Note: Descriptions are shown in the official language in which they were submitted.
CA 02508871 2008-03-12
-1-
CRYSTALLINE FORM F OF ATORVASTATIN HEMI-CALCIUM SALT
The present invention is directed to a crystalline form of Atorvastatin
calcium, processes for the
preparation thereof and pharmaceutical compositions comprising this
crystalline form.
The present invention relates to a crystalline form of Atorvastatin calcium.
Atorvastatin calcium
is known by the chemical name, [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-
dihydroxy-5-(1-
methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1 H-pyrrole-1 -heptanoic acid
calcium salt (2:1).
Atorvastatin has the following formula:
HO
CO 2H
OH F
N
H I I
N O
Atorvastatin calcium is an orally-active hypocholesterolaemic, a liver-
selective HMG-CoA
reductase inhibitor. Processes for the preparation of Atorvastatin calcium are
described in US-
A-5,273,995, US-A-5,298,627, US-A-6,087,51 1, US-A-6,274,740, WO-A-97/03960,
WO-A-
02/059087, WO-A-02/072073, and in the publications by P.L. Brower et al. in
Tetrahedron
Letters (1992), vol. 33, pages 2279-2282, K.L. Baumann et al. in Tetrahedron
Letters (1992),
vol. 33, pages 2283-2284 and A. Graul et al. in Drugs of the Future (1997),
vol. 22, pages 956-
968.
This calcium salt (2:1) is desirable since it enables Atorvastatin calcium to
be conveniently
formulated. The processes in the above mentioned patents and publications
result in the
preparation of amorphous Atorvastatin calcium.
The preparations of Atorvastatin calcium (2:1) described in WO-A-97/03958 and
WO-A-
97/03959 result in the isolation of crystalline Atorvastatin calcium with the
polymorphic forms III,
and I, II, and IV, respectively. WO-A-01/36384, WO-A-02/41834 and WO-A-
02/43732 claim the
preparation of crystalline Atorvastatin calcium with the polymorphic forms
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-2-
V to XII, whereas WO-A-02/051804 claims the polymorphic forms A, B1, B2, C, D
and E.
However, there is still a need to produce Atorvastatin calcium in a
reproducible, pure and
crystalline form to enable formulations to meet exacting pharmaceutical
requirements and
specifications. Furthermore, it is economically desirable that the product is
stable for
extended periods of time without the need for specialized storage conditions.
Surprisingly, there has now been found a novel crystalline form of
Atorvastatin calcium salt
(2:1), herein designated as Form F. This novel form of the present invention
can be prepared
in ecological friendly solvents and has a good thermal stability combined with
good solubility
characteristics.
Accordingly, the present invention is directed to the polymorphic Form F of
Atorvastatin
calcium salt (2:1).
Therefore, the present invention is directed to a crystalline polymorph F of
[R-(R*,R*)]-2-(4-
fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-
[(phenylamino)carbonyl]-1 H-
pyrrole-1-heptanoic acid calcium salt (2:1) which exhibits a characteristic X-
ray powder
diffraction pattern with characteristic peaks expressed in d-values (A) at
24.3 (s), 10.2 (s),
8.6 (s), 4.57 (vs), 4.26 (m); wherein (vs) = very strong intensity; (s) =
strong intensity;
(m) = medium intensity.
More particularly, the crystalline polymorph F exhibits a characteristic X-ray
powder
diffraction pattern with characteristic peaks expressed in d-values (A) at
32.3 (w), 24.3 (s), 16.5 (m), 13.0 (w), 11.4 (m), 10.2 (s), 8.6 (s), 7.0 (m),
6.4 (m), 5.16 (m),
4.96 (m), 4.57 (vs), 4.26 (m), 3.95 (m), 3.67 (m), 3.48 (m), 3.20 (w).
The abbreviations in brackets mean: (vs) = very strong intensity; (s) = strong
intensity;
(m) = medium intensity; (w) = weak intensity.
