Note: Descriptions are shown in the official language in which they were submitted.
Le A36 492-:~L. CA 02508788 2005-06-06
-1-
3-PYRROLYL UREA DERIVATIVES AND THEIR USE AS ANTIVIRAL
AGENTS
The invention relates to substituted pyrroles and to processes for preparing
them, to their
use for the treatment and/or prophylaxis of diseases and also to their use for
producing
medicaments for the treatment and/or prophylaxis of diseases, particularly for
use as
antiviral agents, in particular against cytomegaloviruses.
DE-A 197 17 898 describes substituted pyrroles as additives for photographic
recording
material.
WO 99/23091 describes aromatic heterocyclic compounds as anti-inflammatory
agents
which may among other things also be suitable for treating viral infections.
Distamycin derivatives (four pyrroles linked via amide or urea groups) are
described inter
alia in Possati, L. et al., Clinical & Experimental Metastasis 1999, 17(7),
575-582, Manetti,
F. et al., Journal of Computer-Aided Molecular Design 2000, 14(4), 355-368 and
Turpin, J.
A. et al., Expert Opinion on Therapeutic Patents 2000, 10(12), 1899-1909 as
anti-HIV
active compounds.
Although structurally different agents with antiviral activity are on the
market,
development of resistance is a regular possibility. New agents for better and
effective
therapy are therefore desirable.
It is an object of the present invention, therefore, to provide new compounds
having equal
or improved antiviral action for the treatment of viral infectious diseases in
humans and
animals.
Surprisingly it has been found that the substituted pyrroles described in the
present
invention are highly active antivirally.
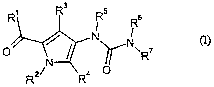
The present invention provides compounds of the formula
CA 02508788 2005-06-06
-2-
R~ R3 RS s
R
O ~ ~ N N~ ~ ~l)~
~R
N
R2~ Ra O
in which
Rl is -OR8 or -NR9Rlo,
R2 is hydrogen, C1-C6-alkyl or aryl,
it being possible for alkyl RZ to be substituted by 0, l, 2 or 3 substituents
R2n
selected independently of one another from the group consisting of halogen,
hydroxyl, C1-C6-alkoxy, hydroxycarbonyl, C1-C6-alkoxycarbonyl, C1-C6-
alkylcarbonyloxy, amino, C~-C6-alkylamino, aminocarbonyl, CI-C6-
alkylaminocarbonyl, C3-C8-cycloalkyl, 5- to 10-membered heterocyclyl, C6-Clo-
aryl,
phenoxy and 5- to 10-membered heteroaryl,
in which cycloalkyl, heterocyclyl, aryl or heteroaryl R2'1 may be substituted
by 0, 1, 2 or 3 substituents selected independently of one another from the
group consisting of halogen, hydroxyl, oxo, vitro, cyano, trifluoromethyl,
trifluoromethoxy, C~-C6-alkyl, C,-C6-alkoxy, hydroxycarbonyl, CI-C6-
alkoxycarbonyl, amino, C~-C6-alkylamino, aminocarbonyl, C1-C6-
alkylaminocarbonyl and phenyl,
it being possible for aryl R2 to be substituted by 0, l, 2 or 3 substituents
R2-2
selected independently of one another from the group consisting of halogen,
hydroxyl, vitro, cyano, trifluoromethyl, trifluoromethoxy, CI-C6-alkyl, Cl-C6-
alkoxy, hydroxycarbonyl, C1-C6-alkoxycarbonyl, amino, C1-C6-alkylamino,
aminocarbonyl, C,-C6-alkylaminocarbonyl, C3-Cg-cycloalkyl, 5- to 10-membered
heterocyclyl, C6-Coo-aryl and 5- to 10-membered heteroaryl,
R3 and R4 independently of one another are hydrogen or C1-C6-alkyl,
RS and R6 independently of one another are hydrogen or C1-C6-alkyl,
CA 02508788 2005-06-06
R7 is 3- to 12-membered carbocyclyl,
it being possible for the carbocyclyl to be substituted by 0, 1, 2, 3, 4 or S
substituents selected independently of one another from the group consisting
of
halogen, hydroxyl, C1-C6-alkyl and C1-C6-alkoxy,
R8 is hydrogen or C1-C6-alkyl,
it being possible for alkyl Rg to be substituted by 0, 1, 2 or 3 substituents
Rg-1
selected independently of one another from the group consisting of hydroxyl,
amino, C1-C6-alkoxy, C1-C6-alkylamino, aminocarbonyl, Cl-C6-
alkylcarbonylamino,
C3-Cg-cycloalkyl, 5- to 10-membered heterocyclyl, C6-Coo-aryl and 5- to 10
membered heteroaryl,
in which cycloalkyl, heterocyclyl, aryl or heteroaryl R8-~ may be substituted
by 0, 1, 2 or 3 substituents selected independently of one another from the
group consisting of halogen, hydroxyl, vitro, cyano, C1-C6-alkyl, C1-C6-
alkoxy, hydroxycarbonyl, C1-C6-alkoxycarbonyl, amino, CI-C6-alkylamino,
aminocarbonyl and C1-C6-alkylaminocarbonyl,
R9 is hydrogen or C1-C6-alkyl,
it being possible for alkyl R9 to be substituted by 0 or 1 substituent R9-1
selected
from the group consisting of hydroxyl, C~-C6-alkoxy, hydroxycarbonyl, C1-C6-
alkoxycarbonyl, amino, CI-C6-alkylamino, aminocarbonyl, C1-C6-
alkylaminocarbonyl, C3-C8-cycloalkyl, 5- to 10-membered heterocyclyl, C6-Clo-
aryl
and 5- to 10-membered heteroaryl,
in which cycloalkyl, heterocyclyl, aryl or heteroaryl R9-1 may be substituted
by 0, 1, 2 or 3 substituents selected independently of one another from the
group consisting of halogen, hydroxyl, vitro, cyano, C~-C6-alkyl, C1-C6-
alkoxy, hydroxycarbonyl, CI-C6-alkoxycarbonyl, amino, C~-C6-alkylamino,
aminocarbonyl and C1-C6-alkylaminocarbonyl,
and
CA 02508788 2005-06-06
-4-
R1° is hydrogen, C1-C6-alkyl, C3-C8-cycloalkyl, 5- to 10-membered
heterocyclyl, C6-
Coo-aryl or 5- to 10-membered heteroaryl,
it being possible for alkyl Rl° to be substituted by 0, 1, 2 or 3
substituents Rlo-i
selected independently of one another from the group consisting of halogen,
hydroxyl, C1-C6-alkoxy, hydroxycarbonyl, C~-C6-alkoxycarbonyl, amino; C1-C6-
alkylamino, aminocarbonyl, C1-C6-alkylaminocarbonyl, C3-C8-cycloalkyl, 5- to
10-
membered heterocyclyl, C6-C 1 °-aryl and 5- to 10-membered heteroaryl,
in which cycloalkyl, heterocyclyl, aryl or heteroaryl Rlo-1 may be substituted
by 0, 1, 2 or 3 substituents selected independently of one another from the
group consisting of halogen, hydroxyl, oxo, vitro, cyano, trifluoromethyl,
trifluoromethoxy, C1-C6-alkyl, C~-C6-alkoxy, hydroxycarbonyl, C~-C6-
alkoxycarbonyl, amino, C1-C6-alkylamino, aminocarbon~l and C1-C6-
alkylaminocarbonyl,
it being possible for cycloalkyl, heterocyclyl, aryl or heteroaryl Rl°
to be substituted
by 0, 1, 2 or 3 substituents Rlo-2 selected independently of one another from
the
group consisting of halogen, hydroxyl, vitro, cyano, trifluoromethyl,
trifluoromethoxy, C1-C6-alkyl, C1-C6-alkoxy, hydroxycarbonyl, C1-C6-
alkoxycarbonyl, amino, C1-C6-alkylamino, aminocarbonyl and C1-C6-
alkylaminocarbonyl,
or
R9 and R1° together with the nitrogen atom to which they are attached
form a 4- to 8-
membered heterocycle which may contain up to two further heteroatoms
from the series N, O and/or S,
it being possible for the heterocycle to be substituted by 0, 1, 2 or 3
substituents
selected independently of one another from the group consisting of halogen,
hydroxyl, C1-C6-alkyl, C1-C6-alkoxy, hydroxycarbonyl, C1-C6-alkoxycarbonyl,
amino, C1-C6-alkylamino, aminocarbonyl and C1-C6-alkylaminocarbonyl,
and their salts, their solvates and the solvates of their salts.
CA 02508788 2005-06-06
-5-
Compounds of the invention are the compounds of the formula (I) and their
salts, solvates
and solvates of the salts, compounds referred to below as exemplary
embodiments) and
their salts, solvates and solvates of the salts, where the compounds which are
encompassed
by formula (I) and are referred to below are not already salts, solvates and
solvates of the
salts.
The compounds of the invention may, depending on their structure, exist in
stereoisomeric
forms (enantiomers, diastereomers). The invention therefore provides the
enantiomers or
diastereomers and their respective mixtures. From such mixtures of enantiomers
and/or
diastereomers it is possible to isolate the stereoisomerically pure
constituents in a known
manner.
Where the compounds of the invention can occur in tautomeric forms the present
invention
embraces all tautomeric forms.
Salts preferred for the purposes of the present invention are physiologically
acceptable salts
of the compounds of the invention. Also embraced, however, are salts which,
though not
themselves suitable for pharmaceutical applications, can nevertheless be used,
for example,
for isolating or purifying the compounds of the invention.
Physiologically acceptable salts of the compounds of the invention embrace
acid addition
salts of mineral acids, carboxylic acids and sulfonic acids, examples being
salts of
hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid,
methanesulfonic acid,
ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid,
naphthalenedisulfonic acid,
acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid,
malic acid, citric
acid, fiunaric acid, malefic acid and benzoic acid.
Physiologically acceptable salts of the compounds of the invention also
embrace salts of
customary bases, such as, by way of example and preferably, alkali metal salts
(e.g. sodium
and potassium salts), alkaline earth metal salts (e.g. calcium and magnesium
salts), and
ammonium salts derived from ammonia or organic amines having 1 to 16 carbon
atoms, such
as, by way of example and preferably, ethylamine, diethylamine, triethylamine,
ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine,
dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-
methylmorpholine,
arginine, lysine, ethylenediamine and N-methylpiperidine.
CA 02508788 2005-06-06
-6-
Solvates refer for the purposes of the invention to those forms of the
compounds of the
invention which in the solid or liquid state form a complex through
coordination with
solvent molecules. Hydrates are one specific form of the solvates, in which
the
coordination takes place with water.
For the purposes of the present invention the substituents, unless otherwise
specified, have
the following definition:
Alkyl per se and "alk" and "alkyl" in alkoxy alkylamino alkylcarbonyl
alkylcarbonyloxy
and alkoxycarbonyl are a linear or branched alkyl radical having generally 1
to 6 ("C1-C6-
alkyl"), preferably 1 to 4, more preferably 1 to 3 carbon atoms, by way of
example and
preferably methyl, ethyl, n-propyl, isopropyl, tert-butyl, n-pentyl and n-
hexyl.
Alkoxv is, by way of example and preferably, methoxy, ethoxy, n-propoxy,
isopropoxy,
tent-butoxy, n-pentoxy and n-hexoxy.
Alkylamino is an alkylamino radical having one or two alkyl substituents
(chosen
independently of one another), by way of example and preferably methylamino,
ethylamino,
n-propylamino, isopropylamino, tent-butylamino, n-pentylamino, n-hexylamino,
N,N
dimethylamino, N,N diethylamino, N ethyl-N methylamino, N methyl-N n-
propylamino, N
isopropyl-N n-propylamino, N t-butyl-N methylamino, N ethyl-N n-pentylamino
and N n-
hexyl-N methylamino. C1-C3-alkylamino is for example a monoalkylamino radical
having 1
to 3 carbon atoms or a dialkylamino radical having 1 to 3 carbon atoms per
alkyl substituent.
Alkylcarbonyl is, by way of example and preferably, acetyl and propanoyl.
Alkylcarbonyloxy is, by way of example and preferably, methylcarbonyloxy,
ethylcarbonyloxy, n-propylcarbonyloxy, isopropylcarbonyloxy, tert-
butylcarbonyloxy, n-
pentylcarbonyloxy and n-hexylcarbonyloxy.
Alkoxycarbonyl is, by way of example and preferably, methoxycarbonyl,
ethoxycarbonyl,
n-propoxycarbonyl, isopropoxycarbonyl, tert-butoxycarbonyl, n-pentoxycarbonyl
and
n-hexoxycarbonyl.
Alkylaminocarbonyl is an alkylaminocarbonyl radical having one or two alkyl
substituents
(chosen independently of one another), by way of example and preferably
CA 02508788 2005-06-06
methylaminocarbonyl, ethylaminocarbonyl, n-propylaminocarbonyl,
isopropylaminocarbonyl, tert-butylaminocarbonyl, n-pentylaminocarbonyl, n-
hexylaminocarbonyl, N,N dimethylaminocarbonyl, N,N diethylaminocarbonyl, N
ethyl-N
methylaminocarbonyl, N methyl-N n-propylaminocarbonyl, N isopropyl-N n-
propylaminocarbonyl, N tert-butyl-N methylaminocarbonyl, N ethyl-N n-
pentylaminocarbonyl and N n-hexyl-N methylaminocarbonyl. C~-C3-
alkylaminocarbonyl is
for example a monoalkylaminocarbonyl radical having 1 to 3 carbon atoms or a
dialkylaminocarbonyl radical having 1 to 3 carbon atoms per alkyl substituent.
Alkylcarbonylamino is, by way of example and preferably, methylcarbonylamino,
ethylcarbonylamino, n-propylcarbonylamino, isopropylcarbonylamino, tert-
butylcarbonylamino, n-pentylcarbonylamino and n-hexylcarbonylamino.
Air 1 is a mono- to tricyclic aromatic, carbocyclic radical having generally 6
to 14 carbon
atoms; by way of example and preferably, phenyl, naphthyl and phenanthrenyl.
5- to 10-membered heteroaryl for the purposes of the invention is generally an
aromatic,
mono- or bicyclic radical having 5 to 10 ring atoms and up to 5 heteroatoms
from the series
S, O and/or N. Preference is given to 5- to 6-membered heteroaryls having up
to 4
heteroatoms. The heteroaryl radical may be attached via a carbon atom or
heteroatom. By
way of example and preferably mention may be made of the following: thienyl,
furyl,
pyrrolyl, thiazolyl, oxazolyl, imidazolyl, pyridyl, pyrimidyl, pyridazinyl,
indolyl, indazolyl,
benzofuranyl, benzothiophenyl, quinolinyl and isoquinolinyl.
C cloal 1 is a cycloalkyl group having generally 3 to 8, preferably 3 to 6,
carbon atoms,
by way of example and preferably cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl and
cycloheptyl.
3- to 12-membered carbocyclyl is a mono- or polycyclic, carbocyclic radical
having 3 to 12
ring atoms. 3- to 10-membered, especially 4- to 8-membered, carbocyclyl are
preferred.
Mono- or bicyclic carbocyclyl is preferred. The carbocyclyl radicals may be
saturated or
partly unsaturated. Saturated carbocyclyl radicals are preferred. By way of
example and
preferably mention may be made of the following: cyclopropyl, cyclobutyl,
cyclopentyl,
cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, cycloheptenyl,
cyclooctyl,
cyclooctenyl, cyclononyl, cyclononenyl, bicyclo[2.1.1]hexyl,
bicyclo[2.2.1]heptyl,
CA 02508788 2005-06-06
_g_
bicyclo[3.2.1]octyl, bicyclo[2.2.2]octyl, bicyclo[3.2.2]nonyl,
bicyclo[3.3.1]nonyl,
bicyclo[3.3.2]decyl, bicyclo[4.3.1]decyl, adamant-1-yl, adamant-2-yl,
bicyclo[2.2.1]heptenyl,
bicyclo[2.2.2]octenyl and bicyclo[3.2.2]nonenyl.
5- to 10-membered heterocyclyl is for the purposes of the invention a mono- or
bicyclic,
saturated or partly unsaturated heterocycle having up to three heteroatoms
from tie series
N, O and/or S, which is attached via a ring carbon atom or a nitrogen atom of
the
heterocycle. By way of example and preferably mention may be made of the
following:
tetrahydrofuryl, dihydrofuryl, imidazolidinyl, thiolanyl, dioxolanyl,
pyrrolidinyl, pyrrolinyl,
tetrahydropyranyl, dihydropyranyl, piperidinyl, piperazinyl, morpholinyl,
thiomorpholinyl,
7-oxabicyclo[2.2.1]heptanyl and 7-oxabicyclo[2.2.1]kept-5-enyl.
A 4- to 8-membered heterocycle having at least one ring nitrogen atom is for
the purposes
of the invention a saturated or partly unsaturated, monocyclic heterocycle
which may
contain up to two further heteroatoms from the series N, O and/or S and is
attached via a
ring nitrogen atom of the heterocycle. Preference is given to a 5- to 7-
membered, saturated,
monocyclic N-heterocycle which may contain a second nitrogen atom or an oxygen
atom as
a further heteroatom. By way of example and preferably mention may be made of
the
following: pyrrolidinyl, pyrrolinyl, oxazolidinyl, thiazolidinyl, piperidinyl,
piperazinyl,
morpholinyl, thiomorpholinyl, hexahydroazepinyl, hexahydro-1,4-diazepinyl,
octahydroazocinyl.
