Note: Descriptions are shown in the official language in which they were submitted.
CA 02485433 2000-11-24
1 r
Preparation of non-crystalline and crystalline dihydrate forms of azithromycin
BACKGROUND OF THE INVENTION
1. Field of the Invention.
Azithromycin is the USAN generic name of the azalide 9-deoxo-9a-aza-9a-methyl-
9a-homoerythromycin A, which systematic name is 1-oxa-6-azacyclopentadecan-15-
one,13-((2,6-dideoxy-3-C-methyl-1-3-0-methyl-alpha-L-ribo-hexopyranosyl)-oxy)-
2-
ethyl-3,4,10-trihydroxy-3,5,6,8,10,12,14-heptamethyl-11-((3,4,6-trideoxy-3-
(dimethyl-
amino)-beta-D-xylo-hexopyranosyl)oxy). It is a semisynthetic macrolide that
shows an
excellent antimicrobial activity against gram-positive and some cases of gram-
negative
bacteria (H.A. Kirst, G.D. Sides, Antimicrob. Agents. Chemother. 1989, 33,
1419-1422).
Clinical use of this macrolide is broadening its application to the treatment
of
opportunistic infections (F. Lecomte, Rev. Med. Interne 1998, 19(4), 255-61;
S.
Alvarez-Elcoro, Mayo Clin. Proc. 1999, 74(6), 613-34; J. Schater, Lancet,
1999,
354(9179), 630-35).
2. Description of the Prior Art.
Figure 1 shows the different synthetic routes to azithromycin 1. The names of
the
intermediates displayed in Figure 1 are gathered in the following table.
Intermediate Name
1 Azithromycin
2 Erythromycin A oxime
3 6,9-iminoether
4 9,11 -iminoether
5 Azaerythromycin A
6 Azaerythromycin 11,12-hydrogenorthoborate
7 Azitliromycin 11,12-hydrogenorthoborate
The following table summarizes the patents, articles, authors and applicants
that
describe the different synthetic paths (A, B, C, D, E) towards azithromycin 1.
CA 02485433 2000-11-24
2
Route Patents Articles Author Applicant
A a) US 4,328,334 = J. Chem. Soc. Perkin Trans
= US 4,517,359 1, 1986, 1881
= J. Chem. Res., 1988, 132 S. Djokic PLIVA
= Idem miniprint., 1988, 1239
B b) US 4,474,768 G.M. Bright PFIZER
C c) US 5,686,587
d) EP 0,699,207 B.V. Yang PFIZER
e) ES 2,104,386
D f) US 5,869,629 = J.Org.Chem, .1997, 62, '(21),
g) EP 0,827,965 7479 - 7481 M. Bayod ASTUR PHARMA
h) ES 2,122,905 = Magn. Reson. Chem, 1998,
36, 217-225
E i) EP 0,879,823 W. Heggie HOVIONE
The structural elucidation studies carried out with azithromycin 1 have shown
the
existence of two different crystalline forms: hygroscopic monohydrate and non-
hygroscopic dihydrate, being the latter preferred for manufacturing
formulations used in
therapeutical treatments, as it is described in EP 0,298,650.
Azithromycin dihydrate is easily distinguishable. from hygroscopic
azithromycin by
means of the following differentiative assays:
a) The dihydrate form keeps its percentile water content constant at values
(4.5-5%)
which are very close to the theoretical value (4.6%).
b) The differential calorimetry analysis (DSC) of azithromycin dihydrate
reveals the
presence of a single endotherm which may vary between 115 and 13 5 C, with an
energy absorbed during the process which ranges between 27 and 34 cal/g.
c) Each crystalline form presents its own characteristic X-Ray Diffraction
spectrum.
d) The infrared spectra in KBr of both crystalline forms present clear
differences:
azithrom cin dihydrate azithrom cin monoh drate
v (cm"' v (cm")
3560 and 3496 (2 sharp bands) 3500 (wide band)
1344 Does not present any
1282 and 1268 (2 sharp bands 1280
1083 Does not present any
Two other synthesis, affording azithromycin 1 as a form that should differ
from the
crystalline ones previously mentioned, have also been described. In these
cases,
azithromycin is obtained by simple evaporation to dryness. However, in these
documents
there is no reference to the crystalline state of the azithromycin thus
obtained.
