Note: Descriptions are shown in the official language in which they were submitted.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
PROCESS FOR PREPARING COMBINATION PHARMACEUTICAL
FORMULATIONS USING SUPERCRITICAL FLUIDS
CROSS-REFERENCE TO RELATED PATENT APPLICATIONS
[0001] This patent application claims the benefit of U.S. Provisional Patent
Application
No. 60/435,054, filed December 19, 2002.
FIELD OF THE INVENTION
[0002] This invention pertains to a process for combining two or more active
pharmaceutical ingredients including, for example, anti-infective and
anticancer agents,
using a supercritical fluid to obtain a blended, dry powder pharmaceutical
formulation:
BACKGROUND OF THE INVENTTON
[0003] Pharmaceuticals containing a combination of two or more active
pharmaceutical
ingredients, especially anti-infective agents, are commercially available in a
dry powder
form. Anti-infective agents as well as many other active agents are not stable
for extended
periods of time in an aqueous solution which requires the preparation of such
actives as a
solid powder.
[0004] Combination anti-infectives are typically produced by milling of the
active
agents and excipients and blending the dry solid components to form the
finished drug
product. However, the use of milling and blending techniques has several
significant
limitations. Most significantly, the mechanical equipment used to accomplish
the milling
and blending operations is in direct contact with the drug product components
which can
result in contamination from pyxogens and/or particular matter. Such
contaminants
compromise the sterility required for pharmaceutical products that are
administered
parenterally. Other drawbacks include, for example, the need for specialized
ventilation
equipment to collect dust produced during milling, the difficulty in obtaining
blend
uniformity, and the degradation of the active ingredients and excipients
caused by high
shear milling. Moreover, the potential segregation of the components of the
blended
powder during its transfer from blender to the filling line and during vial
filling may
eventually lead to content non-uniformity in the final blended drug product.
[0005] An alternative to the use of traditional milling and blending
procedures to
produce combination drug products is spray drying. The spray drying process
involves the
dissolution of active agents in a suitable cosolvent (which may be a single
solvent or two or
more solvents combined together) followed by spraying of the solution in a
heated chamber.
However, spray drying has several significant limitations. Stability issues
exist with the
solution or dispersion of the active agents formed before spraying. In
addition, the high
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
2
temperatures used during the process can cause degradation of the drugs. Spray
drying also
gives low yields of the final product and often requires the use of a
secondary drying step to
ensure removal of cosolvent from the powder.
[0006] Thus, there remains a need for an efficient process for producing
sterile
combination pharmaceutical drug products in a powder form that exhibit good
blend
uniformity.
[0007] The invention provides such a process for preparing sterile
pharmaceutical
formulations in a dry powder form that contain two or more active
pharmaceutical
ingredients in a homogenous blend. These and other advantages of the
invention, as well as
additional inventive features, will be apparent from the description of the
invention
provided herein.
BRIEF SUMMARY OF THE INVENTION
[0008] The invention provides a process for preparing a pharmaceutical
formulation
containing two or more active pharmaceutical ingredients by (a) contacting two
or more
active ingredients with a supercritical fluid, and (b) separating the active
ingredients from
the supercritical fluid to yield a dry powder precipitate containing the
active ingredients.
The invention further provides a supercritical fluid solution comprising a
supercritical fluid
and two or more active pharmaceutical ingredients.
[0009] The invention further relates to a process for preparing a
pharmaceutical
formulation containing a combination of two anti-infective agents comprising:
(a) contacting two anti-infective agents with supercritical carbon dioxide to
form
a supercritical carbon dioxide solution;
(b) spraying the supercritical carbon dioxide solution through a nozzle; and
(c) recovering the precipitate in a powder form containing the combination of
anti-infective agents.
[0010] In another embodiment, the invention is directed to a process for
preparing a
pharmaceutical formulation containing two or more active pharmaceutical
ingredients
comprising:
(a) combining two or more active ingredients with a solvent to form a
solution;
(b) contacting the solution with a supercritical fluid; and
(c) recovering the precipitate in a powder form.
[001I] The invention fiu-ther includes a process for preparing a
pharmaceutical
formulation containing a combination of two anti-infective agents comprising:
(a) combining two anti-infective agents with a solvent to form a solution;
(b) contacting the solution with supercritical carbon dioxide; and
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
3
(c) recovering the precipitate in a powder form containing the combination of
anti-
infective agents.
[0012] In another embodiment, the present invention provides a process for
preparing a
combination product containing two or more substances comprising:
(a) contacting two or more desired substances with a supercritical fluid to
form a
supercritical fluid solution; and
(b) separating the substances from the supercritical fluid solution to yield a
powder precipitate.
[0013] The invention is further directed to a process for preparing a
combination
product containing two or more substances comprising:
(a) combining two or more substances with a solvent to form a solution;
(b) mixing the solution with a supercritical fluid; and
(c) recovering the precipitate in a powder form.
BRIEF DESCRIPTION OF THE DRAWINGS
[0014] Figure 1 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations containing two or more active pharmaceutical ingredients using
the RESS
technique.
[0015] Figure 2 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations containing two or more active pharmaceutical ingredients using
the SAS
technique.
[0016] Figure 3 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations containing two or more active pharmaceutical ingredients using
the GAS
technique.
[0017] Figure 4 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations according to Examples 1-2,12, 18 and 20-21.
[0018] Figure 5 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations according to Example 19.
[0019] Figure 6 is a schematic of the apparatus for the recrystallization of
pharmaceutical
formulations according to Examples 3-11 and 13-17.
DETAILED DESCRIPTION OF THE INVENTION
[0020] The present invention is directed to a process for preparing
pharmaceutical
formulations containing two or more active pharmaceutical ingredients using
supercritical
fluid technology.
[0021] A "supercritical fluid" is a fluid at or above its critical pressure
(P~~ and critical
temperature (T~) simultaneously. Thus, a fluid above its critical pressure and
at its critical
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
4
temperature is in a supercritical state. A fluid at its critical pressure and
above its critical
temperature is also supercritical. As used herein, an "antisolvent" is a
supercritical fluid.
[0022] As used herein, supercritical fluids also encompass both near
supercritical fluids
and subcritical fluids. A "near supercritical fluid" is above but close to its
critical pressure
(P~) and critical temperature (T~) simultaneously. A "subcritical fluid" is
above its critical
pressure (P~) and close to its critical temperature (T~).
[0023] Any suitable supercritical fluid may be used in the process of the
present w
invention. The supercritical fluid should be compatible with the active agents
that are
dissolved in or contacted with the supercritical fluid in the
recrystallization processes
detailed herein.
[0024] Typical supercritical fluids and their critical properties (i.e.,
critical temperature,
critical pressure, and critical density) are listed in Table I .
TABLE 1
Fluid T (C) P~MPa) p~
/cm3
ethylene 9.3 5.04 0.22
xenon 16.6 5.84 0.12
carbon dioxide31. I 7.3 0.47
8
ethane 32.2 4.88 0.20
nitrous oxide36.5 7.17 0.45
propane 96.7 4.25 0.22
ammonia 132.5 11.28 0.24
n-butane 152.1 3.80 0.23
n-pentane 196.5 3.37 0.24
isopropanol 235.2 4.76 0.27
methanol 239.5 8.10 0.27
toluene 318.6 4.11 0.29
water 374.2 22.05 0.32
[0025] Carbon dioxide is preferably utilized used as the supercritical fluid
for producing
pharmaceutical formulations containing two or more active agents according to
the present
invention. The use of supercritical carbon dioxide in pharmaceutical
processing is further
described in Subramaniam et al., J. Pharm. Sci. 1997: 86, 8, which is
incorporated herein by
reference.
[0026] Other suitable supercritical fluids, also referred to as antisolvents,
useful in the
present invention include water, ammonia, nitxogen, nitrous oxide, methane,
ethane,
ethylene, propane, butane, n-pentane, benzene, methanol, ethanol,
isopropanol,l-propanol,
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
isobutanol,l-butanol, monofluoromethane, trifluoromethane,
chlorotrifluoromethane,
monofluoromethane, hexafluoroethane, l,I-difluoroethylene, 1,2-
difluoroethylene,toluene,
pyridine, cyclohexane, m-cresol, decalin, cyclohexanol, xylene, tetralin,
aniline, acetylene,
chlorotrifluorosilane, xenon, sulfur hexafluoride and combinations thereof.
[0027] Any combination of two or more active pharmaceutical ingredients may be
used
in the present invention. Preferably, two or more anti-infectives are combined
in
pharmaceutical formulations of the present invention. More preferably, two
anti-infectives
are combined.
[0028] Some examples of anti-infectives suitable for use including macrolide
antibiotics
such as clarithromycin, erythromycin, and azithromycin, anthracycline
antibiotics such as
doxorubicin and daunorubicin, camptothecin and its analogs such as topotecan
and
irenotecan, and quinolone antibiotics such as ciprofloxacin, ofloxacin,
levofloxacin,
clinafloxacin, and moxifloxacin. Cephalosporins may also be used such as, for
example,
cefotaxime, ceftriaxone, ceftazidime, and cefepime.
