Note: Descriptions are shown in the official language in which they were submitted.
CA 02511750 2005-07-21
3005-31 CA
DIBENZODIAZEPINONE ANALOGUES, PROCESSES FOR THEIR PRODUCTION
AND THEIR USE AS PHARMACEUTICALS
FIELD OF THE INVENTION
This invention relates to dibenzodiazepinone analogues, represented by a
naturally produced farnesylated dibenzodiazepinone referred to as Compound 1
and its
chemical derivatives, their pharmaceutically acceptable salts, solvates and
prod rugs,
and to methods for obtaining the compounds. One method of obtaining Compound 1
is
by cultivation of a strain of a Micromonospora sp., i.e., 046-EC011 or
[S01]046. One
method of obtaining the derivatives involves post-biosynthesis chemical
modification of
Compound 1. The present invention further relates to the use of
dibenzodiazepinone
analogues, and their pharmaceutically acceptable salts, solvates and prod rugs
as
pharmaceuticals, in particular to their use as inhibitors of cancer cell
growth,
mammalian lipoxygenase, and for treating acute and chronic inflammation, and
to
pharmaceutical compositions comprising a dibenzodiazepinone analogue, or a
pharmaceutically acceptable salt, solvate or prodrug thereof.
BACKGROUND OF THE INVENTION
The euactinomycetes are a subset of a large and complex group of Gram-
positive bacteria known as actinomycetes. Over the past few decades these
organisms, which are abundant in soil, have generated significant commercial
and
scientific interest as a result of the large number of therapeutically useful
compounds,
particularly antibiotics, produced as secondary metabolites. The intensive
search for
strains able to produce new antibiotics has led to the identification of
hundreds of new
species.
Many of the euactinomycetes, particularly Streptomyces and the closely related
Saccharopolyspora genera, have been extensively studied. Both of these genera
produce a notable diversity of biologically active metabolites. Because of the
commercial significance of these compounds, much is known about the genetics
and
physiology of these organisms. Another representative genus of
euactinomycetes,
Micromonospora, has also generated commercial interest. For example, U.S.
Patent
No. 5,541,181 (Ohkuma et al.) discloses a dibenzodiazepinone compound,
specifically
1
CA 02511750 2005-07-21
3005-31 CA
5-farnesyl-4,7,9-trihydroxy-dibenzodiazepin-11-one (named "BU-4664L"),
produced by
a known euactinomycetes strain, Micromonospora sp. M990-6 (ATCC 55378). ECO-
4601 (Compound 1 ) and novel Micromonospora sp. strains 046-EC011 and [S01]046
are disclosed in CA 2,466,340.
i i
ECO-4601 (Compound 1 )
Although many biologically active compounds have been identified from
bacteria,
there remains the need to obtain novel compounds with enhanced properties.
Thus,
there exists a considerable need to obtain pharmaceutically active compounds
in a
cost-effective manner and with high yield. The present invention solves these
problems
by providing new therapeutic compounds and methods to generate these novel
compounds by post-biosynthetic chemical modifications.
SUMMARY OF THE INVENTION
In another aspect, the invention relates to dibenzodiazepinone analogues as
defined below and represented by Compound 1 and derivatives of Compound 1, and
to
pharmaceutical compositions comprising a dibenzodiazepinone analogue or a
pharmaceutically acceptable salt, solvate or prodrug thereof, together with a
pharmaceutically acceptable carrier. In one embodiment, the dibenzodiazepinone
analogue is represented by a compound of Formula I as defined below, or an
ether,
ester, N-alkylated and N-acylated derivative, or a pharmaceutically acceptable
salt,
solvate or prod rug of a compound of Formula I. In another embodiment, the
dibenzodiazepinone analogue is represented by a compound of Formula ll as
defined
below, or an ether, ester, N-alkylated and N-acylated derivative, or a
pharmaceutically
acceptable salt, solvate or prod rug of a compound of Formula II. In a further
embodiment, the dibenzodiazepinone analogue is represented by any one of
Compounds 1 to 98 as defined below, or an ether, an ester, an N-alkylated or N-
acylated derivative, or a pharmaceutically acceptable salt, solvate of prodrug
of any one
2
CA 02511750 2005-07-21
3005-31 CA
of Compounds 1 to 98. In a further embodiment, the dibenzodiazepinone analogue
is
represented by any one of Compounds 1 to 7, 9 to 12, 14, 17, 18, 46, 63, 64,
67, 77,
78, 80, 82 to 85, 87, 89, 92, and 95 to 98 as defined below, or an ether, an
ester, an N-
alkylated or N-acylated derivative, or a pharmaceutically acceptable salt,
solvate of
prodrug of any one of Compounds 1 to 7, 9 to 12, 14, 17, 18, 46, 63, 64, 67,
77, 78, 80,
82 to 85, 87, 89, 92, and 95 to 98.
The invention further encompasses a dibenzodiazepinone analogue obtained by
a method comprising: a) cultivating a Micromonospora sp. strain selected from
strains
[S01]046 and 046-EC011, wherein the cultivation is performed under aerobic
conditions in a nutrient medium comprising at least one source of carbon atoms
and at
least one source of nitrogen atoms; and b) isolating a farnesyl
dibenzodiazepinone
from the bacteria cultivated in step (a).
The invention further encompasses a dibenzodiazepinone analogue obtained by
a method comprising: a) cultivating Micromonospora sp. strain selected from
strains
[S01]046 and 046-EC011, wherein the cultivation is performed under aerobic
conditions in a nutrient medium comprising at least one source of carbon atoms
and at
least one source of nitrogen atoms; b) isolating a farnesyl dibenzodiazepinone
from the
bacteria cultivated in step (a) and c) chemically modifying the compound
isolated in (b).
In one embodiment the dibenzodiazepinone analogue is a compound of Formula I.
In
another embodiment, the dibenzodiazepinone analogue is a compound of Formula
II. In
a further embodiment, the dibenzodiazepinone analogue is selected from
Compounds
2 to 98. In another embodiment the dibenzodiazepinone analogue is selected
from
Compounds 2 to 12, 14, 17, 18, 46, 63, 64, 67, 77, 78, 80, 82 to 85, 87, 89,
92, and 95
to 98.
The invention further encompasses a process for making a dibenzodiazepinone
compound, comprising cultivation of a Micromonospora sp. strain selected from
strains
046-EC011 and [S01]046, in a nutrient medium comprising at least one source of
carbon atoms and at least one source of nitrogen atoms, isolation and
purification of
the compound. In a subclass of this embodiment, the process further comprises
the
step of chemically modifying the isolated compound.
In one embodiment, the cultivation occurs under aerobic conditions.
In another embodiment, the carbon atom and nitrogen atom sources are chosen
3
CA 02511750 2005-07-21
3005-31 CA
from the components shown in Table 1.
In another embodiment, the cultivation is carried out at a temperature ranging
from 18°C to 40°C. In a further embodiment, the temperature
range is 18°C to 29°C.
In another embodiment, the cultivation is carried out at a pH ranging from 6
to 9.
The invention further encompasses a method for making a dibenzodiazepinone
compound, comprising chemically modifying the farnesyl dibenzodiazepinone
Compound 1, and optionally isolating and purifying the dibenzodiazepinone
compound
produced. In one embodiment, the chemical modification step comprises at least
one
step selected from N-alkylations, N-acylations, O-alkylations, O-acylations,
and
modifications of the double bonds of the farnesyl side chain including,
hydrogenation,
electrophilic additions (e.g., epoxidation, dihydroxylation, hydration,
hydroalkoxylation,
hydroamidation, and the like), and double bond cleavage, like ozonolysis, and
reduction
of the ozonolysis product. In a subclass of this embodiment, the farnesyl side
chain
modification reaction is partial (one or two double bonds modified) or
complete (all three
double bonds are modified).
The invention further encompasses a method of treating a diseases selected
from pre-cancerous or cancerous conditions, inflammation, autoimmune diseases,
infections, neurodegenerative diseases and stress, the method comprising
administering a compound of Formula I to a mammal in need of such treatment.
In one
embodiment, the compound is a compound selected from Compounds 1 to 98. In
another embodiment, the invention provides a method for treating a diseases
selected
from pre-cancerous or cancerous conditions, inflammation, autoimmune diseases,
infections, neurodegenerative diseases and stress, the method comprising
administering a compound of Formula I to a mammal in need of such treatment,
with
the proviso that the compound of Formula I is not Compound 1, or alternatively
is not
Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses a method of inhibiting the growth of a
cancer
cell, the method comprising contacting the cancer cell with a compound of
Formula I,
such that growth of the cancer cell is inhibited. In one embodiment, the
compound is a
compound selected from Compounds 1 to 98. In another embodiment, the invention
provides a method of inhibiting the growth of a cancer cell, the method
comprising
contacting the cancer cell with a compound of Formula I such that growth of
the cancer
4
CA 02511750 2005-07-21
3005-31 CA
cell is inhibited, with the proviso that the compound of Formula I is not
Compound 1, or
alternatively is not Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses a method of inhibiting the growth of a
cancer
cell in a mammal, the method comprising administering a compound of Formula I
to a
mammal comprising a cancer cell, such that growth of the cancer cell is
inhibited in the
mammal. In one embodiment, the compound is a compound selected from Compounds
1 to 98. In another embodiment, the invention provides a method of inhibiting
the
growth of a cancer cell in a mammal, the method comprising administering a
compound
of Formula I to the mammal such that growth of the cancer cell is inhibited,
with the
proviso that the compound of Formula I is not Compound 1, or alternatively is
not
Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses a method of treating a pre-cancerous or
cancerous condition in a mammal, comprising the step of administering to the
mammal
a therapeutically effective amount of a compound of Formula I, such that a pre-
cancerous or cancerous condition is treated. In one embodiment, the compound
is a
compound selected from Compounds 1 to 98. In another embodiment, the invention
provides a method of treating a pre-cancerous or cancerous condition in a
mammal, the
method comprising the step of administering to the mammal a therapeutically
effective
amount of a compound of Formula I such that a pre-cancerous or cancerous
condition
is treated, with the proviso that the compound of Formula I is not Compound 1,
or
alternatively is not Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses the use of a compound of Formula I, or a
pharmaceutically acceptable salt, solvate or prodrug thereof as an antitumor
agent for
the treatment of a pre-cancerous or cancerous condition in a mammal. In one
embodiment, the compound is a compound selected from Compounds 1 to 98. In
another embodiment, the invention provides use of a compound of Formula I, or
a
pharmaceutically acceptable salt or prodrug thereof as an antitumor agent for
the
treatment of a pre-cancerous or cancerous condition in a mammal, with the
proviso that
the compound of Formula I is not Compound 1, or alternatively is not Compound
2, or
alternatively is neither Compound 1 nor 2.
The invention further encompasses the use of a compound of Formula I, or a
pharmaceutically acceptable salt or prod rug thereof as an antineoplastic
agent for the
CA 02511750 2005-07-21
3005-31 CA
treatment of a proliferative disorder in a mammal. In one embodiment, the
compound
is a compound selected from Compounds 1 to 98. In another embodiment, the
invention provides use of a compound of Formula I, or a pharmaceutically
acceptable
salt or prodrug thereof as an antineoplastic agent for the treatment of a
proliferative
disorder in a mammal, with the proviso that the compound of Formula I is not
Compound 1, or alternatively is not Compound 2, or alternatively is neither
Compound
1 nor 2.
The invention further encompasses the use of a compound of Formula I, or a
pharmaceutically acceptable salt or prodrug thereof in the preparation of a
medicament
for the treatment of a pre-cancerous or cancerous condition in a mammal. In
one
embodiment, the compound is a compound selected from Compounds 1 to 98. In
another embodiment, the invention provides use of a compound of Formula I, or
a
pharmaceutically acceptable salt or prodrug thereof in the preparation of a
medicament
for the treatment of a pre-cancerous or cancerous condition in a mammal, with
the
proviso that the compound of Formula I is not Compound 1, or alternatively is
not
Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses a commercial package comprising a
compound of Formula I, or a pharmaceutically acceptable salt or prodrug
thereof,
together with instructions for use in the treatment of a neoplasm or a pre-
cancerous or
cancerous condition. In one embodiment, the compound is a compound selected
from
Compounds 1 to 98. In one embodiment, the invention provides a commercial
package
comprising a compound of Formula I, or a pharmaceutically acceptable salt or
prod rug
thereof, together with instructions for use in the treatment of a neoplasm or
a pre-
cancerous or cancerous condition, with the proviso that the compound of
Formula I is
not Compound 1, or alternatively is not compound 2, or alternatively is
neither
Compound 1 nor 2.
The invention further encompasses the use of a compound of Formula I, or a
pharmaceutically acceptable salt or prodrug thereof as a peripheral
benzodiazepine
receptor (PBR) binding agent for the treatment of a condition involving the
PBR. The
peripheral benzodiazepine receptor (PBR) is a well-characterized receptor
known to be
directly involved in diseases states. The PBR is involved in the regulation of
immune
responses. These diseases states include tumors, inflammatory diseases (such
as
rheumatoid arthritis and lupus), parasitic infections and neurodegenerative
diseases
6
CA 02511750 2006-05-04
3005-31 CA
(such as Alzheimer's, Huntington's and Multiple Sclerosis). See, for example,
Casellas
et al (2003), Current Med. Chem., vol 10, 1563-1572; Miettinen et al (1995),
Cancer
Res., vol 55, 2691; Junck et al (1989), J. Ann. Neurol., vol 26, 752; Gavish
et al (1993),
Clin. Neuropharm., vol 16, no 5, 401-417; Papadopoulos et al (2003), Ann.
Pharm. Fr.,
vol 61, 30-50; Vowinckel et al (1997), J. Neurosci. Res., vol 50, 345; Gelhert
et al
(1997), Neurochem. Int., vol 31, 705; Lacor et al (1999), Brain Res., vol 815,
70). In
one embodiment, the compound for use in the treatment of a condition involving
the
PBR is a compound selected from Compounds 1 to 98. In another embodiment, the
compound for use in the treatment of a condition involving the PBR is a
compound of
Formula I with the proviso that the compound of Formula I is not Compound 1,
or
alternatively is not Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses a method of reducing inflammation in a
mammal, comprising administering to a mammal having inflammation a
therapeutically
effective amount of a compound of Formula I, such that the inflammation is
reduced. In
one embodiment, the compound is a compound selected from Compounds 1 to 98. In
another embodiment, the compound for use as an anti-inflammatory is a compound
of
Formula I with the proviso that the compound of Formula I is not Compound 1,
or
alternatively is not Compound 2, or alternatively is neither Compound 1 nor 2.
The invention further encompasses the use of a compound of Formula I, or a
pharmaceutically acceptable salt or prodrug thereof as a 5-Lipooxygenase (5-
LO)
inhibitor for the treatment of a condition involving the 5-LO enzyme. 5-
Lipoxygenase (5-
LO) catalyzes the oxidative metabolism of arachidonic acid to 5-
hydroxyeicosatetraenoic acid (5-HETE), the initial reaction leading to
formation of
leukotrienes. Eicosanoids derived from arachidonic acid by the action of
lipoxygenases
or cyclooxygenases have been found to be involved in acute and chronic
inflammatory
diseases (i.e. asthma, multiple sclerosis, rheumatoid arthritis, ischemia,
edema) as well
in neurodegeneration (Alzheimer's disease), aging and various steps of
carcinogenesis,
including tumor promotion, progression and metastasis. Soberman et al (2003),
J. Clin.
Invest., vol 111, no 8, 1107-1113. In one embodiment, the compound for use in
the
treatment of a condition involving the 5-LO enzyme is a compound selected from
Compounds 1 to 98. In another embodiment, the compound for use as in the
treatment
of a condition involving the 5-LO enzyme is a
7
CA 02511750 2006-05-04
3005-31 CA
compound of Formula I with the proviso that the compound of Formula I is not
Compound 1, or alternatively is not Compound 2, or alternatively is neither
Compound
1 nor 2.
Leukotriene, Cysteinyi (CysLT~) receptor is involved in inflammation and
CysLT~-
selective antagonists are used as treatment for bronchial asthma. CysLT~ and 5-
LO
were known to be upregulated in colon cancer. See, for example, Lynch et al
(1999),
Nature, 399(57387), 789; Nielsen et al (2003), Adv. Exp. Med. Biol., vol 525,
201-4. In
one embodiment, the invention provides a compound of Formula I for treatment
of a
condition involving the Leukotriene, Cysteinyl (CysLT~) receptor. In one
embodiment,
the compound is a compound selected from Compounds 1 to 98. In another
embodiment, the compound for use in the treatment of a condition involving
CysLT~
receptor is a compound of Formula I with the proviso that the compound of
Formula I is
not Compound 1, or alternatively is not Compound 2, or alternatively is
neither
Compound 1 nor 2.
Cyclooxygenase-2 (COX-2) enzyme is known to be produced only in response to
injury or infection. It produces prostaglandins involved in inflammation and
the immune
response. Elevated levels of COX-2 in the body have been linked to cancer.
See, for
example, Wikstrom et al (2003), Biochem. Biophys. Res. Commun, vol 302, no 2,
330-
335. In one embodiment, the invention provides a compound of Formula I for
treatment
of a condition involving the COX-2 enzyme. In one embodiment, the compound is
a
compound selected from Compounds 1 to 98. In another embodiment, the compound
for use as in the treatment of a condition involving the COX-2 enzyme is a
compound of
Formula I with the proviso that the compound of Formula I is not Compound 1,
or
alternatively is not Compound 2, or alternatively is neither Compound 1 nor 2.
AcyICoA-Cholesterol Acyltransferase (ACAT) is known to convert cholesterol to
cholesteryl esters and is involved in the development of artheriosclerosis.
See, for
example, Kusunoki et al (2001), Circulation, 2604-2609. In one embodiment, the
invention provides a compound of Formula I for treatment of a condition
involving
AcyICoA-Cholesterol Acyltransferase. In one embodiment, the compound is a
compound selected from Compounds 1 to 98. In another embodiment, the compound
for use as in the treatment of a condition involving AcyICoA-Cholesterol
Acyltransferase
is a compound of Formula I with the proviso that
8
CA 02511750 2005-07-21
3005-31 CA
the compound of Formula I is not Compound 1, or alternatively is not Compound
2, or
alternatively is neither Compound 1 nor 2.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1: shows the in vitro anti-inflammatory activity of Compound 1. Graph
shows
percent inhibition of 5-lipoxygenase activity plotted against the Log pM
concentration of
Compound 1 and NDGA. Graph shows the ECSO of Compound 1 to be 0.93pM.
Figure 2: shows inhibition of tumor growth resulting from administration of 10
to 30
mg/kg of Compound 1 to C6 glioblastoma-bearing mice one day after tumor cell
inoculation.
Figure 3: shows inhibition of tumor growth resulting from administration of 20-
30 mg/kg
of Compound 1 to glioblastoma-bearing mice ten days after tumor cell
inoculation.
Figure 4: shows micrographs of tumor sections from mice bearing glioblastoma
tumors
and treated with saline or Compound 1. The cell density of tumor treated with
ECO-
4601 appears decreased and nuclei from tumor cells are larger and pycnotic
suggesting a cytotoxic effect.
Figure 5: inhibition of tumor growth resulting from administration of 20 to 75
mg/kg of
Compound 2 to C6 glioblastoma-bearing mice from day 11 to day 20 of treatment.
Figure 6: shows the survival of mice xenografted with orthotopic C6 glioma
tumor,
treated daily with vehicle (squares) or Compound 1 (circles). Daily treatment
with the
compound of Formula I led to an increase survival of 7 days resulting in a 29%
increase
in life span.
Figure 7: shows the antitumor efficacy of Compound 1 against human prostate
tumor
(PC3) xenografts in male Harlan nude mice.
Figure 8: shows the antitumor efficacy of Compound 1 against human prostate
tumor
(PC3) xenografts on individual male Harlan nude mice at day 22 of treatment.
Figure 9: shows the antitumor efficacy of Compound 1 against human breast
tumor
(MDA-MB-231 ) xenografts in female Harlan nude mice.
Figure 10: shows the antitumor efficacy of Compound 1 against human breast
tumor
(MDA-MB-231 ) xenografts on individual female Harlan nude mice at day 21 of
treatment.
9
CA 02511750 2005-07-21
3005-31 CA
Figure 11: shows the mean (~SD) plasma concentrations of Compound 1 in Swiss
mice
following 30 mg/kg intravenous (iv), intraperitoneal (ip), subcutaneous (sc)
and oral (po)
administrations.
Figure 12: shows the mean (~SD) plasma concentrations of Compounds 1 and 2 in
CD-
1 mice following 30 mg/kg intravenous (iv) and intraperitoneal (ip)
administrations.
Figure 13: shows the mean concentration of Compound 1 in various tissues, 30
minutes after 30mg/kg intravenous (iv), intraperitoneal (ip) and subcutaneous
(sc)
administrations.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to novel dibenzodiazepinone analogues herein
referred as the compounds of Formula I, which include Compound 1 and
derivatives of
Compound 1. Compound 1 is isolated from strains of actinomycetes,
Micromonospora
sp. 046-EC011 and [S01]046. These organisms were deposited on March 7, 2003,
and December 23, 2003, respectively, with the International Depositary
Authority of
Canada (IDAC), Bureau of Microbiology, Health Canada, 1015 Arlington Street,
Winnipeg, Manitoba, Canada R3E 3R2, under Accession Nos. IDAC 070303-01 and
I DAC 231203-01, respectively.
The invention further relates to pharmaceutically acceptable salts, solvates
and
prodrugs of dibenzodiazepinone compounds, and to methods for obtaining such
compounds. One method of obtaining the compound is by cultivating
Micromonospora
sp. strain 046-EC011 or [S01]046, or a mutant or a variant thereof, under
suitable
Micromonospora culture conditions, preferably using the fermentation protocol
described hereinbelow, and by optional chemical modification of the compound
obtained by isolation from the fermentation procedure.
The invention also relates to a method for producing novel dibenzodiazepinone
compounds of Formula II, by chemical modification of the farnesyl
dibenzodiazepinone
obtained from fermentation and isolation.
The present invention also relates to pharmaceutical compositions comprising a
compound of Formula I and its pharmaceutically acceptable salts, solvates and
derivatives. Compounds of Formula I are useful as pharmaceuticals, in
particular for
CA 02511750 2005-07-21
3005-31 CA
use as an inhibitor of cancer cell growth, and mammalian lipoxygenase.
The following detailed description discloses how to make and use the
compounds of Formula I and compositions containing these compounds to inhibit
tumor
growth and/or specific disease pathways.
Accordingly, certain aspects of the present invention relate to pharmaceutical
compositions comprising the dibenzodiazepinone compounds of the present
invention
together with a pharmaceutically acceptable carrier, and methods of using the
pharmaceutical compositions to treat diseases, including cancer, and chronic
and acute
inflammation, autoimmune diseases, and neurodegenerative diseases.
Definitions
All technical and scientific terms used herein have the same meaning as
commonly understood by one of ordinary skill in the art to which this
invention belongs.
For convenience, the meaning of certain terms and phrases used in the
specification,
examples, and appended claims, are provided below.
As used herein, the term "farnesyl dibenzodiazepinone" refers to Compound 1,
namely 10-farnesyl-4,6,8-trihydroxy-5,10-dihydrodibenzo[b,e][1,4]diazepin-11-
one, also
referred to as ECO-4601.
As used herein, the terms "compound(s) of the invention", "dibenzodiazepinone
analogues)", "dibenzodiazepinone compounds)", and equivalent expressions refer
to a
class of dibenzodiazepinone compounds containing a farnesyl moiety or being
derived
from a farnesyl moiety, and pharmaceutically acceptable salts, solvates and
prod rugs
thereof. The term includes Compound 1 and derivatives produced by chemical
modification. The term includes a compound of Formula I or II, a compound
selected
from Compounds 1 to 98, or the exemplified compounds of the present invention,
Compounds 1 to 12, 14, 17, 18, 46, 63, 64, 67, 77, 78, 80, 82 to 85, 87, 89,
92, and 95
to 98, or a pharmaceutically acceptable salt, solvate or prod rug of any of
the above
compounds. As used herein, the term "dibenzodiazepinone analogues" includes
compounds of this class that can be used as intermediates in chemical
syntheses and
variants containing different isotopes than the most abundant isotope of an
atom (e.g,
D replacing H,'3C replacing'2C, etc). The compounds of the invention are also
11
CA 02511750 2005-07-21
3005-31 CA
sometimes referred as "active ingredients".
As used herein, the term "chemical modification" refers to one or more steps
of
modifying a dibenzodiazepinone compound, referred to as "starting material",
by
chemical synthesis. Preferred compounds for use as starting materials in a
chemical
modification process are Compounds 1 to 98, more preferably Compounds 1, 2,
and
46. Examples of chemical modification steps include N-alkylations, N-
acylations, O-
alkylations, O-acylations, aromatic halogenation, and modifications of the
double bonds
of the farnesyl side chain including, hydrogenation, electrophilic additions
(e.g.,
epoxidation, dihydroxylation, hydration, hydroalkoxylation, hydroamidation,
and the
like), and double bond cleavage like ozonolysis, and reduction of ozonolysis
product.
Farnesyl side chain modification reaction can be partial (one or two double
bonds
modified) or complete (three double bonds modified). Chemical modification
steps are
also defined in the Schemes of Section IIIB, and exemplified in Examples 4 to
9 and
Example 15.
The term "ether" refers to a dibenzodiazepinone analogue obtained by the
replacement of a hydrogen atom from an alcohol by an R' replacement group by
an O-
alkylation reaction as defined in Scheme 1 (a) below. More particularly, the
term ether
encompasses ethers of the alcohols in positions 4, 6, and 8 (see Exampes 3-9
for atom
numbering).
The term "ester" refers to a dibenzodiazepinone analogue obtained by the
replacement of a hydrogen atom from an alcohol by a C(O)R" replacement group
by an
O-acylation reaction as defined in Scheme 1 (b) below. The term ester also
encompasses ester equivalents including, without limitation, carbonate,
carbamate, and
the like. More particularly, the term "ester" encompasses esters of the
alcohols in
positions 4, 6, and 8 (see Exampes 3-9 for atom numbering).
The term "N-alkylated derivative" refers to a dibenzodiazepinone analogue
obtained by the replacement of a hydrogen atom of an amine by an R replacement
group by an N-alkylation reaction as defined in Scheme 2(a) below. More
particularly,
the term "N-alkylated derivative" encompasses derivatives of the amine in
position 5
(see Exampes 3-9 for atom numbering).
The term "N-acylated derivative" refers to a dibenzodiazepinone analogue
obtained by the replacement of a hydrogen atom of an amine by a C(O)R
replacement
12
CA 02511750 2005-07-21
3005-31 CA
group by an N-acylation reaction as defined in Scheme 2(b) below. The term N-
acylated
derivative further encompasses amide equivalents such as, without limitation,
urea,
guanidine, and the like. More particularly, the term "N-acylated derivative"
encompasses derivatives of the amine in position 5 (see Exampes 3-9 for atom
numbering).
The term "receptor" refers to a protein located on the surface or inside a
cell that
may interact with a different molecule, known as a ligand, to initiate or
inhibit a
biological response.
As used herein, the term "ligand" refers to a molecule or compound that has
the
capacity to bind to a receptor and modulate its activity.
As used herein, the terms "binder", "receptor binder" or "binding agent"
refers to
a compound of the invention acting as a ligand. The binding agent can act as
an
agonist, or an antagonist of the receptor. An agonist is a drug which binds to
a receptor
and activates it, producing a pharmacological response (e.g. contraction,
relaxation,
secretion, enzyme activation, etc.). An antagonist is a drug which counteracts
or blocks
the effects of an agonist, or a natural ligand. Antagonism can be competitive
and
reversible (i.e. it binds reversibly to a region of the receptor in
competition with the
agonist.) or competitive and irreversible (i.e. antagonist binds covalently to
the receptor,
and no amount of agonist can overcome the inhibition). Other types of
antagonism are
non-competitive antagonism where the antagonist binds to an allosteric site on
the
receptor or an associated ion channel.
As used herein, the term "enzyme inhibitor" or "inhibitor" refers to a
chemical that
disables an enzyme and inhibits it from performing its normal function.
As used herein, abbreviations have their common meaning. Unless otherwise
noted, the abbreviations "Ac", "Me", "Et", "Pr", "i-Pr", "Bu", and "Ph",
respectively refer to
acetyl, methyl, ethyl, propyl (n- or iso-propyl), iso-propyl, butyl (n-, iso-,
sec- or tert-
butyl) and phenyl. Abbreviations in the specification correspond to units of
measure,
techniques, properties or compounds as follows: "RT" means retention time,
"min"
means minutes, "h" means hour(s), "pL" means microliter(s), "mL" means
milliliter(s),
°'mM" means millimolar, "M" means molar, "mmole" means millimole(s),
"eq" means
molar equivalent(s). "High Pressure Liquid Chromatography" and "High
Performance
Liquid Chromatography" are abbreviated HPLC.
13
CA 02511750 2005-07-21
3005-31 CA
The term "alkyl" refers to linear, branched or cyclic, saturated hydrocarbon
groups. Examples of alkyl groups include, without limitation, methyl, ethyl, n-
propyl,
isopropyl, n-butyl, pentyl, hexyl, heptyl, cyclopentyl, cyclohexyl,
cyclohexylmethyl, and
the like. Alkyl groups may optionally be substituted with substituents
selected from
acyl, amino, acylamino, acyloxy, carboalkoxy, carboxy, carboxyamido, cyano,
halo,
hydroxyl, nitro, thio, alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl,
aryl, heteroaryl,
alkoxy, aryloxy, sulfinyl, sulfonyl, oxo, guanidino and formyl.
The term "C~_nalkyl", wherein n is an integer from 2 to 12, refers to an alkyl
group
having from 1 to the indicated "n" number of carbons. The C~_"alkyl can be
cyclic or a
straight or branched chain.
The term "alkenyl" refers to linear, branched or cyclic unsaturated
hydrocarbon
groups containing, from one to six carbon-carbon double bonds. Examples of
alkenyl
groups include, without limitation, vinyl, 1-propene-2-yl, 1-butene-4-yl, 2-
butene-4-yl, 1-
pentene-5-yl and the like. Alkenyl groups may optionally be substituted with
substituents selected from acyl, amino, acylamino, acyloxy, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, nitro, thio, alkyl, alkenyl, alkynyl,
cycloalkyl,
heterocycloalkyl, aryl, heteroaryl, alkoxy, aryloxy, sulfinyl, sulfonyl,
formyl, oxo and
guanidino. The double bond portions) of the unsaturated hydrocarbon chain may
be
either in the cis or trans configuration.
The term "C2_~alkenyl", wherein n is an integer from 3 to 12, refers to an
alkenyl
group having from 2 to the indicated "n" number of carbons. The C2_nalkenyl
can be
cyclic or a straight or branched chain.
The term "alkynyl" refers to linear, branched or cyclic unsaturated
hydrocarbon
groups containing at least one carbon-carbon triple bond. Examples of alkynyl
groups
include, without limitation, ethynyl, 1-propyne-3-yl, 1-butyne-4-yl, 2-butyne-
4-yl, 1-
pentyne-5-yl and the like. Alkynyl groups may optionally be substituted with
substituents selected from acyl, amino, acylamino, acyloxy, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, nitro, thio, alkyl, alkenyl, alkynyl,
cycloalkyl,
heterocycloalkyl, aryl, heteroaryl, alkoxy, aryloxy, sulfinyl, sulfonyl,
formyl, oxo and
guanidine.
14
CA 02511750 2005-07-21
3005-31 CA
The term "C2_nalkynyl", wherein n is an integer from 3 to 12, refers to an
alkynyl
group having from 2 to the indicated "n" number of carbons. The C2_~alkynyl
can be
cyclic or a straight or branched chain.
The term "cycloalkyl" or "cycloalkyl ring" refers to an alkyl group, as
defined
above, further comprising a saturated or partially unsaturated carbocyclic
ring in a
single or fused carbocyclic ring system having from three to fifteen ring
members.
