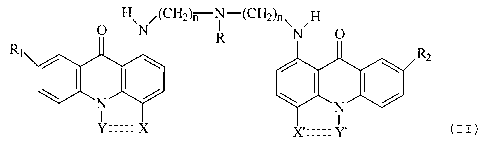Note: Descriptions are shown in the official language in which they were submitted.
CA 02525550 1997-04-09
61351-399D
- 1 -
ACRIDONE-DERIVED COMPOUNDS
USEFUL AS ANTIRETROVIRAL AGENTS
S
This application is a divisional application of
copending application 2,250,863, filed April 9, 1997.
Field of the Invention
The present invention relates to acridone-
derived compounds which are bisimidazoacridones,
bistriazoloacridones and hybrid molecules having both
imidazoacridone and triazoloacridone moieties. These
compounds are useful as anti-neoplastic and anti-
retroviral agents.
Background of the Invention
A number of acridine-based compounds which
exhibit high antitumor activity recently have been
reported. Cholody W.M., et al. (1990) described 5-
[(Aminoalkly)amino]imidazo[4,5,1-de]acridin-6-ones as a
novel class of antineoplastic agents (J. Med. Chem. 33:49-
52 (1990). 8-Substituted 5-[(aminoalkyl)amino,]-6H-v-
triazolo[4,5,1-de]acridin-6-ones have also been described
as potential antineoplastic agents (J. Med. Chern. 33:2852-
2856 (1990)). The synthesis of chromophobe modified
antineoplastic imidazaoacridones and their activity
against murine leukemias has been described (J. Med. Chem.
35:378-382 (1992)). Lapps, D.B., et al. described 2-
(aminoalkyl)-5-nitropyrazolo[3,4,5-kl]acridones as a new
class of anticancer agents (J. Med. Chem. 35:4770-4778
(1992) ) .
The above compounds have a tetracyclic planar
chromophore bearing one side chain containing an
(aminoalkyl)amino residue as a common structural feature.
It is believed that DNA is the primary target for these
compounds and that they bind to DNA by intercalation.
Bifunctional compounds also have been studied as
potential.antitumor agents based upon the ability of
acridones and other planar aromatic compounds to interact
CA 02525550 1997-04-09
- 2 -
0
with DNA by intercalation. Chen, T.K., et al. (1978)
studied diacridines as bifunctional intercalators (J: Med.
Chem. 21:868-874 (1978)). Gaugain, B., et al. (1978)
described the synthesis and conformational properties of
an ethidium homodimer and an acridine ethidium heterodimer
(Biochemistry 17:5071-5078 (1978)). Sinha, B. K., et al.
(Biochemistry 17:5071-5078 (1977)) described the synthesis
and antitumor properties of bis(quinaldine) derivatives
(J. Med. Chem. 20:1528-1531 (1977)). Rogues, B.P., et al.
(1979) described the antileukemic activity of
0 pyridocarbazole dimers (Biochem. Pharmacol 28:1811-1815
(1979)). Pelaprat, D. et al. (1980) described 7H-
pyridocarbazole dimers as potential antitumor agents (J.
Med. Chem. 23:1336-1343 (1980)). Brana, M.F., et al.
(1993) described bis-naphtalimides as a class of antitumor
15 agents (Anti-Cancer Drug Design 8:257-268 (1993)).
The rationale for the strong binding of
bifunctional intercalators containing two aromatic ring
systems joined by a suitable linker to nucleic acids has
been presented (Canellakis, E.S., et al. Biochem Biophys.
20 Acta 418:277-283 (1976). It was found that although such
compounds exhibit high affinity for DNA, this strong
binding with DNA by intercalation is generally not related
to antitumor activity.
Many factors, such as physicochemical
25 characteristics of the planar chromophores, nature of the
linking chain (its length, rigidity and ionization state),
position of attachment, and other factors, strongly
influence the binding with DNA and the biological action
of these compounds. Additionally, it was found that there
30 is no direct correlation between DNA-binding affinity and
cytotoxicity.
Since structure-activity relationships in the
group of bifunctional intercalators in relation to their
in vivo antitumor action remain unclear, it is not
35 possible to predict structures that will show such
CA 02525550 1997-04-09
- 3 -
0
activity. Small structural modifications can drastically
change biological properties of the agent. Accordingly, a
goal exists to find other compounds with high
antineoplastic activity, especially selectively directed
towards specific tumors.
