Note: Descriptions are shown in the official language in which they were submitted.
CA 02479104 2004-08-26
IPT-14.51
METHODS AND APPARATUSES FOR CHARACTERIZING
REFOLDING AND AGGREGATION OF BIOLOGICAL MOLECULES
Related applications
This application claims the benefit of priority to U.S. Provisional Patent
Application
No. 60/498,561, filed on August 28, 2003, which application is hereby
incorporated by
reference in its entirety.
Background of the invention
Recent advances in genomics research provide an opportunity for rapid progress
in the
identification of novel drug targets. The complete genomic sequences for a
number of
microorganisms are already available. However, knowledge of the complete
genomic
sequence is only the first step in a long process toward discovery of a viable
drug target.
Targeted approaches to drug discovery may involve a variety of steps including
annotation of
the genomic sequence to identify open reading frames (ORFs), determination of
the essentiality
of the protein encoded by the ORF, and determination of the mechanism of
action of the gene
product. In addition to increasing the speed with which novel drug targets are
identified; it is
also important to make parallel advances in screening the potential drug
targets in order to
identify drugs which modulate the function of the target.
New technologies are required to facilitate the transition from gene sequence
(or
genomics) to gene function (or functional genomics). Classification of
proteins of unknown
function based on nucleotide or amino acid homology with proteins of known
function may be
difficult. While conservation between amino acid sequences generally indicates
a conservation
of structure and function, specific changes at key residues can lead to
significant variation in
the biochemical, biophysical, and/or functional properties of a protein.
To facilitate the study of proteins, it is important to have the proteins
available in a
reasonably stable form. In addition, characterization of proteins and
identification of drugs
requires the identification of molecules that interact with the proteins.
Therefore, methods for
identifying conditions that stabilize proteins and methods for identifying
molecules that bind to
the proteins are highly desirable.
Typical screening techniques are target-specific. In other words, it is
necessary to
develop custom assays for a given target which is extremely time-consuming.
Furthermore,
existing non-specific screening techniques do not provide sufficiently rich
data and typically
1
CA 02479104 2004-08-26
require additional screening using target-specific techniques or labeling of
targets (e.g. with
fluorescent probes).
In structural proteomics, x-ray crystallography is a powerful technique for
solving the
three-dimensional structure of a protein. A key step in this technique is
protein crystallization.
Increasingly researchers are interested in setting crystal screens at
progressively higher rates,
thus demanding an effective and efficient means for pre-selecting conditions
favorable to
crystallization. Therefore, new and improved methods for pre-selection of
conditions, such as
characterization of proteins using biophysical and biochemical means, are
highly desirable.
Outside of drug discovery, high throughput experimentation is becoming more
commonplace as a discovery tool. For example, combinatorial chemistry, a high
throughput
technique common in drug discovery, is emerging as an important tool in
material discovery in
the chemical and electronics industries. The demand for high throughput
discovery in all of the
above areas of applications requires high throughput detection, including real-
time monitoring,
and screening for desired properties.
Light scattering is recognized in the art as an effective and sensitive means
for detection
and characterization of small particles, particularly those in solution. In
applications such as
those mentioned above where formation of small particles naturally takes
place, process status
can be monitored by means of light scattering. However, commercially available
light
scattering instruments can measure only one sample at a time and are typically
expensive. To
use a number of such instruments to monitor a multiplicity of samples renders
the cost
prohibitive. Therefore, there is a great need for methods and apparatus that
permit monitoring
of light scattering of a multiplicity of samples essentially simultaneously.
Summary of the invention
In one aspect, the invention provides, a method for characterizing aggregation
of a
plurality of biological samples, comprising:
a) providing a plurality of biological samples, wherein each composition
comprises at least one biological molecule;
b) exposing the plurality of biological samples to one or more light sources;
and
c) determining the amount of light scattered by said plurality of biological
samples
upon exposure to said one or more light sources, thereby characterizing
aggregation of said biological samples.
2
CA 02479104 2004-08-26
In one embodiment, the light source may be one or more lasers. For example,
the
plurality of biological samples may be exposed to a plurality of lasers or the
beam of one or
more lasers may be split in order to expose the plurality of biological
samples to the laser light
essentially simultaneously. In an alternative embodiment, the light source may
be one or more
non-laser lights, such as for example, a light emitting diode (LED), a white
light source, a
monochromatic light source, an incandescent light source, a Xenon-arc lamp, a
tungsten-
halogen lamp, an ultraviolet light source, a luminescent light source, and a
low intensity light
source having an intensity in a range of 1.5 to 2.0 ~.W/mm2. In an exemplary
embodiment, the
non-laser light is a plurality of light emitting diodes (LEDs). In various
embodiments,
determining the amount of light scattered may comprise detecting the amount of
non-scattered
light, detecting the amount of scattered light, or both.
In another embodiment, the light source may be passed through an optical
filter, such
as, for example, a monochromator or a polarizing filter, before exposure to
the plurality of
biological samples.
In various embodiments, the plurality of biological samples comprises at least
about 2,
3, 4, 5, 10, 1 S, 20, S0, 100, 200, 500, 100, or more biological samples. In
exemplary
embodiments, the plurality of biological samples comprises at least about 96,
384, 1536
biological samples, for example, as available in various configurations of
standard microtiter
plates. In another embodiment, the plurality of biological samples are
contained in a plurality
of wells of a microtiter plate.
In another embodiment, one or more biological samples comprises at least one
biological molecules, such as, for example, a polynucleotide or a polypeptide.
In another
embodiment, one or more biological samples comprises a mixture of biological
molecules,
such as, for example, a mixture of polypeptides, a mixture of polynucleotides,
or a mixture of
polypeptides and polynucleotides. In other embodiments, the plurality of
samples may
comprise at least one biological molecule in a plurality of test conditions,
at least one mixture
of biological molecules in a plurality of test conditions, a plurality of
biological molecule in
one or more test conditions, a plurality of biological molecule in a plurality
of test conditions,
etc.
In another embodiment, the methods as described herein may further comprise
determining the aggregation rate (kagg) of said one or more biological
samples. In other
3
CA 02479104 2004-08-26
embodiments, characterizing aggregation may comprise determining one or more
of the
following: the aggregation state of the biological sample, the aggregation
kinetics of the
biological sample, or the aggregation dynamics of the biological sample. In
other
embodiments, the methods comprise characterizing aggregation of said plurality
of biological
samples as a function of time and/or temperature.
In another embodiment, the methods as described herein may comprise preparing
the
plurality of compositions in an automated fashion.
In another embodiment, the methods described herein comprise comparing a
property
of aggregation of at least one biological sample in at least one test
condition to a property of
aggregation of said biological sample in a reference condition. A property of
aggregation for at
least one biological sample may be determined, for example, in at least about
2, 5, 10, 20, 50,
100, or more test conditions. In an exemplary embodiment, a property of
aggregation for a
plurality of biological samples is determined in a plurality of test
conditions. Exemplary test
conditions, include, for example, differences as compared to a reference
condition in one or
more of the following: a biochemical condition, pressure, electric current,
time, concentration
of the biological molecule, and presence of a test compound. Exemplary,
biochemical
conditions, include, for example, pH, ionic strength, salt concentration,
oxidizing agent,
reducing agent, detergent, glycerol, metal ions, salt, cofactor concentration,
ligand
concentration, and/or coenzyme concentration. In an exemplary embodiment, a
test condition
comprises the presence of one or more potential ligands of a biological
molecule in a biological
sample.
In another embodiment, the methods described herein may further comprise
bringing
the temperature of said plurality of biological samples to one or more end
temperatures before
determining the amount of light scattered. In one embodiment, characterizing
aggregation of at
least one biological sample may be determined at one or more end temperatures,
optionally, as
a function of time. In another embodiment, characterizing aggregation of a
plurality of
biological samples may be determined over a range of end temperatures. In an
exemplary
embodiment, characterizing aggregation of at least one biological sample may
be determined
over a range of end temperatures by essentially simultaneously bringing a
plurality of
biological samples comprising a biological molecule to a plurality of end
temperatures. In
another exemplary embodiment, characterizing aggregation of at least one
biological sample
4
CA 02479104 2004-08-26
may be determined over a range of end temperatures by sequentially bringing a
biological
sample to a plurality of end temperatures. In various embodiments, the range
of end
temperatures may be sequentially increased over time. In certain embodiments,
characterizing
aggregation of at least one biological sample may be determined, for example,
at about 2, 5,
10, 20, 50, or more end temperatures. In another embodiment, a plurality of
biological samples
may be exposed to a temperature gradient to allow characterizing aggregation
of said plurality
of biological samples as a function of temperature.
In various embodiments of the invention, the extent of unfolding of one or
more
biological molecules in a biological sample may be determined in addition to
characterizing
aggregation of one or more biological molecules in the biological sample: The
extent of
unfolding a biological molecule may be determined, for example, by
fluorescence emission,
circular dichroism, or differential scanning calorimetry. In certain
embodiments, the invention
further comprises determining the rate of unfolding (k") and the rate of
aggregation (kagg) of
one or more biological molecules in one or more biological samples. In another
embodiment,
the methods described herein may further comprise determining the temperature
of unfolding
(Tm) of said one or more biological molecules.
In another embodiment, the invention provides methods for predicting optimal
conditions for crystallization, purification, folding, and/or refolding; high
throughput screening
of target molecules; high throughput study of kinetics and/or dynamics of
unfolding; kinetics
and/or dynamics of aggregation; kinetics and/or dynamics of both unfolding and
aggregation;
dynamics of folding; and biophysical characterization of biological samples.
In exemplary
embodiments, such methods involve characterizing biological samples under a
variety of
physical and biochemical conditions, and determining, for example, molecular
configuration
and conformation, solubility, structural stability, etc.
In another embodiment, the invention provides methods for identifying a
condition in
which a biological molecule has a different stability relative to its
stability in a reference
condition or relative to other conditions under study. In an exemplary
embodiment, the
invention comprises (a) providing a composition comprising a biological
molecule in a test
condition; (b) bringing the temperature of the composition to an end
temperature; (c)
determining the extent of aggregation of the biological molecule in the
composition as a
function of time over a period extending past the time point at which the
temperature of the
5
CA 02479104 2004-08-26
solution attains the end temperature; (d) obtaining a characteristic of
aggregation of the
biological molecule in the test solution from the extent of aggregation
obtained as a function of
time in (c); and (e) comparing the characteristic of aggregation obtained in
(d) with the
characteristic of aggregation of the biomolecule in the reference condition,
wherein a different
characteristic of aggregation of the biological molecule in the test condition
relative to the
reference condition indicates that the test condition is a condition in which
the biological
molecule has a different stability relative to its stability in the reference
condition. In various
embodiments, the end temperature may be lower, lower than, or substantially
equivalent to, the
aggregation temperature of the biological molecule in the reference condition.
The method can
be used to identify conditions in which a biological molecule has a higher
stability relative to
its stability in a reference condition and to compare relative efficacies of
different conditions in
stabilizing the biological molecule. The biological molecule can he a protein.
In an exemplary
embodiment, determining the extent of aggregation of the biological molecule
in the
composition as a function of time is conducted essentially only when the
temperature of the
composition is at the end temperature. The characteristic of aggregation may
be the rate of
unfolding (k) or the rate of aggregation (kagg), respectively. The method may
comprise first
determining the aggregation temperature of the biological molecule in the
reference solution.
In certain embodiments, the temperature of step (b) can be lower than the
aggregation
temperature of the biological molecule in the reference solution by at least
about 5°C.
The test condition can differ from the reference condition in one or more of
the
following: a biochemical condition, pressure, electric current, time,
concentration of the
biological molecule, and presence of a test compound. The test condition can
differ from the
reference condition in a biochemical condition selected ft°om the group
consisting of pH, ionic
strength, salt concentration, oxidizing agent, reducing agent, detergent,
glycerol, metal ions,
salt, cofactor concentration, ligand concentration and coenzyme concentration.
The test
condition can comprise a potential ligand of the biological molecule not known
to bind to the
biological molecule, and wherein a lower kagg of the biological molecule in
the test condition
relative to the reference condition indicates that the potential ligand
interacts with the
biological molecule.
The method can comprise determining the extent of unfolding of the biological
molecule by fluorescence emission, e.g., with 4,4'-dianilino-l,l-binaphthyl-
5,5-disulfonic acid
6
CA 02479104 2004-08-26
(bis-ANS). The method can comprise determining the extent of aggregation of
the biological
molecule by measuring absorption of ultraviolet light, absorption of visible
light, changes in
turbidity, or changes in the polar properties of light.
The method may further comprise increasing the temperature of the composition
after
step (c) and repeating steps (c) to (e) at the higher temperature. In another
embodiment, the
composition forms a temperature gradient and the method comprises determining
the extent of
aggregation of the biological molecule in at least two locations of the
gradient.
The invention also provides a method for identifying a condition in a
plurality of
conditions in which a biological molecule has a higher stability relative to
its stability in the
other conditions, comprising (a) providing a plurality of compositions each
comprising
essentially the same biological molecule in a plurality of different
conditions; (b) changing the
temperature of the composition; (c) determining the extent of aggregation of
the biological
molecule in the compositions as a function of time over a period extending
past the time point
at which the temperature was changed; (d) obtaining the kaggs of the
biological molecule in the
test conditions from the extent of aggregation obtained as a function of time
in (c); and (e)
comparing the kaggs obtained in (d) with each other, respectively, wherein the
test condition in
which the lCagg 1S the highest among the plurality of test conditions is a
condition in which the
stability of the biological molecule is higher relative to its stability in
the other test conditions.
Step (e) may further comprise comparing the kaggs obtained in (d) with the
kagg, respectively, of
the biological molecule in the reference condition. The plurality of test
conditions can
comprise at least 5, 10, or 100 test conditions. The plurality of compositions
can be in a
plurality of wells of a microwell plate and the method can be conducted in an
automated
manner. In one embodiment, step (b) may involve bringing the temperature of
the
compositions to a temperature that is slightly lower than the aggregation
temperature of the
biological molecule in the reference condition.
