Note: Descriptions are shown in the official language in which they were submitted.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-1-
DELIVERY OF IMMUNE RESPONSE MODIFIER COMPOUNDS
BACKGROUND
There has been a major effort in recent years, with significant successes, to
discover new drug compounds that act by stimulating certain key aspects of the
immune system, as well as by suppressing certain other aspects (see, e.g.,
U.S. Pat.
Nos. 6,039,969 and 6,200,592). These compounds, referred to as immune response
modifiers (IRMs), appear to act through basic immune system mechanisms known
as
toll-like receptors to induce selected cytokine biosynthesis and may be used
to treat a
wide variety of diseases and conditions. For example, certain IRMs may be
useful for
treating viral diseases (e.g., human papilloma virus, hepatitis, herpes),
neoplasias (e.g.,
basal cell carcinoma, squamous cell carcinoma, actinic keratosis), and TH2-
mediated
diseases (e.g., asthma, allergic rhinitis, atopic dermatitis), and are also
useful as vaccine
adjuvants. Many of the IRM compounds are small organic molecule
imidazoquinoline
amine derivatives (see, e.g., U.S. Pat. No. 4,6~9,33~), but a number of other
compound
classes are known as well (see, e.g., U.S. Pat. No. 5,446,153) and more are
still being
discovered. Other IRMs have higher molecular weights, such as
oligonucleotides,
including CpGs (see, e.g., U.S. Pat. No. 6,194,353). In view of the great
therapeutic
potential for IRMs, and despite the important work that has already been done,
there is
a substantial ongoing need for new means of controlling the delivery and
activity of
IRh~ls in order to expand their uses and therapeutic benefits.
SUM1VIARY
It has now surprisingly been found that immune response modifiers (IRMs) of
the invention can be attached to macromolecular support materials and,
importantly,
that they retain biological activity even while they remain attached to such
material.
This ability to form biologically active IRM-support complexes allows for a
tremendous range of useful applications where one may not wish to release all
of the
IRM compound to be effective.
For example, in contrast to eluting drug from a coated surface or delivering
drug
from a formulation, the IRMs here can be active while attached to, e.g.,
implantable
medical devices, particles, beads, polymers, and other supports, substrates,
and matrix
materials. This approach can be used, e.g., to help reduce systemic absorption
through
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-2-
dermal, mucosal and other tissues, such as through the GI tract, respiratory
tract, eyes,
rectum, bladder, vagina, etc., as well as to maintain extended deposition of
the lRM at
an intended site of action, such as implanted in a solid tumor mass. As an
illustration,
immune system dendritic cells can be removed from a patient and activated ex
vivo in
the presence of a desired antigen by being placed in contact with a material
lined with
attached IRMs (e.g., container walls, beads, mesh, etc.). The activated
dendritic cells
can then be conveniently returned to the patient for therapeutic use, leaving
the IRM
behind so as to avoid systemic exposure.
Moreover, not only has it been found that the lRMs are still biologically
active
when attached to a support complex, but surprisingly, the cytokine induction
profile of
the 1RM can be altered in potentially desirable ways by virtue of such
attachment. It
has been found that attachment of some II~Ms actually modifies the cytokine
induction
profile in favor of interferon a, which may be important for certain
therapeutic uses.
The IRM may be covalently or non-covalently bound, preferably covalently
bound, to the macromolecular support material. Attachment of an IBM to a
macromolecular support material provides for the localized biological activity
of the
II~M and typically prevents, or at least reduces the occurrence of, the
systemic
distribution of the 1RM.
Accordingly, the present invention provides an IRM-support complex having an
II~M compound attached to a macromolecular support material. In some
embodiments,
the II~M compound may be covalently attached to the macromolecular support
material. In this context, "macromolecular support material" refers to organic
materials, inorganic materials, and combinations thereof that are generally
biologically
inactive relative to the biology being targeted by the IRM. The macromolecular
support material is typically of a size and chemical nature to prevent the
engulfment or
penetration of the macromolecular material into cells, although this is not a
necessary
limitation. For certain embodiments, the macromolecular support material
preferably
has an average largest dimension of at least 1 nanometer (nm). In some
embodiments,
the macromolecular support material may be part of a gel, a foam, a sponge, a
fiber, or
a bead.
In another aspect of the invention, an IRM-support complex that includes an
immune response modifier is attached to a polymer. In certain embodiments, the
immune response modifier is covalently attached to the polymer. In certain
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-3-
embodiments, the polymer is a bioadhesive polymer. In certain embodiments, the
present invention provides a medical article coated with the IRM-support
complex that
includes a polymer.
In another aspect, the present invention also provides a medical article
(i.e.,
medical device such as an implantable device) including an IRM-support
complex,
wherein the IRM-support complex includes an immune response modifier attached
to a
macromolecular support material. hl some embodiments, the medical article may
be a
stmt, a shunt, an artificial valve, a suture, a surgical clip, a surgical
staple, an
indwelling catheter, a dental implant, an orthopedic implant, a surgical
prosthetic, an
implantable vascular access port, an artificial heart, a ventricular assist
pump, a blood
oxygenator, a blood filter, a hemodialysis unit, a hemoperfusion unit, a
conduit tube
within a heart lung machine, a tube within a dialysis apparatus, a tube within
a
plasmapheresis unit, an artificial pancreas, an artificial liver, an
artificial lung, an
intraocular lens, or a contact lens.
In another aspect, the present invention provides a medical article having
disposed thereon an II~Irl, with the proviso that the medical article is not a
periochip.
Typically, the medical article is a stem.
In another aspect, the present invention also provides a stmt, shunt, or valve
having a surface with an immune response modifier attached thereto. W some
embodiments, the immune response modifier may be covalently attached to the
surface
of the scent, shunt, or valve.
In another aspect, the present invention also provides a polymer including an
immune response modifier attached thereto to form an I»M-macromolecular
support
complex. In some embodiments, the immune response modifier may be covalently
attached. In some embodiments, a medical article may be coated with the
polymer. In
some embodiments, the polymer may be a hydrogel.
In another aspect, the present invention also provides a method of making an
IRM-support complex by attaching an immune response modifier to a
macromolecular
support material. In some embodiments, the immune response modifier may be
covalently attached to the macromolecular support material.
In another aspect, the present invention also provides a method of treating a
viral infection in a subject by administering to the subject an IRM-support
complex
having an IRM compound attached to a macromolecular support material. In some
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-4-
embodiments, the IRM-support complex may be administered orally, nasally,
ocularly,
vaginally, transcutaneously, or rectally.
In another aspect, the present invention also provides a method of treating an
atopic immune response in a subject by administering to the subject an IRM-
support
complex having an IRM compound attached to a macromolecular support material.
In
some embodiments, the IRM-substrate may be administered orally, nasally,
vaginally,
ocularly, transcutaneously, or rectally.
In another aspect, the present invention also provides a method of treating
solid
tumors in a subj ect by administering to the subj ect an IRM-support complex
having an
IRM compound attached to a macromolecular support material. In some
embodiments,
the IRM-substrate may be administered orally, nasally, vaginally, ocularly,
transcutaneously, or rectally.
In another aspect, the present invention also provides a method of preventing
restenosis in a subject by implanting into the subject a stmt having a surface
with an
immune response modifier associated therewith (preferably, attached thereto,
and more
preferably, covalently attached thereto).
In another aspect, the present invention also provides a method of modifying
the
cytokine induction profile of an IRM by attaching the IRl~I to a
macromolecular
support complex. In some embodiments, the cytokine induction profile may be
modified in favor of interferon ~ induction.
In another aspect, the present invention also provides a method of reducing
systemic adsorption of an immune response modifier in a subject by
administering to
the subject an IRM-support complex, the IRM-support complex including the
immune
response modifier attached to a macromolecular support material.
In another aspect, the present invention also provides an IRM-support complex
(or a formulation thereof) including a first immune response modifier that is
attached to
a macromolecular support and a second immune response modifier that is not
attached
to the macromolecular support material. The formulation (i.e., composition)
can
include a solvent and/or be in the form of a gel.
In another aspect, the present invention also provides a method of activating
dendritic cells by permitting the cells contact an IRM compound attached to a
support
complex.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-5-
In another aspect, the present invention also provides a method of treating
cervical dysplasia in a subject by applying to the cervix an IRM-support
complex
comprising an lRM compound attached to a macromolecular support material.
In another aspect, the present invention also provides a method of treating
bladder cancer in a subject by applying to the bladder an IRM support complex
comprising an IRM compound attached to a macromolecular support material.
The present invention also provides a method of making an IRM-support
complex, wherein the method involves attaching an immune response modifier to
a
macromolecular support material. Attaching the immune response modifier can
involve covalently attaching it to the macromolecular support material. The
method
can also involve modifying the IRM to include an alkoxysilane moiety. The IRM-
modified alkoxysilane is then attached to a silicon-containing support
material.
In some embodiments of the present invention, the 1RM compound may be an
agonist of at least one TLR, preferably an agonist of TLR6, TLR7, or TLRB. The
IRM
may also in some cases be an agonist of TLR9. In some embodiments of the
present
invention, the IRh~! compound may be a small molecule irnrnune response
modifier
(e.g., molecular weight of less than about 1000 daltons).
In some embodiments of the present invention, the IRliiI compound may
comprise a 2-aminopyridine fused to a five-membered nitrogen-containing
heterocyclic
ring, or a 4-aminopyrimidine fused to a five-membered nitrogen-containing
heterocyclic ring.
In some embodiments of the present invention, the IR~I compound may be
imidazoquinoline amines including, but not limited to, substituted
imidazoquinoline
amines such as, for example, amide substituted imidazoquinoline amines,
sulfonamide
substituted imidazoquinoline amines, urea substituted imidazoquinoline amines,
aryl
ether substituted imidazoquinoline amines, heterocyclic ether substituted
imidazoquinoline amines, amido ether substituted imidazoquinoline amines,
sulfonamido ether substituted imidazoquinoline amines, urea substituted
imidazoquinoline ethers, thioether substituted imidazoquinoline amines, and 6-
, 7-, 8-,
or 9-aryl or heteroaryl substituted imidazoquinoline amines;
tetrahydroimidazoquinoline amines including, but not limited to, amide
substituted
tetrahydroimidazoquinoline amines, sulfonamide substituted
tetrahydroimidazoquinoline amines, urea substituted tetrahydroimidazoquinoline
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-6-
amines, aryl ether substituted tetrahydroimidazoquinoline amines, heterocyclic
ether
substituted tetrahydroimidazoquinoline amines, amido ether substituted
tetrahydroimidazoquinoline amines, sulfonamido ether substituted
tetrahydroimidazoquinoline amines, urea substituted tetrahydroimidazoquinoline
ethers, and thioether substituted tetrahydroimidazoquinoline amines;
imidazopyridine
amines including, but not limited to, amide substituted imidazopyridine
amines,
sulfonamide substituted imidazopyridine amines, urea substituted
imidazopyridine
amines, aryl ether substituted imidazopyridine amines, heterocyclic ether
substituted
imidazopyridine amines, amido ether substituted imidazopyridine amines,
sulfonamido
ether substituted imidazopyridine amines, urea substituted imidazopyridine
ethers, and
thioether substituted imidazopyridine amines; 1,2-bridged imidazoquinoline
amines;
6,7-fused cycloalkylimidazopyridine amines; imidazonaphthyridine amines;
tetrahydroimidazonaphthyridine amines; oxazoloquinoline amines;
thiazoloquinoline
amines; oxazolopyridine amines; thiazolopyridine amines; oxazolonaphthyridine
amines; thiazolonaphthyridine amines; and 11~ imidazo dimers fused to pyridine
amines, quinoline amines, tetrahydroquinoline amines, naphthyridine amines, or
tetrahydronaphthyridine amines. ~Tarious combinations of II~Is can be used if
desired.
In some embodiments, the IRM compound may be a purine, imidazoquinoline
amide, benzimidazole, 1H imidazopyridine, adenine, or a derivative thereof.
The term "comprises" and variations thereof do not have a limiting meaning
where these terms appear in the description and claims,
As used herein "a " "an " "the " "at least one " and "one or more" are used
7 7 7 9 7
interchangeably. Thus, for example, an IIZM-support complex comprising "an"
I12M
compound can be interpreted to mean that the complex includes at least one IRM
compound.
Also herein, the recitations of numerical ranges by endpoints include all
numbers subsumed within that range (e.g., 1 to 5 includes 1, 1.5, 2, 2.75, 3,
3.80, 4, 5,
etc.).
The above summary of the present invention is not intended to describe each
disclosed embodiment or every implementation of the present invention. The
description that follows more particularly exemplifies illustrative
embodiments. In
several places throughout the application, guidance is provided through lists
of
examples, which examples can be used individually and in various combinations.
In
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
each instance, the recited list serves only as a representative group and
should not be
interpreted as an exclusive list.
BRIEF DESCRIPTION OF THE DRAWINGS
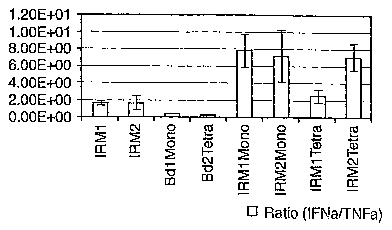
Figure 1. Figure lA is a graphical representation of the IFNa, /TNFa, ratio
produced by human peripheral blood mononuclear cells incubated overnight with
IRM1
or IRM2 bound to monomeric avidin beads or tertrameric avidin beads. Figure 1B
is a
graphical representation of the IFNa and TNFoc produced by human peripheral
blood
mononuclear cells incubated overnight with IRM1 or IRM2 bound to monomeric
avidin beads or tertrameric avidin beads.
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS OF THE
INVENTION
The present invention is directed to the attaclnnent of cytokine inducing
and/or
suppressing immune response modifiers (IRMs) to macromolecular support
materials
to form ll~M-support complexes. The IRMs retain biological activity following
such
attachment to a macromolecular support material. IRM-support complexes allow
for
the localized delivery of an IRM to a desired location in the body of a
subject and
typically prevent, or at least reduce the occurrence of, the systemic
distribution of the
IRM.
As used herein, "macromolecular support material" is a macromolecular
material that is itself generally biologically inactive relative to the
biology being
targeted by the IR1VI. Herein, tlus definition of macromolecular support
materials
excludes bacteria and viruses, for example. The macromolecular support
material may
be of a size and chemical nature to prevent the engulfment or penetration of
the
macromolecular material into cells, in which case the IRM-support complex
retains an
extracellular location. Alternatively, the macromolecular support material may
be of a
size and chemical nature to allow engulfinent by cells. For example, the
macromolecular support material may be of a size and chemical nature to allow
selective deposition in solid tumors on the basis of the tumor's increased
vascular
permeability. For certain embodiments, the macromolecular support material
preferably
has an average largest dimension of at least 1 nanometer (nm) (although
materials
having as an average largest dimension of 0.1 nm or even smaller can be used),
more
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
_g_
preferably, at least 10 nm, even more preferably, at least 100 nm, and even
more
preferably, at least 1 micron.
Typically, the macromolecular support material is in the form of a solid
(i.e., a
solid support such as particles, fibers, membranes, films), but can also be in
the form of
a polymeric gel, sponge, or foam, for example. A macromolecular support
material can
be made of a variety of materials, including substrates made of ceramic,
glassy,
metallic, or polymeric materials, or combinations of materials. The terms
"substrate,"
"support material," or "support," may also be used herein to refer to a
macromolecular
support material.
In an IRM-support complex an IRM is attached to a macromolecular support
material. As used herein, the term "attached" includes both covalent bonding
and non-
covalent chemical association (e.g., ionic bonding and hydrogen bonding) of an
immune response modifier with a macromolecular support material. Non-covalent
association is preferably by specific, high affinity protein-ligand
interaction, as
opposed to nonspecific hydrogen bonding. Preferably, the immune response
modifiers
are attached to a macromolecular support material by means of covalent
bonding. The
terms "coupled," "conjugated," "bonded," or "immobilized" may also be used
herein to
represent "attached." As used herein, "attached" excludes mere coating of the
macromolecular support material with an IRM.
