Note: Descriptions are shown in the official language in which they were submitted.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
MODIFTED RELEASE DOSAGE FORMS WITH TWO CORES AND AN OPENING
CROSS-REFERENCE TO RELATED APPLICATIONS
This is a continuation-in-part of PCT Application Nos. PCT/US02/31129,
filed September 28, 2002; PCT/LTS02/31117, filed September 28, 2002;
PCT/LTS02/31062, filed September 28, 2002; PCTlLTS02/31024, filed September
28,
2002; and PCT/LJS02/31163, filed September 28, 2002, which are each
continuations-in-part of USSN 091966,939, filed September 28, 2001; USSN
09/966,509, filed September 28, 2001; USSN 09/966,497, filed September 28,
2001;
USSN 09/967,414, filed September 28, 2001; and USSN 09/966,450, filed
io September 28, the disclosures of all of the above being incorporated herein
by
referexice in their entirety.
Field of the Invention
This invention relates to dosage forms providing modified xelease of active
ingredient contained therein. The dosage forms comprise two or more cores
surrounded by a shell having one or more openings. The opening or openings are
distal to at least one of the cores. Preferably, the opening or openings are
proximal
to at least one core and distal to at least another core.
Background of the Invention
Modified release pharmaceutical dosage forms have long been used to
z~b optimize drug delivery and enhance patient compliance, especially by
reducing the
number of doses of medicine the patient must take in a day. In some instances,
it is
also desirable for a dosage form to deliver more than one drug at different
rates or
times. Modified release dosage forms should ideally be adaptable so that
release
rates and profiles can be matched to physiological and. chronotheraheutic
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 2 -
requirements. Because the onset and duration of the therapeutic efficacy of
drugs
vary widely, as do their absorption, distribution, metabolism, and
elimination, it is
often desirable to modify the release of different drugs in different ways, or
to have
a first dose of drug (active ingredient) immediately released from the dosage
form,
while a second dose of the same or a different drug is released in a modified,
e.g.
delayed, pulsatile, repeat action, controlled, sustained, prolonged, extended,
or
retarded manner.
Well known mechanisms by which a dosage form (or drug delivery system)
can deliver drug at a controlled rate (e.g. sustained, prolonged, extended or
retarded
Zo release) include diffusion, erosion, and osmosis. It is often practical to
design
dosage forms that use a combination of the above mechanisms to achieve a
particularly desirable release profile for a particular active ingredient. It
will be
readily recognized by those skilled in the art that a dosage form construct
which
offers multiple compartments, such as for example multiple core portions
and/or
z5 multiple shell portions, is particularly advantageous for its flexibility
in providing a
number of different mechanisms for controlling the release of one or more
active
ingredients.
An important obj ective of modified release dosage forms is to provide a
desired blood concentration versus time (pharmacokinetic, or PK) profile for
the
z o drug. Fundamentally, the PK profile for a drug is governed by the rate of
absorption
of the drug into the blood, and the rate of elimination of the drug from the
blood. To
be absorbed into the blood (circulatory system), the drug must first be
dissolved in
the g.i. fluids. For those relatively rapidly absorbed drugs whose dissolution
in g.i.
fluids is the rate limiting step in drug absorption, controlling the rate of
dissolution
25 (i.e. drug release from the dosage form) allows the formulator to control
the rate of
drug absorption into the circulatory system of a patient. The type of PK
profile, and
correspondingly, the type of dissolution or release profile desired, depends
on,
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 3 -
among other factors, the particular active ingredient and physiological
condition
being treated.
One particularly desirable PK profile is achieved by a dosage form that
delivers a delayed release dissolution profile, in which the release of one or
more
s doses of drug from the dosage form is delayed for a pre-determined time
after
contacting of the dosage form by a liquid medium, such as for example, after
ingestion by the patient. The delay period ("lag time") can be followed either
by
prompt release of the active ingredient ("delayed burst"), or by sustained
(prolonged,
extended, or retarded) release of the active ingredient ("delayed then
sustained").
so U.S. Patent No. 5,464,633, for example, discloses delayed-release dosage
forms in
which an external coating layer was applied by a compression coating process.
The
coating level ranged from 105 percent to 140 percent of the weight of the core
in
order to yield product with the desired time delayed profile.
One particularly desirable type of delayed release PK profile is obtained
15 from a "pulsatile" release profile, in which for example, a first dose of a
first drug is
delivered, followed by a delay period ("lag time") during which there is
substantially
no release of the first drug from the dosage form, followed by either prompt
or
sustained release of a subsequent dose of the same drug. In one particularly
desirable type of pulsatile drug delivery system, the first dose is released
essentially
z o immediately upon contacting of the dosage form with a liquid medium. In
another
particularly desirable type of pulsatile drug delivery system, the delay
period
corresponds approximately to the time during which a therapeutic concentration
of
the first dose is maintained in the blood. Pulsatile delivery systems are
particularly
useful for applications where a continuous release of drug is not ideal.
Examples of
z 5 this are drugs exhibiting first pass metabolism by the liver, drugs that
induce
biological tolerance, i.e. the therapeutic effect decreases with continuous
presence of
the drug at the site of action, and drugs whose efficacy is influenced by
circadian
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 4 -
rhythms of body functions or diseases. One typical pulsatile dosage form
design
contains the first dose of drug in an exterior coating, or shell, while
subsequent
doses of drug are contained in underlying layers of subcoatings, or a central
core.
PCT Publication No. W099/62496, for example, discloses a dosage form
comprising an immediate-release dose of drug contained within an overcoat
applied
onto the surface of the semipermeable membrane of an osmotic dosage form. U.S.
Patent Nos. 4,857,330 and 4,801,461, disclose dosage forms comprising an
exterior
drug coat that surrounds a semipermeable wall, which in turn surrounds an
internal
compartment containing a second dose of drug, and comprises exit means for
so connecting the interior of the dosage form with the exterior environment of
use.
These dosage forms are designed to release drug immediately from the exterior
coating, followed by a relatively short delay period, followed by a sustained
release
of drug from the internal compartment.
U.S. Patent No. 4,576,604, for example, discloses an osmotic device (dosage
form) comprising a drug compartment surrounded by a wall (coating) having an
passageway therein. The wall may comprise an immediate release dose of drug,
and
the inner drug compartment may comprise a sustained release dose of drug. U.S.
Patent No. 4,449,983 discloses another osmotic device comprising two
separately
housed drugs that are separately dispensed from the device. The device
comprises
a o two compartments, one for each drug, separated by a partition. Each
compartment
has an orifice for communicating with the exterior of the device. U.S. Patent
No.
5,738,874, discloses a 3-layer pharmaceutical compressed tablet capable of
liberating one or more drugs at different release rates, in which an immediate
release
dose of active may be contained in a compressed coating layer, and in one
a 5 embodiment, the outer compressed coating layer may function via an erosion
mechanism to delay release of a second dose of active ingredient contained in
the
core. Systems such as these are limited by the amount of drug which may be
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 5 -
incorporated into the exterior coating or shell, which is in turn limited by
the
achievable thickness of the exterior coating or shell.
Another design for a pulsatile delivery system is exemplified in U.S. Patent
No. 4,865,849, which describes a tablet able to release active substances at
s successive times, comprising a first layer containing a portion of the
active
substance, a water soluble or water gellable barrier layer which is interposed
between the first layer and a third layer containing the remaining portion of
active
substance, and the barrier layer and third layer are housed in an insoluble,
impermeable casing. The casing can be applied by various methods such as
so spraying, compression, or immersion, or the tablet parts can be inserted
into a pre-
formed casing. Multilayer compressed tablets in stacked layer configurations
necessessarily require an impermeable partial coating (casing) in order to
provide a
pulsatile release profile. These systems suffer from the complexity and high
cost of
assembling multiple, separate compartments comprising multiple, different
i5 compositions.
Dosage forms have been previously designed with multiple cores housed in a
single shell for the purpose of allowing flexability in a dosing regimen. PCT
Publication No. WO00/18447, for example, describes a multiplex drug delivery
system suitable for oral administration containing at least two distinct drug
dosage
a o packages, which exhibit equivalent dissolution profiles for an active
agent when
compared to one another and when compared to that of the entire multiplex drug
delivery unit, and substantially enveloped by a scored compressed coating that
allows the separation of the multiplex drug delivery system into individual
drug
dosage packages. In this example, two immediate-release compartments are
a 5 enveloped by a scored extended-release compartment. Active ingredient may
be
contained in only the extended release compartment, or additionally in the two
immediate release compartments. The multiplex drug delivery systems of this
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 6 -
example are prepared by press coating the extended-release compartment to
substantially envelop the immediate release compartments.
Improved dosage forms for providing modified release of active ingredient
are described herein. The dosage forms comprise at least one active ingredient
and
s at least two cores surrounded by a shell, wherein the shell comprises one or
more
openings that are distal to one of the cores. Preferably, the opening or
openings are
proximal or connected to at least one core, but distal from at least another
core. In
this manner, at least one core communicates with the exterior of the dosage
form
through an opening, while at least another core therein does not. Upon contact
with
1 o a liquid medium, active ingredient, which may be present in one or more of
the
cores, in the shell, or portions or combinations thereof, is released from the
dosage
form in a modified fashion.
Summary of the Invention
The invention provides a dosage form comprising at least one active
i5 ingredient, a first core, and a second core, said first and second cores
being
surrounded by a shell, wherein the shell comprises one or more openings and
provides for modified release of at least one active ingredient upon contact
of the
dosage form with a liquid medium, at least one of the first or second cores
being
distal to the opening or openings.
2 o The invention also provides a dosage form comprising a first core
containing
a pharmaceutically effective dose of a Frst active ingredient, and a second
core
containing a pharmaceutically effective dose of a second active ingredient,
said first
and second cores each being surrounded by a shell, wherein the shell comprises
a
plurality of openings and provides for modified release of the second active
~ s ingredient upon contact of the dosage form with a liquid medium, wherein
the
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
second core is located distal to all the openings, and all the openings are
proximal to
only the first core.
Brief Description of the Drawings
Figures lA and 1B depict a dosage form according to the invention.
Figures 2A, 2B, and 2C depict another dosage form according to the
invention.
Figures 3A, 3B, and 3C depict another dosage form according to the
invention.
to Figures 4A, 4B, and 4C, depict another dosage form according to the
invention.
Figures SA, SB, and SC depict another dosage form according to the
invention.
~ Figure 6 shows various openings according to the invention.
Detailed Description of the Invention
As used herein, the term "dosage form" applies to any solid object, semi-
solid, or liquid composition designed to contain a specific pre-determined
amount
(dose) of a certain ingredient, for example an active ingredient as defined
below.
z o Suitable dosage forms may be pharmaceutical drug delivery systems,
including
those for oral administration, buccal administration, rectal administration,
topical or
mucosal delivery, or subcutaneous implants, or other implanted drug delivery
systems; or compositions for delivering minerals, vitamins and other
nutraceuticals,
oral care agents, flavorants, and the like. Preferably the dosage forms of the
present
a s invention are considered to be solid, however they may contain liquid or
semi-solid
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
_ g _
components. In a particularly preferred embodiment, the dosage form is an
orally
administered system for delivering a pharmaceutical active ingredient to the
gastro-
intestinal tract of a human.
Suitable active ingredients for use in this invention include for example
pharmaceuticals, minerals, vitamins and other nutraceuticals, oral care
agents,
flavorants and mixtures thereof. Suitable pharmaceuticals include analgesics,
anti-
inflammatory agents, antiarthritics, anesthetics, antihistamines,
antitussives,
antibiotics, anti-infective agents, antivirals, anticoagulants,
antidepressants,
antidiabetic agents, antiemetics, antiflatulents, antifungals, antispasmodics,
appetite
to suppressants, bronchodilators, cardiovascular agents, central nervous
system agents,
central nervous system stimulants, decongestants, oral contraceptives,
diuretics,
expectorants, gastrointestinal agents, migraine preparations, motion sickness
products, mucolytics, muscle relaxants, osteoporosis preparations,
polydimethylsiloxanes, respiratory agents, sleep-aids, urinary tract agents
and
15 mixtures thereof.
Suitable oral care agents include breath fresheners, tooth whiteners,
antimicrobial agents, tooth mineralizers, tooth decay inhibitors, topical
anesthetics,
mucoprotectants, and the like.
Suitable flavorants include menthol, peppermint, mint flavors, fruit flavors,
2 o chocolate, vanilla, bubblegum flavors, coffee flavors, liqueur flavors and
combinations and the like.
