Note: Descriptions are shown in the official language in which they were submitted.
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR
PROTEIN INHIBITORS AND USES THEREOF
Background Of The Invention
[0001] The cystic fibrosis transmembrane conductance regulator protein (CFTR)
is a cAMP-
activated chloride (Cl) channel expressed in epithelial cells in mammalian
airways,
intestine, pancreas and testis. CFTR is the chloride-channel responsible for
cAMP-mediated
Cl- secretion. Hormones, such as a (3-adrenergic agonist, or a toxin, such as
cholera toxin,
leads to an increase in cAMP, activation of cAMP-dependent protein kinase, and
phosphorylation of the CFTR Cl- channel, which causes the channel to open. An
increase in
cell Ca2+ can also activate different apical membrane channels.
Phosphorylation by protein
kinase C can either open or shut Cl- channels in the apical membrane. CFTR is
predominantly located in epithelia where it provides a pathway for the
movement of Cl- ions
across the apical membrane and a key point at which to regulate the rate of
transepithelial
salt and water transport. CFTR chloride channel function is associated with a
wide spectrum
of disease, including cystic fibrosis (CF) and with some forms of male
infertility, polycystic
kidney disease and secretory diarrhea.
[0002] The hereditary lethal disease cystic fibrosis (CF) is caused by
mutations in CFTR.
Observations in human cystic fibrosis (CF) patients and CF mouse models
indicate the
functional importance of CFTR in intestinal and pancreatic fluid transport, as
well as in male
fertility (Grubb et al., 1999, Physiol. Rev. 79:S193-S214; Wong, P.Y., 1997,
Mol. Hum.
Reprod. 4:107-110). However, the mechanisms remain unclear by which defective
CFTR
produces airway disease, which is the principal cause of morbidity and
mortality in CF
(Pilewski et al., 1999, Physiol. Rev. 79:S215-S255). Major difficulties in
understanding
airway disease in CF include the inadequacy of CF mouse models, which manifest
little or
no airway disease, the lack of large animal models of CF, and the limited
availability of
human CF airways that have not been damaged by chronic infection and
inflammation.
High-affinity, CFTR-selective inhibitors have not been available to study
airway disease
mechanisms in CF or to create the CF phenotype in large animal models.
[0003] High-affinity CFTR inhibitors also have clinical applications in the
therapy of
secretory diarrheas and cystic kidney disease, and in inhibiting male
fertility. The
compounds diphenylamine-2-carboxylate (DPC) and 5-nitro-2-(3-phenylpropyl-
amino)benzoate (NPPB) inhibit CFTR at high concentrations but are non-specific
in their
inhibitory action (Cabantchik et al., 1992, Am. J. Physiol. 262:C803-C827;
McDonough et
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
al., 1994, Neuron 13:623-634; Schultz et al., 1999, Physiol. Rev. 79:S109-
S144). The best
CFTR inhibitor available for electrophysiological and other cell-based
studies,
glibenclamide, is used at concentrations of >100 M (Sheppard et al., 1992, J.
Gen. Physiol.
100:573-591; Hongre et al, 1994, Pflugers Arch. 426:284-287). However, at this
concentration glibenclamide also inhibits other Cl- transporters as well as K+
channels
(Edwards et al., 1993, Br. J. Pharmacol. 110:1280-1281; Rabe et al., 1995,
Pflugers Arch.
429:659-662; Yamazaki et al., 1997, Circ. Res. 81:101-109). Effective small
molecule
inhibitors of other ion transport proteins are known, but no small molecules
with specific
CFTR inhibitory ability suitable for therapy of secretory diseases have been
available.
[0004] There is accordingly a need for CTFR inhibitor compounds and methods of
using
such compounds for development of animal models useful in the study and
treatment of CF
and the treatment and control of secretory disorders. The present invention
addresses these
needs, as well as others, and overcomes deficiencies found in the background
art.
Summary Of The Invention
[0005] The invention provides compositions, pharmaceutical preparations and
methods for
inhibition of cystic fibrosis transmembrane conductance regulator protein
(CFTR) that are
useful for the study and treatment of CFTR-mediated diseases and conditions.
The
compositions and pharmaceutical preparations of the invention may comprise one
or more
thiazolidinone compounds or derivatives, and may additionally comprise one or
more
pharmaceutically acceptable carriers, excipients and/or adjuvants. The methods
of the
invention comprise, in certain embodiments, administering to a patient
suffering from a
CFTR-mediated disease or condition, an efficacious amount of a thiazolidinone
compound
or derivative. In other embodiments the invention provides methods of
inhibiting CFTR that
comprise contacting cells in a subject with an effective amount of a
thiazolidinone
compound or derivative. In addition, the invention features a non-human animal
model of
CFTR-mediated disease which model is produced by administration of a
thiazolidinone
compound or derivative to a non-human animal in an amount sufficient to
inhibit CFTR.
[0006] These and other objects and advantages of the invention will be
apparent from the
detailed description below.
Brief Description Of The Drawings
[0007] The invention will be more fully understood by reference to the
following drawings,
which are for illustrative purposes only.
2
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00081 FIG. 1A is a schematic representation of a screening technique used for
detection of
CFTR inhibitors. CFTR was maximally stimulated by multiple agonists in stably
transfected
epithelial cells co-expressing human CFTR and a yellow fluorescent protein
(YFP) having
Cl+/r sensitive fluorescence. After addition of a test compound, F influx was
induced by
adding an r containing solution.
[0009] FIG. 1B is a graphical illustration of representative fluorescence data
from individual
wells using the screening technique of FIG. 1A, showing controls (no
activator, no test
compound), inactive compounds and active CFTR inhibitor compounds.
[0010] FIG. 1C shows chemical structures of CFTR inhibitors identified by the
screening
technique of FIG. 1A.
[0011] FIG. 1D shows chemical structures of Ring 2 of the thiazolidinone
derivatives
having the greatest CFTR inhibitory activity. The complete thiazolidinone
derivative
structure is shown in FIG. 1 C. Relative potencies were: 0.2 (CFTR;nh-020),
0.3 (CFTR;nh-
029), 1.0 (CFTR1nh-172), 0.2 (CFTR1nh-185), 0.1 (CFTRinh-214) and 0.1 (CFTR1nh-
236).
[00121 FIG. 2A is a graphical representation of relative fluorescence versus
time using the
screening technique of FIG. 1A for the CFTR inhibitor 3-[(3-
trifluoromethyl)phenyl]-5-[(4-
carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (referred to herein as
CFTRi,h- 172) at
several concentrations.
[00131 FIG. 2B is a graphical representation of the time course of inhibition
showing
CFTR-mediated I" transport rates at different times after addition of 2 M
CFTRinh- 172. The
inset is a graphical representation of the time course of inhibition reversal
showing r
transport rates at different times after washout of 1 gM CFTR;nh-172. Mean
SE from three
sets of experiments.
[0014] FIG. 2C is a graphical representation of inhibition of CFTR after
stimulation by
different agonists, including benzoflavone and benzimidazolone UCCF compounds
(UCCF-
029 (2-(4-pyridinium)benzo[h]-4H-chromen-4-one bisulfate) and UCCF-853
(Galietta et al.
2001 J. Biol. Chem. 276:19723-19728)), genistein, CPT-cAMP, 8-methoxypsoralen
(8-
MPO), 8-cyclopentyl-1,3-dipropylxanthine (CPX) (all 50 M) ( SE from three
sets of
experiments). Filled bars show agonist, and open bars show agonist with 5 M
CFTR;h-172.
[0015] FIG. 3A is a graphical representation of CFTR;nh-172 inhibition of
short-circuit
current in permeabilized FRT cells expressing human CFTR. CFTR was stimulated
by 100
gM CPT-cAMP.
[00161 FIG. 3B graphically provides a summary of dose-inhibition data for
CFTR;nh-172
(circles) and glibenclamide (squares) (SE, three sets of experiments).
3
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[0017] FIG. 3C graphically illustrates CFTR;,,h-172 inhibition of short-
circuit current in
primary culture of (non-permeabilized) human bronchial epithelial cells.
Inhibitor was added
in apical bathing solution (left panel) or basolateral and then apical
solutions (right panel).
[0018] FIG. 3D is a graphical representation of whole-cell patch clamp of CFTR-
expressing
FRT cells showing membrane currents elicited at +80 mV (open circles) and -100
mV
(closed circles). CFTR was stimulated by 5 gM forskolin followed by addition
of 2 gM
CFTR;,,h-172.
[0019] FIG. 3E is a graphic illustration showing that alternate stimulation
was interrupted
(a-c) to apply graded membrane potentials.
[0020] FIG. 3F is a graphical representation of current-voltage relationships
under basal
conditions (control, open circles), after forskolin stimulation (filled
circles), and following
addition of 0.2 M CFTRh,h-172 giving -50% inhibition (open triangles).
[0021] FIG. 4A is a graphical representation of UTP- (100 M) stimulated Cat+-
dependent
Cl" secretion measured in short-circuit current measurements on airway
epithelial cells in the
absence and presence of 5 M of CFTR,,,h-172.
[0022] FIG. 4B is a graphical representation of volume-activated Cl- current
(hypotonic 250
mosM/kg H2O) measured in whole-cell patch clamp experiments on FRT cells.
Currents
were recorded in the absence and presence of 5 gM CFTR;,,h-172.
[0023] FIG. 4C is a graphical representation of 3H-vincristine accumulation in
9HTEo-/Dx
cells with upregulated MDR- 1 expression. Intracellular vincristine was
measured with and
without verapamil (100 M) or CFTR;,,h-172 (5 M) (SE, n=3).
[0024] FIG. 4D is a graphical illustration showing a representative membrane
potential
recording from a pancreatic (3 cell (INS-1) perfused extracellularly with
CFTR;,,h-172,
diazoxide (100 M), and glibenclamide (10 M).
[0025] FIG. 4E is a graphical representation of averaged changes in membrane
potential
(AmV) caused by maneuvers indicated in Fig. 4D (SE, n=4).
[0026] FIG. 5A is a photograph of isolated mouse ileal loops at six hours
after lumenal
injection of 1 gg cholera toxin without (top) and with (middle)
intraperitoneal injection of
CFTR;,,h-172 (150 g/kg). A saline control (no cholera toxin, bottom) is shown
for
comparison.
[0027] FIG. 5B graphically illustrates ileal loop weight at six hours, with a
mean SE (n=6-
8 mice) with 14-16 loops studied. For the inactive analog, the 4-carboxyphenyl
group in
CFTR;,,h-172 was replaced by 3-methoxy-4-methoxyvinylphenyl (SE, 6-8 mice per
group,
p < 0.001, ANOVA).
4
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[0028] FIG. 5C graphically illustrates the ratio of weight of entire small
intestine at six
hours after oral gavage before vs. after luminal fluid removal (SE, 4 mice per
group, p <
0.001).
[0029] FIG. 5D is a graphical illustration showing a representative CFTRinh-
172 inhibition
short-circuit current after amiloride addition and stimulation by forskolin
(20 M) in isolated
rat colonic mucosa. CFTRih-172 added to serosal and then mucosal surfaces as
indicated
(n=4).
[0030] FIG. 6 is a schematic showing synthesis of 14C-labeled CFTRinh-172. 14C
was
incorporated into the thiazolidinone core using 14C-labeled Br-acetic acid as
starting
material.
[0031] ' FIG. 7 is a set of graphs showing the results of pharmacokinetic
analysis of CFTRinh-
172 in rats following a single intravenous bolus infusion of 50 Ci 14C-
labeled CFTRinh-172.
Data shown as mean SE (n=3-6 rats) for serum radioactivities. Fitted curve
corresponds to
a 2-compartment model with redistribution halftime 0.14 hr, elimination half-
time 10.3 hr,
maximum serum concentration 3.2 g/mL, area-undercurve 3.8 g.hr/mL, volume of
distribution 1.2 L, and clearance 99 mL/hr.
[0032] FIG. 8 is a set of graphs showing organ distribution of 14C-labeled
CFTRinh-172 after
bolus infusion. The results in panel A are from mice given a single
intravenous bolus
infusion of 2 Ci 14C-labeled CFTRinh-172, sacrificed at indicated times, and
organs
harvested for measurement of 14C-radioactivity, with data presented as total
organ 14C-
radioactivity at indicated times (except for skeletal muscle where reported as
per gram
tissue) after infusion (mean SE, 4 mice per time point). The results in
panel B are from rats
given a bolus infusion of 50 Ci 14C-labeled CFTRinh-172 and total organ
CFTRinh-172
measured at 60 min after infusion (3 rats).
[0033] FIG. 9 is a set of photographs showing the results of analysis of
CFTRinh-172
metabolism by thin layer chromatography of fluids and liver homogenate from
mice infused
with 14C-labeled CFTRinh- 172 as in Fig. 8, panel A. 14C-CFTRinh-172 standards
were 1, 3
and 6 nCi (left panel), and 10, 30 and 60 nCi (right panel). Film was exposed
for
autoradiography for 48 hr (left panel) and 12 hr (right panel).
[0034] FIG. 10 is a set of graphs providing the results of characterization of
the mouse
closed-intestinal loop model. Panel A: Intestinal loops were injected with 200
L buffer and
loop weight measured at indicated times (mean SEM, 4 mice per time point).
Inset (lower)
% absorption at 30 min with and without CFTRinh-172 (20 gg I.P., n=4). Inset
(top)
Chemical structure of CFTRinh-172. Panel B: Time course of cholera toxin-
induced fluid
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
secretion in mouse closed-loop model. Dashed line shows control (saline-
injected) loops.
Data for injected loops (1 gg cholera toxin/loop) as mean SEM (4-6 mice).
[0035] FIG. 11 is a set of graphs showing CFTR1nh-172 inhibition of intestinal
fluid
secretion after cholera toxin in mice. Panel A: Dose-response for inhibition
of fluid
accumulation in mouse loop model. Mice were given single doses of CFTR1nh-172
by
intraperitoneal injection and loop weight (mean SEM, 4-6 mice per dose)
measured at 6 hr.
Dashed line indicates average weight in saline-injected control loops of same
mice. Panel B:
Persistence of CFTR1nh-172 inhibition. Mice were injected with 20 gg CFTR1h-
172 (I.P.) at
indicated times before or after cholera toxin administration (4-6 mice per
time point). Panel
C: Time course of plasma 14C-CFTR1õ h-172 radioactivity after i.v injection
(tail vein, left
ordinate) and oral administration (CFTR1h-172 in TPGS, right ordinate). Data
shown as
counts per min per tiCi injected (4 mice). Panel D: 14C-CFTR1nh-172
accumulation in
gastrointestinal organs at 6 hr after i.v. and oral 14C-CFTR1nh-172
administration (4 mice).
Panel E:, Inhibition of cholera toxin-induced fluid secretion by orally-
administered CFTR1nh-
172 (200 g in TPGS) in mouse open-loop model. Data shown as ratio of weight
of entire
small intestine 6 hr after oral gavage before vs. after luminal fluid removal
(mean SEM, 4
mice per group, * p < 0.01). Panel F: CFTR1nh-172 permeability across Caco-2
monolayers
(mean SEM, 18 inserts) with Papp = 16 x 10"6 cm/s.
[0036] FIG. 12 is a set of graphs showing CFTR1nh-172 inhibition of cholera
toxin (Panel A)
and STa toxin (Panel B) induced fluid secretion in rat closed-loop model. Data
shown as
mean SEM (4 rats per group), * p < 0.01.
[0037] FIG. 13 is a set of graphs showing CFTR1rih-172 inhibition of forskolin-
and STa
toxin-stimulated short-circuit current in mouse ileum (Panel A) and human
colon (Panel B).
STa toxin shown as inset. Data are representative of studies of 5 mice and 2
sets of human
tissues. CFTR1nh-172 added to both sides of tissue. Amiloride (10 M) was
present in the
apical solutions.
[0038] FIG. 14 is a set of graphs showing short-circuit analysis of CFTR1h-172
inhibition of
Cl- secretion in T84 colonic epithelial cells. Panel A: Data shown as
representative traces
from experiments on 5-12 inserts per condition. CFTR1h-172 added to both sides
of cell
layers. CFTR agonists include forskolin (left), 8-Br-cGMP (middle), and
CFTRact-16 (right).
