Note: Descriptions are shown in the official language in which they were submitted.
CA 02524900 2011-04-04
25771-1846
6-Cyclylmethyl- and 6-alkylmethyl-substituted pyrazolopyrimidines
The invention relates to novel 6-cyclylmethyl- and 6-alkylmethyl-substituted
pyrazolepyrimidines,
process for their preparation and their use for producing medicaments for
improving perception,
concentration, learning and/or memory.
Inhibition of phosphodiesterases modulates the levels of the cyclic
nucleotides 5'-3' cyclic
adenosine monophosphate (CAMP) and 5'-3' cyclic guanosine monophosphate
(cGMP). These
cyclic nucleotides (cAMP and cGMP) are important second messengers and
therefore play a
central role in cellular signal transduction cascades. Each of them
reactivates inter alia, but not
exclusively, protein kinases. The protein kinase activated by cAMP is called
protein kinase A
(PKA), and the protein kinase activated by cGMP is called protein kinase G
(PKG). Activated
PKA and PKG are able in turn to phosphorylate a number of cellular effector
proteins (e.g. ion
channels, G-protein-coupled receptors, structural proteins). It is possible in
this way for the second
messengers cAMP and cGMP to control a wide variety of physiological processes
in a wide
variety of organs. However, the cyclic nucleotides are also able to act
directly on effector
molecules. Thus, it is known, for example, that cGMP is able to act directly
on ion channels and
thus is able to influence the cellular ion concentration (review in: Wei et
al., Prog. Neurobiol.,
1998, 56: 37-64). The phosphodiesterases (PDE) are a control mechanism for
controlling the
activity of cAMP and cGMP and thus in turn these physiological processes. PDEs
hydrolyse the
cyclic monophosphates to the inactive monophosphates AMP and GMP. At least 21
PDE genes
have now been described (Exp. Opin. Investig. Drugs 2000, 9, 1354-3784). These
21 PDE genes
can be divided on the basis of their sequence homology into 11 PDE families
(for proposed
nomenclature, see http://depts.washington.edu/pde/Nomenclature.html.).
Individual PDE genes
within a family are differentiated by letters (e.g. PDEIA and PDE1B). If
different splice variants
within a gene also occur, this is then indicated by an additional numbering
after the letters (e.g.
PDEIA1).
Human PDE9A was cloned and sequenced in 1998. The amino acid identity with
other PDEs does
not exceed 34% (PDE8A) and is never less than 28% (PDE5A). With a Michaelis-
Menten constant
(Km) of 170 nM, PDE9A has high affinity for cGMP. In addition, PDE9A is
selective for cGMP
(Km for cAMP = 230 M). PDE9A has no cGMP binding domain, suggesting
allosteric enzyme
regulation by cGMP. It was shown in a Western blot analysis that PDE9A is
expressed in humans
inter alia in testes, brain, small intestine, skeletal muscle, heart, lung,
thymus and spleen. The
highest expression was found in the brain, small intestine, heart and spleen
(Fisher et al., J. Biol.
Chem., 1998, 273 (25): 15559-15564). The gene for human PDE9A is located on
chromosome
21g22.3 and comprises 21 exons. To date, 4 alternative splice variants of
PDE9A have been
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-2-
identified (Guipponi et al., Hum. Genet., 1998, 103: 386-392). Classical PDE
inhibitors do not
inhibit human PDE9A. Thus, IBMX, dipyridamole, SKF94120, rolipram and
vinpocetine show no
inhibition on the isolated enzyme in concentrations of up to 100 M. An IC50
of 35 .iM has been
demonstrated for zaprinast (Fisher et al., J. Biol. Chem., 1998, 273 (25):
15559-15564).
Murine PDE9A was cloned and sequenced in 1998 by Soderling et al. (J. Biol.
Chem., 1998, 273
(19): 15553-15558). This has, like the human form, high affinity for cGMP with
a Km of 70 nM.
Particularly high expression was found in the mouse kidney, brain, lung and
heart. Murine PDE9A
is not inhibited by IBMX in concentrations below 200 M either; the IC50 for
zaprinast is 29 M
(Soderling et al., J. Biol. Chem., 1998, 273 (19): 15553-15558). It has been
found that PDE9A is
strongly expressed in some regions of the rat brain. These include olfactory
bulb, hippocampus,
cortex, basal ganglia and basal forebrain (Andreeva et al., J. Neurosci.,
2001, 21 (22): 9068-9076).
The hippocampus, cortex and basal forebrain in particular play an important
role in learning and
memory processes.
As already mentioned above, PDE9A is distinguished by having particularly high
affinity for
cGMP. PDE9A is therefore active even at low physiological concentrations, in
contrast to PDE2A
(Km = 10 M; Martins et al., J. Biol. Chem., 1982, 257: 1973-1979), PDE5A (Km
= 4 M; Francis
et al., J. Biol. Chem., 1980, 255: 620-626), PDE6A (Km = 17 M; Gillespie and
Beavo, J. Biol.
Chem., 1988, 263 (17): 8133-8141) and PDE11A (Km = 0.52 M; Fawcett et al.,
Proc. Nat. Acad.
Sci., 2000, 97 (7): 3702-3707). In contrast to PDE2A (Murashima et al.,
Biochemistry, 1990, 29:
5285-5292), the catalytic activity of PDE9A is not increased by cGMP because
it has no GAF
domain (cGMP-binding domain via which the PDE activity is allosterically
increased) (Beavo et
al., Current Opinion in Cell Biology, 2000, 12: 174-179). PDE9A inhibitors may
therefore lead to
an increase in the baseline cGMP concentration.
WO 98/40384 discloses pyrazoleopyrimidines which are PDE1, 2 and 5 inhibitors
and can be
employed for the treatment of cardiovascular and cerebrovascular disorders and
disorders of the
urogenital system.
CH 396 924, CH 396 925, CH 396 926, CH 396 927, DE 1 147 234, DE 1 149 013, GB
937,726
describe pyrazoleopyrimidines which have a coronary-dilating effect and which
can be employed
for the treatment of disturbances of myocardial blood flow.
US 3,732,225 describes pyrazoleopyrimidines which have an antiinflammatory and
blood glucose-
lowering effect.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-3-
DE 2 408 906 describes styrylpyrazoleopyrimidines which can be employed as
antimicrobial and
antiinflammatory agents for the treatment of, for example, oedema.
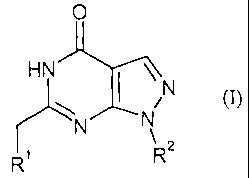
The present invention relates to compounds of the formula
0
HN \N
(I),
N N
R R2
in which
R' is C,-C8-alkyl, C2-C6-alkenyl, C2-C6-alkynyl or C3-C8-cycloalkyl,
where C,-C8-alkyl is optionally substituted by oxo, and
where C,-C8-alkyl, C2-C6-alkenyl, C2-C6-alkynyl and C3-C8-cycloalkyl are
optionally
substituted by up to 3 radicals independently of one another selected from the
group of
C,-C6-alkyl, C,-C6-alkoxy, hydroxycarbonyl, cyano, amino, nitro, hydroxy, C,-
C6-
alkylamino, halogen, trifluoromethyl, trifluoromethoxy, C6-C,o-
arylcarbonylamino, C,-C6-
alkylcarbonylamino, C,-C6-alkylaminocarbonyl, C,-C6-alkoxycarbonyl, C6-C10-
arylamino-
carbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, C,-C6-
alkylsulphonylamino,
C,-C6-alkylsulphonyl, C,-C6-alkylthio,
where
C,-C6-alkyl, C,-C6-alkoxy, C,-C6-alkylamino, C6-C,o-arylcarbonylamino, C,-C6-
alkyl-
carbonylamino, C,-C6-alkylaminocarbonyl, C,-C6-alkoxycarbonyl, C6-C10-aryl-
aminocarbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, C,-C6-
alkylsulphonylamino, C,-C6-alkylsulphonyl and C,-C6-alkylthio are optionally
substituted by one to three radicals independently of one another selected
from the
group of hydroxy, cyano, halogen, trifluoromethyl, trifluoromethoxy, hydroxy-
carbonyl and a group of the formula -NR3R4,
where
R3 and R4 are independently of one another hydrogen or C,-C6-alkyl,
or
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-4-
R3 and R4 together with the nitrogen atom to which they are bonded are 5- to 8-
membered heterocyclyl,
R2 is phenyl or heteroaryl, where phenyl is substituted by 1 to 3 radicals and
heteroaryl is
optionally substituted by 1 to 3 radicals in each case independently of one
another selected
from the group of CI-C6-alkyl, CI-C6-alkoxy, hydroxycarbonyl, cyano,
trifluoromethyl,
trifluoromethoxy, amino, nitro, hydroxy, CI-C6-alkylamino, halogen, C6-CIQ-
arylcarbonylamino, C I -C6-alkylcarbonylamino, CI-C6-alkylaminocarbonyl, CI-C6-
alkoxycarbonyl, C6-CI0-arylaminocarbonyl, heteroarylaminocarbonyl,
heteroarylcarbonylamino, CI-C6-alkylsulphonylamino, CI-C6-alkylsulphonyl and
CI-C6-
alkylthio,
where CI-C6-alkyl, CI-C6-alkoxy, CI-C6-alkylamino, C6-Clo-arylcarbonylamino,
CI-C6-
alkylcarbonylamino, CI-C6-alkylaminocarbonyl, CI-C6-alkoxycarbonyl, C6-Clo-
arylaminocarbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, CI-C6-
alkylsulphonylamino, CI-C6-alkylsulphonyl and CI-C6-alkylthio are optionally
substituted by one to three radicals independently of one another selected
from the
group of hydroxy, cyano, halogen, trifluoromethyl, trifluoromethoxy, hydroxy-
carbonyl and a group of the formula -NR3R4,
where
R3 and R4 have the meanings indicated above,
and the salts, solvates and/or solvates of the salts thereof.
Compounds of the invention are the compounds of the formula (I) and the salts,
solvates and
solvates of the salts thereof; the compounds which are encompassed by formula
(I) and have the
formulae mentioned hereinafter and the salts, solvates and solvates of the
salts thereof, and the
compounds which are encompassed by formula (I) and are mentioned hereinafter
as exemplary
embodiments and the salts, solvates and solvates of the salts thereof, where
the compounds which
are encompassed by formula (I) and are mentioned hereinafter are not already
salts, solvates and
solvates of the salts.
The compounds of the invention may, depending on their structure, exist in
stereoisomeric forms
(enantiomers, diastereomers). The invention therefore relates to the
enantiomers or diastereomers
and respective mixtures thereof. The sterically pure constituents can be
isolated in a known
manner from such mixtures of enantiomers and/or diastereomers.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-5-
Salts which are preferred for these purposes of the invention are
physiologically acceptable salts of
the compounds of the invention.
Physiologically acceptable salts of the compounds (I) include acid addition
salts of mineral acids,
carboxylic acids and sulphonic acids, e.g. salts of hydrochloric acid,
hydrobromic acid, sulphuric
acid, phosphoric acid, methanesulphonic acid, ethanesulphonic acid,
toluenesulphonic acid,
benzenesulphonic acid, naphthalenedisulphonic acid, acetic acid, propionic
acid, lactic acid,
tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic
acid.
Physiologically acceptable salts of the compounds (I) also include salts of
conventional bases such
as, by way of example and preferably, alkali metal salts (e.g. sodium and
potassium salts), alkaline
earth metal salts (e.g. calcium and magnesium salts) and ammonium salts
derived from ammonia
or organic amines having 1 to 16 C atoms, such as, by way of example and
preferably, ethylamine,
diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine,
diethanolamine, tri-
ethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine,
dibenzylamine, N-methyl-
morpholine, dehydroabietylamine, arginine, lysine, ethylenediamine and
methylpiperidine.
Solvates refers for the purposes of the invention to those forms of the
compounds which form, in
the solid or liquid state, a complex by coordination with solvent molecules.
Hydrates are a specific
form of solvates in which the coordination takes place with water.
In addition, the present invention also encompasses prodrugs of the compounds
of the invention. The
term "prodrugs" encompasses compounds which themselves may be biologically
active or inactive
but are converted (for example by metabolism or hydrolysis) into compounds of
the invention
during their residence time in the body.
For the purposes of the present invention, the substituents have the following
meaning, unless
specified otherwise:
C,-C8-Alkyl is a straight-chain or branched alkyl radical having 1 to 8,
preferably 1 to 6,
particularly preferably 1 to 5, carbon atoms. Preferred examples include
methyl, ethyl, n-propyl,
isopropyl, 2-butyl, 2-pentyl and 3-pentyl.
C -C6-Alkenyl is a straight-chain or branched alkenyl radical having 2 to 6,
preferably 2 to 4 and
particularly preferably having 2 to 3, carbon atoms. Preferred examples
include vinyl, allyl, n-prop-l-
en- l -yl and n-but-2-en- 1 -yl.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-6-
C -C6-A1 l is a straight-chain or branched alkynyl radical having 2 to 6,
preferably having 2 to
4 and particularly preferably having 2 to 3 and carbon atoms. Preferred
examples include ethynyl, n-
prop-1-yn-2-yl, n-prop-l-yn-3-yl and n-but-2-yn-1-yl.
C,-C6-Alkoxy is a straight-chain or branched alkoxy radical having 1 to 6,
preferably 1 to 4,
particularly preferably having 1 to 3 carbon atoms. Preferred examples include
methoxy, ethoxy,
n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
C,-C6-Alkoxycarbonyl is a straight-chain or branched alkoxycarbonyl radical
having 1 to 6,
preferably 1 to 4 and particularly preferably 1 to 3, carbon atoms. Preferred
examples include
methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and
tert-butoxycarbonyl.
C,-C6-Alkylamino is a straight-chain or branched mono- or dialkylamino radical
having 1 to 6,
preferably 1 to 4 and particularly preferably having 1 to 3 carbon atoms.
Preferred examples include
methylamino, ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-
pentylamino and
n-hexylamino, dimethylamino, diethylamino, di-n-propylamino, diisopropylamino,
di-t-butylamino,
di-n-pentylamino, di-n-hexylamino, ethylmethylamino, isopropylmethylamino, n-
butylethylamino
and n-hexyl-i-pentylamino.
C,-C6-Al lcarbonylamino is an alkylcarbonyl radical linked via an amino group,
where the alkyl
radical may be straight-chain or branched and comprises 1 to 6, preferably I
to 4 and particularly
preferably 1 to 3, carbon atoms. Preferred examples include
methylcarbonylamino,
ethylcarbonylamino, n-proplcarbonylamino, isopropylcarbonylamino, tert-
butylcarbonylamino,
n-pentylcarbonylamino and n-hexylcarbonylamino.
C,-C6-Alkylaminocarbonyl is a mono- or dialkylamino radical linked via a
carbonyl group, where
the alkyl radicals may be identical or different, are straight-chain or
branched and each comprise 1
to 6, preferably 1 to 4 and particularly preferably 1 to 3, carbon atoms.