Especially, the crystalline polymorph F exhibits a characteristic X-ray powder
diffraction
pattern with characteristic peaks expressed in d-values (A) and in 20 as given
in Table 1 (vs
= very strong intensity, s = strong intensity, m = medium intensity, w = weak
intensity).
CA 02508871 2007-08-20
-3-
Table 1: d-spacings and 20 angles for Form F.
d-spacing [10' m] Angle (20] Intensity
32.3 .7
24.3 3.7 s
16.5 5.4 m
13.0 .8
11.4 7.7 m
10.2 8.7 s
8.6 10.2 s
7.0 12.6 m
4 13.8 m
5.16 17.2 m
.96 17.9 m
.57 19.4 s
.26 20.8 m
3.95 22.5 m
3.67 24.2 m
3.48 25.5 m
3.20 27.8
Brief description of the drawings
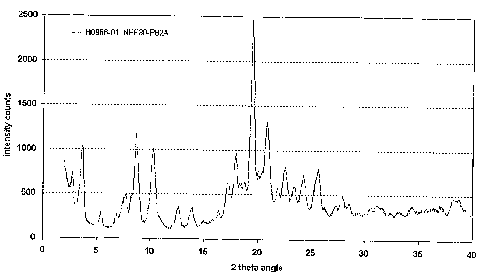
Figure 1 is a characteristic X-ray powder diffraction pattern for Form F.
Figure 2 is a characteristic13C CP-MAS solid state NMR spectrum of Form F.
Figure 3 is a characteristic X-ray powder diffraction pattern for Form F
essentially free of
residual organic solvent.
Figure 4 is a characteristic 13C CP-MAS solid state NMR spectrum of Form F
essentially free of
residual organic solvent.
The polymorphic Form F of Atorvastatin calcium is especially characterized by
a powder X-ray
diffraction pattern substantially as depicted in Figure 1.
Furthermore, the crystalline polymorph F exhibits a characteristic 13C CP-MAS
solid state NMR
spectrum with chemical shifts in parts per million, with peak intensity in
arbitrary units in
parentheses at 188.2 (2.5), 184.3 (2.2), 177.4 (2.8), 167.5 (2.9), 162.6
(2.4), 161.0 (3.9),
139.8 (6.5), 138.2 (4.3), 135.8 (5.1), 134.1 (4.0), 132.0 (8.4), 131.2 (7.3),
130.5 (14.0), 129.0
(9.5), 128.0 (6.6), 127.2 (4.6), 125.5 (2.9), 124.0 (4.9), 123.5 (4.7), 122.8
(4.8), 122.1 (6.2),
CA 02508871 2007-08-20
-4-
120.7 (5.4), 117.6 (4.1), 116.6 (4.1), 115.2 (3.6), 112.8 (1.6), 72.9 (4.8),
71.7 (5.2), 69.4
(6.5), 67.1 (4.9), 63.2 (0.7), 46.3 (10.4), 44.1 (12.6), 40.6 (7.6), 36.4
(0.7), 32.1 (3.8), 31.1 (1.3),
28.2 (5.3), 27.4 (9.0), 25.8 (11.2), 22.5 (3.7), 20.9 (4.2), 20.0 (4.8).
The polymorphic Form F of Atorvastatin calcium is especially characterized by
a 13C CP-
MAS solid state NMR spectrum as depicted in Figure 2. Furthermore, the present
invention
is directed to processes for the preparation of Form F of Atorvastatin
calcium.