Halogen is fluorine, chlorine, bromine and iodine.
An * symbol on a carbon atom means that the compound, in terms of the
configuration at
this carbon atom, is in enantiopure form, by which is meant for the purposes
of the present
invention an enantiomeric excess of more than 90% (> 90% ee).
Preference is given for the purposes of the present invention to compounds of
the formula
(I)
in which
Rl is -OR8 or -NR9Rlo,
R2 is hydrogen or C~-C4-alkyl,
CA 02508788 2005-06-06
-9-
it being possible for alkyl RZ to be substituted by 0 or 1 substitutent RZ'1
selected
from the group consisting of hydroxyl, C1-C6-alkoxy, C1-C6-alkylcarbonyloxy,
C,-
C6-alkylaminocarbonyl, C3-C7-cycloalkyl, 5- to 6-membered heterocyclyl,
phenyl,
phenoxy and 5- to 6-membered heteroaryl,
in which cycloalkyl, heterocyclyl, phenyl or heteroaryl R2'' may be
substituted by 0, 1, 2 or 3 substituents selected independently of one another
from the group consisting of halogen, hydroxyl, oxo, nitro, cyano,
trifluoromethyl, trifluoromethoxy, C1-C6-alkyl, C~-C6-alkoxy,
hydroxycarbonyl, C~-C6-alkoxycarbonyl, amino, C1-C6-alkylamino,
aminocarbonyl, C1-C6-alkylaminocarbonyl and phenyl,
R3 and R4 are hydrogen,
RS and R6 are hydrogen,
R' is 6- to 8-membered carbocyclyl,
it being possible for carbocyclyl R' to be substituted by 0, 1, 2, 3 or 4
substituents
selected independently of one another from the group consisting of C1-C6-
alkyl,
Rg is C1-C4-alkyl,
it being possible for alkyl R8 to be substituted by 0, 1 or 2 substituents
R8'1 selected
independently of one another from the group consisting of hydroxyl, amino, C1-
C6-
alkoxy, C1-C6-alkylamino, aminocarbonyl, C1-C6-alkylcarbonylamino, pyridyl,
1,2,4-triazol-1-yl and pyrazol-1-yl,
R9 is hydrogen or CI-C6-alkyl,
it being possible for alkyl R9 to be substituted by 0 or 1 substituent R9'1
selected
from the group consisting of hydroxyl, C1-C6-alkoxy and amino,
and
R'° is hydrogen, CI-C6-alkyl, C3-C6-cycloalkyl or phenyl,
CA 02508788 2005-06-06
-10-
it being possible for alkyl Rl° to be substituted by 0 or 1 substituent
Rl°-~ selected
from the group consisting of hydroxyl, C~-C6-alkoxy, C1-C6-alkylamino, C5-C~-
cycloalkyl, 5- to 6-membered heterocyclyl, phenyl and 5- to 6-membered
heteroaryl,
in which cycloalkyl, heterocyclyl, phenyl or heteroaryl Rl°-I may be
substituted by 0, l, 2 or 3 substituents selected independently of one'another
from the group consisting of halogen, hydroxyl, vitro, cyano,
trifluoromethyl, trifluoromethoxy, C1-C6-alkyl, C1-C6-alkoxy,
hydroxycarbonyl, C~-C6-alkoxycarbonyl, amino, Ci-C6-alkylamino,
aminocarbonyl and C1-C6-alkylaminocarbonyl,
or
R9 and R1° together with the nitrogen atom to which they are attached
form a 5- to 6-
membered heterocycle which may contain up to two further heteroatoms
from the series N, O and/or S,
and their salts, their solvates and the solvates of their salts.
Preference for the purposes of the present invention is also given to
compounds of the
formula (I),
in which
R' is -ORg or -NR9Rlo,
R2 is hydrogen or C1-C4-alkyl,
it being possible for alkyl RZ to be substituted by 0 or 1 substituent R2-~
selected
from the group consisting of methoxy, diethylaminocarbonyl, cyclopropyl,
phenyl,
phenoxy and pyridyl,
in which phenyl R2-~ may be substituted by 0, 1 or 2 substituents selected
independently of one another from the group consisting of fluorine, chlorine,
vitro, cyano, trifluoromethyl, methyl, methoxy and methyloxycarbonyl,
R3 and R4 are hydrogen,
CA 02508788 2005-06-06
-11-
RS and R6 are hydrogen,
R' is bicyclo[2.2.1]heptyl,
it being possible for bicyclo[2.2.1]heptyl to be substituted by 0, l, 2, 3 or
4 methyl
groups,
R8 is C1-C3-alkyl,
it being possible for alkyl R8 to be substituted by 0 or 1 substituent R8'1
selected
independently of one another from the group consisting of hydroxyl,
dimethylamino, aminocarbonyl, methylcarbonylamino, pyridyl, 1,2,4-triazol-1-yl
and pyrazol-1-yl,
R9 is hydrogen,
and
R1° is hydrogen, CI-C4-alkyl, cyclopropyl or cyclopentyl,
it being possible for alkyl Rl° to be substituted by 0 or 1 substituent
Rlo-1 selected
from the group consisting of hydroxyl, methoxy, dimethylamino, phenyl, pyridyl
and imidazol-1-yl,
in which phenyl Rl°'~ may be substituted by 0, 1 or 2 methoxy
substituents,
and their salts, their solvates and the solvates of their salts.
Preference is given for the purposes of the present invention to compounds of
the formula
in which
R' is -OR8 or -NR9Rlo,
RZ is hydrogen or C1-C3-alkyl,
CA 02508788 2005-06-06
' -12-
it being possible for alkyl R2 to be substituted by 0 or 1 substituent RZ-I
selected
from the group consisting of CS-C~-cycloalkyl, 5- to 6-membered heterocyclyl,
phenyl and 5- to 6-membered heteroaryl,
in which cycloalkyl, heterocyclyl, phenyl or heteroaryl RZ-~ may be
substituted by 0, 1, 2 or 3 substituents selected independently of one-another
from the group consisting of halogen, hydroxyl, vitro, cyano,
trifluoromethyl, trifluoromethoxy, CI-C6-alkyl, CI-C6-alkoxy,
hydroxycarbonyl, C,-C6-alkoxycarbonyl, amino, C1-C6-alkylamino,
aminocarbonyl and C~-C6-alkylaminocarbonyl,
R3 and R4 are hydrogen,
RS and R6 are hydrogen,
R' is 6- to 8-membered carbocyclyl,
it being possible for carbocyclyl R' to be substituted by 0, l, 2, 3 or 4
substituents
selected independently of one another from the group consisting of CI-C6-
alkyl,
R8 is CI-C4-alkyl,
it being possible for alkyl Rg to be substituted by 0, 1 or 2 substituents Rg-
I selected
independently of one another from the group consisting of hydroxyl, amino and
CI-
C6-alkoxy,
R9 is hydrogen or CI-C6-alkyl,
it being possible for alkyl R9 to be substituted by 0 or 1 substituent R9-I
selected
from the group consisting of hydroxyl, CI-C6-alkoxy and amino,
and
RI° is hydrogen or CI-C6-alkyl,
it being possible for alkyl RI° to be substituted by 0 or 1 substituent
RIO-I selected
from the group consisting of CS-C7-cycloalkyl, 5- to 6-membered heterocyclyl,
phenyl and 5- to 6-membered heteroaryl,
CA 02508788 2005-06-06
-13-
in which cycloalkyl, heterocyclyl, phenyl or heteroaryl Rlo-i may be
substituted by 0, 1, 2 or 3 substituents selected independently of one another
from the group consisting of halogen, hydroxyl, nitro, cyano,
trifluoromethyl, trifluoromethoxy, CI-C6-alkyl, C1-C6-alkoxy,
hydroxycarbonyl, C1-C6-alkoxycarbonyl, amino, C~-C6-alkylamino,
aminocarbonyl and CI-C6-alkylaminocarbonyl,
and their salts, their solvates and the solvates of their salts.
Preference for the purposes of the present invention is also given to
compounds of the
formula (I),
in which
Rl is -OR8 or -NR9Rlo,
R2 is hydrogen or benzyl,
R3 and R4 are hydrogen,
RS and R6 are hydrogen,
R' is bicyclo[2.2.1]heptyl,
it being possible for bicyclo[2.2.1]heptyl to be substituted by 0, 1, 2, 3 or
4 methyl
groups,
R8 is methyl or ethyl,
R9 is hydrogen,
and
R'° is hydrogen or benzyl,
and their salts, their solvates and the solvates of their salts.
Preference for the purposes of the present invention is also given to
compounds of the
formula (I) in which RI is -ORg and Rg is methyl or ethyl.
CA 02508788 2005-06-06
-14-
Preference for the purposes of the present invention is also given to
compounds of the
formula (I) in which RI is -NR9R1°, R9 is hydrogen and Rl° is
hydrogen or benzyl.
Preference for the purposes of the present invention is also given to
compounds of the
formula (I) in which R2 is hydrogen.
Preference for the purposes of the present invention is also given to
compounds of the
formula (I) in which R3, R4, RS and R6 are hydrogen.
Preference for the purposes of the present inventions is also given to
compounds of the
formula (I) in which R' is adamantyl.
Preference for the purposes of the present inventions is also given to
compounds of the
formula (I) in which R' is bicyclo[2.2.1]heptyl, it being possible for
bicyclo[2.2.1]heptyl to
be substituted by 0, 1, 2, 3 or 4 methyl groups.
Preference here is given to compounds of the formula (I) in which R' is
bicyclo[2.2.1]heptyl, bicyclo[2.2.1]heptyl being substituted by 3 methyl
groups.
Particular preference is given here to compounds of the formula (I) in which
R' is 1,7,7-
trimethylbicyclo[2.2.1]hept-2-yl.
Very particular preference is given here to compounds of the formula (I) in
which R' is
(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl, (1 S,2R,4S)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-yl, (1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-
2-yl or
(1S,2S,4S)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl, and mixtures thereof,
particularly the
mixtures of enantiomers (1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-yl and
(1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]kept-2-yl and also of the enantiomers
(1R,2R,4R)-
1,7,7-trimethylbicyclo[2.2.1]kept-2-yl and (1S,2S,4S)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-
yl.
Upmost preference is given here to compounds of the formula (I) in which R' is
(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl.
The invention further provides a process for preparing the compounds of the
formula (I),
where
CA 02508788 2005-06-06
- I 5 -
according to process [A]
compounds of the formula
z
f O
(IIa), ,
0 ~O
in which
R' is -ORg,
R8 is the optionally substituted alkyl indicated for R8 in formula (I), and
R2, R3 and R4 are as defined above,
are reacted in the first stage with a reducing agent,
in the second stage optionally with compounds of the formula
I0 X'-RS IIIII_
in which
RS is as defined above and
X1 is halogen, preferably bromine or chlorine,
and in the third stage, in the presence of a carbonic acid derivative, with
compounds of the
formula
(IV)~
H
in which
R6 and R' are as defined above,
to give compounds of the formula
CA 02508788 2005-06-06
-16-
R~ R3 Rs s
I R
O ~ ~ N N' 7 (Ia)~
1~ R
N
R2~ Ra O
in which
R' is -ORg,
R8 has the definition as in formula (IIa), and
R2, R3, R4, R5, R6 anti R' are as defined above,
or
according to process :B]
compounds of the formula (Ia)
in which
R8 is methyl or ethyl,
are reacted in the pre: ence of bases to give compounds of the formula
R~ R3 Rs s
I R
O ~ ~ N N~ ~ ~b)~
~R
N
R2~ Ra O
in which
R' is -ORg,
I S R8 is hydrogen, and
Rz, R3, R4, R5, R6 and R' are as defined above,
or
CA 02508788 2005-06-06
- 17 -
according to process [C]
compounds of the formula (Ib) are reacted with compounds of the formula
R'-H (V),
in which
Rl is as defined above,
in the presence of dehydrating reagents to give compounds of the formula (I),
or
according to process [D]
compounds of the formula
1
t
O ,O_
(IIb),
in which
Rl is -NR9R~°, and
R2, R3, R4, R9 and Rl° are as defined above,
are reacted in the first stage with a reducing agent,
in the second stage optionally with compounds of the formula (III)
and in the third stage, in the presence of a carbonic acid derivative, with
compounds of the
formula (IV)
to give compounds of the formula
CA 02508788 2005-06-06
-18-
R~ R3 RS s
l R
O ~ ~ N N\ 7 (Ic)~
~R
N
R2~ Ra O
in which
Rl is -NR9Rl°, and
R2, R3, R4, R5, R6, R7, R9 and R'° are as defined above,
or
according to process [E]
compounds of the formula
1
f R5 Rs
O N N \ 7 (Id),
R
O
in which
RI, R3, R4, R5, R6 and R' are as defined above,
are reacted with compounds of the formula
XZ-R2 (VIII),
in which
R2 is as defined above, and
Xz is halogen, preferably bromine or chlorine,
to give compounds of the formula (I).
Formula (I) embraces the compounds (Ia), (Ib), (Ic) and (Id).
CA 02508788 2005-06-06
-19-
Formula (II) embraces the compounds (IIa) and (IIb).
The compounds of the formula (III), (IV), (V) and (VIII) are known or can be
synthesized
by known processes from the corresponding reactants.
The following applies to processes [A] and [D]:
I st stage:
The reaction takes place in general in inert solvents, preferably in a
temperature range from
0°C up to the reflux of the solvents under atmospheric pressure up to 3
bar.
Reducing agents are, for example, palladium on active carbon and hydrogen,
formic
acid/triethylamine/palladium on active carbon, zinc, zinclhydrochloric acid,
iron,
iron/hydrochloric acid, iron(II) sulfate/hydrochloric acid, sodium sulfide,
sodium disulfide
sodium dithionite, ammonium polysulfide, sodium borohydride/nickel chloride,
tin
dichloride, titanium trichloride or Raney nickel and aqueous hydrazine
solution, preference
being given to Raney nickel and aqueous hydrazine solution.
Inert solvents are, for example, ethers such as diethyl ether, methyl tert-
butyl ether, 1,2-
dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene
glycol
dimethyl ether, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-
butanol or
tent-butanol, hydrocarbons such as benzene, xylene, toluene, hexane,
cyclohexane or
petroleum fractions, or other solvents such as dimethylformamide,
dimethylacetamide,
acetonitrile or pyridine, and, in the case of water-miscible solvents,
mixtures thereof with
water; a preferred solvent is methanol, ethanol, isopropanol or, in the case
of Raney nickel
and aqueous hydrazine solution, tetrahydrofuran.
2nd stage:
The reaction takes place in general in inert solvents, optionally in the
presence of a base,
preferably in a temperature range from -20°C to 40°C under
atmospheric pressure.
Bases are, for example, amides such as sodium amide, lithium
hexamethyldisilazide,
potassium hexamethyldisilazide, lithium diisopropylamide, or other bases such
as sodium
hydride, DBU or diisopropylethylamine, preferably sodium amide, lithium
hexamethyldisilazide, potassium hexamethyldisilazide or lithium
diisopropylamide.
CA 02508788 2005-06-06
-20-
Inert solvents are, for example, ethers, such as diethyl ether, methyl tert-
butyl ether, 1,2-
dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene
glycol
dimethyl ether, hydrocarbons such as benzene, ethylbenzene, xylene, toluene,
preferably
tetrahydrofuran ortoluene.
3rd stage:
The reaction takes place in general in inert solvents, preferably in a
temperature range from
room temperature up to 40°C under atmospheric pressure.
Carbonic acid derivatives are, for example, N,N-carbonyldiimidazole, phosgene,
diphosgene, triphosgene, phenyl chloroformate or 4-nitrophenyl chloroformate,
preference
being given to N,N-carbonyldiimidazole.
Inert solvents are, for example, halogenated hydrocarbons such as methylene
chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
dichloroethane or trichloroethylene, ethers such as diethyl ether, methyl tent-
butyl ether,
1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or
diethylene glycol
dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane,
cyclohexane or
petroleum fractions, or other solvents such as ethyl acetate, acetone,
dimethylformamide,
dimethylacetamide, 2-butanone, dimethyl sulfoxide, acetonitrile or pyridine,
and, in the
case of water-miscible solvents, mixtures thereof with water, preference being
given to
dimethyl sulfoxide.
The following applies to process [B]:
The reaction takes place in general in inert solvents, preferably in a
temperature range from
0°C up to the reflux of the solvents under atmospheric pressure.
Bases are, for example, alkali metal hydroxides such as sodium, lithium or
potassium
hydroxide, or alkali metal carbonates such as cesium carbonate, sodium or
potassium
carbonate, preferably sodium hydroxide.
Inert solvents are, for example, halogenated hydrocarbons such as methylene
chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
dichloroethane or trichloroethylene, ethers such as diethyl ether, methyl tert-
butyl ether,
CA 02508788 2005-06-06
-21-
1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethylether or
diethylene glycol
dimethyl ether, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-
butanol or
tent-butanol, hydrocarbons such as benzene, xylene, toluene, hexane,
cyclohexane or
petroleum fractions, or other solvents such as dimethylformamide,
dimethylacetamide,
dimethyl sulfoxide, acetonitrile or pyridine, or mixtures of solvents with
water; preference
as solvent is given to a mixture of ethanol and water.