Preferred examples include
methylaminocarbonyl, ethylaminocarbonyl, n-propylaminocarbonyl,
isopropylaminocarbonyl, tert-
butylaminocarbonyl, n-pentylaminocarbonyl, n-hexylaminocarbonyl,
dimethylaminocarbonyl,
diethylaminocarbonyl, di-n-propylaminocarbonyl, diisopropylaminocarbonyl, di-t-
butylaminocarbonyl, di-n-pentylaminocarbonyl, di-n-hexylaminocarbonyl,
ethylmethylaminocarbonyl, isopropylmethylaminocarbonyl, n-
butylethylaminocarbonyl and n-hexyl-
i-pentylaminocarbonyl. A further possibility in the case of a dialkylamino
radical is for the two
alkyl radicals to form together with the nitrogen atom to which they are
bonded a 5- to 8-
membered heterocyclyl.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-7-
C6-C-Arylaminocarbonyl is an arylamino radical linked via a carbonyl group.
Preferred examples
include phenylaminocarbonyl and naphthylaminocarbonyl.
C6-C-Arylcarbonylamino is an arylcarbonyl radical linked via an amino group.
Preferred
examples include phenylcarbonylamino and naphthylcarbonylamino.
C,-C6-Alkylsulphonylamino is a straight-chain or branched alkylsulphonylamino
radical having 1 to
6, preferably I to 4 and particularly preferably having I to 3, carbon atoms.
Preferred examples
include methylsulphonylamino, ethyl sulphonylamino, n-propylsulphonylamino,
isopropyl-
sulphonylamino, tert-butylsulphonylamino, n-pentylsulphonylamino and n-
hexylsulphonylamino.
C,-C6-Alkylsulphonyl is a straight-chain or branched alkylsulphonyl radical
having 1 to 6,
preferably 1 to 4 and particularly preferably having 1 to 3, carbon atoms.
Preferred examples
include methylsulphonyl, etylsulphonyl, n-propylsulphonyl, isopropylsulphonyl,
tert-
butylsulphonyl, n-pentylsulphonyl and n-hexylsulphonyl.
C,-C6-Al lthio is a straight-chain or branched alkylthio radical having 1 to
6, preferably I to 4
and particularly preferably having 1 to 3, carbon atoms. Preferred examples
include methylthio,
ethylthio, n-propylthio, isopropylthio, tert-butylthio, n-pentylthio and n-
hexylthio.
Halogen is fluorine, chlorine, bromine and iodine. Fluorine, chlorine, bromine
are preferred, and
fluorine and chlorine are particularly preferred.
Heteroary] is an aromatic, mono- or bicyclic radical having 5 to 10 ring atoms
and up to 5
heteroatoms from the series S, 0 and/or N. 5- to 6-membered heteroaryls having
up to 4 heteroatoms
are preferred. The heteroaryl radical may be bonded via a carbon or nitrogen
atom. Preferred
examples include thienyl, fury], pyrrolyl, thiazolyl, oxazolyl, imidazolyl,
tetrazolyl, pyridyl, pyridyl
N-oxide, pyrimidinyl, pyridazinyl, indolyl, indazolyl, benzofuranyl,
benzothiophenyl, quinolinyl and
isoquinolinyl.
Heteroarylaminocarbonyl is a heteroarylamino radical linked via a carbonyl
group. Preferred
examples include thienylaminocarbonyl, furylaminocarbonyl,
pyrrolylaminocarbonyl,
thiazolylaminocarbonyl, oxazolylaminocarbonyl, imidazolylaminocarbonyl,
tetrazolylaminocarbonyl,
pyridylaminocarbonyl, pyrimidinylaminocarbonyl, pyridazinylaminocarbonyl,
indolylaminocarbonyl,
indazolylaminocarbonyl, benzofuranylaminocarbonyl,
benzothiophenylaminocarbonyl,
quinolinylaminocarbonyl and isoquinolinylaminocarbonyl.
Heteroarylcarbonylamino is a heteroarylcarbonyl radical linked via an amino
group. Preferred
examples include thienylcarbonylamino, furylcarbonylamino,
pyrrolylcarbonylamino,
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-8-
thiazolylcarbonylamino, oxazolylcarbonylamino, imidazolylcarbonylamino,
tetrazolylcarbonylamino, pyridylcarbonylamino, pyrimidinylcarbonylamino,
pyridazinyl-
carbonylamino, indolylcarbonylamino, indazolylcarbonylamino,
benzofuranylcarbonylamino,
benzothiophenylcarbonylamino, quinolinylcarbonylamino and
isoquinolinylcarbonylamino.
3- to 8-membered cycloalkyl stands for saturated and partially unsaturated
nonaromatic cycloalkyl
radicals having 3 to 8, preferably 3 to 6 and particularly preferably 5 to 6,
carbon atoms in the ring.
Preferred examples include cyclopropyl, cyclobutyl, cyclopentyl,
cyclopentenyl, cyclohexyl and
cyclohexenyl.
5- to 8-membered heterocyclyl is a mono- or polycyclic heterocyclic radical
having 5 to 8 ring
atoms and up to 3, preferably 2, heteroatoms or hetero groups from the series
N, 0, S, SO, SO2.
or bicyclic heterocyclyl is preferred. Monocyclic heterocyclyl is particularly
preferred. N
and 0 are preferred as heteroatoms. The heterocyclyl radicals may be saturated
or partially
unsaturated. Saturated heterocyclyl radicals are preferred. 5- to 7-membered
heterocyclyl radicals
are particularly preferred. Preferred examples include oxetan-3-yl, pyrrolidin-
2-yl, pyrrolidin-3-yl,
pyrrolinyl, tetrahydrofuranyl, tetrahydrothienyl, pyranyl, piperidinyl,
thiopyranyl, morpholinyl,
perhydroazepinyl.
When radicals in the compounds of the invention are optionally substituted,
unless otherwise
specified substitution by up to three identical or different substituents is
preferred.
The compounds of the invention may also be in the form of tautomers as shown
by way of example
below:
O OH
HN rN N N N
2 N2
R R R R
A further embodiment of the invention relates to compounds of the formula (I)
in which
R` is C,-C8-alkyl, C2-C6-alkenyl, C2-C6-alkynyl or C3-C8-cycloalkyl, which are
optionally
substituted by up to 3 radicals independently of one another selected from the
group of
C,-C6-alkyl, C,-C6-alkoxy, hydroxycarbonyl, cyano, amino, nitro, hydroxy, C,-
C6-
alkylamino, halogen, C6-C,o-arylcarbonylamino, C,-C6-alkylcarbonylamino, C,-C6-
alkyl-
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-9-
aminocarbonyl, CI-C6-alkoxycarbonyl, C6-CIo-arylaminocarbonyl, heteroaryl-
aminocarbonyl, heteroarylcarbonylamino, C1-C6-alkylsulphonylamino, C,-C6-
alkylsulphonyl and CI-C6-alkylthio,
where
CI-C6-alkyl, C,-C6-alkoxy, CI-C6-alkylamino, C6-CIO-arylcarbonylamino, C,-C6-
alkylcarbonylamino, C,-C6-alkylaminocarbonyl, C,-C6-alkoxycarbonyl, C6-CIO-
arylaminocarbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, C,-C6-
alkylsulphonylamino, CI-C6-alkylsulphonyl and CI-C6-alkylthio are optionally
substituted by a radical selected from the group of hydroxy, cyano, halogen,
hydroxycarbonyl and a group of the formula -NR3R4,
where
R3 and R4 are independently of one another hydrogen or CI-C6-alkyl,
or
R3 and R4 together with the nitrogen atom to which they are bonded are 5- to 8-
membered heterocyclyl,
R2 is phenyl or heteroaryl, where phenyl is substituted by 1 to 3 radicals and
heteroaryl is
optionally substituted by 1 to 3 radicals in each case independently of one
another selected
from the group of CI-C6-alkyl, CI-C6-alkoxy, hydroxycarbonyl, cyano,
trifluoromethyl,
amino, nitro, hydroxy, C1-C6-alkylamino, halogen, C6-CIO-arylcarbonylamino, C1-
C6-
alkylcarbonylamino, C I -C6-alkylaminocarbonyl, C I -C6-alkoxycarbonyl, C6-CIo-
arylaminocarbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, CI-C6-
alkylsulphonylamino, CI-C6-alkylsulphonyl, CI-C6-alkylthio,
where C,-C6-alkyl, CI-C6-alkoxy, C,-C6-alkylamino, C6-CIO-arylcarbonylamino,
C,-C6-
alkylcarbonylamino, CI-C6-alkylaminocarbonyl, CI-C6-alkoxycarbonyl, C6-C,o-
arylaminocarbonyl, heteroarylaminocarbonyl, heteroarylcarbonylamino, C,-C6-
alkylsulphonylamino, CI-C6-alkylsulphonyl and CI-C6-alkylthio are optionally
substituted by a radical selected from the group of hydroxy, cyano, halogen,
hydroxycarbonyl and a group of formula -NR3R4,
where
R3 and R4 have the meanings indicated above,
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-10-
and the salts, solvates and/or solvates of the salts thereof.
A further embodiment of the invention relates to compounds of the formula (I)
in which
R' is C,-C5-alkyl or C3-C6-cycloalkyl, which are optionally substituted by up
to 3 radicals
independently of one another selected from the group of C,-C4-alkyl, C,-C4-
alkoxy, tri-
fluoromethyl, hydroxycarbonyl, cyano, amino, hydroxy, C,-C4-alkylamino,
fluorine,
chlorine, bromine, C6-C1o-arylcarbonylamino, C,-C4-alkylcarbonylamino, C,-C4-
alkyl-
aminocarbonyl, C,-C4-alkoxycarbonyl, C6-C,o-arylaminocarbonyl,
heteroarylaminocarbonyl, heteroarylcarbonylamino, C,-C4-alkylsulphonylamino,
C1-C4-
alkylsulphonyl, C,-C4-alkylthio,
where C,-C4-alkyl and C,-C4-alkoxy are optionally substituted by a radical
selected from
the group of hydroxy, cyano, fluorine, chlorine, bromine, hydroxycarbonyl and
a
group of the formula -NR3R4,
where
R3 and R4 are independently hydrogen or C,-C4-alkyl,
or
R3 and R4 together with the nitrogen atom to which they are bonded are 5- to
6-membered heterocyclyl,
R2 is phenyl, pyrimidyl, pyridyl N-oxide or pyridyl, where phenyl is
substituted by 1 to 3
radicals and pyrimidyl, pyridyl N-oxide and pyridyl are optionally substituted
by 1 to 3
radicals in each case independently of one another selected from the group of
C,-C4-alkyl,
C,-C4-alkoxy, hydroxycarbonyl, cyano, trifluoromethyl, amino, hydroxy, C,-C4-
alkyl-
amino, fluorine, chlorine, bromine, C6-C1o-arylcarbonylamino, C,-C4-
alkylcarbonylamino,
C,-C4-alkylaminocarbonyl, C,-C4-alkoxycarbonyl, C6-C,o-arylaminocarbonyl,
heteroaryl-
aminocarbonyl, heteroarylcarbonylamino, C,-C4-alkylsulphonylamino, C,-C4-
alkylsulphonyl, C,-C4-alkylthio,
where C,-C4-alkyl and C,-C4-alkoxy are optionally substituted by a radical
selected from
the group of hydroxy, cyano, fluorine, chlorine, bromine, hydroxycarbonyl and
a
group of the formula -NR3R4,
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-11-
where
R3 and R4 have the meanings indicated above,
and the salts, solvates and/or solvates of the salts thereof.
A further embodiment of the invention relates to compounds of the formula (I)
in which
R' has the meanings indicated above, and
R2 is phenyl, pyridyl N-oxide or pyridyl, where phenyl is substituted by 1 to
3 radicals and
pyridyl and pyridyl N-oxide are optionally substituted by 1 to 3 radicals in
each case
independently of one another selected from the group of methyl, ethyl, 2-
propyl,
trifluoromethyl, methoxy, ethoxy, fluorine and chlorine,
and the salts, solvates and/or solvates of the salts thereof.
A further embodiment of the invention relates to compounds of the formula (I),
in which
R' is C,-C5-alkyl or C5-C6-cycloalkyl, which are optionally substituted by up
to 3 radicals
independently of one another selected from the group of C,-C4-alkyl, fluorine,
trifluoromethyl, hydroxy, phenylcarbonylamino, C,-C4-alkylcarbonylamino, C,-C4-
alkylaminocarbonyl or phenylaminocarbonyl, and
R2 is phenyl, pyridyl N-oxide or pyridyl, where phenyl is substituted by 1 to
3 radicals and
pyridyl and pyridyl N-oxide are optionally substituted by 1 to 3 radicals in
each case
independently of one another selected from the group of methyl, ethyl, 2-
propyl,
trifluoromethyl, methoxy, ethoxy, fluorine and chlorine,
and the salts, solvates and/or solvates of the salts thereof.
A further embodiment of the invention relates to compounds of the formula (I),
in which
R' is C,-C5-alkyl or C5-C6-cycloalkyl, which are optionally substituted by up
to 3 radicals
independently of one another selected from the group of C,-C4-alkyl, fluorine,
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-12-
trifluoromethyl, hydroxy, phenylcarbonylamino, C1-C4-alkylcarbonylamino, C1-C4-
alkylaminocarbonyl or phenylaminocarbonyl, and
R2 is phenyl, pyridyl N-oxide or pyridyl, where phenyl is substituted by one
radical and
pyridyl and pyridyl N-oxide are optionally substituted by one radical in each
case
independently of one another selected from the group of methyl, ethyl, 2-
propyl,
trifluoromethyl, methoxy, ethoxy, fluorine and chlorine,
and the salts, solvates and/or solvates of the salts thereof.
A process for preparing compounds of the invention of the formula (I) has also
been found,
characterized in that either
[A] compounds of the formula
O
H 2 N N
H2N N 2 (11),
R
in which
R2 has the meanings indicated above,
are converted by reaction with a compound of the formula
R~ (IIIa),
in which
R' has the meanings indicated above,
and
Z is chlorine or bromine,
in an inert solvent and in the presence of a base, initially into compounds of
the formula
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-13-
O
H2N -1) O I \N
N N (IV),
R' H Rz
in which
R' and R2 have the meanings indicated above,
and then cyclized in an inert solvent in the presence of a base to compounds
of the formula
(I),
or
[B] compounds of the formula (II) are reacted with a compound of the formula
O
R' O R5 (IIIb),
in which
R' has the meanings indicated above,
and
R5 is methyl or ethyl,
in an inert solvent and in the presence of a base, with direct cyclization to
(I),
or
[C] compounds of the formula
NC
rN
H2N R2 (V),
in which
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-14-
R2 has the meanings indicated above,
are converted initially by reaction with a compound of the formula (IIIa) in
an inert solvent
and in the presence of a base into compounds of the formula
NC
O )CIN" N
N (VI),
R Fi R2
in which
R' and R2 have the meanings indicated above,
and the latter are cyclized in a second step in an inert solvent and in the
presence of a base
and of an oxidizing agent to (I),
and the resulting compounds of the formula (I) are where appropriate reacted
with the appropriate
(i) solvents and/or (ii) bases or acids to give their solvates, salts and/or
solvates of the salts.
Suitable for the first step of process [A] and of process [C] are inert
organic solvents which are not
changed under the reaction conditions. These preferably include ethers such
as, for example, diethyl
ether, dioxane, tetrahydrofuran or glycol dimethyl ether, or toluene or
pyridine. It is likewise possible
to employ mixtures of the solvents mentioned. Tetrahydrofuran, toluene or
pyridine are particularly
preferred.