[0029] Other suitable anti-infective agents include (3-lactam antibiotics
(e.g., cefotetan,
aztreonam), penicillins (e.g., amoxicillin, piperacillin), aminoglycosides
(e.g.,
streptomycin), and sulfonamides (e.g., trimethoprim/sulfamethoxazole). Further
anti-
infective agents and classes thereof that may be used include, without
limitation,
carbapenems, bacitracin, gramicidin, mupirocin, chloramphenicol,
thiamphenicol, fusidate
sodium, lincomycin, clindamycin, novobiocin, polymyxins, rifamycins,
spectinomycin,
tetracyclines, vancomycin, teicoplanin, streptogramins, anti-folate agents
including
sulfonamides, trimethoprim and its combinations and pyrimethamine, synthetic
antibacterials including nitrofurans, methenamine mandelate and methenamine
hippurate,
nitroimidazoles, fluoroquinolones, isoniazid, ethambutol, pyrazinamide, para-
aminosalicylic
acid (PAS), cycloserine, capreomycin, ethionamide, prothionamide, thiacetazone
and
viomycin. Specific anti-infectives that are suitable include, without
limitation, amikacin,
netilmicin, fosfomycin, gentamicin, and teicoplanin.
[0030] Preferably the anti-infectives useful in the present invention include
ampicillin
sodium, sulbactam sodium, ticarcillin disodium, clavulanate potassium,
quinupristin,
dalfopristin, piperacillin sodium, tazobactam, imipenem and cilastatin.
[0031] Most preferably, pharmaceutical drug products containing two anti-
infective
active ingredients are produced according to the invention. The following
combinations of
anti-infective agents are preferably used: ampicillin sodium/sulbactam sodium
(marketed
under the brand name Unasyn~ by Pfizer); ticarcillin disodium/clavulanate
potassium
(marketed under the brand name Timentin~ by GlaxoSmithKline);
quinupristin/dalfopristin
(marketed under the brand name Synercid~ by Aventis); piperacillin
sodium/tazobactam
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
6
sodium (marketed under the brand name Zosyn~ by Lederle Pharmaceutical); and,
imipenem/cilastatin (marketed under the brand name Primaxin~ by Merck).
[0032] In another embodiment, the present invention includes pharmaceutical
drug
products containing two or more anticancer agents. Preferably, pharmaceutical
drug
products containing two anticancer active ingredients are produced. The
following
anticancer agents are preferably used etoposide, paclitaxel, cisplatin,
sarcolysine, alkylating
agents, bleomycin, busulfan, docetaxel, carboplatin, doxorubicin, vincristine,
fluorouracil,
methotrexate, vinorelbine, cyclophosphamide, etoposide, ifosfamide, mesna,
gemcitabine
hydrochloride, irinotecan hydrochloride, 5-fluorouracil, platinoids, and
vinorelbine tartarate.
[0033] More preferably, the following combinations of two anticancer agents
are
preferably used: etoposide/paclitaxel; altretaminelcisplatin;
altretamine/sarcolysine;
altretamine/alkylating agents; bleomycin/cisplatin; busulfan/docetaxel;
busulfan/carboplatin; cisplatin/doxorubicin; cisplatin/vincristine;
cisplatin/fluorouracil;
cisplatin/methotrexate; cisplatin/vinorelbine; cyclophosphamide/etoposide;
etoposide/ifosfamide; ifosfamide/mesna; gemcitabine hydrochloride/cisplatin;
gemcitabine/paclitaxel; irinotecan hydrochloride/5-fluorouracil;
paclitaxel/platinoids;
vinorelbine tartarate/platinoids; vinorelabine tartrate/paclitaxel;
paclitaxel/cisplatin; and
toposide/cisplatin.
[0034] Other types of active pharmaceutical ingredients that may be combined
according to the present invention include the following classes of drugs:
anxiolytic (e.g.,
diazepam), antidepressant (e.g., fluoxetine), anesthetic (e.g., midazolam),
antiviral (e.g.,
ganciclovir), protease inhibitor (e.g., saquinavir), chemotherapeutic (e.g.,
mesna, paclitaxel,
cisplatin), anti-inflammatory (e.g., naproxen, ketorolac), antimalarial (e.g.,
mefloquine),
antihypertensive (e.g., enalapril, lisinopril), antiseborheic (e.g.,
isotretinoin), calcium
channel blocker (e.g., diltiazem, nifedipine), lipase inhibitor (e.g.,
orlistat), antiparkinson
(e.g., tolcapone), antiarthritic (e.g., mycophenolate mofetil), and
thrombolytic agent (e.g.,
streptokinase). Also contemplated within the scope of the present invention
are additional
classes of drugs that are administered to a patient to obtain a desired
therapeutic effect.
[0035] The pharmaceutical ingredients useful in the present invention may be
any
known or hereafter discovered pharmacologically active ingredient, and may be
a
compound that occurs in nature, a chemically modified naturally occurring
compound, or a
compound that is chemically synthesized. The ingredient will typically be
chosen from the
generally recognized classes of pharmacologically active ingredients,
including, buff not
necessarily limited to, the following: analgesic ingredients; anesthetic
ingredients;
antiarthritic ingredients; respiratory drugs, including antiasthmatic
ingredients; anticancer
ingredients, including antineoplastic drugs; anticholinergics;
anticonvulsants;
antidepressants; antidiabetic ingredients; antidiarrheals; antihelminthics;
antihistamines;
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
7
antihyperlipidemic ingredients; antihypertensive~ingredients; anti-infective
ingredients such
as antibiotics and antiviral ingredients; antiinflammatory ingredients;
antimigraine
preparations; antinauseants; antiparkinsonism drugs; antipruritics;
antipsychotics;
antipyretics; antispasmodics; antitubercular ingredients; antiulcer
ingredients; anxiolytics;
appetite suppressants; attention deficit disorder (ADD) and attention deficit
hyperactivity
disorder (ADHD) drugs; cardiovascular preparations including calcium channel
Mockers,
CNS °ingredients; beta-blockers and antiarrhythmic ingredients; central
nervous system
stimulants; cough and cold preparations, including decongestants; diuretics;
genetic
materials; herbal remedies; hormonolytics; hypnotics; hypoglycemic
ingredients;
immunosuppressive ingredients; leukotriene inhibitors; mitotic inhibitors;
muscle relaxants;
narcotic antagonists; nicotine; nutritional ingredients, such as vitamins,
essential amino
acids and fatty acids; ophthalmic drugs such as antiglaucorna ingredients;
parasyrnpatholytics; psychostimulants; sedatives; steroids; sympathomimetics;
tranquilizers;
and vasodilators including general coronary, peripheral and cerebral.
[0036] The pharmaceutical ingredient may also be a biomolecule, e.g., a
molecular
moiety selected from the group consisting of DNA, RNA, antisense
oligonucleotides,
peptidyl drugs, i.e., peptides, polypeptides and proteins (including
fluorescent proteins),
ribosomes and enzyme cofactors such as biotin. Biomolecules (as well as other
ingredients)
may be radioactively tagged or otherwise labeled for diagnostic purposes, as
will be
discussed in further detail below.
[0037] Suitable pharmacologically active peptides will generally although not
necessarily have a molecular weight of at least 300 Da, and preferably at
least 800 Da.
Examples of such peptides which may be substantially stable in the extended
release
formulations over the intended period of release, and which may therefore be
used in the
compositions of this invention, are oxytocin, vasopressin, adrenocorticotropic
hormone
(ACTH), epidermal growth factor (EGF), prolactin, luteinizing hormone,
follicle
stimulating hormone, luliberin or luteinizing hormone releasing hormone
(LHRH), insulin,
somatostatin, glucagon, interferon, gastrin, tetragastrin, pentagastrin,
urogastrone, secretin,
calcitonin, enkephalins, endorphins, kyotorphin, taftsin, thymopoietin,
thymosin,
thymostimulin, thymic humoral factor, serum thymic factor, tumour necrosis
factor, colony
stimulating factors, motilin, bombesin, dinorphin, neurotensin, cerulein,
bradykinin,
urokinase, kallikrein, substance P analogues and antagonists, angiotensin II,
nerve growth
factor, blood coagulation factors VII and IX, lysozyme chloride, renin,
bradykinin,
tyrocidin, gramicidines, growth hormones, melanocyte stimulating hormone,
thyroid
hormone releasing hormone, thyroid stimulating hormone, parathyroid hormone,
pancreozymin, cholecystokinin, human placental lactogen, human chorionic
gonadotropin,
protein synthesis stimulating peptide, gastric inhibitory peptide, vasoactive
intestinal
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
peptide, platelet derived growth factor, growth hormone releasing factor, bone
morphogenic
protein, and synthetic analogues and modifications and pharmacologically
active fragments
thereof. Peptidyl drugs also include synthetic analogs of LHRH, e.g.,
buserelin, deslorelin,
fertirelin, goserelin, histrelin, leuprolide (leuprorelin), lutrelin,
nafarelin, tryptorelin, and
pharmacologically active salts thereof.
