Note: Descriptions are shown in the official language in which they were submitted.
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
PHARMACEUTICAL DOSAGE FORM CAPABLE OF MAINTAINING STABLE
DISSOLUTION PROFILE UPON STORAGE
FIELD OF THE INVENTION
[0001] The present invention relates to gelatin capsules filled with a fill
material
comprising a selective COX-2 inhibitory drug of low water solubility.
BACKGROUND OF THE INVENTION
[0002] Gelatin, a mixture of water-soluble proteins derived from collagen by
hydrolysis, is widely used in the pharmaceutical and food industries, among
others. One
major application of gelatin is in preparation of both hard and soft gelatin
capsules. Such
capsules are desirable for, i~zter alia, their versatility (they may contain
thug formulations
in solid, semi-solid, or liquid form) and for their rapid dissolution
characteristics.
Unfortunately, drug dosage forms containing gelatin in an outer layer (e.g.
liquid or
powder filled into a gelatin capsule) can exhibit a drop in dissolution rate
over time. This
drop in dissolution rate can lead to undesirable and unacceptable alterations
in irz vitro
dissolution profile and in bioavailability, especially for drugs of low water
solubility or.
drugs whose absorption is dissolution-rate limited. Such changes in
dissolution profile
are thought to result from cross-linking of gelatin occurring in gelatin
capsule shells.
[0003] Singh et al., Alteration in Dissolution Characteristics of Gelatin-
Containin~ Formulations, Pharmaceutical Techfzology, April 2002, hereby
incorporated
by reference herein but not admitted to be prior art, describes reports
suggesting that
several agents including glycerine, glycine, and hydroxylamine hydrochloride,
when
incorporated into fill contents of gelatin capsules, can limit gelatin cross-
linking.
Unfortunately, existing methods directed at the problem of gelatin cross-
linking in
capsule shells are less than satisfactory, especially in situations where
longer shelf life and
stability through real life storage, shipping and handling conditions are
desired; pursuit of
adequate solutions to the problem of gelatin capsule cross-linking is
therefore desired.
[0004] If a pharmaceutical dosage form comprising a fill material in a gelatin
capsule could be prepared which dosage form is capable of providing stable
drug
dissolution rate, even after storage under stressed conditions, a significant
advance in the
oral delivery of drugs, especially drugs of low water solubility or drugs
whose absorption
is dissolution-rate limited, would result.
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
SUMMARY OF THE INVENTION
[0005] There is now provided in the present invention a pharmaceutical dosage
form comprising a fill material sealed in a gelatin capsule shell, the fill
material
comprising (a) a selective COX-2 inhibitory drug of low water solubility, and
(b) an
amine agent comprising at least one pharmaceutically acceptable primary or
secondary
amine.
[0006] Desirably, the amine agent in the dosage form is present at a
concentration
sufficient to inhibit cross-linking of the gelatin and/or pellicle formation
in the capsule
shell.
[0007] The dosage form of the present invention is especially useful for
dosage
forms with liquid fill materials and for dosage forms with soft gelatin
capsules
[0008] The term "pellicle" herein refers to a relatively water-insoluble
membrane
formed in a gelatin capsule shell wherein the membrane tends to be thin,
tough, and
rubbery. It is now understood that one mechanism underlying pellicle formation
is gelatin
cross-linking. Gelatin cross-linking and pellicle formation result in reduced
dissolution
rates. Accordingly, quantification of dissolution rate of a first capsule
within a reasonably
short time after capsule preparation and of a second capsule after storage
under stressed
conditions (e.g. four weeks at 40°C and ~5% relative humidity in a
closed container) as
described herein provides one means of assessing pellicle formation and/or
gelatin cross-
linking. The term "within a reasonably short time after capsule formation"
means within a
period of time such that substantial cross-linking and/or pellicle formation
is unlikely to
' have yet occurred, for example within one week, dependent upon storage
condition during
that period.
[0009] The term "pellicle resistant" herein means that such a gelatin capsule
so
described has a reduced tendency to form, or exhibits slowed, delayed or
reduced
formation of a pellicle upon storage under stressed conditions. Similarly,
"inhibition of
cross-linking" (or "inhibition of pellicle formation") herein means a slowed,
delayed or
reduced formation of gelatin cross-links (or pellicle formation) by comparison
with an
amount a similar capsule lacking only agent as provided herein.
[0010] Pharmaceutical dosage forms according to the present invention have
been
found to exhibit an unexpected and surprisingly substantial reduction in cross-
linking of
gelatin in the capsule shell and pellicle formation. As a result, such dosage
forms are
2
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
capable of consistently meeting desired in vitro dissolution criteria, even
after storage
under stressed conditions. This invention represents a significant improvement
over
conventional dosage forms and conventional gelatin capsule shells.
BRIEF DESCRIPTION OF THE DRAWINGS
[0011] Figure 1 is a graph showing Tier I dissolution rate of Formulation 30
following storage at 25° C as described in Example 3.
[0012] Figure 2 is a graph showing Tier I dissolution rate of Formulation 30
following storage at 40° C as described in Example 3.
[0013] Figure 3 is a graph showing Tier II dissolution rate of Formulation 30
following storage at 25° C as described in Example 3.
[0014] Figure 4 is a graph showing Tier II dissolution rate of Formulation 30
following storage at 40° C as described in Example 3.
[0015] Figure 5 is a graph showing Tier I dissolution rate of Formulation 19
following storage at 25° C as described in Example 3.
[0016] Figure 6 is a graph showing Tier I dissolution rate of Formulation 19
following storage at 40° C as described in Example 3.
[0017] Figure 7 is a graph showing Tier II dissolution rate of Formulation 19
following storage at 40° C as described in Example 3.
DETAILED DESCRIPTION OF THE INVENTION
[0018] In one embodiment, the present invention provides a dosage form
comprising a fill material sealed in a gelatin capsule shell, the fill
material comprising (a)
a selective cyclooxygenase-2 inhibitory drug of low water solubility and (b)
an amine
agent comprising at least one pharmaceutically acceptable primary or secondary
amine
wherein the amine agent is present in an amount sufficient fo inhibit cross-
linking and/or
pellicle formation in the gelatin capsule shells upon storage.
Gelatin cross-linkin~'pellicle formation, and drug dissolution.
[0019] Without being bound by theory, the inventors' believe that gelatin
cross-
linking can result from a process by which amino acid residues of gelatin
covalently bond
to form an insoluble material. The process can be the result of low levels of
aldehydes
coming into contact with the gelatin. Cross-linking of a gelatin capsule can
impact
product performance by delaying the release of the formulation (containing the
active
3
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
compound) from the capsule shell. The delay in release can, in turn, affect
the rate of
absorption of the compound into the blood stream and clinical onset of action.