Generally suitable bases are alkali metal hydrides such as, for example,
sodium hydride, or cyclic
amines such as, for example, piperidine, pyridine, dimethylaminopyridine
(DMAP) or C,-C4-
alkylamines such as, for example, triethylamine. Sodium hydride, pyridine
and/or dimethyl-
aminopyridine are preferred.
The base is generally employed in an amount of from 1 mol to 4 mol, preferably
from 1.2 mol to
3 mol, in each case based on I mol of the compounds of the general formula
(II) or (V).
In one variant, the reaction is carried out in pyridine to which a catalytic
amount of DMAP is added.
It is also possible where appropriate to add toluene.
The reaction temperature can generally be varied within a relatively wide
range. It is generally in a
range from -20 C to +200 C, preferably from 0 C to +100 C.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-15-
Solvents suitable for the cyclization in the second step of processes [A] and
[C] are the usual organic
solvents. These preferably include alcohols such as methanol, ethanol,
propanol, isopropanol,
n-butanol or tert-butanol, or ethers such as tetrahydrofuran or dioxane, or
dimethylformamide or
dimethyl sulphoxide. Alcohols such as methanol, ethanol, propanol, isopropanol
or tert-butanol are
particularly preferably used. It is likewise possible to employ mixtures of
the solvents mentioned.
Bases suitable for the cyclization in the second step of processes [A] and [C]
are the usual inorganic
bases. These preferably include alkali metal hydroxides or alkaline earth
metal hydroxides such as,
for example, sodium hydroxide, potassium hydroxide or barium hydroxide, or
alkali metal carbonates
such as sodium or potassium carbonate or sodium bicarbonate, or alkali metal
alcoholates such as
sodium methanolate, sodium ethanolate, potassium methanolate, potassium
ethanolate or potassium
tert-butanolate. Potassium carbonate, sodium hydroxide and potassium tert-
butanolate are particularly
preferred.
When carrying out the cyclization, the base is generally employed in an amount
of from 2 mol to
6 mol, preferably from 3 mol to 5 mol, in each case based on I mol of the
compounds of the general
formula (IV) or (VI).
Oxidizing agents suitable for the cyclization in the second step of process
[C] are, for example,
hydrogen peroxide or sodium borate. Hydrogen peroxide is preferred.
The cyclization in processes [A], [B] and [C] is generally carried out in a
temperature range from 0 C
to +160 C, preferably at the boiling point of the particular solvent.
The cyclization is generally carried out under atmospheric pressure. However,
it is also possible to
carry out the process under elevated pressure or under reduced pressure (e.g.
in a range from 0.5 to
5 bar).
Solvents suitable for process [B] are the alcohols mentioned above for the
second step of processes
[A] and [C], with preference for ethanol.
Bases suitable for process [B] are alkali metal hydrides such as, for example,
sodium or potassium
hydride, or alkali metal alcoholates such as, for example, sodium methanolate,
ethanolate,
isopropoxide or potassium tert-butoxide. Sodium hydride is preferred.
The base is employed in an amount of from 2 mol to 8 mol, preferably from 3
mol to 6 mol, in each
case based on I mol of the compounds of the formula (II).
The compounds of the formula (H) are known or can be prepared for example by
initially condensing
ethoxymethylenemalonoxnitrile with hydrazine derivatives of the formula (VII)
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-16-
R2-NH-NH2 (VII),
in which
R2 has the meanings indicated above,
in an inert solvent to give pyrazoleecarbonitriles of the formula (V), and
then reacting the latter with
one of the oxidizing agents mentioned above, preferably hydrogen peroxide, in
the presence of
ammonia [cf., for example, A. Miyashita et al., Heterocycles 1990, 31,
1309ff].
The compounds of the formulae (lHa), (IlIb) and (VII) are commercially
available, known from the
literature or can be prepared in analogy to processes known from the
literature.
The process of the invention can be illustrated by way of example by the
following formula scheme:
Scheme
OC2H5 NH NC
NC Y---- + HN' a )CN
CN RZ H2N R2
H2O2/NH3
O
O
HN I iN RCOOC2H5 H2N
N N I N
R RZ H 2 N N 2
R
Further processes for preparing pyrazoleo[3,4-d]pyrimidin-4-ones are known and
can likewise be
employed for synthesizing the compounds of the invention (see, for example: P.
Schmidt et al.,
Helvetica Chimica Acta 1962, 189, 1620ff.).
The compounds of the invention show a valuable range of pharmacological and
pharmacokinetic
effects which could not have been predicted. They are distinguished in
particular by inhibition of
PDE9A.
They are therefore suitable for use as medicaments for the treatment and/or
prophylaxis of diseases
in humans and animals.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-17-
For the purposes of the present invention, the term "treatment" includes
prophylaxis.
It has surprisingly been found that the compounds of the invention are
suitable for producing
medicaments for improving perception, concentration, learning or memory.
The compounds of the invention can, by reason of their pharmacological and
pharmacokinetic
properties, be employed alone or in combination with other medicaments for
improving
perception, concentration, learning and/or memory.
The compounds of the invention are particularly suitable for improving
perception, concentration,
learning or memory after cognitive impairments like those occurring in
particular in
situations/diseases/syndromes such as mild cognitive impairment, age-
associated learning and
memory impairments, age-associated memory losses, vascular dementia,
craniocerebral trauma,
stroke, dementia occurring after strokes (post stroke dementia), post-
traumatic dementia, general
concentration impairments, concentration impairments in children with learning
and memory
problems, Alzheimer's disease, Lewy body dementia, dementia with degeneration
of the frontal
lobes, including Pick's syndrome, Parkinson's disease, progressive nuclear
palsy, dementia with
corticobasal degeneration, amyotrophic lateral sclerosis (ALS), Huntington's
disease, multiple
sclerosis, thalamic degeneration, Creutzfeld-Jakob dementia, HIV dementia,
schizophrenia with
dementia or Korsakoff's psychosis.
The in vitro effect of the compounds of the invention can be shown with the
following biological
assays:
PDE inhibition
Recombinant PDE1C (GenBank/EMBL Accession Number: NM005020, Loughney et al. J
Biol.
Chem. 1996 271, 796-806), PDE2A (GenBank/EMBL Accession Number: NM 002599,
Rosman
et al. Gene 1997 191, 89-95), PDE3B (GenBank/EMBL Accession Number: NM 000922,
Miki et
al. Genomics 1996, 36, 476-485), PDE4B (GenBank/EMBL Accession Number:
NM_002600,
Obernolte et al. Gene. 1993, 129, 239-247), PDE5A (GenBank/EMBL Accession
Number:
NM_001083, Loughney et al. Gene 1998, 216, 139-147), PDE7B (GenBank/EMBL
Accession
Number: NM_018945, Hetman et al. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 472-
476), PDE8A
(GenBank/EMBL Accession Number: AF_056490, Fisher et al. Biochem. Biophys.
Res. Commun.
1998 246, 570-577), PDE9A (Fisher et al., J. Biol. Chem, 1998, 273 (25): 15559-
15564), PDE10A
(GenBank/EMBL Accession Number: NM_06661, Fujishige et al. JBiol Chem. 1999,
274, 18438-
45), PDE11A (GenBank/EMBL Accession Number: NM_016953, Fawcett et al. Proc.
Natl. Acad.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-18-
Sci. 2000, 97, 3702-3707) were expressed in Sf9 cells with the aid of the
pFASTBAC baculovirus
expression system (GibcoBRL).
The test substances are dissolved in 100% DMSO and serially diluted to
determine their in vitro
effect on PDE 9A. Typically, serial dilutions from 200 M to 1.6 M are
prepared (resulting final
concentrations in the assay: 4 M to 0.032 pM). 2 pL portions of the diluted
substance solutions
are introduced into the wells of microtiter plates (Isoplate; Wallac Inc.,
Atlanta, GA). Then 50 L
of a dilution of the PDE9A preparation described above are added. The dilution
of the PDE9A
preparation is chosen so that less than 70% of the substrate is converted
during the subsequent
incubation (typical dilution: 1:10000; dilution buffer: 50 mM Tris/HC1 pH 7.5,
8.3 mM MgC12,
1.7 mM EDTA, 0.2% BSA). The substrate, [8-3H] guanosine 3',5'-cyclic phosphate
(1 Ci/pL;
Amersham Pharmacia Biotech., Piscataway, NJ) is diluted 1:2000 with assay
buffer (50 mM
Tris/HCI pH 7.5, 8.3 mM MgC12, 1.7 mM EDTA) to a concentration of 0.0005
Ci/gL. The
enzyme reaction is finally started by adding 50 pL (0.025 Ci) of the diluted
substrate. The assay
mixtures are incubated at room temperature for 60 min and the reaction is
stopped by adding 25 p1
of a PDE9A inhibitor (e.g. the inhibitor from preparation example 1, final
concentration 10 M)
dissolved in assay buffer. Immediately thereafter, 25 pL of a suspension
containing 18 mg/mL
Yttrium Scintillation Proximity Beads (Amersham Pharmacia Biotech.,
Piscataway, NJ) are added.
The microtiter plates are sealed with a film and left to stand at room
temperature for 60 min. The
plates are then measured for 30 s per well in a Microbeta scintillation
counter (Wallac Inc.,
Atlanta, GA). IC50 values are determined from the graphical plot of the
substance concentration
versus the percentage inhibition.
Representative examples of the inhibiting effect of the compounds of the
invention on PDE9A are
listed by means of the IC50 values in Table 1:
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-19-
Table 1
Example IC50 [nM]
2 38
12
11 5
34 12
37-1 60
38 13
39-1 30
The in vitro effect of test substances on recombinant PDE3B, PDE4B, PDE7B,
PDE8A, PDElOA
and PDE11A is deteremined in accordance with the assay protocol described
above for PDE 9A
5 with the following adaptations: [5',8-3H] adenosine 3',5'-cyclic phosphate
(1 Ci/ L; Amersham
Pharmacia Biotech., Piscataway, NJ) is used as substrate. Addition of an
inhibitor solution to stop
the reaction is unnecessary. Instead, the incubation of substrate and PDE is
followed immediately
by addition of the yttrium scintillation proximity beads as described above
and thus the reaction is
stopped. To determine a corresponding effect on recombinant PDE1C, PDE2A and
PDE5A, the
protocol is additionally adapted as follows: with PDE1C, additionally 10-7 M
calmodulin and
3 mM CaC12 are added to the reaction mixture. PDE2A is stimulated in the assay
by adding 1 M
cGMP and is assayed with a BSA concentration of 0.01%. The substrate employed
for PDE1C and
PDE2A is [5',8-'H] adenosine 3',5'-cyclic phosphate (1 Ci/ L; Amersham
Pharmacia Biotech.,
Piscataway, NJ), and for PDE5A is [8_3 H] guanosine 3',5'-cyclic phosphate (1
Ci/ L; Amersham
Pharmacia Biotech., Piscataway, NJ).
Long-term potentiation
Long-term potentiation is regarded as a cellular correlate of learning and
memory processes. The
following method can be used to determine whether PDE 9 inhibition has an
influence on long-
term potentiation:
Rat hippocampi are placed at an angle of about 70 degrees to the cutting blade
(chopper). 400 pm-
thick slices of the hippocampus are prepared. The slices are removed from the
blade using a very
soft, thoroughly wetted brush (marten hair) and transferred into a glass
vessel with cold nutrient
solution (124 mM NaCl, 4.9 mM KCI, 1.3 mM MgSO4 x 7 H2O, 2.5 mM CaC12
anhydrous, 1.2
mM KH2PO4, 25.6 mM NaHCO3, 10 mM glucose, pH 7.4) gassed with 95% 02/5% CO2.
During
the measurement, the slices are kept in a temperature-controlled chamber under
a 1-3 mm-high
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-20-
liquid level. The flow rate is 2.5 ml/min. The preliminary gassing takes place
under a slightly
elevated pressure (about 1 atm) and through a microneedle in the prechamber.
The slice chamber is
connected to the prechamber in such a way that a minicirculation can be
maintained. The
minicirculation is driven by the 95% 02/5% CO2 flowing out through the
microneedle. The freshly
prepared hippocampus slices are adapted in the slice chamber at 33 C for at
least 1 hour.
The stimulus level is chosen so that the focal excitatory postsynaptic
potentials (fEPSP) are 30%
of the maximum excitatory postsynaptic potential (EPSP). A monopolar
stimulation electrode
consisting of lacquered stainless steel, and a constant-current biphasic
stimulus generator (AM
Systems 2100) are used for local stimulation of the Schaffer collaterals
(voltage: 1-5 V, pulse
width of one polarity 0.1 ms, total pulse 0.2 ms). Glass electrodes
(borosilicate glass with filament,
1-5 MOhm, diameter: 1.5 mm, tip diameter: 3-20 m), filled with normal
nutrient solution, are
used to record the excitatory postsynaptic potentials (fEPSP) from the stratum
radiatum. The field
potentials are measured versus a chlorinated silver reference electrode
located at the edge of the
slice chamber using a DC voltage amplifier. The field potentials are filtered
through a low-pass
filter (5 kHz). The slope of the fEPSPs (fEPSP slope) is determined for the
statistical analysis of
the experiments. The recording, analysis and control of the experiment takes
place with the aid of a
software program (PWIN) which was developed in the Department of
Neurophysiology. The
formation of the average fEPSP slopes at the respective time points and
construction of the
diagrams takes place with the aid of the EXCEL software, with automatic data
recording by an
appropriate macro.
Superfusion of the hippocampus slices with a 10 M solution of the compounds
of the invention
leads to a significant increase in the LTP.
The in vivo effect of the compounds of the invention can be shown for example
as follows:
Social recognition test
The social recognition test is a learning and memory test. It measures the
ability of rats to
distinguish between known and unknown members of the same species. This test
is therefore
suitable for examining the learning- or memory-improving effect of the
substances of the
invention.
Adult rats housed in groups are placed singly in test cages 30 min before the
start of the test. Four
min before the start of the test, the test animal is put in an observation
box. After this adaptation
time, a juvenile animal is put in with the test animal and the absolute time
for which the adult
animal inspects the young one is measured for 2 min (trial 1). All behaviours
clearly directed at the
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-21-
young animal are measured, i.e. anogenital inspection, pursuit and fur care,
during which the old
animal was no further than 1 cm from the young animal. The juvenile is then
removed, and the
adult is treated with a compound of the invention or vehicle and subsequently
returned to its own
cage. The test is repeated after a retention time of 24 hours (trial 2). A
diminished social
interaction time compared with trial 1 indicates that the adult rat remembers
the young animal.
The adult animals receive intraperitoneal injections either at a fixed time
interval (e.g. 1 hour)
before trial 1 or directly following trial I either with vehicle (10% ethanol,
20% Solutol, 70%
physiological saline) or 0.1 mg/kg, 0.3 mg/kg, 1.0 mg/kg or 3.0 mg/kg compound
of the invention
dissolved in 10% ethanol, 20% Solutol, 70% physiological saline. Vehicle-
treated rats show no
reduction in the social interaction time in trial 2 compared with trial 1.