Attachment of an II~I~ to a anacromolecular support material, such as solid
supports (e.g., particles, fibers, aazd the life) as well as colloids, and
polymeric foams,
sponges, and gels, provides for the localized biological activity of the
IRIVI. Also, the
II~M can be attached as a side group to a polymer, and the polymer coated onto
any
desired surface. Although the IRM may eventually detach from the
macromolecular
support material (e.g., through biodegradation of a polymer to which the IRM
is
attached, for example), the Il~M does not detach during a suitable period of
use while it
is active (although it may of course also be active after detachment). Such
attachment
of an IRM to a macromolecular support material can be used to reduce the
occurrence
of, or prevent, the systemic absorption of the IRM, and can minimize the
systemic side
effects sometimes observed with the systemic administration of an IRM. Also,
such
attachment of an IRM to a substrate can serve to limit or focus the effect of
the IRM to
a localized region for a desired duration, and if the support material can be
removed,
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-9-
the lRM can then be easily removed at will along with it. This provides very
important
control over where and how long the IRM is applied.
For certain embodiments (e.g., medical articles such as stems and other
implantables or extracorporeal devices), the IRM may elute from a coating on
the
article upon contact with a bodily fluid. In such embodiments, the IRM may be
incorporated into a coating material, e.g., a polymeric material, using any of
a variety
of mechanisms, which may or may not include attachment of the IRM to the
macromolecular support material as defined herein.
One or more IRMs can be attached to a solid support or other macromolecular
support material. Also, one 1RM can be attached to multiple solid supports or
other
macromolecular support materials.
The substrate having the IRM attached thereto can be used in a variety of
medical applications, which can be therapeutic, prophylactic (e.g., as a
vaccine
adjuvant), or diagnostic. As used herein, "treating" a condition or a subject
includes
therapeutic, prophylactic, and diagnostic treatments.
For example, in certain embodiments9 II~I~Is can be attached to the surfaces
of
various medical devices or implants, such as, for example9 scents, shunts,
artificial
valves, sutures, surgical clips and staples, indwelling catheters, prosthesis,
including
dental and orthopedic implants and GORE-TEX surgical prosthetics, implantable
vascular access ports artificial hearts, ventricular assist pumps,
extracorporeal devices
such as blood oxygenators, blood filters, hemodialysis units, hemoperfusion
units,
conduit tubes within heart lung machines, tubes of a dialysis apparatus and
plasmapheresis units, hybrid artificial organs such as pancreas or liver and
artificial
lungs, and the like. For example, an IRM, such as an imidazoquinolin-4-amine,
can be
attached to an arterial stmt for use in interventional cardiology to prevent
restenosis.
As such, it is anticipated that the IRM would preferentially activate
precursor
plasmocytoid dendritic cells (pDC cells) of the blood to stimulate INF'a,
formation, for
a localized antiproliferative effect, with reduced systemic distribution of
the IRM.
An IRM can be attached to a macromolecular support complex and used in
wound dressings, wound paclcing materials, wound sealants, sutures, and
surgical clips
to promote healing and/or reduce the occurrence of scarring.
An IRM can be attached to a substrate used anywhere within the body,
including soft tissue such as muscle or fat, hard tissue such as bone, or a
cavity such as
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-10-
the periodontal, gastrointestinal, oral, vaginal, rectal, nasal, bladder,
airway, uterus,
corpus cavernosum, ocular, or a pocket such as a periodontal pocket or the cul-
de-sac
of the eye. Thus, the compositions of the present invention can be used to
treat
disorders such as respiratory disorders, gastrointestinal disorders,
urological
dysfunction, impotence, uterine dysfunction, and premature labor.
An IRM-support complex can be applied to the vagina or uterus to treat vaginal
infections, such as, e.g., herpes or papilloma virus. For example, one or more
IRMs
can be attached to (as opposed to blended or dissolved in) a macromolecular
support
material, such as a material incorporated into a gel or foam, for application
within the
vagina or uterus.
An IRM-support complex can be applied to the nasal cavity. For example, one
or more IRMs can be attached to a macromolecular support material, such as a
gel,
foam, or spray for application to the nasal passages and/or sinuses.
An IRM-support complex can be applied to the eye to treat, e.g., viral
infections, such as herpes. For example, one or more II~IVIs can be attached
to a
macromolecular support material for inclusion in an ophthalmic preparation for
application to the eye and/or to ophthalmic devices, such as intraocular
lenses and
contact lenses.
An IRM-support complex can be delivered to the gastrointestinal tract to treat
gastrointestinal disorders. For example, one or more IRI~s can be attached to
a
macromolecular support material, such as a bead, gel or foam, for delivery to
the
gastrointestinal tract, delivered orally or rectally.
An IRM can also be attached to a macromolecular support material, such as a
polymer, for the formation of a compound depot in the body of a subject to
promote the
long term, localized effect of the IRM.
In some embodiments, an IRM can be attached to a macromolecular support
material, such as an oligomer, a polymer, a bead, a tissue culture flask, a
tissue culture
plate, a microtiter plate, or a column, for use in, e.g., ex-vivo treatment of
immune
cells, experiment testing, or a diagnostic assay in which an IRM is a
component. For
example, use of an IRM-support complex can enhance cellular contact with an
IRM,
can facilitate the removal of an IRM from a diagnostic assay, can allow for
the
concentrated delivery of an IRM, and can assist in the conservation of lRM
reagents.
An IRM support complex can line the interior surface of an in vivo or ex vivo
passage
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-11-
or container through which blood or other cell-containing fluid travels in
order to
activate immune cells without requiring systemic delivery. For example,
dendritic cells
can be removed from the body and matured in the presence of antigen and IRM-
support
complex before being delivered to the body for therapeutic use.
The methods, materials, and articles of the present invention may be
applicable
for any suitable subject. Suitable subjects include, but are not limited to,
animals such
as, but not limited to, humans, non-human primates, rodents, dogs, cats,
horses, pigs,
sheep, goats, cows, or birds.
In some applications, e.g., with an associated antigen, an IRM can be attached
to a solid support material (e.g., a particle) or other macromolecular support
material
accompanied by a specific immunizing antigen on the same solid support
material (e.g.,
particle). Alternatively, an IRM can be attached to a first solid support
material (e.g.,
particle) or other macromolecular support material while the immunizing
antigen is
attached to a second solid support material (e.g., particle) or other
macromolecular
support material. These embodiments allow for the copresentation of an antigen
and an
II~M.
Suitable Inzfnune Res~a~nse 111~dij~e~~s:
Immune response modifiers ("IRMs") of the present invention include
compounds act on the irr~nune system by inducing and/or suppressing cytokine
biosynthesis. IRMs possess potent invnunostimulating activity including, but
not
limited to, antiviral and antitumor activity, and can als~ down-regulate other
aspects of
the immune response, for example shifting the immune response away from a TH2
immune response, which is useful for treating a wide range of TH2 mediated
diseases.
IRMs can also be used to modulate humoral immunity by stimulating antibody
production by B cells. Further, various II~Ms have been shown to be useful as
vaccine
adjuvaxlts (see, e.g., U.S. Pat. Nos. 6,083,505 and 6,406,705, and
International
Publication No. WO 02/24225).
In particular, certain IRMs effect their immunostimulatory activity by
inducing
the production and secretion of cytokines such as, e.g., Type I interferons,
TNF-a, IL-1,
IL-6, IL-8, IL-10, IL-12, MIP-1, and/or MCP-l, and can also inhibit production
and
secretion of certain TH2 cytokines, such as IL-4 and IL-5. Some IRMs are said
to
suppress IL-1 and TNF (see, e.g., International Publication No. WO 00/09506).
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-12-
For some embodiments, preferred IRMs are so-called small molecule IRMs,
which are relatively small organic compounds (e.g., molecular weight under
about 1000
daltons, preferably under about 500 daltons, as opposed to large biologic
protein,
peptides, and the like).
Although not bound by any single theory of activity, some IRMs are known to
be agonists of at least one Toll-like receptor (TLR). IRMs that are agonists
for TLRs
selected from 6, 7, 8, and 9 may be particularly useful for certain
applications. Some
small molecule IRMs are agonists of TLRs such as 6, 7, and 8, while
oligonucleotide
IRM compounds are agonists of TLR9, and perhaps others. Thus, in some
embodiments, the IRM that is attached to a macromolecular support material may
be a
compound identified as an agonist of one or more TLRs.
For example, without being bound to any particular theory or mechaiusm of
action, IRM compounds that activate a strong cytotoxic lymphocyte (CTL)
response
may be particularly desirable as vaccine adjuvants, especially for therapeutic
viral
and/or cancer vaccines because a therapeutic effect in these settings is
dependent on the
activation of cellular immunity. For example, studies have shovan that
activation of T
cell immunity in a given patient has a significant positive effect on the
prognosis of the
patient. Therefore the ability to enhance T cell immunity is believed to be
critical to
producing a therapeutic effect in these disease settings.
~0 IRI~I compounds that are TLR 8 agonists may be particularly desirable for
use
with therapeutic cancer vaccines because antigen presenting cells that express
TLRB
have been shown to produce IL-12 upon stimulation through TLRB. IL-12 is
believed
to play a significant role in activation of CTLs, which are important for
mediating
therapeutic efficacy as described above.
IRM compounds that are TLR 7 agonists and/or TLR 9 agonists may be
particularly desirable for use with prophylactic vaccines because the type I
interferon
induced by stimulation through these TLRs is believed to contribute to the
formation of
neutralizing TH1-like humoral and cellular responses.
IRM compounds that are both TLR 7 and TLR 8 agonists may be particularly
desirable for use with therapeutic viral vaccines andlor cancer vaccines
because TLR7
stimulation is believed to induce the production of type I IFN and activation
of innate
cells such as macrophages and NIA cells, and TLR8 stimulation is believed to
activate
antigen presenting cells to initiate cellular adaptive immunity as described
above.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-13-
These cell types are able to mediate viral clearance and/or therapeutic growth
inhibitory
effects against neoplasms.
IKM compounds that are non-TLR 7 agonists, and do not induce substantial
amounts of interferon alpha, may be desirable for use with certain vaccines
such as
bacterial vaccines because TLR7 induces type I IfN production, which down-
regulates
the production of IL-12 from macrophages and DCs. IL-12 contributes to the
subsequent activation of macrophages, NK cells and CTLs, all of which
contribute to
anti-bacterial immunity. Therefore the induction of anti-bacterial immunity
against
some kinds of bacteria may be enhanced in the absence of IFNa.
For purposes of the present application, one way to determine if an IRM
compound is considered to be an agonist for a particular TLR is if it
activates an
NFkB/luciferase reporter construct through that TLR from the target species
more than
about 1.5 fold, and usually at least about 2 fold, in TLR transfected host
cells such as,
e.g., HEK293 or Namalwa cells relative to control transfectants. For
information
regarding TLR activation, see, e.g., International Publication Nos. WO
03/04.3573 and
WO 03/043588, U.S. Patent application Serial Nos. 101777,310, 10/732,563,
10/732,796, and 10/788,731, U.S. Patent Publication No. US2004./0014779, and
the
other IRIVI patents and applications disclosed herein.
Preferred IRM compounds include a 2-aminopyridine fused to a five-membered
nitrogen-containing heterocyclic ring.
Certain IRMs are small organic molecules (e.g., molecular weight under about
1000 Daltons, preferably under about 500 Daltons, as opposed to large biologic
protein,
peptides, and the like) such as those disclosed in, for example, U.S. Pat.
Nos.
4,689,338; 4,929,624; 4,988,815; 5,037,986; 5,175,296; 5,238,944; 5,266,575;
5,268,376; 5,346,905; 5,352,784; 5,367,076; 5,389,640; 5,395,937; 5,446,153;
5,482,936; 5,693,811; 5,741,908; 5,756,747; 5,939,090; 6,039,969; 6,083,505;
6,110,929; 6,194,425; 6,245,776; 6,331,539; 6,376,669; 6,451,810; 6,525,064;
6,545,016; 6,545,017; 6,558,951; 6,573,273; 6,656,938; 6,660,735; 6,660,747;
6,664,260; 6,664,264; 6,664,265; 6,667,312; 6,670,372; 6,677,347; 6,677,348;
6,677,349; 6,683,088; European Patent 0 394 026; U.S. Patent Publication Nos.
2002/0016332; 2002/0055517; 2002/0110840; 2003/0133913; 2003/0199538; and
2004/0014779; and International Patent Publication Nos. WO 02/102377 and WO
03/103584. .
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-lq.-
Examples of classes of small molecule lRM compounds include, but are not
limited to, compounds having a 2-aminopyridine fused to a five-membered
nitrogen-
containing heterocyclic ring. Such compounds include, for example,
imidazoquinoline
amines including, but not limited to, substituted imidazoquinoline amines such
as, for
example, amide substituted imidazoquinoline amines, sulfonamide substituted
imidazoquinoline amines, urea substituted imidazoquinoline amines, aryl ether
substituted imidazoquinoline amines, heterocyclic ether substituted
imidazoquinoline
amines, amido ether substituted imidazoquinoline amines, sulfonamido ether
substituted imidazoquinoline amines, urea substituted imidazoquinoline ethers,
thioether substituted imidazoquinoline amines, and 6-, 7-, 8-, or 9-aryl or
heteroaryl
substituted imidazoquinoline amines; tetrahydroimidazoquinoline amines
including, but
not limited to, amide substituted tetrahydroimidazoquinoline amines,
sulfonamide
substituted tetrahydroimidazoquinoline amines, urea substituted
tetrahydroimidazoquinoline amines, aryl ether substituted
tetrahydroimidazoquinoline
amines, heterocyclic ether substituted tetrahydroimidazoquinoline amines,
amido ether
substituted tetrahydroimidazoquinoline amines, sulfonamido ether substituted
tetrahydroimidazoquinoline amines, urea substituted tetrahydroimidazoquinoline
ethers, and thioether substituted tetrahydroimidazoquinoline amines;
imidazopyridine
amines including, but not limited to, amide substituted imidazopyridine
amines,
sulfonamide substituted imidazopyridine amines, urea substituted
imidazopyridine
amines, aryl ether substituted imidazopyridine amines, heterocyclic ether
substituted
imidazopyridine amines, amido ether substituted imidazopyridine amines,
sulfonamido
ether substituted imidazopyridine amines, urea substituted imidazopyridine
ethers, and
thioether substituted imidazopyridine amines; 1,2-bridged imidazoquinoline
amines;
6,7-fused cycloalkylimidazopyridine amines; imidazonaphthyridine amines;
tetrahydroimidazonaphthyridine amines; oxazoloquinoline amines;
thiazoloquinoline
amines; oxazolopyridine amines; thiazolopyl-idine amines; oxazolonaphthyridine
amines; thiazolonaphthyridine amines; and 1H imidazo dimers fused to pyridine
amines, quinoline amines, tetrahydroquinoline amines, naphthyridine amines, or
tetrahydronaphthyridine amines.
Preferred IRM compounds comprise a 2-aminopyridine fused to a five-
membered nitrogen-containing heterocyclic ring.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-15-
Additional examples of small molecule IRMs said to induce interferon (among
other things), include purine derivatives (such as those described in U.S.
Pat. Nos.
6,376,501, and 6,028,076), imidazoquinoline amide derivatives (such as those
described in U.S. Pat. No. 6,069,149), 1H imidazopyridine derivatives (such as
those
described in Japanese Patent Application No. 9-255926), benzimidazole
derivatives
(such as those described in U.S. Pat. No. 6,387,938), derivatives of a 4-
aminopyrimidine fused to a five membered nitrogen containing heterocyclic ring
(such
as adenine derivatives described in U.S. Pat. Nos. 6,376,501; 6,028,076 and
6,329,381;
and in International Publication No. WO 02/08595), and certain 3-~3-D-
ribofuranosylthiazolo[4,5-d]pyrimidine derivatives (such as those described in
U.S.
Patent Publication No. 2003/0199461). 1H imidazopyridine derivatives (such as
those
described in U.S. Pat. No. 6,518,265 and European Patent Application EP No.
1 256 582)) are said to inhibit TNF and IL-1 cytokines.
Examples of small molecule IRMs that comprise a 4-aminopyrimidine fused to
a five-membered nitrogen-containing heterocyclic ring include adenine
derivatives
(such as those described in U. S. Pat. Nos. 6,376,501; 6,028,076 and
6,329,381; and in
International Publication No. WO 02/08595).