CA 02485433 2000-11-24
3
Patent Applicant Priority Procedure
(Author)
= WO 94/26758 PFIZER May 19,1993 Methylene chloride evaporation
a) US 5,686,587 (B.V. Yang)
b) EP 0,699,207
c) ES 2,104,386
= BE 892,357 PLIVA Mar. 3, 1981 Chloroform evaporation
= US 4,517,359 (S. Djokic)
In the following table are summarized the different procedures for the
preparation
of both crystalline forms of azithromycin 1.
Crystalline form Patent (Author) Applicant
Priority Procedure
HYGROSCOPIC a) EP 0,101,186 PFIZER July 19, 1982 Recrystallization from
MONOHYDRATE b) US 4,474,768 (G.M. Biight) ethanol/water
HYGROSCOPIC c) EP 0,298,650 PFIZER July 9, 1997 Recrystallization from
MONOHYDRATE (D. Allen) ethanol/water
NON- d) EP 0,298,650 PFIZER Recrystallization from
HYGROSCOPIC e) WO 89/00576 July 9, 1997 THF / petroleum ether/
DIHYDRATE f) ES 2,038,756 ~' ~1~) water
= Recrystallization from
acetone/water
NON- g) CN 1,093,370 Faming = Recrystallization from
HYGROSCOPIC (Chem. Abs. Zhuanli.... Dec. 10, 1993 other solvents
DIHYDRATE 29525q,124,1996) (Q. Song) (methanol, DMF,
acetonitrile, dioxane,
... ) and water
NON- CHEMO- Recrystallization from
HYGROSCOPIC h) EC 95-1389 TECNICA May, 1995 acetone/ water
DIHYDRATE SINTYAL
NON- i) EP 0,827,965 ASTUR Recrystallization from
HYGROSCOPIC j) ES 2,122,905 PHARMA July 11, 1996 acetone/ water
DIHYDRATE k) US 5,869,629 (M.Bayod)
NON- Precipitation from a base
HYGROSCOPIC 1) EP 0,941,999 HOVIONE Mar. 13, 1998 neutralized acid solution of
DIHYDRATE (W=Heggie) azithromycin in
acetone/ water
Crystalline form Article Author Date Procedure
Two recrystallizations:
J. Chem. Res., 1. Precipitation from a
NON- = 1988, 132 S.Djokic May, 1988 base neutralized acid
HYGROSCOPIC m) idem miniprint., (PLIVA) (received solution of azithromycin in
DIHYDRATE 1988, 1239, June 4, 1987) acetone/ water.
2. From ethyl ether:
NON- = J. Org. Chem, M.Bayod Nov., 1997 Recrystallization from
HYGROSCOPIC 1997, 62, (21), (ASTUR- (received acetone/water
DIHYDRATE 7479 - 7481 PHARMA) May 1, 1997)
HYGROSCOPIC = J. Org. Chem, M.Bayod Nov., 1997 Recrystallization from
MONOHYDRATE 1997, 62, (21), (ASTUR (received ethanol/water
7479 - 7481 PHARMA) May 1, 1997)
CA 02485433 2007-05-14
3a
SUMMARY OF INVENTION
In a first aspect, the present invention provides a method of preparing 9-
deoxo-9a-aza-9a-methyl-9a-homoerythromycin A (Azithromycin) in a crystalline
dihydrate form, the method comprising:
dissolving Azithromycin in tert-butanol to form a solution; and
combining the solution with water; thereby crystallizing Azithromycin as the
crystalline dihydrate form.