They have consequently
forgotten that they have already had contact with the young animal.
Surprisingly, the social inter-
action time in the second run after treatment with the compounds of the
invention is significantly
reduced compared with those treated with vehicle. This means that the
substance-treated rats have
remembered the juvenile animal and thus the compounds of the invention display
an improving
effect on learning and memory.
The present invention further relates to the use of the compounds of the
invention for the treatment
and/or prophylaxis of disorders, in particular of the aforementioned
disorders.
The present invention further relates to the use of the compounds of the
invention for producing a
medicament for the treatment and/or prophylaxis of disorders, in particular of
the aforementioned
disorders.
The present invention further relates to a method for the treatment and/or
prophylaxis of disorders,
in particular of the aforementioned disorders, using an effective amount of
the compounds of the
invention.
The present invention further relates to medicaments comprising at least one
compound of the
invention and one or more other active ingredients, in particular for the
treatment and/or
prophylaxis of the aforementioned disorders.
The compounds of the invention may have systemic and/or local effects. They
can for this purpose
be administered in a suitable way, such as, for example, by the oral,
parenteral, pulmonary, nasal,
sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival or otic
route or as implant or
stent.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-22-
The compounds of the invention can be administered in suitable administration
forms for these
administration routes.
Administration forms suitable for oral administration are those which function
according to the
state of the art and deliver the compounds of the invention in a rapid and/or
modified way, and
which contain the compounds of the invention in crystalline and/or amorphized
and/or dissolved
form, such as, for example, tablets (uncoated or coated tablets, for example
with coatings which
are resistant to gastric juice or dissolve slowly or are insoluble and which
control the release of the
compound of the invention), tablets which rapidly disintegrate in the mouth,
or films/wafers,
films/lyophilisates, capsules (for example hard or soft gelatin capsules),
sugar-coated tablets,
granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
Parenteral administration can take place with avoidance of an absorption step
(e.g. intravenous,
intraarterial, intracardiac, intraspinal or intralumbar) or with inclusion of
an absorption (e.g.
intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal).
Administration
forms suitable for parenteral administration are, inter alia, injection and
infusion preparations in
the form of solutions, suspensions, emulsions, lyophilisates or sterile
powders.
Examples suitable for other administration routes are medicinal forms for
inhalation (inter alia
powder inhalators, nebulizers), nasal drops, solutions, sprays; tablets for
lingual, sublingual or
buccal administration, films/wafers or capsules, suppositories, preparations
for the ears or eyes,
vaginal capsules, aqueous suspensions (lotions, shaking mixtures), lipophilic
suspensions,
ointments, creams, transdermal therapeutic systems (such as, for example,
patches), milk, pastes,
foams, dusting powders, implants or stents.
The compounds of the invention can be converted into the stated administration
forms. This can
take place in a manner known per se by mixing with inert, non-toxic,
pharmaceutically suitable
excipients. These excipients include, inter alia, carriers (for example
microcrystalline cellulose,
lactose, mannitol), solvents (e.g. liquid polyethylene glycols), emulsifiers
and dispersants or
wetting agents (for example sodium dodecyl sulphate, polyoxysorbitan oleate),
binders (for
example polyvinylpyrrolidone), synthetic and natural polymers (for example
albumin), stabilizers
(e.g. antioxidants such as, for example, ascorbic acid), colours (e.g.
inorganic pigments such as, for
example, iron oxides) and masking tastes and/or odours.
The present invention further relates to medicaments which comprise at least
one compound of the
invention, normally together with one or more inert, non-toxic,
pharmaceutically suitable
excipients, and to the use thereof for the aforementioned purposes.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-23-
It has generally proved advantageous on parenteral administration to
administer amounts of about
0.00 1 to 10 mg/kg of body weight per day to achieve effective results. The
amount per day on oral
administration is about 0.005 to 3 mg/kg of body weight.
It may nevertheless be necessary to deviate from the stated amounts, in
particular as a function of
body weight, administration route, individual behaviour towards the active
ingredient, type of
preparation and time or interval over which administration takes place. Thus,
it may in some cases
be sufficient to make do with less than the aforementioned minimum amount,
whereas in other
cases the stated upper limit must be exceeded. Where larger amounts are
administered, it may be
advisable to divide them into a plurality of single doses over the day.
The percentage data in the following tests and examples are, unless indicated
otherwise,
percentages by weight; parts are parts by weight. Solvent ratios, dilution
ratios and concentration
data for liquid/liquid solutions are in each case based on volume.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-24-
Abbreviations:
BSA bovine serum albumin
DCI direct chemical ionization (in MS)
DMSO dimethyl sulphoxide
EDTA ethylenediaminetetraacetic acid
ESI electrospray ionization (in MS)
Fp. melting point
h hour(s)
HPLC high pressure, high performance liquid chromatography
LC-MS coupled liquid chromatography-mass spectroscopy
min minute(s)
MS mass spectroscopy
NMR nuclear magnetic resonance spectroscopy
R, retention time (in HPLC)
Tris tris(hydroxymethyl)aminomethane
TLC thin-layer chromatography
LC-MS methods:
Method 1
MS apparatus type: Micromass ZQ; HPLC apparatus type: TSP P4000, TSP AS300,
TSP UV3000;
column: Grom-Sil 120 ODS-4 HE, 50 x 2 mm, 3.0 m; eluent A: water + 250 l of
50% strength
formic acid / 1, eluent B: acetonitrile + 250 l of 50% strength formic acid /
1; gradient: 0.0 min 0%
B -> 0.2 min 0% B -> 2.9 min 70% B -+ 3.1 min 90% B -* 4.5 min 90% B; oven: 50
C; flow rate:
0.8 ml/min; UV detection: 210 nm.
Method 2
Instrument: Micromass Platform LCZ with HPLC Agilent Series 1100; column: Grom-
Sil 120
ODS-4 HE, 50 mm x 2.0 mm, 3 m; eluent A: 1 1 of water + 1 ml of 50% strength
formic acid,
eluent B: 1 1 of acetonitrile + 1 ml of 50% formic acid; gradient: 0.0 min
100% A -* 0.2 min 100%
A -> 2.9 min 30% A 3.1 min 10% A - 4.5 min 10% A; oven: 55 C; flow rate: 0.8
ml/min; UV
detection: 208-400 nm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-25-
Method 3
MS apparatus type: Micromass ZQ; HPLC apparatus type: Waters Alliance 2790;
column: Grom-
Sil 120 ODS-4 HE, 50 x 2 mm, 3.0 gm; eluent B: acetonitrile + 0.05% formic
acid, eluent A: water
+ 0.05% formic acid; gradient: 0.0 min 5% B -* 2.0 min 40% B -> 4.5 min 90% B -
5.5 min
90% B; oven: 45 C; flow rate: 0.0 min 0.75 ml/min --> 4.5 min 0.75 ml/min -f
5.5 min
1.25 ml/min; UV detection: 210 nm.
Method 4
Instrument: Micromass Quattro LCZ, with HPLC Agilent Series 1100; colunn: Grom-
Sil 120
ODS-4 HE, 50 mm x 2.0 mm, 3 m; eluent A: 1 1 water + 1 ml 50% strength formic
acid, eluent B:
1 1 acetonitrile + I ml 50% strength formic acid; gradient: 0.0 min 100% A ->
0.2 min 100% A ->
2.9 min 30% A --> 3.1 min 10% A -> 4.5 min 10% A; oven: 55 C; flow rate: 0.8
ml/min; UV
detection: 208-400 nm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-26-
Starting compounds:
Example 1A
5-Amino- l -(2,6-dimethylphenyl)-1 H-pyrazolee-4-carbonitrile
NC
H 2 N N-iN
H 3 C / CH3
~ I
3.0 g (17.3 mmol) of 2,6-dimethylphenylhydrazine hydrochloride are suspended
with 2.1 g
(17.3 mmol) of ethoxymethylenemalononitrile in 40 ml of ethanol, and 7.3 ml
(52.1 mmol) of
triethylamine are added. The reaction mixture is heated to reflux for 3 h,
during which a clear
solution forms. After cooling to room temperature, diethyl ether is added. The
triethylammonium
chloride which precipitates is filtered off. The solvent is removed in vacuo,
and the residue is
purified by preparative HPLC (YMC gel ODS-AQ S 5/15 m; eluent A: water,
eluent B:
acetonitrile; gradient: 0 min 30% B, 5 min 30% B, 50 min 95% B). 2.3 g (62% of
theory) of the
product are obtained as yellow crystals.
LC-MS (Method 1): Rt = 2.77 min.
MS (ESI pos): m/z = 213 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-27-
Example 2A
-Amino- l -(2,3 -dimethylphenyl)-I H-pyrazolee-4-carbonitrile
NC
H2N NON
H 3 C H3C
In analogy to the preparation of Example IA, 2.08 g (56% of theory) of the
desired product are
obtained starting from 3 g (17.4 mmol) of 2,3-dimethylphenylhydrazine
hydrochloride, 2.12 g
(17.4 mmol) of ethoxymethylenemalononitrile and 7.3 ml (52.1 mmol) of
triethylamine.
LC-MS (Method 1): R, = 2.79 min.
5 MS (ESI pos): m/z = 213 (M+H)+.
Example 3A
5-Amino-l-(4-methylphenyl)-I H-pyrazolee-4-carbonitrile
NC
H 2 N NON
I
CH3
In analogy to the preparation of Example IA, 2.16 g (57% of theory) of the
desired product are
obtained starting from 3 g (18.9 mmol) of 4-methylphenylhydrazine
hydrochloride, 2.3 g
(18.9 mmol) of ethoxymethylenemalononitrile and 7.9 ml (56.7 mmol) of
triethylamine.
LC-MS (Method 2): R, = 3.0 min.
MS (ESI pos): m/z = 199 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-28-
Example 4A
5-Amino-l -(2,6-dichlorophenyl)-1 H-pyrazolee-4-carbonitrile
NC
H2N NON
CI CI
In analogy to the preparation of Example IA, 2.9 g (83% of theory) of the
desired product are
obtained starting from 3 g (14.1 mmol) of 2,6-dichlorophenylhydrazine
hydrochloride, 1.7 g
(14.1 mmol) of ethoxymethylenemalononitrile and 5.8 ml (42.2 mmol) of
triethylamine after
purification by column chromatography (mobile phase dichloromethane/methanol
98:2).
LC-MS (Method 3): Rt = 2.8 min.
MS (ESI pos): m/z = 253 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.82 (s, 2H), 7.59 (m, 2H), 7.69 (m, 1H), 7.80
(s, 1H) ppm.
Example 5A
5-Amino- l -(2,5-dichlorphenyl)-1 H-pyrazolee-4-carbonitrile
NC
H2N N,N
CI
CI
In analogy to the preparation of Example IA, 2.2 g (51% of theory) of the
desired product are
obtained starting from 3 g (16.9 mmol) of 2,5-dichlorophenylhydrazine, 2.0 g
(16.9 mmol) of
ethoxymethylenemalononitrile and 7.1 ml (50.8 mmol) of triethylamine.
LC-MS (Method 2): Rt = 3.2 min.
MS (ESI pos): m/z = 253 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-29-
Example 6A
-Amino-l-(2-nitrophenyl)-I H-pyrazol ee-4-c arbonitrile
NC
H2N NiN
NO2
In analogy to the preparation of Example IA, 1.9 g (53% of theory) of the
desired product are
5 obtained starting from 3 g (15.8 mmol) of 2-nitrophenylhydrazine
hydrochloride, 1.93 g
(16.9 mmol) of ethoxymethylenemalononitrile and 6.6 ml (47.6 mmol) of
triethylamine.
LC-MS (Method 2): Rt = 2.8 min.
MS (ESI pos): m/z = 230 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.87 (s, 2H), 7.72 (m, 1H), 7.77 (s, 1H), 7.78
(m, 1H), 7.88
(m, 1H), 8.16 (dd, 1H) ppm.
Example 7A
5-Amino- l -(3-fluorophenyl)-1 H-pyrazolee-4-carbonitrile
NC
H 2 N ON
6F
In analogy to the preparation of Example IA, 1.5 g (31% of theory) of the
desired product are
obtained starting from 4 g (24.6 mmol) of 3-fluorophenylhydrazine
hydrochloride, 3 g (24.6 mmol)
of ethoxymethylenemalononitrile and 10.3 ml (73.8 mmol) of triethylamine.
LC-MS (Method 2): Rt = 2.9 min.
MS (ESI pos): m/z = 203 (M+H)+
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-30-
'H-NMR (300 MHz, DMSO-d6): 6 = 6.81 (s, 2H), 7.28 (m, 1H), 7.36 (m, 2H), 7.57
(m, 1H), 7.80
(s, 1 H) ppm.
Example 8A
5-Amino- l -(2-methylphenyl)-1 H-pyrazolee-4-carbonitrile
NC
H2N N,N
CH3
10.2 g (64.4 mmol) of 2-methylphenylhydrazine hydrochloride are suspended with
7.8 g
(64.4 mmol) of ethoxymethylenemalononitrile in 100 ml of methanol, and 26.9 ml
(193.3 mmol) of
triethylamine are added. The reaction mixture is heated to reflux overnight,
during which a clear
solution forms. The solution is subsequently distilled off under reduced
pressure, and the crude
product is purified by column chromatography (silica gel, mobile phase
dichloromethane). 10.8 g
(85% of theory) of the desired product are obtained.
LC-MS (Method 2): Rt = 3.10 min.
MS (ESI pos): m/z = 199 (M+H)+.
Example 9A
5 -Amino- l -(2-ethylphenyl)-1 H-pyrazolee-4-carbonitrile
NC
NO
H2N N
CH3
In analogy to the preparation of Example IA, 3.05 g (83.5% of theory) of the
desired product are
obtained starting from 3.0 g (17.0 mmol) of 2-ethylphenylhydrazine
hydrochloride, 2.12 g
(17.0 mml) of ethoxymethylenemalononitrile and 7.1 ml (51.1 mmol) of
triethylamine.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-31-
m.p.: 130 C
MS (ESI pos): m/z = 213 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.0 (t, 3H), 2.35 (q, 2H), 6.4 (s, 2H), 7.2-7.5
(m, 4H), 7.7 (s,
1 H) ppm.
Example 10A
5-Amino-l-(2-trifluoromethylphenyl)-1 H-pyrazolee-4-carbonitrile
NC
NO
H2N N
L,,CF3
In analogy to the preparation of Example IA, 5.02 g (76.9% of theory) of the
desired product are
obtained starting from 4.8 g (25.9 mmol) of 2-trifluoromethylphenylhydrazine
hydrochloride,
3.16 g (25.9 mmol) of ethoxymethylenemalononitrile and 7.2 ml (51.7 mmol) of
triethylamine.
m.p.: 190 C
MS (ESI pos): m/z = 253 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.6 (s, 2H), 7.5 (d, 1H), 7.7-8.0 (m, 4H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-32-
Example 11A
5-Amino-I-(2-fluorophenyl)-1 H-pyrazolee-4-carbonitrile
NC
ti
H2N NF
In analogy to the preparation of Example IA, 5.13 g (88% purity, 84% of
theory) of the desired
product are obtained starting from 5.0 g (30.8 mmol) of 2-
fluorophenylhydrazine hydrochloride,
3.27 g (26.7 mmol) of ethoxymethylenemalononitrile and 11.3 ml (81.3 mmol) of
triethylamine.