While
'mild' cross-linking does not necessarily have a significant impact on release
of the
formulation from the dosage form, 'severe' cross-linking can have a
significant impact.
When cross-linking is severe, it can lead to a delay of release of formulation
from the
dosage form in humans, potential bioequivalence problems, and a potential
delay in
clinical onset of action.
[0020] Dosage forms of the present invention exhibit decreased gelatin cross-
linking (and pellicle formation) and, therefore, when placed in an in vitro
dissolution
assay, are capable of advantageously exhibiting less dissolution rate change
during storage
under stressed conditions than conventional dosage forms. Dosage forms
according to the
present invention also exhibit more uniform inter-dosage form drug dissolution
rate than
standard dosage forms.
[0021] In one embodiment of the present invention wherein the fill material
further comprises at least one substance that promotes cross-linking of
gelatin when in
contact therewith (the substance being the drug itself or an excipient
substance, and the
substance acting independently or in combination with one or more other
substances to
promote said cross-linking); upon (a) immediately testing a first dosage form
in a first in
vitro dissolution assay; (b) storing a second dosage form which is identical
to the first
dosage form in a closed container maintained at 40 °C and 75°Io
relative humidity for a
period of four weeks and, after said storage; (c) testing the second dosage
form in a
second in vitro dissolution assay which is identical to the first in vitro
dissolution assay;
the amount of drug dissolved at 45 minutes in the second dissolution assay is
within ~ 15
percent and preferably within ~ 10 percent of the amount of drug dissolved at
45 minutes
in the first dissolution assay.
[0022] Because gelatin cross-linking may lead to delayed dissolution, storage
time-dependent delays in dissolution profile may be a good indicator of
gelatin cross-
linking during such storage. There are a number of ifz vitro dissolution
assays suitable for
determining dissolution profile. Indeed, one skilled in the art is able to
design additional
assays or modifications thereof. Two dissolution-type test methods were
developed and
set forth herein and designated the "Tier I" and "Tier II "tests.
[0023] In the Tier I test, a dosage form is placed in a TJSP apparatus II with
a
4
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
rotating paddle with a paddle speed of 50 rpm in 900 mL of O.O1N HCl + 1%
Tween 80.
Samples are typically withdrawn at 15, 30, 45, 60 and 90 minutes and assayed
for drug
content by HPLC.
[0024] The Tier II test employs the addition of the enzyme pepsin to the
media.
Pepsin in the human stomach digests cross-linked gelatin. The appropriate
amount of
pepsin added to the media (750,000 units/L) was determined and reported in
Collaborative Development of Two-Tier Dissolution Testing for Gelatin Capsules
and
Gelatin-Coated Tablets using Enzyme-Containing Media, Stimuli to the Revision
Process,
Pharmacopeial Forum, Vol. 25, No. 5, Sept.-Oct. 1998. The Tier II drug release
test
designed in this way is expected to produce a drug release profile that is a
reasonable
approximation of the drug release profile in humans.
[0025] An 'initial' drug release profile is determined for each dosage form
within
a reasonably short time after formation (i.e. dosage form before the
formulation is
exposed to conditions which might result in gelatin cross-linking, such as
temperature or
relative humidity). A subsequent profile is determined for samples pulled at
subsequent
time points. A change from initial to subsequent Tier I profile (i.e. a delay
in dissolution)
is presumptively attributed to gelatin cross-linking. When such a change is
reduced in the
Tier II assay (containing pepsin), this reduction is deemed further evidence
of gelatin
cross-linking upon storage.
Fill material
Selective cyclooxygenase-2 inhibitory drug.
[0026] Dosage forms of the invention comprise a selective cyclooxygenase-2
inhibitory drug, also referred to herein as a selective COX-2 inhibitory drug.
Preferably,
the COX-2 inhibitory drug is a drug of low water solubility (e.g. having a
room
temperature solubility in water of not more than about 10 mg/ml and more
preferably not
more than about 1 mg/ml). A preferred selective COX-2 inhibitory drug useful
herein, or
to which a salt or prodrug useful herein is converted ifi vivo, is a compound
of formula (I)
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
1
~X)n R
4
R ~ AwRs
O
R2/S O
(I)
wherein:
A is a substituent selected from partially unsaturated or unsaturated
heterocyclyl
and partially unsaturated or unsaturated carbocyclic rings, preferably a
heterocyclyl group selected from pyrazolyl, furanonyl, isoxazolyl, pyridinyl,
cyclopentenonyl and pyridazinonyl groups;
X is O, S or CH2;
nis0orl;
Rl is at least one substituent selected from heterocyclyl, cycloalkyl,
cycloalkenyl
and aryl, and is optionally substituted at a substitutable position with one
or
more radicals selected from alkyl, haloalkyl, cyano, carboxyl,
alkoxycarbonyl, hydroxyl, hydroxyalkyl, haloalkoxy, amino, alkylamino,
arylamino, nitro, alkoxyalkyl, alkylsulfinyl, halo, alkoxy and alkylthio;
R2 is methyl, amino or aminocarbonylalkyl;
R3 is one or more radicals selected from hydrido, halo, alkyl, alkenyl,
alkynyl,
oxo, cyano, carboxyl, cyanoalkyl, heterocyclyloxy, alkyloxy, alkylthio,
alkylcarbonyl, cycloalkyl, aryl, haloalkyl, heterocyclyl, cycloalkenyl,
aralkyl,
heterocyclylalkyl, acyl, alkylthioalkyl, hydroxyalkyl, alkoxycarbonyl,
arylcarbonyl, aralkylcarbonyl, aralkenyl, alkoxyalkyl, arylthioalkyl,
aryloxyalkyl, aralkylthioalkyl, aralkoxyalkyl, alkoxyaralkoxyalkyl,
alkoxycarbonylalkyl, aminocarbonyl, aminocarbonylalkyl,
alkylaminocarbonyl, N-arylaminocarbonyl, N-alkyl-N-arylaminocarbonyl,
alkylaminocarbonylalkyl, carboxyalkyl, alkylamino, N-arylamino, N-
aralkylamino, N-alkyl-N-aralkylamino, N-alkyl-N-arylamino, aminoalkyl,
alkylaminoalkyl, N-arylaminoalkyl, N-aralkylaminoalkyl, N-alkyl-N-
aralkylaminoalkyl, N-alkyl-N-arylaminoalkyl, aryloxy, aralkoxy, arylthio,
aralkylthio, alkylsulfinyl, alkylsulfonyl, aminosulfonyl, alkylaminosulfonyl,
6
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
N-arylaminosulfonyl, arylsulfonyl and N-alkyl-N-arylaminosulfonyl, R3
being optionally substituted at a substitutable position with one or more
radicals selected from alkyl, haloalkyl, cyano, carboxyl, alkoxycarbonyl,
hydroxyl, hydroxyalkyl, haloalkoxy, amino, alkylamino, arylamino, nitro,
alkoxyalkyl, alkylsulfinyl, halo, alkoxy and alkylthio; and
R4 is selected from hydrido and halo.
