Note: Descriptions are shown in the official language in which they were submitted.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
1-Phenylallcyl-piper azines
DESCRIPTION
The invention relates to 1-phenylallcyl-piperazines having affinity for
serotoninergic
receptors, to pharmaceutical compositions containing them and to uses for such
compounds and compositions.
In mammals, micturition (urination) is a complex process that requires the
integrated
action of the bladder, its internal and external sphincters, the musculature
of the pelvic
floor, 'and neurological control over these muscles at three levels (in the
bladder wall or
sphincter itself, in the autonomic centres of the spinal cord and in the
central nervous
system at the level of the pontine micturition centre (PMC) in the brainstem
(pons) under
the control of the cerebral cortex) (De Groat, Neurobiology of I~conti~ence,
Ciba
Foundation Symposium 151:27, 1990). Micturition results from contraction of
the
detrusor muscle, which consists of interlacing smooth-muscle f byes, under the
control of
the parasympathetic autonomic system originating from the sacral spinal cord.
A simple
voiding reflex is triggered by sensory nerves for pain, temperature and
distension that run
from the bladder to the sacral spinal cord. However, sensory tracts from the
bladder reach
the PMC too, generating nerve impulses that normally suppress the sacral
spinal
suppression of cortical inhibition of the reflex arc, and relaxing the muscles
of the pelvic
floor and external sphincter. Finally, the detrusor muscle contracts and
voiding occurs.
Abnormalities of lower-urinary tract function, e.g., dysuria, incontinence and
enuresis, are
common in the general population. Dysuria includes urinary frequency, nocturia
and
urgency, and may be caused by cystitis (including interstitial cystitis),
prostatitis or
benign prostatic hyperplasia (BPH) (which affects about 70% of elderly males),
or by
neurological disorders. Incontinence syndromes include stress incontinence,
urgency
incontinence, overflow incontinence and mixed incontinence. Enuresis refers to
the
involuntary passage of urine at iught or during sleep.
Previously, treatment of neuromuscular dysfunction of the lower urinary tract
involved
administration of compounds that act directly on the bladder muscles, such as
flavoxate, a
spasmolytic drug (Ruffman, J. I~t. Med. Res. 16:317, 1988) which is also
active on the
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
PMC (Guarneri et al., Drugs of Today, 30:91, 1994), or anticholinergic
compounds such
as oxybutynin (Andersson, Dy°ugs 36:477, 1988) and tolterodine
(Nilvebrant, Life Sci.
68(22-23): 2549, 2001). The use of al-adrenergic receptor antagonists for the
treatment
of BPH is common too, but is based on a different mechanism of action (Lepor,
Urology,
42:483, 1993). However, treatments that involve direct inhibition of the
pelvic
musculature (including the deti~usor muscle) may have unwanted side effects,
such as
incomplete voiding or accommodation paralysis, tachycardia and dry mouth
(Andersson,
D~~ugs 35:477, 1988). Thus, it would be preferable to utilize compounds that
act via the
central nervous system to affect for, example, the sacral spinal reflex and/or
the PMC
inhibition pathways in a manner that restores normal functioning of the
micturition
mechanism.
LTS 5346896 discloses 5-HT1A binding agents which may be used in the treatment
of CNS
disorders, such as, for example, anxiety. EP 0924205 discloses aryl piperazine
compounds that bind to 5-HT1A receptors. .
SUMMARY OF THE INVENTION
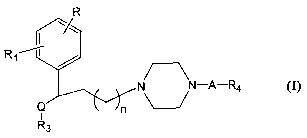
The present invention provides compounds of formula I:
R
-A-R4
wherein
R represents hydrogen or one or more substituents selected from the group
consisting of (C1-C6)-alkyl, (C1-C6)-alkoxy, (C1-C6)-alkylthio, hydroxy, halo,
(C2-C6)-
allcenyl, (C2-C6)-alkynyl, (C1-C6)-haloalkyl, (C1-C6)-haloalkoxy, (C1-C6)-
hydroxyalkyl,
allcoxyallcyl, vitro, amino, (Cl-C6)-aminoalkyl, (C1-C6)-alkylamino-(C1-C6)-
alkyl, (C1-
C6)-alkylamino, di-(Cl-C6)-alkylamino, acylamino, (C1-C6)-alkylsulphonylamino,
aminosulphonyl, (C1-C6)-alkylaminosulphonyl, cyano, aminocarbonyl, N-(Cl-C6)-
allcylaminocarbonyl, N, N-di-(C1-C6)-alkylaminocarbonyl, (C1-C6)-
alkoxycarbonyl, (C1-
C6)-allcylcarbonyl, allcylcarbonylallcyl, formyl, allcanoyloxyalkyl, (C1-C6)-
I
R3
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
allcylaminocarbonylamino, (C1-C6)-allcylsulphinyl, (C1-C6)-alkylsulphonyl, and
N, N-di-
(C1-C~)-allcylaminosulphonyl groups;
Rl represents a member selected from the group consisting of hydrogen,
cycloallcyl, aryl, aryloxy, aralltyl, arallcoxy, heterocyclic, heterocycloxy,
heterocycloalkyl
and heterocycloallcoxy groups, each group being optionally substituted with
one or more
substituent R, defined as above;
Q represents -C(O)- or -CH(OR2)- where R2 represents a member selected
from the group consisting of hydrogen, (C1-C6)-alkyl, (C2-C6)-alkenyl, (C2-C6)-
allcynyl
and cycloallcyl groups, wherein each group is optionally substituted with one
or more
groups selected from RS and R6, where RS is selected from the group consisting
of halo,
(C1-C6)-allcoxy, (C1-C6)-haloalkoxy, cyano, (C1-C6)-allcoxycarbonyl, (C1-C6)-
allcylcarbonyl, allcoxyallcyl, aminocarbonyl, N-(C1-C6)-alkylaminocarbonyl,
N,N-di-(C1-
C6)-allcylaminocarbonyl groups and R6 is selected from the group consisting of
aryl,
heteroaryl, aryloxy, heteroaryloxy, arylalkoxy, and heteroarylalkoxy groups,
each
optionally substituted with R, or Ra represents -C(O)- (C1-C6)-alkyl, -C(O)O-
(C1-C6)-
allcyl, -C(O)NR~R$ or -C(S)NR~RB wherein R~ and R8 are independently hydrogen
or (C1-
C6)-allcyl;
R3 represents hydrogen or a (C1-C6)-alkyl, (C2-C6)-alkenyl, (CZ-C6)-
allcynyl, cycloallcyl, aryl or heterocycle group, each group being optionally
substituted
with one or more substituent R or Rl, defined as above;
R4 represents an aryl or heterocyclic group, each being optionally
substituted with one or more substituent R, defined as above;
A represents a bond or (CH2)"; and
n = 1 or 2,
or an enantiomer, optical isomer, diastereomer, N-oxide (e.g., N-
piperazine oxide), crystalline form, hydrate, solvate or pharmaceutically
acceptable salt
thereof.
As referred to in the definition of R6, aryl, heteroaryl, aryloxy,
heteroaryloxy, arylalkoxy and heteroarylallcoxy group may be optionally
substituted with
one or more substituents selected from the group consisting of, (C1-C6)-alkyl,
(C1-C6)-
allcoxy, (C1-C6)-allcylthio, hydroxy, halo, (CZ-C6)-allcenyl, (CZ-C6)-alkynyl,
(C1-C6)-
haloallcyl, (C1-C6)-haloalkoxy, (C1-C6)-hydroxyallcyl, allcoxyalkyl, vitro,
amino, (Cl-
C6)aminoallcyl, (C1-C6)-allcylamino(C1-C6)-allcyl, (C1-C6)-alkylamino, di(C1-
C6)-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
alleylamino, acylamino, (C1-C6)-alleylsulphonylamino, aminosulphonyl, (C1-C6)-
alleylaminosulphonyl, cyano, aminocarbonyl, N-(C1-C6)-alkylaminocarbonyl, N, N-
di-
(C1-C6)-alleylaminocarbonyl, (C1-C6)-alleoxycarbonyl, (C1-C6)-alleylcarbonyl,
formyl,
alleylcarbonylalkyl, alka~ioyloxyalkyl, (C1-C6)-alkylaminocarbonylamino, (C1-
C6)-
alleylsulphinyl, (C1-C6)-alleylsulphonyl, and N, N-di-(C1-C6)-
alkylaminosulphonyl groups.
In preferred embodiments, Q represents -CH(ORa)-, where R~ is defined as
above.
In a preferred embodiment, the invention provides compounds of formula I as
defined above, with the proviso that the substituents of formula I are not
such that
simultaneously R= hydrogen or (C1-C6)-alkyl, Rl = halogen, Q= -C(O)- or -
CH(ORa)-
where R2 = hydrogen, R3 = cycloalleyl or alkyl and R4 = phenyl substituted
with a
member selected from the group consisting of (C1-C6)-alkyl, (C1-C6)-alkoxy and
(C1-C6)-
haloalleoxy groups, A is a bond and n =1.
In another preferred embodiment, the invention provides compounds of formula I
as defined above, with the proviso that the substituents of formula I are not
such that
simultaneously Q = -CH(OR~) where RZ = H; R3 = cycloalkyl; R = 2-fluoro, Rl =
H, R4 =
2-methoxyphenyl or 2-(2,2,2-trifluoroethoxy)-phenyl, A = bond and n= 1.
Also preferred is an embodiment wherein the invention provides compounds of
formula I as defined above with the proviso that the substituents of formula I
are not such
that simultaneously Q= -C(O)- or -CH(ORa)- where Ra = hydrogen; Rl = H, phenyl
or
phenyl substituted with halo, (C1-C6)-alkyl or (C1-C6)-alleoxy; R = H, (C1-C6)-
alleyl, (C1-
C6)-alkoxy, halo, haloalkyl, vitro, amino, (C1-CG)-alkylamino or di-(C1-C6)-
alkylamino;
R4 is an unsubstituted aryl, unsubstituted heteroaryl or an aryl or heteroaryl
group
substituted with one or more substituent selected from the group consisting of
(C1-C6)-
alleyl, (C1-C6)-alkoxy, halo, (C1-C6)-haloalkyl, vitro, amino, (C1-C6)-
alkylamino, di-(C1-
C6)-alleylamino, hydroxy, (C1-C6)-hydroxyalkyl, -CONR7R8, wherein R~ and Rg
are
independently hydrogen or (CI-C6)-alleyl, and -NHSOa-(CI-C6)-alkyl groups; A
is a bond;
and R3 represents unsubstituted aryl, unsubstituted heteroaryl, or aryl or
heteroaryl
substituted with one more substituent selected from group consisting of (Cl-
C6)-alkyl,
(C1-C6)-alleoxy, halo, (C1-C6)-haloalkyl, vitro, amino, (C1-C6)-alkylamino, di-
(C1-C6)-
alleylamino, phenyl, halophenyl, (Cl-C6)-alleylphenyl and (C1-C6)-
alleoxyphenyl groups.
Also preferred is an embodiment wherein the invention provides compounds of
formula I as defined above with the proviso that the substituents of formula I
are not such
that simultaneously Q= -C(O)- or -CH(ORZ)- where Ra = hydrogen; Rl = H or
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
unsubstituted cycloallcyl or cycloalkyl substituted with (C1-C6)-alkyl; R = H,
(C1-C6)-
allcyl, (C1-C6)-all{oxy, halo, (C1-C6)-haloallcyl, (C1-C6)-alklythio, (CZ-C6)-
alkenyl or (Ca-
C6)-allcynyl; R4 is an unsubstituted aryl, unsubstituted heteroaryl, or an
aryl or heteroaryl
substituted with one to three substituents selected from the group consisting
of (C1-C6)-
alkyl, (C1-C6)-allcoxy, halo, (C1-C6)-haloalkyl, (C1-Cs)-alklythio, (C2-C6)-
alkenyl and (C2-
C~)-allcynyl groups; A is a bond; and R3 represents unsubstituted phenyl,
unsubstituted
naphthyl or unsubstituted cycloallcyl, or phenyl, naphthyl or cycloallcyl
substituted with
one to three substituents selected from the group consisting of (C1-C6)-alkyl,
(C1-C6)-
alkoxy, halo, (C1-C6)-haloalkyl, (C1-C6)-alklythio, (C2-C6)-alkenyl and (C2-
C6)-alkynyl
groups.
In each of the preferred embodiments, it is further preferred that Q is -
CH(OR2)-.
Compounds of formula I can exist as four stereoisomers, which may be present
in
racemic mixtures or in any other combination. Racemic mixtures can be
subjected to
enantiomeric enrichment, to yield compositions enriched in a particular
enantiomer, or
resolved to a composition comprising a single enantiomer. Enantiomeric
enrichment can
be expressed as ee (enantiomeric excess) as defined below.
Some of the preferred compounds according to the invention are described in
the
examples.
The invention also includes metabolites of the foregoing compounds having the
same type of activity, hereinafter referred to as active metabolites.
The present invention also contemplates prodrugs which are metabolized in the
body to generate any of the foregoing compounds.
In another embodiment, the present invention provides pharmaceutical
compositions comprising the foregoing compounds, enantiomers, diastereomers, N-
piperazine oxides, crystalline forms, hydrates, solvates or pharmaceutically
acceptable
salts of such compounds of formula I, in admixture with pharmaceutically
acceptable
diluents or carriers such as those disclosed.
In another embodiment, the invention provides intermediates useful in the
synthesis of compounds of formula I. Some of these are included in the claims.
Yet another embodiment is a method for reducing the frequency of bladder
contractions due to bladder distension in a mammal (such as a human) in need
thereof by
administering an effective amount of at least one compound of the present
invention to
reduce the frequency of bladder contractions due to bladder distension to the
mammal.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
Yet another embodiment is a method for increasing urinary bladder capacity in
a
maanmal (such as a human) in need thereof by administering an effective amount
of at
least one compomid of the present invention to increase urinaay bladder
capacity to the
mammal.
Yet another embodiment is a method for treating disorders of the urinary tract
in a
mammal (such as a human) in need thereof by administering an effective amount
of at
least one compound of the present invention to ameliorate at least one
condition among
urinary urgency, overactive bladder, increased urinary frequency, decreased
urinary
compliance (decreased bladder storage capacity), cystitis (including
interstitial cystitis),
incontinence, urine lealcage, enuresis, dysuria, urinary hesitancy and
difficulty in
emptying the bladder.
In yet other embodiments, the invention provides for methods of treating the
above disorders, by administering a compound of formula I in combination with
other
agents such as, for example, one or more additional SHT1A antagonist,
antimuscarinic
drugs, al-adrenergic antagonists, inhibitors of the cyclooxygenase enzyme,
which may
inhibit both COX1 and COX~ isozymes or which may, alternatively, be selective
for
COX2 isozyme, a,nd NO donor derivatives thereof.
In yet other embodiments, the invention provides a method for treating a
mammal
suffering from a central nervous system (CNS) disorder due to serotoninergic
dysfunction
by administering an effective amount of at least one compound of the present
invention to
treat the CNS disorder. Such dysfunctions include, but are not limited to,
anxiety,
depression, hypertension, sleeplwake cycle disorders, feeding, behaviour,
sexual
dysfunction and cognition disorders in mammals (particularly in humans)
associated with
stroke, injury, dementia, and originated by neurological development,
attention-deficit
hyperactivity disorders (ADHD), drug addiction, drug withdrawal, irritable-
bowel
syndrome and symptoms caused by withdrawal or partial withdrawal from the use
of
nicotine or tobacco.
In yet another embodiment, the invention provides a method for treating a
disorder due to serotoninergic dysfunction by delivering a compound of the
invention to
the environment of a 5-HT1A serotoninergic receptor, for example, to the
extracellular
medium (or by systemically or locally administering to a mammal possessing
such a 5-
HT1A receptor) an amount of a compound of the invention effective in the
treatment of
said disorder due to serotoninergic dysf~ulction.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
In a preferred embodiment, the invention provides methods for treating a
mammal
(including a human) suffering from a urinary tract disorder by administering
at least one
compound of the invention to the environment of a 5-HT1A receptor in an amount
effective to increase the duration of bladder quiescence. More highly
preferred is where
the increase in the duration of bladder quiescence is accomplished with little
or no effect
(e.g., decrease or increase ) on micturition pressure.
Compounds
The invention relates to compowids of formula I as disclosed above. The
invention includes the enantiomers, diastereoisomers, N-piperazine oxides,
crystalline
forms, hydrates, solvates or pharmaceutically acceptable salts of these
compounds, as
well as active metabolites of these compounds having the same type of
activity.
The term "haloalkyl" includes alkyl groups substituted by a single halogen
atom
(monohaloallcyl) and those substituted by more than one halogen atom
(polyhalaalkyl).
Examples of the latter are trifluoromethyl and 2,2,2-trifluoroethyl groups.
The term
haloallcoxy is to be interreted correspondingly., Preferred haloalkoxy groups
include
trifluoromethoxy and 2,2,2-trifluoroethoxy groups.
The term "aryl", alone or in combination, refers to a carbocyclic aromatic
system
containing one, two or three rings wherein such rings may be attached together
in a
pendent manner or may be fused. The term "aryl" includes aromatic radicals
such as
phenyl, naphthyl, tetrahydronaphthyl, indane and biphenyl.
The terms "heterocyclic" and "heterocyclo" refer to saturated, partially
saturated
and unsaturated heteroatom-containing ring-shaped radicals, where the
heteroatoms may
be selected from nitrogen, sulphur and oxygen. Examples of saturated
heterocyclic
radicals include saturated heteromonocylic groups containing 1 to 4 nitrogen
atoms (e.g.,
pyrrolidinyl, imidazolidinyl, piperidino, piperazinyl); saturated
heteromonocyclic groups
containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms (e.g., morpholinyl);
saturated
heteromonocyclic groups containing 1 to 2 sulphur atoms and 1 to 3 nitrogen
atoms (e.g.,
thiazolidinyl). Examples of partially saturated heterocyclic radicals include
dihydrothiophene, dihydropyran, dihydrofuran and dihydrothiazole.
The terms "heterocyclo" and "heterocyclic" encompass the term "heteroaryl,"
which refers to unsaturated heterocyclic radicals. Examples of "heteroaryl"
radicals
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
include unsaturated 5 to 6 membered heteromonocyclic groups containing 1 to 4
nitrogen
atoms, for example, pyrrolyl, pyrrolinyl, imidazolyl, pyrazolyl, 2-pyridyl, 3-
pyridyl, 4-
pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, triazolyl (e.g., 4H-1,2,4-
triazolyl, 1H-1,2,3-
triazolyl, 2H-1,2,3-triazolyl) tetrazolyl (e.g., 1H-tetrazolyl, 2H-
tetrazolyl); unsaturated
condensed heterocyclic groups containing 1 to 5 nitrogen atoms, for example,
indolyl,
isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, indazolyl,
benzotriazolyl,
tetrazolopyridazinyl (e.g., tetrazolo[1,5-b]pyridazinyl); unsaturated 3 to 6-
membered
heteromonocyclic groups containing an oxygen atom, for example, pyranyl, 2-
furyl, 3-
f~uyl; unsaturated 5 to 6-membered heteromonocyclic groups containing a
sulphur atom,
for example, 2-thienyl, 3-thienyl; unsaturated 5- to 6-membered
heteromonocyclic groups
containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms, for example,
oxazolyl,
isoxazolyl, oxadiazolyl (e.g., 1,2,4-oxadiazolyl, 1,3,4-oxadiazolyl, 1,2,5-
oxaciiazolyl);
unsaturated condensed heterocyclic groups containing 1 to 2 oxygen atoms and 1
to 3
nitrogen atoms (e.g., benzoxazolyl, benzoxadiazolyl); unsaturated 5 to 6-
membered
heteromonocyclic groups containing 1 to 2 sulphur atoms and 1 to 3 nitrogen
atoms, for
example, thiazolyl, thiadiazolyl (e.g., 1,2,4-thiadiazolyl, 1,3,4-
thiadiazolyl, 1,2,5-
thiadiazolyl); unsaturated condensed heterocyclic groups containing 1 to 2
sulphur atoms
and 1 to 3 nitrogen atoms (e.g., benzothiazolyl, benzothiadiazolyl) and the
lilce. The term
"heteroaryl" also refers to radicals where heterocyclic radicals are fused
with aryl
radicals. Examples of such fused bicyclic radicals include benzofuran,
benzothiophene,
and the lilce. Said "heterocyclic group" may have 1 to 3 substituents such as,
for example
and without limitation, lower allcyl, hydroxy, oxo, amino and lower
allcylamino.
Preferred heterocyclic radicals include five to ten membered fused or unfused
radicals.
Examples of heteroaryl radicals include benzofuryl, 2,3-dihydrobenzofuryl,
benzothienyl,
indolyl, dihydroindolyl, chromanyl, benzopyran, thiochromanyl, benzothiopyran,
benzodioxolyl, benzodioxanyl, pyridyl, thienyl, thiazolyl, oxazolyl, fwyl, and
pyrazinyl.
The term "cycloallcyl" refers to saturated carbocyclic radicals having three
to ten
carbon atoms. Preferred cycloallcyl radicals are "lower cycloallcyl" radicals
having three
to seven carbon atoms. Examples include radicals such as cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl and cycloheptyl. A most preferred cycloalkyl group is
cyclohexyl.
The term "acyl", whether used alone, or within a term such as "acylamino",
denotes a radical provided by the residue after removal of hydroxyl from a
carboxylic
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
acid. Preferred acyl groups are allcanoyl groups, such as acetyl.
A "metabolite" of a compound disclosed herein is a derivative of a compound
wluch is formed when the compound is metabolised. The term "active metabolite"
refers
to a biologically active derivative of a compound that is formed when the
compound is
metabolised. The term "metabolised" refers to the sum of the processes by
which a
particular substance is changed in the living body. All compounds present in
the body are
manipulated by enzymes within the body in order to derive energy and/or to
remove them
from the body. Specific enzymes produce specific structural alterations to the
compound.
Cytochrome P450, for example, catalyses a variety of oxidative and reductive
reactions.
Uridine diphosphate glucuronyltransferases, for example, catalyse the transfer
of an
activated glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols,
carboxylic
acids, amines and free sulphhydryl groups. Further information on metabolism
may be
obtained from The Phas~macologieal Basis of Therapeutics, 9~' Edition, McGraw-
Hill
(1996), pages 11-17.
The metabolites of the compounds disclosed herein can be identified either by
administration of compounds to a host and analysis of tissue samples from the
host, or by
incubation of compounds with hepatic cells or other in vit~~o systems such as
cytochromes
or microsomes, and analysis of the resulting compounds. Both methods are well
known in
the art.
As used herein, the term "stereoisomer" refers to a compound made up of the
same atoms bonded by the same bonds but having different three-dimensional
structures
wluch are not interchangeable. The three-dimensional structures are called
configurations. As used herein, the term "enantiomer" refers to two
stereoisomers whose
molecules are nonsuperimposable mirror images of one another. As used herein,
the term
"optical isomer" is equivalent to the term "enantiomer". Compounds that are
stereoisomers of one another, but are not enantiomers of one another, are
called
diastereoisomers. The terms "racemate" or "racemic mixture" refer to a mixture
of equal
parts of enantiomers. The term "chiral center" refers to a carbon atom to
which four
different groups are attached. The term "enantiomeric enrichment" as used
herein refers
to the increase in the amount of one enantiomer as compared to the other. A
convenient
method of expressing the enantiomeric enrichment achieved is the concept of
enantiomeric excess, or "ee", which is found using the following equation:
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
ee-E1-E~*100
El+E2
wherein E1 is the amount of the first enantiomer and E2 is the amount of the
second
enantiomer. Thus, if the initial ratio of the two enantiomers is 50:50, such
as is present in
a racemic mixture, and an enantiomeric enrichment sufficient to produce a
final ratio of
50:30 is achieved, the ee with respect to the first enantiomer is 25%.
However, if the
final ratio is 90:10, the ee with respect to the first enantiomer is 80%.
According to one
embodiment of the invention, an ee of greater.than 90% is preferred, an ee of
greater than
95% is most preferred and an ee of greater than 99% is most especially
preferred.
Enantiomeric enrichment is readily determined by one of ordinary skill in the
art using
standard techniques and procedures, such as high performance liquid
chromatography
with a chiral column. Choice of the appropriate chiral column, eluent and
conditions
necessary to effect separation of the enantiomeric pair is within the
lcnowledge of one of
ordinary skill in the art. In addition, the enantiomers of compounds of
formula I can be
resolved by one of ordinary skill in the art using standard techniques well
known in the
art, such as those described by J. Jacques, et al., "Enantiomers, Racemates,
and
Resolutions", John Wiley and Sons, Inc., 1981. Examples of resolutions include
recrystallization techniques or chiral chromatography.
Diastereisomers differ in both physical properties and chemical reactivity. A
mixture of diastereomers can be separated into enantiomeric pairs based on
solubility,
fractional crystallization or chromatographic properties, e.g., thin layer
chromatograph,
column chromatography or HPLC.
Purification of complex mixtures of diastereomers into enantiomers typically
requires two steps. In a first step, the mixture of diastereomers is resolved
into
enantiomeric pairs, as described above. In a second step, enantiomeric pairs
are further
purified into compositions enriched for one or the other enantiomer or, more
preferably
resolved into composition comprising pure enantiomers. Resolution of
enantiomers
typically requires reaction or molecular interaction with a chiral agent,
e.g., solvent or
column matrix. Resolution may be achieved, for example, by converting the
mixture of
enantiomers, e.g., a.racemic mixture, into a mixture of diastereomers by
reaction with a
pure enantiomer of a second agent, i.e., a resolving agent. The two resulting
diasteromeric products can then be separated. The separated diastereomers are
then
reconverted to the pure enantiomers by reversing the initial chemical
transformation.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
11
Resolution of enantiomers can also be accomplished by differences in their non-
covalent binding to a chirak substance, e.g., by chromatography on homochiral
absorbants. The noncovalent binding between enantiomers and the
chromatographic
adsorbant establishes diastereomeric complexes, leading to differential
partitioning in the
mobile and bound states in the chromatographic system. The two enantiomers
therefore
move through the chromatographic system, e.g, column, at different rates,
allowing for
their separation.
