Note: Descriptions are shown in the official language in which they were submitted.
CA 02507669 2005-05-30
WO 2004/052836 2 PCT/FR2003/003542
"Procédé de préparation de diastéréoisomêres et
d'énantiomêres de la 4-hydroxyisoleucine et de ses dérivês"
L'invention a pour objet un procédé de prêparation de
diastérêoisoméres et d'énantiomères de la 4-hydroxyisoleucine
et de ses dérivés, ce terme couvrant les analogues pouvant
être obtenus par le procêdê de l'invention. Elle vise en
particulier la préparation de la (2S, 3R, 4S)-4-
hydroxyisoleucine (4-OH-iLeu en abrégé).
La 4-OH-iLeu est un produit naturel isolé de la graine de
fénugrec, qui répond â la formule A .
O
OH
OH NHZ
A
25 Ce produit est actif en particulier contre le diabète de type
II, mais les quantitês que l'on peut obtenir par extraction
sont insuffisantes pour suppléer aux besoins des populations
atteintes par ce type de diabète. On mesure donc l'intêrêt
d'une synthèse totale qui permettrait de remêdier à ce manque.
Plusieurs procédés ont été proposés â ce jour, mais ils se
sont toutefois révélés non exploitables à l'échelle
industrielle.
Les inventeurs ont rêussi à surmonter ce problème et à
développer un procédé comportant un nombre réduit d'étapes,
grâce au choix de produits réactionnels et de conditions -
opératoires déterminés.
Ce procédé permet d'obtenir .les diastéréoisomères et les
yJïh
énantiomères de la 4-hydroxyisoleucine et de ses dérivés avec
des rendements élevés. En particulier, la 4-OH-iLeu est
CA 02507669 2005-05-30
WO 2004/052836 2 PCT/FR2003/003542
obtenue avec des rendements pouvant dépasser 40~.
Avantageusement ce procédé permet également de synthétiser des
dérivês de la 4-hydroxyisoleucine.
L'ïnvention a donc pour but de fournir un procédé économique
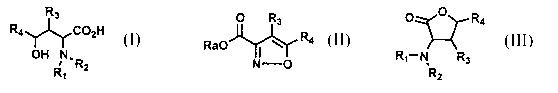
de synthêse d'acides aminés a de formule générale I
R3
R~~G02H
1OH IN~R
R
1
dans laquelle R1 et RZ représentent
.un atome d'hydrogène, ou
.l'un de R1 ou Rz représente un atome d'hydrogène et l'autre
substituant est un radical Ra, un groupe acyle -CORa, notamment
acétyle, ou encore un groupe fonctionnel -C~ORa, -SOZRa, -N (Ra,
Rb) , Ra et Rb, identiques ou différents, étant un radical alkyle
linéaire ou ramifié en Cl-C12, 1e cas échéant substitué, un
groupe aryle à un ou plusieurs cycles aromatïques, comportant
5 à 8C, le cas échéant substitué, ou aralkyle, 1e substituant
alkyle et le groupe aryle étant comme défini ci-dessus, ou
. R1 et RZ représentent taus deux un substituant tel que défini
ci-dessus,
caractérisê en ce qu'il comprend la réduction d'un dérivë
d'isoxazole de formule II
O R3 ,
Ra0 i ~ Ra.
N 0
5 i~
CA 02507669 2005-05-30
WO 2004/052836 3 PCT/FR2003/003542
dans laquelle
. Ra est tel que défini ci-dessus, et
. R3 représente un atome d'hydrogène ou Ra,et
. R4 présente I°es significations de Ra, à l'exception d'un
atome d'hydrogêne,
dans des conditions conduisant directement aux dérivês de
formule I,ou à l'obtention d'au moins une lactone de structure
III
R4
R~_N Rs
R2
TO
sous forme (s) racémique (s) , ou d' un mélange énantiomériquement
enrichi suivie de l'ouverture, en conditions basiques, dans un
solvant aprotique ou protique, de la lactone ou des lactones
recherchées et, si nécessaire, de la séparation de la forme
recherchée.
Une méthode de choix pour l'ouverture du cycle Iactone
comprend l'utilisation de LiOH dans THF.
Selon un mode préféré de réalisation de l'invention, ladite
lactone de structure III est obtenue par rêduction dudit
dérivé d'isoxazole de formule II, conduisant à l'obtention
d'un mélange renfermant 4 lactones L-Z, L-2, L-3 et L-4 .
CA 02507669 2005-05-30
WO 2004/052836 4 PCT/FR2003/003542
° ° R4 0 ° . ,, R~ o ° . ,, R4 ° °
R~
R~-N Ft3 R~~N R3 R~-N R3 R~--N R3
Rz Rz Rz Rz
L-1 L-2 L-3 L-4
On notera que dans 1e cas oû R3 représente un atome d'hydrogène
dans l'isoxazole de formule II, on introduit ultérïeurement un
groupement Ra au niveau des produits intermédiaires obtenus.
Selon une variante de réalisatïon, on procède à la séparation
de la lactone ou des lactones recherchées sous forme racémique
ou énantiomériquement pure.
Selon les catalyseurs et les conditions utilisées, on peut
favoriser l'obtention de l'une des lactones et/ou de l'un des
énantiomères. Des exemples sont donnés à titre illustratif
dans la partie expërimentale.
Conformëment à l'invention, les différentes lactones dans
lesquelles R~ etfou R2 représentent un atome d'hydrogène
peuvent être substituées notamment alkylêes, carbamylées,
sulfonylées, acylées, notamment acétylées. On utilise à cet
effet, en particulier, un agent d'alkylation, de
carbamylation, de sulfonylation ou d'acylation approprié,
avantageusement de l'anhydride acétique pour synthétiser les
dérivés acétylés.
Selon une variante d'obtention des dérivés d'acides aminés a
?5 de structure I de l'invention, on procède â la réduction d'une
isoxa~ole de formule IT dans laquelle ORa représente un groupe
hydrogênolysable, tel que le groupe benzyle. Cette étape de
rêduction est réalisée gin' ~.r~r~i.lieu basique lorsque Ra est
différent d'un groupe benzyle.
CA 02507669 2005-05-30
WO 2004/052836 5 PCT/FR2003/003542
Les produits intermédiaires formés lors de l'étape de
réduction du dérivé d'isoxazole de formule II peuvent être
isolés si on le souhaite. Comme indiqué ci-dessus en rapport
avec les lactones, les produits dans lesquels R1 et/ou Rz
~ représentent un atome d'hydrogène peuvent être substitués
notamment alkylêes, carbamylées, sulfonylées, acylêes,
notamment acétylées. On utilise à cet effet, en particulier un
agent d'alkylation, de carbamylation, de sulfonylation ou
d'acylatïon approprié, avantageusement de l'anhydride acétique
pour synthétiser les dérivés acétylés. I1 est important de
noter qu'en fonction du catalyseur utilisé, il est possible
d'enrichir le produit en une forme diastéréoisomère et/ou
ënantiomére donnée.