In one embodiment, the invention provides a method for identifying a condition
in
which a biological molecule has a different stability relative to its
stability in a reference
condition, comprising: (a) providing a composition comprising a biological
molecule in a test
condition; (b) increasing the temperature of the composition over time; (c)
determining the
extent of unfolding and aggregation of the biological molecule in an
essentially simultaneous
manner during the increase in temperature; (d) obtaining a characteristic of
unfolding and
7
CA 02479104 2004-08-26
aggregation of the biological molecule in the test condition from the extent
of unfolding and
aggregation obtained in (c); and (e) comparing the characteristic of unfolding
and aggregation
obtained in (d) with that of the biological molecule in the reference
condition; wherein a,
different characteristic of aggregation of the biological molecule in the test
condition relative to
the reference condition indicates that the test condition is a condition in
which the biological
molecule has a different stability relative to its stability in the reference
condition. The
characteristic of unfolding and aggregation can be the temperature of
unfolding (Tm) and the
temperature of aggregation (Tags), respectively. The extent of unfolding can
be determined by
bis-ANS fluorescence and the extent of aggregation can be determined by light
scattering. The
composition can be alternatively exposed a UV light and a light source for
light scattering
during the increase in temperature. The UV light and light source for light
scattering can be
computer controlled to be switched on and off alternatively for fluorescence
and Light
scattering, respectively. In exemplary embodiments, a light source for light
scattering may be
one or more of the following: a laser, a light emitting diode (LED), a cluster
of LEDs, a white
light source, a monochromatic light source, an incandescent light source, a
Xenon-arc lamp, a
tungsten-halogen lamp, an ultraviolet light source, a luminescent light
source, and/or a low
intensity light source with an intensity in a range of 1.5 to 2.0 ~W/mm2.
In another embodiment, the invention provides methods for identifying a
condition
among a plurality of conditions in which a biological molecule has a higher
stability relative to
its stability in the other conditions, comprising: (a) providing a plurality
of compositions
comprising essentially the same biological molecule in a plurality of
different test conditions;
(b) increasing the temperature of the plurality of compositions over time; (c)
determining the
extent of unfolding and aggregation of the biological molecule in the
plurality of compositions
in an essentially simultaneous manner during the increase in temperature; (d)
obtaining the Tm
and Tags of the biological molecule in each of the test conditions from the
extent of unfolding
and aggregation obtained in (c); and (e) comparing the Tms and Taggs obtained
in (d) with one
another, wherein the test condition in which the k" or kagg is the lowest
among the plurality of
test conditions is a condition in which the stability of the biological
molecule is higher relative
to its stability in the other test conditions. The temperature of the
plurality of compositions can
be increased essentially simultaneously over time.
CA 02479104 2004-08-26
In another aspect, the invention provides a method for identifying conditions
that
facilitate refolding of one or more biological molecules, comprising: a)
exposing one or more
biological samples to one or more test conditions, wherein the biological
samples comprise at
least one denatured biological molecule; b) exposing said one or more
biological samples to
one or more light sources; c) characterizing the aggregation of said one or
more biological
samples by determining the amount of light scattering by said one or more
biological samples,
thereby characterizing the refolding of the biological molecule in the
biological sample.
In another aspect, the invention provides a method for identifying a modulator
of
aggregation of one or more biological molecules, comprising: a) exposing one
or more
biological samples to denaturing conditions in the presence of one or more
test compounds,
wherein each biological sample comprises at least one biological molecule; b)
exposing said
one or more biological samples to one or more light sources; and c)
characterizing the
aggregation of said one or more biological samples by determining the amount
of light
scattering by said one or more biological samples, wherein a change in the
amount of light
scattering by said one or more biological samples in the presence of the test
compound as
compared to the amount of light scattering by said one or more biological
samples in the
absence of the test compound is indicative of a modulator of protein
aggregation.
Also within the scope of the invention are computer readable media and
databases
comprising the results of the methods of the invention. Kits and apparatuses
are also provided.
The invention further provides an apparatus for measuring an extent of
aggregation in
at least one molecular sample. In an exemplary embodiment, the apparatus
comprises a light
source positioned to illuminate the molecular sample; a sample container for
containing the
molecular sample; a light guide positioned in an optical path between the
light source and the
sample container to direct light from the light source into the sample
container; a scattered light
detector positioned to receive the light passing through the molecular sample
and scattered
from the molecular sample at an angle from the optical path of the light
entering the sample
from the light guide, the scattered light detector producing a signal
proportional to the received
scattered light; and a processor in communication with the scattered light
detector to receive
and process the signal from the scattered light detector to determine the
extent of aggregation
in the at least one molecular sample.
9
CA 02479104 2004-08-26
In one embodiment, the light guide is positioned at an angle with respect to
an optical
path between the at least one molecular sample and the detector, such that the
angle is less than
45° and preferably in a range from 15° to 30°.
In one embodiment, the detector can be one of a photomultiplier and a charged-
couple
device (CCD). The apparatus can include a luminescence detector positioned to
receive
fluorescence emanating from the at least one molecular sample, the
luminescence detector
producing a signal proportional to the received fluorescence, the processor
receiving and
processing the signal from the luminescence detector to determine an extent of
unfolding in the
molecular sample. A switch can select which detector the processor can receive
the signal
from.
In one embodiment, the apparatus includes a luminescent light source to
illuminate the
molecular sample. The detector can receive fluorescence emanating from the
illuminated
sample resulting from the illumination by the luminescent light source and can
produce a signal
proportional to the received fluorescence. The processor can determine an
extent of unfolding
in the molecular sample based on the signal received from the detector. A
switch can
selectively operate the luminescent light source.
In one embodiment the sample container includes an array of sample wells, each
sample well being sized to contain one of the molecular samples. The sample
wells can be
spatially separated from each other to inhibit cross-contamination and can be
optically isolated
to inhibit scattered light from one sample from illuminating other samples.
The light guide can
be a collimator positioned in the optical path between the light source and
the sample container
to substantially collimate light from the light source into the sample wells.
The collimator can
be an array of optical fibers, wherein the optical fibers are each optically
aligned with a
respective sample well within the sample container.
In one embodiment, the apparatus can include aneans for selectively directing
and/or
occluding light from the light source to at least one of the sample wells. In
one embodiment,
the apparatus can include means for selectively directing or occluding
scattered light from at
least one of the sample wells to the scattered light detector.
In one embodiment, the apparatus can include a heating element for heating the
sample
container. The heating element can be configured to create a temperature
gradient across the
sample container and can be configured to selectively heat at least one
selected sample well,
CA 02479104 2004-08-26
such that the at least one selected sample well is heated to a temperature
distinct from other
sample wells.
The light source can be one of a number of sources, including, for example, a
laser, a
light emitting diode (LED), a cluster of LEDs, a white light source, a
monochromatic light
source, an incandescent light source, a Xenon-arc lamp, a tungsten-halogen
lamp, an ultraviolet
light source and a luminescent light source. The light source can be a low
intensity light source
having an intensity in a range of 1.5 to 2.0 ~,W/mm2. A monochromator can be
positioned in
the optical path between the light source and the molecular sample to
illuminate the molecular
sample with monochromatic light.
In one embodiment, an apparatus measures an extent of aggregation in a
plurality of
molecular samples. The apparatus can include a sample container for containing
the molecular
samples; a light source positioned to illuminate selected ones of the
molecular samples; a
scattered light detector positioned to receive the light passing through the
selected ones of the
molecular samples and scattered from the selected ones of the molecular
samples, the scattered
light detector producing a signal proportional to the received scattered
light; and a processor in
communication with the scattered light detector to receive and process the
signal from the
scattered light detector to determine the extent of aggregation in the
selected ones of the
molecular samples.
The apparatus can include a collimator positioned in an optical path between
the light
source and the sample container to substantially collimate light from the
light source into the
molecular samples. The collimator can be an array of optical fibers that can
each be optically
aligned with a respective molecular sample within the sample container. The
collimator can be
positioned at an angle with respect to an optical path between the molecular
samples and the
detector, wherein the angle is less that 45° and preferably in a range
from 15° to 30°.
The sample container can include an array of sample wells, each sample well
being
sized to contain one of the molecular samples and each sample well being
optically isolated
from other sample wells of the array to inhibit scattered light from the
molecular sample in the
sample well from illuminating the molecular sample in the other sample wells.
The apparatus
can further include optical directing means for selectively directing light
from the light source
to at least one of the molecular samples, wherein the optical directing means
can include
11
CA 02479104 2004-08-26
micro-electromechanical devices selectively controlling movements of an array
of directing
optics to form an optical path between the light source and the at least one
molecular sample.
Means for selectively occluding the optical path between th.e light sourc.P
and the
molecular samples, means for selectively directing scattered light from the
molecular samples
to the scattered light detector, or means for selectively occluding light from
the molecular
samples to the scattered light detector can be provided. The light source can
be one of a laser,
a light emitting diode (LED), a cluster of LEDs, a white light source, a
monochromatic light
source, an incandescent light source, a Xenon-arc lamp, a tungsten-halogen
lamp, an ultraviolet
light source, or a luminescent light source. The light source can be a low
intensity light source
with an intensity in a range of 1.~ to 2.0 ~W/mm2.
An apparatus for measuring an extent of aggregation in a plurality of
molecular samples
can include an array of sample wells, each sample well being sized to contain
one of the
molecular samples; a light source positioned to illuminate selected ones of
the sample wells; a
light guide positioned in an optical path between the light source and the
sample container to
direct light from the light source into the sample wells; a light detector
positioned to receive at
least one of scattered light and fluorescence from the molecular samples in
the selected ones of
the sample wells, the light detector producing a signal proportional to the
received light; and a
processor in communication with the light detector to receive and process the
signal from the
light detector to determine the extent of aggregation in the molecular samples
in the selected
ones of the sample wells when the received light is scattered light and to
determine the extent
of unfolding in the molecular samples in the selected ones of the sample wells
when the
received light is fluorescence.
In one embodiment, the light source can include a low intensity light source
and/or a
luminescent light source. The light emitted from the low intensity light
source can pass
through the selected ones of the sample wells and be scattered by the
molecular sample to be
received as scattered light at the detector. The detector can receive
fluorescence emanating
from the molecular samples in the selected ones of the sample wells that have
been illuminated
by the luminescent light source. A switch can selectively operate the low
intensity light source
and the luminescent light source.
In one embodiment, the detector can include a scattered light detector to
receive light
from the light source passing through the selected ones of the sample wells
and scattered by the
12
CA 02479104 2004-08-26
molecular samples and a fluorescence detector to receive fluorescence
emanating from the
molecular samples in the selected ones of the sample wells illuminated by the
light source. A
switch can be operated to select between the processor receiving the signal
from scattered light
detector and the processor receiving the signal from the luminescence
detector.
An apparatus for measuring an extent of aggregation in a plurality of
molecular samples
and/or an extent of unfolding in a plurality of molecular samples can include
an array of
sample wells, each sample well being sized to contain one of the molecular
samples; a first
light source positioned to illuminate selected ones of the sample wells; a
second light source
positioned to illuminate the same, or other selected ones of the sample wells;
a light guide
positioned in an optical path between the light sources and the sample
container to direct light
from the light sources into the sample wells; a light detector positioned to
receive light from
the first light source passing through the selected ones of the sample wells
and scattered by the
molecular sample and to receive fluorescence emanating from the molecular
samples in the
selected ones of the sample wells being illuminated by the second light
source, the light
detector producing a signal proportional to the received light; and a
processor in
communication with the light detector to receive and process the signal from
the light detector
to determine the extent of aggregation in the molecular samples in the first
selected ones of the
sample wells when the received light is scattered light and to determine the
extent of unfolding
in the molecular samples in the second selected ones of the sample wells when
the received
light is fluorescence.
In one embodiment, the first light source can be a low intensity light source.
A switch
can be included such that the first light source and the second light can be
selectively operated.
The detector can include a scattered light detector and a fluorescence
detector. A switch can be
included to select between the processor receiving the signal from the
scattered light detector
and the processor receiving the signal from the fluorescence detector.
Brief description of the figures
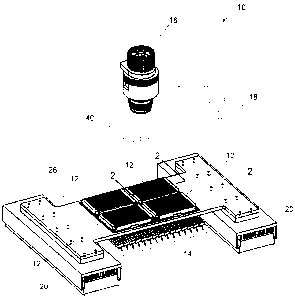
Figure 1 shows an illustrative isometric view of an apparatus for measuring
aggregation
in molecular samples.
Figure 2 shows an illustrative cross-sectional view taken along the line 2-2
in Figure 1.
Figure 3 shows an illustrative cross-section view, corresponding to that of
Figure 2, of
an alternative embodiment of the apparatus of Figure 1.
13
CA 02479104 2004-08-26
Figure 4 illustrates types of scattering by macromolecules.
Figures SA and B show the light scattering and fluorescence of a protein as a
function
of time.
Figure 6 shows the fluoresence of the protein as a function of time at 55
°C.
Figure 7 shows an example of a selective illumination pattern which minimizes
or
inhibits cross-talk.
Figure 8 shows a flow chart of an exemplary high throughput method for
analyzing
protein refolding conditions in a 384 well plate.
Figure 9 shows the results of an experiment as outlined in Figure 8. A single
protein
was analyzed in 16 different test conditions and conditions that facilitated
refolding of the
protein were identified by measuring protein aggregation (intensity) as a
measure of time. The
circled curves indicate transitiorn curves for proteins detected in a folded
state.
Figure 10 shows the a plot of protein aggregation (intensity) as a function of
temperature for refolded gaf domain of PDE10 in different conditions as
indicated.