Chiral resolving columns axe well known in the art and are commercially
available
(e.g., from MetaChem Technologies Inc., a division of ANSYS Technologies,
Inc., Lake
Forest, CA). Enantiomers can be analyzed and purified using, for example,
chiral
stationary phases (CSPs) for HPLC. Chiral HPLC columns typically contain one
form of
an enantiomeric compound immobilized to the surface of a silica packing
material. For
chirak resolution to occur, there must be at least three points of
simultaneous interaction
between the CSP and one analyte enantiomer, with one or more of these
interactions
being stereochemically dependent.
D-phenylglycine and L-leucine are Type I CSPs and use combinations of p-p
interactions, hydrogen bonds, dipole-dipole interactions, and steric
interactions to achieve
chiral recognition. To be resolved on a Type I column, analyte enantiomers
must contain
functionality complementary to that of the CSP so that the analyte undergoes
essential
interactions with the CSP. The sample should preferably contain one of the
folkowing
functional groups: p-acid or p-base, hydrogen bond donor andlor acceptor, or
an amide
dipole. Derivatization is sometimes used to add the interactive sites to those
compounds
lacking them. The most conunon derivatives involve the formation of amides
from
amines and carboxylic acids.
The MetaChiral ODMTM is a type II CSP. The primary mechanisms for the
formation of solute-CSP complexes is through attractive interactions, but
inclusion
complexes also play an important role. Hydrogen bonding, pi-pi, and dipole
staclcing are
important for chiral resolution on the MetaChirakTM ODM. Derivatization is
often
necessary when the solute molecule does not contain the groups required for
sokute-
column interactions. Derivatization, usually to benzykamides, is also required
of some
strongly polar molecules lilce amines and carboxylic acids, which would
otherwise
interact too strongly with the stationary phase through non-stereo- specific
interactions.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
12
In certain embodiments, formula I set forth above may include a proviso that
excludes compowids represented by the generic formula disclosed in U.S. Patent
No.
5,346,896.
In certain embodiments, formula I set forth above may include a proviso that
excludes compounds represented by the generic formula disclosed in U.S. Patent
No.
6,358,958.
In certain embodiments, formula I set forth above may include one or more
proviso that excludes compounds represented by the generic formulas disclosed
in both
U.S. Patent No. 5,346,896 and U.S. Patent No. 6,358,958.
In certain embodiments, compounds represented by formula I exclude compounds
within formula I that are with the generic formula disclosed in U.S. Patent
No. 5,346,896.
Preferred groups that R represent are a hydrogen or halogen atom or (C1-C6)-
alkoxy, (C1-C6)-haloalkoxy, N,N-di-(C1-C6)-aminocarbonyl or cyano group. A
preferred
haloallcoxy the R is a polyhaloalkoxy, more preferably prefereably
trifluoromethoxy. A
preferred halogen atom that R represents is a fluorine atom. The preferred
position for
the aforementioned atoms and groups is on the 2-position of the phenyl group
to which
they are attached.
A preferred group that Rl represents is a hydrogen atom.
Also preferred is where simultaneously, R represents one or more member
selected from the groups consisting hydroxy, (C1-C6)-haloallcoxy, (C1-C6)-
hydroxyalkyl,
allcoxyallcyl, (Cl-C6)-aminoallcyl, (C1-C6)-alkylamino-(C1-C6)-alkyl ,
acylamino, (C~-C6)-
allcylsulphonylamino, aminosulphonyl, (Cl-C6)-alkylaminosulphonyl, cyano,
aminocarbonyl, N-(C1-C6)-alkylaminocarbonyl, N, N-di-(C1-C6)-
alkylaminocarbonyl,
(C1-Cg)-alkoxycarbonyl, (C1-C6)-allcylcarbonyl, alkylcarbonylalkyl, formyl,
allcanoyloxyalkyl, (C1-C6)-allcylaminocarbonylamino, (C1-C6)-alkylsulphinyl,
(C1-C6)-
allcylsulphonyl, and N, N-di-(Cl-C6)-alkylaminosulphonyl groups; and Rl
represents a
member selected from the group consisting of unsubstituted aryloxy, arallcyl,
aralkoxy,
heterocycloxy, heterocycloalkyl and heterocycloalkoxy groups, or a member
selected
from the group consisting of aryloxy, aralkyl, aralkoxy, heterocycloxy,
heterocycloalkyl"
heterocycloallcoxy, aryl, heterocyclic and cycloalkyl groups substituted with
one or more
substituent selected from the group consisting of R represents hydrogen or one
or more
substituents selected from the group consisting of (C1-C6)-alkylthio, hydroxy,
(Ca-C6)-
allcenyl, (C2-C6)-allcynyl, (C1-C6)-haloalkoxy, (C1-C6)-hydroxyalkyl,
allcoxyalkyl, (C1-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
13
C6)-aminoalkyl, (C1-Cg)-alkylamino-(C1-C6)-alkyl, acylamino, (C1-C6)-
allcylsulphonylamino, aminosulphonyl, (C1-C6)-alkylaminosulphonyl, cyano,
aminocarbonyl, N-(C1-C6)-allcylaminocarbonyl, N, N-di-(C1-C6)-
alkylaminocarbonyl,
(C1-C6)-allcoxycarbonyl, (C1-C6)-allcylcaxbonyl, allcylcarbonylallcyl, formyl,
allcanoyloxyallcyl, (C1-C6}-allcylaminocarbonylamino, (Cl-C6)-alkylsulphinyl,
(C1-C6)-
allcylsulphonyl, and N, N-di-(C1-C6)-alkylaminosulphonyl groups.
Preferred groups that Q represents are -C(O)- and -CH(ORa)- where R~
represents
a hydrogen atom or (C1-C6)-allcyl, (C2-C6)-alkenyl, (C2-C6)-alkynyl, -C(O)-
(C1-C6)-alkyl,
-C(O)O-(C1-C6)-alkyl, -C(O)NR~R$ or -C(S)NR~R$ wherein R~ and Rg are
independently
hydrogen or (Cl-C6)-alkyl;
Preferred groups that R3 represents are a hydrogen atom or a (C1-C6)-allcyl,
(C~-
C6)-allcenyl, (Ca-C6)-allcynyl, cycloalkyl, aryl or heterocycle group. Also
preferred is
where R3 represents hydrogen or a (C1-C6)-allcyl, (CZ-C6)-alkenyl, (C2-C6)-
alkynyl, each
group being optionally substituted with one or more substituent R or RI,
defined as above.
More preferably, R3 represents a cyclohexyl group.
Preferred groups that R4 represents are an aryl or heterocyclic group, each
being
optionally substituted with one or more substituent selected from the group
consisting of
halogen atom or (C1-C6)-allcoxy or (Cl-C6)-haloalkoxy groups. A preferred
halogen atom
that is a substitutent on R4 is fluorine. A preferred alkoxy group that is a
substitutent on
Rø is a methoxy group. A preferred haloalkoxy group that is a sustitutent on
R4 is a
polyhaloalkoxy group, most preferably a trifluoroethoxy group. A preferred
aryl group
that R4 represents is a phenyl group. A preferred heterocyclic group that R4
represents is
a bicyclic heterocyclic group. More preferably R4 represents a bicyclic
heteroaryl group,
most prefereably a 2,3-dihydro-1,4-benzodioxinyl group.
Also preferred is where R4 represents an aryl or heterocyclic group,
substituted
with one or more substituent selected from the group consisting of (C1-C6)-
haloalkoxy,
allcoxyalkyl, (C1-C6)-aminoalkyl, (C1-C6)-alkylamino-(C1-C6)-alkyl, acylamino,
aminosulphonyl, (C1-C6)-alkylaminosulphonyl, cyano, (C1-C6)-allcoxycarbonyl,
(C1-C6)-
alkylcarbonyl, allrylcarbonylallcyl, formyl, alkanoyloxyalkyl, (Cl-C6)-
allcylaminocarbonylamino, (C1-C6)-alkylsulphinyl, (Cl-C6)-alkylsulphonyl, and
N, N-di-
(C1-C6)-alkylaminosulphonyl groups.
A prefereably represents a bond.
n is prefereably 1.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
14
Also preferred are compounds of formula I wherein, simultaneously, R
represents
a hydrogen or halogent atom or (C1-C~)-allcoxy, (C1-C6)-haloallcoxy, N,N-di-
(C1-C6)-
aminocarbonyln or cyano group; Rl represents is a hydrogen atom, Q represents -
C(O)- or
-CH(OR2)- where R2 represents a hydrogen atom or (Cl-C6)-alkyl, (CZ-C6)-
alkenyl, (C2-
C6)-alkynyl, -C(O)- (C1-C6)-alkyl, -C(O)O-(C1-Cg)-alkyl, -C(O)NR~RB or -
C(S)NR~RB
wherein R~ and R8 are independently hydrogen or (Cl-C6)-alkyl group; R3
represents a
hydrogen atom or a (C1-C6)-alkyl, (CZ-C6)-alkenyl, (C2-C6)-alkynyl,
cycloalkyl, aryl or
heterocycle group; R4 represents are an aryl or heterocyclic group, each being
optionally
substituted with one or more substituent selected from the group consisting of
halogen
atom or (C1-C6)-alkoxy or (Cl-C6)-haloallcoxy groups; A represents a bond; and
n =2.
Also preferred are compounds of formula I represented by the formula
Compounds of formula I can be separated into diastereomeric pairs by, for
example, by separation by TLC. These diastereomeric pairs are referred to
herein as
diastereoisomer with upper TLC Rf; and diastereoisomer with lower TLC Rf. The
diastereoisomers can further be enriched for a particular enantiomer or
resolved into a
single enantiomer using methods well known in the art, such as those described
herein.
SYNTHESIS OF THE COMPOUNDS OF THE INVENTION
The compounds of the invention are generally prepared according to the
following
schemes:
O . R3 Scheme 1 O R
R3 3
I ~ ~ I \ ~-O~ -~ I \ ~-CHO + HN~ -B
R
R ~~) R O~R a ~2) R I3) U')
a
HO R3 O R3
I \ U _B ~ I \ ~JN_B
R ~R
O~Rz=H)
Groups B, R are the same as groups A-R4, and (R+ Rl ) respectively, as given
in the
general formula I. R2 and R3 are the same as given in the general formula and
Ra is a
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
lower alkyl group.
Starting material (1) is treated with a base, preferably potassium tert-
butoxide,
followed by allcylation with 2-bromoacetaldehyde dialkyl acetal or other
carbonyl
protected 2-haloacetaldehyde (e.g., the R~ alkyl groups can also be joined in
a cycle to
give a dioxolane or dioxane ring). Other alternative and appropriate bases to
carry out the
condensation include lithium amides, sodium hydride, sodium hydroxide,
potassium
hydroxide, potassium carbonate, cesium carbonate and the like with the aid or
not of
phase transfer catalysts.
The reaction is preferably carried out in a solvent such as dimethyl
sulphoxide or
toluene at a temperature of 0°C to reflux.
The use of 3-bromopropionaldehyde dialkyl acetal or other carbonyl protected 3-
halopropionaldehyde allows to obtain, by following the same reaction
conditions
described above in Scheme 1, compound I having n = 2 as foreseen in the
general
formula.
Treatment of (2) with an acid, such as hydrochloric acid or p-toluene-
sulphonic
acid or trifluoroacetic acid in a suitable organic solvent, achieves aldehyde
(3). Generally,
the reaction is conducted in a erotic solvent, such a mixture of aqueous acid
and acetone
or tetrahydrofiuan, at temperatures of from about 5 ° to 75 ° C
preferably at ambient
temperature. A preferred and alike method consists of carrying out the
reaction in a
mixuture of aqueous trifluoroacetic acid in a chlorinated solvent at ambient
temperature.
Aldehyde (3) is coupled with the desired aryl piperazine (4) by reductive
amination procedure to prepare (5). The reaction is preferably conducted at
ambient
temperature in a non-reactive solvent such as dichloroethane or methylene
chloride or
chloroform in the presence of sodium triacetoxyborohydride and is
substantially complete
in one to 24 hours (see for example A. F. Abdel-Magid, et al., J. Org. Chem.,
61, 3849
(1996)) or it can be conducted in a erotic solvent (e.g., methanol) with the
aid of sodium
cyanoborohydride optionally in the presence of molecular sieves.
Reduction of (5) to the alcohol (I) is readily accomplished using a reducing
agent
such as sodium borohydride or, diisobutylaluminum hydride or other aluminum or
boron
hydride or other reduction method to carry out the conversion lcetone to
alcohol very well
known to those skilled in the art, to prepare the hydroxy compound (I). The
reaction is
preferably conducted in an organic solvent such as methanol or methylene
chloride or
tetrahydrofuran at temperatures of from about -20 ° C to ambient
temperature.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
16
Scheme 2
O
R
H3C~N~R3 +
~M
O A
HsCi l6) L7)
Starting material (1) is either commercially available or can be prepared by
coupling the proper Weinreb amide (6) [See Nahm and Weinreb, Tetrahedron
Lett., 22,
3815, (1981)] with (7); as described in Scheme 2 above, where M is a metallic
salt, such
as lithium or magnesium halide.
The reaction is preferably carried out under nitrogen atmosphere, in ail
aprotic
solvent, such as tetrahydrofuran, at ambient or lower temperatures down to -
78°C.
Alternatively an ester of structure R3COOalkyl can be treated with a
substituted
benzylmagnesium chloride or benzylmagnesium bromide or lithium derivative
under
standard conditions well known in the art to provide the ketone of structure
(1).
An alternative route to obtain compounds (1) consists in reacting the
appropriate
arylaldehyde with an allcylnitro derivative in a nitroaldol fashion,
dehydration of the
resultant nitro alcohol followed by double bond reduction afford a 2-nitro(2
Ak)-
phenethyl derivative, which can undergo Nef reaction to yield the wished lceto
derivative
1. This lcind of pathway is well documented in the experimental part and in
the literature.
Preferred and alike way of synthesis of (1) is the palladium catalysed
coupling of
an acyl halide with a compound (7) where M is Zn halide.
More specifically, the compounds of formula (5) can be prepared following the
procedure described in Scheme 3. All substituents, unless otherwise indicated,
are as
previously defined. The reagents and starting materials are readily available
to one of
ordinaay slcill in,the art.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
17
Scheme 3
O
p ~ ~ Ste~ ~ Step C
COCI Ste A O
R O Et
R ~Et (9)
O HN N-B O
4
( ) I ~ VN-B
Step D
R O R
(10) (5)
In Scheme 3, step A, for example, cyclohexanecarbonyl chloride is added to a
mixture of the suitable benzylzinc chloride or bromide and a proper palladium
catalyst,
e.g., dichlorobis(triphenylphosphine)-palladium (II) stirred at 0°C in
a solvent such as
tetralrydrofuran. Afterwards, stirring is continued at ambient temperature for
4-24 h. Then
the reaction is quenched for example with an aqueous saturated solution of
ammonium
chloride. Usual worlc-up procedure by extraction provide the lcetone (8).
I~etone (8) can
be purified by techniques well known in the art, such as flash chromatography
on silica
gel with a suitable eluent, such as ethyl acetatelhexane to provide the
purified material.
Alternatively, the crude lcetone (8) can be carried on to step B.
In Scheme 3, step B, ketone (8) is alkylated with bromoacetaldehyde diethyl .
acetal under conditions well known in the art to provide compound of structure
(9). For
example, lcetone (8) is dissolved in a suitable organic solvent, such as
dimethyl
sulphoxide or toluene and treated with a slight excess of a suitable base,
such as
potassium tert-butoxide. The reaction is stirred for about 15 to 30 minutes at
a
temperature of between 0°C and the reflux temp. of the solvent and
bromoacetaldehyde
diethyl acetal is added dropwise to the reaction. One of ordinary skill in the
art would
readily appreciate that bromoacetaldehyde dimethyl acetal, bromoacetaldehyde
ethylene
acetal and the lilce may be used in place of the corresponding diethyl acetal.
In Scheme 3, step C, compound (9) is hydrolyzed under acidic conditions to
provide aldehyde (10) in a manner analogous to the procedure described in
Scheme I.
More specifically, for example, compound (9) is dissolved in a suitable
organic solvent,
such as dichloromethane and treated with a suitable acid, such as aq.
trifluoroacetic acid.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
18
The reaction mixture is stirred for about 1 to 6 hours at room temperature.
The reaction
mixture is then diluted with the same solvent, washed with brine, the organic
layer is
separated, dried over anhydrous sodium sulphate, filtered and concentrated
under vacuum
to provide aldehyde (10). Aldehyde (10) can be purified by techniques well
known in the
art, such as flash chromatography on silica gel with a suitable eluent, such
as ethyl
acetate/hexane. Alternatively, crude aldehyde (10) can be used directly in
step D.
In Scherr~e 3, step D, aldehyde (10) is reductively aminated, under conditions
well
lcnown in the art, with piperazine (4) to provide the lcetone (5) in a manner
analogous to
the procedure described in Scheme I. More specifically, for example, aldehyde
(10) is
dissolved in a suitable orga~uc solvent, such as methylene chloride. To this
solution is
added about 1.05 or more equivalents of piperazine (4). Acetic acid may
optionally be
added to aid in dissolution of the piperazine (4). Then about 1.4 to 1.5
equivalents of
sodium triacetoxyborohydride is added and the reaction is stirred at room
temperature for
about 3 to 5 hours. The reaction is then quenched by addition of a suitable
base, such as
aqueous sodium carbonate or hydroxide to provide a pH of about 8 to about 12.
The
quenched reaction is then extracted with a suitable organic solvent, such as
methylene
chloride. The organic extracts are combined, washed with brine, dried,
filtered and
concentrated under vacuum to provide the compound of formula (5). This
material can
then be purified by techniques well known in the art, such as flash
chromatography on
silica gel with a suitable eluent, such as ethyl acetate/petroleum ether or
hexane.
O H HO R3 Scheme 4 O R
3
Ste~ ~ Ste
AM I I R (1)
(11) R (12) R (13)
Step C
O R3 O Rs
O Rs
I w ~N N-B St~ \ St~ I ~ W
R
R (5) ~ HN N-B R CHO
V (3) (1 )
(4)
Alternatively, compounds of structure (5) can be prepared following the
procedure
described in Scheme 4. All substituents, unless otherwise indicated, are
defined
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
19
previously. The reagents and starting materials are readily available to one
of ordinary
skill in the art.
In Scheme 4, step A, aldehyde (11) is combined with a suitable organometallic
reagent (12) under conditions well known in the art to provide alcohol. (13).
Examples of
suitable organometallic reagents include Grignard Reagents, allcyl lithium
reagents, alkyl
zinc reagents, and the like. Grignard Reagents are preferred. For examples of
typical
Grignard Reagents and reaction conditions, see J. March, "Advanced Organic
Chemistry:
Reactions, Mechanisms, and Structure", 2nd Edition, McGraw-Hill, pages 836-841
(1977). More specifically, aldehyde (11) is dissolved in a suitable organic
solvent, such as
tetrahydrofuran or toluene, cooled to about -5 °C and treated with
about 1.1 to 1.2
equivalents of a Grignard reagent of formula (12) wherein M is MgCI or MgBr.
The
reaction is stirred for about 0.5 to 6 hours, then quenched, and alcohol (13)
is isolated by
well-known work-up procedure.
In Scheme 4, step B, alcohol (13) is oxidized under standard conditions well
lcnow
in the art, such as those described by J. March, "Advanced Organic Chemistry:
Reactions,
Mechanisms, and Structure", 2nd Edition, McGraw-Hill, pages 1082-1084 (1977),
to
provide lcetone (1). (Ketone (1) is the starting material used in Scheme 1
above.)
The oxidation can also be performed using standard Swern Oxidation conditions
which are well lcnown to one of ordinary skill in the art (Marx,Tidwell - J.
Org. Chem.
49,788 (1984) , or the alcohol (13) is dissolved in a suitable organic
solvent, such as
methylene chloride, the solution cooled with a wet ice-acetone bath, and
treated with 2.5
to 3.0 equivalents of dimethyl sulphoxide. After stirring for about 30
minutes, the reaction
is then treated with about 1.8 equivalents of Pa05. The reaction is stirred
for about 3 hours
and then, preferably, treated over about 30 minutes with about 3.5 equivalents
of a
suitable amine, such as triethylamine. The cooling bath is then removed and
the reaction
is stirred for about 8 to 16 hours. The lcetone (1) is then isolated by
standard extraction
techniques well known in the art.
In Scheme 4, step C, lcetone (1) is treated with a suitable base followed by
addition of the alkene (15), wherein X is a suitable leaving group, to provide
compound
(14). For example, lcetone (1) is combined with an excess of allcene (15) in a
suitable
organic solvent, such as tetrahydrofuran, and cooled with a wet ice acetone
bath.
Examples of suitable leaving groups are Cl, Br, I, tosylate, mesylate, and the
like.
Preferred leaving groups are Cl and Br. About 1.1 equivalents of a suitable
base is added
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
and the reaction is allowed to stir for about 2 hours at room temperature.
Examples of
suitable bases are potassium tert-butoxide, sodium hydride, NaN(Si(CH3)s)a,
LDA,
I~N(Si(CH3)3)2, NaNH2, sodium ethoxide, sodium methoxide and the like.
Potassium tert-
butoxide is the preferred suitable base. The reaction is then quenched with
aqueous acid
and compound (14) is isolated by usual work-up procedure.
In Scheme 4, step D, compound (14) is treated with a suitable oxidizing agent
to
provide aldehyde (3). (Aldehyde (3) is also prepared in Scheme 1) Examples of
suitable
oxidizing agents are ozone, NaI04 /Osmium catalyst, and the like. Ozone is the
preferred
oxidizing agent. Examples of suitable oxidizing reagents and conditions are
described by
J. March, "Advanced Organic Chemistry: Reactions, Mechanisms, and Structure",
2nd
Edition, McGraw-Hill, pages 1090-1096 (1977).
For example, compound (14) is dissolved in a suitable organic solvent, such as
methanol, a small amount of Sudan III is added, and the solution is cooled to
about -20
°C. Ozone is bubbled into the solution for about 4 hours until the
pinlc color turns to a
pale yellow color. Then a reducing agent such as Me2 S or tributylphosphine is
added.
Concentration provides the intermediate dimethyl acetal of aldehyde (3). This
dimethyl
acetal is readily hydrolyzed under standard acidic conditions to provide
aldehyde (3).
Alternatively, direct acidic work-up of the crude reaction mixture provides
aldehyde (3).
Alternatively, aldehyde (3) can be obtained directly by ozonolysis of (14) in
a non-acetal
forming solvent, such as methylene chloride.
In Scheme 4, step E, aldehyde (3) is reductively aminated under conditions
analogous to those described above in Scheme 3, step D, to provide compound
(5).
(Compound 5 is also prepared in Scheme I)
Scheme 5
O R3
O R3 O R3
Step A Step B
CHO ~ I W ~ V -B ~ ~ N~ -g
R (3) HNV B R R (5)
(4) (15)
Scheme 5 provides an alternative synthesis for the preparation of ketone (5).
All
substituents, unless otherwise indicated, are as defined previously. The
reagents and
starting materials are readily available to one of ordinary skill in the art.
In Scheme 5, step A, aldehyde .(3) is condensed with piperazine (4) under
standard
{~6y",a'~ ,~u~0~~ 3 CA 02489449 2004-12-14
-21 -
conditions well known in the art to~provide the enamine {15). For example,
about 1.05
equivalents of aldehyde (3) dissolved in a suitable organic solvent, such as
isopropyl acetate
or isopropanol, is added to neat piperazine (4), free base. Additional organic
solvent is added
to produce a slurry and the reaction is stirred for about 1 to 2 hours. The
enamine (15) is then
isolated by standard techniques, such as collection by filtration.
In Scheme 5, step B, the enamine (15) is hydrogenated under conditions well
known
to one of ordinary skill in~the art to provide compound (5). For example,
enamine (15) is
combined with a suitable organic solvent, such as isopropyl alcohol and a
catalytic amount of
5°!° palladium on carbon in a Parr bottle. The mixture is placed
under 50 psi of hydrogen and
shaken for about 2 days at room temperature. The slurry is then filtered to
remove catalyst
and the filtrate is concentrated to provide compound (5).
. R3
H-N N-B
~N\ '/N-B ~ (4) CHO
I (RZ not Ii~
For the synthesis of compounds I where R2 is different than H, the method
given in
Scheme 6 is used. Intermediate ketone (2) is reduced with the same reduction
methods used
above in scheme 1 for compound (5) affording intermediate (16), which is
etherified by
reaction with a base, for example NaH or potassium tert-butoxide or NaNH2 or
LiNH2 or
others in a suitable solvent e.g. tetrahydrofuran, affording the alkoxide,
which is then reacted
in situ with the proper Rz-X with X leaving group (halogen or mesylate or
tosylate) and R2 =
lower alkyl at a temperature of from 0°C to the reflux temperature. The
obtained compounds
(17) can undergo the same reactions described in scheme 1 affording product I
with R2 not H.