Examples of cycloalkyl groups include, without limitation, cyclopropyl,
cyclobutyl,
cyclopentyl, cyclopenten-1-yl, cyclopenten-2-yl, cyclopenten-3-yl, cyclohexyl,
cyclohexen-1-yl, cyclohexen-2-yl, cyclohexen-3-yl, cycloheptyl,
bicyclo[4,3,0]nonanyl,
norbornyl, and the like. Cycloalkyl groups may optionally be substituted with
substituents selected from acyl, amino, acylamino, acyloxy, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, vitro, thio, alkyl, alkenyl, alkynyl,
cycloalkyl,
heterocycloalkyl, aryl, heteroaryl, alkoxy, aryloxy, sulfinyl, sulfonyl and
formyl.
The term "C3_ncycloalkyl", wherein n is an integer from 4 to 15, refers to a
cycloalkyl ring or ring system or having from 3 to the indicated "n" number of
carbons.
The term "heterocycloalkyl", "heterocyclic" or "heterocycloalkyl ring" refers
to a
cycloalkyl group, as defined above, further comprising one to four hetero
atoms (e.g. N,
O, S, P) or hetero groups (e.g. NH, NR", P02, SO, S02) in a single or fused
heterocyclic
ring system having from three to fifteen ring members (e.g. tetrahydrofuranyl
has five
ring members, including one oxygen atom). Examples of a heterocycloalkyl,
heterocyclic or heterocycloalkyl ring include, without limitation,
pyrrolidino,
tetrahydrofuranyl, tetrahydrodithienyl, tetrahydropyranyl,
tetrahydrothiopyranyl,
piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl,
oxetanyl,
thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl,
thiazepinyl,
1,2,3,6-tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-
pyranyl, 4H-pyranyl,
dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl,
dihydrothienyl,
dihydrofuranyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, 3-
azabicyclo[3,1,0]hexanyl, 3-
azabicyclo[4,1,0]heptanyl, 3H-indolyl, and quinolizinyl. The foregoing
heterocycloalkyl
groups, as derived from the compounds listed above, may be C-attached or N-
attached
where such is possible. Heterocycloalkyl, heterocyclic or heterocycloalkyl
ring may
optionally be substituted with substituents selected from acyl, amino,
acylamino,
acyloxy, oxo, thiocarbonyl, imino, carboalkoxy, carboxy, carboxyamido, cyano,
halo,
hydroxyl, vitro, thio, alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl,
aryl, heteroaryl,
CA 02511750 2005-07-21
3005-31 CA
alkoxy, aryloxy, sulfinyl, sulfonyl and formyl.
The term "C3_~heterocycloalkyl", wherein n is an integer from 4 to 15, refers
to an
heterocycloalkyl group having from 3 to the indicated "n" number of atoms in
the cycle
and at least one hetero group as defined above.
The terms "halo" or "halogen" refers to bromine, chlorine, fluorine or iodine
substituents.
The term "aryl" or "aryl ring" refers to common aromatic groups having "4n+2"
electrons, wherein n is an integer from 1 to 3, in a conjugated monocyclic or
polycyclic
system and having from five to fourteen ring atoms. Aryl may be directly
attached, or
connected via a C~_3alkyl group (also referred to as aralkyl). Examples of
aryl include,
without limitation, phenyl, benzyl, phenethyl, 1-phenylethyl, tolyl, naphthyl,
biphenyl,
terphenyl, and the like. Aryl groups may optionally be substituted with one or
more
substituent group selected from acyl, amino, acylamino, acyloxy, azido,
alkythio,
carboalkoxy, carboxy, carboxyamido, cyano, halo, hydroxyl, nitro, thio, alkyl,
alkenyl,
alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, alkoxy, aryloxy,
sulfinyl, sulfonyl and
formyl.
The term "C5_"aryl", wherein n is an integer from 5 to 14, refers to an aryl
group
having from 5 to the indicated "n" number of atoms, including carbon,
nitrogen, oxygen
and sulfur. The C5_naryl can be mono or polycyclic.
The term "heteroaryl" or "heteroaryl ring" refers to an aryl ring, as defined
above,
further containing one to four heteroatoms selected from oxygen, nitrogen,
sulphur or
phosphorus. Examples of heteroaryl include, without limitation, pyridyl,
imidazolyl,
pyrimidinyl, pyrazolyl, triazolyl, tetrazolyl, furyl, thienyl, isooxazolyl,
thiazolyl, oxazolyl,
isothiazolyl, pyrrollyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl,
benzofuranyl,
cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl,
isoindolyl, pteridinyl,
purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl,
benzothiophenyl,
benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinyl, and
furopyridinyl
groups. Heteroaryl may optionally be substituted with one or more substituent
group
selected from acyl, amino, acylamino, acyloxy, azido, alkythio, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, nitro, thio, alkyl, alkenyl, alkynyl,
cycloalkyl,
heterocyclyl, aryl, heteroaryl, alkoxy, aryloxy, sulfinyl, sulfonyl and
formyl. Heteroaryl
may be directly attached, or connected via a C,_3alkyl group (also referred to
as
16
CA 02511750 2005-07-21
3005-31 CA
heteroaralkyl). The foregoing heteroaryl groups, as derived from the compounds
listed
above, may be C-attached or N-attached where such is possible.
The term "C5_nheteroaryl", wherein n is an integer from 5 to 14, refers to an
heteroaryl group having from 5 to the indicated "n" number of atoms, including
carbon,
nitrogen, oxygen and sulphur atoms. The C5_nheteroaryl can be mono or
polycyclic.
The term "amino acid" refers to an organic acid containing an amino group. The
term includes both naturally occurring and synthetic amino acids; therefore,
the amino
group can be but is not required to be, attached to the carbon next to the
acid. A C-
coupled amino acid substituent is attached to the heteroatom (nitrogen or
oxygen) of
the parent molecule via its carboxylic acid function. C-coupled amino acid
forms an
ester with the parent molecule when the heteroatom is oxygen, and an amide
when the
heteroatom is nitrogen. Examples of amino acids include, without limitation,
alanine,
valine, leucine, isoleucine, proline, phenylalanine, tryptophan, methionine,
glycine,
serine, threonine, cysteine, asparagine, glutamine, tyrosine, histidine,
lysine, arginine,
aspartic acid, glutamic acid, desmosine, ornithine, 2-aminobutyric acid,
cyclohexylalanine, dimethylglycine, phenylglycine, norvaline, norleucine,
hydroxylysine,
allo-hydroxylysine, hydroxyproline, isodesmosine, allo-isoleucine,
ethylglycine, beta-
alanine, aminoadipic acid, aminobutyric acid, ethyl asparagine, and N-methyl
amino
acids. Amino acids can be pure L or D isomers or mixtures of L and D isomers.
The compounds of the present invention can possess one or more asymmetric
carbon atoms and can exist as optical isomers forming mixtures of racemic or
non-
racemic compounds. The compounds of the present invention are useful as single
isomers or as a mixture of stereochemical isomeric forms. Diastereoisomers,
i.e.,
nonsuperimposable stereochemical isomers, can be separated by conventional
means
such as chromatography, distillation, crystallization or sublimation. The
optical isomers
can be obtained by resolution of the racemic mixtures according to
conventional
processes, including chiral chromatography (e.g. HPLC), immunoassay
techniques, or
the use of covalently (e.g. Mosher's esters) or non-covalently (e.g. chiral
salts) bound
chiral reagents to respectively form a diastereomeric ester or salt, which can
be further
separated by conventional methods, such as chromatography, distillation,
crystallization
or sublimation. The chiral ester or salt is then cleaved or exchanged by
conventional
means, to recover the desired isomer(s).
17
CA 02511750 2006-05-04
3005-31 CA
The invention encompasses isolated or purified compounds. An "isolated" or
"purified" compound refers to a compound which represents at least 10%, 20%,
50%,
80% or 90% of the mixture by weight, provided that the mixture comprising the
compound of the invention has demonstrable (i.e. statistically significant)
biological
activity including cytostatic, cytotoxic, enzyme inhibitory or receptor
binding action when
tested in conventional biological assays known to a person skilled in the art.
The term "pharmaceutically acceptable salt" refers to nontoxic salts
synthesized
from a compound which contains a basic or acidic moiety by conventional
chemical
methods. Generally, such salts can be prepared by reacting the free acid or
base forms
of these compounds with a stoichiometric amount of the appropriate base or
acid in
water or in an organic solvent, or in a mixture of the two; generally,
nonaqueous media
like ether, ethyl acetate, methanol, ethanol, isopropanol, or acetonitrile are
preferred.
Another method for the preparation of salts is by the use of ion exchange
resins. The
term "pharmaceutically acceptable salt" includes both acid addition salts and
base
addition salts, either of the parent compound or of a prodrug or solvate
thereof. The
nature of the salt is not critical, provided that it is pharmaceutically
acceptable.
Exemplary acids used in acid addition salts include, without limitation,
hydrochloric,
hydrobromic, hydroiodic, nitric, carbonic, sulfuric, sulfonic, phosphoric,
formic, acetic,
citric, tartaric, succinic, oxalic, malic, glutamic, propionic, glycolic,
gluconic, malefic,
embonic (pamoic), methanesulfonic, ethanesulfonic, 2-hydroxyethanesulfonic,
pantothenic, benzenesulfonic, toluenesulfonic, sulfanilic, mesylic,
cyclohexylaminosulfonic, stearic, algenic, ~i-hydroxybutyric, malonic,
galactaric,
galacturonic acid and the like. Suitable pharmaceutically acceptable base
addition salts
include, without limitation, metallic salts made from aluminium, calcium,
lithium,
magnesium, potassium, sodium and zinc or organic salts, such as those made
from
N, N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamine, N-methylglucamine, lysine, procaine and the like. Additional
examples of pharmaceutically acceptable salts are listed in Berge et al
(1977), Journal
of Pharmaceutical Sciences, vol 66, no 1, pp 1-19.
The term "solvate" refers to a physical association of a compound of this
invention with one or more solvent molecules, whether organic or inorganic.
This
physical association includes hydrogen bonding. In certain instances the
solvate will be
18
CA 02511750 2005-07-21
3005-31 CA
capable of isolation, for example when one or more solvent molecules are
incorporated
in the crystal lattice of the crystalline solid. "Solvate" encompasses both
solution-phase
and isolable solvates. Exemplary solvates include hydrates, ethanolates,
methanolates,
hemiethanolates, and the like.
The term "pharmaceutically acceptable prod rug" means any pharmaceutically
acceptable ester, salt of an ester or any other derivative of a compound of
this
invention, which upon administration to a recipient, is capable of providing,
either
directly or indirectly, a compound of this invention or a biologically active
metabolite or
residue thereof. Particularly favored salts or prod rugs are those with
improved
properties, such as solubility, efficacy, or bioavailability of the compounds
of this
invention when such compounds are administered to a mammal (e.g., by allowing
an
orally administered compound to be more readily absorbed into the blood) or
which
enhance delivery of the parent compound to a biological compartment (e.g., the
brain or
lymphatic system) relative to the parent species. As used herein, a prodrug is
a drug
having one or more functional groups covalently bound to a carrier wherein
metabolic or
chemical release of the drug occurs in vivo when the drug is administered to a
mammalian subject. Pharmaceutically acceptable prodrugs of the compounds of
this
invention include derivatives of hydroxyl groups such as, without limitation,
acyloxymethyl, acyloxyethyl and acylthioethyl ethers, esters, amino acid
esters,
phosphate esters, sulfonate and sulfate esters, and metal salts, and the like.
II. Compounds of the invention
In one aspect, the invention relates to novel dibenzodiazepinone analogues,
referred to herein as the compounds of the invention, and to pharmaceutically
acceptable salts, solvates and prod rugs thereof.
The compounds of the invention may be characterized as Compound 1 and
derivatives of Compound 1, by chemical modifications as defined herein.
Compounds 2
to 12, 14, 17, 18, 46, 63, 64, 67, 77, 78, 80, 82 to 85, 87, 89, 92, and 95 to
98 may be
characterized by any one of their physicochemical and spectral properties,
such as
mass and NMR, detailed in Example 4 through Example 9.
In another aspect, the invention relates to dibenzodiazepinone analogues,
represented by Formula I:
19
CA 02511750 2005-07-21
3005-31 CA
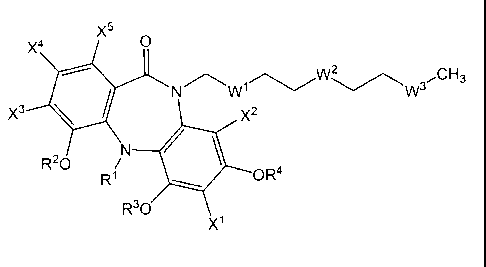
X5
~w~ 3 CH3
w
x~
Formula I
wherein,
W~, W2 and W3 are each independently selected from
CH3 CH3 CH3
H I I H
' - i - i -~- ; _~-H-C-~_ ; '~- \ ~ -~- ; Or
R5 R6 O
the chain from the tricycle terminates at W3, W2 or W' with W3, W2 or W'
respectively
being either -CH=O, -CH(OC~_6alkyl)2, -CH20H, -CH20C~_6alkyl or C(O)OR';
R' is selected from H, C~_~oalkyl, C2_~oalkenyl, C2_~oalkynyl, C6_~oaryl, C5_
~oheteroaryl, C3_~ocYcloalkyl, C3_~oheterocycloalkyl, C(O)H, C(O)C~_~oalkyl,
C(O)C2_
~oalkenyl, C(O)C2_~oalkynyl, C(O)C6_~oaryl, C(O)C5_~oheteroaryl,
C(O)C3_~ocycloalkyl;
C(O)C3_~oheterocycloalkyl or a C-coupled amino acid;
R2, R3, and R4 are each independently selected from H, C~_~oalkyl,
C2_~oalkenyl,
C2_~oalkynyl, C6_~oaryl, C5_~oheteroaryl, C3_~ocYcloalkyl,
C3_~oheterocycloalkyl, C(O)H,
C(O)C~_~oalkyl, C(O)C2_~oalkenyl, C(O)C2_~oalkynyl, C(O)C6_~oaryl,
C(O)C5_~oheteroaryl,
C(O)C3_~ocycloalkyl; C(O)C3_~oheterocycloalkyl or a C-coupled amino acid;
R5 and R6 are each independently selected from H, OH, OC~_6alkyl, NH2, NHC~_
6alkyl, N(C~_6alkyl)2, NHC(O)C~_6alkyl;
R' is selected from H, C~_~oalkyl, C2_~oalkenyl, C2_~oalkynyl, C6_~oaryl, C5_
,oheteroaryl, C3_~ocycloalkyl and C3_,oheterocycloalkyl;
X', X2, X3, X4 and X5 are each H; or
one of X~, X2, X3, X4 or X5 is halogen and the remaining ones are H; and
wherein, when any of R', R2, R3, R4, R5, R6 and R' comprises an alkyl,
alkenyl,
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl group, then the
alkyl, alkenyl,
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl group is optionally
substituted
with substituents selected from acyl, amino, acylamino, acyloxy, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, nitro, thio, C~_6alkyl, C2_~alkenyl,
C2_~alkynyl, C3_
~ocycloalkyl, C3_~oheterocycloalkyl, C6_~oaryl, C5_~oheteroaryl, alkoxy,
aryloxy, sulfinyl,
CA 02511750 2005-07-21
3005-31 CA
sulfonyl, oxo, guanidino and formyl;
and an ester, ether, N-alkylated or N-acylated derivative, or a
pharmaceutically
acceptable salt, solvate or prod rug thereof.
In further aspect, the invention relates to dibenzodiazepinone analogues,
represented by Formula II:
X5
w~ 3 CH3
w
x3
Formula II
wherein,
W', W2 and W3 are each independently selected from
CH3 CH3 CH3
H I I H
_ - i - i -~_ ; _~-H-C-~_ ; -~- \ ~ -~_ ; or
R5 R6 O
the chain from the tricycle terminates at W3, W2 or W' with W3, W2 or W'
respectively
being either -CH=O, -CH(OC~_6alkyl)2, -CH20H, -CH20C~_6alkyl or C(O)OR';
R' is selected from H, C~_~oalkyl, C2_~oalkenyl, C2_~oalkynyl, C6_~oaryl, C5_
~oheteroaryl, C3_~ocYcloalkyl, C3_~oheterocycloalkyl, C(O)H, C(O)C~_~oalkyl,
C(O)C2_
~oalkenyl, C(O)C2_~oalkynyl, C(O)C6_~oaryl, C(O)C5_~oheteroaryl,
C(O)C3_~ocYcloalkyl;
C(O)C3_~oheterocycloalkyl or a C-coupled amino acid;
R2, R3, and R4 are each independently selected from H, C~_~oalkyl,
C2_~oalkenyl,
C2_~oalkynyl, C6_~oaryl, C5_~oheteroaryl, C3_~ocycloalkyl,
C3_~oheterocycloalkyl, C(O)H,
C(O)C~_~oalkyl, C(O)C2_~oalkenyl, C(O)C2_~oalkynyl, C(O)C6_~oaryl,
C(O)C5_~oheteroaryl,
C(O)C3_~ocycloalkyl; C(O)C3_~oheterocycloalkyl or a C-coupled amino acid;
R5 and R6 are each independently selected from H, OH, OC~_6alkyl, NH2, NHC~_
6alkyl, N(C~_6alkyl)2, NHC(O)C~_6alkyl;
R' is selected from H, C~_~oalkyl, C2_~oalkenyl, C2_~oalkynyl, C6_~oaryl, C5_
~oheteroaryl, C3_~ocycloalkyl and C3_~oheterocycloalkyl;
X', X2, X3, X4 and X5 are each H; or
one of X', X2, X3, X4 or X5 is halogen and the remaining ones are H; and
21
CA 02511750 2005-07-21
3005-31 CA
wherein, when any of R', R2, R3, R4, R5, R6 and R' comprises an alkyl,
alkenyl,
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl group, then the
alkyl, alkenyl,
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl group is optionally
substituted
with substituents selected from acyl, amino, acylamino, acyloxy, carboalkoxy,
carboxy,
carboxyamido, cyano, halo, hydroxyl, nitro, thio, C~_6alkyl, C2_~alkenyl,
C2_~alkynyl, C3_
~ocycloalkyl, C3_~oheterocycloalkyl, C6_~oaryl, C5_~oheteroaryl, alkoxy,
aryloxy, sulfinyl,
sulfonyl, oxo, guanidino and formyl;
with the proviso that when W', W2 and W3 are all -CH=C(CH3)-, and R2, R3 and
R4 are all H, then R' is not H;
and an ester, ether, N-alkylated or N-acylated derivative, or a
pharmaceutically
acceptable salt, solvate or prodrug thereof.
In one embodiment, R' is H, and all other groups are as previously disclosed.
In
another embodiment, R' is -CH3, and all other groups are as previously
disclosed. In
another embodiment, R' is C~_~oalkyl, and all other groups are as previously
disclosed.
In a subclass of this embodiment, the alkyl group is optionally substituted
with a
substituent selected from halo, fluoro, C6_~oaryl, and C5_~oheteroaryl. In
another
embodiment, R' is -C(O)C~_~oalkyl, and all other groups are as previously
disclosed. In
another embodiment, R2 is H, and all other groups are as previously disclosed.
In
another embodiment, R3 is H, and all other groups are as previously disclosed.
In
another embodiment, R4 is H, and all other groups are as previously disclosed.
In
another embodiment, R2, R3 and R4 are each H, and all other groups are as
previously
disclosed. In another embodiment, one of R2, R3 and R4 is CH3, the others
being each
H, and all other groups are as previously disclosed. In another embodiment,
two of R2,
R3 and R4 are CH3, the other being H, and all other groups are as previously
disclosed.
In another embodiment, R2, R3 and R4 are each CH3, and all other groups are as
previously disclosed. In another embodiment, R2, R3 and R4 are each H, and W'
is -
CH=C(CH3)-, and all other groups are as previously disclosed. In another
embodiment,
R2, R3 and R4 are each H, and W2 is -CH=C(CH3)-, and all other groups are as
previously disclosed. In another embodiment, R2, R3 and R4 are each H, and W3
is -
CH=C(CH3)-, and all other groups are as previously disclosed. In another
embodiment,
R' is H and R2, R3 and R4 are each H, and all other groups are as previously
disclosed.
In another embodiment, R' is H, each of W', W2, and W3 is -CH=C(CH3)-, and all
other
22
CA 02511750 2005-07-21
3005-31 CA
groups are as previously disclosed. In another embodiment, R~ is H, each of
W', W2,
and W3 is -CH2CH(CH3)-, and all other groups are as previously disclosed. In
another
embodiment, X' is Br, and each of X2, X3, X4 and X5 are H, and all other
groups are as
previously disclosed. In another embodiment, if each of W', W2 and W3 are -
CH=C(CH3)-, and each of R2, R3, and R4 are H, then R' is not H. In further
embodiment, if each of W', W2 and W3 are -CH=C(CH3)-, and each of R2, R3, and
R4
are H, then R' is not CH3. In further embodiment, if each of W~, W2 and W3 are
-
CH=C(CH3)-, and each of R2, R3, and R4 are H, then R' is neither H nor CH3.
The
invention encompasses all esters, ethers, N-alkylated or N-acylated
derivatives, and
pharmaceutically acceptable salts, solvates and prodrugs of the foregoing
compounds.
The following are exemplary compounds of the invention, such named
compounds are not intended to limit the scope of the invention in any way:
0 0
/ / /
~N
/ ~ ~ OH ~ /
.N I
OH ~ Y OH M
HO
Compound 1; Compound 2;
/ /
Compound 3; Compound 4;
0
/ ~ ~ OH
N
N / / /
OH H
Me0
Compound 5; Compound 6;
23
CA 02511750 2005-07-21
3005-31 CA
Compound 7; Compound 8;
0
NY
i i i i i
oA~
OH
Ac0
Compound 9; Compound 10;
i i i i
OAc
Compound 11; Compound 12;
Compound 13; Compound 14;
i i i i
Compound 15; Compound 16;
Compound 17; Compound 18;
24
CA 02511750 2005-07-21
3005-31 CA
/ /
0 0
Compound 19; Compound 20;
0 0
Compound 21; Compound 22;
0 0
N /
O
/ I / ~ ~ OH
OH ~ OH H
HO
Compound 23; Compound 24;
o
~N
O I ~ O O
/ ~ ~ off
N
OH
HO
Compound 25; Compound 26;
0 0
~N ~ ~N
O O O O
I / ~ ~ OH ~ / ~ ~ OH
OH H Y OH H
HO HO
Compound 27; Compound 28;
/ /
0
Compound 29; Compound 30;
CA 02511750 2005-07-21
3005-31 CA
0
N
O
OH
N
OH
HO
Compound 31; Compound 32;
~ ....i.
Compound 33; Compound 34;
Compound 35; Compound 36;
i i i i
Compound 37; Compound 38;
i i
Compound 39; Compound 40;
0 0
i i
~N
OH
OH H Y pH
HO
Compound 41; Compound 42;
26
CA 02511750 2005-07-21
3005-31 CA
i ":....i. a. . . ..
Compound 43; Compound 44;
0
W
H / N
OH H
Compound 45; Compound 46;
/ ,o / off
H
Compound 47; Compound 48;
~o
Compound 49; Compound 50;
0
OOH
~N
OH
NY
OH H
HO
Compound 51; Compound 52;
0
N / /
OH
OH
/ ~ OH
N
OH
HO
Compound 53; Compound 54;
27
CA 02511750 2005-07-21
3005-31 CA
o '
N / /
OH
/ ~ OH
OH
N
OH H
HO
Compound 55; Compound 56;
Compound 57; Compound 58;
/ /
OH ~ OH
OH OH
H
Compound 59; Compound 60;
0
~N
/ ~ ~ OH
N
/ / / / /
OH
HO
Compound 61; Compound 62;
Compound 63; Compound 64;
0
N / / /
/ /
/ ~ ~ off
N
OH
HO
I
Compound 65; Compound 66;
28
CA 02511750 2005-07-21
3005-31 CA
i
H
Compound 67; Compound 68;
i i
i i
Compound 69; Compound 70;
i i
Compound 71; Compound 72;
i i
Compound 73; Compound 74;
° i i
N
OH
N
OH
HO
Ph
Compound 75; Compound 76;
29
CA 02511750 2005-07-21
3005-31 CA
Compound 77; Compound 78;
i i
OCH3 OCH3
Compound 79; Compound 80;
' o
N
OCH3 OCH3 ~ ~ OCH3
OH
H
OH
NY
HO
Compound 81; Compound 82;
i
OCH3 OCH3 OCH3
Compound 83; Compound 84;
i
OCH3
Compound 85; Compound 86;
0
N
OCH3 OCH3 ~ ~ OH
H ~ ~ ~ OH
N
H
OH
HO
Compound 87; Compound 88;
CA 02511750 2005-07-21
3005-31 CA
0
/
N
H
OH
Compound 89; Compound 90;
0
/
NHAc
N
H
OH
Compound 91; Compound 92;
/ OMe
NHAc NHAc
OMe
H
Compound 93; Compound 94;
/OMe
IYOMe
Compound 95; Compound 96;
0
/ /
/
N
OH
Compound 97; and Compound 98;
and an ester, ether, N-alkylated or N-acylated derivative, or a
pharmaceutically
acceptable salt, solvate of prod rug of any one of Compounds 1 to 98.
The invention further provides ethers, esters, N-acylated and N-alkylated
derivatives of any of the foregoing compounds.
Certain embodiments expressly exclude one or more of the compounds of
Formula I. In one embodiment, Compound 1 is excluded. In another embodiment,
31
CA 02511750 2006-05-04
3005-31 CA
Compound 2 is excluded. In a further embodiment, both Compound 1 and Compound
2
are excluded.
Prodrugs of the compounds of Formula I or II include compounds wherein one or
more of the 4, 6 and 8-hydroxy groups, or any other hydroxyl group on the
molecule is
bounded to any group that, when administered to a mammalian subject, is
cleaved to
form the free hydroxyl group. Examples of prodrugs include, but are not
limited to,
acetate, formate, hemisuccinate, benzoate, dimethylaminoacetate and
phosphoryloxycarbonyl derivatives of hydroxy functional groups;
dimethylglycine esters,
aminoalkylbenzyl esters, aminoalkyl esters or carboxyalkyl esters of hydroxy
functional
groups. Carbamate and carbonate derivatives of the hydroxy groups are also
included.
Derivatizations of hydroxyl groups also encompassed, are (acyloxy)methyl and
(acyloxy)ethyl ethers, wherein the acyl group contains an alkyl group
optionally
substituted with groups including, but not limited to, ether, amino and
carboxylic acid
functionalities, or where the acyl group is an amino acid ester. Also included
are
phosphate and phosphonate esters, sulfate esters, sulfonate esters, which are
in
alkylated (such as bis-pivaloyloxymethyl (POM) phosphate triester) or in the
salt form
(such as sodium phosphate ester (-P(O)O-2Na+2)). For further examples of
prodrugs
used in anticancer therapy and their metabolism, see Rooseboom et al (2004),
Phamacol. Rev., vol 56, 53-102. When the prodrug contains an acidic or basic
moiety,
the prodrug may also be prepared as its pharmaceutically acceptable salt.
The compounds of this invention may be formulated into pharmaceutical
compositions comprised of a compound of Formula I or II, in combination with a
pharmaceutically acceptable carrier, as discussed in Section IV below.
III. Methods of Producing Dibenzodiaze~inone Analogues
A. Fermentation
The terms "farnesyl dibenzodiazepinone-producing microorganism" and
"producer of farnesyl dibenzodiazepinone," as used herein, refer to a
microorganism
that carries genetic information necessary to produce a farnesyl
dibenzodiazepinone
compound, whether or not the organism naturally produces the compound. The
terms
32
CA 02511750 2005-07-21
3005-31 CA
apply equally to organisms in which the genetic information to produce the
farnesyl
dibenzodiazepinone compound is found in the organism as it exists in its
natural
environment, and to organisms in which the genetic information is introduced
by
recombinant techniques.
Specific organisms contemplated herein include, without limitation, organisms
of
the family Micromonosporaceae, of which preferred genera include
Micromonospora,
Actinoplanes and Dactylosporangium; the family Streptomycetaceae, of which
preferred
genera include Streptomyces and Kitasatospora; the family Pseudonocardiaceae,
of
which preferred genera are Amycolatopsis and Saccharopolyspora; and the family
Actinosynnemataceae, of which preferred genera include Saccharothrix and
Actinosynnema; however the terms are intended to encompass all organisms
containing genetic information necessary to produce a farnesyl
dibenzodiazepinone
compound. A preferred producer of a farnesyl dibenzodiazepinone compound
includes
microbial strain 046-EC011 or [S01]046, a deposit of which was made
respectively on
March 7, 2003 and December 23, 2003, with the International Depositary
Authority of
Canada (IDAC), Bureau of Microbiology, Health Canada, 1015 Arlington Street,
Winnipeg, Manitoba, Canada R3E 3R2, respectively under Accession No. IDAC
070303-01 and 231203-01.
In one embodiment, ECO-4601 is obtained by cultivating strains of
Micromonospora, namely Micromonospora sp. strains 046-EC011 or [S01]046.
Strains
046-EC011 and [S01]046 were deposited on March 7, 2003, with the International
Depositary Authority of Canada (IDAC), Bureau of Microbiology, Health Canada,
1015
Arlington Street, Winnipeg, Manitoba, Canada R3E 3R2, respectively under
Accession
Nos. 070303-01 and 231203-01. The deposit of the strain was made under the
terms
of the Budapest Treaty on the International Recognition of the Deposit of
Microorganisms for Purposes of Patent Procedure. The deposited strains will be
irrevocably and without restriction or condition released to the public upon
the issuance
of a patent. The deposited strains are provided merely as convenience to those
skilled
in the art and are not an admission that a deposit is required for enablement.
It is to be understood that the present invention is not limited to use of the
particular strains 046-EC011 and [S01]046. Rather, the present invention
contemplates the use of other ECO-4601 producing organisms, such as mutants or
variants of 046-EC011 or [S01]046 that can be derived from this organism by
known
33
CA 02511750 2006-05-04
3005-31 CA
means such as X-ray irradiation, ultraviolet irradiation, treatment with
nitrogen mustard,
phage exposure, antibiotic resistance selection and the like; or through the
use of
recombinant genetic engineering techniques. For examples, see Manual of
Industrial
Microbiology and biotechnology, Demain and Solomon, American Society for
Microbiology,
Washington D.C., 1986; Hesketh et al. (1997), J. Antibiotics, vol 50, no 6,
532-535; and
Hosoya et al. (1998), Antimicrobial Agents and Chemotherapy, vol 42, no 8,
2041-2047).
The farnesyl dibenzodiazepinone compound may be biosynthesized by various
microorganisms. Microorganisms that may synthesize the farnesyl
dibenzodiazepinone
compound include but are not limited to bacteria of the order Actinomycetales,
also
referred to as actinomycetes. Non-limiting examples of members belonging to
the
genera of Actinomycetes include Nocardia, Geodermatophilus, Actinoplanes,
Micromonospora, Nocardioides, Saccharothrix, Amycolatopsis, Kutzneria,
Saccharomonospora, Saccharopolyspora, Kitasatospora, Streptomyces,
Microbispora,
Streptosporangium, and Actinomadura. The taxonomy of actinomycetes is complex
and reference is made to Goodfellow, Suprageneric Classification of
Actinomycetes
(1989); Bergey's Manual of Systematic Bacteriology, Vol. 4 (Williams and
Wilkins,
Baltimore, pp. 2322-2339); and to Embley and Stackebrandt, '"The molecular
phylogeny
and systematics of the actinomycetes," Annu. Rev. Microbiol. (1994) 48:257-
289, for
genera that may synthesize the compounds of the invention.
Farnesyl dibenzodiazepinone-producing microorganisms are cultivated in culture
medium containing known nutritional sources for actinomycetes. Such media
having
assimilable sources of carbon, nitrogen, plus optional inorganic salts and
other known
growth factors, at a pH of about 6 to about 9. Suitable media include, without
limitation,
the growth media provided in Table 1. Microorganisms are cultivated at
incubation
temperatures of about 18 °C to about 40 °C for about 3 to about
40 days.
