Note: Descriptions are shown in the official language in which they were submitted.
CA 02486495 2005-10-19
1
WO 03/104253 PCT/EP03/06172
9-Alpha-Substituted Estratrienes as Selectively Active Estrogens
Field of the Invention
This invention relates to new compounds as pharmaceutical active ingredients,
which have in vitro a higher affinity to estrogen receptor preparations from
rat prostates
than to estrogen receptor preparations from rat uteri and in vivo a
preferential action in
the ovary in comparison to the uterus, their production, their therapeutic use
and
pharmaceutical dispensing forms that contain the new compounds.
The chemical compounds are new, steroidal, tissue-selective estrogens.
Background of the Invention
The efficiency of estrogens in the treatment of hormone-deficiency-induced
symptoms such as hot flashes, atrophy of estrogen target organs and
incontinence, as well
as the successful use of estrogen therapies for prevention of bone mass loss
in peri- and
postmenopausal women, is well documented and generally accepted (Grady et al.
1992,
Ann Intern Med 117: 1016-1037). It is also well documented that estrogen
replacement
therapy in postmenopausal women or in women with ovarian dysfunction that is
caused in
some other way reduces the risk of cardiovascular diseases compared to women
who are
not treated with estrogen (Grady et al., loc. cit.).
In conventional estrogen or hormone replacement therapy (= HRT), natural
estrogens, such as estradiol, and conjugated estrogens that consist of equine
urine are used
either by themselves or in combination with a gestagen. Instead of the natural
estrogens,
derivatives that are obtained by esterification, such as, e.g., 170-estradiol-
valerate, can
also be used.
Because of the stimulating action of the estrogens that are used on the
endometrium, which results in an increase of the risk of endometrial carcinoma
(Harlap,
S. 1992, Am J Obstet Gynecol 166: 1986-1992), estrogen/gestagen combination
CA 02486495 2005-10-19
2
preparations are preferably used in hormone replacement therapy. The
gestagenic
component in the estrogen/gestagen combination avoids hypertrophy of the
endometrium,
but the occurrence of undesirable intracyclic menstrual bleeding is also
linked to the
gestagen-containing combination.
Selective estrogens represent a more recent alternative to the
estrogen/gestagen
combination preparations. Up until now, selective estrogens have been defined
as those
compounds that have an estrogen-like effect on the brain, bones and vascular
system,
owing to their antiuterotropic (i.e., antiestrogenic) partial action, but they
do not have a
proliferative effect on the endometrium.
A class of substances that partially meet the desired profile of a selective
estrogen
is the so-called "Selective Estrogen Receptor Modulators" (SERM) (R. F.
Kauffinan, H.
U. Bryant 1995, DNAP 8 (9): 531-539). In this case, these are partial agonists
of
estrogen receptor subtype "ERa." This substance type is ineffective, however,
with
respect to the therapy of acute postmenopausal symptoms, such as, e.g., hot
flashes. As
an example of a SERM, the raloxifene that was recently introduced for the
indication of
osteoporosis can be mentioned.
DE-A-19906159 describes new compounds as pharmaceutical active ingredients
that have in vitro a higher affinity to estrogen receptor preparations from
rat prostates than
to estrogen receptor preparations from rat uteri and in vivo a preferential
action on bones
in comparison to the uterus, their production, their therapeutic use and
pharmaceutical
dispensing forms that contain the new compounds. The compounds are 16a- and
160-
hydroxy-estra-1,3,5(10)-estratrienes that carry additional substituents in the
steroid
skeleton and can exhibit one or more additional double bonds in the B-, C-
and/or b-
rings.
For the treatment of fertility disorders of women, frequently caused by
ovarian
dysfunction that is caused by surgery, medication, etc., new possible
therapies are also
opened up by the use of new selective estrogens. The in-vitro fertility
treatment is a
process that has been established for more than 20 years. Numerous methods for
treating
ovarian-induced infertility with exogenic gonadotropins are known. By
administration of
CA 02486495 2005-10-19
3
gonadotropins such as FSH (FSH = follicle-stimulating hormone), a stimulation
of the
ovaries, which is to make possible a healthy follicular maturation, is to be
produced.
The follicle is the functional unit of the ovary and has two purposes: it
accommodates the oocytes and provides for the latter the possibility for
growth and for
maturation. Folliculogenesis comprises the development of an ovarian follicle
from a
primordial stage to a continuously increasing antral follicle, which
represents the last
stage before ovulation. Only an optimally developed antral follicle can
release a mature
ovocyte by ovulation.
Patients with ovarian-induced infertility (PCOS = syndrome of polycystic
ovaries)
suffer from a disrupted follicular maturation, which is associated both with
hormonal and
ovulatory disruptions and with inadequately matured ovocytes. The number of
primary
and secondary follicles is approximately twice as high here as in the normal
ovary
(Hughesden et al., Obstet. Gynecol. Survey 37, 1982, pp. 59-77).
There are indications that the early development stages of folliculogenesis
(which
relates to the development of primordial follicles to antral follicles) are
gonadotropin-
independent. It is not clearly explained how great the influence of known
paracrine and
autocrine factors is on early folliculogenesis (Elvin et al., Mol. Cell
Endocrinol. 13, 1999,
pp. 1035-1048; McNatty et al., J. Reprod. Fertil. Suppl. 54, 1999, pp. 3-16).
Gonadotropins such as FSH are mainly involved in the last development stages
of
folliculogenesis in follicular maturation, i.e., in the development of the
early antral
follicle to a mature follicle that can undergo ovulation.
The in-vivo and in-vitro infertility is preferably treated with gonadotropins
(FSH
and antiestrogens) (White et al., J. Clin. Endocrinol. Metab. 81, 1996, pp.
3821-3824). In
in-vitro fertilization treatment, oocytes are removed from preovulatory antral
follicles to
be able to mature in vitro into an ovocyte that can be fertilized. After
fertilization and
preembryonal development, one to three embryos are implanted in the uterus of
the
woman.
In many respects, the treatment with exogenic gonadotropins is accompanied by
numerous risks and side effects. The greatest risk consists in an
overstimulation of the
CA 02486495 2005-10-19
4
ovaries, which in severe cases can represent a serious danger to life (OHSS =
Ovarian
Hyperstimulation Syndrome). Other side effects are the high costs of the in-
vitro fertility
treatment that must be paid by the couples. Negative side effects such as
weight gain,
bloatedness, nausea, vomiting and an as yet unknown long-term risk of
developing cancer
are attributed to the gonadotropin treatment.
One method to avoid the above-mentioned drawbacks and risks is to ensure the
maturation and stimulation in vivo of follicular growth in the case of ovarian-
induced
infertility with a suitable active ingredient before treatment with exogenic
gonadotropins
begins.
Estrogen Receptor Beta (ERa)
Several years ago, estrogen receptor P (ERF3) was discovered as a second
subtype
of the estrogen receptor (Kuiper et at. (1996), Proc. Natl. Acad. Sci. 93:
5925-5930;
Mosselman, Dijkema (1996) Febs Letters 392: 49-53; Tremblay et al. (1997),
Molecular
Endocrinology 11: 353-365). The expression pattern of ER(3 differs from that
of the ERa
(Kuiper et al. (1996), Endocrinology 138: 863-870). ER(3 thus predominates
over ERa in
the rat prostate, while ERa predominates over ERI3 in the rat uterus. The
highest
concentrations of ER(3 and mRNA were found in the ovaries (Couse et al.
Endocrinology
138, 1997, pp. 4612-4613).
Other organ systems with comparatively higher ER(3-expression comprise the
bones (Onoe, Y. et al., 1997, Endocrinology 138: 4509-4512), the vascular
system
(Register, T. C., Adams, M. R. 1998, J. Steroid Molec Biol 64: 187-191), the
urogenital
tract (Kuiper, G. J. M. et al. 1997, Endocrinology 138: 863-870), the
gastrointestinal tract
(Campbell-Thopson 1997, BBRC 240: 478-483), as well as the testis (Mosselmann,
S. et
al. 1996 FEBS Lett. 392, 49-53) including the spermatides (Shugrue et al.
1998, Steroids
63: 498-504). The tissue distribution suggests that estrogens regulate organ
functions via
ER(3. The fact that ER(3 is functional in this respect also follows by studies
in ERa-
(ERKO) or ERI3-(c3ERKO)-knockout mice: ovariectomy produces bone mass loss in
ERKO-mice, which can be eliminated by estrogen substitution (Kimbro et al.
1998,
CA 02486495 2005-10-19
Abstract OR7-4, Endocrine Society Meeting, New Orleans). Estradiol in the
blood
vessels of female ERKO mice also inhibits vascular media and smooth muscle
cell
proliferation (lafrati, M. D. et al. 1997, Nature Medicine 3: 545-548). These
protective
actions of estradiol are carried out in the ERKO mouse presumably via ERR.
The fact that ERa and ERP have a functionally different action was confirmed
after successful production of aERKO and j3ERKO mice. ERa consequently plays
an
important role in the adult uterus, in mammary gland tissue, in the negative
regulation of
the gonadotropin activity, while ER(3 is mainly bonded in the processes of
ovarian
physiology, especially that of folliculogenesis and ovulation (Couse et al.,
Endocrine
Reviews 20, 1999, pp. 358-417).
