Note: Descriptions are shown in the official language in which they were submitted.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
NEW COMPOUNDS USEFUL FOR THE TREATMENT OF OBESITY,
TYPE II DIABETES AND CNS DISORDERS
RELATED APPLICATIONS
This application claims priority to Swedish application number 0201925-5,
filed on June
20, 2002, Swedish application number 0202908-0, filed on October l, 2002,
Swedish
application number 0202181-4, filed on July 11, 2002, Swedish application
number
0300357-1, filed on February 10, 2003, U.S. provisional application
60/406,120, filed on
August 26, 2002, U.S. provisional application 60/434,010, filed on December
17, 2002,
and U.S. provisional application 60/464,701, filed on April 23, 2003, the
contents of which
are incorporated herein by reference.
TECHNICAL FIELD
The present invention relates to substituted sulphone and sulphonamide
compounds, to
pharmaceutical compositions comprising these compounds, and to the use of the
compounds for the prophylaxis and treatment of medical conditions relating to
obesity,
type 2 diabetes, and/or disorders of the central nervous system (CNS), to
achieve reduction
of body weight and of body weight gain, as well as for cosmetic use.
BACKGROUND ART
Obesity is a condition characterized by an increase in body fat content
resulting in excess
body weight above accepted norms. Obesity is the most important nutritional
disorder in
the western world and represents a major health problem in all industrialized
countries.
This disorder leads to increased mortality due to increased incidences of
diseases such as
cardiovascular disease, digestive disease, respiratory disease, cancer and
type 2 diabetes.
Searching for compounds, which reduce body weight has been going on for many
decades.
One line of research has been activation of serotoninergic systems, either by
direct
activation of serotonin receptor subtypes or by inhibiting serotonin reuptake.
The exact
receptor subtype profile required is however not known.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Serotonin (5-hydroxytryptamine or 5-HT), a key transmitter of the peripheral
and central
nervous system, modulates a wide range of physiological and pathological
functions,
including anxiety, sleep regulation, aggression, feeding and depression.
Multiple serotonin
receptor subtypes have been identified and cloned. One of these, the 5-HT6
receptor, was
cloned by several groups in 1993 (Ruat, M. et al. (1993) Biochem. Biophys.
Res.
Commun.193: 268-276; Sebben, M. et al. (1994) NeuroReport 5: 2553-2557). This
receptor is positively coupled to adenylyl cyclase and displays affinity for
antidepressants
such as clozapine. Recently, the effect of 5-HT6 antagonist and 5-HT6
antisense
oligonucleotides to reduce food intake in rats has been reported (Bentley,
J.C. et al. (1999)
Br J Pharmac. Suppl. 126, P66; Bentley, J.C. et al. (1997) J. Psychopharmacol.
Suppl.
A64, 255; Woolley M.L. et al. (2001) Neuropharmacology).
Compounds with enhanced affinity and selectivity for the 5-HT6 receptor have
been
identified, e.g. in WO 00/34242 and by Isaac, M. et al. (2000) 6-
Bicyclopiperazinyl-I-
arylsulfonylindoles and 6-Bicyclopipe~idinyl-1-a~ylsulfonylindoles derivatives
as novel,
potent and selective S-HT6 receptor antagonists. Bioorganic & Medicinal
Chemistry
Letters 10: 1719-1721 (2000).
INFORMATION DISCLOSURE
J. Med. Chem. 1970, 13(4), 592-598 describes N-(4-{[2-
(diethylamino)ethyl]amino}-1-
naphthyl)amides; N-{5,6,7,8-Tetrahydro-4-[(3-piperidinopropyl)amino]-1-
naphthyl]amides and related amides and urea derivatives as schistosomicides.
WO 99/42465 discloses sulphonamides derivatives that bind to the 5-HT6
receptor and that
can be used for the treatment of CNS disorders such as anxiety, depression,
epilepsy,
obsessive compulsive disorders, cognitive disorders, ADHD, anorexia and
bulimia
schizophrenia, drug abuse.
WO 01/32646 A1 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders and which inter alia may be used for the
treatment of
eating disorders.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
WO 99/37623 A2 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders and which inter alia may be used for the
treatment of
eating disorders.
WO 99/42465 A3 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders and which inter alia may be used for the
treatment of
eating disorders.
EP 0 815 861 A1 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders.
WO 99/02502 A2 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders and which inter alia may be used for the
treatment of
eating disorders.
WO 98/27081 A1 discloses compounds that bind to the 5-HT6 receptor and that
are used
for the treatment of CNS disorders and which inter alia may be used for the
treatment of
eating disorders.
EP 0701819 discloses compounds that bind to the 5-HT1D receptor and that are
used for
the treatment of CNS disorders and obesity.
US 6,191,141 and WO 01/12629 disclose compounds that bind to the 5-HT6
receptor and
that are used for the treatment of CNS disorders.
DISCLOSURE OF THE INVENTION
It has surprisingly been found that the compounds of formula (I) show affinity
for the 5-
HT6 receptor as antagonists at low nanomolar range. Compounds according to the
invention and their pharmaceutically acceptable salts have 5-HT6 receptor
antagonist,
agonist and partial agonist activity and are believed to be of potential use
in the treatment
or prophylaxis of obesity and type 2 diabetes, to achieve reduction of body
weight and of
body weight gain, as well as in the treatment or prophylaxis of disorders of
the central
nervous system such as anxiety, depression, panic attacks, memory disorders,
cognitive
disorders, sleep disorders, migraine, anorexia, bulimia, binge disorders,
obsessive
compulsive disorders, psychoses, Alzheimer's disease, Parkinson's disease,
Huntington's
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
chorea and/or schizophrenia, Attention Deficit Hyperactive Disorders (ADHD),
drug
abuse. The reduction of body weight and of body weight gain (e.g. treating
body-weight
disorders) is achieved inteY alia by reduction of food intake. As used herein,
the term
"body weight disorders" refers to the disorders caused by an imbalance between
energy
intake and energy expenditure, resulting in abnormal body (e.g., excessive)
weight. Such
body weight disorders include obesity.
Defihitio~es
Unless otherwise stated or indicated, the term "Ci_6 alkyl" (or "CZ_6
alkenyl") denotes a
straight or branched hydrocarbon chain group having from 1 to 6 carbon atoms
(or 2 to 6
carbon atoms). Examples of said lower alkyl include methyl, ethyl, n-propyl,
iso-propyl, n-
butyl, iso-butyl, sec-butyl, t-butyl and straight- and branched-chain pentyl
and hexyl.
Alkenyl groups have one or more double carbon-carbon bonds in the chain.
Unless otherwise stated or indicated, the term "C1_g alkoxy" denotes a
straight or branched
alkoxy group having from 1 to 6 carbon atoms. Examples of said lower alkoxy
include
methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, iso-butoxy, sec-butoxy, t-
butoxy and
straight- and branched-chain pentoxy and hexoxy.
Unless otherwise stated or indicated, the term "C1_6 alkoxyalkyl" denotes a
straight or
branched alkoxyalkyl group having from 1 to 6 carbon atoms. Examples of said
lower
alkoxyalkyl include methoxymethyl, ethoxymethyl, iso-propoxymethyl, n-
butoxymethyl, t-
butoxyethyl and straight- and branched-chain pentoxymethyl.
The expression "C2_6 alkenyl" as used herein refers to straight-chained and
branched
alkenyl groups containing from 2 to 6 carbon atoms. Typical examples include
vinyl, allyl,
2,3-dimethylallyl, 1-butenyl groups, 1-pentenyl, and 1-hexenyl groups.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
The expression "C2_6 alkynyl" as used herein refers to straight-chained and
branched
alkynyl groups containing from 2 to 6 carbon atoms. Typical examples include
ethynyl, 1-
propynyl, 1-butynyl, 1-pentynyl, and 1-hexynyl groups.
Unless otherwise stated or indicated, the term "halogen" shall mean fluorine,
chlorine,
bromine or iodine.
The term "alkylhalide" refers to an alkyl group substituted with one or more
halogen
groups (e.g., F, Cl, Br, I).
The term "C3_~ cycloalkyl" denotes a cyclic alkyl group having a ring size
from C3 to C~,
which can be saturated or partially unsaturated. Examples of said cycloalkyl
include
cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclopentadienyl,
cyclohexyl,
methylcyclohexyl, cyclohexenyl, cyclohexadienyl, and cycloheptyl.
The term "CS_10 cycloalkenyl" denotes a cyclic alkenyl group having a ring
size from CS
to C10. Examples of said cycloalkenyl include 1-cyclopentyl, 2-cyclopentenyl,
1-
cyclohexenyl, 1-cyclohepentyl, 1-cyclooctenyl, 1-cyclononenyl, and 1-
cyclodecenyl
groups.
The term "heterocyclic" refers to a hydrocarbon ring system containing 4 to 8
ring
members that have at least one heteroatom (e.g., S, N, or O) as part of the
ring. It includes
saturated, unsaturated, aromatic, and nonaromatic heterocycles. Suitable
heterocyclic
groups include thienyl, furyl, pyridyl, pyrrolidinyl, imidazolyl, pyrazolyl,
piperidyl,
azepinyl, morpholinyl, pyranyl, dioxanyl, pyridazinyl, pyrimidinyl, and
piperazinyl groups
Unless otherwise stated or indicated, the term "aryl" refers to a hydrocarbon
ring system
having at least one aromatic ring. Examples of aryl groups include phenyl,
cinnamyl,
pentalenyl, indenyl, 1-naphthyl, 2-naphthyl, anthryl and phenanthryl.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
The term "heteroaryl" refers to a hydrocarbon ring system having at least one
aromatic ring
which contains at Ieast one heteroatom such as O, N, or S. Examples of
heteroaryl groups
include furyl, pyrrolyl, thienyl, oxazolyl, imidazolyl, thiazolyl, pyridinyl,
pyrimidinyl,
quinazolinyl, and indolyl groups.
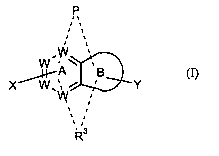
Compounds of Formula (I)
One object of the present invention is a compound having the general formula
(I):
P
~Aw
X W,~~ ~ ' Y
'' ~3
R (I)
or a pharmaceutically acceptable salt thereof, wherein:
W~.
W
Y D Y
wii
ring B is W or ; in which D is a five-membered heterocyclic or
heteroaryl ring, said heteroaryl ring comprising one or two atoms selected
from the group
consisting of nitrogen, sulfur and oxygen, with the proviso that when D
contains an oxygen
atom, D is heteroaryl;
each W is independently N-, -(CH)-, or -C- provided that not more than three
groups W
are N- in both rings A and B together;
P is any one of formula (a), (b) or (c)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
R~ R~ R~
p O \N R2
RZ N ~; O ~S ~, ~~ Y
' , ' ,or ' ;
(a) (b) (c)
wherein x = 0, 1, or 2 and y = 0, 1, or 2;
and P and R3 can be attached to any carbon atom that allows the substitution
in one of
either the A- or B-ring, or when ring A contains at least one nitrogen atom
and P is (c),
then P can also be attached to any nitrogen in ring B that allows the
substitution;
the dashed bonds denote that P and R3, respectively, may be attached to either
the A or B
ring; but each P or R3 may not be simultaneously bound to both rings A and B;
Rl is
(a) C1_6 alkyl,
(b) C 1 _6 alkoxyalkyl,
(c) straight-chained or branched C1_6 hydroxyalkyl,
(d) straight-chained or branched C1_6 alkylhalides,
(e) aryl carbonylmethyl,
(f) C3_~ cycloalkyl, which is optionally partially unsaturated,
(g) C3_~ cycloalkyl-Cl_6 alkyl, wherein the cyclic ring is optionally
partially unsaturated, or
(h) a group Ar;
wherein Ar is
(a) phenyl,
(b) 1-naphthyl,
(c) 2-naphthyl,
(d) arYl_Cl_6 alkyl,
(e) cinnamyl,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(f) a 5 to 7-membered, optionally aromatic, partially saturated or completely
saturated,
mono- or bi-cyclic heterocyclic ring, each containing 1 to 4 heteroatoms,
selected from
oxygen, sulfur, and nitrogen,
(g) a bicyclic ring system comprising at least one heterocyclic ring according
to (f) and a
group Ar,
wherein the group Ar is substituted in one or more positions with
(a) H, X or Y, or
(b) a 5 to 7-membered, optionally aromatic, partially saturated or completely
saturated, heterocyclic ring each containing 1 to 4 heteroatoms selected from
oxygen, nitrogen or sulfur;
RZ is
(a) H,
(b) C 1 _6 alkyl,
(c) C2_6 alkoxyalkyl,
(d) straight or branched Ci_6 hydroxyalkyl, or
(e) straight or branched Cl_6 alkylhalides;
(f) a group Ar,
or Rl and RZ are linked to form a group -CHZCHZOCH2CH2- or
\N
(CH )v
2
wherein v is 0-2,
X and Y are independently
(a) H,
(b) halogen,
(c) Ci_6 allcYl,
(d) CF3,
(e) hydroxy,
(f) C1_6 alkoxy,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(g) C2_6 alkenyl,
(h) phenyl,
(i) phenoxy,
(j) benzyloxy,
(k) benzoyl,
(1) -OCF3,
(m) -CN,
(n) straight or branched C1_6 hydroxyalkyl,
(o) straight or branched Ci_6 alkylhalides,
(p) -NHa,
(r) NR4R5~
(S) -N02a
(t) -CONR4R5,
(u) -NHSOaR4,
(v) NR4COR5,
(x) -SOZNR4R5,
(z) -C(=O)R4,
(aa) -C02R4, or
(ab) -S(O)"R4, wherein n is 0, 1, 2 or 3,
(ac) -S-(Cl_6) alkyl, or
(ad) -SCF3; and
R4 and RS are independently
(a) H,
(b) C1_6 alkyl,
(c) C3-~ cycloalkyl, or
(d) Ar, as defined above for Rl;
alternatively, R4 and RS are linked to form a group -CH20CH2-, -CH2CHZOCH2CHz-
or
(CH2) 3_s;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
R3 is a group selected from any one of
__N_. __N . _N__. O N-R
Rq ~ m ~ N Rq Rq
N ~N N, 6 N
Is R Is N
R m R Rs
__. _. N-R
Ra ~ Rq ~ ,. Ra ~ Ra
N N N N N-Rs
Rs Rs Rs Rs Rs
Oy, .. ~ , im
O O ,.
n Rq
Rq ~~m ~] R ~ ~m
m
Rs~ ~ Rs~NwRs
__ ~ Oy, it
0 0'~ o
n N ~n
N <~ ~ N and N
RG N ~ ~ m
~m R~ Rq ,m Rq~O
s
wherein R3 is optionally substituted on each carbon atom that allows the
substitution with Rq groups, wherein Rq is independently H, or (C1_6) allcyl,
and
wherein two Rq groups can be present on the same carbon atom simultaneously,
wherein
q=1,2,3,4,5or6,
m = 1 or 2, and
n = 0, 1 or 2;
R6 is independently
(a) H,
(b) lineax or branched C1_6 alkyl,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(c) benzyl,
(d) -CHa-CHa-OH, or
(e) -CHZ-CHz-O-C1_6 alkyl;
P and R3 can be attached to the same ring or to different rings of rings A
acid B;
provided that
R~ R~
i
R~ N,O
R2 N ~; S;_ O
when P is (a) or r ~ (b), and P and R3 both are attached to ring A in the
meta- or para-position relative to one another then R3 is selected from any
one of
O '~ ~ O OJ:. __t_.
O
n
Rq N~~m N ~m Rq ~ RAN ~mq
Rs Rs Rs Rs
__N~R -N-R ',,' .\ ',
Rq / Rq ~ Rq
N N_Rs N N
Rs Rs Rs Rs
OW
p
~n ~ O
N ~n N
w
N N a
Rq Rq~~ ~ m R
1 ~m O ~m Rs
0
R~
.,O
wow S~ O
C ~ W Y R? N'~;
when rmg B is w , and P is ' (a), then P and R3 are simultaneously
attached to the same ring A or B;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
R~
. . C ,W Y ~ y ,
when ring B is W , and P is ' , wherein y = 0, then P and R3 are
attached to the different rings of rings A and B;
when the ring system A + B is benzofurane or benzothiophene, and P is
R~
~v
,.
(c)
and attached to position 3 in the A+B ring system, and R3 is a group selected
from any one
of
_ _~__
N N
~Rq
N m
N~ ~
Rs m N
and attached to position 7 in the A+B ring system, then y =1 or 2;
when the ring system A + B is indole, and P is
R~
5~~~" ~Y
,.
' (C)
and P is attached to position 3 in the A+B ring system, and R3 is a group
selected from any
one of
_ \ _. _ N_ _N _ __N
/ Rq ~ Rq ~ / Rq C and
N N N m
N~ ~
Rs Rs Rs m N
and R3 is attached to any one of positions 5, 6 or 7 in the A+B ring system,
then y = 1 or 2;
or
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
W~
~W
i
~w
when ring B is W and R1= Ar is partially saturated bi-cyclic heterocyclic ring
containing a N atom, the N atom in Ar cannot be attached to the S atom in P;
with the proviso that:
when rings A and B are both phenyl, P is any one of formula (a) or (c)
substituted in
position 7 on the naphthalene ring, then R3 is not substituted in position 1
on the
naphthalene ring; and with the proviso that:
when ring D is a pyrrole ring, P is of the formula (c), then R3 is not of the
formula
Ra J m Ra °r Ra
N ~N
Rs Rs Ns
Jm R
substituted in position 3 on the pyrrole ring.
A naphthalene ring has the following position numbers:
P5 P4
P6 / \ Pa
\ I ~
P2
wherein P1-P8 denote the position on the naphthalene ring.
A pyrrole ring, as connected to an A ring, has the following position numbers:
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
P3
.W
W ~ P2
W vj N
i
P~
wherein P1-P3 denote the position on the pyrrole ring.
It is preferred that:
Rl is
(a) C 1 _6 alkyl, or
(e) a group Ar;
Ar is
(a) phenyl,
(b) 1-naphthyl,
(c) 2-naphthyl, or
(f) a 5 to 7-membered, optionally aromatic, partially saturated or completely
saturated,
heterocyclic ring containing 1 to 4 heteroatoms, selected from oxygen,
nitrogen and sulfur,
wherein the group Ar is substituted in one or more positions with
(a) H,
(b) halogen,
(c) Ci-6 alkyl,
(d) -CF3,
(f) Ci_6 alkoxy,
(g) CZ_6 alkenyl (preferably C2_q, alkenyl),
(1) -OCF3,
(m) straight or branched C1_6 hydroxyalkyl,
(n) phenyloxy,
(o) benzyloxy,
(V) NR4COR5,
(x) -SO2~4RSa
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(z) -C(=O)R4,
(ab) -S(O)nR4, wherein n is 0, 1, 2 or 3;
(ac) -S-(C1_6) alkyl, or
(ad) -SCF3;
R2 is
(a) H, or
(b) C i _6 alkyl;
or R1 and R2 are linked to form a group -CH2CH~,OCHaCHa-;
X and Y are H;
R4 and RS are each independently H or Cl_3 alkyl; and
R3 is selected from any one of
_. _.
N
~~m / Rq ~Rq
N N N
Rs Rs Rs
__
n R N-R
Jn s
Rq ~ m Rq ~ RAN ~ m Rq
N ~m N N I s N
Rs Rs R ~d I s
R
wherein R3 can be substituted on each carbon atom that allows the substitution
with
Rq groups, wherein Rq is independently H, or Cl_6 alkyl, and wherein two Rq
groups can be present on the same carbon atom simultaneously, wherein
q =1 or 2,
m=1 or2,
n = 0, and
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
R6 is independently
(a) H,
(b) C1_6 alkyl (preferably Cl_3 alkyl), in particular methyl,
(d) -CHa-CHz-OH, or
(e) -CHZ-CH2-OCH3.
It is especially preferred that R3 is selected from any one of
_. _ . o~''~
N _ O ,,.
Rq~ Ra \~ Ra ~ Rq ~ ~ m
~m
Rs Rs Rs Rs and N.Rs
wherein R3 can be substituted on each carbon atom that allows the substitution
with
Rq groups, wherein Rq is independently H, or C1_~ alkyl, and wherein two Rq
groups
can be present on the same carbon atom simultaneously, wherein
q = 1 or 2,
m = 1 or 2; and
R6 is independently
(a) H,
(b) C1_3 alkyl,
(d) -CH2-CHa-OH, or
(e) -CHZ-CHZ-OCH3.
It is also preferred that R3 is selected from any one of
__ O~ __E_ __
n R N-R
!n s l a
R ~m Ra ~ RAN 1m Ra
a 1
N Jm N N ~ s N
Rs R ~d Rs
wherein R3 can be substituted on each carbon atom that allows the substitution
with
Rq groups, wherein Rq is independently H, or C1_6 alkyl, and wherein two Rq
groups
can be present on the same carbon atom simultaneously, wherein
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
q =1 or 2,
m =1 or 2,
n = 0, and
R6 is independently
(a) H,
(b) C1_3 alkyl,
(d) -CHI-CH2-OH, or
(e) -CHZ-CH2-OCH3.
It is also preferred that R3 is selected from any one of
N N N N N
C>;~C~~C~: ~ C~:
N N N
N N
Rs Rs Rs Rs Rs
-N - -N _ __N_ _ _
c ~c ~< > >; >;
N
Rs Rs Rs N Rs Rs
R6 is independently
(a) H,
(b) C1_3 alkyl,
(d) -CHZ-CH2-OH, or
(e) -CH2-CHa-OCH3.
It is preferred that R6 is H or methyl.
It is also preferred that R3 is piperazine; homopiperazine; 2,6-
dimethylpiperazine; 3,5-
dimethylpiperazine; 2,5-dimethylpiperazine; 2-methylpiperazine; 3-
methylpiperazine;
2,2-dimethylpiperazine; 3,3-dimethylpiperazine; piperidine; 1,2,3,6-tetrahydro-
pyrazine; or
4-pyrrolidin-3-yloxy.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
It is preferred that the groups Y and X are attached to any unsubstituted
carbon atom.
It is preferred that D is pyrrolyl, thienyl or furanyl.
It is preferred that P is
R~
,.
. (c)
wherein Rl, x, and y are as defined in claim 1.
It is also preferred that P is
R'
R' I _
2
Sip p\N R
(a) or ~ (b)
wherein Rl and R2 are as defined in claim 1.
It is preferred that R~ is H.
Another object of the present invention is a compound of the general formula
(II)
R~
x , , R~ (I~
wherein Rl, x, y, X, and Y are as defined in claim 1, and R3 is as defined in
claim 2.
It is preferred that y = 0 and x = 2.
Another object of the present invention is a compound of the general formula
(III)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
R~
S~~~x
~N
Y
_ / 'Y
X
R3 (I~
wherein Rl, x, y, X, and Y are as defined in claim l, and R3 is as defined in
claim 2.
It is preferred that y = 0 and x = 2
Another object of the present invention is a compound of the general formula
(IV)
X N D Y
Rs P
wherein P is of the formula (c), R1, x, y, X, and Y are as defined in claim 1,
and R3 is as
defined in claim 2, and
wherein D is a five-membered heteroaryl ring, said ring comprising one or two
atoms
selected from the group consisting of nitrogen, sulfur and oxygen; and when
the heteroaryl
ring comprises one or two nitrogen atoms, a group R6 is attached at any
nitrogen atom
which allows the substitution.
It is preferred that D is a thiophene and P is attached to the D ring, giving
a skeleton as any
of the following:
N/ ~ ~ Ni ~ N/ ~ S
, S
\ S/ \ w \
It is also preferred that D is pyrrole and P is attached to the nitrogen atom
in the D ring,
giving a skeleton as any of the following:
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H
N/ \ N/ ~ N/ N
~NH
\ ~ N \ ~ \
H
It is also preferred that D is furan and P is attached to the D ring, giving a
skeleton as any
of the following:
N/ N/ ~ N/ O
v
O
\ ~ \ \ ~ \
O
Another object of the present invention is a compound of the general formula
(V)
P
\~
X D Y
R3O
wherein P is of the formula (c) as defined in claim 1, Rl, x, y, X, Y, and R3
are as defined
in claim 1, and
wherein D is a five-membered heteroaryl ring, said heteroaryl ring comprising
one or two
atoms selected from the group consisting of nitrogen, sulfur and oxygen; and
when the
heteroaryl ring comprises one or two nitrogen atoms, a group R6 is attached at
any nitrogen
atom which allows the substitution.
Another object of the present invention is a compound of the general formula
(V)
P
\~
X D Y
a
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein R2 is H, X,
Y, and R3 are as defined in claim l, and
wherein D is a five-membered heteroaryl ring, said heteroaryl ring comprising
one or two
atoms selected from the group consisting of nitrogen, sulfur and oxygen; and
when the
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
heteroaryl ring comprises one or two nitrogen atoms, a group R6 is attached at
any nitrogen
atom which allows the substitution.
Another object of the present invention is a compound of the general formula
(VI)
N ~ P
R3 / S Y
X (V~
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein R2 is H, X
and Y are as defined in claim 1, and R3 is as defined in claim 2.
Another object of the present invention is a compound of the general formula
(VII)
N ~ P
R3 / ~ Y
X (VII)
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein R2 is H, X
and Y are as defined in claim 1, and R3 is as defined in claim 4.
Another object of the present invention is a compound of the general formula
(VIII)
N ~ N P
R3 '~
/ ~Y
(VIII)
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein R2 is H, X,
Y, and R3 are as defined in claim 1.
Another object of the present invention is a compound of the general formula
(IX)
N ~ P
R3 / N Y
X \R7
wherein R' in formula (IX) is:
(a) H,
~) Ci-6 alkyl,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(c) benzyl,
(d) -CHa-CHZ-OH, or
(e) CHZ-CH2-O-CH3, and
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein R2 is H, X,
Y, and R3 are as defined in claim 1.
Another object of the present invention is a compound of the general formula
(X)
N ~ \
R3
N Y
X \P
(X)
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein Ra is H, X,
Y, and R3 are as defined in claim 1.
Another obj ect of the present invention is a compound of the general formula
(XI)
P
/ 'Y
X 3
R (XI).
wherein P is of the formula (a) or (b) as defined in claim 1, preferably
wherein Ra is H, X
and Y are as defined in claim 1, and R3 is as defined in claim 4.
Another object of the present invention is a compound of the general formula
(XII):
P
,,
,,
,,
, ,
W.W ,
Y-H- A
W ~~ ~ O
,,
R3
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
or a pharmaceutically acceptable salt thereof, wherein P and R3 are attached
to the same
ring or to different rings of rings A and B, wherein A, B, Y, P, and R3 are as
defined in
claim 1.
Preferred compounds of the formula (Ilk are
6-Benzenesulfonyl-4-piperazin-1-yl-quinoline hydrochloride;
6-[(2-Fluorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-(1-Naphthylsulfonyl)-4-piperazin-1-ylquinoline hydrochloride;
6-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(3,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(2-Chloro-6-methylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(2-Methyl,4-tent-butyl-phenyl)sulfonyl]-4-piperazin-1-ylquinoline
hydrochloride;
6-[(3,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(2,3-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(4-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
6-[(4-Isopropylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride;
(4-Piperazin-1-yl-6- f [4-(trifluoromethyl)phenyl]sulfonyl}quinoline
hydrochloride;
6-[(4-tent-Butylphenyl)sulfonyl]-4-(1,4-diazepan-1-yl)quinoline hydrochloride;
and
4-(1,4-Diazepan-1-yl)-6-[(4-isopropylphenyl)sulfonyl]quinoline hydrochloride.
Preferred compounds of the formula (III) are
7-(2-Chloro-6-methyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline
hydrochloride;
7-(2-t-Butyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride;
7-(3,4-Dichloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride;
7-(2,4-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride;
7-(2,5-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride;
7-(p-Chloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride;
7-Benzenesulfonyl-1-[1,4]diazepan-1-yl-isoquinoline,hydrochloride;
7-(4-tert-Butyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline,
hydrochloride;
7-(2-Chloro-6-methyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline
hydrochloride;
7-(3,5-Dimethyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
7-(3,4-Dichloro-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline
hydrochloride;
7-(4-Chloro-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline hydrochloride;
7-(3,4-Dimethyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline
hydrochloride;
7-(2-tert-Butyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline
hydrochloride;
7-Benzenesulfonyl-1-piperazin-yl-isoquinoline hydrochloride; and
7-(4-tert-Butyl-benzenesulfonyl-1-piperazin-yl-isoquinoline hydrochloride
Preferred compounds of the formula (1V) are
4-(1,4-Diazepan-1-yl)-2-(phenylsulfonyl)thieno[3,2-c]pyridine hydrochloride;
4-(1,4-Diazepan-1-yl)-2-[(3,4-dichlorophenyl)sulfonyl]thieno[3,2-c]pyridine
hydrochloride;
4-(1,4-Diazepan-1-yl)-2-[4-tent-butylphenylsulfonyl)thieno[3,2-c]pyridine
hydrochloride;
4-(1,4-Diazepan-1-yl)-2-[4-tent-butylphenylsulfonyl)thieno[3,2-c]pyridine
hydrochloride;
4-(1,4-Diazepan-1-yl)-2-[3,4-dimethylphenylsulfonyl)thieno[3,2-c]pyridine
hydrochloride;
2-[(4-Bromophenyl)sulfonyl]-4-(1,4-diazepan-1-yl)thieno[3,2-c]pyridine
hydrochloride;
2-(Phenylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine hydrochloride;
2-(3-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2-c]pyridine
hydrochloride;
2-(4-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2-c]pyridine
hydrochloride;
4-Pip erazin-1-yl-2- ~ [4-trifluoromethyl)phenyl] sulfonyl J thieno [3,2-
c]pyridine
hydrochloride;
2-[[2-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine-2-
sulfonamide
hydrochloride;
2-[(4-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-(1-Naphthyl sulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine hydrochloride;
2-[(3-Fluorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine-2-
sulfonamide
hydrochloride;
2-(Mesitylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine hydrochloride;
2-[(2-Methoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(2,4-Dimethoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(2,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(2,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
2-[(2-Ethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
4-(Piperazinyl)-2-(3-methoxybenzyl-sulfonyl)-thienopyridine hydrochloride;
2-(Benzylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine hydrochloride;
4-Piperazin-1-yl-2- { [4-(trifluoromethyl)b enzyl] sulfonyl} thieno [3,2-
c]pyridine
hydrochloride;
2-[(3-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(2,3-Difluorobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(4-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2- ( [2, 5-bis(Trifluoromethyl)benzyl] sulfonyl} -4-pip erazin-1-ylthieno [3,2-
c]pyridine
hydrochloride;
2-[(4-Methylbenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-~[5-Chloro-2-(trifluoromethyl)benzyl]sulfonyl}-4-piperazin-1-ylthieno[3,2-
c]pyridine
hydrochloride;
2-[(3,5-Dimethoxybenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
2-[(2-Naphthylmethyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride;
4-Piperazin-1-yl-2-~[4-(1,2,3-thiadiazol-4-yl)benzyl]sulfonyl}thieno[3,2-
c]pyridine
hydrochloride;
1-(4-Pyrrolidin-1-ylphenyl)-2-[(4-piperazine-1-ylthieno[3,2-c]pyridin-2-yl)
sulfonyl]
ethanone hydrochloride; and
1-[4-(Diethylamino)phenyl]-2-[(4-piperazine-1-ylthieno[3,2-c]pyridin-2-yl)
sulfonyl]
ethanone hydrochloride.
Also preferred compounds of the formula (IV) are
1-(4-Methylphenylsulphonyl)-4-piperazin-1-yl-1H pyrrolo[3,2-c]pyridine
hydrochloride;
1-(3-Chloro-2-methylphenylsulphonyl)-4-piperazin-1-yl-1H pyrrolo[3,2-
c]pyridine
hydrochloride;
1-(3,4-Dimethoxyphenylsulphonyl)-4-piperazin-1-yl-1H pyrrolo[3,2-c]pyridine
hydrochloride;
4-(4-Piperazin-1-yl-pyrrolo[3,2-c]pyridine-1-sulfonyl)-benzonitrile
hydrochloride;
1-(4,5-Dichloro-thiophene-2-sulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-
c]pyridine
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
1-(2-Chloro-4-fluorophenylsulphonyl)-4-piperazin-1-yl-1H pyrrolo[3,2-
c]pyridine
hydrochloride;
1-Phenylinethanesulfonyl-4-piperazin-1-yl-1H-pyrrolo [3,2-c]pyridine
hydrochloride;
1-(5-Chloro-thiophene-2-sulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine
S hydrochloride;
1-(4-Butyl-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2-c)pyridine
hydrochloride;
1-(4-Phenoxy-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2-c)pyridine
hydrochloride;
1-(Phenylsulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine hydrochloride;
1-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine
hydrochloride;
1-[(4-Methoxyphenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine
hydrochloride;
1-[(2-Methoxy-5-methylphenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-
c]pyridine
hydrochloride; and
4-Piperazin-1-yl-1- f [2-(trifluoromethyl)phenyl]sulfonyl)-1H-pyrrolo[3,2-
c]pyridine
hydrochloride.
Preferred compounds of the formula (Vn are
N-(4-Methylphenyl)-4-(4-methylpiperazin-1-yl)thieno[3,2-c]pyridine-2-
sulfonamide
hydrochloride;
2-Bromo-4-(4-methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-3-sulfonic acid p-
tolylamide
hydrochloride;
4-(4-Methylpiperazin-1-yl)-n-phenylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3-fluoro-5-
trifluoromethyl-phenyl)-
amide hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-chloro-phenyl)-amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-isopropyl-phenyl)-
amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acidp-tolylamide
hydrochloride;
4-(4-Methylpiperazin-1-yl)-N (2-cyclohex-1-en-1-ylethyl) thieno [3,2-c]
pyridine-2-
sulfonamide hydrochloride;
2-(4-(4-Methylpiperazin-1-yl) thieno [3,2-c] pyridin-2-ylsulfonyl)-1,2,3,4-
tetrahydroisoquinoline hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
4-(4-Methylpiperazin-1-yl)-N (2-thien-2-ylethyl) thieno [3,2-e] pyridine-2-
sulfonamide
hydrochloride;
4-(4-Methylpiperazin-1-yl)-N [1-(1-naphthyl) ethyl] thieno [3,2-c] pyridine-2-
sulfonamide
hydrochloride;
4-(4-Methylpiperazin-1-yl)-N (4-hexylphenyl) thieno [3,2-c] pyridine-2-
sulfonamide
hydrochloride;
N (3-Chlorobenzyl)-4-(4-methylpiperazin-1-yl) thieno [3,2-c] pyridine-2-
sulfonamide;
4-(4-Methylpiperazin-1-yl)-N [1-(4-fluorophenyl) ethyl] thieno [3,2-cJ
pyridine-2-
sulfonamide hydrochloride;
N (2,3-Difluorobenzyl)-4-(4-methylpiperazin-1-yl) thieno [3,2-c] pyridine-2-
sulfonamide
hydrochloride;
4-(4-Methylpiperazin-1-yl) N (4-chloro-2, 5-dimethoxyphenyl) thieno [3,2-cJ
pyridine-2-
sulfonamide hydrochloride;
2-Bromo-4- (4-methylpiperazin-1-yl)-N (2-cyclohex-1-en-1-ylethyl) thieno [3,2-
c]
1 S pyridine-3-sulfonamide hydrochloride;
2-Bromo-4- (4-methylpiperazin-1-yl)-N [(1,5~-1-(2-naphthyl) ethyl] thieno [3,2-
cJ
pyridine-3-sulfonamide hydrochloride;
2-Bromo-4- (4-methylpiperazin-1-yl)-N [1-(4-fluorophenyl) ethyl] thieno [3,2-
c] pyridine-
3-sulfonamide hydrochloride;
2-Bromo-4- (4-methylpiperazin-1-yl)-N (2,4,5-trimethoxyphenyl) thieno [3,2-cJ
pyridine-
3-sulfonamide;
N-(3,4-Dichlorophenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(2,4-Difluorophenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride; '
4-Piperazin-1-yl-N-[-3-(trifluoromethyl)phenyl]thieno [3,2-c]pyridine-2-
sulfonamide
hydrochloride;
N-(3-Ethylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(3,4-Dimethoxyphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(4-Bromo-2-methylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
2-(4-Piperazin-1-yl-thieno [3,2-c]pyridine-2-sulfonyl)-1,2,3,4-tetrahydro-
isoquinoline
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (2-thiophen-2-yl-ethyl)-
amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-chloro-2,5-dimethoxy-
phenyl)-
amide hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid phenethyl-amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (2,6-diethyl-phenyl)-
amide
hydrochloride;
. 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3-phenyl-propyl)-
amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3,3-diphenyl-propyl)-
amide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid [2-(5-methoxy-1H-indol-
3-yl)-
ethyl]-amide hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid 4-trifluoromethyl-
benzylamide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid benzyl-ethyl-amide
hydrochloride;
N-(3-Ethylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-3-sulfonamide
hydrochloride;
N-(4-Isopropylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-3-sulfonamide
hydrochloride;
N-(4-Methylphenyl)-4-(pyrrolidin-3-yloxy)thieno [3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(4-Methylphenyl)-4-(piperidin-4-yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(2,3-Difluorobenzyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-(3-Chlorobenzyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid phenylamide
hydrochloride;
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-tert-butyl-phenyl)-
amide
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
4-(4-Methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-2-sulfonic acid phenylamide
hydrochloride;
4-(4-Methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-2-sulfonic acid (3-chloro-
phenyl)-amide
hydrochloride;
2-Bromo-4-(4-methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-3-sulfonic acid
phenylamide
hydrochloride;
4-(4-Methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-3-sulfonic acid (4-
methylphenyl)-amide
hydrochloride; and
N-Phenyl-7-piperazin-1-ylthieno[2,3-c]pyridine-2-sulfonamide hydrochloride.
Preferred compounds of the formula (VII) are
N-(4-methylphenyl)-4-piperazin-1-ylfuro[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-phenyl-4-piperazin-1-ylfuro[3,2-c]pyridine-2-sulfonamide hydrochloride; and
N-phenyl-7-piperazin-1-ylfuro[2,3-c]pyridine-2-sulfonamide hydrochloride.
A preferred compound of the formula (VIII) is
4-Piperazin-1-yl-thiazolo[4,5-c]pyridine-2-sulfonic acid phenylamide
hydrochloride.
Preferred compounds of the formula (IX) are
N-(4-methylphenyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine-2-sulfonamide
hydrochloride;
N-phenyl-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine-2-sulfonamide
hydrochloride; and
N-phenyl-7-piperazin-1-yl-1H-pyrrolo[2,3-c]pyridine-2-sulfonamide
hydrochloride.
Preferred compounds of the formula (X) are
4-Chloro-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl]benzenesulfonamide
hydrochloride;
4-Methoxy-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl]benzenesulfonamide
hydrochloride;
5-Ghloro-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl]thiophene-2-sulfonamide
hydrochloride;
4-Chloro-N-[4~(piperidin-3-yloxy)-1-naphthyl]benzenesulfonamide hydrochloride;
4-Methoxy-N-[4-(piperidin-3-yloxy)-1-naphthyl]benzenesulfonamide
hydrochloride;
5-Fluoro-2-methyl-N-[4-(piperidin-4-yloxy)-1-naphthyl]benzenesulfonamide
hydrochloride;
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
5-Chloro-N-[4-(piperidin-4-yloxy)-1-naphthyl]thiophene-2-sulfonamide
hydrochloride;
4-Chloro-N-{4-[(3S)-pyrrolidin-3-yloxy]-1-naphthyl~benzenesulfonamide
hydrochloride;
and
4-Chloro-N- f 4-[(3R)-pyrrolidin-3-yloxy]-1-naphthyl]benzenesulfonamide
hydrochloride.