Table 1
Examples of Fermentation Media for Compound 1 Production
Component QB MA KH RM JA FA XX CL
pH*' 7.2 7.5 7 6.85 7.3 7.0 7.0 7.0
Glucose 12 10 10 I
~ 10
34
CA 02511750 2005-07-21
3005-31 CA
Table 1
Examples of Fermentation Media for Compound 1 Production
Component QB MA KH RM JA FA XX CL
Sucrose 100
Cane molasses 15
Corn starch 30
Soluble starch10 25
Potato dextrin 20 40 20 20
Corn steep 5
solid
Corn steep 5 15
liquor
Dried yeast 2
Yeast extract 5 8.34
Malt extract 35
PharmamediaT"'10 15
Glycerol 30 20
NZ-Amine A 5 10
Soybean powder 15
Fish meal 10
Bacto-peptone 2.5 5
MgS04.7H20 1
CaC03 4 1 2 2 3 2
NaCI 5
(NH4)2 S04 2 2
KZ S04 0.25
MgC12.6H20 10
NaZHP04 3
Casamino acid 0.1
Proflo oilT"'4 0.05
(mL/L)
Silicon defoamer
oil (mL/L) 0.3
MOPS 21
Trace element
solution *z 2
ml/L
Unless otherwise indicated all the ingredients are in g/L.
*' The pH is to adjusted as marked prior to the addition of CaC03.
*ZTrace elements solution contains: ZnCl2 40 mg; Fe C136Hz0 (200 mg); CuCl2
2H20 (10 mg);
MnC12.4H20; Na2B40,.10Hz0 (10mg); (NH4)6 M0~024.4H20 (10 mg) per litre.
The culture media inoculated with a farnesy dibenzodiazepinone-producing
microorganism may be aerated by incubating the inoculated culture media with
agitation, for example, shaking on a rotary shaker, a shaking water bath, or
in a
fermentor. Aeration may also be achieved by the injection of air, oxygen or an
appropriate gaseous mixture to the inoculated culture media during incubation.
Following cultivation, the farnesyl dibenzodiazepinone compound can be
extracted and
isolated from the cultivated culture media by techniques known to a person
skilled in
CA 02511750 2005-07-21
3005-31 CA
the art and/or disclosed herein, including for example centrifugation,
chromatography,
adsorption, filtration. For example, the cultivated culture media can be
optionally
acidified and mixed with a suitable organic solvent such as methanol, ethanol,
n-
butanol, ethyl acetate, n-butyl acetate or 4-methyl-2-pentanone. The organic
layer can
be separated from the mycelial cake for example, by centrifugation and
decantation or
filtration. The mycelial cake is further optionally extracted with an organic
solvent, and
the organic extracts combined. The organic layer is further optionally
treated, for
example by: aqueous washings, precipitation, filtration and the like, followed
the
removal of the solvent, for example, by evaporation to dryness under vacuum.
The
resulting residue can optionally be reconstituted with for example water,
ethyl ether,
ethanol, ethyl acetate, methanol or a mixture thereof, and re-extracted in a
two-phase
system with a suitable organic solvent such as hexane, carbon tetrachloride,
methylene
chloride or a mixture thereof. After removal of the solvent, the compound can
be
further purified by the use of standard techniques such as normal and reverse-
phase
liquid chromatography, crystallization, sublimation, adsorption, mass
exclusion
chromatography, and the like.
8. Chemical Modifications:
The farnesyl dibenzodiazepinone Compound 1 is biosynthesized by
microorganisms and isolated as described herein, and in Canadian patent
2,466,340.
Compound 1 is subjected to random and/or directed chemical modifications to
form
compounds that are derivatives or structural analogues. Such derivatives or
structural
analogues having similar functional activities are within the scope of the
present
invention. The farnesyl dibenzodiazepinone may be modified by one or more
chemical
modification steps, using methods known in the art and described herein.
Examples of
chemical modifications procedures are also provided in Examples 4 to 9 and
Example
15.
Dibenzodiazepinone analogues that are derivatives of Compound 1, for example
those identified herein as the compounds of Formula II and Compounds 2 to 98,
are
generated by standard organic chemistry approaches. General principles of
organic
chemistry required for making and manipulating the compounds described herein,
including functional moieties, reactivity and common protocols are described,
for
36
CA 02511750 2006-05-04
3005-31 CA
example, in "Advanced Organic Chemistry," 4t" Edition by Jerry March (1992),
Wiley-
Interscience, USA. In addition, it will be appreciated by one of ordinary
skill in the art
that the synthetic methods described herein may use a variety of protecting
groups,
whether or not they are explicitly described. A "protecting group" as used
herein means
a moiety used to block one or more functional moieties such as reactive groups
including oxygen, sulfur or nitrogen, so that a reaction can be carried out
selectively at
another reactive site in a poiyfunctional compound. General principles for the
use of
protective groups, their applicability to specific functional groups and their
uses are
described for example in T. H. Greene and P. G. M. Wuts, Protective Groups in
Organic
Synthesis, 3~d Edition, John Wiley & Sons, New York (1999).
Alcohols and phenols are protected with, for example: silyl ethers (TMS:
trimethylsilyl, TIPS: triisopropylsilyl), acetals (MOM: methyloxymethyl, BOM:
benzyloxymethyl), esters (acetate, benzoyl) and ethers (Bn: benzyl). Alcohols
are
deprotected by conditions such as: TBAF (tetrabutylammonium fluoride) for
silyl ethers,
aqueous acid catalysis for acetals and esters, saponification for esters, and
hydrogenolysis for Bn and BOM. Amine is protected using standard amino acid
protecting groups, for example, carbamates (such as t-butyl (BOC) and benzyl
(CBZ)),
fluorene derivatives (such as FMOC: N-(9-fluorenylmethoxycarbonyl)-), etc.
Amine is
deprotected by conditions such as: acid hydrolysis for BOC, hydrogenolysis for
CBZ, or
base treatment for FMOC. All protection and deprotection conditions are
demonstrated
in the Greene et al reference above.
Those skilled in the art will readily appreciate that many synthetic chemical
processes may be used to produce derivatives of Compound 1. The following
schemes
are exemplary of the routine chemical modifications that may be used to
produce
compounds of Formula II. Any chemical synthetic process known fio a person
skilled in
the art providing the structures described herein may be used and are
therefore
comprised in the present invention.
Scheme 1: Alcohol(s) modifications (O-alkylations and O-acylations)
37
CA 02511750 2005-07-21
3005-31 CA
~~OR'
~~OH (a
I
(b\ ~ O R"
O
wherein, R' and R" are each selected from alkyl, alkenyl, alkynyl, cycloalkyl,
heterocycloalkyl, aryl, or heteroaryl; or R"C(O)- is HC(O)- or a C-coupled
amino acid.
In Scheme 1, Phenols in positions 4, 6 and 8 (for position numbers, see
Example
3) are independently alkylated (to produce an ether) or acylated (to produced
an ester).
In Scheme 1 (a), allkylation is accomplished with an alkylating agent such as
R'X is a
diazoalkane, or with a R'X reagent, wherein X is a suitable leaving group such
as Br, I
and trifluoromethane sulfonate in the presence of a base, preferably, a
diazoalkane is
used. When R' is aryl or heteroaryl, the reaction may further need the use of
a catalyst,
such as copper salts (Ullman ether synthesis, Jerry March, supra). In Scheme 1
(b), a
phenolic alcohol is converted to ester when reacted with an activated
carboxylic acid
(R"C(O)X) such as an acid halide, anhydride, N-hydroxysuccinimide ester, or a
carboxylic acid activated by a coupling agent (e.g.: EDC (1-(3-
dimethylaminopropyl)-3-
d'iisopropylethylcarbodiimide hydrochloride); or HATU (O-(7-azabenzotriazol-1-
yl)-
N,N,N;N'-tetramethyluronium hexafluorophosphate)) with a base (e.g., pyridine
or N,N-
diisopropylethylamine (DIPEA)) and optional acatalysts such HOBt (1-
hydroxybenzotriazole hydrate) and/or DMAP (4-(dimethylamino)pyridine). The
same
reactions may be accomplished on alcohols formed by farnesyl modification
reactions
(Scheme 3).
Scheme 1 is used to obtain, for example, Compounds 4 to 12 and 35 to 39 from
Compound 1, and Compound 15 from Compound 13; and to produce any of the
Compounds of Formula I or II comprising an O-alkyl or O-acyl group.
Scheme 2: Amine modifications (N-alkylations and N-acylations)
/R
~N
(a~
~N H O
I (b~ ~
~N R
I
I
38
CA 02511750 2006-05-04
3005-31 CA
wherein, R is selected from alkyl, alkenyl, alkynyl, cycloalkyl,
heterocycloalkyl, aryl, and
heteroaryl; or RC(O)- is HC(O)- or a C-coupled amino acid.
In Scheme 2, amine group in position 5 (for position, see Example 3) is
optionally alkyiated or acylated. in Scheme 2(a), an amine is alkylated using
an RX
alkylating agent such as dialkyl sulfates and alkyl halides, preferably in the
presence of
a base (e.g., sodium bicarbonate, pyridine and the like). When R is an aryl or
a
heteroaryl group, the alkylation reaction with an aryl iodide may further need
a catalyst,
such as copper (for an example, see Plater et al. 2000, J. Chem. Soc., Perkin
Trans. 1,
2695-2701). In Scheme 2(b), an amine is acylated when reacted with an
activated
carboxylic acid such as an acid halide, anhydride, N-hydroxysuccinimide ester,
or a
carboxylic acid activated by a coupling agent (see Scheme 1 ) in the presence
of a base
like DIPEA, and optional use of a catalyst, such as DMAP or HOBt.
Scheme 2 is used to prepare, for example, Compounds 2, 3, 13, 14, 60 to 77
and 98 from Compound 1, and Compound 78 from Compound 46; and to produce any
of the Compounds of Formula I or II comprising an N-alkyl or N-acyl group.
Scheme 3: Double bonds) modifications
(~ ~ OMe (~ ~ O
I s_ y
O OMe
H H
(b) (C) H (e) H
(d) (h)
Ry H Rz
OH
S'' w
H R" H H H H H H
wherein R" and R'' are each selected from H, OH and OC~_salkyl, provided that
at least
one of R" or Ry is OH; RZ is selected from halogen, OH, OC~_salkyl, and
NHC(O)C~_s
alkyl.
In Scheme 3, double bond is modified by: (a) epoxidation; (b) epoxide ring
opening (dihydroxylation, hydration, or hydroxyalkoxylation product) (c)
direct
dihydroxylation, hydration, or hydroxyalkoxylation; (d) hydrogenation; (e)
electrophilic
39
CA 02511750 2005-07-21
3005-31 CA
addition; (f) ozonolysis; (g) hydrolysis of the acetal produced in (f); and
(h) reduction of
the aldehyde produced in (g). In (a), epoxides are obtained from the reaction
of double
bonds with oxidizing agents such as peracids (e.g., mCPBA: 4-chloroperbenzoic
acid).
In (b), the epoxide obtained in (a) is opened by nucleophiles. In basic
conditions
epoxides will preferentially open to give the residual OH at the most hindered
position
(Ry). In acidic conditions, the compound having the residual OH at the R"
position will
be formed as the major product. In (b), the diol (dihydroxylation product: R"
and Ry are
each OH) is obtained from hydrolysis of the epoxide in acidic or basic aqueous
conditions, preferably acidic. In (b), also alcoholysis of the epoxide
(hydroxyalkoxylation
product) is accomplished in basic (RX is OC~_6alkyl and R'' is OH, as major)
or acidic (R"
is OH and Ry is OC~_6alkyl, as major) conditions in a C~_6alkyl alcohol as
solvent,
preferably acidic conditions. In (b), hydration product (R" is H and Ry is OH,
as the
major component) is obtained from the opening of the epoxide by a hydride
source (e.g.
lithium aluminium hydride (LAH)). In (c), the diol (R" and R'' are each OH) is
obtained
from the dihydroxylation of the double bond in oxidizing conditions (e.g.:
osmium
tetroxide, potassium permanganate, N-methylmorpholine-N-oxide, and the like).
In (c),
hydration product (RX is OH and Ry is H, as major) is obtained from the
oxidative
cleavage (NaOH/hydrogen peroxide) of the intermediate formed by hydroboration
of the
double bond (e.g., using 9-BBN (9-borabicyclo[3,3,1]nonane), and the like). In
(d),
hydrogenation is carried out using a hydrogen source (e.g. hydrogen, formic
acid) and a
catalyst (such as rhodium, platinum, or palladium). In (e), electrophilic
addition to the
double bond is achieved by the formation of a carbocation from addition of a
proton in
acidic conditions (e.g., p-toluene sulfonic acid, alkyl sulfate/NaHC03/MeOH,
and the
like), and trapping of the carbocation with an alcohol (C~_6alkyl alcohol,
hydroalkoxylation), water (hydration) or another electron rich atom (e.g., an
halogen or
a nitrite, which is subsequently hydrolyzed to give an amide). In (f), an
acetal is
obtained by the reaction of the double bond with a controlled quantity of
ozone and the
use of a dialkyl sulfide (e.g., Me2S) to decompose the ozonide at the end.
When the
ozonolysis is done in an alcohol, e.g. methanol, the dialkyl actetal is
obtained. In (g), an
aldehyde is obtained by the hydrolysis of the acetal obtained in (f). In (h),
the aldehyde
obtained from (g) is reduced to alcohol by a reducing agent [H] such as sodium
borohydride (NaBH4), sodium cyanoborohydride (NaBH3CN) or LAH.
Scheme 3 is used to obtain, for example: in (a) Compounds 16 from Compound
CA 02511750 2005-07-21
3005-31 CA
1, and Compounds 23, 24 and 26 from Compound 42, Compounds 25, 27 and 29 from
Compound 41, Compounds 28, 30 and 31 from Compound 40, and Compounds 32, 33
and 34 respectively from Compounds 45, 44 and 43; in (b) Compounds 53 to 59
respectively from Compounds 16 to 22; in (c) Compounds 53 to 59 from Compound
1;
in (d) Compounds 40 to 46 from Compound 1, and Compound 78 from Compound 2; in
(e) Compounds 79 to 81 from Compound 2, Compounds 82 to 84 and 88 to 93 from
Compound 1, Compounds 85 to 87 from Compound 14; in (f) Compounds 94 to 96
from Compound 1; in (g) Compounds 47, 49 and 51 from Compound 1; and in (h)
Compounds 48, 50 and 52 respectively from Compounds 47, 49 and 51. Schemes 3
(a)-(g) are also used to produce any Compound of Formula I or II comprising a
modified
farnesyl group.
Scheme 4: Aromatic substitutions
~~H ~~X
wherein, X is selected from F, CI, Br and I.
In Scheme 4, the aryl group is modified (when one of X~ to X5 is halo in
Formula
I or II) by aromatic substitutions, such as halogenation, including
bromination,
chlorination, fluorination, and iodination. Halogenating agents include
bromine, N-
haloamides (e.g, N-bromosuccinimide (NBS), tetraalkylammonium polyhalides),
chlorine, chlorinated cyclohexadienes, N-chloroamines, chlorodimethylsulfonium
chloride, sulfur monochloride/aluminum chloride/thionyl chloride, iodine
chloride,
iodine/oxidizing agent (e.g, nitric acid, iodic acid, sulfur trioxide, etc),
silver(II) fluoride,
cesium fluoroxysulfate, and the like.
Scheme 4 is used to prepare, for example, Compound 97 from Compound 1;
and to produce any of the Compounds of Formula I or II comprising a halogen
group on
the aromatic ring.
Prodrugs are prepared by routine chemical modifications such as described in
Jerry March, supra, including esterification and alkylation reactions, i.e.,
use of
activated acids or mixed anhydrides (acyl halides, use of coupling reagents,
etc), and
41
CA 02511750 2006-05-04
3005-31 CA
by the use of alkylating agents (R-X, wherein X is a leaving group, such as
diazo, and R
is the desired group). Phosphate prodrugs are prepared by phosphorylation, for
example, by a procedure such as described in U.S. patent 5,561,122 (Pettit et
an and in
Hwang and Cole (2004), Org. Lett., vol 6, no 10, 1555-1556 ((POM)2phosphate
triester
from (POM)2phosphoryl chloride).
IV. Pharmaceutical compositions comprising the compounds of the invention
The invention provides a pharmaceutical composition comprising a compound of
Formula I, or a pharmaceutically acceptable salt, solvate or prodrug thereof,
in
combination with a pharmaceutically acceptable carrier. The pharmaceutical
composition comprising a dibenzodiazepinone analogue is useful for treating
diseases
and disorders associated with uncontrolled cellular growth and proliferation,
such as a
neoplastic condition. The pharmaceutical composition is also useful in
treating other
diseases and disorders, including inflammation, autoimmune diseases,
infections,
neurodegenerative diseases and stress. The pharmaceutical composition
comprising a
dibenzodiazepinone analogue may be packaged into a convenient commercial
package
providing the necessary materials, such as the pharmaceutical composition and
written
instructions for its use in treating a neoplastic condition, in a suitable
container.
The compounds of the present invention, or pharmaceutically acceptable salts,
solvates or prodrugs thereof, can be formulated for oral, sublingual,
intranasal,
intraocular, rectal, transdermal, mucosal, topical or parenteral
administration for the
therapeutic or prophylactic treatment of neoplastic and proliferative diseases
and
disorders. Parenteral modes of administration include without limitation,
intradermal,
subcutaneous (s.c., s.q., sub-Q, Hypo), intramuscular (i.m.), intravenous
(i.v.),
intraperitoneal (i.p.), intra-arterial, intramedulary, intracardiac, intra-
articular (joint),
intrasynovial (joint fluid area), intracerebral or intracranial, intraspinal,
intracisternal, and
intrathecal (spinal fluids). Any known device useful for parenteral injection
or infusion of
drug formulations can be used to effect such administration. For oral and/or
parental
administration, compounds of the present invention can be mixed with
conventional
pharmaceutical carriers and excipients and used in the form of solutions,
emulsions,
tablets, capsules, soft gels, elixirs, suspensions, syrups, wafers and the
like. The
42
CA 02511750 2006-05-04
3005-31 CA
compositions comprising a compound of the present invention will contain from
about
0.1 % to about 99.9%, about 1 % to about 98%, about 5% to about 95%, about 10%
to
about 80% or about 15% to about 60% by weight of the active compound.
The pharmaceutical preparations disclosed herein are prepared in accordance
with standard procedures and are administered at dosages that are selected to
reduce,
prevent, or eliminate cancer. (See, e.g., Remington's Pharmaceutical Sciences,
Mack
Publishing Company, Easton, PA; and Goodman and Gilman, Pharmaceutical8asis of
Therapeutics, Pergamon Press, New York, NY, for a general description of the
methods
for administering various agents for human therapy).
As used herein, the term "unit dosage" refers to physically discrete units
suitable
as unitary dosages for human subjects and other mammals, each unit containing
a
predetermined quantity of dibenzodiazepinone analogue calculated to produce
the
desired therapeutic effect, in association with a suitable pharmaceutically
acceptable
carriers. In one embodiment, the unit dosage contains from 10 to 3000 mg of
active
ingredient. In another embodiment, the unit dosage contains 20 to 1000 mg of
active
ingredient. The compositions of the present invention can be delivered using
controlled
(e.g., capsules) or sustained release delivery systems (e.g., bioerodable
matrices).
Exemplary delayed release delivery systems for drug delivery that are suitable
for
administration of the compositions of the invention are described in U.S.
Patent Nos
4,452,775 (issued to Kent), 5,039,660 (issued to Leonard), and 3,854,480
(issued to
Zaffaroni).
The pharmaceutically-acceptable compositions of the present invention comprise
one or more compounds of the present invention in association with one or more
non-
toxic, pharmaceutically-acceptable carriers andlor diluents .and/or adjuvants
and/or
excipients, collectively referred to herein as "carrier" materials, and if
desired other
active ingredients. Pharmaceutically acceptable carriers include, for example,
solvents,
vehicles or medium such as saline, buffered saline, dextrose, water, glycerol,
ethanol,
propylene glycol, polysorbate 80 (Tween-80T""), polyethylene) glycol 300 and
400
(PEG 300 and 400), PEGylated castor oil (E.g. Cremophor EL), poloxamer 407 and
188, hydrophobic carriers, and combinations thereof. Hydrophobic carriers
include, for
example, fat emulsions, lipids, PEGylated phopholids, polymer matrices,
biocompatible
polymers, lipospheres, vesicles, particles, and liposomes. The term
specifically
43
CA 02511750 2005-07-21
3005-31 CA
excludes cell culture medium.
Excipients or additives included in a formulation have different purposes
depending, for example on the nature of the drug, and the mode of
administration.
Examples of generally used excipients include, without limitation: stabilizing
agents,
solubilizing agents and surfactants, buffers, antioxidants and preservatives,
tonicity
agents, bulking agents, lubricating agents, emulsifiers, suspending or
viscosity agents,
inert diluents, fillers, disintegrating agents, binding agents, wetting
agents, lubricating
agents, antibacterials, chelating agents, sweetners, perfuming agents,
flavouring
agents, coloring agents, administration aids, and combinations thereof.
The compositions may contain common carriers and excipients, such as
cornstarch or gelatin, lactose, sucrose, microcrystalline cellulose, kaolin,
mannitol,
dicalcium phosphate, sodium chloride and alginic acid. The compositions may
contain
crosarmellose sodium, microcrystalline cellulose, sodium starch glycolate and
alginic
acid.
Formulations for parenteral administration can be in the form of aqueous or
non-
aqueous isotonic sterile injection solutions, suspensions or fat emulsions,
comprising a
compound of this invention, or a pharmaceutically acceptable salt or prodrug
thereof.
The parenteral form used for injection must be fluid to the extent that easy
syringability
exists. These solutions or suspensions can be prepared from sterile
concentrated
liquids, powders or granules. The compounds can be dissolved in a carrier such
as a
solvent or vehicle, for example, polyethylene glycol, propylene glycol,
ethanol, corn oil,
benzyl alcohol, glycofurol, N,N-dimethylacetamide, N-methylpyrrolidone,
glycerine,
saline, dextrose, water, glycerol, hydrophobic carriers, and combinations
thereof.
Excipients used in parenteral preparations also include, without limitation,
stabilizing agents (e.g. carbohydrates, amino acids and polysorbates),
solubilizing
agents (e.g. cetrimide, sodium docusate, glyceryl monooleate,
polyvinylpyrolidone
(PVP) and polyethylene glycol (PEG)) and surfactants (e.g. polysorbates,
tocopherol
PEG succinate, poloxamer and CremophorTM), buffers (e.g. acetates, citrates,
phosphates, tartrates, lactates, succinates, amino acids and the like),
antioxidants and
preservatives (e.g. BHA, BHT, gentisic acids, vitamin E, ascorbic acid and
sulfur
containing agents such as sulfites, bisulfites, metabisulfites, thioglycerols,
thioglycolates
and the like), tonicity agents (for adjusting physiological compatibility),
suspending or
44
CA 02511750 2005-07-21
3005-31 CA
viscosity agents, antibacterials (e.g. thimersol, benzethonium chloride,
benzalkonium
chloride, phenol, cresol and chlorobutanol), chelating agents, and
administration aids
(e.g. local anesthetics, anti-inflammatory agents, anti-clotting agents, vaso-
constrictors
for prolongation and agents that increase tissue permeability), and
combinations
thereof.
Parenteral formulations using hydrophobic carriers include, for example, fat
emulsions and formulations containing lipids, lipospheres, vesicles, particles
and
liposomes. Fat emulsions include in addition to the above-mentioned
excipients, a lipid
and an aqueous phase, and additives such as emulsifiers (e.g. phospholipids,
poloxamers, polysorbates, and polyoxyethylene castor oil), and osmotic agents
(e.g.
sodium chloride, glycerol, sorbitol, xylitol and glucose). Liposomes include
natural or
derived phospholipids and optionally stabilizing agents such as cholesterol.
In another embodiment, the parenteral unit dosage form of the compound can be
a ready-to-use solution of the compound in a suitable carrier in sterile,
hermetically
sealed ampoules or in sterile pre-loaded syringes. The suitable carrier
optionally
comprises any of the above-mentioned excipients.
Alternatively, the unit dosage of the compound of the present invention can be
in
a concentrated liquid, powder or granular form for ex tempore reconstitution
in the
appropriate pharmaceutically acceptable carrier at the time of delivery. In
addition the
above-mentioned excipients, powder forms optionally include bulking agents
(e.g.
mannitol, glycine, lactose, sucrose, trehalose, dextran, hydroxyethyl starch,
ficoll and
gelatin), and cryo or lyoprotectants.
For example, in intravenous (IV) use, a sterile formulation of the compound of
formula I and optionally one or more additives, including solubilizers or
surfactants, can
be dissolved or suspended in any of the commonly used intravenous fluids and
administered by infusion. Intravenous fluids include, without limitation,
physiological
saline, phosphate buffered saline, 5% glucose or Ringer's T"' solution.
In another example, in intramuscular preparations, a sterile formulation of
the
compound of the present invention or suitable soluble salts or prod rugs
forming the
compound, can be dissolved and administered in a pharmaceutical diluent such
as
Water-for-Injection (WFI), physiological saline or 5% glucose. A suitable
insoluble form
of the compound may be prepared and administered as a suspension in an aqueous
CA 02511750 2005-07-21
3005-31 CA
base or a pharmaceutically acceptable oil base, e.g. an ester of a long chain
fatty acid
such as ethyl oleate.
For oral use, solid formulations such as tablets and capsules are particularly
useful. Sustained released or enterically coated preparations may also be
devised.
For pediatric and geriatric applications, suspension, syrups and chewable
tablets are
especially suitable. For oral administration, the pharmaceutical compositions
are in the
form of, for example, tablets, capsules, suspensions or liquid syrups or
elixirs, wafers
and the like. For general oral administration, excipient or additives include,
but are not
limited to inert diluents, fillers, disintegrating agents, binding agents,
wetting agents,
lubricating agents, sweetening agents, flavoring agents, coloring agents and
preservatives.
The oral pharmaceutical composition is preferably made in the form of a unit
dosage containing a therapeutically-effective amount of the active ingredient.
Examples of such dosage units are tablets and capsules. For therapeutic
purposes,
the tablets and capsules which can contain, in addition to the active
ingredient,
conventional carriers such as: inert diluents (e.g., sodium and calcium
carbonate,
sodium and calcium phosphate, and lactose), binding agents (e.g., acacia gum,
starch,
gelatin, sucrose, polyvinylpyrrolidone (Providone), sorbitol, or tragacanth
methylcellulose, sodium carboxymethylcellulose, hydroxypropyl methylcellulose,
and
ethylcellulose), fillers (e.g., calcium phosphate, glycine, lactose, maize-
starch, sorbitol,
or sucrose), lubricants or lubricating agents (e.g., magnesium stearate or
other metallic
stearates, stearic acid, polyethylene glycol, waxes, oils, silica and
colloical silica, silicon
fluid or talc), disintegrants or disintegrating agents (e.g., potato starch,
corn starch and
alginic acid), flavouring, coloring agents, or acceptable wetting agents.
Carriers may
also include coating excipients such as glyceryl monostearate or glyceryl
distearate, to
delay absorption in the gastrointestinal tract.
Oral liquid preparations, generally in the form of aqueous or oily solutions,
suspensions, emulsions, syrups or elixirs, may contain conventional additives
such as
suspending agents, emulsifying agents, non-aqueous agents, preservatives,
coloring
agents and flavoring agents. Examples of additives for liquid preparations
include
acacia, almond oil, ethyl alcohol, fractionated coconut oil, gelatin, glucose
syrup,
glycerin, hydrogenated edible fats, lecithin, methyl cellulose, methyl or
propyl para-
hydroxybenzoate, propylene glycol, sorbitol, or sorbic acid.
46
CA 02511750 2005-07-21
3005-31 CA
For both liquid and solid oral preparations, flavoring agents such as
peppermint,
oil of wintergreen, cherry, grape, fruit flavoring or the like can also be
used. It may also
be desirable to add a coloring agent to make the dosage form more aesthetic in
appearance or to help identify the product. For topical use the compounds of
present
invention can also be prepared in suitable forms to be applied to the skin, or
mucus
membranes of the nose and throat, and can take the form of creams, ointments,
liquid
sprays or inhalants, lozenges, or throat paints. Such topical formulations
further can
include chemical compounds such as dimethylsulfoxide (DMSO) to facilitate
surface
penetration of the active ingredient. For application to the eyes or ears, the
compounds
of the present invention can be presented in liquid or semi-liquid form
formulated in
hydrophobic or hydrophilic bases as ointments, creams, lotions, paints or
powders. For
rectal administration the compounds of the present invention can be
administered in the
form of suppositories admixed with conventional carriers such as cocoa butter,
wax or
other glyceride.
V. Medical Use in the Treatment of Neoplasms
In one aspect, the invention relates to a method for inhibiting growth and/or
proliferation of cancer cells in a mammal. In another aspect, the invention
provides a
method for treating neoplasms in a mammal. Mammals include ungulates (e.g.
sheeps,
goats, cows, horses, pigs), and non-ungulates, including rodents, felines,
canines and
primates (i.e. human and non-human primates). In a preferred embodiment, the
mammal is a human.
Although not wishing to be bound by any particular theory, dibenzodiazepinone
analogues of the present invention may exert their anticancer effects, at
least in part,
through interaction with the peripheral benzodiazepine receptor (PBR). PBR is
an
evolutionarily conserved 18-kDa protein, which is present in all tissues but
highly
expressed in steroid producing tissues and cancers, and has been associated
with
numerous biological functions, including regulation of apoptosis, regulation
of cell
proliferation, and stimulation of steroidogenesis. PBR is a critical component
of the
mitochondrial permeability transition pore (MPTP), a multiprotein complex
located at the
contact site between inner and outer mitochondrial membranes, which is
intimately
involved in the intiation and regulation of apoptosis. Moreover, PBR ligands
have been
47
CA 02511750 2006-05-04
3005-31 CA
shown to modulate MPTP and apoptotic response (see Carayon et al., Blood
(1995)
87(8):3170; Hirsch et al., Experimental Cell. Research (1998) 241 no 2:426-
434; and
Bono et al., Biochem. Biophys. Res. Comm. (1999) 265:457). Several recent
reports
have linked PBR and cancer based on alteration of PBR expression in tumor
cells and
PBR-dependent apoptotic modulations. Some of the highest densities of PBR are
observed in neoplastic tissues and cell lines. Ovarian, hepatic and colon
carcinomas,
adenocarcinoma, glioma and breast cancer cells all show increased PBR
densities
relative to untranformed tissues (see Miettinen et al., Cancer Res. (1995)
55:2691-
2695; Katz et al., Clin. Sci. (1990) 78:155; Katz et al., Oncology (1990)
47:139; and
Venturini et al., Life Sci. (1999) 65:2223).
PBR ligands, both endogenous and synthetic, have been shown to have
antiproliferative and pro-apoptotic properties. For example, the therapeutic
potency of
porphrins for the treatment of skin, bladder and lung cancers are reportedly
linked to
their affinities to PBR, and the sentitivity of tumor cell lines to
photodynamic therapy
reportedly parallel their PBR densities (Verma et al., Mol. Med. (1998) 4(1
):40;
Kupczyk-Subotkowska et al., J. Med. Chem. (1997) 40(11):1726; and Guo et al.,
Cancer Chemother. Pharmacol. (2001 ) 48(2):169). In addition to indirect PBR-
based
anticancer therapies, some PBR ligands have direct anticancer properties,
including
apoptotic and cell cycle inhibitor properties (Wang et al., Proc. Natl. Acad.
Sci. USA
(1984) 81:753; Landau et al., J. Biochem. Pharmacol. (1998) 56:1029; and
Stoebner et
al., Cell Death Differ. (2001) 8(7):747). Similarly, the dibenzodiazepinone
analogues of
the present invention, in particular Compound 1, have been shown to bind to
the PBR
and inhibit cellular proliferation in a panel of different types of tumor cell
lines, including
low and high-grade gliomas. Compound 1 also increases expression of several
genes
involved in the regulation of apoptosis and signal transduction, as well as
genes
involved in steroid biosynthesis. Since human glioblastomas have an increased
density
of PBR compared with normal human brain, Compound 1's anticancer activity is
believed to be via interaction with the PBR. Compound 1 has been shown to
penetrate
into brain tissues.