Observations of RERKO mice provide an indication on a function of ER(3 in the
prostate and bladder: in the case of older male mice, symptoms of prostate and
bladder
hyperplasia occur (Krege, J. H. et al. 1998, Proc Natl Acad Sci 95: 15677-
15682). In
addition, female ERKO mice (Lubahn, D. B. et al. 1993, Proc Natl Acad Sci 90:
11162-
11166) and male ERKO mice (Hess, R. A. et al. 1997, Nature 390: 509-512) as
well as
female PERKO mice (Krege, J. H., 1998, Proc Nat! Acad Sci 95: 15677-15682)
have
fertility disorders. Consequently, the important function of estrogens with
respect to
maintaining testis and ovary functions as well as fertility is confirmed.
It was possible to achieve a selective estrogenic action on specific target
organs by
subtype-specific ligands based on the different tissue or organ distribution
of the two
subtypes of the ERs. Substances with a preference for ER(3 compared to ERa in
the in-
vitro receptor binding test were described by Kuiper et al. (Kuiper et al.
(1996),
Endocrinology 138: 863-870). A selective action of subtype-specific ligands of
the
estrogen receptor on estrogen-sensitive parameters in vivo was not previously
shown.
The object of this invention is therefore to prepare compounds that have in
vitro a
dissociation with respect to the binding to estrogen receptor preparations
from rat
prostates and rat uteri. The compounds are to show in vitro a higher affinity
to estrogen
receptor preparations from rat prostates than to estrogen receptor
preparations from rat
uteri.
CA 02486495 2005-10-19
6
The ERR-specific compounds are to produce in vivo a profertility action in the
ovary. At the same time, the compounds are to exhibit a dissociation with
respect to
ovary action in comparison to uterus action. The compounds according to the
invention
are to have a certain protective action against hormone-deficiency-induced
bone mass loss
in comparison to uterus-stimulating action.
In the broader sense, a structure-action relationship, which allows for access
to
compounds that have the above-formulated pharmacological profile, is to be
made
available by this invention. The compounds according to the invention are to
produce
enhanced fertility in the ovary while at the same time affecting the uterus
very little in
cases of ovarian-associated infertility.
According to the invention, the object above is achieved by the provision of
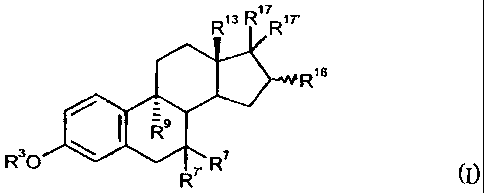
9a-substituted estra-1,3,5(10)-triene derivatives of general formula I
R13 R1 R17'
R16
R9
R3O \ R7. R7
(I)
in which radicals R3, R7, R7', R9, R13, R16 as well as R'7 and R17,
independently of one
another, have the following meaning:
R3 means a hydrogen atom or a group R18
, in which
R18 means a straight-chain or branched-chain, saturated or unsaturated
hydrocarbon radical with up to 6 carbon atoms, a trifluoromethyl
group, an aryl, heteroaryl or aralkyl radical, a substituted aryl,
heteroaryl radical with a methyl, ethyl, trifluoromethyl,
CA 02486495 2005-11-25
7
pentafluoroethyl, trifluoromethylthio, methoxy, ethoxy, nitro,
cyan, halogen (fluorine, chlorine, bromine, iodine), hydroxy,
amino, mono (Ci_8-alkyl)- or di (C1.8-alkyl)amino, whereby both
alkyl groups are identical or different, di(aralkyl)amino, whereby
both aralkyl groups are identical or different, carboxyl,
carboxyalkoxy, Cl-C20-acyl or CI-C2o--acyloxy groups as
substituents, an acyl radical COR19, in which R19 is a straight-chain
or branched-chain hydrocarbon radical with up to 10 carbon atoms
that is saturated or unsaturated in up to three places and partially or
completely halogenated, or
R18 means a group R20SO2, in which
zo 2122 21 z2
R is an R R N group, whereby R and R, independently
of one another, mean a hydrogen atom, a C1-C5 -alkyl
radical, a group C(O)R23, in which R23 represents a straight-
chain or branched-chain hydrocarbon radical with up to 10
carbon atoms that is saturated or unsaturated in up to three
places and is partially or completely halogenated, a
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or
cycloheptyl group, a C4-C 15 -cycloalkylalkyl radical with 3
to 7 carbon atoms in the cycloalkyl portion and with an
alkyl portion of up to 8 carbon atoms or an aryl, heteroaryl
or aralkyl radical, or a substituted aryl, or heteroaryl radical,
with a methyl, ethyl, trifluoromethyl, pentafluoroethyl,
trifluoromethylthio, methoxy, ethoxy, nitro, cyano, halogen
(fluorine, chlorine, bromine, iodine), hydroxy, amino,
mono(C1_8-alkyl)- or. di (C1_8-alkyl) amino, whereby both
alkyl groups are identical or different, di (aralkyl)amino,
whereby both aralkyl groups are identical or different,
CA 02486495 2005-10-19
8
carboxyl, carboxyalkoxy, C1_C20-acyl or C 1 -C20-acyloxy
groups as substituents, or, together with the N atom, a
polymethylenimino radical with 4 to 6 C atoms or a
morpholino radical,
R7 and R7', in each case independently of one another, are a hydrogen atom or
a
halogen atom,
R9 is a straight-chain or branched-chain alkenyl or alkinyl radical with 2 to
6
carbon atoms, which can be partially or completely fluorinated, or an
ethinyl- or prop- I-inyl radical,
R13 is a methyl group or an ethyl group,
R16 is a hydroxy group or a group R18O-, R20SO2- or OC(O)R23 with R18, R20
and R23 in each case in the meaning that is indicated under R3,
R17 and R17 , in each case independently of one another, are a hydrogen atom
or a
halogen atom,
R16 can in each case be in a- or a-position.
According to a variant of the invention, gonatriene derivatives are preferred,
in
which R7 and R7' are a hydrogen atom, R9 is a vinyl, ethinyl or prop- I-inyl
group, R16 is a
hydroxy group, and R17 and R17' in each case are a hydrogen atom.
In addition, the following combinations of halogen substitution, preferably
fluorine, in C-atoms 7 and 17 are preferred: 7-mono or 7-di and R17 as well as
R17' , in
each case a hydrogen, 17-mono or 17-di and R7 as well as R7', in each case a
hydrogen as
well as 7-mono/17-mono, 7-mono/17-di, 7-di/17-mono, 7-di/17-di. The 7a-
position or
the 173-position is preferred in the monofluorine compounds.
Another variant of the invention in particular calls for compounds in which
R16
stands for a group R18O- or R20SO2-O- with R18 and R20 in each case in the
meaning that
is indicated under R3.
Preferred according to this invention are the following compounds:
9a-Vinyl-estra-1,3,5(10)-triene-3,16a-diol
CA 02486495 2005-10-19
9
9a-Allyl-estra-1,3,5(10)-triene-3,16a-diol
18a-Homo-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diol
18a-Homo-9a-allyl-estra-1,3,5(10)-triene-3,16a-diol
3 -Methoxy-9a-vinyl-estra-1,3,5 (10)-trien- l 6a-ol
9a-Allyl-3 -methoxy-estra-1,3,5(1 0)-trien-16a-ol
18a-Homo-3-methoxy-9a-vinyl-estra-1,3,5(l 0)-trien- l 6a-ol
1 8 a-Homo-9a-allyl-3 -methoxy-estra-1,3,5(1 0)-trien-16a-ol
9a-(2' ,2 `-Difluorovinyl)-estra-1,3,5(1 0)-triene-3,16a-diol
9a-(2' ,2`-Difluorovinyl)-3-methoxy-estra-1,3,5(10)-trien-16a-ol
16a-Hydroxy-9a-vinyl-estra-1,3,5(l 0)-trien-3yl-sulfamate
9a-Allyl-16a-hydroxy-estra-1,3,5(l 0)-trien-3yl-sulfamate
18a-Homo- l 6a-hydroxy-9a-vinyl-estra-1,3,5(l 0)-trien-3yl-sulfamate
18 a-Homo-9a-allyl- l 6a-hydroxy-estra-1, 3, 5 (10)-trien-3 yl-sulfamate
9a-Vinyl-estra-1,3,5(10)-triene-3,16a-diyl-disulfamate
9a-Allyl-estra-1,3,5(1 0)-triene-3,16a-diyl-disulfamate
18a-Homo-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diyl-disulfamate
18a-Homo-9a-allyl-estra-1,3,5(10)-triene-3,16a-diyl-disulfamate
16a-Hydroxy-9a-vinyl-estra-1,3,5(10)-trien-3y1-(N-acetyl)-sulfamate
9a-Allyl- l 6a-hydroxy-estra-1,3,5(10)-trien-3y1-(N-acetyl)-sulfamate
18a-Homo- 16a-hydroxy-9a-vinyl-estra-1,3,5(l 0)-trien-3y1-(N-ac etyl)-
sulfamate
18a-Homo-9a-allyl- l 6a-hydroxy-estra-1,3,5(10)-trien-3y1-(N-acetyl)-sulfamate
9a-(Prop-(Z)-enyl)-estra-1,3,5(1 0)-triene-3,16a-diol
9a-(n-Propyl)-estra-1,3,5(10)-triene-3,16a-diol
9a-Ethinyl-estra- 1,3,5 (10)-triene-3,16a-diol
9a-V inyl-estra-1,3,5(1 (1 0)-triene-3,16a-diol-diacetate
18a-Homo-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diol-diacetate
1 6a-V aleroyloxy-9a-vinyl-estra-1,3,5(l 0)-trien-3 -ol
1 6a-Ac etoxy-9a-vinyl-estra-1,3,5(l 0)-trien-3 -o l
CA 02486495 2005-10-19
18a-Homo-16a-acetoxy-9a-vinyl-estra-1,3,5 (10)-trien-3-ol
7a-Fluoro-9a-vinyl-estra-1,3,5(1 0)-triene-3,16a-diol
7a-Fluoro-9a-allyl-estra-1,3,5(1 0)-triene-3,16a-diol
17 (3-Fluoro-9a-vinyl-estra-1,3,5(1 0)-triene-3,16a-diol
17(3-Fluoro-9a-allyl-estra-1,3,5(10)-triene-3,16a-diol
1 8 a-Homo-7a-fluoro-9 a-vinyl-estra-1,3,5(l 0)-triene-3,16a-dio l
1 8 a-Homo-7a-fluoro-9a-allyl-estra-1,3,5(l 0)-triene-3,16a-diol
18a-Homo- l 73-fluoro-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diol
18a-Homo-170-fluoro-9a-a11y1-estra-1,3,5(10)-triene-3,16a-diol
Other possible configurations of this invention will emerge from the
subclaims.