Panel B: (left) CFTR1nh-172 inhibition of forskolin-stimulated short-circuit
current after
basolateral permeabilization with amphotericin B (250 gg/mL). Representative
of
experiments on 6 inserts. (middle) Average dose-response for CFTR1nh-172
inhibition of
forskolin-stimulated (circles) and 8-Br-cGMP-stimulated (triangles) short-
circuit current in
6
CA 02500498 2011-01-27
WO 2004/028480 PCT/US2003/031005
permeabilized vs. non-permeabilized T84 cells (mean SEM, 6-12 inserts).
(right) CFTR;,,h-
172 inhibition of forskolin-stimulated short-circuit current in the presence
of high K" (68
mM) in the basolateral solution with low Cl" in-the apical solution.
Representative of 4
experiments.
[0039] Before the present invention is described, it is to be understood that
this invention is
not limited to particular embodiments described, as such may, of course, vary.
It is also to be
understood that the terminology used herein is for the purpose of describing
particular
embodiments only, and is not intended to be limiting, since the scope of the
present
invention will be limited only by the appended claims.
[0040] Unless defined otherwise, all technical and scientific terms used
herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Although any methods and materials similar or equivalent to
those
described herein can be used in the practice or testing of the present
invention, the
preferred methods and materials are now described. All publications mentioned
herein
disclose and describe the methods and/or materials in connection with which
the
publications are cited.
[0041] It should be noted that, as used herein and in the appended claims, the
singular forms
"a", "an", and "the" include plural referents unless the context clearly
dictates otherwise.
Thus, for example, reference to "an inhibitor" includes a plurality of such
inhibitors, and
reference to "the cell" includes reference to one or more cells and
equivalents thereof known
to those skilled in the art, and so forth.
[0042] The publications discussed herein are provided solely for their
disclosure prior to
the filing date of the present application. Nothing herein is to be construed
as an admission
that the present invention is not entitled to antedate such publication by
virtue of prior
invention. Further, the dates of publication provided may be different from
the actual
publication dates that may need to be independently confirmed.
[0043] The definitions used herein are provided for reason of clarity, and
should not be
considered as limiting. The technical and scientific terms used herein are
intended to have
the same meaning as commonly understood by those of ordinary skill in the art
to which the
invention pertains.
7
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Detailed Description Of The Invention
[0044] The invention is based on the discovery of thiazolidinone compounds and
derivatives
that are high-affinity CFTR inhibitors. The structure of the compounds and
derivatives of the
invention, as well as pharmaceutical formulations and methods of use are
described in more
detail below.
DEFINITIONS
[0045] A "cystic fibrosis transmembrane conductance regulator protein-mediated
condition
or symptom" or "CFTR-mediated condition or symptom" means any condition,
disorder or
disease, or symptom of such condition, disorder, or disease, that results from
activity of
cystic fibrosis transmembrane conductance regulator protein (CFTR), e.g.,
activity of CFTR
in ion transport. Such conditions, disorders, diseases, or symptoms thereof
are treatable by
inhibition of CFTR activity, e.g., inhibition of CFTR ion transport. CFTR
activity has been
implicated in, for example, intestinal secretion in response to various
agonists, including
cholera toxin (see, e.g., Snyder et al. 1982 Bull. World Health Organ. 60:605-
613; Chao et
al. 1994 EMBO J. 13:1065-1072; Kimberg et al. 1971 J. Clin. Invest.50:1218-
1230).
[0046] A "CFTR inhibitor" as used herein is a compound that reduces the
efficiency of ion
transport by CFTR, particularly with respect to transport of chloride ions by
CFTR.
Preferably CFTR inhibitors of the invention are specific CFTR inhibitors,
i.e., compounds
that inhibit CFTR activity without significantly or adversely affecting
activity of other ion
transporters, e.g., other chloride transporters, potassium transporters, and
the like. Preferably
the CFTR inhibitors are high-affinity CFTR inhibitors, e.g., have an affinity
for CFTR of at
least about one micromolar, usually about one to five micromolar.
[0047] "Treating" or "treatment" as used herein covers the treatment of a
disease, condition,
disorder or symptom in a subject, wherein the disease, condition, disorder or
symptom is
mediated by the activity of CFTR, and includes: (1) preventing the disease,
condition, or
disorder, i.e. causing the clinical symptoms of the disease not to develop in
a subject that
may be exposed to or predisposed to the disease, condition, or disorder, but
does not yet
experience or display symptoms thereof, (2) inhibiting the disease, condition
or disorder, i.e.,
arresting or reducing the development of the disease, condition or disorder,
or its clinical
symptoms, or (3) relieving the disease, condition or disorder, i.e., causing
regression of the
disease, condition or disorder, or its clinical symptoms.
[0048] A "therapeutically effective amount" or "efficacious amount" means the
amount of a
compound of the invention that, when administered to a mammal or other subject
in need
8
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
thereof, is sufficient to effect treatment, as defined above, for diseases,
conditions, disorders
or symptoms mediated by the activity of CFTR. The amount of a compound of the
invention
that constitutes a "therapeutically effective amount" will vary depending on
the compound,
the disease and its severity and the age, weight, etc., of the subject to be
treated, but can be
determined routinely by one of ordinary skill in the art having regard to his
own knowledge
and to this disclosure.
[0049] The terms "subject" and "patient" mean a member or members of any
mammalian or
non-mammalian species that may have a need for the pharmaceutical methods,
compositions
and treatments described herein. Subjects and patients thus include, without
limitation,
primate (including humans), canine, feline, ungulate (e.g., equine, bovine,
swine (e.g., pig)),
avian, and other subjects. Humans and non-human animals having commercial
importance
(e.g., livestock and domesticated animals) are of particular interest.
[0050] "Mammal" means a member or members of any mammalian species, and
includes,
by way of example, canines; felines; equines; bovines; ovines; rodentia, etc.
and primates,
particularly humans. Non-human animal models, particularly mammals, e.g.
primate,
murine, lagomorpha, etc. may be used for experimental investigations.
[0051] The term "unit dosage form," as used herein, refers to physically
discrete units
suitable as unitary dosages for human and animal subjects, each unit
containing a
predetermined quantity of compounds of the present invention calculated in an
amount
sufficient to produce the desired effect in association with a
pharmaceutically acceptable
diluent, carrier or vehicle. The specifications for the novel unit dosage
forms of the present
invention depend on the particular compound employed and the effect to be
achieved, and
the pharmacodynamics associated with each compound in the host.
[0052] The term "physiological conditions" is meant to encompass those
conditions
compatible with living cells, e.g., predominantly aqueous conditions of a
temperature, pH,
salinity, etc. that are compatible with living cells.
[0053] A "pharmaceutically acceptable excipient" means an excipient that is
useful in
preparing a pharmaceutical composition that is generally safe, nontoxic and
neither
biologically nor otherwise undesirable, and includes an excipient that is
acceptable for
veterinary use as well as human pharmaceutical use. "A pharmaceutically
acceptable
excipient" as used in the specification and claims includes both one and more
than one such
excipient.
[0054] As used herein, "pharmaceutically acceptable derivatives" of a compound
of the
invention include salts, esters, enol ethers, enol esters, acetals, ketals,
orthoesters,
9
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
hemiacetals, hemiketals, acids, bases, solvates, hydrates or prodrugs thereof.
Such
derivatives may be readily prepared by those of skill in this art using known
methods for
such derivatization. The compounds produced may be administered to animals or
humans
without substantial toxic effects and either are pharmaceutically active or
are prodrugs.
[0055] A "pharmaceutically acceptable salt" of a compound of the invention
means a salt
that is pharmaceutically acceptable and that possesses the desired
pharmacological activity
of the parent compound. Such salts include: (1) acid addition salts, formed
with inorganic
acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid,
phosphoric acid,
and the like; or formed with organic acids such as acetic acid, propionic
acid, hexanoic acid,
cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic
acid, succinic
acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid,
benzoic acid, 3-(4-
hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic
acid,
ethanesulfonic acid, 1,2-ethanedisulfonic acid, 2-hydroxyethanesulfonic acid,
benzenesulfonic acid, 4-chlorobenzenesulfonic acid, 2-naphthalenesulfonic
acid, 4-
toluenesulfonic acid, camphorsulfonic acid, 4-methylbicyclo[2.2.2]oct-2-ene-l-
carboxylic
acid, glucoheptonic acid, 4,4'-methylenebis-(3-hydroxy-2-ene-l-carboxylic
acid), 3-
phenylpropionic acid, trimethylacetic acid, tertiary butylacetic acid, lauryl
sulfuric acid,
gluconic acid, glutamic acid, hydroxynaphthoic acid, salicylic acid, stearic
acid, muconic
acid, and the like; or (2) salts formed when an acidic proton present in the
parent compound
either is replaced by a metal ion, e.g., an alkali metal ion, an alkaline
earth ion, or an
aluminum ion; or coordinates with an organic base such as ethanolamine,
diethanolamine,
triethanolamine, tromethamine, N-methylglucamine, and the like.
[0056] A "pharmaceutically acceptable ester" of a compound of the invention
means an ester
that is pharmaceutically acceptable and that possesses the desired
pharmacological activity
of the parent compound, and includes, but is not limited to, alkyl, alkenyl,
alkynyl, aryl,
heteroaryl, aralkyl, heteroaralkyl, cycloalkyl and heterocyclyl esters of
acidic groups,
including, but not limited to, carboxylic acids, phosphoric acids, phosphinic
acids, sulfonic
acids, sulfinic acids and boronic acids.
[0057] A "pharmaceutically acceptable enol ether" of a compound of the
invention means an
enol ether that is pharmaceutically acceptable and that possesses the desired
pharmacological
activity of the parent compound, and includes, but is not limited to,
derivatives of formula
C=C(OR) where R is hydrogen, alkyl, alkenyl, alkynyl, aryl, heteroaryl,
aralkyl,
heteroaralkyl, cycloalkyl or heterocyclyl.
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[0058] A "pharmaceutically acceptable enol ester" of a compound of the
invention means an
enol ester that is pharmaceutically acceptable and that possesses the desired
pharmacological
activity of the parent compound, and includes, but is not limited to,
derivatives of formula
C=C(OC(O)R) where R is hydrogen, alkyl, alkenyl, alkynyl, aryl, heteroaryl,
aralkyl,
heteroaralkyl, cycloalkyl or heterocyclyl.
[0059] A "pharmaceutically acceptable solvate or hydrate" of a compound of the
invention
means a solvate or hydrate complex that is pharmaceutically acceptable and
that possesses
the desired pharmacological activity of the parent compound, and includes, but
is not limited
to, complexes of a compound of the invention with one or more solvent or water
molecules,
or 1 to about 100, or 1 to about 10, or one to about 2, 3 or 4, solvent or
water molecules.
[0060] A "pro-drug" means any compound that releases an active parent compound
of
formula (I) in vivo when the prodrug is administered to a mammalian subject.
Prodrugs of
the compounds of formula (I) contain functional groups that, under standard
physiological
conditions, are hydrolyzed into the corresponding carboxy, hydroxy, amino or
sulfhydryl
group. Examples of such functional groups include, but are not limited to,
esters (e.g,
acetate, formate and benzoate derivatives) and carbamates (e.g., N,N-
dimethylaminocarbonyl) of hydroxy groups in compounds of formula (I), and the
like.
Additional examples include dipeptide or tripeptide esters of hydroxy or
carboxy groups in
compounds of formula (I), and the like. The preparation of such functional
groups is well
known in the art. For example, a compound of formula (I) having a hydroxy
group attached
thereto (e.g., when XI, X2, X3, Y1, Y2 or Y3 is hydroxy) may be treated with a
carboxylic
acid or a dipeptide having a free carboxy terminus under esterification
conditions well
known in the art to yield the desired ester functional group. Likewise, a
compound of
formula (I) having a free carboxy group attached thereto may be treated with
an alcohol or a
tripeptide containing a hydroxy group such as a serine residue (e.g., -N(H)-
C(H)(CH2OH)-
C(O)-) under esterification conditions well known in the art to produce the
desired ester
functional group. In addition, compounds of formula (I) having a carboxylic
ester group
attached thereto may be treated with a different carboxylic ester under
standard
transesterification conditions to produce compounds of formula (I) with the
desired
functional ester group attached thereto. All such functional groups are
considered to be
within the scope of this invention.
[0061] The term "organic group" and "organic radical" as used herein means any
carbon-
containing group, including hydrocarbon groups that are classified as an
aliphatic group,
cyclic group, aromatic group, functionalized derivatives thereof and/or
various combination
11
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
thereof. The term "aliphatic group" means a saturated or unsaturated linear or
branched
hydrocarbon group and encompasses alkyl, alkenyl, and alkynyl groups, for
example. The
term "alkyl group" means a substituted or unsubstituted, saturated linear or
branched
hydrocarbon group or chain (e.g., C1 to C8) including, for example, methyl,
ethyl, isopropyl,
tert-butyl, heptyl, n-octyl, dodecyl, octadecyl, amyl, 2-ethylhexyl, and the
like. Suitable
substituents include carboxy, protected carboxy, amino, protected amino, halo,
hydroxy,
protected hydroxy, mercapto, lower alkylthio, nitro, cyano, monosubstituted
amino,
protected monosubstituted amino, disubstituted amino, C1 to C7 alkoxy, C1 to
C7 acyl, C1 to
C7 acyloxy, and the like. The term "substituted alkyl" means the above defined
alkyl group
substituted from one to three times by a hydroxy, protected hydroxy, amino,
protected
amino, cyano, halo, trifloromethyl, mono-substituted amino, di-substituted
amino, lower
alkoxy, mercapto, lower alkylthio, carboxy, protected carboxy, or a carboxy,
amino, and/or
hydroxy salt. As used in conjunction with the substituents for the heteroaryl
rings, the terms
"substituted (cycloalkyl)alkyl" and "substituted cycloalkyl" are as defined
below substituted
with the same groups as listed for a "substituted alkyl" group. The term
"alkenyl group"
means an unsaturated, linear or branched hydrocarbon group with one or more
carbon-
carbon double bonds, such as a vinyl group. The term "alkynyl group" means an
unsaturated,
linear or branched hydrocarbon group with one or more carbon-carbon triple
bonds. The
term "cyclic group" means a closed ring hydrocarbon group that is classified
as an alicyclic
group, aromatic group, or heterocyclic group. The term "alicyclic group" means
a cyclic
hydrocarbon group having properties resembling those of aliphatic groups. The
term
"aromatic group" or "aryl group" means a mono- or polycyclic aromatic
hydrocarbon group,
and may include one or more heteroatoms, and which are further defined below.
The term
"heterocyclic group" means a closed ring hydrocarbon in which one or more of
the atoms in
the ring are an element other than carbon (e.g., nitrogen, oxygen, sulfur,
etc.), and are further
defined below.
(0062] "Organic groups" maybe functionalized or otherwise comprise additional
functionalities associated with the organic group, such as carboxyl, amino,
hydroxyl, and the
like, which may be protected or unprotected. For example, the phrase "alkyl
group" is
intended to include not only pure open chain saturated hydrocarbon alkyl
substituents, such
as methyl, ethyl, propyl, t-butyl, and the like, but also alkyl substituents
bearing further
substituents known in the art, such as hydroxy, alkoxy, mercapto, alkylthio,
alkylsulfonyl,
halo, cyano, nitro, amino, carboxyl, etc. Thus, "alkyl group" includes ethers,
esters,
haloalkyls, nitroalkyls, carboxyalkyls, hydroxyalkyls, sulfoalkyls, etc.
12
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00631 The terms "halo group" or "halogen" are used interchangeably herein and
refer to the
fluoro, chloro, bromo or iodo groups. Preferred halogens are chloro and
fluoro.
[00641 The term "haloalkyl" refers to an alkyl group as defined above that is
substituted by
one or more halogen atoms. The halogen atoms may be the same or different. The
term
"dihaloalkyl " refers to an alkyl group as described above that is substituted
by two halo
groups, which may be the same or different. The term "trihaloalkyl" refers to
an alkyl group
as describe above that is substituted by three halo groups, which may be the
same or
different. The term "perhaloalkyl" refers to a haloalkyl group as defined
above wherein each
hydrogen atom in the alkyl group has been replaced by a halogen atom. The term
"perfluoroalkyl" refers to a haloalkyl group as defined above wherein each
hydrogen atom in
the alkyl group has been replaced by a fluoro group.
[0065] The term "cycloalkyl" means a mono-, bi-, or tricyclic saturated ring
that is fully
saturated or partially unsaturated. Examples of such a group included
cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, cis-
or trans-
decalin, bicyclo[2.2.1]hept-2-ene, cyclohex-l-enyl, cyclopent-l-enyl, 1,4-
cyclooctadienyl,
and the like.