Examples of particular IRM compounds include 2-propyl[1,3]thiazolo[4,5-
c]quinolin-4-amine, which is considered predominantly a TLR 8 agonist (and not
a
substantial TLR 7 agonist), 4-amino-ot,~ dimethyl-lII imidazo[4.,5-a]quinoline-
1-
ethanol, which is considered predominantly a TLR 7 agonist (and not a
substantial TLR
8 agonist), and 4-amino-2-(ethoxymethyl)-cx,cx dimethyl-6,7,8,9-tetrahydr~-11I
imidazo[4,5-c]quinoline-1-ethanol, which is a TLR 7 and TLR 8 agonist. In
addition to
its TLR 7 activity (and TLR 6 activity, but low TLR 8 activity), 4-amino- cx,a
dimethyl-lIl imidazo[4,5-c]quinoline-1-ethanol has beneficial characteristics,
including that it has a much lower GNS effect when delivered systemically
compared to
imiquimod. Other examples of specific 1RM compounds include, e.g., N-[4-(4-
amino-
2-butyl-1H imidazo[4,5-c][1,5]naphthyridin-1-yl)butyl]-N'-cyclohexylurea, 2-
methyl-
1-(2-methylpropyl)-1H imidazo[4,5-c][1,5]naphthyridin-4-amine, 1-(2-
methylpropyl)-
1H imidazo[4,5-c][1,5]naphthyridin-4-amine, N-~2-[4-amino-2-(ethoxymethyl)-1H
imidazo[4,5-c]quinolin-1-yl]-1,1-dimethylethyl~methanesulfonamide, N-[4-(4-
amino-
2-ethyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]methanesulfonamide, 2-methyl-1-[5-
(methylsulfonyl)pentyl]-1H imidazo[4,5-c]quinolin-4-amine, N-[4-(4-amino-2-
propyl-
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-16-
1H imidazo[4,5-c]quinolin-1-yl)butyl]methanesulfonamide, 2-butyl-1-[3-
(methylsulfonyl)propyl]-1H imidazo[4,5-c]quinoline-4-amine, 2-butyl-1-{2-[(1-
methylethyl)sulfonyl]ethyl}- 1H imidazo[4,5-c]quinolin-4-amine, N-{2-[4-amino-
2-
(ethoxymethyl)-1H imidazo[4,5-c]quinolin-1-yl]-1,1-dimethylethyl}-N'-
cyclohexylurea, N-{2-[4-amino-2-(ethoxymethyl)-1H imidazo[4,5-c]quinolin-1-yl]-
l,l-dimethylethyl)cyclohexanecarboxamide, N-{2-[4-amino-2-(ethoxymethyl)-1H
imidazo[4,5-c]quinolin-1-yl]ethyl}-N'-isopropylurea. Resiquimod, 4-amino-2-
ethoxymethyl-a,a-dimethyl-1H imidazo[4,5-c]quinoline-1-ethanol, may also be
used
in certain situations where a combination TLR 7 and TLR 8 agonist is desired.
Other IRMs include large biological molecules such as oligonucleotide
sequences. Some IRM oligonucleotide sequences contain cytosine-guanine
dinucleotides (CpG) and are described, for example, in U.S. Pat. Nos.
6,194,388;
6,207,646; 6,239,116; 6,339,068; and 6,406,705. Some CpG-containing
oligonucleotides can include synthetic immunomodulatory structural motifs such
as
those described, for example, in U.S. Pat. Nos. 6,4.26,334 and 6,476,000.
Other IRM
nucleotide sequences lack CpG and are described, for example, in International
Patent
Publication No. ~O 00175304.
Various combinations of IRMs can be used if desired.
~°xerralralaby A~a~alica~icfzs:
IRMs such as imiquimod - a small molecule, imidazoquinoline IRM, marketed
as ALDARA (3M Pharmaceuticals, St. Paul, MN) - have been shown to be useful
for
the therapeutic treatment of warts, as well as ceutain cancerous or pre-
cancerous lesions
(See, e.g., Geisse ct al., J. Am. Acad. Derr~aatol., 47(3): 390-398 (2002);
Shwnack et
al., Arcla. De~matol., 138: 1163-1171 (2002); U.S. Pat. No. 5,238,944 and U.S.
Pat.
Publication No. US2003/0199538.
Conditions that may be treated by administering an IRM-support complex of the
present invention include, but are not limited to:
(a) viral diseases such as, for example, diseases resulting from infection by
an
adenovirus, a herpesvirus (e.g., HSV-I, HSV-II, CMV, or VZV), a poxvirus
(e.g., an
orthopoxvirus such as variola or vaccinia, or molluscum contagiosum), a
picornavirus
(e.g., rhinovirus or enterovirus), an orthomyxovirus (e.g., influenzavirus), a
paramyxovirus (e.g., parainfluenzavirus, mumps virus, measles virus, and
respiratory
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-17-
syncytial virus (RSV)), a coronavirus (e.g., SARS), a papovavirus (e.g.,
papillomaviruses, such as those that cause genital warts, common warts, or
plantar
warts), a hepadnavirus (e.g., hepatitis B virus), a flavivirus (e.g.,
hepatitis C virus or
Dengue virus), or a retrovirus (e.g., a lentivirus such as HIV);
(b) bacterial diseases such as, for example, diseases resulting from infection
by
bacteria of, for example, the genus Escherichia, Enterobacter, Salmonella,
Staphylococcus, Shigella, Listeria, Aerobacter, Helicobacter, Klebsiella,
Proteus,
Pseudomonas, Streptococcus, Chlamydia, Mycoplasma, Pneumococcus, Neisseria,
Clostridium, Bacillus, Corynebacterium, Mycobacterium, Campylobacter, Vibrio,
Serratia, Providencia, Chromobacterium, Brucella, Yersinia, Haemophilus, or
Bordetella;
(c) other infectious diseases, such chlamydia, fungal diseases including but
not
limited to candidiasis, aspergillosis, histoplasmosis, cryptococcal
meningitis, or
parasitic diseases including but not limited to malaria, pneumocystis carnii
pneumonia,
leislnnaniasis, cryptosporidiosis, toxoplasmosis, and trypanosome infection;
and
(d) neoplastic diseases, such as intraepithelial neoplasias, cervical
dysplasia,
actinic keratosis, basal cell carcinoma, squamous cell carcinoma, renal cell
carcinoma,
I~aposi's sarcoma, melanoma, renal cell carcinoma, leukemias including but not
limited
to myelogeous leukemia, chronic lymphocytic leukemia, multiple myeloma, non-
Hodgkin's lymphoma, cutaneous T-cell lymphoma, B-cell lymphoma, and hairy cell
leukemia, and other cancers;
(e) TH2-mediated, atopic diseases, such as atopic dermatitis or eczema,
eosinophilia, asthma, allergy, allergic rhinitis, and ~mmen's syndrome;
(f) certain autoimmune diseases such as systemic lupus erythematosus,
essential
thrombocythaemia, multiple sclerosis, discoid lupus, alopecia areata; and
(g) diseases associated with wound repair such as, for example, inhibition of
keloid formation and other types of scarring (e.g., enhancing would healing,
including
chronic wounds).
Additionally, an IRM-support complex of the present invention may be useful
as a vaccine adjuvant for use in conjunction with any material that raises
either humoral
and/or cell mediated immune response, such as, for example, live viral,
bacterial, or
parasitic immunogens; inactivated viral, tumor-derived, protozoal, organism-
derived,
fungal, or bacterial immunogens, toxoids, toxins; self antigens;
polysaccharides;
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-18-
proteins; glycoproteins; peptides; cellular vaccines; DNA vaccines; autologous
vaccines; recombinant proteins; glycoproteins; peptides; and the like, for use
in
connection with, for example, BCG, cholera, plague, typhoid, hepatitis A,
hepatitis B,
hepatitis C, influenza A, influenza B, parainfluenza, polio, rabies, measles,
mumps,
rubella, yellow fever, tetanus, diphtheria, hemophilus influenza b,
tuberculosis,
meningococcal and pneumococcal vaccines, adenovirus, HIV, chicken pox,
cytomegalovirus, dengue, feline leukemia, fowl plague, HSV-1 and HSV-2, hog
cholera, Japanese encephalitis, respiratory syncytial virus, rotavirus,
papilloma virus,
yellow fever, and Alzheimer's Disease.
Certain IRM-support complexes of the present invention may be particularly
helpful in individuals having compromised immune function. For example,
certain
complexes may be used for treating the opportunistic infections and tumors
that occur
after suppression of cell mediated immunity in, for example, transplant
patients, cancer
patients and HIV patients.
S'a~bst~-ezt~s:
Selection of a macromolecular support material to serve as a substrate for
attachment of an II~M can vary widely within the scope of the invention. A
macromolecular support material can be porous or nonporous, depending on
preferred
final use. A macromolecular support mateuial can be continuous or non-
continuous
depending on ultimate desired usage. A macromolecular support material can be
made
of a variety of materials, which may be organic, inorganic, or combinations
thereof,
including substrates made of ceramic, glassy, metallic, oligomeric or
polymeric
materials, or combinations of materials. For certain embodiments, silicon-
based
materials (e.g., silica-based materials can be used). Thus, herein, the term
"metal"
includes metalloids such as silicon. A macromolecular support material can be
flexible
or inflexible depending on ultimate desired usage.
Exemplary materials include polymeric materials, such as woven and nonwoven
webs (such as fibrous webs), microporous fibers, and microporous membranes.
Also, in certain embodiments, IRMs can be attached to the surface of various
materials, including, but not limited to, particles (e.g., beads), films,
membranes, fibers,
gels, creams, foams, or sponges. Typically, such particles, films, membranes,
fibers,
gels, creams, foams, and sponges are organic polymeric materials.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-19-
Suitable polymers may be natural or synthetic polymers. Synthetic polymers
are preferred. Herein, a polymer includes homopolymers and copolymers. A
copolymer is used to refer to a polymer prepared from two or more monomers,
and
includes terpolymers, tetrapolymers, etc.
Exemplary synthetic polymers include, but are not limited to: polyamides,
polycarbonates, polyalkylenes, polyalkylene glycols, polyalkylene oxides,
polyalkylene
terepthalates, polyvinyl alcohols, polyvinyl ethers, polyvinyl esters,
polyvinyl halides,
polyvinylpyrrolidone, polyglycolides, polysiloxanes, polyurethanes and
copolymers
thereof, alkyl cellulose, hydroxyalkyl celluloses, cellulose ethers, cellulose
esters, nitro
celluloses, polymers of acrylic and methacrylic esters, methyl cellulose,
ethyl cellulose,
hydroxypropyl cellulose, hydroxy-propyl methyl cellulose, hydroxybutyl methyl
cellulose, cellulose acetate, cellulose propionate, cellulose acetate
butyrate, cellulose
acetate phthalate, carboxylethyl cellulose, cellulose triacetate, cellulose
sulphate
sodium salt, poly(methyl methacrylate), poly(ethyl methacrylate), poly(butyl
methacrylate), poly(isobutyl methacrylate), poly(hexyl methacrylate),
poly(isodecyl
methacrylate), poly(lauryl methacrylate), poly(phenyl methacrylate),
poly(methyl
acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), poly(octadecyl
acrylate),
polyethylene, polypropylene, polyethylene glycol), polyethylene oxide),
polyethylene terephthalate), polyvinyl alcohols), polyvinyl acetate),
polyvinyl
chloride), polystyrene, polyamides, polyvinylpyrrolidone, said polymers of
lactic acid
and glycolic acid, polyanhydrides, poly(outho)esters, poly(butic acid),
poly(valeric
acid), and poly(lactide-cocaprolactone). Fluoropolymeric materials can also be
used.
Examples of such materials are disclosed in LJ.S. Pat. Nos. 6,630,047;
6,451,925; and
6,096,428.
Exemplary natural polymers include, but are not limited to: alginate and other
polysaccharides including dextran and cellulose, collagen, chemical
derivatives thereof
(substitutions, additions of chemical groups, for example, alkyl, allcylene,
hydroxylations, oxidations, and other modifications routinely made by those
skilled in
the art), zero, and other prolamines and hydrophobic proteins, copolymers and
mixtures
thereof. Copolymers and mixtures of any of these polymers could be used if
desired.
The support material can be a bioadhesive polymer such as a hydrogel,
including, for example, those described by H. S. Sawhney, C. P. Pathak and J.
A.
Hubell in Macromolecules, (1993) 26:581-587, as well as polyhyaluronic acids,
casein,
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-20-
polysaccharides, keratin, collagen, gelatin, glutin, polyethylene glycol,
crosslinked
albumin, fibrin, polyanhydrides, polyacrylic acids, alginate, chitosan,
poly(methyl
methacrylates), poly(ethyl methacrylates), poly(butyl methacrylate),
poly(isobutyl
methacrylate), poly(hexyl methacrylate), poly(isodecyl methacrylate),
poly(latuyl
methacrylate), poly(phenyl methacrylate), poly(methyl acrylate),
poly(isopropyl
acrylate), poly(isobutyl acrylate), poly(octadecyl acrylate), and cellulose
gums.
Alternatively, polymeric hydrogel materials can be constructed from polyvinyl
alcohol) precursors as disclosed in U.S. Pat. Nos. 4,528,325 and 4,618,649 or
from
poly(methyl methacrylate). Poly(methyl methacrylate) is commercially available
and is
often used in ophthalmic devices such as intraocular lenses, contact lenses,
and the like.
A suitable hydrogel can be natural, synthetic, or a combination thereof. In
some
embodiments, the hydrogel can be thermally responsive to a designed
temperature such
as, for example, a hydrogel as described in U.S. Patent Application Serial
Number
10/626261, filed July 24, 2003. For example, the thermally responsive
hydrogels can
be harden when they are warmed up to body temperature, can be further harden
upon
UV irradiation.
Preferred bioadhesive polymers include crosslinked polymers of acrylic acid.
Suitable examples include acrylic acid polymers crosslinked with allyl sucrose
or allyl
pentaerythriol such as those polymers designated as carbomers. Suitable
carbomers
include, for example, those available as CAP.B~P~L 971P NF Polymer and
CA1ZB(aP~L 974P NF Polymer, available from Noveon, Inc., Cleveland, ~H. ~ther
examples of crosslinked acrylic acid polymers include those crosslinked with
divinyl
glycol such as those designated as polycarbophils. Suitable polycarbophils
include, for
exa~.nple, those available as N~VE~N AA-1 USP Polycarbophil, available from
Noveon, Inc., Cleveland, OH.
Bioadhesive organic polymers are preferred for certain applications of IP'Ms.
For example, if the IRM is to be used for treating cervical dysplasia or
bladder cancer, a
bioadhesive polymer is desired. Advantageously, the adhesive qualities of the
formulation would allow the IRM to be in contact with the biological tissue
allowing
for greater contact time for cytokine induction.
Again, however, for many embodiments described herein it is important to note
that the IRM is not simply dissolved or blended into a formulation from which
it is to
be released, but is attached to the support material by a sufficiently strong
bond (which
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-21-
sometimes may require a covalent bond) so that under the circumstances of
intended
use the IRM is biologically active during use while it is attached to the
support.
Preferably, for certain embodiments (e.g., for bioadhesive polymeric support
materials),
the IEtM is covalently attached to the support material. It should also be
understood,
however, that for each of the uses (e.g., medical articles such as stems or
other
implantable devices or extracorporeal devices) described herein an IRM may be
provided in an unattached, releasable form, or become unattached over time, so
that the
IRM can be released and function in that manner. That is, for example, the IRM
can be
simply dissolved or blended into a macromolecular support material (e.g., as
in a
polymeric coating). Mixtures of the two types can also be used where
desirable.
Gels, creams, films, salves, coatings, sticks, colloids, pastes, and foams
incorporating IItM-support complexes can be applied to a variety of bodily
surfaces,
and among the many uses may include, e.g., wound dressings and wound packing
materials. These sorts of solid, semi-solid, or viscous preparations can serve
to
promote the retention of the llZM compounds on the bodily surface and also to
prevent
the systemic adsorption of the Il~M. bodily surfaces can include, but are not
limited to,
gastrointestinal tract, vagina, uterus, bladder, oral cavity, nasal passages,
periodontal
surfaces, rectum, ocular surfaces, or surfaces of the ear.
Particles can be the substrate to which an IRM is attached. For example,
'~0 particles can be in the form of beads, including, but not lunited to
carbohydrate beads
and latex beads, such as those commercially available from many suppliers,
including,
for example Biorad and Pierce. Various oxide-containing particles (e.g.,
silica
particles) can be used as the substrate as well. The particles can also be in
the form of
microparticles, such as microspheres, microcapsules, etc. Nanoparticles, such
as
quantum dots can be used as well.
Ceramic supports, glass supports, and metallic supports are all known in the
art
and are commercially available or can be prepared by a variety of known
techniques.
Woven and nonwoven webs are useful as substrates having either regular or
irregular physical configurations of surfaces from which the IRMs can extend.