Detailed description of the invention
As used herein, the following terms and phrases shall have the meanings set
forth
below. Unless defined otherwise, all technical and scientific terms used
herein have the same
meaning as commonly understood to one of ordinary skill in the art to which
this invention
belongs.
The singular forms "a," "an," and "the" include plural reference unless the
context
clearly dictates otherwise.
The term "biochemical conditions" encompasses any characteristic of a
physical,
chemical, or biochemical process or reaction. In exemplary embodiments, the
term refers to
conditions including, for example, temperature, pressure, protein
concentration, pH, ionic
strength, salt concentration, time, electric current, potential difference,
concentrations of
cofactor, coenzyme, oxidizing agents, reducing agents, detergents, metal ion,
ligands, or
glycerol.
The term "biophysical characteristics" refer to physical characteristics of a
biological
molecule relevant to the biological function of the molecule, including its
state, solubility,
structure, etc. The term "biophysical characterization" refers to tests or
processes carried out to
determine a sample's biophysical characteristics.
14
CA 02479104 2004-08-26
The term "carrier" encompasses a platform or other object, of any shape, which
itself is
capable of supporting at least two containers. The carrier can be made of any
material,
including, but not limited to, glass, plastic, or metal. Preferably, the
carrier is a multiwell
microplate. The terms microplate and microtiter plate are synonymous. The
carrier can be
removed from the heating element. Each carrier can hold a plurality of
containers.
The term "combinatorial library" refers to a plurality of molecules or
compounds which
are formed by combining, in close to every possible way for a given compound
length, a set of
chemical or biochemical building blocks which may or may not be related in
structure.
Alternatively, the term can refer to a plurality of chemical or biochemical
compounds which
are formed by selectively combining a particular set of chemical building
blocks.
Combinatorial libraries can be constructed according to methods familiar to
those skilled in the
art. For example, see Rapoport et al., Immunology Today 16:4349 (1995);
Sepetov, N. F. et al.,
Proc. Natl. Acad. Sci. U.S.A. 92:5426-5430 (1995); Gallop, M. A. et al., J.
Med. Chem.
9:1233-1251 (194); Gordon, E. M. et al., J. Med. Chem. 37:1385-1401 (1994);
Stankova, M.
et al., Peptide Res. 7:292-298 ( 1994); Erb, E. et al., Proc. Natl. Acad. Sci:
U. S.A. 91:11422-
11426 (1994); DeWitt, S. H. et al., Proc. Natl. Acad. Sci. U.S.A. 90:6909-6913
(1993); Barbas,
C. F. et al., Proc. Natl. Acad. Sci. U.S.A. 89:4457-4461 (1992); Brenner, S.
et al. Proc. Natl.
Acad. Sci. U.S.A. 89:5381-5383 (1992); Lam, K. S. et al., Nature 354:82-84
(1991); Devlin, J.
J. et al., Science 245:404-406 (1990); Cwirla, S. E. et al., Proc. Natl. Acad.
Sci. U.S.A.
87:6378-6382 (1990); Scott, J. K: et al., Science 249:386-390 (1990). In an
exemplary
embodiment, the term "combinatorial library" refers to a diversity chemical
library, as set forth
in U.S. Pat. No. 5,463,564. Regardless of the manner in which a combinatorial
library is
constructed, substantially every different molecule or compound in the library
is catalogued for
future reference.
The term "compound library" refers to a plurality of molecules or compounds
which
were not formed using the combinatorial approach of combining chemical or
biochemical
building blocks. Instead, a compound library is a plurality of molecules or
compounds which
are accumulated and are stored for use in binding assays, such as, for
example, binding assays
between a target molecule and a ligand. Substantially every different molecule
or compound in
the compound library is catalogued for future reference.
CA 02479104 2004-08-26
The term "container" refers to any vessel or chamber, in which the receptor
and
molecule to be tested for binding can be placed. The term "container"
encompasses reaction
tubes (e.g., test tubes, microtubes, vials, etc.). In an exemplary embodiment;
the terra
"container" refers to one or more wells in a multiwell microplate or
microtiter plate. The term
S "sample" refers to the contents of a container.
As used herein, the "folded state" of a protein refers to the native or
undenatured form
of the protein as it is present under physiological conditions, with
secondary, tertiary and/or
quaternary structures intact. Physiological conditions include conditions
similar to the natural
environment of the protein, or conditions under which it is stable after
expression, isolation,
and/or purification, i.e. before exposure to denaturing conditions. Similarly,
the °'unfolded
state" refers to a situation in which the polypeptide has lost elements of its
secondary, tertiary
andlor quaternary structure that are present in its "folded state." It will be
recognized by those
skilled in the art that it is difficult to determine experimentally when a
polypeptide has become
completely unfolded (i.e., when a polypeptide has lost all elements of
secondary, tertiary,
and/or quaternary structure). Thus, the term "unfolded state" as used herein
encompasses
partial or total unfolding.
The term "capable of denaturing" refers to the ability to cause the loss of
secondary,
tertiary, andlor quaternary structure through unfolding, uncoiling, or
untwisting.
The terms "folding," "refolding,"' and "renaturing" refer to the acquisition
of the correct
secondary, tertiary, and/or quaternary structure, of a protein or a nucleic
acid, which affords a
full chemical and/or biological function of the biomolecule.
The term "aggregation" refers to the association of two or more biological
molecules.
In one embodiment, the term is meant to encompass crystallization, native
aggregation, andlor
pathological aggregation. "Crystallization" refers to the aggregation of
molecules in an orderly
fashion such that essentially all molecules are oriented in essentially the
same way. "Native
aggregation" refers to the formation of homo- or hetero- dimers, trimers,
etc., of biological
molecules, especially proteins, which interact to form a multimeric molecule
having a
biological function. "Pathological aggregation" refers to the association of
two or more
biological molecules due to hydrophobic interactions. Pathological aggregates
of proteins
often are not biologically active. In an exemplary embodiment, the term
"aggregation" refers
16
CA 02479104 2004-08-26
to pathological aggregation of biological molecules and excludes
crystallization and native
aggregation.
The term "characterizing aggregation", with reference to a biological sample,
refers to
determining a property of aggregation of one or more biological molecules in
the biological
sample. The term "property of aggregation" is meant to encompass the extent of
aggregation,
aggregation state, aggregation kinetics, and/or aggregation dynamics of a
biological molecule.
The term "extent of aggregation", with reference to a biological molecule,
refers to the
proportion by mass of biological molecules in a biological sample that are in
aggregates
relative to the total mass of the biological molecules in the sample under a
given set of
conditions. The extent of aggregation can be determined by a variety of
methods, such as those
described herein.
"Extent of unfolding" or "extent of denaturation" of a biological molecule
refers to the
extent of unfolding of the biological molecule, i.e., the extent of changes in
its secondary,
tertiary and/or quaternary structure. Extent of unfolding of a biological
molecule also refers to
the proportion of biological molecules in a composition that are partially or
completely
unfolded relative to those that are in their native configuration under
particular conditions. The
extent of unfolding can be determined by a variety of methods, such as those
described herein.
"Characteristics of unfolding" or "characteristics of aggregation" refer to
parameters, or
changes in parameters, that reflect the extent of unfolding or aggregation of
a sample,
respectively. Characteristics of unfolding or aggregation include, for
example, thermal
unfolding or aggregation curves, or portions thereof, e.g., Tm or Tagg - the
transition
temperatures from the unfolding or aggregation curves respectively, and rates
of unfolding (ku)
and aggregation (lc~gg).
A "thermal unfolding curve" is a plot of the physical change associated with
the
unfolding of a protein or a nucleic acid as a function of temperature. See,
for example,
Davidson et al, Nature Structure Biology 2:859 (1995); Clegg, R. M. et al.,
Proc. Natl. Acad.
Sci. U.S.A. 90:2994-2998 (1993). The "midpoint temperature" or "Tm" is the
temperature on a
thermal unfolding curve at which the ratio of folded vs. unfolded protein is
1:1. It is also
referred to as "transition temperature" or "melting temperature". The Tm can
be readily
determined using methods well known to those skilled in the art. See, for
example, Weber, P.
17
CA 02479104 2004-08-26
C. et al., J. Am. Chem. Soc. 11 x:2717-2724 ( 1994); Clegg, R. M. et al.;
Proc. Nati. Acad. Sci.
U.S.A. 90:2994-2998 (1993).
A "thermal aggregation curve" is a plot of physical change associated varith
the
aggregation of a biological molecule as a function of temperature. The
"aggregation transition
temperature", or Tagg, is the temperature on the thermal aggregation curve at
which the ratio of
aggregated vs. unaggregated protein is 1:1. It is also referred to as the
"aggregation
temperature".
The term "fluorescence probe molecule" refers to a fluorophore, which is a
molecule or
a compound capable of binding to an unfolded or denatured receptor and, after
excitement by
light of a defined wavelength, emits fluorescent energy. The term fluorescence
probe molecule
encompasses all fluorophores. More specifically, for proteins, the term
encompasses
fluorophores such as thioinosine, and N-ethenoadenosine, formycin, dansyl
derivatives,
fluorescein derivatives, 6-propionyl-2-(dimethylamino)-napthalene (PRODAN), 2-
anilinonapthalene, and N-arylamino-naphthalene sulfonate derivatives such as 1-
anilinonaphthalene-8-sulfonate (1,8-ANS), 2-anilinonaphthalene-6-sulfonate
(2,6-ANS), 2-
aminonaphthalene-6-sulfonate, N,N-imethyl-2-aminonaphthalene-5-sulfonate, N-
phenyl-2-
aminonaphthalene, N-cyclohexyl-2-aminonaphthalene-6-sulfonate, N-phenyl-2-
aminonaphthalene-6-sulfonate, N-phenyl-N-methyl-2-aminonaphthalene-6-
sulfonate, N-(o-
toluyl)-2-aminonaphthalene-6-sulfonate, N-(m-toluyl)-2-aminonaphthalene-6-
sulfonate, N-(p-
toluyl)-2-aminonaphthalene-6-sulfonate, 2-(p-toluidinyl)-naphthalene-6-
sulfonic acid (2,6-
TNS), 4-(dicyanovinyl) julolidine (DCVJ), 6-dodecanoyl-2-
dimethylaminonaphthalene
( L A U R D A N ) , 6 - h a x a d a c a n o y l - 2 - ( ( ( 2
(trimethylammonium)ethyl)methyl)amino)naphthalenechloride (PATMAN), nile red,
N-
phenyl-1-naphthylamine, l,1-dicyano-2-[6-(dimethylamino) naphthalen-2-
yl]propene (DDNP),
4,4'-diariilino-1,1-binaphthyl-5,5-disulfonic acid (bis-ANS), and DAPOXYLTM
derivatives
(Molecular Probes, Eugene, Oreg.). In an exemplary embodiment, the term refers
to 1,8-ANS
or 2,5-TNS in association with proteins. A "donor fluorophore" is one which,
when excited by
light, will emit fluorescent energy. The energy emitted by the donor
fluorophore is absorbed by
the acceptor fluorophore. The term '°donor fluorophore" encompasses all
fluorophores
including, but not limited to, carboxyfluorescein, iodoa~etamidofluorescein,
and fluorescein
1R
CA 02479104 2004-08-26
isothiocyanate. The term "acceptor fluorophore" encompasses all fluorophores
including, but
not limited to, iodoacetamidoeosin and tetramethylrhodamine.
The term "polypeptide", and the terms "protein" and "peptide" which are used
interchangeably herein, refers to a polymer of amino acids. Exemplary
polypeptides include
gene products, naturally-occurring proteins, recombinant polypeptides,
fragments, and other
equivalents, variants and analogs of the foregoing. Tn certain instances, a
protein may
comprise two or more polypeptide chains that are associated through covalent
or non-covalent
interactions.
The terms "recombinant protein" or "recombinant polypeptide" refer to a
polypeptide
which is produced by recombinant DNA techniques. An example of such techniques
includes
the case when DNA encoding the expressed protein is inserted into a suitable
expression vector
which is in turn used to transform a host cell to produce the protein or
polypeptide encoded by
the DNA.
The term "target molecule" encompasses peptides, proteins, nucleic acids, and
other
biological molecules. The term encompasses both enzymes and proteins which are
not
enzymes. The term encompasses monomeric and multimeric proteins. Multimeric
proteins may
be homomeric or heteromeric. The term encompasses nucleic acids comprising at
least two
nucleotides, such as oligonucleotides. Nucleic acids can be single-stranded,
double-stranded or
triple-stranded. The term encompasses a nucleic acid which is a synthetic
oligonucleotide, a
portion of a recombinant DNA molecule, or a portion of chromosomal DNA. The
term target
molecule also encompasses portions of peptides, proteins, and other receptors
which are
capable of acquiring secondary, tertiary, or quaternary structure through
folding, coiling or
twisting. The target molecule may be substituted with substituents including,
but not limited to,
cofactors, coenzymes, prosthetic groups, lipids, oligosaccharides, or
phosphate groups.
As used herein, the term "target protein" refers to a test molecule which may
be a
peptide, protein or protein complex for which characterization of the
stability and/or
identification of a ligand or binding partner is desired. Target proteins
include without
limitation peptides or proteins known or believed to be involved in the
etiology of a given
disease, condition or pathophysiological state, or in the regulation of
physiological function.
Target proteins may be derived from any living organism, such as a
prokaryotes, virus, and
eukaryotes, including, for example, vertebrates, particularly mammals, and
even more
19
CA 02479104 2004-08-26
particularly humans. For use in the present invention, it is not necessary
that the protein's
biochemical function be specifically identified. Target proteins include
without limitation
receptors, enzymes, oncogene products, tumor suppressor gene products, vital
proteins, and
transcription factors, either in purified form or as part of a complex mixture
of proteins and
other compounds. Furthermore, target proteins may comprise wild type proteins,
or,
alternatively, mutant or variant proteins, including those with altered
stability, activity, or other
variant properties, or hybrid proteins to which foreign amino acid sequences;
e.g., sequences
that facilitate purification (e.g., a tag or fusion), have been added.