Form F can generally be prepared by adding Form A to a ketone solvent,
especially
acetone. It is preferred that the ketone solvent contains as a further solvent
some water. The
amount of water is preferably about 1 to 30% v/v, more preferably about 5 to
20% v/v,
especially about 10 to 20% by volume of the suspension (v/v). It is preferred
that the
suspension is treated at temperatures between 10 and 60 C, preferably at
temperatures of
20 to 40 C, especially for a longer periods of time, like 10 to 40 hours. It
is further preferred
that nucleation of Form F is induced at a temperature of 40 to 60 C,
especially at about
60 C, and subsequent ripening and equilibration is performed at temperatures
between 20
and 40 C. If desired, during the preparation process seeding with Form F can
be carried out.
Form F can, for example, be isolated by filtration and dried in air or in
vacuum. The above
mentioned process can also be carried out using another crystalline form or
the amorphous
form of atorvastatin calcium. Examples of other crystalline forms are Forms I,
II, III, IV, V, VI,
VII, VIII, IX, X, XI, XII, A, B1, B2, C, D and E, which are disclosed and
characterized in the
references given hereinbefore. Preferred forms for this purpose are Form A
(see for
example WO-A-02/051804; last but one paragraph of page 2; page 4, last but one
paragraph to page 5, first paragraph; Examples 2, 8 and 9; Fig. 2) or Form I
(see for
example WO-A-97/03959; table on page 4; page 20, line 9 to page 22, line 11;
Example 1;
Fig. 1). As to Form B1 see for example WO-A-02/051804; the paragraph bridging
pages 2
and 3; page 5, second paragraph; Example 3; and Fig. 3). As to Form B2 see for
example
WO-A-02/051804; page 3, second paragraph; page 5, third paragraph; Example 4;
and Fig.
3.
Form F can also be prepared from Atorvastatin lactone upon subsequent reaction
with NaOH to
form Atorvastatin sodium followed by reaction with CaCIZ in a ketone solvent,
especially in
acetone. It is preferred that the ketone solvent contains as a further solvent
some water. The
amount of water is preferably about 1 to 30% v/v. If desired, during the
preparation process
CA 02508871 2007-08-20
-5-
seeding with Form F can be carried out.
Form F can also be prepared directly from Atorvastatin lactone upon reaction
with a
calcium(II) salt, like Ca(OH)2 or Ca(OAc)2, in a ketone solvent, especially in
acetone. It is
preferred that the ketone solution contains as a further solvent some water.
The amount of
water is preferably about 1 to 30% v/v. If desired, during the preparation
process seeding
with Form F can be carried out.
Form F can also be prepared by adding a concentrated solution of Atorvastatin
calcium in an
organic solvent, like tetrahydrofuran, to a ketone solvent, especially
acetone. It is preferred that
the ketone solution contains as a further solvent some water. The amount of
water is preferably
about 1 to 30% v/v. If desired, during the preparation process seeding with
Form F can be
carried out.
As to the ketone solvent of the preparation processes given above it is
preferred to use C3-C8
ketones, especially acetone.
Another object of the present invention are crystalline forms of atorvastatin
calcium which are
essentially free of residual organic solvent. Preferred are Forms I to XII and
A, B1, B2, C, D,
E and F which are essentially free of residual organic solvent. Highly
preferred are Forms B1,
B2 and F, especially Form F. Forms I to XII and A, B1, B2, C, D, E are
disclosed in the
references given above. The following preparations of forms which are
essentially free of
residual organic solvents can be applied to any form of Atorvastatin calcium.
It is preferred that
the crystalline forms contain less than 0.5% by weight of residual organic
solvent. Highly
preferred are amounts of residual organic solvents of less than 5000 ppm,
especially less than
2000 ppm. Most preferred are amounts of residual organic solvents of less than
400 ppm,
especially less than 200 ppm. Examples of such residual organic solvents are
acetone,
ethylacetate, tetrahydrofuran, ethanol, methanol, acetonitrile and hexane.