The following applies to process [C]:
The reaction takes place in general in inert solvents, optionally in the
presence of a base,
preferably in a temperature range from -70°C to 40°C under
atmospheric pressure.
Suitable dehydrating reagents in this case are, for example, carbodiimides
such as N,N'-
diethyl-, N,N,'-dipropyl-, N,N'-diisopropyl-, N,N'-dicyclohexylcarbodiimide, N-
(3-
dimethylaminoisopropyl)-N'-ethylcarbodiimide hydrochloride (EDC), N-
cyclohexylcarbodiimide-N'-propyloxymethyl-polystyrene (PS-carbodiimide) or
carbonyl
compounds such as carbonyldiimidazole, or 1,2-oxazolium compounds such as 2-
ethyl-5-
phenyl-1,2-oxazolium 3-sulfate or 2-tert-butyl-5-methylisoxazolium
perchlorate, or
acylamino compounds such as 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline, or
propanephosphonic anhydride, or isobutyl chloroformate, or bis(2-oxo-3-
oxazolidinyl)phosphoryl chloride or
benzotriazolyloxytri(dimethylamino)phosphonium
hexafluorophosphate, or O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate (HBTU), 2-(2-oxo-1-(2H)-pyridyl)-1,1,3,3-
tetramethyluronium
tetrafluoroborate (TPTU) or O-(7-azabenzotriazol-1-yl)-N,N,N',N'-
tetramethyluronium
hexafluorophosphate (HATU), or 1-hydroxybenzotriazole (HOBt), or benzotriazol-
1-
yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP), or mixtures of
these,
with bases.
Bases are; for example, alkali metal carbonates, such as sodium or potassium
carbonate, or
sodium or potassium hydrogen carbonate, or organic bases such as
trialkylamines, for
example triethylamine, N-methylmorpholine, N-methylpiperidine, 4-
dimethylaminopyridine or diisopropylethylamine, or DBU, DBN, pyridine;
triethylamine is
preferred.
Preferably the condensation is carried out with carbonyldiimidazole.
CA 02508788 2005-06-06
-22-
Inert solvents are, for example, halogenated hydrocarbons such as methylene
chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
dichloroethane or trichloroethylene, ethers such as diethyl ether, methyl tert-
butylether, 1,2-
dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene
glycol
dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane,
cyclohexane or
petroleum fractions, or other solvents such as ethyl acetate, acetone,
dimethylformamide,
dimethylacetamide, 2-butanone, dimethyl sulfoxide, acetonitrile or pyridine,
and, in the
case of water-miscible solvents, mixtures thereof with water, preference being
given to
dimethylformamide.
The following applies to process [E]:
The reaction takes place in general in inert solvents, in the presence of a
base, preferably in
a temperature range from -20°C to 40°C under atmospheric
pressure.
Bases are, for example, amides such as sodium amide, lithium
hexamethyldisilazide,
potassium hexamethyldisilazide, lithium diisopropylamide, or other bases such
as sodium
hydride, DBU, diisopropylethylamine or potassium tent-butoxide, preference
being given to
potassium tent-butoxide.
Inert solvents are, for example, ethers such as diethyl ether, methyl tent-
butyl ether, 1,2-
dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene
glycol
dimethyl ether, hydrocarbons such as benzene, ethylbenzene, xylene, toluene,
or other
solvents such as dimethylformamide, preference being given to
dimethylformamide.
The compounds of the formula (II) are known or can be prepared by reacting
compounds of
the formula
O R3 O
CI / N+
CI ~ ~ ~ O (VI)~
CI RZ/ R4
in which
R2, R3 and R4 are as defined above,
CA 02508788 2005-06-06
-23-
with compounds of the formula (V).
The reaction takes place in general in inert solvents, optionally in the
presence of a base,
preferably in a temperature range from room temperature to 40°C under
atmospheric
pressure.
Bases are, for example, alkali metal carbonates such as cesium carbonate,
sodium or
potassium carbonate, or potassium tert-butoxide, or other bases such as sodium
hydride,
DBU, triethylamine or diisopropylethylamine, preference being given to
diisopropylethylamine and triethylamine.
Inert solvents are, for example, halogenated hydrocarbons such as methylene
chloride,
trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-
dichloroethane or trichloroethylene, ethers such as diethyl ether, methyl tert-
butylether, 1,2-
dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene
glycol
dimethyl ether, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-
butanol or
tert-butanol, hydrocarbons such as benzene, xylene, toluene, hexane,
cyclohexane or
petroleum fractions, or other solvents such as ethyl acetate, acetone,
dimethylformamide,
dimethylacetamide, 2-butanone, dimethyl sulfoxide, acetonitrile or pyridine,
preference
being given to ethanol and tetrahydrofuran.
The compounds of the formula (VI) are known or can be prepared by reacting
compounds
of the formula
R3
O
CI
CI
(VII),
CI R2 N R4
in which
R2, R3 and R4 are as defined above,
with fuming nitric acid, concentrated nitric acid, nitrating acid or other
proportions of
sulfuric acid and nitric acid, optionally in acetic anhydride as solvent,
preferably in a
temperature range from -60°C to 0°C under atmospheric pressure.
CA 02508788 2005-06-06
-24-
The compounds of the formula (VII) are known or can be synthesized by known
processes
from the corresponding reactants.
The introduction of the substituent R2 by alkylating methods that are known to
the skilled
worker may take place, depending on the substitution pattern of the pyrrole,
at different
points on the synthesis route.
Synthesis scheme 1:
CA 02508788 2005-06-06
- 25 -
O~N=O O
r ~ "- N°~ O r ~ ---_
N
CI " CI
CI C~ CI
CI
HsC CHa
'J HsC CHa
"~~CHa \ Br
lI ~~ ",~CHa
0 ",
H
H
H3C
H3C
H3C CHa
,,uCHa " ~ aC CHa
I N~ ",~CHa
O r N
H
H H
HO
NH2 I i NHZ
HsC CHa HsC CHa
O H O
-~(~ "~~CHa N~ ,"~CHa
O r N '\ H ."~ H ~ O r
NH / NH H H
i
\ / \
CA 02508788 2005-06-06
Synthesis scheme 2:
-26-
o-
~ N03
N -"
N
H
H3C CH3 O
."~ CH3 O
H H.
The compounds of the general formula (I) according to the invention exhibit an
unforeseeable, surprising spectrum of action. They exhibit an antiviral action
on
representatives of the group of Herpes viridae (herpes viruses), particularly
on
cytomegaloviruses (CMV), particularly on the human cytomegalovirus (HCMV).
They are
suitable accordingly for the treatment and prophylaxis of diseases,
particularly of infections
with viruses, especially the abovementioned viruses, and the infectious
diseases caused
thereby. By a viral infection is meant hereinafter not only an infection with
a virus but also
a disease caused by an infection with a virus.
The compounds of the general formula (I) can, on the basis of their particular
properties, be
used for producing medicaments which are suitable for the prophylaxis and/or
treatment of
diseases, particularly viral infections.
Areas of indication which may be mentioned by way of example include the
following:
IS 1) treatment and prophylaxis of HCMV infections in AIDS patients
(retinitis,
pneumonitis, gastrointestinal infections).
CA 02508788 2005-06-06
-27-
2) Treatment and prophylaxis of cytomegalovirus infections in bone-marrow and
organ transplant patients who develop often life-threatening HCMV pneumonitis,
or
encephalitis, and gastrointestinal and systemic HCMV infections.
3) Treatment and prophylaxis of HCMV infections in neonates and infants.
4) Treatment of an acute HCMV infection in pregnant women.
5) Treatment of HCMV infection in immunosuppressed patients in association
with
cancer and cancer therapy.
The compounds of the invention are preferably used for producing medicaments
which are
suitable for the prophylaxis and/or treatment of infections with a
representative of the
group of Herpes viridae, particularly a cytomegalovirus, in particular the
human
cytomegalovirus.
The compounds of the invention, on the basis of their pharmacological
properties, can be
used alone and, if required, in combination with other active ingredients,
particularly active
antiviral ingredients such as gancyclovir or acyclovir, for example, for the
treatment andlor
prevention of viral infections, especially HCMV infections.
The present invention further provides for the use of the compounds of the
invention for
the treatment and/or prophylaxis of diseases, preferably of viral infections,
particularly of
infections with the human cytomegalovirus (HCMV) or with another
representative of the
group of the Herpes viridae.
The present invention further provides for the use of the compounds of the
invention for
the treatment and/or prophylaxis of diseases, especially of the aforementioned
diseases.
The present invention further provides for the use of the compounds of the
invention for
producing a medicament for the treatment and/or prophylaxis of diseases,
especially of the
aforementioned diseases.
The present invention further provides a method for the treatment and/or
prophylaxis of
diseases, especially of the aforementioned diseases, using an antivirally
active amount of
the compounds of the invention.
CA 02508788 2005-06-06
-28-
The compounds of the invention may act systemically and/or locally. For this
purpose they
may be administered in a suitable way, such as, for example, orally,
parenterally,
pulmonally, nasally, sublingually, lingually, buccally , rectally, dermally,
transdermally,
conjunctivally, optically or as an implant or stmt.
For these administration routes it is possible to administer the compounds of
the invention
in suitable administration forms.
Administration forms suitable for oral administration are those which function
according to
the prior art and deliver the compounds of the invention rapidly and/or in
modified form,
and which comprise the compounds of the invention in crystalline and/or
amorphized
and/or dissolved form, such as, for example, tablets (uncoated or coated
tablets, having for
example coatings which are resistant to gastric juice or are insoluble or
dissolve with a
delay and control the release of the compound of the invention), tablets which
disintegrate
rapidly in the mouth, or films/wafers, films/lyophilizates, capsules (for
example, hard or
soft gelatin capsules), sugar-coated tablets, granules, pellets, powders,
emulsions,
suspensions, aerosols or solutions.
Parenteral administration can take place with avoidance of an absorption step
(for example,
intravenously, intraarterially, intracardiacally, intraspinally or
intralumbarly) or with
inclusion of absorption (for example, intramuscularly, subcutaneously,
intracutaneously,
percutaneously or intraperitoneally). Administration forms suitable for
parenteral
administration include injection preparations and infusion preparations in the
form of
solutions, suspensions, emulsions, lyophilizates or sterile powders.
Examples of drug forms suitable for the other administration routes include,
for example,
drug forms for inhalation (including powder inhalers, nebulizers), nasal
drops, nasal
solutions and nasal sprays; tablets, films/wafers or capsules for lingual,
sublingual or
buccal administration, suppositories, preparations for the eyes or ears,
vaginal capsules,
aqueous suspensions (lotions, shaking mixtures), lipophilic suspensions,
ointments,
creams, transdermal therapeutic systems, milk, pastes, foams, dusting powders,
implants or
stems.
The compounds of the invention can be converted into the administration forms
recited.
This can be done in a manner known per se by mixing with inert, nontoxic,
CA 02508788 2005-06-06
-29-
pharmaceutically appropriate excipients. These excipients include, among
others, carriers
(for example, microcrystalline cellulose, lactose, mannitol), solvents (for
example, liquid
polyethylene glycols), emulsifiers and dispersants or wetting agents (examples
being
sodium dodecyl sulfate, polyoxysorbitan oleate), binders (for example,
polyvinylpyrrolidone), synthetic and natural polymers (for example, albumin),
stabilizers
(e.g. antioxidants such as, for example, ascorbic acid), colorants (e.g.,
inorganic pigments
such as, for example, iron oxides) and taste- and/or odor-masking agents.
The present invention further provides medicaments which comprise at least one
compound of the invention, usually together with one or more inert, nontoxic,
pharmaceutically appropriate excipients, and also for their use for the
purposes specified
above.
In general it has proven advantageous in the case of intravenous
administration to
administer amounts of about 0.001 to 10 mg/kg, preferably about 0.01 to 5
mg/kg, of body
weight in order to achieve effective results, and, in the case of oral
administration, the dose
is about 0.01 to 25 mg/kg, preferably 0.1 to 10 mg/kg, of body weight.
It may nevertheless be necessary where appropriate to deviate from the amounts
specified,
specifically as a function of body weight, administration route, individual
response to the
active ingredient, mode of preparation and time or interval at which
administration takes
place. Thus in certain cases it may be sufficient to employ less than the
minimum amount
mentioned above, while in other cases the upper limit mentioned must be
exceeded. In the
event of administration of relatively large amounts it may be advisable to
divide them into
a number of individual doses over the day.
The percentages in the tests and examples below are percentages by weight
unless
otherwise indicated; parts are parts by weight. Solvent ratios, dilution
ratios and
concentration figures of liquid/liquid solutions are based in each case on
volume.
CA 02508788 2005-06-06
-30-
A. Examples
Abbreviations used:
CD3CN Deuteroacetonitrile
DCI direct chemical ionization (in MS)
DCM Dichloromethane
DIEA N,N diisopropylethylamine (Hiinig's base)
DMSO dimethyl sulfoxide
DMF N,N dimethylformamide
EA ethyl acetate (acetic acid ethyl ester)
EI electron impact ionization (in MS)
ESI electrospray ionization (in MS)
h Hour
HPLC high-pressure, high-performance liquid chromatography
conc. Concentrated
LC-MS liquid chromatography-coupled mass spectroscopy
LDA lithium diisopropylamide
m.p. melting point
MS mass spectroscopy
NMR nuclear magnetic resonance spectroscopy
RP-HPLC reverse phase HPLC
RT room temperature
Rt retention time (in HPLC)
THF Tetrahydrofuran
TLC thin-layer chromatography
HPLC and LC-MS methods:
Method 1 (LC-MS):
Instrument: Micromass Quattro LCZ, with HPLC Agilent series 1100; column: Grom-
SIL120 ODS-4 HE, 50 mm x 2.0 mm, 3 ~.m; eluent A: 1 1 water + 1 ml 50% formic
acid,
eluent B: 1 1 acetonitrile + 1 ml SO% formic acid; gradient: 0.0 min 100%A -~
0.2 min
CA 02508788 2005-06-06
-31-
100%A -~ 2.9 min 30%A -~ 3.1 min 10%A --j 4.5 min 10%A; oven: 55°C;
flow rate:
0.8 ml/min; UV detection: 208-400 nm.
Method 2 (LC-MS):
MS instrument type: Micromass ZQ; HPLC instrument type: Waters Alliance 2790;
column: Grom-Sil 120 ODS-4 HE, 50 mm x 2 mm, 3.0 Vim; eluent A: water + 500
~.l 50%
formic acid; eluent B: acetonitrile + 500 ~l 50% formic acidll; gradient: 0.0
min 5%B -~
2.0 min 40%B ~ 4.5 min 90%B ~ 5.5 min 90%B; oven: 45°C; flow rate: 0.0
min
0.75 ml/min ~ 4.5 min 0.75 ml/min 5.5 min -3 5.5 min 1.25 ml/min; UV
detection:
210 nm.
Method 3 (LC-MS):
Instrument: Micromass Platform LCZ with HPLC Agilent series 1100; column: Grom-
SIL120 ODS-4 HE, 50 mm x 2.0 mm, 3 um; eluent A: 1 1 water + 1 ml 50% formic
acid,
eluent B: 1 1 acetonitrile + 1 ml 50% formic acid; gradient: 0.0 min 100%A -~
0.2 min
100%A -~ 2.9 min 30%A -~ 3.1 min 10%A -~ 4.5 min 10%A; oven: 55°C; flow
rate:
0.8 ml/min; UV detection: 208-400 nm.
Method 4 (LC-MS):
MS instrument type: Micromass ZQ; HPLC instrument type: HP 1100 series; UV
DAD;
column: Grom-Sil 120 ODS-4 HE, 50 mm x 2 mm, 3.0 Vim; eluent A: water + 500 ~1
50%
formic acid/l, eluent B: acetonitrile + 500 ~.1 50% formic acid/l; gradient:
0.0 min 0%B -~
2.9 min 70%B -~ 3.1 min 90%B -~ 4.5 min 90%B; oven: 50 °C; flow rate:
0.8 ml/min;
UV detection: 210 nm.
Method 5 (LC-MS):
MS instrument type: Micromass ZQ; HPLC instrument type: Waters Alliance 2795;
column: Merck Chromolith SpeedROD RP-18e, 50 mm x 4.6 mm; eluent A: water +
500 ~1 50% formic acid/l; eluent B: acetonitrile + 500 ~1 50% formic acid/1;
gradient:
0.0 min 10%B~ 3.0 min 95%B~ 4.0 min 95%B; oven: 35°C; flow rate: 0.0
min
1.0 mllmin-~ 3.0 min 3.0 ml/min~ 4.0 min 3.0 ml/min; UV detection: 210 nm.
Method 6 (preparative HPLC):
CA 02508788 2005-06-06
-32-
Column: Nucleosil 100-5 C 18 Nautilus, S Vim, 20 mm x 50 mm, 220 nm, 550 ~1
injection
volume; eluent A: water + 0.3% formic acid, eluent B: acetonitrile; gradient:
0.0 min
10%B, 2.0 min 10%B, 6.0 min 90%B, 7.0 min 90%B, 7.1 min 10%B, 8.0 min 10%B;
flow
rate: 25 ml/min; UV detection: 210 nm.
Method 7 (preparative HPLC):
Column: Waters XTerra Prep MS C18, 5 Vim, 19 mm x 20 mm; injection volume 700
~l;
eluent A: acetonitrile, eluent B: water + 0.1% formic acid; gradient: 0.00 min
10%A,
2.00 min 10%A, 6.00 min 90%A, 7.00 min 90%A, 7.10 min 10%A, 8.00 min 10%A;
flow
rate: 25 ml/min; UV detection: 220 nm.