Hydrocarbon radical R18 is, for example, a methyl, ethyl, propyl, isopropyl,
butyl,
isobutyl, tert.-butyl, pentyl, isopentyl, neopentyl, or hexyl radical.
Alkoxy groups OR'8 in the compounds of general formula I in each case can
contain 1 to 6 carbon atoms, whereby methoxy, ethoxy, propoxy, isopropoxy and
t-
butyloxy groups are preferred.
Representatives of the C1-C5-alkyl radicals R21 and R22 are methyl, ethyl,
propyl,
isopropyl, butyl, isobutyl, tert-butyl, pentyl, isopentyl and neopentyl.
As representatives of straight-chain or branched-chain hydrocarbon radicals
R23
with 1 to a maximum of 10 carbon atoms, for example, methyl, ethyl, propyl,
isopropyl,
butyl, isobutyl, tert-butyl, pentyl, isopentyl, neopentyl, heptyl, hexyl, and
decyl can be
mentioned; methyl, ethyl, propyl and isopropyl are preferred.
As a C3-C7-cycloalkyl group, a cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl or
cycloheptyl group can be mentioned.
A C4-C15-cycloalkylalkyl radical has 3 to 7 carbon atoms in the cycloalkyl
portion;
typical representatives are the cycloalkyl groups that are mentioned directly
above. The
alkyl portion has up to 8 carbon atoms.
As examples of a C4-C15-cycloalkylalkyl radical, the cyclopropylmethyl,
cyclopropylethyl, cyclopentylmethyl, cyclopentylpropyl groups, etc., can be
mentioned.
CA 02486495 2005-10-19
11
In terms of this invention, an aryl radical is a phenyl, 1- or 2-naphthyl
radical; the
phenyl radical is preferred.
Aryl always also includes a heteroaryl radical. Examples of a heteroaryl
radical
are the 2-, 3- or 4-pyridinyl, the 2- or 3-furyl, the 2- or 3-thienyl, the 2-
or 3-pyrrolyl, the
2-, 4- or 5-imidazolyl, the pyrazinyl, the 2-, 4- or 5-pyrimidinyl or 3- or 4-
pyridazinyl
radical.
As substituents that can be present on an aryl or heteroaryl radical, for
example, a
methyl-, ethyl-, trifluoromethyl-, pentafluoroethyl-, trifluoromethylthio-,
methoxy-,
ethoxy-, nitro-, cyano-, halogen- (fluorine, chlorine, bromine, iodine),
hydroxy-, amino-,
mono(C1_8 alkyl) or di(C1_8 alkyl)amino, whereby both alkyl groups are
identical or
different, di(aralkyl)amino, whereby both aralkyl groups are identical or
different,
carboxyl, carboxyalkoxy, C1-C20-acyl or C1-C20-acyloxy groups can be
mentioned.
An aralkyl radical is a radical that contains in the ring up to 14, preferably
6 to 10,
C atoms, and in the alkyl chain 1 to 8, preferably 1 to 4, C atoms. Thus, as
aralkyl
radicals, for example, benzyl, phenylethyl, naphthylmethyl, naphthylethyl,
furylmethyl,
thienylethyl, and pyridylpropyl are suitable.
The alkyl groups or hydrocarbon radicals can be partially or completely
substituted by 1-5 halogen atoms, hydroxy groups or C1-C4-alkoxy groups.
A vinyl or allyl radical is primarily defined with a C2-C6-alkenyl radical.
A C2-C6-alkinyl radical is preferably defined as an ethinyl radical or a prop-
l -inyl
radical.
C1_lo-Acyl radicals mean, for example, acetyl, propionyl, butyryl, valeroyl,
isovaleroyl, pivaloyl, hexanoyl, octyl, nonyl, or decanoyl.
One or two hydroxyl groups at C atoms 3 and 16 can be esterified with an
aliphatic, straight-chain or branched-chain, saturated or unsaturated Ct-C14-
mono- or
polycarboxylic acid or an aromatic carboxylic acid.
Suitable as such carboxylic acids for esterification are, for example:
Monocarboxylic acids: formic acid, acetic acid, propionic acid, butyric acid,
isobutyric acid, valeric acid, isovaleric acid, pivalic acid, lauric acid,
myristic acid, acrylic
CA 02486495 2005-10-19
12
acid, propionic acid, methacrylic acid, crotonic acid, isocrotonic acid, oleic
acid, and
elaidic acid.
Esterification with acetic acid, valeric acid or pivalic acid is preferred.
Dicarboxylic acids: oxalic acid, malonic acid, succinic acid, glutaric acid,
adipic
acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, maleic acid,
fumaric acid,
muconic acid, citraconic acid, and mesaconic acid.
Aromatic carboxylic acids: benzoic acid, phthalic acid, isophthalic acid,
terephthalic acid, naphthoic acid, o-, m- and p-toluic acid, hydratropic acid,
atropic acid,
cinnamic acid, nicotinic acid, and isonicotinic acid.
Esterification with benzoic acid is preferred.
As prodrugs, the esters of the 9a-substituted estratrienes according to the
invention have advantages compared to the unesterified active ingredients with
respect to
their method of administration, their type of action, strength and duration of
action.
Especially the sulfamates of 9a-substituted estratrienes according to the
invention
have pharmacokinetic and pharmacodynamic advantages. Related effects were
already
described in other steroid-sulfamates (J. Steroid Biochem. Molec. Biol, 55,
395-403
(1995); Exp. Opinion Invest. Drugs 7, 575-589 (1998)).
In this patent application, steroids on which the 9a-substituted estra-
1,3,5(10)-
triene skeleton is based are described for the treatment of estrogen receptor
(3-mediated
disorders and conditions as selective estrogens, which have in-vitro
dissociation with
respect to their binding to estrogen receptor preparations from rat prostates
and rat uteri
and which have in vivo preferably a dissociation with respect to ovary action
in
comparison to uterus action. In addition, the compounds have a certain
protective action
against hormone-deficiency-induced bone mass loss.
It was found that the 9a-substituted estra-1,3,5(10)-trienes according to
general
formula I are suitable as selective estrogens for the treatment of various
conditions and
disorders that are characterized by a higher content of estrogen receptor 0
than estrogen
receptor a in the corresponding target tissue or target organ.
The invention also relates to pharmaceutical preparations that contain at
least one
CA 02486495 2005-10-19
13
compound of general formula I (or physiologically compatible addition salts
with organic
and inorganic acids thereof) and the use of the compounds of general formula I
for the
production of pharmaceutical agents, especially for the indications mentioned
below.
The new selective estrogens that are described here can be used as individual
components in pharmaceutical preparations or in combination especially with
gestagens.
Especially preferred is the combination of selective estrogens with ERa-
selective
antiestrogens that are peripherally-selectively active, i.e., that do not pass
through the
blood-brain barriers, as well as with selective estrogen receptor modulators
(SERM). The
ERR-selective compounds according to the invention can be used in particular
for the
production of pharmaceutical agents for treating fertility disorders, for
prevention and
therapy of prostate hyperplasia, for prevention and treatment of hormone-
deficiency-
induced mood swings in women and men and for use in hormone replacement
therapy
(HRT) in men and women.
