Note: Descriptions are shown in the official language in which they were submitted.
CA 02491932 1999-06-17
io NOVEL ANTIHISTAMINIC PIPERIDINE DERIVATIVES AND INTERMEDIATES
FOR THE PREPARATION THEREOF
BACKGROUND OF THE INVENTION
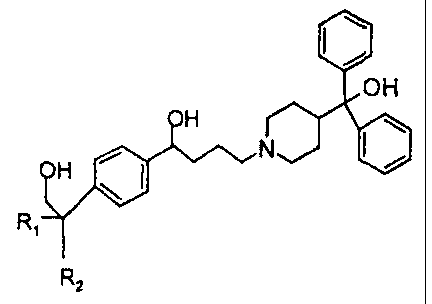
is This invention relates to novel piperidine derivatives of formula (I) and a
process for the preparation thereof.
R
wherein
Rt is H or C~-Csalkyl wherein the C~Csalkyl moiety is straight or branched;
2o RZ is -COOH or -COOalkyl wherein the alkyl moiety has from 1 to 6 carbon
atoms and is straight or branched; or
stereoisomers or pharmaceutically acceptable acid addition salt thereof.
Terfenadine, a-{4-(1,1-dimethylethyl)phenyl]-4-(hydroxydiphenylmethyl)-1-
2s piperidinebutanol, is a known antihistaminic agent which is currently
available
commercially under the name Seidane~ with a recommended dosage of 60 mg b.i.d.
CA 02491932 1999-06-17
-2-
(See PHYSICIAN'S DESK REFERENCE, 52nd Edition, 1998, pp. 1238-1244,
Medical Economics Data, a division of Medical Economics Company, Inc.
Montvale,
New Jersey). Terfenadine is disclosed in U.S. Patent 3,878,217, issued April
15,
1975. Sorken and Heel have provided a review of the pharmacodynamic properties
s and therapeutic efficacy of terfenadine [Drugs 29, 34-56 (1985)].
Terfenadine undergoes extensive (99%) first pass metabolism to two primary
metabolites (fexofenadine) and an inactive dealkylated metabolite.
Fexofenadine,
a.k.a. 4-[1-hydroxy-4-[4-(hydroxydiphenyilmethyl)-1-piperidinyl]butyl-a,a-
dimethyl-
benzeneacetic acid, has been disclosed as an antihistaminic agent having oral
io activity in U.S. Patent No. 4;254,129, issued March 3, 1981. It is
currently available
commercially under the name Allegra~ (See PHYSICIAN'S DESK REFERENCE,
52nd Edition, 1998, pp. 1189-1190, Medical Economics Data, a division of
Medical
Economics Company, Inc. Montvale, New Jersey).
~5 SUMMARY OF THE INVENTION
An object of the present invention is to provide novel piperidine derivatives
of
formula (I) useful for the treatment of allergic disorders. It is a further
object to
provide a process for the preparation of said derivatives and to provide novel
2o intermediates useful for preparation of the same.
Additionally, it is an object of the present invention to provide a method of
treating a patient suffering from an allergic disorder comprising
administering to said
patient an effective antiallergic amount of a compound of formula (I).
Furthermore, it is an object of the present invention to provide a composition
2s comprising an assayable amount of a compound of formula (I) in admixture or
otherwise in association with one or more pharmaceutically acceptable carriers
or
excipients.
Another object of the present invention is to provide novel processes for the
preparation of intermediates useful for the synthesis of fexofenadine and
related
so compounds.
Upon further study of the specification and appended claims, further objects
and advantages of this invention will become apparent to those skilled in the
art.
CA 02491932 1999-06-17
-3-
DETAILED DESCRIPTION OF THE INVENTION
Compounds of the formula (I) can be prepared using techniques and
procedures weA known and appreciated by one of ordinary skill in the art.
As used herein, straight or branched alkyl groups having from 1 to 6 carbon
atoms as referred to herein are methyl, ethyl, n-propyl, isopropyl, n-butyl,
sec-butyl,
isobutyl, tert-butyl, and straight- and branched-chain pentyl and hexyl.
The piperidine derivatives of the formula (I) can form pharmaceutically
acceptable salts. Pharmaceutically acceptable acid addition salts of the
compounds
io of this invention are those of any suitable inorganic or organic acid.
Suitable
inorganic acids are, for example, hydrochloric, hydrobromic, sulfuric, and
phosphoric
acids. Suitable organic acids include carboxylic acids, such as, acetic,
propionic,
gfycolic, lactic, pyruvic, malonic, succinic, fumaric, malic, tartaric,
citric, cyclamic,
ascorbic, malefic, hydroxyrnaleic, and dihydroxymaleic, benzoic, phenylacetic,
4-
is aminobenzoic, 4-hydroxybenzoic, anthranillic, cinnamic, saficyclic, 4-
aminosalicyclic,
2-phenoxybenzoic, 2-acetoxybenzoic, and mandelic acid, sulfonic acids, such
as,
methanesulfonic, ethanesulfonic and ~3-hydroxyethanesuifonic acid. Non-toxic
salts
of the compounds of the above-identified formula formed with inorganic or
organic
bases are also included within the scope of this invention and include, for
example,
2o those of alkali metals, such as, sodium, potassium and lithium, alkaline
earth metals,
for example, calcium and magnesium, light metals of group ItIA, for example,
aluminum, organic amines, such as, primary, secondary or tertiary amines, for
example, cyclohexylamine, ethylamine, pyridine, methylaminoethanol and
piperazine.
The salts are prepared by conventional means, as for example, by treating a
2s piperidine derivative of formula (I) with an appropriate acid or base.
The novel process for preparing the piperidine derivatives of formula (I) is
set
forth in Scheme A. in Scheme A, R1, and R2 are C~- Csalkyl wherein the C~-
Csalkyl
moiety is straight or branched; R3 is H or C,-Csalkyl wherein the C~-Csalkyl
moiety is
straight or branched; and X is CI, Br or I.
CA 02491932 1999-06-17
-4-
SCHEME A
I
X . .O ~ , ,
I + CH30NHCH3 Step.A N
O O
tCzC03 2
O Step B I O X
.O
X --~ N ~ -"
2 + X A1CI3/DCM O
3
O X
Step C
3 ~ N '
O
EtOAc
4
O
Step D OH
.i
KOH/EtOH O
O
S ORS
5 + R~OH O ~ v
HZS04
6
CA 02491932 1999-06-17
-S-
SCHEME A Cont.
O
O'I Step F R3 ~ I
RX ~ V
6 + R O~O R2 s
2
R,02C C02R2
7
O
Step G ~ X
7 + HX '' R3 ~ I
ROH
R,02C 0282
8
I
~ v
Step H O Oh~=J
N
8 + H ~ ~ R3 ~ i
K2C03
H R~02C 0282
I
!JH-=J
Step I N
9 '-'
reduction
R~
I
10 Step .! H _ N O
NaOH/H20 R
OH 11
HOOC
CA 02491932 1999-06-17
Scheme A provides a general synthetic procedure for preparing the
compounds of formula (I).
In step A, a phenylacetyl halide (1) wherein X is GI, Br, or I, is reacted
with N-
O-dimethylhydroxylamine hydrochloride to provide N-methoxy-N-methyl
benzeneacetamide (2).
For example, a suitable phenylacetyl halide (1) is contacted with a molar
excess of potassium carbonate in a suitable solvent such as toluene. Suitable
phenylacetyl halides include phenylacetyl chloride, phenylacetyl bromide or
phenylacetyl iodide. A preferred phenylacetyl halide is phenylacetyl chloride.
A
to molar equivalent of N-O-dimethylhydroxylamine hydrochloride dissolved in
water is
then added. The reaction mixture is stirred for a period of time ranging from
1 to 24
hours at a temperature range of from 0°C to 60°C. A preferred
stirring time is 3
hours. A preferred temperature is 25'C. N-methoxy-N-methyl-benzeneacetamide is
(2) is recovered from the reaction zone by extractive methods as are known in
the
is art.
In step B, N-methoxy-N-methyl-benzeneacetamide (2) is acylated with a
suitable 4-halo-substituted butyrylhalide of the formula
O
X
X '
wherein each X is independently CI, Br or I;
2o under Friedel-Crafts conditions to give a mixture of para, meta substituted
w-halo-a-
keto-benzeneacetamide (3). Surprisingly, the para isomer is readily isolated
by
subsequent crystallization as set forth in step C.
For example, in step B, N-methoxy-N-methylbenzeneacetamide (2) is
contacted with suitable 4-halo-substituted butyrylhalide under the general
2s conditions of a Friedel-Crafts acylation using a suitable Lewis acid.
Examples of
suitable 4-halo-substituted butyrylhalides include 4-chlorobutyrylchloride, 4
bromobutyrylbromide, and the like. A preferred 4-halo-substituted
butyrylhalide is 4
chlorobutyrylchloride. The reaction is carried out in a solvent, such as
carbon
disulfide, 1,2-dichloroethane, n-hexane, acetonitrile, 1-nitropropane,
nitromethane,
so diethyl ether, carbon tetrachloride, methylene chloride, tetrachloroethane
or
nitrobenzene with dichloromethane being the preferred solvent. The reaction
time
CA 02491932 1999-06-17
varies from about 1/2 hour to 25 hours at a temperature range of from
0°C to 40°C.
A preferred stirring time is 6 hours. A preferred temperature is 40°C.
The mixture of
para, meta substituted w-halo-a-keto-benzeneacetamide (3) is recovered from
the
reaction zone by an aqueous quench followed by extractive methods as are known
in
the art.