[0038] Any suitable salts of active pharmaceutical ingredients may be used
including,
for example, sodium, hydrochloride, potassium, mesylate, axetil, phosphate,
succinate,
maleate. Alternatively, the free acid form of active agents may be used.
[0039] In another embodiment, the invention includes a process for preparing
combination products containing two or more substances using supercritical
fluid
technology.
[0040] The substances of interest to be prepared in the combination product
may be any
molecular entity. Those substances that are particularly suited to uses
involving particles
are preferred. The uses for such combination products include cosmetics,
foodstuffs,
polymer technology (including plastics, fibers, biopolymers, etc.), chemical
reagents,
catalysts, energy storage materials, fuel cells, propellants, ceramics,
microelectronics,
photographic film and developer products, colorants (including pigments, dyes,
etc.),
phosphors, powder metallurgy products, ceramics, papermaking technology, etc.
[0041] The following examples of substances useful in preparing combination
products
of interest according to the invention and uses thereof. These examples are
for purposes of
illustration and are not intended to be limiting.
[0042] Catalysts: Generally although not necessarily metal-based, comprised of
a single
metal, a mixture or alloy of two or more metals, or an organometallic complex
(e.g.,
metallocenes, Ziegler-Natta catalysts).
[0043] Ceramics: Generally although not necessarily based on oxides, carbides,
nitrides,
borides, and silicates, including, for example, silicon nitride, silicon
oxynitride, silicon
carbide, tungsten carbide, tungsten oxycarbide, molybdenum carbide, aluminum
oxide,
calcium oxide, magnesium oxide, titanium oxide, aluminum silicates (e.g.,
sillimanite and
mullite), magnesium silicates (forsterite), zirconium silicates (zircon),
magnesium
aluminum oxide (spinel), etc.
[0044] Metals: Industrially or otherwise useful metal particles may be
comprised of any
metal or metallic alloy or composite, e.g., silver, gold, copper, lithium,
aluminum, platinum,
palladium, or the like.
[0045] Semiconductor materials include, but are not limited to, silicon,
silicon dioxide,
other metal oxides, germanium, and silicon-germanium. Semiconductors also
include those
comprised of a first element selected from Group 13 of the Periodic Table of
the Elements
and a second element selected from Group 15 (GaN, GaP, GaAs, GaSb, InN, InP,
InAs,
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
9
InSb, and the like); and those comprised of a first element selected from
Groups 2 and 12 of
the Periodic Table of the Elements and a second element selected from Group 16
(e.g., ZnS,
ZnSe, ZnTe, CDs, CdSe, CdTe, HgS, HgSe, HgTe, MgS, MgSe, MgTe, CaS, Case,
Care,
SrS, SrSe, SrTe, BaS, Base, Bare, and the like).
[0046] Conductive and semiconductive organics are typically conjugated
polymers, for
example, cis and trans polyacetylenes, polydiacetylenes, polyparaphenylenes,
polypyrroles,
polythiophenes, polybithiophenes, polyisothianaphthene, polythienylvinylenes,
polyphenylenesulfide, polyaniline, polyphenylenevinylenes, and
polyphenylenevinylene
derivatives, e.g., poly(2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene vinylene
("MEH-
PPV") (see U.S. Pat. No. 5,189,136 to Wudl et al.), poly(2,5-bischelostanoxy-
1,4-phenylene
vinylene) ("BCHA-PPV") (e.g., as described in International Patent Publication
No. WO
98/27136), and poly(2-N,N-dimethylamino phenylene vinylene)(described in U.S.
Pat. No.
5,604,292 to Stenger-Smith et al.).
[0047] Capacitor materials: Particles useful in capacitors include polyester,
polypropylene, polystyrene, glass, silica, mica, silver mica, aluminum oxide,
tantalum
oxide, and barium titanate.
[0048] Colorants include dyes and pigments. Dyes include azo or "direct" dyes
as well
as disperse dyes and dyes containing reactive groups, e.g., dyes containing
acidic groups
(e.g., carboxylate, phosphonate or sulfonate moieties), basic groups (e.g.,
unsubstituted
amines or amines substituted with 1 or 2 alkyl, typically lower alkyl,
groups), or both. Dyes
may also be luminescent, e.g., from the fluorescein, rhodamine, pyrene and
porphyrin
families. Inorganic pigments include, for example, iron blue, titanium
dioxide, red iron
oxide, strontium chromate, hydrated aluminum oxide, zinc oxide, zinc sulfide,
lithopone,
antimony oxide, zirconium oxide, kaolin (hydrous aluminosilicate), and carbon
black.
[0049] Organic pigments include, without limitation: azo pigments such as azo
lake
pigments, insoluble azo pigments, condensed azo pigments, and chelated azo
pigments;
polycyclic pigments such as phthalocyanine pigments, perylene pigments,
perynone
pigments, anthraquinone pigments, quinacridone pigments, dioxazine pigments,
thin-indigo
pigments, isoindolinone pigments, and quinophthalone pigments; vitro pigments;
nitroso
pigments; and aniline black.
[0050] Energy storage materials: In high voltage systems, examples of suitable
particles
for use in anodes include, but are not limited to, lithium, lithium/aluminum
alloys, caxbon,
graphite, nitrides, and tin oxide. Suitable particles for use in cathodes
include manganese
oxide (spinet), lithium cobalt oxide, lithium nickel oxide, vanadium oxide,
iron oxide,
mixed metal oxides, iron sulfide, copper sulfide, CFx, iodine, sulfur, mixed
metal sulfides,
metal and mixed metal phosphates.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
[0051] Battery Applications: Particles for use as anodes in alkaline battery
applications
include, but are not limited to, zinc and various zinc alloys with, e.g.,
lead, mercury, indium,
tin, etc. Suitable alkaline cathodes include, for example, manganese dioxide,
silver oxide
with graphite and carbon for electronic conduction. Metal hydride battery
electrode
materials axe typically nickel alloys with lanthanum and other trace elements.
[0052] Fuel cells: In direct methanol fuel cells platinum-ruthenium alloy
particles or
particles made from platinum-based alloys in which a second metal is tin,
iridium, osmium,
or rhenium are suitable for use as anodes. Cathodes may be prepared from
platinum
particles.
[0053] Photographic applications: Examples of particles that may be used in
photographic applications include, but are not limited to, silver halides such
as silver
chloride, silver bromide, silver bromoiodide, and dye sensitive variants
thereof.
[0054] Phosphors: Phosphors are normally composed of inorganic luminescent
materials that absorb incident radiation and subsequently emit radiation
within the visible
region ofthe spectrum. Phosphors are preferably capable of maintaining
luminescence (e.g.,
fluorescence) under excitation for a relatively long period of time to provide
superior image
reproduction. Various phosphors include, for example, Y2 03:Eu, ZnS:Ag,
Zn2SiO4:Mn,
ZnO:Zn, and other doped rare earth metal oxides.
[0055] Powder metallurgy products: Examples of suitable powder metallurgy
particles
include tungsten copper, silver tungsten, silver graphite, silver nickel,
tungsten
molybdenum, high density tungsten based heavy metals, tungsten carbide. Other
ferrous and
non-ferrous particles include iron and steel, iron, copper steel, iron nickel
steel, low alloy
steels, sinter hardened steels, and copper infiltrated steels, along with a
variety of bronze,
copper and brass materials.
[0056] Resins: Examples of synthetic resin particles include, without
limitation,
polyester resin particles, polyamide resin particles, polyvinyl chloride resin
particles,
polyurethane resin particles, urea resin particles, polystyrene resin
particles, particles of
styrene-acrylic copolymers (copolymers of styrene and derivatives of
(meth)acrylic acid),
polymethyl methacrylate particles, melamine resin particles, epoxy resin
particles, and
silicone resin particles. A wide variety of other polymeric particles are also
useful, e.g., in
plastics technology, fiber manufacturing, etc.
[0057] Conventional processes using supercritical fluids for producing
pharmaceutical
particles may be used. Examples of preferred supercritical processing
techniques for
recrystallizing pharmaceuticals include Rapid Expansion from Supercritical
Solutions
CRESS), Supercritical Anti-Solvent (SAS), and Gas Antisolvent (GAS). Another
example
of a suitable process fox use in the present invention is the Supercritical
Antisolvent
Precipitation with Enhanced Mass Transfer (SAS-EM) technique.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
11
[0058] Further techniques suitable for preparing combination pharmaceutical
formulations include U.S. Patent 6,620,351, U.S. Patent 5,707,634, U.S. Patent
5,360,478,
U.S. Patent 5,043,280, U.S. Patent 4,582,731, European Patent EP 0 542 314 and
Larson,
K.A., Biotechnol. Prog., 2:73-82 (1986).