As used herein, the term "ligand" refers to an agent that binds a target
protein. The
agent may bind the target protein when the target protein is in its native
conformation, when it
is partially or totally unfolded or denatured, or when it is partially or
totally aggregated.
According to the present invention, a ligand is not limited to an agent that
binds a recognized
functional region of the target protein e.g. the active site of an enzyme, the
antigen-combining
site of an antibody, the hormone-binding site of a receptor, a cofactor-
binding site, and the like.
A ligand can also be an agent that binds any surface or internal sequences or
conformational
domains of the target protein. Therefore, the ligands of the present invention
encompass agents
that in and of themselves may have no apparent biological function, beyond
their ability to bind
to the target protein in the manner described above.
As used herein, the term "test ligand" refers to an agent, comprising a
compound,
molecule or complex, which is being tested for its ability to bind to a target
protein. Test
ligands can be virtually any agent, including without limitation metals,
peptides, proteins,
lipids, polysaccharides, nucleic acids, small organic molecules, and
combinations thereof.
Complex mixtures of substances such as natural product extracts, which may
include more than
one test ligand, can also be tested, and the component that binds the target
protein can be
purified from the mixture in a subsequent step.
The term "test compound" refers to a molecule to be tested by one or more
screening
methods) described herein. Examples of test compounds include, but are not
limited to,
peptides, nucleic acids, carbohydrates, and small molecules. The term is meant
to encompass
both natural compounds (e.g., purified from a biological source) as well as
synthetic
compounds.
CA 02479104 2004-08-26
The terms "multiplicity of molecules," "multiplicity of compounds,"
"multiplicity of
samples", or "multiplicity of containers" refer to at least two molecules,
compounds, samples,
or containers, respectively. The term "multiplicity" is used interchangeably
herein ~~ith
"plurality."
The term "polarimetric measurement" relates to measurements of changes in the
polarization properties of light and fluorescent emission. Circular dichroism
and optical
rotation are examples of polarization properties of light which can be
measured
polarimetrically. Measurements of circular dichroism and optical rotation are
taken using a
spectropolarimeter. "Nonpolarimetric" measurements are those that are not
obtained using a
spectropolarimeter.
The terms "spectral measurement" and "spectrophotometric measurement" refer to
measurements of changes in the absorption of light. Tuxbidity measurements,
measurements of
visible light absorption, and measurement of ultraviolet light absorption are
examples of
spectral measurements.
"Stability" of a biological molecule refers to the ability of the biological
molecule to
resist aggregation and/or unfolding in conditions that tend to unfold or
aggregate biological
molecules. For example, a first protein is more stable than a second protein
if the first protein
is not significantly unfolded or aggregated at a temperature at which the
second protein is
significantly unfolded.
"Kinetics of unfolding" or "unfolding kinetics" or "denaturation kinetics"
refers to the
study of the extent of unfolding as a function of time. "Kinetics of
aggregation" or
"aggregation kinetics" refers to the study of the extent of aggregation as a
function of time.
"Dynamics of unfolding" or "unfolding dynamics" or "denaturation dynamics"
refers to
the study of unfolding or denaturation as a function of environmental
conditions in which a
biological sample is disposed, including biochemical conditions. "Dynamics of
aggregation"
or "aggregation dynamics" refers to study of aggregation as a function of
environmental
conditions in which a biological sample is disposed, including biochemical
conditions.
The terms "thermal change'° and "physical change" encompass the release
of energy in
the form of light or heat, the absorption of energy in the form or light or
heat, changes in
turbidity, and/or changes in the polar properties of light. In exemplary
embodiments, the terms
include, for example, fluorescent emission, fluorescent energy transfer,
absorption of
21
CA 02479104 2004-08-26
ultraviolet or visible light, changes in the polarization properties of light,
changes in the
polarization properties of fluorescent emission, changes in turbidity, and
changes in enzyme
activity. Fluorescence emission can be intrinsic to a protein or can be due to
a fluorescence
reporter molecule (below). For a nucleic acid, fluorescence can be due to
ethidium bromide,
which is an intercalating agent. Alternatively, the nucleic acid can be
labeled with a
fluorophore (below).
Methods of the Invention
The invention is based at least in part on the observation that most proteins
denature
and/or aggregate when they are exposed to a variety of conditions, such as,
for example, a
change in temperature (increase or decrease) or non-physiological conditions.
This unfolding
is a consequence of the protein unfolding to an intermediate hydrophobic rate
which may be
followed by aggregation. Proteins may also aggregate directly from its folded
state and may
also remain in an unfolded state without proceeding to aggregation, depending
on the
environmental conditions. The invention provides methods and apparatus for
studying the
process of aggregation and also for studying the combined processes of
unfolding and
aggregation. Such studies can be summarized with quantitative measures, such
as transition
temperatures or rate constants, comparing the process of aggregation under
different
conditions, leading to, for example, identification of conditions which are
favorable to
stabilizing or crystallizing a particular biological molecule.
Most cellular proteins denature irreversibly, however some of the well studied
proteins
denature largely reversibly: There are tyo reasons for this, proteins such as
ribonuclease,
lysozyme, trypsin, etc., which can be isolated in large quantities and are
normally used in most
in vitro studies, are either secreted proteins, or intracellular proteins
which are very stable and
denature reversibly. Also, reversible unfolding can be analyzed by reversible
thermodynamics,
which is simpler to interpret. However, virtually all cellular proteins
denature irreversibly at
neutral pH and at the very high concentration present inside cells, which,
indicates that
understanding irreversible unfolding is important (Lepock, J.R., Frey, H.E.
and Ritchie, K.P.
(1993). Protein Unfolding in Intact Hepatocytes and Isolated Cellular
Organelles During Heat
Shock. J Cell Biol. 122, 1267-1276). This irreversibility is mainly a
consequence of the
irreversible aggregation that occurs after a reversible unfolding making the
whole unfolding an
irreversible process.
22
CA 02479104 2004-08-26
This three state, reversible-irreversible process can be modeled as:
1
where H represents the native state, U the intermediate partially unfolded
state, A represents
the aggregated state, k2 (or kagg) is the rate constant of aggregation and n
is representative of the
degree of cooperativity during aggregation. Once the protein unfolds,
hydrophobic residues axe
exposed, aggregation occurs and the protein becomes kinetically locked in the
unfolded state.
The unfolding process is described by kl (or k") and can be evaluated by bis
ANS fluorescence
or other methods like circulax dichroism or differential scanning calorimetry.
Light scattering
is an example of a method that can be used to measure the process regulated by
k2. This three
state model can often be simplified to a two state model of the form:
kapp
NOD 2
wherein N represents native state and D represents the denatured aggregated
state and kapP is an
apparent rate constant. This approximation can be made if k2 is of the same
order of magnitude
as ki.
The calculated rate constants of unfolding k" and aggregation kagg are
temperature
dependent following the Arrhenius law:
k=Ae ~T 3
and the logarithmic form is:
In k= ln(A)-Eal(RT) 4
where A is the Arrhenius pre-exponential factor, Ea the activation energy, R
is the
universal gas constant and T is the temperature in kelvin.
With the method and apparatus of the invention, rate constants (k) at
different
temperatures can be measured, which allows calculating the Ea of unfolding and
aggregation respectively. When proteins are in the presence of molecules that
interact
with them, from the variations in the values of Ea's the interaction energy
between the
protein and the interacting molecule can be deduced.
In one embodiment, the invention provides methods and apparatus for
identifying a
condition that changes the stability of a biological molecule relative to its
stability in a
reference condition. Such methods involve characterizing aggregation and/or
unfolding of a
23
CA 02479104 2004-08-26
biological sample in one or more test conditions and/or one or more reference
conditions and
looking for changes in a properly of aggregation and/or unfolding of the
biological sample.
Accordingly, the invention provides methods and apparatus for identifying
conditions that
increase the stability of a biological molecule and conditions that decrease
the stability of a
biological molecule.
The biological molecule can be, e.g.; a peptide, polypeptide, protein
(monomeric or
multimeric); a nucleic acid, e.g., RNA, single, double, or triple stranded
DNA, lipids, sugars,
and combinations thereof. For example, the methods and apparatus of the
invention permit the
identification of conditions that stabilize a protein.
The condition can be, for example, a biochemical condition, pressure, electric
current,
time, concentration of the biological molecule, and presence of a test
compound. A
biochemical condition can be, far example, one relating to pH, ionic strength,
salt
concentration, oxidizing agent, reducing agent, detergent, glycerol, metal
ions, salt, cofactor
concentration, ligand concentration and coenzyme concentration. For example,
the methods
and apparatus of the invention permit the identification of salt
concentrations that affect the
unfolding and/or aggregation kinetics and dynamics of a protein.
In one embodiment, the biochemical condition is a solution comprising a
compound,
such as a compound of a small molecule library, and the methods and apparatus
of the
invention permit the identification of one or more compounds that bind to the
biological
molecule and thereby stabilize it. In an illustrative embodiment, the
biological molecule is a
protein and the method comprises identifying a ligand of the protein. Such a
method can
comprise incubating the protein with different solutions, each comprising a
different potential
ligand and/or concentration of potential ligand. The methods of the invention
compare the
relative affinity of the potential ligands to a given target, or relative
affinity of a given ligand to
a given protein in different test conditions.
In one embodiment, the method comprises (a) providing a composition comprising
a
biological molecule in a test condition; (b) bringing the temperature of the
composition to an
end temperature; and (c) determining the extent of aggregation and/or
unfolding of the
biological molecule as a function of time. In one embodiment, the end
temperature of step (b)
may be lower than the aggregation temperature of the biological molecule in a
reference
condition. The reference condition can be a condition in which the biological
molecule is
24
CA 02479104 2004-08-26
known to be relatively stable. The aggregation temperature of the biological
molecule in the
reference condition can be determined, e.g., by measuring the extent of
aggregation as a
function of increasing temperature, as known in the art and further described
herein The end
temperature can be selected based on the desired rate of experimentation and
the objectives of
the study. The temperature can be about 1°C, 2°C, 5°C,
10°C, 15°C, 20°C, 25°C, or more
degrees lower or higher than the transition aggregation temperature.
In an illustrative embodiment, a protein is provided in a solution A. Under
these
conditions, it is known, or was determined, that the aggregation temperature
is about 50°C.
The protein in mixed in a solution B, the solution with the protein is heated
up to 45°C, and the
extent of aggregation is measured.
In one embodiment, the compositions may be heated to their end temperature
through a
heat shock or by jumping the temperature, i.e., by bringing the temperature to
the end
temperature as fast as possible.
In certain embodiments, the extent of aggregation and/or unfolding is measured
from a
time point preceding the heat shock to the end temperature and continued after
the time point at
which the temperature of the composition attains the end temperature. In other
embodiments,
measurements of the extent of aggregation and/or unfolding are initiated about
1 second, 5
seconds, 10 seconds, 30 seconds, 45 seconds, 1 minute, 2 minutes, 3 minutes, 4
minutes, 5
minutes, 6 minutes, 7 minutes, 8 minutes, 9 minutes, or 10 minutes after the
heat shock is
initiated. In one embodiment, a plurality of essentially identical
compositions are heat shocked
in different tubes or wells of microwell plates and the measurements of extent
of aggregation
are initiated at different time points after the heat shock is initiated. This
can be done, e.g., by
heat shocking the tubes or microwell plates at different points, and starting
the measurements
of the different compositions essentially at the same time. Alternatively, the
compositions are
heat shocked at the same time, and the measurements of each composition are
started after
different intervals of time.
A heat shock can be grade by different methods. For example, a tube at room
temperature may be incubated in an environment that is at the desired end
temperature under
heat conducting conditions. Alternatively, a test solution at the desired end
temperature is
added to the protein that is in a minimum volume at a different temperature.
CA 02479104 2004-08-26
In another embodiment, the temperature is raised gradually, and the extent of
aggregation is measured as a function of time. Measurements may be initiated
at the time the
temperature starts to rise, or at a time prior to, or after, the time the
temperat~~re st~.rts to xise.
The increase in temperature may be conducted, for example, at about one degree
Celsius per
minute.
In another embodiment, unfolding and aggregation are study in parallel as a
function of
time (kinetics) or temperature. To study kinetics, the compositions may be
heated to their final
temperature very rapidly. The extent of aggregation and unfolding are
simultaneously
measured as a function of time. Aggregation and unfolding curves as a function
of time can
then be generated. The aggregation kinetics of the said compositions can be
characterized by
one or more parameters such as rate of unfolding (k,~) and aggregation (kag~).
To study
aggregation in parallel with unfolding as a function of temperature, thermal
unfolding and
aggregation curves can be generated by heating the compositions gradually, for
example, one
degree Celsius per minute, with measurements of extent of aggregation and/or
unfolding
initiated at the time the temperature starts to rise of at a time prior to or
after the time the
temperature starts to rise. The total process of unfolding and aggregation can
be characterized
by the transition temperatures of the two curves, respectively Tm and Tagg.
The extent of unfolding of a biological molecule can be determined according
to a
variety of techniques, such as calorimetry, circular dichroisrn, fluorescence
emission (e.g.,
using intrinsic fluorescence or a fluorescence reporter molecule),
fluorescence energy transfer,
absorbance of ultraviolet or visible light, changes in the polarization
properties of light,
changes in the polarization properties of fluorescence emission, changes in
turbidity, changes
in enzyme activity, chaperone binding, or antibody binding (e.g., using
different antibodies
capable of recognizing the native or denatured state of the biological
molecule). In an
exemplary embodiment, fluorescence emission is used to determine the extent of
reversible
unfolding.