It was previously disclosed that certain
bisimidazoacridones, the closely related
bistriazoloacridones and hybrid molecules containing both
the imidazoacridone and triazoloacridone moieties were
potent but selective antitumor agents (U.S. Pat. No.
5,508,289).
This invention relates to a novel class of
acridine-based compounds and their use as antineoplastic
agents and as antiretroviral agents. It has further been
found that the bismidazocridones, the bistriazoloacridones
and hybrid molecules disclosed in U.S. Pat. No. 5,508,289
are useful a.s antiretroviral agents.
'Summary of the Invention
The present invention relates to compounds
having the following general formula (I):
O HN~~~HZ)n-NON (CH2)n~NH
v
II
~ H2
...... x ::..: r
wherein R1 and R2 are independently -H, -OH, amino,
alkylamino, dialkylamino, alkoxy, alkyl, haloalkyl or
halogen; n is 2 to 6, X and X' are independently -N or
N02; Y and Y' are independently -N or -CH, or -H;~ and the
double-slash represents a double bond or no bond; such
CA 02525550 1997-04-09
- 4 -
that when X or X' is -N, Y or Y' is -CH or -N, and the
double-slash is a double bond, and when X or X' is -NOz, Y
or Y' is -H, and the double slash is no bond.
The present invention also provides a
pharmaceutical composition comprising at least one of the
above compounds and a pharmaceutically acceptable carrier.
The present invention further provides a method
for treating a neoplastic cell growth in a subject in need
of such treatment which comprises administering to the
subject an amount of the pharmaceutical composition above
effective to treat the neoplastic cell growth.
The present invention still further provides a
method for treating retroviral infections in a population of
cells including human cells comprising contacting the cell
population or administering to a subject having retroviral
infected cells an effective amount of at least one compound
of the formula ( I ) .
Also included within the scope of the present
invention is a method for protecting a population of cells
including human cells against retroviral pathogenesis
comprising contacting or treating said cells with an anti-
retroviral effective amount of at least one compound
according to formula ( I ) .
The invention also provides use of a compound of
the formula (I) or a pharmaceutical composition thereof to
inhibit viral replication or neoplastic cell growth, or for
preparing a medicament to inhibit viral cell replication or
neoplastic cell growth.
The invention also provides a commercial package
comprising a compound of the formula (I) or a pharmaceutical
CA 02525550 1997-04-09
- 4a -
composition thereof and associated therewith instructions
for the use thereof to inhibit viral replication or
neoplastic cell growth.
A yet still further aspect of the present
invention provides a method of treating retroviral
infections in a population of cells including human cells
comprising administering and/or contacting the cells of a
subject having retroviral infected cells an effective amount
of at least one compound of formula (II):
O H\N (CH2)n N-(CH2)n~ ~H
R N O
Rz
(II)
CA 02525550 1997-04-09
61351-399D
- 5 -
wherein R is H, alkyl, or a grouping which makes the
compound function as a prodrug; n is 2 to 6; and R1, Rz, X,
Y, X' or Y' and the double dash is as defined hereinabove
for formula ( I ) .
The present divisional application provides uses
of the compounds of the general formula (II) for:
(i) inhibiting viral replication, and (ii) preparing
medicaments for inhibiting viral replication. Also provided
are compositions comprising the compounds of the general
formula (II) and a pharmaceutically acceptable carrier for
(i) and (ii) as defined above. Also provided are commercial
packages comprising the compounds of the general formula
(II) or the above defined compositions and associated
therewith instructions for the use thereof to inhibit viral
replication.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1. Schematic showing the preparation of a
symmetrical compound.
Figure 2. Schematic showing preparation of a
bistriazoloacridone compound.
Figure 3. Anti-tumor activity against human
xenografts in vitro.
Figure 4. Cytoprotective activity of WMC-26.
Figure 5. Cytoprotective activity of WMC-42.
Figure 6. Parallel experiment with uninfected
cells (WMC-26) .
Figure 7. Parallel experiment with uninfected
cells (WMC-42 ) .
CA 02525550 1997-04-09
61351-399D
- 5a -
Figure 8. Inhibition of HIV replication
(NSC-682404).