Alternatively, or in addition, the dibenzodiazepinone analogues of the
invention
may have chemosensitizing or multidrug resistance modulating activity, as has
been
reported for other PBR ligands. For example, a non-cytotoxic dose of PK11195
48
CA 02511750 2006-05-04
3005-31 CA
increased the efficacy of a daunorubicin treatment on human multidrug-
resistant
leukemia cells in vitro and in vivo (Jakubikova et al., Neoplasma (2002)
49(4):231; and
Decaudin et al., Cancer Res. (2002) 62(5):1388). Thus, the dibenzodiazepinone
analogues, like other PBR ligands, may inhibit the expression or activity of
multi-drug
resistance (MDR)-associated protein (MDRP) or rnulti-drug resistance protein-1
(MDR1). "Multi-drug resistance" (MDR) broadly refers to a pattern of
resistance to a
variety of chemotherapeutic drugs with unrelated chemical structures and
different
mechanisms of action. Although the etiology of MDR is multifactorial, the
overexpression of P-glycoprotein (Pgp), a membrane protein that mediates the
transport of MDR drugs, remains the most common alteration underlying MDR in
laboratory models (Childs, S., Imp. Adv. Oncol. (1994) 21-36). Moreover,
expression of
Pgp has been linked to the development of MDR in human cancer, particularly in
the
leukemias, lymphomas, multiple myeloma, neuroblastoma, and soft tissue sarcoma
(Fan., D., et al., Reversal of Multidrug Resistance in Cancer, ed. Kellen,
J.A. (1993)
(CRC, Boca Raton, FL), pp. 93-125). Recent studies showed that tumor cells
expressing MDRP (Cole, S.P., et al., Science (1992) 258:1650-1654) and lung
resistance protein (LRP) (Scheffer, G.L., et al., Nat. Med. (1995)1:5?8-582)
and
mutation of DNA topoisomerase II (Beck, W.T., J. Natl. Cancer Inst. (1989)
81:1683-
1685) also may render MDR.
While the above suggests that the dibenzodiazepinone analogues of the
invention may exert anticancer effects via interaction with the PBR, the
mechanism of
action may also be due, at least in part, to some as yet undefined mechanism
or
pathway. Alternatively or in addition to PBR, the dibenzodiazepinone analogues
of the
present invention may bind to or interact with other cancer-associated
proteins and
polypeptides, including, without limitation, polypeptides encoded by
oncogenes,
polypeptides that induce angiogenesis, proteins involved in metastasizing
and/or
invasive processes, and proteases that regulate apoptosis and the cell cycle.
Regardless of the mechanism of action, the dibenzodiazepinone analogues of the
invention have been demonstrated to exhibit anti-cancer activity both in vitro
and in
vivo. Based on these discoveries, applicants have developed methods for
treating
neoplasms.
As used herein, the terms "neoplasm", "neoplastic disorder", "neoplasia"
"cancer," "tumor" and "proliferative disorder" refer to cells having the
capacity for
49
CA 02511750 2005-07-21
3005-31 CA
autonomous growth, i.e., an abnormal state of condition characterized by
rapidly
proliferating cell growth which generally forms a distinct mass that show
partial or total
lack of structural organization and functional coordination with normal
tissue. The
terms are meant to encompass hematopoietic neoplasms (e.g. lymphomas or
leukemias) as well as solid neoplasms (e.g. sarcomas or carcinomas), including
all
types of pre-cancerous and cancerous growths, or oncogenic processes,
metastatic
tissues or malignantly transformed cells, tissues, or organs, irrespective of
histopathologic type or stage of invasiveness. Hematopoietic neoplasms are
malignant
tumors affecting hematopoietic structures (structures pertaining to the
formation of
blood cells) and components of the immune system, including leukemias (related
to
leukocytes (white blood cells) and their precursors in the blood and bone
marrow)
arising from myeloid, lymphoid or erythroid lineages, and lymphomas (relates
to
lymphocytes). Solid neoplasms include sarcomas, which are malignant neoplasms
that
originate from connective tissues such as muscle, cartilage, blood vessels,
fibrous
tissue, fat or bone. Solid neoplasms also include carcinomas, which are
malignant
neoplasms arising from epithelial structures (including external epithelia
(e.g., skin and
linings of the gastrointestinal tract, lungs, and cervix), and internal
epithelia that line
various glands (e.g., breast, pancreas, thyroid). Examples of neoplasms that
are
particularly susceptible to treatment by the methods of the invention include
leukemia,
and hepatocellular cancers, sarcoma, vascular endothelial cancers, breast
carcers,
central nervous system cancers (e.g. astrocytoma, gliosarcoma, neuroblastoma,
oligodendroglioma and glioblastoma), prostate cancers, lung and bronchus
cancers,
larynx cancers, esophagus cancers, colon cancers, colorectal cancers, gastro-
intestinal
cancers, melanomas, ovarian and endometrial cancer, renal and bladder cancer,
liver
cancer, endocrine cancer (e.g. thyroid), and pancreatic cancer.
The dibenzodiazepinone analogue is brought into contact with or introduced
into
a cancerous cell or tissue. In general, the methods of the invention for
delivering the
compositions of the invention in vivo utilize art-recognized protocols for
delivering
therapeutic agents with the only substantial procedural modification being the
substitution of the dibenzodiazepinone analogue of the present invention for
the
therapeutic agent in the art-recognized protocols. The route by which the
dibenzodiazepinone analogue is administered, as well as the formulation,
carrier or
vehicle will depend on the location as well as the type of the neoplasm. A
wide variety
CA 02511750 2005-07-21
3005-31 CA
of administration routes can be employed. The dibenzodiazepinone analogue may
be
administered by intravenous or intraperitoneal infusion or injection. For
example, for a
solid neoplasm that is accessible, the compound of the invention may be
administered
by injection directly into the neoplasm. For a hematopoietic neoplasm the
compound
may be administered intravenously or intravascularly. For neoplasms that are
not
easily accessible within the body, such as metastases or brain tumors, the
compound
may be administered in a manner such that it can be transported systemically
through
the body of the mammal and thereby reach the neoplasm and distant metastases
for
example intrathecally, intravenously or intramuscularly or orally.
Alternatively, the
compound can be administered directly to the tumor. The compound can also be
administered subcutaneously, intraperitoneally, topically (for example for
melanoma),
rectally (for example colorectal neoplasm) vaginally (for example for cervical
or vaginal
neoplasm), nasally or by inhalation spray (for example for lung neoplasm).
The dibenzodiazepinone analogue is administered in an amount that is
sufficient
to inhibit the growth or proliferation of a neoplastic cell, or to treat a
neoplastic disorder.
The term "inhibition" refers to suppression, killing, stasis, or destruction
of cancer cells.
The inhibition of mammalian cancer cell growth according to this method can be
monitored in several ways. Cancer cells grown in vitro can be treated with the
compound and monitored for growth or death relative to the same cells cultured
in the
absence of the compound. A cessation of growth or a slowing of the growth rate
(i.e.,
the doubling rate), e.g., by 50% or more at 100 micromolar, is indicative of
cancer cell
inhibition (see Anticancer Drug Development Guide: preclinical screening,
clinical trials
and approval; B.A. Teicher and P.A. Andrews, ed., 2004, Humana Press, Totowa,
NJ).
Alternatively, cancer cell inhibition can be monitored by administering the
compound to
an animal model of the cancer of interest. Examples of experimental non-human
animal cancer models are known in the art and described below and in the
examples
herein. A cessation of tumor growth (i.e., no further increase in size) or a
reduction in
tumor size (i.e., tumor volume by least a 58%) in animals treated with the
compound
relative to tumors in control animals not treated with the compound is
indicative of
significant tumor growth inhibition (see Anticancer Drug Development Guide:
preclinical
screening, clinical trials and approval; B.A. Teicher and P.A. Andrews, ed.,
2004,
Humana Press, Totowa, NJ).
The term "treatment" refers to the application or administration of a
51
CA 02511750 2006-05-04
3005-31 CA
dibenzodiazepinone analogue to a mammal, or application or administration of a
dibenzodiazepinone analogue to an isolated tissue or cell line from a mammal,
who has
a neoplastic disorder, a symptom of a neoplastic disorder or a predisposition
toward a
neoplastic disorder, with the purpose to cure, heal, alleviate, relieve,
alter, ameliorate,
improve, or control the disorder, the symptoms of disorder, or the
predisposition toward
disorder. The term "treating" is defined as administering, to a mammal, an
amount of a
dibenzodiazepinone analogue sufficient to result in the prevention, reduction
or
elimination of neoplastic cells in a mammal ("therapeutically effective
amount"). The
therapeutically effective amount and timing of dosage will be determined on an
individual basis and may be based, at least in part, on consideration of the
age, body
weight, sex, diet and general health of the recipient subject, on the nature
and severity
of the disease condition, and on previous treatments and other diseases
present.
Other factors also include the route and frequency of administration, the
activity of the
administered compound, the metabolic stability, length of action and excretion
of the
compound, drug combination, the tolerance of the recipient subject to the
compound
and the type of neoplasm or proliferative disorder. In one embodiment, a
therapeutically effective amount of the compound is in the range of about 0.01
to about
750 mg/kg of body weight of the mammal. In another embodiment, the
therapeutically
effective amount is in the range of about 0.01 to about 300 mg/kg body weight
per day.
In yet another embodiment, the therapeutically effective amount is in the
range of 10 to
about 50 mg/kg body weight per day. The therapeutically effective doses of the
above
embodiments may also be expressed in milligrams per square meter (mg/m2) in
the
case of a human patient. Conversion factors for different mammalian species
may be
found in:Freireich et ai, Quantitative comparison of toxicity of anticancer
agents in
mouse, rat, dog, monkey and man, Cancer Chemoth. Report, 1966, 50(4): 219-244.
When special requirements may be needed (e.g. for children patients), the
therapeutically effective doses described above may be outside the ranges
stated
herein. Such higher or lower doses are within the scope of the present
invention.
To monitor the efficacy of tumor treatment in a human, tumor size and/or tumor
morphology is measured before and after initiation of the treatment, and
treatment is
considered effective if either the tumor size ceases further growth, or if the
tumor is
reduced in size, e.g., by at least 10% or more (e.g., 20%, 30%, 40%, 50%, 60%,
70%,
52
CA 02511750 2005-07-21
3005-31 CA
80%, 90% or even 100%, that is, the absence of the tumor). Prolongation of
survival,
time-to-disease progression, partial response and objective response rate are
surrogate
measures of clinical activity of the investigational agent. Tumor shrinkage is
considered
to be one treatment-specific response. This system is limited by the
requirement that
patients have visceral masses that are amenable to accurate measurement.
Methods of
determining the size of a tumor in vivo vary with the type of tumor, and
include, for
example, various imaging techniques well known to those in the medical imaging
or
oncology fields (MRI, CAT, PET, etc.), as well as histological techniques and
flow
cytometry. For certain types of cancer, evaluation of serum tumor markers are
also
used to evaluate response (eg prostate-specific antigen (PSA) for prostate
cancer, and
carcino-embryonic antigen (CEA), for colon cancer). Other methods of
monitoring
cancer growth include cell counts (e.g. in leukemias) in blood or relief in
bone pain (e.g.
prostate cancer).
The dibenzodiazepinone compound may be administered once daily, or the
compound may be administered as two, three, four, or more sub-doses at
appropriate
intervals throughout the day. In that case, the dibenzodiazepinone compound
contained in each sub-dose must be correspondingly smaller in order to achieve
the
total daily dosage. The dosage unit can also be compounded for delivery over
several
days, e.g., using a conventional sustained release formulation which provides
sustained
release of the dibenzodiazepinone compound over a several day period.
Sustained
release formulations are well known in the art. In this embodiment, the dosage
unit
contains a corresponding multiple of the daily dose. The effective dose can be
administered either as a single administration event (e.g., a bolus injection)
or as a slow
injection or infusion, e.g. over 30 minutes to about 24 hours. The compound
may be
administered as a treatment, for up to 30 days. Moreover, treatment of a
subject with a
therapeutically effective amount of a composition can include a single
treatment or a
series of treatments (e.g., a four-week treatment repeated 3 times, with a 2
months
interval between each treatment). Estimates of effective dosages, toxicities
and in vivo
half-lives for the dibenzodiazepinone compounds encompassed by the invention
can be
made using conventional methodologies or on the basis of in vivo testing using
an
appropriate animal model.
The dibenzodiazepinone compound may be administered in conjunction with or
in addition to known other anticancer treatments such as radiotherapy, or
other known
53
CA 02511750 2005-07-21
3005-31 CA
anticancer compounds or chemotherapeutic agents. Such agents include, but are
not
limited to, 5-flurouracil, mitomycin C, methotrexate, hydroxyurea,
cyclophosphamide,
dacarbazine, mitoxantrone, anthracyclines (Epirubicin and Doxurubicin),
etopside,
pregnasome, platinum compounds such as carboplatin and cisplatin, taxanes such
as
PaclitaxelT"' and DocetaxelT""; hormone therapies such as tamoxifen and anti-
estrogens; antibodies to receptors, such as herceptin and Iressa; aromatase
inhibitors,
progestational agents and LHRH analogues; biological response modifiers such
as IL2
and interferons; multidrug reversing agents such as the cyclosporin analogue
PSC
833.
Toxicity and therapeutic efficacy of dibenzodiazepinone compounds can be
determined by standard pharmaceutical procedures in cell cultures or
experimental
animals. Therapeutic efficacy is determined in animal models as described
above and
in the examples herein. Toxicity studies are done to determine the lethal dose
for 10%
of tested animals (LD10). Animals are treated at the maximum tolerated dose
(MTD):
the highest dose not producing mortality or greater than 20% body weight loss.
The
effective dose (ED) is related to the MTD in a given tumor model to determine
the
therapeutic index of the compound. A therapeutic index (MTD/ED) close to 1.0
has
been found to be acceptable for some chemotherapeutic drugs, a preferred
therapeutic
index for classical chemotherapeutic drugs is 1.25 or higher.
The data obtained from cell culture assays and animal studies can be used in
formulating a range of dosage for use in humans. The dosage of compositions of
the
invention will generally be within a range of circulating concentrations that
include the
MTD. The dosage may vary within this range depending upon the dosage form
employed and the route of administration utilized. For any compound used in
the
method of the invention, the therapeutically effective dose can be estimated
initially
from cell culture assays. A dose may be formulated in animal models to achieve
a
circulating plasma concentration range of the compound. Such information can
be
used to more accurately determine useful doses in humans. Levels in plasma may
be
measured, for example, by HPLC.
Animal models to determine antitumor efficacy of a compound are generally
carried out in mice. Either murine tumor cells are inoculated subcutaneously
into the
hind flank of mice from the same species (syngeneic models) or human tumor
cells are
inoculated subcutaneously into the hind flank of severe combined immune
deficient
54
CA 02511750 2006-05-04
3005-31 CA
(SCID) mice or other immune deficient mice (nude mice) (xenograft models).
Advances in mouse genetics have generated a number of mouse models for the
study of various human diseases including cancer. The MMHCC (Mouse models of
Human Cancer Consortium), sponsored by the National Cancer Institute, provides
disease-site-specific compendium of known cancer models, and provides access
to the
searchable Cancer Models Database, as well as the NCI-MMHCC mouse repository.
Mouse repositories can also be found at: The Jackson Laboratory, Charles River
Laboratories, Taconic, Harlan, Mutant Mouse Regional Resource Centers (MMRRC)
National Network and at the European Mouse Mutant Archive. Such models may be
used for in vivo testing of dibenzodiazepinone compounds, as well as for
determining a
therapeutically effective dose.
In addition to the compounds of the invention, pharmaceutically acceptable
salts,
solvates or prodrugs of said compounds may also be employed in compositions to
treat
or prevent the above-identified disorders.
VI. Method of Inhibiting Lipoxygenase
In another embodiment, the present invention also provides for a method of
treating diseased states, in particular inflammation, caused by the 5-
lipoxygenase
system and/or by the synthesis of the Leukotrienes C4, D4, E4 and F4 as well
as
Leukotriene B4 in mammals, especially in human subjects. This method comprises
administering to a subject an effective amount of a compound of Formula I. The
compound of Formula I may be used alone or in combination with other anti-
inflammatory compounds to treat or prevent disease states related to
inflammation
including pulmonary conditions, inflammation, cardiovascular conditions,
central
nervous system conditions or skin conditions. More specific diseases include
gastritis;
erosive esophagitis; inflammatory bowel disease; ethanol-induced hemorrhagic
erosions; hepatic ischemia; ischemic neuronal injury; noxious agent induced
damage or
necrosis of hepatic, pancreatic, renal, neuronal or myocardial tissue; liver
parenchyma)
damage caused by hepatoxic agents such as CC14 and D-galactosamine; ischemic
renal failure; disease-induced hepatic damage; trauma- or stress-induced cell
damage;
asthma, multiple sclerosis, ischemic reperfusion, edema, rheumatoid arthritis,
viral
CA 02511750 2005-07-21
3005-31 CA
encephalitis, bacterial pneumonia, neurodegeneration, Alzheimer's disease and
glycerol-induced renal failure.
For the method of the invention related to the 5-lipoxygenase system and/or
the
biosynthesis of leukotrienes, a typical effective unit dose of a compound of
Formula I,
or an analogue given orally or parenterally would be from about 5 to about 100
mg/kg of
body weight of the subject with a daily dose ranging from about 15 to about
300 mg/kg
of body weight of the subject.
The inhibition of lipoxygenase enzymes is monitored using methods well known
in the art and as described in the examples herein. A decrease in enzyme
activity by at
least 10%, relative to the activity in the absence of a compound as described
herein is
indicative of effective inhibition of lipoxygenase activity.
The compounds useful according to the invention can be used to reduce or
prevent inflammation. Among the hallmarks of local acute inflammation are
heat,
redness, swelling, pain and loss of function. These changes are induced
largely by
changes in vascular flow and caliber, changes in vascular permeability and
leukocyte
exudation (Robbins et al., "Pathologic Basis of Disease", 6t" Ed., W.B.
Saunders Co.,
Philadelphia, PA). Anti-inflammatory therapy performed using compounds useful
according to the invention can be monitored for success by tracking any of
these
changes. For example, a decrease in swelling (e.g., at least 10% decrease
following
treatment) or reported pain (e.g., a sustained decrease of 1 point or more on
a 1-10
scale reported by the patient, with 10 being the worst pain experienced in
association
with this disorder prior to treatment, and 0 being no pain) can be used to
indicate
successful treatment.
Other measurable hallmarks of inflammation include leukocyte infiltration and
inflammatory cytokine levels. These hallmarks can be monitored by biopsy of
the
affected tissue. A decrease of 10% or more in leukocyte infiltration in fixed,
stained
tissue relative to infiltration in similar tissue prior to treatment can be
used to indicate
successful treatment, as can a decrease of 10% or more in the level of any
given
inflammatory cytokine, relative to the level before treatment. Those skilled
in the art
can readily assay for inflammatory cytokine levels in tissue, blood, or other
fluid
samples. Alternatively, the level of systemic indicators of inflammation such
as C-
reactive protein levels and erythrocyte sedimentation rate can be monitored.
Each of
56
CA 02511750 2005-07-21
3005-31 CA
these has established normal ranges in medicine, and treatment is considered
successful if one or more of such indicators goes from outside the normal
range to
inside the normal range after the initiation of treatment.
In addition to the compounds of this invention, pharmaceutically acceptable
salts, solvates or prodrugs of the compounds of this invention may also be
employed in
compositions to treat or prevent the above-identified disorders.
FX~4MP1 F
Unless otherwise noted, all reagents were purchased from Sigma Chemical Co.
(St. Louis, MO), Aldrich.
All NMR spectra were collected in deuterated solvent on a Varian 500T""
Spectrometer ('H NMR at 500 MHz,'3C NMR at 125 MHz). UV and mass spectra were
collected by Waters 2690 T"" HPLC using a photodiode array detector (PDA, 210-
400nm) coupled to a Waters MicromassT"" ZQT"' mass detector.
Unless otherwise indicated, all numbers expressing quantities of ingredients,
properties such as molecular weight, reaction conditions, molar equivalents
(eq),
percentage of binding and/or inhibition, Gl5o, IC5o and so forth used in the
specification
and claims are to be understood as being modified in all instances by the term
"about".
Accordingly, unless indicated to the contrary, the numerical parameters set
forth in the
present specification and attached claims are approximations. At the very
least, and
not as an attempt to limit the application of the doctrine of equivalents to
the scope of
the claims, each numerical parameter should at least be construed in light of
the
number of significant figures and by applying ordinary rounding techniques.
Notwithstanding that the numerical ranges and parameters setting forth the
broad
scope of the invention are approximations, the numerical values set in the
examples,
Tables and Figures are reported as precisely as possible. Any numerical values
may
inherently contain certain errors resulting from variations in experiments,
testing
measurements, statistical analyses and such.
In the following section, examples describe in detail the chemical synthesis
of
representative compounds of the present invention. The procedures are
illustrations,
and the invention should not be construed as being limited by chemical
reactions and
57
CA 02511750 2006-05-04
3005-31 CA
conditions they express. No attempt has been made to optimize the yields
obtained in
these reactions, and it would be obvious to one skilled in the art that
variations in
reaction times, temperature, solvent and/or reagents could increase the
yields.
In addition, the materials, methods, and examples, including in vitro and in
vivo
efficacy, bioavailabiiity, toxicity and pharmacological properfiies are
illustrative only and
not intended to be limiting. Although methods and materials similar or
equivalent to
those described herein can be used in the practice or testing of the present
invention,
suitable methods and materials are described below. In case of conflict, the
present
specification, including definitions, will control.
EXAMPLE 1: PRODUCTION OF COMPOUND 1 EY FERMENTATION
a) Fermentation procedure
Micromonospora sp. (deposit accession number IDAC 070303-01) was
maintained on agar plates of ISP2 agar (Difco Laboratories, Detroit, MI). An
inoculum
for the production phase was prepared by transferring the surface growth of
the
Micromonospora sp. from the agar plates to 125-mL flasks containing 25 mL of
sterile
medium comprised of glucose 10g, potato dextrin type IV (Sigma) 20 g, yeast
extract 5
g, N Z Amine-A 5 g, 1 g CaC03 made up to one liter with tap water (pH 7.0).
The
culture was incubated at about 28°C for approximately 70-72 hours on a
rotary shaker
set at 250 rpm. Following incubation, 10 mL of culture was transferred to a 2L
baffled
flask containing 600 mL of sterile production medium containing 20 g/L potato
dextrin
type IV (sigma), 30 g/L glycerol, 2.5 g/L Bacto-peptone, 8.34 g/L yeast
extract, 3 g/L
CaC03, pH 7Ø Fermentation broth was prepared by incubating the production
culture
at 28°C in a rotary shaker set at 250 rpm for 5 days.
b) Alternate procedure:
The fermentation was accomplished as a 1 x 10L batch in a 14.5 L fermentor
(BioFlo 110T"" Fermentor, New Brunswick Scientific, Edison, NJ, USA) using an
improved procedure described in CA patent application 2,466,340, filed January
21,
2004.
58
CA 02511750 2005-07-21
3005-31 CA
Micromonospora sp. (deposit accession number IDAC 070303-01 ) was
maintained on agar plates of ISP2 agar (Difco Laboratories, Detroit, MI). An
inoculum
for the production phase was prepared by transferring the surface growth of
the
Micromonospora sp. from the agar plates to 2-L flasks containing 500 mL of
sterile
medium comprised of 10 g glucose, 20 g potato dextrin, 5 g yeast extract, 5 g
NZ-
Amine A, and 1 g CaC03 made up to one liter with tap water (pH 7.0). The
culture was
incubated at about 28°C for approximately 70 hours on a rotary shaker
set at 250 rpm.
Following incubation, 300 mL of culture was transferred to a 14.5 L fermentor
containing 10 L of sterile production medium. Each liter of production medium
was
composed of 20 g potato dextrin, 30 g glycerol, 2.5 g Bacto-peptone, 8.34 g
yeast
extract, 0.3 mL Silicone defoamer oil CChem Service), 0.05 ml Proflo oilT""
(Traders
protein) and 3 g CaC03 made to one liter with distilled water and adjusted to
pH 7Ø
The culture was incubated at 28 °C, with dissolved oxygen (d02)
controlled at 25% in a
cascade loop with agitation varied between 320-600 RPM and aeration set at a
fixed
rate of 0.5 v/v/m.
In addition to the above medium, other preferred media for the production of
Compound
1 by fermentation are provided in Table 1 (QB, MA, KH, RM, JA, FA, CL).
EXAMPLE 2: ISOLATION OF COMPOUND 1
Several isolation procedures were used to purify Compound 1, three different
conditions are exemplified herein.
a) Isolation procedure 1:
500 mL ethyl acetate was added to 500 mL of fermentation broth prepared as
described in Example 1 above. The mixture was agitated for 30 minutes on an
orbital
shaker at 200 rpm to create an emulsion. The phases were separated by
centrifugation
and decantation. Between 4 and 5 g of anhydrous MgS04 was added to the organic
phase, which was then filtered and the solvents removed in vacuo.
An ethyl acetate extract from 2 L fermentation was mixed with HP-20 resin (100
mL; Mitsubishi Casei Corp., Tokyo, Japan) in water (300 mL). Ethyl acetate was
removed in vacuo, the resin was filtered on a Buchner funnel and the filtrate
was
discarded. The adsorbed HP-20 resin was then washed successively with 2 x 125
mL
59
CA 02511750 2005-07-21
3005-31 CA
of 50% acetonitrile in water, 2x125 mL of 75% acetonitrile in water and 2 x
125 mL of
acetonitrile.
Fractions containing Compound 1 were evaporated to dryness and 100 mg was
digested in the 5 mL of the upper phase of a mixture prepared from chloroform,
cyclohexane, methanol, and water in the ratios, by volume, of 5:2:10:5. The
sample
was subjected to centrifugal partition chromatography using a High Speed
Countercurrent Chromatography (HSCC) system (Kromaton Technologies, Angers,
France) fitted with a 200 mL cartridge and prepacked with the upper phase of
this two-
phase system. The HSCC was run with the lower phase mobile and Compound 1 was
eluted at approximately one-half column volume. Fractions were collected and
Compound 1 was detected by TLC of aliquots of the fractions on commercial
Kieselgel
60F25a plates. Compound could be visualized by inspection of dried plates
under UV
light or by spraying the plates with a spray containing vanillin (0.75%) and
concentrated
sulfuric acid (1.5%, v/v) in ethanol and subsequently heating the plate.
Fractions
contained substantially pure Compound 1, although highly colored. A buff-
colored
sample could be obtained by chromatography on HPLC as follows.
6 mg of sample was dissolved in acetonitrile and injected onto a preparative
HPLC column (XterraTM ODS (10~m), 19x150mm, Waters Co., Milford, MA), with a 9
mL/min flow rate and UV peak detection at 300 nm. The column was eluted with
acetonitrile/buffer (5 mM of NH4HC03) according to the following gradient
shown in
Table 2.
Table 2
Preparative HPLC gradient
Time (min) Water (%) Acetonitrile (%)
0 50 50
0 100
0 100
50 50
50 50
Fractions containing Compound 1 were combined, concentrated and lyophilized
to give a yield of 3.8 mg compound.
b) Isolation procedure 2:
CA 02511750 2005-07-21
3005-31 CA
Compound 1 was also isolated using the following alternative protocol. At the
end of the incubation period, the fermentation broth from the baffled flasks
of Example
1 was centrifuged and the supernatant decanted from the pellet containing the
bacterial
mycelia. 100 mL of 100% MeOH was added to the mycelial pellet and the sample
was
stirred for 10 minutes and centrifuged for 15 minutes. The methanolic
supernatant was
decanted and saved. 100 mL of acetone was then added to the mycelial pellet
and
stirred for 10 minutes then centrifuged for 15 minutes. The acetonic
supernatant was
decanted and combined with the methanolic supernatant. Finally, 100 mL of 20%
MeOH/H20 was added to the mycelial pellet, stirred for 10 minutes and
centrifuged for
15 minutes. The supernatant was combined with the acetonic and methanolic
supernatants.
The combined supernatant was added to 400 ml of HP-20 resin in 1000 mL of
water and the organics were removed in vacuo. The resulting slurry was
filtered on a
Buchner funnel and the filtrate was discarded. HP-20 resin was washed
successively
with 2x500mL of 50% MeOH/H20, 2x500mL of 75% MeOH/H20 and 2x500mL of
MeOH.
The individual washes were collected separately and analyzed by TLC as
described above. Those fractions containing Compound 1 were evaporated to near
dryness and lyophilized. The lyophilizate was dissolved in methanol and
injected onto a
preparative HPLC column (XterraTM ODS (10~m), 19x150mm, Waters Co., Milford,
MA)
with a flow rate of 9 mL/min and peak detection at 300 nm.
The column was eluted with acetonitrile/buffer (5 mM of NH4HC03) according to
gradient shown in Table 3.
Table 3
Preparative HPLC gradient
Time (min) Buffer (%) Acetonitrile (%)
0 95 5
15 45 55
20 5 95
30 5 95
35 95 5
Fractions containing Compound 1 were combined, concentrated and lyophilized
to yield about 33.7 mg of compound.
61
CA 02511750 2005-07-21
3005-31 CA
c) Isolation procedure 3:
liters of the whole broth from Example 1 was extracted twice with equal
volumes of ethyl acetate and the two extracts were combined and concentrated
to
dryness. The dried extract was weighed, and for every gram of dry extract, 100
mL of
MeOH-H20 (2:1 v/v) and 100 mL of hexane was added. The mixture was swirled
gently
but well to achieve dissolution. The two layers were separated and the aqueous
layer is
washed with 100 mL of hexane. The two hexane layers were combined and the
combined hexane solution was washed with 100 mL methanol:water (2:1, v/v). The
two
methanol:water layers were combined and treated with 200 mL of EtOAc and 400
mL of
water. The layers were separated and the aqueous layer extracted twice further
with
200 mL portions of EtOAc. The EtOAc layers are combined and concentrated. The
residue obtained (220 mg) was suitable for final purification, either by HSCC
or by
HPLC as described above. This extraction process achieved a ten-fold
purification
when compared with the extraction protocol used in (a) or (b).
EXAMPLE 3: ELUCIDATION OF THE STRUCTURE OF COMPOUND 1
1" 2" 3"
O
1~
11a1/ _ 1~~2/~~ 4~ 5~ 6/7~ 8~ 9' 1 ~ 1' 12'
2
3
5N C28H34N2~4
Mol. Wt.: 462.25
OH
HO
The calculated molecular weight of the major isotope (462.25) and formula
(C28H34N2O4) of Compound 1 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 461.2 and positive ionization gave
an (M+H)+
molecular ion of 463.3. UVmax was determined to be 230nm with a shoulder at
290
nm.
Proton and carbon NMR spectral analysis is shown in Table 4. NMR data were
collected dissolved in MeOH-d4 including proton, carbon and multidimensional
pulse
sequences gDQCOSY, gHSQC, gHMBC, and NOESY. A number of cross peaks in the
62
CA 02511750 2005-07-21
3005-31 CA
2D spectra of Compound 1 are key in the structural determination. For example,
the
farnesyl chain is placed on the amide nitrogen by a strong cross peak between
the
proton signal of the terminal methylene of that chain at 4.52 ppm and the
amide
carbonyl carbon at 170 ppm in the gHMBC experiment. This conclusion is
confirmed
by a cross peak in the NOESY spectrum between the same methylene signals at
4.52
ppm and the aromatic proton signal at 6.25 ppm from one of the two protons of
the tetra
substituted benzenoid ring. Assignment of proton and carbon signals are shown
in
Table 4.
Table 4
'H and'3 C NMR (8H,
ppm) Data
of Compound
1 in MeOH-D4
Assignment'H '3C Group
1 7.15 122.3 CH
2 6.74 121.0 CH
3 6.83 116.9 CH
4 - 146.0 C-O H
4a - 142.0 C
5a - 126.0 C
6 - 148.2 C-O H
7 6.20 100.0 CH
8 - 153.0 C-OH
9 6.25 101.0 CH
9a - 135.0 C
11 - 170.0 C(O)
11 a - 125.0 C
1' 4.52 48.7 CH2
2' 5.35 121.1 CH
3' - 138.5 C
4' 2.03 39.5 CH2
5' 2.08 26.7 CHZ
6' 5.09 124.1 CH
7' - 135.0 C
8' 1.95 39.6 CH2
9' 2.02 26.3 CH2
10' 5.06 124.4 CH
11' - 130.9 C
12' 1.64 24.8 CH3
1 " 1.72 15.5 CH3
2" 1.59 14.9 CH3
3" 1.55 16.5 CH3
Based on the mass, UV and NMR spectroscopy data, the structure of the
compound was determined to be the structure of Compound 1 shown above.