[0027] Dosage forms of the invention are especially useful for selective COX-2
inhibitory drugs having the formula (II):
\ Y\z
~i
Rs ~ ~~
O \ X.
R6 (B)
where RS is a methyl or amino group, R6 is hydrogen or a C1_4 alkyl or alkoxy
group, X' is
N or CRS where R~ is hydrogen or halogen, and Y and Z are independently carbon
or
nitrogen atoms defining adjacent atoms of a five- to six-membered ring that is
optionally
substituted at one or more positions with oxo, halo, methyl or halomethyl
groups, or an
isomer, tautomer, pharmaceutically-acceptable salt or prodrug thereof.
Preferred such
five- to six-membered rings are cyclopentenone, furanone, methylpyrazole,
isoxazole and
pyridine rings substituted at no more than one position.
[0028] Illustratively, dosage forms of the invention are suitable for
celecoxib,
deracoxib, valdecoxib, rofecoxib, etoricoxib, 2-(3,5-difluorophenyl)-3-[4-
(methylsulfonyl)phenyl]-2-cyclopenten-1-one, 2-(3,4-difluorophenyl)-4-(3-
hydroxy-3-
methyl-1-butoxy)-5-[4-(methylsulfonyl)phenyl]-3-(2H)-pyridazinone,
pharmaceutically
acceptable salts and prodrugs thereof. A especially useful prodrug of
valdecoxib for use
in dosage forms of the invention is parecoxib, preferably parecoxib. sodium.
[0029] Dosage forms of the invention are also useful for compounds having the
formula (III):
7
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
R'
R'
(
where X" is O, S or N-lower alkyl; Rs is lower haloalkyl; R~ is hydrogen or
halogen; Rlo
is hydrogen, halogen, lower alkyl, lower alkoxy or haloalkoxy, lower
aralkylcarbonyl,
lower dialkylaminosulfonyl, lower alkylaminosulfonyl, lower
aralkylaminosulfonyl, lower
heteroaralkylaminosulfonyl, or 5- or 6-membered nitrogen-containing
heterocyclosulfonyl; and Rl1 and Rl2 are independently hydrogen, halogen,
lower alkyl,
lower alkoxy, or aryl; and for pharmaceutically acceptable salts thereof.
[0030] A especially useful compound of formula (III) is (S)-6,8-dichloro-2-
(trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid, especially in the form of
a water- .
soluble salt thereof, for example the sodium salt.
[0031] Where the drug is celecoxib, the dosage form typically comprises
celecoxib in a therapeutically and/or prophylactically effective total amount
of about 10
mg to about 1000 mg per dose unit. Where the drug is a selective COX-2
inhibitory, drug
other than celecoxib, the amount of the drug per dose unit is therapeutically
equivalent to
about 10 mg to about 1000 mg of celecoxib.
[0032] It will be understood that a therapeutically and/or prophylactically
effective
amount of a drug for a subject is dependent ifiter alia on the body weight of
the subject.
A "subject" herein to which a therapeutic agent or composition thereof can be
administered includes a human patient of either sex and of any age, and also
includes any
nonhuman animal, especially a domestic or companion animal, illustratively a
cat, dog or
horse.
[0033] Where the subject is a child or a small animal (e.g., a dog), for
example, an
amount of celecoxib relatively low in the preferred range of about 10 mg to
about 1000
mg is likely to be consistent with therapeutic effectiveness. Where the
subject is an adult
human or a large animal (e.g., a horse), therapeutic effectiveness is likely
to require dose
units containing a relatively greater amount of celecoxib. For an adult human,
a
therapeutically effective amount of celecoxib per dose unit in a dosage form
of the present
invention is typically about 10 mg to about 400 mg. Especially preferred
amounts of
8
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
celecoxib per dose unit are about 100 mg to about 200 mg, for example about
100 mg or
about 200 mg.
[0034] For other selective COX-2 inhibitory drugs, an amount of the drug per
dose unit can be in a range known to be therapeutically effective for such
drugs.
Preferably, the amount per dose unit is in a range providing therapeutic
equivalence to
celecoxib in the dose ranges indicated immediately above.
Amine went in the fill material.
[0035] An amine agent in a dosage form of the invention may be any
pharmaceutically acceptable primary or secondary amine compound. The term
"primary
or secondary amine compound" herein includes those primary and secondary
amines
which are pharmaceutically acceptable excipients. Preferably, primary or
secondary
amine compounds of the present invention are compounds that are not
therapeutically or
nutritionally active. Non-limiting examples of suitable primary amine
compounds include
tromethamine (also known and referred to herein as "Tris" or
tris(hydroxymethyl)aminomethane), ethanolamine, ethylenediamine, diethylamine,
ethylene N-methyl-D-glucamine, and amino acids such as L-arginine, L-lysine,
and
guanidine. Non-limiting examples of suitable secondary amine compounds include
diethanolamine, benethamine (i.e., N-phenymethyl)benezeneethanamine),
benzathine (i.e.,
N,N-dibenzylethylenediamine), piperazine, hydrabamine (i.e., N,N-
bis(dehydroabietyl)ethylenediamine), and imidazole. Preferably, the primary or
secondary
amine compound is present in a dosage form of the invention in a total amine
agent
amount of not more than about 10%, preferably not more than about 7%, and more
preferably not more than about 5% of the dosage form on a dry weight basis,
for example
about 0.1% to about 4%. It should be understood that "on a dry weight basis"
means total
weight excepting water weight..
[0036] In a first preferred embodiment, about 50%, preferably at least about
55%,
more preferably at least about 60%, and still more preferably at least about
65% the total
amine agent amount present in a dosage form of the invention is present in the
fill
material.
Sulfite compound in the fill material.
[0037] The dosage form of the present invention may optionally comprise any
9
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
pharmaceutically acceptable sulfite compound. Illustrative pharmaceutically
acceptable
sulfite compounds include sodium metabisulfite, sodium bisulfite, and sodium
thiosulfate
(sodium hyposulfite). One or more sulfite compounds are optionally present in
a
composition of the invention in an amount of not more than about 10%, for
example
about 0.01% to about 5%, and preferably about 0.1% to about 2%, of the dosage
form on
a dry weight basis. The sulfite compound can alternatively or additionally be
present in
the gelatin capsule wall.