Alternatively, compounds of formula I where R2 is not a hydrogen atom, can be
obtained by allcylating compounds of formula I where RZ = H with the same
methods
~N~~NC?ED SHEET:
.' ...... ,
Scheme 6
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
22
described above for alkylating compound 16, limiting this procedure to the
allcylation
with very reactive halogenide or mesylate/tosylate (e.g. benzyl bromides)
which can react
under time/temperature controlled reaction condition, preferably at r.t.
Scheme 7 describes a double functionalization approach to the synthesis.of
Compound (I). This kind of approach can be useful for the synthesis of
libraries of
compounds (I) introducing different piperazine moieties and different R3
groups at the
same time.
Compounds of formula (I), where Ra represents -C(O)Alk, -C(O)OAIk, -
C(O)NR~RB or -C(S)NR~RB can be obtained by allcylation or addition reactions
staz-ting
from compounds of formula (I) where R2 = H. These lcinds of reactions can be
carried
out using proper acyl halides, allcyl chloroformates, isocyanates or
isothiocyanates in
methylene chloride, pyridine or DMF, optionally in the presence of a base such
as TEA or
NaH, or alternatively (e.g., for isothiocyanates) of an acid such as
trifluoroacetic acid, at a
temperature range of r.t. - 80°C.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
23
Scheme 7
_I~ I/,
I / / ~ / ~CHa --~ I / ,CH
CN CN
(19) (20) cHO (21)
.B
I / i CH~ I / CHO ~ I N
/ NJ
0 o R R HN~ B o o (24)
Ra Ra (22) a (23) (4) Ra Ra
TFA, / CHZCIZ Ha0 50% I /
CHO
CN
(26)
HN N-B
(4) ~--/
~N'B
N-B I / NJ
I / J cN (27)
CHO (25) I Ra
/ O
cN o.R (28)
a
/
-B
I / ~ CN
R3 off (1) (19)
In scheme 7 groups B and R are the same as groups A-R4, and (R+ Rl)
respectively, as given in the general formula I; Ra and R3 are the same as
given in the
general formula and Ra is a lower alkyl group or the two Ra groups are linked
forming a
1,3-dioxolanyl or 1,3-dioxanyl group.
A proper commercial benzyl derivative (with X = halogen or
methanesulphonyloxy or p-toluenesulphonyloxy groups) can be reacted, as well
known to
those skilled in the art, to afford the benzyl cyanide (19). These reactants
can be
converted following known alkylation methods into compounds (20) or (28)
respectively
reacting them with allyl halogenides (or allyl mesylates or tosylates) or
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
24
haloallcylaldehydes in their carbonyl protected form (acetals or dioxolanyl
derivatives or
other).
These alkylation reactions can be carried out by the use of bases to generate
the
reactive benzyl carbanions. Example of used bases are lithium diisopropylamide
(LDA)
or tert-butyl litluum or NaH or potassium tent-butoxide or sodium amide or
potassium
amide or others in a proper solvent such as THF or Et20 or DMF or other at a
temperature
ranging from -78°C to the reflux temperature. A preferred method of
alkylation includes
the use of hindered bases such as LDA in the presence of hexamethyl
phosphorous
triamide or DMPU at -78°C - r.t.
Compounds (20) can be in turn reduced by the use of diisobutylaluminum hydride
(DIBAL-H) in a proper solvent (toluene, DMF, CH2Cla or other) at a temperature
ranging
from -78°C to the reflux of the solvent. The so obtained aldehydes (21)
are then carbonyl
protected following methods very well known to those skilled in the art to
give
compounds (22), which can be catalytically osmilated (C. P. Forbes J.C.S.
Perlcin Trans I,
1979, 906-910) or undergo ozonolysis to afford compounds (23). Compounds (23)
can be
reductively aminated as described above to afford compounds (24). Deprotection
by
common methods leads to the aldehydes (25).
Compounds (25) can be alternatively obtained from compounds (20) applying the
osmilation or ozonolysis procedure on them. The cyanopropionaldehydes (26)
thus
obtained are then reductively aminated to compound (27). Repeating the DIBAL-H
reduction described above on these compounds affords the aldehydes (25).
Compounds (26) are also easily obtained from compounds (28) by simple
deprotection of the carbonyl functionality.
The reaction of R3-M (where M is a metallic salt, such as lithium or magnesium
halide) with compounds (25) afford compounds (I). A large number of
organometallics
such as lithium or magnesium derivatives are commercially available or easily
prepared
and can be reacted in a proper solvent such as THF or EtaO or others at -
78°C - reflux.
Ste~eocheyrtistr~
In Schemes l, 6 and 7, compounds I are obtained in syn/anti mixture of
diastereoisomers with ratio depending on the reaction condition used. The
diastereoisomers can be separated by usual techniques known to those skilled
in the art
including fractional crystallization of the bases or their salts or
chromatographic
techniques such as LC or flash chromatography. For both of the
diastereoisomers, the (+)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
enantiomer of formula Ia can be separated from the (-) enantiomer using
techniques and
procedures well known in the art, such as that described by J. Jacques, et
al.,
"Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981.
For
example, chiral chromatography with a suitable organic solvent, such as
ethanol/acetonitrile and Chiralpalc AD pacl~ing, 20 micron can also be
utilized to effect
separation of the enantiomers.
The free bases of formula I, their diastereoisomers or enantiomers can be
converted to the corresponding pharmaceutically acceptable salts under
standard
conditions well known in the art. For example, the free base of formula I is
dissolved in a
suitable organic solvent, such as methanol, treated with one equivalent of
malefic or oxalic
acid for example, one or two equivalents of hydrochloric acid or
methanesulphonic acid
for example, and then concentrated under vacuum to provide the corresponding
pharmaceutically acceptable salt. The residue can then be purified by
recrystallization
from a suitable organic solvent or organic solvent mixture, such as
methanol/diethyl
ether.
The N-oxides of compounds of formula I can be synthesized by simple oxidation
procedures well known to those slcilled in the art. The oxidation procedure
described by
P. Brougham et al. (Synthesis, 1015-1017, 1987), allows the two nitrogen of
the
piperazine ring to be differentiated, enabling both the N-oxides and N,N'-
dioxide to be
obtained.
Combination treatments
In certain embodiments, disorders of the urinary tract are treated by
administering
a compound of formula I in combination with an additional 5-HT1A antagonist or
an
antagonist of one or more additional class of receptors. In preferred
embodiments a
compound of formula I is administered in combination with an antagonist of an
a,l-
adrenergic, or muscarinic receptor.
In further embodiments, lower urinary tract disease is treated by
administering a
compound of formula I in combination with one or more inhibitor of the
cyclooxygenase
enzyme, which may inhibit both COXl and COX2 isozymes or which may,
alternatively,
be selective for COX2 isozyme, and NO donor derivatives thereof.
Examples of antimuscarinic drugs for administration in combination with a
compound of formula I are oxybutynin, tolterodine, darifenacin, and
temiverine.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
26
A compound of formula I may be administered in combination with ocl-adrenergic
antagonists, for the therapy of lower urinary tract symptoms, whether or not
these are
associated with BPH. Preferred al-adrenergic antagonists suitable for
administration in
combination with a compound of formula I are, fox example, prazosin,
doxazosin,
terazosin, alfuzosin, and tamsulosin. Additional al-adrenergic antagonists
suitable for
administration in combination with a compound of formula I are described in
U.S. Patents
No. 5,990,114; 6,306,861; 6,365,591; 6,387,909; and 6,403,594.
Examples of 5-HTIA antagonists that may be administered in combination with a
compound of formula I are found in Leopardi et al., J. Pha~macol. Exp. They.
299: 1027-
1037, 2001 (e.g., Rec 15/3079), U.S. Patent No. 6,071,920, other
phenylpiperazine
derivatives described in WO 99/06383 and pending U.S. Patent Applications
Serial No.
10/266,088 and 10/266,104 filed on October 7, 2002. Additional 5-HT1A
antagonists
include DU-125530 and related compounds described in U.S. Patent No. 5,462,942
and
robalzotan and related compounds described in WO 95/11891.
Examples of selective COX2 inhibitors that may be administered in combination
with a compound of formula I are, without limitation, nimesulide, meloxicam,
rofecoxib,
celecoxib, parecoxib and valdecoxib. Additional examples of selective COX2
inhibitors
are described, without limitation, in US 6,440,963. Examples of non-selective
CO~1-
COX2 inhibitors are, without limitation, acetylsalicylic acid, niflumic acid,
flufenamic
acid, enfenamic acid, meclofenamic acid, tolfenamic acid, thiaprophenic acid,
ibuprofen,
naproxen, lcetoprofen, flurbiprofen, furprofen, indomethacin, acemethacin,
proglumethacin, lcetorolac, diclofenac, etodolac, sulindac, fentiazac,
tenoxicam,
lornoxicam, cymioxicam, ibuproxam, nabumetone, tolmetin, amtolmetin.
Accordingly,
each of the foregoing are non-limiting examples of COX inhibitors that may be
administered in combination with a compound of formula I.
Examples of derivatives of COX inhibitors that may be administered in
combination with a compound of formula I are derivatives of COX inhibitors
bearing
nitrate (nitrooxy) or nitrite groups, such as those given , for example, in WO
98/09948,
able to release NO in vivo.
Pharmaceutical Compositions
The invention fiu~ther provides pharmaceutical compositions comprising a
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
27
compound of formula I or an enantiomer, diastereomer, N-piperazine oxide,
crystalline
form, hydrate, solvate, active metabolite or pharmaceutically acceptable salt
of the
compound. The pharmaceutical composition may also include optional additives,
such as
a pharmaceutically acceptable carrier or diluent, a flavouring, a sweetener, a
preservative,
a dye, a binder, a suspending agent, a dispersing agent, a colorant, a
disintegrator, an
excipient, a diluent, a lubricant, an absorption enhancer, a bactericide and
the like, a
stabiliser, a plasticizer, an edible oil, or any combination of two or more of
said additives.
Suitable pharmaceutically acceptable carriers or diluents include, but axe not
limited to, ethanol, water, glycerol, aloe vera gel, allantoin, glycerine,
vitamin-A and E
oils, mineral oil, phosphate buffered saline, PPG2 myristyl propionate,
magnesium
carbonate, potassium phosphate, vegetable oil, animal oil and sollcetal.
Suitable binders include, but are not limited to, starch, gelatine, natural
sugaxs
such as glucose, sucrose and lactose, corn sweeteners, natural and synthetic
gums such as
acacia, tragacanth, vegetable gum, sodium alginate, carboxymethylcellulose,
polyethylene
glycol, waxes and the lilce.
Suitable disintegrators include, but are not limited to, starch such as corn
starch,
methyl cellulose, agax, bentonite, xanthan gum and the lilce.
Suitable lubricants include, but are not limited to, sodium oleate, sodium
stearate,
magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the
lilce.
Suitable suspending agents include, but are not limited to, bentonite.
Suitable dispersing and suspending agents include, but are not limited to,
synthetic
and natural gums such as vegetable gum, tragacanth, acacia, alginate, dextran,
sodium
carboxymethylcellulose, methylcellulose, polyvinylpyrrolidone and gelatine.
Suitable edible oils include, but are not limited to, cottonseed oil, sesame
oil,
coconut oil and peanut oil.
Examples of additional additives include, but are not limited to, sorbitol,
talc,
stearic acid and dicalcium phosphate.
Unit Dosage Foy~ms
The pharmaceutical composition may be formulated as unit dosage forms, such as
tablets, pills, capsules, boluses, powders, granules, sterile parenteral
solutions, sterile
parenteral suspensions, sterile parenteral emulsions, elixirs, tinctures,
metered aerosol or
liquid sprays, drops, ampoules, autoinjector devices or suppositories. The
unit dosage
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
28
forns may be used for oral, parenteral, intranasal, sublingual or rectal
administration, or
for administration by inhalation or insufflation, transdermal patches, and a
lyophilized
composition. In general, any delivery of active ingredients that results in
systemic
availability of such ingredients can be used. Preferably the unit dosage form
is an oral
dosage form, most preferably a solid oral dosage; therefore the preferred
dosage forms are
tablets, pills and capsules. However, parenteral preparations are preferred
too.
Solid unit dosage forms may be prepared by mixing the active agents of the
present invention with a pharmaceutically acceptable carrier and any other
desired
additives as described above. The mixture is typically mixed until a
homogeneous
mixture of the active agents of the present invention is obtained and the
carrier and any
other desired additives are formed, i.e. the active agents are dispersed
evenly throughout
the composition. In this case, the composition can be formed as dry or moist
granules.
Dosage forms can be formulated as, for example, "immediate release" dosage
forms. "Immediate release" dosage forms are typically formulated as tablets
that release
at least 60%-90% of the active ingredient within 30-60 min when tested in a
drug
dissolution test, e.g., U.S. Pharmacopeia standard <711>. In a preferred
embodiment,
immediate dosage forms release at 75% of active ingredient within about 45
min.
Dosage forms can also be formulated as, for example, "controlled release"
dosage
forms. "Controlled," "sustained," "extended" or "time release" dosage forms
are
equivalent terms that describe the type of active agent delivery that occurs
when the
active agent is released from a delivery vehicle at an ascertainable and
manipulatable rate
over a period of time, which is generally on the order of minutes, hours or
days, typically
ranging from about sixty minutes to about 3 days, rather than being dispersed
immediately upon entry into the digestive tract or upon contact with gastric
fluid. A
controlled release rate can vary as a function of a multiplicity of factors.
Factors
influencing .the rate of delivery in controlled release include the particle
size,
composition, porosity, charge structure, and degree of hydration of the
delivery vehicle
and the active ingredient(s), the acidity of the environment (either internal
or external to
the delivery vehicle), and the solubility of the active agent in the
physiological
environment, i.e., the particular location along the digestive tract. Typical
parameters for
dissolution test of controlled release forms are found in U.S. Pharmacopeia
standard
<724>.
Dosage forms can also be formulated to deliver active agent in multiphasic
stages
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
29
whereby a first fraction of an active ingredient is released at a first rate
and at least a
second fractions of active ingredient is released at a second rate. In a
preferred
embodiment, a dosage form can be formulated to deliver active agent in a
biphasic
tnanner, comprising a first "immediate release phase", wherein a fraction of
active .
ingredient is delivered at a rate set forth above for immediate release dosage
forms, and a
second "controlled release phase," wherein the remainder of the active
ingredient is
released in a controlled release manner, as set forth above for controlled
release dosage
forms.
Tablets or pills can be coated or otherwise prepared so as to form a unit
dosage
form that has delayed and/or sustained action, such as controlled release and
delayed
release unit dosage forms. For example, the tablet or pill can comprise an
inner dosage
and an outer dosage component, the latter being in the form of a layer or
envelope over
the former. The two components can be separated by an enteric layer which
serves to
resist disintegration in the stomach and permits the inner component to pass
intact into the
duodenum or to be delayed in release.
Biodegradable polymers for controlling the release of the active agents
include,
but are not limited to, polylactic acid, polyepsilon caprolactone,
polyhydroxybutyric acid,
polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and
crosslinked or
a~nphipathic block copolymers of hydrogels.
For liquid dosage forms, the active substances or their physiologically
acceptable
salts are dissolved, suspended or emulsified, optionally with the usually
employed
substances such as solubilizexs, emulsifiers or other auxiliaries. Solvents
for the active
combinations and the corresponding physiologically acceptable salts can
include water,
physiological salt solutions or alcohols, e.g. ethanol, propanediol or
glycerol.
Additionally, sugar solutions such as glucose or mannitol solutions may be
used. A
mixture of the various solvents mentioned may be used in the present invention
too.
A transdermal dosage form is contemplated by the present invention too.
Transdermal forms may be'a diffusion transdermal system (transdermal patch)
using
either a fluid reservoir or a drug-in-adhesive matrix system. Other
transdermal dosage
forms include, but are not limited to, topical gels, lotions, ointments,
transmucosal
systems and devices, and iontophoretic (electrical diffusion) delivery
systems.
Transdermal dosage forms may be used for delayed release and sustained release
of the
active agents of the present invention.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
The pharmaceutical compositions and unit dosage forms of the present invention
for parenteral administration, and in particular by injection, typically
include a
pharmaceutically acceptable carrier, as described above. A preferred liquid
carrier is
vegetable oil. Injection may be, for example, intravenous, epidural,
intrathecal,
intramuscular, intraluminal, intratracheal or subcutaneous.
The active agents can also be administered in the form of liposome delivery
systems, such as small unilamellar vesicles, large unilamellar vesicles and
multilamellar
vesicles. Liposomes can be formed from a variety of phospholipids, such as
cholesterol,
steaxylamine or phosphatidylcholines.
The active agents of the present invention may also be coupled with soluble
polymers such as targetable drug carriers. Such polymers include, but are not
limited to,
polyvinylpyrrolidone, pyran copolymers, polyhydroxypropylmethacrylamidophenol,
polyhydroxyethylaspartamidophenol, and polyethylenoxypolylysine substituted
with
palmitoyl residues.
Adnzihist~ation
The pharmaceutical composition or unit dosage forms of the present invention
may be administered by a variety of routes, such as the oral and enteral,
intravenous,
intramuscular subcutaneous, transdermal, transmucosal (including rectal and
buccal) and
by inhalation routes. Oral or transdermal routes are preferred (e.g., solid or
liquid
formulations or skin patches, respectively).
The pharmaceutical composition or unit dosage forms comprising an effective
amount of the present invention may be administered to an animal, preferably a
human, in
need of treatment of neuromuscular dysfunction of the lower urinary tract
described by E.
J. McGuire in "Campbell's UROLOGY", 5~' Ed., 616-638, 1986, W.B. Saunders
Company, and patients affected by any physiological dysfunction related to
impairment
of 5-HTIA receptor function. Such dysfunctions include, without limitation,
central-
nervous-system disorders such as depression, anxiety, eating disorders, sexual
dysfunction, addiction and related problems.
As used herein, the term "effective amount" refers to an amount that results
in
measurable amelioration of at least one symptom or parameter of a specific
disorder. In a
preferred embodiment, the compound treats disorders of the urinary tract, such
as urinary
urgency, overactive bladder, increased urinary frequency, reduced urinary
compliance
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
31
(reduced bladder storage capacity), cystitis (including interstitial
cystitis), incontinence,
urine lealcage, enuresis, dysuria, urinary hesitancy and difficulty in
emptying the bladder,
or central nervous system disorders due to serotonergic dysfunction (such as
anxiety,
depression, hypertension, sleep/walce cycle disorders, feeding behaviour,
sexual function
and cognition disorders in mammals (particularly a human) associated to
stroke, injury,
dementia and due to neurological development, disorders from hyperactivity
related to an
attention deficit (ADHD), drug addiction, drug withdrawal, irritable bowel
syndrome.
The pharmaceutical composition or unit dosage form of the present invention
may
be administered according to a dosage and administration regimen defined by
routine
testing in the light of the guidelines given above in order to obtain optimal
activity while
minimising toxicity or side effects far a particular patient. However, such
fine tuning of
the therapeutic regimen is routine in the light of the guidelines given
herein.
The dosage of the active agents of the present invention may vary according to
a
variety of factors such as underlying disease conditions, the individual's
condition,
weight, sex and age, and the mode of administration. An effective amount for
treating a
disorder can easily be determined by empirical methods known to those of
ordinary skill
in the art, for example by establishing a matrix of dosages and frequencies of
admiiustration and comparing a group of experimental units or subjects at each
point in
the matrix. The exact amount to be administered to a patient will vary
depending on the
state and severity of the disorder and the physical condition of the patient.
A measurable
amelioration of any symptom or parameter can be determined by a person
slcilled in the
art or reported by the patient to the physician. It will be understood that
any clinically or
statistically significant attenuation or amelioration of any symptom or
parameter of
urinary tract disorders is within the scope of the invention. Clinically
significant
attenuation or amelioration means perceptible to the patient and/or to the
physician.
For example, a single patient may suffer from several symptoms of dysuria
simultaneously, such as, for example, urgency and excessive frequency of
urination or
both, and these may be reduced using the methods of the present invention. In
the case of
incontinence, any reduction in the frequency or volume of unwanted passage of
urine is
considered a beneficial effect of the present method of treatment.
The amount of the agent to be administered can range between about 0.01 and
about 25 mg/kg/day, preferably between about 0.1 and about 10 mg/lcg/day and
most
preferably between 0.2 and about 5 mg/lcglday. It will be understood that the
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
32
pharmaceutical formulations of the present invention need not necessarily
contain the
entire amount of the agent that is effective in treating the disorder, as such
effective
amounts can be reached by administration of a plurality of doses of such
pharmaceutical
formulations.
In a preferred embodiment of the present invention, the compounds are
formulated
in capsules or tablets, preferably containing 50 to 200 mg of the compounds of
the
invention, and are preferably administered to a patient at a total daily dose
of 50 to 400
mg, preferably 150 to 250 mg and most preferably about 200 mg, for relief of
urinary
incontinence and dysfunctions under treatment with 5-HT1A receptor ligand.
A pharmaceutical composition for parenteral administration contains from about
0.01 % to about 100% by weight of the active agents of the present invention,
based upon
100% weight of total pharmaceutical composition.
Generally, transdermal dosage forms contain from about 0.01 % to about 100% by
weight of the active agents versus 100% total weight of the dosage form.
The pharmaceutical composition or unit dosage form may be administered in a
single daily dose, or the total daily dosage may be administered in divided
doses. In
addition, co-achninistration or sequential administration of another compound
for the
treatment of the disorder may be desirable. For example, the compounds of the
invention
may be administered in combination with more antimuscarinic, al-adrenergic
antagonist,
5-HT1A receptor antagonist, or COX inhibitors or NO releasing derivatives
thereof, for
the therapy of lower urinary tract symptoms. Examples of antimuscarinics, ocl-
adrenergic
antagonists, 5-HT1A receptor antagonist, COX inhibitors and NO releasing
derivatives
thereof are set forth above, without limitation.
For combination treatment where the compounds are in separate dosage
formulations, the compounds can be administered concurrently, or each can be
administered at separate staggered times. For example, the compound of the
invention
may be administered in the morning and the antimuscarinic compound may be
administered in the evening, or vice versa. Additional compounds may be
administered at
specific intervals too. The order of administration will depend upon a variety
of factors
including age, weight, sex and medical condition of the patient; the severity
and aetiology
of the disorders to be treated, the route of administration, the renal and
hepatic function of
the patient, the treatment history of the patient, and the responsiveness of
the patient.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
33
Determination of the order of administration may be fine-tuned and such fine-
tuning is
routine in the light of the guidelines given herein.
Uses-Methods fog Tt eatment
Without wishing to be bound by theory, it is believed that administration of 5-
HT1A receptor antagonists prevents unwanted activity of the sacral reflex
and/or cortical
mechanisms that control micturition. Thus, it is contemplated that a wide
range of
neuromuscular dysfunctions of the lower urinary tract can be treated using the
compounds
of the present invention, including without limitation dysuria, incontinence
and enuresis
(overactive bladder). Dysuria includes urinary frequency, nocturia, urgency,
reduced
urin_ ary compliance (reduced bladder storage capacity), difficulty in
emptying the bladder,
i.e. a suboptimal volume of urine is expelled during micturition. Incontinence
syndromes
include stress incontinence, urgency incontinence and enuresis incontinence,
as well as
mixed forms of incontinence. Enuresis refers to the involuntary passage of
urine at night
or during sleep.
The compounds of the invention may also be useful for the treatment of central
nervous system disorders due to serotonergic dysfiulction.
The following examples represent typical syntheses of the compounds of formula
I as described generally above. These examples are illustrative only and are
not intended
to limit the invention in any way. The reagents and starting materials are
readily available
to one of ordinary skill in the art.
Example 1 1-[4-Cyclohexyl-3-(2-fluorophenyl)-4-methoxybutyl]-4-[2-(2,2,2-
trifluoroethoxy)-phenyl]-piperazine
1-Cyclohexyl-2-(2-fluorophenyl)ethanone (Compound la)
To a mixture of 36 ml of 2-fluorobenzylzinc chloride (0.5 M sol. in THF) and
0.008 g of dichlorobis(triphenylphosphine)-palladium (II) stirred at
0°C was added
dropwise via a syringe 2.14 ml of cyclohexanecarbonyl chloride. Afterwards,
the reaction
mixture was stirred at r.t, for 4 h, quenched with an aqueous saturated
solution of
ammonium chloride (25 ml), extracted with 20 ml of EtOAc, which was dried
(Na2SO4)
arid evaporated to dryness in vacuo affording 3.52 g of the title compound as
a crude,
which could be used in the following step without further purification.
1H-NMR (CDCl3, b): 1.10-2.05 (m, lOH), 2.47 (tt,lH), 3.77 (s, ZH), 6.97-7.32
(m, 4H)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
34
4-Cyclohexyl-4-oxo-3-(2-fluorophenyl)-butyraldehyde diethyl acetal (Compound
1b)
A solution of 5.02 g of compound la in 136 ml of toluene was heated at reflex
recovering 3 5 ml of toluene by distillation to remove water. Afterwards, 3.18
g of
potassium tert-butoxide was added and stirring at reflex was continued for 30
min.; the
reaction mixture was cooled to 80°G and 4.27 ml of 2-bromoacetaldehyde
diethyl acetal
was added. After 18 h at reflex, the reaction mixture was.cooled to r.t.,
quenched with an
aqueous saturated solution of ammonium chloride (30 ml), extracted with 30 ml
of
EtOAc, which was dried (Na2S04) and evaporated to dryness in vacuo giving a
dude
which was purified by flash chromatography (petroleum ether-EtOAc 92.5:7.5)
affording
2.97 g of the pure title product.