Fibrous
webs are particularly desired because such webs provide large surface areas,
with
nonwoven fibrous webs being preferred due to ease of manufacture, low material
cost,
and allowance for variation in fiber texture and fiber density. A wide variety
of fiber
diameters, e.g., 0.05 micrometer (,um) to 50 micrometers, can be used. Web
thickness
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-22-
can be varied widely to fit the application, e.g., 0.2 micrometer to 10
centimeters (cm)
thick or more.
Fibrous webs can be prepared by methods known in the art, or by modifications
of methods known in the art. Nonwoven webs can be prepared by melt-blowing as
is
known to those skilled in the art. In general, a molten polymeric material is
extruded in
such a way as to produce a stream of melt blown polymer microfibers. The
fibers are
collected on a collection screen, with the microfibers forming a web. The
nonwoven
webs can also optionally include a permeable support fabric laminated to one
or both
sides of the web, or can additionally contain reinforcing fibers.
Exemplary materials useful to prepare nonwoven fibrous webs include
polymers and copolymers of monomers that form fibrous webs. Suitable polymers
include polyalkylenes such as polyethylene and polypropylene, polyvinyl
chloride,
polyamides such as the various nylons, polystyrenes, polyarylsulfones,
polyvinyl
alcohol), polybutylene, polyethylene vinyl acetate), polyacrylates such as
polymethyl
methacrylate, polycarbonate, cellulosics such as cellulose acetate butyrate,
polyesters
such as polyethylene terephthalate), polyimides, and p~lyurethanes such as
polyether
polyurethanes, and combinations thereof.
Nonwoven webs can also be prepared from combinations of co-extruded
polymers such as polyesters and polyalkylenes. Copolymers of the monomers that
provide the above-described polymers are also in eluded within the scope of
the present
invention. Nonwoven webs can also be combined webs, which are an intimate
blend of
fine fibers and crimped staple fibers.
Suitable substrates for attachment of IRMs can also include microporous
membranes, fibers, hollow fibers, or tubes, all of which are lcnown in the
art. The same
materials useful for preparing webs are also suitable for preparing fibers and
membranes.
An exemplary microporous membrane is one made from thermoplastic
polymeric material using a thermally induced phase separation technique that
involves
melt blending a thermoplastic polymer with immiscible liquid at a temperature
sufficient to form a homogeneous mixture, forming an article from the solution
into a
desired shape, cooling the shaped article so as to induce phase separation of
the liquid
and the polymer and to ultimately solidify the polymer, and removing at least
a
substantial portion of the liquid leaving a microporous polymer matrix. This
method
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-23-
and the preferred compositions used in the method are described in detail in
U.S. Pat.
Nos. 4,957,943; 4,539,256; and 4,726,989. Alternatively, polymeric supports
can also
be hydrophobic polyolefin membranes prepared by thermally induced phase
separation
techniques, but also having a hydrophilic polymeric shell interlocked about
such
hydrophobic membrane surfaces.
The support materials having and an IRM associated therewith can include a
combination of materials. For example, they can include a combination of
inorganic
and organic materials or a combination of different organic polymers. This can
occur
by layering the materials, for example. One or more of the materials can be
associated
(e.g., attached) to the particulate support material on the outermost surface
such that an
IRM is masked or hidden from a body's immune system until it reaches its
targeted site
of action. For example, polymers of lactic acid and glycolic acid in the form
of
particles having one or more IRMs attached thereto can have a coating of a
polyalkylene oxide polymer (e.g., polyethylene glycol) thereon (see, e.g.,
Gref et al.,
Colloids and Surfaces ~: l3iointerfaces 18, 301-313, 2000). The polyalkylene
oxide
can function to mask the Il~ from the body's immune system until it reaches
its
targeted site of action.
Steyats:
131ood vessel occlusions are commonly treated by mechanically enhancing
blood flow in the affected vessels, such as by employing a stem. Stems act as
scaffoldings, functioning to physically hold open and, if desired, to expand
the wall of
the passageway. Typically, stems are capable of being compressed, so that they
can be
inserted through small lumens via catheters, and then expanded to a larger
diameter
once they are at the desired location. Examples in the patent literature
disclosing stems
include U.S. Pat. No. 4,733,665 issued to Palmaz, U.S. Pat. No. 4,800,882
issued to
Gianturco, and U.S. Pat. No. 4,886,062 issued to Wiktor.
A stmt useful in the present invention can be any stmt, including a self
expanding stmt, or a stmt that is radially expandable by inflating a balloon
or
expanded by a.n expansion member, or a stmt that is expanded by the use of
radio
frequency which provides heat to cause the stmt to change its size. The stent
can also
be made of any desired material, including a metallic material, a metal alloy
(e.g.,
nickel-titanium) or even polymeric composites. The stmt can have any wire or
cell
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-24-
design. Examples of self expanding wire mesh stems that can be used include
the
coronary WALLSTENT marketed by Schneider, and the SciMED RADIUS stmt
marketed by Boston Scientific Corp. Examples of balloon expandable stents that
can be
used include the MULTILINK stmt by Guidant Corp., the Coronary Stent 5670 by
Medtronic AVE, the NIR stmt by Boston Scientific Corp., the CROSS FLEX stmt by
Cordis, the PAS stmt by Progressive Angioplasty Systems Inc., the V-FLEX PLUS
stmt by Cook, Inc., and the PALMAZ-SCHATZ crown and spiral stems by Cordis,
among others. The vessels in which the stmt of the present invention can be
deployed
include, but are not limited to, natural body vessels such as ducts, arteries,
trachea,
veins, intestines, bile ducts, ureters and the esophagus.
In addition to cardiac applications, the development of cancerous blockages
inside body passageways (e.g., esophagus, bile ducts, trachea, intestine,
vasculature and
urethra, among others) can also be treated with stems, which operate to hold
open
passageways that have been blocked by the cancerous growth or tumors. However,
such stems do not prevent the ingrowth of the cancerous material through the
interstices
of the stmt.
In the present invention, stems are used not only for mechanical intervention
but
also as vehicles for providing biological therapy. Biological therapy can be
achieved
by attaching an IRM to the stmt. Stems that have been medicated by the
attachment of
an IRM provide for the local administration of a therapeutic substance at the
diseased
site. Local delivery of an IRM is a preferred method of treatment because the
substance is concentrated at a specific site and thus smaller total levels of
medication
can be administered in comparison to systemic dosages that often produce
adverse or
even toxic side effects for the subject.
Restenosis is a form of chronic vascular injury leading to vessel wall
thickening
and loss of blood flow to the tissue supplied by the blood vessel. It occurs
in response
to vascular reconstructive procedures, including virtually any manipulation
that
attempts to relieve vessel obstructions, such as the insertion of a stmt.
Restenosis is a
major factor limiting the effectiveness of invasive treatments for vascular
diseases and
has been a major challenge to cardiovascular research for the past 15 years.
According
to 1994 estimates (LJ.S. Heart and Strolce Foundation), over 60 million
Americans have
one or more forms of cardiovascular disease. These diseases claimed
approximately 1
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-25-
million lives in the same year (41 % of all deaths in the United States) and
are
considered the leading cause of death and disability in the developed world.
Systemic therapies that have been investigated for the prevention of
restenosis
include agents directed at treatment of endothelial loss, anti-platelet agents
(e.g.,
aspirin), vasodilators (e.g., calcium channel blockers), antithrombotics
(e.g., heparin),
anti-inflammatory agents (e.g., steroids), agents that prevent vascular smooth
muscle
cell (VSMC) proliferation (e.g., colchicine), and promoters of re-
endothelialization
(e.g., vascular endothelial growth factor). Local treatments that have been
investigated
include local drug delivery (e.g., heparin) and beta and gamma radiation. All
have been
disappointing in human use, primarily because they appear to act on a limited
portion
of the restenotic process. Systemic treatments have also encountered the
additional
problem of achieving adequate absorption and retention of the drug at the site
of the
disease to provide a lasting biological effect, without causing unfavorable
systemic
complications and toxicities.
The inflammatory response induced by coronary angioplasty and stent
placement may play a role in the Bevelopment of neointimal hyperplasia, a
major cause
of restenosis after coronary intervention. Inflammation triggered by vascular
tissue
injury triggers a complex cascade of cellular and biochemical processes such
as
fibroblast and smooth muscle migration and proliferation, apoptosis, and
matrix
synthesis and remodeling. Cytokines are mediators of both the acute and
chronic
inflammatory rasp~nse. Many cytokines such as tumor necrosis factor alpha,
interferon-alpha, interferon-gamma, interleukins 1, 4, 13, and monocyte
chemoattractant protein-l, and nitric oxide have been identified to play a
role in the
inflammatory response after vascular tissue injury. While the exact role that
these and
other cytokines play is not clear, there is evidence to suggest alterations in
the
fibroproliferative responses seen in oral, dermal and vascular tissues can be
accomplished by altering the local cytokine profile in the tissue. For example
interferon-alpha and gamma have been shown to reduce collagen synthesis by
dermal
fibroblasts and hypertrophic scar fibroblasts. In mice, interferon-gamma
reduced the
fibrotic response to an implanted foreign body. Interferon-gamma has also been
shown
to inhibit proliferation of vascular smooth muscle cells in culture and reduce
arterial
restenosis after balloon angioplasty.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-26-
Immune response modifiers (IRMs) include small molecules that trigger the
production of cytokines from antigen presenting cells through, for example,
toll-like
receptor (TLR) pathways such as, for example, TLR 7 and/or 8. Some IRMs can
direct
the innate immune response to produce cytokines, such as IL-12 and interferon-
gamma,
that stimulate a TH1 or cell-mediated response. This TH1 cytokine response can
also
lead to the reduction in cytokines implicated in the TH2 response. The over
expression
of TH2 cytokines have been implicated in atopic and granulomatous conditions.
Therefore, manipulation of the inflammatory reaction in vascular tissue injury
with an
IRM could prevent restenosis following angioplasty and stmt placement.
For stems, as well as other medical devices, particularly implantable and
extracorporeal devices, the IRM may elute from a coating on the article upon
contact
with a bodily fluid. In such embodiments, the IRM may be incorporated into a
coating
material, e.g., a polymeric material, using any of a variety of mechanisms,
which may
or may not include attachment of the IRM to the macromolecular support
material. For
example, the IR1VI can be simply mixed in a polymeric coating material.
I~erein, the terms "medical device" and "medical article" are used
interchangeably and refer generally to any device that has surfaces that can,
in the
ordinary course of their use and operation, contact bodily tissue, organs or
fluids such
as blood. Examples of medical devices include, without limitation, stems, stmt
grafts,
anastomotic connectors, leads, needles, guide wires, catheters, sensors,
surgical
instmunents, angioplasty balloons, wound drains, shunts, tubing, urethral
inserts,
pellets, implants, pumps, vascular grafts, valves, pacemakers, and the like. A
medical
device can be an extracorporeal device, such as a device used during surgery,
which
includes, for example, a blood oxygenator, blood pump, blood sensor, or tubing
used to
carry blood, and the like, which contact blood which is then returned to the
subject. A
medical device can likewise be an implantable device such as a vascular graft,
stmt,
stmt graft, anastomotic connector, electrical stimulation lead, heart valve,
orthopedic
device, catheter, shunt, sensor, replacement device for nucleus pulposus,
cochlear or
middle ear implant, intraocular lens, and the like. Implantable devices
include
transcutaneous devices such as drug injection ports and the lilce.
Such medical devices or medical articles do not include within their scope
transdermal patches or articles used in female hygiene, such as tampons.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
_27_
For certain embodiments, the medical devices include a shunt, an artificial
valve, a suture, a surgical clip, a surgical staple, an indwelling catheter, a
dental implant
(with the proviso that the dental implant is not a periochip inserted into the
periodontal
cavity), an orthopedic implant, a surgical prosthetic, an implantabhe vascular
access
port, an artificial heart, a ventricular assist pump, a blood oxygenator, a
blood filter, a
hemodiahysis unit, a hemoperfusion unit, a conduit tube within a heart lung
machine, a
tube within a dialysis apparatus, a tube within a plasmapheresis unit, an
artificial
pancreas, an artificial liver, an artificial lung, an intraocular lens, or a
contact lens.
Attaclafyaefat to Substf~ates:
IRMs can be attached to a macromolecular support material through either
covalent attachment or non-covalent attachment. Non-covalent attachunent of an
IRM
to a macromolecular support material includes attachment by ionic interaction
or
hydrogen bonding, for example.
One example of a non-covalent attachment included in the present invention is
the well-know biotin-avidin system. Avidin-biotin affinity-based technology
has found
wide applicability in numerous fields of biology and biotechnology since the
pioneering work by Dr. Edward Bayer and Dr. IVIeier Wilchek in the 1970'x. The
affinity constant between avidin and biotin is remarkably high (the
dissociation
constant, I~d, is approximately 10-ls 1'~, see, Green, N., Biocheri~ J, ~9,
599, 1963) and
is not significantly lessened when biotin is coupled to a wide variety of
biomohecules.
Numerous chemistries have been identified for coupling biomolecules to biotin
with
minimal or negligible loss in the activity or other desired characteristics of
the
biomolecule. A review of the biotin-avidin technology can be found in
Applications of
Avidin-Biotin Technology to Affinity-Based Separation, Bayer, et al., J. of
Chromatography, 1990, pgs. 3-11.
Streptavidin, and its functional homolog avidin, are tetrameric proteins,
having
four identical subunits. Streptavidin is secreted by the actinobacterium
Streptomyces
avidinii. A monomer of streptavidin or avidin contains one high-affinity
binding site
for the water-soluble vitamin biotin and a streptavidin or avidin tetramer
binds four
biotin molecules.
Biotin, also known as vitamin H or cis-hexahydro-2-oxo-1H thieno-[3,4]-
imidazole-4-pentanoic acid, is a basic vitamin which is essential for most
organisms
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-28-
including bacteria and yeast. Biotin has a molecular weight of about 244
daltons, much
lower than its binding partners avidin and streptavidin. Biotin is also an
enzyme
cofactor of pyruvate carboxylase, trans-carboxylase, acetyl-CoA-carboxylase
and beta-
methylcrotonyl-CoA carboxylase which together carboxylate a wide variety of
substrates.
Both streptavidin and avidin exhibit extremely tight and highly specific
binding
to biotin which is one of the strongest known non-covalent interactions
between
proteins and ligands, with a molar dissociation constant of 10-15 molar (M)
(N. M.
Green, Advances in Protein Chemistry, Vol. 29, pp. 85-133, 1975), and a tl/2
of ligand
dissociation of 89 days (N. M. Green, Advances in Protein Chemistry, Vol. 29,
pp. 85-
133, 1975). The avidin-biotin bond is stable in serum and in the circulation
(R. D. Wei,
D. H. I~ou, S. L. Hoo, Experientia, Vol. 27, pp. 366-368, 1970). ~nce formed,
the
avidin-biotin complex is unaffected by most extremes of pH, organic solvents
and
denaturing conditions. Separation of streptavidin from biotin requires
conditions, such
as 8M guanidine, pH 1.5, or autoclaving at 121 °C for 10 minutes.
IRMs may be biotinylated using any known methodologies. For example,
I~Ms may be biotinylated chemically, using activated biotin analogues, such as
N-
hydroxysuccinimidobiotin (NHS-biotin), which is commercially available from
Pierce
Chemical Company, Rockford, IL, and requires the presence of a free primary
amino
group on the II~M.
Representative methods for covalent attaching an IRM to a macromolecular
support material include chemical cross linkers, such as heterobifunctional
cross
linking compounds (i.e., "linkers") that react to form a bond between reactive
groups
(such as hydroxyl, amino, amido, or sulfllydryl groups) in a the immune
response
modifier and other reactive groups (of a similar nature) in the support
material. This
bond may be, for example, a peptide bond, disulfide bond, thioester bond,
amide bond,
thioether bond, and the like.
Immune response modifiers may be covalently bonded to a macromolecular
support material by any of the methods lcnown in the art. For example, U.S.
Pat. Nos.
4,722,906, 4,979,959, 4,973,493, and 5,263,992 relate to devices having
biocompatible
agents covalently bound via a photoreactive group and a chemical linking
moiety to the
biomaterial surface. U.S. Pat. Nos. 5,258,041 and 5,217,492 relate to the
attachment of
biomolecules to a surface through the use of long chain chemical spacers. U.S.
