Gly Cys Pro Asn Gln Arg Gly Leu Gln Arg G1n Pro Ser Ser Ser Arg
340 345 350
77
CA 02478138 2004-12-24
Arg Asp Ser Ser Trp Ser Glu Thr Ser Glu Ala Ser Tyr Ser Gly Leu
355 360 365
(2) INFORMATION FOR SEQ ID NO.: 7:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 367
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Murinae gen. sp.
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 7:
Met Tyr Leu Glu Val Ser Glu Arg Gln Val Leu Asp Ala Ser Asp Phe
1 5 10 15
Ala Phe Leu Leu Glu Asn Ser Thr Ser Pro Tyr Asp Tyr Gly Glu Asn
20 25 30
Glu Ser Asp Phe Ser Asp Ser Pro Pro Cys Pro Gln Asp Phe Ser Leu
35 40 45
Asn Phe Asp Arg Thr Phe Leu Pro Ala Leu Tyr Ser Leu Leu Phe Leu
50 55 60
Leu Gly Leu Leu Gly Asn Gly Ala Val Ala Ala Val Leu Leu Ser Gln
65 70 75 80
Arg Thr Ala Leu Ser Ser Thr Asp Thr Phe Leu Leu His Leu Ala Val
85 90 95
Ala Asp Val Leu Leu Val Leu Thr Leu Pro Leu Trp Ala Val Asp Ala
100 105 110
Ala Val Gln Trp Val Phe Gly Pro Gly Leu Cys Lys Val Ala Gly Ala
115 120 125
Leu Phe Asn Ile Asn Phe Tyr Ala Gly Ala Phe Leu Leu Ala Cys Ile
130 135 140
Ser Phe Asp Arg Tyr Leu Ser Ile Val His Ala Thr Gln Ile Tyr Arg
145 150 155 160
78
CA 02478138 2004-12-24
Arg Asp Pro Arg Val Arg Val Ala Leu Thr Cys Ile Val Va1 Trp Gly
165 170 175
Leu Cys Leu Leu Phe Ala Leu Pro Asp Phe Ile Tyr Leu Ser Ala Asn
180 185 190
Tyr Asp Gln Arg Leu Asn Ala Thr His Cys Gln Tyr Asn Phe Pro Gln
195 200 205
Val Gly Arg Thr Ala Leu Arg Val Leu Gln Leu Val Ala Gly Phe Leu
210 215 220
Leu Pro Leu Leu Val Met Ala Tyr Cys Tyr Ala His Ile Leu Ala Val
225 230 235 240
Leu Leu Val Ser Arg Gly Gln Arg Arg Phe Arg Ala Met Arg Leu Val
245 250 255
Val Val Val Val Ala Ala Phe Ala Val Cys Trp Thr Pro Tyr His Leu
260 265 270
Val Val Leu Val Asp Ile Leu Met Asp Val Gly Val Leu Ala Arg Asn
275 280 285
Cys Gly Arg Glu Ser His Val Asp Val Ala Lys Ser Val Thr Ser Gly
290 295 300
Met Gly Tyr Met His Cys Cys Leu Asn Pro Leu Leu Tyr Ala Phe Val
305 310 315 320
Gly Val Lys Phe Arg Glu Gln Met Trp Met Leu Phe Thr Arg Leu Gly
325 330 335
Arg Ser Asp Gln Arg Gly Pro Gln Arg Gln Pro Ser Ser Ser Arg Arg
340 345 350
Glu Ser Ser Trp Ser Glu Thr Thr Glu Ala Ser Tyr Leu Gly Leu
355 360 365
(2) INFORMATION FOR SEQ ID NO.: 8:
(i) SEQUENCE CHARACTERISTICS
79
i i i ii i, ~ i i I
CA 02478138 2004-12-24
(A) LENGTH:
1608
(B) TYPE:nucleic
acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE DNA
TYPE:
(vi) ORIGINAL SOURCE:
(A) ORGANISM: nae gen.
Muri sp.
