Note: Descriptions are shown in the official language in which they were submitted.
CA 02516269 2005-08-16
WO 2004/072060 PCT/EP2004/000993
1
Description
Substituted 3-(benzoylureido)-thiophene derivatives, method for the production
and
use thereof
The invention relates to substituted 3-(benzoylureido)thiophene derivatives
and to
their physiologically tolerated salts and physiologically functional
derivatives.
io
EP 0 300 972 describes benzoylureidothiophenes having pesticidal, specifically
insecticidal and acaricidal, action.
It is an object of the invention to provide compounds which make it possible
to
prevent and treat diabetes mellitus. To this end, the compounds should in
particular
exhibit a therapeutically utilizable blood sugar-reducing action.
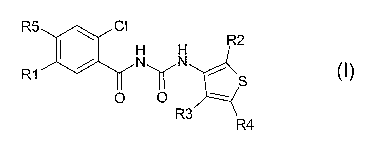
The invention therefore relates to compounds of the formula I
R5 ~ CI
R2
R1 ~ NON
~ S
O O
R3
R4
where
R5 is F, CI or Br;
R1 is H, F, CI, Br;
CA 02516269 2005-08-16
2
R2 is H, F, CI, Br, (C~-C6)-alkyl, CF3, OCF3, N02, CN, O-(C,-Cs)-alkyl, CO-
(C~-C6)-alkyl, COOH, COO(C~-C6)-alkyl, CONH2, CONH(C,-C6)-alkyl,
CON((C~-Cs)-a(kyl)Z, S02-(C~-C6)-alkyl, or the A radical;
R3 is H, (CI-Cs)-alkyl, COO(C~-C6)-alkyl, S02(C~-C6)-alkyl, (C~-C6)-alkyl-
phenyl, phenyl, S02-phenyl, where the phenyl ring may in each case be
up to disubstituted by F, Cl, CN, OH, (C~-C6)-alkyl, O-(C,-C6)-alkyl, CF3,
OCF3, COOH, COO(C~-C6)-alkyl or CONH2;
to R4 is H, (C,-C6)-alkyl, COO(C~-Cs)-alkyl, S02-(C,-Cs)-alkyl, S02-
piperidinyl,
S02-piperazinyl, (C~-C6)-alkylphenyl, where the phenyl ring, the
piperidinyl ring and the piperazinyl ring may be up to disubstituted by F,
CI, CN, OH, (C~-Cs)-alkyl, O-(C~-C6)-alkyl, CF3, OCF3, COOH, COO(C~-
C6)-alkyl or CONH2;
A is a heterocyclic radical of the formula 2a, 2b, 2c or 3;
N_N ~N_N ~_N wN O
~O X ~O~Y N~N
Z
2a
2o X is O or NH;
Y is OH or NH2;
Z is OH, O(C~-C6)-alkyl, NH2, NH(C~-C6)-alkyl, N((C1-C6)-alkyl)2;
and their physiologically tolerated salts.
Particular preference is given to compounds of the formula I where one or more
radicals are defined as follows:
R5 is F, CI or Br;
CA 02516269 2005-08-16
3
R1 is H, F;
R2 is H, F, CI, Br, (C~-C6)-alkyl, CF3, OCF3, N02, CN, O-(C~-C6)-alkyl,
CO(C~-C6)-alkyl, GOOH, COO(C~-C6)-alkyl, CONH2, CONH(C,-C6)-alkyl,
CON((C1-Cs)-alkyl)2, S02-(C~-C6)-alkyl, or the A radical;
R3 is H, (C~-Cs)-alkyl, COO(C~-Cs)-alkyl, SOZ(C~-Cs)-alkyl, (C,-C6)-
alkylphenyl, phenyl, S02-phenyl, where the phenyl ring may in each
io case be up to disubstituted by F or CI;
R4 is H, (C~-C6)-alkyl, COO(C~-C6)-alkyl, S02-(C~-C6)-alkyl, S02-piperidinyl,
S02-piperazinyl, (C~-C6)-aikyiphenyi, where the phenyl ring, the
piperidinyl ring and the piperazinyl ring may be up to disubstituted by F,
i5 CI, CN, OH, (C~-C6)-alkyl, O-(C~-C6)-alkyl, CF3, OCF3, COOH, COO(C,-
C6)-alkyl or CONH2;
A is a heterocyclic radical of the formula 2a, 2b, 2c or 3;
N_N /N_N /N_~ ~N O
X ~O Y ~N N
Z
X isOorNH;
Y is OH or NH2;
Z is OH;
and their physiologically tolerated salts.
so Particular preference is given to compounds of the formula I where one or
more
radicals are defined as follows:
CA 02516269 2005-08-16
4
R5 is F;
R1 is F;
R2 is COOH, COO(C~-C6)-alkyl, CONHZ, CONH(C~-C6)-alkyl, CON((C~-Cs)-
alkyl)Z, or the A radical;
R3 is H, (C,-C6)-alkyl, COO(Ct-C6)-alkyl, SOz(C1-C6)-alkyl, (C~-C6)-alkyl
io phenyl, phenyl, S02-phenyl, where the phenyl ring may in each case be
up to disubstituted by F;
R4 is H, (C~-C6)-alkyl, COO(C~-C6)-alkyl, SOZ-(C1-C6)-alkyl, S02-piperidinyl,
S02-piperazinyl, (C~-Cs)-alkylphenyl, where the phenyl ring, the
i5 piperidinyl ring and the piperazinyl ring may be up to disubstituted by F
or (C1-C6)-alkyl;
A is a heterocyclic radical of the formula 2a or 2b;
O X O Y
20 2a 2b
X is O or NH;
Y is OH or NH2;
and their physiologically tolerated salts.