Therefore, the present invention is directed to a crystalline polymorph F of
[R-(R",R*)]-2-(4-
fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-
[(phenylamino)carbonyl]-1 H-
pyrrole-1-heptanoic acid calcium salt (2:1) which is essentially free of
residual organic solvents
and which exhibits a characteristic X-ray powder diffraction pattern with
characteristic
peaks expressed in d-values (A) at 24.3 (s), 10.2 (s), 8.7 (s), 4.80 (m), 4.56
(vs), 4.00 (m), 3.72
CA 02508871 2007-08-20
-6-
(m); wherein (vs) = very strong intensity; (s) = strong intensity; (m) =
medium intensity.
More particularly, the crystalline polymorph F of [R-(R*,R*)]-2-(4-
fluorophenyl)-beta,delta-
dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1 H-pyrrole-1-
heptanoic acid
calcium salt (2:1) which is essentially free of residual organic solvents
exhibits a characteristic
X-ray powder diffraction pattern with characteristic peaks expressed in d-
values (A) at 31.9 (w),
24.3 (s), 16.3 (w), 13.1 (vw), 12.1 (w), 11.4 (m), 10.2 (s), 8.7 (s), 8.1
(vw), 7.1 (w), 6.9 (w), 6.5
(m), 5.98 (vw), 5.60 (vw), 5.21 (m), 5.00 (m), 4.80 (m), 4.56 (vs), 4.32 (w),
4.23 (m), 4.00 (m),
3.72 (m), 3.48 (m); wherein (vs) = very strong intensity; (s) = strong
intensity; (m) = medium
intensity; (w) = weak intensity; (vw) = very weak intensity.
The crystalline polymorph F of [R-(R*,R")]-2-(4-fluorophenyl)-beta,delta-
dihydroxy-5-(1-
methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1 H-pyrrole-1-heptanoic acid
calcium salt (2:1)
which is essentially free of residual organic solvents is especially
characterized by an X-ray
powder diffraction pattern substantially as depicted in figure 3.
Furthermore, the crystalline polymorph F of [R-(R*,R'')]-2-(4-fluorophenyl)-
beta,deltadihydroxy-
5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic
acid calcium salt
(2:1) which is essentially free of residual organic solvents exhibits a
characteristic 13CP-MAS
solid state NMR spectrum with chemical shifts in parts per million, with peak
intensity in arbitrary
units in parentheses, at 187.7 (1.3), 184.6 (1.7), 177.71, (2.5), 167.5 (3.4),
162.8 (1.7), 161.1
(3.0), 143.6 (1.3), 140.0 (3.3), 139.3 (3.7), 138.0 (4.0), 137.1, (3.4), 136.0
(5.7), 134.1 (8.8),
132.0 (8.7), 131.3, (8.5), 130.2, (13.3), 129.5 (14.0), 127.9, (9.6), 126.8
(3.6), 125.3 (4.4), 123.1
(8.5), 120.5, (3.8), 117.5, (4.5), 115.3 (4.9), 112.8, (1.1), 72.7 (3.9), 71.5
(6.2), 69.3 (9.3), 67.2
(5.8), 46.3, (11.3), 44.4, (12.4), 41.3 (6.2), 40.6, (7.2), 34.7 (0.9), 32.0
(1.4), 30.8 (1.4), 28.2
(6.5), 27.3, (9.5), 25.9 (7.8), 21.1 (4.2), 20.3, (4.3).
The crystalline polymorph F of [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-
dihydroxy-5-(1-
methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1 H-pyrrole-1 -heptanoic acid
calcium salt (2:1)
which is essentially free of residual organic solvents is especially
characterized by a 13C CP-
MAS solid state NMR spectrum substantially as depicted in figure 4.
It is preferred that the crystalline polymorphs F have a water content of up
to 5% by weight
(w/w) (independently whether the polymorphs are essentially free of residual
organic solvents or
not).