The fluorescence emission spectra of many fluorophores are sensitive to the
polarity of
their surrounding environment and therefore are effective probes of phase
transitions for
proteins (i.e., from the native to the unfolded phase). The most studied
example of these
environment dependent fluorophores is 8-anilinonaphthalene-1-sulfonate (1,8-
ANS), for which
it has been observed that the emission spectrum shifts to shorter wavelengths
(blue shifts) as
26
CA 02479104 2004-08-26
the solvent polarity decreases. These blue shifts are usually accompanied by
an increase in the
fluorescence quantum yield of the fluorophore. In the case of ANS, the quantum
yield is 0.002
in water and increases to 0.4 when ANS is bound to serum albumin. ANS may be
excited with
a wavelength near 360 nm and produces a fluorescence emission that may be
measured at 460
nm.
Fluorescence probe molecules are fluorophores that are capable of binding to
an
unfolded or denatured receptor and, after excitement by light of a defined
wavelength, emitting
fluorescent energy, such as UV light. Any fluorophore capable of binding to a
denatured
polypeptide may be used in accordance with the invention, including, for
example, thioinosine,
N-ethenoadenosine, formycin, dansyl derivatives, fluorescein derivatives, 6-
propionyl-2-
(dimethylamino)-napthalene (PRODAN), 2-anilinonaphtalene, and N-arylamino-
naphthalene
sulfonate derivatives such as 1-anilinonaphtalene-8-sulfonate (1,8-ANS), 2-
anilinonaphthalene-
6-sulfonate (2,6-ANS), 2-aminonaphthalene6-sulfonate, N,N-dimethyl-2-
aminonaphthalene-6-
sulfonate, N-phenyl-2-aminonaphthalene, N-cyclohexyl-2-aminonaphthalene-6-
sulfonate, N-
phenyl-2-aminonaphthalene-6-sulfonate, N-phenyl-N-methyl-2-aminonaph-thalene-6-
sulfonate, N-(o-toluyl)-2-aminonaphthalene-6-sulfonate, N-(m-toluyl)-2-
aminonaphthalene-6-
sulfonate, N-(p-toluyl)-2-aminonaphthalene-6-sulfonate, 2-(p-toluidinyl)-
naphthalene-6-
sulfonic acid (2,6-TNS), 4-(dicyanovinyl) julolidine (DCVJ), 6-dodecanoyl-2-
dimethylaminonaphthalene (LAURDAN), 6-hexadecanoyl-2-(((2-trimethylammonium-
ethyl)methyl)amino) naphthalenechlo ride(PATMAN), vile red, N-phenyl-1-
naphthylamine,
1,1-dicyano-2-[6-(dimethylamino) naphthalen-2-yl]propene (DDNP), 4,4'-
dianilino-1,1'-
binaphthyl-5,5'-disulfonic acid (bis-ANS), and DAPOXYLTM derivatives
(Molecular Probes,
Eugene, OR).
Bis ANS is a fluorescent probe that does not bind to most native proteins but
binds to
hydrophobic surfaces of partially denatured proteins with a corresponding
increase in
fluorescence (Cardamone, M. and Puri, N.K. (1992). Spectrofluorometric
assessment of the
surface hydrophobicity of proteins. Biochem. J. 282, 589-593, Semisotnov,
G.V., Rodionova,
N.A., Razgulyaev, O.L., Uvershy, V.N., Gripas, A.F. and Gilmanshin, R.I. Study
of the molten
globule intermediate state in protein folding by a hydrophobic fluorescent
probe.
(Biopolymers, 31, 119-128).
27
CA 02479104 2004-08-26
When using a fluorophore, the fluorophore is added to the composition
comprising the
biological molecule prior to initiating the denaturing process, e.g., prior to
a change in
temperature or addition of a chemical denaturant. For example, bis-ANS can be
added to a
composition comprising a biological molecule, the composition is mixed, the
composition is
subjected to a heat shock to the end temperature, and the fluorescence of the
composition is
measured over time.
Any of the variety of fluorescence emission imaging systems known to those
skilled in
the art may be used to monitor the extent of reversible unfolding of a
biological molecule. For
example, CytoFluor II fluorescence microplate reader (PerSeptive Biosystems,
Framingham,
MA) is an example of a fluorescence imager that may be used in accordance with
the
invention. A Charge Coupled Device Camera ("CCD camera") may also be used to
measure
fluorescence emission.
Intrinsic tryptophan (Trp) fluorescence is an alternative method for
determining the
extent of unfolding of a polypeptide. The intrinsic Trp residues of a
polypeptide may be
excited with light near 280 nm resulting in a fluorescence emission near 3S0
nm. Such Trp
fluorescence excitation may be achieved using a Xenon-Arc lamp, such as the
Biolumin 960
(Molecular Dynamics).
If the biological molecule is a nucleic acid, the extent of unfolding may be
determined
using light spectrophotometry. Unfolding is measured by determining the change
in
hyperchromicity, which is the increase in absorption of light by
polynucleotide solutions due to
a loss of ordered structure, for example, in response to an increase in
temperature.
Fluorescence emission may also be used to measure the extent of unfolding of a
polynucleotide. The nucleic acid may be labeled with ethidium bromide or a
fluorophore and
fluorescence spectrometry may be used to monitor the level of fluorescence
emission.
Fluorescence resonance energy transfer may also be used in accordance with the
invention. In
this approach, the transfer of fluorescent energy, from a donor fluorophore on
one strand of an
oligonucleotide, to an acceptor :fluorophore on the other strand, is measured
by determining the
emission of the acceptor fluorophore. Unfolding diminishes or prevents the
transfer of
fluorescent energy.
The extent of aggregation of a biological molecule can be measured
spectrophotometrically, e.g., through measurements of light scattering or
turbidity, or by
28
CA 02479104 2004-08-26
electron microscopy, velocity sedimentation, centrifugation, or filtration. In
an exemplary
embodiment, the extent of aggregation is measured by determining the optical
density ("OD")
of a sample using ultraviolet or visible light. A higher optical density
denotes larger particles
and thus a greater extent of aggregation of the biological molecule in the
sample.
In an exemplary embodiment, the formation of aggregates is followed by static
light
scattering. This is possible because the intensity of the light scattered is
proportional to the
size of the particles in suspension. The size of these particles increased
from a few nm, which
is the normal size of a protein in solution to sizes on the order of ~.m when
the proteins have
aggregated.
In exemplary embodiments, the light source for light scattering may be one or
more of
the following: a laser (e.g., a monochromatic, intense, well defined beam of
light), a light
emitting diode (LED), a cluster of LEDs, a white light source, a monochromatic
light source,
an incandescent light source, a Xenon-arc lamp, a tungsten-halogen lamp, an
ultraviolet light
source, a luminescent light source, and/or a low intensity light source with
an intensity in a
range of 1.5 to 2.0 ~W/mm2.
Depending on the wavelength of the incident light source and the dimensions of
the
particle, this scattered light can show very characteristic intensity
patterns. Small molecules
scatter light equally in all directions. On the other hand, particles at least
as big as the
wavelength of the incident light scatter more in certain directions than
others.
The Raleigh ratio R(~)=MP(8)K*c describes the absolute intensity scattered at
an angle
8 in excess of the light scattered by the pure solvent. M is the molecular
mass of the scattering
particle and proportional to its size, c is the concentration and P(8) is the
form factor (ratio of
scattered intensity at angle 0 to intensity at angle 0), K is an optical
constant and contains the
refractive index of the solvent, Avogadro's number, the wavelength of the
incident light, and
the specific refractive index increment of the sample molecules. For simple
mathematical
reasons the maximum intensity of the light scattered can be measured at a
90° angle.
Additional information regarding spectrophotornetry and spectrofluorometry can
be
found, e.g., in Bashford, C. L. et al.; Spectrophotometry and
Spectrofluorometry: A Practical
Approach, pp. 91-114, IRL Press Ltd. (1987); Bell, J. E.; Spectroscopy in
Biochemistry, Vol. I,
pp. 155-194, CRC Press (1981); Brand, L, et al., Ann. Rev. Biochem. 41:843
(1972); Ozaki, H.
et al., Nucleic Acids Res. 20:5205-5214 ( 1992); Clegg, RIM. et al., Proc.
Natl. Acad. Sci.
29
CA 02479104 2004-08-26
U.S.A. 90:2994-2998 (1993); Clegg, R. M. et al., Biochemistry 31:4846-4856
(1993); Lee, M.
et al., J. Med. Chem. 36:863-870 (1993); U.S. Pat. Nos. 6,303,322; 5,858,277;
6,270,954;
5,854,204.
A person of skill in the art will recognize that where appropriate, unfolding
and
aggregation can also be measured by other methods, such as non-spectroscopic
methods. For
example, methods for detecting unfolding of biological molecules include
methods which detect
the presence of folded and/or unfolded biological molecules by virtue of
binding of another
molecule to the folded or unfolded biological molecules. Exemplary techniques
include the use
of antibodies which specifically recognize epitopes that are exposed only in a
protein when it is
unfolded or alternatively, which are exposed only in a protein when it is
folded. Such techniques
are further described, e.g., in U.S. patent no. 5,679,582.
The measurements of extent of aggregation and/or unfolding can be conducted
about
every 10 seconds, 20 seconds, 30 seconds, 40 seconds, 50 seconds, 1 minute, 2
minutes, 3
minutes, 4 minutes, S minutes, or more. As little as one or two measurements
may be
sufficient in certain embodiments. In other embodiments, 5, 10, 15, 20, 30, 50
or more
measurements are conducted over time on one or more samples. Measurements may
be
conducted until the maximum aggregation is attained, e.g., when the
fluorescence or light
scattering has attained a maximum and is not further increased with time.
Measurements can be conducted in an automated fashion. For example, one or a
plurality compositions can be incubated in wells of a microwell plate; the
plate is heated; the
plate is then illuminated in an automated fashion at particular time
intervals; and fluorescence
emission or light scattering is measured in an automated fashion over time.
Measurements can
be taken from the top of the plate.
The results of the measurements can then be collected, e.g., in an automated
fashion.
The results can be transmitted to a computer readable medium or a computer.
Analysis of the
results can be conducted on a computer. The computer may further comprise
results obtained
from other assays and may contain reference data.
In embodiments in which fluorescence or light scattering is measured, the
results can be
plotted as fluorescence or light scattering as a function of time ("time scale
method"; see, e.g.,
Fig. 6). The curve, or one or more points thereof, may then be compared to
that of the
biological molecule in different conditions, e.g., in a different salt
concentration. In one
CA 02479104 2004-08-26
embodiment, the curve, or one or more points thereof, is compared with that of
the biological
compound in a reference condition.
In other embodiments, the rate constant of unfolding (k~,) or aggregatior_
(k~,gg) are
determined from the results obtained. k" and kagg can be obtained from the
fitted exponential
growth portion of the curves. A lower k" or kagg Of a biological molecule in a
first condition
relative to that of the biological molecule in a second condition indicates
that the biological
molecule is more stable in the first condition relative to the second
condition.
In another embodiment, the energy of activation (Ea) can be deduced based on
the
Arrhenius law (see above). Fox example, the interaction energy between a
protein and an
interacting molecule can be deduced by determining the change in the value of
Ea's when the
protein is in the presence of the interacting molecule. Other variables that
can be deduced
include the maximum scattered intensity (ImaX). I~ can be used to evaluate
conditions that
induce or prevent aggregation with or without inducing destabilization.
The amount of native protein remaining after exposure to some temperature (T)
for a time (t) is:
N(t)~Noe xap~t 5
where No is the amount of native protein at time t=0.
Making N(t)= No/2 and obtaining ka~, from the experimental fit, the time
required to
obtain 50% aggregation at the temperature T can be calculated as a measure of
stability.
A computer with appropriate algorithm can derive some or all of these
variables for each
biological molecule at each temperature in each condition. A comparison
readily indicates which
conditions provide the most stabilizing effect on the biological molecule.
In an exemplary embodiment, both the extent of unfolding and aggregation are
determined. Measuring both factors may be relevant to complete understanding
of aggregation
kinetics and dynamics, particularly in cases where macromolecules unfold prior
to proceeding to
aggregation. Measuring both parameters is preferably conducted essentially
simultaneously. For
example, a tube or microwell plate may be irradiated alternatively with a UV
light to detect the
extent of unfolding and with another light source to detect light scattering.
In this embodiment;
the fluorescence emission and light scattered are also measured essentially
simultaneously, e.g.,
with a CCD camera. Thus, in such an embodiment, the extent of unfolding and
the extent of
aggregation are essentially determined simultaneously.
31
CA 02479104 2004-08-26
In another embodiment, two identical tubes, or microwell plates, or portions
of microwell
plates, are used. One tube, or microwell plate, or portion thereof, is
illuminated with UV light for
detecting the extent of unfolding, and the other tube, or microwell plate, or
portion thereof, is
illuminated with a light source for detecting the extent of aggregation:
Results can be measured
and processed simultaneously.
After having obtained the measurement of extent of aggregation and/or
unfolding at one
temperature, the temperature can be increased, e.g., jumped or gradually
increased, to another
temperature, e.g., a higher temperature. For example, once maximum levels of
aggregation
andlor unfolding are obtained at a particular temperature, the temperature may
be increased by
1 °C, 2°C, 3°C, 5°C, 7°C, 10°C,
15°C, 20°C, 25°C, or more degrees. Measurements can be
continued when the temperature is jumped or measurements can be interrupted
during the jump
in temperature, and reiterated a certain time after the beginning of the
temperature jump.
In one embodiment, the extent of aggregation and/or unfolding are determined
at several
temperatures essentially simultaneously. For example two essentially identical
compositions
comprising a biological molecule can be exposed to different temperatures. The
essentially
identical compositions can be in different tubes or microwell plates. In one
embodiment, the
compositions are in different wells of a microwell plate. For example, one or
more individual
wells (including e.g., a row of wells or entire plate) can be exposed to one
temperature and
another one or more wells can be exposed to another temperature. Measurements
of the extent of
aggregation and/or unfolding in the tubes or wells exposed to the different
temperatures can be
conducted simultaneously overtime.
