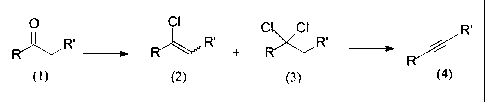Note: Descriptions are shown in the official language in which they were submitted.
CA 02510093 2005-06-16
TITLE OF THE INVENTION
Novel process for the preparation of a-chlorovinyl, a,a -dichloro, and
acetylenes from ketones
FIELD OF THE INVENTION
An improved process to convert ketones to the corresponding a-chlorovinyl and
a,a-dichloro
compounds. More specifically, the present invention is directed at a process
to convert ketones
to corresponding acetylenes with corresponding a-chlorovinyl and a,a-dichloro
compounds as
intermediates.
BACKGROUND OF THE INVENTION
A class of compounds which have considerable synthetic utility are a-
chlorovinyl and a,a-
dichloro compounds. These compounds may then be converted to the corresponding
acetylenes,
furthering their synthetic utility. This includes their use in polymer
chemistry as well in the
preparation of pharmaceuticals. The overall process is depicted in figure 1.
Figure 1
o ci c ci R'
R~,,,, R' + R R' i
ketone, I a-chlorovinyl, 2 a,a-dichloro, 3 acetylene, 4
Examples of commercially useful acetylenes which are valuable for the
preparation of active
pharmaceuticals include 3,3-dimethyl-l-butyne, which has been used as a key
raw material for
terbinafine (5) (for instance, US Patent 5,817,875) and cyclopropylacetylene,
which is a key raw
material for Efavirenz (6) (for instance, US Patent 6,297,410). A box is
placed around the
acetylenic moiety in these pharmaceuticals as depicted in figure 2.
CA 02510093 2005-06-16
Page 2
Figure 2
HCI
FaC
CI 0
NO
H
5, Terbinafine 6, Efavirenz
The use of the highly toxic and corrosive chlorinating agent phosphorus
pentachloride (PC15) for
a transformation of this type is known. For instance, T.L. Jacobs provides a
review in Organic
Reactions, Vol. 5, Chapter 1. However, the PC15 reagent suffers from many
disadvantages.
These include the cost of this reagent coupled with the fact that only two of
the five chlorine
atoms are used. An even greater disadvantage, especially on an industrial
scale, is the fact that
the granular PCl5 tends to sublime and agglomerate to a hard, solid mass upon
storage, which
makes it very difficult to load this reagent into a reactor. A further
disadvantage of using PC15
reacts on exposure to moisture in the atmosphere to generate hydrogen chloride
gas.
Specific examples of the preparation of 3,3-dimethyl-l-butyne (tert-
butylacetylene) from
pinacolone (R = tert-butyl, R' = hydrogen) using PC15 include work done by
P.D. Bartlett and
L.J. Rosen (J. Am. Chem. Soc., 64, p. 543, 1942) and P.J. Kocienski (J. Org.
Chem., 39, p.3285,
1974). Likewise, a description of the process used for the preparation of
cyclopropylacetylene
from cyclopropyl methyl ketone using PC15 is described in Synlett, 1999,
pp.1948 to 1950 by
Schmidt et al.
The isolation of 3,3-dimethyl-l-butyne by the process described by Bartlett
and Rosen and
Kocienski was accomplished using fractional distillation which would require
specialized
equipment for further scale-up. Thus, a more facile and industrially
acceptable process to isolate
the acetylenic compound from the reaction mixture would be advantageous.
A substitute to the PCl5 reagent is described in US Patent 3,715,407 whereby
dichlorophosphoranes having the formula R3PC12 were used. This reagent was
obtained by the
CA 02510093 2005-06-16
Page 3
reaction of phosgene with phosphine oxide having formula R3PO. However, this
method suffers
from the severe disadvantage that it employs phosgene, which is a highly
poisonous gas being
used as a chemical warfare agent. Very recently, the use of this type of
reagent was described in
US Patent 6,207,864 B 1 for the preparation of the key intermediate
cyclopropylacetylene from
cyclopropyl methyl ketone. Briefly cyclopropylacetylene is disclosed in PCT/WO
96/22955 as
an intermediate for a pharmaceutical (Efavirenz), which acts as an inhibitor
of the HIV reverse
transcriptase enzyme. This is a key enzyme for the replication of the Human
Immuno
Deficiency virus that is the cause of Acquired Immune Deficiency (AIDS)
syndrome. The
method again uses dihalotriorganophosphoranes prepared from triorganophosphane
oxides or
triorganophosphane sulfides and a chlorinating agent of which the toxic
phosgene is preferred.