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-7-
Another object of the present invention are processes for the preparation of
crystalline forms
of atorvastatin calcium which are essentially free of residual organic
solvent. Preferred are
processes for the preparation of Forms I to XII and A, B1, B2, C, D, E and F
which are
essentially free of residual organic solvent. Highly preferred are processes
for the
preparation of Forms B1, B2 and F, especially Form F, which are essentially
free of residual
organic solvent. Forms I to XII and A, B1, B2, C, D, E are disclosed in the
references given
above. The following preparations of forms which are essentially free of
residual organic
solvents can be applied to any form of Atorvastatin calcium.
Particularly, the present invention is related to processes for the
preparation of crystalline
forms of atorvastatin calcium essentially free of residual organic solvent by
exposing the
crystalline form of atorvastatin calcium to an atmosphere with a defined
relative air humidity.
A relative air humidity of 5 to 100%, especially 40 to 80%, is preferred. A
corresponding
process for the preparation of Form F is preferred.
More particularly, atorvastatin calcium essentially free of residual organic
solvent can be
prepared by exposure to an inert gas flow with a defined relative air humidity
(to exchange
residual organic solvent with water). A relative air humidity of 5 to 100%,
especially 40 to
80%, is preferred. A corresponding process for the preparation of Form F is
preferred.
For example, Form F can be generally prepared essentially free of residual
organic solvent
by storage of this form in an atmosphere with a relative air humidity of 5 to
100%, preferably
40 to 80%, or by treating this form with a gas stream with a relative air
humidity of 5 to
100%, preferably 40 to 80%.
The powder X-ray diffraction pattern as well as the 13C CP-MAS solid state NMR
spectrum of
Form F essentially free of residual organic solvent may appear with slight
deviations as
compared to Form F containing residual organic solvent, compare Figure 3 and
Figure 4 with
Figure 1 and Figure 2, respectively.
Another object of the present invention are pharmaceutical compositions
comprising an
effective amount of crystalline polymorphic Form F, and a pharmaceutically
acceptable
carrier.
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-8-
The polymorphic Form F may be used as single component or as mixtures with
other
polymorphic forms or the amorphous form of atorvastatin calcium.
As to Atorvastatin calcium it is preferred that it contains 25-100% by weight,
especially 50-
100% by weight of the novel form, based on the total amount of Atorvastatin
calcium.
Preferably, such an amount of the novel polymorphic form of Atorvastatin
calcium is 75-
100% by weight, especially 90-100% by weight. Highly preferred is an amount of
95-100%
by weight.
The compositions of the present invention include powders, granulates,
aggregates and
other solid compositions comprising polymorphic Form F. In addition, the
compositions that
are contemplated by the present invention may further include diluents, such
as
cellulose-derived materials like powdered cellulose, microcrystalline
cellulose, microfine
cellulose, methyl cellulose, ethyl cellulose, hydroxyethyl cellulose,
hydroxypropyl cellulose,
hydroxypropylmethyl cellulose, carboxymethyl cellulose salts and other
substituted and
unsubstituted celluloses; starch; pregelatinized starch; inorganic diluents
like calcium
carbonate and calcium diphosphate and other diluents known to the
pharmaceutical industry.
Yet other suitable diluents include waxes, sugars and sugar alcohols like
mannitol and
sorbitol, acrylate polymers and copolymers, as well as pectin, dextrin and
gelatin.
Further excipients that are within the contemplation of the present invention
include binders,
such as acacia gum, pregelatinized starch, sodium alginate, glucose and other
binders used
in wet and dry granulation and direct compression tableting processes.
Excipients that also
may be present in the solid compositions further include disintegrants like
sodium starch
glycolate, crospovidone, low-substituted hydroxypropyl cellulose and others.
In addition,
excipients may include tableting lubricants like magnesium and calcium
stearate and sodium
stearyl fumarate; flavorings; sweeteners; preservatives; pharmaceutically
acceptable dyes
and glidants such as silicon dioxide.
The dosages include dosages suitable for oral, buccal, rectal, parenteral
(including
subcutaneous, intramuscular, and intravenous), inhalant and ophthalmic
administration.