[0038] In a preferred embodiment, at least about 40%, preferably at least
about
50%, still more preferably at least about 55%, even more preferably at least
about 60%,
and yet more preferably at least about 70% of all sulfite compound present in
a dosage
form of the invention is present in the fill material.
Other excipients.
[0039] Optionally, a fill material according to the invention can comprise any
additional pharmaceutically acceptable e~ccipients. Such excipients can
include, by way
of illustration and not limitation, diluents, disintegrants, dispersants,
binding agents,
adhesives, wetting agents,. lubricants, glidants, crystallization inhibitors,
stabilizers,
antioxidants, substances added to mask or counteract a disagreeable taste or
odor, flavors,
dyes, fragrances, preservatives, and substances added to improve appearance of
the
dosage form.
[0040] Such optional additional components should be physically and chemically
compatible with the other ingredients of the fill material and should not be
deleterious to
the recipient. Importantly, some of the above-listed classes of excipients
overlap each
other.
[0041] Fill material of the present invention optionally further comprises at
least
one pharmaceutically acceptable free radical-scavenging antioxidant. A free
radical-
scavenging antioxidant is to be contrasted with a "non-free radical-scavenging
antioxidant", i.e., an antioxidant that does not possess free radical-
scavenging properties.
Non-limiting illustrative examples of suitable free radical-scavenging
antioxidants include
a-tocopherol (vitamin E), ascorbic acid (vitamin C) and salts thereof
including sodium
ascorbate and ascorbic acid palmitate, butylated hydroxyanisole (BHA),
butylated
hydroxytoluene (BHT), fumaric acid and salts thereof, hypophosphorous acid,
malic acid,
alkyl gallates, for example propyl gallate, octyl gallate and lauryl gallate,
sodium sulfite,
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
sodium bisulfite and sodium metabisulfite. Preferred free radical-scavenging
antioxidants
are alkyl gallates, vitamin E, BHA and BHT. More preferably the at least one
free
radical-scavenging antioxidant is propyl gallate.
[0042] One or more free radical-scavenging antioxidants are optionally present
in
dosage forms of the invention in a total amount effective to substantially
reduce formation
of an addition compound, typically in a total amount of about 0.01% to about
5%,
preferably about 0.01% to about 2.5%, and more preferably about 0.01% to about
1%, by
weight of the fill material.
[0043] Fill material according to the invention optionally comprises one or
more
pharmaceutically acceptable sweeteners. Non-limiting examples of suitable
sweeteners
include mannitol, propylene glycol, sodium saccharin, acesulfame K, neotame
and
aspartame. Alternatively or in addition, a viscous sweetener such as sorbitol
solution,
syrup (sucrose solution) or high-fructose corn syrup can be used and, in
addition to
sweetening effects, can also be useful to increase viscosity and to retard
sedimentation.
[0044] Fill material of the invention optionally comprises one or more
pharmaceutically acceptable preservatives other than free radical-scavenging
antioxidants.
Non-limiting examples of suitable preservatives include benzalkonium chloride,
benzethonium chloride, benzyl alcohol, chlorobutanol, phenol, phenylethyl
alcohol,
phenylmercuric nitrate, thimerosal, etc.
[0045] Fill material of the invention optionally comprises one or more
pharmaceutically acceptable wetting agents. Surfactants, hydrophilic polymers
and
certain clays can be useful as wetting agents to aid in dissolution and/or
dispersion of a
hydrophobic drug such as celecoxib. Non-limiting examples of suitable
surfactants
include benzalkonium chloride, benzethonium chloride, cetylpyridinium
chloride, dioctyl
sodium sulfosuccinate, nonoxynol 9, nonoxynol 10, octoxynol 9, poloxamers,
polyoxyethylene (8) caprylic/capric mono- and diglycerides (e.g., LabrasolTM
of
Gattefosse), polyoxyethylene (35) castor oil, polyoxyethylene (20) cetostearyl
ether,
polyoxyethylene (40) hydrogenated castor oil, polyoxyethylene (10) oleyl
ether,
polyoxyethylene (40) stearate, polysorbate 20, polysorbate 40, polysorbate 60,
polysorbate
80 (e.g., TweenTM 80 of ICI), propylene glycol laurate (e.g., LauroglycolTM of
Gattefosse),
sodium lauryl sulfate, sorbitan monolaurate, sorbitan monooleate, sorbitan
monopalmitate, sorbitan monostearate, tyloxapol, and mixtures thereof.
11
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
[0046] Additionally, fill material of the invention optionally comprise one or
more
pharmaceutically acceptable buffering agents, flavoring agents, colorants,
stabilizers
and/or thickeners. Buffers can be used to control pH of a formulation and can
thereby
modulate drug solubility. Flavoring agents can enhance patient compliance by
making the
dosage form more palatable, and colorants can provide a product with a more
aesthetic
andlor distinctive appearance. Non-limiting examples of suitable colorants
include D&C
Red No. 33, FD&C Red No. 3, FD&C Red No. 40, D&C Yellow No. 10, and C Yellow
No. 6.
Liquid fill material
[0047] In a preferred embodiment, fill material comprising the selective COX-2
inhibitory drug is in the form of a liquid. More preferably, the fill material
is self-
emulsifying upon contact with simulate gastric fluid.
Solvents
[0048] Fill material according to this embodiment comprises at least one
solvent
which is preferably suitable for dissolving the drug and/or any additional
ingredients or
excipients present therein.
i. Glycols and glycol ethers
[0049] A preferred solvent is a glycol or glycol ether. Suitable glycol ethers
include those conforming to formula (X):
R1-O-((CH2)m0)n R2 (X)
wherein Rl and RZ are independently hydrogen or C1_6 alkyl, C1_6 alkenyl,
phenyl or
benzyl groups, but no more than one of Rl and R2 is hydrogen; m is an integer
of 2 to
about 5; and n is an integer of 1 to about 20. It is preferred that one of Rl
and R2 is a C1_4
alkyl group and the other is hydrogen or a C1_4 alkyl group; more preferably
at least one of
Rl and Rz is a methyl or ethyl group. It is preferred that m is 2. It is
preferred that n is an
integer of 1 to about 4, more preferably 2.
[0050] Glycol ethers used as solvents in fill material typically have a
molecular
weight of about 75 to about 1000, preferably about 75 to about 500, and more
preferably
about 100 to about 300. Importantly, the glycol ethers used in fill material
of this
embodiment must be pharmaceutically acceptable and must meet all other
conditions
prescribed herein.