[00661 The term "(cycloalkyl)alkyl" means the above-defined alkyl group
substituted for one
of the above cycloalkyl rings. Examples of such a group include
(cyclohexyl)methyl, 3-
(cyclopropyl)-n-propyl, 5-(cyclopentyl)hexyl, 6-(adamantyl)hexyl, and the
like.
[0067] The term "substituted phenyl" specifies a phenyl group substituted with
one or more-
moieties, and in some instances one, two, or three moieties, chosen from the
groups
consisting of halogen, hydroxy, protected hydroxy, cyano, nitro; mercapto,
alkylthio,
trifluoromethyl, C1 to C7 alkyl, C1 to C7 alkoxy, C1 to C7 acyl, C1 to C7
acyloxy, carboxy,
oxycarboxy, protected carboxy, carboxymethyl, protected carboxymethyl,
hydroxymethyl,
protected hydroxymethyl, amino, protected amino, (monosubstituted)amino,
protected
(monosubstituted)amino, (disubstituted)amino, carboxamide, protected
carboxamide, N-(C1
to C6 alkyl)carboxamide, protected N-( C1 to C6 alkyl)carboxamide, N,N-di(C1
to C6
alkyl)carboxamide, trifluoromethyl, N-(( C1 to C6 alkyl)sulfonyl)amino, N-
(phenylsulfonyl)amino or phenyl, substituted or unsubstituted, such that, for
example, a
biphenyl or naphthyl group results.
[00681 Examples of the term "substituted phenyl" includes a mono- or
di(halo)phenyl group
such as 2-, 3- or 4-chlorophenyl, 2,6-dichlorophenyl, 2,5-dichlorophenyl, 3,4-
dichlorophenyl, 2-, 3- or 4-bromophenyl, 3,4-dibromophenyl, 3-chloro-4-
fluorophenyl, 2-, 3-
or 4-fluorophenyl and the like; a mono or di(hydroxy)phenyl group such as 2,
3, or 4-
13
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
hydroxyphenyl, 2,4-dihydroxyphenyl, the protected-hydroxy derivatives thereof
and the like;
a nitrophenyl group such as 2-, 3- or 4-nitrophenyl; a cyanophenyl group, for
example, 2-, 3-
or 4-cyanophenyl; a mono- or di(alkyl)phenyl group such as 2-, 3- or 4-
methylphenyl, 2,4-
dimethylphenyl, 2-, 3- or 4-(iso-propyl)phenyl, 2-, 3- or 4-ethylphenyl, 2-, 3-
or 4-(n-
propyl)phenyl and the like; a mono or di(alkoxy)phenyl group, for example, 2,6-
dimethoxyphenyl, 2-, 3- or 4-(isopropoxy)phenyl, 2-, 3- or 4-(t-butoxy)phenyl,
3-ethoxy-4-
methoxyphenyl and the like; 2-, 3- or 4-trifluoromethylphenyl; a mono- or
dicarboxyphenyl
or (protected carboxy)phenyl group such as 2-, 3- or 4-carboxyphenyl or 2,4-
di(protected
carboxy)phenyl; a mono- or di(hydroxymethyl)phenyl or (protected
hydroxymethyl)phenyl
such as 2-, 3- or 4-(protected hydroxymethyl)phenyl or 3,4-
di(hydroxymethyl)phenyl; a
mono- or di(aminomethyl)phenyl or (protected aminomethyl)phenyl such as 2-, 3-
or 4-
(aminomethyl)phenyl or 2,4-(protected aminomethyl)phenyl; or a mono- or di(N-
.
(methylsulfonylamino))phenyl such as 2-, 3- or 4-(N-
(methylsulfonylamino))phenyl. Also,
the term "substituted phenyl" represents disubstituted phenyl groups wherein
the substituents
are different, for example, 3-methyl-4-hydroxyphenyl, 3-chloro-4-
hydroxyphenyl, 2-
methoxy-4-bromophenyl, 4-ethyl-2-hydroxyphenyl, 3-hydroxy-4-nitrophenyl, 2-
hydroxy-4-
chlorophenyl and the like.
[0069] The term "(substituted phenyl)alkyl" means one of the above substituted
phenyl
groups attached to one of the above-described alkyl groups. Examples include
such groups
as 2-phenyl-l-chloroethyl, 2-(4'-methoxyphenyl)ethyl, 4-(2',6'-
dihydroxyphenyl)-n-hexyl,
2-(5'-cyano-3'-methoxyphenyl)-n-pentyl, 3-(2',6'-dimethylphenyl)propyl, 4-
chloro-3-
aminobenzyl, 6-(4'-methoxyphenyl)-3-carboxyhexyl, 5-(4'-aminomethylphenyl)-3-
(aminomethyl)pentyl, 5-phenyl-3-oxopent-l-yl, (4-hydroxynapth-2-yl)methyl and
the like.
[0070] As noted above, the term "aromatic" or "aryl" refers to five and six
membered
carbocyclic rings. Also as noted above, the term "heteroaryl" denotes
optionally substituted
five-membered or six-membered rings that have 1 to 4 heteroatoms, such as
oxygen, sulfur
and/or nitrogen atoms, in particular nitrogen, either alone or in conjunction
with sulfur or
oxygen ring atoms. These five-membered or six-membered rings may be fully
unsaturated.
[0071] Furthermore, the above optionally substituted five-membered or six-
membered rings
can optionally be fused to an aromatic 5-membered or 6-membered ring system.
For
example, the rings can be optionally fused to an aromatic 5-membered or 6-
membered ring
system such as a pyridine or a triazole system, and preferably to a benzene
ring.
[0072] The following ring systems are examples of the heterocyclic (whether
substituted or
unsubstituted) radicals denoted by the term "heteroaryl": thienyl, furyl,
pyrrolyl,
14
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
pyrrolidinyl, imidazolyl, isoxazolyl, triazolyl, thiadiazolyl, oxadiazolyl,
tetrazolyl,
thiatriazolyl, oxatriazolyl, pyridyl, pyrimidyl, pyrazinyl, pyridazinyl,
oxazinyl, triazinyl,
thiadiazinyl tetrazolo, 1,5-[b]pyridazinyl and purinyl, as well as benzo-fused
derivatives, for
example, benzoxazolyl, benzthiazolyl, benzimidazolyl and indolyl.
[0073] Substituents for the above optionally substituted heteroaryl rings are
from one to
three halo, trihalomethyl, amino, protected amino, amino salts, mono-
substituted amino, di-
substituted amino, carboxy, protected carboxy, carboxylate salts, hydroxy,
protected
hydroxy, salts of a hydroxy group, lower alkoxy, mercapto, lower alkylthio,
alkyl,
substituted alkyl, cycloalkyl, substituted cycloalkyl, (cycloalkyl)alkyl,
substituted
(cycloalkyl)alkyl, phenyl, substituted phenyl, phenylalkyl, and (substituted
phenyl)alkyl.
Substituents for the heteroaryl group are as heretofore defined, or in the
case of
trihalomethyl, can be trifluoromethyl, trichloromethyl, tribromomethyl, or
triiodomethyl. As
used in conjunction with the above substituents for heteroaryl rings, "lower
alkoxy" means a
C1 to C4 alkoxy group, similarly, "lower alkylthio" means a C1 to C4 alkylthio
group.
[0074] The term "(monosubstituted)amino" refers to an amino group with one
substituent
chosen from the group consisting of phenyl, substituted phenyl, alkyl,
substituted alkyl, C1 to
C4 acyl, C2 to C7 alkenyl, C2 to C7 substituted alkenyl, C2 to C7 alkynyl, C7
to C16 alkylaryl,
C7 to C16 substituted alkylaryl and heteroaryl group. The (monosubstituted)
amino can
additionally have an amino-protecting group as encompassed by the term
"protected
(monosubstituted)amino." The term "(disubstituted)amino" refers to amino
groups with two
substituents chosen from the group consisting of phenyl, substituted phenyl,
alkyl,
substituted alkyl, C1 to C7 acyl, C2 to C7 alkenyl, C2 to C7 alkynyl, C7 to
C16 alkylaryl, C7 to
C16 substituted alkylaryl and heteroaryl. The two substituents can be the same
or different.
[0075] The term "heteroaryl(alkyl)" denotes an alkyl group as defined above,
substituted at
any position by a heteroaryl group, as above defined.
[0076] "Optional" or "optionally" means that the subsequently described event,
circumstance, feature or element may, but need not, occur, and that the
description includes
instances where the event or circumstance occurs and instances in which it
does not. For
example, "heterocyclo group optionally mono- or disubstituted with an alkyl
group" means
that the alkyl may, but need not, be present, and the description includes
situations where the
heterocyclo group is mono- or disubstituted with an alkyl group and situations
where the
heterocyclo group is not substituted with the alkyl group.
[0077] The term "electron-withdrawing group" refers to the ability of a
functional group on
a molecule to draw electrons to itself more than a hydrogen atom would if the
hydrogen
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
atom occupied the same position in the molecule. Examples of electron-
withdrawing groups
include, but are not limited to, halogen groups, -C(O)R groups (where R is
alkyl); carboxylic
acid and ester groups; -NR3+ groups (where R is alkyl or hydrogen); azo;
nitro; -OR and -SR
groups (where R is hydrogen or alkyl); and organic groups (as defined herein)
containing
such electron-withdrawing groups, such as haloalkyl groups (including
perhaloalkyl groups),
and the like.
[0078] Compounds that have the same molecular formula but differ in the nature
or
sequence of bonding of their atoms or the arrangement of their atoms in space
are termed
"isomers." Isomers that differ in the arrangement of their atoms in space are
termed
"stereoisomers." Stereoisomers that are not mirror images of one another are
termed
"diastereomers" and those that are non-superimposable mirror images of each
other are
termed "enantiomers." When a compound has an asymmetric center, for example,
it is
bonded to four different groups, a pair of enantiomers is possible. An
enantiomer can be
characterized by the absolute configuration of its asymmetric center and is
described by the
R- and S-sequencing rules of Cahn and Prelog, or by the manner in which the
molecule
rotates the plane of polarized light and designated as dextrorotatory or
levorotatory (i.e., as
(+) or (-)-isomers respectively). A chiral compound can exist as either an
individual
enantiomer or as a mixture of thereof. A mixture containing equal proportions
of the
enantiomers is called a "racemic mixture."
[0079] The compounds of this invention may possess one or more asymmetric
centers; such
compounds can therefore be produced as individual (R)- or (S)- stereoisomers
or as mixtures
thereof. Unless indicated otherwise, the description or naming of a particular
compound in
the specification and claims is intended to include both individual
enantiomers and mixtures,
racemic or otherwise, thereof. The methods for the determination of
stereochemistry and the
separation of stereoisomers are well-known in the art (see, e.g., the
discussion in Chapter 4
of "Advanced Organic Chemistry", 4th edition J. March, John Wiley and Sons,
New York,
1992).
OVERVIEW
[0080] The invention provides thiazolidinone compositions, thiazolidinone
derivatives
compositions and methods of their use in high affinity inhibition of cystic
fibrosis
transmembrane conductance regulator protein (CFTR) and for the study and
treatment of
CFTR-mediated diseases and conditions. The discovery of the subject
thiazolidinone
compounds and derivatives was based on screening of numerous potential
candidate
16
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
compounds using an assay designed to identify CFTR inhibitors that interact
directly with
CFTR. Without being held to any particular theory or mode of operation, since
multiple
CFTR activators that work on different activating pathways were included in
the studies
leading to identification of the subject compounds, the inhibitory compounds
of the
invention likely effect inhibition by acting at or near the CFTR Cl"
transporting pathway. A
screening of 50,000 diverse compounds identified several 2-thioxo-4-
thiazolidinone
compounds and derivatives as effective CFTR inhibitors. These compounds and
derivatives
are unrelated chemically and structurally to previously known CFTR activators
or to the
previously known CFTR inhibitors DPC, NPPB or glibenclamide. The most potent
CFTR
inhibitor identified from screening had a KI of -300 nM for inhibition of CF
current in
human airway cells. Inhibition was rapid, reversible and CFTR-specific.
[0081] The compositions and methods of the invention will now be described in
more detail.
THIAZOLIDINONE COMPOUNDS AND DERIVATIVES
[0082] The thiazolidinone compounds and derivatives used in the compositions
and methods
of the invention comprise a heterocyclic ring of five or more atoms, including
an aryl
substituted nitrogen, at least one sulfur, oxygen or selenium heteroatom, and
one or more
carbonyl or thiocarbonyl groups associated with the heterocyclic ring. More
specifically, the
subject thiazolidinone compounds and derivatives may have the following
formula (I):
Al
X2 ~A3 (I)
X-~ / N 4 Y1
i =jY2
X3 A
2
Y3
wherein X1, X2 and X3 are independently chosen from hydrogen, an organic
group, a halo
group, a nitro group, an azo group, a hydroxyl group and a mercapto group; Yl,
Y2 and Y3
are independently chosen from hydrogen, an organic group, a halo group, a
nitro group, an
azo group, a hydroxyl group and a mercapto group; Al and A2 are independently
chosen
from oxygen and sulfur, A3 is chosen from sulfur and selenium; and A4
comprises one or
more carbons or heteroatoms and may be present or absent; or a
pharmaceutically acceptable
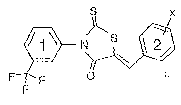
derivative thereof, as an individual stereoisomer or a mixture thereof. Where
A4 is absent the
central heterocyclic ring is, a five membered ring.
17
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[0083] In certain embodiments, the thiazolidinone compounds and derivatives of
formula (I)
above comprise the formula (Ia):
Al
X2 S Y %^/ 2
N (la)
X
X3 A2 Y3
wherein X1, X2 and X3 are independently chosen from hydrogen, an organic
group, a halo
group, a nitro group, an azo group, a hydroxyl group and a mercapto group; Yl,
Y2 and Y3
are independently chosen from hydrogen, an organic group, a halo group, a
nitro group, an
azo group, a hydroxyl group and a mercapto group; and Al and A2 are
independently chosen
from oxygen and sulfur.- In specific embodiments, Xl may be an electron
withdrawing group,
and may comprise a haloalkyl group, dihaloalkyl group, trihaloalkyl group
(e.g.,
trifluoroalkyl group) or a fluoro group. Y2 is independently chosen from the
group consisting
of alkyl, hydroxyl, carboxyl, nitro, carbonate, carbamate, alkoxy,
alkylcarbonyl, and halo
groups, Yl is independently chosen from hydroxyl and bromo groups, and Y3 is
independently chosen from hydrogen and a nitro group.
[0084] The subject thiazolidinone compounds and derivatives of formula (I) in
many
embodiments may comprise 3-aryl-5-arylmethylene-2-thioxo-4-thiazolidinones of
the
formula (Ib)
S
\ /Y 2
X2 Y
/ N I (lb)
X1 I \ \
X3 p Y3
wherein at least one of X1, X2 and X3 is an electron-withdrawing group; and
Y1, Y2 and Y3
are independently chosen from hydrogen, alkyl, hydroxyl, carboxyl, nitro,
carbonate,
carbamate, alkoxy, alkylcarbonyl, and a halo group. In one embodiment Xt is at
a position
selected from 2, 3, or 4; Y2 is at a position selected from 2, 3, or 4; and Yl
and Y3 may be
hydrogen.
[0085] The 3-aryl-5-arylmethylene-2-thioxo-4-thiazolidinones may more
specifically have
the formula (Ic):
18
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
S
Yl
,-~
F3 C0~~X/ N I (Ic)
O Y3
wherein Y1 - Y3 are as described above. In one embodiment the trifluoromethyl
group is at a
position selected from 2, 3, or 4; Y2 is at a position selected from 2, 3, or
4; where Yl and Y3
may be hydrogen in this embodiment.