Selon les conditions opératoires mises en ouvre, désignées ci-
après par C-SH,C-SC,C-SE, ou C-SH suivïe de C-HC ou de C-HE,
ces produits sont diffêrents (voir figure 1).
Ainsi, selon les conditions C-SH, on opéra par exemple dans un
milieu éthanol/eau, auquel on ajoute une solution de NiR dans
l'éthanol et le dérivé d'isoxazole de formule II et on purge à
l'hydrogène.
Le milieu rêactionnel est ensuite agité sous une pression
d'hydrogène de l'ordre de 1 atmosphère à température ambiante,
ce qui canduit aux dérivés IV et V, ces derniers pouvant être
isolés, par exemple par chromatographie sur silice avec un
rendement de l'ordre de 80%.
CA 02507669 2005-05-30
WO 2004/052836 ~ PCT/FR2003/003542
o R3
Ra,O ~, R4
N, O
Rz
tV
O O R4
Ra~O ~ R
3
~ N.
R~ Rz
V
Une variante de l'invention permet d'obtenir les composês de
formules IV et V, directement à partir du composé de structure
vz .
O R3
R4
Ra0
OH O
v~
par réaction avec l' amine de formule NH (R1, R2) , avantageusement
en présence d'un catalyseur acide et d'un agent de
déshydratation.
5 On récupère le mélange des 4 lactones L-1, L-2, L-3 et L-4 et
on ïsole la lactone recherchée si souhaité.
CA 02507669 2005-05-30
WO 2004/052836 % PCT/FR2003/003542
Un exemple de réalisation de l'invention consiste à favoriser
la formation de la lactone L-1 en conduisant la réduction dans
un mélange NiFè/DABCO dans l'éthanol, alors que les produits
C1-C2 sont obtenus directement si la réaction est conduite
dans un système tel que, Pd/C/DABCO dans de l'éthanol ou
Pd/C/triëthylamine dans de 1'éthanol.
En variante, les composés C-1 et C-2
O R3
Ra0'~~~ R4
R~. N,R20
C-1
O R3
Ra0'~~'~(' R'~
R1 N,R20
C-2
peuvent être obtenus selon les conditions C-HC, en soumettant,
â l'issue de l'étape C-SH, V à l'action d'un catalyseur de
réduction et~ dans un solvant, en présence d'une source
d'hydrogène, par exemple le Pd/C dans l'éthanol, en présence
d'hydrogène. On obtient un mélange C-L/C-2 de l'ordre de 70/30
avec un rendement d'environ 5
Le mélange de lactones recherché peut alors être obtenu par la
vois C-CL.
Pour obtenir majoritairement la lactone L-2, on soumet '
avantageusement C-1 dans l'éthanol à l'action NaBH4. La lactone
?5 peut ainsi être obtenue avec un. rendement de l'ordre de 75~,
le restant reprêsentant la~~'l.ac~one L-g .
CA 02507669 2005-05-30
WO 2004/052836 g PCT/FR2003/003542
En opérant avec un mélange d'éthanol et d'eau, auquel on
ajoute une solution de catalyseur, par exemple du Ni.R dans
l'éthanol et C-1, on peut former de manière prépondérante la
lactone L-4. Selon des conditions de traitement avantageuses,
on porte le mélange réactionnel à 0°C, on le purge â
l'hydrogène, puis on le soumet à agitation sous pression
d'hydrogène. Le mélange des 4 lactones L-1, L-2, L-3 et L-4
est obtenu de manière quantitative. La lactone L-4 peut être
isolêe, par exemple par HPLC, avec un rendement de 75ô
environ, le restant étant essentiellement formé par la lactone
L-2.
La lactone L-3 peut être obtenue de manière majoritaire, en
opérant comme indiqué ci-dessus, mais en utilisant C-2. La
lactone L-3 peut être alors isolée, par exemple par HPLC, avec
un rendement de 754 environ, le restant étant essentiellement
formé par la lactone L-1.
En variante, les composés E-1 et E-2 peuvent être obtenus
selon les conditions C-HE.
Ainsi, la synthèse de E-2 peut être réalisée à partir de IV
ou de V, avec des rendements d'au moins 90~. On utilise, avec
avantage, à cet effet,un milieu réactionnel contenant un
catalyseur de réduction homogène, tel que [Ru(p-cym)2C12], un
ligand chiral ou achiral, notamment un ligand tosylê, tel que
TsDPEN (monotosyldiphényléthylènediamine), un solvant
organique, de la triéthylamine et une source d'hydrogène, par
exemple l'isopropanol ou l'acide formique.
Le dérivé E-2 est alors obtenu avec un rendement de l'ordre de
90%,
CA 02507669 2005-05-30
WO 2004/052836 g PCT/FR2003/003542
On peut également synthêtiser E-l ou E-2 à partir,
respectivement, de V et de Iv, par rêduction dans un mélange
éthanoljeau, en présence de NaBH4 et . de CeC13.7H20. Les
produits recherchës sont obtenus avec des rendements de
l'ordre de 95~.
Les lactones L-1 et L-4 sont obtenues de manière majoritaire,
respectivement, par réduction â partir de E-2 et de E-1. De
maniëre préférëe, E-2 est placé dans l'éthanol avec du NiR
sous hydrogène, à pression atmosphêrique. L-~. est obtenue avec
des rendements d'environ 75~, le restant étant constitué par
les autres lactones L-2, L-3 et L-4. Pour obtenir
majoritairement L-4, on opère comme précédemment, mais à
partir de E-1 et le rendement est ëgal à 85%.
Conformément à un mode préféré de réalisation de l'invention,
le dérivé d"isoxazole de formule II est obtenu par réaction
d'une hydroxylamine avec un dérivë d'acide 4-céto-2-hydroxy-2
buténoïque de formule VT .
O R3
RaO ~ R'~
OH O
vf
L'hydroxylamine est utilisée plus spécialement sous forme de
?5 sel et la réactïon est réalisée à température ambiante.
Dans le mode préféré de réalisation de l'invention, le dérivê
d'acide 4-céto-2-hydroxy-2-buténoïque est obtenu par
condensation d'une cétone VII~.~~t d'un dérivê d'oxalate VIII .
0
CA 02507669 2005-05-30
WO 2004/052836 1o PCT/FR2003/003542
O
R4~ Rs
V
O
RaO~ORc
'-'O
Dans ces formules,Rs représente un radical alkyle, tel
qu'éthyle ou méthyle, alkylaryle, vinyle, ou vinyle substitué,
R4 et Ra sont tels que définis ci-dessus, R~ présente les
significations données par Ra et peut étre est identique ou
différent de Ra.
Dans une variante de réalisation de l'étape de condensation,
on utilise, comme cétone, la 2-butanone. Le dérivé de l'acide
4-céto-2-hydraxy-2-butênoïque conduisant à la 4-
hydroxyisoleucine est alors obtenu en mélange avec notamment
un dérivé de l'acide hex-2-ênoïque, ces composés étant séparés
au cours d'une étape ultërieure.