12
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
[0051] Non-limiting examples of glycol ethers that may be used in fill
material of
this embodiment include ethylene glycol monomethyl ether, ethylene glycol
dimethyl
ether, ethylene glycol monoethyl ether, ethylene glycol diethyl ether,
ethylene glycol
monobutyl ether, ethylene glycol dibutyl ether, ethylene glycol monophenyl
ether,
ethylene glycol monobenzyl ether, ethylene glycol butylphenyl ether, ethylene
glycol
terpinyl ether, diethylene glycol monomethyl ether, diethylene glycol dimethyl
ether,
diethylene glycol monoethyl ether, diethylene glycol diethyl ether, diethylene
glycol
divinyl ether, ethylene glycol monobutyl ether, diethylene glycol dibutyl
ether, diethylene
glycol monoisobutyl ether, triethylene glycol dimethyl ether, triethylene
glycol monoethyl
ether, triethylene glycol monobutyl ether, tetraethylene glycol dimethyl
ether, and
mixtures thereof. See for example Flick (1998): Industrial Solvents Handbook,
5th ed.,
Noyes Data Corporation, Westwood, NJ. An especially suitable glycol ether
solvents are
diethylene glycol monoethyl ether, sometimes referred to in the art as DGME or
ethoxydiglycol. It is available for example under the trademark TranscutolTM
of
Gattefosse Corporation.
[0052] Glycols suitable as solvents in fill material include propylene glycol,
1,3-
butanediol and polyethylene glycols. A presently preferred solvent is
polyethylene glycol
(PEG).
[0053] Any pharmaceutically acceptable PEG can be used. Preferably, the PEG
has an average molecular weight of about 100 to about 10,000, and more
preferably about
100 to about 1,000. Still more preferably, the PEG is of liquid grade. Non-
limiting
examples of PEGs that can be used in solvent liquids of this invention include
PEG-200,
PEG-350, PEG-400, PEG-540 and PEG-600. See for example Flick (1998), op. cit.,
p.
392. A presently preferred PEG has an average molecular weight of about 375 to
about
450, as exemplified by PEG-400.
[0054] PEGs such as PEG-400 have many desirable properties as solvents for
poorly water-soluble drugs. In the case of celecoxib, for example, the drug
can be
dissolved or solubilized at a very high concentration in PEG-400, enabling
formulation of
a therapeutically effective dose in a very small volume of solvent liquid.
This is
especially important where the resulting solution is to be encapsulated, as
capsules of a
size convenient for swallowing can be prepared containing a therapeutically
effective dose
even of a drug such as celecoxib having a relatively high dose requirement for
efficacy.
13
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Importantly, ethanol, water, and other excipients identified as co-solvents
hereinbelow or
elsewhere can, if desired, be used as solvents in a fill material of the
invention. Typically,
one or more solvents will be present in a fill material in a total amount of
about 5% to
about 95%, preferably about 10% to about 90% and more preferably about 15% to
about
85%, by weight of the fill material.
Co-solvents.
[0055] A fill material of this embodiment optionally comprises one or more
pharmaceutically acceptable co-solvents. Non-limiting examples of suitable co-
solvents
include additional glycols, alcohols, for example ethanol and n-butanol; oleic
and linoleic
acid triglycerides, for example soybean oil; caprylic/capric triglycerides,
for example
MiglyolTM 812 of Huls; caprylic/capric mono- and diglycerides, for example
CapmulTM
MCM of Abitec; polyoxyethylene caprylic/capric glycerides such as
polyoxyethylene (8)
caprylic/capric mono- and diglycerides, for example LabrasolTM of Gattefosse;
propylene
glycol fatty acid esters, for example propylene glycol laurate;
polyoxyethylene (35) castor
oil, for example CremophorTM EL of BASF; polyoxyethylene glyceryl trioleate,
for
example TagatTM TO of Goldschmidt; lower alkyl esters of fatty acids, for
example ethyl
butyrate, ethyl caprylate and ethyl oleate; and water.
Gelatin capsules
[0056] Any pharmaceutically acceptable gelatin capsules can be used to prepare
a
dosage form of the present invention, including hard and soft gelatin
capsules. Such
capsules can be prepared according to any suitable process.
Hard gelatin capsules
[0057] Non-limiting methods for preparing hard gelatin capsules are described
in
the following patents and/or publications, each of which is hereby
incorporated by
reference herein.
[0058] U.S. Patent No. 3,656,997 to Cordes.
[0059] U.S. Patent No. 4,231,211 to Strampfer et al.
[0060] U.S. Patent No. 4,263,251 to Voegle.
[0061] U.S. Patent No. 4,403,461 to Goutard et al.
[0062] U.S. Patent No. 4,705,658 to Lukas.
[0063] U.S. Patent No. 4,720,924 to Hradecky et al.
14
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
[0064] U.S. Patent No. 4,756,902 to Harvey et al.
[0065] U.S. Patent No. 4,884,602 to Yamamoto et al.
[0066] U.S. Patent No. 4,892,766 to Jones.
[0067] U.S. Patent No. 6,350,468 to Sanso.
[0068] International Patent Publication No. WO 84/00919
to Mackie.
[0069] International Patent Publication No. WO 85/04100
to Kalidindi.
ii. Soft gelatin capsules
[0070] In a preferred embodiment, capsule shells are soft gelatin capsule
shells.
Such shells can be prepared according to any suitable process including but
not limited to
the plate process, vacuum process, or the rotary die process. See, for
example, (1) Ansel
et al. (1995) in Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th
ed.,
Williams & Wilkins, Baltimore, MD, pp. 176-182; and (2) Remin~ton: The Science
and
Practice of Pharmacy, 19th Ed., Mack Publishing Co. Easton. PA, pp. 1646 -
1647, the
above-recited pages of which are hereby incorporated by reference herein.
[0071] Non-limiting examples of suitable methods for preparing soft gelatin
capsules are described in the following patents and publications, each of
which is hereby
incorporated by reference herein.
[0072] U.S. Patent No. 3,592,945 to Pesch.
[0073] U.S. Patent No. 4,609,403 to Wittwer et al.
[0074] U.S. Patent No. 4,744,988 to Brox.
[0075] U.S. Patent No. 4,804,542 to Fischer et al.
[0076] U.S. Patent No. 5,146,758 to Herman.
[0077] U.S. Patent No. 5,254,294 to Wunderlich et al.
[0078] U.S. S Patent No. 6,260,332 to Takayanagi.
[0079] U.S. Patent No. 6,238,616 to Ishikawa et al. and
[0080] International Patent Publication No. WO 92/15828 to Herman.
[0081] As used herein, unless specific context instructs otherwise, the term
"capsule shell" (and "gelatin capsule shell") embraces capsule half shells
(that can
cooperate to form a whole capsule shell) and whole capsule shells (that define
a fill
volume). Such term also embraces soft gelatin capsule shells and hard gelatin
capsules,
irrespective of the process by which such shells are made.