Method 8 (LC-MS):
Instrument: Micromass TOF-MUX-Interface 4-fold parallel injection with HPLC
Waters
600; column: Grom-SIL 120, 50 mm x 2.0 mm, 3.0 Vim; eluent A: 1 1 water + 1 ml
50%
formic acid, eluent B: 1 1 acetonitrile + 1 ml 50% formic acid; gradient: 0.0
min 100%A -~
0.2 min 100%A ~ 2.9 min 30%A ~ 3.1 min 10%A ~ 4.5 min 10%A -~ 4.6 min 100%A
~ 6.5 min 100%A; oven: room temperature; flow rate: 0.8 ml/min; UV detection:
210 nm.
Method 9 (preparative HPLC):
Column: Macherey-Nagel VP 50121 Nucleosil 100-5 C 18 Nautilus, 20 mm x 50 mm;
injection volume 500 ~1; eluent A: acetonitrile, eluent B: water + 0.1% formic
acid;
gradient: 0.00 min 10%A, 2.00 min 10%A, 6.00 min 90%A, 7.00 min 90%A, 7.10 min
I 0%A, 8.00 min 10%A; flow rate: 25 ml/min; UV detection: 220 nm.
Method 10 (LC-MSI:
Instrument: Micromass Platform LCZ with HPLC Agilent series 1100; column: Grom-
SIL120 ODS-4 HE, 50 mm x 2.4 mm, 3 Vim; eluent A: 1 1 water + 1 ml 50% formic
acid,
eluent B: 1 1 acetonitrile + 1 ml 50% formic acid; gradient: 0.0 min 100%A -~
0.2 min
100%A -~ 2.9 min 30%A ~ 3.1 min 10%A ~ 4.5 min 10%A; oven: 55°C; flow
rate:
0.8 ml/min; UV detection: 210 nm.
Method 11 (preparative HPLC):
CA 02508788 2005-06-06
- 33 -
Column material: YMC GEL ODS AQ S 5/15 Vim; eluent: acetonitrile-water,
gradient:
10:90 - > 90:10.
Starting compounds
Example 1 A
2,2,2-trichloro-1-(4-vitro-1H-pyrrol-2-yl)ethanone
-O
g (47 mmol) of 2,2,2-trichloro-1-(1H-pyrrol-2-yl)ethanone are dissolved in
47.2 ml of
acetic anhydride. At -50 to -60°C 2.21 ml (47 mmol) of 90% strength
nitric acid are added
dropwise. The reaction mixture is slowly warmed to 0°C and then stirred
at room
10 temperature for 1 h. The reaction solution is diluted with ethyl acetate
and washed twice
with saturated sodium chloride solution and four times with saturated sodium
hydrogen
carbonate solution. The organic phase is dried with magnesium sulfate and
concentrated by
evaporation under reduced pressure. The evaporation residue is admixed with a
mixture of
10 ml of diethyl ether and 20 ml of cyclohexane and left to stand at
5°C for 48 hours.
Yield: 5.2 g (43% of theory)
MS (ESI~: m/z = 256 (M+H)+
1H-NMR (300MHz, CDCl3): 8 = 9.75 (broad s, 1 H), 7.9 (d, 1 H), 7.8 (d, 1 H)
ppm.
Example 2A
Ethyl 4-vitro-1 H-pyrrole-2-carboxylate
O
\\
N_O_
HaC~O
N
O H
CA 02508788 2005-06-06
-34-
2.57 g (10 mmol) of 2,2,2-trichloro-1-(4-vitro-1H-pyrrol-2-yl)ethanone
(Example lA) are
dissolved under argon in 50 ml of absolute ethanol and 1.39 ml (10 mmol) of
triethylamine
are added. The reaction mixture is stirred at room temperature for 16 hours.
Then 100 ml of
water are added dropwise and the precipitated crystals are filtered off with
suction: 1 g of
product. The mother liquor is concentrated under reduced pressure and the
crystals which
precipitate there are filtered off with suction: 0.5 g of product.
Yield: 1.5 g (71 % of theory)
MS (ESI~: m/z = 185 (M+H)+
1H-NMR (300MHz, DMSO-d6): 8 = 13.1 (broad s, 1H), 8.05 (d, 1H), 7.25 (d, 1H),
4.3 (q,
2H), 1.3 (tr, 3H) ppm.
Example 3A
1-( 1-benzyl-1 H-pyrrol-2-yl)-2,2,2-trichloroethanone
O
-N
CI CI
CI
60.55 g (333.3 mmol) of trichloroacetyl chloride are dissolved in 200 ml of
dichloromethane. Then a solution of 52.4 g (333.3 mmol) of 1-benzyl-1H-pyrrole
in 120 ml
of dichloromethane is added dropwise at room temperature over 1 hour. Argon is
passed
through the flask. The reaction solution is stirred overnight and then
concentrated by
evaporation under reduced pressure. The residue crystallizes from methanol.
Yield: 61.9 g (61 % of theory)
MS (ESI~: m/z = 302 (M+H)+
'H-NMR (200MHz, CDCl3): S = 7.6 (dd, 1H), 7.2-7.4 (m, 3H), 7.15-7.05 (m, 3H),
6.3 (dd,
1 H), 5.6 (s, 2H) ppm.
Example 4A
1-( 1-benzyl-4-vitro-1 H-pyrrol-2-yl)-2,2,2-trichloroethanone
CA 02508788 2005-06-06
- 35 -
O
~CI \ ,
CI
30.26 g (100 mmol) of 1-(1-benzyl-1H-pyrrol-2-yl)-2,2,2-trichloroethanone are
dissolved
in 100 ml of acetic anhydride, the solution is cooled to -40°C and at -
40°C 10 ml
(200 mmol) of 90% strength nitric acid are added dropwise. The reaction
mixture is slowly
warmed to room temperature and subsequently stirred for 1 hour. The reaction
mixture is
poured into 500 ml of ice-water and stirred vigorously for 15 minutes. The
precipitate is
filtered off with suction and suspended in 150 ml of methanol, with stirring,
and filtered off
with suction again. These crystals filtered off with suction are once again
suspended in
50 ml of methanol, with stirring, filtered off with suction and dried under
reduced pressure.
Yield: 25.4 g (73% of theory)
MS (ESI~: m/z = 347 (M+I-~+
1H-NMR (300MHz, CDC13): S = 8.0 (d, 1H), 7.75 (d, 1H), 7.4 (m, 3H), 7.2 (m,
2H), 5.6 (s,
2H) ppm.
Example SA
I 5 1-benzyl-4-vitro-1 H-pyrrole-2-carboxamide
O
0
H
17.38 g (50 mmol) of 1-(1-benzyl-4-vitro-1H-pyrrol-2-yl)-2,2,2-
trichloroethanone are
dissolved in 300 ml of a saturated solution of ammonia in THF (prepared by
passing 8.85 g
CA 02508788 2005-06-06
-36-
(520 mmol) of ammonia gas into 300 ml of THF) and the solution is stirred at
room
temperature for 3 hours. The reaction solution is then concentrated under
reduced pressure,
diethyl ether is added and the mixture is suspended with stirring.
Yield: 11.95 g (98% of theory)
MS (ESI~: m/z = 246 (M+H)+
1H-NMR (200MHz, DMSO-d6): b = 8.3 (d, 1H), 7.9 (broad s, 1H), 7.5 (d, 1H), 7.4-
7.15
(m, 6H), 5.7 (s, 2H) ppm.
Example 6A
1-( 1-phenyl-1 H-pyrrol-2-yl)-2,2,2-trichloroethanone
CI CI
CI
-N
O
Synthesis analogous to Example 3A.
Yield: 75% of theory
MS (DCI+): m/z = 288/290/292 (M+H)+
1H-NMR (300MHz, CDC13): b = 7.7 (dd, 1H), 7.5 (m, 3H), 7.3 (m, 2H), 7.1 (d,
1H), 6.4
(dd, 1 H).
Example 7A
1-(4-vitro-1-phenyl-1 H-pyrrol-2-yl)-2,2,2-trichloroethanone
O
O
CA 02508788 2005-06-06
' -37-
Synthesis analogous to Example 4A.
Yield: 81 % of theory
1H-NMR (200MHz, CDCl3): 8 = 8.1 (d, 1H), 7.85 (d, 1H), 7.6-7.1 (m, about 5H).
Example 8A
N-benzyl-1-phenyl-4-vitro-1H-pyrrole-2-carboxamide
O
Synthesis analogous to Example 5A.
Yield: 48% of theory
LC-MS (Method 4): Rt= 3.6 min, MS (ES+): m/z = 321 (M+H)+
1H-NMR (300MHz, CDC13): 8 = 7.7 (d, 1H), 7.5-7.2 (m, 11H), 4.45 (d, 2H), 2.3
(m, 1H),
1.6-1.8 (m, 2H), 1.3 (m, 2H), 1.1 (m, 1H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (s,
3H), 0.7 (dd,
1 H).
Exemplary embodiments
Example 1
Ethyl4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-
pyrrole-2-carboxylate
rJ CH3
CH3
,,,~~~CH3
HsC~O ~ ~,
H
CA 02508788 2005-06-06
- 38 -
1.47 g (8 mmol) of ethyl 4-vitro-1H-pyrrole-2-carboxylate (Example 2A) are
dissolved in
40 ml of THF and a spatula tip of Raney nickel and 3.2 ml (12 mmol) of 25%
strength
aqueous hydrazine solution are added. After 30 minutes of stirring at room
temperature the
solution is admixed with magnesium sulfate and filtered over kieselguhr, and
the solid
product is washed with ethyl acetate. The filtrate is concentrated by
evaporation under
reduced pressure, the residue is dissolved in 24 ml of absolute DMSO under
argon, and
3.89 g (24 mmol) of N,N-carbonyldiimidazole are added. After 30 minutes of
stirnng at
room temperature, 0.3 ml of water is added to the solution, which is stirred
for 10 minutes.
Then 1.23 g (8 mmol) of R-(+)-bornylamine are added and the solution is
stirred for 1 h.
The reaction solution is diluted with ethyl acetate and washed twice with
saturated sodium
chloride solution. The organic phase is washed with 1N hydrochloric acid and
saturated
sodium hydrogen carbonate solution, dried with magnesium sulfate and
concentrated by
evaporation under reduced pressure. The residue is dissolved in 15 ml of
diethyl ether, and
5 ml of cyclohexane are added with stirnng. The crystals are filtered off with
suction and
subsequently heated under reflux in diethyl ether for 1 hour. After cooling,
the crystals are
filtered off with suction.
Yield: 1.3 g (49% of theory)
MS (ESI~: m/z = 334 (M+H)+
1H-NMR (300MHz, DMSO-d6): 8 = 11.4 (broad s, 1H), 7.85 (s, 1H), 7.0 (d, IH),
6.5 (d,
1H), 6.0 (d, 1H), 4.2 (q, 2H), 3.95 (m, 1H), 2.2 (m, 1H), 1.55 - 1.8 (m, 3H),
1.35-1.1 (m
2H), 1.3 (tr, 3H), 0.9 (s, 3 H), 0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
Example 2
4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-yl]amino}
carbonyl)amino]-1 H-
pyrrole-2-carboxylic acid
O CHs
N \ CHs
,,~CH3
HO ~ ~ H ~'
H
CA 02508788 2005-06-06
-39-
333 mg (1 mmol) of ethyl 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino}-
carbonyl)amino]-1H-pyrrole-2-carboxylate (Example 1) are suspended in 2 ml of
ethanol,
and 0.24 ml (4 mmol) of 45% strength sodium hydroxide solution is added.
Following the
addition of 0.5 ml of water the reaction solution is left to stand at room
temperature for
20 h. The reaction solution is diluted with water and washed with ethyl
acetate. The
aqueous phase is acidified with 1N hydrochloric acid and extracted three times
with ethyl
acetate. The combined extracts are dried with magnesium sulfate and
concentrated by
evaporation under reduced pressure. The residue crystallizes from diethyl
ether.
Yield: 228 mg (75% of theory)
MS (ESI~: m/z = 306 (M+H)+
1H-NMR (200MHz, DMSO-d6): 8 = 12.1 (broad s, IH), 11.4 (broad s, 1H), 7.85 (s,
1H),
7.0 (tr, 1 H), 6.5 (tr, 1 H), 6.0 (d, 1 H), 3.95 (m, 1 H), 2.2 (m, 1 H), 1.55 -
1.8 (m, 3 H), 1.3 5-
1.1 (m 2H), 0.9 (s, 3 H), 0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
Example 3
N-benzyl-4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino } carbonyl)amino]-1 H-pyrrole-2-carboxamide
O CHs
i
CH3
H N ~~ ,,,.CH3
H
H
152 mg (0.5 mmol) of 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 2) are
dissolved in 2 ml
of DMF under argon, 243 mg (1.5 mmol) of N,N-carbonyldiimidazole are added and
the
mixture is stirred at room temperature for 1 h. Then 0.02 ml of water are
added and stirring
is carried out for 30 minutes. Following the addition of 80 mg (0.75 mmol) of
benzylamine
the reaction solution is left to stand at room temperature for 16 h. With
stirnng, first 1 ml
of 1N hydrochloric acid then, slowly, a further 3 ml of water are added
dropwise. The
crystals are filtered off with suction.
Yield: 172 mg (87% of theory)
CA 02508788 2005-06-06
-40-
MS (ESI~: m/z = 395 (M+H)+
1H-NMR (300MHz, DMSO-d6): 8 = 10.95 (broad s, 1H), 8.4 (tr, 1H), 7.8 (s, 1H),
7.2-7.35
(m, 5 H), 6.8 5 (tr, 1 H), 6.6 (tr, 1 H), 5.9 (d, 1 H), 4.4 (d, 2H), 3.95 (m,
1 H), 2.2 (m, 1 H),
1.55 - 1.8 (m, 3H), 1.35-1.1 (m 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (m, 1H),
0.75 (s, 3H)
ppm.
Example 4
Ethyl 1-benzyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-
yl]amino } carbonyl)-
amino]-1 H-pyrrole-2-carboxylate
O . CHs
CH3
,,..CH3
HsC~O ~ ~ H ,,
I .N H
1.67 g (5 mmol) of ethyl 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
yl]amino}-
carbonyl)amino]-1H-pyrrole-2-carboxylate (Example 1) are dissolved in absolute
DMF,
and 0.73 g (6.5 mmol) of potassium tent-butoxide is added. Stirnng is
continued for 5
minutes and thereafter 1.11 g (6.5 mmol) of benzyl bromide are added. After
three hours of
stirring at room temperature, 3 ml of water are slowly added dropwise. The
crystals are
filtered off with suction and washed with a 1:1 mixture of water and methanol.
The crystals
are recrystallized from a mixture of ethyl acetate and methanol.
Yield: 1.2 g (57% of theory)
MS (ESI~: m/z = 424 (M+I-~+
1H-NMR (300MHz, DMSO-d6): 8 = 7.9 (s, 1H), 7.2-7.35 (m, 4H), 7.05 (d, 2H),
6.65 (d,
1 H), 6.05 (d, 1 H), 5.45 (d, 2H), 4.15 (q, 2H), 3.95 (m, 1 H), 2.2 (m, 1 H),
1.55 - 1.8 (m,
3H), 1.35-1.1 (m 2H), 1.2 (tr, 3H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (m, 1H),
0.75 (s, 3H) ppm.
r
CA 02508788 2005-06-06
-41-
Example 5
1-benzyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-yl]amino}
carbonyl)amino]-
1H-pyrrole-2-carboxylic acid
H O CH3 ,
N~ CH3
,,~CH3
HO ~ ~ H''
I _N H
O
211 mg (0.5 mmol) of ethyl 1-benzyl-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]hept-
2-yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylate (Example 4) are suspended
in 1 ml
of ethanol, and 0.24 ml (4 mmol) of 45% strength sodium hydroxide solution and
2 ml of
THF are added. The reaction mixture is stirred at RT for 72 hours, diluted
with water,
acidified with 1N hydrochloric acid and extracted twice with ethyl acetate.
After the
extracts have been dried with magnesium sulfate they are concentrated by
evaporation
under reduced pressure.
Yield: 159 mg (80% of theory)
MS (ESI~: m/z = 396 (M+H)+
'H-NMR (300MHz, DMSO-d6): b = 12.0 (broad s, 1H), 7.9 (s, 1H), 7.2-7.35 (m,
3H), 7.15
(d, 1 H), 7.05 (d, 2H), 6.6 (tr, 1 H), 6.0 (d, 1 H), 5.45 (s, 2H), 3.95 (m, 1
H), 2.2 (m, 1 I~, 1.55
-1.8 (m, 3H), 1.35-1.1 (m 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (m, 1H), 0.75
(s, 3H) ppm.