Figure 9. Inhibition of HIV replication
(NSC-682405) .
Figure 10. Inability of NSC 682405
("temacrazine") to prevent the formation of proviral DNA by
reverse transcription.
Figure 11. Actions of temacrazine in the U1 Assay.
Figure 12. Effect of temacrazine on the
production and processing of viral proteins.
Figure 13. Temacrazine inactivation of the
infectious titer of intact HIV-1 virions.
Figure 14. Failure of temacrazine to non-
specifically inhibit HIV-1 LTR-directed transcription.
Figure 15. Temacrazine effect on mRNA expression
and splicing in HIV-1 infected cells.
CA 02525550 1997-04-09
- 6 -
Figure 16. Temacrazine inhibits HIV-1 specific
mRNA creating a "rev independent-like" phenotype.
Detailed Description of the Invention
As utilized herein, the term "alkyl°, alone or
in combination, means a straight-chain or branched-chain
alkyl radical containing from 1 to about 8, preferably
from 1 to about 6, carbon atoms. Examples of such
radicals include methyl, etlfyl, n-propyl, isopropyl, n-
butyl, isobutyl, sec-butyl, tert-butyl, amyl, isoamyl,
hexyl, octyl and the like. The alkyl radical may be
optionally substituted by substituents which are set forth
hereinbelow. The term "alkoxy", alone or in combination,
means an alkyl ether radical wherein the term alkyl is as
defined above. Examples of suitable alkyl ether radicals
include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy,
iso-butoxy, sec-butoxy, tert-butoxy and the like.
The term "halogen" or "halo" means fluorine,
chlorine, bromine or iodine.
The term "haloalkyl" means an alkyl radical
having the significance as defined above wherein one or
more hydrogens are replaced with a halogen. Examples of
such haloalkyl radicals include chloromethyl, 1-bromethyl,
fluoromethyl, difluoromethyl, trifluoromethyl, 1,1,1-
trifluroethyl and the like.
The possible optional substituents mentioned in
the hereinabove generic description include at least one
alkyl, cycloalkyl, aryl, aralkyl, alkaryl, heteroaryl,
alkoxy, halogen, hydroxy, amino, nitro, cyano, haloalkyl
wherein the optional substituents may also be optionally
substituted and the radicals which are optionally
substituted may be singly or multiply substituted with the
same or different optional substituents.
The present invention relates compounds having
the general formula I: w
CA 02525550 1997-04-09
- 7 _
( 2)n ~N (CH2)n~NH
0 HN~ CH -N--~
II a2
~ ~ r
x ::... r
s "",
(I)
wherein R1 and R2 are each independently -H, -OH, amino,
alkylamino, dialkylamino, alkoxy, alkyl, haloalkyl or a
halogen atom; n is 2 to 6; X and X and X' are
independently -N or -NO2; Y and Y' are independently -N or
-CH, or -H; and the double-slash represents a double bond
or no bond; such that when X or X' is -N, Y or Y' is -CH
or -N, and the double-slash is a double bond, and when X
or X' is -NO2, Y or Y' is -H, and the double slash is no
1 s bond .
In a preferred embodiment, Rl and Ry are each.
independently -H, -OH, -NH2, Cl-C6 alkyl amino, Cl-C6
dialkylamino, Cl-C6-alkyl, fluorine, chlorine or bromine.
A more preferred embodiment includes compounds in which
n=3, Y and Y' are -N, X and X' are -CH, R1 and RZ are H,
and the double dash is a double bond.
The compounds of the present invention may be
employed in the form of free bases or pharmaceutically
acceptable acid addition salts thereof. Examples of
2s suitable acids for salt formation are, methanesulfonic,
sulfuric, hydrochloric, phosphoric, acetic, citric,
lactic, ascorbic, malefic, and the like. In the preferred
embodiment, the compounds of the present invention are
present in the form of methanesulfonates, such as
dimethanesulfonate, or other salts, such as
dihydrochloride, which can be hydrated to variable extent.
Additionally, the compounds of formulas I and II may be
employed in prodrug form. Prodrug forms are known to
those skilled in the art and the most effective such form
can be determined by the skilled artisan.