Although the most suitable route in any given case will depend on the nature
and severity of
the condition being treated, the most preferred route of the present invention
is oral. The
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-9-
dosages may be conveniently presented in unit dosage form and prepared by any
of the
methods well-known in the art of pharmacy.
Dosage forms include solid dosage forms, like tablets, powders, capsules,
suppositories,
sachets, troches and losenges as well as liquid suspensions and elixirs. While
the
description is not intended to be limiting, the invention is also not intended
to pertain to true
solutions of Atorvastatin calcium whereupon the properties that distinguish
the solid form of
Atorvastatin calcium are lost. However, the use of the novel form to prepare
such solutions is
considered to be within the contemplation of the invention.
Capsule dosages, of course, will contain the solid composition within a
capsule which may
be made of gelatin or other conventional encapsulating material. Tablets and
powders may
be coated. Tablets and powders may be coated with an enteric coating. The
enteric coated
powder forms may have coatings comprising phthalic acid cellulose acetate,
hydroxypropylmethyl-cellulose phthalate, polyvinyl alcohol phthalate,
carboxymethylethylcellulose, a copolymer of styrene and maleic acid, a
copolymer of
methacrylic acid and methyl methacrylate, and like materials, and if desired,
they may be
employed with suitable plasticizers and/or extending agents. A coated tablet
may have a
coating on the surface of the tablet or may be a tablet comprising a powder or
granules with
an enteric coating.
Preferred unit dosages of the pharmaceutical compositions of this invention
typically contain
from 1 to 100 mg of the novel Atorvastatin calcium form or mixtures with other
forms of
Atorvastatin calcium (including the amorphous form). More usually, the
combined weight of
the Atorvastatin calcium forms of a unit dosage are from 5 mg to 80 mg, for
example 10, 20
or 40 mg.
The following Examples illustrate the invention in more detail. Temperatures
are given in
degrees Celsius.
Example 1:
277 mg of Atorvastatin calcium Form A are added to 11 ml of a mixture of
acetone and water
(80:20 v/v). This suspension is stirred at ambient temperature for about ten
minutes, leading
to almost complete dissolution of Form A. When the resulting slightly turbid,
opalescent
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-10-
solution is stirred at 40 C for about 14 hours, a white precipitate is formed.
This precipitate is
separated by filtration and dried at 60 C for 2 hours. Yield: 153 mg (55%).
Analysis by
powder X-ray diffraction shows that the obtained sample is Atorvastatin
calcium Form F as
shown in Figure 1. Karl Fischer titration of the sample after X-ray
diffraction reveals a water
content of 2.0 %.
Example 2:
303 mg of Atorvastatin calcium Form A are added to a mixture of 10 ml acetone
and 1 ml of
water. This mixture is stirred at ambient temperature for about 15 minutes
which leads to
almost complete dissolution of the solid. The slightly turbid, opalescent
solution/suspension
is stirred at 40 C for 22 hours. Within this time a thick precipitate is
formed. This suspension
is thoroughly stirred at 50 C for about 15 minutes, then the mixture is cooled
to 20 C while
stirring is continued for another 4 hours. Then the suspension is filtrated
and dried at 80 C
for 3 hours (300 mbar). An X-ray powder diffraction study shows the product to
be
polymorphic Form F.
Example 3:
500 mg of Atorvastatin calcium Form I are suspended in 15 mi of acetone and
water mixture
(80:20 v/v). This suspension is shortly stirred at 60 C giving a clear
solution which becomes
immediately turbid. This turbid suspension is stirred for an additional 16
hours at 40 C. The
resulting precipitate is filtered, washed with 2 ml of the acetone/water
mixture and dried for 1
hour at 50 C/800 mbar. Yield 400 mg (80%). An X-ray powder diffraction study
shows the
product to be polymorphic Form F.