[0086] In some embodiments of the invention, the thiazolidinone compounds of
the
invention may comprise:
S
S N02
\ / N I
CF3 O
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(4-nitrophenyl)methylene]-2-thioxo-4-
thiazolidinone;
S~
N/ \S 0 OH
CF3 0
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(4-oxycarboxyphenyl)methylene]-2-thioxo-
4-
thiazolidinone;
S~
N/ -S C02H
CF3 O
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-
thiazolidinone;
S~'S OH
CF3 0 OH
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(3,4-dihydroxyphenyl)methylene]-2-
thioxo-4-
thiazolidinone;
19
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
S Br
~_S OH
N
&Br
CF3 O
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(3,5-dibromo-4-hydroxyphenyl)methylene]-
2-thioxo-4-
thiazolidinone; and
S Br
~_s OH
N;~
CF3 0 NOa
i.e., 3-[(3-trifluoromethyl)phenyl]-5-[(3-bromo-4-hydroxy-5-
nitrophenyl)methylene]-2-
thioxo-4-thiazolidinone. Alternatively, the trifluoromethyl group in any of
the above recited
compounds maybe position 2 or position 4 of the phenyl ring.
PHARMACEUTICAL PREPARATIONS
[00871 Also provided by the invention are pharmaceutical preparations of the
subject
thiazolidinone compounds described above. The subject compounds can be
incorporated into
a variety of formulations for therapeutic administration by a variety of
routes. More
particularly, the compounds of the present invention can be formulated into
pharmaceutical
compositions by combination with appropriate, pharmaceutically acceptable
carriers,
diluents, excipients and/or adjuvants, and may be formulated into preparations
in solid, semi-
solid, liquid or gaseous forms, such as tablets, capsules, powders, granules,
ointments,
solutions, suppositories, injections, inhalants and aerosols. Preferably, the
formulations are
free of detectable DMSO (dimethyl sulfoxide), which is not a pharmaceutically
acceptable
carrier, diluent, excipient, or adjuvant for non-topical, parenteral
administration or enteral
administration. The formulations may be designed for administration to
subjects or patients
in need thereof via a number of different routes, including oral, buccal,
rectal, parenteral,
intraperitoneal, intradermal, transdermal, intracheal, etc., administration.
[00881 In one embodiment, topical administration (e.g., by transdermal
administration) is of
interest. Topical formulations can be in the form of a transdermal patch,
ointment, paste,
lotion, cream, gel, and the like. Topical formulations may include one or more
of a
penetrating agent, thickener, diluent, emulsifier, dispersing aid, or binder.
Where the
compound is formulated for transdermal delivery, the compound may be
formulated with or
for use with a penetration enhancer. Penetration enhancers, which include
chemical
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
,, .u.,~... .,.ter .mar,, IrAt tL, i6 :k r
penetration enhancers and physical penetration enhancers, facilitate delivery
of the
compound through the skin, and may also be referred to as "permeation
enhancers"
interchangeably. Physical penetration enhancers include, for example,
electrophoretic
techniques such as iontophoresis, use of ultrasound (or "phonophoresis"), and
the like.
Chemical penetration enhancers are agents administered either prior to, with,
or immediately
following compound administration, which increase the permeability of the
skin, particularly
the stratum corneum, to provide for enhanced penetration of the drug through
the skin.
[0089] Compounds that have been used to enhance skin permeability include: the
sulfoxides
dimethylsulfoxide (DMSO) and decylmethylsulfoxide (Clo MSO); ethers such as
diethylene
glycol monoethyl ether, dekaoxyethylene-oleylether, and diethylene glycol
monomethyl
ether; surfactants such as sodium laurate, sodium lauryl sulfate,
cetyltrimethylammonium
bromide, benzalkonium chloride, Poloxamer (231, 182, 184), Tween (20, 40, 60,
80) and
lecithin; the 1-substituted azacycloheptan-2-ones, particularly 1-n-
dodecylcyclazacycloheptan-2-one; alcohols such as ethanol, propanol, octanol,
benzyl
alcohol, and the like; petrolatums, such as petroleum jelly (petrolatum),
mineral oil (liquid
petrolatum), and the like; fatty acids such as C8-C22 and other fatty acids
(e.g., isostearic
acid, octanoic acid, oleic acid, lauric acid, valeric acid); C8-C22 fatty
alcohols (e.g., oleyl
alcohol, lauryl alcohol); lower alkyl esters of C8-C22 fatty acids and other
fatty acids (e.g.,
ethyl oleate, isopropyl myristate, butyl stearate, methyl laurate, isopropyl
myristate,
isopropyl palmitate, methylpropionate, ethyl oleate); monoglycerides of C8-C22
fatty acids
(e.g., glyceryl monolaurate); tetrahydrofurfuryl alcohol polyethylene glycol
ether; 2-(2-
ethoxyethoxy)ethanol; diethylene glycol monomethyl ether; alkylaryl ethers of
polyethylene
oxide; polyethylene oxide monomethyl ethers; polyethylene oxide dimethyl
ethers; di-lower
alkyl esters of C6-C8 diacids (e.g., diisopropyl adipate); ethyl acetate;
acetoacetic ester;
polyols and esters thereof such as propylene glycol, ethylene glycol,
glycerol, butanediol,
polyethylene glycol, and polyethylene glycol monolaurate; amides and other
nitrogenous
compounds such as urea, dimethylacetamide (DMA), dimethylformamide (DMF), 2-
pyrrolidone, N-alkylpyrrolidone, e.g.,'1-methyl-2-pyrrolidone; ethanol amine,
diethanol
amine and triethanolamine; terpenes; alkanones, and organic acids,
particularly salicylic acid
and salicylates, citric acid and succinic acid. Additional chemical and
physical penetration
enhancers are described in, for example, Transdermal Delivery of Drugs, A. F.
Kydonieus
(ED) 1987 CRL Press; Percutaneous Penetration Enhancers, eds. Smith et al.
(CRC Press,
1995); Lenneruas et al., J Pharm Pharmacol 2002;54(4):499-508; Karande et al.,
Pharm Res
2002;19(5):655-60; Vaddi et al., J Pharm Sci 2002 July; 91(7):1639-51; Ventura
et al., J
21
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Drug Target 2001;9(5):379-93; Shokri et al., Int J Pharm 2001;228(1-2):99-107;
Suzuki et
al., Biol Pharm Bull 2001;24(6):698-700; Alberti et al., J Control Release
2001;71(3):319-
27; Goldstein et al., Urology 2001;57(2):301-5; Kiijavainen et al., Eur J
Pharm Sci
2000;10(2):97-102; and Tenjarla et al., Int J Pharm 1999;192(2):147-58.
[0090] Where the compound is formulated with a chemical penetration enhancer,
the
penetration enhancer is selected for compatibility with the compound, and is
present in an
amount sufficient to facilitate delivery of the compound through skin of a
subject, e.g., for
delivery of the compound to the systemic circulation. In one embodiment, the
compound is
formulated with a penetration enhancer other than DMSO.
[0091] In one embodiment, the compound is provided in a drug delivery patch,
e.g., a
transmucosal or transdermal patch, and can be formulated with a penetration
enhancer. The
patch generally includes a backing layer, which is impermeable to the compound
and other
formulation components, a matrix in contact with one side of the backing
layer, which
matrix provides for sustained-release, which may be controlled release, of the
compound,
and an adhesive layer, which is on the same side of the backing layer as the
matrix. The
matrix can be selected as is suitable for the route of administration, and can
be, for example,
and can be a polymeric or hydrogel matrix.
[0092] In pharmaceutical dosage forms, the subject compounds of the invention
may be
administered in the form of their pharmaceutically acceptable derivative, such
as a salt, or
they may also be used alone or in appropriate association, as well as in
combination, with
other pharmaceutically active compounds. The following methods and excipients
are merely
exemplary and are in no way limiting.
[0093] For oral preparations, the subject compounds can be used alone or in
combination
with appropriate additives to make tablets, powders, granules or capsules, for
example, with
conventional additives, such as lactose, mannitol, corn starch or potato
starch; with binders,
such as crystalline cellulose, cellulose derivatives, acacia, corn starch or
gelatins; with
disintegrators, such as corn starch, potato starch or sodium
carboxymethylcellulose; with
lubricants, such as talc or magnesium stearate; and if desired, with diluents,
buffering agents,
moistening agents, preservatives and flavoring agents. Of particular interest
is formulation of
the subject thiazolidinone compounds with a buffering agent, to provide for
protection of the
compound from low pH of the gastric environment. It may also be preferable to
provide an
enteric coating so as to avoid precipitation of the compound while in transit
through the
stomach.
22
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[0094] The subject compounds of the invention can be formulated into
preparations for
injection by dissolving, suspending or emulsifying them in an aqueous or
nonaqueous
solvent, such as vegetable or other similar oils, synthetic aliphatic acid
glycerides, esters of
higher aliphatic acids or propylene glycol; and if desired, with conventional
additives such
as solubilizers, isotonic agents, suspending agents, emulsifying agents,
stabilizers and
preservatives. Solubilizers of particular interest include vitamin E TPGS (d-a-
tocopheryl
polyethylene glycol 1000 succinate), cyclodextrins, and the like.
[0095] The compounds of the invention can be utilized in aerosol formulation
to be
administered via inhalation. The compounds of the present invention can be
formulated into
pressurized acceptable propellants such as dichlorodifluoromethane, propane,
nitrogen and
the like.
[0096] . Furthermore, the subject compounds can be made into suppositories by
mixing with a
variety of bases such as emulsifying bases or water-soluble bases. The
compounds of the
present invention can be administered rectally via a suppository. The
suppository can include
vehicles such as cocoa butter, carbowaxes and polyethylene glycols, which melt
at body
temperature, yet are solidified at room temperature.
[0097] Unit dosage forms for oral or rectal administration such as syrups,
elixirs, and
suspensions may be provided wherein each dosage unit, for example,
teaspoonful,
tablespoonful, tablet or suppository, contains a predetermined amount of the
composition
containing one or more inhibitors. Similarly, unit dosage forms for injection
or intravenous
administration may comprise the inhibitor(s) in a composition as a solution in
sterile water,
normal saline or another pharmaceutically acceptable carrier.
[0098] Depending on the subject and condition being treated and on the
administration
route, the subject compounds may be administered in dosages of, for example,
0.1 gg to 10
mg/kg body weight per day. The range is broad, since in general the efficacy
of a therapeutic
effect for different mammals varies widely with doses typically being 20, 30
or even 40
times smaller (per unit body weight) in man than in the rat. Similarly the
mode of
administration can have a large effect on dosage. The inventors have found
that cholera
toxin-induced intestinal fluid secretion in mice is effectively blocked by a
single
intraperitoneal dose of about 10-20 micrograms with a dosage of about ten
times greater
being effective in rats. Thus, for example, oral dosages may be about ten
times the injection
dose. Higher doses may be used for localized routes of delivery.
[0099] A typical dosage may be a solution suitable for intravenous
administration; a tablet
taken from two to six times daily, or one time-release capsule or tablet taken
once a day and
23
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
containing a proportionally higher content of active ingredient, etc. The time-
release effect
may be obtained by capsule materials that dissolve at different pH values, by
capsules that
release slowly by osmotic pressure, or by any other known means of controlled
release.
[00100] For use in the subject methods, the subject compounds may be
formulated with other
pharmaceutically active agents, including other CFTR-inhibiting agents.
[00101] Pharmaceutically acceptable excipients usable with the invention, such
as vehicles,
adjuvants, carriers or diluents, are readily available to the public.
Moreover,
pharmaceutically acceptable auxiliary substances, such as pH adjusting and
buffering agents,
tonicity adjusting agents, stabilizers, wetting agents and the like, are
readily available to the
public.
[00102] Those of skill in the art will readily appreciate that dose levels can
vary as a function
of the specific compound, the severity of the symptoms and the susceptibility
of the subject
to side effects. Preferred dosages for a given compound are readily
determinable by those of
skill in the art by a variety of means.
[00103] Kits with unit doses of the subject compounds, usually in oral or
injectable doses, are
provided. In such kits, in addition to the containers containing the unit
doses will be an
informational package insert describing the use and attendant benefits of the
drugs in treating
pathological condition of interest. Preferred compounds and unit doses are
those described
herein above.
CONDITIONS AMENABLE TO TREATMENT USING THE CFTR INHIBITORS OF THE
INVENTION
[00104] The CFTR inhibitors disclosed herein are useful in the treatment of a
CFTR-
mediated condition, i.e., any condition, disorder or disease, or symptom of
such condition,
disorder, or disease, that results from activity of CFTR, e.g., activity of
CFTR in ion
transport. Such conditions, disorders, diseases, or symptoms thereof are
amenable to
treatment by inhibition of CFTR activity, e.g., inhibition of CFTR ion
transport.
[00105] In one embodiment, the CFTR inhibitors of the invention are used in
the treatment of
conditions associated with aberrantly increased intestinal secretion,
particularly acute
aberrantly increased intestinal secretion. CFTR activity has been implicated
in intestinal
secretion in response to various agonists, including cholera toxin (see, e.g.,
Snyder et al.
1982 Bull. World Health Organ. 60:605-613; Chao et al. 1994 EMBO J 13:1065-
1072;
Kimberg et al. 1971 J Clin. Invest. 50:1218-1230). Thus CFTR inhibitors of the
invention
24
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
can be administered in an amount effective to inhibit CFTR ion transport and
thus decrease
intestinal fluid secretion.
[00106] Thus, CFTR inhibitors can be used in the treatment of intestinal
inflammatory
disorders and diarrhea, particularly secretory diarrhea. Secretory diarrhea is
the biggest cause
of infant death in developing countries, with about 5 million deaths annually
(Gabriel et al.,
1994 Science 266: 107-109). Several studies, including those using CF mice,
indicate that
CFTR is the final common pathway for intestinal chloride ion (and thus fluid)
secretion in
response to various agonists (Snyder et al., 1982, Bull. World Health Organ.
60: 605-613;
Chao et al., 1994 EMBO. J. 13: 1065-1072; and Kimberg et al., 1971, J. Clin.
Invest. 50:
1218-1230). The mouse models of intestinal fluid secretion used herein
indicate that CFTR
inhibition by systemic administration of the inhibitor at a non-toxic dose
effectively blocked
intestinal fluid secretion induced by cholera toxin (see Examples).
[00107] Diarrhea that may be amenable to treatment using the CFTR inhibitors
of the
invention can result from exposure to a variety of pathogens or agents
including, without
limitation, cholera toxin (Vibrio cholera), E. coli (particularly
enterotoxigenic (ETEC)),
Shigella, Salmonella, Campylobacter, Clostridium difficile, parasites (e.g.,
Giardia,
Entamoeba histolytica, Cryptosporidiosis, Cyclospora), diarrheal viruses
(e.g., rotavirus),
food poisoning, or toxin exposure that results in increased intestinal
secretion mediated by
CFTR.
[00108] Other diarrheas include diarrhea associated with AIDS (e.g., AIDS-
related diarrhea),
and inflammatory gastrointestinal disorders, such as ulcerative colitis,
inflammatory bowel
disease (IBD), Crohn's disease, and the like. It has been reported that
intestinal inflammation
modulates the expression of three major mediators of intestinal salt transport
and may
contribute to diarrhea in ulcerative colitis both by increasing
transepithelial Cl- secretion and
by inhibiting the epithelial NaCl absorption (see, e.g., Lohi et al., 2002,
Am. J. Physiol.
Gastrointest. Liver Physiol. 283(3):G567-75).
[00109] CFTR inhibitors of the invention can also be used in treatment of
conditions such as
polycystic kidney disease, and find further use as male infertility drugs, by
inhibition of
CFTR activity in the testis.
[00110] CFTR inhibitors of the invention can be further screened in larger
animal models
(e.g., the rabbit model described in Spira et al., 1981, Infect. Immun. 32:739-
747.). In
addition, analysis of stool output using live Vibrio cholerae can also be
examined to further
characterize the CFTR inhibitors of the invention.
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
NON-HUMAN ANIMAL MODELS AND HUMAN TISSUE MODELS OF CFTR-DEFICIENCIES
[00111] The CFTR inhibitors of the invention can also be used to generate non-
human animal
models of disease, where the disease is associated with decreased CFTR
function (e.g.,
decreased ion transport). There is increasing evidence that defective fluid
and
macromolecular secretion by airway submucosal glands leads to impaired
mucociliary and
bacterial clearance in CFTR-deficient subjects, particularly in those affected
with cystic
fibrosis (CF); however, functional studies in human airway glands have been
restricted to
severely diseased airways obtained at the time of lung transplantation
(Jayaraman et} al. 2001
Proc. Natl. Acad. Sci. USA 98:8119-8123). Acute CFTR inhibition permits
determination of
the role of CFTR in water, salt and macromolecule secretion by submucosal
glands. High-
affinity CFTR inhibitors permit the pharmacological creation of non-human
animal models
that mimic CFTR-deficiency in humans, e.g., mimics the human CF phenotype. In
particular, large animal models of CFTR deficiency (e.g., CF) find particular
use in
elucidating the pathophysiology of initiation and progression of airway
disease in CF, and in
evaluating the efficacy of CF therapies, e.g., screening candidate agents for
treatment of
CFTR-deficiencies or symptoms thereof.