Another object of the present invention is a process for the preparation of a
compound
above, said method comprising the steps of
(a) Mitsonobu reaction of 4-nitro-1-naphthol with boc-protected 3-
hydroxypyrrolidine or
4-hydroxypiperidine;
(b) reduction of the nitro group in the nitronaphthalene obtained in step (a)
to form an
aminonaphthalene derivative; and
(c) synthesis of a sulfonamide by reacting the aminonaphthalene obtained in
step (b)
with a suitable sulfonyl chloride.
Another object of the present invention is a process for the preparation of a
compound
above, wherein
R1
O~S
P is O ~ , said method comprising the steps of
preparation of the heteroaromatic 5-member ring fused halogen-substituted
pyridine,
reduction of an aromatic vitro group; aromatic nucleophilic substitution with
a thiol via a
diazointermediate; oxidation of the thiol derivative to a sulphone;
introduction of a halogen
atom by electrophilic aromatic substitution; aromatic nucleophilic
substitution of the
halogen with a diamine.
Another object of the present invention is a process for the preparation of a
compound
above, wherein
R~
O=S=O
N-RZ
P is ~ , said method comprising the steps of: preparation of the
heteroaromatic 5-
member ring fused pyridine; introduction of a carboxylic moiety; conversion of
the
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
carboxylic moiety to amine by Curtius rearrangement; reaction of the amine
group with a
sulphonylchloride.
Another object of the present invention is a process for the preparation of a
compound
above, wherein
1
R2 NCR
O=S=O
P is ~ , said method comprising the steps of: preparation of the
heteroaromatic 5-
member ring fused pyridine; introduction of sulfonylchloride moiety by
nucleophilic
addition; reaction of sulphonylchloride moiety with an aniline to obtained a
sulfonamide;
aromatic nucleophilic substitution of the chloro with a diamine.
All diastereomeric forms possible (pure enantiomers, tautomers, racemic
mixtures and
unequal mixtures of two enantiomers) are within the scope of the invention.
Such
compounds can also occur as cis- or trans-, E- or Z double bond isomer forms.
All
isomeric forms are contemplated.
The compounds of the formulae (I) to (XII) may be used as such or, where
appropriate, as
pharmacologically acceptable salts (acid or base addition salts) thereof.
The pharmacologically acceptable addition salts as mentioned above are meant
to comprise
the therapeutically active non-toxic acid and base addition salt forms that
the compounds
are able to form. Compounds that have basic properties can be converted to
their
pharmaceutically acceptable acid addition salts by treating the base form with
an
appropriate acid. Exemplary acids include inorganic acids, such as hydrogen
chloride,
hydrogen bromide, hydrogen iodide, sulphuric acid, phosphoric acid; and
organic acids
such as acetic acid, propanoic acid, hydroxyacetic acid, lactic acid, pyruvic
acid, glycolic
acid, malefic acid, malonic acid, oxalic acid, benzenesulphonic acid,
toluenesulphonic acid,
methanesulphonic acid, trifluoroacetic acid, fumaric acid, succinic acid,
malic acid, tartaric
acid, citric acid, salicylic acid, p-aminosalicylic acid, pamoic acid, benzoic
acid, ascorbic
acid and the like. Exemplary base addition salt forms are the sodium,
potassium, calcium
salts, and salts with pharmaceutically acceptable amines such as, for example,
ammonia,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
alkylamines, benzathine, and amino acids, such as, e.g. arginine and lysine.
The term
addition salt as used herein also comprises solvates which the compounds and
salts thereof
are able to form, such as, for example, hydrates, alcoholates and the like.
For clinical use, the compounds of the invention are formulated into
pharmaceutical formulations for oral, rectal, parenteral or other mode of
administration. Pharmaceutical formulations are usually prepared by mixing the
active substance, or a pharmaceutically acceptable salt thereof, with
conventional
pharmaceutical excipients. The formulations can be further prepared by known
methods such as granulation, compression, microencapsulation, spray coating,
etc.
The formulations may be prepared by conventional methods in the dosage form of
tablets, capsules, granules, powders, syrups, suspensions, suppositories or
injections. Liquid formulations may be prepared by dissolving or suspending
the
active substance in water or other suitable vehicles. Tablets and granules may
be
coated in a conventional manner.
Another object of the present invention is a compound above for use in
therapy:
Another object of the present invention is a compound above, and for the case
when rings
A and B are both phenyl, P is any one of formula (a) or (c) substituted in
position 7 on the
naphthalene ring, and R3 is substituted in position 1 on the naphthalene ring,
for use in the
treatment or prophylaxis of a 5-HT6 receptor related disorder, such as
obesity, type II
diabetes, and/or disorders of the central nervous system, to achieve reduction
of body
weight and of body weight gain.
Another object of the present invention is a compound above for use in the
treatment or
prophylaxis of disorders of the central nervous system.
Another object of the present invention is a compound above, for the case when
rings A
and B are both phenyl, P is any one of formula (a) or (c) substituted in
position 7 on the
naphthalene ring, and R3 is substituted in position 1 on the naphthalene ring,
and for the
case when ring D is a pyrrole ring, P is of the formula (c) and R3 is of the
formula
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Ra ~ m Ra °r Ra
N ~N
Rs Rs Rs
~m
substituted in position 3 on the pyrrole ring, for use in the treatment or
prophylaxis of type
II diabetes.
Another object of the present invention is a compound above, and for the case
when ring D
is a pyrrole ring, P is, of the formula (c) and R3 is of the formula
Ra ~ m Ra °r Ra
~N
Rs Rs R6
~m
substituted in position 3 on the pyrrole ring, for use in the treatment or
prophylaxis of
obesity, to achieve reduction of body weight and of body weight gain.
Another object of the present invention is a pharmaceutical formulation
comprising a
compound above as an active ingredient, in combination with a pharmaceutically
acceptable diluent or carrier.
Another object of the present invention is a pharmaceutical formulation
comprising a
compound above, and for the case when rings A and B are both phenyl, P is any
one of
formula (a) or (c) substituted in position 7 on the naphthalene ring, and R3
is substituted in
position 1 on the naphthalene ring, as an active ingredient, for use in the
treatment or
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
prophylaxis of a 5-HT6 receptor related disorder, such as obesity, type II
diabetes, and/or
disorders of the central nervous system, to achieve reduction of body weight
and of body
weight gain.
Another object of the present invention is a compound above as an active
ingredient, for
use in the treatment or prophylaxis of disorders of the central nervous
system.
Another object of the present invention is a pharmaceutical formulation
comprising a
compound above, for the case when rings A and B are both phenyl, P is any one
of formula
(a) or (c) substituted in position 7 on the naphthalene ring, and R3 is
substituted in position
1 on the naphthalene ring, and for the case when ring D is a pyrrole ring, P
is of the
formula (c) and R3 is of the formula
Ra ~ m Ra ar Ra
N ~N
Rs Rs Rs
~m
substituted in position 3 on the pyrrole ring, as an active ingredient, for
use in the treatment
or prophylaxis of type II diabetes.
Another object of the present invention is a pharmaceutical formulation
comprising a
compound above, and for the case when ring D is a pyrrole ring, P is of the
formula (c) and
R3 is of the formula
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
__ _. __N . _. __.
~m
N ~N
Rs R6 Rs
Jm
substituted in position 3 on the pyrrole ring, as an active ingredient, for
use in the treatment
or prophylaxis of obesity, to achieve reduction of body weight and of body
weight gain.
Another object of the present invention is a method for the treatment or
prophylaxis of a 5-
HT6 receptor related disorder, such as obesity, type II diabetes, andlor
disorders of the
central nervous system, to achieve reduction of body weight and of body weight
gain,
which comprises administering to a subject (e.g., a mammal, a human, a horse,
a dog, or a
cat) in need of such treatment an effective amount of one or more compounds of
any of the
formulae described above, their salt forms or compositions that include the
compounds or
their salt forms, and for the case when rings A and B are both phenyl, P is
any one of
formula (a) or (c) substituted in position 7 on the naphthalene ring, and R3
is substituted in
position 1 on the naphthalene ring.
Another object of the present invention is a method for the treatment or
prophylaxis of
disorders of the central nervous system, which comprises administering to a
subject in need
of such treatment an effective amount of one or more compounds of any of the
formulae
described above, their salt forms or compositions that include the compounds
or their salt
forms.
Another object of the present invention is a method for the treatment or
prophylaxis of type
II diabetes, which comprises administering to a subject in need of such
treatment an
effective amount of one or more compounds of any of the formulae described
above, their
salt forms or compositions that include the compounds or their salt forms, for
the case
when rings A and B are both phenyl, P is any one of formula (a) or (c)
substituted in
position 7 on the naphthalene ring, and R3 is substituted in position 1 on the
naphthalene
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
ring, and for the case when ring D is a pyrrole ring, P is of the formula (c)
and R3 is of the
formula
Ra ~ m Ra °r Ra
N ~N
Rs Rs Rs
~m
substituted in position 3 on the pyrrole ring.
Another object of the present invention is a method for the treatment or
prophylaxis of
obesity, which comprises administering to a subject in need of such treatment
an effective
amound of one or more compounds of any of the formulae described above, their
salt
forms or compositions that include the compounds or their salt forms, and for
the case
when ring D is a pyrrole ring, P is of the formula (c) and R3 is of the
formula
__N_. __N . _. __.
Ra ~ m Ra °r Ra
N ~N
Rs Rs Re
~m
substituted in position 3 on the pyrrole ring.
Another obj ect of the present invention is a method for modulating 5-HT6
receptor
activity, comprising administering to a subject in need thereof an effective
amount of one
or more compounds of any of the formulae described above, their salt forms or
compositions that include the compounds or their salt forms.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Another object of the present invention is the use of one or more compounds of
any of the
formulae described above, their salt forms or compositions that include the
compounds or
their salt forms, and for the case when rings A and B are both phenyl, P is
any one of
formula (a) or (c) substituted in position 7 on the naphthalene ring, and R3
is substituted in
position 1 on the naphthalene ring, for the manufacture of a medicament for
use in the
treatment or prophylaxis of a 5-HT6 receptor related disorder, such as
obesity, type II
diabetes, and/or disorders of the central nervous system, to achieve reduction
of body
weight and of body weight gain.
Another object of the present invention is the use of one or more compounds of
any of the
formulae described above, their salt forms or compositions that include the
compounds or
their salt forms for the manufacture of a medicament for use in the treatment
or
prophylaxis of disorders of the central nervous system.
Another object of the present invention is the use of one or more compounds of
any of the
formulae described above, their salt forms or compositions that include the
compounds or
their salt forms, for the case when rings A and B are both phenyl, P is any
one of formula
(a) or (c) substituted in position 7 on the naphthalene ring, and R3 is
substituted in position
1 on the naphthalene ring, and for the case when ring D is a pyrrole ring, P
is of the
formula (c) and R3 is of the formula
__N_. __ . _. _.
Ra ~ m Ra ° r Ra
N N N
Rs Rs Rs
m
substituted in position 3 on the pyrrole ring, for the manufacture of a
medicament for use
in the treatment or prophylaxis of type II diabetes.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Another object of the present invention is the use of one or more compounds of
any of the
formulae described above, their salt forms or compositions that include the
compounds or
their salt forms, and for the case when ring D is a pyrrole ring, P is of the
formula (c) and
R3 is of the formula
__ _. __N . _. __.
Rq ~ m Rq ° r Rq
Ns _N Ns Ns
R ~ R R
substituted in position 3 on the pyrrole ring, for the manufacture of a
medicament for use
in the treatment or prophylaxis of obesity, to achieve reduction of body
weight and of body
weight gain.
The methods delineated herein can also include the step of identifying that
the subject is in
need of treatment of obesity, type II diabetes, or disorders of the central
nervous system, or
in need of reducing body weight and of body weight gain.
The invention fizrther relates to cosmetic use of one or more compounds of any
of the
formulae described herein, for causing loss of weight, as well as cosmetic
compositions
containing said compounds.
Still further, the invention relates to a non-therapeutic metod for impriving
the bodily
appearance of a mammal, including a human, in which the method comprises
orally
administering to said mammal one or more compounds of any of the formulae
described
herein.
"An effective amount" refers to an amount of a compound that confers a
therapeutic effect
on the treated subject. The therapeutic effect may be objective (i.e.,
measurable by some
test or marker) or subjective (i.e., subject gives an indication of or feels
an effect).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
For clinical use, the compounds ofthe invention are formulated into
pharmaceutical
formulations for oral, rectal, parenteral or other mode of administration.
Usually the
amount of active compounds is between 0.1-95% by weight of the preparation,
preferably
between 0.2-20% by weight in preparations for parenteral use and preferably
between 1
and SO% by weight in preparations for oral administration.
The typical daily dose of the active substance varies within a wide range and
will depend
on various factors such as, for example, the individual requirement of each
patient and the
route of administration. In general, oral and parenteral dosages will be in
the range of 5 to
1000 mg per day of active substance, preferably 50 to 150 mg per day.
Processes for preparation
In a further aspect the invention relates to methods of making compounds of
any of the
formulae herein comprising reacting any one or more of the compounds of the
formulae
delineated herein, including any processes delineated herein. The compounds of
the
formulae above may be prepared by, or in analogy with, conventional methods,
and
especially according to or in analogy with the following methods.
The chemicals used in the above-described synthetic route may include, for
example,
solvents, reagents, catalysts, protecting group and deprotecting group
reagents. The
methods described above may also additionally include steps, either before or
after the
steps described specifically herein, to add or remove suitable protecting
groups in order to
ultimately allow synthesis of the compounds of any of the formulae described
above, their
salt forms, or compositions that include the compounds or their salt forms. In
addition,
various synthetic steps may be performed in an alternate sequence or order to
give the
desired compounds. Synthetic chemistry transformations and protecting group
methodologies (protection and deprotection) useful in synthesizing applicable
compounds
are known in the art and include, for example, those described in R. Larock,
Compf-ehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and
P.G.M. Wuts, PYOtective Groups in Organic Synthesis, 2"a Ed., John Wiley and
Sons
(1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic
Synthesis, John
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia ofReagents for
Organic
Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
The specific examples below are to be construed as merely illustrative, and
not limitative
of the remainder of the disclosure in any way whatsoever. Without further
elaboration, it is
believed that one skilled in the art can, based on the description herein,
utilize the present
invention to its fullest extent. All publications cited herein are hereby
incorporated by
reference in their entirety.
Methods
1H nuclear magnetic resonance (NMR) and 13C NMR were recorded on a Broker
Advance
DPX 400 spectrometer at 400.1 and 100.6 MHz, respectively. All spectra were
recorded
using residual solvent or tetramethylsilane (TMS) as internal standard. IR
spectra were
recorded on a Perkin-Eliner Spectrum 1000 FT-IR spectrophotometer. Ionspray
mass
spectrometry (MS) spectra were obtained on a Perkin-Eliner API 150EX mass
spectrometer. Accurate mass measurements were performed on a Micromass LCT
dual
probe. Preparative HPLC/MS was performed on a Waters/Micromass Platform ZQ
system
equipped with System A: ACE 5 C8 column (19x50mm), eluents: MilliQ water, MeCN
and MilliQ/MeCN/0.1%TFA and system B: Xterra MS C18, S~,m column (19x50mm),
eluents: MilliQ water, MeCN and NH4HC03 (100mM). Analytical HPLC were
performed
on Agilent 1100, column: ACE 3 C8 (system A) or column: YMC-Pack (system B),
eluents: MilliQl0.1%TFA and MeCN. Elemental analyses were performed on a Vario
El
instrument. Preparative flash chromatography was performed on Merck silica gel
60 (230-
400 mesh).
Table 1
R6 R4
J
N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE R R
1 6-Benzenesulfonyl-4-piperazin-1-yl-quinoline~ N
hydrochloride N
2 6-[(2-Fluorophenyl)sulfonyl]-4-piperazin-1-ylquinolineI ~ N
h ~
drochloride
y - N
F
3 6-(1-Naphthylsulfonyl)-4-piperazin-1-ylquinoline~ ~ N
hydrochloride I / / N
4 6-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinolineCI CI C
h
drochloride
I N
y
6-[(3,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinolineI ~ N
h ~
drochlo
ide
y N
r
6 6-[(2-Chloro-6-methylphenyl)sulfonyl]-4-piperazin-1-I ~ N
l
uinolin
h
d
hl
id
y CI N
q
e
y
roc
or
e
7 6-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-ylquinolineCI
h
drochloride
I N
y
8 6-[(2-Methyl,4-tent-butyl-phenyl)sulfonyl]-4-piperazin-1- N
l
uinoline h
drochlorid
y N
q I
y
e
9 6-[(3,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinoline N
h ~
drochloride
y I N
6-[(2,3-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline~ CI N
h ~
drochloride i
y - N
CI
11 6-[(4-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylquinoline N
h
drochloride
y I ~ N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
12 6-[(4-Isopropylphenyl)sulfonyl]-4-piperazin-1-ylquinoline CN\
J
hydrochloride I ~ N
13 (4-Piperazin-1-yl-6-{[4- F F N
F
(trifluoromethyl)phenyl]sulfonyl}quinoline~ w N
hydrochloride i
14 6-[(4-tert-Butylphenyl)sulfonyl]-4-(1,4-diazepan-1-yl)quinoline
hydrochloride
N
15 4-(1,4-Diazepan-1-yl)-6-[(4-isopropylphenyl)sulfonyl]quinoline
hydrochloride w
I N
Scheme 1
O-
i+
O-,N \ \ HzN \ \ S \ \
~ / ~ iii
i n N ()
N () N ( )
R\S/O R\ 'O CI R\ ~O Ra
O// I \ % ~~S I \ \ ~S I \ \ , HCI
(iv) / NJ (v)~ (vi) N~
O
Legend to Scheme 1: i) Hydrogen gas, Pd/C, Methanol; ii) Sodium nitrite,
Sulphuric acid, diverse thiols (Rl-
SH), 3h; iii) meta-chloroperoxybenzoic acid (na-CPBA), dichloromethane
(CHZClz), overnight; iv)
phosphorus oxylchloride (POCl3), acetonitrile (CH3CN), 80 °C, 2h; v)
aliphatic cyclic amines (RZ), 80 °C,
CH3CN; vi) HCl in diethyl ether.
Methods
The assigned structures were confirmed by standard spectroscopical methods and
elemental analysis and/or high resolution MS.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
NMR spectra were obtained on Broker 500 MHz or JEOL 270 MHz spectrometers at
25°C, and the chemical shift values are reported as parts per million
(8). MS spectra were
acquired on a 2690 Separation Module (Waters) with a Platform LCZ (Micromass).
Flash
chromatography was performed on Silica gel 60 (Merck) or LiChroprep RP-18
(Merck).
HPLC analysis were accomplished on a HP Series1100, with a GROM-SIL 100 ODS-0
AB column, 4.6x50mm. The HPLC purifications were performed on preparative
HPLC/
Mass system using YMC Combi prep ODS-AQ column, 56x20 mm, Gilson pumps,
Dynamax UV-1 detector and Finnigan Mass detector. The used eluents were HBO
and
CH3CN, both with 0.1 % TFA. The purity of the compounds was determined by
HPLC.
Elemental analysis was performed at Structural Chemistry Department, Biovitrum
AB,
Stockholm. Melting points, when given, were obtained on a Buchi or a
Gallenkamp
melting point apparatus and are uncorrected.
INTERMEDIATE 1
Synthesis of 6-Amino-quinoline
A suspension of 6-nitro-quinoline (8.7 g, 5 mmol), palladium on charcoal (10
%) (0.1 g) in
methanol (0.2 L) was hydrogenated at room temperature for 24 with stirring.
The catalyst
was filtered and the solvent evaporated to yield a yellow solid.
Crystallisation from ethyl
acetate yielded the pure title compound as a pale yellow solid (3.3 g , 46 %).
MS m/z: 145
[M+H+]. 1H NMR (270 MHz, CHC13-d) 8 ppm 3.89 (s, 2 H) 6.87 (d, J--2.64 Hz, 1
H) 7.14
(dd, J--8.97, 2.64 Hz, 1 H) 7.25 (dd, J--8.44, 4.22 Hz, 1 H) 7.88 (dd, J 7.92,
1.58 Hz, 1 H)
7.90 (d, J--8.97 Hz, 1 H) 8.63 (dd, J 4.22, 1.58 Hz, 1 H).
INTERMEDIATE 2
Synthesis of 6-phenylsulfanyl-quinoline
A solution of sodium nitrite (1 g, 14 mmol) in water (6 rnL) was slowly added
to a stirred
solution of 6-amino-quinoline (1.44 g, 10 mmol) in sulfuric acid (50 %) (8
mL). The
temperature was kept below 5 °C during the addition. The reaction
mixture was poured
into a solution of potassium hydroxide (9 g, 16 mmol) and thiophenol (1 mL, 9
mmol) in
water (30 mL). The reaction mixture was refluxed for 3 h, cooled and extracted
with
diethyl ether. The insoluble material was eliminated by filtration. During
filtration most of
the material was trapped in the solid phase. The filtrate was evaporated and
the residue was
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
purified by column chromatography (SiOa, ethyl acetate:hexane, 1:2) to yield a
colorless
oil (100 mg, 4% PS: the low yield is due to the loss of the material during
the filtration
procedure). MS m/z: 238 [M+H+]. 1H NMR (270 MHz, CD3C1) 8 ppm 7.34 (m, 4 H)
7.42
(m, 2 H) 7.57 (dd, J--8.97, 2.11 Hz, 1 H) 7.67 (d, J--2.11 Hz, 1 H) 7.99 (m, 2
H) 8.84 (dd,
J--4.22, 1.58 Hz, 1 H).
INTERMEDIATE 3
Synthesis of 6-benzenesulfonyl-quinoline 1-oxid
A solution of m-chloroperbenzoic acid (1 g, 5.8 mmol) in DCM (10 mL) was added
to a
stirred solution of 6-phenylsulfanyl-quinoline (0.25 g, 1 mmol) and NaHCO3
(0.5 g) in
DCM (10 mL). The reaction was left stirring over night, washed with water,
NaHC03
solution and evaporated. Trituration of the residue in diethyl ether gave the
pure title
product as a slightly yellow solid (0.14 g, 30 %). MS m/z: 287 [M+H+].
INTERMEDIATE 4
Synthesis of 6-benzenesulfonyl-4-chloro-quinoline
A solution of 6-benzenesulfonyl-quinoline 1-oxid (135 mg, 0.47 mmol) in POC13
(4 mL)
was heated at 90 °C for 2 h after which the solution was poured on ice,
ammonium
hydroxide was added and extraction with DCM. The organic phase was dried
(NaS04), the
volatiles were evaporated and the residue was purified by column
chromatography (SiOa,
ethyl acetate:petroleum ether, 1:1) to yield a white solid (39 mg, 27 %). MS
m/z: 305
[M+H+].
EXAMPLE 1
Synthesis of 6-benzenesulfonyl-4-piperazin-1-yl-quinoline hydrochloride
A solution of 6-benzenesulfonyl-4-chloro-quinoline (35 mg, 0.11 mmol) and
piperazine
(0.5 g, 2.5 mmol) in acetonitrile (2 mL) was heated at 80 °C over
night. The mixture was
extracted with toluene and water. The organic phase was purified by
chromatography on
silica gel eluted with CHCl3 saturated with NH3 (gas). The pure product was
dissolved in
ethyl acetate and HCl (gas) in diethyl ether was added. The resulting oily
residue was
dissolved in methanol and ethyl acetate and evaporated to yield a white solid
(24 mg,
77%). MS m/z: 354 [M+H+]. 1H NMR (270 MHz, CH30H-D4) 8 ppm 3.52 (m, 4 H) 4.13
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(m, 4 H) 7.36 (d, J--7.18 Hz, 1 H) 7.57 (m, 3 H) 8.01 (m, J 12.25, 8.54 Hz, 3
H) 8.28 (d,
J--8.91 Hz, 1 H) 8.63 (d, J--6.68 Hz, 1 H) 8.69 (s, 1 H).
Scheme 2
o~,. o~ o~ o~
N N N CIH
1 1 1
CI CN,(CHz)n CN.(CHz)n ~N.~CI..Iz)n
R
Br \ I % ~ Br / \ i~ R \ S / \ iii; iv \ OSO / \
N \ I N~ I ~ \
N N
2a, n=0
2b, n=1
Legend for Scheme 2: i) NatBuO, Pd(PPh3)4, n-BuOH, BOC-protected diamines; ii)
tart butyl piperazine-1-
carboxylate or tart-butyl 1,4-diazepane-1-carboxylate, triethylamine or KzC03,
DMSO, thiols; iv) TFA,
HZOz, NaOH; iv) HCl.
Method A
Preparation of thiol derivatives
tent-Butyl 4-(6-bromoquinolin-4-yl)-1,4-diazepane-1-carboxylate (0.5 g, 1.23
mmol) was
mixed with the thiol (1 equiv.), NaOtBu (2 equiv.), Pd(PPh3)4 (0.05 equiv.)
and n-BuOH (S
mL) in a reaction tube. N2 (g) was flushed through the mixture for 30 minutes.
The reaction
mixture was heated to 120°C overnight. The precipitate was filtrated
and the reaction
mixture concentrated in vacuo. The residue was dissolved in EtOAc and washed
with H20,
dried (MgS04) and evaporated. Purification by flash chromatography using DCM:
MeOH
98:2 as eluent afforded the title product that was used in the next step
without further
purification.
Method B
Oxidation of thiol derivatives to sulphone derivatives
The appropriate thiophenols derivatives are dissolved in TFA (5 mL) and
stirred for 15
minutes at room temperature. HZOa (2 mL) was added and the reaction was left
stirring
overnight. The reaction mixtures are evaporated and the residues are portioned
between
diethyl ether and water. The layers are separated and the water layer is
extracted with
diethyl ether and made basic by adding NaOH 1M. Extraction with DCM, drying
with
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
MgS04 and evaporation gives the free bases of the products which are dissolved
in
MeOH, excess of HCl/ether (2M) was added and the solvent evaporated. The
residues are
purified on preparative HPLC/MS (Xterra MS C18, S~,m column) using a 10 to 40%
MeCN-water gradient (containing 0.1 % HOAc) over 10 minutes. The pure
fractions are
pooled and lyophilised. The residues are dissolved in MeOH and treated with
excess of
HCl/ether (2M). After evaporation of solvent, a solid is obtained and
triturated with diethyl
ether giving the desired products as HCl-salts.
INTERMEDIATE 5
tert-Butyl4-(6-bromoquinolin-4-yl)piperazine-1-carboxylate
6-Bromo-4-chloroquinoline (5.0 g, 20.6 mmol), tent-butyl-1-piperazine (4.1 g,
22 mmol),
triethylamine (3 mL, 22 mmol) and DMSO (20 mL) were mixed and heated overnight
in
an oil bath at 100°C. The reaction was cooled and diluted with diethyl
ether and washed
with water (Sx), dried (MgS04) and evaporated. The residue was filtered
through a short
column of silica (2.5-5 %) MeOH in CH2C12 and evaporated. Yield 8.02 g. (97
%). Brown
liquid. HPLC 98 %, RT=3.01 (System Al, 10-97 % MeCN over 3 min). 1H NMR (400
MHz, CDCl3) 8 ppm 1.52 (s, 9 H) 3.12-3.17 (m, 4 H) 3.69-3.75 (m, 4 H) 6.86 (d,
J--5.0 Hz,
1 H) 7.72 (dd, J--9.0, 2.26 Hz, 1 H) 7.92 (d, J 8.8 Hz, 1 H) 8.14 (d, J--2.3
Hz, 1 H) 8.73 (d,
J 5.0 Hz, 1 H). MS (ESI+) for C18H22BrN30a m/z 392.2 (M+H+)
EXAMPLE 2
6-[(2-Fluorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriate thiophenol was used and the
reaction was
prolonged with 8 hours. The oxidation step was completed after 24 hours at
ambient
temperature. Purification by column chromatography on silica gel 10-20 % MeOH
in
DCM gave the free amine that was additionally purified by preparative HPLC.
Yield 15
mg (4 %) Yellow solid. HPLC 95 %, RT=2.33 (System Al, 10-97 % MeCN over 3
min).
1H NMR (400 MHz, DMSO-d6) 8 ppm 3.30-3.42 (m, 4 H) 3.51-3.62 (m, 4 H) 7.28 (d,
J--5.27 Hz, 1 H) 7.42 (dd, J--10.29, 8.78 Hz, 1 H) 7.52 (t, J--7.28 Hz, 1 H)
7.77-7.84 (m, 1
H) 8.04-8.21 (m, 3 H) 8.62 (s, 1 H) 8.87 (d, J--5.27 Hz, 1 H) 9.82 (br s, 2
H). MS (ESI+)
for C19H18FN30aS m/z 372.0 (M+H+). HRMS for Ci9H18FN30zS: calcd, 371.1104;
found,
371.1102.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 3
6-(1-Naphthylsulfonyl)-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriate thiophenol was used and the
reaction was
prolonged with 8 hours. The oxidation step was completed after 24 hours at
ambient
temperature. Purification by column chromatography on silica gel 10-20 % MeOH
in
DCM gave the free amine that was converted to the HCl-salt. Yield 14 mg (4 %).
Grey
solid. HPLC 95%, RT=2.54 (System Al, 10-97% MeCN over 3 min).1H NMR (400 MHz,
DMSO-d6) ~ ppm 3.33 (s, 4 H) 4.06 (s, 4 H) 7.38 (d, J 6.78 Hz, 1 H) 7.65 (d, J-
-7.53 Hz, 1
H) 7.69-7.75 (m, 1 H) 7.82 (t, J--7.78 Hz, 1 H) 8.12 (d, J--8.03 Hz, 1 H) 8.21-
8.30 (m, 2 H)
8.38 (d, J--8.03 Hz, 1 H) 8.56 (t, J--8.53 Hz, 2 H) 8.74-8.79 (m, 2 H) 10.05
(s, 2 H). MS
(ESI+) for Ca3HZ1N3O2S m/z 404.4 (M+H+) HRMS for Ca3Ha1N302S: calcd, 403.1354;
found, 403.1365.
EXAMPLE 4
6-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriate thiophenol was used and the
reaction was
prolonged with 8 hours. The oxidation step was completed after 24 hours at
ambient
temperature. Purification by column chromatography on silica gel 10-20 % MeOH
in
DCM gave the free amine which was converted to the HCl-salt giving yellow
solid. Yield
15 mg (3 %). Yellow solid.. 1H NMR (400 MHz, DMSO- d6) 8 ppm 3.35-3.41 (m, 4
H)
4.06-4.15 (m, 4 H) 7.40 (d, J 6.78 Hz, 1 H) 7.93 (d, J--8.53 Hz, 1 H) 8.04
(dd, J 8.53,
2.01 Hz, 1 H) 8.27 (d, J--9.03 Hz, 1 H) 8.32 (d, J--2.01 Hz, 1 H) 8.36-8.42
(m, 1 H) 8.73
(d, J 1.51 Hz, 1 H) 8.82 (d, J--6.53 Hz, 1 H) 9.86 (s, 2 H). MS (ESI+) for Cl9
Hl~ C12 N3
OZ S mlz 422.2 (M+H+). HRMS for C19H1~C1aN302S: calcd, 421.0419; found,
421.0422.
HPLC 95%, RT=2.69 (System Al, 10-97 % MeCN over 3 min)
EXAMPLE 5
6-[(3,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
The oxidation step was completed after 2 hours at ambient temperature.
Purification by
column chromatography on silica gel 10-20 % MeOH in DCM gave the free amine
which
was converted to the HCl-salt giving grey solid. Yield 0.007 g (2 %). Yellow
solid. HPLC
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
90 %, RT=2.57 (System A1, 10-97 % MeCN over 3 min). MS (ESI+) for CZ1H23FN3O2S
m/z 382.2. HRMS for CzlHz3F'Ns02S: calcd, 381.1511; found, 381.1521.
EXAMPLE 6
6-[(2-Chloro-6-methylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriatethiophenol was used and the
reaction was
prolonged with 8 hours. Additional Ha02 (1 mL) was added and the reaction
mixture was
stirred at 50 °C for another 48 hours. Purification by column
chromatography on silica gel
10-20 % MeOH in DCM gave the free amine that was converted to the HCl-salt.
Yield 33
mg (7.5 %). White solid. 1H NMR (400 MHz, DMSO-d6) ~ ppm 2.13 (s, 3 H) 2.98
(s, 4 H)
3.72 (s, 4 H) 7.06 (d, J--6.78 Hz, 1 H) 7.14 (dd, J 11.54, 8.03 Hz, 2 H) 7.23
(t, J--7.78 Hz,
1 H) 7.89 (d, J--8.78 Hz, 1 H) 7.94-8.00 (m, 1 H) 8.24 (s, 1 H) 8.45 (d, J--
6.78 Hz, 1 H)
9.68 (s, 2 H). MS (ESI+) for C2oHzoC1N3O2S m/z 402.2 (M+H+). HRMS for
CaoHaoC1N302S: calcd, 401.965; found, 401.967. HPLC 95 %, RT=2.55 (System Al,
10-97
% MeCN over 3 min).
EXAMPLE 7
6-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriate thiophenol was used and the
reaction was
prolonged for another 8 hours. The oxidation step was completed after 24 h at
ambient
temperature. Purification by column chromatography on silica gel 10-20 % MeOH
in
DCM gave the free amine that was converted to the HCl-salt. Yield 14 mg (3 %).
Yellow
solid. HPLC 95 %, RT=2.66 (System A1, 10-97 % MeCN over 3 min). MS (ESI+) for
C19H18C1N302S m/z 388.2 (M+H+). HRMS for C19H18C1N302S: calcd, 387.0808;
found,
387.0821.
EXAMPLE 8
6-[(2-Methyl,4-tert-butyl-phenyl)sulfonyl]-4-piperazin-1-ylquinoline
hydrochloride
The oxidation step was completed after 2 hours at ambient temperature.
Purification by
column chromatography on silica gel 10-20 % MeOH in DCM gave the free amine
which
was converted to the HCl-salt giving gray solid. Yield 17 mg (4 %). HPLC 95 %,
RT=2.81
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(System Al, 10-97 % MeCN over 3 min). MS (ESI+) for C24H29N302S m/z 424.2
(M+H+).
HRMS for C2q.H29N3~2s: calcd, 423.1980; found, 423.1969.
EXAMPLE 9
6-[(3,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
The oxidation step was completed after 2 hours at ambient temperature.
Purification by
column chromatography on silica gel 10-20 % MeOH in DCM gave the free amine
which
was converted to the HCl-salt. Yield 33 mg (8 %). Yellow solid. 1H NMR (400
MHz,
DMSO- d6) 8 pprn 2.27 (d, J--6.27 Hz, 6 H) 3.34 (s, 4 H) 4.12 (s, 4 H) 7.39
(dd, J 7.40,
2.13 Hz, 2 H) 7.75 (d, J 7.78 Hz, 1 H) 7.81 (s, 1 H) 8.32 (s, 2 H) 8.61 (s, 1
H) 8.78 (d,
J 6.78 Hz, 1 H) 10.18 (s, 2 H). MS (ESI+) for C21Ha3NsOaS m/z 382.2 (M+H~).
HRMS for
C21H23N3~2S: calcd, 381.151 l; found, 381.1519. HPLC 95 %, RT=2.54 (System Al,
10-97
MeCN over 3 min).
EXAMPLE 10
6-[(2,3-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
A total amount of 2.25 mmol, of the appropriate thiophenol was used and the
reaction was
prolonged for another 8 hours. The oxidation step was completed after 24 hours
at ambient
temperature. Purification by column chromatography on silica gel 10-20% MeOH
in DCM
gave the free amine which was converted to the HCl-salt. Yield 15 mg (3 %).
Yellow solid.
1H NMR (400 MHz, DMSO-d6) 8 ppm 3.36 (m, 4 H) 4.10 (m, 4 H) 7.42 (d, J--6.78
Hz, 1
H) 7.75 (t, J--8.03 Hz, 1 H) 8.07 (d, J--8.03 Hz, 1 H) 8.24 (d, J--9.04 Hz, 1
H) 8.33 (dd,
J--13.93, 8.41 Hz, 2 H) 8.70 (s, 1 H) 8.82 (d, J--6.78 Hz, 1 H) 10.00 (s, 2
H). MS (ESI+)
for Cl9HmC12N30aS m/z 422.2 (M+H~). HRMS for C19H1~C1zN302S: calcd, 421.0419;
found, 421.0408. HPLC 95 %, RT=2.50 (System Al,10-97 % MeCN over 3 min).
EXAMPLE 11
6-[(4-text-Eutylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
tent-Butyl 4-(6-[(4-tert-butylphenyl)thio]quinolin-4-yl)piperazine-1-
carboxylate
(0.60 g, 1.3 mmol) was dissolved in TFA (12 mL) and stirred for 30 minutes
before HaOz
(0.65 mL, 6.3 mmol) was added. The mixture was stirred for 2 hours and water
(5 mL) was
added. The mixture was evaporated and the residue was taken up in water and
washed with
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
diethyl ether (2x). The aqueous phase was adjusted to pH 10 with 1 N NaOH and
the
mixture was extracted with CHZC12 (2x), dried (MgS04) and evaporated. The
residue was
diluted with CH2C12 and 1.3 mL 2N HCl in diethyl ether was added under
vigorous stirring
and the mixture was evaporated and washed with diethyl ether (2 x) and
dried.Yield: 0.40
g (69 %). Grey solid. HPLC 95 %, R~=2.77 (System Al, 10-97 % MeCN over 3 min).
tH
NMR (400 MHz, DMSO-d6) 8 pprn 1.23 (s, 9 H) 3.38 (s, 4 H) 4.08 (s, 4 H) 7.39
(d, J--7.03
Hz, 1 H) 7.65 (d, J 8.53 Hz, 2 H) 7.96 (d, J--8.53 Hz, 2 H) 8.25 (d, J--8.78
Hz, 1 H) 8.30-
8.36 (m, 1 H) 8.66 (d, J--1.76 Hz, 1 H) 8.81 (d, J--7.03 Hz, 1 H) 9.85 (br. s,
2 H), MS
(ESI+) for C23Ha~N3OzS m/z 410.4 (M+H+).