Suitable Lewis acids for the acylation reaction described in step B are well
known and appreciated in the art. Examples of suitable Lewis acids are boron
trichloride, aluminum chloride, titanium tetrachloride, boron trifluoride, tin
tetrachloride
and zinc chloride. The selection and utilization of suitable t_ewis acids for
the
to acylation reaction of step B is well known and appreciated by one of
ordinary skill in
the art.
The para-substituted cu-halo-a-keto-benzeneacetamide (3) is purified by
recrystallization techniques as set forth in step C.
For example, the product of the extractive methods as set forth in step B is
is stirred in a suitable organic solvent such as a mixture of heptane/ethyl
acetate (ca.
4:1) and collected. The solid is dissolved in a suitable solvent such as ethyl
acetate
at a temperature range of from 25°C to 76°C. A preferred
temperature is 76°C. The
solution is then contacted with charcoal. This mixture is then filtered and
diluted with
a suitable solvent such as heptane. The resultant slurry is then heated until
a
2o homogenous solution is obtained. Substantially pure para-substituted w-halo-
a-keto-
benzeneacetamide (4) crystallizes upon standing at room temperature.
In step D, the substantially pure para-substituted w-halo-a- keto-
benzeneacetamide (4) is hydrolyzed to give the 4-
(cyclopropylcarbonyl)benzeneacetic acid (5).
2s For example, the substantially pure para-substituted w-halo-a-keto-
benzeneacetamide (4) is contacted with a molar excess of an appropriate base
such
as potassium hydroxide in a suitable solvent such as ethanol. The reactants
are
typically stirred together for a period of time ranging from 1 to 24 hours at
a
temperature range of from 0°C to 78°C. A preferred stirring time
is 18 hours. A
3o preferred temperature is 25°C. The 4-
(cyclopropylcarbonyl)benzeneacetic acid (5) is
recovered from the reaction zone by acidification and extractive methods as
are
known in the art.
In step E, the 4-(cyclopropylcarbonyl)benzeneacetic acid (5) is esterified to
CA 02491932 1999-06-17
_g_
give the corresponding 4-(cyclopropylcarbonyl)benzeneacetic acid ester (6).
For example, the appropriate 4-(cyclopropylcarbonyl)benzeneacetic acid (5) is
reacted with an excess of an appropriate C~-Cs alcohol which is straight or
branched
in the presence of a catalytic amount of mineral acid, such as hydrochloric
acid or
sulfuric acid, hydrochloric acid being preferred, at a temperature range of
from 25°C
to 78°C. The reactants are typically stirred together for a period of
time ranging
from 2 to 72 hours. A preferred stirring time is 24 hours. A preferred
temperature is
25°C. The corresponding 4-(cyclopropylcarbonyl)benzeneacetic acid ester
(6) is
recovered from the reaction zone by basification and extractive methods as are
~o known in the art. It can be purified by silica gel chromatography.
In step F, the appropriate 4-(cyclopropylcarbonyl)benzeneacetic acid ester (6)
is acylated with the appropriate acylating agent to give the corresponding [4
(cyclopropylcarbonyl)phenylJpropanedioic acid diester (7).
For example, the appropriate 4-(cyclopropylcarbonyl)benzeneacetic acid ester
is (6) is reacted with a slight molar excess of a suitable acylating agent.
Suitable
acylating agents include dialkylcarbonates, such as, dimethylcarbonate or
diethylcarbonate; or chloroformates, such as, methyl chloroformate or ethyl
chloroformate. The reaction is typically conducted in a suitable aprotic
solvent in the
presence of a suitable non-nucleophilic base from about 0.5 hour to 7 days and
at a
2o temperature of about 0°C to the reflex temperature of the solvent. A
preferred stirring
time is 3 days. A preferred temperature is 25°C. Suitable solvents for
the acylation
reaction include tetrahydrofuran, dioxane, or tert-butyl methyl ether. A
preferred
solvent is tetrahydrofuran. Suitable non-nucleophilic bases for the acylation
reaction
include inorganic bases, for example, sodium bicarbonate, potassium
bicarbonate, or
2s hydrides, for example, sodium hydride or potassium hydride or alkoxides,
for
example, potassium tert-butoxide. A preferred base is sodium
bis{trimethylsilyi)amide.
The derivative formed upon acylation is optionally alkylated with a suitable
alkylating agent in situ subsequent to the acylation. Suitable alkylating
agents
3o include alkyl halides, such as, iodomethane, chloromethane or bromomethane;
or
dialkylsulfates, such as, dimethylsulfate or diethylsulfate. The reactants are
typically
stirred together for a period of time ranging from 1 to 48 hours at a
temperature
range of from 0°C to 30°C. A preferred stirring time is 24
hours. A preferred
CA 02491932 1999-06-17
-9-
temperature is 25°C.
The corresponding [4-(cyclopropylcarbonyi)phenylJpropanedioic acid diester
(7) is recovered from the reaction zone by extractive methods as are known in
the
art. it can be purified by silica gel chromatography and/or recrystallization.
s While not necessary for utilization in the acylation and subsequent
alkylation in
step F, the keto functionality of the 4-(cyclopropylcarbonyl)benzeneacetic
acid ester
(6) may be protected with a suitable protecting group. The selection and
utilization of
suitable protecting groups for the keto group of structure (6) is well known
by one of
ordinary skill in the art and is described in "Protective Groups in Organic
Synthesis",
~o Theodora W. Greene, Wiley (1981). For example, suitable protecting groups
for the
keto functionality include acyclic ketals such as dimethyl ketal; cyclic
ketals such as
1,3-dioxanes and 1,3-dioxalanes; acyclic dithioketals such as S,S-dimethyl
ketal;
cyclic dithio ketals such as 1,3-dithiane and 1,3-dithiolane derivatives;
acyclic
monothio ketals; cyclic monothio ketals such as 1,3-oxathiolanes.
is In step G, the appropriate [4-(cyclopropylcarbonyl)phenylJpropanedioic acid
diester (7) is ring-opened to give the corresponding [4-(4-halo-1-oxo-
butyl)phenyl]propanedioic acid diester (8).
For example, the appropriate [4-(cyclopropylcarbonyl)phenyl]propanedioic acid
diester (7) is contacted with a suitable hydrogen halide such as hydrogen
chloride,
2o hydrogen bromide, or hydrogen iodide in a suitable organic solvent or in
the absence
of solvent. Suitable organic solvents include alcohol solvents, such as,
ethanol,
methanol, isopropyl alcohol, or n-butanol; hydrocarbon solvents, such as,
benzene,
toluene or xylene; halogenated hydrocarbons, such as chlorobenzene, chloroform
or
methylene chloride or dimethylformamide or acetic acid or dioxane at a
temperature
2s range of from 0°C to 100°C. The absence of solvent is
preferred. The reactants are
typically stirred together for a period of time ranging from 1 hour to 24
hours. A
preferred stirring time is 4 hours to 16 hours. A preferred temperature range
is 60°C
to 80°C. If solvent is present, the [4-(4-halo-1-oxo-
butyl)phenyf]propanedioic acid
diester (8) is recovered from the reaction zone by extractive methods as are
known in
3o the art and subsequent evaporation of the solvent.
In step H, the halo functionality of the appropriate [4-(4-halo-1-oxo-
butyl)phenylJpropanedioic acid diester (8) is alkylated with a-(4-
pyridyl)benzhydrol
(commercially available from A(drich Chemicals) to give the corresponding [4-
[4-[4-
CA 02491932 1999-06-17
-10-
(hydroxydiphenyfmethyl)-1-piperidinyl]1-oxobutyl]phenyl]propanedioic acid
diester
(9).
For example, the alkylation reaction is carried out in a suitable solvent
preferably in the presence of a non-nucleophilic base and optionally in the
presence
of a catalytic amount of an iodide source, such as potassium or sodium iodide.
The
reaction time varies from about 4 hours to 7 days and the reaction varies from
about
25°C to the reflux temperature of the solvent. A preferred stirring
time is 3 days. A
preferred temperature is the reflux temperature of the solvent. Suitable
solvent for
the alkylation reaction include alcohol solvents such as, methanol, ethanol,
isopropyl
~o alcohol, or n-butanol; ketone solvents, such as, methyl isobutyl ketone;
hydrocarbon
solvents, such as, benzene, toluene or xylene, and mixtures thereof with
water;
halogenated hydrocarbons, such as, chlorobenzene or methylene chloride or
dimethylformamide. A preferred solvent is toluene/water (10:4). Suitable non-
nucleophilic bases for the alkyiation reaction include inorganic bases, for
example,
is sodium bicarbonate, potassium bicarbonate, or potassium carbonate or
organic
bases, such as, a trialkylamine, for example, triethylamine, or pyridine, or
an excess
of a-(4-pyridyl)benzhydrol may be used. A preferred base is potassium
carbonate.
The corresponding [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
oxobutyl]phenyl]propanedioic acid diester (9) is recovered from the reaction
zone by
2o extractive methods as are known in the art. it can be purified by silica
gel
chromatography.
While not necessary for the utilization in the aikylation of step H, the keto
functionality of the [(4-halo-1-oxo-butyl)phenyl]propanedioic acid diester (8)
may be
protected with a suitable protecting group. The selection and utilization of
suitable
2s protecting groups for the keto group of structure (8) is well known by one
of ordinary
skill in the art and is described in "Protective Groups in Organic Synthesis",
Theodora
W. Greene, Wiley (1981). For example, suitable protecting groups for the keto
functionality include acyclic ketals such as dimethyl ketal; cyclic ketals
such as 1,3-
dioxanes and 1,3-dioxalanes; acyclic dithioketals such as S,S-dimethyl ketal;
cyclic
3o dithio ketals such as 1,3-dithiane and 1,3-dithiolane derivatives; acyclic
monothio
ketals; cyclic monothio ketals such as 1,3-oxathiolanes.