63
CA 02511750 2005-07-21
3005-31 CA
EXAMPLE 4: DIALKYLSULFATE REACTIONS
a) Synthesis and structural elucidation of Comaounds 2 and 80
1" 2" 3"
O
1'
2 \1 a 1 1N 2~3 4 5 6~7~ 8~ g. 1 ~ 1, 12'
9a g
3
4~4a 5N 5a~ OH C29H36N204
-~ Mol. Wt.: 476.27
OH CH3 6
HO
Compound 2, namely 10-farnesyl-4,6,8-trihydroxy-5-methyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, and
1 " 2.. 3..
O
1' T
\1 a 1 1 2~3, 4' S, 6' 8' g' 1 ~ 1, 12'
2 ga H3C0
9
/4a off C H N O
N 5a 3o 40 2 5
_ Mol. Wt.: 508.29
nu ~ R 7
Compound 80, namely 10-(7-methoxy-3,7,11-trimethyldodeca-2,10-dienyl)-4,6,8-
trihydroxy-5-methyl-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one, were
prepared and
identified as follows:
Preparation:
Compound 1 (500.0 mg) was dissolved in methanol (MeOH, 20 mL) and stirred
with dimethyl sulfate (0.5 mL) and NaHC03 (250 mg) at room temperature for 48
hrs.
The reaction mixture was diluted to 200 mL by adding water and extracted with
ethyl
acetate (EtOAc, 300 mL x 3). The organic layer was separated and dried under
vacuum, re-dissolved in MeOH and filtered through a 0.45 pm 13 mm AcrodiscT""
GHP
syringe filter. The filtrate was subjected for isolation on a Waters HPLC
coupled to a
photodiode array detector. Compound 80 (12.1 mg) and Compound 2 (308.5 mg)
were
isolated by the multiple injections on Nova-PackTM HR 6pm C18 25 x 200 mm
column
(20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-8 min; 30:70-0:100, 8-18 min),
eluting
at 14.5 and 16.8 min, respectively.
Structural elucidation of Compounds 2 and 80:
64
CA 02511750 2005-07-21
3005-31 CA
The calculated molecular weight for the major isotope (476.27) and formula
(C29H36N2O4) of Compound 2 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 475.6 and positive ionization gave
an
(M+Na)+ molecular ion of 499.4. Proton and carbon NMR spectral analysis is
shown in
Table 5. Signals were easily assigned based on Compound 1 structure knowledge.
The
calculated molecular weight for the major isotope (508.29) and formula
(C3oH4oN2O5) of
Compound 80 was confirmed by mass spectral analysis: negative ionization gave
an
(M-H)- molecular ion of 507.3 and positive ionization gave an (M+H)+ molecular
ion of
509.3. The characteristic N-methyl (signal 5), methoxy (signal 7'-OMe) and the
methylene group (6'), from the addition of methanol on the farnesyl chain were
easily
assigned as shown in Table 5.
Table 5
NMR (~, ppm) Data of Compounds 2 and 80 in MeOH-D4
Compound Compound 80
2
Assignment~ Grou
p
H C H
1 7.21 122.1 7.21 CH
2 7.14 127.3 7.14 CH
3 7.02 118.4 7.02 CH
4 - 152.6 - C-OH
4a - 139.3 - C
5-N-Me 2.92 41.1 2.93 N-CH3
5a - 125.4 - C
6 - 154. - C-O H
8
7 6.22 99.6 6.20 CH
8 - 156.8 - C-O H
9 6. 34 101.4 6. 34 C H
9a - 142.0 - C
11 - 168.2 - C(O)
11 a - 133.5 - C
1' 4.83, 4.58 47.7 4.89, 4.57 CH2
2' 5.44 119. 5.42 C H
8
3' - 139.3 - C
4' 2.07 b 39.5 2.06 CHz
5' 2.12 b 26.2 1.42 CHZ
6' 5.10 123.8 1.42 CH (CHZ)a
7' - 135.1 - C
7' OMe N/A N/A 3.13 OCH3
8' 1.95 b 39.8 1.42 CHZ
9' 2.04 b 26.8 1.93 CHZ
10' 5.07 124.3 5.12 CH
11 - 130.8 - C
12' 1.65 24.9 1.68 CH3
CA 02511750 2005-07-21
3005-31 CA
Table 5
NMR (8, ppm) Data of Compounds 2 and 80 in MeOH-D4
Assignment 1 H ompound 2 C Comp H nd 80 Group
1 " 1.78 15.8 1.77 CH3
2" 1.60 15.1 1.10 CH3
3" 1.55 16.6 1.60 CH3
N/A: not applicable, group not present in the molecule
a. CH in Compound 2, CH2 in Compound 80
b. Signals for 4', 5', 8' and 9' are very close; assignment was based on
Compound 1
b) Synthesis and structural elucidation of Compounds 14, 82, 83, 84, 85, and
87
1 " 2.. 3..
O
1'
\ 1 al l 1N 2~3, 4, 5, 6~~, g' 9, 1 ~ 1, 12'
\2
9a g
3 8
~a ~ off C H N O
4 5 N 5a 30 38 2 4
Mol. Wt.: 490.28
nu 6
HO
Compound 14: 10-farnesyl-4,6,8-trihydroxy-5-ethyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one;
1 " 2.. 3,.
O
1 11 10 ~ 2~~ 4' S' 6~ T 8' g~ 10' 11 ~ 12'
\1a
2 H3C0
/a Compound 82: Compound 85:
4 5 N R: H R: Et
OH R~ C29H38N2~5 C31H42N205
Mol. Wt.: 494.28 Mol. Wt.: 522.31
Compound 82: 10-(11-methoxy-3,7,11-trimethyl-2,6-dodecadienyl)-4,6,8-
trihydroxy-
5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one;
Compound 85: 10-(11-methoxy-3,7,11-trimethyl-2,6-dodecadienyl)-4,6,8-
trihydroxy-5-
ethyl-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one;
1,. 2" 3..
O
T 11'
1 11 10 ~2~~ 4' S' 6~ 8' g~ 10' 12'
\ 11a
\2
H3C0 H3C0
3 ~4a Compound 84: Compound 87:
4 5 N R: H R: Et
OH R~ C30H42N2~6 C32H46N2~5
Mol. Wt.: 526.30 Mol. Wt.: 554.34
66
CA 02511750 2005-07-21
3005-31 CA
Compound 84: 10-(7,11-dimethoxy-3,7,11-trimethyl-2-dodecenyl)-4,6,8-trihydroxy-
5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one;
Compound 87: 10-(7,11-dimethoxy-3,7,11-trimethyl-2-dodecenyl)-4,6,8-trihydroxy-
5-
ethyl-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one;
Preparation:
Compound 1 (85.3 mg) was stirred for 72 hrs at room temperature in a mixture
of
diethyl sulfate (2.0 mL) and NaHC03 (99.9 mg) in MeOH (2 mL). The resulting
mixture
was filtered through a 0.45 pm 13 mm AcrodiscT"" GHP syringe filter. The
solution was
purified by preparative HPLC (multiple injections on a NovaPackT"" C-18 25x200
mm
column (20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-8 min; 30:70-0:100, 8-18
min)
to give four major peaks: Compound 14 (20.0 mg with some impurities, RT: 16.6
min),
Compound 82 (5.65 mg, RT: 11.6 min), Compound 84 (2.20 mg, RT: 10.3 min),
Compound 85 (17.54 mg, RT: 14.3 min) and Compound 86 (7.82 mg, RT: 12.6 min)
were obtained. The fraction containing Compound 14 was further purified by
HPLC
using the same column (20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-8 min;
30:70-
0:100, 8-18 min, curve 7), to give substantially pure Compound 14 (13.85 mg,
RT: 17.9
min).
Structural elucidation of Compounds 14, 82, 84, 85 and 87:
The calculated molecular weight of the major isotope (490.28) and formula
(C3oH38N2O4) of Compound 14 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 489.3 and positive ionization gave
an (M+H)+
molecular ion of 491.3. Proton NMR signals were easily assigned based on
Compounds 1 and 2 structures knowledge. The characteristic N-ethyl group (5-N-
Et)
was easily assigned as shown in Table 6.
The calculated molecular weight of the major isotope (494.28) and formula
(C29H38N2O5) of Compound 82 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 493.3 and positive ionization gave
an (M+H)+
molecular ion of 495.4, and a fragment having an (M+H-HOCH3)+ molecular ion of
463.3. Proton NMR signals were easily assigned based on Compound 1 structure
knowledge. The characteristic methoxy group (11'-OMe) and the methylene group
(10'),
from the addition of methanol on the farnesyl chain were easily assigned as
shown in
Table 6.
67
CA 02511750 2005-07-21
3005-31 CA
The calculated molecular weight (526.30) and formula (C3oH42N2O6) of
Compound 84 was confirmed by mass spectral analysis: negative ionization gave
an
(M-H)- molecular ion of 525.3 and positive ionization gave an (M+H)+ molecular
ion of
527.4, and fragments having respectively (M+H-HOCH3)+ and (M+H-HOCH3-HOCH3)+
molecular ion of 495.4 and 463.4. Proton NMR signals were easily assigned
based on
Compound 1 structure knowledge. The characteristic methoxy groups (signals 7'-
OMe
and 11'-OMe) from the addition of two molecules of methanol on the farnesyl
chain
were easily assigned as shown in Table 6. The methylene groups (5', 6', 8', 9'
and 10')
were found to have similar chemical shifts, which is consistent with a
saturated chain.
The calculated molecular weight of the major isotope (522.31 ) and formula
(C3~ H42N205) of Compound 85 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 521.3 and positive ionization gave
an (M+H)+
molecular ion of 523.5, and a fragment having an (M+H-HOCH3)+ molecular ion of
491.3. The characteristic N-ethyl group (5-N-Et), and the methoxy (11'-OMe)
and
methylene (10') groups from the addition of methanol on the farnesyl chain
were easily
assigned as shown in Table 6.
The calculated molecular weight of the major isotope (554.34) and formula
(C32H46N2O6) of Compound 87 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 553.4 and positive ionization gave
an (M+H)+
molecular ion of 555.4, and fragments having respectively (M+H-HOCH3)+ and
(M+H-
HOCH3-HOCH3)+ molecular ion of 523.5 and 491.3. The characteristic N-ethyl
group (5-
N-Et), and methoxy (7'-OMe and 11'-OMe) groups from the addition of two
molecules of
methanol on the farnesyl chain were easily assigned as shown in Table 6. The
methylene groups (5', 6', 8', 9' and 10') were all found to have similar
chemical shifts,
which is consistent with the saturated alkyl group.
Table 6
'H NMR
(8H,
ppm)
Data
of Compounds
14, 82,
84, 85
and 87
in MeOH-D4
Assignment14 82 84 85 87 Group
1 7.20 7.17 7.17 7.20 7.20 CH
2 7.13 6.77 6.76 7.14 7.14 CH
3 7.02 6.85 6.83 7.03 7.03 CH
5-N-Et 3.23, 3.15N/A N/A 3.23, 3.153.24, 3.16 CHZ
(C1 )
5-N-Et 1.07 N/A N/A 1.09 1.09 CH3
(C2)
7 6.22 6.23 6.22 6.22 6.22 CH
9 6.34 6.28 6.27 6.34 6.34 CH
1' 4.58, 4.564.55 4.57 4.82, 4.564.91, 4.54 CHZ
68
CA 02511750 2005-07-21
3005-31 CA
Table 6
'H NMR (8H, ppm) Data of Compounds 14, 82, 84, 85 and 87 in MeOH-D4
Assignment 14 82 84 85 87 Group
2' 5.41 5.37 5.37 5.41 5.38 CH
4' 2.06 2.07 2.04 2.07 2.05 CH2
5' 2.11 2.13 * 2.13 ** CH2
6' 5.10 5.12 * 5.11 ** CH (CHZ)
a
7'-OMe N/A N/A 3.18 N/A 3.17 OCH3
8' 1.95 1.97 * 1.95 ** CHZ
9' 2.04 1.40 * 1.38 ** CH2
10' 5.07 1.40 * 1.38 ** CH (CH2)
b
11'-OMe N/A 3.14 ' 3.11 3.13 3.12 OCH3
12' 1.65 1.10 1.16 1.08 1.14 CH3
1 " 1.77 1.74 1.72 1.78 1.78 CH3
2" 1.60 1.61 1.10 1.10 1.10 CH3
3" 1.55 1.10 1.16 1.08 1.14 CH3
N/A: not applicable, group not present in the molecule
* signal 1.22-1.51 ppm, 10 protons
** signal 1.22-1.49 ppm, 10 protons
a. CH in Compounds 14, 82 and 85, CHZ in Compounds 84 and 87
b. CH in Compound 14, CH2 in Compounds 82, 84, 85 and 87
Alternate procedure, preparation of Compounds 82 and 83:
Compound 1 (107.2 mg) and p-toluenesufonic acid (pTSA, 13.8mg) were stirred
reflux in methanol for 30 minutes. The reaction was filtered and subjected to
Waters
HPLC purification (multiple injections on a NovaPackT"" C-18 25x200 mm column:
20
mL/min, H20/CH3CN gradient 70:30-20:80, 0-4 min; 20:80-0:100, 4-9 min) to give
Compound 82 (8.5 mg, RT 7.2 min) and Compound 83 (4.3 mg, RT 7.7 min).
Structural elucidation of Compound 83 was done as for Compounds 82 and 84.
Mass spectral analysis (ES+: 495.5; ES-: 493.3) confirmed a calculated
molecular
weight of the major isotope (494.28) and formula (C29H38N2O5) as for Compound
82.
Proton NMR analysis showed signals 6' (1.42 ppm, CH2) and 7'-OMe (3.13 ppm,
OCH3)
corresponding to the addition of a methanol molecule on the second double bond
of the
farnesyl group (as for Compound 80).
c) Synthesis and structural elucidation of Compound 63
69
CA 02511750 2005-07-21
3005-31 CA
2
3
~ z'
Compound 63, namely 10-farnesyl-4,6,8-trihydroxy-5-n-propyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified as follows:
Preparation:
Compound 1 (46.7 mg) was stirred for 72 hrs at room temperature in a mixture
of
dipropyl sulfate (0.5 mL) and NaHC03 (46.3 mg) in MeOH (3 mL). The resulting
mixture
was filtered through a 0.45 pm 13 mm AcrodiscT"" GHP syringe filter. The
solution was
purified by preparative HPLC (multiple injections on a NovaPackT"' C-18 25x200
mm
column: 20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-8 min; 30:70-0:100, 8-18
min)
to give substantially pure Compound 63 (18.0 mg, RT: 17.3 min).
Structural elucidation of Compound 63:
The calculated molecular weight of the major isotope (504.30) and formula
(C31H40N2~4) of Compound 63 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 503.4 and positive ionization gave
an (M+H)+
molecular ion of 505.5. Proton NMR signals were easily assigned based on
Compounds 1 and 2 structures knowledge. The characteristic N-Propyl group (5-N-
Pr
(C1 to C3)) was easily assigned as shown in Table 7 below.
d~ Synthesis and elucidation of Compound 98
2
3
2..
O
111' 1'
11a/ _10 2~3, 4. 5. 6~~, g~ 9. 1~1~ 12'
9
4 5N~5a _ C29H33~3N204
off ~ 6 7 Mol. Wt.: 479.29
CD3
Compound 98: 10-Farnesyl-4,6,8-trihydroxy-5-(trideuteriomethyl)-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified according to the
" ~~~ z"
CA 02511750 2005-07-21
3005-31 CA
following procedure:
Preparation:
Compound 1 (121.3 mg) was dissolved in MeOH (3.0 mL), dimethyl sulfate-ds
(150 pL, CDN isotopes Inc.) and NaHC03 (58.1 mg) were added, and the reaction
was
stirred at room temperature overnight. The reaction mixture was filtered and
the filtrate
was subjected to Waters HPLC purification (multiple injections on Nova PackT""
C-18 25
x 200 mm column: 20 mL/min, H20/CH3CN gradient 70:30-20:80, 0-4 min; 20:80-
0:100,
4-9 min) to give Compound 98 (82.7 mg, RT 9.4 min).
Structural elucidation:
The calculated molecular weight of the major isotope (479.29) and formula
(C2gH33D3N2O4) of Compound 98 was confirmed by mass spectral analysis:
negative
ionization gave an (M-H)- molecular ion of 478.5, and positive ionization gave
an
(M+H)+ molecular ion of 480.6. The structure was further confirmed by proton
NMR
spectrum as shown in Table 7 below.
Table 7
'H NMR
(8H, ppm)
Data of
Compounds
63 and
98 in
MeOH-D4
Assign Compound Compound 98 Group
63
1 7.19 7.21 CH
2 7.12 7.14 CH
3 7.01 7.02 CH
5-N-Pr 3.04, 3.15 - CHZ a
(C1 )
5-N-Pr 1.50 N/A CHZ
(C2)
5-N-Pr 0.91 N/A CH3
(C3)
7 6.22 6.22 C H
9 6.34 6.34 CH
1' 4.54, 4.88 4.83, 4.59 CHZ
2' 5.40 5.44 CH
4' 2.07 2.07 CH2
5' 2.12 2.12 CHz
6' 5.09 5.10 CH
8' 1.96 1.95 CHz
9' 2.03 2.03 CHZ
10' 5.07 5.07 CH
12' 1.65 1.65 CH3
1 " 1.78 1.77 CH3
2" 1.60 1.60 CH3
3" 1.55 1.55 CH3
N/A: not applicable, group not present in the molecule
a. CHZ in Compound 63, CD3 in Compound 98.
71
CA 02511750 2005-07-21
3005-31 CA
EXAMPLE 5: ALKYL HALIDE REACTIONS
a) Synthesis and structural elucidation of Comaound 3
1 " 2.. 3"
O
1'
1 11 10 2~3, q. 5. 6~~~ g' g. 1 ~ 1 ~ 12'
\ 11a ~N
2 9a g
~4a ~ ~ $ OH
4 5 N 5a
OH 6 7 C'35H40N2~4
Mol. Wt.: 552.30
HO
Compound 3, namely 5-benzyl-10-farnesyl-4,6,8-trihydroxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one was prepared and identified as follows:
Preparation:
Compound 1 (60.5 mg) was stirred 84 hrs with benzyl chloride (1.8 mL, 120 eq,
Sigma) in presence of two drops of pyridine (Aldrich). The resulting mixture
was directly
subjected to HPLC separation. Purification by multiple injection on a
WatersT"" RCM
Nova-PakT"" HR C18 6pm 60A 25 x 200 mm column (20 mL/min H20/CH3CN 80:20-
30:70, 0-8 min; 30:70-0:100, 8-18 min) gave Compound 3 (46.0 mg) with
retention time
of 17.5 min.
Structural elucidation:
The calculated molecular weight of the major isotope (552.30) and formula
(C35H40N2~4) of Compound 3 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 551.7 and positive ionization gave
an
(M+Na)+ molecular ion of 575.5. Proton NMR signals were easily assigned based
on
Compound 1 structure knowledge. The characteristic N-benzyl group (5-N-alkyl
(C1-
C5)) were assigned as shown in Table 8 below.
b) Synthesis and structural elucidation of Compound 64
72
CA 02511750 2005-07-21
3005-31 CA
1" 2.. 3..
O
1'
2 \1 al l 1N 2~3, 4, 5. 6~~, g' g. 1 ~ 1, 12'
9a g
~4a ~ ~ a OH C32H42N2~4
N 5a Mol. Wt.: 518.31
OH 6 7
HO
Compound 64, namely 10-farnesyl-4,6,8-trihydroxy-5-n-butyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified as follows:
Preparation:
Compound 1 (43.5 mg) was stirred in 1-bromobutane (2.0 mL) with pyridine (50
pL) at 80 °C overnight. The reaction mixture was diluted with MeOH (1.0
mL), filtered
and subjected for Waters HPLC as described above (in a) to give a semi-
purified
Compound 64 (RT: 18.1 min). The semi-purified compound was further purified
using
the same conditions (except with curve 7) to give substantially pure Compound
64 (10.5
mg, RT: 17.9 min).
Structure elucidation:
The calculated molecular weight of the major isotope (518.31 ) and formula
(C32H42N2O4) of Compound 64 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 517.4 and positive ionization gave
an (M+H)+
molecular ion of 519.5. The characteristic N-n-butyl group (5-N-alkyl (C1-C4))
was
easily assigned as shown in Table 8 below.
c) Synthesis and structural elucidation of Compound 67
1 " 2.. 3..
O
1'
\ 1 al l 1N 2~3, 4, 5, 6~~, g' g, 1 ~ 1, 12'
\2
9a g
a OH C34H46N2~4
g 5 N 5a Mol. Wt.: 546.35
OH 6
HO
Compound 67, namely 10-farnesyl-4,6,8-trihydroxy-5-n-hexyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified as follows:
73
CA 02511750 2005-07-21
3005-31 CA
Preparation:
Compound 1 (39.2 mg) was stirred in 1-bromohexane (2.0 mL) with pyridine (50
pL) at 80 °C overnight. The reaction mixture was diluted with MeOH (1.0
mL), filtered
and subjected for Waters HPLC (multiple injections on a NovaPackT"' C-18
25x200 mm
column: 20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-8 min; 30:70-0:100, 8-18
min;
isocratic CH3CN 18-24 minutes) to give substantially pure Compound 67 (14.0
mg, RT:
20.1 min).
Structural elucidation:
The calculated molecular weight (546.35) and formula (C34HasN204) of
Compound 67 was confirmed by mass spectral analysis: negative ionization gave
an
(M-H)- molecular ion of 545.6 and positive ionization gave an (M+H)+ molecular
ion of
547.6. Proton NMR signals were easily assigned based on Compounds 1 and 2
structures knowledge. The characteristic N-n-hexyl group (5-N-alkyl (C1-C6))
was easily
assigned as shown in Table 8 below.
d~ Synthesis and structural elucidation of Compound 77
i 1 " 2.. 3..
4' S' 6~7' $' g' ~ ~ ~ ~ 12'
2
C32H42N2~4
Mol. Wt.: 518.31
Compound 77, namely 10-Farnesyl-4,6,8-trihydroxy-5-sec-butyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified as follows:
Preparation procedure 1:
Compound 1 (26.0 mg) was stirred in 2-bromobutane (4.0 mL) with pyridine (100
pL) under reflux for one hour. The reaction mixture was concentrated in vacuo,
diluted
with MeOH (2.0 mL), filtered and subjected for Waters HPLC as described above
(in a)
to give Compound 77 (1.65 mg, RT: 18.0 min).
Preparation procedure 2:
Compound 1 (104.5 mg) was stirred in 2-bromobutane (5.0 mL) with pyridine
(400 pL) under reflux for two hours. The reaction mixture was concentrated in
vacuo,
74
CA 02511750 2005-07-21
3005-31 CA
diluted with MeOH (4.0 mL), filtered and subjected for Waters HPLC (multiple
injections
on a NovaPackT"" C-18 25x200 mm column: 20 mL/min, H20/CH3CN gradient 70:30-
20:80, 0-4 min; 20:80-0:100, 4-9 min) to give Compound 77 (7.38 mg, RT: 11.2
min).
Structure elucidation:
The calculated molecular weight of the major isotope (518.31 ) and formula
(C32H42N2O4) of Compound 64 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 517.4 and positive ionization gave
an (M+H)+
molecular ion of 519.6. The characteristic N-sec-butyl group (5-N-Alkyl (C1-
C4)) was
easily assigned as shown in Table 8 below.
Table 8
'H
NMR
(8H,
ppm)
Data
of
Compounds
3,
64,
67
and
77
in
MeOH-D4
Assign Compound Compound Compound Compound Group
3 64 67 77
1 7.19 7.20 7.20 7.19 CH
2 7.09 7.13 7.12 7.14 CH
3 6.96 7.02 7.02 7.01 CH
5-N-alkyl(C1 4.34, 4.25 3.18, 3.09 3.18, 3.08 3.29
)
(C2) - 1.46 1.48 1.34
(C3) 7.20 1.35 1.30 086, 0.81
"
(C4) 7.23 0.89 1.30 1.00, 0.96"
(C5)e7.23 N/A 1.30 N/A
(C6) N/A N/A 0.89 N/A
7 6.16 6.22 6.23 6.22 CH
9 6.31 6.34 6.33 6.34, 6.32" CH
1' 4.55, 4.48 4.89, 4.54 4.92, 4.52 4.90, 4.53 CH2
2' 5.44 5.40 5.39 5.41, 5.36 CH
"
4' 9 2.08 2.06 2.06 2.06 CHZ
5' g 2.13 2.12 2.11 2.13 CH2
6' 5.10 5.09 5.09 5.10 CH
8' g 1.94 1.96 1.96 1.97 CHZ
9' 9 2.01 2.04 2.04 2.05 CH2
10' 5.04 5.07 5.07 5.08 CH
12' 1.64 1.65 1.64 1.66 CH3
1" 1.79 1.78 1.78 1.80, 1.79" CH3
2" 1.56 1.60 1.60 1.61 CH3
3" 1.52 1.55 1.56 1.56 CH3
N/A: not applicable, group not present in the molecule
a. CHZ in Compounds 3, 64 and 67, and CH in Compound 77.
b. C in Compound 3, CHZ in Compounds 64, 67 and 77.
c. CH (2H) in Compound 3, CH2 in Compounds 64 and 67, and CH3 in Compound 77.
d. CH (2H) in Compound 3, CH3 in Compounds 64 and 77, and CHZ in Compound 67.
e. CH in Compound 3, CHZ in Compound 67, absent in Compounds 64 and 77.
f. Absent in Compounds 3, 64 and 77.
g. Signals at 4', 5', 8' and 9' are very close; assignment was based on
Compound 1
h. From two different isomers.
CA 02511750 2005-07-21
3005-31 CA
Other N-Alkyl Compounds 60 to 62, 65, 66, and 68 to 76 are also prepared via
this
procedure, by reaction of Compound 1 with the appropriate alkyl halide.
EXAMPLE 6: DIAZOALKANE O-ALKYLATIONS
1 2.. 3..
"
O
1'
1 2/ 6/ ~ 1~
$ '
12'
1a 3 ~ g 1
N 5
2 ~
9a g
3 ~ ~ 8 x Compound 4-OMe6-OH8-OH
4:
as Compound 4-OH6-OMe8-OH
4 \N 5a 5:
5
Compound 4-OH,6-OMe8-OMe
6:
x g 7 Compound 4-OMe6-OMe8-OH
7:
Compound 4-OMe6-OMe8-OMe
8:
x
Compounds 4 and 5: 10-farnesyl-6,8-dihydroxy-4-methoxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one and 10-farnesyl-4,8-dihydroxy-6-methoxy-5,10-
dihydro-dibenzo[b,e][1,4]diazepin-11-one, are monomethylated have a calculated
molecular weight of the major isotope of 476.27 g/mol and a formula of
C29H36N204.
Compounds 6 and 7: 10-farnesyl-4-hydroxy-6,8-dimethoxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one and 10-farnesyl-8-hydroxy-4,6-dimethoxy-5,10-
dihydro-dibenzo[b,e][1,4]diazepin-11-one, are dimethylated and have a
calculated
molecular weight of the major isotope of 490.28 g/mol and a formula of
C3oH38N204.
Compound 8: 10-farnesyl-4,6,8-trimethoxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-
one, is trimethylated and has a calculated molecular weight of the major
isotope of
504.30 g/mol and a formula of C3~H4oN2Oa.
All O-methylated compounds (4 to 8) were prepared and identified according to
the following procedure:
Preparation:
Compound 1 (20.0 mg) in MeOH (2.0 mL) was treated with excess of CH2N2 in
diethyl ether and the mixture stirred overnight. The resulting mixture was
separated by
preparative TLC (Merck Silica gel 60 F25a), using 2.5% MeOH in CHC13 as
eluent. A
mixture of Compounds 4 and 5 (1.0 mg), Compound 6 (0.5 mg), Compound 7 (5.5
mg)
and Compound 8 (3.0 mg) were isolated with Rf value of 0.09, 0.35, 0.39 and
0.92
respectively.
76
CA 02511750 2005-07-21
3005-31 CA
Structural elucidation:
The calculated molecular weights of the major isotopes (mono: 476.27, di:
490.28 and tri: 504.30) and formulae (mono: C29H36N2O4, di: C3oH38N2O4 and
trl:
C3~H4pN2O4) respectively of mono methylated (Compounds 4 and 5), dimethylated
(Compounds 6 and 7) and trimethylated (Compound 8) were confirmed by mass
spectral (MS) analysis. MS of both Compounds 4 and 5 gave a (M-H)- molecular
ion of
475.5 by negative ionization and a (M+Na)+ molecular ion of 499.4 by positive
ionization. MS of Compound 6 gave a (M-H)- molecular ion of 489.5 by negative
ionization and a (M+Na)+ molecular ion of 513.4 by positive ionization. MS of
Compound 7 gave a (M-H)- molecular ion of 489.5 by negative ionization and a
(M+Na)+
molecular ion of 513.4 by positive ionization. MS of Compound 8 gave a (M-H)-
molecular ion of 503.6 by negative ionization and a (M+Na)+ molecular ion of
527.4 by
positive ionization. Proton NMR spectral analysis for Compounds 4 to 8 is
shown in
Table 9. Signals were easily assigned based on comparison with the spectra of
Compound 1.
Table 9
' H NMR
(8H,
ppm)
Data
of Compounds
4, 5,
6, 7
and 8
in MeOH-D4
Assignment4 5 6 7 8 Group
1 7.28 7.17 7.19 7.28 7.29 CH
2 6.91 6.78 6.86 6.92 6.92 CH
3 7.03 6.86 6.79 7.03 7.03 CH
4 3.94 N/D N/D 3.93 3.94 C-X a
N/D N/D N/D N/D 6.95 NH
6 N/D 3.87 3.91 3.87 3.89 C-X b
7 6.28 6.23 6.46 6.33 6.45 CH
8 N/D N/D 3.75 N/D 3.74 C-X
9 6.38 6.34 6.52 6.38 6.57 CH
1' 4.56 4.56 4.61 4.57 4.60 CHZ
2' 5.36 5.36 5.38 5.34 5.31 CH
4' d 2.05 2.05 2.05 2.05 2.04 CHz
5' d 2.10 2.10 2.09 2.09 2.08 CHz
6' 5.09 5.09 5.07 5.08 5.06 CH
8' d 1.96 1.96 1.95 1.95 1.93 CHZ
9' d 2.04 2.04 2.04 2.04 2.03 CH2
12' 1.66 1.66 1.65 1.66 1.65 CH3
10' 5.09 5.09 5.07 5.08 5.06 CH
1 " 1.74 1.74 1.75 1.73 1.73 CH3
2" 1.60 1.60 1.60 1.59 1.59 CH3
3" 1.57 1.57 1.55 1.57 1.55 CH3
N/D: Not determined - not observed
a. X is OCH3 in Compounds 4, 7 and 8; X is OH in Compounds 5 and 6
77
CA 02511750 2005-07-21
3005-31 CA
b. X is OCH3 in Compounds 5, 6, 7 and 8; X is OH in Compound 4
c. X is OCH3 in Compounds 6 and 8; X is OH in Compounds 4, 5 and 7
d. Signals of 4', 5', 8' and 9' are very close; assignement was based on
Compound 1
EXAMPLE 7: ACETIC ANHYDRIDE O-ACYLATIONS
1 2.. 3..
"
O
1'
a 1 1 2~ , 5, 6~ 7, 1 12'
4, g' ~
g. 1,
3
2 ~1
9a g
~ ~ ~ $ X OA
4a Compound 6-OAc8-
9: c
4-OH
5 Compound 6-OAc8-OH
~ 10:
5a 4-OAc
- Compound 6-OH8-OAc
7 11:
4-OAc
Compound 6-OAc8-OAc
12:
4-OAc
Compounds 9, 10 and 11: 6,8-diacetoxy-10-farnesyl-4-hydroxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one; 4,6-diacetoxy-10-farnesyl-8-hydroxy-5,10-
dihydro-
dibenzo[b,e][1,4]diazepin-11-one and 4,8-diacetoxy-10-farnesyl-6-hydroxy-5,10-
dihydro-dibenzo[b,e][1,4]diazepin-11-one, are diacetylated and have a
calculated
molecular weight of the major isotope of 546.27 g/mol and a formula of
C32H38N206.
Compound 12: 4,6,8-triacetoxy-10-farnesyl-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-
one, is triacetylated and has a calculated molecular weight of the major
isotope of
588.28 g/mol and a formula of C34H4oN20~.