1H-NMR (CDCl3, ~: 1.00-2.10 (m, 17H), 2.20-2.52 (m, 2H), 3.30-3.72 (m, 4H),
4.25-
4.45 (m, 2H), 6.90-7.35 (m, 4H)
4-Cyclohexyl-3-(2-fluorophenyl)-4-hydroxybutyraldehyde diethyl acetal upper
TLC Rf
diastereoisomer (Compound lc)
4-Cyclohexyl-3-(2-fluorophenyl)-4-hydroxybutyraldehyde diethyl acetal lower
TLC Rf
diastereoisomer
To a solution of 0.84 g of the compound lb in 25 ml of MeOH stirred at
0°C was added
0.095 g of Na.BH4 and the mixture was stirred at r.t. for 5 h. The solvent was
evaporated
and the reaction crude was taken up with Ha0 (15 ml) and extracted with EtOAc
(2 x
l5ml).The organic layer was separated, washed with brine (2 x 15ml), dried
(Na~,S04) and
evaporated to dryness in vacuo to give a crude which was purified by flash
chromatography (petroleum ether-EtOAc gradient from 92:8 to 85:15) afforded
compound lc (upper Rf) (0.56 g, 63 %) and the corresponding compound with
lower Rf
(4.8%). TLC eluent petroleum ether-EtOAc 9:1.
lc: 1H-NMR (CDCl3, b>: 0.90-1.35 (m, 12H), 1.50-1.95 (m, 5H and OH), 2.00-2.10
(m,
2H), 3.25-3.75 (m, 6H), 4.25 (t, 1H), 6.95-7.30 (m, 3H), 7.40-7.55 (m, 1H)
4-Cyclohexyl-3-(2-fluorophenyl)-4-methoxybutyraldehyde diethyl acetal
(Compound 1 d)
To a solution of 0.514 g of compound lc in 2 ml of anhydrous DMF stirred at
r.t.
was added 0.091 g of 60% NaH. The reaction mixture was stirred at r.t. far 1
h, then
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
0.142 ml of methyl iodide was added and the resulting mixture was stirred at
r.t. for 2 h.
Afterwards, the reaction mixture was poured into water (30 ml), extracted with
2 x 20 ml
of EtOAc, which was washed, dried (NaaS04) and evaporated to dryness in vacuo
affording 0.50 g of the title compound as a crude, which could be used in the
following
step without further purification.
1H-NMR (CDCl3, ~: 0.90-1.40 (m, 12H), 1.50-1.90 (m, SH), 1.92-2.20 (m, 2H),
3.05 (t,
1H), 3.20 (s, 3H), 3.20-3.70 (m, SH), 4.05-4.18 (m, 1H), 6.90-7.20 (m, 3H),
7.40-7.55 (m,
1 H)
4-Cyclohexyl-3-(2-fluorophenyl)-4-methoxybutyraldehyde (Compound 1 e)
A mixture of 0.502 g of the compound ld, 3.5 ml of 50% aq. trifluoroacetic
acid
and 7 ml of CH2Cla was stirred for 2 h at r.t., and then diluted with 8 ml of
CHaCla. The
organic layer was separated, washed with brine (2 x l5ml), dried (Na2S04) and
evaporated to dryness in vacuo to afford a crude (0.365 g), used in the next
step without
further purification.
1H-NMR (CDCl3, d): 0.95-1.40 (m, 6H), 1.41-2.00 (m, SH), 2.65-2.95 (m, 2H),
3.05-3.15
(m, 1H), 3.35, 3.37(2s, 3H), 3.70-3.90 (m, 1H), 6.90-7.25 (m, 3H), 7.40-7.55
(m, 1H),
9.65 (s, 1 H)
1-~4-Cyclohexyl-3-(2-fluoro-phenyl)-4-methoxybutyl~-4-~2-(2,2,2-
trifluoroethoxy)-
phenyl~-piperazine
A mixture of 0.212 g of the compound le, 0.237 g of 1-[2-(2,2,2-
trifluoroethoxy)-
phenyl]-piperazineHCl, 0.24 g of sodium triacetoxyborohydride, 0.11 ml of AcOH
and
6rn1 of CH~Cla was stirred at r.t. for 1 h, kept overnight resting,
allcalinised with 20% aq.
Na2C03, The organic layer was separated, washed with brine (2 x 30m1), dried
(NaaS04)
and evaporated to dryness in vacuo the give a crude (0.46 g), which was
purified by flash
chromatography (petroleum ether - EtOAc 7:3) affording the title compound
(0.25 g;
62%).
1H-NMR (CDCl3, s): 0.95-1.30 (m, 6H), 1.55-2.50 (m, 9H), 2.45-270 (m, 4H),
3.00-3.20
(m, SH), 3.20-3.38 (m, 4H), 435 (q, 2H), 6.85-7.20 (m, 7H), 7.40-7.55 (m, 1H)
Example 2 1-(4-Fluoro-2-methoxyphenyl)-4-[4-oxo-3-(2-trifluoromethoxyphenyl)-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
36
pentyl]-piperazine
1-(2-Trifluoromethoxyphenyl)-propan-2-one (Compound 2a)
A solution of 1.9 g of 1-(2-trifluoromethoxy)-benzaldehyde, 4 ml of EtOH, 1.3
ml of 96%
2-nitroethane and 0.10 ml of n-butylamine was stirred at reflux for 18 h.
Afterwards, it
was diluted with HaO, extracted with EtOAc (2x30 ml), washed with Ha0 (2x30
ml),
brine, dried (NaaS04) ed evaporated in vacuo to afford 2.47 g of an orange
oil, which
was purified by flash chromatography (PE- EtOAc 100:5). Evaporation of the
collected
fractions yielded 1.60 g of 2-vitro-3-(2-trifluoromethoxyphenyl)-prop-2-eve as
a pale
yellow oil.
1H-NMR (CDCl3, ~: 2.35 (s, 3H), 7.30-7.55 (m, 4H), 8.10 (s,lH)
A mixture of 1.6 g of the above compound, 0.024 g of Fe(C104)3, 3.0 g of Fe, 6
ml of
H20 was heated at reflux and stirred for 7.5 h. After overnight resting at
r.t., was added
2.80 ml of 37% HCl , heating for 1 h. After cooling, the mixture was extracted
with
EtOAc (3x40 ml), which was dried (Na2SO4) ed evaporated in vacuo to give the
title
compound (g 1.28) as an orange oil.
1H-NMR (CDCl3, ~: 2.22 (s, 3H), 3.77 (s, 2H), 7.15-7.40 (m, 4H)
4-Oxo-3-(2-trifluoromethoxyphenyl)-pentanal diethyl acetal (Compound 2b)
To a suspension of 1.87 g of 60% NaH oil dispersion in 10 ml of anhydrous DMF
was
added dropwise during 6 min under a nitrogen stream, a solution of compound 2a
in 15
ml of DMF and the reaction mixture was stirred at r.t. for 3 h. After
overnight resting,
was added 0.447 g of 2-bromoacetaldehyde diethyl acetal ( 97 %) in 5 ml of
DMF; the
mixture was stirred at r.t. for 30', then at 80°C for 3 h. Afterwards,
the mixture was
diluted with H20 (250 ml), acidified with HCl 2N, extracted with Et2O (3x50
ml), washed
with Ha0 (40 ml), dried (NaZS04) and evaporated in vacuo, affording a crude
(brownish
oil), which was purified by flash chromatography (PE - EtOAc 100:2) to yield
1.44 g of
compound 2b as a yellowish oil.
1H-NMR (CDCl3, ~: 1.08-1.32 (m, 6H), 1.75-1.95 (m, 1H), 2.08 (s, 3H), 2.35-
2.60 (m,
1H), 3.20-3.80 (m, 4H), 4.20-4.40 (2H), 7.15-7.35 (4H)
4-Oxo-3-(2-trifluoromethoxyhenyl)-pentanal (Compound 2c)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
37
The title compound was obtained following the procedure described for Compound
le but
using as a starting material Compound 2b instead of Compound 1 d. After the
usual work-
up procedure, the title compound was obtained (99%) and used without further
purification in the next step.
1H-NMR (CDCl3, ~: 2.12 (s, 3H), 2.58 (dd, 1H), 3.40 (dd, 1H), 4.61 (dd, 1H),
7.11-7.40
(m, 4H), 9.75 (s, 1H)
1-(4-Fluoro-2-methoxyphenyl)-4-~4-Oxo-3-(2-trifluoromethoxyphenyl)-pentyl~-
piperazme
The title compound was obtained following the procedure described for the
Compound of
Exaanple l, but using as a starting material Compound 2c instead of compound
le and 1-
(4-fluoro-2-methoxyphenyl)-piperazine instead of 1-(2,2,2-
trifluoroethoxyphenyl)-
piperazine. Purification by flash chromatography (PE - EtOAc 7:3) yielded the
title
compound (60%). Oil.
1H-NMR (CDCl3, ~: 1.65-1.85 (m, 1H), 2.10 (s, 3H), 2.25-2.45 (m, 3H), 2.50-
2.70 (m,
4H), 2.85-3.10 (m, 4H), 3.82 (s, 3H), 4.15-4.31 (m, 1H), 6.50-6.68 (m, 2H),
6.78-6.90 (m,
1 H), 7.20-7.3 5 (m, 4H)
Example 3 1-(4-Fluoro-2-methoxyphenyl)-4- [4-hydroxy-3-(2-
trifluoromethoxyphenyl)-pentyl]-piperazine
The title compound was synthesised using the method described for compound 1 c
but
starting from the Compound of Example 2 instead of Compoundlb. After the usual
work-up procedure, the tile compound was isolated (93.1%) and characterized by
LC as a
mixture of diastereomers (RS;SR- RS, RS 78.8 :20.5). LC purity: 98.6%
1H-NMR (CDCl3, ~: 0.95; 1.07 (2d, 3H), 1.80-2.10 (m, 2H), 2.35-2.50 (m, 2H),
2.60-
2.85 (m, 4H), 2.92-3.18 (m, 4H), 3.18-3.35 (m, 1H), 3.80 (s, 3H), 4.00-4.20
(m, 1H),
4.60-6.10 (b, 1H), 6.50-6.70 (m, 2H), 6.78-6.95 (m, 1H), 7.15-7.35 (m, 3H),
7.60-7.75
(m, 1 H)
Example 4 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-oxo-3-(~-
trifluoromethoxyphenyl)-pentyl]-piperazine
The title compound was obtained following the procedure described for for the
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
38
Compound of Example 1, but using as a starting material Compound 2c instead of
Compound le and 1-(2,3-dihydro-1,4-benzodioxin-5-yl)-piperazine instead of 1-
(2,2,2-
trifluoroethoxyphenyl)-piperazine. Purification by flash chromatography (PE -
EtOAc
7:3) yielded the title compound (33%). Oil.
1H-NMR (CDCl3, d~: 1.65-1.85 (m, 1H), 2.10 (s, 3H), 2.25-2.40 (m, 3H), 2.50-
2.70 (m,
4H), 2.90-3.15 (m, 4H), 4.18-4.40 (m, 4H), 6.48-6.65 (m, 2H), 6.70-6.82 (m,
1H), 7.20-
7.3 5 (m, 4H)
Example 5 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- [4-hydroxy-3-(2-
trifluoromethoxyphenyl)-pentyl]-piperazine
The title compound was synthesised using the method described fox Compound 1 c
but
starting from the compound of Example 4 instead of Compoundlb. After the usual
work-
up procedure, the title compound was isolated (92.7%) and characterized by LC
as a
mixture of diastereomers (RS,SR- RS,RS 72.9 :19.4). LC Purity: 92.3
1H-NMR (CDCl3, ~: 0.95; 1.07 (2d, 3H), 1.80 2.15 (m, 2H), 2.30-2.50 (m, 2H),
2.60-
2.85 (m, 4H), 3.00-3.20 (m, 4H), 3.20-3.40 (m, 1H), 4.00-4.15 (m, 1H), 4.15-
4.40 (m,
4H), 4.60-6.20 (b, 1H), 6.45-6.65 (m, 2H), 6.65-6.85 (m, 1H), 7.15-7.30 (m,
3H), 7.60-
7.75 (m, 1 H)
Example 6 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- [4-hydroxy-3-(2-
trifluoromethoxyphenyl)-hexyl]-piperazine
2-Allyl-2-(2-trifluoromethoxyphenyl)acetonitrile (Compound 6a)
To 2.74 ml of a 2M solution of LDA in THF cooled at -78°C was added
dropwise 2-(2-
trifluoromethoxyphenyl)acetonitrile in 20 ml of THF; the mixture was stirred
at the same
temperature for 10 min.. Afterwards, was added a mixture of 0.474 ml of allyl
bromide
and 0.446 g of HMPTA and the reaction was stirred at -78°C for 2 h,
then it was brought
to r.t. by spontaneous heating. After overnight resting, it was quenched with
an aq.
saturated solution of NH4C1 and extracted with EtOAc. The combined extracts
were dried
(Na~S04) and evaporated to dryness. The crude was purified by flash
chromatography
(PE - EtOAc 95:5) affording the title product as a pale yellow oil (1.015 g).
1H-NMR (CDCl3, b>: 2.60 (t, 2H), 4.15-4.26 (m, 1H), 5.16-5.29 (m, 2H), 5.79-
5.91 (m,
1H), 7.28-7.41 (m, 3H), 7.51-7.67 (m, 1H).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
39
2-(2-Trifluoromethoxyphenyl)-pent-4-enal (Compound 6b)
To a solution.of 0.88 g of Compound 6a in anhydrous toluene (50 ml) was added
dropwise at r.t. 4.01 ml of DIBAL-H (2M sol. in toluene) over 10 min. The
reaction
mixture was stirred at r.t. for 2h, diluted with 0.01 N HCl , extracted with
EtOAc (2 x 50
ml); the combined extracts were washed with H20, dried (Na~S04) and evaporated
to
dryness in vacuo. Compound 6b was obtained as a pale yellow oil and used in
the next
step without further purif cation.
1H-NMR (CDCl3, ~: 2.49-2.68 (m, 1H), 2.80-2.95 (m, 1H), 4.02-4.15 (m, 1H),
5.12-5.26
(m, 2H), 5.75-5.85 (m, 1H), 7.28-7.37 (m, 4H), 9.75 (bs ,1H)
2-~1-~3-Butenyl-1-(2-trifluoromethoxyphen 1)~~-1,3-dioxolane (Compound 6c)
A solution of 0.72 g of Compound 6b, 0.052 g ofp-toluenesulphonic acid
monohydrate,
0.328 ml of ethylen glycol in 30 ml of toluene was stirred at reflux for 8h.
Afterwards, the
solvent was removed by evaporation in vacuo, diluted with EtOAc and aq.
NaHC03; the
organic layer was separated, dried on Na2S04 and evaporated to dryness in
vacuo. The
crude was purified by flash chromatography (PE - EtOAc 95:5) affording the
title product
as a pale yellow oil (0.85 g).
1H-NMR (CDCl3, ~: 2.39-2.51 (m, 1H), 2.52-2.79 (m, 1H), 3.46-3.57 (m, 1H)
,3.80-3.92
(m, 4H), 4.88-4.95 (m, 2H), 4.96-5.12 (m, 1H), 5.72-5.81 (m, 1H), 7.21-7.33
(m, 3H),
7.33-7-45 (m, 1H).
3-(1,3-Dioxolan-2-yl~-3-(2-trifluoromethoxyphenyl)~propionaldehyde (Compound
6d)
To a biphasic mixture of 0.31 g of Compound 6c, 10 ml of EtaO and 10 ml of H20
vigorously stirred was added 0.196 ml of Os04 followed by addition of 3.6 g of
NaT04 in
aliquots over a period of 20 min. After 6 h, the organic layer was separated,
the aqueous
layer was extracted with Et20. the combined organic layers were dried (Na2SO4)
and
evaporated to dryness in vacuo. The crude was purified by flash chromatography
( PE -
EtOAc 8) to afford 0.311 g of the title product.
1H-NMR (CDCl3, ~: 2.52-2.69 (m, 1H), 2.88-3.03 (m, l~, 3.81-3.93 (m, 4H), 3.94-
4.15
(m, 1H), 5.03-5.08 (m, 1H), 7.22-7.38 (m, 3H), 7.39-7-55 (m, 1H), 9.76 (bs,
1H).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
1-(2,3-Dihydro-1,4-benzodioxin-5-yl)-4-(3-(1,3-dioxolan-2-yl)-3-(2-
trifluoromethoxyphenyl)-propyl~-piperazine (Compound 6e)
The title compound was obtained following the procedure described for the
compound of
Example 1, but using as a starting material Compound 6d instead of Compound le
and 1-
(2,3-dihydro-1,4-benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-
trifluoroethoxyphenyl)-
piperazine. Purification by flash chromatography (PE - EtOAc l :l) yielded the
title
compound (61 %). Oil.
1H-NMR (CDCl3, ~: 1.80-2.01 (m, 1H), 2.02-2.44 (m, 3H), 2.45-2.71 (m, 4H),
2.92-3.07
(m 4H), 3 .41-3 .61 (m, 1 H), 3 .81-3 . 93 (m, 4H), 4.21-4.3 9 (m, 4H), 5.01-5
.05 (m, 1 H),
6.49-6.57 (m, 2H), 6.71-6.82 (m, 1H), 7.21-7.39 (m, 3H), 7.41-7-58 (m, 1H).
1-(2,3-Dihydro-1,4-benzodioxin-5-yl)-4-~3-formyl-3-(2-trifluoromethoxyphenyl)-
propyl~-
piperazine (Compound 6f)
A mixture of 0.12 g of Compound 6e, 0.005 g of 4-toluenesulphonic acid
monohydrate, 1
ml of Ha0 and 7 ml of dioxane was stirred at reflux for 24 h. Afterwards, the
solvent was
removed by evaporation in vacuo, the residue was diluted with EtOAc and aq.
NaOH; the
organic layer was separated, dried on Na2S04 and evaporated to dryness in
vacuo. The
crude, obtained as a pale yellow oil, was used in the next step without
further purification.
1H-NMR (CDCl3, ~: 1.79-1.99 (m, 1H), 2.25-2.49 (m, 3H), 2.50-2.71 (m, 4H),
2.91-3.12
(m 4H), 4.07-4.15 (m, 1H), 4.16-4.39 (m, 4H), 6.48-6.61 (m, 2H), 6.73-6.86 (m,
1H),
7.20-7.38 (m, 3H), 9.81 (bs, 1H).
4,4-Diethoxy-2-(2-trifluoromethoxyphenyl)-butyronitrile (Compound 6g)
The title compound was synthesised following the procedure reported for
Compound 6a
but using 2-bromoacetaldehyde diethyl acetal instead of allyl bromide. After
spontaneous
heating to r.t. during 2 hours, the reaction misture was refluxed for
additional 2 h. After
the usual worlc-up procedure, the crude was purified by flash chromatography
(PE -
EtOAc 95:5) affording the title product (47.7%) as a pale yellow oil.
1H-NMR (CDCl3, d~: 1.12-1.35 (m, 6H), 2.07-2.27 (m, 2H), 3.50-3.71. (m, 4H),
4.22-4.38
(m 1H), 4.65-4.71 (m, 1H), 7.19-7.48 (m, 3H), 7.49-7.63 (m, 1H).
4-Oxo-2-(2-trifluoromethoxy~henyl)-butyronitrile (Compound 6h)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
41
The title compound was obtained following the procedure described for Compound
le but
using as a starting material Compound 6g instead of Compound 1 d. After the
usual worlc-
up procedure the so obtained title compound was used without further
purification in the
next step.
1H-NMR (CDCl3, ~: 2.98-3.31 (m, 2H), 4.65-4.78 (m 1H), 4.65-4.71 (m, 1H), 7.22-
7.49
(m, 3H), 7.51-7.66 (m, 1H), 9.81 (bs, 1H).
1-~3-Cyano-3-(2-trifluoromethoxyphenyl)-propyl~-4- (2,3-dihydrobenzo-1,4-
dioxin-5-yl)-
piperazine (Compound 6i)
The title compound was obtained following the procedure described for the
compound of
Example 1 but using as a starting material Compound 6h instead of compound le
and 1-
(2,3-dihydro-1,4-benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-
trifluoroethoxyphenyl)-
piperazine. Purification by flash chromatography (PE - EtOAc 1:1) yielded the
title
compound (93 %). Oil. ,
1H-NMR (CDCl3, ~: 1.99-2.14 (m, 1H), 2.49-2.71 (m, 3H), 3.01-3.19 (m, 4H),
4.20-4.38
(m 4H), 4.40-4.55 (m, 1H), 6.49-6.65 (m, 3H), 6.72-6.88 (m, 1H), 7.24-7.41 (m,
3H),
7.52-7.68 (m, 1H).
1-(2,3-Dih dro-1,4-benzodioxin-5-yl)-4-~3-formyl-3-(2-trifluoromethoxyphenyl)-
propyl~-
piperazine (Compound 6fa
To a solution of 0.414 g of Compound 6i in anhydrous CH2Cla (50 ml) was added
dropwise at -78°C 1.2 ml of 1 M DIBAL-H in toluene. The reaction was
allowed to warm
up to and stirred overnight; afterwards, it was diluted with water, extracted
with CH~C12
(2 x 50 ml); the combined extracts were washed with H20, dried (NaaS04) and
evaporated to dryness in vacuo. Purification by flash chromatography (CHzCIz-
MeOH
95:5) yielded the title compound (0.23 g; 55.3%). Oil.
1-~5-(1,4-benzodioxinyl)~-4- f4-hydroxy-3-(2-trifluoromethoxyphenyl)-hexyl~-
piperazine
Into a solution of 0.1 g of Compound 6f in 10 ml of THF cooled at 0-5°C
was dropped a
1M solution of ethylmagnesium bromide in THF (0.888 ml). The reaction mixture
was
allowed to warm up to r.t. and stirred at the same temperature for 3 h.
Afterwards, it was
quenched with an aq, saturated solution of NH4C1, allcalinised and extracted
with EtOAc.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
42
The combined extracts were dried (NaZS04) and evaporated to dryness. The crude
was
purified by flash chromatography (CHaCl2- MeOH/NH3 97:3) affording the title
product
as a yellow glassy oil (84.4 %).
1H-NMR (CDCl3, ~: 0.86-0.99 (m, 3H), 1.21-1.35 (m, 2H), 1.36-1.65 (m, 1H),
1.66-1.89
(m, 1H) 1.90-2.21 (m, 2H), 2.25-2.95 (m, 6H), 2.96-3.27 (m 4H), 3.61-3.80 (m,
1H),
4.21-4.41 (m, 4H), 6.49-6.61 (m, 2H), 6.65-6.86 (m, 1H), 7.15-7.39 (m, 4H).
[M+H]+=481.6
Example 7 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-hydroxy-3-(2-
trifluoromethoxyphenyl)-hex-5-enyl]-piperazine
The title product was obtained by the same procedure described for the
compound of
Example 6 but using Compound 6f and vinylmagnesium bromide (1M in THF) instead
of
ethyl magnesium bromide in THF. The crude was purified by flash chromatography
(CH2Cl2- MeOH/NH3 95:5) affording the title product as a yellow glassy oil
(42.6 %).
1H-NMR (CDCl3, b]: 1.76-1.98 (m, 1H) 1.99-2.28 (m, 1H), 2.29-2.51 (m, 2H),
2.52-2.89
(m 4H), 2.89-3.25 (m, 6H), 4.20-4.43 (m, 4H), 4.65-5.31 (m, 3H), 5.61-5.70 (m,
1H),
6.49-6.62 (m, 2H), 6.70-6.89 (m, 1H), 7.15-7.42 (m, 4H).
[M+H]+=479.5
Example 8 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-hydroxy-5-methyl-3-(2-
trifluoromethoxyphenyl)-hexyl]-piperazine
The title product was obtained by the same procedure described for the
compound of
Example 6 but using Compound 6f and isopropylmagnesium chloride(2 M in THF)
° instead of ethylmagnesium bromide in THF. The crude was purified by
flash
chromatography (CH2Cla- MeOH/NH3 97:3) affording the title product as a yellow
glassy oil (30.9 %).
1H-NMR (CDCl3, ~: 0.78-0.98 (m, 6H) 1.15-1.45 (m, 2H), 1.71-1.91 (m, 2H), 1.92-
2.19
(m, 1H), 2.25-2.51 (m 2H), 2.52-2.95 (m, 4H), 3.01-3.29 (m, 5H), 3.51-3.72 (m,
1H),
4.19-4.40 (m, 4H), 6.47-6.63 (m, 2H), 6.67-6.87 (m, 1H), 7.15-7.41 (m, 4H).
[M+H]+=495.6
Example 9 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-methoxy-3-(2-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
43
trifluoromethoxyphenyl)-5-hexenyl]-piperazine
4,4-Diethoxy-2-(2-trifluoromethoxyphenyl)-butyraldehyde (Compound 9a)
The title compound was prepared following the procedure described for Compound
6f
(alternative method) but starting from Compound 6g instead of Compound 6i. The
crude
was purified by flash chromatography (CH2Cla MeOH 99:1) affording the title
product
as a yellow glassy oil (41.9 %).
1H-NMR (CDCl3, d~: 1.11-1.34 (m, 6H); 1.85-2.04 (m, 1H); 2.41-2.62 (m, 1H);
3.34-3.77
(m, 4H); 4.08-4.19 (m, 1H); 4.39-4.51 (m, 1H); 7.19-7.42 (m, 4H); 9.66 (s,
1H).
6,6-Diethoxy-4-(2-trifluoromethoxyphenyl)-hex-1-en-3-of (Compound 9b)
The title product was obtained by the same procedure described for the
compound of
Example 6 but using vinylmagnesium bromide (1M in THF) instead of ethyl
magnesium bromide in THF and starting from Compound 9a. The crude was purified
by
flash chromatography (PE - EtOAc 4:6). Yield: 63.1 %.