MS (ESI pos): m/z = 203 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.7 (s, 2H), 7.3-7.6 (m, 4H), 7.8 (s, 1H) ppm.
Example 12A
5-Amino-l-(2-chlorphenyl)-1H-pyrazolee-4-carbonitrile
NC
H2N NON
yCI
In analogy to the preparation of Example IA, 4.64 g (78% of theory) of the
desired product are
obtained starting from 5.0 g (27.1 mmol) of 2-chlorophenylhydrazine
hydrochloride, 3.31 g
(27.1 mmol) of ethoxymethylenemalononitrile and 11.3 ml (81.3 mmol) of
triethylamine.
m.p.:135 C
MS (ESI pos): m/z = 219 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.6 (s, 2H), 7.45-7.75 (m, 4H), 7.8 (s, 1H)
ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-33-
Example 13A
5-Amino- l -(2-pyridinyl)-1 H-pyrazolee-4-carbonitrile
NC
NO
H2N N
N
In analogy to the preparation of Example IA, 2.3 g (46.6% of theory) of the
desired product are
obtained starting from 3.0 g (26.7 mmol, 97% purity) of 2-hydrazinopyridine,
3.26 g (26.7 mmol)
of ethoxymethylenemalononitrile and 7.4 ml (53.3 mmol) of triethylamine.
m.p.: 193 C
MS (ESI pos): m/z = 186 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 7.35 (m, 1H), 7.8-8.12 (m, 3H), 8.15 (s, 2H),
8.5 (m, 1H)
ppm.
Example 14A
5-Amino- l -(2-methoxyphenyl)-1 H-pyrazolee-4-carbonitrile
NC
H2N NON
O
~1 CH3
In analogy to the preparation of Example IA, 3.5 g (88% of theory) of the
desired product are
obtained starting from 4.1 g (18 mmol) of 2-methoxyphenylhydrazine
hydrochloride, 2.19 g
(18 mmol) of ethoxymethylenemalononitrile and 10 ml (71.9 mmol) of
triethylamine.
m.p.: 129 C
MS (ESI pos): m/z = 215 (M+H)+
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-34-
'H-NMR (300 MHz, DMSO-d6): 6 = 3.8 (s, 3H), 6.3 (s, 2H), 7.05 (t, 1H), 7.2 (d,
1H), 7.25 (d, 1H),
7.5 (t, 1 H), 7.7 (s, 1 H) ppm.
Example 15A
-Amino- l -(2, 6-dimethylphenyl)-1 H-pyrazolee-4-carboxamide
0
H2N
N
H 2 N Ni
H 3 C / CH3
5
2 g (9.4 mmol) of 5-amino-l-(2,6-dimethylphenyl)-1H-pyrazolee-4-carbonitrile
(Example IA) are
dissolved in 25 ml of ethanol, and a mixture of 20 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia is added. The solution is stirred at room
temperature overnight
and then concentrated to about 15 ml in a rotary evaporator. The oily emulsion
resulting thereby is
taken up in dichloromethane. It is washed several times with water and
saturated sodium
thiosulphate solution. Drying over magnesium sulphate is followed by removal
of the solvent in
vacuo. The residue is purified by preparative HPLC (YMC Gel ODS-AQ S 5/15 m;
eluent A:
water, eluent B: acetonitrile; gradient: 0 min 30% B, 5 min 30% B, 50 min 95%
B). 0.88 g (40% of
theory) of the product is obtained as colourless solid.
LC-MS (Method 2): Rt = 2.6 min.
MS (ESI pos): m/z = 231 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-35-
Example 16A
5-Amino- l -(2,3-dimethylphenyl)-1 H-pyrazolee-4-carboxamide
O
H 2 N
N
H2N Ni
H 3 C H 3 C
In analogy to the preparation of Example 15A, 1.29 g (70% of theory) of the
desired product are
obtained from 1.5 g (7.1 mmol) of 5-amino-1-(2,3-dimethylphenyl)-1H-pyrazolee-
4-carbonitrile
(Example 2A) in a mixture of 25 ml of ethanol, 10 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia.
LC-MS (Method 2): R, = 2.7 min.
MS (ESI pos): m/z = 231 (M+H)+.
Example 17A
5-Amino-l-(4-methylphenyl)-1H-pyrazolee-4-carboxamide
O
H 2 N
/ 7!3N
H2N NCH3
In analogy to the preparation of Example 15A, 1.02 g (47% of theory) of the
desired product are
obtained from 2 g (10.1 mmol) of 5-amino-1-(4-methylphenyl)-1H-pyrazolee-4-
carbonitrile
(Example 3A) in a mixture of 25 ml of ethanol, 20 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-36-
LC-MS (Method 2): Rt = 2.7 min.
MS (ESI pos): m/z = 217 (M+H)+.
Example 18A
5-Amino- l -(2,6-dichlorophenyl)-1 H-pyrazolee-4-carboxamide
O
H2N
N
H 2 N Ni
CI / CI
In analogy to the preparation of Example 15A, 1.6 g (74% of theory) of the
desired product are
obtained from 2 g (7.9 mmol) of 5-amino-l-(2,6-dichlorophenyl)-1H-pyrazolee-4-
carbonitrile
(Example 4A) in a mixture of 25 ml of ethanol, 10 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia by crystallization from the reaction solution.
LC-MS (Method 2): Rt = 2.5 min.
MS (ESI pos): m/z = 271 (M+H)+.
Example 19A
5-Amino- l -(2,5-dichlorophenyl)-1 H-pyrazolee-4-carboxamide
O
H2N
N
H 2 N Ni
CI
CI
In analogy to the preparation of Example 15A, 2.02 g (94% of theory) of the
desired product are
obtained from 2 g (7.9 mmol) of 5-amino-l-(2,5-dichlorophenyl)-1H-pyrazolee-4-
carbonitrile
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-37-
(Example 5A) in a mixture of 25 ml of ethanol, 18 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia by crystallization from the reaction solution.
LC-MS (Method 2): Rt = 2.8 min.
MS (ESI pos): m/z = 271 (M+H)+.
Example 20A
5-Amino- l -(2-nitrophenyl)-1 H-pyrazolee-4-carboxamide
O
H2N
N
H 2 N N
NO2
In analogy to the preparation of Example 15A, 1.4 g (86% of theory) of the
desired product are
obtained from 1.5 g (6.5 mmol) of 5-amino-l-(2-nitrophenyl)-1H-pyrazolee-4-
carbonitrile
(Example 6A) in a mixture of 25 ml of ethanol, 16 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia by crystallization from the reaction solution.
LC-MS (Method 2): Rt = 2.3 min.
MS (ESI pos): m/z = 248 (M+H)+.
Example 21A
5-Amino- l -(2-aminophenyl)-1 H-pyrazolee-4-carboxamide
O
H2N
N
H 2 N N
NH2
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-38-
1.28 g (5.27 mmol) of 5-amino-l-(2-nitrophenyl)-1H-pyrazole-4-carboxamide
(Example 20A) are
introduced into 30 ml of ethyl acetate and stirred with 5.8 g (25.8 mmol) of
tin(II) chloride
dihydrate at 70 C for 16 hours. After cooling to room temperature, the
solution is adjusted to pH
9-10 with saturated sodium bicarbonate solution. The tin salts precipitated
thereby are filtered off
through kieselguhr. The filtrate is extracted with ethyl acetate. The combined
organic phases are
washed with saturated sodium chloride solution. After drying over sodium
sulphate, the solvent is
removed in vacuo. 0.82 g (72% of theory) of the desired product is obtained.
LC-MS (Method 4): R, = 3.0 min.
MS (ESI pos): m/z = 218 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 5.04 (s, 2H), 6.00 (s, 2H), 6.66 (m, 1H), 6.89
(m, 1H), 7.03
(m, 2H), 7.92 (s, 1 H) ppm.
Example 22A
5-Amino-1 -(3-fluorophenyl)-1 H-pyrazole-4-carboxamide
0
H2N
N
H2N Ni
F
In analogy to the preparation of Example 15A, 1.1 g (75% of theory) of the
desired product are
obtained from 1.3 g (6.4 mmol) of 5-amino-l-(3-fluorophenyl)-1H-pyrazole-4-
carbonitrile
(Example 7A) in a mixture of 25 ml of ethanol, 10 ml of 30% strength hydrogen
peroxide and
40 ml of 25% strength ammonia by crystallization from the reaction solution.
LC-MS (Method 2): Rt = 2.6 min.
MS (ESI pos): m/z = 221 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-39-
Example 23A
-Amino-l-(2-methylphenyl)-1 H-pyrazole-4-carboxamide
0
H2N
H2N IN
k,CH3
300 ml of 96% strength sulphuric acid are cautiously added to 40.0 g (201.8
mmol) of 5-amino-l-
(2-methylphenyl)-1H-pyrazole-4-carbonitrile (Example 8A) while cooling in ice.
The mixture is
5 then heated to 40 C and stirred for 2 hours at this temperature. After
cooling, it is poured into 2 1
of ice-water and cautiously neutralized with 50% strength sodium hydroxide
solution. After
extraction with ethyl acetate three times (2 1 each time) the combined organic
phases are washed
with saturated sodium chloride solution and dried over sodium sulphate, and
the solvent is distilled
off under reduced pressure. 36.0 g (82% of theory) of product (purity >90%)
are obtained and are
employed without further purification in subsequent reactions.
LC-MS (Method 1): Rt = 2.14 min.
MS (ESI pos): m/z = 217 (M+H)+.
Example 24A
5 -Amino- l -(2-ethylphenyl)-1 H-pyrazole-4-carboxamide
0
H2N
N'
H2N N
CH3
In analogy to the preparation of Example 15A, 2.58 g (87% of theory) of the
desired product are
obtained from 2.75 g (12.8 mmol) of 5-amino-l-(2-ethylphenyl)-1H-pyrazole-4-
carbonitrile
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-40-
(Example 9A) in a mixture of 106 ml of ethanol, 27 ml of 30% strength hydrogen
peroxide and
133 ml of 25% strength ammonia after chromatography on silica gel (mobile
phase
dichloromethane with 0-10% methanol).
m.p.: 147 C
MS (ESI pos): m/z = 231 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.0 (t, 3H), 2.4 (q, 2H), 5.95 (s, 2H), 6.3
(broad d, 2H), 7.2-
7.5 (m, 4H), 7.8 (s, 1H) ppm.
Example 25A
5-Amino- l -(2-trifluoromethylphenyl)-1 H-pyrazole-4-carboxamide
O
H 2 N
1
H2N NON
L-CF3
In analogy to the preparation of Example 15A, 4.01 g (87% of theory) of the
desired product are
obtained from 5.0 g (19.8 mmol) of 5-amino-l-(2-trifluoromethylphenyl)-1H-
pyrazole-4-
carbonitrile (Example 10A) in a mixture of 195 ml of ethanol, 49 ml of 30%
strength hydrogen
peroxide and 244 ml of 25% strength ammonia after chromatography on silica gel
(mobile phase
dichloromethane with 0-10% methanol).
m.p.: 186 C
MS (ESI pos): m/z = 271 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.1 (s, 2H), 7.0 (broad d, 2H), 7.45-8.0 (m,
5H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-41-
Example 26A
5-Amino-l-(2-fluorophenyl)-1 H-pyrazole-4-carboxamide
O
H 2 N
Dol H 2N NN
F
In analogy to the preparation of Example 15A, 3.89 g (81% of theory) of the
desired product are
obtained from 5.0 g (21.9 mmol, 89% purity) of 5-amino-l-(2-fluorophenyl)-1H-
pyrazole-4-
carbonitrile (Example 11A) in a mixture of 173 ml of ethanol, 43 ml of 30%
strength hydrogen
peroxide and 216 ml of 25% strength ammonia after chromatography on silica gel
(mobile phase
dichloromethane with 0-10% methanol).
m.p.: 181 C
MS (ESI pos): m/z = 221 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 6.2 (s, 2H), 7.0 (broad d, 2H), 7.3-7.6 (m,
4H), 7.9 (s, 1H)
PPM-
Example 27A
5-Amino- l -(2-chlorophenyl)-1 H-pyrazole-4-carboxamide
O
H 2 N
H2N NON
Cl
In analogy to the preparation of Example 15A, 3.93 g (79% of theory) of the
desired product are
obtained from 4.6 g (21.0 mmol) of 5-amino-l-(2-chlorophenyl)-1H-pyrazole-4-
carbonitrile
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-42-
(Example 12A) in a mixture of 159 ml of ethanol, 39 ml of 30% strength
hydrogen peroxide and
198 ml of 25% strength ammonia after chromatography on silica gel (mobile
phase
dichloromethane with 0-10% methanol).
m.p.: 166 C
MS (ESI pos): m/z = 237 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 6.1 (s, 2H), 7.0 (broad d, 2H), 7.4-7.7 (m,
4H), 7.85 (s, 1H)
ppm.
Example 28A
5-Amino- l -(2-pyridinyl)-1 H-pyrazole-4-carboxamide
O
H 2 N
H2N NON
N
In analogy to the preparation of Example 15A, 2.28 g (90% of theory) of the
desired product are
obtained from 2.3 g (12.4 mmol) of 5-amino-l-(2-pyridinyl)-1H-pyrazole-4-
carbonitrile (Example
13A) in a mixture of 90 ml of ethanol, 23 ml of 30% strength hydrogen peroxide
and 113 ml of
25% strength ammonia after chromatography on silica gel (mobile phase
dichloromethane with
0-10% methanol).
m.p.: 218 C
MS (DCI): m/z = 204 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 7.1 (broad d, 2H), 7.3 (dd, 1H), 7.5 (s, 2H),
7.85 (d, 1H), 7.95
(s, I H), 8.0 (dd, I H), 8.45 (d, I H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-43-
Example 29A
5-Amino- l -(2-methoxyphenyl)- I H-pyrazole-4-carboxamide
O
H 2 N
H2N NON
01" CH3
In analogy to the preparation of Example 15A, 2.61 g (70% of theory) of the
desired product are
obtained from 3.5 g (16.0 mmol, 98% purity) of 5-amino-l-(2-methoxyphenyl)-1H-
pyrazole-4-
carbonitrile (Example 14A) in a mixture of 172 ml of ethanol, 34 ml of 30%
strength hydrogen
peroxide and 137 ml of 25% strength ammonia after chromatography on silica gel
(mobile phase
dichloromethane with 0-10% methanol).
m.p.: 191 C
MS (ESI pos): m/z = 233 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 3.8 (s, 3H), 5.9 (s, 2H), 7.0 (broad s, 2H),
7.05-7.55 (m, 4H),
7.8 (s, 1H) ppm.
Example 30A
5-Amino-l-(2-ethoxyphenyl)-1 H-pyrazole-4-carbonitrile
NC
H2N NON
ON~I/CH3
In analogy to the preparation of Example IA, 2.9 g (59% of theory) of the
desired product are
obtained starting from 4.0 g (21.2 mmol) of 2-ethoxyphenylhydrazine
hydrochloride, 2.5 g
(21.2 mmol) of ethoxymethylenemalononitrile and 8.8 ml (63.6 mmol) of
triethylamine.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-44-
LC-MS (Method 1): R, = 2.32 min.