All acetylated compounds (9 to 12) were prepared and identified according to
the
following procedure:
Preparation:
Compound 1 (120.5 mg) was stirred overnight with acetic anhydride (720 pL, 29
eq, Aldrich) in presence of 6 drops of pyridine (Aldrich). The reaction
mixtures
submitted to HPLC separation. Purification by multiple injection on a Waters
TM RCM
Nova-Pak HRT"" C18, 6pm, 60A 25 x 200 mm column (20 mL/min H20/CH3CN 80:30-
70:75, 0-8 min; 30:70-0:100, 8-18 min and HPLC run for 20 min) gave Compound
11
(11.4 mg), Compound 10 (9.2 mg), Compound 9 (11.4 mg), Compound 12 (91.2 mg)
with retention time of 16.2, 17.6, 18.0 and 18.5 min, respectively.
Structural elucidation:
The calculated molecular weights of the major isotopes (di: 546.27 and tri:
588.28) and formulae (di: C32H38N206 and tri: C34H40N2O7) respectively of
diacetylated
(Compounds 9, 10 and 11 ) and triacetylated (Compound 12) were confirmed by
mass
78
CA 02511750 2005-07-21
3005-31 CA
spectral (MS) analysis. MS of Compound 9 gave a (M-H)- molecular ion of 545.6
by
negative ionization and a (M+Na)+ molecular ion of 569.4 by positive
ionization. MS of
Compound 10 gave a (M-H)- molecular ion of 545.6 by negative ionization and a
(M+Na)+ molecular ion of 569.5 by positive ionization. MS of Compound 11 gave
a (M-
H)- molecular ion of 545.5 by negative ionization and a (M+Na)+ molecular ion
of 569.4
by positive ionization. MS of Compound 12 gave a (M-H)- molecular ion of 587.6
by
negative ionization and a (M+Na)+ molecular ion of 611.5 by positive
ionization. Proton
NMR spectral analysis for Compounds 9 to 12 is shown in Table 10. Signals were
easily assigned based on comparison with the spectra of Compound 1.
Table 10
'H NMR (8H, ppm) Data of Compounds 9, 10, 11 and 12 in CDC13
Assignment 9 10 11 12 Group
1 7.27 7.72 7.75 7.75 CH
2 6.67 6.99 7.02 7.10 CH
3 6.67 7.15 7.15 7.16 CH
4 N/D 2.42 2.42 2.41 C-X a
6.45 5.90 6.29 6.15 NH
6 2.36 2.41 N/D 2.40 C-X b
7 6.82 6.52 6.15 6.96 CH
8 2.28 N/D 2.25 2.27 C-X
9 6.95 6.67 6.55 6.84 CH
1' 4.60 4.57 4.57 4.58 CHZ
2' 5.43 5.38 5.41 5.42 CH
4' d 2.08 2.06 2.05 2.06 CHZ
5' d 2.09 2.08 2.09 2.09 CHz
6' 5.10 5.09 5.09 5.10 CH
8' d 1.98 1.97 1.98 1.98 CHZ
9' d 2.08 2.06 2.05 2.06 CHz
10' 5.10 5.09 5.09 5.10 CH
12' 1.69 1.69 1.69 1.68 CH3
1 " 1.72 1.70 1.71 1.72 CH3
2" 1.61 1.60 1.60 1.61 CH3
3" 1.60 1.60 1.60 1.60 CH3
N/D: Not determined - not observed
a. X is OAc in Compounds 10, 11 and 12; X is OH in Compound 9
b. X is OAc in Compounds 9, 10 and 12; X is OH in Compound 11
c. X is OAc in Compounds 9, 11 and 12; X is OH in Compound 10
d. Signals of 4', 5', 8' and 9' are very close; assignement was based on
Compound 1
EXAMPLE 8: FARNESYL SIDE CHAIN MODIFICATIONS
a) Synthesis and structural elucidation of Compound 46
79
CA 02511750 2005-07-21
3005-31 CA
1" 2" 3"
O
1'
1 11 10 2' 3, 4, 5, g~ ~, g~ 9, 10' 11' 12'
\ 11a ~N
2 I \ ~ 9a 9
~4a ~ ~ $ OH
4 5N 5a
CzsHaoN2~a
OH H 6 ~ Mol. Wt.: 468.30
Compound 46: 10-(3,7,11-trimethyldodecyl)-4,6,8-trihydroxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified according to the
following procedure:
Preparation:
A solution of Compound 1 (51.1 mg in 3.0 mL MeOH) was stirred under
hydrogen gas overnight in presence of platinum oxide (Pt02,10 mg, 0.4 eq) as a
catalyst. The reaction mixture was filtered and purified by direct preparative
HPLC using
a Phenomenex SynergiT"" MAX RP 21.2 x 200 mm column (20 mL/min, H20/CH3CN
gradient 30:70-30:70, 0-2 min; 30:70-0:100, 2-20 min). Fractions having a
retention
time of 12.8 min were combined to give 45.2 mg of Compound 46.
Structural elucidation:
Calculated molecular weight of the major isotope (468.30) and formula
(C28H4oN204) were confirmed by mass spectral analysis. Compound 46 mass
spectra
gave a (M-H)- molecular ion of 467.4 by negative ionization and a (M+H)+
molecular ion
of 469.4 by positive ionization. Proton NMR spectral analysis of Compound 46
is shown
in Table 11 below. Signals were easily assigned based on Compound 1 structure
knowledge. As expected, aliphatic proton signals at positions 2'-11' all have
very close
chemical shifts ranging from about 1 to 1.75 ppm (integrating for 17 protons),
methyl
protons at positions 12' and 1 "-3" are all very close as well (shifts 0.8-
0.95 ppm,
integrating for 12 protons). These signals are also complex from the fact that
2
diastereomers of positions 3' and 7' are present in the mixture, and in
different
proportions. Labile protons were not observed since NMR was done in deuterated
methanol.
b) Synthesis and structural elucidation of Compound 78
CA 02511750 2005-07-21
3005-31 CA
1 " 2.. 3..
O
1'
1 11 10 2' 3, 4. 5, 6~ ~, g~ g, 10' 11' 12'
11a
2 i \ ~9a g
~aa ~ ~ $ off CzsHazNzOa
N 5a Mol. Wt.: 482.31
OH ~ H3
Compound 78, namely 10-(3,7,11-trimethyldodecyl)-4,6,8-trihydroxy-5-methyl-
5,10-
dihydro-dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified as
follows:
Preparation:
A solution of Compound 2 (23.7 mg) in MeOH (2.0 mL) was stirred under
hydrogen gas overnight in presence of platinum oxide (Pt02, 10 mg, catalyst)
as a
catalyst. The reaction mixture was filtered and concentrated in vacuo to give
21.6 mg of
Compound 78.
Structural elucidation:
The calculated molecular weight (482.31 ) and formula (C29H42N2O4) of
Compound 78 was confirmed by mass spectral analysis: negative ionization gave
an
(M-H)- molecular ion of 481.3 and positive ionization gave an (M+H)+ molecular
ion of
483.3. The farnesyl olefinic protons on the NMR spectra were replaced by
aliphatic
proton signals in the region of around 0.76-1.86 ppm, integrating for 17
protons, 3CH,
7CH2. The characteristic N-methyl group (5-N-Me) was easily assigned as shown
in
Table 11 below.
Table 11
1H NMR (bH,
ppm) Data
of Compounds
46 and 78
in CD30D
Assignment Compound 46 Compound 78 Group
1 7.15 7.18 CH
2 6.76 7.12 CH
3 6.84 7.01 CH
5-N-Me N/A 2.96, 2.95 CH3
b
7 6.24 6.23 CH
9 6.26 6.33, 6.35 CH
b
1' 4.16, 3.99 4.42, 3.86 CH2
2'-11' a '- 1.00-1.75 '-' 0.76-1.863CH, 7CH2 (17H)
12' and 1"-3" - 0.8-0.95 - 0.87-1.00 4CH3 (12H)
a
N/A: not applicable, group not present in the molecule
a. Signals are very close.
b. Mixture of isomers.
81
CA 02511750 2005-07-21
3005-31 CA
c) Synthesis and elucidation of Compounds 17 and 18 by epoxidation
1 " 2" 3.'
1'
2~ 4' S, 6' g' 9, 10~ 12'
N
2
O
3 ~ \8
OH
CzsH3aNz~s
Mol. Wt.: 478.25
6
HO Compound 17
1 " 2" 3..
1'
2~ 4' S~ g~ 8' 9, 10' 12'
2 O
9
8 OH
C28H34N2~5
Mol. Wt.: 478.25
OH H 6
HO Compound 18
Compound 17: 10-(3,7,11-trimethyl-6,7-epoxydodeca-2,10-dienyl)-4,6,8-
trihydroxy-
5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one, and
Compound 18: 10-(3,7,11-trimethyl-10,11-epoxydodeca-2,10-dienyl)-4,6,8-
trihydroxy-
5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one, were prepared and identified
according
to the following procedure:
Preparation:
A mixture of Compound 1 (24.0 mg) and 3-chloroperbenzoic acid (mCPBA, 7.8
mg, 0.9 eq) in THF (1.0 mL) were stirred overnight at room temperature. The
reaction
mixture was diluted with MeOH (1.0 mL) and subjected to purification on Waters
HPLC
using a Photodiode Array detector. The mixture was purified by multiple
injections on a
Waters TM RCM Nova-PakT"" C-18 25 x 200 mm column (20 mL/min, H20/CH3CN
gradient 80:20-30:70, 0-8 min; 30:70-0:100, 8-20 min). Pure Compound 17 (2.11
mg)
and Compound 18 (1.68 mg) were obtained by concentration in vacuo of the
combined
fractions respectively having retention time 11.2 min and 10.6 min.
Structural elucidation:
Calculated molecular weights of the major isotopes (478.25) and formulae
(C28H34N2O5) were confirmed by mass spectral analysis. Compound 17 mass
spectra
gave a (M-H)- molecular ion of 477.3 by negative ionization and a (M+H)+
molecular ion
82
CA 02511750 2005-07-21
3005-31 CA
of 479.4 by positive ionization. Compound 18 mass spectra gave a (M-H)-
molecular ion
of 477.3 by negative ionization and a (M+H)+ molecular ion of 479.4 by
positive
ionization. Proton NMR spectral analysis of Compounds 17 and 18 is shown in
Table
12. Signals were easily assigned based on Compound 1 structure knowledge. As
expected, epoxide protons signals were shifted upfield, compared to the alkene
protons
of Compound 1 (from 5.09 to 2.75 ppm for Compound 17, and from 5.06 to 2.73
ppm
for Compound 18). Exchangeable protons were not observed as NMR was done in
deuterated methanol.
Table 12
1H NMR (8H, ppm) Data of Compounds 17 and 18 in CD30D
Assignment Compound 17 Compound 18 Group
1 7.17 7.18 CH
2 6.77 6.77 CH
3 6.85 6.86 CH
7 6.22 6.23 CH
9 6.27 6.27 CH
1' 4.61, 4.54 4.55 CH2
2' 5.42 5.37 CH
4' 2.17 2.08 a CH2
5' 1.62, 1.42 2.13 a CH2
6' 2.75 5.16 CH
8' 1.62, 1.42 2.08 a CH2
9' 2.09 1.60 CH2
10' 5.09 2.73 CH
12' 1.67 1.26 CH3
1" 1.77 1.74 CH3
2" 1.26 1.64 CH3
3" 1.60 1.20 CH3
a. Signals are very close, and are interchangeable
d) Synthesis and structural elucidation of Compound 89 and 92
i 1" i2~~ i 3
1 11 10 1' 2~3, 4. 5, 6' T g' 9. 1 ~ ~ ~ 12'
\1a ~N
2 ~ 9a g X
~4a / ~ ° off Compound 89 Compound 92
4 5H 5a\ / X: OH X: NHAc
OH 6 77 C'28H36N2~5 C30H39N3~5
Mol. Wt.: 480.26 Mol. Wt.: 521.29
Compound 89, namely 10-(7-hydroxy-3,7,11-trimethyldodeca-2,10-dienyl)-4,6,8-
trihydroxy-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one, and
83
CA 02511750 2005-07-21
3005-31 CA
Compound 92, namely 10-(7-acetamido-3,7,11-trimethyldodeca-2,10-dienyl)-4,6,8-
trihydroxy-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-one, were prepared and
identified
as follows:
Preparation:
Compound 1 (20.0 mg) was dissolved in CH3CN (2.0 mL) and water (50 pL) and
pTSA (56.0 mg) was added. The solution was stirred under reflux for 30 min.
The
reaction mixture was filtered and the filtrate subjected to Waters HPLC
purification
(multiple injections on Nova-PackT"' C-18 25 x 200 mm column: 20 mL/min,
H20/CH3CN gradient 70:30-20:80, 0-4 min; 20:80-0:100, 4-9 min), to give
Compound
89 (0.73 mg, RT 10.0 min) and Compound 92 (0.33 mg, RT 10.5 min).
Structure elucidation of Compounds 89 and 92:
The calculated molecular weight of the major isotope (480.26) and formula
(C28H36N2O5) of Compound 89 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 479.8 and positive ionization gave
an (M+H-
H20)+ molecular ion of 464.1. The characteristic side chain signal (signal 6')
aliphatic
methylene was easily assigned as shown in Table 13 below.
The calculated molecular weight of the major isotope (521.29) and formula
(C3pH3gN3O5) of Compound 92 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 522.8 and positive ionization gave
an (M+H)+
molecular ion of 522.9. The characteristic side chain (signal 6') aliphatic
methylene and
the acetamide (signal 7'-NHAc) were easily assigned as shown in Table 13
below.
Table 13
'H NMR (bH, ppm) Data
of Compounds
89 and
92 in CD30D
Assignment Compound Compound 92 Group
89
1 7.18 7.17 CH
2 6.77 6.77 CH
3 6.86 6.85 CH
7 6.23 6.22 C H
9 6.29 6.31 CH
1' 4.56 4.54 CHZ
2' 5.39 5.40 CH
4' 2.05 2.05 CH2
5', 6', 8', 1.49-1.27 N/A 4 (CHZ)
9' a
5', 6', 8' - 1.77-1.40 b 3 (CHZ)
7'-X - 1.92 X
9' - 1.93 CH2
84
CA 02511750 2005-07-21
3005-31 CA
Table 13
'H NMR (8H, ppm) Data of Compounds 89 and 92 in CD30D
Assignment Compound 89 Compound 92 Group
10' 5.13 5.12 CH
12' 1.68 1.67 CH3
1 " 1.73 1.71 CH3
2" 1.14 1.23 CH3
3" 1.61 1.59 CH3
N/A: not applicable, group not present in the molecule
a. Signals 5', 6', 8' and 9' of Compound 89 are all very close.
b. Signals 5', 6' and 8' of Compound 92 are all very close.
c. In Compound 89, X is OH, in Compound 92, X is NHC(O)CH3.
e) Synthesis and structural elucidation of Compounds 95 and 96 by ozonolysis
1"
O O
1 11a 1 1N 1' 2~3' 4' S~ 6' OMe 1 11a 1 1N%'~OMe
9a g OMe
OMe
8
~4a 3 ~4a ~ OH
4 5H 4 5H 5a
OH OH 6 7
Compound 95 Compound 96
C22H26N2~6 ~'17H18N2~6
Mol. Wt.: 414.18 Mol. Wt.: 346.12
Compound 95, namely 10-(6,6-dimethoxy-3-methyl-2-hexenyl)-4,6,8-trihydroxy-
5,10-
dihydro-dibenzo[b,e][1,4]diazepin-11-one, and
Compound 96, namely 10-(6,6-dimethoxyethyl)-4,6,8-trihydroxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, were prepared and identified as follows:
Preparation:
Compound 1 (201.2 mg) was dissolved in MeOH (3.0 mL) and 03 (ozone) was
bubbled in the solution for 2 min at -80 °C (dry ice/acetone). Dimethyl
sulfide (146 ml)
was added and the reaction mixture was warmed up and stirred at room
temperature
for 24 hrs. The reaction mixture was filtered and the filtrate subjected to
purification on
a Waters Auto-Purification System (multiple injections on YMC-Pack ODS-AQ
column
20 x 250 mm: 20 mL/min, H20/CH3CN gradient: 75:25 isocratic 3 min, 75:25-5:95,
3-30
min; 5:95-0:100, 30-31 min and 100% CH3CN isocratic for 5 min), to give
Compound 95
(0.96 mg, RT 12.4 min) and Compound 96 (1.23 mg, RT 8.7 min).
Structural elucidation of Compounds 95 and 96:
The calculated molecular weight of the major isotope (414.18) and formula
CA 02511750 2005-07-21
3005-31 CA
(C22H2sN20s) of Compound 95 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 413.4 and positive ionization gave
an
(M+Na)+ molecular ion of 437.6. The characteristic farnesyl side chain proton
NMR
signals (7' to 11', and 2", 3") were replaced by an aliphatic carbon (signal
6') and two
methoxy groups (6'-OMe's), easily assigned as shown in Table 14 below.
The calculated molecular weight of the major isotope (346.12) and formula
(C17H1gN2O6) of Compound 96 was confirmed by mass spectral analysis: negative
ionization gave an (M-H)- molecular ion of 345.2 and positive ionization gave
an
(M+Na)+ molecular ion of 369.3. The characteristic farnesyl side chain proton
NMR
signals were replaced by aliphatic carbon (signal 2') and two methoxy groups
(2'-
OMe's), easily assigned as shown in Table 14 below.
Table 14
'H NMR (8H,
ppm) Data
of Compounds
95 and
96 in CD30D
Assignment Compound 95 Compound 96 Group
1 7.18 7.15 CH
2 6.77 6.77 CH
3 6.86 6.86 CH
7 6.23 6.24 CH
9 6.27 6.41 CH
1' 4.58 4.64 CH2
2' 5.35 4.72 CH
2'-OMe N/A 3.40 2 x OCH3
4' 2.07 N/A CH2
5' 1.66 N/A CHZ
6' 4.22 N/A CH
6'-OMe 3.37 N/A 2 x OCH3
1" 1.74 N/A CH3
N/A: not applicable, group not present in the molecule
EXAMPLE 9: AROMATIC SUBSTITUTION REACTION
Synthesis and structural elucidation of Compound 97 by bromination
1., 2" 3"
O
1'
2 \1a11 1N 2~3, 4~ 5, 6~7, g' 9, 10~ 1, 12'
I ~ ~ 9a 9
/4a ~ ~ 8 off C2aHssBrN204
4 5N 5a
Mol. Wt.: 540.16 (M)
off ~ 6 ~ Mol. Wt.: 542.16 (M+2)
HO
86
CA 02511750 2005-07-21
3005-31 CA
Compound 97: 10-(farnesyl)-7-bromo-4,6,8-trihydroxy-5,10-dihydro-
dibenzo[b,e][1,4]diazepin-11-one, was prepared and identified according to the
following procedure:
Preparation:
Compound 1 (116.0 mg) and N-bromosuccinimide (NBS, 45.5 mg) were
dissolved in tetrahydrofuran (THF, 3.0 mL) and stirred at room temperature for
4 days.
The reaction mixture was filtered and subjected to Waters HPLC purification
(Nova-
PackT"" C-18 25 x 200 mm column: 20 mL/min, H20/CH3CN gradient 80:20-30:70, 0-
8
min; 30:70-0:100, 8-18 min) to give Compound 97 (13.6 min) together with some
impurities. The semi-purified sample was further purified by HPLC (SymmetryT"'
C-18
25 x 100 mm column: 20 mL/min, H20/CH3CN gradient 70:30-30:70, 0-15 min), to
give
Compound 97 (9.5 mg, RT 13.0 min).
Structural elucidation:
The calculated molecular weight of the major isotopes (540.16 and 542.16) and
formula (C28H33BrN204) of Compound 97 was confirmed by mass spectral analysis:
negative ionization gave (M-H)- molecular ions of 539.2 and 541.1, and
positive
ionization gave (M+H)+ molecular ions of 541.3 and 543.2. The presence of the
two
molecular ions in each mass spectrum confirmed the presence of a bromine group
in
the molecule. The structure was further confirmed by the absence of the
aromatic (7)
signal in the proton NMR spectrum as shown in Table 15 below.
Table 15
'H NMR (8H,
ppm) Data
of Compound
97 in CD30D
Assignment Compound 97 Group
1 7.18 CH
2 6.79 CH
3 6.87 CH
9 6.49 CH
1' 4.56 CHZ
2' 5.34 CH
4' 2.06 CHz
5' 2.09 CHZ
6' 5.10 CH
8' 1.96 CHZ
9' 2.04 CHZ
10' 5.08 CH
12' 1.66 CH3
87
CA 02511750 2005-07-21
3005-31 CA
Table 15
'H NMR (8H, ppm) Data of Compound 97 in CD30D
Assignment Compound 97 Group
1 " 1.74 CH3
2" 1.60 CH3
3" 1.57 CH3
EXAMPLE 10: PHARMACOLOGICAL ACTIVITY PROFILE
Compound 1 and Compounds 2 to 12 and Compound 46 were tested for binding
against a variety of enzymes and/or receptors. The enzymes or receptors used
in these
assays were known to be involved in anticancer activity of known compounds, as
well
as other diseases, or related to such enzymes or receptors.
A. Enzymes and Receptors:
5-Lipoxygenase (5-LO) catalyzes the oxidative metabolism of arachidonic acid
to
5-hydroxyeicosatetraenoic acid (5-HETE), the initial reaction leading to
formation of
leukotrienes. Eicosanoids derived from arachidonic acid by the action of
lipoxygenases
or cycloxygenases have been found to be involved in acute and chronic
inflammatory
diseases (i.e. asthma, multiple sclerosis, rheumatoid arthritis, ischemia,
edema) as well
in neurodegeneration (Alzheimer's disease), aging and various steps of
carcinogenesis,
including tumor promotion, progression and metastasis. The aim of this study
was to
determine whether Compound 1, is able to block the formation of leukotrienes
by
inhibiting the enzymatic activity of human 5-LO.
Acyl CoA-Cholesterol Acyltransferase (ACAT) converts cholesterol to
cholesteryl
esters and is involved in the development of artherioscerosis.
Cyclooxygenase-2 (COX-2) enzyme is made only in response to injury or
infection. It produces prostaglandins involved in inflammation and the immune
response. Elevated levels of COX-2 in the body have been linked to cancer.
The peripheral benzodiazepine receptor (PBR or PBenzR) is a well-
characterized receptor known to be directly involved in diseases states. PBR
is involved
in the regulation of immune responses. These diseases states include
inflammatory
diseases (such as rheumatoid arthritis and lupus), parasitic infections and
neurodegenerative diseases (such as Alzheimer's, Huntington's and Multiple
Sclerosis).
This receptor is known to be involved in anticancer activity of known
compounds.
88
CA 02511750 2005-07-21
3005-31 CA
Leukotriene, Cysteinyl (CysLT~) is involved in inflammation and CysLT~-
selective
antagonists are used as treatment for bronchial asthma. CysLT, and 5-LO were
found
to be upregulated in colon cancer.
GABAA, the Central Benzodiazepine Receptor (CBenzR or CBR) is involved in
anxiolitic activities.
B. General Procedures:
The procedures used were based on known assays: ACAT (from rat; Ref: Largis
et al (1989), J. Lipid. Res., vol 30, 681-689), COX-2 (human; Ref: Riendeau et
al
(1997), Can. J. Physiol. Pharmacol., vol 75, 1088-1095 and Warner et al
(1999), Pro.
Natl. Acad Sci. USA, vol 96, 7563-7568), 5-LO (human; Ref: Carter et al (1991
), J.
Pharmacol. Exp. Ther., vol 256, no 3, 929-937, and Safayhi et al (2000),
Planta Medica,
vol 66, 110-113), PBR (from rat; Le Fur et al (1983), Life Sci. USA, vol 33,
449-457),
CysLT~ (human; Martin et al (2001 ), Biochem. Pharmacol., vol 62, no 9, 1193-
1200)
and CBR (from rat; Damm et al (1978), Res. Comm. Chem. Pathol. Pharmacol., vol
22,
597-600 and Speth et al (1979), Life Sci., vol 24, 351-357).
C. Binding assay of Compound 1 on 5-LO:
Human peripheral blood mononuclear cells (PMNs) were isolated through a
Ficoll-Paque density gradient. PMNs were stimulated by addition A23187 (30 pM
final
concentration). Stimulated PMNs were adjusted to a density of 5 x106 cells/mL
in HBBS
medium and incubated with the vehicle control (DMSO), Compound 1 (at final
concentrations of 0.1, 0.5, 1, 2.5, 5 and 10 pM) and NDGA as positive control
(at final
concentrations of 3, 1, 0.3, 0.1 and 0.03 pM) for 15 minutes at 37°C.
Following
incubation, samples were neutralized with NaOH and centrifuged. Leukotriene B4
content was measured in the supernatant using an Enzyme Immunosorbant Assay
(EIA) assay. The experiment was performed in triplicate.
Results shown in Figure 1 demonstrated that Compound 1 inhibited the activity
of human 5-LO with an apparent ICSO = 0.93 pM (versus 0.1 pM for the positive
control
NDGA) and therefore displays anti-inflammatory properties.
D. Percentage inhibition or binding of Compounds 1-12 and 46:
Binding assays were done for each of Compounds 1-12 and 46 using ACAT,
COX-2, 5-LO, PBR and CysLT~ enzymes. The procedures used are based on the
respective references mentioned above and the conditions are summarized in
Tables
16 (enzyme assays) and 17 (radioligand receptor assays).
89
CA 02511750 2005-07-21
3005-31 CA
Table 16
Enzyme Assays Conditions
Source Substrate Pre-I a I b
ACAT ° Wistar rat hepatic 12.7 pM ['4C]palmitoyl CoA
15min/37°C 10min/37°C
microsomes
a Human recombinant
COX-2 insect Sf21 cells 0.3 pM arachidonic acid 15min/37°C
5min/37°C
5-LO a Human PBML cells Arachidonic acid 15min/37°C 15min/37°C
a. Pre-Incubation Time/Temperature
b. Incubation Time/Temperature
c. Incubation buffer: 0.2 M phosphate buffer (pH 7.4 at 25°C); Method:
Quantitation of
['4C]cholesterol ester by column chromatography.
d. Incubation buffer: 100mM Tris-HCI, pH 7.7, 1 mM glutathione, 1 pM hematin,
500pM phenol;
Method: EIA quantitation of PGE2.
e. Incubation buffer: HBSS (Hank's balanced salt solution); Method: EIA
quantitation of LTB4.
Table 17
Radioligand Binding Assays Conditions a
Source Ligand I b Non-spec
ligand
PBR ° Wistar rat heart 0.3 nM [3H]PK-11195 15min/25°C
Dipyridamole f
a Human recombinant s ° Leukotriene D4
CysLT, CHO-K1 cells 0.3 nM [ H]leukotriene D4 30min/25 C
CBR a Wistar rat brain 1 nM [3H]flunitrazepam 60min/25°C Diazepam
"
a. Ouantitation Method: Radioligand binding
b. Incubation Time/Temperature
c. Incubation buffer: 50mM Tris-HCI, pH 7.5, 10mM MgCl2 at 25°C.
d. Incubation buffer: 50mM Tris-HCI, pH 7.4, 5mM CaCl2, 5mM MgClz, 100Ng/mL
bacitracin, 1mM
benzamidine, 0.1 mM PMSF.
e. Incubation buffer: 50mM Na-K phosphate, pH 7.4 at 25°C.
f. Non specific ligand: 100NM, Kp: 2.3nM, Bmax. 0.17 pmol/mg protein, Specific
binding: 90%
g. Non specific ligand: 0.3pM, Kp: 0.21 nM, BmaX: 3 pmol/mg protein, Specific
binding: 93%
h. Non specific ligand: 10pM, Kp: 4.4nM, Bmax~ 1.2 pmol/mg protein, Specific
binding: 91
Binding Assays were done at constant concentration of the compound, in 1
DMSO as vehicle, and are specified below each enzyme/receptor type in Table
18. The
results are expressed in Table 18 as percentage inhibition. Significance was
obtained
when a result was ?50% binding or inhibition (underlined).
Table 18
Percentage of inhibition or binding activity
Compound ACAT COX-2 5-LO PBR CysLT, CBR
(10 pM) (4 pM) (4 pM) (1 NM) (4 pM) (10 pM)
1 90 96 99 80 92 39
2 51 92 93 65 75 22
CA 02511750 2005-07-21
3005-31 CA
Table 18
Percentage of inhibition or binding activity
ACAT COX-2 5-LO PBR CysLT, CBR
Compound (10 pM) (4 pM) (4 pM) (1 pM) (4 pM) (10 pM)
3 63 76 72 11 59 10
4 65 78 98 92 64 12
60 63 98 68 72 21
6 54 45 71 75 24 14
7 95 26 63 65 15 21
8 40 19 -13 55 13 1
9 77 44 96 32 70 10
90 45 97 86 67 5
11 71 57 97 39 74 20
12 83 30 86 39 33 -24
46 8 95 65 -1 71 27
All of the exemplified Compounds 1-12 and 46 possessed inhibition and/or
binding activity. None of them significantly bound the central benzodiazepine
receptor
(CBR), which demonstrated that selectivity for the peripheral receptor was
present.
PBR binding studies using multiple dilutions indicated that Compound 1 had an
inhibition concentration (IC5o) value of 0.291 pM and an inhibition constant
(Ki) of 0.257
pM, compared to the binding results above, which showed an IC50 above 10 pM in
the
inhibition of CBR.
Also treatment of animals with Compound 1 resulted in an increased expression
of several genes involved in steroid biosynthesis, cholesterol
transport/metabolism,
signal transduction and apoptosis, which is consistent with Compound 1 acting
as a
PBR ligand.
EXAMPLE 11. IN VITRO PROFILING OF THE COMPOUNDS OF THE INVENTION
(a) In vitro anticancer activity of the compounds of Formula I against four
cell lines:
In vitro cytotoxic activities of exemplified Compounds are shown in Table 19,
along with hemolytic activity of each compound. Compounds were tested in four
cell
lines: HT-29 (colorectal carcinoma), SF268 (CNS), MDA-MB-231 (mammary gland
adenocarcinoma) and PC-3 (prostate adenocarcinoma). Procedures used for each
series of tests are described below.
91
CA 02511750 2005-07-21
3005-31 CA
Table 19
In vitro
Cytotoxic
Activities
and Hemolysis
Compound HT-29 SF-268 PC-3 MDA-MB-231 Hemolysis
No: (GlSO (Glen NNI) (Glso NM) (Gleo uM) (E~so Ng/mL)
NNI)
1 a~b 11.2 / 1.96 /1.55 1.95 / 3.761.79 / 3.18 7.6
9.33
2 b 0.65 0.12 0.45 0.24 5.12
3 a 7.3 5.73 5.36 6.32 > 64
4 a 14.7 4.97 5.86 11.3 > 64
a 14.4 13.4 15.6 20.5 > 64
6 a > 30 18.9 19.0 24.6 > 64
7a 14.1 18.5 14.6 17.4 > 64
9 a 12.6 1.88 1.44 2.48 > 64
a 13.0 2.02 1.35 1.55 > 64
11 a 16.0 5.79 5.35 7.72 9.8
12 a 9.33 1.95 1.2 2.79 > 64
14 b 2.04 0.76 1.15 2.16 43.9
17 b > 30 13.4 18.7 >30 35.0
18 b > 30 7.45 > 30 > 30 > 64
46 a 4.26 0.72 0.90 0.59 13.9
63 b 2.57 0.89 1.25 2.27 > 64
64 b 2.5 0.56 1.14 1.39 > 64
67 b 2.44 0.53 1.33 1.92 > 64
77 b 13.9 3.31 17.1 5.62 60.9
78 b 0.29 0.07 0.23 0.24 9.89
80 b 1.43 0.33 1.80 1.02 > 64
82 b 23.6 4.75 13.4 11.0 > 64
83 b 19.6 9.74 13.2 6.71 12.4
84 b 21.5 3.49 16.4 23.5 > 32
85 b 1.89 1.73 1.08 2.19 > 64
87 b 1.83 0.91 1.39 2.40 > 64
89 b >30 13.7 13.5 25.3 >64
92 b >30 13.5 16.6 11.1 >64
97 b 2.02 2.04 1.19 2.02 15.1
98 b 0.69 0.16 0.82 0.51 4.5
a. Results
obtained
by method
(1 )
below
b. Results obtained
by method
(2) below
c. Hemol ysis is lysis of
measured SRBC
as the
concentration
necessary
to achieve
50% hemo
(Amph otericin
B : 4
pg/mL)
Method (1):
Cytotoxic activities were determined in vitro for Compounds 1, 3-7, 9-12 and
46
to determine the concentration of each compound needed to obtain a 50%
inhibition of
cell proliferation (Gl5o). The Gl5o value emphasizes the correction for the
cell count at
time zero and, using the seven absorbance measurements [time zero, (Tz),
control
growth, (C), and test growth in the presence of drug at the five concentration
levels
92
CA 02511750 2005-07-21
3005-31 CA
(Ti)], GlSO is calculated as [(Ti-Tz)/(C-Tz)] x 100 = -50, which is the drug
concentration
resulting in a 50% reduction in the net DNA increase in control cells during
the drug
incubation.