1H-NMR (CDCl3, ~: 1.04-1.32 (m, 6H); 1.93-2.09 (m, 1H); 2.01-2.39 (m, 1H);
2.51 (bs,
1H); 3.28-3.75 (m; SH); 4.18-4.36 (m, 2H); 5.01-5.22 (m, 2H); 5.67-5.87 (m,
1H); 7.14-
7.43 (m, 4H).
4-Methoxy-3-(2-trifluoromethoxyphenyl)-but-3-enal diethyl acetal (Compound 9c)
The title compound was synthesised as described for Compound ld using as a
starting
material Compound 9b instead of Compound 1 c.The crude was used in the next
step
without further purification.
'H-NMR (CDCl3, d>: 1.01-1.42 (m, 6H); 1.86-2.04 (m, 1H); 2.24-2.43 (m, 1H);
3.24 (s,
3H); 3.30-3.79 (m, 6H); 4.13-4.28 (m, 1H); 4.98-5.17 (m, 2H); 5.50-5.71 (m,
1H); 7.13-
7.29 (m, 3H); 7.31-7.48 (m, 1H).
4-Methoxy-3-(2-trifluoromethoxyphenyl)-but-3-enal (Compound 9d)
The title compound was obtainedlfollowing the procedure described for Compound
le but
using as a starting material Compound 9c instead of compound.ld. After the
usual work-
up procedure the so obtained title compound was used without further
purification in the
next step.
1H-NMR (CDCl3, d>: 2.58-2.76 (m, 1H); 2.81-3.06 (m, 1H); 3.24 (s, 3H); 3.59-
3.72 (m,
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
44
1H); 3.73-3.91 (m, 1H); 4.99-5.17 (m, 2H); 5.43-5.67 (m, 1H); 7.13-7.41 (m,
4H); 9.61-
9.69 (m, 1H).
1-~5-(2,3-Dihydro-1,4-benzodioxinyl)~-4-~4-methoxy-3-(2-
trifluoromethoxyphenyl)-5-
hexenyl~-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 9d instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
yield after flash chromatography (PE - acetone 6:4) was 34.5%.
1H-NMR (CDCl3, ~: 1.74-1.93 (m, 1H); 2.09-2.42 (m, 3H); 2.48-2.71 (m, 4H);
2.89-3.28
(m, 4H); 3.18-3.31 (m, 4H); 3.54-3.68 (m, 1H); 4.17-4.38 (m, 4H); 4.96-5.30
(m, 2H);
5.47-5.68 (m, 1H); 6.47-6.63 (m, 2H); 6.71-6.85 (m, 1H); 7.12-7.31 (m, 3H);
7.32-7.48
(rn, 1H).
Example 10 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-methoxy-3-phenyl)-
heptyl]-
piperazine
4-Oxo-3-phenylheptanal diethyl acetal (Compound l0a)
The title compound was prepared following the method described for Comp~und 2b
but
using 1-phenyl-2-pentanone instead of Compound 2a. The crude was purified by
flash
chromatography (EtOAc-PE 95:5). Yield: 59.2 %.
1H-NMR (CDCl3, ~: 0.78-0.88 (m, 3H), 1.10-1.31 (m, 8H), 1.42-1.72 (m, 2H),
2.38-2.50
(m, 3H), 3.31-3.90 (m, 4H), 4.18-4.35 (m, 1H), 7.05-7.42 (m, SH).
4-Hydroxy-3-phen lheptanal diethyl acetal (Compound lOb)
The title compound was obtained following the procedure described for Compound
1 c but
using as a starting material Compound l0a instead of Compound lb. After the
usual
worlc-up procedure, the crude was purified by flash chromatography (EtOAc-PE
2:8).
Yield: 73.3 %.
1H-NMR (CDCl3, b~: 0.70-0.82 (m, 3H), 0.90-1.48 (m, 12H), 2.10-2.57 (m, 2H),
3.32-
3.94 (m, 4H), 4.08-4.30 (m, 1H), 5.18-5.35 (m, 1H), 7.05-7.42 (m, SH).
4-Methox -3-phenylheptanal dieth 1 acetal (Compound lOc)
The title compound was obtained following the procedure described for Compound
ld,
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
but using as a starting material Compound 1 Ob instead of Compound 1 c. The
title product
was used in the next step without further purification.
1H-NMR (CDCl3, ~: 0.70-0.82 (m, 3H), 1.02-1.48 (m, 12H), 2.02-2.15 (m, 1H),
2.95-
3.01 (m, 1H), 3.20-3.80 (m, 7H), 4.15-4.35 (m, 1H), 7.05-7.42 (m, SH).
4-Methoxy-3-phenylheptanal (Compound lOd)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound lOc instead of compound lc. The
title product
was used in the next step without further purification.
1H-NMR (CDCl3, d): 0.70-1.59 (m, 7H), 2.81-2.95 (m, 2H), 3.22-3.61 (m, 5H),
7.05-7.42
(m, SH). 9.75 (s, 1H)
1- ~5-(2, 3-Dihydro-1,4-benzodioxinyl)~-4-~(4-methoxy-3-phenyl)-heptyl~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound lOd instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
yield after flash chromatography (EtOAc-PE-MeOH/NH3 1:1:0.2 to 8:2:0.2) was
39.5 %.
IH-NMR (CDCl3, b>: 0.78-0.91 (m, 3H), 1.20-1.48 (m, 4H),1.90-2.07 (m, 2H),
2.19-2.33
(m, 2H), 2.50-2.68 (m, 4H), 2.80-2.91 (m, 1H), 2.99-3.12 (m, 4H), 3.18-3.30
(m, 1H),
3.35 (s, 3H), 4.20-4.38 (m, 4H), 6.48-6.62 (m, 2H), 6.78 (s, 1H), 7.25-7.33
(m, SH).
Example 11 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-methoxy-3-phenyl)-
pentyl]-
piperazine
4-Oxo-3-phenylpentanal diethyl acetal (Compound 11 a)
The title compound was prepared using the method described for Compound 2b but
using
1-phenylacetone (commercially available) instead of Compound 2a. The crude was
used
in the next step without further purification. Yield: 93 %.
'H-NMR (CDCl3, s): 1.10-1.25 (m, 6H), 1.82-2.17 (m, 4H), 2.32-2.50 (m, 1H),
3.30-3.70
(m, 4H), 3.82 (t, 1H), 4.23-4.33 (m, 1H), 7.15-7.39 (m, SH).
4-Hydroxy-3-phenylpentanal diethyl acetal (Compound 1 lb)
The title compound was obtained following the procedure described for Compound
lc but
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
46
using as a starting material Compound l la instead of Compound lb. After the
usual
worlc-up procedure, the crude was used in the next step without further
purification.
Yield: 60 %.
1H-NMR (CDCl3, ~: 1.00-1.32 (m, 9H), 2.05-2.15 (m, 1H),1.89-2.17 (m, 2H), 2.68-
2.81
(m, 1H), 3.28-3.71 (m, 4H), 3.82-4.02 (m, 1H), 4.15-4.26 (m, 1H), 7.12-7.41
(m, SH).
4-Methoxy-3-phenylpentanal diethyl acetal (Compound 11 c)
The title compound was obtained following the procedure described for Compound
1 d,
but using as a starting material Compound 11b instead of compound lc. The
crude was
purified by flash chromatography (EtOAc 5-PE 95).
4-Methoxy-3-phenylpentanal (Compound l ld)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compoundl 1 c instead of compound 1 d. The
title product
was used in the next step~without further purification.
1-~5-(2,3-Dihydro-1,4-benzodioxinyl)~-4-~(4-methoxy-3-phenyl)-pentyl~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 9d instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
yield after flash chromatography (EtOAc -PE - MeOH/NH3 8:2:0.1 to 8:2:0.3 was
15.5%.
1H-NMR (CDCl3, d>: 1.04 (d, 3H), 1.88-2.05 (m, 2IT),2.18-2.31 (m, 2H), 2.50-
2.68 (m,
4H), 2.73-2.85 (m, 1H), 2.97-3.12 (m, 4H), 3.30 (s, 3H), 3.42-3.50 (m, 1H),
4.18-4.38 (m,
4H), 6.48-6.62 (m, 2H), 6.71-6.82 (t, 1H), 7.15-7.33 (m, SH).
Example 12 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-propoxy-3-phenyl)-
heptyl]-
piperazine
4-Propoxy-3-phenylheptanal (Compound 12a)
The title compound was obtained following the procedure described for Compound
1 d,
but using as a starting material Compound 1 Ob instead of compound 1 c. The
crude was
purified by flash chromatography (EtOAc 5-PE 95).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
47
4-Propoxy-3-phenylheptanal (Compound 12b)
The title compound was obtained following the procedure described for Compound
1 d,
but using as a starting material Compound 12a instead of compound 1 c. The
crude was
used in the next step without further purification.
1-~5-(2,3-Dihydro-1,4-benzodioxinyl)~-4-((4-methoxy-3-phen 1)-heptyl~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 12b instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
yield after flash chromatography (EtOAc-PE-MeOHlNH3 4:6:0.1 to EtOAc-MeOH/NH3
97:3 was 9.5%.
IH-NMR (CDCl3, ~: 0.72-0.92 (m, 6H), 1.15-1.61 (m, 6H), 1.89-2.08 (m, 2H),
2.18-2.31
(m, 2H), 2.50-2.68 (m, 4H), 2.78-2.92 (m, 1H), 2.97-3.12 (m, 4H), 3.28-3.43
(m, 3H),
4.18-4.38 (m, 4H), 6.48-6.62 (m, 2H), 6.71-6.82 (t, 1H), 7.15-7.33 (m, SH).
Example 13 1-[3-(Z-Cyanophenyl)-4-cyclohexyl-4-oxobutyl]-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
2-(2-Cyclohexyl-2-oxoethyl)-benzonitrile (Compound 13a)
To a solution of 0.47 g of 2-tolunitrile in 4 ml of THF was added 0.535 ml of
1,3-
dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone(DMPU) and the mixture was
cooled at
-78°C; 2.22 ml of a 2M sol. of LDA in THF was dropped during 5 min.,
then the reaction
mixture was stirred at the same temperature for 15 min. followed by dropwise
addition of
0.757 g of N-methyl-N-methoxycyclohexanecarboxamide in 4 ml of THF. After 1 h
stirring at -78°C, the reaction mixture was quenched with a 10% aq.
sol. of NH4Cl. The
temperature was allowed to raise at r.t. and the mixture was extracted with
EtOAc
(2x20m1), washed with 30 ml of brine, dried on NaaSO~ and evaporated to
dryness in
vacuo. The crude was purified by flash chromatograophy (PE - EtOAc 85:15 to
1:1) to
afford 0.34 g of the title compound.
1H-NMR (CDCl3, ~: 1.10-2.05 (m,IOH); 2.45-2.602 (m,lH); 4,00 (m,2H); 7.20-7.43
(m,2H); 7.48-7.70 (m,2H);
3-(2-Cyanophenyl)-4-cyclohexyl-4-oxobutyraldehyde diethyl acetate (Compound
13b)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
48
The title compound was prepared using the method described for Compound 2b but
using
Compound 13 a instead of Compound 2a. The crude was purified by flash
chromatography (toluene-EtOAc 97:3). Yield: 39.1%.
1H-NMR (CDCl3, a): 1.05-1.90 (m,lSH); 1.90-2.05 (m,2H); 2.32-2.60 (m,2H); 3.20-
3.70
(m,4H); 4.30 (t,lH); 4.55 (t,lH); 7.30-7.45 (m,2H); 7.55 (dd,lH); 7.68 (dd,lH)
3-(2-Cyanophenyl)-4-cyclohexyl-4-oxobutyraldehyde (Compound 13c)
The title compound was obtained following the procedure described for Compound
1 d,
but using as a starting material Compound 13b instead of compound lc. The
crude was
used in the next step without further purification.
1H-NMR (CDCl3, ~: 1.00-1.90 (m,lOH); 2.05-2.15 (m,lH);2.35-2.50 (m,lH); 2.70
(dd, l H); 3 .45 (dd, l H); 4. 8 5 (dd, l H); 7.25 (dd, l H); 7.3 0-7.40 (m, l
H); 7. 50-7.60 (m, l H);
7.75 (dd, l H)
1-(3-(2-Cyanophenyl)-4-cyclohexyl-4-oxobutyll-4-~5-(2,3-dihydro-1,4-
benzodioxinyl)~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 13c instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :EtOAc 6:4) to afford the title
compound
(79.5%).
1H-NMR (CDCl3, ~: 1.10-2.10 (m,llH); 2.20-2.50 (m, 4H); 2.50-2.75 (m,4H); 2.92-
3.20
(m,4H); 4.20-4.38 (m,4H); 4.55 (t,lH); 6.48-6.65 (m,2H); 6.70-6.85 (m,lH);
7.30-7.45
(m,2H); 7.45-7.60 (m,lH); 7.65-7.75 (m,lH)
Example 14 (RS,SR)-1-[3-(2-Cyanophenyl)-4-cyclohexyl-4-hydroxybutyl]-4-[5-(2,3-
dihydro-1,4-benzodioxinyl)]-piperazine
The title compound was synthesised using the method described for compound 1 c
but
starting from the Compound of Example 14 instead of Compoundlb. After the
usual
worlc-up procedure, the crude was purified by flash chromatography (PE - EtOAc
-
NH3/MeOH 65:35:3) affording the title compound (70.5%).
1H-NMR (CDCl3, d~: 0.80-1.40 (m,7H); 1.45-1.80 (m,SH);1.85-2.05 (m,lH); 2.20-
2.50
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
49
(m,2H); 2.50-2.80 (m,4H); 2.95-3.20 (m,4H); 3.30-3.50 (m,lH); 3.50-3.65
(m,lH); 4.20-
4.40 (m,4H); 4.40-5.90 (bs,lH); 6.50-6.67 (m,2H);6.70-6.85 (m,lH); 7.20-7.40
(m,lH);
7.50-7.68 (m,2H); 7.93-8.08 (m,lH)
Example 14a (RS)-1-f3-(2-Cyanophenyl)-4-cyclohexyl-4-hydroxybutyl~-4-~5-(2,3-
dihydro-1,4-benzodioxinyl)~-piperazine (enantiomer at Rt = 30.298 min.)
This compound was obtained from the Compound of Example 14 resolving by chiral
column chromatography using Chiralpak AD (0.46x25 cm), eluting with n-hexane-
EtOH
95:5 (flow = 1 ml/min; detector LTV 254 nm).
Example 14b (SR)-1-~3-(2-Cyanophenyl)-4-cyclohexyl-4-hyclioxybutyl~-4-~5-(2,3-
dihydro-1,4-benzodioxinyl)~-piperazine (enantiomer at Rt = 34.834 min)
This compound was obtained from the Compound of Example 14 resolving by chiral
column chromatography using Chiralpak AD (0.46x25 cm), eluting with n-hexane-
EtOH
95:5 (flow = 1 ml/min; detector LTV 254 nm).
Example 15 1-[3-(2-Cyanophenyl)-4-cyclohexyl-4-oxobutyl]-4-(4-fluoro-2-
methoxyphenyl)-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 13c instead of Compound le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :EtOAc 6:4) to afFord the title
compound
(82 %).
IH-NMR (CDCl3) 8:1.00-2.10 (m,llH); 2.15-2.75 (m,BH); 2.75-3.15 (m,4H); 3.90
(s,3H); 4.55 (t,lH); 6.50-6.70 (m,2H); 6.80-6.95 (m,lH); 7.30-7.42 (m,2H);
7.45-7.60
(m, l H); 7.60-7.72 (m, l H)
Example 16 1-[3-(2-Cyanophenyl)-4-cyclohexyl-4-hydroxybutyl]-4-(4-fluoro-2-
methoxyphenyl)-piperazine
The tilde compound was synthesised using the method described for Compound 1 c
but
starting from the Compound of Example 14 instead of Compoundlb. After the
usual
worlc-up procedure, the crude was purified by flash chromatography (PE - EtOAc
-
NH3/MeOH 65:35:3) affording the title compound (51.3 %).
1H-NMR (CDCl3, b>: 0.90-1.20 (m,6H); 1.40-1.85 (m,SH);1.90-2.10 (m,2H); 2.20-
2.45
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
(m,2H); 2.50-2.85 (m,4H); 2.90-3.15 (m,4H); 3.32-3.50 (m,lH) 3.50-3.65 (m,lH);
3.95
(s,3H); 4.60-5.20 (bs,lH); 6.55-6.68 (m,2H); 6.80-6.92 (m,lH); 7.28-7.36
(m,lH); 7.45-
7.68 (m,2H);7.95-8.05 (m,lH)
Example 17 1-(4-cyclohexyl-4-methoxy-3-phenylbutyl)-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
4,4-Dimethoxy-2-phenylbutyronitrile (Compound 17 a)
The title compound was synthesised following the procedure reported for
Compound 6b
but using 2-bromoacetaldehyde dimethyl acetal instead of allyl bromide and
phenylacetonitrile instead of 2-trifluoromethoxyphenylacetonitrile. After
spontaneous
heating to r.t. during 2 hours, the reaction mixture was refluxed for
additional 2 h. After
the usual work-up procedure, the crude was purified by flash chromatography
(PE-EtOAc
9:1 ) affording the title product (72.1 %) as a pale yellow oil.
1H-NMR (CDCl3, ~: 2.02-2.36 (m, 2H); 3.39 (d, 6H); 3.76-4.01 (m, 1H); 4.41-
4.54 (m,
1H); 7.30-7.48 (m, 5H).
4,4-Dimethoxy-2-phenylbutyraldehyde(Compound 17b)
The title compound was obtained following the procedure described for Compound
6f
(alternative method) but using as a starting material Compound 17a instead of
compound
6i. After the usual work-up procedure the crude was purified by flash
chromatography
(CH2C12 - EtOAc 95:5 ) to afford the title compound (73.2 %).
1H-NMR (CDCl3, ~: 1.83-2.02 (m, 1H); 2.39-2.58 (m, 1H); 3.32 (d, 6H); 3.66-
3.81 (m,
1H); 4.23-4.38 (m, 1H); 7.07-7.48 (m, 5H); 9.61-9.70 (m, 1H).
4-Cyclohexyl-4-hydroxy-3-phenylbutyraldehyde dimethyl acetal (Compound 17c)
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 17b instead of compound 6f
and
cyclohexylmagnesium.chloride (2M sol. in THF) instead of ethylmagnesium
chloride.
The crude was purified by flash chromatography (CHaCl2 - Acetone 9:1)
affording the
title product. (55 %).
1H-NMR (CDCl3, ~: 0.97-2.09 (m, 12H); 2.27-2.45 (m, 1H); 2.88-3.03 (m, 1H);
3.31 (d,
6H); 3.41-3.53 (m, 1H); 4.11-4.22 (m, 1H); 7.21-7.43 (m, 5H).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
51
The OH signal was not detectable.
4-Cyclohexyl-4-methoxy-3-phenylbutyraldehyde dimethyl acetal (Compound 17d)
The title compound was synthesised as described for Compound ld using as a
starting
material Compound 17c instead of Compound 1 c. After EZO extraction, the crude
was
purified by flash chromatography (PE - EtOAc 8:2) affording the title product.
(71.4 %).
1H-NMR (CDCl3, ~: 0.98-1.37 (m, 6H); 1.49-1.98 8m, 6H); 2.17-2.33 (m, 1H);
2.82-3.02
(m, 2H); 3.21 (dd, 6H); 3.37 (s, 3H); 3.94-4.08 (m, 1H); 7.17-7.39 (m, SH).
4-Cyclohexyl-4-methoxy -3-phenylbutyraldehyde (Compound 17e)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 17d instead of compound 1 d. The
title product
was used in the next step without further purification.
1H-NMR (CDCl3, ~: 0.93-1.86 (m, 12H); 2.61-2.79 (m, 2H); 3.01-3.16 (m, 1H);
3.31 (s,
3H); 3.41-3.59 (m, 1H); 7.15-7.39 (m, SH); 9.53-9.61 (m, 1H).
1-(4-Cyclohexyl-4-methoxy-3-phenylbutyl)-4-~5-(2,3-dihydro-1,4-benzodioxinyl)~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 17e instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :MeaCO 75:25) to afford the
title
compound (77.4 %).
1H-NMR (CDCl3, ~: 0.97-1.31 (m, 6H); 1.48-1.99 (m, 6H); 2.07-2.28 (m, 3H);
2.42-2.67
(m, 4H); 2.71-2.90 (m, 1H); 2.92-3.26 (m, SH); 3.3 (s, 3H); 4.17-4.38 (m, 4H);
6.45-6.64
(m, 2H); 6.66-6.84 (m, 1H); 7.12-7.34 (m, SH).
Example 18 1-(4-Cyclohexyl-4-methoxy-3-phenylbutyl)-4-(4-fluoro-2-
methoxyphenylj-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 17e instead of Compound le and 1-(4-fluoro-2-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
52
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2C0 75:25) to afford the
title
compound (79.8 %).
1H-NMR (CDCl3, b): 0.97-1.37 (m, 6H); 1.46-1.98 (m, 6H); 2.07-2.31 (m, 3H);
2.42-2.71
(m, 4H); 2.74-2.80 (m, 1H); 2.81-3.18 (m, SH); 3.39 (s, 3H); 3.81 (s, 3H);
6.49-6.68 (m,
2H); 6.77-6.92 (m, 1H); 7.13-7.38 (m, SH).
Example 19 1-(4-Cyclohexyl-4-ethoxy-3-phenylbutyl)-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
4-Cyclohexyl-4-ethoxy-3-phenylbutyraldehyde dimethyl acetal (Compound 19a)
The title compound was synthesised as described for Compound 1 d using as a
starting
material Compound 17c instead of Compound 1 c and ethyl iodide instead of
methyl
iodide. After E20 extraction, the crude was purified by flash chromatography
(PE -
EtOAc 8:2) affording the title product. (50.7 %).
1H-NMR (CDCl3, ~: 0.91-1.34 (m, 9H); 1.42-1.99 (m, 6H); 2.14-2.34 (m, 1H);
2.80-2.94
(m, 1 H); 3.00-3.11 (m, 1 H); 3.22 (d, 6H); 3.41-3.57 (m, 2H); 3.92-4.08 (m, 1
H); 7.14-
7.35 (m, SH).
4-Cyclohexyl-4-ethoxy-3-phenylbutyraldehyde (Compound 19b)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 19a instead of Compound 1 d. The
title product
was used in the next step without further purification.
1H-NMR (CDCl3, b~: 0.91-1.39 (m, 8H); 1.48-1.88 (m, 6H); 2.57-2.89 (m, 2H);
3.08-3-20
(m, 1H); 3.23-3.40 (m, 1H); 3.41-3.61 (m, 2H); 7.13-7.38 (m, SH); 9.57-9.66
(m, 1H).
1-(4-Cyclohexyl-4-ethoxy-3-phenylbutyl)-4-~5-(2,3-dihydro-1,4-benzodioxinyl)~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 19b instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2CO 8:2) to afford the title
compound (60.6 %).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
53
1H-NMR (CDCl3, ~: 0.97-1.32 (m, 9H); 1.42-1.99 (m, 6H); 2.04-2.36 (m, 3H);
2.46-2.69
(m, 4H); 2.71-2.90 (m, 1H); 2.94-3.21 (m, SH); 3.26-3.61 (m, 2H); 4.17-4.39
(m, 4H);
6.48-6.74 (m, 2H); 6.68-6.83 (m, 1H); 7.14-7.37 (m, SH).
Example 20 1-(4-Cyclohexyl-4-ethoxy-3-phenylbutyl)-4-(4-fluoro-2-
methoxyphenyl)-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 19b instead of Compound le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2C0 75:25) to afford the
title
compound (73 %).
1H-NMR (CDCl,3, ~: 0.96-1.40 (m, 9H); 1.44-1.99 (m, 6H); 2.05-2.34 (m, 3H);
2.42-
2.69 (m, 4H); 2.74-2.90 (m, 1H); 2.92-3.16 (m, SH); 3.21-3.60 (m, 2H); 4.83
(s, 3H);
6.52-6.68 (m,2H); 6.78-6.93 (m, 1H); 7.12-7.36 (m, SH).
Example 21 1-(4-Allyloxy-4-cyclohexyl-3-phenylbutyl)-4- [5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
4-Allyloxy-4-cyclohexyl-3-phenylbutyraldehyde dimethyl acetal (Compound 21a)
The title compound was synthesised as described for Compound 1 d using as a
starting
material Compound 17c instead of Compound 1 c and allyl bromide instead of
methyl
iodide. The reaction mixture was stirred for 8 h at r.t. and 5 h at
45°C. EtaO extraction
and purification by column chromatography (PE - EtOAc 85:15) yielded the title
compound (48.5 %).
1H-NMR (CDCl3, ~: 0.94-1.38 (m, 6H); 1.51-2.01 (m, 6H); 2.16-2.34 (m, 1H);
2.82-3.01
(m, 1H;) 3.07-3.19 (m, 1H); 3.21 (d, 6H); 3.72-3.88 (m, 1H); 3.90-4.07 (m,
2H); 5.04-
5.32 (m, 2H); 5.77-6.00 (m, 1H); 7.16-7.37 (m, SH).
4-Allyloxy-4-cyclohexyl-3-phenylbutyraldehyde (Compound 21b)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 21 a instead of compound 1 d. The
title product
was used in the next step without further purification (99.3%).