Pat.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-29-
Nos. 5,002,582 and 5,263,992 relate to the preparation and use of polymeric
surfaces,
wherein polymeric agents providing desirable properties are covalently bound
via a
photoreactive moiety to the surface. Others have used photochemistry to modify
the
surfaces of biomedical devices, e.g., to coat vascular grafts. (See, e.g.,
Kito, H. et al.,
ASAIO Journal 39:M506-M511, 1993; and Clapper, D. L., et al., Trans. Soc.
Biomat.
16:42, 1993). Cholakis and Sefton synthesized a polymer having a polyvinyl
alcohol
(PVA) backbone and heparin bioactive groups. The polymer was coupled to
polyethylene tubing via nonlatent reactive chemistry, and the resultant
surface was
evaluated for thromboresistance in a series of in vitro and in vivo assays.
(See,
Cholalcis, C. H. and M. V. Sefton, J. Biomed. Mater. Res. 23:399-415, 1989;
and
Cholakis, C. H., et. al., J. Biomed. Mater. Res. 23:417-441, 1989). Also,
I~inoshita et
al. disclose the use of reactive chemistry to generate polyacrylic acid
backbones on
porous polyethylene, with collagen molecules being subsequently coupled to
carboxyl
moieties on the polyacrylic acid backbones. (See I~inoshita, Y., et al.,
Biomaterials
14:209-215, 1993).
IRMs could be attached to macromolecular supports in a similar fashion to the
methods described in LJ.S. Pat. Nos. 5,200,4715,344,701, 5,4869358, 5,510,421,
and
5,907,016. These patents disclose macromolecular supports having biologically
active
agents covalently bound via the reaction of a nucleophilic-functional group on
the
biologically active agent with an azlactone functional group on the
macromolecular
support. In a preferred embodiment, the IRM can be attached to a
macromolecular
support material using a linking group. The linking group can be any suitable
organic
linking group that allows the substrate to be covalently coupled to the immune
response
modifier moiety while preserving an effective amount of IRM activity. In some
embodiments, the linking group may be selected to create sufficient space
between the
active core of the immune response modifter moiety and the substrate that the
substrate
does not interfere with a biologically effective interaction between the
active core and
the T cells that results in IRM activity such as cytokine production.
The linking group includes a reactive group capable of reacting with a
reactive
group on the substrate to form a covalent bond. Suitable reactive groups
include those
discussed in Hermanson, G. (1996), Biocor jugate Techniques, Academic Press,
Chapter 2 "The Chemistry of Reactive Functional Groups", 137-166. For example,
the
linking group may react with a primary amine (e.g., an N-hydroxysuccinimidyl
ester or
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-3 0-
an N-hydroxysulfosuccinimidyl ester); it may react with a sulfhydryl group
(e.g., a
maleimide or an iodoacetyl), or it may be a photoreactive group (e.g. a phenyl
azide
including 4-azidophenyl, 2-hydroxy-4-azidophenyl, 2-nitro-4-azidophenyl, and 2-
nitro-
3-azidophenyl). The linking group may also be an alkoxysilyl group (e.g., a
triethyoxysilyl group) that can be covalently coupled to an IRM. The
alkoxysilyl group
can then be covalently coupled to a silicon-containing support material such
as silica,
which may be in the form of particles.
The substrate includes a chemically active group accessible for covalent
coupling to the linking group. A chemically active group accessible for
covalent
coupling to the linking group includes groups that may be used directly for
covalent
coupling to the linking group or groups that may be modified to be available
for
covalent coupling to the linking group. For example, suitable chemically
active groups
include, but are not limited to, primary amines and sulfhydryl groups.
Typically, attachment may occur by reacting an immune response modifier with
a crosslinker and then reacting the resulting intermediate with a substrate.
Many
crosslinkers suitable for preparing bioconjugates are known and many are
commercially available. See for example, I-Iermanson, Ca. (1996) ~i~eoy ju~-
cate
Techfaiques, Academic Press.
Attachment also may occur, for example, according to the method shown in
Reaction Scheme I in which the substrate is linked to the IRM moiety through
Rl. In
step (1) of Reaction Scheme I a compound of Formula III is reacted with a
heterobifunctional crosslinker of Formula IV to provide a compound of II. RA
and RB
each contain a functional group that is selected to react with the other. For
example, if
R~ contains a primary amine, then a heterobifunctional crosslinker may be
selected in
which RB contains an amine-reactive functional group such as an N-
hydroxysulfosuccinimidyl ester. RA and RB may be selected so that they react
to
provide the desired linker group in the conjugate.
Methods for preparing compounds of Formula III where RA contains a
functional group are lcnown. See, for example, U.S. Pat. Nos. 4,689,338;
4,929,624;
5,268,376; 5,389,640; 5,352,784; 5,494,916; 4,988,815; 5,367,076; 5,175,296;
5,395,937; 5,741,908; 5,693,811; 6,069,149; 6,194,425; 6,331,539; 6,451,810;
6,525,064; 6,541,485; 6,545,016; 6,545,017; 6,573,273; 6,656,938; 6,660,735;
6,660,747; 6,664,260; 6,664,264; 6,664,265; 6,667,312; 6,670,372; 6,677,347;
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-31-
6,677,348; 6,677,349; 6,683,088; and International Publication WO 03/103584.
Many
heterobifunctional crosslinkers are known and many are commercially available.
See
for example, Hermanson, G. (1996), Bioconjugate Techniques, Academic Press,
Chapter 5 "Heterobifunctional Cross-Linkers", 229-285. The reaction generally
can be
carried out by combining a solution of the compound of Formula III in a
suitable
solvent such as N,N-dimethylformamide with a solution of the
heterobifunctional
cross-linker of Formula IV in a suitable solvent such as N,N-
dimethylformamide. The
reaction may be run at ambient temperature. The product of Formula II may then
be
isolated using conventional techniques.
In step (2) of Reaction Scheme I a compound of Formula II that contains
reactive group ~A is reacted with the substrate to provide the IRM-couples
substrate of
Formula I. In one embodiment the reaction can be carried out by combining a
solution
of the compound of Formula II in a suitable solvent such as dimethyl sulfoxide
with the
substrate. The reaction may be run at ambient temperature or at a reduced
temperature
(approximately 4°C). If ~A is a photoreactive group such as a phenyl
aide then the
reaction mixture will be exposed to long wave UV light for a length of time
adequate to
effect cross-linking (e.g., 10-20 minutes). The average number of immune
response
modifier moieties per substrate surface area may be controlled by adjusting
the amount
of compound of~Formula II used in the reaction.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-32-
Reaction Scheme I
NHz NHz
N>--Rz + RB ZA ~~~ N ~ N>---Rz
R3 N R3
R4 RA R4
III IV II Z
A
~z)
Y
NHz
N W N
Rz
R ~ N
3
R R~ Substrate
4
n
Alternatively, a compound of Formula II may be synthesized without using a
heterobifunctional crosslinker. So long as the compound of Formula II contains
the
reactive group ZA, it may be reacted with the substrate using the method of
step (2)
above to provide an IRI~I-coupled substrate.
The R groups can be hydrogen or organic groups that can optionally include
various substitutions. They can include alkyl groups, alkenyl groups,
including
haloalkyl groups, aryl groups, heteroaryl groups, heterocyclyl groups, and the
like.
For example, preferred R2 groups include hydrogen, alkyl groups having 1 to 4
carbon atoms (i.e., methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl,
isobutyl, tert-
butyl, and cyclopropylmethyl), and alkoxyalkyl groups (e.g., methoxyethyl and
ethoxymethyl). Preferably R3 and R4 are independently hydrogen or methyl or R3
and
R4 join together to form a benzene ring, a pyridine ring, a 6-membered
saturated ring or
a 6-membered saturated ring containing a nitrogen atom. One or more of these
preferred substituents, if present, can be present in the compounds of the
invention in
any combination.
As used herein, the terms "alkyl", "alkenyl" and the prefix "alk-" include
straight chain, branched chain, and cyclic groups, i.e. cycloalkyl and
cycloalkenyl.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-33-
Unless otherwise specified, these groups contain from 1 to 20 carbon atoms,
with
alkenyl groups containing from 2 to 20 carbon atoms. Preferred groups have a
total of
up to 10 carbon atoms. Cyclic groups can be monocyclic or polycyclic and
preferably
have from 3 to 10 ring carbon atoms. Exemplary cyclic groups include
cyclopropyl,
cyclopentyl, cyclohexyl, cyclopropylmethyl, and adamantyl.
The term "haloalkyl" is inclusive of groups that are substituted by one or
more
halogen atoms, including perfluorinated groups. This is also true of groups
that include
the prefix "halo-". Examples of suitable haloalkyl groups are chloromethyl,
trifluoromethyl, and the like.
The term "aryl" as used herein includes carbocyclic aromatic rings or ring
systems. Examples of aryl groups include phenyl, naphthyl, biphenyl, fluorenyl
and
indenyl. The teen "heteroaryl" includes aromatic rings or ring systems that
contain at
least one ring hetero atom (e.g., O, S, I~. Suitable heteroaryl groups include
furyl,
thienyl, pyridyl, quinolinyl, isoquinolinyl, indolyl, isoindolyl, triazolyl,
pyrrolyl,
tetrazolyl, imida~olyl, pyrazolyl, oxa~olyl, thia~olyl, ben~ofuranyl,
benzothiophenyl,
carbazolyl, ben~oxa~olyl, pyrimidinyl, ber~imidazolyl, quinoxalinyl,
ben~othia~olyl,
naphthynidinyl, isoxazolyl, isothia~olyl, puninyl, quina~olinyl, and so on.
"Heterocyclyl" includes non-aromatic rings or ring systems that contain at
least
one ring hetero atom (e.g., O, S, I~ and includes all of the fully saturated
and partially
'~0 unsaturated derivatives of the above mentioned heteroaryl groups. Exemplaa-
y
heterocyclic groups include pyrrolidinyl, tetrahycliofuranyl, morpholinyl,
thiomorpholinyl, piperidinyl, piperazinyl, thia~olidinyl, isothia~olidinyl,
and
imidazolidinyl.
The aryl, heteroaryl, and heterocyclyl groups can be unsubstituted or
substituted
~5 by one or more substituents independently selected from the group
consisting of alkyl,
alkoxy, methylenedioxy, ethylenedioxy, alkylthio, haloallcyl, haloalkoxy,
haloalkylthio,
halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy,
arylthio,
arylalkoxy, arylalkylthio, heteroaryl, heteroaryloxy, heteroarylthio,
heteroarylalkoxy,
heteroarylalleylthio, amino, allcylamino, dialkylamino, heterocyclyl,
heterocycloallcyl,
30 alkylcarbonyl, allcenylcarbonyl, alkoxycarbonyl, haloalkylcarbonyl,
haloallcoxycarbonyl, allcylthiocarbonyl, arylcarbonyl, heteroarylcarbonyl,
. aryloxycarbonyl, heteroaryloxycarbonyl, arylthiocarbonyl,
heteroarylthiocarbonyl,
alkanoyloxy, alkanoylthio, alkanoylamino, arylcarbonyloxy, arylcarbonythio,
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-34-
alkylaminosulfonyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl,
aryldiazinyl,
alkylsulfonylamino, arylsulfonylamino, arylalkylsulfonylamino,
alkylcarbonylamino,
alkenylcarbonylamino, arylcarbonylamino, arylalkylcarbonylamino,
heteroarylcarbonylamino, heteroarylalkycarbonylamino, alkylsulfonylamino,
alkenylsulfonylamino, arylsulfonylamino, arylalkylsulfonylamino,
heteroarylsulfonylamino, heteroarylalkylsulfonylamino,
alkylaminocarbonylamino,
alkenylaminocarbonylamino, arylaminocarbonylamino,
arylalkylaminocarbonylamino,
heteroarylaminocarbonylamino, heteroarylalkylaminocarbonylamino and, in the
case of
heterocyclyl, oxo. If other groups are described as being "substituted" or
"optionally
substituted", then those groups can also be substituted by one or more of the
above-
enumerated substituents.
In Reaction Scheme I the IRM is attached to the substrate through a linking
group at the Nl nitrogen of the imidazole ring. Alternatively the linking can
occur at
different positions on the ring system. Examples of which are shown below for
imidazoquinoline amines, imidazonaphthyridine amines and imidazopyridine
amines
respectively.
NHS NHS
NHZ
N
N / y Rz N s N~ Ra N ~ N R
N ~ R3 ' ~ ~~ 2
R1 a R1 a
R1 R1 R1 R1a
Substrate Substrate Substrate
> >
The attachment is effected using the method of Reaction Scheme I starting with
an IRM containing reactive group RA at the desired attachment point.
The IRM-support complexes of the present invention can be incorporated into a
wide variety of formulations, including, for example gels, creams,
dispersions, or
solutions. Such formulations can include solvents (e.g., propylene glycol,
sorbitol,
polyethylene glycol, hexylene glycol, dipropylene glycol), oils (e.g., mineral
oil,
vegetable oils, fatty acid triglycerides, isopropyl mysristate, isopropyl
palmitate, and
isostearic acid), emulsifiers (polysorbate 60, sorbitan monostearate,
polyglyceryl-4
oleate, polyoxyethylene(4) lauryl ether, poloxamers, and sorbitan trioleate),
preservatives (e.g., methylparaben and propyl paraben), neutralizers (e.g.,
sodium
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-35-
hydroxide), etc. Examples of suitable formulations are disclosed in U.S. Pat.
No.
6,245,776 and U.S. Patent Publication No. 2003/0199538. If the formulation is
a gel,
for example, it can be a preformed gel or formed at the application site.
An amount of an IRM-support complex effective for a given therapeutic or
prophylactic application is an amount sufficient to achieve the intended
therapeutic or
prophylactic application. The precise amount of IRM-support complex used will
vary
according to factors known in the art including but not limited to the
physical and
chemical nature of the IRM compound, the nature of the macromolecular support
material, the intended dosing regimen, the state of the subject's immune
system (e.g.,
suppressed, compromised, stimulated), the method of administering the IRM
compound, and the species to which the formulation is being administered.
Accordingly it is not practical to set forth generally the amount that
constitutes an
amount of IRM support complex effective for all possible applications. Those
of
ordinary skill in the art, however, can readily determine the appropriate
amount with
due consideration of such factors.
The dosing regimen may depend at least in part on many factors known in the
art including but not limited to the physical and chemical nature of the
II~1~I compound,
the nature of the macromolecular support material, the amount of III being
administered, the state of the subject's immune system (e.g., suppressed,
compromised,
stimulated), the method of administeung the IPLM-support complex, and tlae
species to
which the formulation is being administered. Accordingly it is not practical
to set forth
generally the dosing regimen effective for all possible applications. Those of
ordinary
skill in the art, however, can readily determine the appropriate amount with
due
consideration of such factors.
EXAMPLES
The following examples have been selected merely to further illustrate
features,
advantages, and other details of the invention. It is to be expressly
understood,
however, that while the examples serve this purpose, the particular materials
and
amounts used as well as other conditions and details are not to be construed
in a matter
that would unduly limit the scope of this invention.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-36-
IRM Compounds
The IRM compounds used in the examples are shown in Table 1.
Table 1
Compound Chemical Name Reference
IRMI N'-[2-(4-amino-2-butyl-1H imidazo[4,5-Example
28
c]quinolin-1-yl)ethyl]-5-(2-oxoperhydrothieno[3,4-U.S. Pat.