Preference is further given to compounds of formula I in which at least one of
the
radicals R2, R3 and R4 is not hydrogen.
CA 02516269 2005-08-16
The invention relates to compounds of the formula I, in the form of their
racemates,
racemic mixtures and pure enantiomers, and also to their diastereomers and
mixtures
thereof.
5 The alkyl radicals in the substituents R2, R3 and R4 may be either straight-
chain or
branched.
When radicals or substituents can occur more than once in the compounds of the
formula I, for example A, they may all each independently be defined as
specified,
io and be the same or different.
As a consequence of their higher water solubility compared to the starting or
basic
compounds, pharmaceutically tolerated salts are particularly suitable for
medical
applications. These salts have to have a pharmaceutically tolerated anion or
cation.
i5 Suitable pharmaceutically tolerated acid addition salts of the compounds
according to
the invention are salts of inorganic acids such as hydrochloric acid,
hydrobromic acid,
phosphoric acid, metaphosphoric acid, nitric acid and sulfuric acid, and also
of
organic acids, e.g. acetic acid, benzenesulfonic acid, benzoic acid, citric
acid,
ethanesulfonic acid, fumaric acid, gluconic acid, glycolic acid, isethionic
acid, lactic
2o acid, lactobionic acid, malefic acid, malic acid, methanesulfonic acid,
succinic acid, p-
toluenesulfonic acid and tartaric acid. Suitable pharmaceutically tolerated
basic salts
are ammonium salts, alkali metal salts (such as sodium and potassium salts),
alkaline
earth metal salts (such as magnesium and calcium salts), trometamol (2-amino-2-
hydroxymethyl-1,3-propanediol), diethanolamine, lysine or ethylenediamine.
Salts having a pharmaceutically unacceptable anion, for example
trifluoroacetate, are
likewise encompassed by the scope of the invention as useful intermediates for
the
preparation or purification of pharmaceutically tolerated salts and/or for use
in
nontherapeutic, for example in vitro, applications.
The term "physiologically functional derivative" used herein refers to any
physiologically tolerated derivative of a compound of the formula I according
to the
invention, e.g. an ester which is able, on administration to a mammal, e.g. a
human,
CA 02516269 2005-08-16
6
to (directly or indirectly) form a compound of the formula I or an active
metabolite
thereof.
The physiologically functional derivatives also include prodrugs of the
compounds
according to the invention, for example as described in H. Okada et al., Chem.
Pharm. Bull. 1994, 42, 57-61. Such prodrugs can be metabolized in vivo to a
compound according to the invention. These prodrugs may or may not be active
themselves.
io The compounds according to the invention can also exist in different
polymorphous
forms, for example as amorphous and crystalline polymorphous forms. All
polymorphous forms of the compounds according to the invention are encompassed
by the scope of the invention and are a further aspect of the invention.
i5 All references given below to "compound(s) of formula I" refer to
compounds) of the
formula I as described above, and also to their salts, solvates and
physiologically
functional derivatives as described herein.
In this context, an aryl radical is a phenyl, naphthyl, biphenyl,
tetrahydronaphthyl,
zo alpha- or beta-tetralone, indanyl or indan-1-onyl radical.
The terms "heterocyclic ring" and "heterocyclic radical" used here relate to
heteroaryl
radicals and heterocycloalkyi radicals which derive from 3 to 10 membered
carbon
rings in which one or more carbon atoms have been replaced by one or more
atoms
25 selected from the group of oxygen, sulfur and nitrogen.
Suitable "heterocyclic rings" and "heterocyclic radicals" are acridinyl,
azocinyl,
benzimidazolyl, benzofuryl, benzothienyl, benzothiophenyl, benzoxazolyl,
benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl,
benzisothiazolyl,
so benzimidazalinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, quinazolinyl,
quinolinyl, 4H-
quinolizinyl, quinoxalinyl, quinuclidinyl, chromanyl, chromenyl, cinnolinyl,
decahydroquinolinyl, 2H,6H-1,5,2-dithiazinyl, dihydrofuro[2,3-b)-
tetrahydrofuran, furyl,
furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1 H-indazolyl, indolinyl,
indolizinyl,
indolyl, 3H-indolyl, isobenzofuranyi, isochromanyi, isoindazolyl,
isoindolinyi,
CA 02516269 2005-08-16
7
isoindolyl, isoquinolinyl (benzimidazolyl), isothiazolyl, isoxazolyl,
morpholinyl,
naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-
oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl,
oxazolidinyl,
pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl,
phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl,
pteridinyl, purynyi,
pyranyl, pyrazinyl, pyroazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl,
pyridooxazole,
pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl,
pyrrolidinyl, pyrrolinyl,
2H-pyrrolyl, pyrrolyl, tetrahydrofuranyl, tetrahydroisoquinolinyl,
tetra~hydroquinolinyl,
6H-1,2,5-thiadazinyl, thiazolyl, 1,2,3-thiadiazoiyl, 1,2,4-thiadiazolyl, 1,2,5-
thiadiazolyl,
io 1,3,4-thiadiazolyl, thienyl, triazolyl, tetrazolyl and xanthenyl.
Pyridyl is either 2-, 3- or 4-pyridyl. Thienyl is either 2- or 3-thienyl.