Example 4:
1.58 g of Atorvastatin calcium Form A are suspended in 44.2 g of a mixture of
acetone and
water (80:20), heated to 60 C and stirred for 3 hours. The white suspension
formed is slowly
cooled to 40 C, stirred for an additional 18 hours, slowly cooled to 20 C and
stirred for an
additional 2 hours. The suspension was filtered and dried at ambient
temperatures with dry
nitrogen. Yield 1.192 g. An X-ray powder diffraction study shows the product
to be
polymorphic Form F. Further analysis by 13C CP-MAS solid state NMR
spectroscopy reveals
a solid state NMR spectrum as shown in Figure 2. analysis by Karl Fischer
titration reveals a
water content of 1.3%, and analysis by GC-head space chromatography reveals an
acetone
content of 0.7%.
CA 02508871 2005-05-27
WO 2004/050618 PCT/US2003/038090
-11-
Example 5:
250 mg of Atorvastatin calcium Form F as obtained in example 4 was placed in a
U-shaped
glass tube in a oven at 60 C. This glass tube was purged with a nitrogen flow
with a relative
air humidity of ca. 50% for a period of 16 hours. Analysis by Karl Fischer
titration reveals a
water content of 2.5%, and analysis by GC-head space chromatography reveals an
acetone
content of less than 0.05%.
Example 6:
250 mg of Atorvastatin calcium Form F as obtained in example 4 was placed in a
desiccator
over a saturated NaCI solution at room temperature, i.e. in an atmosphere with
a relative air
humidity of 75%, for 44 hours. Analysis by Karl Fischer titration reveals a
water content of
2.7%, and analysis by GC-head space chromatography reveals an acetone content
of less
than 0.05%. Further analysis by X-ray powder diffraction and 13C CP-MAS solid
state NMR
spectroscopy shows that the crystal structure of Atorvastatin Form F was
essentially retained
under the given conditions, see Figures 3 and 4, respectively.
X-ray powder diffraction measurements were performed on a Philips 1710 powder
X-ray
diffractometer using Cu Ka radiation (Cu Kai and Cu Ka2 at a ratio of 2, k of
Cu K(Xi =
1.54060, and X of Cu Ka2 = 1.54447). The X-ray source is operated at 45 kV and
45 mA.
Spectra are recorded at a step size of 0.02 with a counting time of 2.4
seconds per step.
The accuracy of the 2 theta values of conventionally recorded powder X-ray
diffraction
patterns is generally +/- 0.2 . For sample preparation, about 40 mg of
substance was
prepared into circular shaped quartz sample holders of 0.5 mm depth and 10 mm
width. The
13C CP-MAS solid state NMR spectra were recorded on a Bruker Avance-600 NMR
spectrometer operating at 600 MHz proton resonance frequency. Samples were
filled into 4
mm rotors without further pretreatment. Bruker xwinnmr version 3.1 was used to
acquire the
spectra. All spectra were recorded with variable-amplitude (linear ramp) cross
polarization
from the protons and using high-power proton decoupling (100 KHz field
strength using the
XiX decoupling scheme). The MAS frequency was set to 15 KHz and stabilized to
within 5
Hz. 3072 transients with 4096 complex data points each (acquisition time 41
ms) were
added. The recycle delay was set to 3 sec leading to a measurement time of
about 3 hours.
The data analysis was carried out using MATLAB version 6.5. The first 200
complex data
points of the acquired signal (FID) were Fourier transformed using a cosine-
squared window
CA 02508871 2007-08-20
-12-
function and zero filling to 32768 data points leading to an optimized signal-
to-noise ratio and
spectral resolution. The digital resolution in the frequency domain is 1.53
Hz. The spectra were
referenced to an external standard using uniformly13C/15N labeled alanine by
setting the
resonance frequency of the Ca, to 51.9 ppm. Under these measurement conditions
and above
described data evaluation the accuracy of the NMR shifts as presented is +/-
0.05 ppm.