[0059] These supercritical fluid processes involve the use of a solvent, also
referred to
as a solvent, in preparing the pharmaceutical combinations. Examples of
solvents useful in
the processes of the present invention include, without limitation, the
following: water,
hydrocarbons, including aliphatic alkanes such as hexane, heptane, decalin,
octane, etc.,
cyclic alkanes such as cyclohexane, and aromatic hydrocarbons such as benzene,
cumene,
pyridine, pseudocumene, cymene, styrene, toluene, xylenes,
tetrahydronaphthalene and
mesitylene; halogenated compounds such as carbon tetrachloride and
chlorinated,
fluorinated and brominated hydrocarbons such as chloroform, bromoform, methyl
chloroform, chlorobenzene, o-dichlorobenzene, chloroethane, 1,1-
dichloroethane, 1,2-
dichloroethane, tetrachloroethane, epichlorohydrin, trichloroethylene and
tetrachloroethylene; ethers such as diethyl ether, diisopropyl ether,
diisobutyl ether,
diglyme, 1,4-dioxane, 1,3-dioxolane, dimethoxymethane, furan and
tetrahydrofuran;
aldehydes such as methyl formate, ethyl formate and furfural; ketones such as
acetone,
diisobutyl ketone, cyclohexanone, methyl ethyl ketone, N-methyl-2-pyrrolidone
and
isophorone; amides such as dimethyl formamide and dimethyl acetamide; alcohols
such as
ethanol, isopropanol, n-propanol, t-butyl alcohol, cyclohexanol, 1-hexanol, 1-
octanol and
firifluoroethanol; polyhydric alcohols such as 1,3-propanediol, glycerol,
ethylene glycol,
propylene glycol, and low molecular weight (typically less than 400)
polyethylene glycol;
amines, including cyclic amines such as pyridine, piperidine, 2-
methylpyridine, morpholine,
etc., and mono-, di- and tri-substituted amines such as trimethylamine,
dimethylamine,
methylamine, triethylamine, diethylamine, ethylamine, n-butylamine, t-
butylamine,
triethanolamine, diethanolamine and ethanolamine, and amine-substituted
hydrocarbons
such as ethylene diamine, diethylene triamine; carboxylic acids such as acetic
acid,
trifluoroacetic acid and formic acid; esters such as ethyl acetate, isopentyl
acetate,
propylacetate, etc.; lactams such as caprolactam; nitrites such as
acetonitrile, propane nitrite
and adiponitrile; organic nitrates such as nitrobenzene, nitroethane and
nitromethane; and
sulfides such as carbon disulfide.
(0060] The solvent may optionally be a lipidic material including, but not
limited to, the
following: phospholipids such as phosphorylated diacyl glycerides, and
particularly
phospholipids selected from the group consisting of diacyl
phosphatidylcholines, diacyl
phosphatidylethanolamines, diacyl phosphatidylserines, diacyl
phosphatidylinositols, diacyl
phosphatidylglycerols, diacyl phosphatidic acids, and mixtures thereof,
wherein each acyl
group contains about 10 to about 22 carbon atoms and is saturated or
unsaturated; fatty
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
12
acids such as isovaleric acid, valeric acid, caproic acid, enanthic acid,
caprylic acid,
pelargonic acid, capric acid, lauric acid, myristic acid, palmitic acid,
stearic acid, arachidic
acid, behenic acid, lignoceric acid, oleic acid, linoleic acid, linolenic
acid, and arachidonic
acid; lower fatty acid esters comprising esters of the foregoing fatty acids,
wherein the
carboxylic acid group of the fatty acid is replaced with an ester moiety --
(CO)--OR wherein
R is a C1_3 alkyl moiety optionally substituted with one or two hydroxyl
groups; fatty
alcohols corresponding to the aforementioned fatty acids, wherein the
carboxylic acid group
of the fatty acid is replaced by a-CH20H group; glycolipids such as
cerebroside and
gangliosides; oils, including animal oils such as cod liver oil and, menhaden
oil, and
vegetable oils such as babassu oil, castor oil, corn oil, cotton seed oil,
linseed oil, mustard
oil, olive oil, palm oil, palm kernel oil, peanut oil, poppyseed oil, rapeseed
oil, safflower oil,
sesame oil, soybean oil, sunflower seed oil, tong oil or wheat germ oil; and
waxes, i.e.,
higher fatty acid esters, including animal waxes such as beeswax and shellac,
mineral waxes
such as montan, petroleum waxes such as microcrystalline wax and paraffin, and
vegetable
waxes such as caxnauba wax.
[0061] In the RESS process, the two or more active pharmaceutical ingredients
are
dissolved in a supercritical fluid, preferably carbon dioxide, to form a
homogenous solution.
Other excipients may optionally be added to the supercritical fluid. The
active agents and
optional excipients may be added to the supercritical fluid simultaneously or
other suitable
order. The resulting solution is then passed through an orifice or nozzle into
a chamber.
Preferably, the pressure in the chamber is atmospheric. By spraying the
homogenous
solution through an orifice or nozzle, the solution is depressurized rapidly
resulting in the
vaporization of the carbon dioxide or other supercritical fluid. The active
agents and
optional excipients are recrystallized as a uniform mixture in dry powder
form.
[0062] RESS can be used if the active pharmaceutical ingredients to be
precipitated axe
soluble in the supercritical fluid, such as supercritical caxbon dioxide. If
the active agents
are not readily soluble in the supercritical fluid, the active agents may be
first dissolved in a
cosolvent system and then added to the supercritical fluid. The cosolvent may
be a single
solvent or two or more solvents combined together. Alternatively, the
cosolvent may be
added to the supercritical fluid initially followed by the addition of the
active agents to the
mixture of the supercritical fluid and cosolvent. When a cosolvent is
required, the cosolvent
used generally has a higher dielectric constant than the supercritical fluid
(e.g., supercritical
carbon dioxide), but is miscible in the supercritical fluid.
(0063] Examples of suitable solvents and cosolvents include acetone, methanol,
ethanol,
propanol, butanol, tetrahydrorfuran, methylene chloride, chloroform, toluene,
dimethylsufloxide, N,N-dimethylformamide, cyclohexanone, butrylactone, water,
and
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
13
combinations thereof. Other suitable solvents include those compounds known in
the art in
which the active pharmaceutical ingredients to be blended can be dissolved.
[0064] A typical flow diagram of a RESS process for recrystallization using
carbon
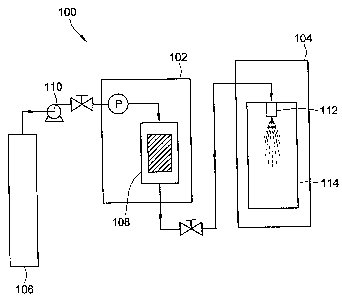
dioxide as the supercritical fluid is shown in FIG 1. The RESS apparatus 100
generally
includes an extraction unit 102 and precipitation unit 104. Carbon dioxide is
transferred
from storage tank 106 to high-pressure vessel 108, optionally using pump 110.
The
temperature and pressure in high-pressure vessel 108 are maintained such that
the carbon
dioxide exists in a supercritical state. The active pharmaceutical ingredients
are then added
to high-pressure vessel 108 to form a homogenous solution of the carbon
dioxide in which
the active agents are dissolved. Alternatively, the active pharmaceutical
ingredients may be
added to the high-pressure vessel 108 initially followed by the addition of
supercritical
carbon dioxide to form a homogeneous solution. The homogenous solution is
sprayed
through nozzle 112 into vessel 114, preferably under atmospheric pressure
conditions.
Alternatively, pressures greater than atmospheric pressure may be used. The
supercritical
carbon dioxide is vaporized and the active agents precipitate from the
solution in the form
of a dry powder. The carbon dioxide may either be collected for possible reuse
or
discarded. The solid precipitate is collected from vessel 114 for further
processing.
[0065] The upstream and downstream temperatures and pressures in the RESS
process
may be modified to obtain the desired morphology of the precipitated drug
product. In
addition, the shape of the nozzle employed may be altered to transition
between fibers and
particles. A smaller length-to-nozzle diameter ratio (L/D) typically results
in the formation
of particles.
[0066] Another suitable process for recrystallization according to the present
invention
is the SAS process. The SAS technique is well-suited for precipitation of
active agents that
are only slightly soluble in the supercritical fluid of interest, such as
supercritical carbon
dioxide.
[0067] In the SAS process, the active pharmaceutical ingredients and optional
excipients are dissolved in a solvent. The solvent may be any suitable liquid
containing one
or moxe solvents in which the active agents are dissolved. The solvent is also
miscible in
the supercritical fluid. Examples of solvents suitable for use in the SAS
method include
those solvents discussed herein that may be used in the RESS process as
cosolvents as well
as other solvents in which the active agents can be dissolved.
[0068j The solution containing the active agents is then contacted with a
supercritical
fluid (e.g., supercritical carbon dioxide). Preferably, mixing is carried out
by spraying the
solution through a nozzle into a chamber filled with the supercritical fluid.
The supercritical
fluid acts as an anti-solvent to extract out the cosolvent. The active agents
and optional
excipients form a precipitate upon contact with the supercritical fluid which
is recovered.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
I4
The precipitate from the SAS process is a uniformly mixed dry powder
containing the
active pharmaceutical ingredients and any optional excipients.