Furyl is either 2- or
3-fury!.
i5 Also included are the corresponding N-oxides of these compounds, for
example
1-oxy-2-, 3- or 4-pyridyl.
Also included are singly or multiply benzofused derivatives of these
heterocycles.
2o The compounds) of the formula (i) can also be administered in combination
with
further active ingredients.
The amount of a compound of formula I which is required in order to achieve
the
desired biological effect is dependent upon a series of factors, for example
the
25 specific compound selected, the intended use, the mode of administration
and the
clinical condition of the patient. The daily dose is generally in the range
from 0.3 mg
to 100 mg (typically from 3 mg and 50 mg) per day per kilogram of bodyweight,
for
example 3-10 mg/kg/day. An intravenous dose may, for example, be in the range
from 0.3 mg to 1.0 mg/kg and may advantageously be administered as an infusion
of
3o from 10 ng to 100 ng per kilogram per minute. Suitable infusion solutions
for these
purposes may, for example, contain from 0.1 ng to 10 mg, typically from 1 ng
to
mg, per milliliter. Individual doses may contain, for example, from 1 mg to 10
g of
the active ingredient. Ampules for injections may therefore contain, for
example, from
1 mg to 100 mg, and single dose formulations which can be administered orally,
for
CA 02516269 2005-08-16
8
example tablets or capsules, may contain, for example, from 1.0 to 1000 mg,
typically
from 10 to 600 mg. The compounds of formula I may be used for therapy of the
abovementioned conditions as the compounds themselves, although they are
preferably in the form of a pharmaceutical composition with an acceptable
carrier.
The carrier of course has to be acceptable, in the sense that it is compatible
with the
other constituents of the composition and is not damaging to the health of the
patient.
The carrier may be a solid or a liquid or both and is preferably formulated
with the
compound as a single dose, for example as a tablet, which may contain from
0.05 to
95% by weight of the active ingredient. Further pharmaceutically active
substances
io may likewise be present, including further compounds of formula I. The
pharmaceutical compositions according to the invention may be produced by one
of
the known pharmaceutical methods which consist essentially of mixing the
ingredients with pharmacologically acceptable carriers and/or excipients.
Pharmaceutical compositions according to the invention are those which are
suitable
for oral, rectal, topical, peroral (for example sublingual) and parenteral
(for example
subcutaneous, intramuscular, intradermal or intravenous) administration,
although the
most suitable mode of administration depends in each individual case on the
nature
and severity of the condition to be treated and on the type of the compound of
2o formula I used in each case. Coated formulations and coated slow-release
formulations are also encompassed by the scope of the invention. Preference is
given to acid- and gastric fluid-resistant formulations. Suitable gastric
fluid-resistant
coatings include cellulose acetate phthalate, polyvinyl acetate phthalate,
hydroxypropylmethylceilulose phthalate and anionic polymers of methacryiic
acid and
methyl methacrylate.
Suitable pharmaceutical compounds for oral administration may be in the form
of
separate units, for example capsules, cachets, lozenges or tablets, each of
which
contains a certain amount of the compound of formula I; as powder or granules;
as
3o solution or suspension in an aqueous or nonaqueous liquid; or as an oil-in-
water or
water-in-oil emulsion. These compositions may, as already mentioned, be
prepared
by any suitable pharmaceutical method which includes a step in which the
active
ingredient and the carrier (which may consist of one or more additional
ingredients)
are brought into contact. In general, the compositions are prepared by uniform
and
CA 02516269 2005-08-16
9
homogeneous mixing of the active ingredient with a liquid and/or finely
divided solid
carrier, after which the product is shaped if necessary. For example, a tablet
can be
produced by compressing or shaping a powder or granules of the compound,
optionally with one or more additional ingredients. Compressed tablets can be
prepared by tableting the compound in free-flowing form, for example a powder
or
granules, optionally mixed with a binder, lubricant, inert diluent andlor one
(or more)
surfactants/dispersants in a suitable machine. Shaped tablets can be prepared
by
shaping the pulverulent compound moistened with an inert liquid diluent in a
suitable
machine.
io
Pharmaceutical compositions which are suitable for peroral (sublingual)
administration include lozenges which contain a compound of formula I with a
flavoring, customarily sucrose, and gum arabic or tragacanth, and pastilles
which
include the compound in an inert base such as gelatin and glycerol or sucrose
and
Z5 gum arabic.
Suitable pharmaceutical compositions for parenteral administration include
preferably
sterile aqueous preparations of a compound of formula I which are preferably
isotonic
with the blood of the intended recipient. These preparations are preferably
2o administered intravenously, although the administration may also be
subcutaneous,
intramuscular or intradermal as an injection. These preparations can
preferably be
produced by mixing the compound with water and making the solution obtained
sterile and isotonic with the blood. The injectable compositions according to
the
invention generally contain from 0.1 to 5% by weight of the active compound.
Suitable pharmaceutical compositions for rectal administration are preferably
in the
form of single dose suppositories. These can be prepared by mixing a compound
of
formula I with one or more conventional solid carriers, for example cocoa
butter, and
shaping the resulting mixture.
Suitable pharmaceutical compositions for topical use on the skin are
preferably in the
form of an ointment, cream, lotion, paste, spray, aerosol or oil. Useful
carriers include
petroleum jelly, lanolin, polyethylene glycols, alcohols and combinations of
two or
more of these substances. The active ingredient is generally present in a
CA 02516269 2005-08-16
concentration of from 0.1 to 15% by weight of the composition, preferably from
0.5 to
2%.