Example 6
N,1-dibenzyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-
yl]amino} carbonyl)amino]-1 H-pyrrole-2-carboxamide
CA 02508788 2005-06-06
-42-
p CH3
CH3
\ ~ H N ,"~ CH3
H ~,
H
39.5 mg (0.1 mmol) of 1-benzyl-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 5) are
dissolved in
0.5 ml of DMF under argon, then 48 mg (0.3 mmol) of N,N-carbonyldiimidazole
are added
and the mixture is stirred at room temperature for 30 minutes. Then 0.036 ml
of water is
added and stirnng is continued for 30 minutes at RT. Then 16 mg (0.15 mmol) of
benzylamine are added and the reaction solution is left to stand at RT for 24
hours. The
reaction solution is filtered and purified by preparative HPLC (column:
Nucleosil 100-5 C
18 Nautilus, 5 ~.m, 20 x 50 mm, wavelength: 220 nm, 600 pl injection volume,
gradient: A
= water + 0.3% formic acid, B = acetonitrile, 0 min = 10% B, 2 min = 10% B, 6
min = 90%
B, 7 min = 90% B, 7.1 min = 10% B, 8 min = 10% B, flow rate 25 ml/min).
Concentration
of the product fractions by evaporation under reduced pressure yields 26 mg of
product.
Yield: 26 mg (54% of theory)
MS (ESI~: m/z = 485 (M+I~+
1H-NMR (200MHz, DMSO-d6): 8 = 8.5 (tr, 1H), 7.9 (s, 1H), 7.2-7.35 (m, 8H), 7.1-
7.05
(m, 3H), 6.6 (d, 1 H), 6.0 (d, 1 H), 5.5 (s, 2H), 4.35 (d, 2H), 3.95 (m, 1 H),
2.2 (m, 1 H), 1.55
-1.8 (m, 3H), 1.35-1.1 (m 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (m, 1H), 0.75
(s, 3H) ppm.
Example 7
1-Benzyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxamide
CA 02508788 2005-06-06
- 43 -
HsC CHs
H ,,,.CH3
H N n~",
N
H
HZN I ~ O
-N- ,
O
1.00 g (4.1 mmol) of 1-benzyl-4-nitro-1H-pyrrole-2-carboxamide (Example SA) is
dissolved under argon protection in 20 ml of tetrahydrofuran, and then a
spatula tip of
Raney nickel is added. With ice cooling, 784 mg (6.1 mmol) of a 25% solution
of
hydrazine in water are added via a syringe. Stirnng is continued for 1 h until
the evolution
of hydrogen is at an end. The reaction mixture is diluted with dichloromethane
and filtered
over kieselguhr. Washing of the solid, drying of the filtrate with magnesium
sulfate, and
concentration yield an oily residue. This residue is taken up in 30 ml of DMSO
under
argon, and 1.98 g (12.2 mmol) of 1,1'-carbonyldiimidazole are added. After 1 h
of stirring
at RT, two drops of water are added in order to destroy excess imidazole
reagent. 625 mg
(4.1 mmol) of (R)-(+)-bornylamine are added to the solution. After 3 days of
stirring at RT
the reaction mixture is purified by preparative HPLC separation (into 3
portions, RP18,
gradient: 30% acetonitrile/70% water - > 95% acetonitrile/5% water).
Concentration of the
product fractions yields, after drying under reduced pressure (4 mbar,
60°C), the target
product.
Yield: 813 mg (51% of theory)
Optical rotation: [a]DZO = + 9° (c = 0.28 g/100 ml, CHC13)
MS (ESI+): m/z = 395 (M+H)+
'H-NMR (300MHz, CDC13): 8= 7.15 - 7.35 (m, SH), 6.85 (d, 1H), 6.5 (d, 1H),
5.95 (s,
1 H), 5.3 - 5.6 (m, 4H), 4.75 (d, 1 H), 4.0 (m, 1 H), 2.3 (m, 1 H), 1.6 - 1.8
(m, 2H), 1.3
m(2H), 1.0 (m, 1H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (s, 3H), 0.7 (dd, 1H)
CA 02508788 2005-06-06
-44-
Example 8
1-benzyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-yl]amino }
carbonyl)amino]-
1H-pyrrole-2-carboxamide and 1-benzyl-4-[({[(1S,2R,4S)-1,7,7-
trimethylbicycle[2.2.1]-
hept-2-yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxamide
O CHs
N~ CH3
H H
~CH3
Synthesis analogous to Example 7.
Amine used: (1RS)-bornylamine (enantiomer mixture)
Yield: 55% of theory
Example 9
1-benzyl-4-[( { [( 1 R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-yl]amino}
carbonyl)amino]-
1H-pyrrole-2-carboxamide and 1-benzyl-4-[({[(1S,2S,4S)-1,7,7-
trimethylbicyclo[2.2.1]-
hept-2-yl]amino} carbonyl)amino]-1 H-pyrrole-2-carboxamide
NH2
O
N~ O CHs
N~N CH
H H s
CH3
1 S Synthesis analogous to Example 7.
Amine used: (1RS)-isobornylamine (enantiomer mixture)
Yield: 32% of theory
m. p.: 130°C
Example 10
CA 02508788 2005-06-06
- 45 -
4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-yl] amino }
carbonyl)amino]-1 H-
pyrrole-2-carboxamide
O CHa
CH3
H2N
,,,~CH3
O H H
5.28 g (17.3 mmol) of 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 2) are
dissolved in
69 ml of DMF under argon. Then 8.4 g (51.9 mmol) of N,N-carbonyldiimidazole
are
added. After 1 h of stirnng at RT, 18.1 ml (242 mmol) of 25% strength aqueous
ammonia
solution are added dropwise with ice cooling. After 1 h of stirnng at RT the
reaction
solution is diluted with water and extracted three times with ethyl acetate.
The combined
organic phases are washed twice with saturated sodium chloride solution, dried
with
magnesium sulfate and concentrated. The crystalline residue is stirred up with
ethyl acetate
and the crystals are filtered off with suction.
Yield: 3.48 g (66% of theory)
Example 11
1-(2-phenyloxyethyl)-4-[({ [(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]kept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid
CA 02508788 2005-06-06
-46-
O CHs
N_,I/ CH3
,...CH3
HO
O
1.62 g (3.56 mmol) of ethyl 1-(2-phenyloxyethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethyl-
bicyclo[2.2.1]kept-2-yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylate
(Example 27)
are dissolved in 7.1 ml of ethanol and 14.3 ml of THF, and 1.7 ml (28.5 mmol)
of 45%
strength sodium hydroxide solution are added. The mixture is stirred at RT
overnight and
then diluted with 1N hydrochloric acid and extracted four times with ethyl
acetate. The
combined organic phases are washed with saturated sodium chloride solution,
dried with
magnesium sulfate and concentrated by evaporation under reduced pressure. This
gives a
solid foam which is used directly for the next synthesis.
Yield: 1.75 g (quantitative)
MS (ESI~: m/z = 426 (M+H)+
'H-NMR (200MHz, DMSO-d6): 8 = 7.9 (s, 1 H), 7.25 (tr , 2H), 7.2 (d, l H), 6.85-
6.95 (m,
3 H), 6.6 (d, 1 H), 6.05 (d, 1 H), 4.6 (tr, 2H), 4.2 (tr, 2H), 3 .95 (m, 1 H),
2.2 (m, 1 H), 1.5 5-
1.8 (m, 3H), 1.35-1.1 (m 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 11-~, 0.75
(s, 3H) ppm.
Example 12
1-methyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-yl]amino}
carbonyl)amino]-
1 H-pyrrole-2-carboxylic acid
HO
3
~'h3
CA 02508788 2005-06-06
-47-
Synthesis analogous to Example 11.
Yield: 1.16 g (quantitative)
MS (ESI~: m/z = 320 (M+H)+
' H-NMR (200MHz, DMSO-d6): 8 = 7.9 (s, 1 H), 7.05 (d, l H), 6.5 (d, 1 H), 6.05
(d, 1 H),
3.95 (m, 1H), 3.75 (s, 3H), 2.2 (m, 1H), 1.55-1.8 (m, 3H), 1.35-1.1 (m 2H),
0.9 (s, 3H),
0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
Example 13
1-butyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]kept-2-yl] amino}
carbonyl)amino]-
1 H-pyrrole-2-carboxylic acid
H3
~H3
HO
O
1~
Synthesis analogous to Example 11.
Yield: 1.09 g (quantitative)
MS (ESI~: m/z = 362 (M+H)+
'H-NMR (300MHz, DMSO-d6): 8 = 7.85 (s, 1H), 7.1 (d,lH), 6.5 (d, 1H), 6.0 (d,
1H), 4.2
(tr, 2H), 3.95 (m, 1H), 2.2 (m, 1H), 1.55-1.8 (m, 5H), 1.35-1.1 (m, 4H), 0.9
(s, 3H), 0.85 (s,
3 H), 0.8 (d, 1 H), 0.75 (s, 3 H) ppm.
Example 14
1-(cyclopropylmethyl)-4-[({ [(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]kept-2-
yl]amino}-
carbonyl)amino]-1H-pyrrole-2-carboxylic acid
CA 02508788 2005-06-06
-48-
O CH3
CH3
,,,~CH3
HO j''~~
H
Synthesis analogous to Example 11.
Yield: 1.66 g (quantitative)
MS (ESI~: m/z = 360 (M+H)+
' H-NMR (3 OOMHz, DMSO-d6): 8 = 7.8 S (s, 1 H), 7.1 (d, l H), 6.5 (d, 1 H),
6.0 (d, 1 H), 4.1
(dd, 2H), 3.95 (m, 1H), 2.2 (m, 1H), 1.55-1.8 (m, 3H), 1.35-1.1 (m, 3H), 0.9
(s, 3H), 0.85
(s, 3H), 0.8 (d, 1H), 0.75 (s, 3H), 0.45 (q, 2H), 0.3 (q, 2H) ppm.
Example 15
1-[2-(diethylamino)-2-oxoethyl]-4-[( { [( 1 R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1 ]-hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid
O CHs
N \ CHa
,,..CH3
HO
-N
O H
O
'N'
ICH3 CI H3
Synthesis analogous to Example 11.
Yield: 0.72 g (quantitative)
MS (ESI~: m/z = 419 (M+H)+
'H-NMR (300MHz, DMSO-d6): 8 = 7.85 (s, 1 H), 7.1 (d, l H), 6.5 (d, 1 H), 6.0
(d, 1 H), 5.1
(s, 2H), 3.95 (m, 1H), 3.35 (q, 2H), 3.25 (q, 2H), 2.2 (m, 1H), 1.55-1.8 (m,
3H), 1.35-1.1
(m, 2H), 1.2 (tr, 3H), 1.05 (tr, 3H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 1H),
0.75 (s, 3H) ppm.
Example 16
CA 02508788 2005-06-06
-49-
1-(2-methoxyethyl)-4-[({ [(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid
CH3
CH3
,,,~CH3
H '
~CH3
Synthesis analogous to Example 11.
Yield: l .l g (quantitative)
MS (ESI~: m/z = 364 (M+I-~+
1H-NMR (300MHz, DMSO-d6): 8 = 7.85 (s, 1 H), 7.1 (d, l H), 6.5 (d, 1 H), 6.0
(d, 1 H), 4.4
(tr, 2 H), 3 .95 (m, 1 H), 3 .5 5 (tr, 2H), 3 .2 (s, 3 H), 2.2 (m, 1 H), 1.5 5-
1. 8 (m, 3 H), 1.3 5-1.1
(m, 2H), 0.9 (s, 3I-~, 0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
Example 17
1-(2-phenylethyl)-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]kept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid
Hs
~H3
HO
Synthesis analogous to Example 11.
Yield: 1.64 g (quantitative)
MS (ESI~: m/z = 410 (M+I~+
'H-NMR (200MHz, DMSO-d6): 8 = 7.85 (s, 1H), 7.15-7.35 (m, 5H), 7.1 (d,lH),
6.55 (d,
1 H), 6.0 (d, 1 H), 4.4 (tr, 2H), 3.95 (m, 1 H), 2.9 (tr, 2H), 2.2 (m, 1 H),
1.55-1'.8 (m, 3H),
1.35-1.1 (m, 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
CA 02508788 2005-06-06
-50-
Example 18
N-benzyl-1-phenyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-
yl] amino } carbonyl)amino]-1 H-pyrrole-2-carboxamide
HsC CHa
i ,,,.CH3
H
H N~~",
N
\\ H
HN ~ ~ O
-N
O
Synthesis analogous to Example 7 from Example 8A.
Yield: 34% of theory
LC-MS (Method 1): Rt= 4.3 min, MS (ES+): m/z = 471 (M+H)+
1H-NMR (300MHz, CDCl3): 8 = 7.15-7.45 (m, 11H), 6.60 (d, 1H), 5.95 (m, 1H),
5.35 (d,
1 H), 4.40 (d, 1 H), 4.1 (m, 1 H), 2.3 (m, 1 H), 1.6-1.8 (m, 2H), 1.3 (m, 2H),
1.1 (m, 1 H), 0.9
(s, 3H), 0.85 (s, 3H), 0.8 (s, 3H), 0.7 (dd, 1H).
Example 19
2-hydroxyethyl 1-benzyl-4-[({ [(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-
2-yl]amino}-
carbonyl)amino]-1 H-pyrrole-2-carboxylate
H3
~H3
HO~p
62.9 mg (0.16 mmol) of 1-benzyl-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 5) are
dissolved in
0.5 ml of DMF, and 77.35 mg (0.48 mmol) of N,N-carbonyldiimidazole are added.
The
CA 02508788 2005-06-06
-51 -
mixture is stirred at RT for 1 h, diluted with water and extracted twice with
ethyl acetate.
The combined organic phases are washed with saturated sodium chloride
solution, dried
with magnesium sulfate and concentrated by evaporation under reduced pressure.
The
evaporation residue is dissolved in 0.5 ml (9 mmol) of ethylene glycol under
argon.
Following the addition of 0.02 ml (0.36 mmol) of triethylamine the mixture is
stirred at
100°C for 1 h. The reaction mixture is diluted with a little methanol
and purified in 3
portions by preparative HPLC (Method 6). The fractions comprising product are
concentrated by evaporation under reduced pressure.
Yield: 11.6 mg (17% of theory)
MS (ESI~: mlz = 440 (M+I~+
1H-NMR (200 MHz, DMSO-db): 8 = 7.95 (s, 1H), 7.2-7.4 (m, 4H), 7.1 (d, 1H), 6.7
(d, 1H),
6.1 (d, 1 H), 5.5 (s, 2H), 4.85 (tr, 1 H), 4.1 (tr, 2H), 3.95 (m, 1 H), 3.6
(q, 2H), 2.2 (m, 1 H),
1.55-1.8 (m, 3H), 1.35-1.1 (m, 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 1H),
0.75 (s, 3H) ppm.
Example 20
2-(acetylamino)ethyl-1-benzyl-4-[( f [(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]kept-2-
yl] amino } carbonyl)aminoJ-1 H-pyrrole-2-carboxylate
O CHa
O N \ CHs
H C N ,,~~CH3
3
O ~ ~ H ,
O
59.3 mg (0.15 mmol) of 1-benzyl-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 5) are
dissolved in
0.5 ml of DMF under argon, and 73 mg (0.45 mmol) of N,N-carbonyldiimidazole
are
added. After 1 h of stirring at RT the mixture is diluted with water and
extracted with ethyl
acetate. The organic phase is washed with saturated sodium chloride solution,
dried with
magnesium sulfate and concentrated by evaporation under reduced pressure. To
the
evaporation residue there are added 0.2 ml of N-(2-hydroxyethyl)acetaxnide and
0.02 ml of
CA 02508788 2005-06-06
-52-
triethylamine. The reaction mixture is stirred at 100°C for 1 h,
cooled, admixed with 0.4 ml
of methanol, and purified by preparative HPLC (Method 6). The fractions
comprising
product are concentrated by evaporation under reduced pressure.
Yield: 24.1 mg (33% of theory)
MS (ESI~: m/z = 481 (M+I-~+
1H-NMR (300MHz, DMSO-d6): 8 = 7.95 (s, 2H), 7.2-7.4 (m, 4H), 7.1 (d, 1H), 6.7
(d, 1H),
6.05 (d, 1 H), 5.45 (s, 2H), 4.1 (tr, 2H), 3.95 (nn, 1 H), 3.3 (q, 2H), 2.2
(m, 1 H), 1.8 (s, 3 H),
1.55-1.8 (m, 3H), 1.35-1.1 (m, 2H), 0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 1H),
0.75 (s, 3H) ppm.
CA 02508788 2005-06-06
-53-
In the same way as for Example 20 it is possible to prepare Examples 21 to 26
from the
following table.