Therefore, an industrially acceptable and general process, which overcomes the
deficiencies of
the prior art, was required for the conversion of ketones to the corresponding
a-chlorovinyl and
a,a-dichloro compounds and, optionally, their further conversion to their
corresponding
acetylenes.
To overcome the difficulties associated with prior art processes for these
reactions, alternative
chlorinating reagents were examined. No reaction was obtained when ketones
having general
formula 1(figure 3) were treated with phosphorus oxychloride (POC13) in the
absence a base.
Addition of a base produced an unidentified phosphonate intermediate which did
not undergo
elimination. Surprisingly, the addition of a copper catalyst such as cuprous
chloride to the
phosphorus oxychloride and base mixture efficiently converted the ketone to a
mixture of a-
chlorovinyl (2) and a,a-dichloro (3) compounds as depicted in figure 3. These
results were
similar to the results obtained using phosphorus pentachloride.
Figure 3
0 POC13, Base Ci CI CI
+ R~R
R, R
Copper catalyst
ketone, 1 a-chlorovinyl, 2 a,a-dichloro, 3
CA 02510093 2005-06-16
Page 4
In figure 3, R is a cyclic or acyclic C, to C6 alkyl, aryl, or C7 to Clo
aralkyl. R' is hydrogen,
cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to C10 aralkyl. Most preferably,
R is a tert-butyl,
phenyl, or cyclopropyl.
SUMMARY OF THE INVENTION
According to one aspect of the invention there is provided a process for the
conversion of a
ketone of general formula (1)
0
R'
R
(1)
wherein R is a cyclic or acyclic C, to C6 alkyl, aryl, or C7 to Clo aralkyl
and R' is hydrogen,
cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to Clo aralkyl, to a-
vinylchloride of formula (2)
and/or an a,a-dichloro of formula (3)
CI CI CI
R)~ R, R~~ R1
(2) (3)
wherein the process comprises using phosphorus oxychloride in the presence of
a trialkylamine
base and a catalyst.
According to another aspect of the invention there is provided a process for
the conversion of a
ketone (1)
0
R'
(1)
CA 02510093 2005-06-16
Page 5
wherein R is a cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to Clo aralkyl
and R' is hydrogen,
cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to C10 aralkyl, to an a-
vinylchloride (2) and a,a -
dichloro (3)
CI CI CI
R' R~'C R'
(2) (3)
wherein the process comprises using phosphorus oxychloride in the presence of
a trialkylamine
base and a catalyst in an inert organic solvent.
Preferably, the trialkylamine base is selected from the group consisting of
triethylamine,
tripropylamine and tributylamine. Most preferably, the base is triethylamine.
Preferably, the trialkylamine base and ketone have an equivalent ratio ranging
from 0.05:1 to
5:1. More preferably, the trialkylamine base and ketone have an equivalent
ratio ranging from
0.1:1 to 1:1. Most preferably, the trialkylamine base and ketone have an
equivalent ratio of
0.2:1.
Preferably, the catalyst is selected from the group consisting of cuprous
acetate and cuprous
chloride. More preferably, the catalyst is cuprous chloride.
Preferably, the inert organic solvent is selected from the group consisting of
C5 to Clo cyclic and
acyclic hydrocarbon. More preferably, the inert organic solvent is selected
from the group
consisting of hexane, heptane and cyclohexane. Most preferably, the inert
organic solvent is
hexane.
According to yet another aspect of the invention there is provided a process
for the isolation of an
acetylene of general formula (4)
CA 02510093 2005-06-16
Page 6
R'
~
R
(4)
wherein R is a cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to Clo aralkyl
and R' is hydrogen,
cyclic or acyclic C1 to C6 alkyl, aryl, or C7 to Clo aralkyl, comprising the
steps of:
(i) treating an a-chlorovinyl (2) and/or an a,a-dichloro (3)
Ci CI CI
R~ R' R~~ R'
(2) (3)
compound in an alkyl sulfoxide or sulfone solvent in the presence of a strong
base; and
(ii) extracting the resulting acetylene (4) by using an organic extraction
solvent.
Preferably, the alkyl sulfoxide solvent for step (i) is dimethylsulfoxide.