1H-NMR (CDCl3, ~: 0.93-1.41 (m, 6H); 1.47-2.01 (m, 6H); 2.62-2.91 (m, 1H);
3.14-3.29
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
54
(m, 1H); 3.41-3.60 (m, 1H); 3.71-4.03 (m, 2H); 5.01-5.32 (m, 2H); 5.73-5.98 (m
,1H);
7.08-7.41 (m, SH); 9.56-9.69 (rn,lH).
1-(4-Allylox -4-cyclohexyl-3-phenylbutyl)-4- [5-(2,~-dihydro-1,4-
benzodioxinyl)]-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 21b instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :MeaCO 7:3) to afford the title
compound (64.1 %).
1H-NMR (CDCl3, b~: 0.98-1.40 (m, 6H); 1.48-1.98 (m, 6H); 1.88-2.00 (m, 2H);
2.08-
2.31 (m, 3H); 2.39-2.71 (m, 4H); 2.2.78-2.94 (m, 1H); 2.96-3.21 (m, SH); 3.72-
4.06 (m,
2H); 4.68-4.87 (m, 4H); 5.05-5.34 (m, 2H); 5.81-6.02 (m, 1H); 6.47-6.63 (m,
2H); 6.80-
6.88 (m, 1H); 7.11-7.37 (m, SH).
[M+H]+= 491
Example 22 1-(4-Allyloxy-4-cyclohexyl-3-phenylbutyl)-4-(4-fluoro-2-
methoxyphenyl)-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 21b instead of Compound le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2C0 7:3) to afford the title
compound (77.1 %).
1H-NMR (CDCl3, b~: 0.98-1.41 (m, 6H); 1.47-2.01 (m, 6H); 2.09-2.28 (m, 3H);
2.41-2.70
(m, 4H); 2.79-2.92 (m, 1H); 2.93-3.09 (m, 4H); 3.11-3.22 (m, 1H); 3.77-3.89
(m, 4H);
3.39-4.08 (m, 1H); 5.07-5.34 (m, 2H); 5.79-6.01(m, 1H); 6.51-6.68 (m, 2H);
6.69-6.92
(m, 1H); 7.13-7.37 (m, SH).
[M+H]~= 481
Example 23 1-(4-Cyclohexyl-3-phenyl-4-propargyloxybutyl)-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
4-Cyclohexyl-3-phenyl-4-propargyloxybutyraldehyde dimethyl acetal (Compound
23a)
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
The title compound was synthesised as described for Compound 1 d using as a
starting
material Compound 17c instead of Compound 1 c and propargyl bromide instead of
methyl iodide. The reaction mixture was stirred for 8 h at r.t. and 5 h at
45°C. Et20
extraction and purification by column chromatography (PE - EtOAc 85:15)
yielded the
title compound (50 %).
1H-NMR (CDCl3, ~: 0.90-1.41 (m, 6H); 1.48-1.74 (m, SH); 1.75-1.89 (m, 1H);
1.90-2.04
(m, 1H); 2.18-2.37 (m, 1H); 2.38-2.44 (m, 1H); 2.88-3.04 (m, 1H); 3.21 (d,
6H); 3.90-
4.17 (m, 3H); 7.12-7.37 (m, SH).
4-Cyclohexyl-3-phenyl-4-propargyloxy -3-phenylbutyraldehyde (Compound 23b)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 23a instead of Compound ld. The
title product
was used in the next step without further purification (99 %).
1H-NMR (CDCl3, ~: 0.81-1.41 (m, 6H); 1.49-1.90 (m, SH); 2.39-2.51 (m, 1H);
2.66-2.88
(m, 1H); 2.89-3.08 (m, 1H); 3.31-3.42 (m, 1H); 3.43-6.59 (m, 1H); 3.97-4.19
(m, 2H);
7.12-7.39 (m, SH); 9.57-9.69 (m, 1H).
1-(4-Cyclohexyl-3-phenyl-4-propargyloxybutyl)-4-r5-(2,3-dihydro-1,4-
benzodioxinyl)~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 23b instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2CO 7:3) to afford the title
compound (67.1 %).
1H-NMR (CDCl3, b): 0.98-1.39 (m, 6H); 1.48-1.99 (m, 6H); 2.11-2.29 (m, 3H);
2.39-2.46
(m, 1H); 2.47-2.71 (m, 4H); 2.82-3.96 (m, 1H); 2.97-3.12 (m 4H); 3.17-3.29 (m,
1H);
3.95-4.16 (m, 2H); 4.17-4.38 (m, 4H); 6.48-6.72 (m, 2H); 6.69-6.83 (m, 1H);
7.12-7.35
(m; SH)
[M+H]+=489
Example 24 1-(4-Cycloheayl-3-phenyl-4-propargyloxybutyl)-4-(4-fluoro-2-
methoxyphenyl)-piperazine
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
56
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 23b instead of Compou~id le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2C0 8:2) to afford the title
compound (66.2 %).
1H-NMR (CDCZ3, ~: 0.99-1.41 (m, 6H); 1.51-2.00 (m, 6H); 2.11-2.29 (m, 3H);
2.39-2.46
(m, 1H); 2.47-2.70 (m, 4H); 2.78-3.12 (rn, 5H); 3.13-3.29 (m, 1H); 3.81 (s,
3H); 3.96-
4.17 (m, 2H); 6.51-6.67 (m, 2H); 6.79-7.94 (m, 1H); 7.11-7.34 (m, 5H).
[M+H]+=479
Example 25 1-(4-Cyclohexyl-3-phenyl-4-propoxybutyl)-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
4-Cyclohexyl-3-phenyl-4-propoxybutyraldehyde dimethyl acetal (Compound 25a)
The title compound was synthesised as described for Compound 1 d using as a
starting
material Compound 17c instead of Compound lc and propyl bromide instead of
methyl
iodide. The reaction mixture was stirred for 8 h at r.t. and 5 h at
45°C. EtaO extraction
and purification by column chromatography (PE - EtOAc 85:15) yielded the title
compound (32.7 %).
1H-NMR (CDCl3, ~: 0.91 (t, 3H); 0.99-1.32 (m, 6H); 1.45-1.98 (m, lOH); 2.19-
2.38 (m,
1H); 2.83-2.99 (m, 1H); 3.01-3.10 (m, 1H); 3.16-3.29 (m, 5H); 3.31-3.5 (m,
1H); 3.91-
4.08 (m, 1H); 7.13-7.34 (m, 5H).
4-Cyclohexyl-3-phenyl-4-propoxybutyraldehyde (Compound 25b)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 25a instead of compound 1 d. The
title product
was used in the next step without further purification (99.3%).
IH-NMR (CDCl3, b]: 0.80-0.95 (m, 3H); 0.96-1.37 (m, 6H); 1.41-1.88 (m, 7H);
2.57-3.09
(m, 3H); 3.11-3.59 (m, 3H); 7.11-7.39 (m, 5H); 9.10-9.15 (m, 1H).
1-(4-Cyclohexyl-3-phenyl-4-propoxybutyl)-4-~5-(2,3-dihydro-1,4-benzodioxinyl)~-
piperazme
The title compound was prepared using the method described for the Compound of
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
57
Example 1 but using Compound 25b instead of Compound le and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :MeaCO 8:2) to afford the title
compound (50.7 %).
1H-NMR (CDCl3, b~: 0.93 (t, 3H); 0.99-1.37 (m, 6H); 1.44-1.98 (m, 8H); 2.10-
2.31 (m,
3H); 2.41-2.69 (m, 4H); 2.72-2.90 (m, 1H); 2.95-3.18 (m, SH); 3.20-3.98 (m,
2H); 4.19-
4.37 (m, 4H); 6.48-6.67 (m, 2H); 6.69-6.83 (m, 1H); 7.11-7.36 (m, SH).
[M+H]+=493 .
Example 26 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-(4-hydroxy-3-phenyl)-
hexylpiperazine
4-Oxo-2-phenylbutyronitrile(Compound 26a)
The title compound was obtained following the procedure described for Compound
1 a but
using as a starting material Compound 17a instead of compound 1 d. After the
usual work-
up procedure, the so obtained title compound was used without further
purification in the
next step.
1H-NMR (CDCl3, S): 2.94-3.29 (m, 2H), 4.31-4.45 (m, 1H), 7.30-7.48 (m, SH),
9.78 (bs,
1 H).
1-(3-Cyano-3-phenylpropyl)-4- (2,3-dihydrobenzo-1,4-dioxin-5-yl)-piperazine
(Compound 26b)
The title compound was obtained following the procedure described for the
compound of
Example 1 but using as a starting material Compound 26a instead of compound le
and 1-
(2,3-dihydro-1,4-benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-
trifluoroethoxyphenyl)-
piperazine. Purification by flash chromatography (PE - EtOAc 4:6) yielded the
title
compound (85 °l°). Oil.
1H-NMR (CDCl3, S): 2.01-2.29 (m, 2H), 2.31-2.72 (m, 6H), 3.02-3.22 (m, 4H),
4.03-4.18
(m, 1H), 4.19-4.38 (m, 4H), 6.50-6.62 (m, 2H), 6.73-6.85 (m, 1H), 7.31-7.42
(m, SH).
1-(2,3-Dihydrobenzo-1,4-dioxin-5-yl)-4-(3-formyl-3-phenylpropyl)-piperazine
Compound 26c)
The title compound was obtained following the procedure described for Compound
6f
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
58
(alternative method) but using as a starting material Compound 26b instead of
Compound
6h. Purification by flash chromatography (CHzCl2-MeOH 95:5) yielded the title
compound
(60 %). Oil.
1H-NMR (CDCl3, ~: 1.88-2.02 (m, 1H), 2.30-2.51 (m, 3H), 2.52-2.98 (m, 4H),
2.99-3.31
(m, 4H), 3.63-3.77 (m, 1H), 4.20-4.41 (m, 4H), 6.48-6.67 (m, 2H), 6.68-6.85
(m, 1H),
7.21-7.43 (m, SH), 9.79 (bs, 1H).
1-~5-(2, 3-Dihydro-1,4-benzodioxinyl)~-4-(4-hydroxy-3-phenylhexyl)-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
.
The crude was purified by flash chromatography (CH2Cla-MeOH/NH3 97:3)
affording
the title product as a yellow glassy oil (22.6 %).
1H-NMR (CDCl3, b~: 0.79-1.01 (m, 3H) 1.02-1.79 (m, 4H), 1.80-1.98 (m, 1H),
1.99-2.24
(m, 1H), 2.26-2.96 (m 6H), 2.98-3.33 (m, 4H), 3.41-3.79 (m, 1H), 4.18-4.38 (m,
4H),
6.45-6.68 (m, 2H), 6.69-6.87 (m, 1H), 7.19-7.38 (m, SH).
[M+H]+=397.4
Example 27 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenyl)-
heptyl]-
piperazine
The title compound was obtained together with the compound.of Example 12 as
the main
impurity. It was purified by flash chromatography (EtOAc- MeOH/NH3 95:5).
1H-NMR (CDCl3, ~: 0.78-0.92 (m, 3H), 1.15-1.3 (m, SH), 1.80-2.08 (m, 2H), 2.28-
2.40
(m, 2H), 2.52-2.83 (m, SH), 3.02-3.18 (m, 4H), 3.65-3.79 (m, 1H), 4.16-4.32
(m, 4H),
6.48-6.62 (m, 2H), 6.71-6.82 (t, 1H), 7.15-7.33 (m, SH).
Example 28 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylhex-5-
enyl]-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
vinylmagnesium chloride (1M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CH2C12-MeOH/NH3 97:3)
affording
the title product as a yellow glassy oil (66 %).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
59
1H-NMR (CDCl3, ~: 1.35-1.85 (br, 1H), 1.86-2.28 (m, 2H), 2.30-2.91 (m, 7H),
2.98-3.25
(m, 4H), 3.43-3.81 (m, 1H), 4.19-4.40 (m, 4H), 4.90-5.35 (m, 2H), 5.66-5.89
(m, 1H),
6.47-6.69 (m, 2H), 6.71-6.85 (m, 1H), 7.14-7.42 (m, SH).
[M+H]+=3 95 .3
Example 29 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-(4-hydroxy-5-methyl-3-
phenyl)-hexyl]-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
isopropylmagnesium chloride (2M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CH2Cl2- MeOH/NH3 98:2)
affording
the title product as a white solid (35 °f°).
1H-NMR (CDCl3, ~: 0.73-0.95 (m, 6H), 1.30-1.48 (m, 1H), 1.76-1.95 (m, 1H),
1.96-2.21
(m, 1H), 2.22-2.48 (m, 2H), 2.49-2.95 (m, SH), 2.96-3.28 (m, 4H), 3.52-3.73
(m, 1H),
4.19-4.41 (m, 4H), 5.02-5.68 (bs,~lH), 6.49-6.63 (m, 2H), 6.75-6.87 (m, 1H),
7.15-7.39
(m, SH).
[M+H]+=411.7
Example 30 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-(4-hydroxy-3-phenyl)-
pentyl]-
piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
methylmagnesium bromide (3M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CH2C12- MeOH/NH3 99:1)
affording
the title product as a white solid (42 °f°) characterized as a
7:3 (RS,RS)-(RS,SR) mixture.
1H-NMR (CDCl3, ~: 0.85-1.15 (m, 3H), 1.41-1.67 (m, 2H), 1.74-1.95 (m, 1H),
1.96-2.24
(m, 1H), 2.25-3.29 (m, lOH), 3.81-3.99 (m, 1H), 4.19-4.39 (m, 4H), 6.50-6.65
(m, 2H),
6.72-6.87 (m, 1H), 7.14-7.41 (m, SH).
[M+H]+=3 83.6
Example 31 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylhept-5-
ynyl)-piperazine
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and 1-
propynylmagnesium bromide (0.5 M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CHaCl2- MeOH/NH3 99:1)
affording
the title product as a pale yellow solid (35 %).
IH-NMR (CDCl3, ~: 1.72-1.89 (m, 3H), 1.91-2.21 (m, 2H), 2.30-2.50 (m, 2H),
2.51-2.82
(m, 4H), 2.83-3.24 (m, SH), 3.51-3.73 (m, 1H), 4.20-4.41 (m, 4H), 4.42-4.61
(m, 1H),
6.48-6.63 (m, 2H), 6.73-6.82 (m, 1H), 7.20-7.39 (m, SH).
[M+H]+=407.4
Example 32 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylhept-5-
enyl)-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and 1-
propenylmagnesium bromide (0.5 M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CH2C1~- MeOH/NH3 99:1)
affording
the title product as a pale yellow solid (83 %).
1H-NMR (CDCl3, a): 1.31-2.31 (m, 8H), 2.32-2.91 (m, 6H), 2.92-3.28 (m, 4H),
4.17-4.33
(m, 4H), 5.23-5.75 (m, 2H), 6.48-6.63 (m, 2H), 6.71-6.84 (m, 1H), 7.12-7.39
(m, SH).
[M+H]+=409.6
Example 33 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylhex-5-
ynyl)-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
1-ethynyl magnesium bromide (0.5 M THF sol.) instead of ethylmagnesium
chloride.
The crude was purified twice, first by flash chromatography (CH2C1~,- MeOH/NH3
99:1)
followed by preparative LC affording the title product as a white solid (8 %).
IH-NMR (CDCl3, d~: 1.11-1.99 (br, 1H), 2.01-2.25 (m, 2H), 2.27-2.31 (m, 1H),
2.34-2.37
(m, 2H), 2.61-2.82 (m, 4H), 2.85-3.22 (m, SH), 4.18-4.32 (m, 4H), 6.47-6.62
(m, 2H),
6.74-6.86 (m, 1H), 7.19-7.41 (m, SH).
[M+H]+=393.7
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
61
Example 34 1-[5-(2,3-Diliydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylhept-6-
enyl)-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
allylmagnesium bromide (1 M THF sol.) instead of ethylmagnesium chloride.
The crude was purified by flash chromatography (CH2Cl2- MeOH/NH3 99:1)
affording
the title product as a brownish oil (27 %).
1H-NMR (CDCl3, ~: 1.41-1.72 (br, 1H),1.73-2.25 (m, 4H), 2.26-2.50 (m, 2H),
2.51-2.91
(m, SH), 3.03-3.24 (m, 4H), 3.78-3.92 (m, 1H), 4.20-4.39 (m, 4H), 4.92-5.17
(m, 2H),
5.73-5.95 (m, 1H), 6.51-6.64 (m, 2H), 6.67-6.84 (m, 1H), 7.11-7.40 (m, SH).
[M+H]+=409.7
Example 35 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-6-methyl-3-
phenylhept-5-enyl)-piperazine
The title compound was obtained following the~procedure described for the
compound of
Example 6 but using as a starting material Compound 26c instead of Compound 6f
and
2-methyl-1-propenylmagnesium bromide (0.5 M THF sol.) instead of
ethylmagnesium
chloride. The crude was doubly purified by flash chromatography (CH2Cla-
MeOH/NH3
99:1) followed by preparative LC affording the title product as a white solid
(10 %).
1H-NMR (CDCl3, ~: 1.12-1.85 (m, 8H), 1.87-2.02 (m, 1H), 2.03-2.29 (m, 1H),
2.30-2.91
(m, 6H), 2.93-3.21 (m, 4H), 4.17-4.35 (m, 4H), 4.36-4.48 (m, 1H), 4.96-5.22
(m, 1H),
6.48-6.62 (m, 2H), 6.75-6.85 (m, 1H), 7.12-7.38 (m, SH).
[M+H]+=423.8
Example 36 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-6-methyl-3-
phenyl)-heptyl]-piperazine
To a solution of 0.708 ml of 2 M isobutyl magnesium chloride (in THF) in 3 ml
of THF
was added 0.226 g of LiCl04, The mixture was stirred at r.t. for 1 h;
afterwards, 0.13 g of
Compound 26c in 3 ml of THF was added dropwise. The reaction mixture was
allowed to
stir at r.t. for 3 h then quenched with an aq. saturated solution of NH4Cl,
alkalinised and
extracted with EiOAc. The combined extracts were dried (Na2S04) and evaporated
to
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
62
dryness. The crude was purified by flash chromatography (CHaCl2- MeOH 99:1)
affording the title product as an ivory white solid (54.6 %).
1H-NMR (CDCl3, ~: 0.68-1.01 (m, SH), 1.12-1.38 (m, 2H), 1.65-1.85 (m, 1H),
2.03-2.29
(m, 1H), 2.30-2.52 (m, 1H), 2.53-2.83 (m, 2H), 2.84-3.47 (m, 9H), 3.73-3.92
(m, 1H),
4.18-4.39 (m, 4H), 6.49-6.65 (m, 2H), 6.67-6.87 (m, 1H), 7.10-7.40 (m, SH).
[M+H]+=425.2
Example 37 1-(5-(2,3-dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-phenylbutyl]-
piperazine
The title compound was obtained together with the compound of Example 36.
Purification by flash chromatography afforded the title product as an oil.
1H-NMR (CDCl3, b): 1.36-1.64 (m, 1H), 1.81-2.05 (m, 2H), 2.50-2.73 (m, 4H),
2.75-2.93
(m, 2H), 2.94-3.27 (m, 4H), 3.62-3.72 (m, 2H), 4.20-4.39 (m, 4H), 6.10-6.45
(br, 1H),
6.46-6.62 (m, 2H), 6.70-6.83 (m, 1H), 7.13-7.37 (m, SH).
[M+H]+=369.7
Example 38 (RS,SR)-1-j5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4-hydroxy-3-
phenylpentyl)-piperazine
The title compound was obtained together with the compound of Example 11.
Purification by flash chromatography afforded the title product as an oil,
characterized as
the pure (RS,SR) diastereomer.
1H-NMR (CDCl3, ~: 0.85-1.12 (m, 3H), 1.80-2.21 (m, 3H), 2.32-2.45 (m, 2H),
2.46-2.83
(m, SH), 2.95-3.17 (m, 4H), 3.92-4.01 (m, 1H), 4.18-4.37 (m, 4H), 6.45-6.61
(m, 2H),
6.72-6.85 (m, 1H), 7.11-7.38 (m, SH).
[M+H]+=3 83.6
Example 39 1-(4-Cyclohexyl-3-(2-dimethylaminocarbonylphenyl)-4-oxobutyl]-4-(5-
(2,3-dihydro-1,4-benzodioxinyl)]-piperazine
2-(2-Cyclohexyl-2-oxoethyl)-N,N-dimethylbenzamide (Compound 39a)
The title compound was obtained as described for Compound 13a. after the usual
work-up
procedure, the crude was purified by flash chromatography (PE - EtOAc 1:1) to
afford
the title compound (34.6%).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
63
1H-NMR (CDCl3, ~: 1.11-1.50 (m, SH); 1.61-1.98 (m, 6H); 2.84 (d,3H); 3.04 (d,
3H);
3.81-4.02 (m, 2H); 7.11-7.42 (m, SH).
[M+H]+= 274
4-Cyclohexyl-3-(2-dimethylaminocarbonyl)-4-oxobutyraldehyde dimethyl acetale
(Compound 39b)
The title compound was prepared using the method described for Compound 2b but
using Compound 39a instead of Compound 2a. The crude was purified by flash
chromatography (PE - Me2C0 75:25). Yield: 21.3 %.
1H-NMR (CDCl3, b): 0.94-1.49 (m, SH); 1.51-1.83 (m, SH); 1.84-2.01 (m, 1H);
1.36-1.69
(m, 2H); 2.88 (s, 3H); 3.18 (s, 3H); 3.31 (d, 6H); 4.12-4.34 (m, 2H); 7.12-
7.39 (m, 4H).
4-Cyclohexyl -3-(2-dimetlrylaminocarbonyl)-4-oxobutyraldehyde (Compound 39c)
The title compoluzd was obtained following the procedure described for
Compound 1 d,
but using as a starting material Compound 39b instead of Compound lc. The
crude was
used in the next step without further purification.
1H NMR (CDCl3, d~: 0.90-2.12 (m, 12 H); 2.60-3.32 (m, 8H); 4.39-4.58 (m, 1H);
7.04-
7.51 (m, 4H); 9.63-9.72 (m, 1H)
1-~4-Cyclohexyl-3-(2-dimethylaminocarbonylphenyl)-4-oxobutyl~-4- ~5-(2,3 -
dihydro-1,4-
benzodioxinyl)~-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 39c instead of Compound 1e and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :Me2CO 6:4) to afford the title
compound
(65 %).
'H-NMR (CDCl3, ~: 1.03-2.01 (m, 11H); 2.23-2.42 (m, 3H); 2.44-2.72 (m, SH);
2.91 (s,
3H); 3.97-3.13 (m, 4H); 3.19 (s, 3H); 4.13-4.38 (m, SH); 6.48-6.63 (m, 2H);
6.70-6.84
(m, 1H); 7.13-7.39 (m, 4H).
[M+H]+=520
EXample 40 1-[4-Cyclohexyl-3-(2-dimethylaminocarbonylphenyl)-4-hydroxybutyl]-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
64
4-j5-(2,3-dihydro-1,4-benzodioxinyl)]-piperazine
The title compound was synthesised using the method described for Compound 1 c
but
starting from the Compound of Example 39 instead of Compoundlb. After the
usual
work-up procedure, the crude was purified by flash chromatography (PE - Me2C0-
NH3JMeOH 7:3:0.2) affording the title compound (65.2 %).
[M+H]+=522.45
Example 41 1-[4-Cyclohexyl-3-(2-dimethylaminocarbonylphenyl)-4-oxobutyl]-4-(4-
fluoro-2-methoxyphenyl) piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 39c instead of Compound le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :MezCO 6:4) to afford the title
compound
(64.5 %).
1H-NMR (CDCl3, ~: 1.03-2.02 (m,11H); 2.17-2.37 (m, 3H); 2.41-2.73 (m, SH);
2.94 (s,
3H); 2.95-3.12 (m, 4H); 3.17 (s, 3H); 3.85 (s, 3H); 4.11-4.27 (m, 1H); 6.51-
6.69 (m, 2H);
6.78-6.92 (m, 1H); 7.12-7.41 (m, 4H).
[M+H]+=510
Example 42 1-[4-Cyclohexyl-3-(2-dimethylaminocarbonylphenyl)-4-hydroxybutyl]-
4-(4-fluoro-2-methoxyphenyl)
The title compound was synthesised using the method described fox Compound lc
but
starting from the Compound of Example 41 instead of Compoundlb. After the
usual
work-up procedure, the crude was purified by flash chromatography (PE - MeaCO-
NH3JMeOH 75:25:0.2) affording the title compound (64.2%).
[M+H]+=512.6
Example 43 1-[3-(2-Cyanophenyl)-4-oxopentyl]-4-[5-(2,3-dihydro-1,4-
benzodioxinyl)]-piperazine
3-(2-Cyanophenyl)-4-oxopentanaldehyde diethyl acetal (Compound 43a)
The title compound was prepared using the method described for Compound 2b but
using 1-(2-cyanophenyl)-propan-2-one ( R.A. Bruce, Org. Prep. Proc. Int. 407-
412, 1999)
instead of Compound 2a. The crude was purified by flash chromatography (PE-
EtOAc
8:2). Yield: 13%.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
1H-NMR (CDCl3, b): 1.10-1.29 (m, 6H), 1.87-2.04 (m, 1H), 2.14 (s, 3H), 2.42-
2.59 (m,
1H), 3.31-3.71 (m, 4H), 4.28-4.43 (m, 2H), 7.30-7.41 (m, 2H), 7.51-7.72 (m,
2H).
3-(2-Cyanophenyl)-4-oxopentanaldehyde (Compound 43b)
The title compound was obtained following the procedure described for Compound
1 d,
but using as a starting material Compound 43b instead of Compound lc. The
crude was
used in the next step without further purification.