EXAMPLE 12
6-[(4-Isopropylphenyl)sulfonyl]-4-piperazin-1-ylquinoline hydrochloride
4-Isopropylthiophenol (0.152 g, 1.0 mmol) was added dropwise to a suspension
of te~t-
butyl 4-(6-bromo-quinolin-4-yl)-piperazine-1-caxboxylate (0.2 g, 0.51 mmol),
Na-t-
butoxide (0.192 g, 2.0 mmol) and Pd[P(Ph)3]4 (0.030 g, 0.025 mmol) in ethanol
(3 mL) at
90°C and the mixture was stirred for 18 h. The mixture was diluted with
THF and filtered
through a plug of silica and evaporated. The crude product was dissolved in
TFA (5 mL)
and stirred for 1S minutes before 30 % H202 (1 mL) was added. The mixture was
stirred
for 2 hours and evaporated. The residue was dissolved in water and washed with
CHZCl2
(2x) and 2 N NaOH was added until pH reached 10 and the mixture was extracted
with
CHaCIa (3x), dried (MgS04) and evaporated. The crude product was purified by
preparative HPLC 5-95 water/acetonitrile collecting on m/z 395.2. After
evaporation the
free amine was dissolved in CHZC12 and and excess of HCl in diethyl ether was
added and
the mixture was evaporated. Yield 0.015 g (7 %). 1H NMR (400 MHz, DMSO-d6) 8
ppm
1.17 (d, J 7.03 Hz, 6 H) 2.90-3.02 (m, 1 H) 3.34-3.42 (m, 4 H) 4.03-4.12 (m, 4
H) 7.39 (d,
J--6.78 Hz, 1 H) 7.51 (d, J--8.28 Hz, 2 H) 7.96 (d, J--8.53 Hz, 2 H) 8.26 (d,
J--9.03 Hz, 1
H) 8.29-8.36 (m, 1 H) 8.66 (s, 1 H) 8.80 (d, J--6.78 Hz, 1 H) 9.85-9.97 (m, 2
H). HPLC
95%, RT=2.65 (System Al, 10-97% MeCN over 3 min).
EXAMPLE 13
4-Piperazin-I-yl-6-{[4-(trifluoromethyl)phenyl]sulfonyl}quinoline
hydrochloride
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
4-Trifluoromethylthiophenol (0.178 g, 1.0 mmol) was added dropwise to a
suspension of
tent-butyl4-(6-bromo-quinolin-4-yl)-piperazine-1-carboxylate (0.2 g, O.S1
mmol), Sodium-
t-butoxide (0.192 g, 2.0 mmol) and Pd[P(Ph)3]4 (0.030 g, 0.025 mmol) in
ethanol (3 mL) at
90°C and the mixture was stirred for 18h. The mixture was diluted with
THF and filtered
S through a plug of silica and evaporated. The crude product was dissolved in
TFA (S mL)
and stirred for 1S minutes before 30 % HaOz (1 mL) was added. The mixture was
stirred
for 2 hours and evaporated. The residue was dissolved in water and washed with
CHZCl2
(2x) and 2 N NaOH was added until pH reached 10 and the mixture was extracted
with
CH2C12 (3x) dried (MgS04) and evaporated. The crude was purified by
preparative HPLC
S-9S water/acetonitrile collecting on m/z 421.1. After evaporation the free
amine was
dissolved in CH2Cl2 and excess of HCl in diethyl ether was added and the
mixture was
evaporated. Yield 0.024 g (10%). 1H NMR (400 MHz, DMSO-d6) 8 ppm 3.33-3.40 (m,
4
H) 4.13-4.21 (m, 4 H) 7.42 (d, J--7.03 Hz, 1 H) 8.02 (d, J--8.53 Hz, 2 H) 8.25-
8.35 (m, 3
H) 8.37-8.53 (m, 1 H) 8.76 (d, J--1.76 Hz, 1 H) 8.80 (d, J--7.03 Hz, 1 H) 9.95-
10.OS (m, 2
1 S H). Yellow oil. HPLC 9S %, RT=2.66 (System A1, 10-97% MeCN over 3 min).
INTERMEDIATE 6
tart-Butyl 4-(6-bromoquinolin-4-yl)-1,4-diazepane-1-carboxylate
6-Bromo-4-chloroquinoline (3.S g, 14.5 mmol) was reacted with tart-butyl 1,4-
diazepane-
1-caxboxylate (3.7 g, 18.8 mmol) and K2C03 (4 g, 29 mmol) in DMSO at 100
°C
overnight. After cooling the mixture was poured into water and extracted with
DCM. The
organic layer was washed with water, dried (MgS04) and evaporated. The residue
was
purified by flash chromatography using a gradient of EtOAc:hexane 1:1 to 2:1
giving 2.1 g
(36 %) of yellow oil. 1H NMR (400 MHz, CDC13) 8 ppm 1.47 (d, J S.S Hz, 9 H)
2.08-2.16
2S (m, 2 H) 3.35-3.44 (m, 4 H) 3.60-3-73 (m, 4 H) 6.87 (d, J--S.S Hz, 1 H)
7.69 (dd, J--9.0,
2.0 Hz, 1 H) 7.88 (d, J--S.S Hz, 1 H) 8.16 (s, 1 H) 8.65 (d, J S.0 Hz, 1 H).
MS (ESI+) for
C19H24BrN3O2 m~Z 406.4 (M+H)+. HRMS (EI) calcd for C19Hz4BrN3: 405.1 OS2,
found
405.1045.
INTERMEDIATE 7
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
tert-Butyl-4{3-[(4-tert-butylphenyl)thio]quinolin-5-yl}-1,4-diazepane-1-
carboxylate
(General Method A)
The compound was prepared from tent-butyl 4-(6-bromoquinolin-4-yl)-1,4-
diazepane-1-
carboxylate (0.5 g, 1.23 mmol) and p-tent-butylbenzenethiol (0.2 g, 1.23
mmol).Yield: 0.27
g (44 %) of the title compound. HPLC 89 %, RT: 3.76 min (5-99 % MeCN
containing 0.1
TFA over 3 min).
INTERMEDIATE 8
tert-Butyl-4{3-[(4-isopropylphenyl)thio]quinolin-5-yl}-1,4-diazepane-1-
carboxylate
(General Method A)
The compound was prepared from tert-butyl 4-(6-bromoquinolin-4-yl)-1,4-
diazepane-1-
carboxylate (0.5 g, 1.23 mmol) and 4-isopropylbenzenethiol (0.19 g, 1.23
mmol). Yield:
0.27 g (46 %) of the title compound that was used in the next step without
further
purification. HPLC 89 %, RT: 3.67 min (5-99 % MeCN containing 0.1 % TFA over 3
min); MS (ESI+) for C2sH35N30zS m/z 478.2 (M+H)+.
EXAMPLE 14
6-[(4-tert-Butylphenyl)sulfonyl]-4-(1,4-diazepam-1-yl)quinoline hydrochloride
(Geheral Method B)
The compound was synthesized from tert-butyl-4 f 3-[(4-tert-
butylphenyl)thio]quinolin-5-
yl}-1,4-diazepane-1-carboxylate (0.27 g, 0.55 mmol).
Yield: 20 mg (8 %) of the title compound.; 1H NMR (270 MHz, DMSO-d6) 8 ppm
1.25 (s,
9 H) 2.31 (br s, 2 H) 3.25 (br s, 2 H) 3.49 (br s, 2 H) 4.11 br s, 2 H) 4.26
(br s, 2 H) 7.16 (d,
J 7.1 Hz, 1 H) 7.65 (d, J--8.2 Hz, 2 H) 7.94 (d, J--8.2 Hz, 2 H) 8.19 (d, J--
8.7 Hz, 1 H)
8.26 (d, J--8.7 1 H) 8.62 (d, J--6.3 Hz,1 H) 8.75 (s, 1 H) 9.65 (br s, 2 H);
MS (ESI+) for
Ca4HasN3~aS m/z 424.2 (M+H)+. HPLC 93%, RT: 2.79 min (5-99 % MeCN over 3 min).
EXAMPLE 15
4-(1,4-Diazepam-1-yl)-6-[(4-isopropylphenyl)sulfonyl]quinoline hydrochloride
(General Method B)
The compound was prepared from tert-Butyl-4{3-[(4-
isopropylphenyl)thio]quinolin-5-yl}-
1,4-diazepane-1-carboxylate (0.27 g, 0.57mmo1). Yield: 15 mg (6 %) of the
title
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
compound.; 1H NMR (270 MHz, DMSO-d6) b ppm 1.16 (d, J--6.9 Hz, 6 H) 2.31 (br
s, 2 H)
2.96 (m, 1 H) 3.25 (br s, 2 H) 3.49 (br s, 2 H) 4.11 (br s, 2 H) 4.26 (br s, 2
H) 7.16 (d,
J--6.9 Hz, 1 H) 7.51 (d, J--8.2 Hz, 2 H) 7.94 (d, J--8.2 Hz, 2 H) 8.18 (d, J
8.7 Hz, 1 H)
8.30 (d, J--8.4 Hz, 1 H) 8.62 (d, J--6.1 Hz, 1 H) 8.75 (s, 1 H) 9.62 (br s, 2
H); MS (ESI+)
for C23H2~N3O2S m/z 410.4 (M+bI)+. HpLC 93 %, RT: 2.70 min (5-99 % MeCN over 3
min).
Table 2
R' R~
~ ~N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE - R gl
16 7-(2-Chloro-6-methyl-benzenesulfonyl)-1-piperazin-1-yl-~ N
I
iso
uinoline h
drochloride
q C~ N
y
17 7-(2-t-Butyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline~ N
h I ~
drochloride
y N
18 7-(3,4-Dichloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinolineci
~
h ci
drochloride
I N
y
19 7-(3,4-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline
h
drochloride
y
20 7-(2,5-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline~
N
h ~
drochloride
y _~ N
21 7-(p-Chloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinolinec
h
drochloride
I N
y
22 7-Benzenesulfonyl-1-[1,4]diazepan-1-yl-isoquinolin,hydrochloride~
N
N
23 7-(4-tert-Butyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]- N
isoquinoline, hydrochloride
N
24 7-(2-Chloro-6-methyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-~ N
isoquinoline hydrochloride
N
25 7-(3,5-Dimethyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-~ N
isoquinoline hydrochloride
I
_
N
26 7-(3,4-DickToro-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-~ ~N
i '
isoquinoline hydrochloride c
I
~ N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
27 7-(4-Chloro-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinolinei
N
hydrochloride ~
N--
28 7-(3,4-Dirnethyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]- N
isoquinoline hydrochloride
N
29 7-(2-tert-Butyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-
isoquinoline hydrochloride
,-
N
30 7-Benzenesulfonyl-1-piperazin-yl-isoquinoline~ N
hydrochloride N
31 7-(4-tert-Butyl-benzenesulfonyl-1-piperazin-yl-isoquinoline N
hydrochloride N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Scheme 3
O
N ~ ~ Br
i
Lancaster
O i
\\O
N CI N
, ii N ~ ~ Br ~i
CNJ E , > N
w w Br N w ~ gr
I r i
O~O
O
CN, iii iii
JN
N w w S w
I , , I , R S
~R
IV, V IV; V
R
Legend to Scheme 3: i) POC13; ii) KZCO3, DMF, BOC-diamines; iii) Nat-BuO,
thiophenols, Pd(PPh3)4; n-
BuOH; iv) TFA, H202; v) HCl in diethyl ether.
INTERMEDIATE 9
7-Bromo-1-Chloroisoquinoline
To phosphorus oxychloride (46.6 mL, 0.5 mol) at room temperature was added,
portionwise, 7-bromo-1-hydroxyisoquinoline (11.2 g, 0.05 mol). The mixture was
heated
to 100 °C for 90 min with rapid stirring. On cooling to room
temperature, the mixture was
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
poured, cautiously onto ice/water (200 mL). Dropwise addition of aqueous
ammonia raised
the pH=8 and the resulting precipitate was collected by filtration, washing
with cold water.
The solid was dried under reduced vacuum at 45 °C for 12 h. 13.86 g
(115 %) Beige solid
isolated. 1H NMR (DMSO-d6) 8 8.4 (s, 1 H), 8.34-8.38 (d, J = 6 Hz, 1 H), 8.03-
8.07 (m, 2
H), 7.91-7.96 (d, J = 6 Hz, 1 H); HPLC: 96%; LCMS: 242,244,246.
Nucleophilic displacement of Chlorine
INTERMEDIATE 10
4-(7-Bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, tent-butyl ester
To a suspension of 7-bromo-1-chloroisoquinoline (3.14 g, 1 3 mmol) in DMSO (20
mL) at
room temperature was added either carboxylic acid tert-butyl (BOC)piperazine
(7.23 g,
38.8 mmol) or BOC-homopiperazine (7.77 g, 38.8 mmol) and then potassium
carbonate
(5.36 g, 39 mmol). The mixture was heated to 110 °C for 24 h. On
cooling, the mixture
was poured onto ice/water (50 mL) and extracted with ethyl acetate (3 x 50
mL). The
combined organic phases were washed with water 50 mL) and brine (50 mL).
Before
drying over anhydrous sodium sulfate. Removal of solvent under reduced
pressure gave
crude product. Purification was performed by applying the crude material to a
plug of
silica in a filter funnel and eluting with heptane/ethyl acetate (2:1) and
gave 2.73 g (54 %)
yellow oil.1H NMR (CDCl3) 8 8.20-8.22 (m, 1 H), 8.13-8.18 (d, J= 6 Hz, 1 H),
7.65-7-71
(dd, J= 12, 3 Hz, 1 H), 7.59-7.65 (d, J=12 Hz, 1 H), 7.21-7.25 (m, 1 H), 3.64-
3.73 (m, 4
H), 7.27-7.36 (m, 4 H), 1.49 (s, 9 H); LCMS: 392,394,395.
INTERMEDIATE 11
4-(7-Bromo-isoquinoline-1-yl)-[1,4]diazepane-1-carboxylic acid, tent-butyl
ester
2.75 g (52 %) yellow oil isolated 1H NMR (CDC13) S 8.19-8.24 (m, 1 H), 8.06-
8.12 (d, J=
9 Hz, 1 H), 7.54-7.68 (m, 2 H), 7.11 (m, 1 H), 3.47-3.74 (m, 8 H), 1.98-2.16
(m, 2 H), 1.48
(s, 9 H); LCMS: 406,407,408.
Palladium-catalysed aryl thiol coupling
To 7-bromo-1-chloroisoquinoline (1 mmol) in butan-1-of (20 mL) at room
temperature
was added sodium tent-butoxide (481 mg, 5 mmol), thiol (1.5 mmol) and tetrakis
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
triphenylphosphine palladium (60 mg, catalytic). The mixture was heated to 120
°C for 16
h. On cooling to room temperature, the mixture was filtered through silica
eluting with
THF. Removal of solvent under reduced pressure gave the crude product which
was used
without further purification in the subsequent step.
INTERMEDIATE 12
4-[7-(2-Ghloro-6-methyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic
acid tert-butyl ester
A mixture of 4-(7-bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, text-
butyl ester
(0.5 g, 1.3 mmol), 2-chloro-6-methyl-thiophenol (0.206 g, 1.3 mmol), NatBuO
(0.44 g, 4.5
mmol), Pd(PPh3)4 (74 mg, 0.065 mmol) in nBuOH (10 mL) was heated at 110
°C, 3h. The
reaction mixture was filtered. The filtrate was concentrated and the residue
was dissolved
in ethyl acetate. The organic phase was washed with water (50 mL x 3),
separated and
dried (MgS04), filtered. The volatiles were evaporated and the residue was
purified by
flash column chromatography (Si02, n-heptane: ethyl acetate 8:2) to give 530
mg of the
title compound as colourless oil (yield 86.4 %). 1H NMR (CDCl3) S 8.05 (d,
1H), 7.65 (d,
1H), 7.20-7.45 (m, SH), 7.15 (d, 1H), 3.26-3.40 (m, 4H), 3.10-3.20 (m, 4H),
2.5 (s, 3H),
1.38 (s, 9H).
INTERMEDIATE 13
4-[7-(2-t butyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-carboxylic acid
tert-
butyl ester
A mixture of 4-(7-bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, tent-
butyl ester
(0.5 g, 1.3 mmol), 2-t-butyl-thiophenol (0.216 g, 1.3 mmol), Nat-Bu0 (0.44 g,
4.5 mmol),
Pd(PPh3)4 (74 mg, 0.065 mmol) in n-BuOH (10 mL) was heated at 110 °C,
3h. The
reaction mixture was filtered. The filtrate was concentrated and the residue
was dissolved
in ethyl acetate. The organic phase was washed with water (50 mL x 3),
separated and
dried (MgS04), filtered. The volatiles were evaporated and the residue was
purified by
flash column chromatography (SiOz, n-heptane: ethyl acetate 8:2) to give 440
mg of the
title compound as colorless oil (yield 71 %). 1H NMR (CDCI~) 8 8.00-8.10 (m,
2H), 7.15-
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
7.65 (m, 7H), 3.60-3.70 (m, 1H), 3.30-3.45 (m, 4H), 3.05-3.20 (m, 3H), 1.55
(s, 9H), 1.50
(s, 9H).
INTERMEDIATE 14
4-[7-(3,4-Dichloro-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-carboxylic
acid tert-
butyl ester
A mixture of 4-(7-bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, test-
butyl ester
(0.5 g, 1.3 mmol), 3,4-dichloro-thiophenol (165 uL, 1.3 mmol), NatBuO (0.44 g,
4.5
mmol), Pd(PPh3)4 (74 mg, 0.065 mmol) in n-BuOH (10 mL) was heated at 110
°C, 3h. The
reaction mixture was filtered. The filtrate was concentrated and the residue
was dissolved
in ethyl acetate. The organic phase was washed with water (50 mL x 3),
separated and
dried (MgS04), filtered. The volatiles were evaporated and the residue was
purified by
flash column chromatography (Si02, n-pentane:ethyl acetate 9.5:0.5-X8:2) to
give 230 mg
of the title compound as colorless oil (yield 36 %). 1H NMR (CDCl3) 8 8.10-
8.20 (m, 2H),
7.90 (bs, 1H), 7.65-7.75 (m, 2H), 7.10-7.55 (m, 3H), 3.50-3.65 (m, 4H), 3.20-
3.30 (m, 4H),
1.50 (s, 9H).
INTERMEDIATE 15
4-[7-(3,4-Dimethyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-carboxylic
acid tert-
butyl ester
A mixture of 4-(7-bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, tart-
butyl ester
(0.5 g, 1.3 mmol), 3,4-dimethyl-thiophenol (175 uL, 1.3 mmol), NatBuO (0.44 g,
4.5
mmol), Pd(PPh3)4 (74 mg, 0.065 mmol) in n-BuOH (10 mL) was heated at 110
°C, 3h. The
reaction mixture was filtered. The filtrate was concentrated and the residue
was dissolved
in ethyl acetate. The organic phase was washed with water (50 mL x 3),
separated and
dried (MgS04), filtered. The volatiles were evaporated and the residue was
purified by
flash column chromatography (SiOa, n-pentane:ethyl acetate 9.5:0.5-X8:2) to
give 260 mg
of the title compound as colorless oil (yield 44 %). 1H NMR (CDC13) 8 8.00-
8.10 (m, 2H),
7.55-7.65 (m, 3H), 7.40-7.50 (m, 1H), 7.10-7.30 (m, 2H), 3.30-3.40 (m, 4H),
3.10-3.20 (m,
4H), 2.30 (s, 3H), 2.25 (s, 3H), 1.50 (s, 9H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 16
4-[7-(3,5-Dimethyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-carboxylic
acid tart
butyl ester
A mixture of 4-(7-bromo-isoquinoline-1-yl)-piperazine-1-carboxylic acid, tent-
butyl ester
S (O.S g, 1.3 mmol), 3,S-dimethyl-thiophenol (180 mg, 1.3 mmol), NatBuO (0.44
g, 4.S
mmol), Pd(PPh3)4 (74 mg, 0.065 mmol) in nBuOH (10 mL) was heated at 110
°C, 3h. The
reaction mixture was filtered. The filtrate was concentrated and the residue
was dissolved
in ethyl acetate. The organic phase was washed with water (SO mL x 3),
separated and
dried (MgS04), filtered. The volatiles were evaporated and the residue was
purified by
flash column chromatography (SiOa, n-pentane:ethyl acetate 9.8:0.2-X8:2) to
give 380 mg
of the title compound as colourless oil (yield 6S %). 1H NMR (CDC13) S 8.OS-
8.10 (m,
1H), 7.80-7.85 (m. 1H), 7.60-7.75 (m, 1H), 7.17-7.25 (m, 1H), 7.10 (bs, 2H),
7.00 (bs, 1H),
3.40-3.50 (m, 4H), 3.10-3.20 (m, 4H), 2.25 (bs, 6H), 1.50 (s, 9H).
INTERMEDIATE 17
4-[7-(p-Chloro-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-carboxylic acid
tent
butyl ester
A mixture of 4-(7-bromo-isoquinoline-I-yl)-piperazine-1-carboxylic acid, tent-
butyl ester
(O.S g, 1.3 mmol), p-chloro-thiophenol (188 mg, 1.3 mmol), NatBuO (0.44 g, 4.S
mmol),
Pd(PPh3)4 (74 mg, 0.065 mmol) in nBuOH (10 mL) was heated at 110 °C,
3h. The reaction
mixture was filtered. The filtrate was concentrated and the residue was
dissolved in ethyl
acetate. The organic phase was washed with water (SO mL x 3), separated and
dried
(MgS04), filtered. The volatiles were evaporated and the residue was purified
by flash
column chromatography (Si02, n-pentane:ethyl acetate 9.S:O.S -~ 8:2) to give
300 mg of
2S the title compound as colourless oil (yield 50 %). 1H NMR (CDCl3) 8 8.OS-
8.1 S (m, 2H),
7.60-7.70 (m, 2H), 7.40-7.50 (m, 2H), 7.15-7.30 (m, 3H), 3.45-3.SS (m, 4H),
3.10-3.15 (m,
4H), I.SO (s, 9H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 18
4-(7-Phenylsulfanyl-isoquinoline-1-yl)-piperazine-1-carboxylic acid, tent
butyl ester
LCMS : 422,423 .
INTERMEDIATE 19
4-[7-(4-tent-Butyl-phenylsulfanyl)-isoquinoline-1-yl]-piperazine-1-carboxylic
acid,
tent-butyl ester
LCMS: 478,479.
INTERMEDIATE 20
4-(7-Phenylsulfanyl-isoquinoline-1-yl)-[I,4]diazepane-1-carboxylic acid, tart-
butyl
ester
MS: 368,369,370.
INTERMEDIATE 21
4-[7-(4-tent-Butyl-phenylsulfanyl)-isoquinoline-1-yl)-[1,4]diazepane-1-
carboxylic acid,
tent-butyl ester
MS : 424,425,426.
INTERMEDIATE 22
4-[7-(2-Chloro-6-methyl-phenylsulfanyl)-isoquinoline-1-yl]-[1,4] diazep ane-1-
carboxylic acid, tent-butyl ester
MS: 416,417,418.
INTERMEDIATE 23
4-[7-(3,4-Dimethyl-phenyIsulfanyl)-isoquinoline-I-yl]-[1,4] diazepane-I-
carboxylic
acid, tent-butyl ester
MS: 396,397,398.
INTERMEDIATE 24
4-[7-(3,4-Dichloro-phenylsulfanyl)-isoquinoline-I-yl]-[1,4) diazepane-1-
carboxylic
acid, tart-butyl ester
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
MS: 436,437438.
INTERMEDIATE 25
4-[7-(4-Chloro-phenylsulfanyl)-isoquinoline-I-yl]-(1,4]diazepane-1-carboxylic
acid,
tent-butyl ester
MS: 402,404.
INTERMEDIATE 26
4-(7-(3,4-Dimethyl-phenylsulfanyl)-isoquinoline-1-yl]-[1,4] diazepane-1-
carboxylic
acid, tart-butyl ester
LCMS: 464,465,466.
INTERMEDIATE 27
4-[7-(2-tent-Butyl-phenylsulfanyl)-isoquinoline-1-yl]-[1,4]diazepane-1-
carboxylic acid,
test-butyl ester
LCMS: 492,493,494.
BOC deprotection and oxidation of thiols to sulphone derivatives
Each thiol (0.2-1.14 mmol) was dissolved in trifluoroacetic acid (1.5 mL) at 0
°C and
stirred for I S mins at this temperature. To this was added 33% aqueous
hydrogen peroxide
solution (5-100 mL). The resulting mixture was stirred at room temperature for
90 min and
then treated with sodium hydroxide solution (1M, 25 mL). Extraction of this
mixture with
ethyl acetate (3 x 50 mL) was followed by the washing of the combined organic
layers
with brine (SOmL). The organic extracts were dried over anhydrous sodium
sulfate and
then the solvent was removed under reduced pressure. The crude product was
purified by
preparative LCMS. Treatment of the purified material with HCl/Ether (1M, 1 mL)
gave the
final product as a white solid.
EXAMPLE 16
7-(2-Chloro-6-methyl-benzenesuIfonyl)-1-piperazin-I-yI-isoquinoline
hydrochloride
A mixture of 4-[7-(2-chloro-6-methyl-phenylsulfanyl)-isoquinolin-1-yl]-
piperazine-1-
carboxylic acid test-butyl ester (160 mg, 0.340 mmol), HZOa (30% in water, 200
uL),
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
trifluoroacetic acid (2 mL) was heated at 50 °C, 2h. A water solution
of NaOH (1N) was
added (pH = 14), ethyl acetate was added and the organic phase was separated,
dried
(MgS04), filtered. The filtrated was concentrated and the residue was purified
by flash
column chromatography (SiOa, dichloromethane:methanol 8:2) to lead to 77 mg of
the
product compound as free base (yield 56 %). The free base was converted into
hydrochloride by treatment with HCl in diethyl ether. 1H NMR (CH30H-d4) 8 8.88
(bs,
1H), 8.05-8.20 (m. 3H), 7.65 (d, 1H), 7.35-7.55 (m, 3H), 3.85-3.95 (m, 4H),
3.00-3.15 (m,
4H), 2.95 (s, 3H).
EXAMPLE 17
7-(2-t Butyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride
A mixture of 4-[7-(2-tbutyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic acid
tent-butyl ester (273 mg, 0.571 mmol), Ha02 (30% in water, 1 mL),
tnfluoroacetic acid (3
mL) was heated at 50 °C, 2h. The reaction was continued overnight at 35
°C. A water
solution of NaOH (1N) was added (pH =14), ethyl acetate was added and the
organic
phase was separated, dried (MgS04), filtered. The filtrated was concentrated
and the
residue was purified by flash column chromatography (SiOa,
dichloromethane:methanol
8:2) to lead to 50 mg of the title compound as free base (yield 56 %). The
free base was
converted into hydrochloride by treatment with HCl in diethyl ether. 1H NMR
(CH30H-d4)
8 8.55 (d, 1H), 8.25 (d, 1H), 7.95-8.10 (m, 3H), 7.55 (d, 1H), 7.55-7.65 (m,
2H), 7.4 (d,
1H), 3.60-3.75 (m, 4H), 3.40-3.50 (m, 4H), 1.55 (s, 9H).
EXAMPLE 18
7-(3,4-Dichloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride
A mixtuxe of 4-[7-(3,4-dichloro-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic
acid test-butyl ester (230 mg, 0.47 mmol), H20a (30% in water, 0.5 mL),
trifluoroacetic
acid (1.5 mL) was heated at 50 °C, 2h. The reaction was continued
overnight at 35 °C. A
water solution of NaOH (1N) was added (pH = 14), ethyl acetate was added and
the
organic phase was separated, dried (MgS04), filtered. The filtxated was
concentrated and
the residue was purified by flash column chromatography (Si02,
dichloromethane:methanol 9:1) The free base was converted into hydrochloride
by
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
treatment with HCl in diethyl ether to obtained 45 mg of the title compound.
1H NMR
(CH30H-d4) 8 8.75-8.85 (m, 1H), 8.10-8.30 (m, 4H), 7.90-8.00 (m, 1H), 7.75-
7.85 (m,
1H), 7.50-7.60 (m, 1H), 3.85-3.90 (m, 4H), 3.50-3.70 (m, 4H).
EXAMPLE 19
7-(3,4-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride
A mixture of 4-[7-(3,4-dimethyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic
acid test-butyl ester (260 mg, 0.58 mmol), H20a (30% in water, 0.5 mL),
trifluoroacetic
acid (1.5 mL) was heated at 50 °C, 2h. The reaction was continued
overnight at 35 °C. A
water solution of NaOH (1N) was added (pH = 14), ethyl acetate was added and
the
organic phase was separated, dried (MgS04), filtered. The filtrated was
concentrated and
the residue was purified by flash column chromatography (Si02,
dichloromethane:methanol 9:1) The free base was converted into hydrochloride
by
treatment with HCl in diethyl ether to obtained 20 mg of the title compound.
1H NMR
(CH30H-d4) ~ 8.75-8.80 (m, 1H), 8.10-8.25 (m, 3H), 7.70-7.85 (m, 2H), 7.60-
7.70 (m,
1H), 7.35-7.40 (m, 1H), 3.90-4.00 (m, 4H), 3.55-3.65 (m, 4H), 2.35 (bs, 6H).
EXAMPLE 20
7-(2,5-Dimethyl-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride
A mixture of 4-[7-(2,5-dimethyl-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic
acid tent-butyl ester (380 mg, 0.846 mmol), Hz02 (30% in water, 0.5 mL),
trifluoroacetic
acid (3 mL) was heated at 50 °C, 2h. The reaction was continued
overnight at 35 °C. A
water solution of NaOH (1N) was added (pH =14), ethyl acetate was added and
the
organic phase was separated, dried (MgS04), filtered. The filtrated was
concentrated and
the residue was purified by flash column chromatography (SiOa,
dichloromethane:methanol 9.8:0.2-X9.5:0.5) The free base was converted into
hydrochloride by treatment with HCl in diethyl ether to obtained 120 mg of the
title
compound. 1H NMR (CH3OH-d4) 8 8.75-8.80 (m, 1H), 8.25-8.30 (m, 1H), 8.05-8.20
(m,
2H), 7.60-7.70 (m, 3H), 7.30-7.35 (m, 1H), 4.00-4.10 (m, 4H), 3.60-3.70 (m,
4H), 2.30-
1.35 (bs, 6H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 21
7-(p-Chloro-benzenesulfonyl)-1-piperazin-1-yl-isoquinoline hydrochloride
A mixture of 4-[7-(p-chloro-phenylsulfanyl)-isoquinolin-1-yl]-piperazine-1-
carboxylic
acid text-butyl ester (297 mg, 0.65 mmol), H2O2 (30% in water, 0.5 mL),
trifluoroacetic
acid (3 mL) was heated at 50 °C, 2h. The reaction was continued
overnight at 35 °C. A
water solution of NaOH (1N) was added (pH =14), ethyl acetate was added and
the
organic phase was separated, dried (MgS04), filtered. The filtrated was
concentrated and
the residue was purified by flash column chromatography (Si02,
dichloromethane:methanol 9.5:0.5-X9.0:1.0) The free base was converted into
hydrochloride by treatment with HCl in diethyl ether to obtained 70 mg of the
title
compound. 1H NMR (CH30H-d4) b 8.75-8.85 (m, 2H), 8.10-8.25 (m, 3H), 8.00-8.08
(m,
2H), 7.60-7.68 (m, 3H), 3.85-3.95 (m, 4H), 3.55-3.65 (m, 4H).
EXAMPLE 22
7-Benzenesulfonyl-1-[1,4]diazepam-1-yl-isoquinoline hydrochloride
30 mg. 1H NMR (DMSO-d6) 8 9.3 (s, 1 H), 8.58 (s, 1 H), 8.26-8.30 (d, J= 9 Hz,
1 H), 8.1-
8.13 (m, 2 H), 8.01-8.06 (d, J= 6 Hz, 1 H), 7.6-7.76 (m, 3 H), 7.53-7.58 (d,
J= 6 Hz, 1 H),
3.70-3.90 (m, 4H) 3.58-3.66 (m, 2 H), 3.29-3.40 (m, 2H); LCMS: 368,369 HPLC:
98 %.
EXAMPLE 23
7-(4-tent-Butyl-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline
hydrochloride
Isolated 69 mg. 1H NMR (DMSO-d6) 8 9.2 (s, 1 H), 8.65 (s, 1 H), 8.09-8.15 (d,
J= 6 Hz, 1
H), 8.03-8.08 (d, J= 15 Hz, 1 H), 7.89-7.96 (d, J= 9 Hz, 2 H), 7.60-7.67 (d,
J= 9 Hz, 2
H), 7.33-7.38 (d, J= 9 Hz, 1 H), 4.01-4.09 (m, 2 H), 3.83-3.91 (m, 2 H), 3.43-
3.52 (m, 2
H), 3.23-3.33 (m, 2 H), 1.25 (s, 9 H); LCMS: 424,425, HPLC: 97 %.
EXAMPLE 24
7-(2-Chloro-6-methyl-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline
hydrochloride
Isolated 27 mg 1H NMR (DMSO-d6) 8 8.81 (s, 1 H), 8.28 (m, 1 H), 8.10-8.18 (d,
J= 6 Hz,
1 H), 7.94-8.08 (m, 2 H), 7.45-7.62 (m, 3 H), 7.36-7.42 (d, J= 6 Hz, 1 H),
3.75-3.86 (m, 2
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H), 3.41-3.51 (m, 2 H), 3.18-3.32 (m, 2 H), 2.86 (s, 3 H), 2.14-2.19(m 2 H);
LCMS:
416,418 HPLC: 98 %.
EXAMPLE 25
7-(3,S-Dimethyl-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline
hydrochloride
Isolated 62 mg.'H NMR (DMSO-d6) 8 9.35 (s, 1 H), 8.67 (m, 1 H), 8.00-8.18 (m,
3 H),
7.58-7.69 (m, 2 H), 7.45-7.41 (d, J= 6 Hz, 1 H), 7.30-7.35 (m, 1 H), 4.06-4.14
(m, 2 H),
3.86-3.97 (m, 2 H), 3.42-3.52 (m, 2 H), 3.23-3.31 (m, 2 H), 2.33 (s, 6 H) 2.23-
2.25 (m 2
H); LCMS: 436,438, HPLC: 95 %.
EXAMPLE 26
7-(3,4-Dichloro-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline,
hydrochloride
Isolated 11 mg. 1H NMR (CD30D) 8 8.88 (m, 1 H), 8.23-8.29 (d, J= 12 Hz, 1 H),
8.13-
8.16 (d, J= 3 Hz, 1 H), 8.04-8.10 (d, J= 9 Hz, 1 H), 7.88-7.94 (d, J= 9 Hz, 1
H), 7.79-
7.84 (d, J= 6 Hz, 1 H), 7.67-7.73 (d, J= 9 Hz, 1 H), 7.42-7.46 (d, J= 6 Hz, 1
H), 4.28
4.35 (m, 2 H), 4.09-4.16 (m, 2 H), 3.69-3.75 (m, 2 H), 2.33-2.46 (m, 2 H);
LCMS:
368,369; HPLC: 97 %.
EXAMPLE 27
7-(4-Chloro-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline, hydrochloride
Isolated 41 mg. 1H NMR (DMSO-d6) b 9.27 (s, 1 H), 8.68 (m, 1 H), 7.99-8.17 (m,
5 H),
7.66-7.75 (d, J= 9 Hz, 2 H), 7.33-7.39 (d, J= 6 Hz, 1 H), 4.03-4.11 (m, 2 H),
3,83-3.93
(m, 2 H), 3.43-3.53 (m, 2 H), 3.23-3.32 (m, 2 H), 2.19-2.30 (m, 2 H); LCMS:
402,404;
HPLC: 98 %.
EXAMPLE 28
7-(3,4-Dimethyl-benzenesulfonyl)-1-[1,4]diazepan-1-yl]-isoquinoline,
hydrochloride
Isolated 10 mg. 1H NMR (DMSO-d6) ~ 9.41 (s, 1 H), 8.67 (s, 1 H), 8.00-8.16 (m,
3 H),
7.76-7.82 (m, 1 H), 7.68-7.77 (d, J= 9 Hz, 1 H), 7.32-7.42 (d, J= 9 Hz, 2 H),
3.99-4.40
(m, 4 H), 3.49 (m, 2 H), 3.33 (m, 2 H), 2.29 (s, 3 H), 2.25 (s, 3 H); LCMS:
396,397;
HPLC: 92 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 29
7-(2-tent-Butyl-benzenesulfonyl)-1-[1,4]diazepam-1-yl]-isoquinoline,
hydrochloride
Isolated S mg. 1H NMR (DMSO-d6) 8 9.27 (s, 1 H), 8.52 (s, 1 H), 8.06-8.16 (m,
2 H),
S 7.97-7.97 (m, 2 H), 7.72-7.78 (m, 1 H), 7.61-7.70 (m, 1 H), 7.40-7.45 (d, J=
9 Hz, 2 H),
3.69-3.99 (m, 4 H), 3.43 (s, 2 H), 3.25 (s, 2 H), 2.05-2.26(m, 2 H);1.52 (s, 9
H); LCMS:
424,425; HPLC: 90 %.
EXAMPLE 30
7-Benzenesulfonyl-I-piperazin-yl-isoquinoline, hydrochloride
Isolated 10 mg. 1H NMR (DMSO-d~) b 9.04 (s, 1 H), 8.65 (s, 1 H), 8.12-8.16 (d,
J= 6 Hz,
1 H), 7.98-8.OS (m, S H), 7.58-7.72 (m, 2 H), 7.32-7.36 (d, J= 6 Hz, 1 H),
3.98-4.04 (m, 4
H), 3.80-3.86 (m, 4 H); LCMS: 354,355; HPLC: 98 %.
1 S EXAMPLE 31
7-(4-tart-Butyl-benzenesulfonyl-1-piperazin-yl-isoquinoline, hydrochloride
Isolated 10 mg. 1H NMR (DMSO-d6) 8 9.33 (s, 1 H), 8.57 (s, 1 H), 8.24-8.29 (d,
J= 9 Hz,
1 H), 8.11 (m, 2 H), 7.91-7.97 (d, J= 9 Hz, 2 H), 7.60-7.66 (d, J= I2 Hz, 2
H), 7.52-7.57
(d, J= 6 Hz, 1 H), 3.59-3.68 (m, 4 H), 3.29-3.40 (m, 4 H), 1.24 (s, 9 H);
LCMS: 410,411
HPLC: 90 %.
Table 3
R~
HN'S~~ O
O
/ /
R3
2S
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE NAME R R
32 4-Chloro-N-[4-(pyrrolidin-3-yloxy)-1- _ O ~
naphthyl]benzenesulfonamide hydrochloride ~ HN~'
CI
33 4-Methoxy-N-[4-(pyrrolidin-3-yloxy)-1- O~
naphthyl]benzenesulfonamide hydrochloride - ~ HN~'
OMe
34 5-Chloro-N-[4-(pyrrolidin-3-yloxy)-1- O\
naphthyl]thiophene-2-sulfonamide hydrochloride ,~ ~ HN
$ CI
35 4-Chloro-N-[4-(piperidin-3-yloxy)-1- -
O
naphthyl]benzenesulfonamide hydrochloride
/
CI
26 4-Methoxy-N-[4-(piperidin-3-yloxy)-1- -
O
naphthyl]benzenesulfonamide hydrochloride
/
OMe N
H
37 5-Fluoro-2-methyl-N-[4-(piperidin-4-yloxy)-1- F ~ ~,
O
naphthyl]benzenesulfonamide hydrochloride ~ ,
N
H '
3~ 5-Chloro-N-[4-(pipexidin~4-yloxy)-1- CI
/ / O,.
naphthyl]thiophene-2-sulfonamide hydrochloride
S
NJ
H
39 4-Cbloro-N-{4-[(3S)-pyrrolidin-3-yloxy]-1- CI I ~ O
naphthyl}benzenesulfonamide hydrochloride -'
N
H
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
40 4-Chloro-N-{4-[(3R)-pyrrolidin-3-yloxy]-1-
naphthyl}benzenesulfonamide
hydrochloride N
H
Scheme 4
O; N+.O- O,.N+,O NH
z
\ \ i \ \ ii \ \ iii
/ / / / / /
OH O O
[ N [ N
n~ n~
boc boc
R1 R1
HN~S'O HN~S'O
\ iv I \ \
O O
nN [ nH CIH
boc
Legend to Scheme 4: (i) tart-butyl 3-hydroxypyrrolidine-1-carboxylate or tart-
butyl 4-hydroxypiperidine-1-
carboxylate, PPh3, DEAD, THF; (ii) Hz(g), Pd/C, MeOH; (iii) Rl-SOZ-Cl,
pyridine, CHZCIz; (iv) HCl in
diethyl ether
General method C
Mitsonobu reaction of 4-vitro-1-naphthol with boc-protected 3-
hydroxypyrrolidine and 4-
hydroxypiperidine 4-Nitro-1-naphthol (1 equiv.) was dissolved in THF (3
mL/mmol), tert-
butyl 3-hydroxypyrrolidine-1-carboxylate (2 equiv.) was added followed by PPh3
(2
equiv.). The solution was kept under NZ-atmosphere and cooled with ice-bath.