Compounds were dissolved at 10 mM in DMSO. Dilution in vehicle to
concentrations of 30, 10, 3, 1 and 0.3 pM were prepared immediately before
assays.
Depending on the cell line's growth characteristics, 4000-10000 cells were
plated in two
96-wells pates (day 0) and incubated for 16 hours. The following day,
propidium iodide
ws added to one of the two plates and fluorescence measured (Tz). Test
compounds
were added to the second plate, as well as vehicle control, and cells further
incubated
for 96 hours. Each compound was tested at each concentration and in
triplicates. The
equivalent cell number was determined after adding propidium iodide by
measuring the
signal by fluorescence (C for control). Gl5o results were calculated using the
formula
above and are shown in Table 19.
Compound 2 has an unexpected increase in cytotoxic activity compared to
parent Compound 1. A fifty fold increase of activity was observed against HT-
29 cell
line. Cytotoxic activity (GlSO) of Compound 2 for the other three strains was
outside the
expected range of concentrations used in the first tests. The second test
showed
nanomolar activities for Compound 2, a 100-fold increase in potency.
Method (2):
In vitro cytotoxic activities (GlSO) of Compounds 1, 2, 14, 17, 18, 63, 64,
67, 77,
78, 80, 82 to 85, 87, 89, 92 and 96 to 98, were determined using propidium
iodide (PI)
as being the concentration of drug resulting in 50% growth inhibition, and by
using the
following method.
Two 96-well plates were seeded in duplicate with each cell line at the
appropriate
inoculation density (HT29: 3,000; SF268: 3,000; PC-3: 3,000; and MDA-MB-231:
7,500
cells) and according to the technical data sheet of each cell line (rows A-G,
75 pL of
media per well). Row H was filled wih medium only (150 pL, negative control-
medium).
The plates were incubated at appropriate temperature and C02 concentration for
24
h rs.
Test Compounds were prepared as 15X stock solutions in appropriate medium
and corresponding to 450, 45, 0.45, 0.045, and 0.0045 pM (prepared the day of
the
experiment). An aliquot of each was diluted 7.5-fold in appropriate test
medium to give
93
CA 02511750 2005-07-21
3005-31 CA
a set of six 2X concentration solutions (60, 6, 0.6, 0.06, 0.006, and 0.0006
pM). A 75 pL
aliquot of each concentration was added to each corresponding well (rows A to
F) of
the second plate. Row G was filled with 75 pL of medium/0.6% DMSO (negative
control-cells). The second plate was incubated at appropriate temperature and
C02
concentration for 96 hrs.
First Plate: PI (30 pL, 50 pg/mL) was added to each well of the first plate
without
removing the culture medium. The plate was centrifuged (Sorvall Legend-RT,
swinging
bucket) at 3500 rpm/10 min. Fluorescence intensity (Thermo, Varioskan, ~eX:
530 nm;
hem: 620 nm) was measured to give the first measurement, dead cells (DC at To;
before
freezing). Two round of Freeze (-80°C)/Thaw (37°C) were done.
Fluorescence intensity
was determined to give the second measure, total cells (TC at To; after
freeze/thaw)
Second plate was processed as the first one, except there were three rounds of
freeze/thaw instead of two. First measurement gave the treated dead cells
value (TDC),
and the second measurement gave the treated total cells value (TTC). Both
values
were collected for each treated well and control (CTC and CDC).
Each value (DC, TC, TDC, TTC, CTC and CDC) was corrected by removing the
background value (medium only) to give the value (FUpC(T=0)~ FUTC(T=0), FUTDC,
FUTTC~
FUcTC and FUcDC) used in the calculation of the T/C (%) (Treated/Control) for
each
concentration. T/C (%) for each concentration is calculated using the
following formula:
(FUTTC - FUTDC) - (FUTC(T=0) - FUDC(T=0)) x 1 OO
T/C (%)= FUTC - FUDc
(FUCTC - FUCDC) - ( (T=0) (T=0))
The Gl5o value emphasizes the correction for the cell count at time zero for
cell
survival. The T/C values are transposed in a graph to determine GlSO values,
the
concentration at with the T/C is 50%.
(b) Anticancer activity profiling of Compound 2 against 36 cell lines (ICSO):
Culture conditions and activity evaluations were done as indicated for
Compound
1 in Example 12 (a) below. Results were expressed as the concentration of drug
which
inhibits 50% of the cell growth (IC5o). The low micromolar to nanomolar levels
of IC5o
values shown in Table 20 demonstrated a pharmacologically relevant cytotoxic
activity
of Compound 2 against a variety of 36 tumor types including melanomas,
pancreatic,
lung, colon, gastric, bladder, renal, CNS, head and neck, prostate, uterus,
ovarian and
breast carcinomas.
94
CA 02511750 2005-07-21
3005-31 CA
Table 20
In vitro profiles of Compound 2 (ICSO)
# Type Cell line Origin Histology in nude ICSO (NM)
mice
1 Bladder T24 ATCC transitional cell 0.127
ca
2 Bladder 1218L Xenograft urothelial adeno 0.166
ca
3 Colon HCT116 NCI adeno ca, pd 0.156
4 Colon HT29 NCI adeno ca, pd 0.223
CNS 498NL Xenograft glioblastoma 0.176
6 CNS SF268 NCI nd 0.010
7 Gastric 251 L Xenograft adeno ca, pd 0.105
8 Head & 536L Xenograft hypopharynx ca 0.181
Neck
9 Lung 1121 L Xenograft large cell ca 0.125
Lung 289L Xenograft adeno ca 1.553
11 Lung 526L Xenograft adeno ca 0.104
12 Lung 629L Xenograft adeno ca 0.164
13 Lung 529L Xenograft large cell ca, 0.127
14 Lung H460 NCI large cell ca 0.366
Mammary 401 NL Xenograft pap adeno ca, wd 0.194
16 Mammary MCF7 NCI mamma ca 0.276
17 Melanoma 276L Xenograft mm, amelanotic 1.948
18 Melanoma 394NL Xenograft mm, amelanotic,pd 0.020
19 Melanoma 462NL Xenograft mm, amelanotic 0.978
Melanoma 514L Xenograft mm, melanotic 0.110
21 Melanoma 520L Xenograft mm, melanotic 0.085
22 Ovarian 1619L Xenograft adeno ca, and 0.579
23 Ovarian 899L Xenograft pap serous ca, and 0.238
24 Ovarian OVCAR3 NCI adeno ca, and 0.139
Pancreas 1657L Xenograft adeno ca, and 1.777
26 Pancreas PANC1 ATCC nd 0.125
27 Prostate 22RV1 ATCC adeno ca, and 0.142
28 Prostate DU145 NCI adeno ca, and 0.158
29 Prostate LNCAP DSMZ adeno ca, and 0.485
Prostate PC3M NCI adeno ca, pd 0.114
31 Pleuramesot.1752L Xenograft pleuromesothelioma 1.503
32 Renal 1781 L Xenograft renal ca 0.172
33 Renal 393NL Xenograft hypernephroma, wd 0.527
34 Renal 486L Xenograft hypernephroma, pd 1.144
Renal 944L Xenograft hypernephroma 0.230
36 Uterus 1138L Xenograft carcinosarcoma, wd 0.139
Mean of all cell 0.407
lines:
ca=carcimoma; pd=poorly differentiated; pap=papillary; md=moderately
differentiated; wd=well differentiated;
mm=malignant melanoma; nd=not determined
EXAMPLE 12. IN VITRO ANTICANCER ACTIVITY OF COMPOUND 1
a) Human and animal tumor cell lines from various tissues (ICSO):
CA 02511750 2005-07-21
3005-31 CA
Culture conditions: The cell lines listed in Table 21 were used to
characterize the
cytotoxicity of Compound 1 against human and animal tumor cell lines. These
cell lines
were shown to be free of mycoplasma infection and were maintained in the
appropriate
media (Table 21 ) supplemented with 10% heat-inactivated fetal bovine serum
and 1
penicillin-streptomycin, under 5% C02 at 37°C. Cells were passaged two
to three times
per week. Viability was examined by staining with 0.25% trypan blue and only
flasks
where cell viability was >95% were used for this study.
Cell lines amplification and plating: Tumor cells were seeded (1-3 x 103 cells
per 100
~L) in 96-well flat bottom microtiter plates and incubated at 37°C and
5% C02 for 16 hrs
before treatment in drug-free medium supplemented with 10% fetal bovine serum.
Evaluation of inhibitory activity on cell proliferation: Cells were incubated
for 96 hrs with
6 logo-fold concentrations of the test substance starting at 10pg/ml (20 pM).
The test
substance stock solution (5 mg/mL) was initially diluted at 1/70 fold in
medium
supplemented with serum. Other concentrations were then obtained from 1/10
fold
successive dilutions in the same supplemented medium. Cell survival was
evaluated 96
hours later by replacing the culture media with 150 pL fresh medium containing
10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer, pH 7.4. Next, 50 pL
of 2.5
mg/mL of 3-(4,5-dimethylthiazo-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in
phosphate buffer solution, pH 7.4, was added. After 3-4h of incubation at
37°C, the
medium and soluble MTT was removed, and 200 pL of dimethylsulfoxide was added
to
dissolve the precipitate of reduced MTT followed by addition of 25 pL glycine
buffer (0.1
M glycine plus 0.1 M NaCI, pH 10.5). The absorbance was determined at 570 nm
with a
microplate reader. Results were expressed as the concentration of drug which
inhibits
50% of the cell growth (IC5o). The low micromolar levels of IC5o values shown
in Table
21 demonstrated a pharmacologically relevant cytotoxic activity of Compound 1
against
a variety of tumor types such as leukemias, melanomas, pancreatic and breast
carcinomas.
Table 21
In vitro Antitumor activity of Compound 1, ICSO (Ng/mL)
Cell lines Type Origin Source Culture medium IC5°
_ ( pg/m L)
K562 Leukemia Human ATCC RPMI1640 8.6
myelogeneous
P388 Leukemia Mouse ATCC RPM11640 10.9
96
CA 02511750 2005-07-21
3005-31 CA
Table 21
In vitro Antitumor activity of Compound 1, ICSO (pgimL)
ICSo
Cell linesType Origin Source Culture medium ~NgImL)
183 Leukemia Human ATCC RPMI 1640 2.7
B16 (F10) Melanoma Mouse ATCC RPMI 1640 11.4
SK-MEL Melanoma Human ATCC RPMI 1640 14.0
28
SK-MEL Melanoma (expressingHuman ATCC RPMI 1640 14.3
28
VEGF)
SK-MEL-1 Melanoma Human ATCC EMEM 1% non-essential14.1
amino acid 1 %
Sodium
puryvate
Panc 96 Pancreatic carcinomaHuman ATCC RPMI 1% Sodium 12.5
puryvate
Panc 10.05Pancreatic carcinomaHuman ATCC RPMI 1 % Sodium 14.2
puryvate Insulin
MCF-7 Breast Human ATCC RPM11640 9.7
adenocarcinoma
b) Human cell lines from the U. S. NCI panel (Gl5o):
A study measuring the in vitro antitumor activity of Compound 1 was performed
by the National Cancer Institute (National Institutes of Health, Bethesda,
Maryland,
USA) against panel of human cancer cell lines in order to determine the
Compound 1
concentrations needed to obtain a 50% inhibition of cell proliferation (GlSO).
The
operation of this unique screen utilizes 50 different human tumor cell lines,
representing
leukemia, melanoma and cancers of the lung, colon, brain, ovary, breast,
prostate, and
kidney.
Culture conditions and plating: The human tumor cell lines of the cancer-
screening
panel were grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM
L-glutamine. For a typical screening experiment, cells were inoculated into 96
well
microtiter plates in 100 NL at plating densities ranging from 5,000 to 40,000
cells/well
depending on the doubling time of individual cell lines (Table 22). After cell
inoculation,
the microtiter plates were incubated at 37°C, 5% C02, 95% air and 100%
relative
humidity for 24 h prior to addition of experimental drugs. After 24 h, two
plates of each
cell line were fixed in situ with TCA, to represent a measurement of the cell
population
for each cell line at the time of drug addition (Tz).
Evaluation of inhibitory activity on cell .proliferation: Compound 1 was
provided as a
lyophilized powder with an estimated purity of 95+%. The compound was stored
at
97
CA 02511750 2005-07-21
3005-31 CA
20°C until day of use. Compound 1 was solubilized in dimethyl sulfoxide
at 400-fold the
desired final maximum test concentration. At the time of drug addition, an
aliquot of
frozen concentrate was thawed and diluted to twice the desired final maximum
test
concentration with complete medium containing 50pg/mL gentamicin. Additional
four,
10-fold or'/Z log serial dilutions were made to provide a total of five drug
concentrations
plus control. Aliquots of 100 pl of these different drug dilutions were added
to the
appropriate microtiter wells already containing 100 pl of medium, resulting in
the
required final drug concentrations (8.0 x 10-5 M to 8.0 x 109 M).
Following drug addition, the plates were incubated for an additional 48 h at
37°C,
5% C02, 95% air, and 100% relative humidity. For adherent cells, the assay was
terminated by the addition of cold TCA. Cells were fixed in situ by the gentle
addition of
50 pl of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for
60
minutes at 4°C. Supernatants were discarded, and the plates were washed
five times
with tap water and air-dried. Sulforhodamine B (SRB) solution (100 pl) at 0.4%
(w/v) in
1 % acetic acid was added to each well, and plates were incubated for 10
minutes at
room temperature. After staining, unbound dye was removed by washing five
times with
1 % acetic acid and the plates were air-dried. Bound stain was subsequently
solubilized
with 10 mM trizmaT"" base, and the absorbance was read on an automated plate
reader
at a wavelength of 515 nm. For suspension cells, the methodology was the same
except that the assay was terminated by fixing settled cells at the bottom of
the wells by
gently adding 50 pl of 80% TCA (final concentration, 16% TCA).
The growth inhibitory activity of Compound 1 was measured by NCI utilizing the
GlSO value, rather than the classical ICSO value. The Gl5o value emphasizes
the
correction for the cell count at time zero and, using the seven absorbance
measurements [time zero, (Tz), control growth, (C), and test growth in the
presence of
drug at the five concentration levels (Ti)], Gl5o is calculated as [(Ti-Tz)/(C-
Tz)] x 100 = -
50, which is the drug concentration resulting in a 50% reduction in the net
protein
increase (as measured by SRB staining) in control cells during the drug
incubation.
Results: Compound 1 shows a significant antitumor activity against several
types of
tumor as revealed by the NCI screening. Results of the screen are shown in
Table 22,
and more detailed results of activity against gliomas are shown in Example
12(c) (Table
23).
98
CA 02511750 2005-07-21
3005-31 CA
Table 22
In vitro tumor growth
inhibition of Compound
1 GlSO (pM)
Inoculation Density
Cell Line Type Origin GlSO
(NM)
(cells/well)
CCRF-CEM Leukemia Human 40,000 1.08
K-562 Leukemia Human 5,000 1.43
RPM I-8226 Leu kem is Human 20,000 3.15
A5491ATCC Non-Small Cell LungHuman 7,500 9.10
EKVX Non-Small Cell LungHuman 20,000 0.23
HOP-62 Non-Small Cell LungHuman 10,000 8.29
NCI-H226 Non-Small Cell LungHuman 20,000 2.00
NCI-H23 Non-Small Cell LungHuman 20,000 2.02
NCI-H460 Non-Small Cell LungHuman 7,500 13.60
NCI-H522 Non-Small Cell LungHuman 20,000 3.44
COLO 205 Colon Human 15,000 12.70
HCT-116 Colon Human 5,000 2.92
HCT-15 Colon Human 10,000 9.73
HT29 Colon Human 5,000 20.70
SW-620 Colon Human 10,000 2.72
SF-268 CNS Human 15,000 4.94
SF-295 CNS Human 10,000 12.70
SF-539 CNS Human 15,000 0.0075
SNB-19 CNS Human 15,000 2.90
SNB-75 CNS Human 20,000 7.71
U251 CNS Human 7,500 2.19
LOX IMVI Melanoma Human 7,500 4.53
M14 Melanoma Human 15,000 4.57
SK-M EL-2 Melanoma Human 20,000 25.0
SK-MEL-28 Melanoma Human 10,000 11.6
SK-MEL-5 Melanoma Human 10,000 7.80
UACC-257 Melanoma Human 20,000 2.31
UACC-62 Melanoma Human 10,000 1.55
IGR-OV1 Ovarian Human 10,000 3.11
OVCAR-3 Ovarian Human 10,000 13.50
OVCAR-4 Ovarian Human 15,000 9.67
OVCAR-5 Ovarian Human 20,000 2.81
OVCAR-8 Ovarian Human 10,000 2.65
SK-OV-3 Ovarian Human 20,000 4.00
786-0 Renal Human 10,000 6.99
A498 Renal Human 25,000 22.30
ACHN Renal Human 10,000 3.10
CAKI-1 Renal Human 10,000 15.20
RXF 393 Renal Human 15,000 7.71
SN12C Renal Human 15,000 3.85
UO-31 Renal Human 15,000 19.70
DU-145 Prostate Human 10,000 3.56
MCF7 Breast Human 10,000 10.10
NCI/ADR-RESBreast Human 15,000 18.30
99
CA 02511750 2005-07-21
3005-31 CA
Table 22
In vitro tumor
growth inhibition
of Compound
1 GlSO (pM)
Cell Line Type Origin Inoculation DensityG15 ~pM)
(cells/well)
MDA-MB-2311ATCCBreast Human 20,000 2.72
HS 578T Breast Human 20,000 2.76
MDA-MB-435 Breast Human 15,000 15.30
BT-549 Breast Human 20,000 0.11
T-47D Breast Human 20,000 0.77
The results indicate that Compound 1 was effective against most of the human
tumor
cell lines that have been assayed in the NCI screening panel suggesting a
broad
anticancer activity against several types of human cancer.
c) Human and animal glioma cell lines (ICSO):
The anticancer activity of Compound 1 was evaluated using a panel of glioma
cancer cell lines shown in Table 23, and the 50% inhibition of cell
proliferation (ICSO)
was determined.
Culture conditions: The cell lines listed in Table 23 were shown to be free of
mycoplasma infection and were maintained on DMEM medium supplemented with 10%
heat-inactivated fetal bovine serum and 1 % penicillin-streptomycin, under 5%
C02 at
37°C. Cells were passaged once a week. Prior to use the cells were
detached from the
culture flask by treating with trypsin for five to ten minutes. The cells were
counted with
a Neubauer glass slide and viability assessed by 0.25% trypan blue exclusion.
Only
flasks with >95% cell viability, were used in the study.
Cell lines amplification and plating: Cells, 5 x 103 cells per well in 100 pL
drug-free
medium supplemented with 10% fetal bovine serum, were plated in 96-well flat
bottom
microtiter plates and incubated at 37°C for 48 hrs before treatment.
Evaluation of inhibitory activity on cell proliferation: Cells (in triplicate
wells) were
incubated 96 hrs with medium containing different concentrations of Compound
1,
starting at 5.0 pg/ml (10 pM). The compound was used in a solution of 1 % DMSO
in D-
MEM or RPMI media (or other equivalent media). The concentrations of Compound
1
were as follows: 10 pM (5.0 pg/ml), 1 pM (0.50 pg/ml), 0.5 pM (0.25 pg/ml),
0.1 pM
(0.050 pg/ml), 0.5 pM (0.025 pg/ml), 0.01 pM (0.0050 pg/ml), 0.001 pM (0.00050
100
CA 02511750 2005-07-21
3005-31 CA
pg/ml). Negative controls were cells treated with vehicle alone (1 % DMSO in
culture
medium). Positive controls were cells treated with 4 to 6 increasing
concentrations of
cisplatin (CDDP) (data not shown). The optical density was measured before
incubation (time 0) and following 96 hrs of incubation with test compound in
order to
measure the growth rate of each cell line.
At the end of the cell treatment, cell culture media was replaced with 150 pl
of
fresh medium containing 10 mM of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid
buffer, pH 7.4. Then 50 pl of 2.5 mg/ml of 3-(4,5-dimethylthiazo-2-yl)-2,5-
diphenyltetrazolium bromide in PBS pH 7.4, were added to each well and the
culture
plates incubated for 4 hrs at 37°C. The resulting supernatant was
removed and
formazan crystals were dissolved with 200 pl of DMSO followed by 25 pl of
glycine
buffer (0.1 M glycine plus 0.1 M NaCI, pH 10.5). The optical density was read
in each
well using a single wavelength spectrophotometer plate reader at 570 nm.
Results
were expressed as the concentration of drug, which inhibits 50% of the cell
growth
(IC5o). Each of the cell lines was tested in at least 3 independent
experiments.
Results shown in Table 23 confirmed the activity of Compound 1 against
different brain cancer cell lines including gliosarcoma, which is the most
malignant form
of type IV glioblastoma multiform. Gliosarcomas are a mixture of glial and
endothelial
cells and are resistant to any chemotherapy.
Table 23
Compound 1 in vitro antitumor activity against Glioma cell lines (ICSO)
Cell linesType Origin Source IC5 (x 10's
M)
9L Gliosarcoma Rat ATCC 6.82 2.90
GHD Astrocytoma Human ATCC 6.29 2.98
U 373 Astrocytoma Human ATCC 3.83 1.37
GL26 Glioblastoma Human ATCC 8.93 1.10
C6 Glioblastoma Rat ATCC 4.28 2.82
DN OligodendrogliomaHuman ATCC 3.26 0.93
GHA OligodendrogliomaHuman ATCC 1.78 0.84
EXAMPLE 13: IN VIVO EFFICACY OF COMPOUNDS 1 AND 2
a) In vivo efficacy of Compounds 1 and 2 in a glioma model:
The aim of this study was to test whether Compound 1 administered by the i.p.
route prevents or delays tumor growth in C6 glioblastoma cell-bearing mice,
and to
101
CA 02511750 2005-07-21
3005-31 CA
determine an effective dosage regimen.
Animals: A total of 60 six-week-old female mice (Mus musculus nude mice),
ranging
between 18 to 25 g in weight, were observed for 7 days before treatment.
Animal
experiments were performed according to ethical guidelines of animal
experimentation
(Charte du comite d'ethique du CNRS, juillet 2003) and the English guidelines
for the
welfare of animals in experimental neoplasia (WORKMAN, P., TWENTYMAN, P.,
BALKWILL, F., et al. (1998). United Kingdom Coordinating Committee on Cancer
Research (UKCCCR) Guidelines for the welfare of animals in experimental
neoplasia
(Second Edition, July 1997; British Journal of Cancer, 77:1-10). Any dead or
apparently
sick mice were promptly removed and replaced with healthy mice. Sick mice were
euthanized upon removal from the cage. Animals were maintained in rooms under
controlled conditions of temperature (23~2°C), humidity (45~5%),
photoperiodicity (12
hrs light / 12 hrs dark) and air exchange. Animals were housed in
polycarbonate cages
(5/single cage) that were equipped to provide food and water. Animal bedding
consisted
of sterile wood shavings that were replaced every other day. Food was provided
ad
libitum, being placed in the metal lid on the top of the cage. Autoclaved tap
water was
provided ad libitum. Water bottles were equipped with rubber stoppers and
sipper
tubes. Water bottles were cleaned, sterilized and replaced once a week. Two
different
numbers engraved on two earrings identified the animals. Each cage was labeled
with a
specific code.
Tumor Cell Line: The C6 cell line was cloned from a rat glial tumor induced by
N-
nitrosomethyurea (NMU) according to Premont et al. (Premont J, Benda P, Jard
S., (3H]
norepinephrine binding by rat glial cells in culture. Lack of correlation
between binding
and adenylate cyclase activation. Biochim Biophys Acta. 1975 Feb 13;381
(2):368-76.)
after series of alternate culture and animal passages. Cells were grown as
adherent
monolayers at 37°C in a humidified atmosphere (5% C02, 95% air). The
culture
medium was DMEM supplemented with 2 mM L-glutamine and 10% fetal bovine
serum. For experimental use, tumor cells were detached from the culture flask
by a 10
min treatment with trypsin-versen. The cells were counted in a hemocytometer
and their
viability assessed by 0.25% trypan blue exclusion.
Preparation of the Test Article: For the test article, the following procedure
was followed
for reconstitution (performed immediately preceding injection). The vehicle
consisted of
a mixture of benzyl alcohol (1.5%), ethanol (8.5%), propylene glycol (27%),
PEG 400
102
CA 02511750 2005-07-21
3005-31 CA
(27%), dimethylacetamide (6%) and water (30%). The vehicle solution was first
vortexed in order to obtain a homogeneous liquid. 0.6 mL of the vortexed
vehicle
solution was added to each vial containing the test article (Compound 1 ).
Vials were
mixed thoroughly by vortexing for 1 minute and inverted and shaken vigorously.
Vials
were mixed again prior to injection into each animal.
Animal Inoculation with tumor cells: Experiment started at day 0 (Do). On Do,
mice
received a superficial intramuscular injection of C6 tumor cells (5 x 105
cells) in 0.1 mL
of DMEM complete medium into the upper right posterior leg.
Treatment regimen and Results:
First series of experiments:
In a first series of experiments, treatment started 24 hrs following
inoculation of
C6 cells. On the day of the treatment, each mouse was slowly injected with 100
pL of
test or control articles by the i.p. route. For all groups, treatment was
performed until the
tumor volume of the saline-treated mice (group 1 ) reached approximately 3 cm3
(around
day 16). Mice of group 1 were treated daily with a saline isosmotic solution
for 16 days.
Mice of group 2 were treated daily with the vehicle solution for 16 days. Mice
of group 3
were treated daily with 10 mg/kg of Compound 1 for 16 days. Mice of group 4
were
treated every two days with 30 mg/kg of Compound 1 and received 8 treatments.
Mice
of group 5 were treated every three days with 30 mg/kg of Compound 1 and
received 6
treatments. Measurement of tumor volume started as soon as tumors became
palpable
(>100 mm3; day 11 post-inoculation) and was evaluated every second day until
the end
of the treatment using callipers. As shown in Table 24 and Figure 2, the mean
value of
the tumor volume of all Compound 1 treated groups (6 mice/group) was
significantly
reduced as demonstrated by the one-way analysis of variance (Anova) test
followed by
the non-parametric Dunnett's multiple comparison test comparing treated groups
to the
saline group. An asterisk in the P value column of Table 24 indicates a
statistically
significant value, while "ns" signifies not significant.
Table 24
Compound 1 in vivo antitumor efficacy against C6 glioblastoma
Treatment Treatment Tumor volume (mm ) % Inhibition P value
regimen (mean ~ SEM)
Saline Q1 x 16 3,004.1 ~ 249.64 - -
Vehicle solution Q1 x 16 2,162.0 ~ 350.0 28.0% >0.05 ns
103
CA 02511750 2005-07-21
3005-31 CA
Compound 1 (10 Q1 x 16 1,220.4 283.46 59.4% <0.01
mglkg)
Compound 1 (30 Q2 x 8 1,236.9 233.99 58.8% <0.01
mg/kg)
Compound 1 (30 Q3 x 6 1,184.1 221.45 60.6% <0.01
mglkg)
Second series experiments:
In a second series of experiments, treatment started at day 10 following
inoculation of C6 cells when tumors became palpable (around 100 to 200 mm3).
Treatment was repeated daily for 5 consecutive days. On the day of the
treatment,
each mouse was slowly injected with 100 pL of Compound 1 by i.p. route. Mice
of
group 1 were treated daily with saline isosmotic solution. Mice of group 2
were treated
daily with the vehicle solution. Mice of group 3 were treated daily with 20
mg/kg of
Compound 1. Mice of group 4 were treated daily with 30 mg/kg of Compound 1.
Mice
were treated until the tumor volume of the saline-treated control mice (group
1 ) reached
around 4 cm3. Tumor volume was measured every second day until the end of the
treatment using callipers. As shown in Table 25 and Figure 3, the mean value
of the
tumor volume of all Compound 1 treated groups (6 mice/group) was significantly
reduced as demonstrated by the one-way analysis of variance (Anova) test
followed by
the non-parametric Dunnett's multiple comparison test comparing treated groups
to the
saline group. An asterisk in the P value column of Table 25 indicates a
statistically
significant value, while "ns" signifies not significant.
Histological analysis of tumor sections showed pronounced morphological
changes between tumors from Compound 1-treated mice and those from mice in the
control groups. In tumors from mice treated with Compound 1 (20 - 30 mg/kg),
cell
density was decreased and the nuclei of remaining tumor cells appeared larger
and
pycnotic while no such changes were observed for tumors from vehicle-treated
mice
(Figure 4).
Table 25
Compound 1 in vivo antitumor efficacy against C6 glioblastoma
Treatment Treatment Tumor volume (mm')oho InhibitionP value
_ regimen (mean SEM)
Saline Q1 x 5 4,363.1 614.31 - -
Vehicle solution Q1 x 5 3,205.0 632.37 26.5% >0.05
ns
Compound 1 (20 Q1 x 5 1,721.5 374.79 60.5% <0.01
mg/kg)
Compound 1 (30 Q1 x 5 1,131.6 525.21 74.1% <0.01
mglkg)
104
CA 02511750 2005-07-21
3005-31 CA
Antitumor efficacy of Compound 2 against C6 Glioblastoma:
Antitumor efficacy of Compound 2 against rat glioblastoma tumor (C6)
xenografts in
female swiss nude mice was accomplished as described above. The results and
dosage regimen are summarized in Figure 5. Significant efficacy was shown
following
intravenous administration, at a dosage regimen of 75 mg/kg (qd5/2/qd5).
b) In vivo efficacy of Compound 1, against orthotopic C6 glioma tumor
xenograft:
The antitumor activity of Compound 1 was further tested in a orthotopic C6
glioma tumor xenograft model in mice. CD1 female nude mice (6 weeks of age)
were
grafted intra-cerebrally with 5 x 104 (volume of 10 microliters) rat C6 glioma
cells (day
0). Treatment was initiated 24 h after tumor cell implantation. Compound 1 was
administered intraperitoneally (i.p.) at a concentration of 30 mg/kg (volume
of 10
mL/Kg) on days 1, 2 and 3 followed by i.p. injections of 10 mg/kg on days 4
and 5 and 9
to 38. Vehicle (30% PEG; 30% PG; 40% H20) was injected in a volume of 10 mL/Kg
using the same route and schedule.
Body weight of animals was monitored every other day and the effect of
Compound 1 on the growth of intracerebral glioma tumors was evaluated by mouse
survival and percentage increase in life span (%ILS, expressed as mean
survival time
of treated animals minus that of control animals over the mean survival time
of the
control group). By criteria established by the National Cancer Institute,
increases in life
span exceeding 25% indicate that the drug has significant antitumor activity
(Plowman
et al. (1997) Human tumor xenografts models in NCI drug development. In:
Theicher
BA (ed) Anticancer drug development guide: prescreening, clinical trials and
approval.
Human press, Totowa, pp 101-125). Statistical analysis of mouse survival was
performed by Kaplan-Mayer analysis. Daily treatment with Compound 1 led to an
increase survival of 7 days resulting in a 29% increase in life span (see
Figure 6).
c) In vivo efficacy of Compound 1, against murine P388 leukemia model:
The anticancer activity of Compound 1 was further tested in a murine P388
leukemia model in mice.