Examples of suitable gastrointestinal agents include antacids such as calcium
carbonate, magnesium hydroxide, magnesium oxide, magnesium carbonate,
aluminum hydroxide, sodium bicarbonate, dihydroxyaluminum sodium carbonate;
a 5 stimulant laxatives, such as bisacodyl, cascara sagrada, danthron, senna,
phenolphthalein, aloe, castor oil, ricinoleic acid, and dehydrocholic acid,
and
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
_ g _
mixtures thereof; H2 receptor antagonists, such as famotadine, ranitidine,
cimetadine, nizatidine; proton pump inhibitors such as omeprazole or
lansoprazole;
gastrointestinal cytoprotectives, such as sucraflate and misoprostol;
gastrointestinal
prokinetics, such as prucalopride, antibiotics for H. pylori, such as
clarithromycin,
amoxicillin, tetracycline, and metronidazole; antidiarrheals, such as
diphenoxylate
and loperamide; glycopyrrolate; antiemetics, such as ondansetron, analgesics,
such
as mesalamine.
In one embodiment of the invention, the active ingredient may be selected
from bisacodyl, famotadine, ranitidine, cimetidine, prucalopride,
diphenoxylate,
to loperamide, lactase, mesalamine, bismuth, antacids, and pharmaceutically
acceptable salts, esters, isomers, and mixtures thereof.
In another embodiment, the active ingredient is selected from analgesics,
anti-inflammatories, and antipyretics, e.g. non-steroidal anti-inflammatory
drugs
(NSAIDs), including propionic acid derivatives, e.g. ibuprofen, naproxen,
15 ketoprofen and the like; acetic acid derivatives, e.g. indomethacin,
diclofenac,
sulindac, tolinetin, and the like; fenamic acid derivatives, e.g. mefanamic
acid,
meclofenamic acid, flufenamic acid, and the like; biphenylcarbodylic acid
derivatives, e.g. diflunisal, flufenisal, and the like; and oxicams, e.g.
piroxicam,
sudoxicarn, isoxicam, meloxicam, and the like. In one particular embodiment,
the
z o active ingredient is selected from propionic acid derivative NSAID, e.g.
ibuprofen,
naproxen, flurbiprofen, fenbufen, fenoprofen, indoprofen, ketoprofen,
fluprofen,
pirprofen, carprofen, oxaprozin, pranoprofen, suprofen, and pharmaceutically
acceptable salts, derivatives, and combinations thereof. In another particular
embodiment of the invention, the active ingredient may be selected from
z 5 acetaminophen, acetyl salicylic acid, ibuprofen, naproxen, ketoprofen,
flurbiprofen,
diclofenac, cyclobenzaprine, meloxicam, rofecoxib, celecoxib, and
pharmaceutically
acceptable salts, esters, isomers, and mixtures thereof.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 10 -
In another embodiment of the invention, the active ingredient may be
selected from upper respiratory agents, such as pseudoephedrine,
phenylpropanolamine, chlorpheniramine, dextromethorphan, diphenhydramine,
astemizole, terfenadine, fexofenadine, loratadine, desloratadine, cetirizine,
mixtures
thereof and pharmaceutically acceptable salts, esters, isomers, and mixtures
thereof.
Examples of suitable polydimethylsiloxanes, which include, but are not
limited to dimethicone and simethicone, are those disclosed in United States
Patent
Nos. 4,906,478, 5,275,822, and 6,103,260, the contents of each is expressly
incorporated herein by reference. As used herein, the term "simethicone"
refers to
1 o the broader class of polydimethylsiloxanes, including but not limited to
simethicone
and dimethicone.
The active ingredient or ingredients are present in the dosage form in a
therapeutically effective amount, which is an amount that produces the desired
therapeutic response upon oral administration and can be readily determined by
one
15 skilled in the art. In determining such amounts, the particular active
ingredient
being administered, the bioavailability characteristics of the active
ingredient, the
dosing regimen, the age and weight of the patient, and other factors must be
considered, as known in the art. Typically, the dosage form comprises at least
about
1 weight percent, for example, the dosage form comprises at least about 5
weight
~ o percent, say at least about 20 weight percent of a combination of one or
more active
ingredients. In one embodiment, a core comprises a total of at least about 25
weight
percent (based on the weight of the core) of one or more active ingredients.
The active ingredient or ingredients may be present in the dosage form in any
form. For example, the active ingredient may be dispersed at the molecular
level,
z 5 e.g. melted or dissolved, within the dosage form, or may be in the form of
particles,
which in turn may be coated or uncoated. If an active ingredient is in the
form of
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 11 -
particles , the particles (whether coated or uncoated) typically have an
average
particle size of about 1-2000 microns. In one embodiment, such particles are
crystals having an average particle size of about 1-300 microns. In another
embodiment, the particles are granules or pellets having an average particle
size of
about 50-2000 microns, for example about 50-1000 microns, say about 100-800
microns. In certain embodiments in which one or more active ingredients are in
the
form of particles, the active ingredient particles are contained within one or
more
cores of the dosage form.
Each core may be any solid form. As used herein, "core" refers to a material
to which is at least partially enveloped or surrounded by another material.
Preferably,
a core is a self contained unitary object, such as a tablet or capsule.
Typically, a
core comprises a solid, for example, a core may be a compressed or molded
tablet,
hard or soft capsule, suppository, or a confectionery form such as a lozenge,
nougat,
caramel, fondant, or fat based composition. In certain other embodiments, a
core or
i5 a portion thereof may be in the form of a semi-solid or a liquid in the
finished
dosage form. For example a core may comprise a liquid filled capsule, or a
semisolid fondant material. In embodiments in which a core comprises a
flowable
component, such as a plurality of granules or particles, or a liquid, the core
preferrably additionally comprises an enveloping component, such as a capsule
z o shell, or a coating, for containing the flowable material. In certain
particular
embodiments in which a core comprises an enveloping component, the shell or
shell
portions of the present invention are in direct contact with the enveloping
component of the core, which separates the shell from the flowable component
of
the core.
~5 The dosage form comprises at least two cores, e.g. a first core and a
second
core. The dosage form may comprise more than two cores. The cores may have the
same or different compositions, comprise the same or different active
ingredients,
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 12 -
excipients (inactive ingredients that may be useful for conferring desired
physical
properties to the dosage core), and the like. One or more cores may be
substantially
free of active ingredient. The cores may even comprise incompatible
ingredients
from one another.
Each core is completely surrounded by, or embedded in, the shell. A portion
of the shell, referred herein as the "interior wall" separates the first and
second
cores. The distance between the first and second cores, i.e. thickness of the
interior
wall, may vary depending upon the desired release characteristics of the
dosage
form, or practical considerations related to the manufacturing process. In
certain
to embodiments, the distance between the first and second cores within the
dosage
form, i.e. the thickness of the interior wall, is on the order of the
thickness of the
shell proximal to the core that is distal to the opening or openings. For
example, the
thickness of the interior wall may be from about 10% to about 200% of the
thickness
of a core.
15 Each core may have one of a variety of different shapes. Each core may
have the same or different physical dimensions, shape, etc. as the other
cores. For
example the first and second cores may have different diameters or
thicknesses. For
example, a core may be shaped as a polyhedron, such as a cube, pyramid, prism,
or
the like; or may have the geometry of a space figure with some non-flat faces,
such
a o as a cone, truncated cone, cylinder, sphere, torus, or the like. In
certain
embodiments, a core has one or more major faces. For example, in embodiments
wherein a core is a compressed tablet, the core surface typically has opposing
upper
and lower faces formed by contact with the upper and lower punch faces in the
compression machine. In such embodiments the core surface typically further
a 5 comprises a "belly-band" located between the upper and lower faces, and
formed by
contact with the die walls in the compression machine. A core may also
comprise a
multilayer tablet.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 13 -
In one embodiment at least one core is a compressed tablet having a hardness
from about 2 to about 30 kp/cm2, e.g. from about 6 to about 25 kp/cma.
"Hardness"
is a term used in the art to describe the diametral breaking strength of
either the core
or the coated solid dosage form as measured by conventional pharmaceutical
s hardness testing equipment, such as a Schleuniger Hardness Tester. In order
to
compare values across different size tablets, the breaking strength must be
normalized for the area of the break. This normalized value, expressed in
kp/cmz, is
sometimes referred in the art as tablet tensile strength. A general discussion
of tablet
hardness testing is found in Leiberman et al., PhaYmaceutical Dosage Forms -
to Tablets, Volume 2, 2°d ed., Marcel Dekker Inc., 1990, pp. 213 - 217,
327 - 329. In
another embodiment, all the cores in the dosage form comprise a compressed
tablet
having a hardness from about 2 to about 30 kp/cm2, e.g. from about 6 to about
25
kp/cm2.
The first and second cores may be oriented side by side. For example, in the
~.5 case of cores that are compressed tablets, their belly bands are adjacent
to and in
contact with the interior wall. See for example Figure lA, which depicts a
cross-
sectional view of a dosage form according to the invention comprising two side-
by-
side cores that are compressed tablets. Alternatively, the cores may be
oriented one
on top of the other such that their upper or lower faces are adjacent to and
in contact
2 o with the interior wall. See for example, Figure SA, which shows a cross-
sectional
view of another dosage form according to the invention comprising "upper and
lower" cores. The thickness of the shell may vary among various locations
around
the dosage form. For example, in embodiments where the cores have different
sizes
from one another, the shell may, as a result, have a smaller thickness around
one
2 5 core than the other. In embodiments where one or more cores have a
different shape
than that of the surrounding shell surface, the shell thickness will be
different around
certain portions of a core than around certain other portions. In embodiments
where
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 14 -
the shell comprises more than one portion, the shell portions may have
different
thickness from one another at corresponding locations. In embodiments where
the
.cores are positioned asymmetrically within the dosage form, the shell
thickness will
vary accordingly. This may be exploited to adjust the relative onset or rate
of
release of active ingredient from the two cores. For example, active
ingredient
contained in a smaller core could be released after the release of active
ingredient
from a larger core has begun, due to the relative thinness of the shell around
the
larger core. In another example, active ingredient contained in a first,
elongated,
core could begin to be released sooner than active ingredient from a second,
more
Zo symmetrically shaped core due to the relative thinness of the shell
proximal to the
elongated portion of the first core.
Exemplary core shapes that may be employed include tablet shapes formed
from compression tooling shapes described by "The Elizabeth Companies Tablet
Design Training Manual" (Elizabeth Carbide Die Co., Inc., p. 7 (McI~eesport,
Pa.)
(incorporated herein by reference) as follows (the tablet shape corresponds
inversely
to the shape of the compression tooling):
1. Shallow Concave.
2. Standard Concave.
3. Deep Concave.
4. Extra Deep Concave.
5. Modified Ball Concave.
6. Standard Concave Bisect.
7. Standard Concave Double
Bisect.
8. Standard Concave European
Bisect.
9. Standard Concave Partial
Bisect.
10. Double Radius.
11. Bevel & Concave.
12. Flat Plain.
13. Flat-Faced-Beveled Edge
(F.F.B.E.).
3 0 14. F.F.B.E. Bisect.
15. F.F.B.E. Double Bisect.
16. Ring.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 15 -
17. Dimple.
18. Ellipse.
19. Oval.
20. Capsule.
s 21. Rectangle.
22. Square.
23. Triangle.
24. Hexagon.
25. Pentagon.
26. Octagon.
27. Diamond.
28. Arrowhead.
29. Bullet.
30. Shallow Concave.
is 31. Standard Concave.
32. Deep Concave.
33. Extra Deep Concave.
34. Modified Ball Concave.
35. Standard Concave Bisect.
36. Standard Concave Double
Bisect.
37. Standard Concave European
Bisect.
38. Standard Concave Partial
Bisect.
39. Double Radius.
40. Bevel & Concave.
2 s 41. Flat Plain.
42. Flat-Faced-Beveled Edge
(F.F.B.E.).
43. F.F.B.E. Bisect.
44. F.F.B.E. Double Bisect.
45. Ring.
a o 46. Dimple.
47. Ellipse.
48. Oval.
49. Capsule.
50. Rectangle.
35 51. Square.
52. Triangle.
53. Hexagon.
54. Pentagon.
S5. Octagon.
40 56. Diamond.
57. Arrowhead.
58. Bullet.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 16 -
59. Barrel.
60. Half Moon.
61. Shield.
62. Heart.
63. Almond.
64. House/Home
Plate.