Transdermal administration is also possible. Suitable pharmaceutical
compositions
for transdermal applications may be in the form of single plasters which are
suitable
for long-term close contact with the epidermis of the patient. Such plasters
advantageously contain the active ingredient in an optionally buffered aqueous
solution, dissolved and/or dispersed in an adhesive or dispersed in a polymer.
A
suitable active ingredient concentration is from approx. 1 % to 35%,
preferably from
io approx. 3% to 15%. A particular means of releasing the active ingredient is
by
electrotransport or iontophoresis, as described, for example, in
Pharmaceutical
Research, 2(6): 318 (1986).
Further useful active ingredients for combination products are as follows:
All antidiabetics mentioned in the Rote Liste 2001, chapter 12. They can be
combined with the compounds of the formula I according to the invention, in
particular for synergistic enhancement of the action. The active ingredient
combination can be administered either by separately administering the active
ingredients to the patient or in the form of combination products in which a
plurality of
2o active ingredients are present in one pharmaceutical preparation. Most of
the active
ingredients listed hereinbelow are disclosed in USP Dictionary of USAN and
International Drug Names, US Pharmacopeia, Rockville 2001.
Antidiabetics include insulin and insulin derivatives, for example Lantus~
(see
www.lantus.com) or HMR 1964, fast-acting insulins (see US 6,221,633), GLP-1
derivatives, for example those disclosed in WO 98/08871 of Novo Nordisk A/S,
and
orally active hypoglycemic active ingredients.
The orally active hypoglycemic active ingredients preferably include
sulfonylureas,
biguanidines, meglitinides, oxadiazolidinediones, thiazolidinediones,
glucosidase
inhibitors, glucagon antagonists, GLP-1 agonists, potassium channel openers,
for
3o example those disclosed in WO 97/26265 and WO 99/03861 of Novo Nordisk AIS,
insulin sensitizers, inhibitors of (fiver enzymes which are involved in the
stimulation of
gluconeogenesis and/or glycogenolysis, modulators of glucose uptake, compounds
which alter lipid metabolism such as antihyperlipidemic active ingredients and
antilipidemic active ingredients, compounds which reduce food intake, PPAR and
CA 02516269 2005-08-16
11
PXR agonists and active ingredients which act on the ATP-dependent potassium
channel of the beta cells.
In one embodiment of the invention, the compounds of the formula I are
administered
s in combination with an HMGCoA reductase inhibitor such as simvastatin,
fluvastatin,
pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a cholesterol absorption inhibitor, for example,
ezetimibe,
to tiqueside, pamaqueside.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a PPAR gamma agonist, for example, rosiglitazone,
pioglitazone,
JTT-501, GI 262570.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with PPAR alpha agonist, for example, GW 9578, GW 7647.
In one embodiment of the invention, the compounds of the formula I are
administered
2o in combination with a mixed PPAR alpha/gamma agonist, for example, GW 1536,
AVE 8042, AVE 8134, AVE 0847, or as described in PCT/US 11833, PCT/US 11490,
DE10142734.4.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a fibrate, for example, fenofibrate, clofibrate,
bezafibrate.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with an MTP inhibitor, for example, implitapide, BMS-201038, R-
103757.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with bile acid absorption inhibitor (see, for example, US
6,245,744 or
US 6,221,897), for example, HMR 1741.
CA 02516269 2005-08-16
12
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a CETP inhibitor, for example, JTT-705.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a polymeric bile acid adsorbent, for example,
cholestyramine,
colesevelam.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with an LDL receptor inducer (see US 6,342,512), for example,
io HMR1171, HMR1586.
(n one embodiment of the invention, the compounds of the formula I are
administered
in combination with an ACAT inhibitor, for example, avasimibe.
i5 In one embodiment of the invention, the compounds of the formula I are
administered
in combination with an antioxidant, for example, OPC-14117.
Ln one embodiment of the invention, the compounds of the formula I are
administered
in combination with a lipoprotein lipase inhibitor, for example, NO-1886.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with an ATP-citrate lyase inhibitor, for example, SB-204990.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a squalene synthetase inhibitor, for example, BMS-188494.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a lipoprotein(a) antagonist, for example, CI-1027 or
nicotinic acid.
so In one embodiment of the invention, the compounds of the formula I are
administered
in combination with a lipase inhibitor, for example, orlistat.
In one embodiment of the invention, the compounds of the formula I are
administered
in combination with insulin.
CA 02516269 2005-08-16
13
In one embodiment, the compounds of the formula I are administered in
combination
with a sulfonylurea, for example, tolbutamide, glibenclamide, glipizide or
glimepiride.
In one embodiment, the compounds of the formula I are administered in
combination
with a biguanide, for example, metformin.
In yet another embodiment, the compounds of the formula I are administered in
combination with a meglitinide, for example, repaglinide.
In one embodiment, the compounds of the formula I are administered in
combination
with a thiazolidinedione, for example, troglitazone, ciglitazone,
pioglitazone,
io rosiglitazone or the compounds disclosed in WO 97/41097 of Dr. Reddy's
Research
Foundation, in particular 5-[[4-[(3,4-dihydro-3-methyl-4-oxo-2-
quinazolinylmethoxy]-
phenyl]methyl]-2,4-thiazolidinedione.
In one embodiment, the compounds of the formula I are administered in
combination
with an a-glucosidase inhibitor, for example, miglitol or acarbose.
In one embodiment, the compounds of the formula I are administered in
combination
with an active ingredient which acts on the ATP-dependent potassium channel of
the
beta cells, for example, tolbutamide, glibenclamide, glipizide, glimepiride or
repaglinide.