Amount
Ex. Structure Molar MS Retention Reactant [mgJ
No. mass (EI): time [mint Ex. No. (Yield [%
(M+H)+ (Method) of theo
H O CH~H3
\ N
21 0~0 / \ f",~-~ CH 475.58 476 3.39 (1) 5 22.3 (31)
I .N
O
/
CHa H- ~''O H~H~
N N \ CHa
22 H'°~ ~'~-° ~ ~ "~'' 480.64 481 2.4 (1) 5 9.2 (13)
N
H
O N .O CH~H
~CH3
23 HZN~o / ~ H° 452.55 453 2.91 (1) 5 24.2 (36)
I N H
O
N H .O CH~H
i N~(~/ .CH3
24 ~ I o / \ H'' 486.61 487 2.97 (1) 5 21.2 (29)
H
O
/
H ,O CH~H
N~\/ CHs
25 ' ~N~° / \ H'' 489.62 490 3.17 (1) 5 27.9 (38)
N H
O
H~H~
CH3
26 ~ J'~° / ~ H'' 490.60 491 3.01 (1) 5 27.3 (37)
N N H
O
/
CA 02508788 2005-06-06
-54-
Example 27
Ethyl 1-(2-phenyloxyethyl)-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1
]hept-2-
yl] amino } carbonyl)amino]-1 H-pyrrole-2-carboxylate
O CHs
CH3 '
H3C~0 ~ ~ H ,,, .~~~CH3
I H
1.67 g (5 mmol) of ethyl 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
yl]amino}-
carbonyl)amino]-1 H-pyrrole-2-carboxylate (Example 1 ) are dissolved in 10 ml
of absolute
DMF, and 1.07 g (9.5 mmol) of potassium tert-butoxide are added. After 5
minutes of
stirnng at RT, 1.91 g (9.5 mmol) of 1-bromoethyl 2-phenyl ether are added. The
reaction
mixture is stirred at RT overnight and then a further 600 mg (3 mmol) of 1-
bromoethyl 2-
phenyl ether and 336 mg (3 mmol) of potassium tert-butoxide are added. The
reaction
mixture is stirred at RT overnight and then slowly 3.5 ml of water and 0.5 ml
of methanol
are added dropwise. The crystals which form are filtered off with suction and
washed with
water/methanol (1:1 mixture) and a little methanol.
Yield: 1.87 g (83% of theory)
MS (ESI~: m/z = 454 (M+H)+
1H-NMR (300MHz, DMSO-d6): 8 = 7.9 (s, 1H), 7.25 (tr, 2H), 7.2 (d,lH), 6.85-
6.95 (m,
3 H), 6.65 (d, 1 H), 6.0 (d, 1 H), 4.6 (tr, 2H), 4.15-4.25 (m, 4H), 3.95 (m, 1
H), 2.2 (m, 1 H),
1.55-1.8 (m, 3H), 1.35-1.1 (m, 2H), 1.25 (tr, 3H), 0.9 (s, 3H), 0.85 (s, 3H),
0.8 (d, 1H),
0.75 (s, 3H) ppm.
CA 02508788 2005-06-06
-55-
In the same way as for Example 27 it is possible to prepare Examples 28 to 30
from the
following table.
Amount
Ex. Structure Molar MS RetentionReactan[mg]
No. mass (EI): time [min]t Yield
[%
(M+H)+Method) Ex. of theo
No.
H~p CHI"
N CH
a
28 "3c~o / \ H 389.54 390 4.1 (4) 1 1.17
(60)
N H
O
CH3
H~o CH~H
N CH
3
29 "3c~o / \ H- 424.54 425 2.6 (1) 1 36.8
(43)
N H
O
i
N
H~o CH~H3
N CH
I
30 "3c~o / \ H. 387.52 388 4.05 (4) 1 1.67
(86)
N H
O
Example 31
S 1-(3,4-difluorobenzyl)-4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino)-
carbonyl)amino]-1 H-pyrrole-2-carboxamide
O CHs
CH3
,,.v CH3
H2N
-N
O H
F
F
CA 02508788 2005-06-06
-56-
60.9 mg (0.2 mmol) of 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxamide (Example 10) are dissolved
in
0.5 ml of DMF under argon, and 8.8 mg (0.22 mmol) of sodium hydride (in 60%
form) are
added. After 1 h of stirnng at RT, the reaction solution is added under argon
to a solution
of 45.5 mg (0.22 mmol) of 3,4-difluorobenzyl bromide in 0.2 ml of DMF and
shaken at RT
overnight. Following filtration the reaction mixture is purified by
preparative HPLC
(Method 7). The fractions comprising product are concentrated by evaporation
under
reduced pressure.
Yield: 25 mg (29% of theory)
MS (ESI~: m/z = 431 (M+H)+
1H-NMR (400MHz, DMSO-d6): 8 = 7.9 (s, 1H), 7.5 (broad s, 1H), 7.35 (q, 1H),
7.15 (ddd,
1 H), 7.1 (d, 1 H), 6.95 (m, 1 H), 6.8 (broad s, 1 H), 6.6 (d, 1 H), 6.0 (d, 1
H), 5 .5 (s, 1 H), 3 .95
(m, 1 H), 2.2 (m, 1 H), 1.7 (m, 1 H), 1.6 (m, 2H), 1.3 (m, 1 H), 1.15 (m, 1
H), 0.9 (s, 3 H), 0. 8 5
(s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
In the same way as for Example 31 it is possible to prepare Examples 32 to 59
from the
following table.
Amount
Ex. Structure MolarMS (EI):RetentionReactant[mg]
No. mass (M+I~+ time Ex. (Yield
[min] No. [%
(Method) of theory])
CH3
,O H.... CH3
--N+ CH3
O
~ H\/O
32 ~ ~ NH 439.51440 2.94 10 18 (20)
N (1)
O
NH2
CA 02508788 2005-06-06
-S7-
Amount
Ex. Structure Molar MS (EI): Retention Reactant [mg]
No. mass (M+I~+ time [min] Ex. No. (Yield [%
(Method) of theory])
CH3 CH3
H""
CH3
H O
O
33 ~ NH 388.51 389 2.66 (1) 10 5 (6)
N
O
NH2
CH3 CH3
H~
CH3
H\ /O
34 ~ ~ NH 412.51 413 2.96 (1) 10 29 (35)
N
F
O
NHz
\\ Hn" CH3CH s
H~O
35 ~ ~ NH 419.53 420 2.86 (1) 10 27 (32)
N
O
N HZ
CH3 CH
H~~"
CH3
H\ /O
36 ~ ~ N ~ NH 424.54 42S 2.93 (1) 10 2S (29)
H3C-O
O
NHZ
CA 02508788 2005-06-06
-58-
Amount
Ex. Structure Molar MS (En: Retention Reactant [mg]
No. mass (M+I~+ time [min] Ex. No. (Yield [%
(Method) of theory])
HaC~ CH3CH
O Hn" s
CH3
O ~ H" O
37 ~ ~ NH 452.55 453 2.9 (1) 10 33 (36)
N
O
NHZ
w CH
H.", 'CH3
O CHs
H O
38 ~ 452.60 453 3.15 (1) 10 40 (44)
NH
N
O
NHz
C H3
Hn" CH3
CH3
H\ /O
39 ~ ~ N'YH 408.54 409 3.02 (1) 10 28 (34)
N
H3C
O
NHZ
CH3
H.." CH3
CH3
H\ /O
40 ~ N'YH 422.57 423 3.19 (1) 10 25 (30)
~N
/ o
NHZ
CA 02508788 2005-06-06
-59-
Amount
Ex. --Structure -- -Molar- MS-(E~:- --Retention- Reactant [mg]
No. mass (M+I~+ time [min] Ex. No. (Yield [%
(Method) of theory])
H"" CH3 CH3
CH3
H~O
41 o~~N+ \ ~ NH 439.51 440 3.02 (1) 10 21 (24)
- N ~
O
NHZ
CH CHI
H",.
O ~~~~CH3
H3C\ H\ /O
'ISO
42 ~ \ ~ NH 452.55 453 3 (1) 10 22 (24)
N
O
NHZ
CH3
H ". CH3
CH3
\ H~O
43 ~ o \N ~ NH 452.55 453 3.05 (1) 10 1.1 (1)
0
~CH3
O
NHZ
H",. CH' CH3
CH3
O
44 N~ \ ~ ~ ~ 419.53 420 2.94 (1) 10 19 (23)
~N
O
NHZ
H,". CH3 CH3
CH3
H~O
45 ~ ~ N~ ~N ~ '~NYH 492.62 493 2.94 ( 1 ) 10 1.6 (2)
0
0
NHz
CA 02508788 2005-06-06
-60-
Amount
Ex. --qtr-ucture --Molar M~-(En:- Retention- -Reactant [mg]
No. mass (M+I~+ time [min] Ex. No. (Yield [%
(Method) of theory])
CH3 CH3
Hi",
CH3
N~O ,
H
46 ~ \ ~ NH 395.50 396 2.14 (1) 10 12 (15)
N
O
NHZ
CH3
H"" CH3
CH3
N\ CH3 H~O
47 O ~ ~ NH 413.52 414 2.77 (1) 10 15 (18)
N
H3C
O
NHz
H.", CH3 CH3
~CH3
F F H~O
~F
48 ~ ~ NH 462.51 463 3.22 ( 1 ) 10 43 (46)
~N
O
NHZ
H"" CH' CH3
CH3
H\ /O
49 ~ ~ N'YH 412.51 413 3.82 (8) 10 18 (22)
F \ I N
O
NH2
CA 02508788 2005-06-06
-61-
Amount
Ex. Structure _ .Molar. _MS_(EI):_RetentionReactant[mg]
No. mass(M+I~+ time Ex. ('Yield
[min] No. [%
(Method) of theory])
CH
CH3
Hn.,
CI
N 0
H -
~
~
50 \ 428.96429 3.95 10 20 23
~ (8) (
.
N
O
NHZ
CH
3
H..., CH3
CH3
N O
H
~
51 ' 408.54409 3.88 10 16 2
\ 8 ( O)
~ ( )
N
HC \
3
0
NHZ
CH' CH
3
H....
CH3
N~O
H
~
52 H3C \ 408.54409 3.98 10 18 (22)
N ~ NH (8)
O
N HZ
CH
3
H~". CH3
F CHs
N\ /O
F 'YH
~
53 \ 430.50431 3.87 10 20 (23)
~ NH (8)
N
O
NHZ
CA 02508788 2005-06-06
-62-
Amount
Ex. Structure Molar MS (EI): Retention Reactant [mg]
No. mass (M+I~+ time [mint Ex. No. (Yield [%
(Method) of theory])
CH3
Hn" CH3
CH3
N_ 'C
~H
54 ~ j N ~ NH 428.96 429 3.94 (8) 10 19 (22)
CI
O
NHz
d-i3C'' CH3
CH3
55 HzN N- H 462.51 463 4 (8) 10 24 (26)
I
O
O ,CH3
~~. ~(O
"3C CH3
CH3
> ...
56 HzN N- H 390.48 391 3.3 (3) 10 14.8 (49)
I
O
0 11 CHa
O
"3C CH3
CH3
57 H N ~ ~ H 348.44 349 2.57 (4) 10
z wN
O
OH
CA 02508788 2005-06-06
- 63 -
Amount
Ex. -Structure -- Molar_ MS-(EI):- Retention- Reactant [mg]
No. mass (M+I~+ time [min] Ex. No. (Yield [%
(Method) of theory])
-13C CH3
CH3 ,
I
5g HZN N~ H 412.51 413 2.15 (5) 10 5.9 (7)
O
F
CH3
H,s" CH3
CH3
O
F
59 \ ~ ~ 462.51 463 2.41 (5) 10 7.7 (6)
F ~N \
F
O
NHZ
Example 60
1-benzyl-N-(pyri din-3 -ylmethyl)-4- [( { [( 1 R,2 S,4R)-1,7, 7-
trimethylbicyclo [2.2.1 ] hept-2-
yl]amino}carbonyl)aminoJ-1 H-pyrrole-2-carboxamide
O CHs
CH3
,,.~CH3
N I
N
O H
N~ I ~
39.5 mg (0.1 mmol) of 1-benzyl-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]kept-2-
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic acid (Example 5) are
dissolved
together with 48.6 mg (0.3 mmol) of N,N-carbonyldiimidazole in 0.4 ml of DMF
and the
CA 02508788 2005-06-06
-64-
solution is left to stand at RT for 1 h. Then 0.0036 ml of water is added to
the reaction
solution, which is shaken for 30 minutes. FollowW g the ad ition of 16.2 mg
(0.15 mmol)
of 3-picolylamine it is shaken at RT overnight. Following filtration, the
reaction solution is
purified by preparative HPLC (Method 9). The fractions comprising product are
concentrated by evaporation under reduced pressure.
Yield: 21 mg (43% of theory)
MS (ESI~: m/z = 486 (M+H)+
~ H-NMR (200MHz, DMSO-d6): 8 = 8.5 5 (tr, 1 H), 8.45 (d, 1 H), 8.4 (dd, 1 H),
7.9 (s, 1 H),
7.6 (d tr, 1 H), 7.3 5-7.2 (m, 4H), 7.05-7.15 (m, 3 H), 6.65 (d, 1 H), 6.0 (d,
1 H), 5.5 (s, 1 H),
4.4 (d, 2H), 3.95 (m, 1H), 2.2 (m, 1H), 1.55-1.8 (m, 3H), 1.35-1.1 (m 2H),
1.25 (tr, 3H),
0.9 (s, 3H), 0.85 (s, 3H), 0.8 (d, 1H), 0.75 (s, 3H) ppm.
In the same way as for Example 60 it is possible to prepare Examples 61 to 150
from the
following table.
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. mass (M+I~+ time [min] Ex. No. [mg] (Yield
(Method) [% of
theo
~c cH
~Cii'3
61 \ N ' N H ~ H 408.54 409 3 ( 1 ) 2 27 (66)
O
H3C
H
c~3
N
N~
H
62 N ~ N~ H 395.50 396 2.22 (1) 2 24 (60)
H
O
iN
,~~3~ c~3
N
'~'~H
N~
H
63 N ~ N~ H 360.50 361 2.89 (1) 2 15 (41)
H
O
CH3
CA 02508788 2005-06-06
- 65 -
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -- mass--(M-+-H)~ time-[mi-n-]- -Ex: ~To. [mg]-(Yield
(Method) [% of
theory )
CI~
- \ 3
N,
64 ~ ~ N H H 464.61 465 3.05 ( 1 ) 5 ~i 6 (34)
I
0
H C CH~ -
N
N,
H
65 N ~ ~~ H 438.57 439 2.9 (1) 5 32 (73)
~N
J O
HO_
H~ 3C CFA
N-
N,
H
66 N ~ N~ H 452.60 453 3.08 (1) 5 29 (64)
o ~ \
CH3
CHF~3
H
N
H~
67 N ~ ~ ~ 450.62 451 3.33 (1) 5 31 69
N' ( )
0
CH3
H 3
N
N.
68 H3C~N~N ~ N~ H H 465.64 466 2.43 (1) 5 33 (71)
CH3 0
H
N
N,
H
69 H ~,N ~ N~ H 408.54 409 3.04 (1) 5 34 (83)
3 \
O
/
CA 02508788 2005-06-06
-66-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. mass (M+H)+ time [min] Ex. No. [mg] (Yield
(Method) [% of
theo
H~ 3C CHI
N ~ a
N
7p H C,NH3 ~ N H H 422.57 423 3.07 (1) 5 1.8 (4)
3
/
H p CH~H
I N~ CHI
N I ~ H'
H
71 0 ~ 514.67 515 3.39 (1) 7A 36 (70)
o~
H~ 3C CH'
N ~ s
N
~N ~ N H H
72 ~ 494.63 495 3.1 (1) 7A 16 (32)
0
_ o
H ~ 3C CH~
N ~ a
N
H
N ~ O H
73 ~ 515.65 516 2.7 (1) 7A 36 (70)
0
~ ~N \ o
H~ 3C CHI
N ~ s
N~
H
N ~ \~ H
74 j ~ 468.59 469 2.97 (1) 7A 29 (62)
HO O
O
CA 02508788 2005-06-06
-67-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -m-ass ---(M+H)+- time--[m-in] -Ex~ No.---[mg] (Yield
(Method) [% of
theo )
H- \ aC CHH~
N
N~
H
N I N~ H ,
75 of o 482.62 483 3.14 (1) 7A 35 (73)
i
CH3 O
H~(~~C CHIi~
N~(~/ ' 3
N.
H
N ~ \~ H
76 ~ ( ~ 500.64 501 7A
0
_ o
1 /
C H~3
-'H
N
N
H
N ~ \~ H
77 ~ 480.65 481 3.38 (1) 7A 34 (71)
0
CH3 O
H " 'C CHHa
N
N~
H
H I \)
78 H3C'N~N ~ " 495.66 496 2.5 (1) 7A 36 (73)
C"a o
CA 02508788 2005-06-06
-68-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. --mass---(M+-H)~-time-[min] -Ex.-No.-- [mg] (Yield
(Method) [% of
theo )
H ~ 3C CHH3
3
N~
H
HZN ~_N~ H
79 424.54 425 3.03 (1) 7A 28 (66)
0
0
H3C
H~ CHI-i~
N
N
H
80 H3C~N [ N, H 438.57 439 3.1 (1) 7A 30 (68)
0
_ o
1
H~ 3C CHI
N
CH3 ~ H
,N ~ ~ H
81 H~o 1 ~ 452.60 453 3.12 (1) 7A 3.5 (8)
0
0
H C CH
N
82 H \ "~ 422.57 423 3.13 (1) 8A 28 (66)
N ~ N~ H
p CHI
H ~3C CHI
N~ . s
N~
H
83 ~ [ N H 409.53 410 2.31 (1) 8A 27 (66)
CH3
,N
CA 02508788 2005-06-06
-69-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. mass (M+H)+ time [mint Ex. No. [mg] (Yield
(Method) [% of
theo
i "~ C"~H
N CH3
N I ~ H'
84 ~ H 450.62 451 3.35 (1) 9A 32 (71
o ~ )
CH3
H3C
H~O CHhI
N \ 3
N.
H
85 ( w N ~ N~ " 464.65 465 3.4 (1) 9A 35 (75)
0
~CH
3
H3C
H
c~3
N
N~
86 ~ ~ ~ H H 430.59 431 3.03 (1) 9A 13 (30)
'N
O
CH3
H3C
H
\O CHl.l~a
N
N.