Dans une autre variante préférée de réalisation de l'étape de
condensation, la cétone utilisée est l'acétone (R4=RS=CH3), ce
qui conduit au dérivé d'acide 4-cêto-2-hydroxy-2-buténoïque de
formule VI, dans laquelle R3 est un atome d'hydrogène et R4 ,
représente CH3. Ce composé est ensuite, fonctionnalisé,
notamment par réaction d'alkylation, en présence de bases et
d'un agent alkylant.
CA 02507669 2005-05-30
WO 2004/052836 lZ PCT/FR2003/003542
Dans encore une autre variante prëférêe, l'acide 4-cêto-2-
hydroxy-2-buténoïque de formule VI (R3=R4=CH3) est obtenu en
opérant selon la réaction de Baylis-Hillmann, en faisant
réagïr la méthylvinylcétone sur un glyoxalate IX, suivïe soit
d'une êtape d'isomérïsation, soit de réduction' de la double
liaison puis de l'oxydation de la fonction OH.
O
H ~ORa
O
ix
Le produit de condensation formê est isomérisé en composé X,
en prêsence de catalyseurs de métaux de transition.
O
OH O
X
Les produits intermédiaires suivants sont des produits
nouveaux et, à ce titre, entrent dans le champ de l'invention:
il s'agit des produits de formules IV et V, dans lesquels l'un
de R~ et R2 représente H, l'autre étant différent de H,ceux
ZO correspondant à C-1 et C-~, tels que dêfinis ci-dessus, quel
que soit R1 et R2, et les composés E-1 et E-2 dans lesquels les
substituants sont tels que définis ci-dessus en rapport avec
les composés IV et V.
5 dans lesquels R représente R1 ou R~, et les produits E-1' et E-
2' , dans lesquels R représent~er.R.l ou R2, mais diffère de H.
CA 02507669 2005-05-30
WO 2004/052836 Z~ PCT/FR2003/003542
L'invention vise tout particulièrement l'obtention de la 4-OH-
iLeu de formule A selon un procédé comprenant les étapes de
a) synthèse d'un ester d'acide peut-2-ènoïque de formule X
O
~O
OH O
soit par réaction de la 2-butanone avec l'oxalate d'éthyle,
soit par condensation de la méthylvinylcétone avec le
glyoxalate d'éthyle, suivie, sans purification, d'une
réaction d'isomérisation ou d'une séquence
réduction/oxydation;
b) l'ester d'acide pent-2-ènoï.que obtenu réagit avec de
l'hydroxylamine pour former le dërivê d'isoxazole de
formule XT,
O
N - O
X
c) la rêduction du dérivé d'isoxazole obtenu pour conduire à
aux lactones 1-1 à 1-4,
CA 02507669 2005-05-30
WO 2004/052836 13 PCT/FR2003/003542
O O ...~~ ~ O
,:
HZN .:% H2N H2N '~ HZN
1-'I I-2 I-3 1-4
d) la séparation de la lactone 1-1 à 1-4 sous forme
racémique, suivie de
e) la séparation de l'énantiomère, conduisant au composé A
par ouverture de la lactone, et de
f) l'ouverture du cycle lactone.
D'autres caractéristiques et avantages de l'invention seront
donnés dans les exemples qui suivent en se reportant aux
figures 1 et 2, qui représentent, respectivement, les
schémas réactionnels illustrant les variantes opératoires
pour obtenir à partir du dërivê d'isoxazole de formule TII .
- les lactones L-~. à L-4,
- les lactones 1-~. à 1-4,
Exemple 1 . Synthèse des dérivés de l'acide peut-2-ènoique de
f orFnul e X
Par fonctionnalisation d'un produit de condensatïon d'un anion
dérivé de la butanone avec le diêthyloxalate
Une solution d'êthanolate de sodium est préparée en faisant
réagir du sodium métallique (6,05 g, 260,00 mmol, 1,2 éq) dans
de l'éthanol anhydre (360 mL) à. température ambiante jusqu'à
consommation totale du sodium métallique. De la butanone
(20,00 mL, 220,00 mmol, 1,0 éq) est ensuite additionnée goutte
CA 02507669 2005-05-30
WO 2004/052836 2~ PCT/FR2003/003542
à goutte à température ambiante. Après 1 heure de réaction à
température ambiante, du diéthyloxalate (60,00 mL, 440,00
mmol, 2,0 éq) est additionné avec un goutte à goutte rapide à
température ambiante. Après 5 minutes de réaction, 1e milieu
réactionnel est concentré puis séché sous vide. Le brut
réactionnel est dilué avec une solution aqueuse saturée en
NaCl (800 mL), puis la phase aqueuse est extraite avec de
l'acétate d'éthyle (3x900 mL). La phase aqueuse est ensuïte
diluée dans de l'acétate d'éthyle (900 mL). La phase aqueuse
est acidifiée jusqu'â pH 6 avec une solution de HC1 1N, sous
vive agitation magnétique. La phase organique est séparée et
la phase aqueuse est extraite à l'acétate d'éthyle (3x900 mL).
Les phases organiques sont rassemblées, séchées sur MgS04 puis
concentrées sous vide. Le brut réactionnel est séché sous vide
pour donner avec un rendement isolé de 30 % un mélange 90 . 10
de 2-hydroxy-3-mêthyl-4-oxo-pent-2-ènoate d'éthyle et de 2-
hydroxy-4-oxo-hex-2-énoate d'éthyle, ainsi qu'un produit de
structure non déterminée dont la réactivité est identique à
celle du produit X (m=11,4 g),
~ Le dérivé de l'acide héxénoïque formé est séparé du composé
X par lavage à NaCl(sat), puis extraction à l'acétate
d'éthyle.
Le produit X est récupéré après acidifïcation de la phase
?5 aqueuse à pH 6, puis extraction à l'acétate d'éthyle.
~ Rendement en composé X après lavages . 30 ~.
Par fonctionnalisation d'un produit de condensation de l'anion
dérivé de l'acétone avec Le dséthyloxalate
0
L'acëtone a ëtë condensée sur le dïêthylaxalate en milieu
basique. Les groupements âzofë et le méthyle sont introduits
ultérieurement.
CA 02507669 2005-05-30
WO 2004/052836 15 PCT/FR2003/003542
Le composé de formule VI dans laquelle Ra - CHz CH3, R3 - H et
R4 _ CH3 est fonctionnalisé avec le chlorhydrate de
l'hydroxylamïne pour donner le composé IV dans lequel Ra - CHz
CH3, Rz = H, Rz = OH R3 - H et R4 =CH3, qui est ensuite soumis à
une réaction de méthylation pour conduire au méme composé,
mais avec R3 - CH3.