In step I, the appropriate [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidiny(]-1-
oxobutyl]phenyl]propanedioic acid diester (9) is reduced selectively to the
CA 02491932 1999-06-17
-11-
corresponding 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-benzeneacetic acid ester (10). This is accomplished by
utilizing a
suitable selective reducing agent. A suitable selective reducing agent is a
reagent or
combination of reagents which will selectively reduce only one ester of the
propanedioic acid diester functionality to the corresponding hydroxymethyl
moiety
while not reducing the second ester of the propanedioic acid diester
functionality.
Suitable selective reducing agents include lithium tri-tert-
butoxyaluminohydride or the
combination of a suitable silane and a suitable titanocene-based catalyst.
For example, the appropriate [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
to oxobutyl]phenyl]propanedioic acid diester (9) is contacted with a suitable
selective
reducing agent such as lithium tri-tent-butoxyaluminohydride in a suitable
solvent
such as tetrahydrofuran, diethyl ether, or dioxane. A preferred solvent is
tetrahydrofuran. The reactants are typically stirred together for a period of
time
ranging from 0.5 hours to 168 hours at a temperature range of from 0°C
to 65°C. A
is preferred stirring time is 48 hours. A preferred temperature is
25°C.
Catalytic reduction may also be employed in the preparation of 4-[1-hydroxy-4-
[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-(hydroxymethyl)-
benzeneacetic acid
ester (10) from an appropriate [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-
1-
oxobutyl]phenyl]propanedioic acid diester (9), using, for example, a suitable
2o titanocene-based catalyst in which a suitable silane, such as,
polymethylhydrosiloxane, serves as the stoichiometric reductant. Suitable
titanocene-based catalysts include the active catalytic species commonly known
as
"Cp2TiH". It is well known by one of ordinary skill in the art that the active
catalytic
species "Cp2TiH" may be generated, for example, by the addition of 2
equivalents of
2s ethyl magnesium bromide to 1 equivalent of Cp2TiCl2 in a suitable solvent
such as
tetrahydrofuran.
For example, the catalytic reduction is carried out in a suitable solvent such
as
tetrahydrofuran or diethyl ether or dioxane at temperatures ranging from about
25°C
to the reflux temperature of the solvent. A preferred temperature for use with
the
3o catalytic reduction is 65°C. The reaction time varies from about 8
hours to 24 hours.
A preferred stirring time is 18 hours. The 4-[1-hydroxy-4-[4-
(hydroxydiphenytmethyl)-
1-piperidinyl]butyl]-a-(hydroxymethyl)-benzeneacetic acid ester (10) is
recovered
from the reaction zone after work-up with Bu4NF and utilization of extractive
CA 02491932 1999-06-17
-12-
methods as are known in the art. It can be purified by silica gel
chromatography.
In addition, a chiral catalytic reduction may also be employed in the
preparation of enantiomerically pure 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-
1-
piperidinyl]butylJ-o-(hydroxymethyl)-benzeneacetic acid ester (10) from an
appropriate [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
oxobutyl]phenyl]propanedioic acid diester (9), using an appropriate chiral
titanocene
system, such as, for example, is described in Journal of the American Chemical
Society, 116, 11667-11670 (1994).
As one skilled in the art would appreciate, the j4-[4-[4-
io (hydroxydiphenylmethyl)-1-piperidinyl]-1-oxobutyl]phenyl]propanedioic acid
diester
(9) wherein the keto group is protected must be reacted with an appropriate
deprotecting reagent prior to the reduction reaction described in step I. The
selection
and utilization of appropriate deprotecting reagents is well known by one of
ordinary
skill in the art and is described in "Protective Groups in Organic Synthesis",
Theodora
is W. Greene, Wiley (1981). For example, cleavage of a dimethylketal
protecting group
on the keto functionality of the [4-[4-[4-(hydroxydiphenylmethyl)-1-
piperidinyl]-1
oxobutylJphenylJpropanedioic acid diester (9) can be achieved by using
iodotrimethylsilane or dilute acid as is known in the art.
In step J, the appropriate 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-
2o piperidinyl]butyl]-a-(hydroxymethyl)-benzeneacetic acid ester (10) is
optionally
hydrolyzed to the corresponding 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-'!-
piperidinyl]butyl]-a-(hydroxymethyl)-benzeneacetic acid (11).
For example, hydrolysis may be achieved using methods known in the art
such as potassium carbonate in methanol, methanolic ammonia, potassium
2s carbonate, potassium hydroxide, calcium hydroxide, sodium hydroxide,
magnesium
hydroxide, sodium hydroxide/pyridine in methanol, potassium cyanide in ethanol
and
sodium hydroxide in aqueous alcohols, with sodium hydroxide being preferred.
The
reaction is typically carried out in an aqueous lower alcohol solvent, such as
methanol, ethanol, isopropyl alcohol, n-butanol, 2-ethoxyethanol or ethylene
glycol or
3o pyridine. A preferred solvent is a mixture of
tetrahydrofuranlmethanollwater (3:2:1).
The reaction is typically carried out at temperatures ranging from room
temperature to
the reflux temperature of the solvent. A preferred temperature is 65°C.
The
reactants are typically stirred together for a period of time ranging from 1
to 24 hours.
CA 02491932 1999-06-17
-13-
A preferred stirring time is 4' hours. The 4-j1-hydroxy-4-[4-
(hydroxydiphenylmethyt)-
1-piperidinyl]butyl]-a-(hydroxymethyl)-benzeneacetic acid (11) is recovered
from the
reaction zone by acidification and extractive methods as are known in the art.
Of course it is understood that the compound of formula (I) may exist in a
s variety of stereoisomers. The compound has more than one chiral center. For
example, the benzylic carbon to which the carboxyl, hydroxymethyl and methyl
groups attach may exist in the (R) or the (S) form. In addition, the benzylic
carbon to
which the hydroxy, hydrogen and alkyl amino groups attach may exist in the (R)
or
the (S) form. It is further understood that the present invention encompasses
those
io compounds of formula (I) in each of their various structural and stereo
isomeric
configurations as individual isomers and as mixtures of isomers.
An alternative novel process for the preparation of 4-
(cyclopropylcarbonyl)benzeneacetic acid is set forth in Scheme B. This
compound is
useful for the synthesis of compounds of formula (I) as well as fexofenadine
and
is related compounds. In Scheme B, R~ is C~-Csalkyl and the C~-Csatkyl moiety
is
straight or branched.
CA 02491932 1999-06-17
-ia-
Scheme B
O
SteQ..~ ~ x
\ \
C02R~ COzR~
2
Sty
C02H
O -
steel \
C02H
In step a of Scheme B, the appropriate benzeneacetic acid ester (1) wherein
R~ is Ct-Csalkyl and the Ct-C6alkyl moiety is straight or branched, is
acylated with a
suitable 4-halo-substituted butyrylhalide of the formula
O
J
X
wherein each X is independently CI, Br, or 1, under Friedei-Crafts conditions
to give a
mixture of the corresponding para, meta substituted c~-halo-a-keto-
benzeneacetic
acid ester (2) wherein X is Ci, Br, or I
io For example, in step a, the appropriate benzeneacetic acid ester {1) is
contacted with a 4-halo-substituted butyrylhalide under the general conditions
of a
Friedel-Crafts acylation using a suitable Lewis acid. Examples of suitable 4-
halo-
substituted butyrylhalides include 4-chlorobutyrylchloride, 4-
bromobutyrylbromide,
and the like. A preferred 4-halo-substituted butyrylhalide is 4-
chlorobutyrylchloride.
~s The reaction is carried out in a solvent, such as carbon disulfide, 1,2-
dichloroethane,
CA 02491932 1999-06-17
-I5-
n-hexane, acetonitrile, 1-nitropropane, nitromethane, diethyl ether, carbon
tetrachloride, methylene chloride, tetrachloroethane or nitrobenzene with
dichloromethane being the preferred solvent. The reaction time varies from
about
1I2 hour to 25 hours at a temperature range of from 0°C to 40°C.
A preferred stirring
s time is 6 hours. A preferred temperature is 40°C. The mixture of
para, meta
substituted c~,~-halo-a-keto-benzeneacetic acid ester (2) is recovered from
the
reaction zone by an aqueous quench followed by extractive methods as are known
in
the art.
Suitable Lewis acids for the acylation reaction described in step a are well
Io known and appreciated in the art. Examples of suitable Lewis acids are
boron
trichloride, aluminum chloride, titanium tetrachloride, boron trifluoride, tin
tetrachloride
and zinc chloride. The selection and utilization of suitable Lewis acids for
the
acylation reaction of step a is well known and appreciated by one of ordinary
skill in
the art.
Is In step b of Scheme B, the mixture of meta- and para-substituted w-halo-a-
keto-benzeneacetic acid ester (2) is hydrolyzed to give a mixture of meta- and
para-
substituted (cyclopropylcarbonyl)benzeneacetic acid (3).
For example, the mixture of meta- and para-substituted w-halo-a-keto-
benzeneacetic acid (2) is contacted with a molar excess of an appropriate base
such
2o as lithium hydroxide or potassium hydroxide in a suitable solvent such as
ethanol.
The reactants are typically stirred together for a period of time ranging from
1 to 24
hours at a temperature range of from 0°C to 78°C. A preferred
stirring time is 18
hours. A preferred temperature is 25°C. The meta- and para-substituted
.
(cyclopropylcarbonyl)benzeneacetic acid (3) is recovered from the reaction
zone by
25 acidification and extractive methods as are known in the art.
Surprisingly, the substantially pure para isomer is readily isolated by
subsequent crystallization as set forth in step c of Scheme B.
For example, the product of the extractive methods as set forth in step b is
dissolved in a suitable organic solvent such as a mixture of heptane/ethyl
acetate
30 (ca. 4:1) with heating to reflux. The solution is treated with charcoal and
filtered.