65. Parallelogram.
66. Trapezoid.
67. Figure 8/Bar
Bell.
l0 68. Bow Tie.
69. Uneven Triangle.
The cores may be prepared by any suitable method, including for example
compression or molding, and depending on the method by which they are made,
typically comprise active ingredient and a variety of excipients. The cores
may be
prepared by the same or different methods. For example, a first core may be
prepared by compression, and a second core may be prepared by molding, or both
cores may be prepared by compression.
In embodiments in which one or more cores, or portions thereof are made by
2 o compression, suitable excipients include fillers, binders, disintegrants,
lubricants,
glidants, and the like, as known in the art. In embodiments in which a core is
made
by compression and additionally confers modified'release of an active
ingredient
contained therein, such core preferably further comprises a release-modifying
compressible excipient.
2s Suitable fillers for use in making a core or core portion by compression
include water-soluble compressible carbohydrates such as sugars, which include
dextrose, sucrose, maltose, and.lactose, sugar-alcohols, which include
mannitol,
sorbitol, maltitol, xylitol, starch hydrolysates, which include dextrins, and
maltodextrins, and the like, water insoluble plastically deforming materials
such as
a o microcrystalline cellulose or other cellulosic derivatives, water-
insoluble brittle
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 17 -
fracture materials such as dicalcium phosphate, tricalcium phosphate and the
like
and mixtures thereof.
Suitable binders for making a core or core portion by compression include
dry binders such as polyvinyl pyrrolidone, hydroxypropylmethylcellulose, and
the
like; wet binders such as water-soluble polymers, including hydrocolloids such
as
acacia, alginates, agar, guar gum, locust bean, carrageenan,
carboxymethylcellulose,
tars, gum arabic, tragacanth, pectin, xanthan, gellan, gelatin, maltodextrin,
galactomannan, pusstulan, laminarin, scleroglucan, inulin, whelan, rhamsan,
zooglan, methylan, chitin, cyclodextrin, chitosan, polyvinyl pyrrolidone,
cellulosics,
to sucrose, starches, and the like; and derivatives and mixtures thereof.
Suitable disintegrants for making a core or core portion by compression,
include sodium starch glycolate, cross-linked polyvinylpyrrolidone, cross-
linked
carboxymethylcellulose, starches, microcrystalline cellulose, and the like.
Suitable lubricants for making a core or core portion by compression include
15 long chain fatty acids and their salts, such as magnesium stearate and
stearic acid,
talc, glycerides and waxes.
Suitable glidants for making a core or core portion by compression, include
colloidal silicon dioxide, and the like.
Suitable release-modifying excipients for making a core or core portion by
a o compression include swellable erodible hydrophillic materials, insoluble
edible
materials, pH-dependent polymers, and the like
Suitable swellable erodible hydrophilic materials for use as release-
modifying excipients for making a core or core portion by compression include:
water swellable cellulose derivatives, polyalkalene glycols, thermoplastic
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 18 -
polyalkalene oxides, acrylic polymers, hydrocolloids, clays, gelling starches,
and
swelling doss-linked polymers, and derivitives, copolymers, and combinations
thereof. Examples of suitable water swellable cellulose derivatives include
sodium
carboxymethylcellulose, cross-linked hydroxypropylcellulose, hydroxypropyl
cellulose (HPC), hydroxypropylmethylcellulose (HPMC),
hydroxyisopropylcellulose, hydroxybutylcellulose,hydroxyphenylcellulose,
hydroxyethylcellulose (HEC), hydroxypentylcellulose,
hydroxypropylethylcellulose,
hydroxypropylbutylcellulose, hydroxypropylethylcellulose. Examples of suitable
polyalkalene glyclols include polyethylene glycol. Examples of suitable
to thermoplastic polyalkalene oxides include poly (ethylene oxide). Examples
of
suitable acrylic polymers include potassium methacrylatedivinylbenzene
copolymer,
polymethylinethacrylate, CARBOPOL (high-molceular weight cross-linked acrylic
acid homopolymers and copolymers), and the like. Examples of suitable
hydrocolloids include alginates, agar, guar gum, locust bean gum, kappa
is carrageenan, iota carrageenan, tara, gum arabic, tragacanth, pectin,
xanthan gum,
gellan gum, maltodextrin, galactomannan, pusstulan, laminarin, scleroglucan,
gum
arabic, inulin, pectin, gelatin, whelan, rhamsan, zooglan, methylan, chitin,
cyclodextrin, chitosan. Examples of suitable clays include smectites such as
bentonite, kaolin, and laponite; magnesium trisilicate, magnesium aluminum
silicate,
2 o and the like, and derivatives and mixtures thereof. Examples of suitable
gelling
starches include acid hydrolyzed starches, swelling starches such as sodium
starch
glycolate, and derivatives thereof. Examples of suitable swelling cross-linked
polymers include cross-linked polyvinyl pyrrolidone, cross-linked agar, and
cross-
linked carboxymethylcellose sodium.
~5 Suitable insoluble edible materials for use as release-modifying excipients
for making a core or core portion by compression include water-insoluble
polymers,
and low-melting hydrophobic materials. Examples of suitable water-insoluble
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 19 -
polymers include ethylcellulose, polyvinyl alcohols, polyvinyl acetate,
polycaprolactones, cellulose acetate and its derivatives, acrylates,
methacrylates,
acrylic acid copolymers; and the like and derivatives, copolymers, and
combinations
thereof. Suitable low-melting hydrophobic materials include fats, fatty acid
esters,
phospholipids, and waxes. Examples of suitable fats include hydrogenated
vegetable oils such as for example cocoa butter, hydrogenated palm kernel oil,
hydrogenated cottonseed oil, hydrogenated sunflower oil, and hydrogenated
soybean
oil; and free fatty acids and their salts. Examples of suitable fatty acid
esters include
sucrose fatty acid esters, mono, di, and triglycerides, glyceryl behenate,
glyceryl
io palmitostearate, glyceryl monostearate, glyceryl tristearate, glyceryl
trilaurylate,
glyceryl myristate, GlycoWax-932, lauroyl macrogol-32 glycerides, and stearoyl
macrogol-32 glycerides. Examples of suitable phospholipids include
phosphotidyl
choline, phosphotidyl serene, phosphotidyl enositol, and phosphotidic acid.
Examples of suitable waxes include carnauba wax, spermaceti wax, beeswax,
i5 candelilla wax, shellac wax, microcrystalline wax, and paraffin wax; fat-
containing
mixtures such as chocolate; and the like.
Suitable pH-dependent polymers for use as release-modifying excipients for
making a core or core portion by compression include enteric cellulose
derivatives,
for example hydroxypropyl methylcellulose phthalate, hydroxypropyl
2 o methylcellulose acetate succinate, cellulose acetate phthalate; natural
resins such as
shellac and zero; enteric acetate derivatives such as for example
polyvinylacetate
phthalate, cellulose acetate phthalate, acetaldehyde dimethylcellulose
acetate; and
enteric acrylate derivatives such as for example polymethacrylate-based
polymers
such as poly(methacrylic acid, methyl methacrylate) 1:2, which is commercially
z s available from Rohm Pharma GmbH under the tradename EUDRAGIT S, and
poly(methacrylic acid, methyl methacrylate) l:l, which is commercially
available
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 20 -
from Rohm Pharma GmbH under the tradename EUDRAGIT L, and the like, and
derivatives, salts, copolymers, and combinations thereof.
Suitable pharmaceutically acceptable adjuvants for making a core or core
portion by compression include, preservatives; high intensity sweeteners such
as
aspartame, acesulfame potassium, sucralose, and saccharin; flavorants;
colorants;
antioxidants; surfactants; wetting agents; and the like and mixtures thereof.
In embodiments wherein one or more cores are prepared by compression, a
dry blending (i.e. direct compression), or wet granulation process may be
employed,
as known in the art. In a dry blending (direct compression) method, the active
Zo ingredient or ingredients, together with the excipients, are blended in a
suitable
blender, than transferred directly to a compression machine for pressing into
tablets.
In a wet granulation method, the active ingredient or ingredients, appropriate
excipients, and a solution or dispersion of a wet binder (e.g. an aqueous
cooked
starch paste, or solution of polyvinyl pyrrolidone) are mixed and granulated.
15 Alternatively a dry binder may be included among the excipients, and the
mixture
may be granulated with water or other suitable solvent. Suitable apparatuses
for wet
granulation are known in the art, including low shear, e.g. planetary mixers;
high
shear mixers; and fluid beds, including rotary fluid beds. The resulting
granulated
material is dried, and optionally dry-blended with further ingredients, e.g.
adjuvants
a o and/or excipients such as for example lubricants, colorants, and the like.
The final
dry blend is then suitable for compression. Methods for direct compression and
wet
granulation processes are known in the art, and are described in detail in,
for
example, Lachman, et al., The Theory and Practice of Industrial Pharmacy,
Chapter
11 (3rd ed. 19g6).
25 The dry-blended, or wet granulated, powder mixture is typically compacted
into tablets using a rotary compression machine as known in the art, such as
for
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 21 -
example those commercially available from Fette America Inc., Rockaway, NJ, or
Manesty Machines LTD, Liverpool, UI~. In a rotary compression machine, a
metered volume of powder is filled into a die cavity, which rotates as part of
a "die
table" from the filling position to a compaction position where the powder is
compacted between an upper and a lower punch to an ejection position where the
resulting tablet is pushed from the die cavity by the lower punch and guided
to an
ejection chute by a stationary "take-ofd bar.
In one embodiment, at least one core is prepared by the compression
methods and apparatus described in copending U.S. Patent Application Serial
No.
l0 09/966,509, pages 16-27, the disclosure of which is incorporated herein by
reference. Specifically, the core is made using a rotary compression module
comprising a fill zone, compression zone, and ejection zone in a single
apparatus
having a double row die construction as shown in Figure 6 of U.S. patent
application
Serial No. 09/966,509. The dies of the compression module are preferably
filled
i5 using the assistance of a vacuum, with filters located in or near each die.
Cores made by compression may be single or mufti-layer, for example bi-
layer, tablets.
A shell surrounds the cores. The shell is continuous and completely
surrounds the cores. The shell may be a single, unitary coating, or the shell
may
a o comprise multiple portions, e.g. a first shell portion and a second shell
portion. In
certain embodiments the shell or shell portions are in direct contact with a
core or
core portion. In certain other embodiments, the shell or shell portions are in
direct
contact with a subcoating or enveloping component which substantially
surrounds a
core or core portion. In embodiments in which the shell comprises a first and
25 second shell portion, at least a first shell portion comprises openings
therein. In
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 22 -
embodiments, in which multiple shell portions are employed, the shell portions
may
have the same or different compositions and shapes from one another.
In certain embodiments the dosage form comprises a first shell portion and a
second shell portion that are compositionally different. As used herein, the
term
"compositionally different" means having features that are readily
distinguishable by
qualitative or quantitative chemical analysis, physical testing, or visual
observation.
For example, the first and second shell portions may contain different
ingredients, or
different levels of the same ingredients, or the first and second shell
portions may
have different physical or chemical properties, different functional
properties, or be
to visually distinct. Examples of physical or chemical properties that may be
different
include hydrophylicity, hydrophobicity, hygroscopicity, elasticity,
plasticity, tensile
strength, crystallinity, and density. Examples of functional properties which
may be
different include rate andlor extent of dissolution of the material itself or
of an active
ingredient therefrom, rate of disintegration of the material, permeability to
active
i5 ingredients, permeability to water or aqueous media, and the like. Examples
of
visual distinctions include size, shape, topography, or other geometric
features,
color, hue, opacity, and gloss.
In one embodiment, the first core is surrounded by a first shell portion, and
the second core is surrounded by a second shell portion. For example, in one
2 o particular such embodiment, the first and second cores may contain the
same active
ingredient in the same amount, and may be essentially identical in size,
shape, and
composition, while the first and second shell portions are have different
dissolution
properties, and confer different release profiles to the active ingredient
portions
contained in the first and second cores.
z 5 In another embodiment, the first and second cores are oriented side by
side,
for example as two compressed tablets with their belly bands adjacent to and
in
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 23 -
contact with the interior wall. The upper faces of both cores may be in
contact with
a first shell portion, and the lower faces of both cores may be in contact
with a
second shell portion. In certain other embodiments in which the first and
second
cores are compressed or molded tablets oriented one on top of the other such
that
their upper or lower faces are adj acent to and in contact with the interior
wall, one
core may be entirely surrounded by a first. shell portion, and the other core
may be
entirely surrounded by a second shell portion.