A therapeutic product that contains an estrogen and a pure antiestrogen for
simultaneous, sequential or separate use for the selective estrogen therapy of
perimenopausal or postmenopausal conditions is already described in EP-A 0 346
014.
Because of their dissociation of action in the ovary in comparison to the
action of
the uterus, the substances and the pharmaceutical agents that contain them are
especially
suitable for the treatment in the case of ovarian dysfunction that is caused
by surgery,
medication, etc., such as female infertility for stimulation of
folliculogenesis for treatment
by itself in terms of enhanced fertility, for supporting in-vitro fertility
treatment (IVF) in
connection with an in-vivo treatment and for treatment of ovarian-induced
disorders in
later age ("late fertility") as well as for treatment of hormone-deficiency-
induced
symptoms.
The substances are also suitable for therapy of ovarian diseases such as
polycystic
ovarian syndrome, POF (premature ovarian failure) syndrome, and ovulation
disorders.
Finally, the compounds of general formula I can be used in connection with
selective estrogen receptor modulators (SERM) or raloxifene, specifically in
particular for
use in hormone replacement therapy (HRT) and for treatment of gynecological
disorders.
CA 02486495 2005-10-19
14
The substances are also suitable as individual components for the treatment of
perimenospausal and postmenopausal symptoms, in particular hot flashes, sleep
disturbances, irritability, mood swings, incontinence, vaginal atrophy and
hormone-
deficiency-induced mental disorders. The substances are also suitable for
hormone
substitution and for the therapy of hormone-deficiency-induced symptoms in
ovarian
dysfunction that is caused by surgery, medication, etc.
In addition, the substances can also be used to prevent hormone-deficiency-
induced bone mass loss and osteoporosis, to prevent cardiovascular system
diseases, in
particular vascular diseases such as arteriosclerosis, high blood pressure and
to prevent
hormone-deficiency-induced neurodegenerative diseases, such as Alzheimer's
disease, as
well as hormone-deficiency-induced impairment of memory and learning capacity.
In addition, the substances can be used as active ingredients in preparations
for
treating inflammatory diseases and diseases of the immune system, in
particular
autoimmune diseases, such as, e.g., rheumatoid arthritis, multiple sclerosis,
lupus,
Crohn's disease and other inflammatory intestinal diseases, inflammatory
diseases of the
skin, such as psoriasis, as well as for treating endometriosis.
In addition, the substances are effective against inflammatory diseases of the
respiratory system, the lungs and bronchial tubes, such as, e.g., asthma.
The medication is suitable for therapy and prophylaxis of estrogen-deficiency-
induced diseases both in women and in men.
In men, the compounds are especially suitable for therapy of hormone-
deficiency-
induced bone mass loss and osteoporosis, for prevention of cardiovascular
diseases, in
particular vascular diseases such as arteriosclerosis, high blood pressure and
for
prevention of hormone-deficiency-induced neurodegenerative diseases, such as
Alzheimer's disease, as well as hormone-deficiency-induced impairment of
memory and
learning capacity, and are suitable for prevention and therapy of prostate
hyperplasia.
The substances can be used for prophylaxis and therapy of age-related
dysfunctions or diseases of men. In particular, they can be used for
prevention and
treatment of an age-related drop of androgens, such as testosterone and DHEA,
as well as
CA 02486495 2005-10-19
of the growth hormone.
In addition, the medication can be used for treating inflammatory diseases and
diseases of the immune system, in particular autoimmune diseases in men, such
as, e.g.,
rheumatoid arthritis, MS (multiple sclerosis) and Crohn's disease and other
inflammatory
intestinal diseases, as well as inflammatory diseases of the respiratory
system, the lungs,
and the bronchial tubes. The amount of a compound of general formula I that is
to be
administered fluctuates within a wide range and can cover any effective
amount. On the
basis of the condition that is to be treated and the type of administration,
the amount of
the compound that is administered can be 0.01 g/kg - 100 mg/kg of body
weight,
preferably 0.04 pg/kg - 1 mg/kg of body weight, per day.
In humans, this corresponds to a dose of 0.8 gg to 8 g, preferably 3.2 g to
80 mg,
daily.
According to the invention, a dosage unit contains 1.6 g to 2000 mg of one or
more compounds of general formula I.
The compounds according to the invention and the acid addition salts are
suitable
for the production of pharmaceutical compositions and preparations. The
pharmaceutical
compositions or pharmaceutical agents contain as active ingredients one or
more of the
compounds according to the invention or their acid addition salts, optionally
mixed with
other pharmacologically or pharmaceutically active substances. The production
of the
pharmaceutical agents is carried out in a known way, whereby the known and
commonly
used pharmaceutical adjuvants as well as other commonly used vehicles and
diluents can
be used.
As such vehicles and adjuvants, for example, those are suitable that are
recommended or indicated in the following bibliographic references as
adjuvants for
pharmaceutics, cosmetics and related fields: Ullmans Encyklopadie der
technischen
Chemie [Ullman's Encyclopedia of Technical Chemistry], Volume 4 (1953), pages
1 to
39; Journal of Pharmaceutical Sciences, Volume 52 (1963), page 918 ff., issued
by
Czetsch-Lindenwald, Hilfsstoffe fair Pharmazie and angrenzende Gebiete
[Adjuvants for
Pharmaceutics and Related Fields]; Pharm. Ind., Issue 2, 1961, p. 72 and ff.:
Dr. H. P.
CA 02486495 2005-10-19
16
Fiedler, Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik and angrenzende
Gebiete
[Dictionary of Adjuvants for Pharmaceutics, Cosmetics and Related Fields],
Cantor KG,
Aulendorf in Wurttemberg 1971.
The compounds can be administered orally or parenterally, for example
intraperitoneally, intramuscularly, subcutaneously or percutaneously. The
compounds can
also be implanted in the tissue.
For oral administration, capsules, pills, tablets, coated tablets, etc., are
suitable. In
addition to the active ingredient, the dosage units can contain a
pharmaceutically compatible
vehicle, such as, for example, starch, sugar, sorbitol, gelatin, lubricant,
silicic acid, talc, etc.
For parenteral administration, the active ingredients can be dissolved or
suspended
in a physiologically compatible diluent. As diluents, very often oils with or
without the
addition of a solubilizer, a surfactant, a suspending agent or an emulsifying
agent are used.
Examples of oils that are used are olive oil, peanut oil, cottonseed oil,
soybean oil, castor oil
and sesame oil.
The compounds can also be used in the form of a depot injection or an implant
preparation, which can be formulated so that a delayed release of active
ingredient is made
possible.
As inert materials, implants can contain, for example, biodegradable polymers,
or
synthetic silicones such as, for example, silicone rubber. In addition, for
percutaneous
administration, the active ingredients can be added to, for example, a patch.
For the production of intravaginal systems (e.g., vaginal rings) or
intrauterine
systems (e.g., pessaries, coils, IUDs, Mirena(R) that are loaded with active
compounds of
general formula I for local administration, various polymers are suitable,
such as, for
example, silicone polymers, ethylene vinyl acetate, polyethylene or
polypropylene.
To achieve better bio-availability of the active ingredient, the compounds can
also
be formulated as cyclodextrin clathrates. For this purpose, the compounds are
reacted with
a-, (3-, or y-cyclodextrin or derivatives of the latter (PCT/EP95/02656).
According to the invention, the compounds of general formula I can also be
encapsulated with liposomes.
CA 02486495 2005-10-19
17
Methods
Estrogen Receptor Binding Studies:
The binding affinity of the new selective estrogens was tested in competitive
experiments with use of 3H-estradiol as a ligand to estrogen receptor
preparations from
rat prostates and rat uteri. The preparation of prostate cytosol and the
estrogen receptor
test with prostate cytosol was carried out as described by Testas et al.
(1981) (Testas, J. et
al., 1981, Endocrinology 109: 1287-1289).
The preparation of rat uterus cytosol as well as the receptor test with the ER-
containing cytosol were basically performed as described by Stack and Gorski,
(1985)
(Stack, Gorski 1985, Endocrinology 117, 2024-2032) with some modifications as
described in Fuhrmann et al. (1995) (Fuhrmann, U. et al. 1995, Contraception
51: 45-52).
The substances that are described here have higher binding affinity to the
estrogen
receptor of rat prostates than to estrogen receptors of rat uteri. In this
case, it is assumed that
ERJ3 predominates in the rat prostates over ERa, and ERa predominates in rat
uteri over
ERj3. Table 1 shows that the ratio of the binding to prostate and uterus
receptors
qualitatively coincides with the quotient of relative binding affinity (RBA)
to human ER(3
and ERa of rats (according to Kuiper et al. (1996), Endocrinology 138: 863-
870) (Table 1).
CA 02486495 2005-10-19
18
Table 1
Estrogen Structure hERu hERP ER(3/ Rat uterus Rat prost. prost.