Diethylazodicarboxylate (DEAD; 2 equiv.) was added dropwise. The ice-bath was
removed after 10 min and the reaction mixture was stirred at ambient
temperature
overnight. The solvent was evaporated and the residue was re-dissolved in
EtOAc. The
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
formed precipitate was collected by filtration. The solution was concentrated
in vacuo and
purified by flash chromatography (Si02, EtOAc:iso-hexane 2:8~EtOAc)
Ge>zeral method D
Reduction of nitronaphthalene derivatives
To a solution of corresponding nitronaphthalenes (1 equiv.) (prepared by
General method
A) in MeOH (2 mL/mmol), was added Pd/C (10%) and the reaction mixture was
stirred
overnight under hydrogen (1 atm). The reaction mixture was filtered and the
filtrate was
concentrated ih vacuo to give corresponding aminonaphthalene derivatives.
Ge>zeral method E
Reaction of aminonaphthalene derivatives with sulfonyl chlorides
To a solution of the axninonaphthalene derivatives (1 equiv.) in CHZCIa (8
mL/mmol) was
added pyridine (3 equiv.) followed by the corresponding sulfonyl chloride (1.2
equiv.). The
mixtures were stirred at ambient temperature overnight, washed with HCl (1M)
(2 mL) and
dried (MgS04). The volatiles were eliminated under vacuo and gave the crude
product
which were purified by flash chromatography (SiOa, EtOAc:iso-hexane 1:4) to
give
desired sulfonamide.
Ge>zeral method F
Deprotection of boc-group
The sulfonamide derivatives (prepared by General Method C) were dissolved in a
small
amount of MeOH and treated with an excess of HCl in diethyl ether (1M).
Stirring at
ambient temperature overnight resulted in a precipitate which were collected
by filtration
giving the title compounds as its hydrochloride salts.
Geheral method G
3-Hydroxypyrrolidine (lequiv.) was dissolved in MeOH (1mL/mmol) and cooled on
ice-
bath. (BOC)20 (l.lequiv.) was added and the mixture was stirred for 2 h at
ambient
temperature. Pyridinelwater (10/lOmL) was added and the mixture was stirred
overnight.
Evaporation of solvents and co-evaporation with toluene provided the desired
boc-
protected 3-hydroxypyrrolidine.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 28
tent-Butyl 3-[(4-vitro-1-naphthyl)oxy]pyrrolidine-1-carboxylate (General
Method C)
The crude product was purified by column chromatography. The compound was
prepared
S from 4-vitro-1-naphthol (2.85 g, 1 S.I mmol). The material thus obtained was
dissolved in
small amount of EtOAc and i,ro-hexane was added. The formed solid was
collected by
filtration and triturated with MeOH to give the pure title compound 4.6g
(8S%). HPLC
99%, RT=2.70 (System A1, 10-97% MeCN over 3 min). 1H NMR (400 MHz, CDC13)
8 ppm 1.46 (d, J--7.03 Hz, 9 H) 2.26-2.38 (m, 2 H) 3.60-3.81 (m, 4 H) 5.20 (br
s, 1 H) 6.76
(d, J--8.53 Hz, 1 H) 7.58 (t, J 7.53 Hz, 1 H) 7.73 (t, J--7.53 Hz, 1 H) 8.34
(dd, J--20.33,
8.78 Hz, 2 H) 8.75 (d, J 9.04 Hz, 1 H). MS (ESI+) for Ci9Ha2N2Os m/z 376.2
(M+NH)~,
359.2 (M+H)+, 303.2 (M-tBu)+. HPLC 99%, RT=2.78 min (System BI, 10-90% MeCN
over 3 min).
1 S INTERMEDIATE 29
tent-Butyl 3-[(4-amino-1-naphthyl)oxy]pyrrolidine-1-carboxylate (General
Method D)
The compound was prepared from intermediate 1 (2.6 g, 7.2 mmol), Yield : 2.1g
(87 %) of
the title compound as purple solid. HPLC 96 %, RT=I.768 min (System Al, 10-97
MeCN over 3 min). HPLC 9S %, RT=1.604 min (System Bl, 10-90% MeCN over 3 min).
1H NMR (400 MHz, DMSO-d6) 8 ppm 1.37 (d, J--22.59 Hz, 9 H) 2.12 (s, 2 H) 3.43-
3.46
(m, 4 H) 4.96 (s, 1 H) S.SO (s, 2 H) 6.63 (d, J--8.03 Hz, 1 H) 6.81 (d, J--
8.03 Hz, 1 H) 7.41
(dd, J--6.02, 3.01 Hz, 2 H) 7.94-8.01 (m, 2 H). MS (ESI+) for CigH24NaO3 m/Z
329.2
(M+H)~, 273.2 (M-tBu)+, 229.2 (M-Boc)~.
INTERMEDIATE 30
tent-Butyl 3-[(4-{[(4-chlorophenyl)sulfonyl]amino}-1-naphthyl)oxy]pyrrolidine-
1-
carboxylate (GeheYal method E)
The compound was prepared from intermediate 2 (0.2 g, 0.61 mmol). The crude
material
was triturated with CH3CN to give 0.14 g (46 %) of the title compound as a
pale pink solid.
HPLC 97 %, RT=2.703 min (System AI, I O-97 % MeCN over 3 min). 1H NMR (400
MHz;
CDCl3) 8 ppm 1.46 (d, J--4.02 Hz, 9 H) 2.I8-2.30 (m, 2 H) 3.54-3.76 (m, 4 H)
S.OS (br s, 1
H) 6.62-6.65 (m, 1 H) 6.74-6.76 (m, 1 H) 7.17-7.23 (m, 1 H) 7.32 (d, J--9.04
Hz, 2 H)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
7.39-7.45 (m, 2 H) 7.62 (d, J--8.53 Hz, 2 H) 7.68-7.72 (m, 1 H) 8.16-8.19 (m,
l H). MS
(ESI+) for C25Ha~C1NzO5S ~a/z 520.2 (M+NH4)+, 447.0 (M-tBu)+. HPLC 98%, R~-
2.738
min (System B1, 10-90% MeCN over 3 min).
INTERMEDIATE 31
tent-Butyl 3-[(4- f [(4-methoxyphenyl)sulfonyl] amino}-1-
naphthyl)oxy]pyrrolidine-1-
carboxylate (General method E)
The compound was prepared from intermediate 2 (0.2 g, 0.61 mmol), Yield: 0.2g
(66%) of
the title compound as a pink oil. HPLC 98%, RT =2.617 min (System Al, 10-97%
MeCN
over 3 min). IH NMR (400 MHz, CDCl~) 8 ppm 1.45 (d, J--5.02 Hz, 9 H) 2.14-2.31
(m, 2
H) 3.54-3.76 (m, 4 H) 3.79 (s, 3 H) 5.04 (br s, 1 H) 6.60-6.65 (m, 2 H) 6.81
(d, J--8.53 Hz,
2 H) 7.15-7.24 (m, 1H) 7.38-7.44 (m, 2 H) 7.60-7.64 (m, 2 H) 7.71-7.76 (m, 1
H) 8.14-
8.18 (m, 1 H). MS (ESI+) for C26H3oN2O6S m/z 516.4 (M+NH4)+, 443.0 (M-tBu)+,
399.2
(M-Boc)t.
INTERMEDIATE 32
tent-Butyl 3-[(4-~ [(5-chloro-2-thienyl)sulfonyl] amino}-1-
naphthyl)oxy]pyrrolidine-1-
carboxylate (General method E)
The compound was prepared from intermediate 2 (0.2 g, 0.61 mmol), Yield: 0.218
(68%)
ofthe title compound as a yellow solid. HPLC 99%, R~2.777 min (System Bl, 10-
90%
MeCN over 3 min). 1H NMR (400 MHz, CDCl3) S ppm 1.46 (d, J 6.02 Hz, 9 H) 2.16-
2.30
(m, 2 H) 3.55-3.74 (m, 4 H) 5.07 (br s, 1 H) 6.67-6.70 (m, 1 H) 6.75 (d, J
4.02 Hz, 1 H)
6.77 (br s, 1 H) 7.13 (br s, 1 H) 7.28-7.35 (m, 1 H) 7.46-7.48 (m, 2 H) 7.75-
7.79 (m, 1 H)
8.19-8.22 (m, 1 H). MS (ESI+) for C~3HZSC1N20SS2 m/z 526.2 (M+NH4)+, 453.0 (M-
tBu)'~,
409.2 (M-Boc)+. HPLC 99%, R1-=2.767 min (System A1, 10-97% MeCN over 3 min).
INTERMEDIATE 33
tent-Butyl 4-[(4-vitro-1-naphthyl)oxy]piperidine-1-carboxylate (General method
C)
The compound was prepared from 4-vitro-1-naphthol (2 g, 10.6 mmol). The
material
obtained after flash chromatography was not pure according to NMR.
Recrystallization
from EtOAc/iso-hexane gave 2.3g (62%) of the title compound as a yellow solid.
HPLC
98%, RT=2.842 min (System Al, 10-97% MeCN over 3 min 1H NMR (400 MHz, CDC13)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
8 ppm 1.48 (s, 9 H) 1.99 (m, 4 H) 3.54 (m, 2 H) 3.70 (m, 2 H) 4.87 (m, 1 H)
6.82 (d,
J--9.04 Hz, 1 H) 7.59 (m, 1 H) 7.74 (m, 1 H) 8.38 (d, J--8.53 Hz, 2 H) 8.77
(d, J--8.53 Hz,
1 H). MS (ESI+) for C~oHa4Na05 m/z 373.0 (M+H)+, 390.2 (M+NH4)+, 317.0 (M-
tBu)+.
HPLC 98%, RT=2.973 min (System B l, IO-90% MeCN over 3 min).
INTERMEDIATE 34
tent-Butyl 4-[(4-amino-1-naphthyl)oxyjpiperidine-1-carboxylate (General Method
C)
The compound was prepared from intermediate 6 (2.3 g, 7.0 mmol), Yield: 2g
(95%) as
pink oil. HPLC 94%, RT=2.885 min (System B1, 10-90% MeCN over 3 min). 1H NMR
(400 MHz, CDC13) S ppm 1.46 (s, 9 H) 1.80-1.98 (m, 4 H) 3.35-3.41 (m, 2 H)
3.46 (s, 3 H)
3.69-3.75 (m, 2 H) 3.88 (br s, 1 H) 4.50-4.54 (m, 1 H) 7.45-7.50 (m, 2 H) 7.79-
7.81 (m, 1
H) 8.22-8.24 (m, 1 H). MS (ESI+) for C2oHa6N203 m/z 343.2 (M+H)+. HPLC 94%,
R~-=2.735 min (System A1, 10-97% MeCN over 3 min).
INTERMEDIATE 35
tent-Butyl 4-[(4-f [(4-chlorophenyl)sulfonyljamino}-I-naphthyl)oxyjpiperidine-
1-
carboxylate (GeneYal method E)
The compound was prepared from intermediate 7 (0.25 g, 0.73 rnmol), Yield:
0.29g (77%).
HPLC 98%, RT=2.906 min (System A1, 10-97% MeCN over 3 min). 1H NMR (400 MHz,
CDC13) ~ ppm 1.47 (s, 9 H) 1.88 (m, 2 H) 1.98 (m, 2 H) 3.47 (m, 2 H) 3.68 (m,
2 H) 4.69
(m, 1 H) 6.61 (s, 1 H) 6.70 (d, J--8.03 Hz, 1 H) 7.17 (d, J 8.03 Hz, 1 H) 7.32
(m, 2 H) 7.43
(m, 2 H) 7.62 (m, 2 H) 7.70 (m, 1 H). MS (ESI+) for Ca6H2gC1N2O5S m/z 534.0
(M+NH4)+,
461.2 (M-tBu)+. HPLC 98%, RT=2.843 min (System B1, 10-90% MeCN over 3 min).
INTERMEDIATE 36
tart-Butyl 4-[(4- j [(4-methoxyphenyl)sulfonylj amino}-1-
naphthyl)oxyjpiperidine-1-
carboxylate (GefZeYal method E)
The compound was prepared from intermediate 7 (0.25 g, 0.73 mmol). Yield:
0.218 (56%)
of the title compound as pink solid. HPLC 100%, RT=2.755 min (System Al, I O-
97%
MeCN over 3 min). rH NMR (400 MHz, CDC13) 8 ppm 1.47 (s, 9 H) 1.86-2.01 (m, 4
H)
3.43-3.49 (m, 2 H) 3.65-3.71 (m, 2 H) 3.79 (s, 3 H) 4.65-4.70 (m, 1 H) 6.58
(s, 1 H) 6.69
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(d, J--8.53 Hz, 1 H) 6.83-6.79 (m, 2 H) 7.17 (d, J--8.03 Hz, 1 H) 7.39-7.44
(m, 2 H) 7.61-
7.64 (m, 2 H) 7.75-7.77 (m, 1 H) 8.21-8.24 (m, 1 H). MS (ESI+) for Ca~H3aNz06S
m/z
530.2 (M+NH4)+, 457.2 (M-tBu)+, 413.4 (M-Boc)+. HPLC 99%, RT=2.668 min (System
B1, 10-90% MeCN over 3 min).
INTERMEDIATE 37
tent-Butyl 4-[(4-{ [(5-fluoro-2-methylphenyl)sulfonyl] amino}-1-
naphthyl)oxy]piperidine-1-carboxylate (Geheral method E)
The compound was prepared from tent-butyl 4-[(4-amino-1-
naphthyl)oxy]piperidine-1-
carboxylate (0.25 g, 0.73 mmol). Yield: 0.24 g (64 %) of the title compound as
a pink
solid. HPLC 99 %, RT=2.809 min (System B1, 10-90% MeCN over 3 min). 1H NMR
(400
MHz, CDC13) 8 ppm 1.46 (s, 9 H) 1.83-1.98 (m, 4 H) 2.54 (s, 3 H) 3.42-3.48 (m,
2 H)
3.63-3.69 (m, 2 H) 4.64-4.68 (m, 1 H) 6.64-6.68 (m, 2 H) 7.03 (d, J=8.53 Hz, 1
H) 7.10
(m, 1 H) 7.22 (dd, J=8.53, 5.02 Hz, 1 H) 7.44-7.48 (m, 2 H) 7.55 (dd, J=8.53,
2.51 Hz, 1
H) 7.83-7.86 (m, 1 H) 8.24 (m, 1 H). MS (ESI+) for CZ~H31FN205S m/z 532.2
(M+NH4)+,
459.2 (M-tBu)+, 415.2 (M-Boc)+. HPLC 100 %, RT=2.877 min (System Al, 10-97%
MeCN over 3 min).
INTERMEDIATE 38
tart-Butyl4-[(4-{[(5-chloro-2-thienyl)sulfonyl]amino[-1-
naphthyl)oxy]piperidine-1-
carboxylate (GeneYal method E)
The compound was prepared from tent-butyl 4-[(4-amino-1-
naphthyl)oxy]piperidine-1-
carboxylate (0.25g, 0.73mmo1). Yield: 0.25 g (65 %) of the title compound as a
pink solid.
HPLC 98 %, RT=2.827 min (System B1, 10-90% MeCN over 3 min). 1H NMR (400 MHz,
CDC13) 8 ppm 1.47 (s, 9 H) 1.86-2.03 (m, 4 H) 3.45-3.51 (m, 2 H) 3.66-3.72 (m,
2 H)
4.69-4.74 (m, 1 H) 6.66 (s, 1 H) 6.74-6.76 (m, 2 H) 7.14 (d, J=4.02 Hz, 1 H)
7.30 (d,
J=8.03 Hz, 1 H) 7.45-7.50 (m, 2 H) 7.76-7.79 (m, 1 H) 8.25-8.28 (m, 1 H) MS
(ESI+) for
Ca4H2~C1N205Sz xn/z 540.4 (M+NH4)+, 467.2 (M-tBu)+. HPLC 99 %, RT=2.910 min
(System Al, 10-97 % MeCN over 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 39
tent Butyl (3R)-3-hydroxypyrrolidine-1-carboxylate (General method G)
The compound was prepared from (3R)-3-hydroxypyrrolidine (5 g, 57.4 mmol).
Yield: 9.6
g (90 %) of the title compound. 1H NMR (400 MHz, CDC13) 8 ppm 1.43 (s, 9 H)
1.90-1.98
(m, 2 H) 3.27-3.47 (m, 4H) 4.40 (br s, 1H).
INTERMEDIATE 40
tent-Butyl (3S)-3-hydroxypyrrolidine-1-carboxylate (General method G)
The compound was prepared from (3S)-3-hydroxypyrrolidine (S g, 57.4 mmol).
Yield: 8 g
(86 %) of the title compound. 1H NMR (400 MHz, CDC13) 8 ppm 1.40 (s, 9 H) 1.86-
1.91
(m, 2 H) 3.24-3.42 (m, 4H) 4.36 (br s, 1H).
INTERMEDIATE 41
tent-Butyl (3S)-3-[(4-vitro-1-naphthyl)oxy]pyrrolidine-1-carboxylate (General
method
C)
The compound was prepared from tart-butyl (3R)-3-hydroxypyrrolidine-1-
carboxylate
(3.56 g, 19 mmol) and 4-vitro-1-naphthol (3 g, 15.9 mmol). Yield: 5 g (88 %)
of the title
compound as yellow oil. 1H NMR (400 MHz, CDC13) 8 ppm 1.45 (d, J=7.03 Hz, 9 H)
2.22-2.38 (m, 2 H) 3.54-3.83 (m, 4 H) 5.18 (br s, 1 H) 6.74 (d, J=8.53 Hz, 1
H) 7.56 (t,
J=7.78 Hz, 1 H) 7.71 (t, J=7.78 Hz, 1 H) 8.29 (d, J=8.53 Hz, 1 H) 8.33 (d,
J=8.53 Hz, 1 H)
8.72 (d, J=8.53 Hz, 1 H). MS (ESI+) for C19H2~N205 m/z 376.2 (M+ NH4)~, 303.2
(M-
tBu)~. HPLC 100 %, RT=2.768 min (System A1, 10-97% MeCN over 3 rnin).
INTERMEDIATE 42
tent Butyl (3R)-3-[(4-vitro-1-naphthyl)oxy]pyrrolidine-1-carboxylate (General
method
C)
The compound was prepared from test -butyl (3S)-3-hydroxypyrrolidine-1-
carboxylate
(3.56 g, 19 mmol) and 4-vitro-1-naphthol (3 g, I5.9 mmol). Yield: 2.8 g (49 %)
of the title
compound as yellow oil. 1H NMR (400 MHz, CDCl3) S ppm 1.46 (d, J=7.03 Hz, 9 H)
2.26-2.38 (m, 2 H) 3.55-3.81 (m, 4 H) 5.20 (br s, 1 H) 6.77 (d, J=8.53 Hz, I
H) 7.59 (t,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
J=7.53 Hz, 1 H) 7.72-7.76 (m, 1 H) 8.32 (d, J=8.03 Hz, 1 H) 8.37 (d, J=8.53
Hz, 1 H) 8.76
(d, J=8.53 Hz, 1 H). HPLC 95 %, RT=2.775 min (System A1, 10-97 % MeCN over 3
min).
INTERMEDIATE 43
tent-Butyl (3S)-3-[(4-amino-1-naphthyl)oxy]pyrrolidine-1-carboxylate (Geheral
method
D)
The compound was prepared from tent -butyl (3S)-3-[(4-vitro-1-
naphthyl)oxy]pyrrolidine-
1-carboxylate (5 g, 14 mmol). Yield: 3.5 g (76 %) of the title compound as
dark pink solid.
1H NMR (400 MHz, CDC13) 8 ppm 1.46 (d, J=14.56 Hz, 9 H) 2.08-2.13 (m, 1 H)
2.27-2.30
(m, J=13.05 Hz, 1 H) 3.54-3.77 (m, 4 H) 3.88 (br s, 2 H) 4.96 (br s, 1 H) 6.65-
6.70 (m, 2
H) 7.45-7.51 (m, 2 H) 7.79-7.81 (m, 1 H) 8.15-8.19 (m, 1H). MS (ESI+) for
C19H24N2~3
m/z 329.2 (M+H)+, 273.2 (M-tBu)+, 229.2 (M-Boc)+. HPLC 95 %, RT=1.854 min
(System
Al, 10-97 % MeCN over 3 min).
INTERMEDIATE 44
tent-Butyl (3R)-3-[(4-amino-1-naphthyl)oxy]pyrrolidine-1-carboxylate (Geheral
method D)
The compound was prepared from tent -butyl (3R)-3-[(4-vitro-1-
naphthyl)oxy]pyrrolidine-
1-carboxylate (2.8 g, 7.8 mmol). Yield: 1.8 g (72 %) of the title compound as
dark pink
solid. 1H NMR (400 MHz, CDC13) 8 ppm 1.46 (d, J=14.56 Hz, 9 H) 2.07-2.14 (m, 1
H)
2.27-2.30 (m, 1 H) 3.54-3.77 (m, 4 H) 3.93 (br s, 2 H) 4.96 (br s, 1 H) 6.65-
6.70 (m, 2 H)
7.45-7.51 (m, 2 H) 7.79-7.81 (m, 1 H) 8.16-8.18 (m, 1H). MS (ESI+) for
C19H24N~03 m/z
329.2 (M+H)+, 273.2 (M-tBu)+, 229.2 (M-Boc)+. HPLC 94 %, RT=1.751 min (System
Al,
10-97% MeCN over 3 min).
INTERMEDIATE 45
tent-Butyl (3S)-3-[(4-{[(4-chlorophenyl)sulfonyl]amino}-1-
naphthyl)oxy]pyrrolidine-1-
carboxylate (General method E)
The compound was prepared from tart-butyl (3S)-3-[(4-vitro-1-
naphthyl)oxy]pyrrolidine-
1-carboxylate (0.3 g, 0.9 mmol) and 4-chloro-phenylsulfonylchloride (0.23 g,
1.1 mmol).
Yield: 0.23 g (50 %) of the title compound. 1H NMR (400 MHz, CDCl3) 8 ppm 1.46
(d,
J=4.52 Hz, 9 H) 2.15-2-34 (m, 2 H) 3.54-3.74 (m, 4 H) 5.05 (br s, 1 H) 6.62-
6.71 (m, 2 H)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
7.17-7.23 (m, 1 H) 7.33 (d, J=8.53 Hz, 1 H) 7.39-7.43 (m, 2 H) 7.62-7.64 (m, 2
H) 7.65-
7.70 (m, J=6.02 Hz, 1 H) 8.18 (d, J=8.53 Hz, 1 H) MS (ESI+) for C2sH2~C1N205S
m/z
520.2 (M+NH4)+, 447.0 (M-tBu)+. HPLC 100 %, RT=2.772 min (System Al, 10-97
MeCN over 3 min).
INTERMEDIATE 46
tent Butyl (3R)-3-[(4-{[(4-chlorophenyl)sulfonyl)amino-1-
naphthyl)oxy]pyrrolidine-
1-carboxylate (General method E)
The compound was prepared from tent-butyl (3R)-3-[(4-vitro-1-
naphthyl)oxy]pyrrolidine-
1-carboxylate (0.3 g, 0.9 mmol) and 4-chloro-phenylsulfonylchloride (0.23 g,
1.1 mmol).
Yield: 0.4 g (87 %) of the title compound. 1H NMR (400 MHz, CDC13) 8 ppm 1.46
(d,
J=4.52 Hz, 9 H) 2.15-2-34 (m, 2 H) 3.54-3.74 (m, 4 H) 5.05 (br s, 1 H) 6.60-
6.66 (m, 2 H)
7.17-7.23 (m, l H) 7.33 (d, J=8.53 Hz, 1 H) 7.39-7.43 (m, 2 H) 7.62-7.64 (m, 2
H) 7.65-
7.70 (m, J=6.02 Hz, 1 H) 8.18 (d, J=8.53 Hz, 1 H) MS (ESI+) for CasH2~C1N205S
m/z
520.2 (M+NH4)+, 447.0 (M-tBu)~. HPLC 100 %, R~r=2.769 min (System Al, 10-97
MeCN over 3 min).
EXAMPLE 32
4-Chloro-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl]benzenesulfonamide hydrochloride
(General method F)
The compound was prepared from intermediate 3 (0.13 g, 0.26 mmol). The solid
was
further purified by trituration with diethyl ether giving 0.1 lg (95%) of the
title compound
as white solid. HPLC 98%, R-r=1.810 min (System Al, 10-97% MeCN over 3 min).
1H
NMR (400 MHz, DMSO-d6) 8 ppm 2.21-2.26 (m, 2 H) 3.32-3.37 (m, 2 H) 3.48-3.50
(m, 2
H) 5.28 (br s, l H) 6.91-6.98 (m, 2 H) 7.44-7.50 (m, 2 H) 7.56-7.64 (m, 4 H)
7.88-7.90 (m,
1 H) 8.20-8.23 (m, 1 H). MS (ESI+) for CaoH19C1Na03S m/z 401.2 (M+H)+. HPLC
98%,
RT=1.651 min (System B1, 10-90% MeCN over 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 33
4-Methoxy-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl]benzenesulfonamide
hydrochloride
(General method F)
The compound was prepared from intermediate 4 (0.18 g, 0.36 rnmol), Yield:
0.12 g
(76%) of the title compound as a white solid. HPLC 100%, RT=1.490 min (System
B 1, 10-
90% MeCN over 3 min). 1H NMR (400 MHz, DMSO-d6) 8 ppm 2.20-2.25 (m, 2 H) 3.31-
3.53 (m, 4 H) 3.78 (s, 3 H) 5.27 (br s, 1 H) 6.90-6.97 (m, 2 H) 7.00 (d, J--
8.53 Hz, 2 H)
7.43-7.48 (m, 2 H) 7.57 (d, J--8.53 Hz, 2 H) 7.93-7.96 (m, 1 H) 8.19-8.22 (m,
1 H) 9.63 (br
s, 2 H). MS (ESI+) for C~1H22NaO4S m/z 409.2 (M+H)+. HPLC 100%, RT=1.639 min
(System Al, 10-97% MeCN over 3 min).
EXAMPLE 34
S-Chloro-N-[4-(pyrrolidin-3-yloxy)-1-naphthyl] thiophene-2-sulfonamide
hydrochloride (General method F)
The compound was prepared from intermediate 5 (0.20 g, 0.39 mmol), Yield: 0.14
g (80%)
of the title compound as a pale white solid. HPLC 99%, R~I.651 min (System B
1, 10-
90% MeCN over 3 min). IH NMR (400 MHz, DMSO-d6) b ppm 2.23-2.26 (m, 2 H) 3.28-
3.39 (m, 2 H) 3.40-3.56 (m, 2 H) 5.32 (br s, 1 H) 6.99 (d, J--8.53 Hz, 1 H)
7.13 (d, J--8.03
Hz, 1 H) 7.15 (d, J--4.02 Hz, 1 H) 7.26 (d, J--4.02 Hz, 1 H) 7.48-7.52 (m, 2
H) 7.92 (dd,
J--6.53, 3.01 Hz, 1 H) 8.25 (dd, J--6.53, 3.01 Hz, 1 H) 9.60 (s, 1 H). MS
(ESI+) for
Ci$Hj~C1Nz03S2 m/z 409.2 (M+H)+. HPLC 99%, RT=1.818 min (System A1, IO-97%
MeCN over 3 min).
EXAMPLE 35
4-Chloro-N-[4-(piperidin-3-yloxy)-1-naphthyl]benzenesulfonamide hydrochloride
(General method F)
The compound was prepared from intermediate 8 (0.26 g, 0.50 mmol), Yield: 0.12
g
(53%) of the title compound as a white solid. HPLC 100%, RT=I.872 min (System
Al, 10-
97% MeCN over 3 min). 1H NMR (400 MHz, DMSO-d6) 8 ppm I.95-1.99 (m, 2 H) 2.14-
2.19 (m, 2 H) 3.11 (br s, 2 H) 3.26 (br s, 2 H) 4.84 (br s, 1 H) 6.92-6.99 (m,
2 H) 7.44-7.51
(m, 2 H) 7.57-7.65 (m, 4 H) 7.91 (d, J--7.53 Hz, 1 H) 8.17 (d, J 7.03 Hz, 1 H)
8.94 (br s, 1
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H) 9.05 (br s, 1 H) 10.11 (s, 1 H). MS (ESI+) for CZZH21C1N203S m/z 415.2
(M+H)+.
HPLC 99%, RT=1.657 min (System B1, 10-90% MeCN over 3 min).
EXAMPLE 36
4-Methoxy-N-[4-(piperidin-3-yloxy)-1-naphthylJbenzenesulfonamide hydrochloride
(Geheral methad F)
The compound was prepared from intermediate 9 (0.19 g, 0:37 mmol), Yield: 0.15
g (90%)
of the title compound as a white solid. HPLC 97%, RT=1.508 min (System Bl, 10-
90%
MeGN over 3 min). 1H NMR (400 MHz, DMSO-d6) 8 ppm 1.96 (m, 2 H) 2.16 (m, 2 H)
3.10 (m, 2 H) 3.26 (m, J--6.02 Hz, 2 H) 3.78 (s, 3 H) 4.82 (m, 1 H) 6.95 (q, J-
-8.20 Hz, 1
H) 7.01 (d, J--9.04 Hz, 2 H) 7.47 (m, 2 H) 7.57 (m, 2 H) 7.96 (m, 1 H) 8.16
(m, 1 H) 8.96
(s, 1 H) 9.07 (s, 1 H) 9.81 (s, 1 H). MS (ESI+) for Cz2H24N2O4S m/z 413.4
(M+H)+. HPLC
97%, R~1.713 min (System Al, 10-97% MeCN over 3 min).
EXAMPLE 37
5-Fluoro-2-methyl-N-[4-(piperidin-4-yloxy)-Z-naphthyl]benzenesulfonamide
hydrochloride (General method F)
The compound was prepared from intermediate 10 (0.24 g, 0.47 mrnol).
Yield: 0.21 g (9 9 %) of the title compound as an off white solid. HPLC 100 %,
RT=1.823
min (System Al, 10-97 % MeCN over 3 min). 1H NMR (400 MHz, CH30H-d4) 8 ppm
2.13 (m, 4 H) 2.42 (s, 3 H) 3.18 (m, 2 H) 3.37 (m, 2 H) 4.83 (m, 1 H) 6.81 (d,
J--8.53 Hz, 1
H) 6.97 (d, J--8.03 Hz, 1 H) 7.10 (m, 1 H) 7.24 (m, J--8.53, 5.52 Hz, 1 H)
7.35 (m, 3 H)
7.83 (m, 1 H). MS (ESI+) for Cz2H23FNaOsS m/z 415.2 (M+H)~. HPLC 96 %,
RT=1.628
min (System B1, 10-90% MeCN over 3 min).
EXAMPLE 38
5-Chloro-N-[4-(piperidin-4-yloxy)-1-naphthyl]thiophene-2-sulfonamide
hydrochloride (General method F)
The compound was prepared from tent-butyl 4-[(4-{[(5-fluoro-2 methylphenyl)-
sulfonyl]amino}-1-naphthyl)oxy]piperidine-1-carboxylate (0.24 g, 0.46 mmol).
Yield: 0.16
g (76 %) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-ds) S
ppm
1.96-2.03 (m, 2 H) 2.16-2.22 (m, 2 H) 3.09-3.14 (m, 2 H) 3.25-3.31 (m, 2 H)
4.86-4.89 (m,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
1 H) 7.04-7.11 (m, 2 H) 7.16 (d, J--4.02 Hz, 1 H) 7.26 (d, J--4.02 Hz, 1 H)
7.48-7.53 (m, 2
H), 7.92-7.94 (m, 1 H) 8.19-8.21 (m, 1 H) 9.06 (br s, 1 H) 10.36 (br s, 1 H).
MS (ESI+) for
Ci9Hi9C1Nz03S2 m/z 423.0 (M+H)+. HPLC 99 %, RT=1.861 min (System Al, 10-97
MeCN over 3 min).
EXAMPLE 39
4-Chloro-N-{4-[(3S)-pyrrolidin-3-yloxyJ-1-naphthyl}benzenesulfonamide
hydrochloride (General method F)
The compound was prepared from tart-butyl (3S)-3-j(4- f j(4-
chlorophenyl)sulfonyl]amino}-1-naphthyl)oxy]pyrrolidine-1-carboxylate (0.22 g,
0.44
mmol). Yield: 0.15 g (78 %) of the title compound as a yellow solid. 1H NMR
(400 MHz,
CH30H-d4) 8 ppm 2.37-2.49 (m, 2 H) 3.52-3.56 (m, 2 H) 3.61-3.71 (m, 2 H) 5.37
(br s, 1
H) 6.87 (d, J--8.03 Hz, 1 H) 7.14 (d, J--8.03 Hz, 1 H) 7.38-7.47 (m, 4 H) 7.61
(d, J 8.53
Hz, 2 H) 7.83 (d, J--8.03 Hz, 1 H) 8.22 (d, J--8.03 Hz, 1 H). MS (ESI+) for
CaoHi9C1NzO3S
m/z 403.2 (M+H)~. HPLC 100 %, Rz =1.826 min (System A1, 10-97 % MeCN over 3
min).
EXAMPLE 40
4-Chloro-N-{4-[(3R)-pyrrolidin-3-yloxyJ-I-naphthyl}benzenesulfonamide
hydrochloride (General method F)
The compound was prepared from tart-butyl (3R)-3-[(4-{j(4-
chlorophenyl)sulfonyl]amino}-1-naphthyl)oxy]pyrrolidine-1-carboxylate (0.37 g,
0.74
mmol). Yield: 0.27 g (82 %) of the title compound as a off white solid. 1H NMR
(400
MHz, CH30H-d4) 8 ppm 2.37-2.49 (m, 2 H) 3.52-3.56 (m, 2 H) 3.61-3.71 (m, 2 H)
5.37
(br s, 1 H) 6.88 (d, J=8.53 Hz, 1 H) 7.15 (d, J=8.03 Hz, 1 H) 7.39-7.47 (m, 4
H) 7.60-7.62
(m, 2 H) 7.83 (d, J=7.53 Hz, 1 H) 8.22 (d, J=8.03 Hz, 1 H). MS (ESI+) for
C2oHisClNaO3S
m/z 403.2 (M+H)+. HPLC 100 %, RT=1.815 min (System Al, 10-97 % MeCN over 3
min).