2-Hydroxy-4-oato-gent-2-énoate d'éthyle
IO
Dans un ballon tricol de 2 litres, muni d'une ampoule à
addition et d'un agitateur à palette est préparé une solution
d'éthanolate de sodium, en faisant rêagir du sodium métallique
(7,74 g, 340,00 mmol, 1,2 éq) dans de l'éthanol anhydre (800
mL) à température ambiante jusqu'â consommation totale du
sodium métallique. Une solution de diéthyloxalate (37,20 mL,
280,00 mmol, 1,0 éq) dans l'acétone (10,30 mL, 280,00 mmol,
1,0 éq) est ensuite additionnée goutte à goutte â température
ambiante. Le milieu réactionnel est maintenu sous vive
agitation pendant 2 heures. Le milieu réactionnel est ensuite
concentré sous vide. Le brut réactionnel est dilué dans de
l'eau (200 mL) . On y ajoute dé la glace (100 g) , puis de
l'acide sulfurique concentré (28 mL) par petites portions
jusqu'à obtention d'une solution limpide orange. La phase
aqueuse ainsi obtenue est extraite avec de l'acétate d'éthyle
(3x300 mL). Les phases organiques sont rassemblëes, séchées
sur MgS04 puis concentrées sous vide. Le brut réactionnel est
Bêché sous vide pour donner quantitativement le produit
attendu (m=44,71 g).
Par rêaction de Baylis-Hillmann de l.a méthylvinylcêtone sur le
glyoxalate d'éthyle
CA 02507669 2005-05-30
WO 2004/052836 16 PCT/FR2003/003542
Le composé de condensation est soumis à une étape de réduction
de la double liaison, suivie, sans purification, de
l'oxydation de la fonction hydroxyle.
CONDENSATION
A une solution de méthylvinylcétone (5 mL, 50 mmol, 1 éq) dans
du dioxane anhydre (30 mL) est a~outêe une solution de
glyoxalate d'éthyle à 50 % dans le toluène (14,2 mL, 60 mmol,
l,2 éq), puis du DABCO (600 mg, 0,09 ëq). Le mêlange
rêactionnel est agitê à température ambiante pendant 24 h. I1
est ensuite neutralisé par addition d'une solution de HCl 10
(20 mL), et extrait â l'acétate d'éthyle (2x30 mL). Les phases
25 organiques sont rassemblées, séchées sur MgS04 puis concentrées
sous vide. Le produit de rêaction est recueilli avec un
rendement supérïeur à 90
REDUCTION
Dans un monocol de 250m1, placé sous argon, 1a cétone a,(3
insaturée (8g, 4,65.102 mol) est solubilisée dans 200m1
d'éthanol, puis le Pd/CaC03 (1,6g, 0.2eq) est introduit dans le
mélange. Le système est purgé à l'hydrogêne et agité en
permanence sous pression d'hydrogëne, à température ambiante
pendant 3h30.
Le milieu réactionnel est filtré sur Cëlite~ et le filtrat est
concentré sous pression réduite.
Le milieu réactionnel aïnsi obtenu est engagé directement sans
purification dans l'étape d'oxydation.
OXYDATION
Dans un monocol de 250m1 flambé, placé sous argon, une
solution. de DMSO (2.6m1, ~~~,'7..,~,a0'2 mol) dans le CH~Clz (120m1)
est refroidie à -60~C puis l'anhydride trifluoroacêtique
CA 02507669 2005-05-30
WO 2004/052836 1~ PCT/FR2003/003542
(6.42m1, 3,3.10-2mo1) est ajouté.. Aprës 10 minutes d'agitation
à -60°C, la solution d'alcool (2g, 1,15.102 mol) dilué dans
un minimum de CH2C12 (l2ml) est ajoutée goutte à goutte.
Le milieu réactionnel est agitê à -60~C pendant 2h, puis la
triéthylamine (7.85m1, 7,5.10-2 mol) est ajouté goutte à
goutte.
Le système est agité à -60°C pendant 2h supplémentaires, puis
laissê remonter à température ambiante.
On ajoute une solution tampon (25m1) de KC1 0.2M+ NaOH, pH=12.
Préparation du tampon . 25m1 KC1 0,2M (373mg +25m1 H20)+ 6m1
NaOH 0,2M (2m1 NaOH 1M + 8m1 H20) .
On extrait la phase aqueuse avec du CH2C12 (2x 20m1) , puis la
15 phase organique est séchée sur MgS04, reconcentrêe sous
pression réduite, et chromatographiée sur colonne de
silice(systëme . Hexane/acétate d'êthyle 7/3)
Le produit X (1,5g) est isolé avec un rendement de 75~.
Analyses
COMPOSÉ X
2-Hydroxy-3-znéthyl-4-oxo-gent-2-ênoate d'éthy~.e
~5 CgHy2~4
CCM : Rf = 0,4 (AcOEt / hexane 20 . 80).
RMN zH (CDC13, 200 MHz) ~ (ppzn) . 1, 36 (s, 6H) , 1, 97 (s, 3H) ,
2,23 (s, 3H), 4,23 (m, 4H).
RMN 13C (CDC13, 50 MHz) 8 (ppm)~'n". 11, 2; 13, 7; 25,4; 61, 7; 106, 8;
162,9; 168,4; 200,5.
CA 02507669 2005-05-30
WO 2004/052836 1$ PCT/FR2003/003542
IR (v en cm-z) . 3452 (OH) , 3054, 2987, 1731 (C=O) , 1264, 742,
703.
sM (IC) m/z . [M+H] + - 173 .
Téb = 98 °C ; 0,5 mbar
Huile incolore
2-Hydroxy-4-oxo-hex-2-énoate d'éthyle
C8H1,2~4
CCM : Rf = 0,4 (AcOEt / hexane 20 . 80).
RMN 1H (CDC13, 200 MHz) 8 (ppm) . 1, 11 (t, 3J = 7, 6 H3, 3H) ,
l, 31 (t, 3J = 7, 1 H3, 3H) , 2, 47 (q, 3J ~ 7, 6 Hz, 3H) , 4, 28 (q,
3J = 7, 1 Hz, 3H) , 6, 3l (s, 1H) .
RMN 13C (CDC13, 75 MHz) 8 (ppm) . 8,37; 13,8; 34,1; 62,3; 101,2;
162,0; 165,7; 200,5.
IR (v en czii 1) . 3452 (OH) , 3054, 2987, 1739 (C=O) , 1264, 742,
706.
SM {IC) m/z . [M+H] + - 173 .
Huile incolore
2-Hydroxy-4-oxo-peut-2-ènoate d'éthyle
C~Hia04
CCM . Rf = 0,5 (AcOEt / hexane 50 . 50).
CA 02507669 2005-05-30
WO 2004/052836 ?g PCT/FR2003/003542
RbQ1' ''H (CDCL3, 300 MHz) 8 (ppm) . Z, 35 (t, 3J = 7, 2 Hz, 2H) ,
2, 24 (s, 3H) , 4, 32 (q, t, 3J = 7, 2 Hz, 2H) , 6, 36 (s, 1H) .
RMt~ ~3C (CDC13. 50 MHz) ô (ppm) . 13,7; 27,2; 62,2; 101,8;
161,7; 166,7; 199,8.
TR (v en cm ~) . 3561 (OH) , 2987, 1739 (C=O) , 1643 (C=C) , 1602,
1465, 1419, 1370, 1269, 1212, 1119, 1018, 910, 776, 732.
SM (ZC) m/z . [M+NH4] + - 176 .