[0069] The supercritical fluid may optionally be contacted one or more
cosolvents prior
to the addition of the solution containing the active agents.
[0070] A typical flow diagram of a SAS process for recrystallization using
supercritical
carbon dioxide is shown in FIG. 2. In the SAS apparatus 200, the active
pharmaceutical
ingredients are dissolved ima solvent system in vessel 214. Excipients may
optionally be
dissolved in a solvent along with the active agents. Carbon dioxide is
transferred from
vessel 202 to high-pressure vessel 204, optionally using pump 206, wherein
carbon dioxide
is maintained in a supercritical state. The solvent solution containing the
active agents is
transferred from vessel 214, optionally using pump 208, and sprayed through
nozzle 2I0
into high-pressure vessel 204. The precipitate containing a powder blend of
active agents is
recovered from high-pressure vessel 204 for further processing. The resulting
mixture of
the solvents and supercritical carbon dioxide is then transferred to low-
pressure tank 212 for
recovery of the solvent and carbon dioxide and reuse of these process streams.
[0071] In the GAS process, supercritical carbon dioxide is added to a solution
of the
desired active pharmaceutical ingredients dissolved in an organic cosolvent.
The
supercritical carbon dioxide and organic solvent are miscible whereas the
solid active agents
have limited solubility in carbon dioxide. Thus, the carbon dioxide acts as an
antisolvent to
precipitate solid crystals of the active agents.
[0072] A typical flow diagram of a GAS process for recrystallization using
supercritical
carbon dioxide is shown in FIG. 3. In the GAS apparatus 300, the active
pharmaceutical
ingredients are dissolved in a solvent in vessel 302. Excipients may
optionally be dissolved
in the solvent along with the active agents. The solution in which the active
agents are
dissolved is transferred to a vessel 304 in the precipitator 306 using pump
308. Carbon
dioxide stored in a supercritical state in vessel 310 is rapidly transferred
to vessel 304 using
pump 312. Alternatively, carbon dioxide may be stored as either a gas or
liquid well below
its critical temperature and critical pressure and then rendered supercritical
before
combining the carbon dioxide with the dissolved active agents. Upon contact
with the
supercritical carbon dioxide, the dissolved active agents in the solution 312
in vessel 304 are
crystallized as particles 314 containing a blend of the active agents and
optional excipients.
The particles are recovered for further processing to yield a suitable
pharmaceutical
formulation.
[0073] The supercritical fluid may optionally be contacted with one or more
solvents
prior to the addition of the solution containing the active agents.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
[0074] Other suitable processes known to those persons of ordinary skill in
the art that
involve a supercritical fluid, preferably supercritical carbon dioxide, may be
used to
recrystallize combinations of active pharmaceutical ingredients in a dry
powder form.
[0075] The uniform blend of active pharmaceutical agents recrystallized using
a
supercritical fluid according to the process of the present invention is a
powder, also
referred to herein as a dry powder. The precipitated powder typically contains
about 10%
or less (by weight) of the solvent in which the active agents are dissolved
prior to
crystallization. Preferably, the dry powder contains 5% or less solvent (by
weight) and,
most preferably, 2% or less (by weight) solvent.
(0076] The pharmaceutical formulations produced according to the present
invention
may optionally contain pharmaceutically acceptable excipients such as, for
example,
carriers, additives, and diluents. Pharmaceutical formulations for parenteral
administration
may contain, for example, alkylene glycols such as propylene glycol,
polyalkylene glycols
such as polyethylene glycol, oils of vegetable origin, hydrogenated
naphthalenes, acid or
basic buffers, and the like.
[0077] Other examples of suitable excipients for pharmaceutical dosage forms
prepared
by the present invention include lactose, dextrose, sucrose, sorbitol,
mannitol, starches, gum
acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate,
microcrystalline
cellulose, polyvinylpyrrolidone, cellulose, sterile water, syrup, and methyl
cellulose.
Pharmaceutical formulations can additionally include: lubricating agents such
as talc,
magnesium stearate, and mineral oil; wetting agents; emulsifying and
suspending agents;
preserving agents such as methyl- and propylhydroxy-benzoates; sweetening
agents; and
flavoring agents.
[0078] The pharmaceutical active ingredients used in the present invention may
be
premixed with one or more pharmaceutically acceptable excipients before the
active agents
are contacted with a supercritical fluid according to the inventive processes.
When
premixed with the active agents, the excipients must be compatible with the
cosolvent
systems and supercritical fluids that are employed.
[0079] Alternatively, a uniform blend of two or more active pharmaceutical
ingredients
obtained by recrystallization using a supercritical fluid may be contacted
with one or more
pharmaceutically acceptable excipients to produce a pharmaceutical
formulation.
(0080] The following examples further illustrate the invention but, of course,
should not
be construed as in any way limiting its scope.
EXAMPLES
[0081] The examples demonstrate the process for producing pharmaceutical
formulations using a supercritical fluid according to the present invention.
Examples 1-11
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
16
describe pharmaceutical formulations containing sodium sulbactam and sodium
ampicillin.
Examples 12-21 describe pharmaceutical formulations containing etoposide and
paclitaxel.
The samples of Examples 1-21 are prepared by one of the following three
processes.
[0082] Process 1
[0083] The flow diagram for process 1 is shown in FIG. 4. In apparatus 400,
the active
pharmaceutical ingredients are dissolved in a cosolvent to form a drug
solution 402 in
vessel 404. After closing vessel 404, antisolvent from vessel 406 is slowly
added to the
drug solution 402 in vessel 404. The antisolvent may optionally be pumped to
vessel 404
using pump 408. The temperature of the antisolvent may optionally be adjusted
with cooler
410 andlor heater 412. Temperature is steadily maintained during the
antisolvent addition
to the drug solution 402, for example by thermocouple 414. Due to the
antisolvent addition,
pressure in vessel 404 rises steadily. The antisolvent addition rate may also
be monitored by
recording the rate of increase of pressure in vessel 402 using pressure
monitor 416.
Antisolvent diffuses into the drug solution 402 while the cosolvent diffuses
into the
antisolvent, resulting in the precipitation of the active agents as a powder.
A magnetic
stirrer 418 is employed to obtain uniform mixing in vessel 404. After the
pressure reaches a
desired value, a pressure control valve 420 in the outlet line is opened to
control the
pressure in vessel 402. The antisolvent may optionally be filtered through
filter 422.
Antisolvent flow rate from vessel 404 is maintained constant for a period of
time to remove
any residual cosolvent present in the precipitated powder.
[0084] Preferably, the temperature of vessel 404 is maintained at a
temperature
appropriate to avoid substance degradation, if any. Additional stabilizing
agents may be
added to maintain pH of the solution in case of aqueous solutions. Further
substances may
be added to protect any substance degradation caused during the solution
preparation or
processing.
[0085] Process 2
[0086] The flow diagram for process 2 is shown in FIG. 5. In apparatus 500,
the active
pharmaceutical ingredients axe dissolved in a cosolvent in the solution feed
vessel 502 and
stirred, making a uniform solution. A flow of antisolvent contained in vessel
504 (e.g., CO2
cylinder) is maintained at a desired temperature and pressure using
antisolvent pump 506 to
the particle production vessel 508. The antisolvent flow is monitored with
flow meter 510.
Optionally, the antisolvent may be cooled by heat exchanger 512 or heated by
heat
exchanger 514 prior to contact with the drug solution. The solution is
dispersed using
solution pump 516 into vessel 508 as a fine stream through a capillary nozzle.
The
antisolvent effect precipitates the substance combination as a powder. Powder
is then
filtered and the antisolvent/cosolvent mixture is allowed to exit vessel 508.
The
antisolvent/cosolvent mixture is fiuther separated in solvent collection
vessel 518 and may
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
17
be recycled. The particle production vessel 508 may optionally be jacketed to
permit
contact with a cooling or heating coil to maintain the desired temperature in
vessel 508.
[0087) Process 3
[0088) The flow diagram for process 3 is shown in FIG 6. In apparatus 600, the
active
pharmaceutical ingredients are dissolved in a cosolvent in the solution feed
vessel 602 and
stirred making it a uniform solution. Antisolvent is contained in vessel 604
which may
optionally be a COz cylinder. The flow of antisolvent is maintained at a
desired temperature
and pressure from vessel 604 using pump 606 into the particle production
vessel 608. The
antisolvent flow is monitored with flow meter 610. Optionally, the antisolvent
may be
cooled by heat exchanger 612 or heated by heat exchanger 614 prior to contact
with the
drug solution. A solid surface 616 is vibrated at a desired frequency. The
drug solution
from vessel 602 is applied using solution pump 618 onto the vibrating surface
616 which
results in uniform atomization. Antisolvent effect precipitates the substance
combination as
a powder. The powder is filtered using a stainless steel filter 620 and the
antisolvent/solvent mixture is allowed to exit vessel 608 into the solvent
collection vessel
622 and may be recycled. The particle production vessel 608 may optionally be
jacketed to
permit contact with a cooling or heating coil to maintain the temperature in
vessel 608.