CA 02525550 1997-04-09
_ g _
The present invention also relates to
pharmaceutical compositions comprising at least one of the
formula (I) and/or formula (II), and a pharmaceutically
acceptable carrier. The carrier must be "acceptable" in
the sense of being compatible with the other ingredients
of the formulation and not deleterious to the recipient
thereof. The compound may be formulated with one or more
pharmaceutically acceptable diluents or carriers, and
optionally, any other ingredients which may be therapeutic
per se, which are synergistic with the compound of the
I0 present invention. The concentration of the compound
present in the formulation will depend upon the choice of
carrier as well as the results desired.
Examples of suitable pharmaceutical carriers
include lactose, sucrose, starch, talc, magnesium
IS stearate, crystalline cellulose, methyl cellulose,
carboxymethyl cellulose, glycerin, sodium alginate, gum
arabic, powders, saline, water, among others. The choice
of carrier will depend upon the route of administration.
The formulations may conveniently be presented in unit
20 dosage and may be prepared by methods well-known in the
pharmaceutical art, by bringing the active compound into
association with a carrier or diluent, as a suspension or
solution, and optionally one or more accessory
ingredients, e.g. buffers, flavoring agents, surface
25 active agents, and the like.
For intravenous, intramuscular, subcutaneous, or
intraperitoneal administration, the compound is combined
with a sterile aqueous solution which is preferably
isotonic with the blood of the recipient. Such
30 formulations may be prepared by dissolving solid active
ingredient in water containing physiologically compatible
substances such as sodium chloride, glycine, and the like,
and having a buffered pH compatible with physiological
conditions to produce an aqueous solution, and rendering
35 said solution sterile. The formulations may be present in
CA 02525550 1997-04-09
_ g _
unit or multi-dose containers such as sealed ampoules or
vials.
For oral administration, the formulation may be
presented as capsules, tablets, powders, granules or a
suspension, with conventional additives such as lactose,
mannitol, corn starch or potato starch; with binders such
as crystalline cellulose, cellulose derivatives, acacia,
corn starch or gelatins; with disintegrators such as corn
starch, potato starch or sodium carboxymethyl-cellulose;
and with lubricants such as talc or magnesium stearate.
Formulations suitable for parenteral
administration conveniently comprise a sterile aqueous
preparation of the active compounds which is preferably
made isotonic. Preparations for injections may also be
formulated by suspending or emulsifying the compounds in
non-aqueous solvent, such as vegetable oil, synthetic
aliphatic acid glycerides, esters of higher aliphatic
acids or propylene glycol.
The present invention further provides a method
for treating a neoplastic cell growth in a subject in need
of such treatment which comprises administering to the
subject an amount of the pharmaceutical composition above
effective to treat the neoplastic cell growth.
The term "treatment" includes the partial or
total inhibition of neoplastic cell growth or retroviral
growth, proliferation and/or spread, as well as the
partial or total destruction of the neoplastic cells or
retroviruses and/or retrovirally-infected cells. The term
"subject" includes a human or animal subject diagnosed as
having cancer or a retroviral infection.
The administration may be affected by means
known to those skilled in the art such as oral, rectal,
topical intravenous, subcutaneous, intramuscular, or
intraperitoneal routes of administration.
The dosage form and amount can be readily
established by reference to known antineoplastic treatment
CA 02525550 1997-04-09
- 1~ -
or prophylactic regimens. In general, however, the dosage
of the compound will be within the range of about 0.1
~g/kg to about 100 mg/kg. The actual dose will depend
upon the route of administration, the pharmacokinetic and
toxicological properties of the individual compound, as
well as the results desired.
The compounds of the present invention of
formulas I and II are useful in treating retroviral
infections in a population of cells including human and
animal cells comprising contacting the cell population or
administration to a subject having retrovirally infected
cells an effective amount of at least one compound of the
formulas I and/or II.
The compounds of the present invention including
those of formulas I and II may be functional in many
retroviral systems to curb the spread of infective virus.
Other retroviruses which may be inhibited by the compounds
of the present invention include, but are not limited to,
HTLV-I, HTLV-II, BLV, EIAV, FIV, SIV, STLV and Visna
virus.
Initial experiments involving compounds of
formulas I and II revealed that these substances inhibited
both the integration and the disintegration steps in an in
vitro experiment which utilized the full length purified
HIV-1 integrase. These compounds and several others were
examined in an anti-viral assay involving peripheral blood
lymphocytes which had been infected with HIV-1.