[0082] The terms "sealed capsule shell", "sealed in a capsule shell", "sealing
in
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
the capsule shell" and the like are meant to denote a whole capsule shell that
defines a fill
volume, that such fill volume can contain a fill material, that such fill
material is enclosed
in the whole capsule shell, and that such enclosure affords the fill material
more than a de
minimis amount of protection from the atmosphere outside of the whole capsule
shell.
Utility
[0083] Dosage forms of the invention are useful in treatment and prevention of
a
very wide range of disorders mediated by COX-2, including but not restricted
to disorders
characterized by inflammation, pain and/or fever. Such dosage forms are
especially
useful as anti-inflammatory agents, such as in treatment of arthritis, with
the additional
benefit of having significantly less harmful side effects than compositions of
conventional
NSAIDs that lack selectivity for COX-2 over COX-1. In particular, dosage forms
of the
invention have reduced potential for gastrointestinal toxicity and
gastrointestinal
irritation, including upper gastrointestinal ulceration and bleeding by
comparison with
compositions of conventional NSA)Ds. Thus dosage forms of the invention are
particularly useful as an alternative to conventional NSA)Ds where such NSAIDs
are
contraindicated, for example in patients with peptic ulcers, gastritis,
regional enteritis,
ulcerative colitis, diverticulitis or with a recurrent history of
gastrointestinal lesions;
gastrointestinal bleeding, coagulation disorders including anemia such as
hypoprothrombinemia, hemophilia or other bleeding problems; kidney disease; or
in
patients prior to surgery or patients taking anticoagulants.
[0084] Contemplated dosage forms are useful to treat a variety of arthritic
disorders, including but not limited to rheumatoid arthritis,
spondyloarthropathies, gouty
arthritis, osteoarthritis, systemic lupus erythematosus and juvenile
arthritis.
[0085] Such dosage forms are useful in treatment of asthma, bronchitis,
menstrual
cramps, preterm labor, tendonitis, bursitis, allergic neuritis,
cytomegalovirus infection,
apoptosis including HIV-induced apoptosis, lumbago, liver disease including
hepatitis,
skin-related conditions such as psoriasis, eczema, acne, burns, dermatitis and
ultraviolet
radiation damage including sunburn, and post-operative inflammation including
that
following ophthalmic surgery such as cataract surgery or refractive surgery.
[0086] Such dosage forms are useful to treat gastrointestinal conditions such
as
inflammatory bowel disease, Crohn's disease, gastritis, irritable bowel
syndrome and
ulcerative colitis.
16
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
[0087] Such dosage forms are useful in treating inflammation in such diseases
as
migraine headaches, periarteritis nodosa, thyroiditis, aplastic anemia,
Hodgkin's disease,
sclerodoma, rheumatic fever, type I diabetes, neuromuscular junction disease
including
myasthenia gravis, white matter disease including multiple sclerosis,
sarcoidosis,
nephrotic syndrome, Behcet's syndrome, polymyositis, gingivitis, nephritis,
hypersensitivity, swelling occurring after injury including brain edema,
myocardial
ischemia, and the like. '
[0088] Such dosage forms are useful in treatment of ophthalmic disorders,
including without limitation inflammatory disorders such as endophthalmitis,
episcleritis,
retinitis, iriditis, cyclitis, choroiditis, keratitis, conjunctivitis and
blepharitis, inflammatory
disorders of more than one part of the eye, e.g., retinochoroiditis,
iridocyclitis,
iridocyclochoroiditis (also known as uveitis), keratoconjunctivitis,
blepharoconjunctivitis,
etc.; other COX-2 mediated retinopathies including diabetic retinopathy;
ocular
photophobia; acute trauma of any tissue of the eye including postsurgical
trauma, e.g.,
following cataract or corneal transplant surgery; postsurgical ocular
inflammation;
intraoperative miosis; corneal graft rejection; ocular, for example retinal,
neovascularization including that following injury or infection; macular
degeneration;
cystoid macular edema; retrolental fibroplasia; neovascular glaucoma; and
ocular pain.
[0089] Such dosage forms are useful in treatment of pulmonary inflammation,
such as that associated with viral infections and cystic fibrosis, and in bone
resorption
such as that associated with osteoporosis.
[0090] Such dosage forms are useful for treatment of certain central nervous
system disorders, such as cortical demential including Alzheimer's disease,
neurodegeneration, and central nervous system damage resulting from stroke,
ischemia
and trauma. The term "treatment" in the present context includes partial or
total
inhibition of dementias, including Alzheimer's disease, vascular dementia,
multi-infarct
dementia, pre-senile dementia, alcoholic dementia and senile dementia.
[0091] Such dosage forms are useful in treatment of allergic rhinitis,
respiratory
distress syndrome, endotoxin shock syndrome and liver disease.
[0092] Such dosage forms are useful in treatment of pain, including but not
limited to postoperative pain, dental pain, muscular pain, and pain resulting
from cancer.
For example, such dosage forms are useful for relief of pain, fever and
inflammation in a
17
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
variety of conditions including rheumatic fever, influenza and other viral
infections
including common cold, low back and neck pain, dysmenorrhea, headache,
toothache,
sprains and strains, myositis, neuralgia, synovitis, arthritis, including
rheumatoid arthritis,
degenerative joint diseases (osteoarthritis), gout and ankylosing spondylitis,
bursitis,
burns, and trauma following surgical and dental procedures.
[0093] Such dosage forms are useful for treating and preventing
inflammation-related cardiovascular disorders, including vascular diseases,
coronary
artery disease, aneurysm, vascular rejection, arteriosclerosis,
atherosclerosis including
cardiac transplant atherosclerosis, myocardial infarction, embolism, stroke,
thrombosis
including venous thrombosis, angina including unstable angina, coronary plaque
inflammation, bacterial-induced inflammation including Chlamydia-induced
inflammation, viral induced inflammation, and inflammation associated with
surgical
procedures such as vascular grafting including coronary artery bypass surgery,
revascularization procedures including angioplasty, stmt placement,
endarterectomy, or
other invasive procedures involving arteries, veins and capillaries.
[0094] Such dosage forms are useful in treatment of angiogenesis-related
disorders in a subject, for example to inhibit tumor angiogenesis. Such dosage
forms are
useful in treatment of neoplasia, including metastasis; ophthalmological
conditions such
as corneal graft rejection, ocular neovascularization, retinal
neovascularization including
neovascularization following injury or infection, diabetic retinopathy,
macular
degeneration, retrolental fibroplasia and neovascular glaucoma; ulcerative
diseases such
as gastric ulcer; pathological, but non-malignant, conditions such as
hemangiomas,
including infantile hemaginomas, angiofibroma of the nasopharynx and avascular
necrosis
of bone; and disorders of the female reproductive system such as
endometriosis.