[0089] Examples 1-11 demonstrate the process for producing pharmaceutical
formulations containing sodium sulbactam and sodium ampicillin using a
supercritical fluid
according to the present invention. For each of Examples 1-11, the ratio of
sodium
sulbactam to sodium ampicillin used was 1:2.
EXAMPLE 1
[0090] The desired substance combination of sodium sulbactam and sodium
ampicillin
was made into an aqueous solution and processed using process I . A compressed
carbon
dioxide/ethanol mixture was used as the antisolvent. The concentrations of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 200 mg/ml and 400
mg/ml,
respectively. About 15 mL of the solution was dispensed in the vessel. The
temperature of
the precipitation vessel was maintained at about 35 °C. A 20 ml high
pressure vessel was
used as vessel 402 in Figure 4, and pressure of the system was maintained at
100 bar. The
antisolvent flow consisted of 12g/min of carbon dioxide and 1.5 ml/min (at
atmospheric
conditions) of ethanol. After the pressure reached 100 bar, antisolvent flow
was maintained
for 60 minutes. At the end of 60 minutes, the ethanol flow was stopped and
carbon dioxide
flow was maintained for an additional I S minutes. The vessel was opened and
the powder
material was collected, weighed, labeled and stored.
EXAMPLE 2
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
18
[0091] The desired substance combination of sodium sulbactam and sodium
ampicillin
was made into an aqueous solution and processed using process 1. Compressed
carbon
dioxide/ethanol mixture was used as the antisolvent. The concentration of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 200 mg/ml and 400
mg/ml,
respectively. About 15 ml of the solution was dispensed in the vessel. The
temperature of
the precipitation vessel was maintained at about 60 °C. A 20 ml high
pressure vessel was
used as vessel 402 in Figure 4, and pressure of the system was
maintained°at 100 bar. The
antisolvent flow consisted of 12g/min of carbon dioxide and 1.5 ml/min (at
atmospheric
conditions) of ethanol. After the pressure reached 100 bar, antisolvent flow
was maintained
for 60 minutes. At the end of 60 minutes, the ethanol flow was stopped and
carbon dioxide
flow was maintained for an additional 15 minutes. The vessel was opened and
the powder
material was collected, weighed, labeled and stored.
EXAMPLE 3
[0092] Sodium sulbactam and sodium ampicillin were dissolved in an acetate
buffer
(pH=7.0) by dissolving 92.21mg of sodium acetate trihydrate per ml of water
and 18.4
microliter of acetic acid per ml of water. 50 ml of the abovementioned
solution was mixed
with 450 ml of methanol and the resultant solution was processed using process
3.
Compressed carbon dioxide was used as the antisolvent. The concentrations of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 8 mg/ml of water
and 16
mg/ml of water, respectively. The temperature of the precipitation vessel was
maintained at
about 35 °C. A 500 ml high pressure vessel was used as the particle
production vessel 608
in Figure 6, and pressure of the system was maintained at 100 bar. The
antisolvent flow
consisted of SOg/min of carbon dioxide. The solution flow rate was maintained
at 0.5
ml/min for 150 minutes. A 200 micron capillary tube was used to apply the
solution on to
the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz was
used at
50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide flow was maintained for 60 additional minutes. At the end of 60
minutes, the
system was depressurized. The vessel was opened and the powder material was
collected,
weighed, labeled and stored.
EXAMPLE 4
[0093] Sodium sulbactam and sodium ampicillin were dissolved in distilled
water. The
resultant solution was processed using process 3. Compressed carbon
dioxide/ethanol
mixture was used as the antisolvent. Concentration of sodium sulbactam and
sodium
ampicillin in the aqueous solution were 80 mg/ml of water and 160 mg/ml of
water
respectively. The temperature of the precipitation vessel was maintained at
about 35 °C. A
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
19
500 ml high pressure vessel was used as the particle production vessel 608 in
Figure 6.
Pressure of the system was maintained at 100 bar. The antisolvent flow
consisted of
SOg/min of carbon dioxide and 6 ml/min of ethanol . The solution flow rate was
maintained
at 0.5 ml/min for 45 minutes. A 200 micron capillary tube was used to apply
the solution on
to the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz
was used at
50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped and carbon dioxide flow was continued for additional
30 minutes
and the system was depressurized. The vessel was opened and the powder
material was
collected, weighed, labeled and stored.
EXAMPLE 5
[0094] Sodium sulbactam and sodium ampicillin was dissolved in an acetate
buffer
(pH=7.0) by dissolving 92.21 mg of sodium acetate trihydrate per ml of water
and 18.4
microliter of acetic acid per ml of water. 50 ml of the above mentioned
solution is mixed
with 450 ml of methanol and the resultant solution was processed using process
3.
Compressed carbon dioxide was used as the antisolvent. The concentration of
sodium
sulbactam and sodium arnpicillin in the aqueous solution were 8 mg/ml of water
and 16
mg/ml of water respectively. The temperature of the precipitation vessel was
maintained at
about 35 °C. A 500 ml high pressure vessel was used as the particle
production vessel 608
in Figure 6. Pressure of the system was maintained at_100 bar. The antisolvent
flow
consisted of SOg/min of carbon dioxide. The solution flow rate was maintained
at 0.5
ml/min for 150 minutes. A 200 micron capillary tube was used to apply the
solution on to
the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz was
used at
50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide flow was maintained for 90 additional minutes. At the end of 60
minutes, the
system was depressurized. The vessel was opened and the powder material was
collected
for further processing.
EXAMPLE 6
[0095] Sodium sulbactam and sodium ampicillin was dissolved in distilled
water. The
resultant solution was processed using process 3. Compressed carbon
dioxide/ethanol
mixture was used as the antisolvent. Concentration of sodium sulbactam and
sodium
ampicillin in the aqueous solution were 160 mg/ml of water and 320 mg/ml of
water
respectively. The temperature of the precipitation vessel was maintained at
about 35 °C. A
500 ml high pressure vessel was used as the particle production vessel 608 in
Figure 6.
Pressure of the system was maintained at 100 bar. The antisolvent flow
consisted of
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
SOg/min of carbon dioxide and 6 ml/min of ethanol. The solution flow rate was
maintained
at 0.5 ml/min for 60 minutes. A 200 micron capillary tube was used to apply
the solution on
to the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz
was used at
50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped; carbon dioxide flow was continued for additional 30
minutes;
and the system was depressurized. The vessel was opened and-the powder
material was
collected for further processing.
EXAMPLE 7
[0096] Sodium sulbactam and sodium Ampicillin was dissolved in an acetate
buffer
(pH=7.0) by dissolving 92.21 mg of sodium acetate trihydrate per ml of water
and 18.4
microliter of acetic acid per ml of water. 50 ml of the above mentioned
solution is mixed
with 450 ml of methanol and the resultant solution was processed using process
3.
Compressed carbon dioxide was used as the antisolvent. The concentrations of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 8 mg/ml of water
and 16
mg/ml of water, respectively. The temperature of the precipitation vessel was
maintained at
about 35 °C. A 500 ml high pressure vessel was used as the particle
production vessel 608
in Figure 6, and pressure of the system was maintained at 100 bax. The
antisolvent flow
consisted of SOg/min of carbon dioxide. The solution flow rate was maintained
at 0.5
ml/min for 60 minutes. A 200 micron capillary tube was used to apply the
solution onto the
vibrating surface. A Branson 900 BCA system with a frequency of 20kHz was used
at 50%
amplitude to vibrate the atomizing surface. After the end of solution flow,
carbon dioxide
flow was maintained for 60 additional minutes. At the end of 60 minutes, the
system was
depressurized. The vessel was opened and the powder material was collected for
further
processing.
EXAMPLE 8
[0097] Sodium sulbactam and sodium ampicillin were dissolved in distilled
water. The
resultant solution was processed using process 3. Compressed carbon
dioxide/ethanol
mixture was used as the antisolvent. Concentration of sodium sulbactam and
sodium
ampicillin in the aqueous solution were 160 mg/ml of water and 320 mg/ml of
water
respectively. The temperature of the precipitation vessel was maintained at
about 35 °C. A
500 ml high pressure vessel was used as the particle production vessel 608 in
Figure 6.
Pressure of the system was maintained at 100 bar. The antisolvent flow
consisted of
SOg/min of carbon dioxide and 6 ml/min of ethanol . The solution flow rate was
maintained
at 0.5 ml/min for 45 minutes. A 200 micron capillary tube was used to apply
the solution on
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
21
to the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz
was used at
50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped and carbon dioxide flow was continued for additional
30 minutes
and the system was depressurized. The vessel was opened and the powder
material was
collected for further processing.
EXAMPLE 9
[0098] Sodium sulbactam and sodium ampicillin was dissolved in distilled
water. The
resultant solution was processed using process 3. Compressed carbon
dioxide/ethanol
mixture~was used as the antisolvent. The concentrations of sodium sulbactam
and sodium
ampicillin in the aqueous solution were 160 mg/ml of water and 320 mg/ml of
water,
respectively. The temperature of the precipitation vessel was maintained at
about 35 °C. A
500 ml high pressure vessel was used as the particle production vessel 608 in
Figure 6.