Preferably, the strong base is potassium tert-butoxide.
Preferably, the organic extraction solvent is a organic solvent of C5 to Clo.
Preferably, the
organic solvent of C5 to Cto is heptane.
Preferably, the a-vinylchloride is 2-chloro-3,3-dimethyl-l-butene. Preferably,
the a,a-dichloro
compound is 2,2-dichloro-3,3-dimethylbutane. Preferably, the acetylene is 3,3-
dimethyl-l-
butyne, 1-phenyl-l-propyne, or phenylacetylene.
According to yet another aspect of the invention there is provided a process
for the preparation of
an intermediate in the preparation of terbinafine comprising the steps of:
(i) preparing 3,3-dimethyl-2-chloro-l-butene and 3,3-dimethyl-2,2-
dichlorobutane
using the process as described previously;
CA 02510093 2005-06-16
Page 7
(ii) dehydrohalogenation of the 3,3-dimethyl-2-chloro-l-butene and 3,3-
dimethyl-2,2-
dichlorobutane using a strong base in an alkyl sulfoxide or sulfone solvent;
and
(iii) isolating the resulting 3,3-dimethyl-l-butyne by extraction in an
organic
extraction solvent wherein said solvent is a hydrocarbon being from 5 to 10
carbon atoms.
Preferably, the strong base is potassium tert-butoxide. Preferably, the
alkylsulfoxide is
dimethylsulfoxide.
Preferably, the phosphorus oxychloride is added stepwise. Preferably, the
phosphorus
oxychloride and the base are added stepwise.
In another aspect of this invention, the a-chlorovinyl and/or a,a-dichloro
compounds are
converted to their corresponding acetylenes.
DETAILED DESCRIPTION OF THE INVENTION
The use of the readily available and inexpensive reagent phosphorus
oxychloride, instead of
phosphorus pentachloride, offers numerous advantages on an industrial scale.
For instance, it is
much less difficult to handle since it is a non-volatile liquid. Also,
relative to phosphorus
pentachloride, it uses two of the three available chlorine atoms present on
the molecule and is
less expensive. The use of these reagents for this type of transformation is
unprecedented in
chemical literature.
It has been discovered that the effective stoichiometries of phosphorus
oxychloride are 0.5
equivalents to 1.5 equivalents, more preferably 0.7 to 0.9 equivalents, most
preferably 0.8
equivalents.
The preferred catalysts are salts of copper (I) and copper (II) such as
cuprous acetate or cuprous
chloride, most preferably cuprous chloride. The preferred stoichiometry of
base is 0.05 to 0.6
equivalents, more preferably 0.1 to 0.3 equivalents, most preferably 0.2
equivalents.
CA 02510093 2005-06-16
Page 8
The reaction is typically conducted at 70 to 110 C, more preferably at 80 to
100 C, even more
preferably at 90 to 95 C. The reaction can be conducted neat or in the
presence of a non-reactive
solvent such as a C5 to C10 cyclic or acyclic hydrocarbon, for instance
hexane, heptane or
cyclohexane. When conducted in the presence of an inert solvent, the most
preferred solvent is a
C5 to C10 hydrocarbon, for instance heptane.
For the reaction, the ketone, base, copper catalyst and, optionally the
solvent, are mixed
whereupon the phosphorus oxychloride reagent is added portionwise over time.
This is done
since a strongly exothermic reaction may occur which could pose a safety
hazard. For instance,
in the preparation of 3,3-dimethyl-2-chloro-l-butene and 3,3-dimethyl-2,2-
dichlorobutane from
pinacolone using the phosphorus oxychloride, triethylamine and cuprous
chloride combination, a
delayed exothermic reaction was noted which could have possibly led to runaway
reaction
conditions on further scale-up. It was discovered that by using a portionwise
mode of addition of
the phosphorus oxychloride reagent, these conditions were avoided and the
reaction was
intrinsically safe. In another embodiment, the base together with the
phosphorus oxychloride
reagent are added in a portionwise manner.
In another aspect of this invention, the a-chlorovinyl and a,a-dichloro
compounds are converted
to the acetylenic compound by treatment with a strong base, preferably a metal
alkoxide, most
preferably tert-butoxide, in alkyl sulfoxide and sulfone solvents such as
dimethyl sulfoxide and
sulfolane, most preferably dimethylsulfoxide. This process is depicted in
figure 4 below.