No.
d]imidazol-4-yl)pentanamide 6,451,810
B1
IRM2 Nl-(6- f [4-(4-amino-2-butyl-1H imidazo[4,5-
c]quinolin-1-yl)butyl]amino- 6-oxohexyl)-6-(~5-
[(3aR,4R,6aS)-[2-oxoperhydrothieno[3,4-
d]imidazol-4-yl)pentanoyl~ amino)hexanamide
Preparation of IRM2
1-(4-Aminobutyl)-2-butyl-l~I imidazo[4,5-a]quinolin-4-amine (46 mg, 0.148
mmol) was dissolved with heating in anhydrous N,N-dimethylformamide
(approximately 5 milliliters (mL)). The resulting solution was allowed to cool
to
ambient temperature. EZ-LIl~II~ Sulfo-NHS-LC-LC-Biotin (Pierce, 2 x 50
milligram
(mg) vials, 0.149 millimole (mmol)) was added to the solution. Each vial was
rinsed
with anhydrous N,N-dimethylfonnamide (approximately 1 mL) and the rinse was
added to the reaction mixture. The reaction mixture was allowed to stir at
ambient
temperature for about 90 minutes. The reaction mixture was concentrated under
reduced pressure at 60°C. The residue was purified by flash
chromatography (2 x 15
cm column of Si~Z eluting with 10:2:0.25 chloroform:methanol:water) to provide
a
colorless glass. The glass was dissolved in methanol and then concentrated to
provide
Nl-(6- f [4-(4-amino-2-butyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]amino- 6-
oxohexyl)-6-( ~5-[(3aR,4R,6aS)-[2-oxoperhydrothieno[3,4-d]imidazol-4-
yl)pentanoyl~ amino)hexanamide as a white foam.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-37-
Preparation of N [4-(4-Amino-2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-N'-
[3-
(triethoxysilyl)propyl]urea
NH2
N W N
I ~ N
O
Sid O~
N N~/ O-
H \\
O
Into a flask was placed 1-(4-aminobutyl)-2-propyl-1H imidazo[4,5-c]quinolin-
4-amine (100 mg, 0.336 mmol; which can be prepared using the methods disclosed
in
U.S. Pat. No. 6,069,149) and 5 mL anhydrous dimethyl sulfoxide (DMSO). The
mixture was stirred until the solid was completely dissolved. To the solution
was
slowly added 3-(triethoxysilyl) propyl isocyanate (~3.2 mg, 0.336 mmol) in
DMSO
(1.5 mL) at room temperature. After the addition, the reaction solution was
stirred
overnight. The reaction solution was sampled and analyzed by NMR. The spectra
showed the desired addition product,1~ [4-(4-amino-2-propyl-1~1 imidazo[4.,5-
c]quinolin-1-yl)butyl]-N'-[3-(triethoxysilyl)propyl]urea, at 100°1o
conversion. The
sample was also analyzed by liquid chromatography, the spectrum showed a
single
product peak with the disappeaa-ance of the starting materials.
The reaction was repeated using 15 mL of anhydrous tetrahydrofuran (THF) in
place of the DMSO. Analysis of the resulting product by NMR showed 97%
conversion of the starting material to the desired addition product.
Preparation of N [3-(4-Amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-
c]pyridin-
1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea
NH2 /
N ~ N O
I ~ N~ O
Si: O
N~ N~ O
H \\
O
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-3 8-
Into a flask was placed 1-(3-aminopropyl)-2,-ethoxymethyl-6,7-dimethyl-1H
imidazo[4,5-c]pyridin-4-amine (100 mg, 0.36 mmol; Example 21 in U.S. Pat. No.
6,545,016) and 5 mL anlrydrous dimethyl sulfoxide (DMSO). The mixture was
stirred
until the solid was completely dissolved. To the solution was slowly added 3-
(triethoxysilyl) propyl isocyanate (89.1 mg, 0.36 mmol) in DMSO (1.5 mL) at
room
temperature. After the addition, the reaction solution was stirred overnight.
The
reaction solution was sampled and analyzed by NMR. The spectra showed the
desired
addition product, N [3-(4-amino-2-ethoxyrnethyl-6,7-dimethyl-1H imidazo[4,5-
c]pyridin-1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea, at 100% conversion.
The
sample was also analyzed by liquid chromatography, the spectrum showed a
single
product peak with the disappearance of the starting materials.
Preparation of 2-ethoxymethyl-1-((3-~2-hydroxy-3-[3-
(trimethoxysilyl)propoxy]propyl~amino))propyl-6,7-dimethyl-1H imidazo[4,5-
c]pyridine-4-amine
~! H~
H ~ N
s
H
,~~''Si-p
O ~O
H' ~~H
Into a flask was placed 1-(3-aminopropyl)-2-ethoxymethyl-6,7-dimethyl-1H=
imidazo[4,5-c]pyridin-4-amine (10 mg, 0.036 mrnol; Example 21 in U.S. Pat. No.
6,545,016) and 2.5 mL anhydrous tetrahydrofuran. The mixture was stirred until
the
solid was completely dissolved. To the solution was slowly added 3-
glycidoxypropyltrimethoxysilane (8.51 mg, 0.036 mmol) at room temperature.
After
the addition, the reaction solution was stirred overnight. The reaction
solution was
sampled and analyzed by NMR. The spectra showed the desired addition product,
2-
ethoxymethyl-1-((3-{2-hydroxy-3-[3-
(trimethoxysilyl)propoxy]propyl}amino))propyl-
6,7-dimethyl-1H imidazo[4,5-c]pyridine-4-amine, at 100% conversion.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-39-
Preparation of N f 2-[4-amino-7-(6-aminohexyloxy)-2-ethoxyrnethyl-1H
imidazo[4,5-
c] quinolin-1-yl]-1,1-dimethylethyl} methanesulfonamide
NHZ /
N ~ N O
~N
HzN O ~ ~ _ ~O
H ,S~
O
Part A
A mixture of triethyl orthoformate (92 mL, 0.55 mol) and 2,2-dimethyl-[1,3]-
dioxane-4,6-dione (75.3 grams (g), 0.522 mol) (Meldrum's acid) was heated at
55°C
for 90 minutes and then cooled to 45°C. A solution of 3-
benzyloxyaniline (100.2 g,
0.5029 mol) in methanol (200 mL) was slowly added to the reaction over a
period 45
minutes while maintaining the reaction temperature below 50°C. The
reaction was then
heated at 45°C for one hour, allowed to cool to room temperature, and
stirred
overnight. The reaction mixture was cooled to 1°C, and the product was
isolated by
filtration and washed with cold ethanol (approximately 4.00 mL) until the
filtrate was
colorless. 5-{ [(3-benzyloxy)phenylimino]methyl}-2,2-dimethyl-[ 1,3]-dioxane-
4,6-
dione (170.65 g) was isolated as a tan, powdery solid.
Part B
A mixture of 5-~[(3-benzyloxy)phenylimino]methyl}-2,2-dimethyl-[1,3]
dioxane-4,6-dione (170.65 g, 0.483 mol) and I?~WTHERM A (800 mL) was heated to
2~ 100°C and then slowly added to a flash containing D~WTHERM A (1.3 L,
heated at
210°C) over a period of 40 minutes. During the addition, the reaction
temperature was
not allowed to fall below 207°C. Following the addition, the reaction
was stirred at
210°C for one hour, and then allowed to cool to ambient temperature. A
precipitate
formed, which was isolated by filtration, washed with diethyl ether (1.7
liters (L)) and
acetone (0.5 L), and dried in an oven to provide 76.5 grams (g) of 7-
benzyloxyquinolin-
4-0l as a tan powder.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-40-
Part C
A mixture of 7-benzyloxyquinolin-4-of (71.47 g, 0.2844 mol) and propionic
acid (700 mL) was heated to 125°C with vigorous stirring. Nitric acid
(23.11 mL of 16
M) was slowly added over a period of 30 minutes while maintaining the reaction
temperature between 121 °C and 125°C. After the addition, the
reaction was stirred at
125°C for 1 hour then allowed to cool to ambient temperature. The
resulting solid was
isolated by filtration, washed with water, and dried in an oven for 1.5 days
to provide
69.13 g of 7-benzyloxy-3-nitroquinolin-4-of as a grayish powder.
Part D
N,N Dimethylformasnide (100 mL) (DMF) was cooled to 0°C, and
phosphorous
oxychloride (27.5 mL, 0.295 mol) was added dropwise. The resulting solution
was
stirred for 25 minutes and then added dropwise to a mixture of 7-benzyloxy-3-
nitroquinolin-4-of (72.87 g, 0.2459 mol) in DMF (400 mL). Following the
addition, the
reaction was heated at 100°C for 5 minutes, cooled to ambient
temperature, and poured
into ice water with stirring. A tan precipitate formed, which was isolated by
filtration
and dissolved in dichloromethane. The resulting solution was dried over
magnesimn
sulfate, filtered, and concentrated under reduced pressure to yield 72.9 g of
7-
ben~:yloxy-4-chloro-3-nitroquinoline as a light brown solid.
Pant E
Triethylamine (12.8 mL, 92.0 mmol) and 1,2-diamino-2-methylpropane (5.29
mL, 50.6 mmol) were added sequentially to a solution of 7-benzyloxy-4-chloro-3-
nitroquinoline (14.5 g, 46.0 mmol) in dichloromethane (400 mL). The reaction
mixture
was stirred overnight and then concentrated under reduced pressure. The
residue was
partitioned between water (200 mL) and dichloromethane (300 mL). The organic
layer
was washed with brine, dried over sodium sulfate, and then concentrated under
reduced
pressure to provide crude product as a brown solid. The crude product was
passed
through a layer of silica gel (eluting sequentially with chloroform and 96:4
chloroform:methanol) to provide 12.4 g of (2-amino-2-methylpropyl)(7-benzyloxy-
3-
nitroquinolin-4-yl)amine as a yellow solid.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-41-
Part F
Under a nitrogen atmosphere, a solution of (2-amino-2-methylpropyl)(7-
benzyloxy-3-nitroquinolin-4-yl)amine (12.4 g, 33.9 mmol) in dichloromethane
(400
mL) was cooled to 0°C. Triethylamine (9.43 mL, 67.8 mmol) and
methanesulfonic
anhydride (5.90 g, 33.9 mmol) were sequentially added, and the reaction was
stirred at
ambient temperature for two hours. An analysis by HPLC indicated that the
reaction
was incomplete, and additional methanesulfonic anhydride (1.4 g, 8.0 mmol) was
added. The reaction was stirred for an additional 90 minutes, and additional
methanesulfonic anhydride (0.7 g, 4 imnol) was added. The reaction was stirred
for an
additional three hours, and saturated aqueous sodium bicarbonate (200 mL) was
added.
A precipitate began to form in the organic layer, which was separated and
concentrated
under reduced pressure to provide a yellow solid. The solid was triturated
with water
(200 mL) with heating, isolated by filtration, washed with water (3 x 100 mL)
and
diethyl ether (3 x 50 mL), and dried overnight under vacuum to provide 14.8 g
of N
[1,1-dimethyl-2-(3-utro-7-benzyloxyquinolin-4-ylamino)ethyl]methanesulfonamide
as
a yellow powder.
Part CJ
N [1,1-I~imethyl-2-(3-nitro-7-benzyloxyquinolin-4-
yla~nino)ethyl]methanesulfonamide (14..8 g, 33.3 rrnmol) was mixed with
acetonitrile
(300 mL) and added to a Pan flask9 5°/~ platinum on carbon (2 g) was
added. The
reaction was flushed with nitrogen and placed under hydrogen pressure (40
pounds per
square inch (psi), 2.8 x 105 Pascals (Pa)) for 5.5 hours with the hydrogen
replaced after
two hours. An analysis by TLC indicated the presence of starting material.
Additional
acetonitrile (200 mL) and 5% platinum on carbon (2 g) were added, and the
reaction
was placed under hydrogen pressure overnight. The reaction mixture was
filtered
through a layer of CELITE filter aid, and the filter cake was washed with
acetonitrile.
The filtrate was concentrated under reduced pressure. Toluene and
dichloromethane
were added and removed under reduced pressure twice to yield 12.6 g of N [2-(3-
amino-7-benzyloxyquinolin-4-ylamino)-1,1-dimethylethyl]methanesulfonamide as a
solid.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-42-
Part H
Under a nitrogen atmosphere, a solution of N [2-(3-amino-7-benzyloxyquinolin-
4-ylamino)-l,l-dimethylethyl]methanesulfonamide (12.6 g, 30.4 mmol) in
dichloromethane (300 mL) was cooled to approximately 0°C; triethylamine
(4.23 mL,
30.4 mmol) was added. Ethoxy acetyl chloride (3.33 mL, 30.4 mmol) was added
dropwise, and the reaction was stirred at ambient temperature for 1.5 hours.
The
volatiles were removed under reduced pressure, and the residue was dissolved
in
ethanol (300 mL). Triethylamine (13 mL) was added, and the reaction was heated
at
reflux ovenught and allowed to cool to ambient temperature. The volatiles were
removed under reduced pressure. The residue was dissolved in dichloromethane
(300
mL), and the resulting solution was washed with water (2 x 100 mL) and brine,
dried
over sodium sulfate, filtered, and concentrated under reduced pressure to
provide a
brown oil. The oil was purified by column chromatography on silica gel
(eluting with
97.5:2.5 chloroform:methanol) to provide 12.4 g of N [2-(7-benzyloxy-2-
ethoxymethyl-1H imidazo[4,5-c]quinolin-1-yl)-1,1-
dimethylethyl]methanesulfonamide
as a beige solid.
Part I
A solution ofN [2-(7-benzyloxy-2-ethoxymethyl-1H imidazo[4,5-c]quinolin-1-
yl)-1,1-dimethylethyl]methanesulfonamide (9.3~ g, 1~.5 mmol) in ethanol (150
mL)
was added to a Parr vessel containing 10% palladium on carbon (0.~3 g). The
reaction
was placed under hydrogen pressure (50 psi, 3.4 x 105 Pa) over two nights.
Starting
material remained as evidenced by a TLC analysis, and additional 10% palladium
on
carbon (1.02 g) was added. The reaction was continued for an additional eight
hours.
The reaction mixture was filtered through a layer of CELITE filter aid, and
the filter
cake was washed with ethanol and methanol. The filtrate was concentrated under
reduced pressure, and the residue was several times dissolved in toluene and
concentrated under reduced pressure to yield a yellow powder, which was dried
under
high vacuum to provide 7.37 g of N [2-(2-ethoxymethyl-7-hydroxy-1H imidazo[4,5-
c]quinolin-1-yl)-l,l-dimethylethyl]methanesulfonamide as a yellow solid.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-43-
Part J
Under a nitrogen atmosphere, cesium carbonate (9.18 g, 28.2 mmol) was added
in a single portion to a solution of N [2-(2-ethoxymethyl-7-hydroxy-1H
imidazo[4,5-
c]quinolin-1-yl)-1,1-dimethylethyl]methanesulfonamide (7.37 g, 18.8 mmol) in
DMF.
A solution of tef°t-butyl 6-iodohexylcarbamate (6.75 g, 20.6 mmol)
in DMF
(approximately 100 mL) was added. The reaction mixture was heated overnight at
65°C and then concentrated under reduced pressure to provide an orange
oil. The oil
was partitioned between water (300 mL) and dichloromethane (300 mL). The
organic
layer was washed sequentially with water (x 2) and brine, dried over sodium
sulfate,
filtered, and then concentrated under reduced pressure. The residue was
dissolved in
dichloromethane (100 mL), washed sequentially with water (x 10) and brine,
dried over
sodium sulfate, filtered, and then concentrated under reduced pressure to
provide 10.85
g of crude product as a yellow foam. The crude product was purified by column
chromatography on silica gel (eluting sequentially with 95:5 and 92.5:7.5
dichloromethane:methanol) to provide 8.5 g of ter-t-butyl ~6-[2-ethoxymethyl-1-
(2-
methanesulfonylamino-2-methylpropyl)-lI~ imidazo[4,5-c]quinolin-1-
yloxy]hexyl; carbamate as a white solid.
Part K
3-Chloroperoxybenzoic acid (4..23 g of 60%, 14.4 mmol) was added in a single
portion to a solution of test-butyl ~6-[2-ethoxymethyl-1-(2-
methanesulfonylamino-2-
methylpropyl)-1H imidazo[4,5-c]quinolin-1-yloxy]hexyl~carbamate (8.5 g, 14.4
mmol)
in chloroform (200 mL). The reaction mixture was stirred for several hours and
then
washed sequentially with 1°f° sodium carbonate (x 2) and brine.
The organic layer was
dried over sodium sulfate, filtered, and then concentrated under reduced
pressure to
provide 9.20 g of teJ°t-butyl ~6-[2-ethoxymethyl-1-(2-
methanesulfonylamino-2-
methylpropyl)-5-oxido-1H imidazo[4,5-c]quinolin-1-yloxy]hexyl}carbamate as a
orange solid.
Part L
Ammonium hydroxide (20 mL) andp-toluenesulfonyl chloride (2.74 g, 14.4
mmol) were added sequentially with rapid stirnng to a mixture of the material
from
Part K in dichloromethane (150 mL), and the reaction was stirred for two
hours. The
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-44-
organic layer was then washed with saturated aqueous sodium bicarbonate (2 x)
and
brine, dried over sodium sulfate, filtered, and concentrated under reduced
pressure to
provide tent-butyl {6-[4-amino-2-ethoxymethyl-1-(2-methanesulfonylamino-2-
methylpropyl)-1H imidazo[4,5-c]quinolin-1-yloxy]hexyl}carbamate as ared solid.