Upon cooling, the resultant solid is collected and recrystallized from a
suitable
organic solvent such as ethyl acetate/heptane. Substantially pure para-
substituted
(cyclopropylcarbonyl)benzeneacetic acid (4) crystallizes upon standing at room
CA 02491932 1999-06-17
-16-
temperature.
As shown previously herein, 4-(cyclopropylcarbonyl)benzeneacetic acid has
utility as an intermediate in the synthesis of compounds of formula (I). 4-
(Cyclopropylcarbonyl)benzeneacetic acid may also be used as an intermediate in
the
process of preparing compounds of formula (7) as shown in Scheme C. Compounds
of formula (7) include fexofenadine and related compounds.
CA 02491932 1999-06-17
-17-
SCHEME C
step a ste b
2
step c-
3 R
H
4
step d O
5 step a ,
O
~h a.
OH
s~P ~ ~ OH i ( . a a
O
Rz 7
J
CA 02491932 1999-06-17
-t 8-
In step a of Scheme C, the 4-(cyclopropylcarbonyl)benzeneacetic acid (1) is
esterified to give the corresponding 4-(cyclopropylcarbonyl)benzeneacetic acid
ester
(2) wherein R~ is C1-Csalkyl and the C,-Csalkyl moiety is straight or
branched, under
conditions as set forth in Scheme A, Step E.
In step b of Scheme C, the 4-(cyclopropylcarbonyl)benzeneacetic acid ester
(2) is alkylated with a suitable alkylating agent to provide a corresponding
alkylated
[4-(cyclopropylcarbonyl)phenyl]benzeneacetic acid ester (3) wherein R~ is as
previously defined in step a and R2 and R3 are each independently C~-Csalkyl
wherein the C~-C6alkyl moiety is straight or branched.
to For example, the reaction is typically conducted in a suitable aprotic
solvent in
the presence of a suitable non-nucleophilic base. Suitable solvents for the
alkylation
reaction include diglyme, tetrahydrofuran, dioxane, or tart-butyl methyl
ether. A
preferred solvent is diglyme. Suitable non-nucleophilic bases for the
alkylation
reaction include sodium bis(trimethylsilyl)amide, inorganic bases, for
example,
is sodium bicarbonate, potassium bicarbonate, or hydrides, for example, sodium
hydride or potassium hydride or alkoxides, for example, potassium tart-
butoxide. A
preferred base is potassium tart-butoxide. Suitable alkylating agents include
alkyl
halides, such as, iodomethane, chloromethane or bromomethane; or
dialkylsulfates,
such as, dimethylsulfate, or diethylsulfate. The reactants are typically
stirred together
2o for a period of time ranging from 1 to 48 hours at a temperature range of
from 0°C to
80°C.
In step c of Scheme C, the appropriate alkylated [4-
(cyclopropylcarbonyl)phenyl]benzeneacetic acid ester (3) is ring opened to
provide
the corresponding [4-(4-halo-1-oxo-butyl)phenyi] benzene acetic acid ester (4)
2s wherein R~, RZ and R3 are as previously defined in step b and X is CI, Br
or I. The
reaction occurs under conditions set forth in step G of Scheme A.
In step d, the appropriate [4-(4-halo-1-oxo-butyl)phenyl] benzene acetic acid
ester (4) is alkylated with a-(4-pyridyl)benzhydrol to produce a [4-[4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-oxobutyl]phenyl]benzeneacetic acid
ester
30 (5) wherein R~, RZand R3 are as previously defined in step b, under
conditions that
were previously disclosed in United States Patent No. 4,254,129, the
disclosure of
which is herein incorporated by reference.
In step a of Scheme C, the appropriate [4-[4-[4-(hydroxydiphenylmethyl)-1-
CA 02491932 1999-06-17
-19-
piperidinyl]-1-oxobutyl]phenyl]benzeneacetic acid ester {5) is reacted with a
suitable
reducing agent to produce a 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-
piperidinyl]butyl]benzeneacetic acid ester (6) wherein R~, R2 and R3 are as
previously
defined in step b, under conditions that were previously disclosed in United
States
Patent No. 4,254,129. Suitable reducing agents include, for example, sodium
borohydride or potassium borohydride. Catalytic reduction using, for example,
Raney nickel, palladium, platinum, or rhodium catalysts, may also be employed
in
step a of Scheme C.
In step f of Scheme C, the 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-
io piperidinyl]butyl]benzeneacetic acid ester (6) is optionally hydrolyzed to
produce the
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]benzeneacetic
acid (7)
wherein R2 and R3 are as previously defined in step b, under conditions that
were
previously disclosed in United States Patent No. 4,254,129.
While not necessary for utilization in alkylation steps b and d, the keto
is functionality of the 4-(cyclopropylcarbonyl)benzeneacetic acid ester (6)
may be
protected with a suitable protecting group. The selection and utilization of
suitable
protecting groups for the keto group of structure (6) is well known by one of
ordinary
skill in the art and is described in "Protective Groups in Organic Synthesis",
Theodora
W. Greene, Wiley (1981). For example, suitable protecting groups for the keto
2o functionality include acyclic ketals such as dimethyl ketal; cyclic ketals
such as 1,3-
dioxanes and 1,3-dioxalanes; acyclic dithioketals such as S,S-dimethyl ketal;
cyclic
dithio ketals such as 1,3-dithiane and 1,3-dithiolane derivatives; acyclic
monothio
ketals; cyclic monothio ketals such as 1,3-oxathiolanes.
The following examples present typical syntheses as described in Schemes A
2s B and C. Starting materials for use in Schemes A, 'B and C are readily
available to
one of ordinary skill in the art. These examples are understood to be
illustrative only
and are not intended to limit the scope of the present invention in any way.
As used
herein (throughout the specification), the following terms have the indicated
meanings: "g" refers to grams; "mmol" refers to millimoles; "mL refers to
milliliters;
30 "bp" refers to boiling point; "mp" refers to melting point; "°C"
refers to degrees
Celsius; "mm Hg" refers to millimeters of mercury; "NL" refers to microliters;
"Ng"
refers to micrograms; and "pM" refers to micromolar.
CA 02491932 1999-06-17
-20-
EXAMPLES OF SYNTHESIS SET I=ORTH IN SCHEME A
Step A: Preparation of N-Methoxy-N-methyl-benzeneacetamide
Dissolve potassium carbonate (100 g, 720 mmol) in water (100 mL). Add a
solution of phenylacetyl chloride (50 g, 320 mmol) in toluene (250 mL). Then
add a
solution of N-0-dimethylhydroxylamine hydrochloride (32 g, 330 mmoi) in water
(100
mL) dropwise over one hour. After three hours, carefully add 10% hydrochloric
acid
(250 mL) and fert-butyl methyl ether (125 mL). Separate the organic phase,
wash
with 10% hydrochloric acid and saturated sodium hydrogen carbonate solution.
Dry
io over anhydrous magnesium sulfate and concentrate to give the title compound
(55 g,
95%).
Step B: Preparation of f4-L-Chloro-1-oxobutyl)1-N-methoxy-N-methyl
benzeneacetamide
is Cool a slurry of aluminum chloride (87 g, 650 mmol) in methylene chloride
(100 mL) with an ice bath. Add 4-chlorobutyryl chloride (51 g, 360 mL)
dropwise over
0.5 hours. Add N-methoxy-N-methyl-benzeneacetamide (53 g, 300 mmol) over 0.5
hours. Allow the resultant solution to warm to room temperature. Then heat at
reflux
for 6 hours. Cool the solution to room temperature. Pour the solution into ice
(1 L)
2o and add methylene chloride (1 L). Separate the organic phase. Extract the
aqueous
phase with methylene chloride (2 x 500 mL). Dry the combined organics over
anhydrous magnesium sulfate and concentrate to provide a solid which contains
a
ca. 1:1 mixture of the title compound and the meta isomer.
2s Step C' Crystallization of ~4-(4-Chloro-1-oxobutvl)1-N-methoxy-N-methyl-
benzeneacetamide
Slurry the solid obtained in step B in heptane/ethyl acetate (ca. 4:1 ) and
then
collect it. Dissolve the solid in hot ethyl acetate and treat the resultant
solution with
ca. 5 g of charcoal. Filter through diatomaceous earth and add heptane (60
mL).
3o Heat the slurry until a homogenous solution is obtained. Allow the solution
to stand
overnight at room temperature. Filter the resultant crystalline solid and wash
with
heptane to provide purified [4-(4-chloro-1-oxobutyl)j-N-methoxy-N-methyl-
benzeneacetamide (20 g). Allow the mother liquor to stand for 5 days and
collect a
CA 02491932 1999-06-17
-21-
second crop of the crystalline solid (3.0 g) to obtain a total of 23 g (28%)
of the purified
[4-(4-chloro-1-oxobutyl)]-N-methoxy-N-methyl-benzeneacetamide.
Step D: Preparation of 4-(Cyclo~ropylcarbonyl)benzeneacetic acid
s Add purified [4-(4-chloro-1-oxobutyl)]-N-methoxy-N-methyl-benzeneacetamide
(9.4 g, 330 mmol) to a solution of potassium hydroxide (22.0 g) in ethanol
(160 mL).
Stir 18 hours. Pour the solution into dilute hydrochloric.acid (30 mL of
concentrated
hydrochloric acid in 500 mL of water). Extract the solution with three 500-mL
portions of ethyl acetate. Dry the combined organic phases over anhydrous
io magnesium sulfate and concentrate to give the title compound (6.4 g, 95%).
Step E' Preparation of 4-(cyclopropylcarbonyl) benzeneacetic acid, ethyl ester
Dissolve 4-(cyclopropylcarbonyl)benzeneacetic acid in ethanol (150 mL)
containing concentrated sulfuric acid (10 drops). Stir 24 hours. Add
triethylamine
is (2 mL) and concentrate the solution. Dissolve the residue in water/ethyl
acetate.