Figure 4
cl C CI 1.Strong base R'
~ R' R'
R R alkyl sulfoxide or
sulfone solvent acetylene, 4
a-chlorovinyl, 2 a,a-dichloro, 3 2. Extraction in solvent
CA 02510093 2005-06-16
Page 9
The advantage of this type of elimination process is that it allows the
convenient isolation of the
acetylenic compound in a cyclic or acyclic C5 to Clo hydrocarbon solvent, most
preferably
hexane or heptane. Thus, the reaction is quenched using water and the
acetylenic compound is
extracted into the C5 to Clo hydrocarbon solvent. The residual by-products and
alkyl sulfoxide or
sulfone solvent are readily removed from the C5 to Clo hydrocarbon solvent
containing the
acetylene compound by washing with water and / or brine. This process greatly
simplifies some
of the processes of the prior art, for instance the process described by P.J.
Kocienski (J. Org.
Chem., 39, p.3285, 1974) for the isolation of 3,3-dimethyl-l-butyne, which was
accomplished
using fractional distillation of the compound from the dimethylsulfoxide
solvent.
The following examples exemplify various aspects of the invention.
Example 1
Preparation of 3,3-Dimethyl-l-butyne from pinacolone
Part A: Preparation of 3,3-dimethyl-2-chloro-l-butene and 3,3-dimethyl-2,2-
dichlorobutane
A round bottom flask was charged with cuprous chloride (14.7 g, 0.100 mol),
pinacolone (200.00
g, 1.997 mol), and (40.42 g triethylamine, 0.399 mol) and the mixture was
stirred and heated
under a nitrogen atmosphere to 75 C. The flask is then charged with a portion
of the phosphorus
oxychloride (60.7 g, 0.396 mol) and heated to 90 to 95 C and maintained at
this temperature for
about 2 hours whereupon the mixture was cooled to 70 to 75 C and another
portion of
phosphorus oxychloride (30.40 g, 0.198 mol) was added (note: in this case, non-
portionwise
addition of the phosphorus oxychloride led to a delayed exotherm, see
Disclosure section). The
reaction mixture was then re-heated to 90 to 95 C and maintained at this
temperature for about 2
hours. Similarly, two additional portions of phosphorus oxychloride (2 X 60.70
g, 2 X 0.396
mol; total phosphorus oxychloride = 1.386 mol) were added at which point H NMR
indicated
that the reaction was complete. The temperature was decreased to 55 C and
heptanes (400 mL)
was added followed by the slow addition of water (200 mL) while controlling
the temperature
between 50 to 60 C. This process took about 1 hour whereupon the mixture was
cooled to 20 to
25 C and the layers were split. The organic layer was washed with water (200
mL) and brine
CA 02510093 2005-06-16
Page 10
(300 mL). The weight of the organic layer was 500 g and it was comprised of
130 g of 3,3-
dimethyl-2-chloro-l-butene (55.0% yield from pinacolone) and 71 g of 3,3-
dimethyl-2,2-
dichlorobutane (23.0% yield from pinacolone). The overall yield was 78% from
pinacolone.
Part B: Preparation of 3,3-dimethyl-l-butyne from 3,3-dimethyl-2-chloro-l-
butene and 3,3-
dimethyl-2,2-dichlorobutane
A round bottom flask was charged with potassium tert-butoxide (157 g, 1.40
mol) and
dimethylsulfoxide (235 mL) and the temperature increased to 42 C. The mixture
was cooled
with stirring and under nitrogen to about 20 C whereupon a 195 g portion of
the solution from
part "A" above was added dropwise while maintaining the temperature below 30
C. H NMR
analysis of this solution demonstrated that it contained about 50.7 g of 3,3-
dimethyl-2-chloro-l-
butene (0.427 moL) and 27.7 g of 3,3-dimethyl-2,2-dichlorobutane (0.179 moL).
After a period
of time, a further portion of potassium tert-butoxide (17.5 g, 0.156 mol) was
added and the
reaction maintained at room temperature. Water was added while keeping the
temperature below
30 C and the layers separated. The organic layer was sequentially washed with
water (3 X 235
mL) and brine (1 x 235 mL). This provided a pale, light brown solution
weighing 306 g of
which 11% (w/w) was 3,3-dimethyl-l-butyne (33.7 g, 53% yield).