[0095] Such dosage forms are useful in prevention and treatment of benign and
malignant tumors and neoplasia including cancer, such as colorectal cancer,
brain cancer,
bone cancer, epithelial cell-derived neoplasia (epithelial carcinoma) such as
basal cell
carcinoma, adenocarcinoma, gastrointestinal cancer such as lip cancer, mouth
cancer,
esophageal cancer, small bowel cancer, stomach cancer, colon cancer, liver
cancer,
bladder cancer, pancreas cancer, ovary cancer, cervical cancer, lung cancer,
breast cancer,
skin cancer such as squamous cell and basal cell cancers, prostate cancer,
renal cell
carcinoma, and other known cancers that effect epithelial cells throughout the
body.
18
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Neoplasias for which dosage forms of the invention are contemplated to be
particularly
useful are gastrointestinal cancer, Barrett's esophagus, liver cancer, bladder
cancer,
pancreatic cancer, ovarian cancer, prostate cancer, cervical cancer, lung
cancer, breast
cancer and skin cancer. Such dosage forms can also be used to treat fibrosis
that occurs
with radiation therapy. Such dosage forms can be used to treat subjects having
adenomatous polyps, including those with familial adenomatous polyposis (FAP).
Additionally, such dosage forms can be used to prevent polyps from forming in
subjects
at risk of FAP.
[0096] Such dosage forms inhibit prostanoid-induced smooth muscle contraction
by inhibiting synthesis of contractile prostanoids and hence can be of use in
treatment of
dysmenorrhea, premature labor, asthma and eosinophil-related disorders. They
also can
be of use for decreasing bone loss particularly in postmenopausal women (i.e.,
treatment
of osteoporosis), and for treatment of glaucoma.
[0097] Preferred uses for dosage forms of the invention are for treatment of
rheumatoid arthritis and osteoarthritis, for pain management generally
(particularly post-
oral surgery pain, post-general surgery pain, post-orthopedic surgery pain,
and acute flares
of osteoarthritis), for treatment of Alzheimer's disease, and for colon cancer
chemoprevention.
[0098] Besides being useful for human treatment, dosage forms of the invention
are useful for veterinary treatment of companion animals, exotic animals, farm
animals,
and the like, particularly mammals. More particularly, dosage forms of the
invention are
useful for treatment of COX-2 mediated disorders in horses, dogs and cats.
EXAMPLES
[0100] The following non-limiting examples are provided for illustrative
purposes
only and are not to be construed as limitations.
Example 1
[0101] Three fill formulations, F1 - F3, were prepared as shown in Table 1.
One
ml of each fill formulation were filled into each of several standard (no
primary or
secondary amine) soft gelatin capsules (R.P. Scherer).
19
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Table 1. Composition of fill formulations F1- F3
Com onent F1 F2 F3
Celecoxib 200 278 270
PEG400 271 337 334
Tween80 217 195 194
Oleic Acid 61 80 78
PVP 47 - _
Ethanol 113 - -
Hydroxypropyl 38 74 74
meth lcellulose ("HPMC")
Water 26 - 10
Pro 1 allate 1 2 2
Tromethamine 26 - 5
Dimethylamino-ethanol - 34 33
"DMAE")
Total 1000 1000 ~ 1000
[0102] Filled capsules were placed in a sealed container and stored at 40
°C and
75% relative humidity for a period of up to 24 weeks. At various times during
storage,
capsules were removed from the closed container and evaluated, by visual
inspection, for
presence or absence of pellicle formation (i.e. cross-linking). Each evaluated
capsule was
assigned a numerical indicator based on any pellicle observed according to the
following
scale: (1) = no pellicle; (2) = thin, incomplete pellicle; (3) = thin,
complete pellicle; (4) _
strong, complete pellicle which inhibits compression of capsule; and (5)
thick, strong, and
severe pellicle. Pellicle formation observations are shown in Table 2.
Table 2. Pellicle formation after storage for up to 24 weeks at 40 °C
and 75 %
relative humidity
Time F1 F2 F3
(weeks)
0 1 1 1
2 3 1
4 1 3 2
6 3 3
8 1 4 3
12 1 - -
24 1 - -
[0103] As shown in Table 2, capsules containing Fill Formulation Fl
(comprising
tromethamine in an amount of about 3% by weight of the fill material)
exhibited no
pellicle formation during storage for a period of six months. By contrast,
capsules
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
containing Fill Formulation F2 (no primary or secondary amine compound) or F3
(0.5%
tromethamine) exhibited pellicle formation by two and four weeks of storage,
respectively.
Example 2
[0104] A test material comprising PEG 400 and 414 p,g/ml formaldehyde was
prepared. Four aliquots, A1 - A4, of the test material were drawn and placed
in separate
vials. Individually, one component selected from glycine, tromethamine,
ethanolamine
(or no additional component) was added to each vial in an amount of 5 mglml,
as shown
in Table 3, to form test samples A1 - A4, respectively.
Table 3. Composition of Test Samples A1- A4
Test Sam A1 A2 A3 A4
le
Ali not Al A2 A3 A4
Additional None TromethamineEthanolamineGlycine
com onent
[0105] Each of the test samples were stored at room temperature for a period
of
three days. After three days of storage, formaldehyde concentration in each
sample was
measured using HPLC. Amount of formaldehyde present in each sample (% weight
of
original amount) is shown in Table 4.
Table 4. Amount of formaldehyde present in Test Samples Al - A4 after storage
Test Sam A1 A2 A3 A4
le
Formaldehyde100 19.6 17.8 61.9
content
[0106] These data show that the primary amines tromethamine and ethanolamine
reduced formaldehyde levels upon storage to a greater extent than did glycine.
Without
being bound by theory, formaldehyde is believed to be a chemical which causes
andlor
promotes gelatin cross-linking.
Example 3.
[0107] The cross-linking behavior of two soft gelatin formulations was
investigated over a 6 month period. As shown below (Table 5), Formulation 30
(the
control lot) contains dimethylaminoethanol ("DMAE") and no sulfite.
Formulation 19
(the test lot) was similar to the Formulation 30, except that Formulation 19
additionally
comprises sodium metabisulfite in the fill material.