In a second aspect, the present invention provides a method of preparing 9-
deoxo-9a-aza-9a-methyl-9a-homoerythromycin A (Azithromycin) in a crystalline
dihydrate form, the method comprising:
dissolving Azithromycin in tert-butanol to form a solution; and
combining the solution with a mixture of water and petroleum ether, thereby
crystallizing Azithromycin as the crystalline dihydrate form.
In a third aspect, the present invention provides a method of preparing 9-
deoxo-9a-aza-9a-methyl-9a-homoerythromycin A (Azithromycin) in a crystalline
dihydrate form, the method comprising:
hydrolyzing 9-deoxo-9a-aza-11,12-desoxy-9a-methyl-9a-homoerythromycin
A 11, 1 2-hydrogenorthoborate in an organic solvent by contacting the 9-deoxo-
9a-aza-
11, 1 2-desoxy-9a-methyl-9a-homoerythromycin A 11,12-hydrogenorthoborate with
a
dilute acid at room temperature and at a pH range between 2 and 4, thereby
producing
Azithromycin;
dissolving the Azithromycin in tert-butanol to form a solution; and
combining the solution with water, thereby crystallizing Azithromycin as the
crystalline dihydrate form.
In a fourth aspect, the present invention provides a method of preparing 9-
deoxo-9a-aza-9a-methyl-9a-homoerythromycin A (Azithromycin) in a crystalline
dihydrate form, the method comprising:
CA 02485433 2007-05-14
3b
hydrolyzing 9-deoxo-9a-aza- 11, 1 2-desoxy-9a-methyl-9a-homoerythromycin
A 11,12-hydrogenorthoborate in an organic solvent by contacting the 9-deoxo-9a-
aza-
11, 1 2-desoxy-9a-methyl-9a-homoerythromycin A 11, 1 2-hydrogenorthoborate
with a
dilute acid at room temperature and at a pH range between 2 and 4, thereby
producing
Azithromycin;
dissolving the Azithromycin in tert-butanol to form a solution; and
combining the solution with a mixture of water and petroleum ether, thereby
crystallizing Azithromycin as the crystalline dihydrate form.
The organic solvent used in the method of the third and fourth aspect of the
present invention can be ethyl acetate, acetonitrile, methanol, or ethanol.
Furthermore, the dilute acid used in the method of the third and fourth aspect
of the
present invention can be dilute sulfuric acid, dilute hydrochloric acid, or
dilute oxalic
acid.
BRIEF DESCRIPTION OF THE DRAWINGS
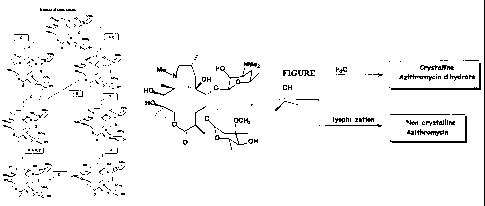
FIG. 1 shows the different synthetic routes to azithromycin.
FIG. 2 shows the infrared spectra of non-crystalline azithromycin and
crystalline
azithromycin dihydrate recorded in FT-IR Nicolet Impact 410 Instrument.
FIG. 3 shows the thermograms of non-crystalline azithromycin and crystalline
azithromycin dihydrate obtained scanning between 20 and 300 C, under nitrogen
with
a heating rate of 5 C/min.
FIG. 4 shows X-ray diffraction spectra of non-crystalline azithromycin and
crystalline
azithromycin dihydrate recorded on a Philips PW1710 diffractometer.
CA 02485433 2000-11-24
4
DESCRIPTION OF THE INVENTION.
First, the present invention provides a series of new procedures for the
preparation
of azithromycin 1:
= A procedure for the preparation of its crystalline dihydrate form,
characterized by
crystallization. of azithromycin from a mixture of tert-butanol / water: In
this
procedure crystalline azithromycin monohydrate is dissolved in tert-butanol
and, after
water addition, is allowed to crystallize for a period of 48-72 hours.