In one embodiment, the compounds of the formula I are administered in
combination
with more than one of the abovementioned compounds, for example in combination
with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide
and
metformin, insulin and a sulfonylurea, insulin and metformin, insulin and
troglitazone,
insulin and lovastatin, etc.
In a further embodiment, the compounds of the formula I are administered in
combination with CART modulators (see "Cocaine-amphetamine-regulated
transcript
influences energy metabolism, anxiety and gastric emptying in mice" Asakawa,
A,
et al., M.: Hormone and Metabolic Research (2001}, 33(9), 554-558), NPY
antagonists, e.g. naphthalene-1-sulfonic acid {4-[(4-aminoquinazolin-2-
ylamino)-
3o methyl]cyclohexylmethyl}amide hydrochloride (CGP 71683A)), MC4 agonists
(e.g.
1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2-(3a-benzyl-2-methyl-
3-oxo-2,3,3a,4,6,7-hexahydropyrazolo[4,3-c]pyridin-5-yl)-1-(4-chlorophenyl)-2-
oxo-
ethyl]amide; (WO 01/91752)), orexin antagonists (e.g. 1-(2-methylbenzoxazol-6-
yl)-
3-[1,5]naphthyridin-4-ylurea; hydrochloride (SB-334867-A)), H3 agonists
CA 02516269 2005-08-16
14
(3-cyclohexyl-1-(4,4-dimethyl-1,4,6,7-tetrahydroimidazo[4,5-c)pyridin-5-
yi)propan-
1-one oxalic acid salt (WO 00/63208)); TNF agonists, CRF antagonists (e.g.
[2-methyl-9-(2,4,6-trimethylphenyl)-9H-1,3,9-triazafluoren-4-yl)dipropylamine
(WO 00166585)), CRF BP antagonists (e.g. urocortin), urocortin agonists, ~i3
agonists
(e.g. 1-(4-chloro-3-methanesulfonylmethy!phenyl)-2-[2-(2,3-dimethyl-1 H-indol-
6-yloxy)ethylamino)ethanol hydrochloride (WO 01/83451)), MSH (melanocyte-
stimulating hormone) agonists, CCK-A agonists (e.g. {2-[4-(4-chloro-2,5-
dimethoxyphenyl)-5-(2-cyclohexylethyl)thiazol-2-ylcarbamoyl)-5,7-dimethylindol-
1-
yl}acetic acid trifluoroacetic acid salt (WO 99115525)), serotonin reuptake
inhibitors
io (e.g, dexfenfluramine), mixed serotoninergic and noradrenergic compounds
(e.g.
WO 00/71549), 5HT agonists e.g. 1-(3-ethylbenzofuran-7-yl)piperazine oxalic
acid
salt (WO 01109111 ), bombesin agonists, galanin antagonists, growth hormone
(e.g.
human growth hormone), growth hormone-releasing compounds (6-benzyloxy-1-(2-
diisopropylaminoethylcarbamoyl)-3,4-dihydro-1 H-isoquinoline-2-carboxylic acid
tert-
butyl ester (WO 01 /85695)), TRH agonists (see, for example, EP 0 462 884),
uncoupling protein 2 or 3 modulators, leptin agonists (see, for example, Lee,
Daniel W.; Leinung, Matthew C.; Rozhavskaya-Arena, Marina; Grasso, Patricia.
Leptin agonists as a potential approach to the treatment of obesity. Drugs of
the
Future (2001 ), 26(9), 873-881 ), DA agonists (bromocriptine, Doprexin),
lipaselamylase inhibitors (e.g. WO 00/40569), PPAR modulators (e.g. WO
00!78312),
RXR modulators or TR-[3 agonists.
In one embodiment of the invention, the other active ingredient is leptin;
see, for
example, "Perspectives in the therapeutic use of leptin", Salvador, Javier;
Gomez-
Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001 ),
2(10), 1615-1622.
In one embodiment, the other active ingredient is dexamphatamine or
amphetamine.
In one embodiment, the other active ingredient is fenfluramine or
dexfenfluramine.
so In another embodiment, the other active ingredient is sibutramine.
In one embodiment, the other active ingredient is orlistat.
In one embodiment, the other active ingredient is mazindol or phentermine.
CA 02516269 2005-08-16
In one embodiment, the compounds of the formula I are administered in
combination
with dietary fiber materials, preferably insoluble dietary fiber materials
(see, for
example, Carob/Caromax~ (Zunft H J; et al., Carob pulp preparation for
treatment of
hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-6.)
Caromax is a carob-containing product supplied by Nutrinova, Nutrition
Specialties 8~
Food Ingredients GmbH, Industriepark Hochst, 65926 Frankfurt/Main)).
Combination
with Caromax~ is possible in one preparation or by separate administration of
compounds of the formula I and Caromax~. Caromax~ can also be administered in
the form of foodstuffs, for example, in bakery products or muesli bars.
to
It will be appreciated that any suitable combination of the compounds
according to
the invention with one or more of the abovementioned compounds and optionally
one
or more further pharmacologically active substances is regarded as being
covered by
the scope of protection of the present invention.