87 N ~ ~~ H H 451.61 452 2.6 (1) 9A 28 (62)
'N
O
i N CH3
H3C
H-~ CHFIa
N
N~
88 N ~ N~ H H 404.55 405 2.88 (1) 9A 25 (62)
0
HO~
CH3
CA 02508788 2005-06-06
-70-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass ---(M+H)~---time-(min] -Ex~ No~ -[mg] (Yield
(Method) [% of
theo ])
H3C
H ~ CH~3
N
N,
H
89 N ~ N~ H 418.58 419 3.06 (1) 9A 9.8 (23)
0 0
i
CH3 CH3
3C Chl3
H
N
N,
H
90 N ~ N~ H 416.61 417 3.34 (1) 9A 32 (77)
0
CH3 CH3
H C
H_// CHhh
N ~
N.
91 H3C~N~N ~ N~ H H 431.62 432 2.43 (1) 9A 23 (53)
cH, o
~CH
3
H3C
H~ CF~3
N
N,
92 H N ~ ~~ H H 360.50 361 2.94 (1) 9A 25 (69)
~N
O
CH3
H3C CH3
O ,,~~CH3
~N ,
93 H C,N I N H ~ 374.53 375 3.01 (1) 9A 6.1 (16)
3
0
CH3
CA 02508788 2005-06-06
-71 -
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. mass-- -(M+H)+- time-[min] -Ex.-No. [mg] (Yield
(Method) [% of
theory
CH3
H_(/ '~~~CH3
~N
N~
94 ,NH3 ~ \ H H 388.55 389 3.06 (1) 9A 6.8 (18)
HsC ,N
O
CH3
CH3
i N /!C CHl
CH3
" ~ ~ H~ 448.61 449 3.27 1 l0A 26 58
95 ~ H ( ) ( )
0
H3C
N~ CHF
- \ 3
96 H ~ H~ ' 462.63 463 3.32 (1) l0A 30 (65)
J H
C
H3C
H O CH3
N'1 ,,~~CH3
N~
97 °~l ~ \ H 428.57 429 2.94 (1) l0A 3.9 (9)
~N N~ H
O
OC CH3
,,..CHs
\Ni
H
98 N ~ \~ H 449.60 450 2.52 (1) l0A 28 (62)
'N
O
iN
H3C
CH3
~~.CH~
99 H ~ \ "~ 402.54 403 2.79 (1) l0A 16 (40)
'N N H
HO Jr O
CA 02508788 2005-06-06
-72-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass--(M+H)+-t-ime-(min] -Ex:-No~ -(mg] (Yield
(Method) [% of
theo )
H3C
CH3
H
N ~~~CH3
N.
100 N I \~ H H 416.56 417 2.97 (1) l0A 27 (65)
~N
CH3
H3C
H O CH3
N-~ ~~ CH3
N~
101 N ~ \~ H H 414.59 415 3.25 (1) l0A 26 (63)
'N
O
CH3
H3C
H~ CI-'1
N-\
N
102 H c, N ~ ~ " H 429.61 430 2.33 (1) l0A 31 (72)
3 N~
CH3 O
HsC CHs
O
H l/ ~''~CH3
'~N
N~
103 ~ \> H 358.48 359 2.85 (1) l0A 27 (75)
H2N N H
O
H3C
O CH3
,'~~CH3
104 H \ H~ 372.51 373 2.92 (1) l0A 6.3 (17)
HaC.N I NJ H
O
H3C CH3
'~CH3
105 ~H3 I \ "~ 386.54 387 2.97 (1) l0A 4.7 (12)
HsC~N N~ H
O
CA 02508788 2005-06-06
_ 73 _
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. --mass- -(M+H)~-time-[min] --Ex.-No. - [mg] (Yield
(Method) [% of
theo
H3
~J/O CHa
i I N \ .CHa
~N ~ ~ "~ 452.60 453 3.1 1 12A 34 75
106 ~ ( ) ( )
H
O
O~CH3
HsC CHa
CHI
~N.
107 r", t ~~ " H 466.62 467 3.16 (1) 12A 35 (75)
I~
0
O~C~"~s
HaC CH3
O
N' \ ,CHs
N,
108 N I ~~ H H 453.58 454 2.36 (1) 12A 35 (77)
'N
O
i N O'CH3
H3C
CH3
H~ CH3
--~N
N,
109 N ~ ~ H H 406.52 407 2.63 (1) 12A 14 (34)
HO~ O
O'CH~
H3C
O CHa
H //
N~ ,.~CH3
N,
I H
110 N N~ H 499.66 500 2.71 (1) 13A 38 (76)
0
~ ~N /
CA 02508788 2005-06-06
-74-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. ---mass --(M+H)+- -time--[mint - ER.-No.- [mg] (Yield
(Method) (% of
theo
H3C
CH3
O
H~ wCHa
N
N~
H
I11 N I N~ H 452.60 453 2.98 (1) 13A 32 (71)
HO- O
O CHs
H~ CH3
\N
N.
H
N I \~ H
112 ~N 464.65 465 3.41 (1) 13A 38 (82)
0
CH3
H3C CH3
O
J/ CHI
N.
H
113 N I H~ H 424.54 425 3.04 (4) 23 (54)
I~
H'~.o
HsC CHs
CH3
'''~
N
H
114 N ~ N~ H 485.63 486 2.56 (4) 5 30 (62)
0
N
CA 02508788 2005-06-06
- 75 -
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass-----(M+I-~~--time-(min] Ex-. No.--[mg] (Yield
(Method) [% of
then )
CH
~~CH3
-\N ,
H
N I y H
11 S N 514.67 515 3.58 (4) 5 30 (58)
0
H~C.o
HaC CHa
N .,CHs
N
116 ~ N I ~ H H 471.60 472 5 23.2 (49)
.N I o
H3C
CH3
H Jf
N~ ,.~CHa
N.
H
117 N N H 464.61 465 3.43 (4) 7A 33 (71)
0
_ o
1 /
HaC CHa
O
H.JC ,wCH3
N
N~
H
118 N I N> H 492.66 493 3.65 (4) 7A 35 (?1)
0
_ o
1 /
CH3
N-\ .CHa
N~
N I ~~ H H
119 ~ ~ 515.65 516 2.65 (4) 7A 32 (62)
0
0
N
CA 02508788 2005-06-06
-76-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. mass (M+H)+ time [min] Ex. No. [mg] (Yield
(Method) [% of
theo ]
HaC CHa
O
CH
H ~~
120 ~ ~ 544.69 545 3.62 (4) 7A 28 (51)
0
~ o
H o-0 1 1
3
CHI
O CH3
H- \ . CHa
N
N.
H
121 ~ ~ N I N> " 501.63 502 7A 20.1 (40)
0
N
O
OH3C CH3
N- \ .CHs
N.
N I ~~ H H
122 ~ 532.69 533 2.49 (4) 7A 36 (68)
0
N O
O aC CHa
H //
N~
CH3
123 N I N> H 482.62 483 3.09 (4) 7A 27 (56)
HO~ O
O
,,
a
CA 02508788 2005-06-06
_ 77 _
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass---(M+I~+--- time-[min] - Ex=-No: --[mg]- (Yield
(Method) [% of
theo ])
HsC CHs
CHs
N.
H H ,
124 ~ ~ 480.65 481 3.63 (4) 7A 34 (71)
H3C CH~
O
HaC CHs
',CHs
N~
H
125 N I ~~ H 409.53 410 2.23 (4) 8A 20 (49)
~N
O CHs
i
N
O sC CHs
N~ ,..CHs
N I ~ H ~~
126 N 438.57 439 3.21 (4) 8A 28 (64)
CHs
HsC,O
OHsC CHs
CH3
- \N .
H
127 N I N~ H 426.56 427 2.15 (4) 8A 23 (54)
O CHs
N
H3C CH3 --.
H~ CH3
\N
N.
128 N ~ N~ H H 400.56 401 3.32 (4) 9A 26 (65)
0
CHs
CA 02508788 2005-06-06
_ 7g _
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. ---mass - -(M+H)~-- time-(min] - Ex. No. [mg] (Yield
(Method) [% of
theo
HsC CHs
O
N CH
N~
129 ,"V 1 ~~ H H 428.62 429 3.61 (4) 9A 29 (68)
N
O
"CH
3
O3C CH3
H- \ CHs
N
N~
130 N ~ N~ H H 451.61 452 2.54 (4) 9A 25 (55)
0
NJ CH3
HaC CHs
H~ CHI
-~'~N
N~
H
131 N N H 480.65 481 3.59 (4) 9A 30 (62)
0
i cH,
H3C,0
H3
~/0 CHa
H \ . CHa
N
N~
132 ~N~N I N~ " H 472.63 473 2.94 (4) 9A 26 (55)
HN'
~J O
~CHa
H3C CH3
N- \O CHs
H J ~ Hy
133 N N~ H 468.64 469 2.4 (4) 9A 24 (51 )
0
CH3
N
CA 02508788 2005-06-06
-79-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass --(M+H)~ - time-[min] Ex~ No.- [mg] (Yield
(Method) [% of
theo )
CH3
O CHs
N~ ."wvCH~
Nr
134 NH' ~ N H H 418.58 419 2.95 (4) 9A k4 (33)
HO- O
~CH
3
H3C
CI-1'3
~H
N
N~
135 N ~ \~ H H 416.61 417 3.58 (4) 9A 16 (38)
N
H3C_ _CH~
CH3
H3C
-(/O CI-h3
~H
N
N
136 H ~ \, H 398.55 399 3.2 (4) l0A 25 (63)
~N N H
O
CH
H~ CHt
N_
N~
137 N ~ \~ H H 426.60 427 3.52 (4) l0A 24 (56)
N
O
CH3
H ~ CHf~s
N
N.
138 N ~ \~ H H 449.60 450 2.45 (4) l0A 27 (60)
~N
O
~ NJ
CA 02508788 2005-06-06
-80-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass - -(M+I~~-time-[min) -Ex~ No. -[mg) (Yield
(Method) [% of
theo )
OaC CH3
CHI
N
H
N I \~ H
139 ~ 478.63 479 3.49 (4) l0A 30 (63)
.~ o
I
H3C.0
HaC CH3
O
// CH3
'~'~N ~
H
140 N I N~ " 466.63 467 2.33 (4) l0A 27 (58)
0
N
03C CH3
H-~ ~,,v CH3
N
141 N I \ "~ H 414.59 415 3.47 (4) l0A 11 (27)
H3C~CH~
H3C
CH3
O
H //
N~ ,.~ CH3
N~
142 N I \ H ~ 453.58 454 2.27 (4) 12A 30 (66)
'N
O
I
NJ CH3
H~ aC CH!-~
N ~ 3
N
H
N I N~ H
143 0 ~ 482.62 483 3.26 (4) 12A 32 (66)
I / O~CH3
H3C.0
CA 02508788 2005-06-06
-81 -
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass---(M+H)~-- time-[min] Ex.-No~-- [mg] (Yield
(Method) (% of
theo )
03C CH3
// ~~CHs
H I ~ ~H y
144 N N~ " 470.61 471 2.18 (4) 12A 31 (66)
0
O~CH3
\ N
H3C CH3
O
H //
N-''~ .~.vCHs
N,
H
N I O H
145 N 499.66 500 2.65 (4) 13A 36 (72)
N
CH3
H~ CH'
N \ . 3
N.
H
N I O H
146 ~N 528.69 529 3.64 (4) 13A 38 (72)
0
i
H3C,0
CH3
N- \O CHI-h
3
N.
I H
147 N N~ H 516.69 517 2.47 (4) 13A 48 (93)
0
N
H3C
H~ CI~
N
N.
H
148 ~ w N ~ N~ o " 521.70 522 3.07 (1) 11A 37 (71)
o ~r
N
CH3
CA 02508788 2005-06-06
-82-
Ex. Structure Molar MS (EI): Retention Reactant Amount
No. -mass--(M-+-H)~--time-[min] Ex: No:---[-mgJ-(Yield
(Method) [% of
theo ])
CH3
O CH3
N.// ~ CH3
149 ~ ~ N ~ ~ ~H, 408.54 409 3.7 (4) 8A 16.6 (41)
H
O CHa
CH3
H p CH3
N
1 Ha ~ . CHs
HN ~ ~ H.
150 p N H 422.57 423 3.8 (3) 13A 25.5 (60)
i
Example 151
1-Ethyl-4-[( { [( 1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]hept-2-yl] amino}
carbonyl)amino]-
1 H-pyrrole-2-carboxamide
rJ CH3
CH3
H N i,~~, ,,..CH3
z
H
H3C~
40.0 mg (0.13 mmol) of 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino}-
carbonyl)amino]-1H-pyrrole-2-carboxamide (Example 10) are dissolved in 1 ml of
DMF,
and 22.1 mg (0.20 mmol) of potassium tent-butoxide are added. After 5 minutes
10 ~1
(0.20 mmol) of bromoethane
Example 151
1-Ethyl-4-[( { [(1 R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1 ]kept-2-yl]amino}
carbonyl)amino]-
1 H-pyrrole-2-carboxamide
CA 02508788 2005-06-06
-83-
O CHs
CH3
,,,~CH3
H2N ~ ~ H .,,
-N
O
H C
3
40.0 mg (0.13 mmol) of 4-[({[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]kept-2-
yl]amino}-
carbonyl)amino]-1H-pyrrole-2-carboxamide (Example 10) are dissolved in 1 ml of
DMF,
and 22.1 mg (0.20 mmol) of potassium tent-butoxide are added. After 5 minutes
10 ~.1
(0.20 mmol) of bromoethane are added dropwise and the mixture is left with
stirring at RT
overnight. Subsequently the reaction mixture is purified by RP-HPLC. A solid
is obtained.
Yield: 11 mg (25% of theory)
LC-MS (Method 10): Rt = 3.31 min, MS (ESI~: m/z = 333 (M+H)+
IH-NMR (300MHz, DMSO-d6): 8 = 7.79 (s, 1H), 7.26 (bs, 1H), 6.95 (d, 1H), 6.77
(bs,
1 H), 6.51 (d, 1 H), 5.95 (d, 1 H), 4.24 (q, 2H), 3.87-3.98 (m, 1 H), 2.14-
2.28 (m, 1 H), 1.54-
1.77 (m, 3H), 1.09-1.33 (m, SH), 0.90 (s, 3H), 0.84 (s, 3H), 0.73 (s, 3H),
0.69-0.78 (m,
1 H).
Example 152
1-propyl-4-[({ [( 1 R,2S,4R)-1,7,7-trimethylbicyclo [2.2.1 ]hept-2-yl]amino }
carbonyl)amino]-
1 H-pyrrole-2-carboxamide
O CHs
CH3
,,..CH3
H H..,,
H
3.5 mg (0.01 mmol) of 18-crown-6 are dissolved in 0.5 ml of DMF and then 40.0
mg
(0.13 mmol) of 4-[( f [(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-
CA 02508788 2005-06-06
- 84 -
yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxamide (Example 10) and 17.3 mg
(0.15 mmol) of potassium tert-butoXide are added:- Subsequently -a solufion -
of -10-~l
(0.16 mmol) of 1-bromopropane in 0.5 ml of DMF is added dropwise and the
mixture is
left with stirring at RT overnight. Subsequently the reaction mixture is
purified by RP-
HPLC. A solid is obtained.
Yield: 14 mg (31 % of theory)
LC-MS (Method 1 ): Rt = 2.75 min, MS (ESI~: m/z = 347 (M+H)+
1H-NMR (300MHz, CDCl3): 8 = 6.83 (d, 1H), 6.46 (d, 1H), 5.84 (s, 1H), 5.46
(bs, 2H),
4.73 (d, 1H), 4.29 (t, 2H), 4.03-4.12 (m, 1H), 2.30-2.42 (m, 1H), 1.68-1.85
(m, 3H), 1.61-
1.66 (m, 1H), 1.25-1.37 (m, 2H), 1.02-1.13 (m, 1H), 0.94 (s, 3H), 0.88 (t,
3H), 0.86 (s, 3H),
0.84 (s, 3H), 0.74 (dd, 1H).
Example 153
4- f [(1-adamantylamino)carbonyl]amino}-1-benzyl-1H-pyrrole-2-carboxamide
H O
N
H2N~ ~ ~~ \H
N-
40 mg (0.16 mmol) of 1-benzyl-4-vitro-1H-pyrrole-2-carboxamide (Example SA)
are
dissolved in 1 ml of THF, and a spatula tip of Raney nickel and then 10 ~l
(0.24 mmol) of
hydrazine hydrate are added. The reaction mixture is stirred vigorously at RT
for 1 h. It is
filtered over kieselguhr and the solid product is washed with ethyl acetate.
The filtrate is
washed with sodium chloride solution, then dried over magnesium sulfate and
freed from
the solvent under reduced pressure. The residue obtained is dissolved in 1 ml
of THF.
Following the addition of 35 mg (0.20 mmol) of adamantyl isocyanate the
mixture is
stirred at RT for 1 h. The reaction mixture is purified by RP-HPLC. A solid is
obtained.
Yield: 58 mg (90% of theory)
CA 02508788 2005-06-06
-85-
LC-MS (Method 5): Rt = 2.23 min, MS (ESI~: m/z = 393 (M+H)+
'H-NMR (300MHz, DMSO-db): 8 = 7.81 (s, 1H), 7.35 (bs, 1H), 7.1?-7.32 (m, 3H),
7.06-
7.13 (m, 2H), 7.00 (d, 1 H), 6.80 (bs, 1 H), 6.5 5 (d, 1 H), 5 .62 (s, 1 H),
5.51 (s, 2H), 2.00 (m,
3H), 1.89 (m, 6H), 1.61 (m, 6H).