= A procedure for the preparation of its crystalline dihydrate form,
characterized by
crystallization of azithromycin from a mixture of tert-butanol / petroleum
ether /
water. In this procedure, crystalline azithromycin monohydrate is dissolved in
tert-
butanol and added to a mixture of petroleum ether and water. This solution is
allowed to crystallize for a period of 48-12 hours.
= A procedure for the preparation of non-crystalline azithromycin by means of
lyophilization of solutions of azithromycin in tert-butanol (2-methyl-2-
propanol).
= A procedure for the preparation of non-crystalline azithromycin by means of
evaporation of solutions of azithromycin in aliphatic alcohols (preferably
ethanol or
isopropanol).
Secondly, the present invention describes the characterization of non-
crystalline
azithromycin and its unambiguous differentiation from the crystalline forms
(dihydrate
and monohydrate) using the following techniques:
/ Infrared Spectroscopy
/ Differential Scan Calorimetry (DSC)
/ X-Ray Diffraction
/ Hygroscopicity
/ Crystallinity test by means of polarized light microscopy
The procedures which are the object of the present invention are advantageous
over previously described methods, essentially at industrial scale:
/ Lyophilization is a technique that guarantees excellent results concerning
homogeneity, purity and consistency of analytical data of different batches.
/ The crystallization procedures, which are characterized by slow crystal
growth, greatly improve the homogeneity and particle distribution of different
CA 02485433 2000-11-24
batches. This minimizes the presence of the non-crystalline fraction (detected
by X-Ray and DSC) that is always present in crystalline azithromycin
dihydrate obtained by the methods reported in the literature and above cited.
The differences observed between crystalline azithromycin dihydrate and its
non-
5 crystalline form, using the techniques previously mentioned, are shown
below:
1. Infrared Spectra (KBr), recorded in a FT-IR Nicolet Impact 410
Instrument, of
both azithromycin forms are clearly different. Fig. 2 reproduces the spectra
which
most significative bands are summarized in the following table:
C stalline azithromycin dihydrate Non-crystalline azithromycin
v crri 1) v (cm-1
3561 and 3496 (2 sharp bands) 3500 (wide band)
1344 Does not present an1282, 1269 and 1251 3 sharp bands) 1280 and 1257 2
sharp bands)
1083 Does not present any
2. DSC. In Fig. 3 are shown the thermograms obtained scanning between 20 and
300'C, under nitrogen with a heating rate of 5*C / min. The thermogram of the
non-
crystalline form does not present any melting peak, what clearly
differentiates it from
the one corresponding to crystalline azithromycin dihydrate.
3. X-Ray Diffraction Spectra were recorded on a Philips PW 1710
diffractometer. As
the spectrum corresponding to non-crystalline azithromycin (Fig. 4) is
characterized
by the absence of defined maxima, this solid is considered to be amorphous.
4. Hygroscopicity. Two different samples of non-crystalline azithromycin
containing 3%
water were kept under an atmosphere over 75% relative humidity. After 8 hours,
water content in the first sample was 5.3%, while the second one contained
9.9%
water after 72 hours. Non-crystalline azithromycin is thus moderately
hygroscopic.
5. Crystallinity tests (polarized light microscopy) carried out with non-
crystalline
azithromycin were negative, as their particles do not show birefringence.
EXPERIMENTAL PART
= Preparation of 9-deoxo-9a-aza-11,12-desoxy-9a-homoerythromycin A 11,12-
hydrogenorthoborate.
CA 02485433 2000-11-24
6
89 g of 9-deoxo-6-desoxy-6,9-epoxy-9,9a-dihydro-9a-aza-homoerythromycin A are
dissolved in 450 ml of methanol and cooled down between -5 and -10 C. While
keeping the temperature in the specified interval 16 portions of 2.2 g each of
sodium
borohydride are added. Temperature and stirring conditions are maintained for
two
additional hours and the bulk of the reaction is allowed to reach 20 C. After
20 h, the
methanol is evaporated to dryness. The residue is dissolved in 500 ml of
methylene
chloride and 750 ml of water and shaked for 30 min. The organic phase is
separated and
the aqueous phase is extracted with 250 ml of methylene chloride. The organic
phases
are combined, filtered over celite, dried with anhydrous sodium sulphate and
concentrated to dryness to yield 85 g of 9-deoxo-9a-aza-11,12-desoxy-9a-
homoerythromycin A 11,12-hydrogenorthoborate.