RBA* RBA* ERa ER(RBA) ER(RBA) ER/uterus
ER
Estradiol 100 100 1 100 100 1 Ch- Estrone 60 37 0.6 3 2 0.8
17a-Estra- 58 11 0.2 2.4 1.3 0.5
diol
Estriol 14 21 1.5 4 20 5
5-Andro- 6 17 3 0.1 5 50
stene-diol
Genisteine " 5 36 7 0.1 10 100
Coumes- 94 185 2 1.3 24 18
trol o
*Cited from: Kuiper et al. (1996), Endocrinology 138: 863-870
Table 2 shows the results for 4 of the 9a-vinyl-estra-1,3,5(10)-triene-3,16a-
diol
derivatives (compounds 1; 2; 4; 5) according to the invention.
CA 02486495 2005-10-19
19
Table 2
Compound RBA RBA
Rat Uterus Rat Prostate
9a-Vinyl-estra-1,3,5(10)- 1.2 100
3,16a-diol (1)
9a-Vinyl-estra-1,3,5(10)- 2 200
17F-3,16a-diol (2)
9a-Di-F-Vinyl-estra- 0.2 4
1,3,5(10)-3,16a-diol (4)
9a-Di-F-Vinyl-estra- 0.2 6
1,3,5(10)-13-Methyl-3,16a-
diol (5)
Compounds 1; 2; 4; 5 according to the invention show a higher binding affinity
to
the estrogen receptor of rat prostates than to the estrogen receptor of rat
uteri.
In addition, the predictability of the prostate-ER versus the uterus-ER test
system
was confirmed with respect to tissue-selective action by in-vivo studies.
Substances with
a preference for prostate-ER are dissociated in vivo preferably with respect
to ovary and
uterus action as well as pituitary gland action in favor of action on the
ovary.
Studies for Dissociation of Action of the Ovary/Uterus and Pituitary Gland
The studies with respect to the action on uterus growth and ovulation
(indirect
effect by influencing the secretion of pituitary gland hormones) are performed
on adult
female rats (body weight of 220-250 g). The substances are subcutaneously
administered
four times on four consecutive days. The first administration is carried out
in the
metestrus. One day after the last administration, the autopsy is carried out.
The number
of ovocytes in the tube (effect on the ovulation) as well as the uterus weight
are
determined.
CA 02486495 2005-10-19
While estradiol produces a dose-dependent ovulation inhibition and an increase
in
uterus weight with an ED50 of 0.004 mg/kg of body weight, substance 1
according to the
invention up to a dose of 0.4 mg/kg of body weight does not exert any effect
on ovulation
and uterus weight.
Ovary Studies:
The substances were tested in vivo on hypophysectomized juvenile rats. In a
modification of this operative method, a GnRH antagonist is administered to
the animals.
It is examined whether the substance stimulates follicular proliferation
(maturation) in
the ovary. The ovary weight is the measurement parameter.
In each case, five animals (body weight 40-50 g) are assigned randomly to the
treatment groups. The animals are fed as much as they want with a standard
diet
(altromin) in Makrolon cages in air-conditioned rooms with a lighting program
(10 hours
of darkness, 14 hours of light) and are given acidified tap water to drink.
For the s.c
administration, the test substance as well as the control substance (estradiol
E2) are
dissolved in benzylbenzoate/castor oil (1+4 v/v).
Juvenile female rats are either hypophysectomized on day 0 and subcutaneously
treated (administration 1 x daily) from day 1 to day 4 with estradiol,
compound 1 or 2
according to the invention, or subcutaneously treated (administration 1 x
daily) with a
vehicle (castor oil/benzyl benzoate). In the modified version of the method,
0.5
mg/animal/day of Cetrorelix is administered to the animals simultaneously with
compound 2 or the vehicle and the control substance estradiol over four days
of treatment.
In both cases, the animals are sacrificed 24 hours after the last
administration, and the
ovary weight is determined.
0.5 mg/animal/day of compound 1 that is administered subcutaneously over 4
days
produces a comparable increase in ovary weight in hypophysectomized animals
like
estradiol with a dose of 0.1 mg/animal/day. The vehicle does not produce any
effect.
Substance 1 according to the invention thus shows a clear dissociation of
action in
the ovary in comparison to the uterus action and the pituitary gland action
and is
CA 02486495 2005-10-19
21
excellently suited for the preferred indication, the treatment of female
infertility, because
of its follicle-stimulating action.
In the GnRH antagonist-treated animals, concentrations of 0.1 and 0.3
mg/animal/day of compound 2 in the ovary already show the same action as the
dose of 1
mg/animal/day of estradiol that is used (Fig. 1). Even lower dosages (0.01,
0.03
mg/animal/day) show an ovary action and can eliminate the antagonistic effect
of
Cetrorelix (Fig. 2).
Ovar
180
= + Cetrorelix
0 160-
140-
120-
a) 100-
U 80
Li. 60
20
0
Veh Veh Sub 3 Sub 2 Sub 2 Sub 2
1 mg 0,1 mg 0,3 mg 1,0 mg
Fig. 1: Change in the ovary weight under the influence of a GnRH antagonist in
the
treatment with estradiol (Sub3) or various dosages of compound 2
[Key to Fig. 1:]
Ovar = Ovary
Feuchtgewicht (%) = Moist weight (%)
0,1 mg = 0.1 mg
0,3 mg=0.3mg
1,0 mg= 1.0 mg
CA 02486495 2005-10-19
22
Ovar
180
160 + Cetrorelix
o
01
140
120
a) 100
U 80
-
U
20
0
Veh Veh Sub 2 Sub 2 Sub 2 Sub 2
0,003mg 0,01 mg 0,03mg 0,1 mg
Fig. 2: Positive effect of compound 2 in low dosage on the ovary weight during
a
combination treatment with the GnRH antagonist Cetrorelix
[Key to Fig. 2:]
Ovar = Ovary
Feuchtgewicht (%) = Moist weight (%)
0,003 mg = 0.003 mg
0,01 mg = 0.01 mg
0,03 mg = 0.03 mg
0,1 mg = 0.1 mg
Substance 2 according to the invention thus also shows a clearly positive
action on
the ovary by stimulating the follicular maturation and therefore is also
suitable for the
treatment of female sub- or infertility.
CA 02486495 2005-10-19
23
Production of the Compounds According to the Invention
For the production of the compounds of general formula I according to the
invention, primarily two synthesis strategies that can generally be applied
are used.
On the one hand, in particular 3,16-protected derivatives of estra-1,3,5(10)-
triene-
3,164-diols, but also optionally the free diols, can be used for modifications
of individual
positions of the steroid skeleton.
On the other hand, correspondingly modified estrone analogs, which can be
obtained in large numbers in known ways [for a typical synthesis process, see
J. Chem.
Soc. Perk. 1, 1973, 2095 for C(9); Steroids 54, 1989, 71 for C(7)], include a
flexible
access to the compounds according to the invention by transposition of oxygen
functionality (Z. Chem. 1970, 221) from C(17) to C(16).
For the case of the 3-methyl ether, after the ketone is converted into a
sulfonyl
hydrazone, the formation of the C(16)-C(17) olefin (Z. Chem. 1970, 10, 221 ff;
Liebigs
Ann. Chem. 1981, 1973 ff), in which hypobromide is stored in a regio-/stereo-
controlled
way, is carried out in the simplest case by reaction with phenyl
sulfonylhydrazide, in a
degradation reaction. Reductive dehalogenation and removal of the protective
group of
C(3) yield the 160-alcohol, which can be converted according to known methods
into the
16a-epimer.
Another variant for the introduction of the hydroxyl group at C-atom 16 is in
the
hydroboration of the 16(17)-double bond with sterically exacting boranes. It
is known of
this reaction that it results in 16-oxidized products (Indian J. Chem. 1971,
9, 287-8).
Consequently, the reaction of estra-1,3,5(10),16-tetraenes with, for example,
9-
borabicyclo[3.3.1]nonane after oxidation with alkaline hydrogen peroxide
yields 16a-
hydroxyestratrienes. To a lesser extent, the epimeric 160-hydroxy steroids are
formed in
this reaction. After the cleavage of the 3-methoxy group, estra-1,3,5(10)-
triene-3,16a-
diols are obtained. By inversion of the configuration at C-atom 16, e.g., by
Mitsunobu
reaction (synthesis 1980, 1), in turn the 160-hydroxyestratrienes are
obtained.
CA 02486495 2005-10-19
24
For further production possibilities of the C(16)-C(17) olefinic intermediate
stage,
see also DE 199 06 159 Al.
The introduction of fluorine substituents is carried out via nucleophilic
substitution reactions of hydroxyl groups with fluoroamine reagents (Org.
React. 1974,
21, 158-173). If the hydroxyl groups are converted into the corresponding
tosylates in
advance, then the fluorinated compounds are obtained by reaction with tetra-n-
butylammonium fluoride Q. Chem. Res. (M) 1979, pp. 4728-4755). Fluorine
compounds
are also accessible by reaction of corresponding alcohols with diethylamino
sulfur
trifluoride (DAST) (US 3 976 691). Geminal difluorine compounds are produced,
for
example, by reaction of carbonyl compounds with sulfur tetrafluoride (US 3 413
321) or
diethylamino sulfur trifluoride (DAST) (US 3 979 691).