1-~3- 2-C anophenyl)-4-oxopenttyl~-4-~5-(2,3-dihydro-1,4-benzodioxinyl)~-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 43b instead of Compound 1e and 1-(2,3-dihydro-1,4-
benzodioxin-5-yl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :EtOAc 6:4) to afford the title
compound
(43.8 %).
1H NMR (CDCl3, b): 1.75-1.92 (m, 1H), 2.21 (s, 3H), 2.30-2.41 (m, 3H), 2.41-
2.69 (m,
4H), 2.92-3.12 (m, 4H), 4.19-4.40 (m, SH), 6.48-6.62 (m, 2H), 6.71-6.82 (t,
1H), 7.30-
7.43 (m, 2H), 7.51-7.72 (m, 2H).
Example 44 1-[4-Cyclohexyl-3-(2-trifluoromethoxyphenyl)-4-oxobutyl]-4-(4-
indolyl)-piperazine
1-Cyclohexyl-2-(2-trifluoromethoxyphenyl)ethanone (Compound 44a)
A mixture of 1.26 g of 2-trifluoromethoxybenzyl chloride, Zn powder (0.59 g)
and 1,2-
DME was refluxed for 3 h, cooled at r.t.; afterwards, it was filtered and was
added 0.002
g of dichlorobis(triphenylphosphine)-palladium (II) followed by 0.72 ml of
cyclohexanecarbonyl chloride to the filtrate stirred at r.t.. Afterwards, the
reaction
mixture was stirred at reflux for 4 h, cooled at r.t.. After the usual work-up
procedure
(see Compound 1 a), the crude was purified by flash chromatography ( methyl
tert-butyl
ether- PE 96:4) to afford 0.22 g of the title compound.
IH-NMR (CDCl3, ~: 1.10-2.00 (m, lOH); 3.80 (s, 3H); 7.18-7.40 (m, 4H);
4-Cyclohexyl-4-oxo-3-(2-trifluoromethoxyphenyl)-butyraldehyde diethyl acetal
(Compound 44b)
To a solution of 0.22 g of Compound 44a in 1 ml of DMSO was added potassium
tert-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
66
butoxide (0.091 g) at r.t.. After 15 min 0.12 ml of 2-bromoacetaldehyde
diethyl acetal
was added and the reaction mixture was heated at 50°C for 5h.
Afterwards, it was cooled
to r.t., diluted with water and extracted with methyl tart-butyl ether, which
was dried
(Na2S04) and evaporated to dryness in vacuo giving a crude which was purified
by flash
chromatography (methyl tart-butyl ether- PE 93:7) affording 0.092 g of the
pure title
product.
1H-NMR (CDCl3, ~: 1.00-2.10 (m,l7H); 2.10-2.45 (m, 2H); 3.75 (q,4H); 4.32
(t,lH);
4,50 (t,lH); 7.10-7.40 (mz 4H)
4-Cyclohexyl-4-oxo-3-(2-trifluoromethoxyphenyl)-butyraldehyde (Compound 44c)
0.098 of Compound 44b, 1.1 ml of 1N HCl and 5 ml of acetone were stirred at
r.t. for 4h.
Evaporation and extraction with CH~Cl2 afforded the title compound, which was
used in
the next step without further purification.
1H-NMR (CDCl3, b~: 0.80-1.95 (m,9H); 1.95-2.15 (m,lH); 2.25-2.45 (m,lH); 2.52
(dd, l H); 3.40 (dd, l H); 4.80 (dd, l H); 7.10-7.40 (m,4H); 9.75 (s, l H)
1-~4-Cyclohexyl-3-(2-trifluoromethoxyphenyl)-4-oxobutyl~-4-(4-indolyl)-
piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 44c instead of Compound le and 1-(4-indolyl)-
piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-piperazine. The crude
was purified
by flash chromatography (PE :EtOAc 6:4) to afford the title compound (53 %).
1H-NMR (CDCl3, b>: 1.00-2.10 (m,l 1H); 2.20-2.50 (m,4H); 2.50-2.80 (m,4H);
3.15-3.40
(m,4H); 4.50 (t,lH); 6.50 (d,lH); 7.60 (dd,lH); 7.007.20 (m,3H); 7.20-7.35
(m,4H); 8.15
(s,1 H)
Example 45 (RS,SR) 1-[4-Acetoxy-4-cyclohexyl-3-(2-fluorophenyl)-butyl]-4-(2-
methoxyphenyl)-piperazine
1-Cyclohexyl-2-(2-fluorophenyl)ethanone (Compound 45a)
To a mixture of 36 ml of 2-fluorobenzylzinc chloride (0.5 M sol. in.THF) and
0.008 g of
dichlorobis(triphenylphosphine)-palladium (II) stirred at 0°C was added
dropwise via a
syringe 2.14 ml of cyclohexanecarbonyl chloride. Afterwards, the reaction
mixture was
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
67
stirred at r.t. for 4 h, quenched with an aqueous saturated solution of
ammonium chloride
(25 ml), extracted with 20 ml of EtOAc, which was dried (Na2S04) and
evaporated to
dryness in vacuo affording 3.52 g of the title compound as a crude, which
could be used
in the following step without further purification.
1H-NMR(CDCl3, ~): 1.10-2.05 (m, lOH), 2.47 (tt,lH), 3.77 (s, 2H), 6.97-7.32
(m, 4H)
4-Cyclohexyl-4-oxo-3-(2-fluorophenyl)-butyraldehyde diethyl acetal (Compound
45b)
A solution of 5.02 g of compound 45a in 136 ml of toluene was heated at reflux
recovering 35 ml of toluene by distillation to remove water. Afterwards, 3.18
g of
potassium tent-butoxide was added and stirring at reflux was continued for 30
min.; the
reaction mixture was cooled to 80°C and 4.27 ml of 2-bromoacetaldehyde
diethyl acetal
was added. After 18 h at reflux, the reaction mixture was cooled to r.t.,
quenched with an
aqueous saturated solution of ammonium chloride (30 ml), extracted with 30 ml
of
EtOAc, which was dried (Na2S04) and evaporated to dryness in vacuo giving a
crude
which was purified by flash chromatography (petroleum ether-EtOAc 92.5:7.5)
affording
2.97 g of the pure title product.
1H NMR (CDCl3, b): 1.00-2.10 (m, 17H), 2.20-2.52 (m, 2H), 3.30-3.72 (m, 4H),
4.25-
4.45 (m, 2H), 6.90-7.35 (m, 4H)
4-Cyclohexyl-4-oxo-3-(2-fluorophenyl)-butyraldehyde (Compound 45c)
A mixture of 1.12 g of the compound 45b, 9 ml of 50% aq. trifluoroacetic acid
and 18 ml
of CHaCl2 was stirred for 2 h at r.t., and then diluted with 10 ml of CH2Cla.
The organic
layer was separated, washed with brine (2x15m1), dried (NaaS04) and evaporated
to
dryness in vacuo to afford a crude (0.88 g), used in the next step without
further
purification.
1H NMR (CDCl3, 8): 0.90-2.10 (m, lOH), 2.25-2.70 (m, 2H), 3.12-3.52 (m, 1H),
4.60-
4.80 (m, 1H), 6.95-7.40 (m, 4H), 9.75 (s, 1H)
1-~4-Cyclohexyl-3-(2-fluorophenyl)-4-oxobutyl~-4-(2-methoxyphenyl)-piperazine
(Compound 45d)
A mixture of 0.88 g of the compound 45c, 0.84 g of 1-(2-methoxyphenyl)-
piperazine'HCI,
1.06 g of sodium triacetoxyborohydride and 33 ml of CH2C12 was stirred at r.t.
for 1 h,
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
68
kept overnight resting, alkalinised with 20% aq. Na2C03, The organic layer was
separated,
washed with brine (2 x 30m1), dried (Na2S04) and evaporated to dryness ih
vacuo the
give a crude (1.46 g) which was used in the next step without further
purification. A
sample was purified by flash chromatography (petroleum ether-EtOAc 6:4)
affording a
pure sample.
1H-NMR (CDCl3, 8): 1.05-2.00 (m, 11H), 2.20-2.44 (m, 4H), 2.45-2.72 (m, 4H),
2.90-
3.20 (m, 4H), 3.85 (s, 3H)
(RS,SR)-1-(4-Cyclohexyl-3-(2-fluorophenyl)-4-hydroxybutyl~-4-(2-
methoxypheriyl)-
piperazine (Compound 45e)
To a solution of 1.46 g of Compound 45d in 33 ml of MeOH stirred at 0°C
was added
0.19 g of NaBH4 and the mixture was stirred at r.t. for 4 h. The solvent was
evaporated
and the reaction crude was taken up with H2O and extracted with EtOAc. The
organic
layer was separated, washed with brine (2 x l5ml), dried (Na2S04) and
evaporated to
dryness in vacuo to give a crude which was purified by sequential flash
chromatography
(petroleum ether-EtOAc-2 N ammonia in methanol 75:25:2; petroleum ether-EtOAc-
2 N
ammonia in methanol 80:20:2) affording 0.82 g of Compound 45e (upper TLC Rf ;
eluentpetroleum ether-EtOAc-2 N ammonia in methanol 70:30:2).
1H-NMR (CDCl3, b): 0.80-1.40 (m, 6H), 1.50-1.82 (m, 4H),1.85-2.10(m, 3 H),
2.21-2.45
(m, 2H), 2.52-2.85 (m, 4H), 2.98-3.26 (m, 4H), 3.28-3.42 (m, 1H), 3.50-3.60
(m, 1H),
3.85 (s, 3H), 6.80-7.30 (m, 7H), 7.62-7.80 (m, 1H); OH peals not detectable
(1R,2S) 1-Cyclohexyl-4-~4-(2-methoxyphenyl)-piperazin-1-yl~-2-(2-fluorophenyl)-
butan-
1-0l (Compound 45eA)
This compound was obtained by chiral column chromatography on Compound 45e
using
Chiralpak AD (0.46x25 cm), eluting with n-hexane-EtOH 95:5 (flow = 0.5 ml/min;
detector IJV 247 nm).
(1S,2R) 1-Cyclohexyl-4-~4-(2-methox phenyl)-piperazin-1-yl~-2-(2-fluorophenyl)-
butan-
1-0l (Compound 45eB)
This compound was obtained by chiral column chromatography on Compound 45e
using
Chiralpalc AD (0.46x25 cm), eluting with n-hexane-EtOH 95:5 (flow = 0.5
ml/min;
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
69
detector LTV 247 nm).
The absolute stereochemistry of Compounds 45eB, in form of its salts with
hydrogen bromide, was determined by single crystal x-ray diffraction, as
follows.
Single crystal X-ray diffraction experiment:
A needle shape single crystal was selected for X-ray diffraction analysis and
mounted on a glass fiber. The data were collected on Rigalcu Rapid cylinder
shape image
plate X-ray area detector with detector aperture = 45.0 x 25.6 cm. It was
controlled by a
Windows 2000 based PC computer with Rapid Auto version 1.06 software (Rigaku,
2000), at low temperature (-120°K), with Micromax-002 micro-Confocal
mirrors
CuKa radiation [~,(CuKa ) =1.5405]. Indexing was performed from three 30
oscillations frames that were exposed for 360 seconds. All reflections were
measured in
five image groups with six frames in each group; the exposure time was 160
seconds per
degree. Among them, five groups of images were at angles phi = 0°,
90°, 180°, 270° with
chi=50°and phi=0° with chi=0° all frames were delta omega
= 30°, and which makes the
ZBmax =136.3°. The sample/detector distance was 12.74 cm. The data
reduction
program, Rapid Auto version 1.06 (Rigaku, 2000), determined the Laue group was
-l,
and total 7,986 reflections were integrated for structure solution and
refinements.
Single crystal results:
The structure was solved by direct methods, using SIR92 (Altomare et al.
1994).
All calculations were performed using the CrystalStructure 3.0 (MSC/Rigaku,
2002;
Watkin et al., 1996, Carruthers and Watkin, 1979) crystallographic software
package. The
trial solution obtained 38 nohydrogen atoms in the asymmetrical unit. Least
squares
refinement included all nonhydrogen atomic coordinates and anisotropic thermal
parameters. The final cycle of full-matrix least-squares refinement on F was
based on
6,297 reflections with I > 36(I), converged with agreement factors: R=0.071,
S=2.224,
Rw=0.073. The absolute configuration was determined by using the calculated
Flack x
parameter, which was 0.00 with esd = 0.04. Expected values are 0.0 (within 3
esd's) for
correct and +1.0 for inverted absolute structure.
References:
Altomare, A., Cascarano, G., Giacovazzo, C. Gualgliardi, A., Burla, M.,
Palidori, G., and
Camalli, M., (1994) SIR92, J. Appl. Cryst., 27, 435.
Carruthers, J.R. and Watkin, D.J. (1979), Acta Cryst, A35, 698-699.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
Rigalcu (2000), Rapid Auto, Rigaku Corporation, Tolcyo, Japan.
Rigalcu and Rigalcu/MSC, (2000-2002), Crystal Structure Analysis Software,
CrystalStructure Version 3.00, Rigaku/MSC, 9009 New Trails Drive, The
Woodlands,
TX, USA 77381-5209. Rigalcu, 3-9-12 Altishima, Tokyo 196-8666, Japan.
Watlcin, D.J., Prout, C.I~. Carruthers, J.R. & Betteridge, P.W., CRYSTALS
Issue 10,
Chemical Crystallography Laboratory, Oxford, UK.
(RS,SR) 1-(4-Acetoxy-4-cyclohexyl-3-(2-fluorophenyl)-butyl-4-(2-methoxyphenyl)-
piperazine '
To a solution of 0.135 g of Compound 45e and 0.043 ml of TEA in 5 ml of CHaCla
stirred at 0-5°C, was added 0.021 ml of acetyl chloride. Afterrwards,
the reaction mixture
was stirred at r.t. for 4h, washed with 5% aq. NaHC03 5%(1x10m1), H2O
(2x15m1), dried
on Na~,S04 and evaporated in vacuo to afford the title product (0,143 g)
1H-NMR (CDCl3, ~): 5:0.80-1.25 (m,SH); 1.25-1.50 (m,lH); 1.50-1.90 (m, 7H);
1.95
(s,3H); 2.10-2.40 (m,2H); 2.40-2.65 (m,r4H); 2.90-3.15 (m, 4H); 3.38-3.55
(m,lH); 3.85
(m, 3H); 5.05 (t,lH); 6.80-7.25 (m,7H); 7.30-7.45 (m,lH)
Example 46 (RS,SR) 1-[4-Cyclohexyl-3-(2-fluorophenyl)-4-
methoxycarbonyloxybutyl]-4-(2-methoxyphenyl)-piperazine
To a solution of 0.112 g of Compound 45e in 0.8 ml of pyridine stirred at
0°C, was added
0.022m1 of methyl chloroformate. The reaction mixture was stirred at r.t. for
4 days and at
40°C for 5 h. Additional 0.045 ml of methyl chloroformate was added,
heating at 40°C
for 4 h.. After 3 days at r.t., methyl chloroformate (0.045 ml) was added and
the mixture
stirred at r.t. for 6h. After cooling, it was poured into water and extracted
with
EtOAc(2x15m1),washed with 2x15m1 of H20, dried (Na~S04) and evaporated to
dryness
in vacuo. The crude was purified by flash chromatography to (PE - EtOAc-
NH3lMeOH
75:25:2.5) to yield 0.022g of the title product.
1H-NMR (CDCl3, 8): 0.80-2.05 (m,13H); 2.10-2.40 (m,2H); 2.40-2.75 (m,4H); 2.90-
3.20
(m,4H); 3.38-3.60 (m,lH); 3.70 (m,3H); 3.85 (m,3H); 4.88 (t,lH); 6.80-7.25
(m,7H);
7.3 8-7.50 (m, l H)
Example 47 (RS,SR) 1-[4-Cyclohexyl-4-ethylaminocarbonyloxy-3-(2-fluorophenyl)-
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
71
butyl]-4-(2-methoxyphenyl)-piperazine
To a solution of 0.126 g of Compound 45e in 0.5 ml of pyridine stirred at
0°C, was added
0.113 of ethyl isocyanate. The reaction mixture was stirred at r.t. for 24 h
and at 50°C for
3 h. After cooling, it was poured into water and extracted with Et20,washed
with H20,
dried (Na2S0~.) and evaporated to dryness in vacuo to afford 0.108g of the
title product.
IH-NMR (CDCl3, ~): 0.90-1.40(m, 9H); 1.60-2.10 (m,7H); 2.10-2.40 (m, 2H); 2.50-
2.65
(m, 4H); 2.95-3.30 (m, 6H); 3.40-3.55 (m,lH); 3.85 (s, 3H); 4.50 (t,lH); 4.90
(t,lH);
6.80-7.22 (m,7H); 7.32-7.45 (m,lH)
Example 48 (RS,SR) 1-[4-Aminocarbonyloxy-4-cyclohexyl-3-(2-fluorophenyl)-
butyl]-4-(2-methoxyphenyl)-piperazine
To a solution of 0.124 g of Compound 45e in 5 ml of CH2Cla was added potassium
cyanate. To the suspension stirred at r.t. was added 0.087 ml of
trifluoroacetic acid. After
24 h at r.t. and 5 h at 40°C, additional trifluoroacetic acid was added
(0,17 ml). After 6 h
at 40°C, the reaction misture was cooled, evaporated to dryness,
diluted with water and
NaOH 2 N, extracted with EtOAC; the extracvt was washed with HaO, dried
(Na2S04)
and evaporated to dryness in vacuo. The crude was purified by flash
chromatography to
(PE - EtOAc-NH3/MeOH 75:25:2.5) to yield 0.064 g of the title product.
1H-NMR (CDCl3, 8): 0.90-1.50 (m,6H); 1.50-2.05 (m,7H); 2.10-2.40 (m,2H); 2.50-
2.70
(m,4H); 2.95-3.15 (m,4H); 3.40-3.55 (m,lH); 3.85 (s,3H); 4.45 (s,2H); 4.85-
4.95 (m,lH);
6.80-7.26 (m,7H);7.32-7.45 (m,lH)
Example 49 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4- (4=hydroxy-5,5-dimethyl-3-
phenyl)-hexyl]-piperazine
The title compound was synthesised following the procedure described for the
compound
of Example 36 but using tert-butylmagnesium chloride (1N in THF) instead of
isobutylmagnesium chloride, The mixture was stirred at r.t. for 3 h. The crude
was
purified by flash chromatography (CH2Cl2- MeOH 99:1) affording the title
product as an
brownish solid (15 %).
1H-NMR (CDCl3, d~: 0.88 (m, 9H), 1.31-1.48 (m, 2H), 1.73-1.97 (m, 1H), 1.96-
2.21 (m,
1H), 2.22-2.48 (m, 2H), 2.49-2.97 (m, 5H), 2.99-3.28 (m, 4H), 3.61-3.73 (m,
1H), 4.19-
4.41 (m, 4H), 6.49-6.63 (m, 2H), 6.75-6.87 (m, 1H), 7.15-7.44 (m, 5H).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
72
[M+H]+=425.7
Example 50 1-(4-Fluoro-2-methoxyphenyl)-4-[(4-hydroxy-3-phenyl)-hept-5-ynyl]-
piperazine
1-(3-Cyano-3-phenylpropyl)-4- (4-fluoro-2-methoxyphenyl)-piperazine (Compound
50a)
. The title compound was obtained following'the procedure described for the
compound of
Example 1 but using as a starting material Compound 26a instead of Compound le
and 1-
(4-fluoro-2-methoxyphenyl)-piperazine instead of 1-(2,2,2-
trifluoroethoxyphenyl)-
piperazine. Purification by flash chromatography (PE - EtOAc 4:6) yielded the
title
compound (90 %). Oil.
1H-NMR (CDCl3, b) 1.94-2.29 (m, 2H); 2.36-2.79 (m, 6H); 2.97-3.14 (m, 4H);
3.85 (s,
3H); 4.01-4.12 (m, 1H); 6.54-6.71 (m, 2H); 6.82-7.97 (m, 1H); 7.28-7.47 (m,
5H)
1-(4-Fluoro-2-methoxyphenyl)-4-(3-formyl-3-phenylpropyl)-piperazine (Compound
50b)
The title compound was obtained following the procedure described for Compound
6f
(alternative method) but using as a starting material Compound 50a instead of
Compound
6h. Purification by flash chromatography (CHaCl2-MeOH 95:5) yielded the title
compound (55 °1°). Oil.
1H-NMR (CDCl3, b~: 1.81-2.00 (m, 1H); 2.24-2.48 (m, 3H); 2.49-3.82 (m, 4H);
3.87-3.19
(m, 4H); 3.61-3.73 (m, 1H); 3.84 (s; 3H); 6.53-6.71 (m, 2H); 6.80-6.94 (m,
1H); 7.12-
7.45 (m, 5H); 9.68-9.79 (m, 1H).
1-(4-Fluoro-2-methoxyphenyl)-4-~(4-hydroxy-3-phenyl)-hept-5-ynyl~-piperazine
The title compound was obtained following the procedure described for the
compound of
Example 36 but using as a starting material Compound 50b instead of Compound
26c and
using 1-propynylmagnesium bromide (0.5 N in THF) instead of a solution of
isobutylmagnesium chloride,. The crude was purified by flash chromatography
(CHaCl2-
MeOH 99:1 ) affording the title product as a yellow glassy oil (22.6 %).
1H-NMR (CDCl3, ~: 0.84-1.17 (m, 3H), 1.40-1.70 (m, 2H), 1.74-1.90 (m, 1H),
1.93-2.22
(m, 1H), 2.25-3.29 (m, lOH), 3.80 (s, 3H), 3.84-3.99 (m, 1H), 6.50-6.65 (m,
2H), 6.70-
6.90 (m, 1H), 7.14-7.38 (m, 5H).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
73
[M+H]+=397.2
Example 51 (E,Z)-1-(4-Fluoro-2-methoxyphenyl)-4-[(4-hydroxy-3-phenyl)-kept-5-
enyl]-piperazine (1:1 mixture)
The title compound was obtained following the procedure described for the
compound of
Example 36 but using as a starting material Compound SOb instead of Compound
26c and
using 1-propenylmagnesium bromide (0.5 N in THF) instead of a solution of
isobutylmagnesium chloride..
The crude was purified by flash chromatography (CH2Cl2- MeOH 99:1) affording
the
title compound as a yellow glassy oil (30 %).
1H-NMR (CDCl3, b~: 1.17-1.73 (m, 4H), 1.92-2.51 (m, 2H), 2.52-3.42 (m, 11H),
3.83 (s,
3H), 4.22-4.65 (m, 1H), 5.28-5.75 (m, 2H), 6.50-6.64 (m, 2H), 6.72-6.91 (m,
1H), 7.13-
7.41 (m, SH).
[M+H]+=399.2
Further purification by preparative LC-MS chromatography afforded the
isolation of the
following compounds:
Example 52 (E,Z)-1-(4-fluoro-2-methoxyphenyl)-4-[(4-hydroxy-3-phenyl)-hept-5-
enyl]-piperazine (5:95 mixture)
1H-NMR (CDCl3, ~: 1.24-1.43 (m, 3H), 1.74-1.91 (m, 2H), 1.92-3.12 (m, 12H),
3.82 (s,
3H), 4.41-4.55 (m, 1H), 5.18-5.65 (m, 2H), 6.48-6.59 (m, 2H), 6.71-6.91 (m,
1H), 7.13-
7.43 (m, SH).
[M+H]+=399.2
Example 53 (E)-1-(4-Fluoro-2-methoxyphenyl)-4-[(4-hydroxy-3-phenyl)-hept-5-
enyl]-piperazine (RS,RS:RS,SR 9:1 mixture)
1H-NMR (CDCl3, ~: 1.44-1.59 (m, 4H), 1.71-2.03 (m, 2H), 2.32-3.15 (m, 11H),
3.84 (s,
3H), 4.09-4.12 (m, 1H), 5.15-5.23 (m, 1H), 5.34-5.49 (m, 1H), 6.48-6.60 (m,
2H), 6.70-
6.92 (m, 1H), 7.13-7.43 (m, SH).
[M+H]+=399.2
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
74
Example 54 1-(5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-hydroxy-5-methyl-3-
phenyl)-hex-5-enyl]-piperazine (RS,RS:RS,SR 6:4)
The title compound was synthesised following the procedure described. for the
compound
of Example 36 but using isopropenylmagnesium bromide (0.5 N in THF) instead a
solution of isobutylmagnesium chloride, The mixture was stirred at r.t. for 3
h. The crude
was purified by flash chromatography (CH2Cl2- MeOH 99:1 ) affording the title
product
as an brownish solid (38 %).
1H-NMR (CDCl3, ~: 1.65-1.79 (m, 3H), 2.03-2.12 (m, 1H), 2.32-2.48 (m, 1H),
2.57-2.78
(m, 1H), 2.79-3.42 (m, 12H), 4.21-4.45 (m, 4H), 4.65-4.95 (m, 2H), 6.49-6.58
(m, 1H),
6.60-6.65 (m, 1 H), 6.72-6.79 (m, 1 H), 7.19-7.41 (m, SH).
[M+H]+=409.6
Example 55 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-hydroxy-6-methyl-3-
phenyl)-hept-6-enyl]-piperazine
The title compound was synthesised following the procedure described for the
compound
of Example 36 but using 2-methylallylmagnesium chloride (0.5 N in THF) instead
a
solution of isobutylmagnesium chloride, The mixture was stirred at r.t. for 3
h. The crude
was purified by flash chromatography (CH~,C12 - MeOH 99:1) affording the title
product
as an brownish solid (48%).