[00112] Inhibition of CFTR ion transport can be manifested in airway and
pancreatic
disorders, as well as infertility in males. For example, inhibition of CFTR
channels in the
lungs and airways influences airway surface fluids leading to accumulation of
mucus, which
in turn plugs airways and collects heavily on the lung walls, providing a
prime environment
for infection to occur, which in turn can lead to chronic lung disease. This
same phenomenon
occurs in the pancreas, where the accumulated mucus disrupts the exocrine
function of the
pancreas and prevents essential food-processing enzymes from reaching the
intestines.
[00113] , Such non-human animal models can be generated by administration of
an amount of
a CFTR inhibitor effective to decrease CFTR activity in ion transport. Of
particular interest
is the use of the CFTR inhibitors of the invention to induce the cystic
fibrosis (CF)
phenotype in a non-human animal. Administration of an amount of a CFTR
inhibitor
effective to inhibit CFTR receptors in, for example, lung effectively mimics
the CFTR defect
found in CF. Routes of delivery for CFTR inhibitor are discussed in detail
above. Depending
on the non-human animal used, the subject compounds may be administered in
dosages of,
for example, 50 to 500 g/kg body weight one to three times a day by an
intraperitoneal,
subcutaneous, or other route to generate the non-human animal models. Oral
dosages may be
up to about ten times the intraperitoneal or subcutaneous dose. '
26
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00114] Non-human animal models of CFTR-associated disease can be used as
models of any
appropriate condition associated with decreased CFTR activity. Such conditions
include
those that are associated with CFTR mutations, which mutations result in
abnormalities in
epithelial ion and water transport. These abnormalities can in turn be
associated with
derangements in airway mucociliary clearance, as well as in other mucosal
epithelia and
ductal epithelia. Conditions that can be pharmacologically modeled by inducing
a CFTR-
deficient phenotype in a non-human animal include, without limitation, cystic
fibrosis
(including atypical CF), idiopathic chronic pancreatitis, vas deferens
defects, mild
pulmonary disease, asthma, and the like. For a review of disorders associated
with impaired
CFTR function, see, e.g., Noone et al. Respir Res 2 328-332 (2001). CFTR
inhibitor-
generated non-human animal models can also serve as models of microbial
infection (e.g.,
bacterial, viral, or fungal infection, particularly respiratory infections) in
a MR-deficient
subject. In one embodiment of particular interest, the CFTR inhibitors of the
invention are
used to pharmacologically induce the cystic fibrosis (CF) phenotype.
[00115] Animals suitable for use in the production of the animal models of the
invention
include any animal, particularly a mammal, e.g., non-human primates (e.g.,
monkey,
chimpanzee, gorilla, and the like), rodents (e.g., rats, mice, gerbils,
hamsters, ferrets, and the
like), lagomorphs, swine (e.g., pig, miniature pig), equine, canine, feline,
and the like. Large
animals are of particular interest.
[00116] The CFTR inhibitors can also be contacted with isolated human tissue
to create ex
vivo models of disease. Such tissue is contacted with an amount of a CFTR
inhibitor
effective to decrease CFTR activity in the tissue, which may be for as little
as 15 minutes, or
as much as two hours or more. Human tissues of interest include, without
limitation, lung
(including trachea and airways), liver, pancreas, testis, and the like.
Physiological,
biochemical, genomic or other studies can be carried out on the inhibitor-
treated tissue to
identify novel therapeutic target molecules that are important in the
pathophysiology of a
disease. For example, isolated tissue from humans without CF can be exposed to
inhibitor
sufficient to induce the CF phenotype and such studies can be carried out to
identify novel
therapeutic target molecules that are important in the pathophysiology of CF.
SYNTHESIS OF THE COMPOUNDS OF THE INVENTION
[00117] Compounds of the invention may be prepared according to methods known
to one
skilled in the art, or by the methods similar to those disclosed in US
5,326,770 and US
27
CA 02500498 2011-01-27
WO 2004/028480 PCTIUS2003/031005
6,380,186, or by methods similar to the method described below.
1001181 It is understood that in the following description, combinations of
substituents and/or
variables of the depicted formulae are permissible only if such contributions
result in stable
compounds.
[00119] It will also be appreciated by those skilled in the art that in the
process described
below the functional groups of intermediate compounds may need to be protected
by suitable
protecting groups. Such functional groups include hydroxy, amino, mercapto and
carboxylic
acid. Suitable protecting groups for hydroxy include trialkylsilyl or
diarylalkylsilyl (e.g.,
t-butyldimethylsilyl, t-butyldiphenylsilyl or trimethylsilyl),
tetrahydropyranyl, benzyl, and
the like. Suitable protecting groups for amino, amidino and guanidino include
t-butoxycarbonyl, benzyloxycarbonyl, and the like. Suitable protecting groups
for mercapto
include -C(O)-R (where R is alkyl, aryl or aralkyl), p-methoxybenzyl, trityl
and the like.
Suitable protecting groups for carboxylic acid include alkyl, aryl or aralkyl
esters.
[001201 Protecting groups may be added or removed in accordance with standard
techniques,
which are well-known to those skilled in the art and as described herein.
[001211 The use of protecting groups is described in detail in Theodora W.
Greene, Peter G.
M. Wuts, Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley-
Interscience. The
protecting group may also be a polymer resin such as a Wang resin or a 2-
chlorotrityl
chloride resin.
[001221 It will also be appreciated by those skilled in the art, although such
protected
derivatives of compounds of formula (I), as described above (e.g., in the
Overview and in
Thiazolidinone Compounds and Derivatives), may not possess pharmacological
activity as
such, they may be administered to a mammal and thereafter metabolized in the
body to form
compounds of the invention which are pharmacologically active. Such
derivatives may
therefore be described as "prodrugs". All prodrugs of compounds of formula (1)
are included
within the scope of the invention.
[00123] The following Reaction Schemes illustrate methods to make compounds of
the
invention. It is understood that one of ordinary skill in the art would be
able to make the
compounds of the invention by similar methods or by methods known to one
skilled in the
art. In general, starting components may be obtained from sources such as
Aldrich, or
synthesized according to sources known to those of ordinary skill in the art
(see, e.g., Smith
and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and
Structure,
5th edition (Wiley Interscience, New York)). Moreover, the various substituted
groups (e.g.,
28
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
X1, X2, X3, Y1, Y2 and Y3, etc.) of the compounds of the invention may be
attached to the
starting components, intermediate components, and/or final products according
to methods
known to those of ordinary skill in the art.
[00124] In the following Reaction Schemes, R represents an alkyl or aralkyl
group and W
represents a halogen atom, such as Cl, Br or I.
[00125] The following Reaction Scheme 1 is directed to the preparation of
compounds of
formula (1), which are compounds of the invention as described above (e.g., in
the Overview
and in Thiazolidinone Compounds and Derivatives), where A4 is absent, and Al,
A2, A3, X1,
X2, X3, Y1, Y2, and Y3 are as described above (e.g., in the Overview and in
Thiazolidinone
Compounds and Derivatives).
REACTION SCHEME 1
X2 Al
X2
1. HA3---"'~ 2 + X1 1 1. Base \ N A3
OR 2. Acid Xl
NCAI
X3 A2
(a) (b) (c)
Y3 Al
Knoevenagel Yi
2. (c + Y O condensation XZ A3
2
2 I~~ j ~~ \ N I-1Y
YJ X1 Y3
X3 Az
(d) (1)
[00126] In general, compounds of formula (1) are prepared by first treating a
compound of
formula (a) with 1 equivalent of a base, such as NaOH, at ambient temperature.
Compound
of formula (b), dissolved in an appropriate solvent such as THF, is then added
to the reaction
mixture. The resulting reaction mixture is then stirred for a period of time
of between about
1 hour to about 24 hours. An acid, such as HCl, is then added to the reaction
mixture. The
resulting reaction mixture is then stirred for a period of time of between
about 1 hour to
about 24 hours. The compound of formula (c) is then isolated from the reaction
mixture by
standard isolation and purification techniques. The compound of formula (c) is
then treated
with a compound of formula (d) under standard Knoevenagel condensation
conditions to
yield the desired product of formula (1).
29
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00127] Alternatively, compounds of formula (1) can be prepared according to
the following
Reaction Scheme 2 wherein Al, A2, A3, Yl, Y2, and Y3 are as described above
(e.g., in the
Overview and in Thiazolidinone Compounds and Derivatives), and W is halo:
REACTION SCHEME 2
Al y3 Al
' 0 Knoevenagel Y1
1. HN + condensation ~-'A3
HN Y2
A2 Y2 1 A Y3
2
(e) (f) (g)
Al Y
~ Ullmann X2\ Di
X3 ~A3 I 2. (g) + X1 condensation X ~N J 2
W 1 Y3
A2
(h) (1)
[00128] In general, the compounds of formula (1) can be prepared by first
treating a
compound of formula (e) with a compound of formula (f) under standard
Knoevenagel
condensation conditions, such as under reflux in the presence of catalytic
amount of
piperidine in glacial acetic acid, an alcohol or another appropriate solvent.
The compound of
formula (g) is then isolated from the reaction mixture by standard isolation
and purification
techniques. The compound of formula (g) is then treated with a compound of
formula (h)
under standard Ullmann condensation conditions, such as in the presence of Cu
or Cu20 or
CuO at elevated temperatures, to yield the desired product of formula (1).
CA 02500498 2011-01-27
WO 2004/028480 PCT/US2003/031005
[00129] Alternatively, compounds of formula (1) can be prepared according to
the following
Reaction Scheme 3 wherein A1, A2, A3, Y1, Y2, and Y3 are as described above
(e.g., in the
Overview and in Thiazolidinone Compounds and Derivatives) and W is halo.
REACTION SCHEME 3
Al X A,
Ullmann
1. ~-'A3 II condensation x2 N 3
HN + X, X W
A2 3 x3 A2
(e) (h) (c)
Y3 Y
Knoevenagel X 3 Y
2. (c) + condensation z z
z yJ i 3 Az Y3
(d) (1) ,
[00130] In this reaction scheme, the first step is the Ullmann condensation
between the
compound of formula (e) and the compound of formula (h) to produce the
compound of
formula (c), which then undergoes Knoevenagel condensation with a compound of
formula
(d) to yield the desired product of formula (1).
[00131] The starting compound of formula (e) can be purchased from different
chemical
suppliers or synthesized according to methods known to one skilled in the art,
or by the
methods similar to those disclosed in F. C. Brown et. al., J. Am. Chem. Soc.,
78, 384-388
(1956); R. E. Strube, Organic Synthesis, CV 4, 6; K. S. Markley and E. E.
Reid, J. Am.
Chem. Soc., 52, 2137-2141.
[00132] In a similar manner as described above, synthesis of 3-[(3-
trifluoromethyl)phenyl]-5-
[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (referred to herein as
CFTR;,,h-172)
(see Fig. 1 C) and analogs with different positions of the trifluoromethyl and
carboxy
substituents (see, e.g., Fig. 1D) was accomplished by Knoevenagel condensation
of 2-
thioxo-3-[a-trifluoromethyl-4-phenyl]-4-thiazolidinone (a=2, 3 or 4) with b-
carboxybenzaldehyde (b=2, 3 or 4) in the presence of piperidine. The
precipitate was
filtered, washed with ethanol, dried and recrystallized 2-3 times from ethanol
to give bright
yellow crystals (70-85% yields). Structures were confirmed by 'H-NMR. Purity
was > 99%
as judged by thin layer chromatography and HPLC.
31
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
EXAMPLES
[00133] The following examples are put forth so as to provide those of
ordinary skill in the
art with a complete disclosure and description of how to make and use the
present invention,
and are not intended to limit the scope of what the inventors regard as their
invention nor are
they intended to represent that the experiments below are all or the only
experiments
performed. Efforts have been made to ensure accuracy with respect to numbers
used (e.g.
amounts, temperature, etc.) but some experimental errors and deviations should
be accounted
for. Unless indicated otherwise, parts are parts by weight, molecular weight
is weight
average molecular weight, temperature is in degrees Centigrade, and pressure
is at or near
atmospheric.
[00134] The synthesis of compounds of the invention are exemplified with but
not limited to
the following examples.
Synthetic Example
Synthesis of 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-
thioxo-4-thiazolidinone
[00135] A. To a stirred solution of 3-trifluromethylanilne (1.6 g, 10 mmol)
and triethylamine
(1 g, 10 mmol) in ethyl acetate (10 mL) was added dropwise carbon disulfide
(0.8 g, 10
mmol) during a 30-minute period. A mild exothermic reaction, which began when
the
addition was about half complete, was easily controlled by intermittent use of
ice bath. After
stirring overnight, the thick yellow slurry was filtered and the precipitate
was washed with
50 mL of diethyl ether and air-dried to give 3 g (89 %) of a pale yellow
dithiocarbamate
solid, m.p. 92-95 C (dec.).
[00136] B. Sodium chloroacetate (prepared from chloroacetic acid (0.064 g,
0.46 mmol) in
0.6 mL of NaHCO3 solution, pH 8-9) was stirred and cooled to 5-10 C and the
dithiocarbamate (0.3 g, 0.9 mmol) was added over a period of ten minutes.
Stirring was
continued while the flask was allowed to warm to ambient temperature. After 2
hours of
stirring, the solution was cooled to 10 C and acidified with concentrated
hydrochloric acid
and the reaction mixture was heated to 90-95 C for 30 minutes. The resulting
precipitate was
filtered, washed with water and recrystallised from ethanol to give 0.103 g of
2-thioxo-3 -(3
trifluoromethylphenyl)-4-thiazolidinone, as shiny crystals in 83% yield, m.p.
177-178 C, 1H
NMR (300 MHz, CDC13): 5 4.18 (s, 2H, CH2), 7.40 (d, 1H, phenyl, J= 8.0 Hz),
7.48 (s, 1H,
phenyl), 7.64 (t, I H, phenyl, J= 8.0 Hz), 7.72 (d, I H, phenyl, J= 7.6 Hz)
ppm.
32
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00137] C. A mixture of 2-thioxo-3-(3-trifluoromethylphenyl)-4-thiazolidinone
obtained
above (0.1 g, 0.36 mmol) and 4-carboxybenzaldehyde (0.054 g, 0.36 mmol) in
absolute
alcohol (1 mL) and piperidine (1 drop) was stirred at reflux for 30 minutes.
The yellow
precipitate was filtered, washed with ethanol, dried and recrystallised from
ethanol to yield
0.108 g (73%) of the title compound as yellow crystalline solid, m.p.:180-182
C, 1H NMR
(300 MHz, DMSO-d6): S 7.78 (d, 2H, carboxyphenyl, J= 8.2 Hz), 7.80-8.00 (m,
5H,
trifluoromethylphenyl and CH), 8.07 (d, 2H, carboxyphenyl; J= 8.31 Hz), 13.20
(s, 1H,
COOH, D20 exchangable) ppm.
[00138] D. In a similar manner as described above, the following compounds
were prepared:
3-[(3-trifluoromethyl)phenyl] -5-[(3-carboxy-4-hydroxyphenyl)methylene]-2-
thioxo-4-
thiazolidinone;
3-[(3-trifluoromethyl)phenyl] -5-[(3,4,5-trihydroxyphenyl)methylene] -2-thioxo-
4-
thiazolidinone;
3-[(3-trifluoromethyl)phenyl] -5-[(2,3,4-trihydroxyphenyl)methylene]-2-thioxo-
4-
thiazolidinone;
3 - [ (3 -trifluoromethyl-4-fluoro)phenyl] -5 - [ (3 -c arb oxy-4-
hydroxyphenyl)methyl ene] -2-
thioxo-4-thiazolidinone; and
3-[(4-fluoro-3-trifluoromethyl)phenyl] -5-[(4-carboxyphenyl)methylene] -2-
thioxo-4-
thiazolidinone.
[00139] The following materials and methods were used in the examples that
follow.
Cell lines, Mice and Compounds
[00140] Fischer rat thyroid (FRT) cells coexpressing human wildtype CFTR and
the halide
indicator YFP-H148Q were generated as described previously (Galietta et al.