Part M
A solution of the material from Part L in hydrochloric acid in ethanol (50
'rnL of
4.25 M) was heated to reflux and then allowed to cool to ambient temperature.
The
reaction mixture was purged with nitrogen for approximately 1 hour and then
concentrated under reduced pressure. The residue was dissolved in water and
then
washed with chloroform (x 2). The pH of the aqueous layer was adjusted with
ammonium hydroxide and then the aqueous layer was extracted with chloroform (x
3).
The combined extracts were washed with brine, dried over sodium sulfate,
filtered, and
then concentrated under reduced pressure to provide 6.86 g of N f 2-[4-amino-7-
(6-
aminohexyloxy)-2-ethoxymethyl-1H imidazo[4.,5-c]quinolin-1-yl]-1,1-
dimethylethyl~methanesulfonamide as a tan solid.
Example 1
IRM1 and IRM2, each containing a biotin moiety, were coupled to
ULTIZALI~TI~ immobilized monomeric avidin beads (item number 53146, PIERCE
Biotechnology, 3737 N. Meridian Road, P.~. Box 117, Rockford IL) and
ULTRALIIVI~ immobilized I~TEUTRAV~IN tetrameric avidin beads (item nmnber
53140, PIERCE Biotechnology, Rockford, IL) in the following mariner. An
aliquot
(3.48 ~.L) of a 50 mM IRM stock solution in dimethyl sulfoxide was added to 1
mL of
the bead suspension. The resulting suspension was allowed to incubate at
ambient
temperature for 4 hours. The beads were allowed to settle and a portion (400
~,L) of the
supernatant was removed. The beads were then washed using the following
procedure.
Phosphate buffered saline (1.4 mL) was added, the suspension was mixed by
vortexing,
the beads were allowed to settle, and then supernatant (1.4 mL) was removed.
The
wash procedure was repeated 5 times and then the beads were resuspended to 1
mL to
provide a solids concentration of 50%.
Twenty microliters (20 ~,L) of each of each compound (0.7 ~M IRM in 50%
slurry) was added to 1 ml of human peripheral blood mononuclear cells (HPBMC)
(2 x
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-45-
106 cells/ml) in RPMI complete media and incubated overnight. 1:1 dilution
duplicates
and 1:2 dilution duplicates were assayed for IFNa and TNFa concentrations by
ELISA. Results are shown in Figure 1. PBS represents the PBS buffer control;
BdlMono represents the monomeric avidin bead control (50% slurry); Bd2Tetra
represents the tetrameric avidin bead control (50% slurry); IRM1 represents
0.7 ~,M
unbound IRM1; IRM2 represents 0.7 ~,M unbound IRM2'; IRMIMono represents IRM1
bound on Bd1 (0.7 ~,M IRM in 50% slurry); IRM2Mono represents IRM2 bound on
Bdl (0.7 ~.M IRM in 50% slurry); IRMITetra represents IRM1 bound on Bd2 (0.7
pM
IRM in 50% slurry); and IRM2Tetra represents lRM2 bound on Bd2 (0.7 ~,M IRM in
50% slurry).
The values shown correspond to the average concentration from the
quadruplicate experiments (dilution factor was used to adjust). Standard
deviations are
shown.
Example 2
Preparation of IRM Grafted Nanopai-ticles
A dispersion of SiO2 particles (0.4.9 g of 2327, 20 nanometers (mn) ammonium
stabilized colloidal silica sol, 41 % solids; Nalco, Naperville, IL) was
placed in a 5 mL
vial. The dispersion was diluted with 0.2 g of deionized water and 0.5 g of
DMSO. To
the stirred dispersion was added 33 mg of N [3-(4-amino-2-ethoxymethyl-6,7-
dimethyl-lII imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-
(triethoxysilyl)propyl]urea in 2
g of DMSO. After the addition, the dispersion was placed in an ultrasonic bath
at 40°C
for 2 hours. The vial was then capped and placed in an oven at ~0°C for
24 hours. The
resulting dispersion was analyzed by liquid chromatography. The spectrum
showed a
broad peak with different retention time compared to that of the starting IRM
silane.
The dispersion was centrifuged to remove the solvents.
Example 3
Preparation of IRM Grafted Nanoparticles
A dispersion of Si02 particles (0.49 g of 2327, 20 mn ammonium stabilized
colloidal silica sol, 41 % solids; Nalco, Naperville, IL) was placed in a 5 mL
vial. The
dispersion was diluted with 0.2 g of deionized water and 0.5 g of DMSO. To the
stirred
dispersion was added 33 mg ofN [3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-46-
imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea in 2 g of
DMSO.
After the addition, the dispersion was placed in an ultrasonic bath at
40°C for 2 hours.
The vial was then capped and placed in an oven at 80°C for 24 hours.
The vial was
cooled to room temperature and to the vial was added PEG triethoxysilane (12.4
mg,
0.0248 mmol available from GELEST, INC., Morrisville, PA). After the addition,
the
vial was capped and placed in an ultrasonic bath for 2 hours. The vial was
then placed
in an oven at 80°C for 24 hours. The dispersion was then centrifuged to
remove the
solvents.
Example 4
Preparation of IRM Grafted Nanoparticles
The procedure of Example 3 was repeated except that the amount of N [3-(4-
amino-2-ethoxyrnethyl-6,7-dimethyl-1F1 imidazo[4,5-c]pyridin-1-yl)propyl]-N'-
[3-
(triethoxysilyl)propyl]urea was reduced from 33 mg to 17 mg.
Example 5
Preparation of IIZM Grafted Nanoparticles
The procedure of Example 3 was repeated except that the amount of N [3-(4-
amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-
(triethoxysilyl)propyl]urea was reduced from 33 mg to 8.5 mg.
Example 6
Preparation of II~M Grafted Nanoparticles
The procedure of Example 3 was repeated except that the amount of PEG
triethoxysilane was increased from 12.4 mg to 31.0 mg.
Example 7
Preparation of IRM Grafted Nanoparticles
The procedure of Example 2 was repeated except that 34 mg of N [4-(4-amino-
2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-N'-[3-
(triethoxysilyl)propyl]urea was
used in lieu ofN [3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-
c]pyridin-
1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-47-
Example 8
Preparation of IRM Grafted Nanoparticles
The procedure of Example 3 was repeated except that 34 mg of N [4-(4-amino-
2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-N'-[3-
(triethoxysilyl)propyl]urea was
used in lieu ofN [3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-
c]pyridin-
1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea.
Example 9
Preparation of IRM Grafted Nanoparticles
The procedure of Example 8 was repeated except that the amount of N [4-(4-
amino-2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-N'-[3-
(triethoxysilyl)propyl]urea was reduced from 34 mg to 17 mg.
Example 10
Preparation of II~~I Grafted Nanoparticles
The procedure of Example 8 was repeated except that the amount of 1!T [4-(4-
amino-2-propyl-lII imidazo[4,5-c]quinolin-1-yl)butyl]-N'-[3-
(triethoxysilyl)propyl]urea was reduced from 34 mg to 8.5 mg.
Example 11
Preparation of I12Ie4 Grafted Nanoparticles
The procedure of Example 8 was repeated except that the amount of PEG
triethoxysilane was increased from 12.4 mg to 31.0 mg.
Example 12
Preparation of an IRM Grafted Fluoropolymer Film
Glass microscope slides (5.1 cm by 7.6 cm) were cleaned with acetone and
distilled water. Qne surface of the glass substrate was coated with a THF
solution
containing N [3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-c]pyridin-
1-
yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea at 1 percent by weight (wt-%),
and a piece
of fluoropolymer Teflon FEP film, a copolymer of tetrafluoroethylene and
hexafluoropropylene, available from E.I DuPont de Nemours and Company, (5.1 cm
by
'7.6 cm at 1 mil thickness) was subsequently laminated onto the coated
substrate in a
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-48-
manner that assured good surface contact. The laminated sample was placed
under a
photoreactor consisting of 6 germicidal lamps (G15T8 bulb, 15W, available from
General Electric Company, Nela Park, Cleveland, OH). The laminated sample was
placed 7.6 -10 cm away from the bulbs with fluoropolymer film facing the UV
lamps.
The sample was subjected to UV irradiation for 10 min. After irradiation, the
treated
fluoropolymer film was removed and immersed in THF for 2 hours. The film was
removed from the THF and subjected to further washing with THF. The film was
dried
under a stream of N2 gas. The grafted film was sampled and analyzed by ESCA:
Surface Composition:
Controlled sample FEP F C O N
68 32
Treated sample FEP 28 58 6 9
Example 13
Preparation of an IRM Grafted Fluoropolymer Film
Glass microscope slides (5.1 cm by 7.6 cm) were cleaned with acetone and
distilled water. One surface of the glass substrate was coated with a methanol
solution
containing 10 wt-% of 3-aminopropyl triethoxysilane and 5 wt-% of n-phenyl
propyl
triethoxy silane (both are available form GELEST, INC. 11 Steel Rd. East
Morrisville,
PA), and a piece of fluoropolymer Teflon FEP film a copolymer of
tetrafluoroethylene
and hexafluoropropylene, available from E.I DuPont de Nemours and Company,
(5.1
cm by 7.6 cm at 1 mil thickness) was subsequently laminated onto the coated
substrate
in a manner that assured good surface contact. The laminated sample was placed
under
a photoreactor consisting of 6 G15T8 bulbs. The laminated sample was placed
7.6 - 10
cm away from the bulbs with fluoropolymer film facing the UV lamps. The sample
was subj ected to UV irradiation for 3 min. After irradiation, the treated
fluoropolymer
film was removed and immersed in methanol for 2 hours to remove any residual
primers. The film was removed from the methanol and subjected to further
washing
with methanol. The resulting triethoxyoxysilane grafted FEP film was then
coated with
a solution ofN [4-(4-amino-2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-1V'-
[3-
(triethoxysilyl)propyl]urea in DMSO (1 wt-%) and subsequently subjected to
heat
treatment in an oven at 50 °C overnight. The grafted fluoropolymer film
was then
thoroughly washed with THF and methanol.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-49-
Example 14
Preparation of an IRM Self Assembled Monolayer
A glass microscope slide was cleaned with Hz02/concentrated sulfuric acid in a
1:3 mixture and subsequently washed with distilled water. The cleaned glass
slide was
immersed in a solution of N [3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H
imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-(triethoxysilyl)propyl]urea in DMSO
(1 wt-
%) for 30 min. The slide was removed and heated at 80 °C for 30 min.
Finally the
slide was rinsed with methanol to remove the excess N [3-(4-amino-2-
ethoxymethyl-
6,7-dimethyl-1H imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-
(triethoxysilyl)propyl]urea
solution.
Example 15
IRMs were covalently coupled to gold particles to form nanometer-sized IRM-
gold conjugates through a two-step reaction: the gold surface was
functionalized with
carbonate by reacting with thiol carbonate; the carbonate functional group was
then
coupled to the primary amine group of an IRM catalyzed by a carbodiimide.
Briefly, ten micro-liters of 100 mM solution of mercaptoacetic acid (catalog
no.
10,900-2, Aldrich, Milwaukee, WI) were added to one mL of colloidal gold
particles
solution (approximately 10 nanomolar (nM), catalog no.: 154015, average size =
40
nm, from ICN Biomedicals Inc., Aurora, OH). Under a nitrogen atmosphere, the
mixture was shaken at 3 Hz for 3 hours (hr) at room temperature.
Twenty micro-liters of 10 mg/L PBS buffer (pH 7.~) solution of an
imidazoquinoline IRM compound (4-amino-2-ethoxymethyl-1H imidazo[4,5-
c]quinoline-1-ethanamine, disclosed in U.S. Pat. No. 6,069,149), ~0
microliters of
freshly made 50 milligrams per liter (mg/L) PBS buffer solution of 1-ethyl-3-
(3-
dimethylaminopropyl carbodiimide) (EDC, HCl salt, Pierce, Rockford, IL), and
one
drop of approximately 1N HCI, were then added to the mixture. The final
mixture was
shaken at 3 Hz at room temperature for another 12 hours (hr) followed by
centrifugation at 14,000 revolutions per minute (rpm) for 30 minutes (min).
After
removing the supernatant, the precipitant was washed with 0.5 mL of PBS buffer
twice
before being redispersed in 1 millilter (mL) of PBS. A Field Emission SEM
micrograph showed that, the modified particles were well separated and
distributed.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-50-
The infrared spectrum showed that there was a significant increase at the -NH-
signal,
indicating the successful coupling of IRM to the colloidal gold.
Example 16
Similarly, a gold conjugate was also made with 10 nm colloidal gold (catalog
number: 154011, ICN Biomedicals).
Example 17
IRM-gold particles was also made from avidin-biotin or anti biotin-biotin
coupling: reacting the commercially available gold-strepavidin (Amersham
Biosciences, Nanoprobes, Inc. Stoney Brook, NY) or anti-biotin Nanogold Fab'
conjugate (Nanoprobes, Inc. Stoney Brook, N~ with the biotin complex of
Example
29 of U.S. Pat. No. 6,451,810, which is comparable to the uncomplexed IRM in
stimulating TNF release, but superior in IL-6 stimulation.
Example 18
An IRI~I conjugate ~f ferritin, a metaloprotein containing 4.000 to 5000 Fe3+
ions, was synthesized through direct coupling between the carboxyl group of
[(4-
amino-1-isobutyl-1H imidazo[4,5-c]quinolin-2-yl)methoxy]acetic acid and the
primary
amine of ferritin catalyzed by 1-ethyl-3-(3-dimethylaminopropyl) carbodiunide
(EDC).
Five milliliters of a solution of [(4-amino-1-isobutyl-1FI imidazo[4,5-
c]quinolin-2-yl)methoxy]acetic acid in pH 7.4 PBS buffer (0.4 g/L) was added
to a
mixture of 3 ml of 50 g/L of ferritin in pH 7.4 PBS buffer solution from ICN
Biochemedicals Inc., Aurora, ~H, 2 mL of 20 mM EDC, and 10 drops of 1N HCl.
After a 5-second vortex mixing, the mixture was allowed to react overnight.
The
mixture was then eluted through a size-exclusion liquid chromatography (PD-10)
column. The brown-colored fraction was collected. The average ratio of [(4-
amino-1-
isobutyl-1H imidazo[4,5-c]quinolin-2-yl)methoxy]acetic acid to ferritin in the
conjugate was determined to be 0.6 (M/M~, based on the UV spectrum measurement
of
[(4-amino-1-isobutyl-1H imidaz[4,5-c]quinolin-2-yl)methoxy]acetic acid in the
initial
solution and the eluted solution. The recovery rate of ferritin was 95% after
passing
through the column. The eluted fraction was verified by HPLC, which showed a
single
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-51-
peak. No significant lost of iron was observed during the modification. The
conjugate
showed biological activities in a test with RAW cells.
Example 19
An IRM was covalently immobilized onto functionalized superparamagnetic
particles using a modified protocol based on the manufacturer's suggested
protocol.
Briefly, one hundred milligrams of freeze-dried DYNABEADS M-270 Epoxy (from
Dynal Biotect, Lake Success, NY, containing approximately 6.7 x 109 beads) was
suspended in 7 mL of de-ionized water. After being vortexed for 30 seconds and
incubated for 10 minutes, the mixture was centrifuged at 3000 Gravity (G) for
10
minutes (min) and the supernatant was discarded.
Three milliliters of a freshly prepared solution (0.4 g/L) of 1-(4-aminobutyl)-
2-
butyl-1H imidazo[4,5-a]quinolin-4-amine (which can be prepared using the
methods
disclosed in U.S. Fat. No. 6,069,149) in carbonate-bicarbonate buffer (0.1 M,
pH 9.4)
and 5 mL of 4 M ammonium sulfate in de-ionized water were added to the beads.
The
mixture was vortexed for 30 seconds and then placed on a shaker operating at
10 Hz at
room temperature for 24 hours.
The mixture was centrifuged at 30006 for 10 min. The supernatant was
removed and the IRM concentration was determined by UV absorption at 247 nm.
The
beads were washed with 7 mL of methanol 3 times and 7 ml of Dulbecco's FBS 3
times. The II~M content in the modified beads was calculated by subtracting
the
amount of IRM found in the supernatant and washes from the amount of IL~M that
was
initially combined with the beads.
Example 20
An IRM was covalently immobilized onto nanosized superparamagnetic
particles using the following procedure.