Table 4
R4 3
R
N j S RZ
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE R R3 R
41 N-(4-Methylphenyl)-4-(4-methylpiperazin- ~ H CN\
JlO
1-yl)thieno[3,2-c]pyridine-2-sulfonamide ~ S:N I i N
hydrochloride ' H CH3
42 2-Bromo-4-(4-methyl-piperazin-1-yl)- Br ~ ~N~
O
thieno[3,2-c]pyridine-3-sulfonic acid p- ~ S:N I i N
tolylamide hydrochloride H CH3
43 4-(4-Methylpiperazin-1-yl)-N- N ~ H ~N~
O
phenylthieno[3,2-c]pyridine-2- ~ S: ~ i
sulfonamide hydrochloride H CH3
44 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2- F H CN\
sulfonic acid (3-fluoro-5-trifluoromethyl- , O Jl
O, , ~ N
phenyl)-amide hydrochloride ~S'N ~ CF3
H
45 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2- ~ CI H CN\
JJ1O
sulfonic acid (4-chloro-phenyl)-amide ~ S:N ~ i N
hydrochloride H
46 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2- H CN\
sulfonic acid (4-isopropyl-phenyl)-amide , p ~ JJ1
0, . ~ N
hydrochloride ~S'N
H
47 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2- ~ H CN\
,O
sulfonic acidp-tolylamide hydrochloride ~ s:N ~ i N
H
48 4-(4-Methylpiperazin-1-yl)-N (2- H __a
0: :o -~O CND
cyclohex-1-en-1-ylethyl) thieno [3,2-c] ~s'N
pyridine-2-sulfonamide hydrochloride H I
49 2-(4-(4-Methylpiperazin-1-yl) thieno [3,2- p S O H __
c] pyridin-2-ylsulfonyl)-1,2,3,4-
tetrahydroisoquinoline hydrochloride I
50 4-(4-Methylpiperazin-1-yl)-N (2-thien-2- O ~ H __a
ylethyl) thieno [3,2-c] pyridine-2- ~ S-N I s
sulfonamide hydrochloride H I
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
51 4-(4-Methylpiperazin-1-yl)-No; ;o H
[1-(1- S
naphthyl) ethyl] thieno ~N
[3,2-c] pyridine-2- '''
H
sulfonamide hydrochloride~ ~ j
52 4-(4-Methylpiperazin-1-yl)-N~n-hexyl H
(4- o --N
P
I
hexylphenyl) thieno [3,2-c]s
pyridine-2- w
N
H
N
sulfonamide hydrochloride (
53 N (3-Chlorobenzyl)-4-(4-methylpiperazin-o-s'~ c~ H --a
~
I
1-yI) thieno [3,2-c] pyridine-2-sulfonamideH N
~ I
54 4-(4-Methylpiperazin-1-yl)-No; :o H
[1-(4-
s.
fluorophenyl) ethyl] thieno~ H i ~ F N
[3,2-c] D
C
pyridine-2-sulfonamide N
hydrochloride I
55 N (2,3-Difluorobenzyl)-4-(4-o:S ~ H
~
methylpiperazin-1-yl) " F i ~
thieno [3,2-c]
pyridine-2-sulfonamide F
hydrochloride
56 4-(4-Methylpiperazin-1-yl)-No H
(4-chloro-2,
5-dimethoxyphenyl) thienoc~
[3,2-c]
pyridine-2-sulfonamide H I
hydrochloride
57 2-Bromo-4- (4-methylpiperazin-1-yl)-NBr o
(2-cyclohex-1-en-1-ylethyl) S~N
thieno [3,2-c]
pyridine-3-sulfonamide H j
hydrochloride
58 2-[2-Bromo-4-(4-methyl-piperazin-1-Br p; ;O
:~SN~~ CN
yl)-benzo[b]thiophene-3-sulfonyl]-1 ' l\v/~~
/
,2,3,4-tetrahydro-isoquinoline j
59 2-Bromo-4- (4-methylpiperazin-1-yl)-NBr
o: :o
[1-(4-fluorophenyl) ethyl] ~s'H
thieno [3,2-c] F
pyridine-3-sulfonamide j
hydrochloride
60 2-Bromo-4- (4-methylpiperazin-1-yl)-NBr o c
(4-chloro)-(2, 5-dimethoxyphenyl)
thieno N
[3,2-c] pyridine-3-sulfonamide " I
hydrochloride
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
61 N-(3,4-Dichlorophenyl)-4-piperazin-1-~CI H
o: : I
S
ylthieno[3,2-c]pyridine-2-sulfonamide~;
'N ~ CI
H
hydrochloride
62 N-(2,4-Difluorophenyl)-4-piperazin-1-~ F H
0
ylthieno[3,2-c]pyridine-2-sulfonamide
H F
hydrochloride
63 4-Piperazin-1-yl-N-[-3- ~ H
's o ~ I
(trifluoromethyl)phenyl]thieno[3,2-~ N CF3
H
c]pyridine-2-sulfonamide
hydrochloride
64 N-(3-Ethylphenyl)-4-piperazin-1-i H
o: : I
ylthieno[3,2-c]pyridine-2-sulfonamide~'s'N
H
hydrochloride
65 N-(3,4-Dimethoxyphenyl)-4-piperazin-1-~ O, H
0
~ I
~
ylthieno[3,2-c]pyridine-2-sulfonamideS'N
O
H
hydrochloride
66 N-(4-Bromo-2-methylphenyl)-4-piperazin-~ Br H
o: : I
1-ylthieno[3,2-c]pyridine-2-sulfonamide
H
hydrochloride
67 2-(4-Piperazin-1-yl-thieno[3,2-c]pyridine-p ,O H
S C
D
2-sulfonyl)-1,2,3,4-tetrahydro-isoquinoline~N I ~ N
'~
hydrochloride
68 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-~ H
O
I
sulfonic acid (2-thiophen-2-yl-ethyl)-s
~ S N
amide hydrochloride
69 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-o c~ H
sulfonic acid (4-chloro-2,5-dimethoxy-'s w I N
. 'N
phenyl)-amide hydrochloride"
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
70 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-~ H N
I C~
,
sulfonic acid phenethyl-amide~ N N
hydrochloride
71 4-Piperazin-1-yl-thieno H N
[3,2-c]pyridine-2- o
lf o ~ I
i
id
2
6
di
h
l
h
l
id
su ;s N
on N
c ac
(
,
-
et
y
-p
eny
)-am
e
hydrochloride H
72 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-~ H N
:: I
sulfonic acid (3-phenyl-propyl)-amide~;s'N N
hydrochloride
73 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-~ H N
:: I
S'
~
sulfonic acid (3,3-diphenyl-propyl)-amide~; N
N
hydrochloride
74 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-o,, H N
sulfonic acid [2-(5-methoxy-1H-indol-3-~N N
yl)-ethyl]-amide hydrochloride~o I ~ I
75 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-o;so I w H N
CF3
~
sulfonic acid 4-trifluoromethyl-H N
benzylamide hydrochloride
76 4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-o,~ H N
N
sulfonic acid benzyl-ethyl-amideI ~ N
~
hydrochloride
77 N-(3-Ethylphenyl)-4-piperazin-1-H ors o N
N
ylthieno[3,2-c]pyridine-3-sulfonamide I ~ N
hydrochloride
78 N-(4-Isopropylphenyl)-4-piperazin-1-H ~ N
ylthieno[3,2-c]pyridine-3-sulfonamide \ N~ N
I ~
hydrochloride
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
79 N-(4-Methylphenyl)-4-(pyrrolidin-3- [?] H y
O
yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride I / ~SO~
N N
H H
80 N-(4-Methylphenyl)-4-(piperidin-4- [?] H
O'y
yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride ~ / ~SOZ
NJ
H
81 N-(2,3-Difluorobenzyl)-4-piperazin-1- [?] H N
ylthieno[3,2-c]pyridine-2-sulfonamide ~ F ~N~
hydrochloride ~ /
~F
NH-SOZ
82 N-(3-Chlorobenzyl)-4-piperazin-1- [?] H N
ylthieno[3,2-c]pyridine-2-sulfonamide ~ C~ CN
hydrochloride
NH-SOZ
Scheme 5
Br /S\ O ~ Br /S\ a OH ~f--a Br /S\ a N3 -_
H 0 O
O CI CI_S ~ CI R1-S'O CI R1 O R3
S
iii Br / I N iv ~ Br ~ ~ \N v ~ Br0' S ~ ~N-'---~' Br O. S I ~N vif _ BrOes \N
S a S a a e- S I a
Iv
CI CI R3
CIS ~ ~ wN , vyNHS ~ I ~N viiyNKS~ ~ I
O S a O S ~ O S
Legend to Scheme 5: i) Malonic acid, pyridine, piperidine, heat; ii)
ethylchloroformate, acetone, NaN3 -10
°C; iii) diphenyl ether, 220 °C; iv) POCl3, heat; v) gas SOZ, n-
BuLi, N-chlorosuccinimide, CHZC12; vi) R'-
NH2, pyridine; vii) HR3, KZC03, DMSO, heat.
INTERMEDIATE 47
(2E)-3-(5-Bromothien-2-yl) acrylic acid
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Malonic acid (44.40 g, 426.7 mmol) was added to a mixture of S-bromothiophene-
2-
carbaldehyde (50 g, 261.7 mmol), piperidine (2.84 mL) and pyridine (150 mL).
The
mixture was refluxed for 1 h at 80°C and than at 100 °C over
night. The volatiles were
evaporated and the residue was dissolved in water and acidified with
hydrochloric acid (pH
2). The crude product was crystallized in ethanol. Yield: 55.24 g (90.5 %).1H
NMR (270
MHz, CH30H-d4) 8 ppm 6.14 (d, J--15.83 Hz, 1 H) 7.11-7.16 (m, 2 H) 7.68 (d, J--
16.36
Hz, 1 H); MS 233.1 (M - H)+; Purity (HPLC) 94 %.
INTERMEDIATE 48
(2~-3-(5-Bromothien-2-yl) acryloyl azide
Thionyl chloride (1.04 mL) was added to a solution of (2E~-3-(5-bromothien-2-
yl) acrylic
acid (1.04 g, 4.46 mmol) in chloroform (20 mL) and the mixture was refluxed
for 2h at
75°C and than used in the next step. The above solution was added drop
wise to a stirred
suspension of sodium azide (0.58 g, 8.93 mmol), dioxane (3 mL) and water (3
mL) in an
ice bath. After 10 min a precipitate appeared which was filtered off and
washed with water.
The residue was dissolved in dichloromethane, dried with MgSO4, filtered and
the solvent
was removed to afford: 0.96 g (83.4 %). 1H NMR (270 MHz CH3OH-d4) 8 ppm 6.20
(d,
J--15.57 Hz, 1 H) 7.15-7.25 (m, 2 H) 7.80 (d, J--15.57 Hz, 1 H); MS 258.1 (M -
H)+;
Purity (HPLC) 65 %.
INTERMEDIATE 49
2-Bromothieno [3,2-c] pyridin-4 (SIB-one
A solution of (2~-3-(5-bromothien-2-yl) acryloyl azide (18.00 g, 69.7 mmol)
solved in
dichloromethane (100 mL) was added dropwise to diphenyl ether (90 mL) at 150
°C. The
temperature was increased to 220°C for lh. The mixture was cooled to
room temperature
followed by the addition of ether. The solid precipitated and was separated by
filtration.
Yield: 13.58 g (84.6 %). 1H NMR (270 MHz, DMSO-d6) b ppm 6.82 (d, J 7.13 Hz, 1
H)
7.27 (d, J--6.86 Hz, 1 H) 7.54 (s, 1 H) 11.55 (s, 1 H); MS 230.1 (M - H)+;
Purity (HPLC)
92 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 50
2-Bromo-4-chloro-thieno [3,2-c] pyridine
Phosphorus oxychloride (4.08 g, 26.6 mmol) was added dropwise to 2-bromothieno
[3,2-c]
pyridin-4 (51~-one (2.04 g, 8.87 mmol) at 0 °C. The mixture was heated
at 135 °C for
2.Sh, then carefully poured over ice water. The precipitated was collected by
filtration and
dried to yield 1.78 g (80.7 %) of title product. 1H NMR (270 MHz, CH30H-dø) 8
ppm
7.67 (d, 1 H) 7.88 (dddd, J--6.33 Hz, 2 H) 8.19 (d, J--5.54 Hz, 1 H); MS 248.0
(M - H)~;
Purity (HPLC) 100 %.
INTERMEDIATE 51 and INTERMEDIATE 52
4-Chlorothieno [3,2-c] pyridine-2-sulfonyl chloride and 2-bromo-4-chlorothieno
[3,2-
c] pyridine-3-sulfonyl chloride
n-Butyl lithium (1.5 mL, 2.4 mmol) was added to 2-bromo-4-chlorothieno [3,2-c]
pyridine
(0.5 g, 2 mmol) dissolved in dry THF (15 ml) at -78°C under nitrogen.
The mixture was
stirred for 40 min. The above solution was added to a dry ether saturated with
S02 (gas) at
-78°C. The mixture was warmed to room temperature, followed by the
addition of ether.
The precipitate was separated by filtration. The two title products were
obtained and taken
to the next step without further purification as follows: N-chlorosuccinimide
(2.07 g, 10.3
mmol) was added to [(4-chlorothieno [3,2-c] pyridin-2-yl) sulfonyl] lithium
and [(2-bromo-
4-chlorothieno [3,2-c] pyridin-3-yl) sulfonyl] lithium in dichloromethane (150
mL) at 0 °C.
The mixture was heated at 60 °C for 2h, extracted with water (3 x 50
mL). The organic
phase was separated, dried with MgS04, filtrated and the volatiles were
eliminated by
vacuum distillation. The crude products were used in the next step without
further
purification.
INTERMEDIATE 53 and INTERMEDIATE 54
4-Chloro-2-thieno [3,2-c] pyridine-2- sulfonic acid p-tolylamide and 2-bromo-4-
chloro-thieno[3,2-c]pyridine-3-sulfonic acid p-tolylamide
p-Toludine (30 mg, 2.87 mmol) was added to a solution of 4-chlorothieno [3,2-
c] pyridine-
2-sulfonyl chloride and 2-bromo-4-chlorothieno [3,2-c] pyridine-3-sulfonyl
chloride (0.07
g, 0.26 mmol) in dichloromethane and pyridine (0.19 mL). The reaction was
stirred at
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
room temperature for 2h. The solvent was removed and the crude mixture was
taken to the
next step without further purification.
EXAMPLE 41 and EXAMPLE 42
4-(4-Methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-2-sulfonic acid p-tolylamide
hydrochloride and 2-bromo-4-(4-methyl-piperazin-1-yl)-thieno[3,2-c]pyridine-3-
sulfonic acid p-tolylamide hydrochloride
A mixture of 4-chloro-2-thieno [3,2-c] pyridine-2- sulfonic acid p-tolylamide
and 2-
bromo-4-chloro-thieno[3,2-c]pyridine-3-sulfonic acid p-tolylamide (70 mg, 0.21
rnmol) in
DMSO (2 mL), 1-methyl piperazine (0.344 mL, 3.1 mmol) and KzC03 (28.5 mg, 0.21
mmol) was heated to 100°C over night. The reaction mixture was
dissolved in water and
extracted with ethyl acetate (3 x 10 mL). The organic layers were collected
and the solvent
was removed. The products were purified by HPLC to afford 1.9 mg of 4-(4-
Methyl-
piperazin-1-yl)-thieno[3,2-c]pyridine-2-sulfonic acid p-tolylamide. The free
base was
converted into the hydrochloride salt by treatment with HCl in ether: 1H NMR
(270 MHz,
Methanol-d4) ~ ppm 2.26 (s, 3H) 2.98 (s, 3 H) 3.40-3.55 (m, 8 H) 7.02-7.10 (m,
6 H) 7.55
(d, J--5.81 Hz, 1 H) 7.69 (s, 1 H) 8.13 (d, J--5.81 Hz, 1 H); LC-MS 403 (M +
H)+; Purity
(LC-MS) 92 % and 3.8 mg 2-bromo-4-(4-methyl-piperazin-1-yl)-thieno[3,2-
c]pyridine-3-
sulfonic acid p-tolylamide. The free base was converted into the hydrochloride
salt by
treatment with HCl in ether: 1H NMR (270 MHz, Methanol-d4) 8 ppm 2.21 (s, 1H)
3.00 (d,
3H) 3.50-3.77 (m, 8 H) 7.00-7.10 (m, 6 H) 7.63 (d, J--5.81 Hz, 1 H) 8.19 (d, J
5.81 Hz, 1
H); LC-MS 481 (M + H)+; Purity (LC-MS) 98 %.
Reaction of sulfonyl chloride with amines (Method H)
To a solution of the amine (1.3 equiv.) and pyridine (8 equiv.) in DCM was
added the
sulfonyl chloride (1 equiv.) and the reaction mixture was stirred over night.
After addition
of TrisamineTM (ca 2 equiv.), the mixture was gently shaken for additional 3
h. The
suspension was then filtered through a short silica plug by the aid of DCM and
ethyl
acetate. The solvent was evaporated, and the residue was dissolved in DCM and
washed
with 1 M aqueous HCl (2 times). The combined organic phases were dried
(MgS04),
filtered, and the solvent was removed to give the sulfonamide product. In
cases of low-
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
purity material, the products were purified by silica gel flash
chromatography. The
products are used in the next step (Procedure B).
Coupling with aromatic amines (Method I)
To the reaction mixtures from the Method H, dissolved in DMSO (2mL), amines
(IS
equiv.) and KaCO3 (1 equiv.) are added. The reactions are stirred at
100°C fox 24 and than
concentrated. The products axe purified by LC-MS. The solvents are removed
under
vacuum by SpeedVac and purified by preparative LC/MS. The products that were
not pure
enough (Purity ~0%) were purified by preparative chromatography using
acetonitrile-
water gradients containing 0.1 % triflouroacetic acid. After HPLC analysis
fractions that
were >_ 90% pure were collected and concentrated. Deprotection of the amine in
the
piperazine was performed by first dissolving the substance in methanol and
adding
portions of 1M HCl/ether. The reactions are analyzed by TLC. The solvents were
concentrated under vacuum by a SpeedVac.
1S
Deprotection of SOC-group (Method L)
The sulfone or sulfonamide derivative (prepared by Methods H and I) were
dissolved in a
small amount of MeOH/DCM 1:1 and treated with an excess of 1 M HCl in diethyl
ether.
Stirring at ambient temperature overnight resulted in a precipitate which were
collected by
filtration to give the products as their corresponding hydrochloride salts.
EXAMPLE 43
4-(4-Methylpiperazin-1-yl)-N-phenylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
2S The synthesis was preformed essentially as described in Method H-L. Yield:
8.1 mg (33.8
%). 1H NMR (270 MHz, CH3OH-d4) ~ ppm 8.I3 (d, J S.BI Hz, 1 H) 7.67 (s, 1 H)
7.54 (d,
J--5.81 Hz, 1 H) 7.SS-7.53 (m, SH) 2.97 (s, 3 H) (4H obscured by solvent
signal); LC-MS
389 (M - H)~; Purity (HPLC) 100 %.
EXAMPLE 44
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3-fluoro-5-
trifluoromethyl-
phenyl)-amide hydrochloride
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
4-[2-(3-Fluoro-5-trifluoromethyl-phenylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-
piperazine-
1-carboxylic acid tert-butyl ester (0.235 mmol, 1 equiv.) was used as the
thienopyridine in
Method H-L. Yield: 25.7 mg HPLC: tR 3.395 (System: 5% to 50 % ACN in 3 min,
C8),
Purity: 100 %, LC/MS: tR=1.375 (System: 30% to 60% ACN in 1.5 min, Hypersil
BDS),
Purity: 99 %. MS: 461 (M+1) 1H NMR (270 MHz, CH30H-d4) 8 ppm 3.47 (m, 4 H)
3.53
(s, 1 H) 3.87 (m, 4 H) 7.21 (m, 1 H) 7.29 (m, 1 H) 7.33 (s, 1 H) 7.66 (d, J--
6.33 Hz, 1 H).
EXAMPLE 45
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-chloro-phenyl)-amide
hydrochloride
4-Chloro-thieno[3,2-c]pyridine-2-sulfonic acid (3-fluoro-5-trifluoromethyl-
phenyl)-amide
(0.208 mmol, 1 equiv.) was used as the thienopyridine in Method H-L. Yield:
7.2 mg
HPLC: tR= 3.039 (System: 5 % to 50 % ACN in 3 min, C8), Purity: 100%, LC/MS:
tR=
0.905 (System: 30 % to 60 % ACN in 1.5 min, Hypersil BDS), Purity: 97%. MS:
409
(M+1). 1H NMR (270 MHz, CH3OH-d4) 8 ppm 3.50 (m, 4 H) 3.91 (m, 4 H) 7.25 (m, 4
H)
7.71 (dd, J 6.33, 0.53 Hz, 1 H) 7.96 (d, J 0.79 Hz, 1 H) 8.04 (d, J--6.33 Hz,
1 H).
EXAMPLE 46
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-isopropyl-phenyl)-
amide
hydrochloride
4-Chloro-thieno[3,2-c]pyridine-2-sulfonic acid (4-isopropyl-phenyl)-amide
(0.201 mmol, 1
equiv.) was used as the thienopyridine in Method H-L. Yield: 6.9 mg HPLC: tR=
3.255
(System: 5 % to 50 % ACN in 3 min, C8), Purity: 95%, LC/MS: tR= 1.255 (System:
30%
to 60 % ACN in 1.5 rnin, Hypersil BDS), Purity: 98 %. MS: 417 (M+1). 1H NMR
(270
MHz, CH30H-d4) 8 ppm 1.18 (d, J--6.86 Hz, 6 H) 2.83 (m, 2 H) 3.52 (m, 4 H)
4.00 (m, 4
H) 7.14 (m, 3 H) 7.75 (d, J--6.60 Hz, 1 H) 8.02 (m, 1 H).
EXAMPLE 47
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acidp-tolylamide
hydrochloride
To a solution of 4-chloro-thieno[3,2-c]pyridine-2-sulfonyl chloride (0.640 g,
2.39 mmol) in
DCM (20 mL) was added pyridine (1.9 mL, 23.9 mmol) followed byp-tolylamine (
0.307
g, 2.86 mmol). The reaction mixture was stirred at room temperature for 16
hours. The
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
mixture was concentrated and re-dissolved in DMSO (10 mL), piperazine-1-
carboxylic
acid text-butyl ester (1.34 g, 7.17 mmol) andK2C03 (0.989 g, 7.17 mmol) were
added. The
mixture was stirred at 100 °C for 16 hours and then concentrated. The
crude reaction
mixture was dissolved in EtOAc (100 mL) and washed with brine (2 x SO mL). The
S organic phase was dried (Na2S04) and concentrated. The crude intermediate
was purified
by column chromatography on silica using EtOAc/h-pentane (1:1) as eluent. The
intermediate was dissolved in EtOAc/MeOH and diethyl ether saturated with HCl
(g) was
added. The mixture was stirred at room temperature for 16 hours. The
precipitate was
collected by filtration and washed with diethyl ether/n-pentane to give 0.475
g of the crude
I O product. Purification by preparative reversed phase HPLC gave 0.133 g of
the pure
product: 1H NMR (DMSO-d6, 2S °C, 270.17 MHz) 8 10.61 (br s, 1H), 9.23
(br s, 2H), 8.13
(d, J = 5.80 Hz, 1H), 7.91 (s, 1H); 7.67 (d, J = 5.80 Hz, 1H), 7.09-7.07 (rn,
4H), 3.68-3.59
(m, 4H), 3.33-3.22 (m, 4H), 2.20 (s, 3H) ; m/z (posES~ 399 (M+H).
1 S EXAMPLE 48
4-(4-Methylpiperazin-1-yl) N (2-cyclohex-1-en-1-ylethyl) thieno [3,2-c]
pyridine-2-
sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield:
25.6 mg 1H
NMR (270 MHz, DMSO-d6) b ppm 10.49-10.48 (m, 1H) 8.23-7.95 (m, 3H) 7.72-7.71
(m,
20 1H) 5.34-5.33 (m, 1H) 4.14-4.11 (m, 2H) 3.53-3.51 (m, 2H) 3.29-3.25 (m, 2H)
2.98-2.97
(m, 2H) 2.85 (s, 3H) 2.04-1.81 (m, 4H) 1.57-1.15 (m, 8H); LC-MS 420.17 (M -
H)+; Purity
(LC-MS) 97 %.
EXAMPLE 49
25 2-(4-(4-Methylpiperazin-1-yl) thieno [3,2-c) pyridin-2-ylsulfonyl)-1,2,3,4-
tetrahydroisoquinoline hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield:
1S.S mg 1H
NMR (270 MHz, DMSO-d6) 8 ppm 10.49-10.48 (m, 1H) 8.17-8.15 (m, 1H) 7.86-7.85
(m,
IH) 7.70-7.65 (m, 2H) 7.28-7.12 (m, 3H) 3.97-3.94 (m, 2H) 3.87-3.85 (m, 2H)
3.25-3.18
30 (m, 2H) 2.84 (s, 3H) 1.67-1.65 (m, 2H) 3.51-3.34 (6H obscured by solvent
signal); LC-MS
428. I3 (M - H)+; Purity (LC-MS) 99 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 50
4-(4-Methylpiperazin-1-yl) N (2-thien-2-ylethyl) thieno [3,2-c] pyridine-2-
sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield:
29.5 mg 1H
NMR (270 MHz, DMSO-d6) 8 ppm 10.28-10.27 (m, 1H) 8.34-8.33 (m, 1H) 8.19-8.17
(m,
1H) 8.01-8.00 (m, 1H) 7.71-7.69 (m, 1H) 7.31-7.30 (m, 1H) 6.91-6.87 (m, 1H)
4.13-4.10
(m, 2H) 3.53-3.51 (m, 2H) 3.29-3.25 (m, 2H) 3.17-3.16 (m, 2H) 2.99-2.95 (m 4H)
2.86 (s,
3H); LC-MS 422.09 (M - H)+; Purity (LC-MS) 99%.
EXAMPLE 51
4-(4-Methylpiperazin-1-yl) N [1-(1-naphthyl) ethyl] thieno [3,2-c] pyridine-2-
sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. 20.1 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm 10.28-10.27 (m, 1H) 8.85-8.84 (m, 1H) 8.02-8.01 (m,
1H)
7.67-7.60 (m, 4H) 7.53-7.50 (m, 2H) 7.39-7.36 (m, 2H) 4.72-4.70 (m, 1H) 3.87-
3.84 (m,
1H) 3.72-3.70 (m, 1H) 3.23-3.13 (m, 4H) 2.84 (s, 3H) 1.42-1.40 (m 3H); LC-MS
466.15
(M - H)+; Purity (LC-MS) 99 %.
EXAMPLE 52
4-(4-Methylpiperazin-1-yl) N (4-hexylphenyl) thieno [3,2-c] pyridine-2-
sulfonamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. 8.0 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm 10.39-10.38 (m, 1H) 8.16-8.15 (m, 1H) 7.90-7.89 (m,
1H)
7.66-7.65 (m, 1H) 7.09-7.08 (m, 4H) 4.00-3.98 (m, 2H) 3.51-3.48 (m, 2H) 3.26-
3.22 (m,
2H) 2.85 (s, 3H) 2.49-2.45 (m, 2H) 1.48-1.46 (m, 2H) 1.24-1.22 (m, 8H) 0.82-
0.81 (m,
3H); LC-MS 472.20 (M - H)+; Purity (LC-MS) 98 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 53
N (3-Chlorobenzyl)-4-(4-methylpiperazin-1-yl) thieno [3,2-c] pyridine-2-
sulfonamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. 30.7 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm 10.25-10.24 (m, 1H) 8.78-8.77 (m, 1H) 8.18-8.17 (m,
1H)
7.91-7.90 (m, 1H) 7.68-7.67 (m, 1H) 7.26-7.19 (m, 3H) 4.21-4.20 (m, 2H) 4.08-
4.05 (m,
2H) 3.54-3.51 (m, 2H) 3.28-3-23 (m, 2H) 2.87 (s, 3H) 2.84-2.60 (2H obscured by
solvent
signal); LC-MS 436.08 (M - H)~; Purity (LC-MS) 94 %.
EXAMPLE 54
4-(4-Methylpiperazin-1-yl) N [1-(4-fluorophenyl) ethyl] thieno [3,2-c]
pyridine-2-
sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. 32.9 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm IO.I6-10.15 (m, IH) 8.75-8.73 (m, 1H) 7.63-7.62 (rn,
2H)
7.25-7.24 (m, 2H) 6.91-6.88 (m, 2H) 4.58-4.55 (m, 1H) 4.02-3.95 (m, 2H) 3.55-
3.53 (m,
2H) 3.25-3-21 (m, 2H) 2.68 (s, 3H) 1.31-1.30 (m, 3H) 2.70-2.64 (2H obscured by
solvent
signal); LC-MS 434.12 (M - H)+; Purity (LC-MS) 92 %.
EXAMPLE 55
N (2,3-Difluorobenzyl)-4-(4-methylpiperazin-1-yl) thieno [3,2-c] pyridine-2-
sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. 26.7 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm 10.36-10.35 (m, 1H) 8.82-8.81 (m, 1H) 7.96-7.95 (m,
1H)
7.69-7.68 (m, 1H) 7.27-7.10 (m, 2H) 4.26-4.25 (m, 2H) 4.11-4.08 (m, 2H) 3.54-
3.52 (m,
2H) 3.28-3-24 (m, 2H) 2.68 (s, 3H) 2.86-2.60 (2H obscured by solvent signal);
LC-MS
438.10 (M - H)+; Purity (LC-MS) 93 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 56
4-(4-Methylpiperazin-1-yl) N (4-chloro-2, 5-dimethoxyphenyl) thieno [3,2-c]
pyridine-2-sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L.14.6 mg 1H
NMR
(270 MHz, DMSO-d6) 8 ppm 10.27-10.26 (m, 1H) 10.14-10.13 (m, 1H) 8.18-8.17 (m,
1H)
7.83-7.82 (m, 1H) 7.69-7.68 (m, 1H) 7.09-7.07 (m, 2H) 4.00 (s 2H) 3.76-3.75
(m, 2H)
3.51-3.48 (m, 2H) 3.24-3.22 (m, 2H) 2.85 (s, 3H); LC-MS 482.08 (M - H)+;
Purity (LC
MS) 95 %.
EXAMPLE 57
2-Bromo-4- (4-methylpiperazin-1-yl)-N (2-cyclohex-1-en-1-ylethyl) thieno [3,2-
c]
pyridine-3-sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. 4.6 mg 1H
NMR
(270 MHz, DMSO-d6) S ppm 10.30-10.29 (m, 1H) 8.31-8.27 (m, 2H) 7.89-7.88 (m,
1H)
5.27-5.26 (m, 1H) 3.68-3.52 (m, 4H) 3.05-3.04 (m, 2H) 2.88-2.87 (m, 3H) 2.04-
2.03 (m,
2H) 2.77-1.81 (m, 2H) 1.54-1.15 (m, lOH); LC-MS 498.08 (M - H)+; Purity (LC-
MS) 93
%.
EXAMPLE 58
2-[2-Bromo-4-(4-methyl-piperazin-1-yl)-benzo[b]thiophene-3-sulfonyl]-1
,2,3,4-tetrahydro-isoquinoline hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 3.7
mg 1H
NMR (270 MHz, DMSO-d6) b ppm 10.30-10.29 (m, 1H) 9.24-9.22 (m, 1H) 8.09-8.08
(m,
1H) 7.67-7.35 (m, 7H) 4.69-4.66 (m, 1H) 2.86-2.85 (m, 3H) 1.50-1.48 (m, 3H)
3.23-2.51
(8H obscured by solvent signal); LC-MS 544.06 (M - H)+; Purity (LC-MS) 92 %.
EXAMPLE 59
2-Bromo-4- (4-methylpiperazin-1-yl)-N [1-(4-fluorophenyl) eth-2-yl] thieno
[3,2-c]
pyridine-3-sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 4.3
mg 1H
NMR (270 MHz, DMSO-d6) 8 ppm 10.35-10.34 (m, 1H) 9.16-9.14 (m, 1H) 8.23-8.22
(m,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
1H) 7.80-7.79 (m, 1H) 7.26-7.25 (m, 1H) 4.55-4.52 (m, 1H) 3.54-3.52 (m, 2H)
2.88-2.87
(m, 3H) 1.38-1.36 (m, 3H) 3.17-2.83 (6H obscured by solvent signal); LC-MS
512.04 (M -
H)+; Purity (LC-MS) 93 %.
EXAMPLE 60
2-Bromo-4- (4-methylpiperazin-1-yl)-N (4-chloro)-(2, 5-dimethoxyphenyl) thieno
[3,2-c] pyridine-3-sulfonamide hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 0.7
mg, LC-
MS 561.91(M - H)+; Purity (LC-MS) 95%.
EXAMPLE 61
N-(3,4-dichlorophenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
3,4-Dichloroaniline (0.49 mmol), was dissolved in acetonitrile (1 mL) and
pyridine (0.440
mL, 4.03 mmol) was added to a solution of 4-chlorothieno [3,2-c] pyridine-2-
sulfonyl
chloride (0.445 mmol) dissolved in acetonitrile (1 mL). The reaction was
shaken for lh,
controlled with HPLC and the solvent was removed. The crude product was used
in the
next step without further purification. To the reaction mixture from the
previous step,
dissolved in DMSO (1mL), piperazine (15 equiv.) and KaC03 (1 equiv.) was
added. The
reaction was stirred at 100 °C for 24 and than concentrated. The
product was purified by
LC-MS and gave 5.8 mg (2.6 %) of the title product. 1H NMR (270 MHz, CH3OH-d4)
8
ppm 8.07-8.05 (m, 2 H) 7.73 (d, J--6.60 Hz, 1 H) 7.46-7.41 (m, 2 H) 7.16 (dd,
J--8.71, 2.38
Hz, 1 H) 3.94-3.90 (m, 4 H) 3.92 (m, 4 H) 3.53-3.50 (m, 4 H); LC-MS 443 (M -
H)+;
Purity (HPLC) 95%.
EXAMPLE 62
N-(2,4-Difluorophenyl)-4-piperazin-1-ylthieno [3,2-c]pyridine-2-sulfonamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 4.3
mg (2.1
%). 1H NMR (270 MHz, CH30H-d4) 8 ppm 8.22 (s, 1 H) 8.07-8.01 (rn, 2H) 7.79-
7.72 (m,
1H) 7.55-7.47 (m, 1H) 7.04-6.94 (m, 2H) 4.00-3.96 8m, 4H) 3.53-3.43 (m, 4H)
2.66 (s,
1H); LC-MS 411 (M - H)+; Purity (HPLC) 98 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 63
4-Pip erazin-1-yl-N-[-3-(trifluoromethyl)phenyl] thieno [3,2-c] pyridine-2-
sulfon amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 2.6
mg (1.2
%). 1H NMR (270 MHz, CH30H-d4) 8 ppm 8.05 (d, J--6.60 Hz, 2 H) 7.81-7.60 (m,
3H)
7.50-7.47 (m, 2H) 3.94-3.90 (m, 4H) 3.56-3.49 (m, 4H); LC-MS 443 (M - H)+;
Purity
(HPLC) 99 %.
EXAMPLE 64
N-(3-Ethylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield: 1.4
mg (0.7
%). 1H NMR (270 MHz, CH30H-d4) 8 ppm 8.35 (s, 1H) 7.58-6.92 (m, 7H) 3.54-3.44
(m,
2H) 3.01-2.95 (m, 4H) 2.66 (s, 1H) 2.18-2.01 (m, 3H)); LC-MS 403 (M - H)+;
Purity
(HPLC) 100 %.
EXAMPLE 65
N-(3,4-Dimethoxyphenyl)-4-piperazin-1-ylthieno [3,2-cjpyridine-2-sulfonamide
hydrochloride
Tha synthesis was preformed essentially as described in Method H-L. Yield: 7.7
mg (3.6
%). 1H NMR (270 MHz, CH30H-d4) ~ ppm 8.04 (d, J 6.60 Hz, 1 H) 7.77-7.75 /m,
2H)
6.85-6.83 (m, 2H) 6.68-6.83 (m, 1H) 3.87-3.85 (m, 4H) 3.77-3.75 (m, 6H) 3.49-
3.45 (m,
4H) 2.65 (s, 1H); LC-MS 435 (M - H)+; Purity (HPLC) 98 %.
EXAMPLE 66
N-(4-Bromo-2-methylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L. Yield:
12.2 mg (5.3
%). 1H NMR (270 MHz, CH30H-d4) 8 ppm 8.07 (d, J 6.33 Hz, 1 H) 7.86-7.79 (m,
2H)
7.40 (d, J--1.58 Hz, 1 H) 7.30-7.29 (m, 1H) 7.08 (d, J--8.71 Hz, 1 H) 3.96-
3.92 (m, 4H)
3.53-3.SI (4H) 2.66 (s, 1H) 2.11 (s, 3H); LC-MS 467 (M - H)+; Purity (HPLC) 90
%.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 67
2-(4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonyl)-1,2,3,4-tetrahydro-
isoquinoline
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(3,4-
dihydro-1 H-isoquinoline-2-sulfonyl)-thieno [3,2-c]pyridin-4-yl]-piperazine-1-
carboxylic
acid test-butyl ester (0.235 mmol, 1 equiv.). Yield: 4.0 mg. LC/MS: tR= 0.801
(System: 30
to 60 % ACN in 1.5 min, Hypersil BDS), Purity: 92%. MS: 415 (M+1) 1H NMR (270
MHz, DMSO-d6) 8 ppm 2.87 (t, J--5.81 Hz, 2 H) 3.30 (s, 4 H) 4.43 (s, 2 H) 7.15
(m, 4 H)
7.70 (d, J--5.54 Hz, I H) 8.12 (s, 1 H) 8.18 (d, J--5.54 Hz, 1 H) 6 aliphatic
protons were
obscured by the water-peak in the spectra and so could not be analyzed.
EXAMPLE 68
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (2-thiophen-2-yl-ethyl)-
amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(2-
thiophen-2-yl-ethylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-
carboxylic acid te~t-
butylester (0.235 mmol, 1 equiv.). Yield: 8.7 mg. LC/MS: tR 0.430 (System: 30
% to 60
ACN in 1.5 min, Hypersil BDS), Purity: 93 %. MS: 409 (M+1) 1H NMR (270 MHz,
DMSO-d6) 8 ppm 2.25 (s, 1 H) 2.75 (s, 1 H) 2.96 (t, J 6.99 Hz, 1 H) 3.16 (q, J-
-6.51 Hz, 1
H) 3.31 (s, 4 H) 3.70 (s, 4 H) 6.90 (m, 1 H) 7.32 (t, J--5.54 Hz, 1 H) 7.70
(d, J--5.81 Hz, 1
H) 8.04 (d, J--1.85 Hz, 1 H) 8.18 (d, J--5.54 Hz, 1 H) 8.36 (m, 1 H) 9.05 (s,
1 H).
EXAMPLE 69
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (4-chloro-2,5-dimethoxy-
phenyl)-amide hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(4-chloro-
2,5-dimethoxy-phenylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-
carboxylic acid
test-butyl ester (0.112 mmol, 1 equiv.) was used as the thienopyridine in
Method C. Yield:
14.7 mg. LC/MS: tR 0.610 (System: 30 % to 60 % ACN in 1.5 min, YMC), Purity:
92 %.
MS: 469 (M+1) 1H NMR (270 MHz, DMSO- d6) 8 ppm 3.17 (s, 1 H) 3.27 (s, 4 H)
3.38 (s,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
3 H) 3.58 (d, J--4.22 Hz, 4 H) 3.77 (s, 3 H) 7.08 (s, 1 H) 7.69 (d, J--5.81
Hz, 1 H) 7.81 (s, 1
H) 8.16 (d, J 5.81 Hz, 1 H) 9.07 (s, 1 H) 10.17 (s, 1 H).
EXAMPLE 70
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid phenethyl-amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-(2-
phenethylsulfamoyl-thieno[3,2-c]pyridin-4-yl)-piperazine-1-carboxylic acid
test-butyl ester
(0.112 mmol, 1 equiv.). Yield: 3.8 mg. LC/MS: tR= 0.410 (System: 30% to 60%
ACN in
1.5 min, YMC), Purity: 91 %. MS: 403 (M+1) 1H NMR (270 MHz, CH30H-d4) 8 ppm
1.44 (d, J--7.13 Hz, 2 H) 3.51 (d, J 4.75 Hz, 4 H) 3.54 (s, 2 H) 3.94 (m, 4 H)
7.05 (m, 4 H)
7.16 (m, l H) 7.62 (s, 1 H) 7.72 (d, J 6.60 Hz, 1 H) 8.00 (d, J--6.60 Hz, 1
H).
EXAMPLE 71
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (2,6-diethyl-phenyl)-
amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(2,6-
dethyl-phenylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-carboxylic
acid tent-butyl
ester (0.112 mmol, 1 equiv.). Yield: 9.0 mg. LC/MS: tR 0.830 (System: 30 % to
60
ACN in 1.5 min, YMC), Purity: 92 %. MS: 431 (M+1) 1H NMR (270 MHz, DMSO-D6) ~
ppm 0.96 (t, J--7.52 Hz, 6 H) 2.25 (m, 1 H) 2.43 (s, 2 H) 2.75 (t, J 1.72 Hz,
1 H) 3.26 (s, 4
H) 3.62 (s, 4 H) 7.09 (s, 1 H) 7.12 (s, 1 H) 7.23 (m, 1 H) 7.73 (d, J 5.81 Hz,
1 H) 7.77 (s, 1
H) 8.19 (d, J 5.81 Hz, 1 H) 9.04 (s, 1 H) 9.95 (s, 1 H).
EXAMPLE 72
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3-phenyl-propyl)-amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(3-phenyl-
propylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-carboxylic acid tent-
butyl ester
(0.112 mmol, 1 equiv.). Yield: 13.0 mg. LC/MS: tR= 0.726 (System: 30 % to 60 %
ACN in
1.5 min, YMC), Purity: 91 %. MS: 417 (M+1) 1H NMR (270 MHz, CH30H-d4) 8 ppm
1.82 (m, 2 H) 2.63 (m, 2 H) 3.04 (t, J--6.86 Hz, 2 H) 3.55 (s, 4 H) 4.09 (s, 4
H) 7.16 (m, 4
H) 7.82 (d, J 6.60 Hz, 1 H) 8.05 (d, J--6.33 Hz, 1 H) 8.14 (s, 1 H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 73
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid (3,3-diphenyl-propyl)-
amide
hydrochloric acid
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(3,3-
diphenyl-propylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-carboxylic
acid tert-
butyl ester (0.112 mmol, 1 equiv.). Yield: 14.4 mg. LC/MS: tR=1.109 (System:
30 % to 60
ACN in 1.5 min, YMC), Purity: 93 %. MS: 493 (M+1) 1H NMR (270 MHz, DMSO-d6)
8 ppm 2.20 (m, 2 H) 2.80 (m, 2 H) 3.29 (s, 4 H) 3.67 (d, J--5.01 Hz, 4 H) 4.01
(m, 1 H)
7.14 (m, 8 H) 7.71 (d, J--5.81 Hz, 1 H) 7.95 (s, 1 H) 8.18 (d, J--5.81 Hz, 1
H) 8.27 (m, 2 H)
9.13 (s, 2 H).