Wash the organic phase with saturated sodium hydrogen carbonate solution, dry
over anhydrous magnesium sulfate and concentrate. Purify by silica gel
chromatography (400 mL silica gel, 20% ethyl acetate/heptane as eluent) to
give the
title compound (7.0 g, 88%).
Step E' Preparation of 4-(cyclopropylcarbonyl) benzeneacetic acid, methyl
ester
Dissolve 4-(cyclopropylcarbonyl)benzeneacetic acid in methanol (100 mL)
containing concentrated sulfuric acid (10 drops). Stir 24 hours. Add
triethylamine (2
mL) and concentrate the solution. Dissolve the residue in water/ethyl acetate.
Wash
2s the organic phase with saturated sodium hydrogen carbonate solution, dry
over
anhydrous magnesium sulfate and concentrate. Purify by silica gel
chromatography
(400 mL silica gel, 20% ethyl acetate/heptane as eluent) to give the title
compound
(4.6 g, 68%).
3o Step F' Preparation of f4-(cyclopropylcarbonyl)phenyllmethyl-propanedioic
acid.
diethyl ester
Dissolve 4-(cyclopropylcarbonyl)benzeneacetic acid, ethyl ester (6.5 g, 28
mmol) and diethylcarbonate (4.0 g, 34 mmol) in tetrahydrofuran (100 mL). Add
62
CA 02491932 1999-06-17
-22-
mL (62 mmol) of a 1.0 molar solution of sodium bis(trimethylsilyl)amide in
tetrahydrofuran over 0.5 hours. Stir 28 hours. Add iodomethane (5.3 g, 35
mmoi).
Stir 2 days. Add water and ethyl acetate. Wash organic phase with brine, dry
over
anhydrous magnesium sulfate and concentrate. Purify by flash chromatography
(200
g silica gel, ethyl acetate/heptane as eluent) to give the title compound
(1.43 g, 17%).
Stea F: Preparation of [4-(cyclopropylcarbonyl)phenyllmethvl-propanedioic
acid,
dimeth~,l ester
Dissolve 4-(cyciopropylcarbonyl)benzeneacetic acid, methyl ester (1.5 g, 6.0
io mmol) and dimethylcarbonate (930 mg, 10.3 mmol) in tetrahydrofuran (10 mL).
Add
20 mL (20 mmol) of a 1.0 molar solution of sodium bis(trimethylsilyl)amide in
tetrahydrofuran over 0.5 hours. Stir 3 days. Add iodomethane (5.3 g, 35 mmol).
Stir
24 hours. Add water and ethyl acetate. Wash organic phase with brine, dry over
anhydrous magnesium sulfate and concentrate. Purify by flash chromatography
(50
is g silica gel, ethyl acetate/heptane as eiuent) to give the title compound
(315 mg,
16%).
Additionally, the following compounds can be prepared by the synthetic
procedure
depicted in Step F:
20 [4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, ethyl methyl
diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, methyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, butyl methyl diester
[4-(cyclopropylcarbonyl)phenylJmethyl-propanedioic acid, methyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, hexyl methyl diester
2s [4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, ethyl propyl
diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, butyl ethyl diester
[4-(cyclopropyicarbonyl)phenyl]methyl-propanedioic acid, ethyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, ethyl hexyl diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, dipropyl ester
30 [4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, butyl propyl
diester
[4-(cyclopropylcarbonyl)phenyljrnethyl-propanedioic acid, pentyl propyl
diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, hexyl propyf diester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, dibutyl ester
CA 02491932 1999-06-17
-23-
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, butt'! pentyl diester
[4-(cyclopropylcarbonyl)phenyljmethyl-propanedioic acid, butyl hexyl diester
[4-(cyclopropylcarbonyl)phenyljmethyl-propanedioic acid, dipentyi ester
[4-(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, hexyl pentyl diester
s [4-(cyclopropylcarbonyl)phenylJmethyl-propanedioic acid, dihexyl ester
[4-(cyclopropylcarbonyl)phenyljethyl-propanedioic acid, diethyl ester
[4-(cyclopropylcarbonyl)phenylethyl-propanedioic acid, dimethyl ester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, ethyl methyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, methyl propyl diester
io [4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, butyl methyl
diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, methyl pentyl diester
[4-(cyclopropylcarbonyl)phenyljethyl-propanedioic acid, hexyl methyl diester
[4-(cyclopropylcarbonyl)phenyljethyl-propanedioic acid, ethyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, butyl ethyl diester
is [4-(cyclopropylcarbonyl)phenylethyl-propanedioic acid, ethyl pentyl diester
[4-(cyclopropylcarbonyl)phenylJethyl-propanedioic acid, ethyl hexyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, dipropyl ester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, butyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, pentyl propyl diester
2o j4-(cyclopropylcarbonyl)phenyljethyl-propanedioic acid, hexyl propyl
diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, dibutyl ester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, butyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, butyl hexyl diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, dipentyl ester
is [4-(cyclopropyicarbonyl)phenyl]ethyl-propanedioic acid, hexyl pentyl
diester
[4-(cyclopropylcarbonyl)phenyl]ethyl-propanedioic acid, dihexyl ester
[4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, diethyl ester
[4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, dimethyl ester
[4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, ethyl methyl diester
30 [4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, methyl propyl
diester
[4-(cyclopropylcarbonyl)phenylJpropyl-propanedioic acid, butyl methyl diester
[4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, methyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, hexyl methyl diester
CA 02491932 1999-06-17
-24-
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, ethyl propyl diester
[4-(cyclopropylcarbonyl)phenyljpropyl-propanedioic acid, butyl ethyl diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, ethyl pentyl diester
(4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, ethyl hexyl diester
s [4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, dipropyl ester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, butyl propyl diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, pentyl propyl diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, hexyl propyl diester
[4-{cyclopropylcarbonyl)phenyl]propyl-propanedioic acid, dibutyl ester
io [4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, butyl pentyl
diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, butyl hexyl diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, dipentyl ester
[4-{cyclopropylcarbony))phenyl]propyl-propanedioic acid, hexyl pentyl diester
[4-(cyclopropylcarbony))phenyl]propyl-propanedioic acid, dihexyl ester
is [4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, diethyl ester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, dimethyl ester
[4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, ethyl methyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, methyl propyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, butyl methyl diester
20 [4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, methyl pentyl
diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, hexyl methyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, ethyl propyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, butyl ethyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, ethyl pentyl diester
2s [4-(cyclopropylcarbonyl)phenyl]butyl-propanedioic~acid, ethyl hexyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, dipropyl ester
[4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, butyl propyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, pentyl propyl diester
[4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, hexyl propyl diester
30 (4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, dibutyl ester
[4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, butyl pentyl diester
[4-(cyclopropylcarbony))phenyl]butyl-propanedioic acid, butyl hexyl diester
[4-(cyclopropylcarbonyl)phenyljbutyl-propanedioic acid, dipentyl ester
CA 02491932 1999-06-17
-25-
[4-(cyclopropylcarbonyl)phenyl]butyl-propanedioic acid, hexyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]butyl-propanedioic acid, dihexyl ester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, diethyl ester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, dimethyl ester
' s [4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, ethyl methyl
diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, methyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, butyl methyl diester
[4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, methyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, hexyl methyl diester
io [4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, ethyl propyl
diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, butyl ethyl diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, ethyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, ethyl hexyl diester
[4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, dipropyl ester
is [4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, butyl propyl
diester
[4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, pentyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, hexyl propyl diester
[4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, dibutyl ester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, butyl pentyl diester
20 [4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, butyl hexyl
diester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, dipentyl ester
[4-(cyclopropylcarbonyl)phenyl]pentyl-propanedioic acid, hexyl pentyl diester
[4-(cyclopropylcarbonyl)phenyljpentyl-propanedioic acid, dihexyl ester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, diethyl ester
2s [4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, dimethyl ester
[4-(cyclopropylcarbony!)phenyl]hexyl-propanedioic acid, ethyl methyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, methyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, butyl methyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, methyl pentyl diester
30 [4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, hexyl methyl
diester
[4-(cyciopropylcarbonyl)phenyl]hexyl-propanedioic acid, ethyl propyl diester
[4-(cyclopr opylcarbonyl)phenyl]hexyl-propanedioic acid, butyl ethyl diester
[4-(cyclopropylcarbonyl)phenyljhexyl-propanedioic acid, ethyl pentyi diester
CA 02491932 1999-06-17
-26-
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, ethyl hexyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, dipropyl ester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, butyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, pentyl propyl diester
s [4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, hexyl propyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, dibutyl ester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, butyl pentyl diester
[4-(cyclopropylcarbonyl)phenyl)hexyl-propanedioic acid, butyl hexyl diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, dipentyl ester
io [4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, hexyl pentyl
diester
[4-(cyclopropylcarbonyl)phenyl]hexyl-propanedioic acid, dihexyl ester
Step G' Preparation of f4-(4-chloro-1-oxobutyl)phenyllmethyl-propanedioic
acid,
diethyl ester
is Bubble hydrogen chloride gas through a solution of [4-
(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, diethyl ester
(570 mg, 2.0 mmol) in ethanol (4 mL) for 5 minutes. Heat the solution to
reflux.
After 18 hours, cool the solution to room temperature and bubble nitrogen
through
the solution for 1 hour. Add ethyl acetate and water to the residue. Dry the
organic
2o phase over anhydrous magnesium sulfate and concentrate. Dissolve the crude
product in ethanol (50 mL) and bubble hydrogen chloride gas through the
solution for
minutes. Heat the solution to reflux. After stirring 2 days, concentrate the
solution. Add water and ethyl acetate to the residue. Dry the organic phase
over
anhydrous magnesium sulfate and concentrate. Purify by flash chromatography
(150
2s g silica gel, 20% ethyl acetatelheptane as eluent) to give the title
compound (409 mg,
59%).