For synthesis of the 9a-substituted 17(3-fluoroestra-1,3,5(10)-triene-3,16-
diols
according to the invention, 17-oxo-estral,3,5(10)-trienes are converted into
the 17,17-
difluoroestra-1,3,5(10)-trienes (US 3 976 691). The thus accessible 17,17-
difluoroestra-
1,3,5(10)-trienes are converted by treatment with aluminum oxide into 17-
fluoroestra-
1,3,5(10),16-tetraene (US 3 413 321). Another possibility for the production
of fluoro-
olefins exists in the reaction of the corresponding ketones with diethylamino
sulfur
trifluoride (DAST) in the presence of polar catalysts, such as fuming sulfuric
acid (US 4
212 815). The reaction of 17-fluoroestra-1,3,5(10),16-tetraenes with boranes
and
subsequent oxidation with alkaline hydrogen peroxide yields the 17(3-
fluoroestra-
1,3,5(10)-trien-16a-ols (Org. React. 1963, 13, 1-54).
Access to the 9a-alkenyl- or 9a-alkinyl-substituted estra-1,3,5(10)-triene-
3,16a-
diols according to the invention is carried out first from the 3,16-dihydroxy-
estra-
1,3,5(10)-trienes that are protected in 3- and 16-position. By reaction with
trimethyl silyl
cyanide in the presence of lithium perchlorate, the regio- and stereoselective
introduction
of a 9a-cyano grouping (Synlett, 1992, 821-2) is carried out. After the
protective groups
are cleaved, the 9a-cyan compound is converted by reduction first into a 9a-
formyl
compound and then by a Wittig reaction (Org. React. Vol. 14, 270) into the 9a-
alkenyl-
or 9a-alkinyl-substituted compound.
CA 02486495 2005-10-19
The estratriene sulfamates according to the invention are accessible in a way
that
is known in the art from the corresponding hydroxy steroids by esterification
with
sulfamoyl chlorides in the presence of a base [Z. Chem. 15, 270-272 (1975);
Steroids 61,
710-717 (1996)].
Subsequent acylation of the sulfamate group results in the (N-acyl)sulfamates
according to the invention. For the (N-acyl)sulfamates, pharmacokinetic
advantages were
already detected (cf. DE 195 40 233 Al).
The regioselective esterification of polyhydroxylated steroids with N-
substituted
and N-unsubstituted sulfamoyl chlorides is carried out after partial
protection of those
hydroxyl groups that are to remain unesterified. Silyl ethers have turned out
to be
protective groups with selective reactivity that is suitable for this purpose
since these silyl
ethers are stable under the conditions of sulfamate formation, and the
sulfamate group
remains intact when the silyl ether(s) is (are) again cleaved for regeneration
of the
(residual) hydroxyl group(s) still contained in the molecule (Steroids 61, 710-
717 (1996)).
The production of the sulfamates according to the invention with an additional
hydroxyl group in the molecule is also possible in that the starting material
is suitable
hydroxy-steroid ketones. First, depending on the goal, one or more hydroxyl
groups that
are present are subjected to sulfamoylation. Then, the sulfamate groups
optionally can be
converted with a desired acyl chloride in the presence of a base into the (N-
acyl)sulfamates in question. The now present oxosulfamates or oxo-(N-
acyl)sulfamates
are converted by reduction into the corresponding hydroxysulfamates or hydroxy-
(N-
acyl)sulfamates (Steroids 61, 710-717 (1996)). Sodium borohydride and the
borane-
dimethyl sulfide complex are considered as suitable reducing agents.
The examples below are used for a more detailed explanation of the invention.
Analogously to the degradation of the 9a-vinyl grouping, other compounds of
general formula I can be obtained with use of reagents that are homologous to
the
reagents that are described in the examples.
Etherification and/or esterification of free hydroxy groups is carried out
according
to the methods that are common to one skilled in the art.
CA 02486495 2005-10-19
26
Example 1
9a-Vinylestra-1,3,5(10)-triene-3,16a-diol
Stage 1
9a-Cyano-3-methoxy-estra-1, 3, 5(10)-trien-16a--yl-acetate
A solution of 2.21 g (9.73 mmol) of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
in 80 ml of methylene chloride is added drop by drop while being stirred to a
suspension
that consists of 2.13 g (6.49 mmol) of 3-methoxy-estra-1,3,5(10)-trien-16a-yl-
acetate,
2.07 ml (16.54 mmol) of trimethylsilyl cyanide and 0.14 g of lithium
perchlorate in 100
ml of methylene chloride. The reaction mixture is green in color. After 1 hour
of
reaction time at room temperature, the mixture is mixed with sodium
bicarbonate
solution. The separated organic phase is washed with water and concentrated by
evaporation in a vacuum. The product mixture is chromatographed on silica gel
(cyclohexane/ethyl acetate, 6/1). 0.44 g (21%) of 9a-cyano-3-methoxy-estra-
1,3,5(10)-
trien-16a-yl-acetate is obtained.
Stage 2
9a-Cyano-3-hydroxy-estra-1, 3, 5(10)-trien-] 6a-yl-acetate
7.51 g (50.1 mmol) of sodium iodide and 8.87 ml (70.14 mmol) of
trimethylchlorosilane are added to a solution that consists of 0.59 g (1.67
mmol) of 9a-
cyano-3-methoxy-estra-1,3,5(10)-trien-16a-yl-acetate in 30 ml of acetonitrile
while being
stirred in an argon atmosphere. After about 3 hours at 60-70 C, the reaction
is completed.
The reaction solution is added to sodium hydrogen sulfite solution and
extracted with
ethyl acetate. The organic phase is washed several times with water, dried on
MgSO4 and
concentrated by evaporation in a vacuum to the dry state.
The crude product is chromatographed on silica gel (cyclohexane/ethyl acetate,
4/1). 0.43 g (76%) of product is obtained.
CA 02486495 2005-10-19
27
Stage 3
3,1 6a-Dihydroxyestra-1, 3, 5(10)-triene-9 carbonitrile
At room temperature, 0.43 g (1.27 mmol) of 9a-cyano-3-hydroxy-estra-1,3,5(10)-
trien-16a-yl-acetate is stirred for 2 hours with 1.0 g (7.24 mmol) of
potassium carbonate
in 40 ml of methanol (1 % water). Then, the methanol is distilled off in a
vacuum, and the
organic residue is dissolved in methylene chloride. The organic phase is
washed with
water and concentrated by evaporation. 3.5 g (93%) of 9a-cyano-estra-1,3,5(10)-
triene-
3,16a-diol is obtained.
Stage 4
3,1 6a-Dihydroxyestra-1, 3, 5(10)-triene-9 carbaldehyde
A suspension that consists of 100 mg (0.34 mmol) of 3,16a-dihydroxyestra-
1,3,5(10)-triene-9 carbonitrile in 40 ml of toluene is cooled to about -20 C
while being
stirred. After 0.9 ml (1.35 mmol) of diisobutylaluminium hydride is added, the
reaction is
mixed after about 10 minutes with sodium bicarbonate solution, filtered over
Celite, and
the filtering adjuvant is extracted again with ethyl acetate. The combined
organic phases
are washed with water. By concentration by evaporation of the solution in a
vacuum,
84.6 mg of a light yellow foam is obtained. The product that is contained in
the mixture
corresponds to a yield of about 52% of theory and is used without further
chromatographic working-up in the next stage.
Stage 5
9a- Vinylestra-1, 3,5(l 0)-triene-3,16a-diol
Under inert-gas atmosphere, 3.1 g (7.9 mmol) of triphenylmethyl phosphonium -
iodide and 0.24 g (8 mmol) of sodium hydride (80% in paraffin oil) in 20 ml of
DMSO in
an ultrasound bath are brought to reaction at about 55 C. After 10 minutes, 80
mg (0.16
mmol, about 60%) of 3,16a-dihydroxyestra-1,3,5(10)-triene-9 carbaldehyde is
added to
the solution, and the mixture is allowed to react for 60 more minutes at about
55 C in an
ultrasound bath. After water is added, it is extracted with ethyl acetate. The
collected
CA 02486495 2005-10-19
28
organic phases are washed with water, and the organic phase is concentrated by
evaporation in a vacuum.
The crude product is purified by column chromatography on silica gel
(cyclohexane/ethyl acetate, 2/1) and subsequent recrystallization from
chloroform. Yield:
24 mg (50%), melting point 88-95 C.