Pressure of the system was maintained at 100 bar. The antisolvent flow
consisted of
SOg/min of carbon dioxide and 6 ml/min of ethanol. The solution flow rate was
maintained
at 1.0 ml/min for 30 minutes. A 200 micron capillary tube was used to apply
the solution
onto the vibrating surface. A Branson 900 BCA system with a frequency of 20kHz
was used
at 50% amplitude to vibrate the atomizing surface. After the end of solution
flow, carbon
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped; carbon dioxide flow was continued for additional 30
minutes;
and the system was depressurized. The vessel was opened and the powder
material was
collected, weighed, labeled and stored.
EXAMPLE 10
[0099] Sodium sulbactam and sodium ampicillin was dissolved in distilled water
in an
icepack. The resultant solution was processed using process 3. Compressed
carbon
dioxide/ethanol mixture was used as the antisolvent. The concentrations of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 40 mg/ml of water
and 80
mg/ml of water, respectively. The temperature of the solution syringe pump was
maintained
at 23 °C. The temperature of the precipitation vessel was maintained at
about 35 °C. A 500
ml high pressure vessel was used as the particle production vessel 608 in
Figure 6. Pressure
of the system was maintained at 100 bar. The antisolvent flow consisted of 1
OOg/min of
carbon dioxide and 10 ml/min of ethanol. The solution flow rate was maintained
at 0.5
mllmin for 90 minutes. A 200 micron capillary tube was used to apply the
solution onto the
vibrating surface. A Branson 900 BCA system with a frequency of 20kHz was used
at 50%
amplitude to vibrate the atomizing surface. After the end of solution flow,
carbon
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
22
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped; carbon dioxide flow was continued for additional 60
minutes;
and the system was depressurized. The vessel was opened and the powder
material was
collected, weighed, labeled and stored.
EXAMPLE 11
[00100] Sodium sulbactam and sodium ampicillin was dissolved in distilled
water in an
icepack. The resultant solution was processed using process 3. Compressed
carbon
dioxide/ethanol mixture was used as the antisolvent. The concentrations of
sodium
sulbactam and sodium ampicillin in the aqueous solution were 40 mg/ml of water
and 80
mg/ml of water, respectively. The temperature of the solution syringe pump was
maintained
at 23 C. The temperature of the precipitation vessel was maintained at about
35 °C. A 500
ml high pressure vessel was used as the particle production vessel 608 in
Figure 6. Pressure
of the system was maintained at 100 bar. The antisolvent flow consisted of
100g/min of
carbon dioxide and 10 ml/min of ethanol. The solution flow rate was maintained
at 0.5
ml/min for 90 minutes. A 200 micron capillary tube was used to apply the
solution on to the
vibrating surface. A Branson 900 BCA system with a frequency of 20 kHz was
used at 50%
amplitude to vibrate the atomizing surface. After the end of solution flow,
carbon
dioxide/ethanol flow was maintained for 60 additional minutes. At the end of
60 minutes,
ethanol flow was stopped; carbon dioxide flow was continued for additional 30
minutes;
and the system was depressurized. The vessel was opened and the powder
material was
collected, weighed, labeled and stored.
[00101] The sample of Example 10 exhibited the most homogenous crystal
structure
which demonstrates blend uniformity. The chemical recovery of drug product was
also
maximized in Example 10, specifically about 58% recovery of sodium sulbactam
and about
93% recovery of sodium ampicillin.
[00102] As demonstrated by the results of Examples 1-11, the process of the
invention
can be used to produce pharmaceutical formulations comprising sodium sulbactam
and
sodium arnpicillin using a supercritical fluid in accordance with the present
invention.
[00103] Examples 12-21 demonstrate the process for producing pharmaceutical
formulations containing etoposide and paclitaxel using a supercritical fluid
according to the
present invention. The ratio of paclitaxel and etoposide used in the samples
of Examples
12-21 and the cosolvent used to produce these samples are set forth in Table
2.
Table 2: Ratios of etoposide/paclitaxel, solvent and antisolvent
Example Etoposide/PaclitaxelSolvent Antisolvent
Ratio
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
23
12 1:1 Methanol Compressed C02
13 1:1 Methanol Compressed C02
14 1:1 Methanol Compressed COa
15 2:1 Methanol Compressed COZ
16 1:1 Methanol Compressed C02
17 1:1 Methanol Compressed COa
18 1:1 Methanol Compressed COZ
19 1:1 Methanol Compressed COa
20 1:1 Methanol Compressed C02
21 2:1 Methanol Compressed COZ
EXAMPLE 12
[00104] Etoposide and paclitaxel were made into a solution in methanol and
processed
using process 1. Compressed carbon dioxide was used as the antisolvent. O.Sg
of etoposide
and O.Sg of paclitaxel were dissolved in 25 ml of methanol and dispensed in to
the vessel.
The temperature of the precipitation vessel was maintained at about 35
°C. A 103 ml high
pressure vessel was used as vessel 404 in Figure 3. Pressure of the system was
maintained at
100 bar. A magnetic stirrer at 520 rpm was used to obtain uniform mixing
during
precipitation. The antisolvent flow consisted of Sg/min of carbon dioxide.
Pressure in the
vessel was increased gradually at a rate of 1 bar/min. After the pressure
reached 100 bar,
antisolvent flow was maintained for 180 minutes. The vessel was depressurized,
opened and
powder material was collected, weighed, labeled and stored.
EXAMPLE 13
[00105] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 3. Compressed carbon dioxide was used as the antisolvent. 1.Og
of etoposide
and 1.0 g of paclitaxel were dissolved in 50 ml of methanol. The temperature
of the
precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was used
as the particle production vessel 608 in Figure 6. Pressure of the system was
maintained at
80 bar. The antisolvent flow consisted of SOg/min of carbon dioxide. The
solution flow rate
was maintained at 1.0 ml/min for 58 minutes. A 100 micron capillary tube was
used to
apply the solution on to the vibrating surface. A Branson 900 BCA system with
a frequency
of 20 kHz was used at 20% amplitude to vibrate the atomizing surface. After
the end of
solution flow, carbon dioxide flow was maintained for 60 additional minutes
and the system
was depressurized. The vessel was opened and the powder material was
collected, weighed,
labeled and stored.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
24
EXAMPLE 14
[00106] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 3. Compressed carbon dioxide was used as the antisolvent. O.Sg
of etoposide
and 0.5 g of paclitaxel were dissolved in 50 ml of methanol. The temperature
of the
precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was used
as the particle production vessel 608 in Figure 6. Pressure of the system was
maintained at
80 bar. The antisolvent flow consisted of SOg/min of carbon dioxide. The
solution flow rate
was maintained at 0.5 ml/min for 100 minutes. A 100 micron capillary tube was
used to
apply the solution on to the vibrating surface. A Branson 900 BCA system with
a frequency
of 20 kHz was used at 20% amplitude to vibrate the atomizing surface. After
the end of
solution flow, carbon dioxide flow was maintained for 60 additional minutes
and the system
was depressurized. The vessel was opened and the powder material was
collected, weighed,
labeled and stored.
EXAMPLE 15
[00107] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 3. Compressed carbon dioxide was used as the antisolvent. l.Og
of etoposide
and 0.5 g of paclitaxel were dissolved in 50 ml of methanol. The temperature
of the
precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was used
as the particle production vessel 608 in Figure 6. Pressure of the system was
maintained at
80 bar. The antisolvent flow consisted of SOg/min of carbon dioxide. The
solution flow rate
was maintained at 1.0 ml/min for 50 minutes. A 100 micron capillary tube was
used to.
apply the solution on to the vibrating surface. A Branson 900 BCA system with
a frequency
of 20 kHz was used at 20% amplitude to vibrate the atomizing surface. After
the end of
solution flow, carbon dioxide flow was maintained for 60 additional minutes
and the system
was depressurized. The vessel was opened and the powder material was
collected, weighed,
labeled and stored.
EXAMPLE 16
[00108] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 3. Compressed carbon dioxide was used as the antisolvent. 0.25g
of
etoposide and 0.25 g of paclitaxel were dissolved in 50 ml of methanol. The
temperature of
the precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was
used as the particle production vessel 608 in Figure 6. Pressure of the system
was
maintained at 80 bar. The antisolvent flow consisted of SOg/min of carbon
dioxide. The
solution flow rate was maintained at 1.0 ml/min for 50 minutes. A 100 micron
capillary tube
was used to apply the solution on to the vibrating surface. A Branson 900 BCA
system with
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
a frequency of 20 kHz was used at 20% amplitude to vibrate the atomizing
surface. After
the end of solution flow, carbon dioxide flow was maintained for 60 additional
minutes and
the system was depressurized. The vessel was opened and the powder material
was
collected, weighed, labeled and stored.