Liquide incolore
Exemple 2 . Formation du systême isoxazole XI
Mode opératoire
Dans un ballon bicol de 250 mL, une solution de 20 mmol de
composé X dans un mélange 1/1 d' ëthanol
anhydre/tétrahydrofurane anhydre (volume total - 54 mL) est
préparée. Le mélange est placë sous agitation vigoureuse et
sous argon. 1,6 g d'hydroxylamine chlorhydratée est ajouté par
portions (une dizaine) pendant trois heures. Le mêlange est
laissê â température ambiante pendant vingt sept heures.
Le brut réactionnel est dilué dans 180 mL de dichlorométhane
et 110 mL d'une solution saturée de chlorure de sodium puis la
phase aqueuse est extraite avec du dichlorométhane (2 x 110
mL). Les phases organiques sont rassemblées, Bêchées sur
sulfate de magnêsium puis concentrées sous vide pour donner un
rendement isolé de 80 ~ en composê XI.
CA 02507669 2005-05-30
WO 2004/052836 2o PCT/FR2003/003542
Analyses
Composé XI
RIYR~i 1H (CDC13, 200 I~iz) 8 (ppm) . 1.36 (t, 3H) , 2 . 07 (s, 3H) ,
2.33 (s, 3H) , 4. 37 (q, 2H)
RMN i3C (CDC13, 50 MHz) 8 (ppm) . 7.3, 10.6, 14.1, 61.6, 117..2,
154.7, 160.9, 167.4
SM(TC) m/z . [M+H] ~ - 170
GC/MS tR = 8 , 17 min
Exemple 3 . Synthêse et réduction des intermédiaires du
systëme isoxazole (voir schéma figure 2)
Préparation d'une solution de Nickel de Raney dans l'éthanol
(Solution A)
20~ Une solution commerciale de Nickel de Raney dans l'eau est
centrifugée pendant 5 minutes à la vitesse de 4200
tours/minute.
Le surnageant est éliminê et le solide lavé à l'eau distillée
puis centrifugé à nouveau.
Ce cycle de lavage est répété 5 fois puis l' eau est remplacëe
par de l'éthanol afin d'obtenir, après 5 cycles de lavage et
élimination du surnageant, un volume de Nickel de Raney de 5
mL (~10g) .
Ce volume de Nickel de Raney est alors dispersé dans 50 mL
d'éthanol pour obtenir une solution A de Nickel de Raney dans
l'éthanol.
Mode opératoire d' acétylat~ioi~,~~
CA 02507669 2005-05-30
WO 2004/052836 21 PCT/FR2003/003542
-Synthèse de H-1', H-2' à partir de H-l, H-2
Dans un ballon monocol, H est placé dans l'anhydride acétique
(concentration de 0,45 M) pendant cinq heures à 70 °C.
L'anhydrïde acétique est évaporée sous vide et le brut
réactionnel est filtré sur silice avec un élisant
Hexane/Acétate d'éthyle (8/2).
Le produit H' obtenu est cristallisë à froid dans un mélange
Bther/Hexane avec un rendement de 90 %.
-Synthêse de 1-ï', 1-2' à partir de 1-2, 1-2 et c-1', c-2' à
partix de c-1, c-2
Dans un ballon monocol, 1 ou c est placé dans l'anhydride
acétique (concentration de 0,45 M) pendant une heure à
température ambiante. L'anhydride acétique est évaporée sous
vide et le brut réactionnel est filtrë sur silice avec un
élisant Hexane/Acëtate d'éthyle (8/2). Le produit pur l' ou c'
est isolé à 98%.
Conditions réactionnelles C-SL
Conditions I-1 1-2 I-3 I-4
_
-
NiR/H2O _ _
EtOH/H20 50/50 25 40 10 25
TA
NiR/ H20
EtOH/Hz0 50 j50 40 10 25 35
55C
NiR/ DABCO 60 15 10 15
EtOH
NzR/ HMTA ~0 16 7 17
EtOH
NiR j Et3N
33 30 10 27
EtOH
CA 02507669 2005-05-30
WO 2004/052836 2~ PCT/FR2003/003542
Synthèse des lactones 1-1 à 1-4 avec obtention majoritaire de
1-2 par réduction de XI
Dans un ballon monocol de 5 ml sont introduits un mëlange
êquivolumique d'éthanol et d'eau (1mL), la solution A de
Nickel de Raney dans l'éthanol (100 uL) et XI (30 mg, leq,
1,76.10-4 mol). L'ensemble est refroïdi à 0°C puis purgé à
l'hydrogène.
Le milieu est agité sous pression d'hydrogène (1 atm) pendant
12 heures â température ambiante.
Le brut réactionnel est filtré sur vélite et le mélange des
quatre lactones obtenu de manière quantïtative.
La lactone 1-2 (lactone de la 4-hydroxyisoleucine) est isolée
par HPLC sur colonne de silice avec un rendement de 40%.
Synthèse des lactones 1-1 à 1-4 avec obtention majoritaire de
1-1 par réduction de Xr
Dans un ballon monocol de 5 mL sont introduits l'ëthanol
(1mL), DABCO (10 mg), la solution A de Nickel de Raney dans
l'éthanol (100 ~L) et XI (30 mg, leq, 1,76.10-4 mol).
L'ensemble est porté à 0°C puis purgé à l'hydrogène.
Le milieu est agité sous pression d'hydrogène (1 atm) pendant
48 heures à température ambiante.
Le brut réactionnel est filtré sur vélite et le mélange des
quatre lactones obtenu de maniére quantitative.
La lactone 1-l est isolée par HPLC sur colonne de silice avec
un rendement de
CA 02507669 2005-05-30
WO 2004/052836 23 PCT/FR2003/003542
Synthèse des ~.actones 1-1 à 1-4 avec obtention majoritaire de
1-4 par réduction de XT
Dans un ballon monocol de 5 ml sont introduits un mêlange
équivolumique d'éthanol et d'eau (1mL), la solution A de
Nickel de Raney dans l'éthanol {100 uL) et XI {30 mg, leq,
1,7&.10-4 mol). L'ensemble est portê à 0°C puis purgé à
l'hydrogène.
Le milieu est agité sous pression d'hydrogêne {1 atm) pendant
12 heures à 55°C.
Le brut réactionnel est filtré sur Célite~ et le mélange des
quatre lactones obtenu de manière quantitative.
La lactone 1-4 est isolée par HPLC sur colonne de silice avec
un rendement de 40%.
Les lactones 1-l', 1-2', 1-3' et 1,-4' sont synthétisées par
acétylation des diffêrents bruts obtenus ci-dessus (Voir mode
opératoire d'acétylation en page 21).
20 Conditions rêactionnelles C-SH
Synthêse de H-2 par réduction de COMPOSö XT
Dans un ballon monocol de 5 mL sont introduits 1'êthanol
25 (1mL), l'eau (50 ~L), la solution A de Nickel de Raney dans
l' éthanol {1.00 uL) et composé Xx (30 mg, leq, 1, 76 . 10-4 mol) .
L'ensemble est porté à 0°C puis purgé à l'hydrogène.
Le milieu est agitê sous pression d'hydrogène (latm) pendant
24 heures à tempéxature ambiante.