In one embodiment, the surface of the first or second core is substantially
totally coated with a subcoating. In this embodiment, a shell comprising first
and
to second shell portions is in direct contact with the surface of the
subcoating. As used
herein, "substantially totally covering" means at least about 95 percent of
the surface
area of the core is covered by the subcoating.
The use of subcoatings is well known in the art and disclosed in, for
example, United States Patent Nos. 3,185,626, which is incorporated by
reference
15 herein. Any composition suitable for film-coating a tablet may be used as a
subcoating according to the present invention. Examples of suitable
subcoatings are
disclosed in United States Patent Nos. 4,683,256, 4,543,370, 4,643,894,
4,828,841,
4,725,441, 4,802,924, 5,630,871, and 6,274,162, which are all incorporated by
reference herein. Additional suitable subcoatings include one or more of the
2 o following ingredients: cellulose ethers such as
hydroxypropylmethylcellulose,
hydroxypropylcellulose, and hydroxyethylcellulose; polycarbohydrates such as
xanthan gum, starch, and maltodextrin; plasticizers including for example,
glycerin,
polyethylene glycol, propylene glycol, dibutyl sebecate, triethyl citrate,
vegetable
oils such as castor oil, surfactants such as polysorbate-80, sodium lauryl
sulfate and
25 dioctyl-sodium sulfosuccinate; polycarbohydrates, pigments, and opacifiers.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 24 -
In one embodiment, the subcoating comprises, based upon the total weight of
the subcoating, from about 2 percent to about 8 percent, e.g. from about 4
percent to
about 6 percent of a water-soluble cellulose ether and from about 0.1 percent
to
about 1 percent, castor oil, as disclosed in detail in United States Patent
No. 5,658,
589, which is incorporated by reference herein. In another embodiment, the
subcoating comprises, based upon the total weight of the subcoating, from
about 20
percent to about 50 percent, e.g., from about 25 percent to about 40 percent
of
HPMC; from about 45 percent to about 75 percent, e.g., from about SO percent
to
about 70 percent of maltodextrin; and from about 1 percent to about 10
percent, e.g.,
to from about 5 percent to about 10 percent of PEG 400.
In embodiments in which a subcoating is employed, the dried subcoating
typically is present in an amount, based upon the dry weight of the core, from
about
0 percent to about 5 percent.
In another embodiment, one or more cores, e.g. all the cores, are
15 substantially free of subcoating, and the shell or a shell portion is in
direct contact
with a core surface.
The shell comprises one or more openings therein. The openings provide a
passageway for communication between at least one core, or portion thereof,
and the
exterior of the dosage form. One or more openings may extend completely
through
a o the thickness of the shell, or only partially through the shell. In a
preferred
embodiment, the shell comprises a plurality of openings. In either case, at
least one
core in the dosage form is distal to the opening or openings. That is, at
least one
core does not communicate with the exterior of the dosage form through such an
opening in the shell.
25 Preferably, the opening or openings are proximal to at least one core.
Accordingly, in one embodiment, at least one core is distal to the opening or
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 25 -
openings and at least one core is proximal to the opening or openings. The
opening
or openings are preferably in contact with one or more cores.
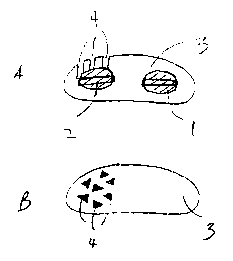
Figure lA depicts a cross-section of a dosage form according to the
invention comprising first and second, side-by-side cores 1, 2 that are
compressed
tablets. The cores are surrounded by a shell 3. Shell 3 comprises a plurality
of
openings 4 in the shape of triangles. Openings 4 are distal to core 1 and
proximal to
core 2. In particular, openings 4 contact core 2. Figure 1B shows a top view
of the
dosage form of Figure 1 A.
Figure 2A depicts a cross-section of another dosage form according to the
1 o invention. This dosage form comprises first and second, side-by-side cores
5, 6
surrounded by a shell having two shell portions 7, 8. Shell portion 7
comprises a
plurality of openings 9, which are distal to core 5 and proximal to and
contacting
core 6. Figure 2B shows a top view of the dosage form of Figure 2A, showing
openings 9 in shell portion 7. Openings 9 are shaped as triangles and circles.
Figure
2C is a bottom view of the dosage form of Figure 2A, showing shell portion 8.
Figure 3A depicts a cross-sectional view of another dosage form according to
the invention. The dosage form comprises four cores 10, 11, 12, and 13, which
are
surrounded by a shell having two shell portions 14, 15. Cores 11 and 13 reside
within shell portion 15, while cores 10 and 12 reside in shell portion 14.
Shell
a o portionl5 comprises a plurality of openings 16, which are distal to cores
10, 1 l and
12, and proximal to only core 13. Openings 16 contact core 13. Figure 3B is a
top
view of the dosage form of Figure 3A, showing openings 16 in shell portion 15.
Figure 3C is a bottom view of the dosage form of Figure 3A, showing shell
portion
14.
2 5 Figure 4A depicts a cross-sectional view of another dosage form according
to
the invention. The dosage form comprises two cores 17, 18 surrounded by shell
19.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 26 -
Shell 19 comprises a plurality of openings 20a, 20b. Openings 20a, 20b axe
distal
to core 17 and proximal to, but not contacting, core 18. In this example,
openings
20a, 20b extend only partially through the shell, and do not contact core 18.
Figure
4B is a top view of the dosage form of Figure 4A, showing openings 20a. Figure
4C
is a bottom view of the dosage form of Figure 4A, showing openings 20b.
In embodiments such as those depicted in Figure 4, in which one or more
openings extend only partially through the shell, and do not contact a core,
the
openings may be in the form of dimples or surface cavities, e.g. debossments
or
intagliations. These partial openings may grow into complete passageways in
Zo contact with a core at some time after exposure of the dosage form to a
suitable
liquid medium, such as for example in-vitro dissolution testing media, or
gastro-
intestinal fluids.
Figure SA is a cross-sectional view of another dosage form according to the
invention. The dosage form comprises first and second cores 21, 22 disposed
one on
top of another. Shell 23 surrounds cores 21, 22 and contains openings 24 that
are
distal to core 21 and proximal to and contacting core 22. Figure SB is a top
view of
the dosage form of Figure SA, showing openings 24, which have three different
shapes 24a, 24b, and 24c. Figure SC is a bottom view of the dosage form of
Figure
SA.
z o Each opening may have dimensions, e.g.. length, width, or diameter, in the
range of about 0.1 % to about 100%, of the diameter of the dosage form, or of
any
dimension (e.g. diameter, length, or width) of a major face of the dosage
form. The
diameter or width of each opening is preferably from about 0.5% to about 5% of
the
diameter of the dosage form, or of any dimension (e.g. diameter, length, or
width) of
z 5 a major face of the dosage form. In certain embodiments the diameter or
width of
the openings may range from about 200 to about 2000 microns. The length of the
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 27 -
openings may range from about 1% to about 100% of the diameter or width of the
dosage form, or of the diameter or width of a major face of the dosage form.
In
certain particular embodiments, the diameter or width of a major face of the
dosage
form is from about 10,000 to about 20,000 microns. In one particular
embodiment,
s the length of the openings is from about 100 to about 20,000 microns. The
depth of
the openings is typically from about 75% to about 100% of the thickness of the
shell
at the location of the openings. In certain embodiments, the thickness of the
shell at
the location of the openings typically ranges from about 20 to about X00
microns,
e.g. from about 100 to about 400 microns. In one particular embodiment, the
depth
to of the openings is from about 75 to about 400 microns. If a plurality of
openings are
present, they are typically spaced from one another by at least about one
half, e.g. at
least about one, times the smallest dimension of the smallest opening. The
openings
may have a variety of shapes, or be arranged in a variety of different
patterns, and
may have similar or different sizes, such as depicted in Figure 6.
15 In one embodiment, the size of the openings is small enough to prevent the
core proximal thereto from being tasted, yet the number of openings is large
enough
to provide communication between a certain percentage of surface area of such
proximal core and the exterior of the dosage form.
The dosage forms of the invention provide modified release of one or more
2 o active ingredients contained therein. The active ingredient or ingredients
may be
found within one or more cores, the shell, or portions or combinations
thereof.
Preferably, one or more active ingredients are contained in one or more cores.
More
preferably, at least one active ingredient is contained in each of the first
and second
cores.
25 Modified release of at least one active ingredient in the dosage form is
provided by the shell, or a portion thereof. As used herein, the term
"modified
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 28 -
release" means the release of an active ingredient from a dosage form or a
portion
thereof in other than an immediate release fashion, i.e., other than
immediately upon
contact of the dosage form or portion thereof with a liquid medium. As known
in
the art, types of modified release include delayed or controlled. Types of
controlled
release include prolonged, sustained, extended, retarded, and the like.
Modified
release profiles that incorporate a delayed release feature include pulsatile,
repeat
action, and the like. As is also known in the art, suitable mechanisms for
achieving
modified release of an active ingredient include diffusion, erosion, surface
area
control via geometry and/or impermeable barners, and other known mechanisms
z o known.
In a preferred embodiment, at least one active ingredient is released from the
first (proximal) core in an immediate release fashion. As used herein,
"immediate
release" means the dissolution characteristics of an active ingredient meets
USP
specifications for immediate release tablets containing the active ingredient.
For
15 example, for acetaminophen tablets, USP 24 specifies that in pH 5.8
phosphate
buffer, using USP apparatus 2 (paddles) at 50 rpm, at least 80% of the
acetaminophen contained in the dosage form is released therefrom within 30
minutes
after dosing, and for ibuprofen tablets, USP 24 specifies that in pH 7.2
phosphate
buffer, using USP apparatus 2 (paddles) at 50 rpm, at least 80% of the
ibuprofen
2 o contained in the dosage form is released therefrom within 60 minutes after
dosing.
See USP 24, 2000 Version, 19 - 20 and 856 (1999).
The composition of the shell may function to modify the release
therethrough of an active ingredient contained in an underlying core. In one
embodiment, the shell may function to delay release of an active ingredient
from an
2 5 underlying core. In another embodiment, the shell may function to sustain,
extend,
retard, or prolong the release of at least one active ingredient from the
second
(distally located) core.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 29 -
In one embodiment, the shell comprises a release modifying moldable
excipient, such as, but not limited to, swellable erodible hydrophilic
materials, pH-
dependent polymers, pore formers, and insoluble edible materials.
In one embodiment, the release-modifying moldable excipient is selected
from hydroxypropylmethylcellulose, polyethylene oxide, ammonio methacrylate
copolymer type B, and shellac, and combinations thereof.
Suitable swellable erodible hydrophilic materials for use as release
modifying moldable excipients include water swellable cellulose derivatives,
polyalkalene glycols, thermoplastic polyalkalene oxides, acrylic polymers,
to , hydrocolloids, clays, gelling starches, and swelling cross-linked
polymers, and
derivitives, copolymers, and combinations thereof. Examples of suitable water
swellable cellulose derivatives include sodium carboxymethylcellulose, cross-
linked
hydroxypropylcellulose, hydroxypropyl cellulose (HPC),
hydroxypropylmethylcellulose (HPMC), hydroxyisopropylcellulose,
15 hydroxybutylcellulose,hydroxyphenylcellulose, hydroxyethylcellulose (HEC),
hydroxypentylcellulose, hydroxypropylethylcellulose,
hydroxypropylbutylcellulose,
hydroxypropylethylcellulose. Examples of suitable polyalkalene glyclols
include
polyethylene glycol. Examples of suitable thermoplastic polyallcalene oxides
include poly (ethylene oxide). Examples of suitable acrylic polymers include
potassium methacrylatedivinylbenzene copolymer, polymethylmethacrylate,
CARBOPOL (high-molceular weight cross-linked acrylic acid homopolymers and
copolymers), and the like. Examples of suitable hydrocolloids include
alginates,
agar, guar gum, locust bean gum, kappa carrageenan, iota carrageenan, tara,
gum
arabic, tragacanth, pectin, xanthan gum, gellan gum, maltodextrin,
galactomannan,
25 pusstulan, laminarin, scleroglucan, gum arabic, inulin, pectin, gelatin,
whelan,
rhamsan, zooglan, methylan, chitin, cyclodextrin, chitosan. Examples of
suitable
clays include smectites such as bentonite, kaolin, and laponite; magnesium
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 30 -
trisilicate, magnesium aluminum silicate, and the like, and derivatives and
mixtures
thereof. Examples of suitable gelling starches include acid hydrolyzed
starches,
swelling starches such as sodium starch glycolate, and derivatives thereof.