MS (ESI pos): m/z = 229 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 1.25 (t, 3H), 4.08 (q, 2H), 6.37 (s, 2H), 7.04
(m, 1H), 7.25 (m,
2H), 7.45 (m, 1H), 7.71 (s, 1H) ppm.
Example 31A
5-Amino-l-(2-ethoxyphenyl)-1 H-pyrazole-4-carboxamide
O
H2N
N
HN Ni
OIn analogy to the preparation of Example 15A, 2.2 g (84% of theory) of the
desired product are
obtained from 2.5 g (10.9 mmol) of 5-amino-l-(2-ethoxyphenyl)-1H-pyrazole-4-
carbonitrile
(Example 30A) in a mixture of 20 ml of ethanol, 10 ml of 30% strength hydrogen
peroxide and
10 ml of 25% strength ammonia.
LC-MS (Method 4): R, = 1.73 min.
MS (ESI pos): m/z = 247 (M+H)+.
Example 32A
cis-Hexahydro-2H-cyclopenta[b]furan-2-one
H
O )zzz~_O
H
32 ml of concentrated sulphuric acid (96% strength) are cooled to -10 C. Then
5.0 g (39.6 mmol)
of 2-cyclopenten-1-ylacetic acid are slowly metered in, and the reaction
mixture is stirred at the
same temperature for 1 h. It is poured into 100 ml of ice-water and extracted
with 100 ml of diethyl
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-45 -
ether. The organic phase is dried over sodium sulphate and the solvent is
cautiously distilled off.
2.9 g of the racemic lactone are obtained in 70% purity (LC-MS) and are
employed further as
crude product.
MS (ESI pos): m/z = 127 (M+H)+.
Example 33A
3 -Hydrazino-4-methylpyridine
HN~NH2
CH3
2.55 ml of a 2.5 M aqueous sodium nitrite solution are added to a solution of
4.0 g (37 mmol) of
3-amino-4-methylpyridine in 19 ml of 6 N hydrochloric acid while cooling in an
ice/salt bath. The
resulting diazonium salt solution is slowly added dropwise at -10 C to -15 C
to a solution of 21 g
(111 mmol) of tin(II) chloride in 26 ml of hydrochloric acid. The solution is
left to stand in a
refrigerator overnight in order to complete the reaction. The precipitated
solid is filtered off with
suction, suspended in 26 ml of water, made basic with concentrated sodium
hydroxide solution and
filtered. The filtrate is extracted ten times with 20 ml of dichloromethane
each time, and the
combined organic phases are dried over sodium sulphate and concentrated.
Drying under high
vacuum results in 1.45 g (31% of theory) of the desired product as a
colourless oil.
MS (ESI pos): m/z = 124 (M+H)+
'H-NMR (400 MHz, DMSO-d6): S = 2.05 (s, 3H), 4.05 (s, 2H), 6.4 (s, 1H), 6.9
(d, 1H), 7.75 (d,
1H), 8.3 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-46-
Example 34A
-Amino-l-(4-methylpyridin-3 -yl)-1 H-pyrazole-4-carbonitrile
NC
\N
H2N -)/: Ni
LJCH3
N
In analogy to the preparation of Example IA, 1.75 g (75% of theory) of the
desired product are
5 obtained starting from 1.44 g (11.7 mmol) of 3-hydrazino-4-methylpyridine
(Example 33A), 1.47 g
(11.7 mmol) of ethoxymethylenemalononitrile and 4.9 ml (35 mmol) of
triethylamine.
MS (ESI pos): m/z = 200 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 2.1 (s, 3H), 6.7 (s, 2H), 7.45 (d, 1H), 7.8 (s,
IH), 8.4 (s, 1H),
8.55 (d, 1H) ppm.
Example 35A
5 -Amino-l-(4-methylpyridin-3 -yl)-1 H-pyrazole-4-carboxamide
O
H 2 N
N
H 2 N Ni
L,CH3
N
In analogy to the preparation of Example 15A, 1.75 g (91% of theory) of the
desired product are
obtained from 1.75 g (8.78 mmol) of 5-amino-l-(4-methylpyridin-3-yl)-1H-
pyrazole-4-carbonitrile
(Example 34A) in a mixture of 105 ml of ethanol, 9.2 ml of 30% strength
hydrogen peroxide and
84 ml of 25% strength ammonia.
MS (ESI pos): m/z = 218 (M+H)+
Le A 36 520-Foreign countries CA 02524900 2005-11-07
- 47 -
'H-NMR (200 MHz, DMSO-d6): 6 = 2.1 (s, 3H), 6.2 (s, 2H), 6.6-7.5 (2 broad s,
2H), 7.45 (d, 1H),
7.9 (s, 1 H), 8.4 (s, 1 H), 8.5 (d, 1 H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-48-
Exemplary embodiments:
Example 1
6-Cyclopentylmethyl- l -(2,6-dimethylphenyl)-1,5 -dihydropyrazolo[3,4-
d]pyrimidin-4-one
0
HN
N N
H3C / CH3
1
0.1 g (0.43 mmol) of 5-amino-l-(2,6-dimethylphenyl)-1H-pyrazole-4-carboxamide
(Example 15A)
is dissolved under argon in 6 ml of absolute ethanol and 0.24 g (1.7 mmol) of
methyl
cyclopentylacetate and 0.17 g (4.34 mmol) of 60% sodium hydride (suspension in
mineral oil) are
added. The reaction mixture is heated to reflux overnight. Cooling to room
temperature is followed
by acidification with concentrated hydrochloric acid. The sodium chloride
precipitated thereby is
filtered off. The filtrate is concentrated in vacuo, and the remaining residue
is purified by
preparative HPLC (YMC Gel ODS-AQ S 5/15 m; eluent A: water, eluent B:
acetonitrile;
gradient: 0 min 30% B, 5 min 30% B, 50 min 95% B). 74 mg (53% of theory) of
the product are
obtained as a colourless solid.
LC-MS (Method 3): Rt = 3.79 min.
MS (ESI pos): m/z = 323 (M+H)+.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-49-
Example 2
6-Cyclopentylmethyl-l -(2,3-dimethylphenyl)-1,5-dihydropyrazolo[3,4-
d]pyrimidin-4-one
0
HN N
N N
LCH3
CH3
0.1 g (0.43 mmol) of 5-amino-1-(2,3-dimethylphenyl)-1H-pyrazole-4-carboxamide
(Example 16A)
is dissolved under argon in 6 ml of absolute ethanol and 0.24 g (1.7 mmol) of
methyl
cyclopentylacetate and 0.17 g (4.34 mmol) of 60% sodium hydride (suspension in
mineral oil) are
added. The reaction mixture is heated to reflux overnight. Cooling to room
temperature is followed
by acidification with concentrated hydrochloric acid. The mixture of sodium
chloride and the
product precipitated thereby is filtered off and washed several times with
water and diethyl ether.
Drying under high vacuum results in 69 mg (49% of theory) of the product as
colourless solid.
LC-MS (Method 3): R, = 3.57 min.
MS (ESI pos): m/z = 323 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 1.17 (m, 2H), 1.48 (m, 2H), 1.59 (m, 4H), 1.87
(s, 3H), 2.19
(m, IH), 2.33 (s, 3H), 2.54 (d, 2H), 7.16 (d, 1H), 7.25 (t, 1H), 7.36 (d, 1H),
8.21 (s, 1H), 12.12 (s,
1 H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-50-
Example 3
6-Cyclopentylmethyl-1-(4-methylphenyl)-1,5-dihydropyrazolo[3,4-d]pyrimidin-4-
one
0
HN
N
N N
CH3
In analogy to the preparation of Example 1, 97 mg (68% of theory) of the
desired product are
obtained as a colourless solid starting from 0.88 g (0.41 mmol) of 5-amino-l-
(4-methylphenyl)-IH-
pyrazole-4-carboxamide (Example 17A), 0.26 g (1.8 mmol) of methyl
cyclopentylacetate and
0.16 g (4.09 mmol) of 60% sodium hydride.
LC-MS (Method 3): R, = 4.09 min.
MS (ESI pos): m/z = 309 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.23 (m, 2H), 1.57 (m, 2H), 1.72 (m, 4H), 2.34
(m, 1H), 2.36
(s, 3H), 2.66 (d, 2H), 7.34 (d, 1H), 7.92 (d, 1H), 8.23 (s, 1H), 12.27 (s, 1H)
ppm.
Example 4
6-Cyc lopentylmethyl- l -(2, 6-dichlorophenyl)-1, 5 -dihydropyrazolo [3 ,4-
d]pyrimidin-4-one
0
HN I \
N N
CI CI
In analogy to the preparation of Example 2, 61 mg (45% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.37 mmol) of 5-amino-l-
(2,6-dichlorophenyl)-
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-51-
1H-pyrazole-4-carboxamide (Example 18A), 0.2 g (1.4 mmol) of methyl
cyclopentylacetate and
0.14 g (3.6 mmol) of 60% sodium hydride.
LC-MS (Method 3): Rt = 3.73 min.
MS (ESI pos): m/z = 363 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 1.15 (m, 2H), 1.49 (m, 2H), 1.60 (m, 4H), 2.21
(m, 1H), 2.57
(d, 2H), 7.60 (m, 2H), 7.69 (m, 1H), 8.41 (s, 1H), 12.51 (s, 1H) ppm.
Example 5
6-Cyclopentylmethyl-l -(2,5-dichlorophenyl)-1,5-dihydropyrazolo[3,4-
d]pyrimidin-4-one
0
HN
N N
CI
CI
In analogy to the preparation of Example 1, 32 mg (23% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.37 mmol) of 5-amino-l-
(2,5-dichlorophenyl)-
1H-pyrazole-4-carboxamide (Example 19A), 0.2 g (1.4 mmol) of methyl
cyclopentylacetate and
0.14 g (3.6 mmol) of 60% sodium hydride.
LC-MS (Method 3): Rt = 4.0 min.
MS (ESI pos): m/z = 363 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.15 (m, 2H), 1.49 (m, 2H), 1.60 (m, 4H), 2.22
(m, IH), 2.55
(d, 2H), 7.16 (d, 1H), 7.31 (m, 1H), 7.32 (m, 2H), 8.23 (s, 1H), 12.39 (s, 1H)
ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-52-
Example 6
1-(2-Aminophenyl)-6-cyclopentylmethyl-1,5 -dihydropyrazolo [3,4-d]pyrimidin-4-
one
0
HN
N
N N
NH2
In analogy to the preparation of Example 1, 61 mg (42% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.46 mmol) of 5-amino-1-(2-
aminophenyl)-1H-
pyrazole-4-carboxamide (Example 21A), 0.19 g (1.4 mmol) of methyl
cyclopentylacetate and
0.18 g (4.6 mmol) of 60% sodium hydride.
LC-MS (Method 4): R, = 3.9 min.
MS (ESI pos): m/z = 310 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 1.17 (m, 2H), 1.45 (m, 2H), 1.56 (m, 4H), 2.19
(m, 1H), 2.52
(d, 2H), 6.12 (s, 2H), 6.64 (m, 1H), 6.90 (m, 1H), 7.05 (m, 2H), 8.25 (s, 1H),
12.47 (s, 1H) ppm.
Example 7
6-Cyc lopentylmethyl- l -(3 -fluorophenyl)-1, 5 -dihydropyrazol o [3 ,4-
d]pyrimidin-4-one
0
HN
N
F
In analogy to the preparation of Example 1, 82 mg (58% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.45 mmol) of 5-amino-1-(3-
fluorophenyl)-1H-
pyrazole-4-carboxamide (Example 22A), 0.26 g (1.8 mmol) of methyl
cyclopentylacetate and
0.18 g (4.5 mmol) of 60% sodium hydride.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
- 53 -
LC-MS (Method 3): R, = 3.74 min.
MS (ESI pos): m/z = 313 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.12 (m, 2H), 1.58 (m, 2H), 1.75 (m, 4H), 2.34
(m, 1H), 2.69
(d, 2H), 7.23 (m, IH), 7.63 (m, I H), 8.00 (m, 2H), 8.31 (s, I H), 12.37 (s, I
H) ppm.
Example 8
6-(2-Cyclopenten-1-ylmethyl)-1-(2-ethylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
0
HN
N
I N N
b,-- CH3
In analogy to the preparation of Example 1, 64 mg (31% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.65 mmol) of 5-amino-1-
(2-ethylphenyl)-1H-
pyrazole-4-carboxamide (Example 24A), 0.27 g (1.95 mmol) of methyl 2-
cyclopenten-l-ylacetate
and 0.13 g (3.2 mmol) of 60% sodium hydride.
m.p.: 146 C
MS (ESI pos): m/z = 321 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 0.95 (t, 3H), 1.45 (m, 1H), 1.95 (m, 1H), 2.1-
2.75 (m, 6H),
3.0 (m, 1H), 5.5-5.8 (m, 2H), 7.25-7.5 (m, 4H), 8.2 (s, 1H), 12.2 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-54-
Example 9
6-(2-Cyclopenten-1-ylmethyl)-1-(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
0
HN !r'~
N N CH3
In analogy to the preparation of Example 1, 44 mg (26% of theory) of the
desired product are
obtained as a colourless solid starting from 0.12 g (0.56 mmol) of 5-amino-l-
(2-methylphenyl)-1H-
pyrazole-4-carboxamide (Example 23A), 0.24 g (1.7 mmol) of methyl 2-
cyclopenten-l-ylacetate
and 0.11 g (2.8 mmol) of 60% sodium hydride.
m.p.: 179 C
MS (ESI pos): m/z = 307 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.45 (m, 1H), 1.95 (m, 1H), 2.1 (s, 3H), 2.1-
2.75 (m, 4H),
3.05 (m, 1H), 5.5-5.8 (m, 2H), 7.3-7.5 (m, 4H), 8.25 (s, 1H), 12.2 (s, 1H)
ppm.
Example 10
6-Cyclohexylmethyl- l -(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN
N CH3
In analogy to the preparation of Example 1, 65 mg (29% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.68 mmol) of 5-amino-l-
(2-methylphenyl)-1H-
pyrazole-4-carboxamide (Example 23A), 0.35 g (2.04 mmol) of ethyl
cyclohexylacetate and
0.136 g (3.4 mmol) of 60% sodium hydride.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-55-
m.p.: 169 C
MS (ESI pos): m/z = 323 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 0.9-1.3 (m, 5H), 1.5-1.9 (m, 6H), 2.1 (s, 3H),
2.45 (d, 2H),
7.3-7.5 (m, 4H), 8.2 (s, 1H), 12.2 (s, 1H) ppm.