Example 154
1-(cyclopropylmethyl)-N (2-furylmethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]kept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-
carboxamide
n
O
H3
HN
SO mg (0.13 mmol) of 1-(cyclopropylmethyl)-4-[({[(1R,2S,4R)-1,7,7-trimethyl-
bicyclo-
[2.2.1]hept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-carboxylic acid are
dissolved in
4 ml of dimethylformamide at RT, and 24 mg (0.19 mmol) of N,N dimethylpyridine-
4-
amine and 98 mg (0.26 mmol) of O-(7-azabenzotriazol-1-yl)-N,N,N',N'-
tetramethyluronium hexafluorophosphate are added. After 10 minutes 25 mg
(0.259 mmol)
of (2-furylmethyl)amine are added dropwise. The mixture is stirred at RT for
16 hours. The
reaction solution is diluted with dimethyl sulfoxide and purified by means of
preparative
HPLC (Method 11 ).
Yield: 50 mg (88% of theory)
LC-MS (Method 1): R~= 3.17 min
MS (ESIpos): m/z = 439 (M+H)+
1H-NMR (300MHz, DMSO-d6) 8 = 0.23-0.31 (m, 2H), 0.37-0.50 (m, 2H), 0.73 (s,
3H),
0.75-0.79 (m, 1H), 0.84 (s, 3H), 0.90 (s, 3H), 1.08-1.33 (m, 3H), 1.53-1.75
(m, 3H), 2.15-
CA 02508788 2005-06-06
- 86 -
2.29 (m, 1H), 3.79-3.98 (m, 1H), 4.09 (dd, 2H), 4.35 (d, 2H), 5.98 (d, 1H),
6.21 (d, 1H),
6.37-6.39 (m, 1 H), b.57 (d, 1 H), 7.00 (d, 1 H), 7.54 (s, 1 H), 7.81 (s, 1
H), 8.34 (t, 1 H).
Example 155
1-(cyclopropylmethyl)-N (1-pyridin-4-ylethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]kept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-
carboxamide
N~
I H O
_rH N~H3C CH3
HN ~H,,,, ,,,.CH3
H
50 mg (0.13 mmol) of 1-(cyclopropylmethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]hept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-carboxylic
acid are
dissolved in 4 ml of dimethylformamide at RT, and 24 mg (0.19 mmol) of N,N
dimethylpyridine-4-amine and 98 mg (0.26 mmol) of O-(7-azabenzotriazol-1-yl)-
N,N,N',N'-tetramethyluronium hexafluorophosphate are added. After 10 minutes
32 mg
(0.26 mmol) of (1-pyridin-4-ylethyl)amine are added dropwise. The mixture is
stirred at
RT for 16 hours. The reaction solution is diluted with dimethyl sulfoxide and
purified by
means of preparative HPLC (Method 11 ).
Yield: 40 mg (61 % of theory)
LC-MS (Method 1): Rt= 2.38 min
MS (ESIpos): m/z = 464 (M+H)+
1H-NMR (300MHz, DMSO-d6) 8 = 0.20-0.26 (m, 2H), 0.32-0.43 (m, 2H), 0.73 (s,
3H),
0.76-0.83 (m, 1H), 0.85 (s, 3H), 0.90 (s, 3H), 0.98-1.32 (m, 3H), 1.50 (d,
3H), 1.56-1.79
(m, 3H), 2.16-2.29 (m, 1H), 3.79-4.19 (m, 3H), 5.15-5.26 (m, 1H), 6.24 (bs,
1H), 6.87 (d,
1 H), 8.01 (d, 1 H), 8.09 (bs, 1 H), 8.85 (d, 1 H).
CA 02508788 2005-06-06
Example 156
_ 87 _
1-(cyclopropylmethyl)-N (1-pyridin-3-ylethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1 ]hept-2-yl]amino } carbonyl)amino]-1 H pyrrole-2-
carboxamide
N
i
O
CH
CH3 N'w~H3C
HN_ ~ ~H~~,, ,,,.CH3
50 mg (0.13 mmol) of 1-(cyclopropylmethyl)-4-[({[(1R,2S,4R)-1,7,7-trimethyl-
bicyclo[2.2.1]kept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-carboxylic acid are
dissolved in 4 ml of dimethylformamide at RT, and 24 mg (0.19 mmol) of N,N
dimethylpyridine-4-amine and 98 mg (0.26 mmol) of O-(7-azabenzotriazol-1-yl)-
N,N,N',N'-tetramethyluronium hexafluorophosphate are added. After 10 minutes
32 mg
(0.26 mmol) of (1-pyridin-3-ylethyl)amine are added dropwise. The mixture is
stirred at
RT for 16 hours. The reaction solution is diluted with dimethyl sulfoxide and
purified by
means of preparative HPLC (Method 11 ).
Yield: 57 mg (95°I° of theory)
LC-MS (Method 1): Rt= 2.48 min
MS (ESIpos): m/z = 464 (M+H)+
'H-NMR (300MHz, DMSO-d6) b = 0.12-0.26 (m, 2H), 0.32-0.43 (m, 2H), 0.73 (s,
3H),
0.76-0.81 (m, 1H), 0.85 (s, 3H), 0.90 (s, 3H), 1.02-1.39 (m, 3H), 1.52 (d,
3H), 1.56-1.74
(m, 3H), 2.16-2.29 (m, 1H), 3.88-4.12 (m, 3H), 5.15-5.28 (m, 1H), 6.23 (bs,
1H), 6.80 (d,
1 H), 6.98 (d, 1 H), 8.00-8.09 (m, 2H), 8.50 (d, 1 H), 8.56 (d, 1 H), 8.80 (d,
1 H) 8.90 (d, 1 H).
Example 157
1-(cyclopropylmethyl)-N [1-(6-methylpyridin-3-y1)ethyl]-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo(2.2.1]kept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-
carboxamide
CA 02508788 2005-06-06
_ gg _
3
;H3
50 mg (0.13 mmol) of 1-(cyclopropylmethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.1]kept-2-yl]amino}carbonyl)amino]-1H-pyrrole-2-carboxylic
acid are
dissolved in 4 ml of dimethylformamide at RT, and 24 mg (0.19 mmol) of N,N-
dimethylpyridine-4-amine and 98 mg (0.26 mmol) of O-(7-azabenzotriazol-1-yl)-
N,N,N',N'-tetramethyluronium hexafluorophosphate are added. After 10 minutes
35 mg
(0.26 mmol) of 1-(6-methylpyridin-3-yl)ethyl]amine are added dropwise. The
mixture is
stirred at RT for 16 hours. The reaction solution is diluted with dimethyl
sulfoxide and
purified by means of preparative HPLC (Method 11).
Yield: 6 mg (10% of theory)
LC-MS (Method 10): Rt= 3.04 min
MS (ESIpos): m/z = 478 (M+I-~+
1H-NMR (300MHz, DMSO-d6) S = 0.12-0.27 (m, 2H), 0.33-0.42 (m, 2H), 0.73 (s,
3H),
0.76-0.83 (m, 1H), 0.85 (s, 3H), 0.90 (s, 3H), 1.01-1.35 (m, 3H), 1.46-1.78
(m, 6H), 2.14-
2.29 (m, 1 H), 2.72 (s, 3H), 3.89-4.11 (m, 3H), 5.13-5.27 (m, 1 H), 6.16 (bs,
1 H), 6.79 (d,
1 H), 6.97 (d, 1 H), 7.95-8.05 (m, 1 H), 8.3 9-8.48 (m, 2H).
Example 158
1-(cyclopropylmethyl)-N [1-(6-methoxypyridin-3-yl)ethyl]-4-[({[(1R,2S,4R)-
1,7,7-
trimethylbicyclo[2.2.1]hept-2-yl]amino}carbonyl)amino]-1H pyrrole-2-
carboxamide
CA 02508788 2005-06-06
-89-
~ Hs
O N
I H O
CH3 N--~H3C CHs
H N ~ \ H.,,, ,,,~C H3
~N~ ,
H
50 mg (0.13 mmol) of 1-(cyclopropylmethyl)-4-[({[(1R,2S,4R)-1,7,7-
trimethylbicyclo[2.2.lJkept-2-yl]amino}carbonyl)aminoJ-1H pyrrole-2-carboxylic
acid are
dissolved in 4 ml of dimethylformamide at RT, and 24 mg (0.19 mmol) of N,N
dimethylpyridine-4-amine and 98 mg (0.26 mmol) of O-(7-azabenzotriazol-1-yl)-
N,N,N;N'-tetramethyluronium hexafluorophosphate are added. After 10 minutes 32
mg
(0.26 mmol) of [1-(6-methoxypyridin-3-yl)ethyl]amine are added dropwise. The
mixture is
stirred at RT for 16 hours. The reaction solution is diluted with dimethyl
sulfoxide and
purified by means of preparative HPLC (Method 11).
Yield: 63 mg (99% of theory)
LC-MS (Method 4): Rt = 3.42 min
MS (ESIpos): m/z = 494 (M+H)+
'H-NMR (300MHz, DMSO-d6) 8 = 0.21-0.43 (m, 4H), 0.73 (s, 3H), 0.75-0.81 (m,
1H),
0.84 (s, 3H), 0.90 (s, 3H), 1.02-1.39 (m, 3H), 1.43 (d, 3H), 1.56-1.76 (m,
3H), 2.09-2.29
(m, 1 H), 3.80-4.11 (m, 3 H), 3.83 (s, 3 H), 4.95-5.16 (m, 1 H), 6.1 S (bs, 1
H), 6.69 (d, 1 H),
6.83 (d, 1 H), 6.96 (d, 1 H), 7.76 (dd, 2H), 7.95 (bs, 1 H), 8.14 (d, 1 H),
8.27 (d, 1 H).
B. Evaluation of the physiological activity
The in vitro action of the compounds of the invention can be shown in the
following
assays:
CA 02508788 2005-06-06
-90-
Anti-HCMV (anti-human cytome~alovirus) cytopatho~enicity tests
The test compounds are used as 50 millimolar (mM) solutions in dimethyl
sulfoxide
(DMSO). Ganciclovir, foscarnet and cidofovir serve as reference compounds.
Following
the addition of 2 ~1 in each case of the 50, 5, 0.5 and 0.05 mM DMSO stock
solutions to
98 ~l portions of cell culture medium in row 2 A-H in duplicate determination,
1:2
dilutions are earned out with 50 ~l portions of medium up to row 11 of the 96-
well plate.
The wells in rows 1 and 12 contain 50 pl of each medium. Then 150 pl portions
of a
suspension of 1 x 104 cells (human prepuce fibroblasts [NHDF]) are pipetted
into the wells
(row 1 = cell control) and a mixture of HCMV-infected and uninfected NHDF
cells (M.O.I.
= 0.001 - 0.002), i.e. 1-2 infected cells per 1000 uninfected cells, is
pipetted into rows
2-12. Row 12 (without substance) serves as virus control. The final test
concentrations are
250 - 0.0005 ~.M. The plates are incubated at 37°C/5% C02 for 6 days,
i.e. until all the
cells are infected in the virus controls (100% cytopathogenic effect [CPE]).
The wells are
then fixed and stained by adding a mixture of formalin and Giemsa's dye (30
minutes),
washed with double-distilled water and dried in a drying oven at 50°C.
Thereafter the
plates are evaluated visually using an overhead microscope (Plaque multiplier
from
Technomara).
The following data can be acquired from the test plates:
CCso (NHDF) = substance concentration in ~M, at which no visible cytostatic
effects on
the cells are evident by comparison with the untreated cell control;
ECSO (HCMV) = substance concentration in pM which inhibits the CPE (cytopathic
effect)
by 50% compared with the untreated virus control;
SI (selectivity index) = CCso (NHDF) ! ECSO (HCMV).
Representative in vitro activity data for the compounds of the invention are
shown in
Table A:
CA 02508788 2005-06-06
-91 -
Table A
HCMV
Example NHDF SI
ECso '
No. CCso [pM] HCMV
[nM]
3 25 60 417
4 12 86 139
6 125 80 1389
7 35 40 875
24 25 5.8 6552
60 50 7 7143
73 38 1.9 20 000
98 50 4 6025
99 50 5.8 8621
131 50 1.9 26 316
133 25 1.9 13 158
The suitability of the compounds of the invention for treating HCMV infections
can be
demonstrated in the following animal model:
HCMV Xeno~raft Gelfoam~ model
Animals:
3-4-week-old female immunodeficient mice (16-18 g), Fox Chase SCID or Fox
Chase
SCID-NOD or SCID beige, are purchased from commercial breeders (Bomholtgaard,
Jackson). The animals are kept under sterile conditions (including bedding and
feed) in
isolators.
Virus ~rowing_
Human cytomegalovirus (HCMV), Davis strain, is grown in vitro on human
embryonic
prepuce fibroblasts (NHDF cells). After the NHDF cells have been infected with
a
CA 02508788 2005-06-06
-92-
multiplicity of infection (M.O.I) of 0.01, the virus-infected cells are
harvested 5-7 days
later and stored in the presence of minimal essential medium (MEM), 10% fetal
calf serum
(FCS) with 10% DMSO at -40°C. After serial ten-fold dilutions of the
virus-infected cells,
the titer is determined on 24-well plates of confluent NHDF cells after vital
staining with
neutral red, or fixing and staining with a formalin-giemsa mixture (as
described under B.).
Preparation of the sponges, transplantation, treatment and evaluation:
Collagen sponges 1 x 1 x 1 cm in size (Gelfoarri ; Peasel & Lorey, Order No.
407534;
K.T. Chong et al., Abstracts of 39th Interscience Conference on Antimicrobial
Agents and
Chemotherapy, 1999, p. 439; P.M. Kraemer et al., Cancer Research 1983, (43):
4822-4827)
are initially wetted with phosphate-buffered saline (PBS), with the trapped
air bubbles
being removed by degassing, and then are stored in MEM + 10% FCS. 1 x 106
virus-
infected NHDF cells (infection with HCMV Davis M.O.I = 0.01) are detached 3
hours after
infection and applied dropwise, in 20 ~l MEM, 10% FCS, to a moist sponge.
Optionally,
after 12-13 hours, 5 ng/~1 basic fibroblast growth factor (bFGF) in 25 ~l of
PBS/0.1
BSAIl mM DTT are applied to the infected sponges, and incubation is earned out
for
1 hour. For the transplantation, the immunodeficient mice are anaesthetized
with Avertin or
an azepromazine/xylazine and ketamine mixture, the fur on the back is removed
using a
dry shaver, the epidermis is opened 1-2 cm and destressed, and the moist
sponges are
transplanted under the dorsal skin. The surgical wound is closed with tissue
glue. 24 hours
after the transplantation, the mice are treated orally with the substance
three times a day
(7.00 h and 14.00 h and 19.00 h) twice a day (8.00 h and 17.00 h), or once a
day (14.00 h)
over a period of 8 days. The daily dose is 3 or 10 or 30 or 100 mglkg of body
weight, the
application volume is 10 ml/kg of body weight. The substances are formulated
as a 0.5%
Tylose suspension optionally with 2% DMSO. 9 days after transplantation and 16
hours
after the last administration of the substance, the animals are painlessly
sacrificed and the
sponge is removed. The virus-infected cells are released from the sponge by
collagenase
digestion (330 U/1.5 ml) and stored in the presence of MEM, 10% fetal calf
serum, 10%
DMSO at -140°C. Evaluation takes place after serial ten-fold dilution
of the virus-infected
cells by determination of the titer on 24-well plates of confluent NHDF cells
after vital
staining with neutral red, or after fixing and staining with a formalin-giemsa
mixture (as
described under B.). The parameter determined is the number of infectious
virus particles
after treatment with the substance, in comparison with the placebo-treated
control group.
CA 02508788 2005-06-06
- 93 -
C. Exemplary embodiments of pharmaceutical compositions
The compounds of the invention can be converted into pharmaceutical
preparations as
follows:
Tablet:
Composition:
100 mg of the compound of Example l, 50 mg of lactose (monohydrate), 50 mg of
corn
starch (native), 10 mg of polyvinylpyrolidone (PVP 25) (BASF, Ludwigshafen,
Germany)
and 2 mg of magnesium stearate.
Tablet weight 212 mg. Diameter 8 mm, radius of curvature 12 mm.
Production:
The mixture of active ingredient, lactose and starch is granulated with a 5%
strength
solution (m/m) of the PVP in water. The granules, after drying, are mixed with
the
magnesium stearate for 5 minutes. This mixture is compressed using a
conventional
tableting press (for tablet format see above). A guideline for the compressive
force used for
compression is 15 kN.
Suspension for oral administration:
Composition:
1000 mg of the compound from Example 1, 1000 mg of ethanol (96%), 400 mg of
Rhodigel (xanthan gum from FMC, Pennsylvania, USA) and 99 g of water.
10 ml of oral suspension correspond to a single dose of 100 mg of the compound
of the
invention.
Production:
The Rhodigel is suspended in ethanol and the active ingredient is added to the
suspension.
The water is added with stirnng. Stirring is carned out for about 6 h until
the swelling of
the Rhodigel is at an end.