2001 J. Biol.
Chem. 276:19723-19728). Cells were plated in 96-well black-walled microplates
(Coming
Costar) at a density of 20,000 cells per well in Coon's modified F12 medium
supplemented
with 5% fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 gg/mL
streptomycin. Assays were done at 48 hours after plating at which time cells
were just
confluent (40,000 cells per well).
[00141] Initial screening was done using a diverse collection of 50,000 drug-
like compounds
from ChemBridge (San Diego, CA) obtained as 10 mM stock solutions in DMSO and
diluted to 100 m1VI in 96-well microplates. Structure-activity analysis was
done on analogs
purchased from ChemBridge and ChemDiv (San Diego, CA).
33
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00142] Wildtype and cystic fibrosis (AF508 homozygous mutant) mice were bred
by the CF
Animal Core facility at University of California, San Francisco (UCSF). Animal
protocols
were approved by the UCSF Committee on Animal Research.
[00143] T84 and Caco-2 cells were obtained from the UCSF cell culture
facility. T84 cells
were cultured in a 1:1 mixture of DMEM and Hams F12 supplemented with 5% fetal
calf
serum, 100 U/mL penicillin, 100 g/mL streptomycin and plated on Snapwell
inserts
(Coming Costar) for growth in a humidified (5% 02 / 95% C02) atmosphere at 37
C. Cells
were used at 10-14 days after plating. Caco-2 cells were cultured in DMEM
containing 10
fetal calf serum, 1 % nonessential amino acids, 100 U/mL penicillin and 100
gg/mL
streptomycin, and cultured on Snapwell inserts. Cells were used at 21-24 days
after plating.
Wildtype mice in a CD1 genetic background were bred as described previously.
Male Wistar
rats (200-250 g) were purchased from Jackson Laboratories. Animal protocols
were
approved by the UCSF Committee on Animal Research. Fragments of human colon
were
obtained freshly at the time of excision surgery and transported in ice-cold
saline for use
within 1 hour after excision.
[00144] Forskolin, 8-bromo cGMP, amiloride, cholera toxin and STa toxin were
purchased
from Sigma Chemical Co. (St. Louis, MO). CFTRact-16 was from ChemBridge (San
Diego,
CA).
Screening Procedures
[00145] Assays were done using a customized screening system (Beckman)
consisting of a 3-
meter robotic arm, CO2 incubator, plate washer, liquid handling workstation,
bar code
reader, delidding station, and two FluoStar fluorescence plate readers (BMG
Labtechnologies, Offenburg, Germany), each equipped with two syringe pumps and
HQ500/20X (500 10 nm) excitation and HQ535/30M (535 d 15 nm) emission
filters
(Chroma). The robotic system was integrated using SAMI version 3.3 software
(Beckman)
modified for two plate readers. Custom software was written in VBA (Visual
Basic for
Applications) to compute baseline-subtracted, normalized fluorescence slopes
(giving halide
influx rates) from stored data files.
[00146] The assay was set-up by loading the incubator (37 C, 90% humidity, 5%
CO2) with
40-60 96-well plates containing the FRT cells, and loading a carousel with 96-
well plates
containing test compounds and disposable plastic pipette tips. To initiate the
assay, each well
of a 96-well plate was washed 3 times in PBS (300 gL/wash), leaving 50 gL PBS.
Ten L of
a CFTR-activating cocktail (5 gM forskolin, 100 M IBMX, 25 M apigenin in
PBS) was
added, and after 5 min one test compound (0.5 L of 1 mM DMSO solution) was
added to
34
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
each well to give 10 gM final concentration. After 10 min, 96-well plates were
transferred to
a plate reader for fluorescence assay. Each well was assayed individually for
CFTR-
mediated r transport by recording fluorescence continuously (200 ms per point)
for 2 s
(baseline) and then for 12 s after rapid (<0.5 s) addition of 160 L of
isosmolar PBS in
which 137 mM Cl" was replaced by I.
Assays of Intracellular [cAMP] And Toxicity
[00147] [cAMP] and phosphatase assays were performed as reported previously
(Galietta et
al. 2001 J. Biol. Chem. 276:19723-19728). Cell toxicity was assessed by the
dihydrorhodamine method at 24 hours after cell incubation with 0-1000 M
inhibitor.
Animal toxicity was assessed by measurement of serum chemistries and
hematology (UCSF
Clinical Laboratory) in mice at 5 days after daily intraperitoneal injections
with 0-100 gg/kg
inhibitor.
MDR -1 Activity
[00148] MDR-1 activity was evaluated by measuring 3H-vincristine accumulation
in an
immortalized human tracheal cell line, 9HTEo-/Dx, in which the endogenous
expression of
MDR-1 was upregulated by selection in increasing concentrations of doxorubicin
(Rasola et
al. 1994 J. Biol. Chem. 269:1432-1436). Cells were seeded in 24-well
microplates (200,000
cells/well). After 48 hours, cells were washed with a solution containing (in
mM): 130 NaCl,
2 KCI, 1 KH2PO4, 2 CaC12, 2 MgCl2, 10 Na-Hepes (pH 7.3) and 10 glucose, and
incubated
for 1 hour at 37 C with 200 L of the same solution containing 3H-vincristine
(0.7 gM; 1
tCi/mL). Cells were then washed three times with ice-cold solution and lysed
in 0.25 M
NaOH. Vincristine content was determined by scintillation counting.
Short-circuit Current Tests Using CFTR-Expressing FRT Cells
[00149] Snapwell inserts containing CFTR-expressing FRT cells or human
bronchial
epithelial cells were mounted in an Ussing chamber system. For FRT cells the
hemichambers were filled with 5 mL of 75 mM NaCl and 75 mM Na gluconate
(apical) and
150 mM NaCl (basolateral) (pH 7.3), and the basolateral membrane was
permeabilized with
250 jig/mL amphotericin B (Galietta et al. 2001 J. Biol. Chem. 276:19723-
19728). For
bronchial epithelial cells and T84 cells, both hemichambers contained a Krebs
bicarbonate
solution. Hemichambers were continuously bubbled with air (FRT cells) or 5%
CO2 in air
(bronchial and T84 cells) and maintained at 37 C. Short-circuit current was
recorded
continuously using a DVC-1000 voltage clamp (World Precision Instruments,
Sarasota,
Florida) using Ag/AgCI electrodes and 1 M KC1 agar bridges.
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Patch-Clamp Analysis of Cl- Channel Activity
[00150] Membrane current was measured in a whole-cell configuration. For
recordings of Cl-
channels, the extracellular (bath) solution contained (in mm): 150 NaCl, 1
CaC12, 1 MgCl2,
glucose, 10 mannitol, 10 TES (pH 7.4), and the intracellular (pipette)
solution contained:
120 CsCl, 1 MgC12, 10 TEA-Cl, 0.5 EGTA, 1 Mg-ATP, 10 Hepes (pH 7.3). CFTR was
activated by forskolin (5 M) in the extracellular solution. The time-course
of membrane
conductance was monitored in response to alternating voltage pulses of -1.00
and +80 mV.
At defined times the protocol was interrupted to generate current-voltage
relationships
(voltage pulses from -100 to +100 mV in 20 mV increments). Volume-sensitive Cl-
channels
were activated by a hypotonic solution (extracellular NaCl decreased to 120
NaCl; 250
mosM/kg). Calcium-sensitive Cl- channels were activated in human bronchial
epithelial cells
by addition of 100 gM UTP to the extracellular solution.
Patch-Clamp Analysis of ATP-Sensitive It Channels
[00151] Membrane potential was recorded in the pancreatic (3 cell line INS-1
in which the
extracellular (bath) solution contained (in mM): 130 NaCl, 2 KCI, 1 KH2PO4, 2
CaCl2, 2
MgCl2, 10 Na-Hepes (pH 7.3) and 10 glucose. The pipette contained (in mM): 140
KCI, 1
CaC12, 2 mM MgC12, 10 EGTA, 0.5 MgATP, 10 K-Hepes (pH 7.3). After achieving
the
whole-cell configuration, the amplifier was switched to current-clamp mode.
Intestinal Fluid Secretion And Short-circuit Current
[00152] In the first of 3 assays, fluid accumulation in ileal loops was
measured (Oi et al. 2002
Proc. Natl. Acad. Sci. USA 99:3042-3046; Gorbach et al. 1971 J. Clin. Invest.
50:881-889).
Mice (age 8-10 weeks, body weight 25-35 g) in a CD1 genetic background (or
AF508
homozygous mice) were starved for 24 hrs and anaesthetized with
intraperitoneal ketamine
(40 mg/kg) and xylazine (8 mg/kg). Body temperature was maintained during
surgery at 36-
38 C using a heating pad. A small abdominal incision was made to expose the
small
intestine and closed ileal loops (length 20-30 mm) proximal to the cecum were
isolated by
sutures. Loops were injected with 100 L of PBS alone or PBS containing
cholera toxin (1
g). In some experiments the inhibitor (150 g/kg) was administered by
intraperitoneal
injection. The abdominal incision was closed with suture and mice were allowed
to recover
from anesthesia. At 6 hours the mice were anesthestized, intestinal loops were
exteriorized,
and loop length and weight were measured after removal of mesentery and
connective tissue.
[00153] In the sealed adult mouse model of secretory diarrhea mice were
gavaged with
cholera toxin (10 g) in 0.1 mL of 7% bicarbonate buffer (or buffer alone)
using a orogastric
feeding needle (Richardson et al. 1986 Infect. Iminun. 54:522-528; Gabriel et
al. 1999 Ana J.
36
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Physiol. 276:G58-G63). Four experimental groups were: control (buffer alone),
cholera-
treated, cholera-treated + inhibitor (150 g/kg intraperitoneal 2 min before
gavage), and
inhibitor alone. After six hours mice were euthanized and the small intestine
(from pylorus
to cecum) was exteriorized and stripped of associated mesenteric and
connective tissues. The
intestine was weighed, then opened longitudinally to remove lumenal fluid (by
blotting), and
weighed again. Fluid accumulation was computed from the ratio in intestinal
weight before
and after lumenal fluid removal. For measurement of short-circuit current,
strips of rat colon
were isolated, stripped of muscle layers by blunt dissection, mounted in
Ussing chambers
(area 0.7 cm), and bathed in oxygenated bicarbonate Ringers solution
containing 10 M
indomethacin. Short-circuit current was measured after inhibition of Na}
current by
amiloride (10 M), followed by stimulation by forskolin (20 M) and subsequent
inhibitor
addition.
Synthesis of 14C-labeled CFTRIõh-172 (Fig. 6)
[00154] The intermediate 2-thioxo-3-(3-trifluoromethylphenyl)-4-thiazolidinone
was
synthesized by dropwise addition of carbon disulfide (0.8 g, 10 mM) to a
stirred solution of
3-trifluromethylaniline (1.6 g, 10 mM) and triethylamine (1 g, 10 mM) in ethyl
acetate (10
mL) over 30 minutes. An ice bath was used to prevent excessive heating during
reaction.
After stirring overnight, the thick yellow slurry was filtered and the
precipitate was washed
with 50 mL of ether and air dried to give 3 g (89 % yield) of a pale yellow
dithiocarbamate
solid (melting point 92-95 C). Na Br-14C-acetate (prepared from Br-14C-acetic
acid
(Amersham), 55 mCi/mmol, 64 mg, 0.46 inM in 0.6 mL of water, pH 8-9 using
NaHCO3)
was stirred and cooled to 5-10 C and dithiocarbamate (0.3 g, 0.9 mM) was added
over 10
minutes. Stirring was continued while the flask was allowed to warm to ambient
temperature. After 2 hours, the solution was cooled to 10 C, acidified with
concentrated
HCI, and heated to 90-95 C for 30 minutes. The resultant precipitate was
filtered, washed
with water and recrystallized from ethanol to give 103 mg of the desired
product as shiny
crystals (83% yield), m.p. 177-178 C; specific activity (14C) 55 mCi/mmol; 1H
NMR (300
MHz, CDC13): 6 4.18 (s, 2H, CH2), 7.40 (d, 1H, phenyl, J= 8.0 Hz), 7.48 (s,
1H, phenyl),
7.64 (t, 1H, phenyl, J= 8.0 Hz), 7.72 (d, 1H, phenyl, J= 7.6 Hz) ppm.
[00155] For synthesis of 2-thioxo-3-(3-trifluoromethylphenyl)-5-[4-
carboxyphenylmethylene]-4- thiazolidinone (14C-5) (14C-CFTRiiih-172), a
mixture of 2-
thioxo-3-(3-trifluoromethylphenyl)-4- thiazolidinone (14C-5) (100 mg, 0.36 mM)
and 4-
carboxybenzaldehyde (54 mg, 0.36 mM) in absolute alcohol (1 mL) and piperidine
(1 drop)
was refluxed for 30 minutes. The yellow precipitate was filtered, washed with
ethanol, dried
37
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
and recrystallized from ethanol to give 108 mg (73% yield) yellow crystals,
m.p. 180-182 C;
specific activity (14C) 54 mCi/mmol; 1H NMR (300 MHz, DMSO-d6): 6 7.78 (d, 2H,
carboxyphenyl, J= 8.2 Hz), 7.80-8.00 (m, 5H, trifluoromethylphenyl and CH),
8.07 (d, 2H,
carboxyphenyl, J= 8.31 Hz), 13.20 (s, 1H, COOH, D20 exchange) ppm.
Purification to
>99.9% was accomplished by repeated recrystallization.
Pharmacokinetic studies
[001561 A bolus of 14C-CFTR;,,h-172 (50 Ci) in PBS containing 3% DMSO
(titrated to pH
7.4 using NaOH) was administered intravenously in rats over 1 min (male
Sprague-Dawley
rats, 360-420 grams) by an indwelling jugular catheter. Blood was collected
from the
catheter at specified times. 14C-Radioactivity was determined in plasma
(isolated by
centrifugation of whole blood at 14,000 g for 10 min) by scintillation
counting (Scintiverse
SE, Fisher, CA) using a LS-6500 Multi-Purpose Scintillation Counter (Beckman).
Pharmacokinetic analysis was done using WinNonLin software (Pharsight). Rats
were
sacrificed by, pentabarbital overdose after collection of the final
blood/tissue samples. All
animal procedures were approved by the UCSF Committee on Animal Research.
Tissue distribution and elimination studies
[001571 A bolus of 14C-CFTRiõh-172 (2 Ci) was administered intravenously over
1 min in
mice (male CD1 mice, 30-35 grams) by tail vein. Mice were sacrificed at 5, 30,
120 and 240
min. Organs were removed, weighed and homogenized in distilled water (10-50
vol %).
Radioactivity was determined by scintillation counting of the homogenates (25-
50 L) and
expressed as total 14C-radioactivity per organ (or per gram tissue for
skeletal muscle). At the
same time blood, urine and bile (from gallbladder or duodenum) were collected
and 14C-
radioactivity was measured and expressed per mL of fluid. Elimination studies
were done by
collections of urine and stool over the first 24 hr after 14C-CFTR;,,h-172
administration.
Tissue distribution studies were also done on rats prepared as for
pharmacokinetic studies.
Analysis of inhibitor metabolism
[001581 Aliquots of bodily fluids (plasma, urine, bile) and liver homogenate
were spotted
onto Silica plates and resolved by thin layer chromatography using a ethyl
acetate : hexane :
methanol (1:1:0.1) solvent system which gave rf - 0.5 for the original
inhibitor.
Autoradiography was performed using Hyperfilm (Amersham) with a Transcreen LE
amplification system (Kodak). 14C-labeled CFTR;,,h-172 standards were included
on all
plates.
38
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Short-circuit current measurements (Examples 7)
[00159] For cell studies, Snapwell inserts containing T84 cell monolayers were
mounted in an
Ussing chamber system (Navicyte, Harvard Apparatus, Holliston, MA).
Hemichambers were
filled with Krebs-bicarbonate solution containing (in mM) NaCl 120, NaHCO3 25,
KH2PO4
3.3, K2HP04 0.8, MgC12 1.2, CaCl2 1.2, glucose 10 (maintained at 37 C) and
continuously
bubbled with 5% CO2 / 95% 02. High K+ buffer contained (in mM) NaCl 65,
KC167.5,
KH2PO4 1.5, CaC12 1, MgCl2 0.5, HEPES 10, glucose 10. Low Cl- buffer contained
(in mM)
Na-gluconate 120, KH2PO4 3.3, K2HP04 0.8, MgC12 1.2, CaC12 1.2, HEPES 10,
glucose 10
(maintained at 37 C) and continuously bubbled with air. For measurements in
mouse colon,
mice were anaesthetized with intraperinoneal ketamine (40 mg/kg) and xylazine
(8 mg/kg).