A portion (0.1 mL) of water-based ferrofluid (EMG 304, Nashua, NH), a water
based dispersion of iron oxide particles with dimensions in the range of 5-15
mn, was
diluted with 4 mL de-ionized water and 20 mL 2-propanol. Under continuous
mechanical stirring, 0.3 mL ammonia (30 wt-%, Aldrich) and 8.5 mg of N [3-(4-
amino-
2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-c]pyridin-1-yl)propyl]-N'-[3-
(triethoxysilyl)propyl]urea was slowly added to the dispersion. The reaction
was
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-52-
allowed to proceed at room temperature for 4 hours under continuous stirnng.
After
the reaction was complete, the IRM-attached magnetic particles were
concentrated
using a magnet.
Example 21
An IRM was covalently attached to core shell superparamagnetic particles using
the following procedure. A portion (1 mL) of water-based silica coated
superparamagnetic particles (50 mg, SiMAG-1, Chemicell Gmbh, Berlin, Germany)
a
water based dispersion of core shell magnetic particles with dimensions in the
range of
100 nm, was diluted with SmL de-ionized water and 15 mL 2-propanol. Under
continuous mechanical stirring, 0.3 mL ammonia (30 wt-%, Aldrich) and 8.5 mg
of N
[3-(4-amino-2-ethoxymethyl-6,7-dimethyl-1H imidazo[4,5-c]pyridin-1-yl)propyl]-
N'-
[3-(triethoxysilyl)propyl]urea was slowly added to the dispersion. The
reaction was
allowed to proceed at room temperature for 4 hours under continuous stirring.
After
the reaction was complete, the II~M-attached magnetic particles were
concentrated
using a magnet.
Example 22
An IRM can be covalently attached to a bioadhieve polymer as follows::
I»Ms can be covalently attached to bioadhesive crosslinked polyners of acrylic
acid through amide or ester formation. An II~M containing a pendant amine or
hydroxyl group is reacted with a free carboxylic acid group on the polymer to
form an
amide or an ester respectively. IRII~I compounds containing pendant amine or
hydroxyl
groups and methods of making them are known. See, for example, U.S. Pat. Nos.
4,689,338; 5,389,640; 5,268,376; 6,451,810; 6,677,349; 6,660,747; 5,352,784;
5,446,153; 6,545,016; 6,194,425; 4,988,815; 5,175,296; 5,395,937; 5,741,908;
and
5,693,81 l; U.S. Patent Publication Nos. 2004/0010007 and 2003/0232852; and
U.S.
Patent Applications Serial No. 10/739787 filed December 18, 2003. Bioadhesive
crosslinked polymers of acrylic acid are commercially available; for example,
CARBOPOL 971P and CARBOPOL 974P, both from Noveon, Inc, Cleveland, OH.
This IRM-bioadhesive polymer complex and other suitable complexes,
designated as (IRM)X(Polymer)y, can be incorporated into a gel, cream, or
solution
formulation. Such formulations can include solvents, oils, emulsifiers, etc.
Examples
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-53-
of suitable formulations are disclosed inU.S. Pat. No. 6,245,776 and U.S.
Patent
Publication No. 2003/0199538.
General examples of formulations of such IRM-bioadhesive polymer complexes
are as follows:
General Example of Solution and Gel Formulations
Component Ingredient Range (wt-%)
(IRM)X(Polymer)y 0.01-8.0
Solvent 0-30
Preservative 0.01-1.0
Water Qs to 100
General Example of Gel Formulations
Component Ingredient Range (wt-%)
(IRI~4)x(POlynler)y0.01-8.0
Solvent 0-30
Preservatives 0.01-1.0
Na~H (20%) Adjust to desired pH
Water Qs to 100
General Examt~le of Cream Formulations
Component Ingredient Range (wt-%)
(IRM)X(Polymer)y 0.01-8.0
~il 1-30
Preservatives 0.01-1.0
Emulsifiers 0.05-5 0
Water Qs to 100
Specific examples of formulations of such IRM-bioadhesive polymer
complexes are as follows:
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-54-
Specific Example of Solution Formulation
Component Function Ingredient Range
(wt-%)
(IRM)X(Polymer)yActive 2.0
Propylene GlycolSolvent 5.0
Methylparaben Preservative0.15
Propylparaben Preservative0.03
Water Diluent 92.82
Specific Example of Gel Formulation
Component Function Ingredient Range
(wt-%)
(IRM)X(Polymer)yActive 0.5
Hexylene GlycolSolvent 15.0
Methylparaben Preservative0.15
Propylparaben Preservative0.03
NaOH (20%) Neutralizer Adjust to pH 4.5
Water Diluent 92.82
~ecific Example of Cream Formulation
Component Function hlgradient Range
(wt-/~)
(IRM)X(Folymer)yActive 5.0
Isostearic AcidOil 25.0
POLOXAMER 188 Emulsifier 1.0
Sorbitan TrioleateEmulsifier 1.0
Methylparaben Preservative0.20
Propylparaben Preservative0.10
NaOH (20%) Neutralizer Adjust to pH 6
Water Diluent Qs to 100
Example 23
An IRM was covalently immobilized in a hydrogel using the following
procedure.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-55-
Preparation of Component A
The pH of a solution (2 mL) of 30% (w/v) human serum albumin (HSA) in 0.1
M pH 9.4 carbonate/bicarbonate buffer was adjusted to approximately 4 by the
dropwise addition of hydrochloric acid (1 mL of 1 normal (I~ HCl). A solution
of N
~2-[4-amino-7-(6-aminohexyloxy)-2-ethoxymethyl-1H imidazo[4,5-c]quinolin-1-yl]-
1,1-dimethylethyl~methanesulfonamide in phosphate buffered saline (3.0 mL of
0.6
mg/mL) and a f~resl~ly-made solution of EDC in phosphate buffered saline (0.2
mL of 3
mg/mL) were added and the mixture was incubated overnight.
Under 2 hertz (Hz) agitation the mixture was dialyzed through a 3,500
molecular weight cut-off membrane (Spectrum Laboratory Inc., Rancho
Domingguez,
CA) at 4°C against 0.1 M carbonate/bicarbonate buffer (pH 9.4) for 120
hours, with
fresh buffer every day. The concentration of the IRM in the dialyzed buffer
was
monitored by UV absorption. No detectable IRM (less than 0.1 ,ug/mL) was found
in
the final buffer. The resulting mixture was concentrated to 15 wt-% HSA by
ultra-
filtration through a 50,000 molecular weight cut-off cellulose ester disc
membrane
(Spectrum Laboratory Inc.).
Preparation of Component E
Polyethylene glycol disuccinimidyl succinate (PEG-SS2, prepared according to
the method of Example 1 in U.S. Pat. No. 5,53,114) was dissolved in sterile
water at
300 mghnL.
Preparation of hydrogel
Component A (1 mL) and Component B (0.5 mL) were combined. A
transparent hydrogel formed in about 10 seconds.
Example 24
Preparation of IRM Grafted Collagen
Collagen fibers (available from Caliochem) were combined with a solution of
N [4-(4-amino-2-propyl-1H imidazo[4,5-c]quinolin-1-yl)butyl]-N'-[3-
(triethoxysilyl)propyl]urea in tetrahydrofuran (0.5-1 wt-%). The mixture was
placed in
an ultrasonic bath at 45°C for 24 hours. The collagen fibers were
removed from the
solution and thoroughly washed with tetrahydrofuran. A sample was analyzed by
Time
of Flight Secondary Ion Mass Spectroscopy (TOF SIMS). The spectra showed the
existence of silicon and of ion fragments of (1-aminobutyl)-2-propyl-1H
imidazo[4,5-
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-56-
c]quinolin-4-amine. Analysis of an untreated collagen fiber did not show
either silicon
or ion fragments of (1-aminobutyl)-2-propyl-1H imidazo[4,5-c]quinolin-4-amine.
Example 25
Preparation of IRM Grafted Collagen
Collagen fibers (available from Caliochem) were combined with a solution of 1-
aminobutyl)-2-propyl-1H imidazo[4,5-c]quinolin-4-amine in tetrahydrofuran (5 g
of 1
wt-%). 1,3-Dicyclohexylcarbodiimide (50 mg) was added and the mixture was
placed
in a in an ultrasonic bath at 45°C for 24 hours. The collagen fibers
were removed from
the solution and thoroughly washed with tetrahydrofuran. A sample was analyzed
by
TOF SIMS. The spectra showed the existence of ion fragments of (1-aminobutyl)-
2-
propyl-1H imidazo[4,5-c]quinolin-4-amine. Analysis of an untreated collagen
fiber did
not show ion fragments of (1-aminobutyl)-2-propyl-1H imidazo[4,5-c]quinolin-4-
amine.
Example 26
Preparation of II~M Grafted Polyethylene terephthlate) Film
Pieces of polyethylene terephthlate) ftlm (available from 3M Company; St.
Paul, MN) were combined with a solution of 1-(4-aminobutyl)-2-propyl-1H
imidazo[4.,5-c]quinolin-4-amine in tetrahydrofuran (2 g of 2.5 wt-°/~).
The mixture was
placed in a in an ultrasonic bath at 45°C for 24. hours. The pieces of
film were removed
from the solution and thoroughly washed with tetrahydroft~ran. Electron
Spectroscopy
of Chemical Analysis (ESCA), 52° with respect to the sample surface, of
a sample
showed the existence of nitrogen at 6-7% of the total composition. Analysis of
an
untreated PET film did not show nitrogen.
Example 27
Preparation of IRM Grafted Acrylate Beads
Oxirane functionalized acrylic beads (160 mg, average 150 ~m in diameter,
from Sigma Chemical, Cat. No. O-76280) were suspended in PBS (2 mL) and
incubated for 30 minutes. A solution of 1-(4-aminobutyl)-2-butyl-1H
imidazo[4,5-
c]quinolin-4-amine in PBS (3 mL of 0.4 g/L) was added. The pH of the mixture
was
brought up to 9 by the addition of 1N sodium hydroxide. The reaction mixture
was
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-57-
shaken (3 Hz) at room temperature for 72 hours. The reaction mixture was then
centrifuged at 3000 G for S min. The concentration of the ZRM in the
supernatant was
determined by UV-Vis absorption at 247 nanometers (nm). The raw coupling ratio
was
determined to be 96.5%. After discarding the supernatant, the beads were
washed with
6 mL of PBS and centrifuged at 3000 G for 2 min. This procedure was repeated
three
times. The beads were further washed 3 times with methanol, followed by 2
additional
washes with 6 mL of Dulbecco's PBS (DPBS). No detectable IRM (less than 0.1
p,g/mL) was found in the final PBS wash. The amount of immobilized IRM was
calculated by subtracting the amount found in the washes from the amount that
was
added to the reaction mixture.
Example 2~
Preparation of IRM Grafted Acrylate Beads
'The procedure of Example 27 was repeated except that 3 mL of a 0.8 g/mL
solution of Il~I was used in lieu of 3 mL of a 0.4 g/mL solution of Il~.
Example 29
Preparation of I12I~ Grafted Acrylate Beads
The procedure of Example 27 was repeated. The beads were further treated
with propylamine (7.7 mg) and then washed fire times with DPBS.
Example 30
Preparation of IRM Grafted Polystyrene Beads
Carboxylate functionalized polystyrene beads (2 mL of P~LYBEAD
Carboxylate solution, 2.6% beads, average 10 pm in diameter, from Polysciences
W c,
Warnngton, PA) were washed and then resuspended in 2 mL of PBS buffer (pH
7.4).
A solution of 1-(4-aminobutyl)-2-butyl-1H imidazo[4,5-c]quinolin-4-amine in
PBS (3
mL of 0.4 g/L) was added and the pH of the mixture was adjusted to pH 4.5 by
addition
of 1N hydrochloric acid. A freshly prepared solution of EDC in deionized water
(200
p.L of 0.3 mg/mL) was added and the mixture was incubated overnight on a
shaker
operating at 3 Hz. The mixture was then centrifuged at 3000 G for 15 min. The
concentration of the IRM in the supernatant was determined by IJV-Vis
absorption at
247 nm. After discarding the supernatant, the beads were washed with 6 mL of
PBS
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-5 8-
and centrifuged at 3000 G for 2 min. This procedure was repeated three times.
The
beads were further washed 3 times with methanol, followed by 2 additional
washes
with 6 mL of Dulbecco's PBS (DPBS). No detectable IRM (less than 0.1 ~,g/mL)
was
found in the final PBS wash. The amount of immobilized IRM was calculated by
subtracting the amount found in the washes from the amount that was added to
the
reaction mixture.
Example 31
Preparation of IRM Grafted Polystyrene Beads
The procedure of Example 30 was repeated except that 3 mL of a 0.8 g/mL
solution of IRM was used in lieu of 3 mL of a 0.4 g/mL solution of IRM.
Example 32
Preparation of IRM Grafted Polystyrene Beads
The procedure of Example 30 was repeated using N {2-[4-amino-7-(6-
aminohexyloxy)-2-ethoxymethyl-11T imidazo[4,5-c]quinolin-1-yl]-1,1-
dimethylethyl}methanesulfonamide in lieu of 1-(4-aminobutyl)-2-butyl-11I
imidazo [4, 5-c] quinolin-4-amine.
Test Data
The beads prepared in Examples 27-31 were tested for their ability to induce
cytolcines in the following manner. Twenty microliters (20 ~,L) of a slurry of
the beads
(80 mg beads/mL PBS) was added to 250 ~L of human peripheral blood mononuclear
cells (5 x 105 cells) in RPMI complete media and incubated overnight. 1:1
dilution
duplicates were assayed for IFNcx and TNFcx concentrations by ELISA. The
results are
shown in the table below where IFN and TNF are reported in picograms/mL and sd
is
the standard deviation. Acrylate C1 beads are beads that were incubated with
PBS
alone. Acrylate C2 beads are beads in which the oxirane functionality was
partially
quenched by incubation with PBS containing 0.396 mg of propylamine. Acrylate
C3
beads are beads in which the oxirane functionality was fully quenched by
incubation
with PBS containing 7.7 mg of propylamine. Polystyrene control beads are beads
that
were incubated with PBS alone.
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-59-
IFNa IFNaAve TNFa TNFa Ave
(1) (2) IFNa sd (1) (2) TNFa sd
Acrylate Beads
High
Dose IRM (Ex 1006 10571031.125.61481.8 848.91165.3316.4
28)
Acrylate Beads
Low
Dose IRM (Ex 1086 11111098.212.3708.3 544.6626.581.9
27)
Acrylate Cl
Beads
PBS Control 0 0 0.0 0.0 8.8 8.1 8.5 0.3
Acrylate C2
Beads
Partially Quenched0 8 4.2 4.2 346.8 278.9. 34.0
312.8
Acrylate C3
Beads
Fully Quenched 0 11 5.4 5.4 102.5 53.3 77.9 24.6
Acrylate Beads
Low
Dose IRM/Quenched
(Ex 29) 1259 12331245.912.9512.1 436.9474.537.6
Polystyrene
Beads
High Dose II~M
(Ex
31) 0 9 4.7 4.7 55.8 61.1 58.4 2.7
Polystyrene
Beads
Low Dose IRM
(Ex 30) 9 0 4.7 4.7 55.5 45.9 50.7 4.8
Polystyrene
Control
Beads 41 0 20.7 20.738.1 31.1 34.6 3.5
The beads prepared in Example 19 were tested for their ability to induce
cytol~ines using the method described above for the beads of Examples 27-31.
The
results are shown in the table below where IFN and TNF are repouted in
picograms/mL
and sd is the standard deviation. Control D~'NABEADS are beads that were
treated
with buffer alone.
IFNa IFNaAve TNFa TNFa Ave
(1) (2) IFNa Sd (1) (2) TNFa sd
IRM on
DYNABEADS
(Ex. 19) 1148.7888.61018.7130.0 33.2 45.8 39.5 6.3
Control
DYNABEADS 5.7 1.4 3.5 2.1 26.1 17.2 21.7 4.5
The particles of Examples 2-1 l, 15, and 16 were tested in a single experiment
using the method described above for the beads of Examples 27-31 and did not
induce
CA 02521529 2005-10-05
WO 2004/091500 PCT/US2004/011062
-60-
significant amounts of either interferon alpha or tumor necrosis factor alpha
at the
concentrations tested.
The complete disclosures of the patents, patent documents and publications
cited herein are incorporated by reference in their entirety as if each were
individually
incorporated. In case of conflict, the present specification, including
definitions, shall
control. Various modifications and alterations to this invention will become
apparent
to those skilled in the art without departing from the scope and spirit of
this invention.
Illustrative embodiments and examples are provided as examples only and are
not
intended to limit the scope of the present invention. The scope of the
invention is
limited only by the claims set forth as follows.