EXAMPLE 74
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid [2-(5-methoxy-1H-indol-
3-yl)-
ethyl]-amide hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-(2-
[2-(5-
methoxy-1 H-indol-3-yl)-ethylsulfamoyl]-thieno[3,2-c]pyridin-4-yl) -piperazine-
1-
carboxylic acid test-butyl ester (0.112 mmol, 1 equiv.). Yield: 6.1 mg. LC/MS:
tR 0.364
(System: 30 % to 60 % ACN in 1.5 min, YMC), Purity: 91 %. MS: 472 (M+1) 1H NMR
(270 MHz, CH30H-d4) b ppm 2.85 (t, J--6.20 Hz, 2 H) 3.48 (t, J--6.20 Hz, 2 H)
3.55 (m, 4
H) 3.80 (s, 3 H) 4.02 (m, 4 H) 6.44 (dd, J 8.71, 2.37 Hz, 1 H) 6.80 (m, 2 H)
6.97 (s, 1 H)
7.64 (s, 1 H) 7.67 (s, 1 H) 7.97 (d, J 6.60 Hz, 1 H).
EXAMPLE 75
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid 4-trifluoromethyl-
benzylamide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(4-
trifluoromethyl-benzylsulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-
carboxylic acid
test-butyl ester (0.112 mmol, 1 equiv.) was used as the thienopyridine in
Method C. Yield:
1.9 mg. LC/MS: tR= 0.771 (System: 30% to 60% ACN in 1.5 min, YMC), Purity:
91%.
MS: 457 (M+1) 1H NMR (270 MHz, CH30H-d~) b ppm 3.54 (m, 4 H) 3.98 (m, 4 H)
4.36
(s, 2 H) 7.49 (m, 4 H) 7.74 (d, J--6.86 Hz, 1 H) 8.02 (s, 1 H) 8.07 (d, J--
6.60 Hz, 1 H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 76
4-Piperazin-1-yl-thieno[3,2-c]pyridine-2-sulfonic acid benzyl-ethyl-amide
hydrochloride
The synthesis was preformed essentially as described in Method H-L from 4-[2-
(benzyl-
ethyl-sulfamoyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-carboxylic acid tent-
butyl ester
(0.112 mmol, 1 equiv.). Yield: 6.4 mg. LC/MS: tR= 0.930 (System: 30 % to 60 %
ACN in
1.5 min, YMC), Purity: 95 %. MS: 417 (M+1) 1H NMR (270 MHz, CH30H-d4) S ppm
1.03(t,J--7.13Hz,3H)3.37(m,2H)3.57(s,2H)3.75(m,2H)4.11 (s,2H)4.50(s,2
H) 5.80 (s, 1 H) 7.32 (m, 5 H) 7.84 (d, J--6.60 Hz, 1 H) 8.07 (d, .I--6.60 Hz,
1 H) 8.14 (s, 1
H).
INTERMEDIATE 55
tent-Butyl-4-(3-{ [(3-ethylphenyl) amino] sulfonyl)thieno [3,2-c] pyridin-4-
yl)piperazine-
1-carboxylate
Prepared from text-butyl 4-[3-(chlorosulfonyl)thieno[3,2-c]pyridin-4-
yl]piperazine-1-
carboxylate (90.0 mg, 0.215 mmol) and 3-ethylaniline (33.9 mg, 0.28 mmol) to
give the
title compound as an off white solid (82.7 mg, 76 %). 1H NMR (400 MHz, CDCl3)
~ 1.03
(t, J = 7.5 Hz, 3 H), 1.48 (s, 9 H), 2.47 (q, J = 7.7 Hz, 2 H), 3.00-3.53 (m,
6 H), 4.02-4.44
(m, 2 H), 6.66 (d, J = 8.0 Hz, 1 H), 6.75 (s, 1 H), 6.66 (d, J = 8.0 Hz, 1 H),
7.02 (t, J = 7.8
Hz, 1 H), 7.67 (d, J = 5.5 Hz, 1 H), 8.24 (s, 1 H), 8.39 (d, J = 5.5 Hz, 1 H),
9.80 (s, 1 H).
MS (ESI+) m/z 503.2 (M+H)+. HPLC 97 %, RT: 3.93 min (5-99 % MeCN over 3 min).
EXAMPLE 77
N-(3-Ethylphenyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-3-sulfonamide
hydrochloride
Prepared from test-butyl 4-(3-{[(3-ethylphenyl)amino]sulfonyl)thieno[3,2-
c]pyridin-4-
yl)piperazine-1-carboxylate (81.1 mg, 0.161 mmol) which afforded 19 mg (98 %)
of the
product as a white solid (38.0 mg, 54 %). IH NMR (400 MHz, CH30H-d4) S 1.09
(t, J=
7.5 Hz, 3 H), 2.51 (q, J= 7.5 Hz, 2 H), 3.59 (br.s, 8 H), 6.89-6.92 (m, 3 H),
7.12-7.14 (m,
1 H), 8.06 (d, J= 6.0 Hz, 1 H), 8.36 (d, J= 6.0 Hz, 1 H), 8.49 (s, 1 H). MS
(ESI+) m/z
403.2 (M+H)+. HPLC 95%, RT: 3.02 min (5-99°1o MeCN over 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 56
tart-Butyl 4-(3-bromothieno [3,2-c] pyridin-4-yl)piperazine-1-Garb oxylate
A mixture of 3-bromo-4-chlorothieno[3,2-c]pyridine (729 mg, 2.93 mmol), tent-
butyl
piperazine-1-carboxylate (1.64 g, 8.80 mmol) and K2C03 (811 mg, 5.87 mmol) in
DMSO
(45 mL) was stirred for 5 days at 100 °C. After addition of HBO and
ethyl acetate, the
layers were separated. The water phase was extracted twice with ethyl acetate,
and the
combined organic phases were washed with water and brine and dried (MgSO4).
After
filtration and removal of the solvent, the residue was purified by silica gel
flash
chromatography (pentane/ethyl acetate, 8:2) to give the product as a white
powder (398
mg, 34 %). HPLC 99%, RT: 3.27 min (5-99% MeCN over 3 min).1H NMR (400 MHz,
CH30H-d4) S 1.48 (s, 9 H), 3.21 (br.s, 4 H), 3.71 (s br., 4 H), 7.61 (d, J=
6.1 Hz, 1 H),
7.72 (s, 1 H), 8.08 (d, J= 5.6 Hz, 1 H). MS (ESI+) m/z 398.2 (M+H)+.
INTERMEDIATE 57
}4-[4-(tent-Butoxycarbonyl)piperazin-1-yl]thieno[3,2-c]pyridin-3-
yl}sulfonyl)lithium
To a suspension of tent-Butyl 4-(3-bromothieno[3,2-c]pyridin-4-yl)piperazine-1-
carboxylate (4.055 g, 10.18 mmol) in diethyl ether (30 mL) at -78 °C
under NZ atmosphere
was added dropwise an 1.6 M solution of h-BuLi in hexanes (9.5 mL, 15.2 mmol).
After 1
h of stirring, a saturated solution of SOa in THF (25 mL) at -78 °C was
transferred via a
cannula to the mixture. The reaction was allowed to gradually increase to
ambient
temperature over night. The solvent was evaporated, and the residue was washed
with
several portions of diethyl ether and then dried under vacuum to give 4.094 g
of an off
white solid consisting of 66 % of the title compound and 34 % of (n-
butylsulfonyl)lithium
as by-product. This mixture was used without any further purification in the
next step. 1H
NMR (400 MHz, CH30H-d4) 8 1.48 (s, 9 H), 3.22 (br.s, 4 H), 3.72 (s br., 4 H),
7.60 (d, J=
5.5 Hz, 1 H), 8.06 (d, J= 5.5 Hz, 1 H), 8.14 (s, 1 H). MS (ESI+) m/z 384.0
(M+H)+.
HPLC RT: 2.62 min (5-99% MeCN over 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 58
tart-Butyl-4-[3-(chlorosulfonyl)thieno [3,2-c] pyridin-4-yl] piperazine-1-carb
oxylate
To a suspension of (~4-[4-(tent-butoxycarbonyl)piperazin-1-yl]thieno[3,2-
c]pyridin-3-
yl}sulfonyl)lithium (2.751 g, 7.06 mmol (3.126 g of the crude product
mixture)) in DCM
(40 mL) at 0 °C was added N chlorosuccinimide (1.338 g, 10.0 mmol).
After 20 minutes,
the temperature was raised to ambient, and the reaction mixture was stirred
for an
additional 2.5 h. The resulting product solution was washed with water, and
the water
phase was extracted with DCM. The combined organic extracts were washed with
brine
and dried over MgS04. After filtration and evaporation of the solvent, the
residue was
washed with several portions of pentane to yield the product as an off white
solid (2.024 g,
69 %). 1H NMR (400 MHz, CH30H-d4) S 1.47 (s, 9 H), 3.11 (br.s, 4 H), 3.2-4.3
(s br., 4
H), 7.68 (d, J= 5.5 Hz, 1 H), 8.45 (d, J= 5.5 Hz, 1 H), 8.60 (s, 1 H). MS
(ESI+) m/z 418.2
(M+H)+. HPLC 92%, RT: 3.76 min (5-99% MeCN over 3 min).
INTERMEDIATE 59
tart-Butyl-4-(3-f [(4-isopropylphenyl)amino]sulfonyl}thieno[3,2-c]pyridin-4-
yl)piperazine-1-carboxylate
Prepared from text-butyl 4-[3-(chlorosulfonyl)thieno[3,2-c]pyridin-4-
yl]piperazine-1-
carboxylate (90.0 mg, 0.215 mmol) and 4-isopropylaniline (37.9 mg, 0.28 mmol)
to give
the title compound as an off white solid (58.3 mg, 52 %). 1H NMR (400 MHz,
CDC13) 8
1.12 (d, J= 7.0 Hz, 6 H), 1.47 (s, 9 H), 2.76 (sept., J= 6.9 Hz, 2 H), 3.01-
3.53 (m, 6 H),
4.04~4..41 (m, 2 H), 6.79 (d, J= 8.5 Hz, 2 H), 6.66 (d, J= 8.5 Hz, 2 H), 7.69
(d, J= 5.5 Hz,
1 H), 8.23 (s, 1 H), 8.40 (d, J= 5.5 Hz, 1 H), 9.90 (s, 1 H). MS (ESI+) m/z
517.2 (M+H)+.
HPLC 97%, RT: 4.01 min (5-99% MeCN over 3 min).
EXAMPLE 78
N-(4-Isopropylphenyl)-4-pip erazin-1-ylthieno [3,2-c] pyridine-3-sulfonamide
hydrochloride
Prepared from tart-butyl 4-(3- f [(4-isopropylphenyl)amino]sulfonyl}thieno[3,2-
c]pyridin-
4-yl)piperazine-1-carboxylate (60.0 mg, 0.116 mmol) which afforded 19 mg (98
%) of the
product as a white solid (25.8 mg, 49 %) according to Method H-L. 1H NMR (400
MHz,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
CH30H-d4) b 1.16 (d, J= 7.0 Hz, 6 H), 2.81 (sept., J= 6.8 Hz, 1 H), 3.59 (s,
br., 8 H), 7.00
(d, J= 8.0 Hz, 2 H), 7.10 (d, J= 8.0 Hz, 2 H), 8.07 (s, br., 1 H), 8.37 (s,
br., 1 H), 8.50 (s, 1
H). MS (ESI+) m/z 417.2 (M+H)+. HPLC 94%, RT: 3.14 min (5-99% MeCN over 3
min).
EXAMPLE 79
N-(4-Methylphenyl)-4-(pyrrolidin-3-yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
4-Chloro-N (4-methylphenyl)thieno[3,2-c]pyridine-2-sulfonamide (60.0 mg, 0.17
rnmol)
in dry DMF (1 mL) and NaH (5,1 mg, 0.21 mmol) was added to pyrrolidin-3-of
(18.5 mg,
0.21 mmol) under nitrogen. The mixture was heated in the microwave at
200°C in 5 min.
The product was purified by preparative HPLC. Yield: 29.9 mg (43.4 %). 1H NMR
(270
MHz, CH30H-d4) 8 ppm 8.09 (s, 1H) 7.71 (d, J 6.93 Hz, I H) 7.46 (d, J--6.93
Hz, 1 H)
7.47-7.44 (m, 4H) 4.67 (d, J 3.22 Hz, 1 H) 3.97 (s, 2 H) 2.26 (s, 3H). LC-MS
390 (M -
H)+; Purity (HPLC) 99 %.
EXAMPLE 80
N-(4-Methylphenyl)-4-(piperidin-4-yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
The synthesis was preformed essentially as described for compound of N-(4-
methylphenyl)-4-(pyrrolidin-3-yloxy)thieno[3,2-c]pyridine-2-sulfonamide
hydrochloride.
Yield: 16.2 mg (22.7 %). 1H NMR (270 MHz, CH30H-d4) 8 ppm 7.81-7.78 (m, 2H)
7.62
(d, J 6.93 Hz, 1 H) 7.13-7.04 (m, 4H) 4.05-3.94 (m, 1H) 3.90-3.88 (m, 2H) 3.63-
3.58 (m,
2H), 2.27 (s, 3H) 2.06-2.03 (m, 2H) 2.01-1.71 (m, 2H); LC-MS 404 (M - H)+;
Purity
(HPLC) 99 %.
EXAMPLE 81
N-(2,3-Difluorobenzyl)-4-piperazin-1-ylthieno[3,2-c]pyridine-2-sulfonamide
hydrochloride
Yield: 74.4 mg (39.2 %). 1H NMR (270 MHz, CH30H-d4) ~ ppm 8.10-8.03 (m, 2H)
7.81
(d, J 6.68 Hz, 1 H) 7.16-7.05 (m, 3H) 4.37 (s, 2 H) 4.11-4.07 (m, 4H) 3.59-
3.53 (m, 4H);
LC-MS 425 (M - H)+; Purity (HPLC) 90 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
. _ ., __ _~ _ _
EXAMPLE 82
N-(3-Chlorob enzyl)-4-piperazin-1-ylthieno [3,2-c] pyridine-2-sulfonamide
hydrochloride
Yield: 74.4 mg (44.7 %). 1H NMR (270 MHz, CH30H-d4) 8 ppm 8.04-8.01 7.85 (d,
J 6.93 Hz, l H) 7.22-7.15 (m, 4H) 4.30 (s, 2 H) 4.14-4.10 (m, 4H) 3.60-3.54
(m, 4H); LC-
MS 423 (M - H)+; Purity (HPLC) 90%.
Table 5
R4
\ QO
S R2
EXAMPLE RZ R4
4-(1,4-Diazepan-1-yl)-2-(phenylsulfonyl)thieno[3,2-c]pyridine ~,
hydrochloride
4-(1,4-Diazepan-1-yl)-2-[(3,4-dichlorophenyl)sulfonyl]thieno[3,2-
c]pyridine hydrochloride
CI
4-(1,4-Diazepan-1-yl)-2-[1-naphthylsulfonyl)thieno[3,2-c]pyridine
hydrochloride
i
4-(1,4-Diazepan-1-yl)-2-[4-tert-butylphenylsulfonyl)thieno[3,2-
c]pyridine hydrochloride
4-(1,4-Diazepan-1-yl)-2-[3,4-dimethylphenylsulfonyl)thieno[3,2-
c]pyridine hydrochloride
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
2-[(4-Bromophenyl)sulfonyl]-4-(1,4-diazepan-1-yl)thieno[3,2-
c]pyridine hydrochloride
Br
89 2-(Phenylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine ~~ N
hydrochloride ~ , ~~
N
9o 2-(3-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2- ~ N
c]pyridine hydrochloride
i N
91 2-(4-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2- N
c]pyridine hydrochloride
' C~
O N
92 4-Piperazin-1-yl-2-{[4-irifluoromethyl)phenyl]sulfonyl}thieno[3,2- \ ~ N
c]pyridine hydrochloride F
FF N
93 2-[[2-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- N
c]pyridine hydrochloride
~'~v c ~
N
94 2-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- ~ ~ N
c]pyridine-2-sulfonamide hydrochloride
cW ~ N
95 2-[(4-tert-Butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- \ ~ N
c]pyridine hydrochloride ~ ~ C
N
2-(1-Naphthyl sulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine N
hydrochloride
~~ C~
N
i
2-[(3-Fluorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine- ~ N
2-sulfonamide hydrochloride
~~~ C ~
N
F
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
2-(Mesitylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine N
hydrochloride
i N
99 2-[(2-Methoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine N
hydrochloride ~ ~'',' C
O N
100 2-[(2,4-Dimethoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- N
c]pyridine hydrochloride
O O N
101 2-[(2,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- N
c]pyridine hydrochloride
N
102 2-[(2,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- N
c]pyridine hydrochloride ~ ,
N
to3 2-[(2-Ethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine N
hydrochloride
'~~~ C ~
N
104 4-(Piperazinyl)-2-(3-methoxybenzyl-sulfonyl)-thienopyridine O N
hydrochloride
~ C~
i N
105 2-(Benzylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine ~ ~ N
hydrochloride
N
los 4-Piperazin-1-yl-2-{[4-(trifluoromethyl)benzyl]sulfonyl}thieno[3,2- I ~ N
c]pyridine hydrochloride
F N
l07 2-[(3-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine Br~ N
hydrochloride
c
N
1os 2-[(2,3-Difluorobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- ~ ~ N
c]pyridine hydrochloride ~ i F
F N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
l09 2-[(4-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine ~ ~ ~ N
hydrochloride Br ~ i ~
N
110 2-{[2,5-bis(Trifluoromethyl)benzyl]sulfonyl}-4-piperazin-1- CF3 I ~ ; ' N
ylthieno[3,2-c]pyridine hydrochloride ' CF3
N
111 2-[(4-Methylbenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine ~ N
hydrochloride
c~
N
112 2-{[5-Chloro-2-(trifluoromethyl)benzyl]sulfonyl}-4-piperazin-1- C~ I ~ % N
ylthieno[3,2-c]pyridine hydrochloride ~ CF3
N
113 2-[(3,5-Dimethoxybenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- ,p I \ ; N
c]pyridine hydrochloride
~p N
114 2-[(2-Naphthylmethyl)sulfonyl]-4-piperazin-1-ylthieno[3,2- ~~ N
c]pyridine hydrochloride
N
i
115 4-Piperazin-1-yl-2-{[4-(1,2,3-thiadiazol-5- N
1 bent 1 sulfon 1 thieno 3 2 c dine h drochloride
Y ) Y ] Y } [ ~ - ]pYri Y
N
N
,N.S
116 1-(4-Pyrrolidin-1-ylphenyl)-2-[(4-piperazine-1-ylthieno[3,2- o ' N
c]pyridin-2-yl) sulfonyl] propanone hydrochloride ~N ~ '
N
117 1-[4-(Diethylamino)phenyl]-2-[(4-piperazine-1-ylthieno[3,2- p ; N
c]pyridin-2-yl) sulfonyl] propanone hydrochloride
J N
11s 1-(4-Bromophenyl)-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-yl) ~ ; N
sulfon 1 ro anone Br I ~
Y]p p
N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
119 ~ ~ N
1-(3-Methoxyphenyl)-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-yl)
sulfonyl] propanone ,O N
tzo 1-Phenyl-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-~ ; N
yl)sulfonyl]pxopanone
N
Scheme 6
ci R4 R4
~ N j w N ii, iii R~, ,~ ~ ~ N
Br ~ ~ Br ~ ~ --~- 'S
S ~ g~ O S
Legend to Scheme 6: i) BOC protected amines (R4), KZCO3, DMSO; ii) thiophenols
(Rl-SH), Cu2(I)O,
DMF; iii) NaOAc, oxone, water; iv) a.TFA, b.HCI, methanol
INTERMEDIATE 60
tent-Butyl 4-(2-bromothieno[3,2-c]pyridin-4-yl)piperazine-1-carboxylate
2-Bromo-4-chlorothieno[3,2-c]pyridine (5.0 g, 20.24 mmol) and I~2CO3 (13.97 g,
101.2
mmol) was stirred in DMSO (20 mL) followed by addintion of test-butyl
piperazine-1-
carboxylate (4.14 g, 22.26 mmol). The reaction mixture was stirred at 100
°C fox 6 days.
The reaction mixture was filtered to eliminate the carbonate and addition of
water (50 mL)
and ethyl was followed. The phases were separated and the aqueous phase was
extracted
with ethyl acetate. The combined organic phases were dried (MgS04) and the
solvent was
evaporated. The crude product was purified by flash chromatography using ethyl
acetate/hexanes (2/8) as eluent to give 2 g of the desired product, yield 25%,
99% pure. 1H
NMR (270 MHz, CDCl3) S 1.48 (s, 9H), 1.52-1.63 (m, 1H), 3.42-3.47 (m, 4H),
3.61-3.64
(m, 4H), 7.22 (dd, J= 5.4, 1 Hz, 1H), 7.35 (d, J=1 Hz, 1H), 8.04 (d, J= 5.4
Hz, 1H). m/z =
398.91 (M+H), bromide pattern.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 61
tent-butyl 4-(2-bromothieno [3,2-c]pyridin-4-yl)-1,4-diazepane-1-carboxylate
The same procedure above intermediate was used starting from 2-bromo-4-
chlorothieno[3,2-c]pyridine (7.5 g, 30.45 mmol), KZC03 (6.7 g, 33.5 mmol) and
tent-butyl
1,4-diazepane-1-carboxylate (21.0 g, 152.2 mmol) in DMSO (30 mL). Purification
by flash
chromatography 3.04 g of the title compound (Yield 25%).HPLC purity 92%; 1H
NMR
(270 MHz, CDC13) 8 1.38 (s, 4.5 H), 1.43 (s, 4.5 H), 1.96-2.11 (m, 2H), 3.36-
3.41 (m, 1H),
3.46-3.51 (m, 1H), 3.65-3.87 (m, 6H), 7.02-7.04 (m, 1H), 7.40-7.42 (m, 1H),
7.94 (d, J=
5.4 Hz, 1 H). m/z = 411.97 (M+H).
Coupling with thiophenols (Method M )
INTERMEDIATE 62
tart-butyl 4-(2-phenylthio)thieno [3,2-c] pyridin-4-yl)-1,4-diazep ane-1-
carboxylate
I5 tent-Butyl 4-(2-bromothieno[3,2-c]pyridin-4-yl)-1,4-diazepane-1-carboxylate
, (0.31 g,
0.752 mmol), pulverized KOH (0.084 g, I.5 mmol) and Cua(I)O (0.1 g, 0.75 mmol)
was
mixed with DMF (1 mL) before the addition of a solution of benzenethiol (.016
g, 1.5
rnmol) in DMF (1 mL). The reaction mixture was heated to 120 °C for I5
h. The reaction
mixture was poured in a silica plug and eluted with chloroform, to give the
crude product.
The crude product was purified by flash chromatography using ethyl
acetate/hexanes (2/8)
as eluent to give 0.21 g of the desired product, yield 64%, 90% pure. 1H NMR
(270 MHz,
CDC13) 8 1.37 (s, 4.5H), 1.43 (s, 4.5 H), I.97-2.10 (m, 2H), 3.36-3.43 (m,
1H), 3.46-3.53
(m, 1H), 3.64-3.73 (m, 2H), 3.78-3.96 (m, 4H), 7.05 (d, J= 5.4 Hz, 1H), 7.22-
7.32 (m, 5H),
7.59-7.63 (m, 1H), 7.97 (d, J= 5.4 Hz, 1H). m/z = 442.15 (M+H).
Oxidation of thio-derivatives (Method 1~
INTERMEDIATE 63
tent-Butyl 4-(2-phenylsulfonyl)thieno[3,2-c]pyridin-4-yl)-1,4-diazepane-1-
carboxylate
A solution of tent-Butyl 4-(2-phenylthio)thieno[3,2-c]pyridin-4-yl)-1,4-
diazepane-1-
carboxylate (0.2I g, 0.48 rnmol) and NaOAc (0.5 g) in ethanol (10 mL), (pH ~5)
followed
by the addition of Oxone (0.64 g, 1.04 mmol) dissolved in water (1 mL). The
reaction
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
mixture was stirred at RT for 16 h. Additional Oxone (4.32 g) in water (1mL)
was added.
Full conversion of the SM was obtained after 8 h. Water (50 mL) and chloroform
(30 mL)
were added. The phases were separated and the aqueous phase was extracted with
chloroform. The combined organic phases were dried over (MgSOa.), the solvent
was
evaporated to give the crude product which was purified by reverse-phase
chromatography
(1090), to give 0.191 g of the desired product as yellow oil (Yield 86%) 98%
pure. 1H
NMR (270 MHz, CDC13) 8 1.28 (s, 4.5 H), 1.38 (s, 4.5 H), 1.99-2.03 (m, 2H),
3.31-3.40
(m, IH), 3.42-3.47 (rn, 1H), 3.63-3.69 (m, 2H), 3.85-3.98 (m, 4H), 7.02 (d, J=
5.4 Hz, 1H),
7.48-7.61 (m, 3H), 7.76-8.01 (m, 3H), 8.06-8.08 (m, 1H). m/z = 474.01 (M+H).
Removal of the t-butyl-carboxylate protecting group (Method O)
EXAMPLE 83
4-(1,4-Diazepan-1-yl)-2-(phenylsulfonyl)thieno[3,2-c]pyridine hydrochloride
tart-Butyl 4-(2-phenylsulfonyl)thieno [3,2-c]pyridin-4-yl)- I ,4-diazepane-1-
carboxylate
(0.165 g, 0.348 mmol) was dissolved in DCM (2 mL) and TFA (1 mL) was added.
The
reaction mixture was stirred for 2 h. The solvent was evaporated. Methanol and
HCl in
ether was added (x 3) to give 0.118 g of the desired HCl salt, yield 85%, 98%
pure. 1H
NMR (270 MHz, CHOH-d~) 8 2.45-2.52 (m, 2H), 3.45-3.52 (m, 2H), 3.70-3.79 (m,
2H),
4.18-4.22 (m, 2H), 4.30-4.40 (m, 2H), 7.62-7.76 (m, SH), 7.9I (d, J = 5.4 Hz,
1H), 8.11
(dd, J= 5.4, 1 Hz, 1H), 8.41 (d, J=1 Hz, 1H). m/z = 374.09 (M+H-HCl).
INTERMEDIATE 64
tent Butyl 4-[2-(4-tart butylphenyl)thio]thieno(3,2-c]pyridin-4-yl)-1,4-
diazepane-1-
carboxylate
The product was prepared according to Method M. Purification by flash
chromatography
using ethyl acetate/hexanes (2/8) as eluent gave 0.035 g, 99% pure. 1H NMR
(270 MHz,
CDC13) b 1.28 (s, 9H), 1.38 (s, 4.5 H), 1.43 (s, 4.5 H), 1.98-2.03 (m, 2H),
3.35-3.41 (m,
1H), 3.46-3.52 (m, 1H), 3.62-3.72 (m, 2H), 3.77-3.93 (4H), 7.04 (d, J= 5.4 Hz,
1H), 7.27-
7.34 (m, 4H), 7.54-7.56 (m, 1H), 7.95 (d, J= 5.4 Hz, 1H). m/z = 498.0 (M+H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 65
tent-Butyl 4-[2-(4-tent-butylphenyl)sulfonyl] thieno [3,2-c] pyridin-4-yl)-1,4-
diazepane-1-
carboxylate
Procedure B from text-butyl 4-[2-(4-tent-butylphenyl)thio]thieno[3,2-c]pyridin-
4-yl)-1,4-
diazepane-1-carboxylate (0.035 g, 0.070 mmol), Oxone (0.17 g, 0.28 mmol),
NaOAc (0.5
g) in EtOH (2 mLfollowed by reversed phase chromatography (40-X70), gave 6 mg
of the
product. Yield 17%, 98% pure.lH NMR (270 MHz, CDC13) 8 1.32 (s, 9H), 1.35 (s,
9H),
2.05-2.15 (m, 2H), 3.45-3.62 (m, 2H), 3.75-4.13 (m, 6H), 7.20-7.27 (m, SH),
7.58 (d, J=
10.8 Hz, 1H), 7.93 (d, J=10.8 Hz, 1H). m/z = 530.0 (M+H).
INTERMEDIATE 66
tent-Butyl 4-[2-(3,4-dimethylphenyl)thio] thien o [3,2-c] pyridin-4-yl)-1,4-
diazep ane-1-
carboxylate
The title compound was obtained according to Method M. Purification by flash
chromatography using ethyl acetate/hexanes (2/8) as eluent gave 0.022 g, 95%
pure. 1H
NMR (270 MHz, CDC13) b 1.38 (s, 4.5 H), 1.43 (s, 4.5 H), 1.96-2.04 (m, 2H),
2.21 (s, 3H),
2.22 (s, 3H), 3.37-3.45 (m, 2H), 3.47-3.50 (m, 2H), 3.77-3.95 (m, 4H), 7.01-
7.12 (m, 3H),
7.16 (s, 1H), 7.53 (dd, J= 5.4, 1 Hz, 1H), 7.94 (d, J= 5.4 Hz, 1H). xn/z =
470.3 (M+H).
INTERMEDIATE 67
tart-Butyl 4-[2-(3,4-dimethylphenyl) sulfonyl] thieno [3,2-c] pyridin-4-yl)-
1,4-diazep ane-
1-carboxylate
Procedure B from test-Butyl 4-[2-(3,4-dimethylphenyl)thio]thieno[3,2-c]pyridin-
4-yl)-1,4-
diazepane-1-carboxylate (0.022 g, 0.047 mmol); OXONE (0.11 g, 0.19 mmol);
NaOAc
(0.5 g) inEtOH (2 mL) followed by reversed phase chromatography (4070), 9 mg
of the
product. Yield 38%, 92% pure.lH NMR (270 MHz, CDCl3) 8 1.35 (s, 9H), 2.08-2.20
(m,
2H), 2.33 (s, 6H), 3.52-3.59 (m, 2H), 3.83-3.88 (m, 2H), 4.08-4.18 (m, 4H),
7.21-7.28 (m,
2H), 7.31-7.35 (m, 1H), 7.73-7.75 (m, 2H), 8.02 (d, J= 5.4 Hz, 1H). m/z =
502.21 (M+H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 68
tart-Butyl 4-[2-(1-naphthyl)thio]thieno[3,2-c]pyridin-4-yl)-1,4-diazepane-1-
carboxylate
The title compound was obtained according to Method M. Purification by flash
chromatography using ethyl acetate/hexanes (2/8) as eluent gave 0.055 g. HPLC
purity 99
%;1H NMR (270 MHz, CDC13) 8 1.37 (s, 4.5 H), 1.43 (s, 4.5 H), 1.89-2.20 (m,
2H), 3.30-
3.40 (m, 1H), 3.43-3.50 (m, 1H), 3.60.3.90 (m, 6H), 6.99 (d, J= 5.4 Hz, 1H),
7.39 (dd, J=
8.1, 1 Hz, 1H), 7.50-7.61 (m, SH), 7.79-7.88 (m, 2H), 7.92 (d, J= 5.4 Hz, 1H),
8.40-8.44
(m, 1H). ii1/z = 498.26 (M+H).
INTERMEDIATE 69
tart-Butyl 4-[2-(1-naphthyl)sulfonyl]thieno[3,2-c]pyridin-4-yl)-1,4-diazepane-
1-
carboxylate
Procedure B from tart-Butyl 4-[2-(1-naphthyl)thio]thieno[3,2-c]pyridin-4-yl)-
1,4-
diazepane-1-carboxylate (0.055 g, 0.112 mmol); Oxone (0.27 g, 0.448 mmol);
NaOAc (0.5
g) in EtOH (2 mL) followed reversed phase chromatography (4070) gave 15 mg of
the
product. Yield 26%, 93% pure. 1H NMR (270 MHz, CDC13) S 1.34 (s, 9H), 2.06-
2.10 (m,
2H), 3.48-3.62 (m, 2H), 3.78-3.86 (m, 2H), 3.95-4.16 (m, 4H), 7.19-7.31 (m,
2H), 7.60-
7.75 (m, 3H), 7.92-7.99 (m, 2H), 8.18 (m, J= 8.1 Hz, 1H), 8.50-8.53 (m, 1H),
8.77-8.80
(m, 1H). m/z = 524.22 (M+H);
EXAMPLE 84
4-(1,4-Diazepan-1-yl)-2-[(3,4-dichlorophenyl)sulfonyl]thieno[3,2-c]pyridine
hydrochloride
tent-Butyl 4- f 2-[(3,4-dichlorophenyl)sulfonyl]thieno[3,2-c]pyridin-4-yl}-1,4-
diazepane-1-
carboxylate was prepared from 3,4-dichlorothiophenol (60 mg, 15%), as a beige
solid, by
the application of the general procedures A and B described above. 1H NMR
(CDCl3) ~
8.27-8.14 (m, 1H), 8.11-8.04 (m, 2H), 7.87-7.80 (m, 1H), 7.67-7.62 (m, 1H),
7.26-7.20 (m,
1H), 4.18-3.98 (m, 4H), 3.87-3.74 (m, 2H), 3.61-3.44 (m, 2H), 2.20-2.00 (m,
2H), 1.33 (s,
9H); MS m/z 542 (M+1).The title compound (50 mg, 95%) was obtained as a beige
solid,
by the application of the general procedure C described above. 1H NMR (270
MHz,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
CH30H-d4) ~ 8.48 (s, 1H), 8.30 (d, J=1.85 Hz, 1H), 8.05 (dd, J= 8.58, 1.98 Hz,
1H),
7.92 (d, J= 6.86 Hz, 1H), 7.83 (d, J= 8.44 Hz, 1H), 7.69 (d, J= 6.86 Hz, 1H),
4.4.41-4.34
(m, 2H), 4.24-4.16 (m, 2H), 3.76-3.69 (m, 2H), 3.51-3.43 (m, 2H), 2.52-2.42
(m, 2H); MS
m/z 442 (M+1).
EXAMPLE 85
4-(1,4-Diazepam-1-yl)-2-[1-naphthylsulfonyl)thieno[3,2-c]pyridine
hydrochloride
The title compound was obtained from tent-butyl 4-[2-(1-
naphthyl)sulfonyl]thieno-[3,2-
c]pyridin-4-yl)-1,4-diazepane-1-carboxylate (15 mg, 0.029 mmol) following
Method O to
give 12 mg of the desired product yield 90 %, 95 % pure. 1H NMR (270 MHz,
CH30H-d4)
~ 2.40-2.50 (m, 2H), 3.45-3.55 (m, 2H), 3.65-3.75 (m, 2H), 4.06-4.26 (m, 2H),
4.27-4.46
(m, 2H), 7.58-7.80 (m, 4H), 7.83-7.86 (m, 1H), 8.06 (d, J= 8.1 Hz, 1H), 8.20
(d, J= 8.1 Hz,
1H), 8.48 (s, 1H), 8.53-8.56 (m, 1H), 8.83-8.86 (m, 1). m/z = 424.06 (M+H-
HCl).
EXAMPLE 86
4-(1,4-Diazepam-1-yl)-2-[4-tent butylphenylsulfonyl)thieno[3,2-c]pyridine
hydrochloride
The title compound was obtained from tent-butyl 4-[2-(4-tert-butylphenyl)-
sulfonyl]thieno[3,2-c]pyridin-4-yl)-1,4-diazepane-1-carboxylate (6 mg, 11.3
mmol)
following Method O to give 4 mg of the desired product, yield 76 %, 88 % pure.
IH NMR
(270 MHz, CH30H-d4) b 1.33 (s, 9H), 2.41-2.47 (m, 2H), 3.41-3.49 (m, 2H), 3.65-
3.78 (m,
2H), 4.15-4.25 (m, 2H), 4.29-4.40 (m, 2H), 7.65-7.70 (m, 3H), 7.90 (d, J= 5.4
Hz, 1H),
8.00-8.04 (m, 2H), 8.37 (s, 1H). m/z = 430.06 (M+H-
EXAMPLE 87
4-(1,4-Diazepam-1-yl)-2-[3,4-dimethylphenylsulfonyl)thieno[3,2-c]pyridine
hydrochloride
The title compound was obtained from tent-butyl 4-[2-(3,4 dimethylphenyl)-
sulfonyl]thieno[3,2-c]pyridin-4-yl)-1,4-diazepane-1-carboxylate (6 mg, 0.012
mmol)
following Method O to give 6 mg of the desired product, yield 88 %, 89 % pure.
1H NMR
(270 MHz, CH30H-d4) 8 2.34 (s, 6H), 2.45-2.55 (m, 2H), 3.42-3.51 (rn, 2H),
3.67-3.76 (m,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
2H), 4.10-4.20 (m, 2H), 3.58-3.70 (m, 2H), 7.39-7.41 (m, 1H), 7.64-7.67 (m,
IH), 7.79-
7.84 (m, 2H), 7.89-7.91 (m, 1H), 8.36 (s, 1H). m/z = 402.07 (M+H-HCl).
EXAMPLE 88
2-[(4-Sromophenyl)sulfonyl]-4-(1,4-diazepam-1-yl)thieno[3,2-c]pyridine
hydrochloride
Trifluoroacetic acid (1 rnL) was added slowly to a solution of tent-butyl 4- f
2-[(4-
bromophenyl)thio]thieno[3,2-c]pyridin-4-yl~-1,4-diazepane-1-carboxylate (26
mg, 0.047
mmol) in CH2C12 at 0 °C. The reaction mixture was allowed to reach room
temperature,
stirred for 40 min and then concentrated in vacuo. The residue was twice re-
dissolved in
MeOH and concentrated in vacuo. The residue was again dissolved in MeOH and an
excess of IM HCl in diethyl ether (4 mL) was slowly added to the solution.
Removal of the
solvents ih vacuo afforded the title compound (21 mg, 91 %) as a yellowish
solid. 1H NMR
(270 MHz, CH30H-d4) S 8.41 (s, 1H), 8.06-7.99 (m, 2H), 7.92 (d, J= 6.86 Hz,
1H), 7.87-
7.80 (m, 2H), 7.66 (d, J= 6.86 Hz, 1H), 4.38-4.31 (m, 2H), 4.22-4.14 (m, 2H),
3.74-3.67
(m, 2H), 3.50-3.42 (m, 2H), 2.51-2.39 (m, 2H); MS m/z 452 (M+1).
EXAMPLE 89
2-(Phenylsulfonyl)-4-piperazin-1-ylthieno[3,2-cJpyridine hydrochloride
To a stirred solution of tent-butyl 4-[2-(phenylthio)thieno[3,2-c]pyridin-4-
yl]piperazine-1
carboxylate (350 mg, 0.819 mmol) in ethanol was added ozone in water solution.
The
reaction was monitored by LCMS. When all starting material was consumed, the
chromatogram showed two major peaks, the product and the N-oxide. After
purification by
preparative HPLC, the resulting Boc-material was treated with HCI in ether .