EXAMPLE 17
[00109] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 3. Compressed carbon dioxide was used as the antisolvent. 1.Og
of etoposide
and l.Og of paclitaxel were dissolved in 50 ml of methanol. The temperature of
the
precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was used
as the particle production vessel 608 in Figure 6. Pressure of the system was
maintained at
80 bar. The antisolvent flow consisted of SOg/min of carbon dioxide. The
solution flow rate
was maintained at 1.0 mllmin for 50 minutes. A 100 micron capillary tube was
used to
apply the solution on to the vibrating surface. A Branson 900 BCA system with
a frequency
of 20 kHz was used at 20% amplitude to vibrate the atomizing surface. After
the end of
solution flow, carbon dioxide flow was maintained for 60 additional minutes
and the system
was depressurized. The vessel was opened and the powder material was
collected, weighed,
labeled and stored.
EXAMPLE 18
[00110] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 1. Compressed carbon dioxide was used as the antisolvent. 1.Og
of etoposide
and 1.Og of paclitaxel were dissolved in 25 ml of methanol and dispensed in to
the vessel.
The temperature of the precipitation vessel was maintained at about 50
°C. A 103 ml high
pressure vessel was used as vessel 402 in Figure 4. Pressure of the system was
maintained at
100 bar. A magnetic stirrer at 520 rpm was used to obtain uniform mixing
during the
precipitation. The antisolvent flow consisted of Sg/min of carbon dioxide.
Pressure in the
vessel was increased gradually at a rate of 1 bar/min. After the pressure
reached 100 bar,
antisolvent flow was maintained for 180 minutes. The vessel was depressurized,
opened and
the powder material was collected, weighed, labeled and stored.
EXAMPLE 19
[00111] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 2. Compressed carbon dioxide was used as the antisolvent. O.Sg
of etoposide
and O.Sg of paclitaxel were dissolved in 50 ml of methanol. The temperature of
the
precipitation vessel was maintained at about 50 °C. A 500 ml high
pressure vessel was used
as vessel 508 in Figure 5. Pressure of the system was maintained at 80 bar.
The antisolvent
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
26
flow consisted of SOg/min of carbon dioxide. The solution flow rate was
maintained at 0.5
ml/min for 100 minutes. A 100 micron capillary tube was used to disperse the
solution
inside the vessel M. After the end of solution flow, carbon dioxide flow was
maintained for
60 additional minutes and the system was depressurized. The vessel was opened
and the
powder material was collected, weighed, labeled and stored. .
EXAMPLE 20
[00112) Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 1. Compressed carbon dioxide was used as the antisolvent. O.Sg
of etoposide
and O.Sg of paclitaxel were dissolved in 25 ml of methanol and dispensed in to
the vessel.
The temperature of the precipitation vessel was maintained at about 50
°C. A 103 ml high
pressure vessel was used as vessel 404 in Figure 4. Pressure of the system was
maintained at
100 bar. A magnetic stirrer at 520 rpm was used to obtain uniform mixing
during the
precipitation. The antisolvent flow consisted of Sg/min of carbon dioxide.
Pressure in the
vessel was increased gradually at a rate of 1 bar/min. After the pressure
reached 100 bar,
antisolvent flow was maintained for 180 minutes. The vessel was depressurized,
opened and
the powder material was collected, weighed, labeled and stored.
EXAMPLE 21
[00113] Etoposide and paclitaxel was made into a solution in methanol and
processed
using process 1. Compressed carbon dioxide was used as the antisolvent. 1.Og
of etoposide
and O.Sg of paclitaxel were dissolved in 25 ml of methanol and dispensed in to
the vessel.
The temperature of the precipitation vessel was maintained at about 50
°C. A 103 ml high
pressure vessel was used as vessel 404 in Figure 4. Pressure of the system was
maintained at
100 bar. A magnetic stirrer at 520 rpm was used to obtain uniform mixing
during the
precipitation. The antisolvent flow consisted of Sg/min of carbon dioxide.
Pressure in the
vessel was increased gradually at a rate of 1 bar/min. After the pressure
reached 100 bar,
antisolvent flow was maintained for 180 minutes. The vessel was depressurized,
opened and
the powder material was collected for further processing.
[00114] The samples produced by Examples 12-21 were tested for chemical
integrity
(using HPLC) and physical characteristics (using XRPD, DSC, TGA, SEM, NMR and
IR).
[00115] HPLC analysis was carried out on each sample. Samples were dissolved
in
ethanol:methanol (50:50) to yield a concentration of 5 mg/mL. This stock
solution was
further diluted in ethanol:water (50:50) to yield a final concentration of 10
~g/ml,.
[00116] Each drug showed a single peak with a retention time of about 6.5 and
8 minutes
for etoposide and paclitaxel, respectively. The amount of recovery of
individual drugs in
each example was calculated based on the average of the experimentally
determined
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
27
concentration for three different samples prepared from each sample in
Examples 1-10. The
individual recoveries of etoposide and paclitaxel (including standard
deviation (SD)) and
combined drug recovery are set forth in Table 3.
Table 3: Concentrations of etoposide and paclitaxel (10 ~.g/mL samples)
Etoposide Paclitaxel EtoposidePaclitaxel
Conc. Conc.
(1~~~) (1~~~)
Example Average SD Average SD % of % of Total
total total Recovery
sample sample
1 6.44 0.47 2.96 0.24 64.45 29.60 94.04
2 3.83 0.32 3.93 0.38 38.34 39.31 77.65
3 4.23 0.22 4.22 0.3 5 42.29 42.21 84. S
0
4 5.07 0.35 3.35 0.43 50.69 33.47 84.17
S 5.71 1.24 3.29 1.29 57.09 32.89 89.98
6 6.87 1.06 2.33 0.76 68.65 23.31 91.96
7 6.40 0.31 2.12 0.11 64.04 21.19 85.23
8 5.87 0.34 2.69 0.11 58.68 26.90 85.58
9 5.84 O.1 S 2.32 0.10 5 8.43 23 .19 81.62
7.25 0.34 0.80 0.24 72.53 8.04 80.56
[00117) As Table 2 illustrates, drug recovery for the samples made in
accordance with
the present invention range from about 77% to about 94%. The overall average
recovery
for the samples of Examples 12-21 combined was 85.51 ~ 5.1 S.
[00118] SEM pictures of individual drugs show irregular particles and acicular
(fibrous)
clumps for paclitaxel and blade-like particles for etoposide. The SEM pictures
of the
combination samples show similarity in particle structure to etoposide and
paclitaxel to
varying extent. XRPD data shows the crystalline nature of the individual drugs
and the
samples prepared according to the invention with many of the samples similar
to the
individual drugs. DSC, IR and NMR data showed similarity between the samples
of
Examples 12-21 and the individual drugs.
[00119] The sample of Example 12 shows the closest similarity to the
individual
components when mixed together in a homogeneous blend. Example 12 provided the
maximum recovery (based on HPLC data), appears to be a uniform blend of
individual
drugs (per SEM), and provides the closest IR spectra to the individual drugs.
XRPD, DSC
and NMR data also demonstrate that the sample of Example 12 is a homogenous,
uniform
blend of the individual etoposide and paclitaxel components.
CA 02510019 2005-06-14
WO 2004/056342 PCT/US2003/040608
28
[00120] As demonstrated by the results of Examples 12-21, the process of the
invention
can be used to produce pharmaceutical formulations comprising etoposide and
paclitaxel by
using a supercritical fluid in accordance with the present invention.
[00121] All references, including publications, patent applications, and
patents, cited
herein are hereby incorporated by reference to the same extent as if each
reference were
individually and specifically indicated to be incorporated by reference and
were set forth in
its entirety herein.
[00122] The use of the terms "a" and "an" and "the" and similar referents in
the context
of describing the invention (especially in the context of the following
claims) are to be
construed to cover both the singular and the plural, unless otherwise
indicated herein or
clearly contradicted by context. The terms "comprising," "having,"
"including," and
"containing" are to be construed as open-ended terms (i.e., meaning
"including, but not
limited to,") unless otherwise noted. Recitation of ranges of values herein
are merely
intended to serve as a shorthand method of referring individually to each
separate value
falling within the range, unless otherwise indicated herein, and each separate
value is
incorporated into the specification as if it were individually recited herein.
All methods
described herein can be performed in any suitable order unless otherwise
indicated herein or
otherwise clearly contradicted by context. The use of any and all examples, or
exemplary
language (e.g., "such as") provided herein, is intended merely to better
illuminate the
invention and does not pose a limitation on the scope of the invention unless
otherwise
claimed. No language in the specification should be construed as indicating
any non-
claimed element as essential to the practice of the invention.
[00123] Preferred embodiments of this invention are described herein,
including the best
mode known to the inventors for carrying out the invention. Variations of
those preferred
embodiments may become apparent to those of ordinary skill in the art upon
reading the
foregoing description. The inventors expect skilled artisans to employ such
variations as
appropriate, and the inventors intend for the invention to be practiced
otherwise than as
specifically described herein. Accordingly, this invention includes all
modifications and
equivalents of the subject matter recited in the claims appended hereto as
permitted by
applicable law. Moreover, any combination of the above-described elements in
all possible
variations thereof is encompassed by the invention unless otherwise indicated
herein or
otherwise clearly contradicted by context.