Example 2
Preparation of Phenylacetylene from acetophenone
Part A: Preparation of a-chlorostyrene from acetophenone
To a stirred mixture of cuprous chloride (0.60 g, 0.0061 mol), triethylamine
(1.3 g, 0.013 mol),
and phosphorus oxychloride (5.2 g, 0.034 mol) in heptanes (15 mL) at 25 C was
added
acetophenone (5.0 g, 0.042 mol). The resulting mixture was heated to 95-100 C
for 20 hours.
The reaction mixture was cooled to 45-50 C and then slowly quenched with water
(ca. 25 mL).
The organic layer was washed with water (10 mL) and brine (10 mL), dried over
(MgSO4), and
evaporated to give 5.4 g of the a-chlorostyrene product (93% yield).
CA 02510093 2005-06-16
Page 11
Part B. Preparation of phenylacetylene from a-chlorostyrene
The vinyl chloride (5.4 g, 0.039 mol) in heptanes (20 mL) mixture produced
above was added
dropwise to a solution of potassium tert-butoxide (7.4 g, 0.066 mol) in
dimethylsulfoxide (20
mL) at a rate such that the temperature was maintained below 30 C. The
reaction mixture was
stirred at 20-25 C for a further 3 hours and quenched by the slow addition of
water (30 mL)
while maintaining the temperature below 30 C. The organic layer was washed
with water (2 X
mL) and brine (1 X 15 mL) and then evaporated to dryness to provide 2-g of
phenyl acetylene
(50% yield).
Example 3
Preparation of 1-Phenyl-l-propyne from propiophenone
Part A: Preparation of 1-chloro-l-phenyl-l-propene from propiophenone
To a stirred mixture of copper (I) chloride (0.60 g, 0.0061 mol),
triethylamine (1.1 g, 0.011 mol)
and phosphorus oxychloride (4.5 g, 0.030 mol) in heptanes (20 mL) was added
propiophenone
(5.0 g, 0.037 mol). The resulting mixture was then cooled to 45-50 C and
slowly quenched with
water (ca. 25 mL) while maintaining the internal temperature below 60 C. The
organic layer was
washed successively with water (20 mL) and brine (20 mL), dried over MgSO4,
and evaporated
to dryness to provide 1-chloro-l-phenyl-l-propene (4.8 g, 86%) as a yellow
oil.
Part B. Preparation of 1-phenyl-l-propyne from 1-chloro-l-phenyl-l-propene
The vinyl chloride compound from part `A' above (4.8 g, 0.030 mol) in heptanes
(20 mL) was
added dropwise to a solution of potassium tert-butoxide (5.8 g, 0.050 mol) in
dimethylsulfoxide
(20 mL) at a rate such that the temperature was maintained below 30 C. The
reaction mixture
was stirred at 20-25 C for a further 2 hours and then quenched by the slow
addition of water (30
mL) while maintaining the temperature below 30 C. The organic layer was washed
with water (2
X 15 mL) and brine (1 X 15 mL) and then dried over Na2SO4 and evaporated to
dryness to
provide 2.7 g of 1-phenyl-l-propyne (77% yield).
CA 02510093 2005-06-16
Page 12
Example 4
Preparation of 2-chloro-2-cyclopropyl- I -propene from cycloprop lY methyl
ketone
A 500 mL round bottom flask was charged with cuprous chloride (1.8 g, 0.018
moL), heptanes
(60 mL), tripropylamine (51 g, 0.356 moL), phosphorus oxychloride (14.4 g,
0.094 moL), and
cyclopropyl methyl ketone and heated with stirring and under nitrogen to 95 C.
After heating for
about 20 hours at this temperature another portion of cuprous chloride (3.2 g,
0.032 moL) was
added and heating was continued. After a further 20 hours, the reaction
mixture was cooled to 60
to 65 C and then slowly quenched with saturated aqueous sodium bicarbonate.
The organic
layer was washed with brine (50 mL). This provided 57 g of an organic layer
which contained,
by H NMR analysis, about 1.9 g (16% yield from cyclopropyl methyl ketone) of 2-
chloro-2-
cyclopropyl-l-propene.
While the foregoing provides a detailed description of a preferred embodiment
of the invention,
it is to be understood that this description is illustrative only of the
principles of the invention
and not limitative. Furthermore, as many changes can be made to the invention
without
departing from the scope of the invention, it is intended that all material
contained herein be
interpreted as illustrative of the invention and not in a limiting sense.