CA 02516269 2005-08-16
16
CH3 ~ \
CH
CH3 O ~N / O~ 3
~ /NJ
O NH CH3 H3C
3
S
I \ CH3 OPC-14117
/ O
JTT-705 CI \
I / ,,, o
00
CI OH
Br
I \ O SB-204990 HO
N \ O CH3
H I
/ ~O/~CH3
I I P
N
NO-1886 O OH
H C OH O CH3
3
H3C O CH3
C I-1027
,O
I \ HaC CH3
O
/ O / CHs
H3C O CH3
BMS-188494 CH3
O O
CH3
O
\ I \ I H OH
-. N O \
O I /
61262570 / I
O \
CH3
O \
\ I I / No
N o O H
CA 02516269 2005-08-16
17
The examples recited hereinbelow serve to illustrate the invention, but
without limiting
it.
In a similar manner, the compounds of the following examples were prepared:
R5 ~ CI
R2
R1
S
O O
R3
R4
Ex. R5 R1 R2 R3 R4 m.p.
1 F F ~ ~ H H 236.1
d 0 0
2c F F ~-~ H H Resin
O NHi
3b F F COOH H F 227.7
\ /
4d F F _N'~ H _S_N~ 219.9
~OH
5 F F COOMe H H
6 F F CONH2 H H
7 F F COOMe H S02Me
8 F F COOMe H
\ / F
9 F F COOMe Me H
F F H COOMe H
11 F F N02 H H
12 F_ F COOH S02Ph H 193.5
13 F F COOH S02iPr H 164.8
14 F F COOMe H Ph 208.6
CA 02516269 2005-08-16
18
15 F F COMB H Ph 204.8
16 F F CONHZ H tert-Butyl >300
17 F F CONH2 H Ph >300
18 F F CONHZ H >300
ci
19 F F -N N-Me H ° n 225.8
-S-NON-Me
O
20 F F - ~ H °- ~ 188.1
N -S N
O
21 F F COOH H Ph 226.5
22 F F CONH2 H 3-Thienyl >300(decomp.)
23 F F CONHz H 2-Thienyt >250(decomp.)
24 F F CONHz H >300
\ / O-Me
25 F F -CN H F >200(decomp.)
\ /
26 F F COMB H H 180(decomp.)
27 F F CONH2 H F 237.2
\ /
28 F F H F H
\ /
29 F F H COOMe COOMe
30 F F H Ph H
31 F F H F_ H
\ /
32 F F H Me H
33 F H ~ N H H 219.8
,N
N
34 F F H H Phenyl 178.7
35 F F COOH H H 214.8
36c F F ~ N H \ / F 206.2
,N
N
37 F H ~ N H \ / F 217.8
N
CA 02516269 2005-08-16
19
The effectiveness of the compounds was tested as follows:
Glycogen phosphorylase a activity test
The effect of compounds on the activity of the active form of glycogen
phosphorylase
(GPa) was measured in the reverse direction by monitoring the synthesis of
glycogen
from glucose 1-phosphate by determining the release of inorganic phosphate.
All
reactions were carried out as duplicate determinations in 96-welt microtiter
plates
io (half area plates, Costar No. 3696), and the change in absorption as a
consequence
of the formation of the reaction product was measured at the wavelength
specified
below in a Multiskan Ascent Elisa Reader (Lab Systems, Finland).
In order to measure the GPa enzyme activity in the reverse direction, the
conversion
of glucose 1-phosphate to glycogen and inorganic phosphate was measured by the
i5 general method of Engers et al. (Engers HD, Shechosky S, Madsen NB, Can J
Biochem 1970 Ju1;48(7):746-754) with the following modifications: human
glycogen
phosphorylase a (for example containing 0.76 mg of protein / ml (Aventis
Pharma
Deutschland GmbH), dissolved in buffer solution E (25 mM (3-glycerophosphate,
pH
7.0, 1 mM EDTA and 1 mM dithiothreitol) was diluted to a concentration of 10
Ng of
2o protein/ml with buffer T (50 mM Hepes, pH 7.0, 100 mM KCI, 2.5 mM EDTA, 2.5
mM
MgC12~6H20) and addition of 5 mg/ml of glycogen. Test substances were prepared
as
a 10 mM solution in DMSO and diluted to 50 NM with buffer solution T. To 10 NI
of
this solution were added 10 NI of 37.5 mM glucose dissolved in buffer solution
T and
5 mg/ml of glycogen, and also 10 NI of a solution of human glycogen
phosphorylase a
2s (10 Ng of protein/ml) and 20 NI of 2.5 mM glucose 1-phosphate. The base
value of the
activity of glycogen phosphorylase a in the absence of test substance was
determined by adding 10 NI of buffer solution T (0.1 % DMSO). The mixture was
incubated at room temperature for 40 minutes and the released inorganic
phosphate
was determined by means of the general method of Drueckes et al. (Drueckes P,
3o Schinzel R, Palm D, Anal Biochem 1995 Sep 1;230(1):173-177) with the
following
modifications: 50 NI of a stop solution of 7.3 mM of ammonium molybdate, 10.9
mM
of zinc acetate, 3.6% of ascorbic acid, 0.9% of SDS are added to 50 NI of the
enzyme
mixture. After 60 minutes of incubation at 45°C, the absorption was
measured at
820 nm. To determine the background absorption, the stop solution was added
CA 02516269 2005-08-16
immediately after the addition of the glucose 1-phosphate solution in a
separate
reaction.
This test was carried out at a concentration of 10 NM of the test substance,
in order to
determine the respective inhibition of glycogen phosphorylase a by the test
substance in vitro.