The ileum was removed, washed with ice-cold Krebs buffer, opened along the
mesenteric
border, and mounted in a micro-Ussing chamber (area 0:7 cm2, World Precision
Instruments,
Sarasota, FL). For measurements in human intestine, colonic fragments were
stripped of
muscle layers by blunt dissection and mounted as described above. Hemichambers
were
filled with oxygenated Ringersbicarbonate solution containing 10 gM
indomethacin. Short-
circuit current was recorded using a DVC- 1000 voltage-clamp (World Precision
Instruments) with Ag/AgCI electrodes and 1 M KCI agar bridges.
Agonists/inhibitors were
added to hemichambers as described below.
In vivo intestinal fluid secretion in mouse and rat models (Examples 5 and 7).
[00160] Mice (age 8-10 weeks, body weight 25- 35 g) in a CD1 genetic
background were
given access to water but not food for 24 hr. Mice were anaesthetized as
described above and
body temperature was maintained during surgery at 36-38 C using a heating pad.
A small
abdominal incision was made to expose the small intestine and closed ileal
loops. (length 20-
30 mm) proximal to the cecum were isolated by sutures. Loops were injected
with 100 L of
PBS alone or PBS containing cholera toxin (1 g). In some experiments CFTR;,,h-
172 (0-200
g) was administered by intraperitoneal injection at specified times before or
after cholera
toxin injection. The abdominal incision was closed with suture and mice were
allowed to
recover from anesthesia. At 6 hours the mice were anesthetized, intestinal
loops were
exteriorized, and loop length and weight were measured after removal of
mesentery and
connective tissue.
[00161] For measurement of enterotoxin-induced fluid secretion in a rat closed-
loop model,
male Wistar rats (body weight 200-250 g) were anesthetized with pentobarbital
sodium (45
mg/kg). Loops (40-60 mm) were isolated and injected with 300 L PBS alone or
PBS
containing cholera toxin (10 g) or STa toxin (0.1 g). In some experiments
CFTRiõ h-172
39
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
(200 g) was given by intraperitoneal injection after cholera toxin or STa
toxin
administration. Loop length and weight were measured at 3 hr (STa) or 6 hr
(cholera toxin).
[00162] In studies of orally administered CFTR;,,h-172, an open-loop mouse
model was used
in which mice were gavaged with 7% bicarbonate buffer or cholera toxin (1 jig
in 7%
bicarbonate buffer) alone and with CFTR;,,h-172 (200 g in vitamin E TPGS, see
below)
using an orogastric feeding needle. After 6 hours the small intestine (from
pylorus to cecum)
was exteriorized and stripped of associated mesenteric and connective tissue.
The intestine
was weighed, opened longitudinally to remove lumenal fluid, and reweighed to
quantify
fluid accumulation.
Caco-2 permeability assay.
[00163] Caco-2 cells were cultured on porous inserts to give monolayer
resistances of 400-
600 SZcm 1. For transport studies culture medium was replaced with an equal
volume of
Hank's buffered salt solution (HBSS) containing 15 mM glucose and 25 mM HEPES
(pH
7.3). After 1 hr CFTR;,,h-172 (25 M) was added to the upper chamber and
plates were
gently rocked at 37 C. At specified times 50 L of solution from the lower
(receiving)
chamber were removed for measurement of CFTR;,,h-172 concentration by UV
absorbance
(385 nm). Apparent permeability (Papp) was calculated from: Papp = dC/dT X
(Vr/ACO),
where dCldT is the rate of increase in CFTR;,,h-172 concentration in the
receiver chamber,
Vr is the volume of the receiver chamber, A is monolayer surface area, and CO
is initial
CFTR;,,h-172 concentration in the donor chamber.
Pharmacokinetic and oral bioavailability studies.
[00164] Mice were anesthetized briefly using halothane and gavaged orally with
14C-labeled
CFTR;,,h-172 (12 Ci) solubilized with vitamin E TPGS (d-a-tocopheryl
polyethylene glycol
1000 succinate, 0.5% w/v) CFTR;h-172 in 10% w/v suspension of TPGS in water).
For
comparison other mice were given 14C-CFTR;,,h-172 (2 Ci) intravenously by
tail vein
infusion. Blood was collected from the tail vein at specified times for
measurement of
plasma 14C radioactivity. At 6 hours mice were killed by pentobarbital
overdose and organs
were removed for measurement of radioactivity in homogenates.
Biological Example 1
Screening of CFTR Inhibitors
[00165] The primary screening technique used to identify the compounds of the
invention
was designed to identify inhibitors of CFTR CY conductance by direct CFTR-
inhibitor
interaction. CFTR was pre-stimulated in CFTR-expressing FRT cells by an
activating
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
cocktail containing forskolin, IBMX and apigenin, as shown schematically in
FIG. IA. The
activation of CFTR by multiple mechanisms (cAMP elevation, phosphodiesterase
inhibition,
and direct CFTR binding) allowed identification of inhibitors that blocked the
CFTR Cl-
transporting pathway directly rather than more proximal step(s) in a signaling
pathway. The
FRT cells co-expressed a yellow fluorescent protein-based Cl- / I- sensor that
provided a
quantitative fluorescence read-out of inhibition potency (See, e.g., Jayaraman
et al., 2000, J.
Biol. Chem. 275:6047-6050; Galietta et al., 2001, Am. J. Physiol. 281:C1734-
C1742.). After
CFTR pre-stimulation and compound addition, cells were subjected to an
inwardly-directed
I- gradient to drive I" influx and produce decreasing fluorescence. Each assay
consisted of
recording baseline fluorescence for 2 seconds, followed by 12 seconds of
continuous
recording of fluorescence after rapid addition of the I" containing solution.
Compounds were
tested separately at 10 M concentration in a 96-well format. utilizing a
fully-automated
high-throughput screening apparatus (see Example 2 below).
[00166] FIG. 1B graphically illustrates representative curves, as relative YFP
fluorescence
versus time, from the primary screen of 50,000 compounds using the assay of
FIG. 1A. As
quantified from the slope of the decreasing fluorescence after I" addition,
49,993 compounds
had no significant effect on the kinetics of I- influx (<10% decrease in
slope). Seven
compounds produced a small decrease in negative slope (10-52 %), nearly all of
which had a
similar core structure consisting of a 2-thioxo-4-thiazolidinone heterocycle
with substituted
phenylmethylene and phenyl moieties (Fig. 1 Q. More than 250 analogs having
thiazolidinone core structure were subsequently screened to identify the most
potent CFTR
inhibitors.
[00167] - FIG. 1D shows the most effective thiazolidinone CFTR inhibitors
identified in the
screening were 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-
thioxo-4-
thiazolidinone (referred to herein as CFTR;nh-172), along with five analogs
having
significant inhibitory potencies. Thus the following compounds were identified
as CFTR
inhibitors: 3 -[(3-trifluoromethyl)phenyl] -5-[(4-carboxyphenyl)methylene] -2-
thioxo-4-
thiazolidinone (CFTR;nh-172); 3-[(3-trifluoromethyl)phenyl]-5-[(4-
nitrophenyl)methylene]-
2-thioxo-4-thiazolidinone(CFTR;nh-020); 3-[(3-trifluoromethyl)phenyl]-5-[(4-
oxycarboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (CFTR;nh-029); 3-[(3-
trifluoromethyl)phenyl] -5-[(3,4-dihydroxyphenyl)methylene] -2-thioxo-4-
thiazolidinone
(CFTR;,,h-185), 3-[(3-trifluoromethyl)phenyl]-5-[(3,5-dibromo-4-
hydroxyphenyl)methylene]-2-thioxo-4-thiazolidinone (CFTR;nh-214) and 3-[(3-
trifluoromethyl)phenyl] -5 - [(3 -bromo-4-hydroxy- 5-nitrophenyl)methyl ene] -
2-thioxo-4-
41
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
thiazolidinone (CFTR;,,h-236). The most effective CFTR inhibitors included one
or more
electron-withdrawing groups, such as a 3-trifluoromethyl group, on ring 1, and
electron-
withdrawing group or polar substituents on ring 2 as discussed above. CFTR;h-
172 was
selected for further analysis. The relative potencies were: 0.2 (CFTR;fh-020),
0.3 (CFTR;,,h-
029), 1.0 (CFTR;,,h-172), 0.2 (CFTR;,,h-185), 0.1 (CFTR,h-214), and 0.1
(CFTR;,,h-236).
[00168] To examine the effect of ring position of the trifluoromethyl and
carboxyl
substituents, 8 analogs of CFTR;,,h-172 were synthesized in which the
substituents were
moved to each unique position on rings 1 (trifluoromethyl) and 2 (carboxy).
Compared to
CFTR;,,h-172 (potency 1.0), the relative inhibitory potencies of the 3-[(a-
trifluoromethyl)phenyl]-5-[(b-carboxyphenyl) methylene]-2-thioxo-4-
thiazolidinone analogs
were: 0.69 (a=2, b=2), 0.70 (2, 3), 0.66 (2, 4), 0.74 (3, 2), 0.90 (3, 3),
0.67 (4, 2), 0.64 (4, 3)
and 0.56 (4, 4).
Biological Example 2
Characterization of CFTR;1h-172
[00169] The level of CFTR inhibition for specific dosages of the subject
thiazolidinone
compounds was determined using the fluorescence assay shown in FIG. IA and
described
above. FIG. 2A shows dose-inhibition data for CFTR;fh-172 as relative YFP
fluorescence
versus time. Significant CFTR inhibition was seen at 0.3-0.6 M concentrations
of this
thiazolidinone compound. FIG. 2B shows that inhibition by CFTRIõ h-172 (shown
graphically
as relative transport rate versus time after addition or washout) was complete
in -10 min (t112
4 min) and was reversed after washout with t1/2 -5 min (inset). The relative
transport rates
illustrated in FIG. 2C show that CFTRiõ h-172 effectively inhibited CFTR
activation by
multiple types of agonists that were not included in the activating cocktail
used for initial
screening. These agonists included genistein, CPT-cAMP, CPX, 8-MPO and the
potent
benzoflavone CFTR activator UCCF-029 (2-(4-pyridinium)benzo[h]4H-chromen-4-one
bisulfate) and the benzimidazolone CFTR activator UCCF-853 (see Galietta, et
al., 2001, J.
Biol. Chem. 276:19723-19728).
[00170] Electrophysiology experiments were also carried out to establish the
inhibitory
potency and specificity of CFTR;,,h-172. Fig. 3A shows the rapid, dose-
dependent inhibition
of short-circuit current in CFTR-expressing FRT cells with CFTR;,,h-172 added
to the
solution bathing the apical cell surface. Fig. 3B shows the average dose-
inhibition
relationships of CFTR;,,h-172 (Kd - 300 nM, Hill coefficient - 1) and
glibenclamide (Kd -
200 M) tested under identical conditions.
42
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
[00171] Similar inhibitory potencies for this thiazolidinone were found in
cells that natively
express wildtype CFTR, including T84 cells and primary cultures of human
bronchial
epithelial cells, as well as in transfected FRT cells expressing G551D-CFTR
and AF508-
CFTR (after low temperature correction). For studies in bronchial cells, the
Na+ channel was
blocked with amiloride so that baseline current is largely CFTR-dependent.
After maximal
CFTR activation by a CPT-cAMP, application of CFTR;,,h-172 from the apical
side inhibited
short-circuit current strongly (Fig. 3C, left). CFTRõh-172 also inhibited
short-circuit current
when added from the basolateral side (Fig. 3C, right).
[00172] Whole-cell membrane currents were measured in CFTR-expressing FRT
cells as
shown in FIG. 3D. Stimulation by 5 gM forskolin produced a membrane current of
381 47
pA/pF (n=4) at +100 mV (total membrane capacitance 21 3 pF). The current-
voltage
relationship was linear as expected for a pure CFTR current (Fig. 3F).
Extracellular
perfusion with 2 gM CFTR;,,h-172 produced a rapid reduction in current at all
membrane
potentials, suggesting voltage-independent CFTR inhibition. The lack of
voltage-dependence
of channel block was confirmed using a lower concentration of CFTR;,,h-172
(0.2 M) to
obtain -50 % inhibition (Fig., 3F).
[00173] The specificity of CFTR;,,h-172 for inhibition of CFTR was also
examined. Two non-
CFTR Cl" channels were studied. CFTR;,,h-172 at 5 gM concentration did not
inhibit Caa+
activated Cl- secretion produced by addition of UTP (100 M) to the apical
bathing solution
in polarized human bronchial epithelial cells (Fig. 4A). Maximal UTP-dependent
short-
circuit currents were 9.9 0.5 jA/cm2 and 10.0 0.2 A/cm2 in the absence and
presence of
CFTR;,,h-172, respectively (SE, n=4). CFTR;h-172 at 5 M also did not block
volume-
activated Cl" currents elicited in FRT cells by extracellular perfusion with a
250 mosM/kg
hypotonic solution (Fig. 4B).
[00174] The activity of a CFTR homolog, the ATP-binding cassette transporter
MDR-1
(multi-drug resistance protein-1), was measured in 9HTEo-/Dx which overexpress
MDR-1
(Rasola et al. 1994 J. Biol. Chenz. 269:1432-1436). Vincristine accumulation,
which is
inversely related to active drug extrusion by MDR-1, was strongly increased by
the MDR-1
inhibitor verapamil (100 M) (Fig. 4C). CFTR;,,h-172 (5 M) did not affect
vincristine
accumulation and thus did not inhibit MDR-1.
[00175] Another homolog of CFTR is the sulphonylurea receptor (SUR) which
regulates the
activity of ATP-sensitive K+ channels (K-ATP channel) (Aguilar-Bryan and Bryan
1999
Endocr. Rev. 20:101-135). SUR1 is expressed in pancreatic P-cells where it
controls
membrane potential and insulin release. Sulphonylureas, like glibenclamide,
cause insulin
43
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
release (and a hypoglycemic response) by blocking K-ATP channels and membrane
depolarization. To determine whether CFTRinh-172 also blocks K-ATP channels,
membrane
potential in a rat pancreatic (3 cell line, INS-1, was measured (Fig. 4D, Fig.
4E). In contrast
to large membrane depolarization caused by glibenclamide, CFTRinh-172 (2 and 5
M) did
not depolarize membrane potential. CFTRinh-172 at 5 M caused a small
hyperpolarization
that was much less than that caused by the K-ATP channel activator diazoxide
(100 M).
Additional studies indicated that CFTRih-172 at 5 M did not block a water
channel
(AQP1), urea transporter (UT-B), Na}/H} exchanger (NHE3) and Cl-/HC03"
exchanger
(AE1).
[00176] Further analysis showed that 5 gM CFTRinh-172 did not affect cellular
cAMP
production or phosphatase activity. In FRT cells, basal cAMP content was 225
22
finol/well, which increased at 30 min after stimulation by 20 M forskolin to
1290 190
finol/well (no inhibitor) and 1140 50 (+CFTRinh-172) (n=3). As judged using
the
dihydrorhodamine assay, CFTRinh-172 was non-toxic to FRT cells after 24 hours
at
concentrations up to 100 M. In mice, intraperitoneal injection of 1000 g/kg
CFTRinh-172
daily for 7 days did not cause overt toxicity. Food and water intake were not
diminished,
and serum electrolyte concentrations, glucose, liver function indices, serum
creatinine,
amylase and hematocrit were not changed. In addition, a single very large
systemic dose of
CFTRinh-172 (10 mg/kg) did not cause overt toxicity.
Biological Example 3
In Vivo Efficacy
[00177] The efficacy of CFTRinh-172 was tested-in vivo using two assays of
cholera toxin-
induced intestinal fluid secretion, and in isolated intestine by short-circuit
analysis. In the
first assay, a series of closed loops of small intestine were created in vivo
and the lumens of
alternate loops were injected with small volumes of saline or saline
containing cholera toxin.
Luminal fluid accumulation was determined after 6 hours. As seen visually in
FIG. 5A, there
was marked fluid accumulation and distention in cholera toxin-treated loops,
whereas
adjacent control (saline) loops remained empty. A single administration of
CFTRinh-172 (150
gg/kg intraperitoneal) prior to cholera toxin infusion effectively prevented
fluid
accumulation in the toxin-treated intestinal loops.