The solution
was centrifugated and the supernatant was removed. Ether was added, then
centrifugated
and decanted (repeated three times) to remove the excess HCl. The remaining
ether was
finally evaporated in a SpeedVac concentrator. Yield 18 %, HPLC purity = 98%,
mlz =
360.0 (M+H). tH NMR (270 MHz, CH30H-d4) 8 ppm 3.56 (m, 4 H) 4.08 (m, 4 H) 7.68
(m, 4 H) 7.77 (dd, J--6.60, 0.79 Hz, 1 H) 8.04 (d, J 6.33 Hz, 1 H) 8.12 (m, 2
H) 8.39 (d,
J--0.79 Hz, 1 H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXfMLE 90
2-(3-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2-c]pyridine
hydrochloride
4-[2-(3-Methoxy-phenylsulfanyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-
1-carboxylic acid tent-butyl ester was obtained from 3-methoxythiophenol (130
pl, 1
S mmol) and tent-Butyl 4-(2-bromothieno[3,2-c]pyridin-4-yl)piperazine-1-
carboxylate (21S
mg, 0.52 mmol). 120 mg, SO%) were obtained by the application of the general
Method M
described above. 1H NMR (270 MHz, CDCl3) 8 1.48 (s, 9 H), 3.42-3.51 (m, 4 H),
3.58-
3.67 (m, 4 H), 3.74 (s, 3 H), 6.76 (dd, J--8.18, 2.38 Hz, 1 H), 6.84-6.92 (m,
2 H), 7.16-7.23
(m, 2 H), 7.51 (s, 1 H), 8.04 (d, J 5.81 Hz, 1 H); MS m/z 4S8 (M+1). The title
compound
was therefore obtained from 4-[2-(3-methoxy-phenylsulfanyl)-thieno[3,2-
c]pyridin-4-yl]-
piperazine-1-carboxylic acid tent-butyl (7 mg, 7%), after triturating with
diethyl ether, as a
beige solid, by the application of the general procedures B and C described
above. 1H
NMR (270 MHz, CD30D) 8 8.31 (s, 1H), 8.09 (d, J = 6.33 Hz, 1H), 7.71-7.62 (m,
2H),
7.59-7.50 (rn, 2H), 7.30-7.23 (m, IH), 3.98-3.92 (m, 4H), 3.87 (s, 3H), 3.54-
3.48 (rn, 4H);
1S MS m/z 390 (M+1).
EXAMPLE 91
2-(4-Methoxy-benzenesulfonyl)-4-piperazin-1-yl-thieno[3,2-c)pyridine
hydrochloride
4-[2-(4-Methoxy-phenylsulfanyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-
1-carboxylic acid tart-butyl ester was obtained from 4-methoxythiophenol (130
ul, 1
mmol) and tent-butyl 4-(2-bromothieno[3,2-c]pyridin-4-yl)piperazine-1-
carboxylate (21S
mg, O.S2 mmol). 100 mg, 42% were isolated by the application of the general
Method M
described above. 1H NMR (270 MHz, CDCl3) 8 1.48 (s, 9 H), 3.40-3.47 (m, 4 H),
3.58-
3.65 (m, 4 H), 3.79 (s, 3 H), 6.83-6.89 (m, 2 H), 7.15 (d, J S.S4 Hz, 1 H),
7.35 (s, 1 H),
2S 7.38-7.43 (m, 2 H), 7.99 (d, J--5.81 Hz, 1 H); MS m/z 458 (M+1).
4-[2-(4-methoxy-phenylsulfanyl)-thieno[3,2-c]pyridin-4-yl]-piperazine-1-
carboxylic acid
tent-butyl ester was obtained (2S mg, 23%) as a clear liquid by the
application of the
general procedure B described above. 1H NMR (270 MHz, CDC13) 8 1.48 (s, 9 H),
3.67-
3.91 (m, 11 H), 7.01 (d, J = 8.97 Hz, 2H), 7.27-7.37 (m, 1H), 7.93 (d, J =
8.97 Hz, 2H),
8.01-8.19 (m, 2H); MS m/z 490 (M+1). The title compound was thereby obtained
following Procedure C): 1H NMR (CD30D) 8 8.25 (s, 1H), 8.09-7.89 (m, 3H), 7.69
(d, J =
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
6.33 Hz, 1H), 7.17-7.10 (m, 2H), 4.00-3.93 (m, 4H), 3.87 (s, 3H), 3.55-3.48
(m, 4H); MS
m/z 390 (M+1).
EXAMPLE 92
4-Piperazin-1-yl-2-~[4-trifluoromethyl)phenyl]sulfonyl]~thieno[3,2-c]pyridine
hydrochloride
2- f [4-(Trifluoromethyl)phenyl]thio}-4-piperazin-1-ylthieno[3,2-c]pyridine
(0.42 mmol)
was dissolved in TFA (1.5 mL) at 0°C, stirred for 15 min and HzOa (100
p,L) was added.
The mixture was stirred at room temperature over night. NaOH (2 M) was added,
extraction with ethyl acetate (3X), washed with brine, dried over NaS04, The
solvent was
removed and the product was purified by preparative HPLC to afford 154.7 mg
(86.2 %).
1H NMR (270 MHz, DMSO-d6) 8 ppm 9.79 (s, 1H) 8.56 (s, 1 H) 8.35 (d, J--8.44
Hz, 2 H)
8.12-8.05 (m, 3H) 7.79 (d, J--6.33 Hz, 1 H) 3.98-3.96 (m, 4H) 3.32-3.31 (m,
4H); LC-MS
428 (M - H)~; Purity (HPLC) 95%
EXAMPLE 93
2-[[2-tent-Butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O. Yield: 10.6 mg (6.3 %)
of 2-[[2
tert-butylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride. 1H NMR
(270 MHz, DMSO-d6) b ppm 9.57 (s, 1 H) 8.42 (s, 1 H) 8.26-8.22 (m, 1H) 8.06-
8.04 (m,
1H) 7.68-7.55 (m, 4H) 3.87-3.86 (m, 4H) 3.34-3.33 (m, 4H) 1.51-1.43 (m, 9H);
LC-MS
400 (M - H)+; Purity (HPLC) 90%.
EXAMPLE 94
2-[(3,4-Dichlorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine-2-
sulfonamide
hydrochloride
The title compound was prepared following Method M-O. Yield: 47.9 mg (22.9 %).
1H
NMR (270 MHz, DMSO-d6) 8 ppm 9.36 (s, 1 H) 8.51 (s, 1 H) 8.38 (d, J--2.11 Hz,
1 H
8.06-7.94 (m, 3H) 7.70-7.68 (m, 1H) 3.81-3.77 (m, 4H) 3.31-3.29 (m, 4H); LC-MS
427 (M
- H)+; Purity (HPLC) 95 %.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 9S
2-[(4-tent Butylphenyl)sulfonyl)-4-piperazin-1-ylthieno[3,2-c)pyridine
hydrochloride
Oxone (O.S2 g, 0.84 mmol) in water (4 mL), buffered to pH ~ 6 with sodium
oxide acetate,
was added to 2-[(4-tart-butylphenyl)thio]-4-piperazin-1-ylthieno[3,2-
c]pyridine (0.42
S mmol) in ethanol (30 mL). The mixture was stirred in room temperature for 2
h and more
oxone (O.S2 g, 0.84 mmol) was added. The reaction was stirred over night.
Water was
added to the mixture, extraction with dichloromethane (2X 20 mL) and the
solvent was
removed. The products were purified by preparative HPLC. Yield: 41.9 mg
(22.0%). 1H
NMR (S00 MHz, CH30H-d4) b ppm 8.38 (s, 1 H) 8.OS-8.01 (m, 3H) 7.80 (d, J--6.59
Hz, 1
H) 7.71-7.69 (m, 2H) 4.15-4.13 (m, 4H) 3.59-3.57 (4H) 1.37-1.33 (m, 9H); LC-MS
416 (M
- H)+; Purity (HPLC) 9S%.
EXAMPLE 96
2-(1-Naphthylsulfonyl)-4-piperazin-1-ylthieno[3,2-c)pyridine hydrochloride
1S The title compound was prepared following Method M-O. Yield: 3.4 mg (0.2
%). 1H
NMR (270 MHz, DMSO-d6) ~ ppm 9.34 (s, I H) 8.82 (s, 1 H) 8.44 (s, 1 H) 8.26-
8.06 (m,
SH) 7.79-7.65 (m, 3H) 3.79-3.78 (m, 4H) 3.32-3.30 (m, 4H); LC-MS 410 (M - H)+;
Purity
(HPLC) 9S%.
EXAMPLE 97
2-[(3-Fluorophenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine-2-
sulfonamide
hydrochloride
2-Bromo-4-chlorothieno[3,2-c]pyridine (190 mg, O.SO mmol) in DMF (1 mL) was
added
to 3-fluorobenzenethiol (9S.S mg, 1.0 mmol), KOH (S6 mg, 0.2 mmol) and Cu20
(71 mg,
2S O.S rnmol) in DMF (1 mL). The reaction was heated to 120°C over
night. The mixture was
filtrated through a silica plug and the solvent was removed. The product was
dissolved in
TFA (I .S mL) at 0°C and the solution were stirred for I S min, H20a
(100 p,L) was added
and the mixture was stirred at room temperature over night. 2M NaOH was added,
extraction with etylacetate, washed with brine and solvent was removed. The
product was
purified by preparative HPLC. Yield: 30.1 mg (16.1%) 1H NMR (270 MHz, DMSO-d6)
8
ppm 9.34 (s, I H) 8.45 (s, 1 H) 8.16 (d, J--5.69 Hz, 1 H) 7.97-7.93 (m, 2H)
7.76-7.62 (m,
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
3H) 3.30 (s, 4 H) (4H obscured by solvent signal); LC-MS 378 (M - H)+; Purity
(HI'LC)
99%.
EXAMPLE 98
2-(Mesitylsulfonyl)-4-piperazin-1-ylthieno(3,2-c]pyridine hydrochloride
The title compound was prepared following Method M-O. Yield: 32.0 mg (16.1 %).
1H
NMR (270 MHz, DMSO-d6) 8 ppm 9.32 (s, 1 H) 8.20-8.14 (m, 2H) 7.66 (d, J--5.69
Hz, 1
H) 7.14 (s, 2 H) 3.29 (s, 4 H) 2.65 (s, 6H) 2.28 (s, 3H) (4H obscured by
solvent signal);
LC-MS 402 (M - H)+; Purity (HPLC) 95%.
EXAMPLE 99
2-[(2-Methoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O.Yield: 14.7 mg (7.6 %).
1H
NMR (270 MHz, DMSO-d6) ~ ppm 9.33 (s, 1 H) 8.24 (s, 1 H) 8.15 (d, J--5.94 Hz,
1 H)
8.00 (dd, J--7.92, 1.48 Hz, 1 H) 7.77-7.68 (m, 2H) 7.28-7.18 (m, 2H) 3.30 (s,
4H) (7H
obscured by solvent signal); LC-MS 390 (M - H)+Purity (HPLC) 99%.
EXAMPLE 100
2-[(2,4-Dimethoxyphenyl)sulfonyl]-4-piperazin-1-ylthieno [3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O. Yield: 42.7 mg (20.5 %).
1H
NMR (270 MHz, DMSO-d6) 8 ppm 9.39 (s, 1 H) 8.37 (s, 1 H) 8.13 (d, J--5.69 Hz,
1 H)
7.68-7.66 (m, 2H) 7.54 (d, J 2.23 Hz, 1 H) 7.19 (d, J--8.66 Hz, 1 H) 3.29 (s,
4 H) (lOH
obscured by solvent signal); LC-MS 420 (M - H)+; Purity (HPLC) 98%.
EXAMPLE 101
2-[(2,4-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O. Yield: 17.8 mg (9.3 %).
1H
NMR (270 MHz, DMSO-d6) ~ ppm 9.32 (s, 1H) 8.27 (s, 1 H) 8.15 (d, J--5.94 Hz, 1
H)
8.00 (d, J--8.17 Hz, 1 H) 7.67 (d, J--5.94 Hz, 1 H) 7.35-7.26 (m, 2H) 3.29 (s,
4H) 2.34 (s,
3H) (7H obscured by solvent signal); LC-MS 388 (M - H)+Purity (HPLC) 98%.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 102
2-[(2,5-Dimethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O. Yield: 16.9 mg (8.8 %).
1H
NMR (270 MHz, DMSO-d6) 8 ppm 9.17 (s, 1 H) 8.29 (s, 1 H) 8.I8-18.15 (m, 1H)
7.94 (s,
1 H) 7.66 (d, J--5.69 Hz, 1 H) 7.47 (d, J--7.67 Hz, 1 H) 7.32 (d, J--8.16 Hz,
1 H) 3.29 (s, 2
H) 2.42 (s, 3 H) (7H obscured by solvent signal); LC-MS 388 (M - H)+; Purity
(HPLC)
99%.
EXAMPLE 103
2-[(2-Ethylphenyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The title compound was prepared following Method M-O. Yield: 22.6 mg (11.2 %).
1H
NMR (270 MHz, DMSO-d6) 8 ppm 9.19 (s, 1 H) 8.32 (s, 1 H) 8.17 (d, J--5.69 Hz,
1 H)
8.08 (d, J--7.92 Hz, 1 H) 7.73-7.66 (m, 2H) 7.52 (t, J--7.67 Hz, 2 H) 3.29 (s,
4 H) 3.00 (q,
J--7.34 Hz, 2 H) 1.10 (m, 3 H) (4H obscured by solvent signal); LC-MS 388 (M -
H)+;
Purity (HPLC) 100%.
Scheme 7
c. c. ci CN~ ci-
o , o
~ Br -'~' y ~ ~ S Li+ ~ w ~ \ ~S - ~ N ~ \ p
i; ii S ~' iii S ~ \ / '~~"8 - R
iv; v
\ /
Legend to Scheme 7: i) nBuLi, diethyl ether; ii) SOZ gas; iii)
benzylbromine(s), DMF, heat; iv) diamine(s),
KzC03, DMF, heat; v) HGl, diethyl ether.
INTERMEDIATE 70
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate
2-Bromo-4-chlorothieno[3,2-c]pyridine (5.00 g, 20.1 mmol) was suspended in dry
ether
(100 ml) and the mixture was cooled to -78 °C under Nz-atmosphere . n-
BuLi (1.6M in
hexane, 15 mL) was added and the reaction mixture was stirred at -78 °C
for 2h. S02 (g)
was then bubbled threw the reaction mixture for lh. After the gas bubbling had
stopped the
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
reaction mixture was stirred fore one more hour at -78 °C and was then
allowed to warm
to room temperature. The precipitate that had formed was filtered and washed
with ether to
give the sulfonate lithium salt (3.59 g, 74 %) that was used in the next step
without further
purification. 1H NMR (270 MHz, DMSO-d6) 8 ppm 7.26 (s, 1 H) 7.99 (d, .I--5.54
Hz, I H)
8.14 (d, J 5.54 Hz, 1 H). MS (M-Li+1) 234.
Benzylation of sulfinate salts (Method P)
To a suspension of lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (100 mg,
0.42 mmol)
in dry DMF (2 mL) was added a benzylbromide (0.83 mmol, 2 equiv.) and the
mixture
heated with stirring for 16 h at 110 °C. Analysis by LCMS showed
desired product and no
starting material remaining. The mixture was treated with polystyrene-
thiophenol (200 mg)
and was rolled for 16 h. The suspension was filtered washing with further DMF
(2mL).
This material was reacted further without purification.
Nucleophilic substitution of chlorine (Method Q)
To a crude solutions of benzylsulfone in DMF (4 mL) are added potassium
carbonate (172
mg, 1.25 mmol) and te~~t-butyl-piperazine-1-carboxylate (155 mg, 0.84 mmol).
The
resulting mixtures are heated for 16 h at 110 °C. LCMS shows desired
compound and no
starting material. The reaction mixtures are filtered and then the solvent
removed under
reduced pressure. The desired compounds are isolated pure following
preparative HPLC.
BOC-deprotection (Method R)
The BOC N-protected piperazine derivatives are dissolved in HCl/ diethyl ether
(1 mL,
1.OM) at room temperature and stirred for 16 h. Removal of the solvent under
reduced
pressure gave the crude hydrochloride salts. Trituration with acetonitrile
gives the desired
compound as a white solid.
INTERMEDIATE 71
tent-Butyl-4-[2-(benzylsulfonyl)thieno[3,2-c]pyridin-4-yl]piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0,44 mmol) was treated with
benzylbromide (0.59 mmol) as described in Method P above and then reacted
further with
tent-butyl-piperazine-1-carboxylate as described in Method Q. Yield 0.009 g (7
% over two
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
steps). 1H NMR (300 MHz, CDC13) 8 8.14 (d, J--5.5 Hz 1 H), 7.27-7.40 (m, 5 H),
7.15-
7.21 (m, 2 H), 4.45 (s, 2 H), 3.50-3.56 (m, 4 H), 3.40-3.45 (m, 4 H), 1.49 (s,
9 H); MS
(ESI+) for C23 H27 N3 04 Sa mlz 474 (M+H)+. HPLC 77%, RT 3.93 min (ACE3 C8
50x4mm, 5-50% acetonitrile in 3 min).
INTERMEDIATE 72
tent-Butyl-4-(2-{ [4-(trifluoromethyl)b enzyl] sulfonyl}thieno [3,2-c] pyridin-
4-
yI)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulflnate (0.44 mmol) was treated with
4-
(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above and
then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method QF. Yield
0.02 g(16 % over two steps). Beige solid. IH NMR (300 MHz, CDC13) 8 8.16 (d, J-
-6 Hzl
H), 7.53-7.61 (d, J 9 Hz 2 H), 7.49 (s, 1 H), 7.26-7.36 (m, 4 H), 4.51 (s, 2
H), 3.49-3.60
(m, 4 H), 3.36-3.49 (m, 4 H), 1.49 (s, 9 H); MS (ESI+) for C24 H26 F3 N3 04 SZ
mlz 542
(M+H)+. HPLC 71 %, RT 4.07min (ACE3 C8 50x4mm, 5-50% acetonitrile in 3 min).
INTERMEDIATE 73
tent-Butyl-4-{2-[(3-bromobenzyl)suIfonyl]thieno[3,2-c]pyridin-4-yl}piperazine-
1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3-
bromobenzylbromide (0.59 mmol) as described in Method P above and then reacted
further with tent-butyl-piperazine-1-carboxylate as described in Method Q.
Yield 0.023 g
(10 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) 8 8.16 (d, J 6 Hz,
1H),
7.50-7.55 (m, 2 H), 7.32-7.40 (m, 2 H), 7.10-7.24 (m, 3 H), 4.44 (s, 2 H),
3.61-3.73 (m, 8
H), 1.50 (s, 9 H); MS (ESI+) for C23 H26 Br N3 04 Sa mlz 554 (M+H)+. HPLC 77
%, RT
4.07min (ACE3 C8 50x4.6mm, 5-50% acetonitrile in 3 min).
INTERMEDIATE 74
tert-Butyl-4-(2-f [3-(trifluoromethyl)benzyl]sulfonyl}thieno[3,2-c]pyridin-4-
yl)piperazine-Z-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3-
(trifluoromethyl)benzylbrornide (0.59 mmol) as described in Method P above and
then
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.023 g (10 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) b 8.14 (d,
J-- 6
Hz, 1 H), 7.85-7.91 (m, 1 H), 7.61-7.72 (m, 2 H), 7.50-7.60 (m, 1 H), 7.12-
7.31 (m, 2 H),
4.74 (s, 2 H), 3.52-3.71 (m, 8 H), 1.50 (s, 9 H); MS (ESI+) for C24 H26 F3 N3
04 Sa mlz
542 (M+H)+. HPLC 85 %, RT 2.13min (YMC ODS AQ, 33x3mm, 10-90% acetonitrile in
3 min).
INTERMEDIATE 75
tart-Butyl-4-(2-~ [2,5-bis(trifluoromethyl)benzyl] sulfonyl}thieno[3,2-
c]pyridin-4-
yl)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
2,5-
bis(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.01 g (4 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) S 8.16 (d, J-
- 5.8
Hzl H), 8.00 (s, 1 H), 7.74-7.85 (m, 3 H), 4.76 (s, 2 H), 3.56-3.64 (m, 4 H),
3.47-3.56 (m,
4 H), 1.49 (s, 9 H); MS (ESI+) for C25 H25 F6 N3 04 SZ m/z 610 (M+H)+. HPLC 73
%,
RT 2.36min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
INTERMEDIATE 76
tart-Butyl4-~2-[(4-methylbenzyl)sulfonyl]thieno[3,2-c]pyridin-4-yl}piperazine-
1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
4-
methylbenzylbromide (0.59 mmol) as described in Method P above and then
reacted
further with test-butyl-piperazine-1-caxboxylate as described in Method Q.
Yield 0.005 g
(3 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) b 8.15 (d, J 6 Hz 1
H),
7.40 (s, 1 H), 7.00-7.16 (m, 4 H), 4.42 (s, 2 H), 3.46-3.60 (m, 4 H), 3.37-
3.46 (m, 4 H),
2.34 (s, 3 H), 1.49 (s, 9 H); MS (ESI+) for C24 H29 N3 04 Sa mlz 488 (M+H)+.
HPLC 69
%, RT 2.06min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 77
tent-Butyl 4.-(2-{ [5-chloro-2-(trifluoromethyl)benzyl] sulfonyl{thieno [3,2-
c]pyridin-4-
yI)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfmate (0.44 mmol) was treated with
5-chloro-
2-(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.019 g (7.5 % over two steps). Beige solid. 1H NMR (300 MHz, CDCI3) S 8.12-
8.14 (m, 1
H), 7.80-7.88 (m, 2 H), 7.47-7.66 (m, 2 H), 4.71 (s, 2 H), 3.74-3.83 (m, 4 H),
3.63-3.72 (m,
4 H), 1.49 (s, 9 H); MS (ESI+) for C24 H25 CI F3 N3 04 Sa mlz 576 (M+H)+. HPLC
74
%, RT 2.30min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
INTERMEDIATE 78
tent-Butyl 4-{2-[(3,4-difluorobenzyl)sulfonyl]thieno[3,2-c]pyridin-4-
yl}piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3,4-
bis(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with test-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.014 g (6 % over two steps). Beige solid. 1H NMR (300 MHz, CDCl3) b 8.17-8.21
(m, 1
H), 7.71 (s, 1 H), 7.07-7.39 (m, 4 H), 4.59 (s, 2 H), 3.55-3.68 (m, 8 H), 1.49
(s, 9 H); MS
(ESI+) for C23 H25 F2 N3 04 SZ m/z 510 (M+H)+. HPLC 64 %, RT 2.02min (YMC ODS
AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
INTERMEDIATE 79
tent-Butyl 4-{2-[(3,5-dimethoxybenzyl)sulfonyl]thieno[3,2-c]pyridin-4-
yl~piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3,5-
dimethoxybenzylbromide (0.59 mmol) as described in Method P above and then
reacted
further with tent-butyl-piperazine-1-carboxylate as described in Method Q.
Yield 0.02 g
(10 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) S 8.14-8.18 (m, 1
H), 7.55
(s, I H), 6.40-6.45 (m, 1 H), 6.26-6.34 (m, 2 H), 4.39 (s, 2 H), 3.54-3.72 (m,
14 H), 1.50 (s,
9 H); MS (ESI+) for C25 H31 N3 06 S2 mlz 534 (M+H)+. HPLC 69 %, RT 1.99min
(YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 104
4-(Piperazinyl)-2-(3-methoxybenzyl-sulfonyl)-thienopyridine
A 1:1 mixture of lithium 4-chloro-thienopyridine-2-sulfinate (0.176 g, 0.734
mmol) and 2-
methoxybenzylbromide (0.295 g, 1.47 mmol) in DMF (5 mL) was heated at 100
°C for 2h.
To the mixture was added Boc-piperazine (546 mg, 2.94 rnmol) and the reaction
was
heated at 110 °C for 1.Sh. The solvent was removed and the crude
product was purified by
preparative HPLC to obtain 17.4 mg of 4-(4-t-butyl-oxy-carbonyl-piperazinyl)-2-
(3-
methoxybenzyl-sulfonyl)-thienopyridine. The boc-protected product was
dissolved in 2
rnL of MeOH and 4 mL of HCl/ether was added to obtain 21.9 mg of 4-
(piperazinyl)-2-(3-
methoxybenzyl-sulfonyl)-thienopyridine. 1T~NMR (CD3OD/Da0 1:1) 8 6.42-6.38 (m,
1H),
7.51-7.48 (m, 1 H), 7.3 9-7.3 S (m, 1 H), 6.92-6.85 (m, 1 H), 6.64-6.59 (m, 1
H), 6.48-6.3 9 (m,
2H), 3.64-3.58 (m, 4H), 3.19-3.13 (m, 4H), 2.96 (s, 3H), 2.92 (s, 2H); MS
(ESI) 404 (M +
H)+; Purity (HPLC, column YMC) 94%.
INTERMEDIATE 80
tent-Butyl-4-[2-(benzylsulfonyl)thieno[3,2-c]pyridin-4-yl]piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
benzylbromide (0.59 mmol) as described in Method P above and then reacted
fiuther with
test-butyl-piperazine-1-carboxylate as described in Method Q. Yield 0.00 g (7
% over two
steps). Beige solid. IH NMR (300 MHz, CDCl3) 8 8.14 (d, J--5.5 Hz 1 H), 7.27-
7.40 (m, 5
H), 7.15-7.21 (m, 2 H), 4.45 (s, 2 H), 3.50-3.56 (m, 4 H), 3.40-3.45 (m, 4 H),
I.49 (s, 9 H);
MS (ESI+) for C23 H27 N3 04 SZ mlz 474 (M+H)+. HPLC 77 %, RT 3.93min (ACE3 C8
SOx4mm, 5-50 % acetonitrile in 3 min).
EXAMPLE 105
2-(Benzylsulfonyl)-4-piperazin-1-ylthieno[3,2-c]pyridine hydrochloride
tent-Butyl 4-[2-(benzylsulfonyl)thieno[3,2-c]pyridin-4-yl]piperazine-1-
carboxylate (O.OI g,
0.02 mmol) was treated as described in Method R to give the desired product as
a white
solid. Yield 0.009 g, (100 %). White solid. 1H NMR (300 MHz, DMSO-d6) 8 8.98
(s, 1 H),
8.13-8.20 (d, J--8 Hzl H), 8.00 (s, 1 H), 7.64-7.71 (m, I H), 7.18-7.35 (m, 5
H), 4.93 (s, 2
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H), 3.60-3.70 (m, 4 H), 3.22.3.34 (m, 4 H); MS (ESI+) for C18 H19 N3 02 S2 .
Cl H m/z
374 (M+H)+. HPLC 90%, RT 2.91min (ACE3 C8 50x4.6mm, 5-50% acetonitrile in 3
min).
INTERMEDIATE 81
tent-Butyl-4-(2-{ [4-(trifluoromethyl)b enzyl] sulfonyl] thieno [3,2-c]
pyridin-4-
yl)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
4-
(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above and
then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.02 g (16 % over two steps). Beige solid. 1H NMR (300 MHz, CDCl3) ~ 8.16 (d,
J--6 Hzl
H), 7.53-7.61 (d, ,I--- 9 Hz 2 H), 7.49 (s, 1 H), 7.26-7.36 (m, 4 H), 4.51 (s,
2 H), 3.49-3.60
(m, 4 H), 3.36-3.49 (m, 4 H), 1.49 (s, 9 H); MS (ESI+) for C24 H26 F3 N3 04 S2
m/z 542
(M+H)+. HPLC 71 %, RT 4.07min (ACE3 C8 50x4mm, 5-50 % acetonitrile in 3 min).
i5
EXAMPLE 106
4-Pip erazin-1-yl-2-{ [4-(trifluoromethyl)b enzyl] sulfonyl] thieno [3,2-c]
pyridine
hydrochloride
tent-Butyl 4-(2- { [4-(trifluoromethyl)benzyl] sulfonyl } thieno [3,2-
c]pyridin-4-yl)pip erazine-
1-carboxylate (0.02 g, 0.03 mmol) was treated as described in Method R to give
the desired
product as a white solid. Yield 0.014 g (100 %) White solid. 1H NMR (300 MHz,
DMSO-
d6) b 9.25 (s, 1 H), 8.12-8.22 (m, 2 H), 7.66-7.77 (m, 4 H), 7.41-7.52 (m, 2
H), 5.12 (s, 2
H), 3.22-3.35 (m, 4 H); MS (ESI+) for C19 H18 F3 N3 02 S2 . Cl H mlz 442
(M+H)+.
HPLC 90 %, RT 3.53min (ACE3 C8 50x4.6mm, 5-50 % acetonitrile in 3 min).
INTERMEDIATE 82
tart-Butyl-4-{2-[(3-bromobenzyl)sulfonyl]thieno [3,Z-c]pyridin-4-yl}piperazine-
1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3-
bromobenzylbromide (0.59 mmol) as described in Method P above and then reacted
further with tart-butyl-piperazine-1-carboxylate as described in Method Q.
Yield 0.023 g
(10 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) 8 8.16 (d, J--6
Hz, 1H),
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
7.50-7.55 (m, 2 H), 7.32-7.40 (m, 2 H), 7.10-7.24 (rn, 3 H), 4.44 (s, 2 H),
3.6I-3.73 (m, 8
H), 1.50 (s, 9 H); MS (ESI+) for C23 H26 Br N3 04 S2 mlz 554 (M+H)+. HPLC 77
%, RT
4.07min (ACE3 C8 50x4.6mm, 5-50 % acetonitrile in 3 min).
EXAMPLE 107
2-[(3-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
tent-Butyl 4-~2-[(3-bromobenzyl)sulfonyl]thieno[3,2-c]pyridin-4-yl~piperazine-
1-
carboxylate (0.023 g, 0.04 mmol) was treated as described in Method R to give
the desired
product as a white solid. Yield 0.013 g (67 %) White solid. 1H NMR (300 MHz,
DMSO
d6) 8 9.19 (s, 1 H), 8.18 (d, J 6 Hz, 1 H), 8.05 (s, 1 H), 7.70 (d, J-- 6 Hz,
1 H), 7.53-7.58
(m, 1 H), 7.43-7.45 (m, 1 H), 7.19-7.32 (m, 2 H), 4.98 (s, 2 H), 3.24-3.35 (m,
4 H); MS
(ESI+) for CI8 H18 Br N3 02 S2 . Cl H mlz 452 (M+H)+. HPLC 90 %, RT 3.30min
(ACE3 C8 50x4.6mm, 5-50 % acetonitrile in 3 min).
INTERMEDIATE 83
tent-Butyl 4-{2-[(3,4-difluorobenzyl)sulfonyl]thieno[3,2-c]pyridin-4-
yl)piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3,4-
bis(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.014 g (6 % over two steps). Beige solid. 1H NMR (300 MHz, CDCl3) 8 8.17-8.21
(m, 1
H), 7.71 (s, 1 H), 7.07-7.39 (m, 4 H), 4.59 (s, 2 H), 3.55-3.68 (m, 8 H), 1.49
(s, 9 H); MS
(ESI+) for C23 H25 F2 N3 04 SZ mlz 510 (M+H)+. HPLC 64 %, RT 2.02 min (YMC ODS
AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
EXAMPLE 108
2-[(2,3-Difluorobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The BOC group was removed from tent-butyl 4-~2-[(3,4-
difluorobenzyl)sulfonyl]thieno[3,2-c]pyridin-4-yl}piperazine-1-carboxylate
using Method
R. Yield 0.068 g (100 %). White solid. 1H NMR (300 MHz, DMSO-d6) S 9.34 (s, 1
H),
8.22 (s, 1 H), 8.18 (d, J--5.5 Hz, 1 H), 7.70 (d, J--5.5 Hz, I H), 7.26-7.35
(m, 1 H), 7.15-
7.22 (m, 1 H), 7.05-7.14 (m, 1 H), 5.08 (s, 2 H), 3.34-3.42 (m, 4 H), 3.25-
3.34 (m, 4 H);
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
MS (ESI+) for C18 H17 F2 N3 02 S2 . Cl H m/z 410 (M+H)+. HPLC 90 %, RT 1.07min
(YMC ODS AQ, 33x3mm, 20-50 % acetonitrile in 1.5 min).
EXAMPLE 109
2-[(4-Bromobenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.42 mmol) was treated with
4-
bromobenzylbromide (0.59 mmol) as described in Method P above and then reacted
further with tent-butyl-piperazine-1-carboxylate as described in Method Q. The
BOC
protecting group was removed using Method R. Yield 0.024 g (12 % over three
steps).
White solid. 1H NMR (300 MHz, DMSO-d6) 8 8.99 (s, 1 H), 8.18 (d, J 5.5 Hz, 1
H), 8.02
(s, 1 H), 7.69 (d, J-- 5.5 Hz, 1 H), 7.53 (d, J-- 8.5 Hz, 2 H), 7.17 (d, J--
8.5 Hz, 2H), 4.95 (s,
2 H), 3.62-3.68 (m, 4 H), 3.27-3.32 (m, 4 H); MS (ESI+) for C18 H18 Br N3 02
S2 . Cl H
m/z 454 (M+H)+. HPLC 90 %, RT 1.24min (YMC ODS AQ, 33x3mm, 20-50
acetonitrile in 1.5 min).
INTERMEDIATE 84
tart-Butyl-4-(2-{[2,5-bis(trifluoromethyl)benzyl]sulfonyl}thieno[3,2-c]pyridin-
4-
yl)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
2,5-
bis(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with tent-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.01 g (4 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) S 8.16 (d, J-
- 5.8
Hzl H), 8.00 (s, 1 H), 7.74-7.85 (m, 3 H), 4.76 (s, 2 H), 3.56-3.64 (m, 4 H),
3.47-3.56 (m,
4 H), 1.49 (s, 9 H); MS (ESI+) for C25 H25 F6 N3 04 S2 m/z 610 (M+H)+. HPLC 73
%,
RT 2.36min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
EXAMPLE 110
2-{ [2,5-Bis(trifluoromethyl)benzyl] sulfonyl}-4-piperazin-1-ylthieno [3,2-
c]pyridine
hydrochloride
The BOC group was removed from tart-butyl 4-(2- f [2,5-(trifluoromethyl)-
benzyl]sulfonyl}thieno[3,2-c]pyridin-4-yl)piperazine-1-carboxylate using
Method R. Yield
0.024 g (100 %). White solid. 1H NMR (300 MHz, DMSO-d6) 8 9.30 (s, 1 H), 8.25
(s, 1
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H), 8.19 (d, J--5.5 Hz, 1 H), 8.00-8.07 (m, 2 H), 7.85 (s, 1 H), 7.69 (d, J--
5.5 Hz, 1 H), 5.22
(s, 2 H), 3.24-3.33 (m, 4 H); MS (ESI+) for C20 H17 F6 N3 02 S2 . Cl H m/z 510
(M+H)+. HPLC 90 %, RT 1.08min (YMC ODS AQ, 33x3mm, 30-60 % acetonitrile in 1.5
min).
INTERMEDIATE 85
tent-Butyl 4-{2-[(4-methylbenzyl)sulfonyl]thieno[3,2-c]pyridin-4-yl}piperazine-
1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
4-
methylbenzylbromide (0.59 mmol) as described in Method P above and then
reacted
further with tent-butyl-piperazine-1-carboxylate as described in Method Q.
Yield 0.005 g
(3 % over two steps). Beige solid. 1H NMR (300 MHz, CDCl3) 8 8.15 (d, J-- 6 Hz
1 H),
7.40 (s, 1 H), 7.00-7.16 (m, 4 H), 4.42 (s, 2 H), 3.46-3.60 (m, 4 H), 3.37-
3.46 (m, 4 H),
2.34 (s, 3 H), 1.49 (s, 9 H); MS (ESI+) for C24 H29 N3 04 Sz mlz 488 (M+H)+.
HPLC 69
%, RT 2.06min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
EXAMPLE 111
2-[(4-Methylbenzyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
The BOC group was removed from test-butyl 4-{2-[(4-
methylbenzyl)sulfonyl]thieno[3,2-
c]pyridin-4-yl}piperazine-1-carboxylate using Method R. Yield 0.05 g (75 %).
White
solid. 1H NMR (300 MHz, DMSO-d6) b 9.18 (s, 1 H), 8.17 (d, J--5.5 Hz, 1 H),
8.01 (s, 1
H), 7.67 (d, J 5.5 Hz, 1 H), 7.38 (s, 1 H), 7.19 (s, 1 H), 7.11 (s, 1 H), 7.00
(s, 1 H), 4.86 (s,
2 H); MS (ESI+) for C19 H21 N3 02 S2 . Cl H m/z 388 (M+H)+.HPLC 90 %, RT 1.65
min
(ACE3 C8 SOx3.Omm, 10-97 % acetonitrile in 3 min).
INTERMEDIATE 86
tent-Butyl 4-(2-f [5-chloro-2-(trifluoromethyl)benzyl]sulfonyl]thieno[3,2-
c]pyridin-4-
yl)piperazine-1-carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
5-chloro-
2-(trifluoromethyl)benzylbromide (0.59 mmol) as described in Method P above
and then
reacted further with tart-butyl-piperazine-1-carboxylate as described in
Method Q. Yield
0.019 g (7.5 % over two steps). Beige solid. 1H NMR (300 MHz, CDCl3) ~ 8.12-
8.14 (m, 1
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H), 7.80-7.88 (m, 2 H), 7.47-7.66 (m, 2 H), 4.71 (s, 2 H), 3.74-3.83 (m, 4 H),
3.63-3.72 (m,
4 H), 1.49 (s, 9 H); MS (ESI+) for C24 H25 Cl F3 N3 04 Sa m/z 576 (M+H)+. HPLC
74
%, RT 2.30min (YMC ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
EXAMPLE 112
2-{[5-Chloro-2-(triouoromethyl)benzyl]sulfonyl}-4-piperazin-1-ylthieno[3,2-
c]pyridine hydrochloride
The BOC group was removed from test-butyl 4-(2- f [5-chloro-2-
(trifluoromethyl)benzyl]sulfonyl}thieno[3,2-c]pyridin-4-yl)piperazine-1-
carboxylate using
Method r. Yield 0.012 g (92 %). White solid. 1H NMR (300 MHz, DMSO-d6) ~ 9.05
(s, 1
H), 8.18-8.23 (m, 2 H), 7.78-7.83 (m, 1 H), 7.72-7.76 (m, 1 H), 7.69 (d, J--
5.5 Hz, 1 H),
7.62-7.65 (m, 1 H), 5.07 (s, 2 H), 3.65-3.72 (m, 4 H), 7.25-7.34 (m, 4 H); MS
(ESI+) for
C19 H17 Cl F3 N3 02 S2 . Cl H m/z 476 (M+H)+. HPLC 90 %, RT 1.65 min (ACE3 C8
SOx3.Omm, 10-97 % acetonitrile in 3 min).
INTERMEDIATE 87
tent-Butyl 4- f 2-[(3,5-dimethoxybenzyl)sulfonyl] thieno [3,2-c] pyridin-4-yl}
piperazine-1-
carboxylate
Lithium 4-chlorothieno[3,2-c]pyridine-2-sulfinate (0.44 mmol) was treated with
3,5-
dimethoxybenzylbromide (0.59 mmol) as described in Method P above and then
reacted
further with tart-butyl-piperazine-1-carboxylate as described in Method Q.
Yield 0.02 g
(10 % over two steps). Beige solid. 1H NMR (300 MHz, CDC13) 8 8.14-8.18 (m, 1
H), 7.55
(s, 1 H), 6.40-6.45 (rn, 1 H), 6.26-6.34 (m, 2 H), 4.39 (s, 2 H), 3.54-3.72
(m, 14 H), 1.50 (s,
9 H); MS (ESI+) for C25 H31 N3 06 Sa mlz 534 (M+H)+. HPLC 69 %, RT 1.99min
(YMC
ODS AQ, 33x3mm, 10-90 % acetonitrile in 3 min).