1H-NMR (CDCl3, b~: 1.68-1.73 (m, 3H), 1.94-2.05 (m, 2H), 2.06-2.18 (m, 1H),
2.19-2.31
(m, 1 H), 2.47-2.51 (m, 1 H), 2.52-3.42 (m, 12H), 4.23-4.44 (m, 4H), 4.62-4.93
(m, 2H),
6.49-6.53 (m, 1H), 6.62-6.65 (m, 1H), 6.77-6.84 (m, 1H), 7.21-7.48 (m, SH).
[M+H]+=423 .6
Example 56 1-(5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-hydroxy-4-(2-thienyl)-3-
phenylbutyl]-piperazine
The title compound was synthesised following the procedure described for the
compound
of Example 36 but using 2-tlv.enylmagnesium bromide (1 M in THF) instead of a
solution
of isobutylmagnesium chloride, The mixture was stirred at r.t. for 3 h. The
crude was
purified by flash~chromatography (CHaCl2- MeOH 99:1) affording the title
product as an
brownish solid (33 %).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
1H NMR (CDCl3, ~: 1.75-2.23 (m, 3H), 2.48-2.53 (m, 2H), 2.54-2.94 (m, 4H),
3.05-3:32
(m, SH), 4.23-4.41 (m, 4H), 5.12-5.21 (m, 1H), 6.50-6.93 (m, SH), 7.11-7.48
(m, 6H).
[M+H]+=451.7
Example 57 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[(4-hydroxy-3-phenyl)octyl]-
piperazine .
The title compound was synthesised following the procedure described for the
compound
of Example 36 but using n-butylmagnesium chloride (2 M in THF) instead of a
solution
of isobutylmagnesium chloride, The mixture was stirred at r.t. for 3 h. The
crude was
purified by flash chromatography (CH2C12- MeOH 99:1) affording the title
product as an
brownish solid (48 %).
1H-NMR (CDCl3, ~: 0.61-0.78 (m, 3H), 1.02-1.31 (m, 6H), 1.94-2.06 (m, 1H),
2.26-2.48
(m, 1H), 2.51-2.61 (m, 2H), 2.62-3.32 (m, lOH), 3.62-3.74 (m, 1H), 4.23-4.44
(m, 4H),
6.47-6.50 (m, 1H), 6.61-6.65 (m, 1H), 6.79-6.84 (m, 1H), 7.21-7.51 (m, SH).
[M+H]+=425.4
Example 58 1-(4-Fluoro-2-methoxyphenyl)-4-[(4-methoxy-3-phenyl)-kept-5-ynyl]-
piperazine
4-Hydroxy-3-phenylhept-5-ynaldehyde dimethyl acetal (Compound 58a)
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 17b instead of compound 6f
and 1-
propynylmagnesiu~.n chloride (2M sol. in THF) instead of ethylmagnesium
chloride. The
crude was purified by flash chromatography (EtOAc - PE 3:7) affording the
title product.
(81 °fo).
1H-NMR (CDCl3, b): 1.78-1.88 (m, 3H), 1.91-2.21 (m, 3H), 2.92-3.08 (m, 1H),
3.20-3.35
(m, 7H), 4.18-4.24 (m, 1H), 7.20-7.39 (m, SH).
4-Methoxy-3-phenylhept-5-ynaldehyde dimethyl acetal (Compound 58b)
The title compound was synthesized as described for Compound 1 d using as a
starting
material Compound 58a instead of Compound lc. After EtOAc extraction, the
crude was
used without further purification in the next step.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
76
4-Methoxy-3-phenylhept-5-ynaldehyde (Compound 58c)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 58b instead of compound ld. The
title product
was used in the next step without further purification.
1-(4-Fluoro-2-methoxyphenyl)-4-~(4-methoxy-3-phenyl)-hept-5-ynyl~-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 58c instead of Compound le and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE-EtOAc 3:7) to afford the title
compound
(17.5 %).
1H-NMR (CDCl3, x:.1.68 and1.83 (2 s, 3H); 1.88-2.05 (m, 2H), 2.15-2.48 (m,
4H), 2.48-
2.78 (m, 4H), 2.81-2.90 (m, 1H); 2.90-3.15 (m, 2H); 3.25 and 3.40 (2 x s, 3H);
3.76 (m,
3H); 3.93-3.98 (m, 1H), 6.48-6.57 (m, 2H), 6.76 (dd, 1H) 7.22-7.48 (m, 5H).
[M+H]+=411.13
Example 59 (E,Z)-1-(4-Fluoro-2-methoxyphenyl)-4-[(4-methoxy-3-phenyl)-kept-5-
enyl]-piperazine (upper TLC rf diastere0mer)
4-Hydroxy-3-phenylhept-5-enaldehyde dimethyl acetal (Compound 59a)
The title compound was obtained following the procedure described for the
compound of
Example 6 but using as a starting material Compound 17b instead of compound 6f
and 1-
propenylmagnesium chloride (2M sol. in THF) instead of ethylmagnesium
chloride.
The crude was purified by flash chromatography (PE - EtOAc 7:3) affording the
title
product. (42 %).
1H-NMR (CDCl3, ~: 1.50 and 1.60 (2 d, 3H), 1.85-2.04 (m, 2H), 2.15-2.38 (m,
1H),
2.78-2.92 (m, 1H), 3.22 and 3.38 (4 x s, 6H), 4.10-4.24 (m, 1H), 4.58 (dd,
1H), 5.22-5.59
(m, 2H), 7.20-7.39 (m, 5H).
4-Methoxy-3-phenylhept-5-enaldehyde dimethyl acetal (Compound 59b)
The title compound was synthesised as described for Compound 1 d using as a
starting
material Compound 59a instead of Compound 1c. After EtOAc extraction, the
crude was
used in the next step without further purification.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
77
1H-NMR (CDCl3, ~: 1.10 and 1.75 (m, 3H), 1.85-2.35 (m, 2H), 2.63-3.05 (m, 1H),
3.12-
3.52 (m, 9H), 4.10-4.24 (m, 1H), 4.58 (dd, 1H), 5.22-5.59 (m, 2H), 7.12-7.39
(m, SH).
4-Methoxy-3-phenylhept-5-enaldehyde (Compound 59c)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 59b instead of compound ld. The
title product
was used in the next step without further purification.
1-(4-Fluoro-2-methoxyphenyl)-4-[(4-methoxy-3-phenyl)-hept-5-enyl~-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using compound 59c instead of Compound 1e and 1-(4-fluoro-2-
methoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :EtOAc 4:6) to afford the title
compound
(13.2 %).
1H-NMR (CDCl3, ~: 1.45 andl.63 (2 d, 3H); 1.68-1.81 (m, 1H), 1.88-2.05 (m,
1H), 2.08-
2.27 (m, 2H), 2.42-2.58 (m, 4H); 2.63-2.75 (m, 1H); 2.88-3.00 (m, 4H), 3.08
and 3.13 (2
x s, 3H); 3.52 and 4.05 (2 d, 1H); 3.75 (s, 3H), 5.05-5.23 (m, 1H), 5.47-5.71
(m, 1H),
6.48-6.57 (m, 2H), 6.76 (m, 1H), 7.05-7.32 (m, SH).
[M+H]+=413.34
Example 60 (E,Z)-1-(4-Fluoro-2-methoxyphenyl)-4-[(4-methoxy-3-phenyl)-hept-5-
enyl]-piperazine (lower TLC rf diastereomer)
The title compound was isolated during the purification step of Example 59.
IH-NMR (CDCl3, b): 1.48 and1.60 (2d, 3H); 1.79-2.12 (m, 2H), 2.18-2.40 (m,
2H), 2.22-
2.78 (m, SH); 2.92-3.13 (m, 4H); 3.25 and 3.30 (2s, 3H); 3.57 and 4.08 (2t,
1H); 3.85 (s,
3H), 5.11-5.23 (m, 1H), 5.39-5.65 (m, 1H), 6.58-6.69 (m, 2H), 6.82-6.91
(m,1H), 7.15-
7.35 (m, SH).
[M+H]+=413.34
Example 61 1-[4-Cyclohexyl-3-(2-methoxymethylphenyl)-4-oxobutyl]-4-(4-fluoro-2-
methoxyphenyl)-piperazine
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
78
2-Methoxymethylbenzyl bromide (Compound 61 a)
A mixture of 1.2 g of 2-methoxymethylbenzyl alcohol ( J.Chem. Soc., 1954, 2819-
2826
), 2.5 g of triphenylphosphine, 3.14 g of tetrabromomethane and 50 ml of
CHaCl2 was
stirred at r.t. for 2 h. Afterwards, the reaction mixture was evaporated to
dryness in vacuo
and purified by flash chromatography (CH~C12) to afford 1.62 g of the title
compound.
1H-NMR (CDCZ3, ~: 3.42 (d, 3H), 4.60 (s, 2H), 4.65 (s, 2H), 7.15-7.30 (m, 3H),
7.22-
7.45 (m, 4H)
1-Cyclohexyl-2-(2-methoxymethylphenyl)ethanone (Compound 61 b)
To a suspension of 1.44 g of Zn(Cu) (prepared as described in Org. Syn. 5,
855) in 5 ml
of anhydrous benzene stirred at r.t. under nitrogen, was added dropwise a
solution of 1.6
g of Compound 61 a and 1.17 ml of N,N-dimethylacetamide in 10 ml of benzene.
The
mixture was stirred at r.t. for 1 h, then at reflux for 4 h. After cooling at
60°C, was added
a solution of 0.073 g of palladium tetrakis triphenylphosphine in 3 ml of
benzene
followed by a solution of 0.55 ml of cyclohexanecarbonyl chloride in 3 ml of
benzene.
The reaction mixture was stirred 2.5 h at r.t.. After overnight resting, the
mixture was
diluted with EtOAc and filtered on a celite panel; the filtrate was washed
with a aq.
saturated solution of ammonium chloride, aq. NaHCO3 and brine, dried and
evaporated to
dryness. The crude was purified by flash chromatography (PE-EtOAc 100 : 4) to
afford 1
g of the title compound.
1H-NMR (CDCl3, ~: 1.10- 2.05 (m, lOH), 2.25-2.48 (m, 1H), 3.40 (s, 3H), 4.50
(s, 2H),
5.18 (s, 2H), , 7.25-7.50 (m, 4H)
4-Cyclohexyl-4-oxo-3-(2-methoxymethylphenyl)-butyraldehyde diethyl acetal
(Compound 61 c)
The title compound was prepared using the method described for Compound 2b but
using
Compound 61b instead of 1-(2-trifluoromethoxyphenyl)-propan-2-one. Usual work-
up
procedure and purification afforded the title compound.
4-CyclohexYl-4-oxo-3-(2-methoxymethylphenyl)-butyraldehyde (Compound 61d)
The title compound was obtained following the procedure described for Compound
1 e,
but using as a starting material Compound 61c instead of compound ld. The
title product
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
79
was used in the next step without further purification.
1-~4-Cyclohexyl-3-(2-methoxymethylphenyl)-4-oxobutyl~-4-(4-fluoro-2-
methoxyphenyl)-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 61 d instead of Compound 1 a and 1-(4-fluoro-2-
inethoxyphenyl)-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography (PE :EtOAc 6:4) to afford the title
compound.
[M+H]+=483.6
Example 62 1-[4-Cyclohexyl-4-hydroxy-3-(2-methoxymethylphenyl)- butyl]-4-(4-
fluoro-2-methoxyphenyl)-piperazine
The title compound was synthesised using the method described fox Compound lc
but
starting from the compound of Example 61 instead of Compoundlb. The title
compound
was isolated after the usual work-up procedure.
[M+H]+=485.5
Example 63 1-[5-(2,3-Dihydro-1,4-benzodioxinyl)]-4-[4-cyclohexyl-3-(2-
methoxymethylphenyl)-4-oxobutyl]-piperazine
The title compound was prepared using the method described for the Compound of
Example 1 but using Compound 61d instead of Compound le and 1-[5-(2,3-dihydro-
1,4-
benzodioxinyl)]-piperazine instead of 1-(2,2,2-trifluoroethoxyphenyl)-
piperazine. The
crude was purified by flash chromatography to afford the title compound.
[M+H]+=493.7
Example 64 1-[4-Cyclohexyl-4-hydroxy-3-(2-methoxymethylphenyl)- butyl]-4-(2,3-
dihydro-1,4-benzodioxinyl)-piperazine
The title compound was synthesised using the method described for Compound lc
but
starting from the compound of Example 63 instead of Compoundlb. The title
compound
was isolated after the usual work-up procedure.
[M+H]+=495.5
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
Example 65 (RS,SR) 1-[4-Cyclohexyl-4-methylaminothiocarbonyloxy-3-(2-
fluorophenyl)-butyl]-4-(2-methoxyphenyl)-piperazine
To a solution of 0.088 g of Compound 45e in 0.6 ml of pyridine stirred at
0°C, was added
0.073 g of methyl isothiocyanate. The reaction mixture was stirred at r.t. fox
24 h and at
100°C for 10 h. After cooling, it was poured into water and extracted
with Et20,washed
with H20, dried (NazSO~) and evaporated to dryness in vacuo. Purification by
flash
chromatography afforded 0.04g of the title product.
[M+H]+=514.4
Example 66: Radioligand binding to recombinant 5-HT1A receptors
A. llletlaod:
Genomic clone coding for the human SHT1A-serotonergic receptor was stably
transfected in a human cell line (HeLa). HeLa cells were grown as monolayers
in
Dulbecco's modified Eagle medium (DMEM), containing 10% foetal bovine serum.,
gentamycin (0.1 mg/ml) and 5% carbon dioxide, at 37°C. The cells were
detached from
the growth flask at 95% confluence by a cell scraper and were lysed in cold 5
mM Tris
and 5 mM EDTA buffer (pH 7.4). The homogenates were centrifuged at 40000 x g x
20
minutes and the pellets were resuspended in a small volume of cold 5 mM Tris
and 5 mM
EDTA buffer (pH 7.4) and immediately frozen and stored at -70°C until
use. On the day
of experiment, the cell membranes were resuspended in incubation buffer: 50 mM
Tris
HCl (pH 7.4), 2.5 mM MgCl2, 10 mM pargyline (Fargin et al., Nature 335, 358-
360,
1988). The membranes were incubated in a final volume of 1 ml fox 30 minutes
at 30°C
with 1 nM [3H]8-OH-DPAT, in the absence or presence of the test compounds. Non-
specific binding was determined in the presence of 10 ~,M 5-HT. Incubation was
stopped
by addition of cold Tris-HC 1 buffer and rapid filtration through a 0.2%-
polyethyleneimine-pretreated Whatman-GF/B or Schleicher-&-Schuell-GF52 filter.
B. Results
The affinities of the tested compounds were evaluated as inhibition of
specific binding of the radioligand to 5-HT1A receptors (ICSO) by using the
non-linear
curve-fitting program Allfit (De Lean et al., Am. J. Physiol. 235, E97-E102
(1978). The
ICSO value was converted to an affinity constant (I~i) by the equation of
Cheng & Prusoff
(Cheng Y. C., Prusoff W. H., Biochem. Pha~naacol. 22, 3099-3108 (1973)).
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
81
The results reported in Table 1 show that the compounds of the invention
tested had a high affinity for the 5-HT1A receptor.
TABLE 1
Binding affinity for SHT1A receptors
Example Ki (nlVl]
1 1.45
8.31
6 3.66
7 7.27
9 1.90
1.68
11 3.65
12 3.01
13 0.53
14 0.401
14a 0.878
14b 0.965
1.69
16 0.90
17 0.64
18 0.81
19 0.69
3.19
21 0.77
22 2.03
23 0.63
24 0.64
1.18
26 2.62
27 1.09
28 2.92
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
82
29 1.08
30 5.43
31 2.71
32 3.57
33 5.03
34 1.67
35 1.47
36 0.56
37 5.03
39 2.92
40 1.84
42 4.87
44 ' 1.14
45 0.54
46 0.72
47 0.85
48 0.36
Example 67 Effects on rhythmic bladder-voiding contractions induced by bladder
filling in anaesthetised rats
A. lllethod:
Female Sprague-Dawley rats weighing 225-275 g (Crl: CD~ (SD) IGS BR,
Charles River Italia) were used. The animals were housed with free access to
food and
water and maintained on a forced 12-hour alternating light-dark cycle at 22-
24°C for at
least one week, except during the experiment. The activity on rhythmic bladder
voiding
contractions was evaluated according to the method of Dray (Dray J.,
Pharmacol.
Methocls,13:157, 1985), with some modifications as in Guarneri (Guarneri,
Pl2armacol.
Res. 27:173, 1993). Briefly, rats were anaesthetised by subcutaneous injection
of 1.25
g/lcg (5 ml/kg) urethane, after which the urinary bladder was catheterised via
the urethra
using PE 50 polyethylene tubing filled with physiological saline. The catheter
was tied in
place with a ligature around the external urethral orifice and was connected
to
conventional pressure transducers (Statham P23 IDIP23 XL). The intravesical
pressure
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
83
was displayed continuously on a chart recorder (Battaglia Rangoni KV 135 with
DCI/TI
a~.nplifier). The bladder was then filled via the recording catheter by
incremental volumes
of warm (37°C) saline until reflex bladder-voiding contractions
occurred (usually 0.8-1.5
ml). For intravenous injection of bioactive compounds, PE 50 polyethylene
tubing filled
with physiological saline was inserted into the jugular vein.
From the cystometrogram, the number of contractions recorded 15 minutes
before (basal values) and after treatment, as well as the mean amplitude of
these
contractions (mean height of the peaks in mmHg), was evaluated.
Since most compounds produced an effect that was relatively rapid in
onset and lead to a complete cessation of bladder contractions, bioactivity
was
conveniently estimated by measuring the duration of bladder quiescence (i.e.,
the length
of the time during which no contractions occured). The niunber of tested
animals showing
a reduction in the number of contractions higher than 30% of that observed in
the basal
period was also recorded.
To compare the potency of the tested compounds for inhibiting the bladder
voiding contractions, equieffective doses that result in the disappearance of
contractions
for a time of 10 minutes (EDlomin) were computed by means of linear regression
using the
least square method. The extrapolated doses which induces a reduction in the
number of
contractions greater than 30% in 50% of the treated rats (EDSO) was evaluated
by the
method of Bliss (Bliss C. L, Qua~~t J. Pharm. Pharmacol. 11, 192-216, 1938).
B. Results
The rapid distension of the urinary bladder in urethane-anaesthetised rats
produced a series of rhythmic bladder-voiding contractions whose
characteristics have
been described (Maggi et al., B~aih Res. 380:83, 1986; Maggi et al., J.
Pha~macol. Exp.
Thei~., 230: 500, 1984). The frequency of these contractions is related to the
sensory
afferent arm of reflex micturition and to the integrity of the micturition
centre, while their
amplitude depends on the function of the reflex efferent arm. In this model
system,
compounds that act mainly on the central nervous system (such as morphine)
cause a
block in voiding contractions, whereas drugs that act at the level of the
detrusor muscle,
such as oxybutynin, lower the amplitude of the bladder contractions.
The results obtained are shown in table 2.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
84
TABLE 2
Effects on rhythmic bladder-voiding contractions after intravenous
administration
Example EDlomin EDso EDso
~ug/kg (frequency) (amplitude)
pg/kg wg~g
Ex. 45 584 127 n.a.
Morphine 50 30
n.a.
Oxybutynin 7770 > 10000 . 240
n.a. = not active; no significant reduction of the height of the pealcs
Data represent the EDiomin values (the extrapolated dose inducing 10
minutes of disappearance of the contractions), the EDso (frequency) values
(the
extrapolated doses inducing a reduction of the number of contractions > 30% in
50% of
treated rats) , and the EDso (amplitude) values (the extrapolated doses
inducing a 30%
reduction of amplitude of the contractions in 50% of treated rats).
The compounds of the present invention inhibited the frequency of
micturition, with no effects on their amplitude.
Example 68 Effect on cystometric parameters in conscious rats after oral
administration
A. Method:
Male Sprague-Dawley rats [Crl: CD~ (SD) IGS BR] of 300-400 g supplied
by Charles River Italia were used. The animals were housed with free access to
food and
water and maintained on a forced 12-hour-light/12-hour-dark cycle at 22-
24°C of
temperature, except during the experiment. To quantify urodynamic parameters
in
conscious rats, cystometrographic studies were performed according to the
procedure
previously reported (Guaxneri et al., Phaxmacol. Res. 24: 175, 1991).
Briefly, rats were anaesthetised by intraperitoneal administration of 3
ml/lcg of Equithensin solution (pentobarbital 30 mg/kg and chloral hydrate 125
mglkg)
and placed in a supine position. An approximately-10-mm-long midline incision
was
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
made in the shaved and cleaned abdominal wall. The urinary bladder was gently
freed
from adhering tissues, emptied and then cannulated via an incision in the
bladder body,
using a polyethylene cannula (0.58-mm internal diameter, 0.96-mm external
diameter)
which had been permanently sutured with sills thread. The cannula was
exteriorised
through a subcutaneous tunnel in the retroscapular area, where it was
connected to a
plastic adapter in order to avoid the rislc of removal by the animal. For drug
testing, the
rats were utilised one day after implantation.
On the day of the experiment, the rats were placed in modified Bollman
cages, i.e., restraining cages that were large enough to permit the rats to
adopt a normal
crouched posture, but naxrow enough to prevent turning around. After a
stabilisation
period of about 20 minutes, the free tip of the bladder cannula was connected
through a
T-shaped tube to a pressure transducer (Statham P23XL) and to a peristaltic
pump
(Gilson minipuls 2) for continuos infusion of a warm (37°C) saline
solution into the
urinary bladder, at a constant rate of 0.1 ml/minute. The intraluminal-
pressure signal
during infusion of saline into the bladder was continuously recorded on a
polygraph
(Rectigraph-8K San-ei with BM614/2 amplifier from Biomedica Mangoni). The
cystometrogram was used to evaluate the urodynamic parameters of bladder
volume
capacity (BVC) and micturition pressure (MP). BVC (ml) was defined as the
volume of
saline infused into the bladder necessary to induce detrusor contraction
followed by
micturition. MP (mmHg) was defined as the maximal intravesical pressure caused
by
contraction during micturition. Basal BVC and MP values were evaluated as mean
of the
values observed in the cystometrograms recorded in an initial period of 30-60
minutes.
Following determination of basal BVC and MP, the infusion was interrupted and
the test
compounds were administered orally by a stomach tube. Bladder infusion was
resumed
and changes in BVC and MP were evaluated from the mean values obtained in the
cystometrograms observed during 1, 2, 3, 4 and 5 hours aftex treatment.
Compounds were
administered in a volume of 2 ml/lcg and groups of control animals received
the same
amount of vehicle (0.5% methocel in water) orally.
Statistical afaalysis
Data were expressed as mean ~ standard error. The percent changes of
BVC and MP vef°sus the basal values, as well as 0 values (difference in
ml or mmHg) of
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
86
BVC and MP (BVC or MP at time "x" minus basal value), were also evaluated for
each
rat/time.
Data were reported as % changes vev~sus basal values.
Statistical analysis on BVC and MP values, as well as on ~ values, was
performed by S.A.S./STAT software, version 6.12. The observed differences
between
vehicle (control) and test treatments were evaluated on d values of BVC and
MP,
whereas the differences between the values at different times versus basal
values were
analyzed on original BVC and MP data.
Example 69 Inhibition of stereotypy (rhythmic forepaw treading) induced by 8-
OH-
DPAT in rats (post-synaptic antagonism)
~1. Method:
The inhibitory effect of 5-HT1A-receptor antagonists on stereotyped
forepaw treading induced in rats by subcutaneous injection of 8-OH-DPAT was
evaluated
by the method of Tricldebank (Tricklebanlc et al., Eu~. J. Pha~macol., 117:
15, 1985) with
minor modifications as described below.
Male Sprague-Dawley rats [Crl: CD° (SD) IGS BR] weighing 150-175 g
from Charles River Italia were used. The animals were housed with free access
to food
and water and maintained on a forced 12-hour-light/12-hour-dark cycle at 22-
24°C of
temperature. On the day of the experiment, the rats were placed singly in
clear plastic
containers, 10-15 minutes before administration of the vehicle or compounds to
be tested.
For evaluation of antagonistic activity after oral administration, the
compounds were
administered 1 and 4 hours before induction of stereotypy by 8-OH-DPAT (1
mg/kg
subcutaneously). Observation sessions last 30 seconds and were begun 3 minutes
after 8-
OH-DPAT treatment and repeated every 3 minutes over a period of 15 minutes.
The appearance of the symptom induced by postsynaptic stimulation of 5-
HT1A receptors was noted, and the intensity was scored using an intensity
scale in which:
0 = absent, 1 = equivocal, 2 = present and 3 = intense. Behavioural scores for
treated rats
were accumulated throughout the observation time (5 observation periods) and
expressed
as mean values of 4 rats/dose. Change in mean values of treated animals in
comparison
with control (vehicle) group, expressed as per-cent inhibition, were used to
quantify the
antagonistic activity.
CA 02489449 2004-12-14
WO 03/106443 PCT/EP03/06289
87
B. Result
The results obtained are shown in table 3.
The compou~lds of the present invention, in particular Ex. 45, showed
potent and long-lasting inhibition of stereotypy induced by 8-OH-DPAT.
TABLE 3
Inhibition of forepaw treading induced by 8-OH-DPAT in rats (post-synaptic
antagonism)
Example Dose % Inhibition
of forepaw
treading
(mg/kg p.o.) 1 h 4 h
Ex. 16 10 40 17
Ex. 31 10 60 18
Ex. 45 10 100 81
Ex. 47 10 5~ 58
Ex. 48 ~ 10 90 74