Table 2: Biological activity
Ex. IC-50 (NM)
1 d 0.03
2c 0.45
3b 0.01
4d 0.11
6 0.70
15 0.17
17 0.03
21 0.01
23 - 0.05
24 0.04
30 1.40
33 0.08
It can be seen from the table that the compounds of the formula I inhibit the
activity of
glycogen phosphorylase a and are thus very suitable for reducing the blood
sugar
level.
i5 The preparation of some examples is described in detail hereinbelow, and
the
remaining compounds of the formula I were obtained in a similar manner:
Experimental section:
Example 1:
2o a) 3-(tert-Butoxycarbonylamino)thiophene-2-carboxylic hydrazide
CA 02516269 2005-08-16
21
0.4 g of hydrazine hydrate is added to a solution of 1.3 g of methyl 3-(tert-
butoxycarbonylamino)thiophene-2-carboxylate in 10 ml of ethanol and the
mixture is
heated to reflux for 5 hours. After the volatile fractions had been removed
under
reduced pressure at 40°C, the remaining oil was purified by column
chromatography
(silica gel, eluent: methylene chloride: methanol = 95:5).
Yield: 740 mg m.p.: 146.5°C
b) tert-Butyl [2-(5-oxo-4,5-dihydro[1,3,4]oxadiazol-2-yl)thiophen-3-
yl]carbamate
4 ml of a 20% toluenic phosgene solution are added dropwise to a solution of
240 mg
of 3-(tert-butoxycarbonylamino)thiophene-2-carboxylic hydrazide in 5 ml of THF
and
io the mixture is stirred at RT. After one hour, the mixture is admixed with
10 ml of water
and, after brief digestion, extracted with ethyl acetate. After the ethyl
acetate phase
has been dried over sodium sulfate, the mixture is concentrated under reduced
pressure and the remaining residue is used further without further
purification.
Yield: 120 mg m.p.: 180°C
i5 c) 5-(3-Aminothiophen-2-yl)-3H-[1,3,4]oxadiazol-2-one hydrochloride
The mixture consisting of 100 mg of tert-butyl [2-(5-oxo-4,5-
dihydro[1,3,4]oxadiazol-2-
yl)thiophen-3-yl]carbamate and 5 ml of a 4 molar HCI solution in dioxane is
stirred for
one hour. Afterwards, the volatile fractions are removed under reduced
pressure and
the residue is stirred with 5 ml of tert-butyl methyl ether and the product is
filtered off
2o with suction and dried under reduced pressure.
Yield: 60 mg m.p.: 211 °C
d) 1-(2-Chloro-4,5-difluorobenzoyl)-3-[2-(5-oxo-4,5-dihydro[1,3,4]oxadiazol-2-
yl)thiophen- 3-yl]urea
5-(3-Aminothiophen-2-yl)-3H-[1,3,4]oxadiazol-2-one hydrochloride (30 mg) is
initially
25 charged in 3 ml of acetonitrile. The equimolar solution of 2-chloro-4,5-
difluorobenzoyl
isocyanate in acetonitrile is added. After 3 hours, the solid is filtered off
with suction
and dried under reduced pressure.
Yield: 50 mg m.p.: 236.1 °C
3o Example 2:
a) tert-Butyl 2-(5-amino[1,3,4]oxadiazol-2-yl)thiophen-3-yl]carbamate
The solution of 192 mg of 3-(tert-butoxycarbonylamino)thiophene-2-carboxylic
hydrazide in 4 ml of acetonitrile is admixed with 0.17 ml of a 5 molar
cyanogen
bromide solution in acetonitrile and 60 mg of potash. The mixture is stirred
at RT for 4
CA 02516269 2005-08-16
22
hours, the solid is filtered off and the filtrate is concentrated under
reduced pressure.
The desired product is then purified by column chromatography (silica gel,
eluent:
methylene chloride:methanol = 95:5).
Yield: 90 mg m.p.: Resin
b) 5-(3-Aminothiophen-2-yl)-[1,3,4]oxadiazol-2-ylamine hydrochloride
tert-Butyl 2-(5-amino[1,3,4]oxadiazol-2-yl)thiophen-3-yl]carbamate (90 mg) are
added
to 5 ml of 4 molar HCI solution in dioxane and the mixture is stirred at RT
for one
hour. After concentration under reduced pressure, the residue is stirred with
tert-butyl
methyl ether and the solid is filtered off with suction and dried under
reduced
io pressure.
Yield: 60 mg m.p.: >250° (decomp.)
c) 1-[2-(5-Amino[1,3,4]oxadiazol-2-yl)thiophen-3-yl]-3-(2-chloro-4,5-
difluorobenzoyl)urea
5-(3-Aminothiophen-2-yl)[1,3,4]oxadiazol-2-ylamine hydrochloride (30 mg) is
initially
charged in 3 ml of acetonitrile. The equimolar solution of 2-chloro-4,5-
difluorobenzoyl
isocyanate in acetonitrile is added and stirred at RT. After 3 hours, the
solid is filtered
off with suction and dried under reduced pressure.
Yield: 25 mg m.p.: Resin
2o Example 3:
a) 3-Amino-5-(4-fluorophenyl)thiophene-2-carboxylic acid
The mixture consisting of 500 mg of methyl 3-amino-5-(4-fluorophenyl)thiophene-
2-
carboxylate, 160 mg of lithium hydroxide, 2 ml of water, 2 ml of THF and 2 m1
of
methanol is stirred at RT for 3 days. After dilution with 15 ml of water,
unhydrolyzed
25 ester is removed by extraction with ethyl acetate. The aqueous phase is
adjusted to
pH 5 using hydrochloric acid and stirred, and the precipitated solid is
filtered off with
suction and dried.