30 Le brut rêactionnel est purifié par chromatographie sur
colonne de silice et H-2 isolé avec un rendement de 80%.
Conditïons rêactionnelles C-SC
CA 02507669 2005-05-30
WO 2004/052836 24 PCT/FR2003/003542
Synthèse de c-1 et c-2 par réduction de COMPOSÉ XI
Conditions r-I e-2
NiR/ DABCO
30 70
EtOH
Pd j C/ DABCO
50 50
EtOH
Pd/ C/ EtaN
~0 30
EtOH
Dans un ballon monocol de 5 mL sont introduits l'éthanol
(1mL), la triéthylamine (50 uL), le palladium sur charbon (6
mg) et composé XI (30 mg, leq, Z, 76. 10-~ mol) .
L'ensemble est porté à 0°C puis purgé â l'hydrogène. Le milieu
est agité sous pression d'hydrogène (1 atm) pendant 48 heures
à tempêrature ambiante.
Le brut réactionnel est filtrê sur célite et le mélange des
deux diastéréoisomères obtenus dans un rapport 70/30.
Les deux diastêréoisomères c-1 et c-2 sont obtenus avec un
rendement de 70~.
>0
Les composés c-1' et c-2' sont synthétisês par acétylation des
différents bruts obtenus ci-dessus (Voir mode opératoire
d'acétylation en page 21). _
5 Conditions rëactionnelles.~C-HL
CA 02507669 2005-05-30
WO 2004/052836 ~~ PCT/FR2003/003542
Synthêse de 1-I', 1-2'. I-3', et 1-4'par réduction de composê
H-2'
Dans un ballon monocol de 5 ml sont introduits un mélange.
êquivolumique d'êthanol et d'eau (300 ~L), la solution A de
Nickel de Raney dans l'êthanol (50 uL) et H-2' (10 mg, leq,
0,6.10'4 mol). L'ensemble est porté à 0°C puis purgé à
l'hydrogène.
Le milieu est agité sous pression d'hydrogëne (1 atm) pendant
12 heures à température ambiante.
Le brut rëactïonnel est filtré sur Cëlite~ et le mélange des
quatre lactones obtenu de manière quantitative.
Les lactônes 1-l', 1-2'., 1-3', et 1-4'sont isolêes par HPLC
sur colonne de silice dans les proportions suivantes .
Conditions 1-1' 1-2' 1-3' 1-4'
NiR j H20
EtOH/H20 50/50 1~ 23 17 56
TA
Conditions réactionnelles C-He
Synthëse de e-2' à partir de H-l' ou H-2'
Dans un tube préalablement flambé sous argon, le milieu
réactionnel contenant 3 mg de [Ru(p-cym)ZCl2j (5 % mol), le
ligand tosylé TsDPEN (1.05 équivalent/Ru), le solvant iPrOH
j5 (136 ~.L),, et la triéthylamine (6.2 ~L) est chauffé à 80 °C
pendant deux heures . Puis le milieu est évaporé sous argon et
laissé reposé à température ambiante.
Le substrat de départ (40 mg), est dissout dans HCOOH/NEt3 (5/2)
..~ - .;~.~.
(92 ~L) et l'ensemble est introduit dans le tube contenant le
catalyseur pendant 17 H 00 â température ambiante. Le brut
CA 02507669 2005-05-30
WO 2004/052836 26 PCT/FR2003/003542
réactionnel est évaporé sous vide et filtré sur silice avec de
l'acétate d'éthyle. Le produit e-2' est ïsolé avec un
rendement de 90
Synthèse de e-l' à partir de H-1.' ou e-2' à partir de H-2'
Le substrat de départ est placé dans un ballon bicol sous
argon dans un mélange éthanol/eau (1/1.5) â -15 °C. 1,5
équivalent de NaBH4 et 1 équivalent de CeCl3 . 7 H20 sont aj outés
et le milieu est agitê pendant 1S minutes. L'ajout de quelques
gouttes d'acétone permet de neutraliser l'excès de NaBH4.
Le brut réactionnel est dilué dans l'ëther et une solution
saturêe de chlorure de sodium puis la phase aqueuse est
extraite trois fois avec de l'éther. Les phases organiques
sont rassemblées, séchées sur sulfate de magnésium puis
concentrées sous vide pour donner un rendement isolé de 95
Conditions réactionnelles C-Hc
Synthêse de c-1 et c-2 à partir de H-2
H-2 est placé sous hydrogène à 40 bars en présence de Pd/C (10
?5 ~ en masse) et d'êthanal (0.1 M) pendant 27h à température
ambiante. Le brut réactionnel est filtrê sur cêlite et évaporé
sous vide. T1 est obtenu avec un rendement de 55 % avec un
rapport c-1/c-2 de 70/30.
Synthèse de c-l' et c-2' à partir de H-2'
H-2' est placé dans l' étha'nôl°lk ~(0 . 1 M) avec 10 ô en masse de
Pd/C sous hydrogène à pression atmosphérique pendant 24 H 00.
CA 02507669 2005-05-30
WO 2004/052836 ~~ PCT/FR2003/003542
Le brut réactionnel est filtré sur cêlite puis évaporé sous
vide. Un mélange 1/1 de c-l' et c-2' est obtenu avec un
rendement de 98 ~. c-l' et c-2' sont séparés par HPLC selon la
méthode décrite précédemment.
Conditions réactionnelles C-eL
Synthêse de l-1' à partir de e-2'
e-2' est placé dans 1 ' éthanol ( 0 . 1 M) avec du nickel de raney
commercial sous hydrogène à pression atmosphérique. Le brut
réactionnel est filtré sur cêlite et évaporé sous vide. 1-l'
est obtenue avec un rendement de 75 %. Les 25 ~ restants sont
un mélange de 1-2', I-3', I-~'.
Synthêse de L-4' à partir de e-l'
e-l' est placé sous hydrogène dans l'éthanol (0.1 M) à
pression atmosphérique ën présence de nickel de raney
commercial pendant 15 H 00. Le brut réactionnel est filtré sur
Célite° et évaporé sous vide. 1-4' est isolé avec un rendement
~5 de 85 ~. Les 15 % restants représentent la lactone acêtylée 7.-
2'.
Conditions rêactionnel3.es C-cL
''>0
Synthëse de ï-2' â partir de c-1.'
c-1' est placé dans l' êthânôJ.'° (0 . 1 M) en présence de NaBH4 (2
équivalents) pendant une heure â 0 °C. Le brut réactionnel est
CA 02507669 2005-05-30
WO 2004/052836 ~8 PCT/FR2003/003542
dilué dans l'acétate d'éthyle et l'eau. La phase aqueuse est
extraite avec l'acétate d'éthyle trois fois. Les phases
organiques sont rassemblées, séchées sur sulfate de magnésium
puis conçentrées sous vide pour donner un rendement isolë de
75 % en lactone acêtylé 1-2'. Les 25 % restants représentent
la lactone 1-4'.