1H-NMR (400 MHz, DMSO-d6, TMS): 9.00 (s, 3-OH); 6.98 (d, J = 8.6 Hz, H-1);
6.49 (dd, J = 8.6/2.7 Hz, H-2); 6.41 (d, J = 2.7 Hz, H-4); 6.25 (dd, J =
17.2/10.5 Hz, -
CH=CH2); 5.00 (dd, 10.5/1.9 Hz, -CH=CH2); 4.47 (d, 4.69 Hz, 16a-OH); 4.45 (dd,
17.2/1.9 Hz, -CH=CH ; 4.24 (m, 163-H); 2.68 (m, H-6); 0.69 (s, H-18)
Example 2
9a-Vinyl-18a-homo-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,16a-Bis[(perhydropyran-2 yl)oxy]-]8a-homo-estra-1, 3,5(l 0)-triene-9-
carbonitrite
1.03 g (2.26 mmol) of 3,16a-bis[(perhydropyran-2-yl)oxy]-18a-homo-estra-
1,3,5(10)-triene, 48.2 mg (0.45 mmol) of lithium perchlorate and 0.71 ml (5.66
mmol) of
trimethylsilyl cyanide are introduced into 10 ml of methylene chloride
(molecular sieve)
and cooled under inert gas to about -70 C while being stirred. Then, 0.77 g
(3.39 mmol)
of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, dissolved in 65 ml of methylene
chloride,
is added in drops within 1 hour. After about 1 hour (heating to room
temperature), the
reaction solution is mixed with sodium bicarbonate solution, and the reaction
products are
extracted with methylene chloride. The crude product that is obtained by
concentration
by evaporation of the organic phases is purified by chromatography. After
chromatography on silica gel (cyclohexane/ethyl acetate, 4/1), 0.74 g (68% of
theory) of
product is obtained.
CA 02486495 2005-10-19
29
Stage 2
3,1 6a-Dihydroxy-18a-homo-estra-1, 3, 5(10)-triene-9-carbaldehyde
1.3 g (2.7 mmol) of 3,16a-bis[(perhydropyran-2-yl)oxy]-18a-homo-estra-
1,3,5(10)-triene-9-carbonitrile is dissolved in 40 ml of toluene and mixed at
room
temperature under inert gas with 7.2 ml (10.8 mmol) of diisobutylaluminum
hydride
solution (1.5 M in toluene). After a reaction time of 30 minutes, a mixture of
30 ml of
methanol and 5 ml of dilute hydrochloric acid (1/1) is added to the reaction
solution. The
reaction solution is concentrated by evaporation under vacuum, and the residue
is taken
up in ethyl acetate. The organic phase that is obtained is extracted with
water and washed
with sodium bicarbonate solution. After the solution is dried, and after
concentration by
evaporation under vacuum, 0.73 g (86% of theory) of yellow crystals is
obtained.
Stage 3
9a- Vinyl- I 8a-homo-estra-1, 3,5(l 0)-triene-3,16a-diol
Under inert gas atmosphere, 13.7 g (34.8 mmol) of triphenylmethyl-phosphonium
iodide and 1.0 g (34.8 mmol) of sodium hydride (about 80% on paraffin oil) in
80 ml of
DMSO is brought to reaction in an ultrasound bath at about 50 C. After 30
minutes, 0.73 g
(2.3 mmol) of 3,16a-dihydroxy-18a-homo-estra-1,3,5(10)-triene-9-carbaldehyde,
dissolved
in 10 ml of DMSO, is added to the reaction solution, and the mixture is
allowed to react in
an ultrasound bath for another 60 minutes. After water is added, it is
extracted with ethyl
acetate, the organic phase is washed with water, dried and concentrated by
evaporation.
The crude product is purified by column chromatography on silica gel
(cyclohexane/ethyl acetate, 2/1) and crystallization from chloroform.
Yield: 0.59 g (81% of theory) after chromatography
Melting point: 214 - 220 C
1H-NMR (400 MHz, DMSO-d6, TMS): 9.00 (s, 3-OH); 6.96 (d, J = 8.6 Hz, H-1);
6.49 (dd, J = 8.6/2.7 Hz, H-2); 6.41 (d, J = 2.7 Hz, H-4); 6.29 (dd, J =
17.2/10.5 Hz, -
CH=CH2); 5.00 (dd, J = 10.5/1.9 Hz, -CH=CH2); 4.48 (d, J = 4.7 Hz, 16a-OH);
4.43 (dd,
J = 17.2/1.9 Hz, -CH=CH2); 4.18 (m, 16(3-H); 2.68 (m, H-6); 0.72 (t, J = 6.8
Hz, H-18a)
CA 02486495 2005-10-19
Example 3
9a-(2' ,2 `-Difluorovinyl)-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,]6a-Bis[(perhydropyran-2 yl)oxyJ-estra-1,3,5(10)-triene-9-carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-estra-1,3,5(10)-triene
analogously to Example 1, stage 1 yields 3,16a-bis[(perhydropyran-2-yl)oxy]-
estra-
1, 3, 5 (10)-triene-9-carbonitrile.
Yield: 58% of theory
Stage 2
3,1 6x-Dihydroxy-estra-1, 3, 5(10)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-estra-1,3,5(10)-triene-9-
carbonitrile analogously to Example 1, stage 2 yields 3,16a-dihydroxy-estra-
1,3,5(10)-
triene-9-c arbaldehyde.
Yield: 83% of theory
Stage 3
9a-(2, 2-Difluorovinyl)-estra-1, 3, 5(10)-triene-3,16a-diol
1.5 ml of dimethoxyethane (molecular sieve), 0.3 ml of pentane and 0.13 ml
(0.77
mmol) of diethyl(difluoromethyl)-phosphonate are introduced into a reaction
flask that
was rendered inert, and cooled to about -75 C. After 0.72 ml (1.07 mmol) of
tert-
butyllithium (1.5 M in pentane) is added and after 30 minutes of reaction
time, 0.14 g
(0.31 mmol) of 3,16a-dihydroxy-estra-1,3,5(10)-triene-9-carbaldehyde,
dissolved in a
mixture of 1.5 ml of dimethoxyethane/0.3 ml of pentane, is added to the
reaction solution.
The reaction solution is refluxed until the reaction is completed. After being
added into
cooled ammonium chloride solution, it is extracted with ethyl acetate. The
organic phase
is concentrated by evaporation under vacuum, the residue is taken up in 5 ml
of methanol
CA 02486495 2005-10-19
31
and mixed with 0.5 ml of dilute hydrochloric acid (1/1). Ethyl acetate is
added to the
reaction solution, the organic phase is washed with sodium bicarbonate
solution and
concentrated by evaporation under vacuum. The crude product that is obtained
is purified
by column chromatography on silica gel (cyclohexane/ethyl acetate, 2/1).
Yield: 22 mg (21% of theory)
'H-NMR (400 MHz, DMSO-d6, TMS): 9.08 (s, 3-OH); 7.10 (d, J = 8.6 Hz, H-1);
6.51 (dd, J = 8.6/2.3 Hz, H-2); 6.41 (d, J = 2.3 Hz, H-4); 4.76 (dd, J =
25.4/10.9 Hz, -
CH=CF2); 4.51 (d, J = 4.69 Hz, 16a-OH); 4.25 (m, 16(3-H); 2.68 (m, H-6); 0.68
(s, H-18)
Example 4
9a-(2 `,2 `-Difluorovinyl)-18a-homo-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,16a-Bis[(perhydropyran-2 yl)oxy]-18a-homo-estra-1,3,5(10)-triene-9-
carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-18a-homo-estra-1,3,5(10)-triene
analogously to Example 1, stage 1 yields 3,16a-bis[(perhydropyran-2-yl)oxy]-
18a-
homoestra-1,3,5(10)-triene-9-carbonitrile.
Yield: 58% of theory.
Stage 2
3,1 6x-Dihydroxy-18a-homo-estra-1, 3, 5(10)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-18a-homo-estra-1,3,5(10)-triene-
9-carbonitrile analogously to Example 1, stage 2 yields 3,16a-dihydroxy-18a-
homo-estra-
1,3,5(10)-triene-9-carbaldehyde.
Yield: 87% of theory
CA 02486495 2005-10-19
32
Stage 3
9a-(2, 2-Diuuorovinyl)-18a-homo-estra-1, 3, 5(10)-triene-3,16a-diol
Reaction of 3,16a-dihydroxy-18a-homo-estra-1,3,5(10)-triene-9-carbaldehyde;
Reaction conditions and execution of the reaction as well as molar ratios as
in the 3ra
stage of 9a-(2,2-difluorovinyl)-estra-1,3,5(10)-triene-3,16a-diol.
The crude product is purified by column chromatography on silica gel
(cyclohexane/ethyl acetate, 2/1) and crystallization from ethyl acetate.
Yield: 12% of theory
Melting point: 225 - 232 C
1H-NMR (400 MHz, DMSO-d6, TMS): 9.06 (s, 3-OH); 7.08 (d, J = 8.6 Hz, H-1);
6.50 (dd, J = 8.6/2.7 Hz, H-2); 6.41 (d, J = 2.7 Hz, H-4); 4.78 (dd, J =
21.5/14.8 Hz, -
CH=CF2); 4.47 (d, J = 4.50 Hz, 16a-OH); 4.18 (m, 16(3-H); 2.68 (m, H-6); 0.72
(t, J =
6.8 Hz, H-18a)
Example 5
170-Fluoro-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,1 6x-Bis[(perhydropyran-2 yl)oxy]-17/3-fluoro-estra-1,3,5(10)-triene-9-
carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-1713-fluoro-estra-1,3,5(10)-
triene
analogously to Example 2, stage 1 yields 3,16a-bis[(perhydropyran-2-yl)oxy]-
17(3-fluoro-
estra-1, 3, 5 (10)-triene-9-carbonitrile.