21
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Table S. Fill material of Formulations 30 and 19 (mg/g)
Com onent Formulation Formulation
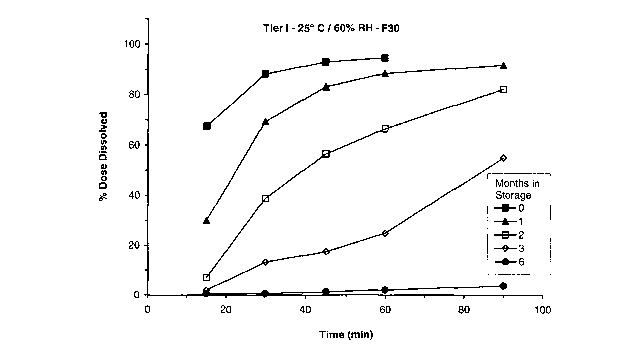
30 19
celecoxib 278 270
PEG 400 337 335
Tween 80 195 195
oleic acid 80 78
HPMC 74 74
DMAE 34 35
ro 1 allate 2 2
w ater - 7
sodium metabisulfite- 4
[0108] Both soft gelatin capsule formulations were placed into non-induction-
sealed hydroxypropyl ethylene bottles and stored at either 25° C and
60% RH or 40° C
and 75% RH. Using such bottles, RH inside the bottles readily equilibrates
with the RH
outside of the bottles (60% or 75%). Periodically, capsules were tested for
degree of
cross-linking of the soft gelatin samples as estimated by the drug release
profile.
[0109] - Formulation 30. The Tier I drug release results for control
Formulation 30
at 25° C / 60% relative humidity ("RH") and 40° C / 75% RH are
shown in Figures 1 and
2 and the Tier II drug release results for the same lot and conditions are
shown in Figures
3 and 4. As early as 1 month of storage, there was a marked~delay in the Tier
I drug
release profile at both temperature conditions. This delay increased with
storage time.
The Tier II drug release profile at 25° C / 60% RH and at 40° C
l 75% RH shows a
significant but markedly reduced delay in release profile.
[0110] Formulation 19. The Formulation 19 Tier I drug release results for the
25°
C / 60% RH condition are shown in Figure 5. No change in the drug release
profile is
observed through 6 months, indicating that no cross-linking has occurred.
Accordingly,
the analogous Tier II test for this sample was not performed. Figure 6
displays the Tier I
results for Formulation 19 at 40° C / 75% RH. No change in drug release
profile is
observed for most of the stability time points with the exception of the 6
month time
point. To determine if the change in drug release profile at 6 months is a
result of cross-
linking, the Tier lI test was performed on this sample. The Tier II results
are displayed in
Figure 7. The Tier I and Tier II results are very similar for this 6 month
sample indicating
that the change in drug release profile is not attributable to cross-linking.
[0111] These data indicate that there was severe cross-linking observed in the
22
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Formulation 30. The change in the Tier II drug release profile (i.e. reduced
delay)
indicates that Tier I delayed release is the result of cross-linking for this
formulation and
further indicates that a significant delay in the drug release profile in
humans would be
likely. The Formulation 19, containing sodium metabisulfite, exhibits no
measurable
cross-linking through 6 months at stringent (40° C / 75% RH) storage
conditions. These
data demonstrate that the addition of sodium metabisulfite to this formulation
significantly reduces the rate of cross-linking and indeed may inhibit cross-
linking
completely. Without being bound by theory, sodium metabisulfite is believed to
inhibit
cross-linking by a process in which sodium metaliisulfite reacts with
aldehydes forming a
bisulfite addition product. Thus, sodium metabisulfite can effectively
scavenges
aldehydes making them unavailable to promote cross-linking in the gelatin.
Example 4.
[0112] Four soft gelatin Celecoxib formulations were prepared as shown in
Table
6 and tested for pellicle formation at 40°C and 75% relative humidity
("RH").
[0113] In absence of sulfite, complete pellicle formation was apparent after
only 2
weeks storage at 40°C / 75% RH (Formulation 30; cross-linking rating
=3).
[0114] At a Tris concentration of 5 mg/g in the formulation (Formulation 20),
delayed pellicle formation but was insufficient to prevent a complete pellicle
formation
(the cross-linking rating =3) upon 1.5 months storage under 40°C / 75%
RH.
[0115] , At a higher Tris concentration in the formulation (26 mg/g,
Formulation
50), gelatin cross-linking is completely prevented upon 6 months storage under
40°C /
75% RH.
[0116] A low sodium metabisulfite (SMB) concentration of 4 mg/g in the
formulation (Formulation 19) appeared sufficient to prevent the pellicle
formation upon 2
months storage under 40°C / 75% RH.
23
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Table 6. Gelatin cross-linking analysis of soft gelatin at 40°C/75% RH
storage
Months Formulation Formulation FormulationFormulation
50
at 40C/mg/ml 30 19 20
75% mg/ml mg/ml mg/ml
RH
Celecoxib Celecoxib Celecoxib Celecoxib
200 278 270 270
PEG400 271 PEG400 337 PEG400 335 PEG400 334
Tween80 217 Tween80 195 Tween80 Tween80 194
195
Oleic acid Oleic acid Oleic acid Oleic acid
61 80 78 78
PVP 47
EtOH 113
HPMC 38 HPMC 74 HPMC 74 HPMC 74
DMAE 34 DMAE 3 5 DMAE 3 3
propyl gallatepropyl gallatepropyl gallatepropyl gallate
1 2 2 2
water 26 water 7 water 10
Tris 26 SMB 4 Tris 5
0 1 1 1 1
0.5 3 1 1
1 1 3 1 2
1.5 3 1 3
2 1 4 1 3
3 1
6 1
Example 5.
[0100] In order to gain insight in to the mechanism by which Tris
(hydroxymethyl
aminomethane) in fill material of a gelatin capsule prevents pellicle
formation, a dosage
form (of Formulation X-60 set forth in Table 7) was prepared and stored under
two
different conditions as shown in Table 8. At the times indicated, capsules
were removed
and Tris content was quantified in the fill material and in the capsule. As
shown in Table
8, upon storage with time, Tris content in the capsules increased and Tris
content in the
fill material decreased in comparison to the initial formulation.
24
CA 02494358 2005-O1-26
WO 2004/010973 PCT/US2003/024043
Table 7. Soft gelatin capsule Formulation X-60
Ingredient Formulation X-60
Celecoxib 200
PEG 400 271
Tween 80 217
Oleic acid 61
Tris 26
Water 26
Propyl gallate 1
PVP-12PF 47
Abs. EtOH 113
HPMC-ES 3 8
Total 1000 mg/g
Fill Volume 0.92 mL
(200 mg drug)
Dosage Form 18 Oblong
soft gelatin
ca sine
Table 8. Tris content in capsule shells following storage of Formulation X-60
Soft gelatin Tris in Tris in
capsule fill shell
Stora a conditions(mg) (mg)
25C/60% RH
T=2 months 18.7 5.3
T=6 moths 17.9 6.0
T=8 months 16.4 6.5
T=10 months 17.6 7.0
40C/75% RH
T=2 months 13.5 10.5
T=6 moths 10.8 11.1
T=8 months 10.0 10.6
T=10 months 10.0 13.3
~6 fng Tris
ih a soft gelatin
capsule at
T=0