Step G' Preparation of (4-(4-chloro-1-oxobutyl)phenyllmethyl-propanedioic
acid.
dimeth ly ester
3o Bubble hydrogen chloride gas through a solution of [4-
(cyclopropylcarbonyl)phenyl]methyl-propanedioic acid, dimethyl ester (315 mg,
1.1
mmol) in ethanol (3 mL) and toluene (9 mL) for 10 minutes. Heat the solution
to
68°C. After 4 hours, cool the solution to room temperature and bubble
nitrogen
CA 02491932 1999-06-17
-27-
through the solution for 1 hour. Add ethyl acetate and water to the residue.
Dry the
organic phase over anhydrous magnesium sulfate and concentrate to give the
title
compound (321 mg, 91 %).
' s Additionally, the following compounds can be prepared by the synthetic
procedure
depicted in Step G:
[4-(4-bromo-1-oxobutyl)phenylJmethyl-propanedioic acid, diethyl ester
[4-(4-iodo-1-oxobutyl)phenylJmethyl-propanedioic acid, diethyl ester
[4-(4-bromo-1-oxobutyl)phenyl]methyl-propanedioic acid, dimethyl ester
io [4-(4-iodo-1-oxobutyl)phenylJmethyl-propanedioic acid, dimethyl ester
Furthermore, compounds derived from all permutations of substituents as set
forth in
the illustrative examples following Step F can be prepared by the synthetic
procedure
depicted in Step G.
t5 Step H~ Preparation of f4-f4-f4-(hydroxydiphenylmethyl)-1-piperidinyli-1-
oxobutvllphenvllmethyl-propanedioic acid, diethyl ester
Mix together [4-(4-chloro-1-oxobutyl)phenyl]methyl-propanedioic acid, diethyl
ester (380 mg, 1.1 mmol), potassium carbonate (450 mg, 3.2 mmol), a-(4-
pyridyl)benzhydrol (500 mg, 1.9 mmol), water (4 mL) and toluene (10 mL). Heat
the
2o mixture to reflux. After 7 days, cool to room temperature. Add ethyl
acetate and
water. Wash the organic phase with brine, dry over anhydrous magnesium sulfate
and concentrate. Purify by flash chromatography (200 g of silica gel, 10%
methanol/chloroform as eluent) to give the title compound (627 mg, 99%).
2s Step H' Preparation of f4-f4-f4-(hydroxydiphenylmethyl)-1-piperidinyll-1-
oxobutyrliphenyilmethyl-propanedioic acid. dimethvl ester
Mix together [4-(4-chloro-1-oxobutyl)phenyl]methyl-propanedioic acid, dimethyl
ester (300 mg, 0.92 mmol), potassium carbonate (350 mg, 2.5 mmol), a-(4-
pyridyl)benzhydrol (500 mg, 1.9 mmol), water (3 mL) and toluene (7 mL). Heat
the
3o mixture to reflux. After 5 days, cool to room temperature. Add ethyl
acetate and
water. Wash the organic phase with brine, dry over anhydrous magnesium sulfate
and concentrate. Purify by flash chromatography (150 g of silica gel, 10%
methanollchloroform as eluent) to give the title compound (387 mg, 76%).
CA 02491932 1999-06-17
~28-
Additionally, compounds derived from all permutations of substituents as set
forth in
the illustrative examples following Step F can be prepared by the synthetic
procedure
depicted in Step H.
s Step i: Preparation of 4-f 1-hydroxy-4-f4-(hydroxydiphenylmethyl)-1-
piperidinyllbutyl]-
a-lhydroxvmethvl)-a-methyl-benzeneacetic acid, etf~yl ester
Dissolve [4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
oxobutyl]phenyl]methyl-propanedioic acid, diethyl ester (515 mg, 0.88 mmol) in
tetrahydrofuran (5 mL) and cool the solution with an ice bath. Add lithium tri-
terf-
to butoxyaluminohydride (10 ml- of a 1 molar solution in tetrahydrofuran, 10
mmol)
portionwise over 20 minutes. After 2 hours, allow the solution to warm to room
temperature. After 48 hours, cool the solution with an ice bath and add a 10%
potassium hydrogen sulfate aqueous solution (10 mL). Wash the organic phase
with
brine, dry over anhydrous magnesium sulfate, and concentrate. Purify by flash
Is chromatography (150 g of silica gel, 5% methanol/chloroform as eluent) to
give the
compound (321 mg, 67%).
Additionally, the following compounds can be prepared by the synthetic
procedure
depicted in Step 1:
20 4-[1-hydroxy-4-[4-(hydroxydiphenylrnethyl)-1-piperidinyl]butylj-a-
(hydroxymethyl)-a-
methyl-benzeneacetic acid, methyl ester
4-[ 1-h yd roxy-4-[4-(hyd roxyd ip henyl m ethyl)-1-pipe rid i nyl]butyl]-a-
(hyd roxymethyl)-a-
methyl-benzeneacetic acid, propyl ester
4-[ 1-hyd roxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butylJ-a-
(hydroxymethyl)-a-
2s methyl-benzeneacetic acid, butyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
methyl-benzeneacetic acid, pentyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
methyl-benzeneacetic acid, hexyl ester
30 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyljbutyl]-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, methyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl)-ar-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, ethyl ester
CA 02491932 1999-06-17
-29-
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl)butyl]-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, propyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, butyl ester
s 4-[1-hydroxy-4-[4-(hydroxydiphenyimethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, pentyl ester
4-[1-hydroxy-4-j4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid, hexyl ester
4- .[1 -hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl)butyl]-a-
(hydroxymethyl)-a-
to propyl-benzeneacetic acid, methyl ester
4-[1 -hydroxy-4-[4-(hydroxydiphenyimethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
propyl-benzeneacetic acid, ethyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
propyl-benzeneacetic acid, propyl ester
1s 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethy!)-a-
propyl-benzeneacetic acid, butyl ester
4-[1-hydroxyl-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
propyl-benzeneacetic acid, pentyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl-1-piperidinyl]butyl]-a-
(hydroxymethy!)-a-
2o propyl-benzeneacetic acid, hexyl ester .
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
{hydroxymethyl)-a-
butyl-benzeneacetic acid, methyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
butyl-benzeneacetic acid, ethyl ester
Zs 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-ar-
butyl-benzeneacetic acid, propyl ester
4-[1-hydroxy- 4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
butyl-benzeneacetic acid, butyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
3o butyl-benzeneacetic acid, pentyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
{hydroxymethyl)-a-
butyl-benzeneacetic acid, hexyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethy()-1-piperidinyl)butyl]-a-
(hydroxymethyl)-a-
CA 02491932 1999-06-17
-30-
pentyl-benzeneacetic acid, methyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyt)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
pentyl-benzeneacetic acid, ethyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
{hydroxymethyl)-a-
s pentyi-benzeneacetic acid, propyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
pentyl-benzeneacetic acid, butyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
pentyl-benzeneacetic acid, pentyl ester
~0 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinylJbutyl]-a-
(hydroxymethyl)-a-
pentyl-benzeneacetic acid, hexyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyi)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid, methyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl}-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
is hexyl-benzeneacetic acid, ethyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinylJbutyl]-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid, propyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid, butyl ester
20 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinylJbutylj-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid, pentyl ester
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid, hexyl ester
2s Step J' Preparation of 4-f1-hvdroxy-4-f4-(hvdroxydiphenylmethvl)-1-
~iperidinvllbutyll-
-~~droxymethy!)-a-methyl-benzeneacetic acid
Dissolve 4-[1-hydroxy-4-[4-(hydroxydiphenyimethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-methyl-benzeneacetic acid, ethyl ester (200 mg, 0.37 mmol)
in
methanol (8 mL) and tetrahydrofuran (12 mL). Add 1.6 mL (1.6 mmol) of a 1
molar
so solution of aqueous sodium hydroxide. After 4 hours, cool the solution to
room
temperature and add 10% hydrochloric acid (ca. 1 mL) dropwise until pH is 5-6.
Concentrate the solution and purify by flash chromatography (50 g of silica
gel,
methanollchloroform gradient elution) to give the title compound (127 mg,
66%).
CA 02491932 1999-06-17
-31-
Additionally, the following compounds can be prepared by the synthetic
procedure
depicted in Step J:
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl)-a-
(hydroxymethyl)-a-
ethyl-benzeneacetic acid
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
propyl-benzeneacetic acid
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)=a-
butyl-benzeneacetic acid
to 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
pentyl-benzeneacetic acid
4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-
(hydroxymethyl)-a-
hexyl-benzeneacetic acid
is EXAMPLES OF SYNTHESIS SET FORTH IN SCHEME B
To a slurry of AIC13 (210 g, 1.6 mol) in methylene chloride (200 mL) cooled in
an ice bath is added 4-chlorobutyryl chloride (121 g) dropwise while
maintaining the
pot temperature below 10°C. After 10 minutes, ethyl benzeneacetic acid
(118 g, 0.72
2o mol) is added dropwise maintaining the pot temperature below 10°C.
The mixture is
stirred at room temperature for 1 hour, then heated to 40°C. After 4
hours, the
solution is cooled to room temperature and poured onto crushed ice (2L).