Example 11
6-Cyclopentylmethyl- l -(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN I \
N N CH3
In analogy to the preparation of Example 1, 43 mg (30% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.46 mmol) of 5-amino-l-(2-
methylphenyl)-1H-
pyrazole-4-carboxamide (Example 23A), 0.237 g (92% purity, 1.39 mmol) of ethyl
cyclopentylacetate and 0.093 g (2.32 mmol) of 60% sodium hydride.
m.p.: 181 C
MS (ESI pos): mlz = 309 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 1.1-1.55 (m, 8H), 2.1 (s, 3H), 2.2 (m, 1H),
2.55 (d, 2H), 7.3-
7.5 (m, 4H), 8.2 (s, IH), 12.15 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-56-
Example 12
6-Cyclopentylmethyl- l -(2-ethoxyphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
!r'~
HN N N
O----/CH3
In analogy to the preparation of Example 1, 73 mg (52% of theory) of the
desired product were
obtained as a colourless solid starting from 0.1 g (0.41 mmol) of 5-amino-l-(2-
ethoxyphenyl)-1H-
pyrazole-4-carboxamide (Example 31A), 0.231 g (1.6 mmol) of ethyl
cyclopentylacetate and
0.162 g (4.1 mmol) of 60% sodium hydride.
LC-MS (Method 3): Rt = 3.5 min.
MS (ESI pos): m/z = 339 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.10 (t, 3H), 1.22 (m, 2H), 1.45 (m, 2H), 1.59
(m, 4H), 1.96
(m, 1H), 2.54 (d, 2H), 4.02 (q, 2H), 7.08 (m, 1H), 7.23 (m, 1H), 7.37 (m, 1H),
7.48 (m, 1H), 8.16
(s, 1H), 12.06 (s, 1H) ppm.
Example 13
6-Cyclopentylmethyl- l -(2-hydroxyphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
0
HN I N OH
4 ml of 1 M boron tribromide solution in dichloromethane are added to 0.2 g
(0.59 mmol) of
6-cyclopentylmethyl-l -(2-ethoxyphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
(Example 12), and the reaction mixture was stirred at room temperature for 1
h. Aqueous
hydrolysis is followed by extraction with dichloromethane. The product is
purified by preparative
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-57-
HPLC (YMC Gel ODS-AQ S 5/15 m; eluent A: water, eluent B: acetonitrile;
gradient: 0 min
30% B, 5 min 30% B, 50 min 95% B). 0.167 g (91% of theory) of the product is
obtained as a
colourless solid.
LC-MS (Method 4): R, = 2.54 min.
MS (ESI pos): m/z = 311 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.17 (m, 2H), 1.42 (m, 6H), 2.19 (m, 1H), 2.54
(d, 2H), 6.93
(m, 1H), 7.04 (m, 1H), 7.32 (m, 1H), 8.18 (s, 1H), 9.92 (s, 1H), 12.12 (s, 1H)
ppm.
Example 14
6-(2-Cyclopenten-l-ylmethyl)-1-[2-(trifluoromethyl)phenyl]-1, 5-dihydro-4H-
pyrazolo [3,4-
d]pyrimidin-4-one
O
HN
N N CF3
In analogy to the preparation of Example 1, 57 mg (29% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.56 mmol) of 5-amino-l-
[2-
(trifluoromethyl)phenyl]-1H-pyrazole-4-carboxamide (Example 25A), 0.233 g
(1.67 mmol) of
methyl 2-cyclopenten-l-ylacetate and 0.111 g (2.78 mmol) of 60% sodium
hydride.
m.p.: 153 C
MS (ESI pos): m/z = 361 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.45 (m, IH), 1.9 (m, 1H), 2.1-2.4 (m, 2H),
2.45-2.7 (m, 2H),
3.0 (m, IH), 5.5-5.8 (m, 2H), 7.6 (d, 1H), 7.75-8.0 (m, 3H), 8.25 (s, 1H),
12.2 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-58-
Example 15
6-(2-Cyclopenten-l-ylmethyl)-1-(2-fluorophenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN !r'~
N
N F
In analogy to the preparation of Example 1, 77 mg (37% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.66 mmol) of 5-amino-l-
(2-fluorophenyl)-1H-
pyrazole-4-carboxamide (Example 26A), 0.279 g (1.99 mmol) of methyl 2-
cyclopenten-l-ylacetate
and 0.133 g (2.78 mmol) of 60% sodium hydride.
m.p.: 163 C
MS (ESI pos): m/z = 311 (M+H)+
'H-NMR (300 MHz, DMSO-d6): b = 1.5 (m, 1H), 1.95 (m, 1H), 2.1-2.45 (m, 2H),
2.45-2.7 (m,
2H), 3.0 (m, IH), 5.6-5.8 (m, 2H), 7.3-7.7 (m, 4H), 8.3 (s, 1H), 12.3 (s, 1H)
ppm.
Example 16
6-(2-Cyclopenten-1-ylmethyl)-1-(2-chlorophenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN ( \
N
N
N ddl
In analogy to the preparation of Example 1, 50 mg (24% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.63 mmol) of 5-amino-l-
(2-chlorophenyl)-1H-
pyrazole-4-carboxamide (Example 27A), 0.266 g (1.90 mmol) of methyl 2-
cyclopenten-l-ylacetate
and 0.127 g (3.17 mmol) of 60% sodium hydride.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-59-
m.p.: 150 C
MS (ESI pos): m/z = 327 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.5 (m, 1H), 1.95 (m, 1H), 2.1-2.4 (m, 2H), 2.5-
2.7 (m, 2H),
3.05 (m, 1H), 5.6-5.8 (m, 2H), 7.5-7.8 (m, 4H), 8.25 (s, IH), 12.2 (s, IH)
ppm.
Example 17
6-(2-Cyclopenten-1-ylmethyl)-1-(2-pyridinyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN !r\,
N
N In analogy to the preparation of Example 1, 76 mg (35% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.74 mmol) of 5-amino-l-
(2-pyridinyl)-1H-
pyrazole-4-carboxamide (Example 28A), 0.31 g (2.21 mmol) of methyl 2-
cyclopenten-l-ylacetate
and 0.147 g (3.69 mmol) of 60% sodium hydride.
m.p.: 239 C
MS (ESI pos): m/z = 294 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 8 = 1.55 (m, 1H), 2.0 (m, 1H), 2.15-2.45 (m, 2H),
2.55-2.75 (m,
2H), 3.15 (m, 1H), 5.65-5.8 (m, 2H), 7.5 (dd, 1H), 8.0 (d, 1H), 8.05 (m, 1H),
8.3 (s, 1H), 8.6 (d,
1 H), 12.3 (s, 1 H) ppm.
Example 18
6-(2-Cyclopenten-1-ylmethyl)-1-(2-methoxyphenyl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-
one
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-60-
O
HN \
N
N N
O~CH3
In analogy to the preparation of Example 1, 82 mg (39% of theory) of the
desired product are
obtained as a colourless solid starting from 0.15 g (0.65 mmol) of 5-amino-1-
(2-methoxyphenyl)-
1H-pyrazole-4-carboxamide (Example 29A), 0.272 g (1.94 mmol) of methyl 2-
cyclopenten-l-
ylacetate and 0.129 g (3.23 mmol) of 60% sodium hydride.
m.p.: 182 C
MS (ESI pos): m/z = 323 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 1.5 (m, 1H), 1.95 (m, 1H), 2.1-2.45 (m, 2H),
2.45-2.75 (m,
2H), 3.05 (m, 1H), 3.0 (s, 3H), 5.6-5.8 (m, 2H), 7.0-7.55 (m, 4H), 8.2 (s,
1H), 12.15 (s, 1H) ppm.
Exemplary embodiments 19 - 31 listed in Table 2 below are obtained just like
the corresponding
starting compounds in analogy to the examples described above:
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-61-
Table 2:
Ex. Structure Yield MS: m/z Rt LC-MS
No. [% of th.] [M+H]+ [min] method
O
19 HN IN 14.1 364 4.05 3
N
N
CI
CI
O
20 HN 29.8 337 3.97 3
N
N N
CH3
H3C
O
21 HN 26.1 337 4.52 3
N
CH3
H3C
0
22 HN N 48.5 363 4.39 3
I N
N
CF3
b
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-62-
Ex. Structure Yield MS: m/z Rt LC-MS
No. [% of th.] [M+H]+ [min] method
0
14.6 398 4.20 3
23 HN rQ
N CI
Ci
CI
0
24 HN\ N 78.7 325 3.88 3 N N
b,_0 /CH3
0
25 HN 28.4 364 4.70 3
N
N N
CI
CI
0
26 HN 48.9 329 4.30 3
N
N N
CI
0
27 HN I 60.1 325 3.79 3
N
N N
O,CH3
Le A 36 520-Foreign countries CA 02524900 2005-11-07
- 63 -
Ex. Structure Yield MS: m!z R, LC-MS
No. [% of th.] [M+H]+ [min] method
0
28 HN 10.5 340 3.61 1
N
N N
tN
O
CH3
0
29 HN I 7.9 324 4.00 4
N
\
N N
LN
CH3
O
30 HN 48.8 339 4.10 4
I
N
r N N
O-CH3
c H3C / 1
0
31 HN! 38.8 343 3.07 1
N N N
3
CI / CH
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-64-
Example 32
6-[(4-Methylcyclohexyl)methyl]-1-(2-methylphenyl)-1,5-dihydropyrazolo[3,4-
d]pyrimidin-4-one
0
HN
N N
C H 3
CH3
150 mg (0.69 mmol) of 5-amino-l-(2-methylphenyl)-1H-pyrazole-4-carboxamide
(Example 23A)
and 130 mg (0.83 mmol) of 2-(4-methylcyclohexyl)acetic acid are mixed with 3
ml of
trimethylsilyl polyphosphate and stirred at 130 C for 3 h. The hot reaction
mixture is added to
20 ml of water and then extracted with dichloromethane (2 x 20 ml). The
combined organic phases
are washed with water (20 ml) and with saturated sodium chloride solution (20
ml) and dried over
sodium sulphate. The solvent is distilled off under reduced pressure, and the
crude product is
purified by preparative HPLC (YMC Gel ODS-AQ S 5/15 m; eluent A: water,
eluent B: aceto-
nitrile; gradient: 0 min 30% B, 5 min 30% B, 50 min 95% B). 182 g (78% of
theory) of the product
are obtained.
LC-MS (Method 3): Rt = 4.09 min.
MS (ESI pos): mlz = 337 (M+H)+
'H-NMR (200 MHz, DMSO-d6): 8 = 0.68-0.90 (5H), 0.99-1.61 (814), 1.98-2.07
(414), 2.16 (d, 1H),
7.19 (d, 114), 7.28-7.51 (m, 3H), 8.26 (s, IH), 10.27 (s, 1H) ppm.
Example 33
6- { [(1,2-cis)-2-Hydroxycyclopentyl]methyl 1-1 -(2-methylphenyl)-1,5-dihydro-
4H-pyrazolo[3,4-
d]pyrimidin-4-one (racemate)
Le A 36 520-Foreign countries CA 02524900 2005-11-07
- 65 -
O
HN
HO N NON
LCH3
200 mg (0.93 mmol) of 5-amino-l-(2-methylphenyl)-1H-pyrazole-4-carboxamide
(Example 23A)
and 525 mg of cis-hexahydro-2H-cyclopenta[b]furan-2-one (approx. 70% pure,
Example 32A) are
dissolved in 10 ml of absolute ethanol under argon, and 315 mg (4.6 mmol) of
sodium ethoxide are
added. The reaction mixture is heated to reflux overnight. Cooling to room
temperature is followed
by hydrolysis with 25 ml of water and then extraction with ethyl acetate (2 x
25 ml). The combined
organic phases are dried over sodium sulphate, and the solvent is distilled
off under reduced
pressure. The crude product is purified by preparative HPLC (YMC Gel ODS-AQ S
5/15 m;
eluent A: water, eluent B: acetonitrile; gradient: 0 min 30% B, 5 min 30% B,
50 min 95% B).
90 mg (30% of theory) of the desired product are obtained.
MS (ESI pos): m/z = 325 (M+H)+
'H-NMR (300 MHz, DMSO-d6): 6 = 1.28-1.74 (7H), 2.07 (s, 3H), 2.55 (dd, 1H),
2.80 (dd, 1H),
3.97 (m, 1H), 4.43 (d, 1H), 7.36 (m, 2H), 7.43 (m, 2H), 8.22 (s, 1H), 12.07
(s, 1H) ppm.
Example 34
6-{[(1,2-trans)-2-Hydroxycyclohexyl]methyl}-1-(2-methylphenyl)-1,5-dihydro-4H-
pyrazolo[3,4-
d]pyrimidin-4-one
O
HN
HO N
N
CH3
200 mg (0.93 mmol) of 5-amino-l-(2-methylphenyl)-1H-pyrazole-4-carboxamide
(Example 23A)
and 583 mg (4.16 mmol) of rac-hexahydro-1-benzofuran-2(3H)one (mixture of the
cis and trans
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-66-
diastereomers; for preparation, see, for example, K.F. Podraza et al., J.
Heterocycl. Chem. 1987,
24, 293-295) are dissolved in 10 ml of absolute ethanol under argon, and 315
mg (4.6 mmol) of
sodium ethoxide are added. The reaction mixture is heated to reflux overnight.
Cooling to room
temperature is followed by hydrolysis with 25 ml of water and then extraction
with ethyl acetate (2
x 25 ml). The combined organic phases are dried over sodium sulphate, and the
solvent is distilled
off under reduced pressure. The crude product is purified by preparative HPLC
(YMC Gel ODS-
AQ S 5/15 m; eluent A: water, eluent B: acetonitrile; gradient: 0 min 30% B,
5 min 30% B,
50 min 95% B). 68 mg (21 % of theory) of the desired product are obtained.
MS (ESI pos): m/z = 339 (M+H)+
'H-NMR (400 MHz, DMSO-d6): S = 0.97 (m, 2H), 1.15 (m, 2H), 2.51 (d, 2H), 1.64
(m, 2H), 1.81
(m, 1H), 2.07 (s, 3H), 2.26 (dd, 1H), 2.99-3.10 (2H), 4.61 (d, 1H), 7.37 (m,
2H), 7.44 (m, 2H), 8.23
(s, 1H), 12.11 (s, 1H) ppm.
Example 35
6-(2-Methylbutyl)-1-(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-
one (racemate)
0
HN I \
N
N N C H 3
H3C 4 / \
CH3 -'
In analogy to the preparation of Example 1, 784 mg (71% of theory) of the
desired product are
obtained as a colourless solid starting from 0.8 g (3.7 mmol) of 5-amino-l-(2-
methylphenyl)-1H-
pyrazole-4-carboxamide (Example 23A), 2.72 g (98% purity, 18.5 mmol) of ethyl
3-methylvalerate
and 0.740 g (24 mmol) of 60% sodium hydride.
m.p.:132 C
MS (ESI pos): m/z = 297 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S = 0.8 (m, 6H), 1.1-1.4 (m, 2H), 1.9 (m, 1H), 2.1
(s, 3H), 2.4
(dd, 1H), 2.55 (dd, 1H), 7.3-7.5 (m, 4H), 8.2 (s, 1H), 12.2 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-67-
Example 35-1
6-(2-Methylbutyl)-1-(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-
one
(enantiomer I)
The racemate from Example 35 (380 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase [based on the chiral selector poly(N-methacryloyl-L-leucine L-
menthylamide), for
the principle of preparation and use, see EP-A-379 917; 380 mm x 100 mm
column, flow rate
100 ml/min, temperature 24 C, mobile phase: isohexane / ethyl acetate 20:80].