Synthèse de 1-4' à partir de c-1'
Dans un ballon monocol de 5 mL sont introduits un mélange
équivolumique d'éthanol et d'eau (300 uL), la solution A de
Nickel de Raney dans l'éthanol (50 uL) et c-1' (10 mg, leq,
0,6.10-' mol). L'ensemble est porté à 0°C puis purgé à
15 l'hydrogène.
Le milieu est agité sous pression d'hydrogène (1 atm) pendant
Z2 heures à température ambiante.
Le brut réactionnel est filtré sur vélite et le mélange des
quatre lactones obtenu de manière quantitatïve.
20 La lactone 1-4' est ïsolée par HPLC sur colonne de silice
avec un rendement de 75~. Les 25~ restants représentent la
lactone ~.-2' .
Synthèse de 1-3' à partir de c-2'
z5
Dans un ballon monocol de 5 mL sont introduits un mélange
équivolumique d' éthanol et d' eau (300 uL) , la solution A de
Nickel de Raney dans l'éthanol (50 uL) et c-2' (10 mg, leq,
0,6.10-4 mol). L'ensemble est porté à 0°C puis purgé à
l'hydrogène.
Le milieu est agité sous pression d'hydrogène (1 atm) pendant
12 heures à température ambiante.
Le brut réactionnel est fi~.ltré sur vélite et le mélange des
quatre lactones obtenu de maniêre quantitative.
CA 02507669 2005-05-30
WO 2004/052836 2g PCT/FR2003/003542
La lactone Z-3' est isolée par HPLC sur colonne de silice
avec un rendement de 75~. Les 25% restants représentent la
lactone I-1':
Conditions réactionnelles I-HH
Synthêse de H-2' à partir de H-2'
Dans un tube préalablement flambé sous argon, le milieu
réactionnel contenant 3 mg de [Ru(p-cym)aClzl (5 % mol), le
ligand tosylé TsDPEN (1.05 équivalent/Ru), le solvant iPrOH
(136 ~L), et la triéthylamine (6.2 ~.L) est chauffé à 80 °C
pendant deux heures . Puis le milieu est évaporé sous argon et
laissé reposé à température ambiante. H-2' est introduit dans
l'êthanol (1.1 M) sur le catalyseur formé et le milieu est
laissé pendant 27 H 00 à température ambiante. Le brut
réactionnel est évaporé sous vide puis filtré sur silice avec
de l'acétate d'éthyle. H-l' est obtenu avec un rendement de 60
Conditions réactionnelles I-cc
Synthèse de c-1' à partir de c-2'
c-2' est placé dans l'éthanol (0.1 M) avec 15 équivalents de
triéthylamine à 80 °C pendant 24 H 00. Le brut réactionnel est
évaporé sous vide et c-1' est isolé avec un rendement de 55 ~.
Conditions HPLC
Séparation des quatre 7.actones ~.-1', l-2', 1-3' et l-4'
CA 02507669 2005-05-30
WO 2004/052836 3~ PCT/FR2003/003542
La séparation des quatre lactones I-L', 1-2', 1-3' et 1-4' est
faite sur HPLC.
HPLC (Gynkotek Gina 50) et colonne Z'ORBAX SIL 4.6 MM ID x 25cm
avec pour éluant un mélange Hexane/Ethanol 95j05 et un débit
de 8 mL/min.
Séparation das deux én:antiomèxes de la lactone 1-2'
La séparation des deux énantiomères est faite sur HPLC
chirale.
HPLC (Shimadzu) et colonne CHIRALPA.K AS avec pour êluant un
mélange Hexane/Ethanol 95j05.
CA 02507669 2005-05-30
WO 2004/052836 ~1 PCT/FR2003/003542
Analyses
Les analyses GCjMS sont toutes faites sur le même type de
matériel.
GC/MS (Shimadzu GCMS-QPS050A)
Colonne SGE CAPILLARY Silice 25m x 0,22mm BPX5 0,25
Gaz vecteur . hélium, débit 29 ml/min ; Pression . l18 kla.
Progra~une
Interface . 260°C
Colonne . 80°C
Dêtecteur . 320°C
2 min â 80°C puis montée en température de 10°C /min
Les analyses HPLC sont toutes faites sur le même type de
matériel.
HPLC (Gynkotek Gina 50j et colonne ZORBAX SIL 4.6 MM 1D x 25cm
Eluant . Hexane/Ethanol 95/05. Débit . 8 mL/min
H-l'
RMN 1H (CDC13, 200 MHz) 8 (ppm) . 1.25 (t, 3H) , 1 . 88 (s, 3H) ,
2.09 (s, 3H), 2.32 (s, 3H), 4.18 (q, 2H), 7.52 (s, 1H)
SM(TC) mjz . [M+H)+ - 214
GC/MS tR ~ 12,15 min
H-2'
CA 02507669 2005-05-30
WO 2004/052836 32 PCT/FR2003/003542
RMN 1H (CDC13, 200 MHz) s (pprn) , 1.33 (t, 3H) , 1.91 (s, 3H) ,
2.10 (s, 3H), 2.26 {s, 3H), 4.37 (q, 2H), 11.85 (s, 1H)
RMN i3C (CDC13, 75 MHz) 8 (ppm) . 13.8, 14.7, 23.5, 29.7, 62,
110.1, 139.1, 164.2, 168.2 , 203.8
SM(ZC) m/z . [M+H] f - 214
GC/MS tR = 12,15 min
H-2
RMN 1H (CDC13, 200 MHz) ~ (ppm) . 1.36 (t, 3H) , 2 . 07 (s, 3H) ,
2.24 (s, 3H), 4.32 (q, 2H), 7.51 (S, 1H)
RMN i3C (CDC13, 75 MHz) 8 (ppm) . 14, 14.9, 29.3, 62, 103,4,
145.3, 165, 202
SM(IC) m/z . [M+H]+ - 172
GC/MS t~ = 9 , 3 8 min.