(xi) SEQUENCE DESCRIPTION: ID NO.:
SEQ 8:
GCAAGTTCCCAACCACAAGTGCCAAAGGCAGAGAAGCAGGCAGCACGAGA CCTGACCCCA60
GCAGCCACAGCCGGAGCACCAGCCAAGCCATGTACCTTGAGGTTAGTGAA CGTCAAGTGC120
TAGATGCCTCGGACTTTGCCTTTCTTCTGGAAAACAGCACCTCTCCCTAC GATTATGGGG180
AAAACGAGAGCGACTTCTCTGACTCCCCGCCCTGCCCACAGGATTTCAGC CTGAACTTTG240
ACAGAACCTTCCTGCCAGCCCTCTACAGCCTCCTCTTCTTGCTGGGGCTG CTAGGCAATG300
GGGCGGTGGCTGCTGTGCTACTGAGTCAGCGCACTGCCCTGAGCAGCACG GACACCTTCC360
TGCTCCACCTGGCTGTAGCCGATGTTCTGCTGGTGTTAACTCTTCCATTG TGGGCAGTGG420
ATGCTGCTGTCCAGTGGGTTTTCGGCCCTGGCCTCTGCAAAGTGGCAGGC GCCTTGTTCA480
ACATCAACTTCTATGCAGGGGCCTTCCTGCTGGCTTGTATAAGCTTCGAC AGATATCTGA540
GCATAGTGCACGCCACCCAGATCTACCGCAGGGACCCCCGGGTACGTGTA GCCCTCACCT600
GCATAGTTGTATGGGGTCTCTGTCTGCTCTTTGCCCTCCCAGATTTCATC TACCTATCAG660
CCAACTACGATCAGCGCCTCAATGCCACCCATTGCCAGTACAACTTCCCA CAGGTGGGTC720
GCACTGCTCTGCGTGTACTGCAGCTAGTGGCTGGTTTCCTGCTGCCCCTT CTGGTCATGG780
CCTACTGCTATGCCCATATCCTAGCTGTTCTGCTGGTCTCCAGAGGCCAG AGGCGTTTTC840
GAGCTATGAGGCTAGTGGTAGTGGTGGTGGCAGCCTTTGCTGTCTGCTGG ACCCCCTATC900
ACCTGGTGGTGCTAGTGGATATCCTCATGGATGTGGGAGTTTTGGCCCGC AACTGTGGTC960
GAGAAAGCCACGTGGATGTGGCCAAGTCAGTCACCTCGGGCATGGGGTAC ATGCACTGCT1020
GCCTCAATCCGCTGCTCTATGCCTTTGTGGGAGTGAAGTTCAGAGAGCAA ATGTGGATGT1080
TGTTCACGCGCCTGGGCCGCTCTGACCAGAGAGGGCCCCAGCGGCAGCCG TCATCTTCAC1140
GGAGAGAATCATCCTGGTCTGAGACAACTGAGGCCTCCTACCTGGGCTTG TAATTCTGGA1200
CTGGAACTGTAGCCTGCGCAGCCCAAGTCCTAACACACTCCAAGTGCTTG TCCTCCTGGT1260
AGTTGGGCTAGCTCGAACTTACCCGTAACTTTGCTGCCAGGATGCACTGA CAGCTCAGCA1320
TATATCCAGCTCTCCTGAGAATCAATCTCAGCAACAAGGACAACACCATT ACTGTGCCTT1380
AGCTGCCATGCCCTATCTTGCTGTTTTAGAACTAGCTGCCTGGAGCCCCA CCGCCCTACT1440
CA 02478138 2004-12-24
AAATTAGCAA GTAGAACTCA GCCATCCCTG TGTGAGAAGA GGGAGAGGCA AATAGCACAG 1500
AGGGCCAGGC GTTGTCAGCA CTGAATGTGC CCATCTCAGT ATCTCAATAT TTGCCCAATT 1560
TTATTTCTAG AAACCTCACT TAAACTTTCA ATAAACAAGG TAATGAGG 1608
81