EXAMPLE 113
2-[(3,5-Dimethoxybenzyl)sulfonyl]-4-piperazin-1-ylthieno [3,2-c]pyridine
hydrochloride
The BOC group was removed from tent-butyl 4-}2-[(3,5-
dimethoxybenzyl)sulfonyl]thieno[3,2-c]pyridin-4-yl}piperazine-1-carboxylate
using
Method R. Yield O.Olg (62%). White solid. 1H NMR (300 MHz, DMSO-d6) 8 9.09 (s,
1
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
H), 8.18 (d, J-- 6.7 Hz, 1 H), 8.02 (s, 1 H), 7.69 (d, J-- 6.7 Hz, 1H), 6.45-
6.48 (m, 1 H),
6.35-6.38 (m, 2 H), 4.84 (s, 2H), 3.61-3.67 (m, 4H), 3.58 (s, 6H), 3.24-3.33
(m, 4H).MS
(ESI+) for CZOH2aN3O4S2 m/Z 434 (M+H)+. HPLC 90 %, RT 1.60 min (ACE3 C8
SOx3.Omm, 10-97 % acetonitrile in 3 min).
EXAMPLE 114
2-[(2-Naphthylmethyl)sulfonyl]-4-piperazin-1-ylthieno[3,2-c]pyridine
hydrochloride
2-(Bromomethyl)naphthalene was used according to Method P-R to give 12.4 mg of
the
desired product. 1H NMR (270 MHz, CH30H-d4) 8 ppm (obscured by CH30H, 4H) 3.70-
3.79 (m, 4 H) 4.95 (s, 2 H) 7.36-7.58 (m, 3 H) 7.70-7.91 (m, 6 H) 8.02 (d, J--
6.60 Hz, 1 H).
MS (M+1) 424.
EXAMPLE 115
4-Piperazin-1-yl-2-{ [4-(1,2,3-thiadiazol-4-yl)benzyl]sulfonyl}thieno [3,2-
c]pyridine
hydrochloride
4-[4-(Bromomethyl)phenyl]-1,2,3-thiadiazole was used according to Method P-R
to give
4.8 mg of the desired product. 1H NMR (270 MHz, CH30H-d4) 8 ppm 3.41-3.50 (m,
4 H)
3.54 (s, 2 H) 3.89-3.98 (m, 4 H) 7.45 (d, J--8.44 Hz, 2 H) 7.75 (d, J 6.33 Hz,
1 H) 7.96-
8.13 (m, 4 H) 9.31 (s, 1 H). MS (M+1) 458.
EXAMPLE 116
1-(4-Pyrrolidin-1-ylphenyl)-2-[(4-piperazine-1-ylthieno[3,2-c]pyridin-2-yl)
sulfonyl]
ethanone hydrochloride
2-Bromo-1-(4-pyrrolidin-1-ylphenyl)ethanone was used according to Method P-R
to give
19.6 mg of the desired product. 1H NMR (270 MHz, CH30H-d4) b ppm 2.02-2.12 (m,
4 H)
2.69 (s, 1 H) 3.37 (t, J--6.73 Hz, 4 H) 3.54 (s, 1 H) 3.56.3.64 (m, 4 H) 4.12-
4.22 (m, 4 H)
6.55 (d, J--8.97 Hz, 2 H) 7.78-7.90 (m, 3 H) 8.03 (d, J--6.86 Hz, 1 H) 8.41
(s, 1 H). MS
(M+1) 471.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 117
1-[4-(Diethylamino)phenyl]-2-[(4-piperazine-1-ylthieno[3,2-c]pyridin-2-yl)
sulfonyl]
ethanone hydrochloride
2-Bromo-1-[4-(diethylamino)phenyl]ethanone ethanone was used according to
Method P-
R to give 9.0 mg of the desired product. 1H NMR (500 MHz, CH30H-d4) b ppm 1.16
(t,
J--7.06 Hz, 6 H) 3.49-3.66 (m, 10 H) 4.14-4.27 (m, 4 H) 7.13 (br.s, 2 H) 7.85
(d, J 6.59
Hz, 1 H) 7.98 (d, J 8.48 Hz, 2 H) 8.03 (d, J--6.59 Hz, 1 H) 8.47 (s, 1 H). MS
(M+1) 473.
EXAMPLE 118
1-(4-Bromophenyl)-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-yl) sulfonyl]
ethanone
2-Bromo-1-(4-bromophenyl)ethanone was used according to method A to give 3.4
mg of
the desired product.1H NMR (270 MHz, CH30H-d4) 8 ppm 3.55 (s, 2 H) 3.56-3.67
(m, 4
H) 4.08-4.26 (m, 4 H) 7.68 (d, J--8.44 Hz, 2 H) 7.79-7.98 (m, 3 H) 8.06 (d, J--
6.60 Hz, 1
H) 8.45 (s, 1 H). MS (M+1) 481.
EXAMPLE 119
1-(3-Methoxyphenyl)-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-yl) sulfonyl]
ethanone
2-bromo-1-(3-methoxyphenyl)ethanone was used according to method A to give 1.0
mg of
the desired product. 1H NMR (270 MHz, CH30H-d4) 8 ppm 3.54 (s, 2 H) 3.55-6.62
(m,
J 10.03 Hz, 4 H) 3.82 (s, 3 H) 4.06-4.18 (m, 4 H) 7.20 (dd, J 8.05, 2.24 Hz, 1
H) 7.34-
7.49 (m, 2 H) 7.57 (d, J--7.39 Hz, 1 H) 7.84 (d, J 6.60 Hz, 1 H) 8.06 (d, J--
6.60 Hz, 1 H)
8.41 (s, 1 H). MS (M+1) 432.
EXAMPLE 120
1-Phenyl-2-[(4-piperazin-1-ylthieno[3,2-c]pyridin-2-yl)sulfonyl]ethanone
2-Bromo-1-phenylethanone was used according to method A to give 1.2 mg of the
desired
product.1H NMR (270 MHz, CH30H-d4) 8 ppm 3.55 (s, 2 H) 3.57-3.66 (m, 4 H) 4.10-
4.24
(m, 4 H) 7.46-4.57 (m, 2 H) 7.66 (t, J--7.39 Hz, 1 H) 7.86 (d, J--6.60 Hz, 1
H) 8.02 (dd,
J--14.12, 6.99 Hz, 3 H) 8.46 (s, 1 H). MS (M+1) 402.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Table 6
O.. . R~
S' O
N
N
TRa
EXAMPLE R R
121 4-Piperazin-I-yl-I-(toluene-4-sulfo N
nyl)-1H-pyrrolo[3,2-c]pyridine hydrochloride
N
I22 1-(3-Chloro-2-methyl-benzenesulfony _ - ~ N
1)-4-piperazin-1-yl-1H-pyrrolo[3,2
c]pyridine hydrochloride I / N
C!
123 1-(3,4-Dimethoxy-benzenesulfonyl)-4 - . CN\
-piperazin-1-yl-IH-pyxrolo[3,2-c]py
ridine hydrochloride ~ / ~. N
~O
~O
124 4-(4-Piperazin-I-yl-pyrrolo[3,2-c]p _ _ C N \
yridine-1-sulfonyl)-benzonitrile hydrochloride
N
CN
125 I-(4,5-Dichloro-thiophene-2-sulfonyl)-4-piperazin-1-yl-IH- CI S N
pyrrolo[3,2-c]pyridine hydrochloride
CI N
126 1-(2-Chloro-4-fluoro-benzenesulfony _ _ ~N~
1)-4-piperazin-I-yl-1H-pyrrolo[3,2- ~ CI
c]pyridine hydrochloride I / N
F
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
127 1-Phenylmethanesulfonyl-4-piperazin-1-yl-1H-pyrrolo [3,2- N
c]pyridine hydrochloride
N
128 1-(5-Chloro-thiophene-2-sulfonyl)-4 CI S CN\
-piperazin-1-yl-1H-pyrrolo[3,2-c]py
ridine hydrochloride N
129 1-(4-Butyl-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2- _ . CN\
c)pyridine hydrochloride
N
130 1-(4-Phenoxy-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2- _ _ CN\
c)pyridine hydrochloride
N
Ph~O
131 1-(Phenylsulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine _ . CN\
hydrochloride
N
132 1-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2- _ _ CN\
c]pyridine hydrochloride
N
CI
133 1-[(4-Methoxyphenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2- _ _ ~N~
c]pyridine hydrochloride
N
~O
134 1-[(2-Methoxy-5-methylphenyl)sulfonyl]-4-piperazin-1-yl-1H- _ . ~N~
pyrrolo[3,2-c]pyridine hydrochloride I ~ O
N
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
135 4-Piperazin-1-yl-1-{[2-(trifluoromethyl)phenyl]sulfonyl}-1H- -- _ F F ~N~
pyrrolo[3,2-c]pyridine hydrochloride ~ F
N
Scheme 8
i
i
N
N O ~ N O ~~ \ ~ \ iii \N ~ \
H ~ \ / -N
N3 O H
CI
R1
iv v N vi N
N ~ \ / \ ~ \
\ / \ IN vii -N
N R4 R4
R4
Legend to Scheme 8: i) Ethylchloroformate, TEA, acetone, NaN3; ii) Bu3N, DCM
and diphenylether; iii)
POC13, NaOH; iv) BOC protected amines (R4), KZCO3, DMSO; v) NH3 gas Na, NH4C1,
THF vi) Sulphonyl
chlorides (Rl), NaH, THF; vii) HCl/diethyl ether, methanol.
INTERMEDIATE 88
(2E)-3-(1-Benzyl-1H-pyrrol-2-yl)acryloyl azide
To a mixture of 1-benzyl-1H-pyrrole-2-carbaldehyde (28.4g, 0.125mo1) and TEA
(13.5
mL, 0.187 mol) in acetone (300 mL) was added ethylchloroformate (17.9 mL, 0.87
mol)
dropwise. The reaction was stirred for 1.5 h after which NaN3 (13 g, 0.200
mol) in H20
(100 mL) was added. After 2h, the reaction was diluted with water and left
overnight. The
acetone was removed and the product was filtered off to afford 21.4 g of a
light brown
solid. This compound was taken to the next step.
INTERMEDIATE 89
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
1-Benzyl-1,5-dihydro-pyrrolo[3,2-c]pyridin-4-one was prepared by the
literature
procedure according to C. Ducrocq; E. Bisangi; J-M, Lhoste; J. Mispelter;
Tetrahedron, Vol 32, pp 773-780, (1976).
To a stirred solution of n-tributylamine (30 mL) in diphenyl ether (150 mL)
heated to 195
°C was slowly added during 30 minutes a solution of the acyl azide
dissolved in DCM
(150 mL). The reaction mixture was stirred at 195 °C for 1 hour and
then cooled to room
temperature. Pentane (1.0 L) and ether (1.0 L) was added to the reaction
mixture and the
precipitate was collected by filtration. The crude solid was triturated with
ether to give 6.89
g (81 %) of the pure product. Purity HPLC >95%; MS (ESI) m/z 225 (m+H); 1H NMR
(DMSO-d6, 25 °C, 270.16) 8 10.84 (br s, 1 H), 7.43-7.14 (m, 6 H), 7.00
(d, J = 7.12 Hz, 1
H), 6.57-6.49 (m, 2 H), 5.83 (s, 2 H).
INTERMEDIATE 90
1-Benzyl-4-chloro-1H-indole
POC13 (3.11 mL, 33.4 mmol) was added to 1-benzyl-1,5-dihydro-4H-pyrrolo[3,2-
c]pyridin-4-one (3.75 g, 16.7 mmol) and the reaction was stirred at
120°C for 2h. NaOH
(1M) was added and the mixture was extracted with DCM three times. The organic
layers
were dried (MgS04), filtered and the solvent was removed. Flash chromatography
(DCM/Heptane/MeOH 4:15:1) gave 1.17 g (29 %) of product. The product was taken
to
the next step.
INTERMEDIATE 91
tert-Butyl 4-(1-benzyl-1H-pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate
A mixture of 1-benzyl-4-chloro-1H-indole (1.17 g, 4.82 mmol), KaC03 (2.0 g,
mmol) and
Boc-piperazine (1.79 g, 9.64 mmol) in DMSO (75 mL) was stirred at 120
°C for 48 h.
Additional of Boc-piperazine (4 equiv.) was added and the reaction was run for
another 48
h. The reaction was diluted with ethyl acetate (200 mL) and the mixture was
washed with
several portions of water. Flash chromatography (DCM/MeOH/Heptane 4:1:15) gave
0.51
g of starting material and 0.38 g of product. 1HNMR (CD30D) b 7.87-7.85 (m,
1H), 7.25-
7.24 (m, 3H), 7.04-6.98 (m, 3H), 6.73-6.71 (m, 1H), 6.53-6.52 (m, 1H), 5.19
(s, 2H), 3.63-
3.59 (m, 8H), 1.47 (s, 9H); MS (ESI) 393 (M + H)+; Purity (HPLC, column ACE)
95%
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 92
tent-Butyl 4-( 1H-pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate
tent-Butyl 4-(1-benzyl-1H-pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate
(383 mg,
0.488 mmol) was dissolved in THF (6 mL) and liquid ammonia (10 mL) in a 30 mL
vial.
Na (67 mg, 2.93 mmol) was added in portions and the reaction turned violet.
After 30 rnin
NH4C1 (sat) was added and the reaction was let to room temperature The THF was
removed and the residue was extracted with DCM. Recrystallization
(DCM/Heptane) gave
112 mg of a white solid. 1HNMR (CD30D) 8 8.66 (s, 1H), 7.89 (d, 1H, J = 5.80
Hz), 7.13-
7.11 (m, 1H), 6.89-6.86 (m, 1H), 6.57-6.56 (m, 1H), 3.67-3.60 (m, 8H), 1.48
(s, 9H); MS
(ESI) 303 (M + H)+; Purity (HPLC, column ACE) 95%.
Method S for sulphonylation: tent-butyl 4-(1H pyrrolo[3,2-c]pyridin-4-
yl)piperazine-1-
carboxylate (total 1.391 mmol, 1 equiv.) dissolved in THF (14 mL) and dispense
to 10 mL
vials with screwcap. A suspension of NaH ( 0.1488 mmol, 1.5 equiv.) in THF (15
mL)
was dispense evenly to the vials containing the solution of tent-butyl 4-(1H
pyrrolo[3,2-
c]pyridin-4-yl)piperazine-1-carboxylate and stired for approximately for 15
min. Different
sulfonylchlorides were dissolved in THF (2 mL) each and added drop-wise to the
reaction
mixtures. The reactions were quenched with MeOH (100 p,L) and PS-Trisamine (3
equiv.)
was added to each vial and shake for 2 hours. The mixtures were filtered and
the filtrates
were concentrate under vacuum. The products that were not pure enough ( Purity
< 90%)
were purified by preparative chromatography using acetonitrile-water gradients
containing
0.1% triflouroacetic acid. After HPLC analysis fractions that were >_ 90% pure
were
collected and concentrated.
Method T BOC deprotection; The Boc-protected compound was dissolved in MeOH (2
mL) and HCL/ether (2 mL) was added. After 45 min the solvent was removed.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 93
tart Butyl-4-[1-(phenylsulfonyl)-1H-pyrrolo[3,2-c]pyridin-4-yl]piperazine-1-
carboxylate
Purification by recrystallization gave 16 mg (S6 %) after Boc-deprotection.
1HNMR (CDC13) 8 8.03-8.01 (m, 1H), 7.89-7.86 (m, 2H), 7.57-7.39 (m, SH), 6.67-
6.64 (m,
1H), 3.55-3.52 (m, 8H), 1.47 (s, 9H); MS (ESI) 443 (M + H)+; Purity (HPLC,
column
ACE) 9S%.
INTERMEDIATE 94
tent-Butyl-4- f 1-[(4-chlorophenyl)sulfonyl]-1H-pyrrolo[3,2-c]pyridin-4-
yl}piperazine-
1-carboxylate
Purification by preparative HPLC gave 4 mg (11 %) after Boc-deprotection.
~HNMR (CDC13) 8 8.03-7.51 (m, 7H), 6.89-6.87 (m, 1H), 3.91-3.66 (m, 8H), 1.47
(s, 9H);
MS (ESI) 377 (M + H)+; Purity (HPLC, column ACE) 95%.
1S
INTERMEDIATE 9S
tent-Butyl-4-{ 1-[(4-methoxyphenyl)sulfonyl]-1H-pyrrolo [3,2-c] pyridin-4-
yl]piperazine-1-carboxylate
Purification by recrystallization (MeOH/Ether) gave 21 mg (67 %) after boc-
deprotection.
1HNMR (CDC13) ~ 8.02-8.00 (m, IH), 7.84-7.80 (m, 2H), 7.48-7.46 (m, 1H), 7.41-
7.38 (m,
1H), 6.92-6.86 (m, 2H), 6.64-6.62 (m, 1H), 3.79 (s, 3H), 3.57-3.52 (m, 8H),
1.48 (s, 9H);
MS (ESA 473 (M + H)~; Purity (HPLC, column ACE) 9S%.
INTERMEDIATE 96
tart Butyl 4-(1-f [2-(trifluoromethyl)phenyl]sulfonyl)-1H-pyrrolo[3,2-
c]pyridin-4-
yl)piperazine-1-carboxylate
Purification by preparative HPLC gave 8.6 mg (25 %) after boc-deprotection.
1HNMR
(CDCl3) 8 8.14-8.11 (m, 1H), 8.01-7.94 (m, 2H), 7.89-7.72 (m, 3H), 7.37-7.34
(m, 1H),
6.89-6.88 (m, 1H), 3.93-3.89 (m, 4H), 3.71-3.67 (m, 4H), 1.47 (s, 9H); MS
(ESI) SI1 (M +
H)~; Purity (HPLC, column ACE) 95%.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
INTERMEDIATE 97
tart-Butyl 4-f 1-[(2-methoxy-5-methylphenyl)sulfonyl]-1H-pyrrolo[3,2-c]pyridin-
4-
yl}piperazine-1-carboxylate
Purification by preparative HPLC gave 10.3 mg (32 %) after boc-
deprotection.1HNMR
(CDC13) 8 7.95-7.92 (m, 2H), 7.74-7.72 (m, 1H), 7.44-7.40 (m, 2H), 6.85-6.77
(m, 2H),
3.92-3.88 (m, 4H), 3.70 (s, 3H), 3.69-3.66 (m, 4H), 2.39 (s, 3H), 1.47 (s,
9H); MS (ESl~
487 (M + H)+; Purity (HPLC, column ACE) 95%.
EXAMPLE 121
4-Piperazin-1-yl-1-(toluene-4-sulfonyl)-1H-pyrrolo[3,2-c]pyridine
hydrochloride
p-Toulenesulfonyl chloride (24.6 mg) was added to ter~t-butyl 4-(IH
pyrrolo[3,2-c]pyridin-
4-yl)piperazine-1-carboxylate the title compound (4.3 mg). LC/MS RT: 1.374
(System 10
till 40% MeCN over 1.5 min, ACE C8), Purity. 91%. MS: 357 (M+1) 1HNIVIR
(CD30D) 8
ppm 2.39 (s, 3 H) 3.48 (m, 4 H) 4.06 (m, 4 H) 7.22 (d, J--3.71 Hz, 1 H) 7.43
(d, J 8.16 Hz,
2 H) 7.95 (m, 5 H).
EXAMPLE 122
1-(3-Chloro-2-methyl-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-
c]pyridine
hydrochloride
3-Chloro-2-methylbenzenesulfonyl chloride (29.0 mg) was added to tent-butyl 4-
(1H
pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate the title compound (6.3
mg). LC/MS
RT: 1.563 (System 10 till 40% MeCN over 1.5 min, ACE C8), Purity. 96%. MS: 392
(M+1).
EXAMPLE 123
1-(3,4-Dimethoxy-b enzenesulfonyl)-4-piperazin-1-yl-1 H-pyrrolo [3,2-c]
pyridine
hydrochloride
3,4-Dimethoxybenzenulfonyl chloride (30.5 mg) was added to tart-butyl 4-(1H
pyrrolo[3,2-c]pyridin-4-y1)piperazine-1-carboxylate the title compound (8.5
mg). LC/MS
RT: 1.284 (System 10 till 40% MeCN over I.5 min, ACE C8), Purity. 92%. MS: 404
(M+1) 1HNMR (CD30D) 8 ppm 3.50 (m, J 4.21 Hz, 2 H) 3.85 (d, J 3.22 Hz, 4 H)
4.10
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
(m, J--3.96 Hz, 2 H) 7.11 (d, J--8.66 Hz, 1 H) 7.23 (d, J--3.46 Hz, 1 H) 7.48
(d, J--1.73 Hz,
1 H) 7.74 (dd, J--8.54, 1.86 Hz, 1 H) 7.92 (s, 2 H) 8.07 (d, J--3.46 Hz, 1 H).
EXAMPLE 124
4-(4-Piperazin-1-yl-pyrrolo[3,2-c]pyridine-1-sulfonyl)-benzonitrile
hydrochloride
4-Cyanobenzenesulfonyl chloride (26.Omg) was added to tent-butyl 4-(1H
pyrrolo[3,2-
c]pyridin-4-yl)piperazine-1-carboxylate the title compound (9.1 mg). LC/MS RT:
1.150
(System 10 till 40% MeCN over 1.5 min, ACE C8), Purity. 93%. MS: 369 (M+1)
1HNMR
(CD30D) 8 ppm 3.50 (m, 4 H) 4.08 (m, 4 H) 7.29 (d, J--3.71 Hz, 2 H) 7.98 (m, 4
H) 8.29
(d, J 8.66 Hz, 2 H).
EXAMPLE 125
1-(4,5-Dichloro-thiophene-2-sulfonyl)-4-piperazin-1-yl-1 H-pyrrolo [3,2-c]
pyridine
hydrochloride
4,5-Dichloro-thiophene-2-sulfonyl chloride (32.4 mg) was added to tart-butyl 4-
(1H
pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate the title compound (0.3
mg). LC/MS
RT: 1.119 (System 10 till 40% MeCN over 1.5 min, ACE C8), Purity. 92%. MS: 418
(M+1).
EXAMPLE 126
1-(2-Chloro-4-fluoro-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-
c]pyridine
hydrochloride
2-Chloro-4-flourobenzenesulfonyl chloride (29.5 mg) was added to test-butyl 4-
(1H
pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate the title compound (2.4
mg). LC/MS
RT: 1.361 (System 10 till 40% MeCN over 1.5 min, ACE C8), Purity. 90%. MS: 396
(M+1) 1HNMR (CD30D) b ppm 3.51 (m, 4 H) 4.08 (m, 4 H) 7.23 (dd, J--3.96, 0.49
Hz, 1
H) 7.47 (m, 1 H) 7.55 (dd, J--8.41, 2.47 Hz, 2 H) 7.62 (d, J 6.93 Hz, 1 H)
7.91 (d, J 7.18
Hz, 1 H) 8.06 (d, J--3.96 Hz, 1 H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 127
1-Phenylmethanesulfonyl-4-piperazin-1-yl-1H-pyrrolo [3,2-c]pyridine
hydrochloride
Phenyl-methanesulfonyl chloride (24.6 mg) was added to tart-butyl 4-(IH
pyrrolo[3,2-
c]pyridin-4-yl)piperazine-1-carboxylate the title compound (0.2 mg). LC/MS RT:
1.007
S (System 10 till 40% MeCN over 1.S min, ACE C8), Purity. 90%. MS: 3S7 (M+1).
EXAMPLE 128
1-(5-Chloro-thiophene-2-sulfonyl)-4-piperazin-1-yl-1 H-pyrrolo [3,2-c]
pyridine
hydrochloride
S-Chlorothiophene-2-sulfonyl chloride (28.0 mg) was added to tent-butyl 4-(1H
pyrrolo[3,2-c]pyridin-4-yl)piperazine-1-carboxylate the title compound (7.2
mg). LC/MS
RT: 1.381 (System 10 till 40% MeCN over 1.S min, ACE C8), Purity. 97%. MS: 483
(M+1).
1 S EXAMPLE 129
1-(4-Butyl-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2-c)pyridine
hydrochloride
4-N-Butylbenzenesulfonylchloride (30.0 mg) was added to text-butyl 4-(1H
pyrrolo[3,2-
c]pyridin-4-yl)piperazine-1-carboxylate the title compound (11.9 mg). LC/MS
RT: 1.904
(System 10 till 40% MeCN over 1.S min, ACE C8), Purity. 9S%. MS: 400 (M+1)
1HNMR
(CD30D) 8 ppm 0.90 (t, ,I--7.18 Hz, 3 H) 1.31 (m, 2 H) 1.SS (m, 2 H) 2.67 (m,
2 H) 3.50
(m, 4 H) 4.09 (m, J--3.96 Hz, 4 H) 7.25 (d, J 3.71 Hz, 2 H) 7.44 (d, J--8.16
Hz, 2 H) 7.91
(m, 2 H) 8.02 (m, 2 H).
EXAMPLE 130
1-(4-Phenoxy-benzenesulfonyl)-4-piperazin-1-yl-1H-pyrrolo(3,2-c)pyridine
hydrochloride
(4-Phenoxy)benzene)sulfonyl chloride (34.7 mg) was added to tent-butyl 4-(1H
pyrrolo[3,2-c]pyridin-4-yI)piperazine-1-carboxylate the title compound (12.8
mg). LC/MS
RT: 1.839 (System 10 till 40% MeCN over 1.S min, ACE C8), Purity. 9S%. MS: 436
(M+1) ~HNMR (CD30D) 8 ppm 3.50 (m, J--3.96 Hz, 4 H) 4.09 (m, J--4.45 Hz, 4 H)
7.OS
(dd, J--8.16, 6.43 Hz, 2 H) 7.26 (m, 2 H) 7.44 (t, J--7.79 Hz, 2 H) 7.90 (m, 3
H) 8.01 (d,
J--3.71 Hz, 2 H) 8.07 (d, J 8.91 Hz, 2 H).
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
EXAMPLE 131
1-(Phenylsulfonyl)-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine hydrochloride
Purification by recrystallization gave 16 mg (56 %) after Boc-deprotection. MS
(ESA
343.1 (M + H)+; Purity (HPLC, column ACE) 94%.
EXAMPLE 132
1-[(4-Chlorophenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine
hydrochloride
Purification by preparative HPLC gave 4 mg (11 %) after Boc-deprotection. MS
(ESI) 377
(M + H)+; Purity (HPLC, column ACE) 96%.
EXAMPLE 133
1-[(4-Methoxyphenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-c]pyridine
. hydrochloride
Purification by recrystallization (MeOH/Ether) gave 21 mg (67 %) after Boc-
deprotection.
MS (ESI) 373 (M + H)+; Purity (HPLC, column ACE) 92%
EXAMPLE 134
1-[(2-Methoxy-5-methylphenyl)sulfonyl]-4-piperazin-1-yl-1H-pyrrolo[3,2-
c]pyridine
hydrochloride
Purification by preparative HPLC gave 10.3 mg (32 %) after Boc-deprotection.
MS (ESI)
387 (M + H)+; Purity (HPLC, column ACE) 95%.
EXAMPLE 135
4-Piperazin-I-yl-1-([2-(trifluoromethyl)phenyl]sulfonyl}-1H-pyrrolo[3,2-
c]pyridine
hydrochloride
Purification by preparative HPLC gave 8.6 mg (25 %) after Boc-deprotection. MS
(ESI)
411 (M + H)+; Purity (HPLC, column ACE) 94%.
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
BIOLOGICAL TESTS
The ability of a compound according to the invention to bind a 5-HT6 receptor,
and to be
pharmaceutically useful, can be determined using in vivo and i~ vitro assays
known in the
art.
(a) 5-HT6 binding Assay
Binding affinity experiment for the 5-HT6 receptor are performed in HEK293
cells
transfected with 5-HT6 receptor using (3H)-LSD as labeled ligand according to
the
general method as described by Boess F.G et al. Neuropharmacology vol. 36(4/5)
713-720,
1997.
Materials
Cell culture
The HEK-293 cell line transfected with the 5-HT6 receptor was cultured in
Dulbeccos
Modified Eagles Medium containing 5 % dialyzed foetal bovine serum, (Gibco BRL
10106-169), 0.5 mM sodium pyruvate and 400 pg/ml Geneticin (G-418) (Gibco
BRL10131-019). The cells were passaged 1:10, twice a week.
Chemicals
The radioligand [3H] LSD 60-240 Ci/mmol, obtained from Amersham Pharmacia
Biotech,
(Buckinghamshire, England) was in ethanol and stored at -20°C. The
unlabelled ligands,
representing different selectivity profiles, are presented in Table 1. The
compounds were
dissolved in 100% DMSO and diluted with binding buffer.
Disposable
Compounds were diluted in Costar 96 well V-bottom polypropylene plates
(Corning Inc.
Costar, NY, USA). Samples were incubated in Packard Optiplate (Packard
Instruments
B.V., Groningen, The Netherlands). The total amount of added radioligand was
measured
in Packard 24-well Barex plates (Packaxd Instruments B.V., Groningen, The
Netherlands)
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
in the presence of Microscint~ 20 scintillation fluid (Packard Bioscience,
Meriden, CT,
USA).
Buffer
The binding buffer consisted of 20 mM HEPES, 150 mM NaCI, 10 mM MgCl2, and 1
mM,
EDTA, pH 7.4.
Methods
Membrane pre amation
Cells were grown to approximately 90% confluence on 24.5 x 24.5 NCJNC culture
dishes.
The medium was aspirated, and after rinsing with ice-cold PBS, the cells were
scraped off
using 25 ml Tris buffer (50 mM Tris-HCI, 1 mM EDTA, 1 mM EGTA, pH 7.4) and a
window scraper. The cells were then broken with a Polytron homogeniser, and
remaining
particulate matter was removed by low-speed centrifugation, 1000x g for 5 min.
Finally,
the membranes were collected by high-speed centrifugation (20 OOOx g),
suspended in
binding buffer, and frozen in aliquots at -70°C.
Radioligand binding
Frozen cell membranes were thawed, immediately rehomogenized with a Polytron
homogenizer, and coupled to SPA wheat germ agglutinin beads (Amersham Life
Sciences,
Cardiff, England) for 30 min under continuous shaking of the tubes. After
coupling, the
beads were centrifuged for 10 minutes at 1000 g, and subsequently suspended in
20 ml of
binding buffer per 96-well plate The binding reaction was then initiated by
adding
radioligand and test compounds to the bead-membrane suspension. Following
incubation
at room temperature, the assay plates were subjected to scintillation
counting.
The original SPA method was followed except for that membranes were prepared
from
HEK293 cells expressing the human 5-HT6 receptor instead of from HeLa cells
(Dinh
DM, Zaworski PG, Gill GS, Schlachter SK, Lawson CF, Smith MW. Validation of
human
5-HT6 receptors expressed in HeLa cell membranes: saturation binding studies,
pharmacological profiles of standard CNS agents and SPA development. The
Upjohn
Company Technical Report 7295-95-064 1995;27 December). The specific binding
of
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
[3H]LSD was saturable, while the non-specific binding increased linearly with
the
concentration of added radioligand. [3H] LSD bound with high affinity to 5-HT6
receptors. The Kd value was estimated to 2.6~ 0.2 nM based on four separate
experiments.
The total binding at 3 nM of [3H] LSD, the radioligand concentration used in
the
competition experiments, was typically 6000 dpm, and the specific binding more
than
70%. S-HT caused a concentration dependent inhibition of [3H] LSD binding with
an over
all average Ki value of 236 nM when tested against two different membrane
preparations.
The inter assay variability over three experiments showed a CV of 10% with an
average K;
values of 173 nM (SD 30) and a Hill coefficient of 0.94 (SD 0.09). The infra
assay
variation was 3% (n=4). Ki values for a limited set of reference compounds
with reported
binding affinities at 5-HT6 receptor are presented in Table 7. All unlabelled
ligands
displaced the specific binding of [3H] LSD in a concentration-dependent
manner, albeit at
different potencies. The rank order of potency for the compounds was
methiothepin (2 nM)
>mianserin (190 nM) .=5-HT (236 nM) >methysergide (482 nM) >mesulergide (1970
nM).
Protein determination
Protein concentrations were determined with BioRad Protein Assay (Bradford MM.
A
rapid and sensitive method for the quantitation of microgram quantities of
protein utilizing
the principle of protein-dye binding. Anal Biochem 1976;72:248-54). Bovine
serum
albumin was used as standard.
Scintillation counting
The radioactivity was determined in a Packard TopCountTM scintillation counter
(Packard
Instruments, Meriden, CT, USA) at a counting efficiency of approximately 20 %.
The
counting efficiency was determined in separate sets of experiments.
Saturation experiments
At least 6 concentrations in duplicates of radioligand (0.1-20 nM of [3H] LSD)
were used
in saturation experiments. The specific binding was calculated as the
difference between
total binding and non-specific binding, which was determined as the binding of
radioligand
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
in the presence of 5 p,M lisuride. BmaX and the dissociation constant, Ka,
were determined
from the non-linear regression analysis using equation 1. L" is the unbound
concentration
of radioligand, and is y is the amount bound.
_ B ~X. Lu
y Lu + Kd (equation 1)
Competition experiments
Total- and non-specific binding of radioligand was defined in eight replicates
of each.
Samples containing test compound were run in duplicate at I I concentrations.
Incubations
were carned out at room temperature for 3 hours. The ICSO value, i.e. the
concentration of
test compound that inhibited 50% of the specific binding of radioligand, was
determined
with non linear regression analysis and the K; value was calculated using the
method of
[Cheng Y.C. Biochem. Pharmacol. 22, 3099-3108, 19735] equation 2.
ICso
Ki = L (equation 2)
1 +-
Ka
L = concentration of radioligand
Ka = Affinity of radioligand
(b) 5-HT6Intriusic Activity Assay
Antagonists to the 5-HT6 receptor were characterized by measuring inhibition
of 5-HT
induced increase in cAMP in HEK 293 cells expressing the human 5-HT6 receptor
(see
Boess et al. (1997) Neuropharmacology 36: 713-720). Briefly, HEK293/5-HT6
cells were
seeded in polylysine coated 96-well plates at a density of 25,000 / well and
grown in
DMEM (Dulbecco's Modif ed Eagle Medium) (without phenol-red) containing 5%
dialyzed Foetal Bovine Serum for 48 h at 37°C in a 5% COa incubator.
The medium was
then aspirated and replaced by 0.1 ml assay medium (Hanks Balance Salt
Solution
containing 20 mM HEPES, 1.5 mM isobutylmethylxanthine and 1 mg/ml bovine serum
albumin). After addition of test substances, 50 pl dissolved in assay medium,
the cells were
incubated for 10 min at 37°C in a 5% COZ incubator. The medium was
again aspirated and
the cAMP content was determined using a radioactive cAMP kit (Amersham
Pharmacia
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Biotech, BIOTRAK RPA559). The potency of antagonists was quantified by
determining
the concentration that caused 50% inhibition of 5-HT (at [5-HT]= 8 times ECso)
evoked
increase in cAMP, using the formula ICso,~°~ ICso/(1+[SHT]/ECso).
The compounds in accordance with the invention have a selective affinity to 5-
HT6
receptors with Ki and ICso,~°,.,. values between 0.5 nM and 5 ~,M or
display a % inhibition
of [3H] LSD >_ 20 % at 50 nM and are antagonists, agonist or partial agonist
at 5-HT6 . The
compounds show good selectivity over 5-HTIa, 5-HT2a, 5-HT2a, 5-HTab, 5-HT2~.
(c) In vivo assay of reduction of food intake
For a review on serotonin and food intake, see Blundell, J.E. and Halford,
J.C.G. (1998)
Serotonin and Appetite Regulation. Implications for the Pharmacological
Treatment of
Obesity. CNS Drugs 9:473-495.
Obese (ob/ob) mouse is selected as the primary animal model for screening as
this mutant
mouse consumes high amounts of food resulting in a high signal to noise ratio.
To further
substantiate and compare efficacy data, the effect of the compounds on food
consumption
is also studied in wild type (C57BL/6J) mice. The amount of food consumed
during 15
hours of infusion of compounds is recorded.
Male mice (obese C57BL/6JBom-Lep b and lean wild-type C57B1/6JBom;
Bomholtsgaard, Denmark) 8-9 weeks with an average body weight of 50 g (obese)
and 25
g (lean) are used in all the studies. The animals are housed singly in cages
at 23~1 °C, 40-
60 % humidity and have free access to water and standard laboratory chow. The
12/12-h
light/dark cycle is set to lights off at 5 p.m. The animals are conditioned
for at least one
week before start of study.
The test compounds are dissolved in solvents suitable for each specific
compound such as
cyclodextrin, cyclodextrin/methane sulfonic acid, polyethylene glycol/methane
sulfonic
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
acid, saline. Fresh solutions are made for each study. Doses of 30, 50 and 100
mg kg Iday 1
are used. The purity of the test compounds is of analytical grade.
The animals are weighed at the start of the study and randomized based on body
weight.
Alzet osmotic minipumps (Model 2001D; infusion rate 8 ~1/h) are used and
loaded
essentially as recommended by the Alzet technical information manual (Alza
Scientific
Products, 1997; Theeuwes, F. and Yam, S.I. Ann. Biomed. Eng. 4(4). 343-353,
1976).
Continuous subcutaneous infusion with 24 hours duration is used. The minipumps
are
either filled with different concentrations of test compounds dissolved in
vehicle or with
only vehicle solution and maintained in vehicle pre-warmed to 37°C
(approx. 1h). The
minipumps are implanted subcutaneously in the necklback region under short
acting
anesthesia (metofane/enflurane). This surgical procedure lasts approximately 5
min. It
takes about 3 h to reach steady state delivery of the compound.
The weight of the food pellets are measured at 5 p.m. and at 8 p. m. for two
days before
(baseline) and one day after the implantation of the osmotic minipumps. The
weigh-in is
performed with a computer assisted Mettler Toledo PR 5002 balance. Occasional
spillage
is corrected for. At the end of the study the animals are killed by neck
dislocation and trunk
blood sampled for later analysis of plasma drug concentrations.
The plasma sample proteins are precipitated with methanol, centrifuged and the
supernatant is transferred to HPLC vials and injected into the liquid
chromatography /mass
spectrometric system. The mass spectrometer is set for electrospray positive
ion mode and
Multiple Reaction Monitoring. A linear regression analysis of the standards
forced through
the origin is used to calculate the concentrations of the unknown samples.
Food consumption for 15 hours is measured for the three consecutive days and
the
percentage of basal level values is derived for each animal from the day
before and after
treatment. The values are expressed as mean ~ SD and ~ SEM from eight animals
per dose
group. Statistical evaluation is performed by Kruskal-Wallis one-way ANOVA
using the
percent basal values. If statistical significance is reached at the level of
p<0.05, Mann-
CA 02486989 2004-11-22
WO 2004/000828 PCT/SE2003/001061
Whitney U-test for statistical comparison between control and treatment groups
is
performed.
The compounds according to the invention show an effect in the range of 5-200
mg/kg.
Table 7. Biological data
In vitro binding at the human 5-HTg receptor
EXAMPLE K; (nM)
human 5-HT6
1 10
11 6.5
20 lo.s
40 7.5
43 4.5
68 13
85 32
131 5
In vivo efficacy data
EXAMPLE % FI reduction* Css, a (uM)
1 12 0.44
11 47.1 0.02
40 44 0.2
*Effect on Food Intake reduction In 0b/ob mice Single administration 50
mg/kg/d measured at steady state