c-1
RMN 1H (CDC13, 300 MHz) ô (ppm) . 1.16 (d, 3H) , 1 .24 (t, 3H) ,
2.17 {s, 3H), 2.92 (m, 1H), 3.53 (d, 1H), 4.16 (q, 2H)
RMN 13C (CDC13, 50 MHz) 8 (ppm) , 13.3, 14.1, 28.8, 50.3, 56.8,
3Q 61, 174.4, 210.2
SM (IC) m/z . [M+H] + - 174
GC/MS t~ = 7 , 5 0 min
CA 02507669 2005-05-30
WO 2004/052836 33 PCT/FR2003/003542
~-z
RMN 1H (CDCl3, 300 MHz) 8 (ppm) . 1.11 (d, 3H) , 1 .25 (t, 3H) ,
2.20 (s, 3H), 2.92 (m, 1H), 3.86 (d, 1H), 4.16 (q, ZH)
RMN 13C (CDC13, 50 MEiz) 8 (ppm) . 10.8, 14.1, 28.2, 49.6, 55.3,
61.2, 174.2, 209.8,
SM(IC) m/z . [M+H] ~ - 174
GC/MS ~R = -7, 60 min
25 c-l'
RMN 1H (CDC7.3, 300 MHz) b (ppm) . 1 .20 (d, 3H) , 1 .23 (t, 3H) ,
2.05 (s, 3H), 2.20 (s, 3H), 3.36 (m, 1H), 4.15 (q, 2H), 4.84
(m, 1H) , 6 . 47 (d, lH)
SM(IC) m/z . [M+H] ~ - 216
GC/MS tR = l1, 02 min
c-2'
RMN ~'H (CDC13, 300 MHz) 8 (ppm) . 1. 16 (d, 3H) , 1 .26 (t, 3H) ,
1.99 (s, 3H), 2.23 (s, 3H), 3.07 (m, 1H), 4.19 (q, 2H), 4.84
(m, 1H) , 6 .31 (d, 1H)
sM ( zc) m/Z . [M+H] '~ - 216
CA 02507669 2005-05-30
WO 2004/052836 34 PCT/FR2003/003542
GC/MS tR = 11,50 min
e- l'
RMN iH (CDC13, 300 MHz) 8 (ppm) . 1 .47 (d, 3H) , 2 . 10 (s, 3H) ,
2.15 (s, 3H) , 4. 92 (q, 1H) , 7.31 (s, 1H)
SM (IC) m/z . [M+H] ~ - 170
e-2'
RMN 1H (CDC13, 300 MHz) ~ (ppm) . 1.23 (d, 3H), 1.29 (t, 3H),
2.03 (s, 3H), 2.08 (s, 3H), 3.64 (s, 1H), 4.22 (q, 2H), 4.58
(m, 1H) , 7.61 (s, 1H)
RMN 13C (CDC13, 75 MHz) b (ppm) . 13.6, 14.1, 19.5, 23, 61.2,
67.5, 121.6, 146.1, 165.2, 170.2
SM (IC) m/z . [M+H] + - 216
e-1
?5
RMr1 ~H (CDC13, 200 MHz) 8 (ppm) . 1.4 (d, 3H) , 1 . 84 (s, 3H) ,
3.39 (s, 3H), 4.79 (q, 1H),
SM(zC) m/z . [M+H]+ - 128
~0
GC/MS t~ ~ 7 , 5 9 min
CA 02507669 2005-05-30
WO 2004/052836 35 PCT/FR2003/003542
l-l'
RMN 1H (CDC13, 200 MHz) 8 (ppm) . 1.22 (d, 3H) , 1.44 .(d, 3H) ,
2.02 (m, 1H) , 2. 08 (s, 3H) , 4.16 (m, 1H) , 4.53 (m, 1H)
SM (IC) m/z . [M+H~ + - 172
HPLC: tR ~ 27 min
L-2'
RMN 1H (CDC13, 200 MHz) b (ppm) , 0.96 (d, 3H) , 1 .46 (d, 3H) ,
2.08 (s, 3H), 2.67 (m, 1H), 4.41 {m, 1H), 4.76 (m, 1H)
SM (TC) m/z . [M+H] '~ - 172
HPLC : tR ~ 2 3 , 5 min
1-3'
RMN 1H (CDC13, 200 MHz) 8 {ppm) . 1 , 16 (d, 3H) , 1 . 32 (d, 3H) ,
2.07 (s, 3H), 2.55 (m, 1H), 3.07 (m, 1H), 4.19 (q, 2H), 4,84
(m, 1H) , 6 . 31 (d, 1H)
SM (IC) m/z . [M+H] + - 172
1-4'
i0
RMN 1H (CDC13, 200 MHz) $ (gpm) . 0.80 (d, 3H) , 1 .38 (d, 3H) ,
2 . 09 (s, 3H) , 2 . 94 (m, 1H)=; 4 ''S& .(m, 1H) , 4 . 70 (m, 1H)
CA 02507669 2005-05-30
WO 2004/052836 36 PCT/FR2003/003542
SM(IC) m/z . [M+Hj~ - 172
HPLC : tR = 19 min
1-1
SM(IC) m/z . [M+HJ t - 130
GC/MS tR = S , 7 min
1-2
SM(IC) m/z . [M+H] * - 130
GC/MS tR = 6, 22 min
~o z-3
SM(IC) miz . [M+H] f - 130
GC/MS tR = 6 , 4 0 min
~5
1-4
SM(IC) zn/z . [M+Hj+ - 130
30 GC/MS t~ = 6 , 6 9 min
CA 02507669 2005-05-30
WO 2004/052836 37 PCT/FR2003/003542
Variante d~obtentiorz â partir du composé X du composé IV (Ri
H, R2 = benzyle, R3 - mêthyle et R4 = méthyle.
Le composé X est placé dans un ballon bicot avec du tamis
moléculaire activé dans l'éthanol anhydre. Le chlorydrate de
la benzylamine est ajouté par portions (une dizaine) pendant
trois heures. Le mélange est agité pendant 24h à température
ambiante. Le brut réactionnel est filtré sur vélite puis dilué
dans du dichlorométhane. La phase organique est lavée avec une
solution saturée d'hydrogénocarbonate de sodium puis avec de
l'eau. La phase organique est séchée sur sulfate de magnêsium
puis concentrée sous vide. Le brut est purifié sur
chromatographie de silice pour donner un rendement de 50 %.
.Analyses
RMN 1H {CDC13, 300 MHz) S {ppm) . 1 .27 (t, 3H) , 1 . 82 (s, 3H) ,
2.17 (s, 3H), 4.27 (q, 2H), 4.32 (d, 2H), 7.31 (m, 5H)
RMN 13C {CDC13, 50 MHz) 8 (ppm) . 13.9, 14.9, 28.6, 48.9, 61.7,
97.3, 127.2, 127.5, 128.6, 137.9, 153.0, 164.2, 199.7
SM(IC) rnJz : [M+H]f = 261
'.5
CA 02507669 2005-05-30
WO 2004/052836 38 PCT/FR2003/003542
Exemple de réduction asymétrique du composé H-2' en composés c-l' et c-2'
~ Préparation du catalyseur
Dans un tube de Schlenk, préalablement purgé (vide-argon), on
place sous argon le catalyseur avec son ligand, dans du
méthanol. On laisse agiter pendant une vingtaine de minutes,
20 jusqu'à l'obtention d'un milieu limpide.
~ Réduction
Le substrat est introduit dans l'autoclave avec un barreau
I5 aimanté et on introduit toujours sous argon la solution
préparée ci-dessus. Le milieu réactionnel est laissé sous
agitation pendant 17 H sous hydrogène à 50 bars.
Le méthanol est évaporé et du diehloromêthane est introduit.
On ajoute du charbon actif et on agite 15 minutes environ. Le
20 milieu est filtré sur Cêlite~ et évaporé. Le brut obtenu est
purifié par chromatographie sur colonne de silice.
Caalysewr proportions Rappoxt en
( /omol) Rendement c~z,/c~2, nanti.omxes
Li and (1.2 e cat) de c-2'
Rh(cod)aBF4 (4
mol
%) 79 % 25/54 90 j10
+ Phane hos
Rh(cod)~BF4 (4
mol
%) 66 % 18/4 66/34
+ Bina
Rh{cod)DipampBF4 ,~~ % 0/40 96/4
4 mol
>5