In another embodiment, a gradient of temperature may be created within an
individual
tube or plate. In an illustrative embodiment, one end of a plate containing a
composition
comprising a biological molecule is at a first temperature and the other end
of the plate is at a
second temperature. A gradient of temperature may be formed between these two
temperatures.
Measurements can be taken at both ends of the plate as well as at locations
between the two ends
for measuring effects at an intermediate temperature.
The methods and apparatus of the invention are easily adaptable to high
throughput
screenings. In an illustrative embodiment, different conditions are tested
simultaneously for one
biological molecule, e.g., in a multiwell plate. All the conditions can be
tested at the same end
temperature. All the conditions can also be tested at a plurality of different
end temperatures.
32
CA 02479104 2004-08-26
For example one 384 well plate contains the same biological molecule in 384
different
conditions, i.e., one condition per well, and is incubated at a first
temperature. A second,
essentially identical plate can be incubated at a different temperature. The
invention also
provides methods for simultaneously evaluating different biological molecules
in one or more
conditions. For example, an assay can comprise evaluating a first protein and
proteins which
differ from the first protein in one or more amino acid differences. In an
illustrative embodiment,
a microwell plate has columns of different biological molecules and rows of
different conditions.
Multiwell plates that can be used exist in numerous formats, e.g., 24 well
plates (4 x 6
array), 96 well plates (8 x 12 arrays), 384 well plates (16 x 24 array), 864
well plates (24 x 36
array), and 1536 well plates (32 x 48 array). Accordingly, the invention
provides methods for
simultaneously evaluating the stability (by the extent of unfolding and/or
aggregation) of at least
about S, 10, 25, 50, 100, 250, 500, 1000, 2500, 5000, 10,000 or more
conditions and/or biological
molecules.
In a particular embodiment, the invention provides methods for identifying
ligands of
biological molecules, such as proteins: A method may comprise providing a
composition
comprising a target protein and a test ligand. In an exemplary embodiment, the
method
comprises heat shocking the temperature of the composition, optionally, to an
end temperature
that is lower than the aggregation temperature of the protein without the test
ligand. The method
may fiuu-ther comprise subjecting the composition to incident illumination
which will result in
scatted light proportional to accumulation of aggregates prior to, at the same
time, and/or after
beginning the heat shock. Scattered Light is detected over time until it
reaches a maximum. The
scattered light intensities can then be plotted as a function of time, and the
curve can be
compared to a curve of the protein that was not incubated with the test
ligand. The rate of
aggregation (ka~) can also be derived from the curve, and compared to the ka~g
of the protein in
the absence of the test ligand. A lower kag~ in the presence of the test
ligand indicates that the
ligand binds to the protein. Using the same method, a library of potential
ligands can readily be
tested. One or more test ligands can be incubated with a target protein and
the measurements of
scattered light can be conducted simultaneously.
In other embodiments, the extent of aggregation and/or unfolding may be
measured as a
function of varying temperature instead of, or in addition to, a function of
time. For example, the
sample may be illuminated with a light source and the light scattering is
measured (e.g., with a
33
CA 02479104 2004-08-26
CCD camera) as the temperature of the compositions are varied (either
increased or decreased) at
a controlled rate. In various embodiments, the temperature may be increased or
decreased in a
continuous or stepwise fashion. Additionally, when using a multiplicity of
samples, the
temperature of essentially all samples may be controlled in bulk or the
temperature of one or
more samples may be controlled separately from other samples. In one
embodiment, the light
source may be a laser. In yet other embodiments, both the extent of unfolding
and the extent of
aggregation are determined to identify ligands binding to a target protein.
In other embodiments, the methods can be used to identify compounds that
modulate the
interaction between a biological molecule and a ligand. For example, the
method may comprise
providing a composition comprising a protein, a ligand, and a test compound.
The composition
is heat shocked, optionally, to a temperature that is lower than the
aggregation temperature of the
protein-ligand complex, and the fluorescence emission or light scattering is
measured as a
function of time. The k" or lcagg of the composition with the test compound
can be determined. A
lower k" or kagg m the presence of the test compound relative to the absence
of the test compound
indicates that the test compound inhibits the interaction between the protein
and the ligand.
The invention also provides methods for identifying a condition in which a
biological
molecule has a difFerent stability relative to its stability in a reference
condition comprising (a)
providing a composition comprising a biological molecule in a test condition;
(b) increasing the
temperature of the composition over time; and (c) determining the extent of
unfolding and
aggregation of the biological molecule in an essentially simultaneous manner
during the increase
in temperature. In one embodiment, identical compositions are present in
different tubes or
microwell plates and one tube or microwell plate is monitored for the extent
of unfolding,
whereas the other tube or microwell plate is monitored for the extent of
aggregation. In an
exemplary embodiment, the extent of unfolding and aggregation are determined
on the same
sample, in an alternative fashion. For example, a sample is alternatively
illuminated with a UV
light (for determining the extent of unfolding) and a light source for light
scattering (for
determining the extent of aggregation) and the fluorescence emission and light
scattering are
detected alternatively with different detectors, or simultaneously with the
same detector, e.g., a
CCD camera. The results can then be analyzed. In one embodiment, the results
are plotted as
the amount of fluorescence and/or light scattered as a function of temperature
("temperature scale
method;" see, e.g., Fig. 5). The curves or at least some points thereof can be
compared to the
34
CA 02479104 2004-08-26
curve or points thereof of the biological molecule in a reference condition,
to determine whether
the test condition stabilized or destabilized the biological molecule relative
to the reference
condition. In another embodiment, the Tm (melting temperature) and T~~ f"a
gregation
temperature") are derived from the sigmoid portion of the curves. A higher Tm
and Tof a
biological molecule in a test condition relative to the Tm and Tai,
relatively, of the biological
molecule in a reference condition indicates that the test condition stabilizes
the biological
molecule. The maximum fluorescence (If) and scattered intensity (Ian) can also
be obtained for
each protein. Ia~ is an indication of the magnitude of the aggregation. Imp is
a measure to
evaluate conditions that may induce or reduce aggregation with or without
protein
destabilization.
This particular embodiment can also be used to screen a library of compounds
to identify
one or more compounds that bind to a target biological molecule. Similarly,
the method can be
adapted to screen for compound which inhibits the interaction between a
biological molecule and
a ligand.
In an exemplary embodiment, the invention provides methods for identifying
conditions
that facilitate refolding of biological molecules, such as proteins. A method
may comprise
providing a composition comprising at least one denatured protein or
polypeptide. The
composition is then incubated in the presence of a variety of test conditions
(e.g., different buffer
conditions, temperatures, etc.) and the amount of aggregation in the sample is
determined using
light scattering. A change in the amount of light scattering in certain
conditions as compared to
other conditions is indicative of conditions that affect the state of folding
of the protein in the
sample. For example, a decrease in the intensity of light scattering is
typically indicative of
decreased aggregation and increased protein folding.
In one embodiment, the method may comprise first exposing a protein to
denaturing
conditions and then transferring the denatured protein to the ane or more test
conditions to
analyze refolding. Denaturation may be carried out by exposing the protein to
any type of
denaturing condition, such as, for example, physical conditions like
temperature, pressure,
magnetic field, etc., and/or chemical conditions like salt, pH, ligands, etc.
The denatured protein
may be purified as necessary prior to exposure to the test refolding
conditions in order to remove
the denaturing conditions or other undesired molecules in the sample.
CA 02479104 2004-08-26
In an exemplary embodiment, the method comprises first incubating the
denatured
protein in the test refolding conditions far a period of time to permit
refolding to occur. .For
example, the protein may be incubated in the test condition for l , 5, 10, 30,
or 60 seconds, or 1,
2, 5, 10, 15, 30, 60 minutes, or 1, 2, 5, 10, 24 hours, or more, prior to
analysis of the folded state
S of the protein. In certain embodiments, analysis of the folded state of the
protein comprises
changing the temperature of the refolded compositions, optionally, to an end
temperature that is
lower than the aggregation temperature of the protein. The method may further
comprise
subjecting the compositions to incident illumination which will result in
scatted light proportional
to accumulation of aggregates prior to, at the same time, and/or after
beginning the heat shock.
Scattered light is detected over time until it reaches a maximum. The
scattered light intensities
can then he plotted as a function of time andlor temperature, and the curve
can be compared to a
curve of the protein in a reference condition. The rate of aggregation (kagg)
can also be derived
from the curve, and compared to the lcagg of the protein in a reference
condition.
In one embodiment, a plurality of potential test conditions may be used to
analyze the
folding of one or more proteins. In an exemplary embodiment, the invention
provides high-
throughput methods for analyzing refolding andlor identifying conditions that
facilitate refolding
of a protein. In one embodiment, the refolding of at least one protein may be
analyzed in the
presence of a variety of test conditions. Alternatively, the refolding of a
plurality of proteins may
be analyzed in the presence of at least one test condition. In yet another
embodiment, the
refolding of a plurality of proteins may be analyzed in the presence of a
plurality of test
conditions. In exemplary embodiments, a plurality of samples may be contained
in a mufti-well
plate, such as, for example, a plate having 96, 384, 1536 wells, or more.
In one embodiment, the methods and apparatus provided herein may be used to
identify
conditions that facilitate refolding of proteins in inclusion bodies.
In yet another embodiment, a database is provided the comprises, among other
things,
information about one or more protein sequences and characteristics of
refolding of the protein
sequences in the presence of at least two test conditions. In yet another
embodiment, the
database comprises a plurality of protein sequences and information about the
folded state of the
proteins in a plurality of test conditions. Such a database may be useful to
develop rules about
conditions that facilitate refolding of proteins in general, conditions that
facilitate refolding of
36
CA 02479104 2004-08-26
proteins sharing certain sequence characteristics, and correlations between
protein sequence and
conditions that facilitate protein refolding.
In an exemplary embodiment, the invention provides a method for identifying
conditions
that facilitate refolding of a protein comprising (a) exposing a biological
sample comprising ax
least one denatured protein to a plurality of test conditions, (b) incubating
the sample to permit
refolding of the denatured proteins, (c) exposing the sample to one or more
light scattering
sources, (d) determining the amount of light scattering by said plurality of
biological samples
upon exposure to said one or more light scattering sources, (e) increasing the
temperature of the
sample comprising the refolded proteins in a controlled manner by a pre-
determined level, and (~
repeating steps (c)-(e) thereby characterizing the aggregation of the protein
and identifying
conditions that facilitate refolding of the protein.
In another embodiment, the invention provides methods for identifying a
modulator of
aggregation of a biological molecule, such as a protein. In an exemplary
embodiment, the
invention provides methods for identifying compounds that inhibit aggregation
of a protein. A
method may comprise exposing one or more biological samples to denaturing
conditions in the
presence of one or more test compounds, wherein each biological sample
comprises at least one
biological molecule. The amount of aggregation of the biological molecule in
the sample is then
determined using light scattering. A change in the amount of light scattering
by the biological
molecule in the presence of a test compound as compared to the amount of light
scattering in the
absence of the test compound is indicative of a modulator of protein
aggregation. For example, a
decrease in the intensity of light scattering in the presence of the test
compound is indicative of a
decreased aggregation and thus permits identification ~f compounds that
inhibit protein
aggregation.
In certain embodiments, methods for analyzing potential modulators of protein
aggregation may further comprise changing the temperature of the biological
sample, optionally,
to an end temperature that is lower than the aggregation temperature of the
protein. The method
may further comprise subjecting the biological sample to incident illumination
which will result
in scatted light proportional to accumulation of aggregates prior to, at the
same time, and/or after
beginning the heat shock. Scattered light is detected over time until it
reaches a maximum. The
scattered light intensities can then be plotted as a function of time and/or
temperature, and the
curve can be compared to a curve of the protein in a reference condition. The
rate of aggregation
37
CA 02479104 2004-08-26
(lcagg) can also be derived from the curve, and compared to the kagg of the
protein in a reference
condition.
In certain embodiments, denaturation may be carried out by exposing the
protein to ax~y
type of denaturing condition, such as, for example, physical conditions like
temperature,
pressure, magnetic field, etc., and/or chemical conditions like salt, pH,
ligands, etc.
In one embodiment, a plurality proteins may be exposed to one or more test
compounds
to identify modulators of protein aggregation. In another embodiment, at least
one protein may
be exposed to a plurality of test compounds to identify modulators of protein
aggregation. In yet
another embodiment, a plurality of proteins may be exposed to a plurality of
test compounds to
identify modulators of protein aggregation. In exemplary embodiment, one or
more proteins
may be exposed to a library of test compounds in accordance with the methods
described herein.
In another embodiment, the invention provides high-throughput methods for
analyzing and/or
identifying compounds that modulate protein aggregation. In exemplary
embodiments, a
plurality of samples may be contained in a mufti-well plate, such as, for
example, a plate having
96, 384, 1536 wells, or more.
In an exemplary embodiment, the methods and apparatus described herein may be
used
to identify compounds that modulate aggregation of biological molecules that
are involved in
disease states. Aggregation of a variety of proteins has been shown to be
correlated with disease
states, including, for example, prions, ~-amyloid, superoxide dismutase,
thioterytin, etc. See e.g.,
Orrell, Neuromuscul Disord 10: 63-68 (2000), Bruening, J. Neurochem 72: 693-
699 (1999),
Hammarstrom, Biochemistry 40: 11453-11459 (2001), Ktabunde, Nat. Struct. Biol.
7: 312-321
(2000), Petrassi, J. Am. Chem. Soc. 122: 2178-2192 (2000), Stathopulos, Proc.
Natl. Acad. Sci.
USA 100: 7021-6 (2003). Accordingly, the methods and compositions described
herein may be
useful for identifying compounds that may be useful for treating and/or
preventing diseases
associated with protein aggregation, such as, for example, neurodegenerative
disorders and/or
diseases.