IR (KBr) v.= 3500, 2980, 2960, 1730, 1470, 1390, 1170, 1090, 1060 cm'
1H-NMR (CDC13) S= 2.21 (NMe2),3.27 (OMe) ppm.
(~artial)
~ C-NMR (CDC13) 8= 180.0 (C=O), 79.63 (ClI), 76.46 (C12) 58.7 (Clo), 57.1(C9),
49.4
(rartial) (OMe), 40.2 (NMe2) ppm
' B-NMR (CDC13) 8= 9.9 ppm ai %= 200 Hz
TLC rf = 0.28 (petroleum ether : ethyl acetate : diethylamine 75:25:10)
developer: ethanol/vanillin (sulphuric acid)
= Preparation of 9-deoxo-9 a-aza-1 1,12-desoxy-9a-methyl-9a-homo-erythromycin
A 11,12-hydrogenorthoborate.
50 g of 9-deoxo-9a-aza-11,12-desoxy-9a-homoerythromycin A 11,12-hydrogenortho-
borate are dissolved in 500 ml of chloroform, and subsequently a mixture of
5.5 ml of
formic acid and 11.75 ml of aqueous 35-40% formaldehyde is added. The reaction
mixture is heated under pressure for 14 hours and subsequently cooled down to
15-20 C.
500 ml of water are added and the mixture is taken to pH=4 by adding 20%
sulphuric
acid. The mixture is shaken for 15 min and the lower organic layer is
separated. The
alkaline aqueous phase is extracted with 2x 100 ml methylene chloride. The
organic
phases are combined and filtered over celite, dried with anhydrous sodium
sulfate and
evaporated to dryness. The residue obtained is washed twice with 250 ml of
ethyl ether,
yielding a dry residue of 29 g of 9-deoxo-9a-aza-11,12-desoxy-9a-methyl-9a-
homo-
erythromycin A 11,12-hydrogenorthoborate.
...._.,-- -.~~.~
CA 02485433 2000-11-24
7
IR (KBr) v,,W= 3500, 1730, 1470, 1390, 1090, 1070 cm''
'H-NMR (CDC13) S= 2.00 (NMeZ), 2.30 (NMe), 3.37 (OMe) ppm.
(~artial)
1 C-NMR (CDC13) S= 179.9 (C=O), 79.40 (C>>), 77.09 (C12), 68.84 (Cy), 64.08
(Cio), 49.36
(partial) (OMe), 40.18 (NMe2), 34.39 (NMe) ppm
"B-1VMR (CDC13) S= 10.1 ppm w 180 Hz
m/e M+= 775.5
TLC rf = 0.38 (petroleum ether : ethyl acetate : diethylamine 75:25:10)
developer: ethanol/vanillin (sulphuric acid)
= Hydrolysis of 9-deoxo-9a-aza-11,12-desoxy-9a-methyl-9a-homo-erythromycin A
11,12-hydrogenorthoborate. Synthesis of 9-deoxo-9a-aza-9a-methyl-9a-homo-
erythromycin A (Azithromycin).