Methyiene
chloride (1 L) is added. The organic phase is separated. The aqueous phase is
extracted with two 1 L portions of methylene chloride. The combined organics
are
2s dried (MgS04) and concentrated. Toluene (1 L) is added and the solution is
concentrated to ca. 500 mL. Ethanol (500 mL) is added. The solution is heated
to
70°C and HCI (g) is bubbled through for 20 minutes. The solution is
sparged with N2
and concentrated. Chromatography through silica gel (2.5 L) using an ethyl
acetate/heptane gradient gives a ~ :1 mixture of meta, para ethyl (4-chloro-1-
30 oxobutyl)benzene acetic acid (119 g). To a slurry of LiOH (23.58, 0.56 mol)
in water
(100 mL) and ethanol (100 mL) is added the 1:1 mixture of meta, para ethyl (4-
chloro-
1-oxobutyl)benzene acetic acid (50g, 0.19 mol). The reaction becomes
exothermic
and is tooted with an ice bath. After 1 hour, the solution is allowed to warm
to room
CA 02491932 1999-06-17
-32-
temperature. After 18 hours, the mixture is concentrated. Water (400 mL) and
conc.
HCI are added (until pH 4). Ethyl acetate (600 mL) is added. The organic phase
is
separated, washed with brine and dried (MgS04). The solution is heated on a
steam
bath and charcoal (ca. 3 g) is added. The mixture is filtered through Celite
and
concentrated. The residue is dissolved in ethyl acetate (300 mL) and heptane
(fi00 mL) with heating. Seed crystals are added. After cooling an organic oil
appears.
Heptane (500 mL) is added and the mixture is heated to reflux, treated with
charcoal
(ca. 5 g), filtered and seeded. A solid forms and is collected. 'H NMR shows
ca. 4:1
mixture of para:meta isomers. Recrystallization from ethyl acetate/heptane
gives 4
io (cyclopropylcarbonyl)benzeneacetic acid (4.3g, 11 %).
EXAMPLES OF SYNTHESIS AS SET FORTH IN SCHEME C
Step a' Preparation of 4-(cyclopropylcarbonyl) benzeneacetic acid. ethyl ester
is See preparation under Step E, Scheme A, disclosed previously herein.
Step b' Preparation of f4-(cycloaropylcarbonvl)1-a-a-dimethvlbenzeneacetic
acid,
ethyl ester
In a 2L, glass, jacketed reactor is loaded the 4-(cyclopropylcarbonyl)
2o benzeneacetic acid, ethyl ester (232 g, 1 mole), diglyme (150 mL) and
methyl
chloride (127 g, 2.5 mole). In a heated addition vessel is loaded potassium
tert-
butoxide (182.4 g, 1.6 mole) and diglyme (1050 mL). The jacket for the,reactor
is set
at -10°C and the addition vessel contents are heated to 60°G.
The base solution is
added to the reactor at a rate which keeps the internal temperature of the
reaction
2s below 25°C. After the base addition, sodium ethoxide as a 21 %
solution in ethanol
(86 g, 0.3 mole) is added to quench any excess methyl chloride. The entire
reaction
mixture is then agitated with toluene (900 mL) and water (1200 mL) containing
sodium bicarbonate (8.4 g). The phases are separated and the organic layer is
washed with additional water (200 mL) to remove any residual potassium
chloride
so salts. Without phase separating, the entire solution is then acidified to
pH=3 with
concentrated HCI. The organic is then stripped of solvents to afford the title
compound.
CA 02491932 1999-06-17
-33-
Step c: Preparation of 4-(4-chloro-1-oxobuty)-a,a-dimethylbenzeneacetic acid
ethyl ester
To a 4L reactor equipped with a gas inlet, overhead stirrer and temperature
control, is charged [4-(cyclopropylcarbonyl)J-a,a-dimethylbenzeneacetic acid,
ethyl
ester (500 g). The oil is heated to 60°C and the head space is
evacuated. HCI is
then added raising the pressure to 10 psig. After 4 hours, the excess HCI is
vented
and the oil is sparged with nitrogen for 5 minutes.
Step d: Preparation of Ethyl 4-f4-f4-(hydroxydiphenylmethyl)-1-piperidinyll-1-
io oxobutylLa-a-dimethylbenzeneacetate hydrochloride
According to the described procedure of Carr et al., United States Patent No.
4,254,129, a mixture of 4.5 g (0.0163 mole) of a,a-Biphenyl-4-
piperidemethanol, 6.1
g (0.0205 mole) of ethyl 4-(4-chloro-1-oxobuty)-a,a-dimethylphenylacetate, 5 g
(0.05
mole) of potassium bicarbonate and 0.05 g of potassium iodide in 50 ml of
toluene is
is stirred and refluxed for 72 hours then filtered. Ether then ethereal
hydrogen chloride
is added to the filtrate, and the resulting precipitate collected and
recrystallized
several times from methanol-butanone and butanone to give ethyl 4-[4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-oxobutyl]-a,a-dimethylbenzeneacetate
hydrochloride. M.P.205.5°-208° C.
Step e' Preparation of Ethyl 4-f4[4-(hydroxydiphenylmethyl)-1-piperidinyll-1-
hydroxybutvll-a a-dimethylbenzeneacetate
According to the described procedure of Carr et al., United States Patent No.
4,254,129, a solution of 5.64 g (0.01 mole) of ethyl 4-[4[4-
(hydroxydiphenylmethyl)-1-
2s piperidinyl]-1-oxobutyl]-a,a-dimethylbenzeneacetate hydrochloride in 200 ml
of
absolute ethanol and 50 ml of methanol and 0.5 g of platinum oxide is
hydrogenated
at about 50 psi for about 1 hour until the infrared shows no evidence of a
ketone
carbonyl function. The solution is filtered and the filtrate concentrated
leaving a
residue which is recrystallized from butanone and methanol-butanone to give
ethyl 4-
[4[4-(hydroxydiphenylmethyl)-1-peperidinylJ-1-hydroxybuty]-a,a-
dimethylbenzeneacetate HCI, M.P. 185°-187°C.
CA 02491932 1999-06-17
-34-
Step f' 4-f4f4-(Hydroxydiphenylmethyl)-1-piperidinyll-1-hydroxybutyll-a.a-
dimethylbenzeneacetic acid
According to the described procedure of Carr et al., United States Patent No.
4,254,129, to a solution of 0.6 g of ethyl 4-[4[4-(hydroxydiphenylmethyl)-1-
piperidinyl]-hydroxybutyl]-a,a-dimethylbenzeneacetate in 20 ml of absolute
ethanol is
added 10 ml of a 50% solution of sodium hydroxide. The mixture is refluxed for
3 1/2
hours and concentrated to a solid after which a minimum amount of methanol to
dissolve the residue is added. 10% Aqueous HCI is added until pH 7 is reached,
the
methanol removed by evaporation and water (25 ml) is added. The resulting
io precipitate is recrystallized from methanolbutanone to give 4-[4[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-hydroxybutyl]-a,a-
dimethylbenzeneacetic
acid, M.P. 195°-197°C.
The piperidine derivative of formula (I) is used according to the present
~s invention as a histamine H~-receptor antagonist and as such will provide
relief of
symptoms associated with histamine-mediated allergic disorders in patients
suffering
therefrom. Histamine-mediated allergic disorders are diseases or conditions
which
have a histamine-mediated allergic component such as, for example, seasonal
allergic rhinitis, perennial rhinitis, idiopathic urticaria, asthma and the
like. Relief of
2o symptoms of an allergic disorder by treatment according to the present
invention
refers to a decrease in symptom severity over that expected in the absence of
treatment and does not necessarily indicate a total elimination of the
disease.
Antihistaminic potential is measured by affinity of a test compound for [3H].
pyrilamine binding sites associated with the antagonist component of H~-
Zs histaminergic receptors in animal brain membranes. Affinity for this
receptor is
indicative of the potential of a test compound to interact with central and
peripheral
H~-histaminergic receptors.
The following method is used to determine the affinity of test compounds for
[3H] pyrilamine binding sites in rat cortex.
3o The brains of young male rats are removed. The cortici are dissected and
stored at -20°C or immediately used. The tissue is homogenized in 10 ml
of ice-cold
50 nM K/NaP04 buffer (pH 7.4) using a Polytron (setting 6 for 15 seconds). The
homogenate is centrifuged at 40,000 g for 15 minutes at 4°C. The pellet
is
CA 02491932 1999-06-17
-3s-
resuspended in the same buffer in order to have 100 mg wet weight/ml buffer.
The
incubation tubes contain 50 mM K/NaP04 buffer, promethazine (2.10-6M final) or
test
compound, 3H pyrilamine (2 nM final) and homogenate (10 mg wet weight per
tube)
in a final volume of 250-1000 ml. After a 30 minute incubation at room
temperature
each incubation is terminated by rapid filtration through Whatman GF/B glass
fiber
filters, presoaked in water when using a Brandel cell harvester, or used as
such
when the filtration is performed with a 96 well Skatron cell harvester. The
filters are
rinsed with 3 x 3 ml 0.9% NaCI (Brandel) or prewetted and rinsed for 10 sec
with
NaCI (Skatron). The filters are either transferred to scintillation vials and
10 ml
io Quicksafe A is added for liquid scintillation spectrometry or a thin layer
of solid
scintillant is melted onto the filters and the filters then counted using a
betaplate beta
counter. Specific binding of 3H pyrilamine is measured as the excess over
blanks
taken in the presence of 2.10-6 M promethazine. Protein content of the
membranes is
determined by the method of Lowry et al., J. Biol. Chem. 193, 265-275 (1951).
is Displacement curves are analyzed using the GraphPad (GraphPad Software,
Inc.) or
similar program to obtain Hili slopes and ICSO values. The K; value is then
determined
with the Cheng-Prusoff equation described by Cheng et al., in Biochem.
Pharmacol.,
22, 3099-3108 (1973), using the Kp for 3H pyrilamine as obtained from previous
saturation experiments performed under the same conditions.