Examples of suitable swelling cross-linked polymers include cross-linked
polyvinyl
pyrrolidone, cross-linked agar, and cross-linked carboxymethylcellose sodium.
Suitable pH-dependent polymers for use as release-modifying moldable
excipients include enteric cellulose derivatives, for example hydroxypropyl
methylcellulose phthalate, hydroxypropyl methylcellulose acetate succinate,
cellulose acetate phthalate; natural resins such as shellac and zero; enteric
acetate
to derivatives such as~for example polyvinylacetate phthalate, cellulose
acetate
phthalate, acetaldehyde dimethylcellulose acetate; and enteric acrylate
derivatives
such as for example polymethacrylate-based polymers such as poly(methacrylic
acid, methyl methacrylate) 1:2, which is commercially available from Rohm
Pharma
GmbH under the tradename EUDRAGIT S, and poly(methacrylic acid, methyl
15 methacrylate) 1:1, which is commercially available from Rohm Pharma GmbH
under the tradename EUDRAGIT L, and the like, and derivatives, salts,
copolymers,
and combinations thereof.
Suitable insoluble edible materials for use as release-modifying moldable
excipients include water-insoluble polymers, and low-melting hydrophobic
z o materials. Examples of suitable water-insoluble polymers include
ethylcellulose,
polyvinyl alcohols, polyvinyl acetate, polycaprolactones, cellulose acetate
and its
derivatives, acrylates, methacrylates, acrylic acid copolymers; and the like
and
derivatives, copolymers, and combinations thereof. Suitable low-melting
hydrophobic materials include fats, fatty acid esters, phospholipids, and
waxes.
25 ' Examples of suitable fats include hydrogenated vegetable oils such as for
example
cocoa butter, hydrogenated palm kernel oil, hydrogenated cottonseed oil,
hydrogenated sunflower oil, and hydrogenated soybean oil; and free fatty acids
and
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 31 -
their salts. Examples of suitable fatty acid esters include sucrose fatty acid
esters,
mono, di, and triglycerides, glyceryl behenate, glyceryl palmitostearate,
glyceryl
monostearate, glyceryl tristearate, glyceryl trilaurylate, glyceryl myristate,
GlycoWax-932, lauroyl macrogol-32 glycerides, and stearoyl macrogol-32
glycerides. Examples of suitable phospholipids include phosphotidyl choline,
phosphotidyl serene, phosphotidyl enositol, and phosphotidic acid. Examples of
suitable waxes include carnauba wax, spermaceti wax, beeswax, candelilla wax,
shellac wax, microcrystalline wax, and paraffin wax; fat-containing mixtures
such
as chocolate; and the like.
to Suitable pore formers for use as release-modifying moldable excipients
include water-soluble organic and inorganic materials. In one embodiment the
pore
former is hydroxypropylmethylcellulose. Examples of suitable water-soluble
organic materials include water soluble polymers including water soluble
cellulose
derivatives such as hydroxypropylmethylcellulose, and hydroxypropylcellulose;
15 water soluble carbohydrates such as sugars, and starches; water soluble
polymers
such as polyvinylpyrrolidone and polyethylene glycol, and insoluble swelling
polymers such as microcrystalline cellulose. Examples of suitable water
soluble
inorganic materials include salts such as sodium chloride and potassium
chloride and
the like and/or mixtures thereof.
2 o In another embodiment, the dosage form is substantially free (i.e. less
than
1% by weight, preferably less than about 0.1% by weight, based upon the shell
weight) of charge control agents. As used herein, the term "charge control
agents"
refers to a material having a charge control function, such as those used for
electrostatic deposition of coatings onto substrates. Such charge control
agents
z5 include metal salicylates, for example zinc salicylate, magnesium
salicylate and
calcium salicylate; quaternary ammonium salts; benzalkonium chloride;
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 32 -
benzethonium chloride; trimethyl tetradecyl ammonium bromide (cetrimide); and
cyclodextrins and their adducts.
Accordingly, in certain embodiments, the dosage form comprises at least two
cores containing the same or different active ingredient surrounded by a shell
comprising a release modifying moldable excipient. The shell also comprises
one or
more openings proximal to a first core but distal to a second core. In this
manner,
the first core communicates with the exterior of the dosage form through the
openings, but the second core does not. Upon contact of the dosage form with a
suitable liquid medium, e.g. in-vitro dissolution media or gastro-intestinal
fluids,
to the liquid medium contacts the first core via the openings, and active
ingredient
contained in the first core is promptly, preferably immediately, released from
the
dosage form. Liquid media cannot, however, initially contact active ingredient
contained in the second core. Release of active ingredient contained in the
second
core is therefore dependent on the nature of the shell. This active ingredient
is
15 released from the dosage form in a modified manner.
In a first preferred embodiment such as described in the preceding paragraph,
a time delay, or lag time precedes release of active ingredient contained in
the
second core. Particularly useful lag times include those of at least about 1
hour, e.g.
at least about 4 hours, say at least about 6 hours. In one such embodiment,
active
~ o ingredient contained in the second core may be released promptly or
substantially
immediately following the lag time, as a delayed burst. In certain such
embodiments
wherein separate doses of the same active ingredient are contained in the
first and
second cores, that particular active ingredient is said to be released from
the dosage
form in a pulsatile manner. In another such embodiment, active ingredient
25 contained in the second core may be released in a controlled, sustained,
prolonged,
or extended manner following the lag time.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 33 -
In a second preferred embodiment such as described in the preceding
paragraphs, one or more active ingredients contained in the second core are
released
in a controlled, sustained, prolonged, or extended manner beginning initially
upon
contact of the dosage for with a liquid medium, without a substantial
preceding lag
time, e.g. release of at least one active ingredients begins within 30
minutes, e.g.
within 15 minutes, say within 10 minutes, of contact of the dosage form with a
liquid medium.
In certain embodiments, the shell itself, e.g. a portion thereof, or an outer
coating thereon may also contain active ingredient. In one embodiment, such
active
to ingredient will be released immediately from the dosage form upon
ingestion, or
contacting of the dosage form with a liquid medium. In another embodiment,
such
active ingredient will be released in a controlled, sustained, prolonged, or
extended
fashion upon ingestion, or contacting of the dosage form with a liquid medium.
In certain preferred embodiments of the invention, the cores, the shell, any
i5 portions thereof, or both are prepared by molding. In particular, the
cores, the shell,
or both may be made by solvent-based molding or solvent-free molding. In such
embodiments, the core or the shell is made from a flowable material optionally
comprising active ingredient. The flowable material may be any edible material
that
is flowable at a temperature between about 37°C and 250°C, and
that is solid, semi-
a o solid, or can form a gel at a temperature between about -10°C and.
about 35°C.
When it is in the fluid or flowable state, the flowable material may comprise
a
dissolved or molten component for solvent-free molding, or optionally a
solvent
such as for example water or organic solvents, or combinations thereof, for
solvent-
based molding. The solvent may be partially or substantially removed by
drying.
25 In one embodiment, solvent-based or solvent-free molding is performed via
thermal setting molding using the method and apparatus described in copending
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 34 -
U.S. patent application Serial No. 09/966,450, pages 57-63, the disclosure of
which
is incorporated herein by reference. In this embodiment, a core or shell is
formed by
injecting flowable form into a molding chamber. The flowable material
preferably
comprises a thermal setting material at a temperature above its melting point
but
below the decomposition temperature of any active ingredient contained
therein.
The flowable material is cooled and solidifies in the molding chamber into a
shaped
form (i.e., having the shape of the mold).
According to this method, the flowable material may comprise solid particles
suspended in a molten matrix, for example a polymer matrix. The flowable
material
to may be completely molten or in the form of a paste. The flowable material
may
comprise an active ingredient dissolved in a molten material in the case of
solvent-
free molding. Alternatively, the flowable material may be made by dissolving a
solid in a solvent, which solvent is then evaporated after the molding step in
the case
of solvent-based molding.
In another embodiment, solvent-based or solvent-free molding is performed
by thermal cycle molding using the method and apparatus described in copending
U.S. patent application Serial No. 09/966,497, pages 27-51, the disclosure of
which
is incorporated herein by reference. Thermal cycle molding is performed by
injecting a flowable material into a heated molding chamber. The flowable
material
a o may comprise active ingredient and a thermoplastic material at a
temperature above
the set temperature of the thermoplastic material but below the decomposition
temperature of active ingredient. The flowable material is cooled and
solidifies in
the molding chamber into a shaped form (i.e., having the shape of the mold).
In the thermal cycle molding method and apparatus of U.S. patent
z s application Serial No. 09/966,497 a thermal cycle molding module having
the
general configuration shown in Figure 3 therein is employed. The thermal cycle
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 35 -
molding module 200 comprises a rotor 202 around which a plurality of mold
units
204 are disposed. The thermal cycle molding module includes a reservoir 206
(see
Figure 4) for holding flowable material to make the core. In addition, the
thermal
cycle molding module is provided with a temperature control system for rapidly
heating and cooling the mold units. Figures 55 and 56 depict the temperature
control system 600.
The mold units may comprise center mold assemblies 212, upper mold
assemblies 214, and lower mold assemblies 210, as shown in Figures 26-28,
which
mate to form mold cavities having a desired shape, for instance of a core or a
shell
to surrounding one or more cores. As rotor 202 rotates, opposing center and
upper
mold assemblies or opposing center and lower mold assemblies close. Flowable
material, which is heated to a flowable state in reservoir 206, is injected
into the
resulting mold cavities. The temperature of the flowable material is then
decreased,
hardening the flowable material. The mold assemblies open and eject the
finished
15 product.
In a particularly preferred embodiment of the invention, the shell is applied
to the dosage form using a thermal cycle molding apparatus of the general type
shown in Figures 28A-C of copending U.S. Application Serial No. 09!966,497
comprising rotatable center mold assemblies 212, lower mold assemblies 210 and
a o upper mold assemblies 214. Cores are continuously fed to the mold
assemblies.
Shell flowable material, which is heated to a flowable state in reservoir 206,
is
injected into the mold cavities created by the closed mold assemblies holding
the
cores. The temperature of the shell flowable material is then decreased,
hardening it
around the cores. The mold assemblies open and eject the finished dosage
forms.
z 5 Shell coating is performed in two steps, each half of the dosage forms
being coated
separately as shown in the flow diagram of Figure 28B of copending U.S.
Application Serial No. 09/966,939 via rotation of the center mold assembly.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 36 -
In particular, the mold assemblies for applying the shell are provided with
two or more cavities to accommodate the desired number of cores in the dosage
form. The cavities are separated by a wall, preferably made of rubber or
metal, and
the overall shape of the cavities conform to the shape of the cores.
In addition, the inside surface of at least one of the mold assemblies
comprises one or more protrusions. Each protrusion, which is adjusted for
shape
and size as desired, masks a small location on an underlying core, leaving
behind an
opening in the shell at the site of the protrusion. The mold assemblies may
comprise
a plurality of protrusions to form a plurality of corresponding openings in
the shell.
Zo In such case, the protrusions are located within the mold assemblies such
that the
resulting openings will be distal to at least one underlying core. For
example, the
protrusions may be located on the inside surface of only one mold assembly,
say the
upper mold assembly, or located within only a portion, i.e., one quadrant of
the
inside surface of one mold assembly.
15 In one embodiment, the compression module of copending U.S. patent
application Serial No. 09/966,509, pp. 16-27 may be employed to make cores.
The
shell may be made applied to these cores using a thermal cycle molding module
as
described above. A transfer device as described in U.S. patent application
Serial No.