22 g of 9-deoxo-9a-aza-11,12-desoxy-9a-methyl-9a-homo-erythromycin A 11,12-
hydrogenorthoborate are dissolved in 250 n-d of acetonitrile to which 125 ml
of water are
subsequently added. 20% sulphuric acid is added to the mixture to take it to
pH=2, and
stirring is maintained for 30 min. The acidic solution is poured into a
mixture of 350 ml
of methylene chloride and 350 ml of water, inmediately adding 48% lime until
pH=9. The
mixture is shaken for 15 min and the lower organic phase is separated. The
alkaline
aqueous phase is extracted with 2x100 ml of methylene chloride. The combined
organic
phases are filtered over celite and evaporated to dryness. The residue is
dissolved in 50
ml of ethanol and 60 ml of water are added over 30 min. Precipitation is
allowed for 2 h,
and the solid is collected by filtration and vacuum-dried at 40 C to yield 15
g of 9-
deoxo-9a-aza-9a-methyl-9a-homo-erythromycin A (Azithromycin).
IR (KBr) v,,.= 3500, 3000, 2970, 1740, 1470, 1380, 1280, 1060 cm''
1H-13Nnt (CDC13) S= 2.31 (NMe.Z), 2.34 (NMe), 3.38 (OMe) ppm
(partial)
' C-NNIR (CDC13) S= 178.9 (C=O), 73.08 (C12), 72.32 (Cõ), 69.88 (C9), 62.43
(Clo), 49.37
(partial) (OMe), 40.23 (1VMe2), 35.92 (NMe) ppm
m/e M+= 749.5
I-IPLC corresponds according to USP XXIII
TLC rf= 0.62 (petroleum ether : ethyl acetate : diethylamine 75:25:10)
developer: ethanol/vanillin (sulphuric acid)
= Preparation of 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin A dihydrate.
Method A.
25 g of crystalline azithromycin monohydrate are dissolved in 130 ml of tert-
butanol
heating at 30'C. This solution, is filtered and 130 ml of water are added over
6 h. The
resulting mixture is taken to pH=i 1 by addition of NaOH 2N, cooled down below
10 C
CA 02485433 2000-11-24
8 and subsequently stirred for 48-72 h. The crystals are collected by
filtration and dried (80
mm Hg / 25 *C) to yield 15 g of azithromycin dihydrate.
IR (KBr) v = 3560, 3496, 1740, 1470, 1380, 1344, 1282, 1268, 1251, 1093 cm'
'H-NMR (CDC13), "C-NMR (CDC13), m/e, TLC and HPLC are identical to those of
the previous
example.
= Preparation of 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin A dihydrate.
Method B.
25 g of crystalline azithromycin monohydrate are dissolved in 50 ml of tert-
butanol
heating at 30 C. This solution is filtered and discharged over a mixture of
500 ml of
petroleum ether and 20 ml of water. The resulting mixture is cooled down below
10 C
and subsequently stirred for 48-72 h. The crystals are collected by filtration
and dried (80
mm Hg / 25 *C) to yield 12 g of azithromycin dihydrate.
IR (KBr),'H-1VMR (CDC13), t'C-NMR (CDC13), m/e, TLC and HPLC are identical to
those of the
previous example.
~ Preparation of non-crystalline 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin
A. Method A.
5 g of crystalline azithromycin monohydrate are dissolved in 25 ml of tert-
butanol
heating at 30 C. This solution is filtered and solidified in a cooling bath.
The solvent is
sublimed at room temperature and 10-2mm Hg. The solid obtained is dried (80 mm
Hg /
40 *C) to yield 5 g of non-crystalline azithromycin.
IR (KBr) v.= 3500, 1740, 1470, 1280, 1268, 1257 cm"'(See Fig. 2)
'H-N1vIR (CDC13),13C-NMR (CDC13), m/e, TLC and HPLC are identical to those of
the previous
example
% H20 (K.F.)= 3.0 %
DSC = See Fig. 3
X-Ray Diffraction = See Fig. 4
= Preparation of non-crystalline 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin
A. Method B.
5 g of crystalline azithromycin monohydrate are dissolved in 25 ml of ethanol.
The
solution is filtered and the solvent evaporated at room temperature and 150 mm
Hg. The
solid obtained is dried (80 mm Hg / 40 *C) to yield 5 g of non-crystalline
azithromycin,
which analytical data are identical to those of the previous example.