In an exemplary embodiment, the invention provides a method for identifying
compounds that inhibit protein aggregation comprising (a) exposing one or more
biological
samples to denaturing conditions in the presence of at least one test
compound, wherein the
biological sample comprises at least one protein, (b) exposing the sample to
one or more light
scattering sources, (c) determining the amount of light scattering by said
plurality of biological
38
CA 02479104 2004-08-26
samples upon exposure to said one or more light scattering sources, (d)
increasing the
temperature of the sample in a controlled manner by a pre-determined level,
and (e) repeating
steps (b)-(d) thereby characterizing the aggregation of the protein and
identifying co~.pounds
that inhibit protein aggregation.
Thus, the invention provides useful methods and apparatus for at least the
following: (i)
to conduct biophysical characterization of protein by generating aggregation
and/or unfolding
curves as a function of time and/or temperature; (ii) to conduct biophysical
characterization of a
protein by generating both unfolding and aggregation curves as a function of
time andlor
temperature; (iii) to characterize protein dynamics by defining precisely
numerical measures such
as the aggregation temperature (Tags) of aggregation, rate of aggregation
(kagg), melting
temperature (Tm) and rate of unfolding (ku) as characteristic biophysical
properties of individual
proteins; (iv) to identify substances or conditions that affect stability of
any individual protein by
shifting, by virtue of their presence, biophysical properties such as Tm,
Tags, k" and kagg; (v) to
identify substances or conditions that without affecting the stability of any
individual protein can
increase the size or number of protein aggregates; (vi) to identify substances
or conditions that
without affecting the stability of any individual protein can decrease the
size or number of
protein aggregates; (vii) to identify substances that prevent protein
aggregation and precipitation;
(viii) to identify substances or conditions that stimulate protein aggregation
and precipitation; (ix)
to measure rates of protein unfolding k" at different temperatures; (x) to
measure rates of protein
aggregation kagg at different temperatures; (xi) to measure the activation
energy of protein
unfolding Eaa; (xii) to measure the activation energy of protein aggregation
Eagga; (xiii) to
measure the interaction energy between a protein and a molecule that interacts
with it.
A person of skill in the art will recognize that denaturing conditions other
than heat can
be used according to the methods of the invention for identifying conditions
that stabilize or
destabilize a biological molecule. For example, a composition comprising a
biological molecule
in a test condition can be subjected to a denaturing agent, such as a
chaotropic agent (e.g., urea
and guanidium), and the extent of aggregation andlor unfolding determined as a
function of time
or temperature. In another embodiment, mechanical denaturation, such as, for
example,
sonication may be used in accordance with the methods and apparatus disclosed
herein. Such
assays will provide information on conditions which stabilize a biological
molecule with respect
to denaturing conditions other than heat.
39
CA 02479104 2004-08-26
Apparatus of the Invention
When light encounters particles in its path, the electric field of the
electromagnetic
radiation displaces the particles, causing them to oscillate around their
equilibrium positions.
The oscillating particles act as secondary sources, re-radiating or scattering
the incident energy.
In elastic scattering, the scattered light is at the same frequency as the
incident radiation. This
phenomenon is the most dominant form of scattering and includes Rayleigh and
Mie scattering.
Inelastic scattering is the phenomenon of the molecules emitting radiation at
their own
characteristic rotational and vibrational frequencies. This includes the
phenomenon of Raman
scattering.
The scattering from molecules arid very tiny particles (<1/10 wavelength) is'
predominantly Rayleigh scattering, which accounts for the blue color for a
clear sky (i-.e. the
blue end of the electromagnetic spectrum is of short wavelength). For particle
sizes larger than
a wavelength, Mie scattering becomes dominant, This phenomenon tends not to be
frequency
dependent resulting typically in white scattered light. Examples of Mie
scattering include the
white glare around the sun when a lot of particulate is present in the air, as
well as mist and
fog.
Fig. 4 shows typical patterns of Rayleigh and Mie scattering. The arrows in
each
pattern represent light scattered from a particle located at the origin of the
arrows, with the
length of an arrow representing the relative intensity of light scattered in
the direction of the
arrow. The incident direction of the light for each pattern is indicated by
arrow 202. Rayleigh
scattering, as indicated by arrows 204, tends to be weakest in the direction
perpendicular to the
incident radiation but is roughly uniform elsewhere. Mie scattering, as
indicated by arrows
206, tends to be strongest in the same direction as the incident light. As
particles become
larger, Mie scattering can result in a narrower dispersion in the direction of
the incident light,
as indicated by arrows 208. This narrowing may pose problems in measuring the
scattered
light.
If particle size (r) is much smaller than the incident light wavelength (~,)
the system
emits radiation as an electric dipole. As known in the art, the Rayleigh ratio
is defined as the
ratio of the scattered light intensity (~ to the incident light intensity (Io)
measured at a given
angle (6) and distance (r) from the scattering volume:
CA 02479104 2004-08-26
1/l0 =RB(1+cos28)lr2 6
Scattering intensity is dependent upon a number of particle and solvent
parameters including
particle polarizability (a), permittivity (~), density (p) and molecular
weight (l~:
RB =1/2(pNAI~ (~c~ l(~hq)),
where NA is Avogadro's number. The strong dependence on ?~ indicates much more
intensive
scattering at short wavelength, e.g.; ultraviolet (UV) and visible light in
contrast to near-
infrared (IR) and IR.
For practical measurements the following form of the Rayleigh equation is
used:
1 16~ZR2
R M + 2AZC 1 + 3~2 g sine 2 , 8
a
where C is the particle concentration in solution, Az is the 2~'d virial
coefficient, indicative of
solute-solvent interaction, Rg is the radius of gyration of the particle; I~
is an optical parameter
equal to 4~n2(drrldC)2/~,~N,~, where n is the solvent refractive index and
(dnldC) is the analyte
specific refractive index increment. The angular dependent portion of the
second term in
Equation 8 arises from interference effects due to multiple scattering from a
single particle.
For particles much smaller than the wavelength of the incident radiation, this
term goes to zero
and the angular dependence of the scattered light vanishes. Under these
conditions, the
absolute molecular weight is determined from the concentration dependence of
the Rayleigh
ratio and angular dependent data need not be obtained.
For larger particles, it is still the concentration dependence that leads to
molecular
weight, but the interference effects, as characterized by the second term of
Equation 8, must be
accounted for. At this point multi-angle instruments may become necessary. As
a rule of
thumb, the size cutoff for angle independent Rayleigh scattering is Rg <
~,I20. Typical
wavelengths used in standard static light scattering (SLS) equipment include
wavelengths in
the range of 600-800 nm, which provides a 30-40 nm upper limit for single-
angle particle
molecular weight determination. However, when considering protein studies, the
native,
folded forms of proteins can be within the above limit, while aggregations of
particles may
typically be larger. For particles sizes r greater than x,/20, incident
radiation can induce
multipole moments and the classical dipole approximation described above
becomes
4l
CA 02479104 2004-08-26
inappropriate. Alternatively, a technique of dynamic light scattering (DLS)
may be used to
obtain the "size" of the particle.
As is known in the art, in DLS one measures the time dependence of the light
scattered
from a very small region of solution. Typical time scales can range from
tenths of
microseconds to milliseconds. Fluctuations in the intensity of the scattered
light are related to
the rate of diffusion of molecules in and out of the region being studied
(Brownian motion).
The measured light signals contain contributi~ns from the slower movement of
large particles
as well as from the faster fluctuations of small size particles.
Signals can be analyzed in terms of an autocorrelation function or a fast
Fourier
transform (FFT) and plotted as maps of intensity vs. time, or intensity vs.
frequency,
respectively. Each mono-dispersed population (particles of a single size)
produces its own
unique correlation function- a single exponential decay:
C(2') =Ae2rr+B, 9
where A and B are instrumental constants. In turn, T = q2 D, where q is a
scattering vector,
q = (4~n1~.)Sin(9/2), 10
and D is a translational diffusion coefficient related to a hydrodynamic
radius, Rh of a spherical
particle and solvent viscosity, r~ by Stokes-Einstein equation:
D= kbTl(3~tr~T). 11
Mixtures of more than one size population produce the sums of exponentials.
The
hydrodynamic radius, Rh characterizes individual mono-dispersed fractions in
the solvent. In
general, proteins are not spherical, and their apparent hydrodynamic size
depends on their
shape (conformation) and mass. Therefore, the apparent hydrodynamic size can
differ
significantly from the true physical size, and may not be a reliable measure
of molecular mass.
DLS data is commonly presented as the fraction of particles versus their
diameters.
Sets of mono-dispersed standards (polystyrene particles of known size) are
typically used to
calibrate the scale of the sizes. The strength of DLS lies in its ability to
analyze samples
containing broad distributions of species of widely differing molecular
masses, (e.g. a native
protein and various sizes of aggregates), and to detect very small amounts of
the higher mass
species (< 0.01 % in many cases).
Light scattering from macroscopic particles can be treated in terms of Mie
scattering
theory. A scattering particle is idealized as a sphere with a particular
geometrical size. This
42
CA 02479104 2004-08-26
sphere redirects incident photons into new directions and so prevents the
forward on-axis
transmission of photons, thereby casting a shadow. The size of the scattering
shadow is called
the effective cross section (a~, cm2) and can be smaller or larger than the
geometrical size of the
scattering particle (~r2, cmz). The effective cross section is related to the
geometrical size by a
proportionality constant referred to as the scattering efficiency Q:, i.e., a
= Q (~t ~ ). The
scattering coefficient ,u (crri l) describes a medium containing many
scattering particles at a
concentration described as a volume density p (cm 3). The scattering
coefficient is the cross-
sectional area per unit volume of medium: ,u = p c~ .
Even though at the molecular level, as described above, incident light photons
are
scattered away from the direction of incidence. The resulting Mie scattering
pattern favors the
incident direction, as shown in Fig. 4. This has to do with constructive and
destructive
interference of scattered light. It is well known in optics that interference
is totally constructive
only in the forward direction. Mie theory provides a mathematically rigorous
algorithm to
describe this anisotropic angular distribution of light scattered by
macromolecules and to
calculate angular distribution as a function of wavelength and refractive
index of the spheres.
Typically, scattered light has a few maxima in angular distribution with
strong preference of
scattering into small angles relative to incident light, e.g., < 45°
from incidence, and preferably
in a range between 15° to 30°, as shown in Fig. 4. The
scattering is stronger if incident light is
polarized in the plane formed by the light source, the scattering object and
the detector: In
practical applications, Mie scattering theory is used to determine particle
size distributions in
the range of 50 nm to 2 mm, diagnostics and imaging of tissues, e.g. density
of lipid
membranes in the cell, size of nuclei, presence of collagen fibers and status
of hydration in the
tissue.
Whichever phenomenon of light scattering is being measured, the light source
may be
critical for obtaining adequate measurements. For Rayleigh and dynamic light
scattering, high
intensity and coherent incident illumination can provide greater sensitivity.
As a result, a laser
light may be chosen as the source of illumination in one embodiment. However,
the use of
individual lasers to illuminate individual samples can quickly become too
costly as the number
of samples increases. The complexity involved in designing such an instrument
can also be
daunting in optics and electronics, resulting in a solution with great
limitations in scalability.
For example, the size of individual laser emitters can limit how closely they
can packed, thus
43
CA 02479104 2004-08-26
limiting the sample density. In one embodiment; non-coherent incident
illumination with
intensities in a range as low as 1.5 - 2.0 wWlmm2 can provide sufficient
illumination to detect
Mie scattering and effectively monitor the aggregation of proteins. At such
low levels of
intensity, inexpensive non-coherent light sources, e.g., light emitting diodes
(LED's) and
filament-based light bulbs, can provide the necessary illumination.
In addition to the above considerations for the light source, the elimination
of crosstalk
can be an important consideration. Crosstalk can occur when reflected light
from one sample
can illuminate one or more adjacent samples. To inhibit crosstalk when
measuring scattered
light from a multiplicity of samples arranged in rows and columns next to each
other, adjacent
samples can be contained in wells that provide optical isolation between the
wells. In one
embodiment, microtiter plates containing arrays of wells can be used. The
walls between the
wells of the microtiter plates can be fabricated of an opaque plastic, e.g.,
black, to optically
isolate the wells from one another. The bottom of the wells can be clear, or
otherwise allow for
the passage of light, so as to allow illumination from the light source to
enter the wells through
the bottom of the wells.
In one embodiment, selective illumination of the samples can be used to
inhibit
crosstalk. For example, in an array of sample wells, a single well can be
illuminated, or wells
in a chosen pattern can be simultaneously illuminated. The illumination
pattern can be chosen
so as to ensure that the light scattered from one illuminated sample does not
add to the
illumination of other illuminated samples. Fig. 7 illustrates an exemplary
illumination pattern
170 for an array of sample wells 22, wherein illuminated sample wells 22a are
alternated with
un-illuminated samples wells 22b, only a portion of which are identified for
clarity. In effect,
the spacing of the illuminated wells can optically isolate the illuminated
wells. Once
measurements of the scattered light from the first illuminated well or pattern
of wells are taken,
another well or pattern of wells can be illuminated. By using selective
illumination, the costs
associated with providing sample wells fabricated with opaque or coated walls
can be avoided.
The light scattered from the illuminated samples can be measured by various
means
known to those skilled in the art, including machine vision, photomultipliers
and CCD's. In
one embodiment, a machine vision system with the appropriate optical
magnification can be
used such that every sample in the experiment can be captured. The image can
be captured
shortly after the samples (or a subset of which) are illuminated. The image
typically can be
44
CA 02479104 2004-08-26
stored for analysis after the experiment. The first stage of analysis
typically can involve image
processing to determine the intensity of the light scattered by each sample.
As previously described, uniform and predictable heating of the samples may be
required to obtain meaningful results. Preferably, the samples in the wells,
including the
control and test samples, can be heated to within 0.5° C of each other,
or can be heated in a
selected pattern with a known temperature distribution. Heating of the samples
can be
provided by a water bath, Pettier heating, and/or other means as may be known
in the art.