09/966,414, pp. 51-57, the disclosure of which is incorporated herein by
reference,
2 o may be used to transfer the cores from the compression module to the
thermal cycle
molding module. Such a transfer device may have the structure shown as 300 in
Figure 3 of copending U.S. Application Serial No. 09!966,939. It comprises a
plurality of transfer units 304 attached in cantilever fashion to a belt 312
as shown in
Figures 68 and 69 of copending U.S. Application Serial No. 09/966,939. The
z s transfer device rotates and operates in sync with the compression module
and the
thermal cycle molding module to which it is coupled. Transfer units 304
comprise
retainers 330 for holding cores as they travel around the transfer device.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 37 -
Each transfer unit comprises multiple retainers for holding multiple cores
side by side. In one embodiment, the between the retainers within each
transfer unit
is adjusted via a cam tracklcam follower mechanism as the transfer units move
around the transfer device. On arnval at the thermal cycle molding module, the
cores grouped together for placement in a single dosage form, which have been
held
within a single transfer unit, are properly spaced from one another and ready
to be
fed into the mold assemblies. The cores may or may not have the same
composition,
as desired. The cores may comprise a single layer or multiple layers.
Alternatively, if cores of the same composition are to be used in the dosage
to forms, the compression module may be equipped with multi-tip compression
tooling. Four-tip tooling, for example, may be used to make four cores within
one
die. The cores may comprise a single layer of multiple layers.
Suitable thermoplastic materials for use in or as the flowable material
include both water soluble and water insoluble polymers that are generally
linear,
i5 not crosslinked, and not strongly hydrogen bonded to adjacent polymer
chains.
Examples of suitable thermoplastic materials include: thermoplastic water
swellable
cellulose derivatives, thermoplastic water insoluble cellulose derivatrives,
thermoplastic vinyl polymers, thermoplastic starches, thermplastic
polyalkalene
glycols, thermoplastic polyalkalene oxides, and amorphous sugar-glass, and the
like,
2 o and derivatives, copolymers, and combinations thereof. Examples of
suitable
thermoplastic water swellable cellulose derivatives include hydroxypropyl
cellulose
(HPC), hydroxypropylmethyl cellulose (HPMC), methyl cellulose (MC). Examples
of suitable thermoplastic water insoluble cellulose derivatrives include
cellulose
acetate (CA), ethyl cellulose (EC), cellulose acetate butyrate (CAB),
cellulose
z5 propionate. Examples of suitable thermoplastic vinyl polymers include
polyvinyl
alcohol (PVA) and polyvinyl pyrrolidone (PVP). Examples of suitable
thermoplastic starches are disclosed for example in U.S. Patent No. 5,427,614.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 38 -
Examples of suitable thermoplastic polyallcalene glycols include polyethylene
glycol. Examples of suitable thermoplastic polyalkalene oxides include
polyethylene oxide having a molecular weight from about 100,000 to about
900,000
Daltons. Other suitable thermoplastic materials include sugar in the form on
an
amorphous glass such as that used to make hard candy forms.
Any film former known in the art is suitable for use in the flowable material.
Examples of suitable film formers include, but are not limited to, film-
forming
water soluble polymers, film-forming proteins, film-forming water insoluble
polymers, and film-forming pH-dependent polymers. In one embodiment, the film-
1 o former for making the core or shell or portion thereof by molding may be
selected
from cellulose acetate, ammonio methacrylate copolymer type B, shellac,
hydroxypropylinethylcellulose, and polyethylene oxide, and combinations
thereof.
Suitable film-forming water soluble polymers include water soluble vinyl
polymers such as polyvinylalcohol (PVA); water soluble polycarbohydrates such
as
i5 hydroxypropyl starch, hydroxyethyl starch, pullulan, methylethyl starch,
carboxymethyl starch, pre-gelatinized starches, and film-forming modified
starches;
water swellable cellulose derivatives such as hydroxypropyl cellulose (HPC),
hydroxypropylmethyl cellulose (HPMC), methyl cellulose (MC),
hydroxyethylmethylcellulose (HEMC), hydroxybutylmethylcellulose (HBMC),
2 o hydroxyethylethylcellulose (HEEC), and hydroxyethylhydroxypropylmethyl
cellulose (HEMPMC); water soluble copolymers such as methacrylic acid and
methacrylate ester copolymers, polyvinyl alcohol and polyethylene glycol
copolymers, polyethylene oxide and polyvinylpyrrolidone copolymers; and
derivatives and combinations thereof.
25 Suitable film-forming proteins may be natural or chemically modified, and
include gelatin, whey protein, myofibrillar proteins, coaggulatable proteins
such as
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 39 -
albumin, casein, caseinates and casein isolates, soy protein and soy protein
isolates,
zero; and polymers, derivatives and mixtures thereof.
Suitable film-forming water insoluble polymers, include for example
ethylcellulose, polyvinyl alcohols, polyvinyl acetate, polycaprolactones,
cellulose
acetate and its derivatives, acrylates, methacrylates, acrylic acid
copolymers; and the
like and derivatives, copolymers, and combinations thereof.
Suitable film-forming pH-dependent polymers include enteric cellulose
derivatives, such as for example hydroxypropyl methylcellulose phthalate,
hydroxypropyl methylcellulose acetate succinate, cellulose acetate phthalate;
natural
Z o resins, such as shellac and zero; enteric acetate derivatives such as for
example
polyvinylacetate phthalate, cellulose acetate phthalate, acetaldehyde
dimethylcellulose acetate; and enteric acrylate derivatives such as for
example
polymethacrylate-based polymers such as poly(methacrylic acid, methyl
methacrylate) 1:2, which is commercially available from Rohm Pharma GmbH
i5 under the tradename, EUDRAGIT S, and poly(methacrylic acid, methyl
methacrylate) 1:1, which is commercially available from Rohm Pharma GmbH
under the tradename, EUDRAGIT L, and the like, and derivatives, salts,
copolymers, and combinations thereof.
One suitable hydroxypropylinethylcellulose compound for use as a
2 o thermoplastic film-forming water soluble polymer is "HPMC 2910", which is
a
cellulose ether having a degree of substitution of about 1.9 and a
hydroxypropyl
molar substitution of 0.23, and containing, based upon the total weight of the
compound, from about 29% to about 30% methoxyl groups and from about 7% to
about 12% hydroxylpropyl groups. HPMC 2910 is commercially available from the
25 Dow Chemical Company under the tradename METHOCEL E. METHOCEL E5,
which is one grade of HPMC-2910 suitable for use in the present invention, has
a
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 40 -
viscosity of about 4 to 6 cps (4 to 6 millipascal-seconds) at 20°C in a
2% aqueous
solution as determined by a Ubbelohde viscometer. Similarly, METHOCEL E6 ,
which is another grade of HPMC-2910 suitable for use in the present invention,
has
a viscosity of about 5 to 7 cps (5 to 7 millipascal-seconds) at 20°C in
a 2% aqueous
solution as determined by a Ubbelohde viscometer. METHOCEL E15, which is
another grade of HPMC-2910 suitable for use in the present invention, has a
viscosity of about 15000 cps (15 millipascal-seconds) at 20°C in a 2%
aqueous
solution as determined by a Ubbelohde viscometer. As used herein, "degree of
substitution" meand the average number of substituent groups attached to a
so anhydroglucose ring, and "hydroxypropyl molar substitution" meand the
number of
moles of hydroxypropyl per mole anhydroglucose.
One suitable polyvinyl alcohol and polyethylene glycol copolymer is
commercially available from BASF Corporation under the tradename KOLLICOAT
IR.
15 As used herein, "modified starches" include starches that have been
modified
by crosslinking, chemically modified for improved stability or optimized
performance, or physically modified for improved solubility properties or
optimized
performance. Examples of chemically-modified starches are well known in the
art
and typically include those starches that have been chemically treated to
cause
a o replacement of some of its hydroxyl groups with either ester or ether
groups.
Crosslinking, as used herein, may occur in modified starches when two hydroxyl
groups on neighboring starch molecules are chemically linked. As used herein,
"pre-gelatinized starches" or "instantized starches" refers to modified
starches that
have been pre-wetted, then dried to enhance their cold-water solubility.
Suitable
a5 modified starches are commercially available from several suppliers such
as, for
example, A.E. Staley Manufacturing Company, and National Starch & Chemical
Company. One suitable film forming modified starch includes the pre-
gelatinized
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 41 -
waxy maize derivative starches that are commercially available from National
Starch & Chemical Company under the tradenames PURITY GUM and FILMSET,
and derivatives, copolymers, and mixtures thereof. Such waxy maize starches
typically contain, based upon the total weight of the starch, from about 0
percent to
about 18 percent of amylose and from about.100% to about 88% of amylopectin.
Other suitable film forming modified starches include the hydroxypropylated
starches, in which some of the hydroxyl groups of the starch have been
etherified
with hydroxypropyl groups, usually via treatment with propylene oxide. One
example of a suitable hydroxypropyl starch that possesses film-forming
properties is
so available from Grain Processing Company under the tradename, PURE-COTE
B790.
Suitable tapioca dextrins for use as film formers include those available from
National Starch & Chemical Company under the tradenames CRYSTAL GUM or
K-4484, and derivatives thereof such as modified food starch derived from
tapioca,
15 which is available from National Starch and Chemical under the tradename
PURITY
GUM 40, and copolymers and mixtures thereof.
Any thickener known in the art is suitable for use in the flowable material of
the present invention. Examples of such thickeners include but are not limited
to
hydrocolloids (also referred to herein as gelling polymers), clays, gelling
starches,
a o and crystallizable carbohydrates, and derivatives, copolymers and mixtures
thereof.
Examples of suitable hydrocolloids (also referred to herein as gelling
polymers) such as alginates, agar, guar gum, locust bean, carrageenan, tara,
gum
arabic, tragacanth, pectin, xanthan, gellan, maltodextrin, galactomannan,
pusstulan,
laminarin, scleroglucan, gum arabic, inulin, pectin, whelan, rhamsan, zooglan,
z 5 methylan, chitin, cyclodextrin, chitosan. Examples of suitable clays
include
smectites such as bentonite, kaolin, and laponite; magnesium trisilicate,
magnesium
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 42 -
aluminum silicate, and the like, and derivatives and mixtures thereof.
Examples of
suitable gelling starches include acid hydrolyzed starches, and derivatives
and
mixtures thereof. Additional suitable thickening hydrocolloids include low-
moisture
polymer solutions such as mixtures of gelatin and other hydrocolloids at water
contents up to about 30%, such as for example those used to make "gummi"
confection forms.
Additional suitable thickeners include crystallizable carbohydrates, and the
like, and derivatives and combinations thereof. Suitable crystallizable
carbohydrates
include the monosaccharides and the oligosaccharides. Of the monosaccharides,
the
Zo aldohexoses e.g., the D and L isomers of allose, altrose, glucose, mannose,
gulose,
idose, galactose, talose, and the ketohexoses e.g., the D and L isomers of
fructose
and sorbose along with their hydrogenated analogs: e.g., glucitol (sorbitol),
and
mannitol are preferred. Of the oligosaccharides, the 1,2-disaccharides sucrose
and
trehalose, the 1,4-disaccharides maltose, lactose, and cellobiose, and the 1,6-
is disaccharides gentiobiose and melibiose, as well as the trisaccharide
raffinose are
preferred along with the isomerized form of sucrose known as isomaltulose and
its
hydrogenated analog isomalt. Other hydrogenated forms of reducing
disaccharides
(such as maltose and lactose), for example, maltitol and lactitol are also
preferred.
Additionally, the hydrogenated forms of the aldopentoses: e.g., D and L
ribose,
2 o arabinose, xylose, and lyxose and the hydrogenated forms of the
aldotetroses: e.g.,
D and L erythrose and threose are preferred and are exemplified by xylitol and
erythritol, respectively.
In one embodiment of the invention, the flowable material comprises gelatin
as a gelling polymer. Gelatin is a natural, thermogelling polymer. It is a
tasteless
25 arid colorless mixture of derived proteins of the albuminous class which is
ordinarily
soluble in warm water. Two types of gelatin - Type A and Type B - are commonly
used. Type A gelatin is a derivative of acid-treated raw materials. Type B
gelatin is
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 43 -
a derivative of alkali-treated raw materials. The moisture content of gelatin,
as well
as its Bloom strength, composition and original gelatin processing conditions,
determine its transition temperature between liquid and solid. Bloom is a
standard
measure of the strength of a gelatin gel, and is roughly correlated with
molecular
weight. Bloom is defined as the weight in grams required to move a half inch
diameter plastic plunger 4 mm into a 6.67% gelatin gel that has been held at
10°C
for 17 hours. In a preferred embodiment, the flowable material is an aqueous
solution comprising 20% 275 Bloom pork skin gelatin, 20% 250 Bloom Bone
Gelatin, and approximately 60% water.
to Suitable xanthan gums include those available from C.P. Kelco Company
under the tradenames KELTROL 1000, XANTROL 180, or K9B310.