[00178] Data from a series of these experiments is summarized graphically in
FIG 5B.
CFTRinh-172 significantly reduced fluid secretion to that in saline control
loops where an
inactive thiazolidinone analog did not inhibit fluid secretion. As suggested
from previous
44
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
data (Gabriel et al. 1994 Science 266:107-109), cholera toxin-treated loops of
intestine from
homozygous AF508-CFTR mice also remained empty, indicating the involvement of
CFTR
in intestinal fluid secretion. In the second assay, intestinal fluid secretion
was induced by
oral administration of cholera toxin (10 g) and CFTRiõ h-172 was administered
systemically.
After six hours there was marked accumulation of fluid as measured by weighing
the entire
small intestine. CFTR1,h-172 administration remarkably reduced intestinal
fluid
accumulation as seen visually and quantified by the ratio of intestinal weight
before vs. after
luminal fluid removal (FIG. 5C).
[00179] Fig. 5D shows CFTRIh-172 inhibition of short-circuit current across
intact rat
colonic mucosa. After inhibition of Na+ current by amiloride, forskolin
produced a prompt
increase in short-circuit current. CFTR;,,h-172 added to the mucosal solution
inhibited short-
circuit current with greater efficacy than when added to the serosal solution,
which may be
related to impaired access to colonic epithelial cells through the residual
submucosal tissue.
,Addition of 5 M CFTR;,,h-172 to the mucosal solution alone reduced short-
circuit current
by > 80%. These results provide electrophysiological evidence for CFTR Cl-
channel
inhibition by CFTR;,,h-172 in intestine.
Biological Example 4
Pharmacokinetic analysis
[00180] Pharmacokinetic analysis in rats was done by serial measurements of
serum 14C
radioactivity after a single intravenous bolus infusion of 14C-labeled
CFTR;,,h-172. The total
amount of inhibitor infused (400 g, -1 mg/kg) was effective as an
antidiarrheal in rats. Fig.
7 shows that the kinetics of serum 14C radioactivity fitted well to a 2-
compartment model
with distribution volume 1.2 L and AUC (area under curve) of 3.8 g-hr/mL. The
half-lives
were 0.14 hr (redistribution) and 10.3 hr (elimination). No 14C-labeled
CFTR;,,h-172 was
detected in plasma at 72 hr after administration or in liver or kidney
homogenates at 14 days
after administration.
[00181] The tissue distribution of 14C-labeled CFTR;,,h-172 was determined
from the
radioactivity of organ homogenates and bodily fluids following a single
intravenous bolus
infusion. Fig. 8, panel A summarizes 14C distributions in the major organs at
indicated times
after CFTR;,,h-172 infusion in mice. 14C radioactivity was observed within 5
min primarily in
liver and kidney, decreasing over time. Little radioactivity was found in
brain, heart, skeletal
muscle or testes. At later times (30-240 min) 14C radioactivity accumulated in
the intestine.
Fig. 3, panel B shows a similar organ distribution of 14C radioactivity in
rats measured at 60
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
min after intravenous bolus infusion, with little radioactivity in brain,
heart and skeletal
muscle. In some experiments, rats were sacrificed at 10 days after infusion of
14C-labeled
CFTR1nh-172 (50 Ci).
[00182] To determine the mechanism of CFTR1nh-172 accumulation in kidney,
liver and
intestine, 14C radioactivity was measured in serum, urine and bile. Average
urine
radioactivity was 4.2 1.2 x 105 cpm/mL in mice over the first 2 hours after
infusion. The
ratios of 14C radioactivity in urine-to-blood were in the range 5-7: 1,
comparable to the ratio
of urine-to-serum osmolalities of -5:1 (1550 mOsm vs. 310 mOsm), suggesting
that
CFTR1nh-172 is cleared by the kidney by glomerular filtration without renal
tubular
absorption or secretion. A renal clearance mechanism for CFTR1nh-172 clearance
was
supported by the approximately parallel kinetics of decreasing 14C
radioactivity in serum,
urine and kidney tissue (data not shown). The possibility of CFTR1nh-172
accumulation in
bile was investigated based on the observation of prompt accumulation of 14C-
radioactivity
in liver and late accumulation in intestine. 14C radioactivity was -9-fold
concentrated in bile
vs. blood at 60 min after administration in mice. To determine whether the
biliary CFTR1nh-
172 was excreted in the stool or returned to the circulation, urine and stool
collections were
done on mice over the first 24 hr after radiolabeled inhibitor infusion. 93
3 % of excreted
radioactivity was found in the urine, supporting a primary renal excretion
mechanism with
enterohepatic circulation.
[00183] To determine whether the 14C radioactivity measured in organs and
fluids
corresponds to intact or chemically-modified CFTRin11-172, thin layer
chromatography and
autoradiography were done on specimens of urine, serum and bile, as well as
supernatants of
liver homogenates prepared by centrifugation. Fig. 9 shows a single spot at rf
-0.5 for the
original CFTR1nh-172 introduced in the bolus infusion. Autoradiography of
fluid and organ
homogenates showed single spots at identical rf, indicating that chemical
modification of
CFTR1nh-172 did not occur.
[00184] CFTR1nh-172 is a weak acid with a pKa of 5.5 as determined by
spectrophotometric
pH titration. At physiological pH -1 % of CFTR1h-172 is present as the
unionized acid
having low polarity and high membrane permeability. The rapid uptake of
CFTR1nh-172 in
cell models described above suggests the feasibility of orally bioavailable
preparations with
the caveat that protection from the low gastric pH may be needed to avoid
precipitation. The
results from these pharmokinetic studies indicate that CFTR1nh-172 is slowly
eliminated in
rodents by renal clearance without chemical modification, and that CFTR1nh-172
is
concentrated in bile and accumulated in intestine. CFTR1nh-172 did not
significantly cross the
46
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
blood-brain barrier and little CFTRiõh-172 accumulation was found in other
vital organs
including heart, lung, skeletal muscle and testes. The slow renal clearance,
intestinal
accumulation, and little blood-brain barrier penetration of CFTRiõ h-172 are
favorable for
antidiarrheal applications.
Biological Example 5
Dose-Response and Duration of Inhibitory Effect of CFTR;fh-172'
[00185] The purpose of this example was to extend the observations above
relating to the
ability of a single intraperitoneal injection of CFTRiõ h-172 to inhibit
cholera toxin-stimulated
fluid secretion in a closed intestinal loop model in mice. Specifically, the
goal of this
example was to measure the dose-response relation and the apparent halftime
for persistence
of the CFTRiõ h-172 inhibitory effect.
[00186] First, the kinetics of intestinal loop fluid absorption and secretion
were determined to
characterize the mouse model. To study absorption, loop fluid content was
measured at
specified times after injection of 200 gL of PBS into individual loops. Fig.
10, panel A
shows rapid fluid absorption with 50% fluid remaining at -25 min.
Intraperitoneal
administration of CFTR;,,h-172 at a dose that strongly inhibited cholera toxin-
induced
intestinal fluid secretion (20 g) did not alter the rate of fluid absorption
(measured at 30
min) compared to controls (Fig 10, panel A, inset). To study secretion,
intestinal loops were
injected with cholera toxin (1 gg in 0.1 mL PBS). Fig. 10, panel B shows a
slow onset of
fluid secretion over 6 hr, in agreement with previous studies in rodent models
(Gorbach et al.
J. Clin. Invest. 1971 50-881-889; Oi et al. Proc. Natl. Acad. Sci. USA 2002
99:3042-3046).
The rapid absorption of fluid in the intestine under normal conditions
suggests that fluid
accumulated in the intestinal lumen after active secretion may be absorbed
rapidly if
secretion is blocked, predicting that CFTR inhibition could be effective in
preventing fluid
accumulation even when administered after cholera toxin.
[00187] Fig. 11, panel A summarizes the results of a CFTR;,,h-172 dose-
response study in
mice in which a single dose of inhibitor was administered by intraperitoneal
injection just
after infusion of cholera toxin into closed intestinal loops. Basal intestinal
fluid content
(dashed line) was near zero as measured in non-cholera toxin injected loops.
CFTRiõ h-172
inhibited fluid accumulation in cholera toxin-injected intestinal loops by -
90%, with 50%
inhibition at -5 gg CFTRiõ h-172 (150 gg/kg). The duration of inhibition was
measured as in
the dose-response study, except that a single 20 g dose of CFTRiõ h-172 was
administered at
different times before or after cholera toxin. Fig. 11, panel B shows
significant inhibition of
47
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
luminal fluid accumulation when CFTRiõh-172 was administered at 3 hr before or
after
cholera toxin. However much less inhibition was seen at 6 hr before cholera
toxin. Taking
into account the 6 hr duration of the cholera toxin challenge study, the ti/2
for persistence of
CFTRiõ h-172 inhibition was - 9-10 hr.
Biological Example 6
Oral Bioavailability of CFTR;,,h-172
[00188] To test the antidiarrheal efficacy of orally administered CFTR1õ h-
172, CFTR;,,h-172
pharmacokinetics in mice was determined, and CFTRiõ h-172 transport across
Caco-2
monolayers was measured. Because CFTRiõ h-172 is a relatively nonpolar weak
acid (pKa
5.5) expected to precipitate in the stomach, oral administration was done
using two agents
used commonly to solubilize drugs for oral administration - Vitamin E TPGS and
cyclodextrin. Measurements were done using 14C-labeled CFTRiõh-172.
[00189] Fig. 11, panel C shows the pharmacokinetics of 14C-CFTRiõ h-172 after
oral vs.
intravenous administration in mice. Intravenous administration produced high
initial serum
concentrations that decreased over -30 min (tissue redistribution), whereas
serum
radioactivity was low just after oral administration, peaked at -60-90 min,
and then declined.
Fig. 11, panel D summarizes the organ distribution of 14C-CFTRiõh-172 at 6 hr
after oral and
intravenous administration, showing accumulation in the gastrointestinal tract
as well as the
liver and kidney. 14C radioactivity was concentrated - 1 0-fold in bile vs.
serum, with little
radioactivity excreted in the stool (<10 % of total excreted radioactivity
over 24 hr),
suggesting that accumulation of CFTRiõ h-172 in intestine is facilitated by
enterohepatic
circulation. Comparison of oral vs. intravenous CFTRiõ h-172 administration
(tissue/serum
content at 4-6 hr) indicated 15-20 % CFTRih-172 oral bioavailability in the
TPGS
preparation.
[00190] Fig. 11, panel F shows a linear increase in the appearance of CFTRiõ h-
172 on the
trans-side of Caco-2 monolayers, giving a deduced CFTRiõh-172 permeability
coefficient of
16 x 10.6 cm/s. This value is in the range found for various orally-
administered drugs (e.g.
pindolol, 36 x 10"6 cm/s, sildenafil, 48 x 10"6 cm/s) (Stenberg et al. J. Med.
Chem. 2001
44:1927-1937.
48
CA 02500498 2005-03-29
WO 2004/028480 PCT/US2003/031005
Biological Example 7
Inhibition of cGMP- and cAMP-Mediated Fluid Secretion
[00191] An in vivo rat intestinal loop model was used to determine the
efficacy of CFTR;,,h-
172 in inhibiting cGMP- and cAMP-mediated fluid secretion, as well as to test
the efficacy
of CFTR,,,h-172 in an alternative animal model. The guanylyl cyclase C
receptor is expressed
in rat enterocytes, permitting STa toxin binding and cytoplasmic cGMP
elevation (Mann et
al. Biochem Biophys Res commun 1997 239:463-466). STa toxin has been found to
cause
fluid secretion in rat ileum after 3 hr (Cohen et al. Am JPhysiol 1989 257:G1
18-123).
CFTR;,,h-172 prevented cholera-toxin induced fluid secretion in rat intestinal
loops (Fig. 12,
panel A) at a dose (600 g/kg) that was effective in mice. For STa toxin-
induced fluid
secretion intestinal loops were injected with STa toxin (0.1 g in 300 L PBS)
and loop
weight measured after 3 hr. Fig. 12, panel B shows -75% inhibition of
intestinal fluid
secretion by CFTR;,,h-172.
[00192] Short-circuit current measurements were done in mouse and human
intestinal
epithelial sheets to assess CFTR;,,h-172 inhibition of transepithelial ion
secretion. Fig. 13,
panel A shows CFTR;,,h-172 dose-dependent inhibition of short-circuit current
in mouse
ileum after stimulation by forskolin or STa toxin (inset). Fifty percent
inhibition was found
at -5 M CFTR;,,h-172 for both cAMP and cGMP-dependent chloride secretion.
Fig. 12,
panel B shows similar CFTR;,,h-172 potency for inhibition of short-circuit
current in human
colon.
[00193] An unexpected observation was that the apparent potency for CFTR;,,h-
172 inhibition
of intestinal short-circuit current (2-5 M) was substantially lower than that
found in
electrophysiological studies done on several cell lines including CFTR-
expressing FRT cells
(0.2- 0.5 M) and Calu-3 cells (0.5 M). Several explanations for this
difference were
considered, including cell-type differences, limited access of CFTRiõ h-172 to
enterocytes in
intact intestine, membrane potential effects (interior-negative cell potential
reducing
intracellular [CFTR1,h-172]), and ATP competition with CFTR;,,h-172.
[00194] Short-circuit current measurements were done on T84 colonic epithelial
cells to
investigate this phenomenon. As shown in representative experiments in Fig.
14, panel A, -3
M CFTR1,h-172 produced 50% inhibition of short-circuit current in non-
permeabilized T84
cell monolayers after stimulation by the cAMP agonist forskolin (left), the
cell permeable
cGMP analog 8-Br-cGMP (middle), or the direct activator of CFTR chloride
conductance
CFTRact-16 identified by high throughput screening. To determine whether the
relative
reduction in CFTR;,,h-172 potency in T84 cells requires an intact cell, short-
circuit current
49
CA 02500498 2011-09-06
measurements were done after permeabilizing the cell basolateral membrane with
amphotericin B and in the presence of a Cl gradient (to generate measurable
currents). Fig.
14, panel B (left) shows substantially greater CFTP,.- h-172 potency for
inhibition of short-
circuit current after permeabilization. Dose-response data summarized in Fig.
14, panel B
(middle) indicate a reduction in apparent KI for CFTRiih-172 inhibition from -
3 to 0.3 M
after cell permeabilization. To test whether the reduced CFTRi,h-172 potency
in intact cells
is due to the interior-negative membrane potential (reducing cytoplasmic vs.
external
[CFTRiõh-172]), short circuit current measurements were done in T84 cells
after
depolarization by a high-K+ basolateral bathing solution. Fig. 14, panel C
shows that
increased CFTRh-172 potency (KI - 0.3 M) was restored in the depolarized
cells,
indicating that cell membrane potential plays a role in CFTRRõ ,-172 potency.
[001951 Based on the data above, the thiazolidine compounds of the invention,
as exemplified
by CFTRiA,-172, can be expected to have antidiarrheal efficacy in enterotoxin
induced
secretory diarrheas caused by enterotoxogenic organisms such as E. coli and
Vibrio cholerae
in cholera, Traveller's and AIDS-complex related diarrheas. CFTR inhibition
may be useful
in adjunct therapy of diarrheas caused by entero-invasive bacterias such as
Clostridium
docile and Salmonella species; however, the mucosal damage produced by these
organisms
would not be reduced by CFTR inhibition. Similarly, CFTR inhibition would not
be
predicted to correct the underlying pathology in inflammatory bowel disease,
but could
reduce the volume of intestinal fluid secretions. Recent evidence suggests
that fluid secretion
caused by viral diarrheas such as rotavirus may involve other mechanisms such
as Cat+-
mediated Cl channels, although the role of CFTR in fluid secretions remains
unknown and
hence testable by use of the compounds of the invention in suitable animal
models.
1001961 In summary, the thiazolidinone CFTR blocker CFTR;,,h-172 prevented
cAMP and
cGMP induced ion/fluid secretion in rodent and human intestine without
affecting intestinal
fluid absorption. Its favorable pharmacological and activity profile support
further
development for antidiarrheal applications.
[001971 While the present invention has been described with reference to the
specific
embodiments thereof, it should be understood by those skilled in the art that
various changes
may be made and equivalents may be substituted. In addition, many
modifications may be
made to adapt a particular situation, material, composition of matter,
process, process step or
steps, to the objective of the present invention.