2o The K; for 4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyljbutylj-a-
(hydroxymethyl)-a-methyl-benzeneacetic acid is 3.6 x 10-' indicating that the
piperidine derivative of formula (I) is useful for the treatment of histamine-
mediated
allergic disorders.
As used herein, the term "patient" refers to an adult person who is suffering
2s from a histamine-mediated allergic disorder. It is understood that for
purposes of the
present invention, the term "adult" refers to a person of 12 years of age or
older who
would typically be treated for allergic disorders with an antihistamine dosage
as
recommended for adults.
The identification of those patients who would benefit from the present
3o invention is well within the ability and knowledge of one skilled in the
art. A clinician
skilled in the art can readily identify, by the use of clinical tests,
physical examination
and medical/family history, those patients who are suffering from an allergic
disorder
that is histamine-mediated.
CA 02491932 1999-06-17
-36-
The quantity of novel compound administered will vary depending on the
mode of administration and can be any effective antiallergic amount. The
quantity of
novel compound administered may vary over a wide range to provide in a unit
dosage an effective amount of from about 0.01 to 20 mg/kg of body weight of
the
patient per day to achieve the desired effect. For example, the desired
antihistamine,
antiallergy and bronchodilator effects can be obtained by administering the
piperidine
compound of formula (I) to a patient in a daily amount of from about 10 mg to
about
50 mg. A preferred daily dose is from about 20 mg to about 40 mg. The most
preferred daily dose is about 30 mg.
~o It is of course understood that the daily dose may be administered to a
patient
according to a dosage regimen in single or multiple dosage units. For example,
a
daily dose may be administered in a regimen requiring one, two, three, or four
unit
doses. Typically, these unit doses will be of equal strength and will be
administered
on a time schedule so that each dose is approximately equally spaced
throughout the
is day. For example, a daily dose requiring a once a day dosage regimen may be
administered about every 24 hours; a daily dose requiring a twice-a-day dosage
regimen may be administered about every 12 hours; a daily dose requiring a
three
times-a-day dosage regimen may be administered about every 8 hours; a daily
dose
requiring a four times-a-day dosage regimen may be administered about every 6
2o hours.
The piperidine derivative of formula (I) can be administered according to the
present invention in any form or mode which makes the compound bioavailable in
effective amounts, including oral and parenteral routes. For example, the
piperidine
derivative of formula (I) can be administered orally, subcutaneously,
transdermally,
2s intranasally, and the like. Oral administration is preferred. One skilled
in the art of
preparing formulations can readily select the proper form and mode of
administration
depending upon the particular characteristics of the compound selected, the
disease
state to be treated, the stage of the disease, and other relevant
circumstances.
The compounds can be administered alone or in the form of a pharmaceutical
3o composition with pharmaceutically acceptable carriers or excipients, the
proportion
and nature of which are determined by the chosen route of administration and
standard pharmaceutical practice. The piperidine derivative of formula (I),
while
effective itself, may be formulated and administered in the form of its
CA 02491932 1999-06-17
-37-
pharmaceutically acceptable acid addition salt for purposes of stability,
convenience
of crystallization, increased solubility and the like. 1n addition, an
individual
polymorph, solvate, or individual stereoisomer of the piperidine derivative of
formula
(I) [i.e., (R,R)-4-[1-hydroxy-4-[4-(hydroxydiphenyfmethyl)-1-
piperidinyl]butyl]-a-
- s (hydroxymethyl)-a-methyl-benzeneacetic acid; (R,S)-4-[1-hydroxy-4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-(hydroxymethyl)-a-methyl
benzeneacetic acid; (S,S)-4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1 -
piperidinyl]butyl]-a-(hydroxymethyl)-a-methyl-benzeneacetic acid and (S,R)-4-
[1
hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-a-(hydroxymethyl)-a-
io methyl-benzeneacetic acid] may be used.
The present invention contemplates compositions comprising the piperidine
derivative of formula (I) in admixture or otherwise in association with one or
more
inert carriers. These compositions are useful, for example, as assay
standards, as
convenient means of making bulk shipments, or as pharmaceutical compositions.
An
is assayable amount of the piperidine derivative of formula (1) is an amount
which is
readily measurable by standard assay procedures and techniques as are well
known and appreciated by those skilled in the art. Assayable amounts of the
piperidine derivative of formula (I) will generally vary from about 0.001
°!o to about
75% of the composition by weight. Inert carriers can be any material which
does not
2o degrade or otherwise covalently react with the piperidine derivative of
formula (I).
Examples of suitable inert carriers are water; aqueous buffers, such as those
which
are generally useful in High Performance Liquid Chromatography (HPLC) analysis
organic solvents, such as acetonitrile, ethyl acetate, hexane and the like;
and
pharmaceutically acceptable carriers or excipients.
2s More particularly, the present invention contemplates a pharmaceutical
composition in solid unit dosage form comprising an amount of the piperidine
derivative of formula (1) from about 15 mg to about 30 mg in admixture with a
pharmaceutically acceptable carrier. As used herein, the term "solid unit
dosage
form" contemplates a solid dosage form for oral administration such as a
tablet,
3o capsule, and the like, as well as solid dosage forms for parenteral
administration
such as a transdermai patch, and the like.
The pharmaceutical compositions are prepared in a manner well known in the
CA 02491932 1999-06-17
-38-
pharmaceutical art. The carrier or excipient may be a solid, semi-solid, or
liquid
material which can serve as a vehicle or medium for the active Ingredient.
Suitable
carriers or excipients are well known in the art. The pharmaceutical
composition may
be adapted for oral or parenteral use and may be administered to the patient
in the
form of tablets, capsules, solutions, suspensions, transdermal patch, and the
like.
The piperidine derivative of formula (I) may be administered orally, for
example, with an inert diluent or with an edible carrier. It may be enclosed
in gelatin
capsules or compressed into tablets. For the purpose of oral therapeutic
administration, the piperidine derivative of formula (I) may be incorporated
with
~o excipients and used in the form of tablets, capsules, elixirs, suspensions,
syrups,
wafers, chewing gums and the like. These preparations should contain at least
4%
of the compound of the invention, the active ingredient, but may be varied
depending
upon the particular form and may conveniently be between about 4% to about 70%
of the weight of the unit. The amount of the compound present in compositions
is
is such that a suitable dosage will be obtained upon administration. Preferred
compositions and preparations according to the present invention are prepared
so
that an oral unit dosage form contains between about 15 mg to about 30 mg.
Most
preferred unit doses for oral administration are those which contain about 15
mg to
about 30 mg.
2o The tablets, pills, capsules, and the like may also contain one or more of
the
following adjuvants: binders such as microcrystalline cellulose, gum
tragacanth or
gelatin; excipients such as starch or lactose, disintegrating agents such as
alginic
acid, PrimogelT"", corn starch, carbonate salts such as sodium bicarbonate or
calcium
carbonate, and the like; lubricants such as magnesium stearate or SterotexT"";
_
2s glidents such as colloidal silicon dioxide; and sweetening agents such as
sucrose or
saccharin may be added or a flavoring agent such as peppermint, methyl
salicylate
or orange flavoring. When the dosage unit form is a capsule, it may contain,
in
addition to materials of the above type, a liquid carrier such as polyethylene
glycol or
a fatty oil. Other dosage unit forms may contain other various materials which
modify
3o the physical form of the dosage unit, for example, as coatings. Thus,
tablets or pills
may be coated with sugar, shellac, or other enteric coating agents. A syrup
may
contain, in addition to the present compounds, sucrose as a sweetening agent
and
certain preservatives, dyes and colorings and flavors. Materials used in
preparing
CA 02491932 1999-06-17
-39-
these various compositions should be pharmaceutically pure and non-toxic in
the
amounts used. Preferred excipients are corn starch, gelatin, lactose,
magnesium
stearate and sodium bicarbonate.
Oral unit dosage forms may be formulated to provide immediate or sustained
release characteristics. These forms may be formulated according to
conventional
techniques and procedures to give the desirable dissolution and
bioavailability
characteristics.
In addition, the piperidine derivative of formula (I) may be incorporated into
a
solution or suspension for oral or parenteral administration. These
preparations
io should contain at least 0.1 % of the piperidine derivative of formula (I),
but may be
varied to be between 0.1 and about 50% of the weight thereof. The amount of
the
piperidine derivative of formula (I) in such compositions is such that a
suitable
dosage will be obtained upon oral or parenteral administration.
~s The solutions or suspensions may also include the one or more of the
following adjuvants: sterile diluents such as water for injection, saline
solution, fixed
oils, .polyethylene glycols, glycerine, propylene glycol or other synthetic
solvents;
antibacterial agents such as benzyl alcohol or methyl paraben; antioxidants
such as
ascorbic acid or sodium bisulfate; chelating agents such as ethylene
2o diaminetetraacetic acid; buffers such as acetates, citrates or phosphates
and agents
for the adjustment of tonicity such as sodium chloride or dextrose.
Transdermal dosage forms for administering the piperidine derivative of
formula (I) can be prepared by conventional techniques well known in the art
of
pharmaceutical science such as by incorporating the piperidine derivative of
formula
2s (I) into various polymeric reservoir matrix materials. These polymeric
matrix
materials may include pressure sensitive acrylic, silicone, polyurethane,
ethylene
vinyl acetate copolymers, polyolefins, and rubber adhesive matrices, medical
grade
silicone fluids, and medical grade silicone elastamers, which are well known
in the art
for forming reservoirs for transdermal delivery of drugs.
so It is further contemplated that the piperidine derivative of formula (I)
according to
the present invention, may be formulated with a variety of other active
ingredients which
CA 02491932 1999-06-17
-40-
are commonly combined with antihistamines, such as a decongestant, including
pseudoephedrine and the like; analgesics such as acetaminophen and the like,
non-
steroidal anti-inflammatory agents such as ibuprofen and the like.