Suitable clays include smectites such as bentonite, kaolin, and laponite;
magnesium trisilicate, magnesium aluminum silicate, and the like, and
derivatives
and mixtures thereof.
15 "Acid-hydrolyzed starch," as used herein, is one type of modified starch
that
results from treating a starch suspension with dilute acid at a temperature
below the
gelatinization point of the starch. During the acid hydrolysis, the granular
form of
the starch is maintained in the starch suspension, and the hydrolysis reaction
is
ended by neutralization, filtration and drying once the desired degree of
hydrolysis is
2 o reached. As a result, the average molecular size of the starch polymers is
reduced.
Acid-hydrolyzed starches (also known as "thin boiling starches") tend to have
a
much lower hot viscosity than the same native starch as well as a strong
tendency to
gel when cooled.
"Gelling starches," as used herein, include those starches that, when
a s combined with water and heated to a temperature sufficient to form a
solution,
thereafter form a gel upon cooling to a temperature below the gelation point
of the
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 44 -
starch. Examples of gelling starches include, but are not limited to, acid
hydrolyzed
starches such as that available from Grain Processing Corporation under the
tradename PURE-SET B950; hydroxypropyl distarch phosphate such as that
available from Grain Processing Corporation under the tradename, PURE-GEL
B990, and mixtures thereof.
Suitable low-melting hydrophobic materials include fats, fatty acid esters,
phospholipids, and waxes. Examples of suitable fats include hydrogenated
vegetable oils such as for example cocoa butter, hydrogenated palm kernel oil,
hydrogenated cottonseed oil, hydrogenated sunflower oil, and hydrogenated
soybean
so oil; and free fatty acids and their salts. Examples of suitable fatty acid
esters include
sucrose fatty acid esters, mono, di, and triglycerides, glyceryl behenate,
glyceryl
palinitostearate, glyceryl monostearate, glyceryl tristearate, glyceryl
trilaurylate,
glyceryl myristate, GlycoWax-932, lauroyl macrogol-32 glycerides, and stearoyl
macrogol-32 glycerides. Examples of suitable phospholipids include
phosphotidyl
15 choline, phosphotidyl serene, phosphotidyl enositol, and phosphotidic acid.
Examples of suitable waxes include carnauba wax, spermaceti wax, beeswax,
candelilla wax, shellac wax, microcrystalline wax, and paraffin wax; fat-
containing
mixtures such as chocolate; and the like.
Suitable non-crystallizable carbohydrates include non-crystallizable sugars
2 o such as polydextrose, and starch hydrolysates, e.g. glucose syrup, corn
syrup, and
high fructose corn syrup; and non-crystallizable sugar-alcohols such as
maltitol
syrup.
Suitable solvents for optional use as components of the flowable material for
making the core or the shell by molding include water; polar organic solvents
such
a s as methanol, ethanol, isopropanol, acetone, and the like; and non-polar
organic
solvents such as methylene chloride, and the like; and mixtures thereof.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 45 -
The flowable material for making cores or the shell by molding may
optionally comprise adjuvants or excipients, which may comprise up to about
30%
by weight of the flowable material. Examples of suitable adjuvants or
excipients
include plasticizers, detackifiers, humectants, surfactants, anti-foaming
agents,
colorants, flavorants, sweeteners, opacifiers, and the like. Suitable
plasticizers for
making the core, the shell, or a portion thereof, by molding include, but not
be
limited to polyethylene glycol; propylene glycol; glycerin; sorbitol; triethyl
citrate;
tribuyl citrate; dibutyl sebecate; vegetable oils such as castor oil, rape
oil, olive oil,
and sesame oil; surfactants such as polysorbates, sodium lauryl sulfates, and
dioctyl-
to sodium sulfosuccinates; mono acetate of glycerol; diacetate of glycerol;
triacetate of
glycerol; natural gums; triacetin; acetyltributyl citrate; diethyloxalate;
diethylmalate; diethyl fumarate; diethylinalonate; dioctylphthalate;
dibutylsuccinate;
glyceroltributyrate; hydrogenated castor oil; fatty acids; substituted
triglycerides
and glycerides; and the like and/or mixtures thereof. In one embodiment, the
15 plasticizer is triethyl citrate. In certain embodiments, the shell is
substantially free
of plasticizers, i.e. contains less than about 1 %, say less than about 0.01 %
of
plasticizers.
In embodiments in which the shell is prepared using a solvent-free molding
process, the shell typically comprises at least about 30 percent, e.g. at
least about 45
a o percent by weight of a thermal-reversible carrier. The shell may
optionally further
comprise up to about 55 weight percent of a release-modifying excipient. The
shell
may optionally further comprise up to about 30 weight percent total of various
plasticizers, adjuvants and excipients. In certain embodiments in which the
shell is
prepared by solvent-free molding, and functions to delay the release of one or
more
a5 active ingredients from an underlying core, the release modifying excipient
is
preferably selected from swellable, erodible hydrophilic materials.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 46 -
In embodiments in which the shell is prepared using a solvent-based molding
process, the shell typically comprises at least about 10 weight percent, e.g.
at least
about 12 weight percent or at least about 15 weight percent or at least about
20
weight percent or at least about 25 weight percent of a film-former. Here, the
shell
may optionally further comprise up to about SS weight percent of a release-
modifying excipient. The shell may again also optionally further comprise up
to
about 30 weight percent total of various plasticizers, adjuvants, and
excipients.
In embodiments in which the shell is applied to the cores by molding, at least
a portion of the shell surrounds the cores such that the shell inner surface
resides
to substantially conformally upon the outer surfaces of the cores. As used
herein, the
term "substantially conformally" means that the inner surface of the shell has
peaks
and valleys or indentations and protrusions corresponding substantially
inversely to
the peaks and valleys of the outer surface of the core. In certain such
embodiments,
the indentations and protrusions typically have a length, width, height or
depth in
15 one dimension of greater than 10 microns, say greater than 20 microns, and
less than
about 30,000 microns, preferably less than about 2000 microns.
The total weight of the shell is preferably about 20 percent to about 400
percent of the total weight of the cores. In embodiments wherein the shell is
prepared by a solvent-free molding process, the total weight of the shell is
typically
ao from about 50 percent to about 400 percent, e.g. from about 75 percent to
about 400
percent, or about 100 percent to about 200 percent of the total weight of the
cores.
In embodiments wherein the shell is prepared by a solvent-based molding
process,
the total weight of the shell is typically from about 20 percent to about 100
percent
of the total weight of the cores.
~ s The thickness of the shell is important to the release properties of the
dosage
form. Advantageously, the dosage forms of the invention can be made with
precise
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 47 -
control over shell thickness, in particular using the thermal cycle or thermal
setting
injection molding methods and apparatus described above. Typical shell
thicknesses
that may be employed are about 50 to about 4000 microns. In certain preferred
embodiments, the shell has a thickness of less than 800 microns. In
embodiments
wherein the shell portion is prepared by a solvent-free molding process, the
shell
portion typically has a thickness of about 500 to about 4000 microns, e.g.
about 500
to about 2000 microns, say about 500 to about 800 microns, or about 800 to
about
1200 microns. In embodiments wherein the, shell portion is prepared by a
solvent-
based molding process, the shell portion typically has a thickness of less
than about
l0 800 microns, e.g. about 100 to about 600 microns, say about 150 to about
400
microns. In a particularly preferred embodiment the dosage form comprises
first
and second cores and first and second shell portions, and at least one of the
shell
portions has a thickness of less than about 800 microns, e.g. about 100 to
about 600
microns, e.g. about 150 to about 400 microns.
15 In embodiments in which the shell is prepared by molding, either by a
solvent-free process or by a solvent-based process, the shell typically is
substantially
free of pores in the diameter range of 0.5 to 5.0 microns, i.e. has a pore
volume in
the pore diameter range of 0.5 to S.0 microns of less than about 0.02 cc/g,
preferably
less than about 0.01 cc/g, more preferably less than about 0.005 cc/g. Typical
2 o compressed materials have pore volumes in this diameter range of more than
about
0.02 cc/g. Pore volume, pore diameter and density may be determined using a
Quantachrome Instruments PoreMaster 60 mercury intrusion porosimeter and
associated computer software program known as "Porowin." The procedure is
documented in the Quantachrome Instruments PoreMaster Operation Manual. The
z s PoreMaster determines both pore volume and pore diameter of a solid or
powder by
forced intrusion of a non-wetting liquid (mercury), which involves evacuation
of the
sample in a sample cell (penetrometer), filling the cell with mercury to
surround the
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 48 -
sample with mercury, applying pressure to the sample cell by: (i) compressed
air (up
to 50 psi maximum); and (ii) a hydraulic (oil) pressure generator (up to 60000
psi
maximum). Intruded volume is measured by a change in the capacitance as
mercury
moves from outside the sample into its pores under applied pressure. The
corresponding pore size diameter (d) at which the intrusion takes place is
calculated
directly from the so-called "Washburn Equation": d= -(4y(cos6)/P) where y is
the
surface tension of liquid mercury, 0 is the contact angle between mercury and
the
sample surface and P is the applied pressure.
Equipment used for pore volume measurements:
i o 1. Quantachrome Instruments PoreMaster 60.
2. Analytical Balance capable of weighing to O.OOOlg.
3. Desiccator.
Reagents used for measurements:
1. High purity nitrogen.
2. Triply distilled mercury.
3. High pressure fluid (Dila AX, available from Shell
Chemical Co.).
4. Liquid nitrogen (for Hg vapor cold trap).
5. Isopropanol or methanol for cleaning sample cells.
6. Liquid detergent for cell cleaning.
Procedure:
The samples remain in sealed packages or as received in the dessicator until
analyslS. The vacuum pump is switched on, the mercury vapor cold trap is
filled
with liquid nitrogen, the compressed gas supply is regulated at 55 psi., and
the
instrument is turned on and allowed a warm up time of at least 30 minutes. The
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 49 -
empty penetrometer cell is assembled as described in the instrument manual and
its
weight is recorded. The cell is installed in the low pressure station and
"evacuation
and fill only" is selected from the analysis menu, and the following settings
are
employed:
Fine Evacuation time: 1 min.
Fine Evacuation rate: 10
Coarse Evacuation time: 5 min.
The cell (filled with mercury) is then removed and weighed. The cell is then
to emptied into the mercury reservoir, and two tablets from each sample are
placed in
the cell and the cell is reassembled. The weight of the cell and sample are
then
recorded. The cell is then installed in the low-pressure station, the low-
pressure
option is selected from the menu, and the following parameters are set:
Mode: Low pressure
Z5 Fine evacuation rate: 10
Fine evacuation until: 200 Hg
Coarse evacuation time: 10 min.
Fill pressure: Contact +0.1
Maximum pressure: 50
a o Direction: Intrusion And Extrusion
Repeat: 0
Mercury contact angle: 140
Mercury surface tension: 4~0
25 Data acquisition is then begun. The pressure vs. cumulative volume-
intruded plot is displayed on the screen. After low-pressure analysis is
complete, the
cell is removed from the low-pressure station and reweighed. The space above
the
mercury is filled with hydraulic oil, and the cell is assembled and installed
in the
high-pressure cavity. The following settings are used:
3 o Mode: Fixed rate
Motor speed: 5
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 50 -
Start pressure: 20
End pressure: 60,000
Direction: Intrusion and extrusion
Repeat: 0
Oil fill length: 5
Mercury contact angle: 140
Mercury surface tension: 480
Data acquisition is then begun and graphic plot pressure vs, intruded volume
is
s o displayed on the screen. After the high pressure run is complete, the low-
and high-
pressure data files of the same sample are merged.
Tn those embodiments in which solvent-free molding is employed, the
flowable material may comprise a thermal-reversible carrier. Suitable thermal-
reversible earners for use in making a core, the shell or both by molding are
i5 thermoplastic materials typically having a melting point below about
110°C, more
preferably between about 20 and about I00°C. Examples of suitable
thermal-
reversible earners for solvent-free molding include thermplastic polyalkalene
glycols, thermoplastic polyalkalene oxides, low melting hydrophobic materials,
thermoplastic polymers, thermoplastic starches, and the like. Preferred
thermal-
z o reversible carriers include polyethylene glycol and polyethylene oxide.