Yield: 260 mg m.p.: 145.2°C (crude)
b) 3-[3-(2-Chloro-4,5-difluorobenzoyt)ureido]-5-(4-fluorophenyl)thiophene-2-
3o carboxylic acid
3-Amino-5-(4-fluorophenyl)thiophene-2-carboxylic acid (50 mg) is initially
charged in
3 ml of acetonitrile. The equimolar solution of 2-chloro-4,5-difluorobenzoyl
isocyanate
in acetonitrile is added and the mixture is stirred at RT. After 3 hours, the
solid is
CA 02516269 2005-08-16
23
filtered off with suction, stirred once more with methanol, filtered off with
suction and
dried under reduced pressure.
Yield: 46 mg m.p.: 227.7°C
Example 4:
a) 1-(5-Chloro-4-nitrothiophene-2-sulfonyl)piperidine and 1-(5-piperidino-4-
nitrothiophene-2-sulfonyl)piperidine
1.7 g of piperidine are added dropwise to the solution of 2.6 g of 5-chloro-4-
nitrothiophene-2-sulfonyl chloride in 8 ml of NMP with stirring and ice
cooling. The
io mixture is stirred at RT for another 30 minutes and diluted with 30 ml of
water, and
the precipitate which forms is filtered off with suction after stirring. The
two products
are separated by column chromatography (silica gel, solvent: ethyl acetate: n-
heptane = 1:1 ).
1-(5-Piperidino-4-nitrothiophene-2-sulfonyl)piperidine: yield: 1.3 g, m.p.:
151.3°C
i5 1-(5-Chloro-4-nitrothiophene-2-sulfonyl)piperidine: Yield: 0.85 g m.p.:
136.4°C
b) 1-(3-Nitro-5-piperidine-1-sulfonylthiophen-2-yl)piperidine-4-carboxylic
acid
The mixture of 310 mg of 1-(5-chloro-4-nitrothiophene-2-sulfonyl)piperidine,
250 mg
of piperidine-4-carboxylic acid and 3 ml of NMP is stirred at 85°C for
1 h, cooled,
diluted with 15 ml of water and stirred. The precipitate is filtered off with
suction,
2o washed with water and recrystallized from isopropanollwater (4:1).
Yield: 310 mg m.p.: 165.8°C
c) 1-[3-Amino-5-(piperidine-1-sulfonyl)thiophen-2-yl]piperidine-4-carboxylic
acid
The solution of 300 mg of 1-(3-nitro-5-piperidine-1-sulfonylthiophen-2-
yl)piperidine-4-
25 carboxylic acid in 20 ml of ethyl acetate is admixed with 1.0 g of tin(II)
chloride and
stirred at 65°C for 8 h. After cooling, it is stirred with 30 ml of
water and filtered off
with suction through Celite, and the ethyl acetate phase is removed, dried and
concentrated under reduced pressure. This crude product is used further
without
further purification.
so Yield: 320 mg(crude) m.p.: Resin
c) 1-[3-[3-(2-Chloro-4,5-diftuorobenzoyl)ureido]-5-(piperidine-1-sulfonyl)-
thiophen-2-yl]piperidine-4-carboxylic acid
1-[3-Amino-5-(piperidine-1-sulfonyl)thiophen-2-yl]piperidine-4-carboxylic acid
(0.2 g of
crude product) is dissolved in 2 ml of acetonitrile and admixed with the
equimolar
CA 02516269 2005-08-16
24
solution of 2-chloro-4,5-difluorobenzoyl isocyanate in acetonitrile, and the
mixture is
stirred at RT. After 3 hours, the solid is filtered off with suction and dried
under
reduced pressure.
Yield: 85 mg m.p.: 219.9°C
Example 36:
a) 5-(4-Fluorophenyl)-2-(1H-tetrazol-5-yl)thiophen-3-ylamine
The mixture consisting of 0.65 g of 3-amino-2-cyano-5-(4-
fluorophenyl)thiophene,
7.5 ml of xylene and 0.93 g of trimethyltin azide is stirred at 130-
140°C for 3 hours.
io Afterwards, the mixture is concentrated at 40°C under reduced
pressure and the
residue is stirred with water with the addition of 1 % trifluoroacetic acid.
The
precipitate is filtered off with suction and dried at 40°C under
reduced pressure.
Yield: 0.6 g m.p.: 217.3°C
c) 1-[4-(4-Fluorophenyl-2-(tetrazol-5-yl)thiophen-3-ylJ-3-(2-chloro-4,5-
i5 difluorobenzoyl)urea
5-(4-Fluorophenyi)-2-(1H-tetrazol-5-yl)thiophen-3-ylamine (0.1 g) is dissolved
in 2 ml
of acetonitrile and admixed with the equimolar solution of 2-chloro-4,5-
difluorobenzoyl isocyanate in acetonitrile and the mixture is stirred at RT.
After 3
hours, the solid is filtered off with suction and dried under reduced
pressure.
2o Yield: 73 mg m.p.: 206.2°C
The compounds of the formula I can be prepared by reacting ureas of the
formula 5
or 3-aminothiophene derivatives of the formula 6 with benzoic acid derivatives
of the
formula 4,
R5 CI H H R2 H R2
H~N N , H~N ~ S
R1 ~ X~ ~S _
O
O R3 R3 R4
R4
where R1 to R5 are each as defined above and X1 may be CI or NCO, with acid
chlorides or anhydrides or benzoyl isocyanates.