Yield: 45% of theory
Stage 2
3,16a-Dihydroxy-17/3 f luoro-estra-1, 3, 5(10)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-17(3-fluoro-estra-1,3,5(10)-
triene-9-carbonitrile analogously to Example 2, stage 2 yields 3,16a-dihydroxy-
17(3-
CA 02486495 2005-10-19
33
fluoro-estra-1,3,5(10)-triene-9-carbaldehyde.
Yield: 83% of theory
Stage 3
17[3-Fluoro-9a-vinyl-estra-1, 3,5(l 0)-triene-3,16a-diol
Reaction of 3,16a-dihydroxy-17(3-fluoro-estra-1,3,5(10)-triene-9-carbaldehyde
analogously to Example 2, stage 3 yields 17(3-fluoro-9a-vinyl-estra-1,3,5(10)-
triene-
3,16a-diol.
The crude product is purified by column chromatography on silica gel
(cyclohexane/ethyl acetate, 2/1) and crystallization from chloroform.
Yield: 51 % of theory
Melting point: 94 - 98 C
'H-NMR (400 MHz, DMSO-d6, TMS): 9.02 (s, 3-OH); 6.97 (d, J = 8.2 Hz, H-1);
6.51 (dd, J = 8.2/2.7 Hz, H-2); 6.42 (d, J = 2.7 Hz, H-4); 6.22 (dd, J =
17.2/10.5 Hz, -
CH=CH2); 5.09 (d, J = 5.5 Hz, 16a-OH); 5.01 (dd, J = 10.5/1.9 Hz, -CH=CH2);
4.45 (dd,
J = 17.2/1.9 Hz, -CH=CH2); 4.35 (dd, J = 55.1/4.7 Hz, H-17a); 4.11 (m, 16(3-
H); 2.68 (m,
H-6); 0.79 (d, J = 1.9 Hz, H-18)
Example 6
17,17-Difluoro-9a-vinyl-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,1 6a-Bis[(perhydropyran-2 yl)oxy]-17,17-difluoro-estra-1,3,5(10)-triene-9-
carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-17,17-difluoro-estra-1,3,5(10)-
triene analogously to Example 2, stage 1 yields 3,16a-bis[(perhydropyran-2-
yl)oxy]-
17,17-difluoro-estra-1,3,5(10)-triene-9-carbonitrile.
Yield: 46% of theory
CA 02486495 2005-10-19
34
Stage 2
3,16a-Dihydroxy-17,17-d fuoro-estra-1, 3, 5(10)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-17,17-difluoro-estra-1,3,5(10)-
triene-9-carbonitrile analogously to Example 2, stage 2 yields 3,16a-dihydroxy-
17,17-
difluoro-estra-1,3,5(10)-triene-9-carbaldehyde.
Yield: 88% of theory
Stage 3
17,1 7-Difluoro-9a-vinyl-estra-1, 3, 5(10)-triene-3,16a-diol
Reaction of 3,16a-dihydroxy-17,17-difluoro-estra-1,3,5(10)-triene-9-
carbaldehyde
analogously to Example 2, stage 3 yields 17,17-difluoro-9a-vinyl-estra-
1,3,5(10)-triene-
3,16a-diol.
The crude product is purified by column chromatography on silica gel
(cyclohexane/ethyl acetate, 2/1) and crystallization from chloroform.
Yield: 75% of theory
1H-NMR (400 MHz, CDC13, TMS): 7.08 (d, J = 8.6 Hz, H-1); 6.63 (dd, J =
8.6/2.7 Hz, H-2); 6.54 (d, J = 2.7 Hz, H-4); 6.23 (dd, J = 17.2/10.5 Hz, -
CH=CH2); 5.08
(dd, J = 10.5/1.9 Hz, -CH=CH2); 4.48 (dd, J = 17.2/1.9 Hz, -CH=CH ; 4.44 (m,
16(3-H);
2.79 (m, H-6); 0.95 (d, J = 1.9 Hz, H-18)
Example 7
9a-(Hex-1 `-enyl)-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,16a-Bis[(perhydropyran-2 yl)oxy]-estra-1,3,5(10)-triene-9-carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-y1)oxy]-estra-1,3,5(10)-triene
analogously to Example 2, stage 1 yields 3,16a-bis[(perhydropyran-2-yl)oxy]-
estra-
1,3, 5 (10)-triene-9-carbonitrile.
CA 02486495 2005-10-19
Yield: 61 % of theory
Stage 2
3,1 6a-Dihydroxy-estra-1, 3,5(l 0)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-estra-1,3,5(10)-triene-9-
carbonitrile analogously to Example 2, stage 2 yields 3,16a-dihydroxy-estra-
1,3,5(l 0)-
triene-9-carbaldehyde.
Yield: 87% of theory
Stage 3
9a-(Hex-l -enyl)-estra-1, 3, 5(10)-triene-3,16a-diol
8.68 g (20 mmol) of pentyltriphenyl-phosphonium bromide + sodium amide (1 g
contains 2.3 mmol of pentyltriphenyl-phosphonium bromide), 0.2 g (0.67 mmol)
of
3,16a-dihydroxy-estra-1,3,5(10)-triene-9-carbaldehyde and 30 ml DMSO are
introduced
into a reaction flask that was rendered inert. The reaction mixture is treated
for about 2
hours in an ultrasound bath at 60 C. After the reaction is completed, water is
added to the
reaction solution. The crude product is isolated by extraction with ethyl
acetate, washing
of the organic phase with water and concentration by evaporation until a dry
state is
reached.
The crude product that is obtained is purified by column chromatography on
silica
gel (cyclohexane/ethyl acetate, 1/1) and crystallization from ethyl acetate.
Yield: 0.18 g (75% of theory) according to chromatography
Melting point: 166 - 168 C
'H-NMR (400 MHz, DMSO-d6, TMS): 8.97 (s, 3-OH); 7.08 (d, J = 8.6 Hz, H-1);
6.49 (dd, J = 8.6/2.7 Hz, H-2); 6.41 (d, J = 2.7 Hz, H-4); 5.73 (d, J = 12.5
Hz, -CH=CH-
CH2-); 5.20 (dt, J = 12.5/7.4 Hz, -CH=CH-CH2-); 4.48 (d, J = 4.7 Hz, 16a-OH);
4.24 (m,
16(3-H); 2.66 (m, H-6); 0.68 (t, J = 7.0 Hz, CH3-CH2-); 0.66 (s, H-18)
CA 02486495 2005-10-19
36
Example 8
9a-(But-1 `-enyl)-estra-1,3,5(10)-triene-3,16a-diol
Stage 1
3,16a-Bis[(perhydropyran-2 yl)oxy]-estra-1,3,5(10)-triene-9-carbonitrile
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-estra-1,3,5(10)-triene
analogously to Example 2, stage 1 yields 3,16a-bis[(perhydropyran-2-yl)oxy]-
estra-
1,3,5 (10)-triene-9-carbonitrile.
Yield: 52% of theory
Stage 2
3,1 6a-Dihydroxy-estra-1, 3, 5(10)-triene-9-carbaldehyde
Reaction of 3,16a-bis[(perhydropyran-2-yl)oxy]-estra-1,3,5(10)-triene-9-
carbonitrile analogously to Example 2, stage 2 yields 3,16a-dihydroxy-estra-
1,3,5(10)-
triene-9-carbaldehyde.
Yield: 87% of theory
Stage 3
9a-(But-1-enyl)-estra-1, 3,5(l 0)-triene-3,16a-diol
8.68 g (20 mmol) of propyltriphenyl-phosphonium bromide + sodium amide (1 g
contains 2.3 mmol of propyltriphenyl-phosphonium bromide), 0.2 g (0.67 mmol)
of
3,16a-dihydroxy-estra-1,3,5(10)-triene-9-carbaldehyde and 30 ml of DMSO are
introduced into a reaction flask that was rendered inert. The reaction mixture
is treated
for about 2 hours in an ultrasound bath at 60 C. After the reaction is
completed, water is
added to the reaction solution. The crude product is isolated by extraction
with ethyl
acetate, washing of the organic phase with water, and concentration by
evaporation until a
dry state is reached.
CA 02486495 2005-10-19
37
The crude product that is obtained is purified by column chromatography on
silica
gel (cyclohexane/ethyl acetate, 1/1).
Yield: 0.16 g (73% of theory) after chromatography
Melting point: 140 -148 C
'H-NMR (400 MHz, DMSO-d6, TMS): 8.98 (s, 3-OH); 7.09 (d, J = 8.6 Hz, H-1);
6.49 (dd, J = 8.6/2.7 Hz, H-2); 6.41 (d, J = 2.7 Hz, H-4); 5.70 (d, J = 12.5
Hz,
-CH=CH-CH2-); 5.19 (dt, J = 12.5/7.4 Hz, -CH=CH-CH2-); 4.47 (d, J = 4.7 Hz,
16(X-OH);
4.24 (m, 16(3-H); 2.66 (m, H-6); 0.66 (s, H-18); 0.57 (t, J = 7.2 Hz, CH3-CH2-
)