Suitable
thermoplastic polyalkylene glycols for use as thermal-reversible carriers
include
polyethylene glycol having molecular weight from about 100 to about 20,000,
e.g.
from about 100 to about 8,000 Daltons. Suitable thermoplastic polyalkalene
oxides
include polyethylene oxide having a molecular weight from about 100,000 to
about
25 900,000 Daltons. Suitable low-melting hydrophobic materials for use as
thermal-
reversible earners include fats, fatty acid esters, phospholipids, and waxes
which are
solid at room temperature, fat-containing mixtures such as chocolate; and the
like.
Examples of suitable fats include hydrogenated vegetable oils such as for
example
cocoa butter, hydrogenated palm kernel oil, hydrogenated cottonseed oil,
3 o hydrogenated sunflower oil, and hydrogenated soybean oil; and free fatty
acids and
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 51 -
their salts. Examples of suitable fatty acid esters include sucrose fatty acid
esters,
1
mono, di, and triglycerides, glyceryl behenate, glyceryl palinitostearate,
glyceryl
monostearate, glyceryl tristearate; glyceryl trilaurylate, glyceryl myristate,
GlycoWax-932, lauroyl macrogol-32 glycerides, and stearoyl macrogol-32
glycerides. Examples of suitable phospholipids include phosphotidyl choline,
phosphotidyl serene, phosphotidyl enositol, and phosphotidic acid. Examples of
suitable waxes which are solid at room temperature include carnauba wax,
spermaceti wax, beeswax, candelilla wax, shellac wax, microcrystalline wax,
and
paraffin wax. Suitable thermoplastic polymers for use as thermal-reversible
Garners
to include thermoplastic water swellable cellulose derivatives, thermoplastic
water
insoluble polymers, thermoplastic vinyl polymers, thermoplastic starches, and
thermoplastic resins, and combinations thereof. Suitable thermoplastic water
swellable cellulose derivatives include include hydroxypropylmethyl cellulose
(HPMC), methyl cellulose (MC), carboxymethylcellulose (CMC), cross-linked
15 hydroxypropylcellulose, hydroxypropyl cellulose (HPC),
hydroxybutylcellulose
(HBC), hydroxyethylcellulose (HEC), hydroxypropylethylcellulose,
hydroxypropylbutylcellulose, hydroxypropylethylcellulose, and salts,
derivatives,
copolymers, and combinations thereof. Suitable thermoplastic water insoluble
polymers include ethylcellulose, polyvinyl alcohols, polyvinyl acetate,
2 o polycaprolactones, cellulose acetate and its derivatives, acrylates,
methacrylates,
acrylic acid copolymers, and the like and derivatives, copolymers, and
combinations
thereof. Suitable thermoplastic vinyl polymers include polyvinylacetate,
polyvinyl
alcohol, and polyvinyl pyrrolidone (PVP). Examples of suitable thermoplastic
starches for use as thermal-reversible carriers are disclosed for example in
U.S.
25 Patent No. 5,427,614. Examples of suitable thermoplastic resins for use as
themal-
reversible carriers include dammars, mastic, rosin, shellac, sandarac, and
glcerol
ester of rosin. In one embodiment, the thermal-reversible carrier for making a
core
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 52 -
by molding is selected from polyalkylene glycols, polyalkaline oxides, and
combinations thereof.
In embodiments in which the shell comprises an active ingredient intended to
have immediate release from the dosage form, the shell is preferably prepared
via
solvent-free molding. In such embodiments a thermal-reversible carrier is
employed
in the flowable material to make the shell, said thermal-reversible carrier
preferably
selected from polyethylene glycol with weight average molecular weight from
about
1450 to about 20000, polyethylene oxide with weight average molecular weight
from about 100,000 to about 900,000, and the like.
to In certain embodiments of the invention, the shell, or a shell portion may
function as a diffusional membrane which contains pores through which fluids
can
enter the dosage form, contact and dissolve active ingredient in the core,
which can
then be released in a sustained, extended, prolonged or retarded manner. In
these
embodiments, the rate of release of active ingredient from an underlying core
i5 portion will depend upon the total pore area in the shell portion, the
pathlength of
the pores, and the solubility and diffusivity of the active ingredient (in
addition to its
rate of release from the core portion itself). In preferred embodiments in
which a
shell portion functions as a diffusional membrane, the release of the active
ingredient from the dosage form may be described as controlled, prolonged,
a o sustained or extended. In these embodiments, the contribution to active
ingredient
dissolution from the shell may follow zero-order, first-order, or square-root
of time
kinetics. In certain such embodiments, the shell portion preferably comprises
a
release modifying moldable excipient comprising a combination of a pore former
and an insoluble edible material, for example a film forming water insoluble
25 polymer. Alternately, in embodiments in which the shell portion is prepared
by
solvent-free molding, described below, the shell portion may comprise a
thermal-
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 53 -
reversible carrier that functions by dissolving and forming pores or channels
through
which the active ingredient may be liberated.
In certain other embodiments, the shell or a shell portion functions as an
eroding matrix from which active ingredient dispersed in the shell is
liberated by the
dissolution of successive layers of the shell surface. In these embodiments,
the rate
of active ingredient release will depend on the dissolution rate of the matrix
material
in the shell or shell portion. Particularly useful matrix materials for
providing
surface erosion include those that first absorb liquid, then swell andlor gel
prior to
dissolving. In certain such embodiments, the hell or shell portion preferably
Zo comprises a release modifying moldable excipient comprising a swellable
erodible
hydrophilic material.
In certain other embodiments, the shell or a portion thereof functions as a
barrier to prevent release therethrough of an active ingredient contained in
an
underlying core. In such embodiments, active ingredient is typically released
from a
s5 portion of the core that is not covered by that portion of the shell, for
example from
a portion of the core in communication with one or more openings in the shell.
Such
embodiments advantageously allow for control of the surface area for release
of the
active ingredient. In certain embodiments for example, the surface area for
release
of active ingredient can be maintained substantially constant over time. In a
a o particularly preferred embodiment, the release of at least one active
ingredient
follows substantially zero-order kinetics. In such embodiments, the shell
preferably
comprises a modified release composition comprising a water insoluble
material, for
example a water insoluble polymer.
In other embodiments, the shell, or a shell portion functions as a delayed
~ 5 release coating to delay release of one or more active ingredients
contained in an
underlying core. In these embodiments, the lag-time for onset of active
ingredient
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 54 -
release may be governed by erosion of the shell, diffusion of active
ingredient
through the shell, or a combination thereof. Tn certain such embodiments, the
shell
preferably comprises a release modifying moldable excipient comprising a
swellable
erodible hydrophilic material.
The following non-limiting example further illustrates the claimed invention.
Example
A dosage form according to the invention providing a double pulse release of
ibuprofen is manufactured by a solvent-free molding process as follows. The
double
to pulse consists of a 200 mg immediate release (IR) ibuprofen followed by a
100 mg
burst release of ibuprofen after a predetermined lag time.
Part A. Preparation of the 100 mg Immediate-Release (IR) Ibuprofen Core
Formulation:
In edient Trade Name Manufacturer M Tablet
Ibuprofen (115 Albemarle Corp. 100.0
microns) Oran ebur , SC
Microcrystalline Avicel pH 101~ FMC Corp. Newark,106.5
cellulose DE
19711
Sodium starch glycolateExplotab Penwest Pharmaceuticals6.0
Co. Patterson,
NJ
Colloidal silicon Cab-O-Sil LM-5~Gabot Corp. Tuscola,0.5
dioxide IL
Total 213.0
Manufacturing process:
Ibuprofen, microcrystalline cellulose and sodium starch glycolate are
delumped through a 30 mesh screen and said ingredients are mixed in a 2 qt. P-
K
blender for 5 minutes. Colloidal silicon dioxide is also delumped through a 30
mesh
a o screen and is added to the aforementioned mixture for blending for another
5
minutes.
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 55 -
A Beta tablet press (Manesty, Liverpool, UK) equipped with round punch
and die unit with a diameter of 0.250" is used to make the first core as a
tablet. The
final blend (from Step 1) is fed into the die and is compressed into a tablet
core
under 2000 lb/in2 of operating pressure. The weight of compressed tablet is
213.0
mg, which contains 100.0 mg of ibuprofen.
Part B. Preparation of the 200 m~ Immediate-Release (IR) Ibuprofen Core
Formulation:
In edients Trade Name Manufacturer M /Tablet
Ibuprofen granules Albemarle Corp. 200.0
(115 Oran ebur , SC
microns
Sodium starch Explotab Penwest Pharmaceuticals12.0
glycolate Co.
Patterson, NJ
Colloidal siliconCab-O-Sil Cabot Corp. Tuscola,1.0
dioxide LM-5 IL
Total 213.0
io Manufacturing process:
Ibuprofen and sodium starch glycolate are delumped through a 30 mesh
screen and said ingredients are mixed in a 2 qt. P-I~ blender for 5 minutes.
Colloidal
silicon dioxide is also delumped through a 30 mesh screen and is added to the
aforementioned mixture for blending for another 5 minutes. Prescreened
(through a
30 mesh screen) ibuprofen and sodium starch glycolate are mixed in a 2 qt. P-
I~
blender for 5 minutes.
The final blend (from Step 1) is fed into the die of the tablet press
described
in Part A and is compressed into a tablet core under 2000 lb/in2 of operating
z o pressure. The weight of compressed tablet is 213.0 mg, which contains
200.0 mg of
ibuprofen.
Part C. Application of the Shell by Solvent-Free Molding
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 56 -
Shell Formulation:
In edient Trade Name Manufacturer M /Tablet
Polyethylene GlycolCarbowax~ Union Carbide Corporation,149.1
8000
Danb , CT 06817-0001
Polyethylene OxidePolyox WSR N-80Union Carbide Corporation,42.6
(MW
200,000 Danb , CT 06817-0001
Hydroxypropyl Methocel K15M The Dow Chemical 63.9
Perm Co.,
Meth lcellulose CR Midland, Michi
an, 48674
Triethyl Citrate Morflex, Inc., 85.2
Greensboro,
North Carolina
27403
Lauroyl Macrogol-32Gelucire SO/13 Gattefosse Corp., 85.2
Gl cerides Westwood, NJ 07675
Manufacturing Process:
A beaker is submersed in a water bath (Ret digi-visc; Antal-Direct, Wayne,
PA 19087) where the water bath temperature is set at 85°C. Polyethylene
glycol
(PEG) 8000 and Gelucire SO113 are added to the beaker and are mixed with a
spatula
until all PEG and Gelucire are melted. Hydroxypropyl methylcellulose is added
to
the molten mixture and is mixed for 10 minutes. Triethyl Citrate is added to
the
molten mixture and is mixed for 2 minutes. Polyethylene Oxide 200,000 is added
to and is mixed for 20 minutes. The shell material is provided in flowable
form.
A laboratory scale thermal cycle molding unit having an overall caplet shape
of dimensions of 0.700" x 0.350" x 0.06", is used to apply first and second
shell
portions to the cores. The molding unit comprises a single mold assembly made
from an upper mold assembly portion comprising an upper mold cavity, and a
lower
mold assembly portion comprising a lower mold cavity. The lower mold assembly
portion is first cycled to a hot stage at 85°C for 30 seconds. The
shell material of
Part C is introduced into the lower mold cavity. Two separate cores prepared
as
described in aforementioned Parts A and B are then inserted into two stations
within
z o the cavity. The stations separate the two cores within the lower mold
cavity by 1
mm. A blank upper mold assembly portion is mated with the lower mold assembly
CA 02500312 2005-03-24
WO 2004/028508 PCT/US2003/008894
- 57 -
portion. The mold assembly is then cycled to a cold stage at 5°C for 60
seconds to
harden the first shell portion. The blank mold assembly portion is removed
from the
lower mold assembly portion. The upper mold assembly portion is cycled to a
hot
stage at 85°C for 30 seconds. The shell material is added to the upper
mold cavity.
\The upper mold cavity comprises a small rod (0.1 mm in diameter x 1 mm in
length) attached to its inner surface that contacts one station for one of the
cores.
The lower mold assembly portion, which has been maintained at 5°C, is
mated with
the upper mold assembly portion in such a way that the first core of Part B
(200 mg
Zo of ibuprofen tablet) is mated with the first core station of the upper mold
assembly.
The upper mold assembly portion is then cycled to a cold stage at 5°C
for 120
seconds to harden the second shell portion. The lower mold assembly portion is
then removed and the finished dosage form, a molded caplet coated with two
halves
of the same shell material, is ejected from the upper mold cavity. The weight
gain
from the shell material (i.e. the difference in weight between the finished
dosage
form and the core) is recorded.