Formulation: Compound 1 was first dissolved in 1 volume of 90% propylene
105
CA 02511750 2005-07-21
3005-31 CA
glycol (PG) followed by the addition of 2 volumes of 45% polyethylene glycol
400 (PEG
400). The volume ratio of PEG 400/PG/water was of 30:30:40. Compound 1 was
injected in a volume of 10 mL/kg.
DBA/2 female mice (6 weeks of age) were injected intraperitoneally (ip) with 1
x
106 P388 murine leukemia cells (day 0). Mice were randomized in 4 groups (10
mice
per group) at Day 1 (D1 ) and treated with the following dose and schedule.
Group1: iv injection of vehicle (PEG/PG formulation) on D1 and D8, daily ip
administration of vehicle from D2 to D7 and from D9 to D10
Group 2: iv injection of Compound 1 in PEG-PG formulation at 50 mg/kg on D1
followed by daily ip administration of Compound 1 in PEG-PG formulation
at 10 mg/kg from D2 to D4 and from D6 to D12
Group 3: Daily ip administration of Compound 1 in PEG-PG formulation at 10
mg/kg from D1 to D4 and from D8 to D14
Mice body weights were recorded twice a week. Lethality and behaviour of
animals were recorded every day. All vehicle control mice died between D8 to
D10
from peritoneal carcinomatosis associated with ascites. Three (3) mice from
group 2
died one day after treatment due to compound toxicity. The remaining seven (7)
died
between days 8 and 12. Mice from group 3 died between days 8 and 12. The
results
(see Table 26) were expressed as percent of mean survival time of treated
animals
over the mean survival time of the control group (treated vs control, T/C%)
and as
increase life span (mean survival time of treated animals minus that of
control animals
over the mean survival time of the control group; ILS%). By NCI criteria, T/C
exceeding
125% and an ILS increase of 25% indicate that the drug has significant
anticancer
activity.
Compared with vehicle-treated mice, %T/C were 133.3% and 138.9% and ILS
33 and 38.9 for groups 2 and 3, respectively. These results indicate a
moderate but
significant enhancement of survival time of P388 IP leukemia bearing mice
treated with
Compound 1.
Table 26
Effect of Compound 1 on survival of DBA/2 mice bearing IP murine leukemia
Mean Survival Median
Gr. Treatment Treatment regimen Days ~ SD survival % TIC % ILS
106
CA 02511750 2005-07-21
3005-31 CA
1 PEG-PG Vehicle IV (D1 and D8) and g ~ 0.8 9 --- ---
IP (D2-D7 and D9-D10)
2 Compound 1 IV 50 mg/kg (D1 ) and 11 ~ 1.5 12 133.3 33
IP 10 mg/kg D2-4, 6-12
IP 10 mg/kg
3 Compound 1 D1-4 and 8-14 12.6 ~ 4.9 12.5 138.9 38.9
d) In vivo efficacy of Compound 1, against human PC3 prosfate cancer model:
The anticancer activity of Compound 1 was further tested in a human PC3
prostate model in mice. HRLN male nude mice (8 weeks of age) were implanted
with 1
mm3 PC3 tumor fragments subcutaneously (sc) in the right flank. Animals were
randomized (ten per group) when tumors reach an average size of 80 - 120 mg
and
treatment began according to Table 27. For these studies, Compound 1 was
formulated
in 5% ethanol, 5% PEG-400 and 15% Polysorbate -80 in dextrose 5% (D5W).
Table 27
Dosing schedule of Compound 1
Gr.N Agent Dose ConcentrationRoute & Dosinggchedule
(mg/kg)(mglmL) volume (mL/kg)
1 10 Cyclophosphamide90 9 ip / 10 qd x5
2 10 D5W - - sc / 5 5/2/5/2/5
3 10 Compound 1 30 6 sc / 5 5/2/5/2/5
4 10 Compound 1 50 10 sc / 5 q3d x7
10 Compound 1 30 6 ip / 5 q3d x7
6 10 Compound 1 100 10 iv / 10 5/2/5/2/5
Tumor measurements were taken twice weekly using callipers and were
converted to tumor mass (in milligrams) using the formula: width2 (mm) x
length (mm) x
0.52. Body weights were also recorded twice weekly. Statistical analysis was
done
using the unpaired two-tailed Student's t test.
%T/C was calculated at day 38 once animals in the control group had to be
sacrificed due to antitumor burden. Intravenous treatment did not result in
activity (likely
due to short half-life and lack of sustained therapeutically effective drug
levels). On the
other hand, subcutaneous administration at 30 mg/kg given from days 1 to 5, 8
to 12
and 15 to 19, or at 50 mg/kg every three days x 7 (days 1, 4, 7, 10, 13, 16
and 19)
where we maintain drug levels at therapeutically effective drug concentrations
for over 8
hours resulted in significant antitumor activity with %T/C of 25.5% and 14.6%,
107
CA 02511750 2005-07-21
3005-31 CA
respectively (P< 0.0001 ).
Figure 7 shows antitumor efficacy results of Compound 1 against human
prostate tumor xenografts. Figure 8 shows antitumor efficacy results on
individual
animals on the 22nd day of treatment.
e) In vivo efficacy of Compound 1, against human MDA-M8-231 breast cancer
model:
The antitumor activity of Compound 1 was further tested in a human MD-MB-231
breast cancer model in mice. HRLN female nude mice (8 weeks of age) were
treated
with 5x106 MDA-MB-231 tumor cells (sc) in the right flank. Animals were
randomized
(ten per group) when tumors reach an average size of 80 - 120 mg and treatment
began according to Table 28. For these studies, Compound 1 was formulated in
5%
ethanol, 5% PEG-400 and 15% Polysorbate 80 in dextrose 5%.
Table 28
Dosing schedule
of Compound
1
Gr N Agent dose Concentration Injected Route Schedule
volume
(mg/kg) (mg/mL) (mL/kg)
1 10 D5W - - 10 iv 5/2/5/2/5
2 10 paclitaxel30 - - iv qod x5
3 10 Vehicle - - 5 sc qd x21
4 10 Compound 100 10 10 iv 5/2/5/2/5
1
10 Compound 30 6 5 sc 5/2/5/2/5
1
6 10 Compound 20 6 3.3 sc qd x21
1
7 10 Compound 50 10 5 sc q3d x7
1
8 10 Compound 30 6 5 ip q3d x 7
1
Tumor measurements were taken twice weekly using calipers and were
converted to tumor mass (in milligrams) using the formula: width2 (mm) x
length (mm) x
0.52. Body weights were also recorded twice weekly. Statistical analysis was
done
using the unpaired two-tailed Student's t test.
%T/C was calculated at day 21 once animals in the control group had to be
sacrificed due to tumor burden. Intravenous treatment did not result in
activity (likely
due to short half-life and lack of sustaining therapeutically effective drug
levels). On
the other hand, subcutaneous administration at 20 mg/kg given everyday for 21
days or
at 30 mg/kg given from days 1 to 5, 8 to 12 resulted in significant antitumor
activity with
%T/Cs of 40% and 35% respectively; P < 0.0001 ). Subcutaneous or
intraperitoneal
108
CA 02511750 2005-07-21
3005-31 CA
administration at 50 and 30 mg/kg respectively every three days x 7 (days 1,
4, 7, 10,
13, 16 and 19) were also effective giving moderate but statistically
significant T/C
values of 68% (P= 0.0019) and 58% (P = 0.0007).
Figure 9 shows antitumor efficacy results of Compound 1 against human breast
tumor xenografts. Figure 10 shows antitumor efficacy results on the 21St day
of
treatment.
EXAMPLE 14: Pharmacokinetic profiles
Compounds 1 and 2 were separately dissolved in ethanol (5%), Polysorbate 80
(15%), PEG 400 (5%) and dextrose (5%) at a final concentration of 6 mg/ml (iv,
ip and
sc administration). For oral administration, Compound 1 was solubilized in
Cremophor~ EL/ Ethanol (50%:50%) at a final concentration of 6 mg/ml. Prior to
dosing, animals (female Crl: CD1 mice; 6 weeks of age, 22-24g) were weighed,
randomly selected and assigned to the different treatment groups. Compound 1
was
administered by the intravenous (iv), subcutaneous (sc), intraperitoneal (ip),
or oral (po)
route to the assigned animals. Compound 2 was administered by the intravenous
(iv),
or intraperitoneal (ip) route to the assigned animals. The dosing volume of
Compounds
1 and 2 was 5 mL per kg body weight. Animals were anesthetized with 5%
isoflurane
prior to bleeding. Blood was collected into microtainer tubes containing the
anticoagulant K2EDTA by cardiac puncture from each of 4 animals per bleeding
timepoint (2 min, 5 min, 15 min, 30 min, 1 h, 2h, 4h and 8h). Following
collection, the
samples were centrifuged and the plasma obtained from each sample was
recovered
and stored frozen (at approximately -80°C) pending analysis. At the 5
min and 30 min
time points, the following organs were harvested from each animal: brain,
lungs,
skeletal muscle, fat tissue, kidneys, spleen, thymus and liver. Tissues were
frozen (at
approximately -80°C) pending analysis. Samples were analysed by
LC/MS/MS.
Standard curve ranged from 25 to 2000 ng/mL with limit of quantitation (LOQ)
<_ 25
ng/mL and limit of detection (LOD) of 10 ng/mL.
Plasma values of Compounds 1 and 2 falling below the limit of quantitation
(LOQ) were set to zero. Mean concentration values and standard deviation (SD)
were
calculated at each timepoints of the pharmacokinetic study (n=4
animals/timepoint).
The following pharmacokinetic parameters were calculated: area under the
plasma
109
CA 02511750 2005-07-21
3005-31 CA
concentration versus time curve from time zero to the last measurable
concentration
time point (AUCo_t), area under the plasma concentration versus time curve
extrapolated to infinity (AUC;nf), maximum observed plasma concentration
(Cmax), time
of maximum plasma concentration (tmax), apparent first-order terminal
elimination rate
constant (ke,), apparent first-order terminal elimination half-life will be
calculated as
0.693/kel (t~,2). The systemic clearance (CL) of Compound 1 after intravenous
administration was calculated using Dose/AUCinf. Pharmacokinetic parameters
were
calculated using KineticaTM 4.1.1 (InnaPhase Corporation, Philadelphia, PA).
Results:
Mean plasma concentrations of Compound 1 following intravenous (iv),
intraperitoneal (ip), subcutaneous (sc), and oral (po) administrations at 30
mg/kg are
presented in Figure 11. Mean plasma concentrations of Compound 2 following iv
and ip
administrations at 30 mg/kg, compared with Compound 1 via the same routes of
administration, are presented in Figure 12. When administered iv, Compound 2
had an
AUC of 92.08 pM~h and an observed CmaX of 105 pg/mL, compared to an AUC of
40.4
pM~h and an observed CmaX of 130 pg/mL for Compound 1. When administered ip,
Compound 2 had an AUC of 58.75 pM~h and an observed CmaX of 5.8 pg/mL,
compared to an AUC of 9.5 pM~h and an observed CmaX of 2.25 pg/mL for Compound
1.
Mean (~ SD) plasma concentrations of Compound 1 following I.V. administration
of a 30 mg/kg dose declined rapidly in a biexponential manner resulting in
very short
half lives (t~,2 a and ~ of 4.6 min and 2.56 h, respectively). The
pharmacokinetics of
Compound 1 following intraperitoneal and subcutaneous administrations, and
Compound 2 following intraperitoneal and intravenous administration, showed a
PK
profile suggestive of slow release. With these routes of administration, the
compound
plasma concentration was sustained and maintained at therapeutically relevant
levels
for over 8 hours. Compound 2 showed a half life (t~,2) of more than 40 hours
following
both IP and IV administrations. Oral administration of Compound 1 resulted in
moderate but sustained drug levels. These data indicated that Compound 1 was
orally
bioavailable at a 5-8% level.
Mean tissue concentrations of Compound 1, 30 min after intravenous (iv),
intraperitoneal (ip) or subcutaneous (sc) administrations at 30 mg/kg are
presented in
110
CA 02511750 2005-07-21
3005-31 CA
Figure 13. The 30 min time point was chosen since plasma concentrations were
similar
with all three routes of administration. Compound 1 is well distributed
following iv and
ip dosing. Surprisingly, although ip and sc administrations resulted in a
similar PK
profile, tissue levels were significantly lower following sc dosing. This
could be
explained by the absence of peak levels following sc administration compared
with iv
and ip administrations.
Acute toxicity studies in CD-1 nulnu mice for Compound 2, using the same
formulation, gave an MTD >_ 50 mg/kg (ip, NOAEL: 30 mg/kg) and ? 100 mg/kg
(iv,
NOAEL: 75 mg/kg), with weight losses of about 7% for several days post-
injection.
Compound 1 had an MTD of 150 mg/kg when administered iv. Acute toxicity
studies
with Compound 46 gave an MTD of 30 mg/kg (ip).
EXAMPLE 15: DIBENZODIAZEPINONE ANALOGS GENERAL PROCEDURES
a) O-Alkylation:
Alkylation Compounds 4-8 are also produced using the procedure presented in
Example 6. Compounds 38 and 39 are also produced using the procedure of
Example
6, by controlling the amount of diazomethane, the reaction temperature and/or
the
reaction time. Compound 38 is also prepared in two steps, from Compound 10,
using
the procedure of Example 6, the resulting mono-methyl-diacetate compound is
subsequently hydrolyzed using aqueous acidic or basic (mild) conditions to
obtain
Compound 38. Compounds 4, 5, 6, 7 and 39 are also prepared in a similar two-
step
procedure, when using the appropriate Compound as starting material, which are
respectively Compounds 9, 11, 35, 37 and 36.
Syntheses of Compounds 6, 7 and 38 by methylation of Compound 1
0 0
' N
\ ~ _N \
Compound 6 HO H / \ OH Compound 7
Me0 _
HO Me0
O
N i i i
Compound 38
HO H OMe
HO
111
CA 02511750 2005-07-21
3005-31 CA
A solution of Compound 1 (1 g) in tetrahydrofuran 50 (ml) is treated with 1.5
equivalents of diazomethane. The mixture is heated under reflux for one hour,
cooled
to room temperature and poured into a mixture of toluene (200 ml) and water
(200 ml).
The layers are separated and the aqueous layer is extracted once more with an
equal
portion of toluene. The combined toluene layers are washed once with 1 N
aqueous
acetic acid and then concentrated to a crude product, which is predominantly a
mixture
of Compounds 6, 7 and 38 with some unchanged starting material and over-
methylated
derivatives. The desired products may be separated and purified by HPLC or
HSCC
chromatography or preparative TLC, using the systems as described in any of
Examples 2 and 4-9 above, to obtain approximately 200 mg of each of Compounds
6, 7
and 38.
b) O-Acylations:
O-acetylated compounds 35-37 are also produced using the procedures
presented in Example 7, using a lower quantity of acetic anhydride, lower
temperature,
and/or shorter reaction times.
Syntheses of Compounds 35, 36 and 37 by esterification of Compound 1.
0 0
i ~ i
Ac0 N / \ Compound 35 Ho N / \ Compound 36
OH H OH
HO Ac0
O
i
Ho H /_ \ oAc Compound 37
HO
To a solution of Compound 1 dissolved in toluene (9 parts) tetrahydrofuran (1
part), cooled in an ice-bath is added 1.1 equivalents of acetic anhydride and
two drops
of boron trifluoride etherate. The reaction is maintained cool in an ice bath
and stirred at
0 °C for 1-2 hours. The reaction mixture is then poured into aqueous 5%
sodium
bicarbonate solution shaken and the toluene layer is removed. The aqueous laer
is re-
extracted with toluene and the combined toluene layers are concentrated to a
mixture
of predominantly Compounds 35, 36 and 37, contaminated with some unchanged
starting material and some diacetates. Compounds 35, 36 and 37 are separated
and
purified by HPLC or HSCC using one of the systems described in Examples 2 and
4-9.
112
CA 02511750 2005-07-21
3005-31 CA
In a typical experiment yields of 25% to 30% are obtained for each of
Compounds 35,
36 and 37.
Compounds 9-12 are also produced using the same procedure, with appropriate
numbers of molar equivalents (2.2 and 3.3).
c) N-alkylations:
N-Alkylations are accomplished using either an alkyl halide (iodide, bromide,
chloride) or another alkylating agent, such as a dialkylsulfate, or an
alkylsulfonate
(triflate, mesylate, tosylate, and the like). Compounds 2, 3, 14, and 60 to 77
are also
produced using the procedures exemplified in Examples 4 and 5.
Syntheses of Compounds 2, 3 and 14 by N-alkylation of Compound 1.
0
\ ~ Compound 2 R = methyl
Compound 3 R = benzyl
Compound 14 R = ethyl
HO
To a solution of Compound 1 (50 mg) dissolved in an excess of the appropriate
alkyl halide (iodomethane for Compound 2, benzyl chloride for Compound 3 or
ethyl
bromide for Compound 14) is added a few drops of pyridine (catalytic amount).
The
reaction mixture is stirred for 72 hours, or until completion, and then
evaporated to
dryness under reduced pressure to obtain Compound 2, 3 or 14 respectively, in
an
essentially pure form in an almost quantitative yield. The crude compound is
further
purified by HPLC or Preparative TLC, using the procedures described in
Examples 2
and 4-9.
Compounds 60 to 77 are also prepared via this procedure, or the procedures of
Example 5, by reaction of Compound 1 respectively with 3-chloro-1-butene, 1-
chloro-2-
methylpropane, crotylchloride, 1-bromopropane, 1-bromobutane, 1-bromo-2-
methylbutane, 2-chloro-2-methylpropane, 1-bromohexane, 1-chlorooctane,
trifuoromethyl iodide, heptafluoro-1-iodopropane, heptafluoro-2-iodopropane, 2-
iodo-
1,1,1-trifluoroethane, bromocyclopropane, 1-chloro-3-phenylpropane, and 2-
bromobutane. Compound 78 is also prepared by this procedure, by reacting
Compound
46 with iodomethane.
Compounds 60 to 77 are also prepared by the procedures of Example 4, by
reaction of Compound 1 with their respective dialkylsulfate (or
alkylsulfonate), which is
either commercially available or can be prepared, for example by the reaction
of the
113
CA 02511750 2005-07-21
3005-31 CA
appropriate alcohol with a activated sulfate or sulfonate (e.g. chloride,
anhydride, and
the like). As an example, 1-hexane triflate is prepared just prior to use by
the reaction of
1-hexanol with trifluoromethanesulfonic anhydride (Tf20) in tetrahydrofuran,
using an
equimolar amount (vs Tf20) of base, such as triethylamine. The reaction is
worked up
by careful treatment with water (containing 1 % triethylamine), extracted with
ether, dried
with magnesium sulfate and concentrated in vacuo. Other examples of procedures
for
the preparation of alkyl sulfates and sulfonates are described in Advanced
Organic
Chemistry, Jerry March, supra (e.g. page 404).
d) N-Acylation:
Synthesis of Compound 13 by N-acetylation of Compound 1.
0
i i i
HO N / ~ off Compound 13
Ac -
HO
To a solution of Compound 1 dissolved in tetrahydrofuran (THF) is added 1.2
equivalents of acetyl chloride and a few drops of pyridine. The reaction
mixture is
allowed to stand at room temperature for 1-2 hours and then evaporated to
dryness
under reduced pressure to obtain a crude mixture containing Compound 13.
Compound
13 is purified using HPLC or preparative TLC plates and the procedure
described in any
one of Examples 2, and 4-9.
e) Peracetylation:
Syntheses of Compound 15 by peracetylation of Compound 1.
0
i i i
AcOA N ~ \ Compound 15
OAc
Ac0
A solution of Compound 1 (100 mg) in acetic anhydride (5 ml) is treated with
pyridine (250 ul). The reaction mixture is allowed to stand overnight at room
temperature and is then diluted with toluene (100 ml). The toluene solution is
washed
well with aqueous 5% sodium bicarbonate solutions, then with water and is
finally
concentrated under reduced pressure to give an essentially pure sample of
Compound
15 in almost quantitative yield.
114
CA 02511750 2005-07-21
3005-31 CA
Epoxidations:
The epoxide compounds of the present invention (e.g., compounds according to
exemplary Compounds 16-22 are made from Compound 1, and Compounds 23 to 34
from the appropriate starting material, by treatment with any of a number of
epoxidizing
reagents such as perbenzoic acid, monoperphthalic acid or more preferably by m-
chloroperbenzoic acid in an inert solvent such as tetrahydrofuran. It will be
appreciated
by one of ordinary skill in the art that slightly greater than one molecule
equivalent of
epoxidizing agent will result in the maximal yield of mono-epoxides, and that
the
reagent, solvent, concentration and temperature of the reaction will dictate
the ratio of
specific mono-epoxides formed. It will also be appreciated that the mono-
epoxides will
be enantiomeric mixtures, and that the di-epoxides and the tri-epoxide can be
prepared
as diastereomers and that the conditions of the reaction will determine the
ratios of the
products. One skilled in the art will appreciate that under most conditions of
reactions
the product will be a mixture of all possible epoxides and that these may be
separated
by standard methods of chromatography. Exemplary approaches to the generation
of
mono-, di-, and tri-epoxides are provided below.
1 ) Mono-epoxides Compounds, 16, 17 and 18 prepared by epoxidation of
Compound 1 (as also shown in Example 8(c)):
0 o
N O ~ ~ \ ~ N i O
\ N / \
Ho H \ off Compound 16 Ho Ho - off Compound 17
HO
O
i i
\ ~ N O
H O /_
\ off Compound 18
HO
To a solution of Compound 1 dissolved in tetrahydrofuran (THF) is added 1.1
equivalents of meta-chloroperbenzoic acid. The reaction is cooled in an ice
bath and
stirred at 0 °C for 1-2 hours. The reaction mixture is then evaporated
to dryness, re-
dissolved in methanol and subjected to liquid chromatography on a column of
SephadexT"~ LH-20 to isolate a mixture of predominantly Compounds 16, 17 and
18,
contaminated with some unchanged starting material and some di- and tri-
epoxides.
Compounds 16, 17 and 18 are separated and purified by HPLC using the system
115
CA 02511750 2005-07-21
3005-31 CA
described in Examples 2 and 4-9. In a typical experiment yields of 15% to 25%
are
obtained for each of Compounds 16, 17 and 18.
2) Synthesis of Compounds 19, 20 and 21 by di-epoxidation of Compound 1:
0 0
\ / \N O O \ / N O O
HO N
HO H OH Compound 19 Ho - off Compound 20
HO
O
i
\ / N O O
HO H / ~ off Compound 21
HO
To a solution of Compound 1 dissolved in tetrahydrofuran (THF) is added 2.3
equivalents of meta-chloroperbenzoic acid. The reaction is cooled in an ice
bath and
stirred at 0 °C for 1-2 hours. The reaction mixture is then evaporated
to dryness, re-
dissolved in methanol and subjected to liquid chromatography on a column of
SephadexTM LH-20 to isolate a mixture of predominantly Compounds 19, 20 and
21,
contaminated with traces of unchanged starting material and some mono- and tri-
epoxides. Compounds 19, 20 and 21 are separated and purified by HPLC using the
system described in Examples 2 and 4-9. In a typical experiment, yields of 15%
to 20%
are obtained for each of Compounds 19, 20 and 21.
3) Synthesis of Compound 22 by tri-epoxidation of Compound 1:
O
\ / N o 0 0
Ho H ~ off Compound 22
HO
To a solution of Compound 1, dissolved in tetrahydrofuran (THF), is added 3.5
equivalents of meta-chloroperbenzoic acid. The reaction is cooled in an ice
bath and
stirred at 0 °C for 1-2 hours. The reaction mixture is then evaporated
to dryness, re-
dissolved in methanol and subjected to liquid chromatography on a column of
SephadexT"" LH-20 to isolate Compound 22 in an essentially pure form in a
yield of
80+%.
116
CA 02511750 2005-07-21
3005-31 CA
4) Syntheses of Compounds 23 and 24 by epoxidation of Compound 42.
0 0
l N O i \ l N i O
Ho N / ~ off Compound 23 Ho H / ~ off Compound 24
H HO
HO
To a solution of Compound 42 dissolved in tetrahydrofuran (THF) is added 1.1
equivalents of meta-chloroperbenzoic acid. The reaction is cooled in an ice
bath and
stirred at 0 °C for 1-2 hours. The reaction mixture is then evaporated
to dryness, re-
dissolved in methanol and subjected to liquid chromatography on a column of
SephadexTM LH-20 to isolate a mixture of predominantly Compounds 23 and 24,
contaminated with some unchanged starting material and some diepoxide.
Compounds 23 and 24 are separated and purified by HPLC or HSCC using one of
the
systems described in Examples 2 and 4-9. In a typical experiment yields of 35%
to 40%
are obtained for each of Compounds 23 and 24.
Compounds 25 and 29 to 34 are prepared using this procedure. In each
procedure, Compound 42 is replaced by the appropriate starting material. More
specifically, Compounds 25 and 29 are prepared using Compound 41 as starting
material; Compounds 30 and 31 are prepared using Compound 40 as starting
material;
and Compounds 32, 33, and 34 are prepared respectively from Compounds 45, 44
and
43.
5) Synthesis of Compound 28 by epoxidation of Compound 40.
O
\ / N o 0
HO N ~ ~ Compound 28
H ~ OH
HO
To a solution of Compound 40 dissolved in tetrahydrofuran (THF) is added 2.2
equivalents of meta-chloroperbenzoic acid. The reaction is cooled in an ice
bath and
stirred at 0 °C for 1-2 hours. The reaction mixture is then evaporated
to dryness, re-
dissolved in methanol and subjected to liquid chromatography on a column of
SephadexTM LH-20 to isolate essentially pure Compound 28 in good yield.
Compounds 26 and 27 are prepared using the same procedure, but using
respectively from Compounds 42 and 41 as starting material, instead of
Compound 40.
117
CA 02511750 2005-07-21
3005-31 CA
g) Epoxide opening:
Syntheses of Compound 53 by opening the epoxide of Compound 16.
0
N H
\ N ~ \o~ Compound 53
HO H OH
HO
A solution of Compound 16 (100 mg) in tetrahydrofuran (50 ml) is treated with
1 N aqueous hydrochloric acid (5 ml). The reaction mixture is stirred
overnight at room
temperature and is then diluted with toluene (100 ml) and water (200 ml). The
toluene
layer is separated and the aqueous layer is extracted with a further 100 ml of
toluene.
The combined toluene layers are washed once more with water (50 ml) and the
separated and dried under vacuum to give the vicinal glycol Compound 53.
The same procedure is used to prepare Compounds 54 to 59, using respectively
Compounds 17 to 22 as starting material.
h) Hydrogenation:
Compounds 40 to 46 (from Compound 1 ) and 78 (from Compound 2) are
produced by catalytic hydrogenation using a source of hydrogen (hydrogen,
formic acid,
and the like), and a catalyst (palladium on charcoal, platinum oxide, Raney-
Nickel, and
the like). Hydrogen uptake is optionally measured or controlled.
1 ) Syntheses of Compounds 40, 41 and 42 by hydrogenation of Compound 1.
Ho \N ~ \ ~~ Compound 40 Compound 41
H
O
N
HO H / ~ off Compound 42
HO
A solution Compound 1 (462 mg) in ethanol (200 ml) with palladium on charcoal
(25 mg of 5%) is shaken in an hydrogenation apparatus in an atmosphere of
hydrogen.
The uptake of hydrogen by the reaction is measured carefully and at the point
where
one millimole of hydrogen has been consumed, shaking is stopped, the vessel is
rapidly
118
CA 02511750 2005-07-21
3005-31 CA
evacuated and the atmosphere is replaced with nitrogen. The catalyst is
removed by
filtration and the filtrate is concentrated to obtain a crude mixture of
Compounds 40, 41
and 42 contaminated by unreacted starting material and minor amounts of over
reduced products. The desired products may be separated and purified by HPLC
or
HSCC chromatography using the systems as described in Examples 2 and 4-9
above,
to obtain approximately 100 mg of each of Compounds 40, 41 and 42.
2) Syntheses of Compounds 43, 44 and 45 by hydrogenation of Compound 1.
:i.
~N-%~OH Compound 43 Hp ~N-~OH Compound 44
H ~ H
HO HO
Compound 45
OH
A solution of Compound 1 (462 mg) in ethanol (200 ml) with palladium on
charcoal (25 mg of 5%) is shaken in an hydrogenation apparatus in an
atmosphere of
hydrogen. The uptake of hydrogen by the reaction is measured carefully and at
the
point where two millimoles of hydrogen has been consumed, shaking is stopped,
the
vessel is rapidly evacuated and the atmosphere is replaced with nitrogen. The
catalyst
is removed by filtration and the filtrate is concentrated to obtain a crude
mixture of
Compounds 43, 44 and 45 contaminated by trace amounts unreacted starting
material
and minor amounts of under and over reduced products. The desired products may
be
separated and purified by HPLC or HSCC chromatography using the systems as
described in Examples 2 and 4-9 above, to obtain approximately 100 mg of each
of
Compounds 43, 44 and 45.
3) Syntheses of Compound 46 by hydrogenation of Compound 1 (also produced
using the procedure of Example 8(a) and 8(b)).
Compound 46
119
CA 02511750 2005-07-21
3005-31 CA
A solution of Compound 1 (462 mg) in ethanol (200 ml) with palladium on
charcoal (25 mg of 5%) is shaken in an hydrogenation apparatus in an
atmosphere of
hydrogen. The uptake of hydrogen by the reaction is measured carefully and at
the
point where three millimoles of hydrogen have been consumed, shaking is
stopped, the
vessel is rapidly evacuated and the atmosphere is replaced with nitrogen. The
catalyst
is removed by filtration and the filtrate is concentrated to obtain an
essentially pure
sample of Compound 46.
Compound 78 is prepared from the same procedure, using Compound 2 as
starting material, instead of Compound 1.
i) Ozonolysis:
Syntheses of Compounds 47, 49 and 51 by ozonolysis of Compound 1.
0 0
N i i ~O \ / N i
off Compound 47 Ho H ~ \ off Compound 49
HO o HO
NCO
Compound 51
N ~ \
HO H OH
HO
A solution of Compound 1 (462 mg) in dry ethyl acetate (200 ml) in an
ozonolysis
apparatus is cooled to below -20°C. A stream of ozone containing oxygen
is passed
into the solution from an ozone generator, which has been precalibrated such
that the
rate of ozone generation is known. To obtain predominantly Compound 47 the
passage
of ozone is halted after 0.9 millimole have been generated. To obtain
predominantly
Compound 49 the ozone passage is halted after 2 millimoles have been generated
and
to obtain Compound 51 as the predominant product 3.3 millimoles of ozone are
generated.
At the completion of the ozonolysis, the reaction mixture is transferred to an
hydrogenation apparatus, 5% palladium on calcium carbonate catalyst (0.2 g) is
added
to the reaction mixture which is maintained at less than -20°C and is
hydrogenated.
When hydrogen uptake is complete the hydrogen atmosphere is replaced with
nitrogen
and the reaction mixture is allowed to come to room temperature, filtered to
remove
catalyst and the filtrate is concentrated. The crude product may be purified
by
120
CA 02511750 2006-05-04
3005-31 CA
chromatography using either HPLC or HSCC with the systems as described in
Examples 2 and 4-9 to give, dependent on the amount of ozone used, Compounds
47,
49 and 51.
Dimethyl acetal compounds, for example, Compounds 94 to 96, are also
produced by ozonolysis in methanol, followed by treatment with dimethyl
sulfide.
Aldehyde Compounds 47, 49 and 51, are obtained by hydrolysis (standard aqueous
acidic conditions) of Compounds 94 to 96.
j) Reduction:
Synthesis of Compound 48 by reduction of the aldehyde of Compound 47.
0
~~H i i OH
<\~)
Compound 48
HO H ~ \ OH
HO
A solution of Compound 47 (50 mg) in isopropanol (5 ml) is cooled in an ice-
salt
bath and sodium borohydride (10 mg) is added and the mixture is stirred for 20
minutes. It is then diluted with water (20 ml) and extracted twice with
toluene (10 ml
portions) at ambient temperature. The combined toluene extracts are filtered
and the
filtrate is concentrated to give Compound 48.
Compounds 50 and 52 are produced by the same procedure, when replacing
Compound 47 by Compounds 51 and 53 respectively as starting material.
While this invention has been particularly shown and described with references
to preferred embodiments thereof, it will be understood by those skilled in
the art that
various changes in form and details may be made therein without departing from
the
scope of the invention encompassed by the appended claims.
121