Referring now to Fig. 1, an isometric view schematically illustrating selected
components of an exemplary apparatus 10 is shown. It can be understood by
those of skill in
the art that different configurations of apparatus 10 can be contemplated,
which configuration
may not be limited by the description and illustrative figures of apparatus 10
provided herein.
Apparatus 10 can be configured to support one or more composition samples. In
the
exemplary embodiment of Fig. 1, the molecular samples can be contained in
microtiter trays 12
that may each have an array of sample wells for the moleeular samples, as
further described in
relation to Fig. 2.
A light source 14, e.g., a laser, an incandescent light source, a Xenon-arc
lamp, or a
tungsten-halogen lamp, can illuminate the samples. For the exemplary
embodiment illustrated
in Fig. 1, the light source can include an array, or cluster, of light
emitting diodes (LED's) that
can selectively illuminate the sample wells in trays 12. The illuminated
samples can scatter the
incoming light such that detector 16 can measure, or otherwise obtain a signal
corresponding to
the intensity of the light from source 14 scattered in the direction of
detector 16 by the
illuminated samples. A processor 18 can receive the signals from detector 16
and can
determine the intensity of scattered light from the illuminated samples. Based
on equations 6
11, processor 18 can determine a measure of the aggregation in the illuminated
samples, as
previously described.
Apparatus 10 can include one or more heaters 20 that may be used to uniformly,
or
selectively heat the sample wells of trays 12. In one embodiment, the heaters
20 can provide a
temperature gradient across the trays 12. In another embodiment, trays 12 may
be contained in
a water bath, which can be heated by heaters 20. As previously described,
heaters 20 can
include other heating means as may be known in the art, e.g., Pettier heaters.
CA 02479104 2004-08-26
Referring now to Fig. 2, there is schematically illustrated a partial cross-
sectional view
of apparatus 10, taken at line 2-2 of Fig. 1. For the exemplary embodiment of
Fig. 2, tray 12
can include an array of sample wells, one of which is labeled 22 in Fig. 2.
The wells can
contain molecular samples, as may be indicated by 24 in Fig. 2. It can be
understood that, in
lieu of tray 12, apparatus 10 can include individual molecular samples that
can be supported on
frame 26.
For the exemplary embodiment of Fig. 2, light source 14 includes an array of
LED
clusters 28, with each cluster having a grouping of LED's 30. Power for the
LED's 30 can be
provided via connectors 32, such that LED's 30 and/or clusters 28 can be
individually and
selectively powered. It can be understood that light source 14 can be an array
of individual
LED's 30, though clustering of LED's 30 may provide cost savings.
As previously noted, light scattering measurements are preferably obtained
with light at
an incident angle of less than 45° and preferably in a range of
15° - 30°. LED's 30 can be
oriented to provide light into sample wells 22 at an incident angle ~, with ~
< 45° and
preferably in a range of 15° - 30°, in order to achieve the dual
purpose of (i) taking
measurements in the range of angle providing maximum intensity of scattered
light and (ii)
avoiding detection of incident illumination. In order to control the beam
geometry and
dimensions of illumination on every sample and in order for the incident angle
to remain
consistent for each of the sample wells 22, a light guide 34 can be provided
for each sample
well 22. The light guides 34 may include parallel bores or passages through
frame 26 which
can be aligned with the chosen incident angle ~. In one embodiment, light
guides 34 can
include optical fibers. Light guide 34 can also contribute to elimination of
crosstalk and can be
configured to project discrete, uniform "spots" of light on each sample at a
roughly constant
location with respect to each sample.
The number of LED's 30 may correspond with the number of sample wells, so that
each LED 30 can illuminate one sample well 22. However, cost and/or size
limitations may
preclude having one light source for each sample well. Furthermore, obviation
of a direct
relationship between the light source and sample density makes the solution
more scalable in
the sense that sample density may be varied without changing the light source.
In the
embodiment of Fig. 2, it can be seen that one LED 30, or a cluster of LED's 28
may illuminate
more than one sample well 22. When it is desired to illuminate selected sample
wells 22,
4fi
CA 02479104 2004-08-26
occluding means can be provided to selectively block light from source 14 from
entering other
sample wells. Such occluding means can include one or more devices, such as
operable
shutters 36 and/or optical switches 38, as shown in Fig. 2, or other devices
as may be known in
the art, e.g., polarizing filters, or liquid crystal arrays. It can be
understood that such occluding
means can be additionally and/or alternatively provided to selectively block
light from other
than the selected sample cells from reaching the detector 16. For example,
shutters 36 may be
positioned between the sample wells 22 and detector 16.
Fig. 3 illustrates a cross sectional view; corresponding with the view of Fig.
2, of an
alternative embodiment of the apparatus 10, wherein light source 14 can
include a single
source of light, 114, though multiple light sources, or arrays of light
sources, rnay also be used.
In the embodiment of Fig. 3, light from source 114 can be selectively directed
to a sample well
22, as indicated by light path 150, by optical directing means 152, which can
utilize reflection,
refraction, diffraction and/or other known light directing methods. For
example, known micro-
electrornechanical (MEMS) devices can control the movements of an array of
directing optics,
e.g., micro-mirrors 154, such that light from a source, such as source 114,
can be directed to a
desired location, such as a selected sample well 22. I1; can also be
understood that MEMS
devices can control the operation of the shutters 36 in the exemplary
embodiment of Fig. 2.
In the illustrative embodiment of Fig. 3, mirror 154a can be seen to be
rotated with
respect to other mirrors 154, such that light from source 114 can follow path
150 into the
selected one of sample wells 22. Alternatively, the device 152 can be
configured to direct
scattered light from a selected sample well 22 to the detector 16. ~ther means
for effecting
such re-direction include mirrors, beam-splitters, fiber optics, lenses, etc.
In another example
embodiment, a single laser can be split into multiple beams to illuminate
multiple samples
directly.
In one embodiment, light source 14 (or alternatively 114) emanates white light
so as to
capture all possible phenomena of scattering. In another embodiment, a
monochrome light
source may be used, or a monochomator, such as a colored filter, can be placed
between the
light source and the samples, or between the samples and the detector 16, as
indicated by filter
40 in Fig. 1. It can be understood that other filter types, including
polarizing filters, may also
be used. In a further exemplary embodiment, light source 14 may include a
luminescent
source, and/or a UV source, as indicated at 156 in Fig. 3. Light source 156
can provide light of
47
CA 02479104 2004-08-26
a defined wavelength such that fluorescence emissions from illuminated samples
can be
measured, as described previously. It can be understood that light source 156
may be spatially
separate from light source 114. For example, light source 156 may be located
on the op~site
side of tray 12 from light source 114.
A switch 158 can permit switching between the sources 14 (114) and 156, such
that
detector 16 can measure both the extent of unfolding and the extent of protein
aggregation.
Where a single detector is used to measure both light scattering and
fluorescence, a filter, such
as filter 40 in Fig. 1, can be controlled by switch 158, or other switching
means, so as to move
into and out of the optical path. In one embodiment, a second detector 16a, as
shown in Fig. 3;
e.g., a fluorescent light detector, can be provided, such that one detector
can measure the extent
of protein aggregation and the other can measure the extent of unfolding of
the biological
molecule, with a switch 160 controlling v~rhich detector is operating. The
switching between
the two light sources and/or between the two detectors can be such that the
extent of unfolding
and the extent of aggregation can be determined essentially simultaneously.
For example,
switching can be done at every second, every tenth of a second, every one
hundredth of a
second, or less.
It can be understood that means for distributing samples to the sample wells
can be
provided. For example, apparatus 10 may be included in a robot or working
station having
robotic arms to manipulate the samples. Apparatus 10 can be provided in a kit
form that can be
easily adapted to such existing equipment.
In another embodiment, the invention provides kits containing one or more
elements or
apparatus necessary for the methods of the invention.
The present invention is further illustrated by the following examples, which
should not
be construed as limiting in any way. The contents of all cited references
including literature
references, issued patents, published and non published patent applications as
cited throughout
this application are hereby expressly incorporated by reference.
The practice of the present invention will employ, unless otherwise indicated,
conventional techniques of cell biology, cell culture, molecular biology,
transgenic biology,
microbiology, recombinant DNA, and immunology, which are within the skill of
the art. Such
techniques are explained fully in the Literature. (See, for example, Molecular
Cloning A
Laboratory Manual, 2nd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring
Harbor
48
CA 02479104 2004-08-26
Laboratory Press: 1989); DNA Cloning, Volumes I and II (D. N. Glover ed.,
1985);
Oligonucleotide Synthesis (M. J. Gait ed., 1984); Mullis et al. U.S. Patent
No: 4,683,195;
Nucleic Acid Hybridization (B. D. Names & S. J. Higgins eds. 1984);
Transcription And
Translation (B. D. Names & S. J. Higgins eds. 1984); (R. I. Freshney, Alan R.
Liss, Inc.,
1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical
Guide To
Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press,
Inc., N.Y.);
Gene Transfer Vectors For Mammalian Cells (J. H. Miller and M. P. Calos eds.,
1987, Cold
Spring Harbor Laboratory); , Vols. 154 and 155 (Wu et al, eds.),
Immunochemical Methods In
Cell And Molecular Biology (Mayer and Walker, eds., Academic Press, London,
1987);
Handbook Of Experimental Immunology, Volumes I-IV (D. M. Weir and C. C.
Blackwell, eds.,
1986) (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986).
Examples
Example 1: Measure of the characteristics of unfoldins and a~gre~ation of a
protein
Multiple identical protein solutions were arranged in iwo-dimensional arrays
of reservoirs
which were incubated at different temperatures. These samples were
alternatively illuminated
with UV light and monochromatic red light. The fluorescence emitted and the
light scattered at
90° were measured from the top of the arrangement. The fluorescence
emitted and the light
scattered intensity data were collected at intervals of time of incubation for
all the temperatures
simultaneously.
Plots of fluorescence and light scattered at 90° at a given time versus
temperature were
automatically built for all the samples, and the temperatures of unfolding Tm
and aggregation Tags
were obtained from the fitted sigmoid portion of the curves.
The results are shown in Fig. SA and B. The red symbols represent the best fit
using a
sigmoid Boltzman function.
In another example, the protein was incubated in S% ethanol; 10% glycerol,
NAD+ or a
control solution and the extent of unfolding was measured at 55 °C. A
plots of fluorescence at
the given temperature versus time was automatically built for all the samples,
and the rate of
unfolding k" was obtained from the fitted exponential grow portion of the
curves for the proteins
jumped to 55°C.
49
CA 02479104 2004-08-26
The results are shown in Fig. 6. 'I hese results show that NAD+ has the
strongest
stabilizing effect on the protein. Glycerol has also a stabilizing effect. On
the contrary, ethanol
has a destabilizing effect or no effect at all on the protein.
Thus, these results show the possibility of simultaneous detection of the
extent of
unfolding and the extent of aggregation of a protein. Such methods can be used
for high
throughput screening of stabilizing conditions and the identification of
compounds, e.g., ligands
that bind to particular biological molecules.
Example 2: Identification of Conditions that Facilitate Protein Refolding
Insoluble proteins were denatured and solubilized using 4 M guanidium
hydrochloride
(GnHCL) followed by denatured protein purification using Ni-NTA affinity
resin. Unfolded
pure proteins at a concentration of ~4 mg/ml were diluted 20-fold in different
refolding
conditions in 384 well plates and incubated over night at 4°C (figure
8). The refolding
conditions contain different pH buffers and compounds known to favor in vitro
protein
refolding (e.g., arginine, divalent ions, DTT, GSH and GSSG, detergents, etc).
Elevating the
temperature caused the refolded proteins to denature. Denaturation happens at
or above the so
called transition temperature (Tm).
Presence of folded protein was screened in a 384 well format by alternatively
illuminating the samples with UV light and monochromatic red light. The
fluorescence emitted
and the light scattered at 90° were measured from the top of the
arrangement. The fluorescence
emitted and the light scattered intensity data were collected at intervals of
time of incubation for
all the temperatures simultaneously.. Proteins which were successfully
refolded show a
transition in aggregation measurement. The temperature which such a transition
is observed at
is called temperature of aggregation (Tag).
Under conditions where the proteins cannot refold, in general they aggregate
which can
be seen as higher initial intensity values in Figure 9.
Folded Gaf domain of PDE10 is expected to bind cAMP and cGMP. The presence of
these ligands increased the Tag of refolded Gaf domain, an indication that
cAMP and cGMP are
binding to the protein (figure 10) and that the protein is folded. Insoluble
urilydate kinase and
denatured PDE 4D were also successfully refolded in at least in one of the
conditions used by
the method.
Insoluble proteins in inclusion bodies can also be refolded using this method.
CA 02479104 2004-08-26
Eauivalents
The present invention provides among other things novel methods and apparatus
for
characterizing the stability of biological molecules. While specific
embodiments of the subject
invention have been discussed, tike above specification is illustrative and
not restrictive. Many
variations of the invention will become apparent to those skilled in the art
upon review of this
specification. The full scope of the invention should be determined by
reference to the claims,
along with their full scope of equivalents, and the specification, along with
such variations.
Unless otherwise indicated, all numbers expressing quantities of ingredients,
reaction
conditions, and so forth used in the specification and claims are to be
understood as being
modified in all instances by the term "about." Accordingly, unless indicated
to the contrary,
the numerical parameters set forth in this specification and attached claims
are approximations
that may vary depending upon the desired properties sought to be obtained by
the present
invention.
All publications and patents mentioned herein are hereby incorporated by
reference in
their entirety as if each individual publication or patent was specifically
and individually
indicated to be incorporated by reference. In case of conflict, the present
application, including
any definitions herein, will control. Also incorporated by reference in its
entirety is WO
03/071269.
51