Example 35-1 is the
enantiomer I which elutes more quickly under these conditions (R, = 15.2 min).
m.p.: 122 C
Example 35-2
6-(2-Methylbutyl)-1-(2-methylphenyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-
one
(enantiomer II)
The racemate from Example 35 (380 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase [based on the chiral selector poly(N-methacryloyl-L-leucine L-
menthylamide), for
the principle of preparation and use, see EP-A-379 917; 380 mm x 100 mm
column, flow rate
100 ml/min, temperature 24 C, mobile phase: isohexane / ethyl acetate 20:80].
Example 35-2 is the
enantiomer II which elutes more slowly under these conditions (Rt = 18.1 min).
m.p.: 122 C
Example 36
1-(2-Methylphenyl)-6-(3, 3 , 3 -tri fluoro-2-methylpropyl)-1,5 -dihydro-4H-
pyrazolo [ 3 ,4-d]pyrimidin-
4-one (racemate)
0
HN
N F3C 3 dCH3
In analogy to the preparation of Example 1, 216 mg (69% of theory) of the
desired product are
obtained as a colourless solid starting from 0.2 g (0.92 mmol) of 5-amino-l-(2-
methylphenyl)-1H-
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-68-
pyrazole-4-carboxamide (Example 23A), 0.852 g (4.62 mmol) of ethyl 3-methyl-
4,4,4-
trifluorobutyrate and 0.129 g (3.24 mmol) of 60% sodium hydride.
m.p.: 160 C
MS (ESI pos): m/z = 337 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 6 = 1.1 (d, 3H), 2.1 (s, 3H), 2.7 (dd, 1H), 2.85-
3.0 (m, 2H), 7.3-
7.5 (m, 4H), 8.3 (s, 1H), 12.4 (s, 1H) ppm.
Example 36-1
1-(2-Methylphenyl)-6-(3,3,3-trifluoro-2-methylpropyl)-1,5-dihydro-4H-
pyrazolo[3,4-d]pyrimidin-
4-one (enantiomer I)
The racemate from Example 36 (180 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase (column: Chiralpak AD, 250 mm x 20 mm; flow rate: 20 ml/min;
temperature:
24 C; mobile phase: isohexane / isopropanol 92:8). Example 36-1 is the
enantiomer I which elutes
more quickly under these conditions (R, = 10.37 min).
m.p.: 154 C
Example 36-2
1-(2-Methylphenyl)-6-(3,3,3-trifluoro-2-methylpropyl)-1,5-dihydro-4H-
pyrazolo[3,4-d]pyrimidin-
4-one (enantiomer II)
The racemate from Example 36 (180 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase (column: Chiralpak AD, 250 mm x 20 mm; flow rate: 20 ml/min;
temperature:
24 C; mobile phase: isohexane / isopropanol 92:8). Example 36-2 is the
enantiomer II which elutes
more slowly under these conditions (Rt = 11.73 min).
m.p.: 153 C
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-69-
Example 37
1-(2-Chlorophenyl)-6-(3 , 3 , 3 -tri fluoro-2-methylpropyl)-1, 5 -dihydro-4H-
pyrazol o [ 3,4-d] pyrimidin-
4-one (racemate)
0
HN
N N CI
F3C CH3
In analogy to the preparation of Example 1, 321 mg (69% of theory) of the
desired product are
obtained as a colourless solid starting from 0.3 g (1.27 mmol) of 5-amino-l-(2-
chlorophenyl)-1H-
pyrazole-4-carboxamide (Example 27A), 1.17 g (6.34 mmol) of ethyl 3-methyl-
4,4,4-
trifluorobutyrate and 0.254 g (6.34 mmol) of 60% sodium hydride.
m.p.: 166 C
MS (ESI pos): m/z = 357 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 8 = 1.1 (d, 3H), 2.7 (dd, 1H), 2.85-3.0 (m, 2H),
7.5-7.8 (m, 4H),
8.3 (s, 1H), 12.4 (s, 1H) ppm.
Example 37-1
1-(2-Chlorophenyl)-6-(3, 3,3 -trifluoro-2-methylpropyl)-1,5-dihydro-4H-
pyrazolo [3,4-d]pyrimidin-
4-one (enantiomer I)
The racemate from Example 37 (240 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase (column: Chiralpak AD, 250 mm x 20 mm; flow rate: 20 ml/min;
temperature:
24 C; mobile phase: isohexane / isopropanol 92:8). Example 37-1 is the
enantiomer I which elutes
more quickly under these conditions (Rt = 11.92 min).
m.p.:220 C
Example 37-2
1-(2-Chlorophenyl)-6-(3,3,3-trifluoro-2-methylpropyl)-1,5-dihydro-4H-
pyrazolo[3,4-d]pyrimidin-
4-one (enantiomer II)
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-70-
The racemate from Example 37 (240 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase (column: Chiralpak AD, 250 mm x 20 mm; flow rate: 20 ml/min;
temperature:
24 C; mobile phase: isohexane / isopropanol 92:8). Example 37-2 is the
enantiomer II which elutes
more slowly under these conditions II (Rt = 12.67 min).
m.p.:218 C
Example 38
6-Cyclopentylmethyl-l -(4-methylpyridin-3-yl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN I \
N N CH3
N
In analogy to the preparation of Example 1, 102 mg (73% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.45 mmol) of 5-amino-l-(4-
methylpyridin-3-
yl)-1H-pyrazole-4-carboxamide (Example 35A), 0.353 g (2.26 mmol) of ethyl
cyclopentylacetate
and 0.09 g (2.26 mmol) of 60% sodium hydride.
m.p.: 206 C
MS (ESI pos): m/z = 310 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 8 = 1.1-1.8 (m, 8H), 2.2 (s, 3H), 2.22 (m, 1H), 2.6
(d, 2H), 7.5 (d,
1H), 8.3 (s, 1H), 8.6 (m, 2H), 12.3 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-71-
Example 39
6-(2-Methylbutyl)-I-(4-methylpyridin-3 -yl)-1, 5 -dihydro-4H-pyrazolo [ 3 ,4-
d]pyrimidin-4-one
(racemate)
0
HN
N N CH3
H3C
CH3 N--
In analogy to the preparation of Example 1, 186 mg (68% of theory) of the
desired product are
obtained as a colourless solid starting from 0.2 g (0.92 mmol) of 5-amino-l-(4-
methylpyridin-3-
yl)-1H-pyrazole-4-carboxamide (Example 35A), 0.677 g (4.6 mmol) of ethyl 3-
methylvalerate and
0.184 g (4.6 mmol) of 60% sodium hydride.
m.p.: 149 C
MS (ESI pos): m/z = 298 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 6 = 0.8 (m, 6H), 1.1-1.4 (m, 2H), 1.9 (m, 1H), 2.2
(s, 3H), 2.4
(dd, 1H), 2.6 (dd, 1H), 7.5 (d, 1H), 8.3 (s, 1H), 8.6 (m, 2H), 12.25 (s, 1H)
ppm.
Example 39-1
6-(2-Methylbutyl)-1-(4-methylpyridin-3-yl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
(enantiomer I)
The racemate from Example 39 (160 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase [based on the chiral selector poly(N-methacryloyl-L-leucine L-
menthylamide), for
the principle of preparation and use, see EP-A-379 917; 380 mm x 75 mm column,
flow rate
100 ml/min, temperature 24 C, mobile phase: isohexane / ethyl acetate 30:70].
Example 39-1 is the
enantiomer I which elutes more quickly under these conditions.
m.p.: 149 C
R, = 7.25 min [chiral selector poly(N-methacryloyl-L-leucine L-menthylamide),
250 mm x 4.6 mm
column; flow rate 1 ml/min; temperature 24 C; mobile phase ethyl acetate].
Le A 36 520-Foreign countriesCA 02524900 2005-11-07
-72-
Example 39-2
6-(2-Methylbutyl)-1-(4-methylpyridin-3-yl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
(enantiomer II)
The racemate from Example 39 (160 mg) is separated into the enantiomers by
HPLC on a chiral
stationary phase [based on the chiral selector poly(N-methacryloyl-L-leucine L-
menthylamide), for
the principle of preparation and use, see EP-A-379 917; 380 mm x 75 mm column,
flow rate
100 ml/min, temperature 24 C, mobile phase: isohexane / ethyl acetate 30:70].
Example 39-2 is the
enantiomer I which elutes more slowly under these conditions.
m.p.: 148 C
R, = 8.0 min [chiral selector poly(N-methacryloyl-L-leucine L-menthylamide),
250 mm x 4.6 mm
column; flow rate 1 ml/min; temperature 24 C; mobile phase ethyl acetate].
Example 40
1-(2-Chlorophenyl)-6-(2-methylpropyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-
4-one
0
HN
N N CI
H 3 C CH3
In analogy to the preparation of Example 1, 57 mg (45% of theory) of the
desired product are
obtained as a colourless solid starting from 0.1 g (0.42 mmol) of 5-amino-l-(2-
chlorophenyl)-1H-
pyrazole-4-carboxamide (Example 27A), 0.344 g (2.96 mmol) of ethyl 3-
methylbutyrate and
0.059 g (1.48 mmol) of 60% sodium hydride.
m.p.: 204 C
MS (ESI pos): m/z = 303 (M+H)+
'H-NMR (200 MHz, DMSO-d6): 6 = 0.9 (d, 6H), 2.05 (m, 1H), 2.45 (d, 2H), 7.5-
7.8 (m, 4H), 8.3
(s, 1H), 12.3 (s, 1H) ppm.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-73-
Example 41
6-(2-Ethylbutyl)-1-(4-methylpyridin-3-yl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
O
HN
N H3C CH3 dCH3
In analogy to the preparation of Example 1, 56 mg (49% of theory) of the
desired product are
obtained as a colourless solid from 0.08 g (0.37 mmol) of 5-amino-l-(4-
methylpyridin-3-yl)-1H-
pyrazole-4-carboxamide (Example 35A), 0.303 g (1.84 mmol) of ethyl 3-
ethylvalerate and 0.074 g
(1.84 mmol) of 60% sodium hydride.
m.p.: 143 C
MS (ESI pos): m/z = 312 (M+H)+
`H-NMR (200 MHz, DMSO-d6): 6 = 0.8 (t, 6H), 1.3 (m, 4H), 1.8 (m, 1H), 2.2 (s,
3H), 2.5 (d, 2H),
7.5 (d, 1H), 8.3 (s, 1H), 8.6 (m, 2H), 12.3 (s, 1H) ppm.
Example 42
6-Cyclopentylmethyl-l-(4-methyl-l-pyridin-3-yl-l -oxide)-1,5-dihydro-4H-
pyrazolo[3,4-
d]pyrimidin-4-one
O
HN
N
N N CH3
,N
O
48 mg (70% purity, 0.195 mmol) of meta-chloroperbenzoic acid are added to a
solution of 40 mg
(0.13 mmol) of 6-cyclopentylmethyl-l-(4-methylpyridin-3-yl)-1,5-dihydro-4H-
pyrazolo[3,4-
d]pyrimidin-4-one (Example 38) in 2 ml of dichloromethane at room temperature
and stirred
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-74-
overnight. The mixture is then stirred at 40 C for 1.5 h until the conversion
is complete according
to a check of the reaction (TCL). For working up, saturated sodium bicarbonate
solution is added,
and the mixture is extracted three times with dichloromethane. The combined
organic phases are
dried over sodium sulphate and concentrated. The crude product is purified by
preparative HPLC.
32 mg (76% of theory) of the desired product are obtained as a colourless
solid.
MS (ESI pos): m/z = 310 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 6 = 1.1-1.8 (m, 8H), 2.2 (s, 3H), 2.22 (m, IH), 2.6
(d, 2H), 7.5 (d,
IH), 8.3 (s, 1H), 8.6 (m, 2H), 12.3 (s, IH) ppm.
Example 43
6-Cyclohexylmethyl-l-(4-methylpyridin-3-yl)-1,5-dihydro-4H-pyrazolo[3,4-
d]pyrimidin-4-one
0
HN ( \
N N CH3
dN'~\
In analogy to the preparation of Example 1, 68 mg (73% of theory) of the
desired product are
obtained as a colourless solid starting from 0.08 g (0.37 mmol) of 5-amino-1-
(4-methylpyridin-3-
yl)-1H-pyrazole-4-carboxamide (Example 35A), 0.32 g (1.84 mmol) of ethyl
cyclohexylacetate and
0.074 g (1.84 mmol) of 60% sodium hydride.
m.p.: 206 C
MS (ESI pos): m/z = 324 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 6 = 0.8-1.3 (m, 6H), 1.5-1.9 (m, 5H), 2.2 (s, 3H),
2.5 (d, 2H), 7.5
(d, 1H), 8.3 (s, 1H), 8.6 (m, 2H), 12.25 (s, 1H) ppm.
The compounds of the invention can be converted into pharmaceutical
preparations in the
following ways:
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-75-
Tablet:
Composition:
100 mg of the compound of the invention, 50 mg of lactose (monohydrate), 50 mg
of maize starch
(native), 10 mg of polyvinylpyrrolidone (PVP 25) (from BASF, Ludwigshafen,
Germany) and
2 mg of magnesium stearate.
Tablet weight 212 mg, diameter 8 mm, radius of curvature 12 mm.
Production:
A mixture of compound of the invention, lactose and starch is granulated with
a 5% strength
solution (m/m) of the PVP in water. The granules are dried and mixed with the
magnesium stearate
for 5 minutes. This mixture is compressed in a conventional tablet press (see
above for format of
the tablet). A guideline compressive force for the compression is 15 kN.
Suspension which can be administered orally:
Composition:
1000 mg of the compound of the invention, 1000 mg of ethanol (96%), 400 mg of
Rhodigel
(xanthan gum from FMC, Pennsylvania, USA) and 99 g of water.
10 ml of oral suspension correspond to a single dose of 100 mg of the compound
of the invention.
Production:
The Rhodigel is suspended in ethanol, and the compound of the invention is
added to the
suspension. The water is added while stirring. The mixture is stirred for
about 6 h until the
swelling of the Rhodigel is complete.
Solution which can be administered orally:
Composition:
500 mg of the compound of the invention, 2.5 g of polysorbate and 97 g of
polyethylene glycol
400. 20 g of oral solution correspond to a single dose of 100 mg of the
compound of the invention.
Le A 36 520-Foreign countries CA 02524900 2005-11-07
-76-
Production:
The compound of the invention is suspended in a mixture of polyethylene glycol
and polysorbate
with stirring. The stirring process is continued until the compound of the
invention has completely
dissolved.
i.v. solution:
The compound of the invention is dissolved in a concentration below the
saturation solubility in a
physiologically tolerated solvent (e.g. isotonic saline, 5% glucose solution
and/or 30% PEG
400 solution). The solution is sterilized by filtration and used to fill
sterile and pyrogen-free
injection containers.
Solution which can be administered intravenously:
Composition:
1 mg of the compound of the invention, 15 g of polyethylene glycol 400 and 250
g of water for
injection.
Production:
The compound of the invention is dissolved together with polyethylene glycol
400 in the water
with stirring. The solution is sterilized by filtration (pore diameter 0.22
gm) and used to fill heat-
sterilized infusion bottles under aseptic conditions. These are closed with
infusion stoppers and
caps.