Although the formulations disclosed hereinabove
are effective and relatively safe medications for treating
HIV infections, the possible concurrent administration of
these formulations with other antiviral medications or
agents to obtain beneficial results is not excluded. Such
other antiviral medications or agents include soluble CD4,
thalidomide, dideoxyinosine, dideoxythymine, zidovudine,
dideoxycytidine, gancyclovir, acyclovir, phosphonoformate,
amatradine, ribavarin, antiviral interferons (e.g. a-
CA 02525550 1997-04-09
- 11 -
interferon, a-interferon, or interleukin-2) or aerosol
pentamidine, and other substances used in anti-HIV
therapy.
Treatment of infected cells to inhibit virus
activity may be for a specific period of time or may be
continuous. Virus activity may be measured by monitoring
levels of viral protein, such as p24, or by measuring
reverse transcriptase levels, or by monitoring virus
protein activity by 355-met pulse-chase labelling and
immunoprecipitation experiments, or by other methods which
are well known by one of skill in the art. {Kayeyama, S.,
et al. (1994), AIDS Res. and Human Retroviruses, 10:735-
745) .
The present invention is described in the
following Experimental Details section, which sets forth
specific examples to aid in an understanding of the
invention, and should not be construed to limit in any way
the invention as defined in the claims which follow
thereafter.
Experimental Details Section
Materials and Methods. All solvents used were
reagent grade. All reagents were obtained either from
Aldrich Chemicals or from Fluka and were used as received.
Melting points were taken on an Electrothermal capillary
melting points apparatus and are uncorrected.
Chemical Synthesis. The compounds of present
invention in which both chromophores are identical were
prepared by the route presented in Figure 1. The
intermediate chloronitroacridone 1 was prepared as
described previously (Capes, D.B., et al. J. Med. Chem.
35:4770-4778 (1992); Lehmstedt, K., et al. Chem. Berichte
70:1526-1538 (1937)), or by procedures analogous thereto.
The acridone is reacted with an equivalent amount of a
suitable piperidine derivative 2 to afford the
bisnitroacridone 3, which is then reacted with formic acid
and stannous chloride to give the final bisimidazoacridone
CA 02525550 1997-04-09
- 12 -
°
4.
Symmetrical drugs of the triazolo series were
prepared according to Figure 2. An acridone is reacted
with an excess of piperazine derivative in such a way that
the product 7 was fornned.
A detailed discussion of the synthesis of the
compounds of the present invention is as follows.
Example 1
1, 4-bis [3 [ (4-vitro (10H) -9-oxo-acridin-1-
yl)amiro]propyl]piperazine (3)
A mixture of 1-chloro-4-vitro- 9(lOH)-
acridinone (1) (2.758, 0.01 mole), 50 mL of DMSO, 1,4-
bis(3-aminopropyl)piperazine (2) (1.0028, 0.005 mole), and
diisopropylethylamine (1.958, 0.015 mole) was stirred at
g0°C for 8 hours. To the reaction mixture 100 mL of 1%
aqueous sodium hydroxide solut~.on was added, stirred for
10 minutes and left overnight in refrigerator.
'Precipitate was collected by filtration, washed with water
and crystallized from DMA to give 2.748 (81%) of yellow
(3 ) , mp 274-279°C (decomp. ) . Anal. (C36H36N8~6 ~ Hz0) C, H, N.
1, 4 -bis [3- f (6-oxo-6H-imidazo [4, 5,1-de] -acridin-5-yl)
amino]propyl]piperazine (4)
2.038 (0.003 mole) of (3) was dissolved in 50 mL
of 85% formic acid. To the solution a solution of 5.78
(0.03 mole) of SnClZ in 6 mL of concentrated hydrochloric
acid was added and the mixture was stirred under reflux
for 36 hours. After cooling the precipitate was filtered
and washed with 50 mL of methanol, transferred into 300 mL
of water, and made alkaline with 10% aqueous sodium
hydroxide. 30 mL of chloroform-methanol (10:1) mixture
was added and stirred vigorously for 2 hours. Undissolved
material was filtered off and the chloroform layer was
separated. 2 g of silica geI was added to the extract and
the solvent was evaporated under vacuo. The gel was then
put on a silica gel column'and eluted with chloroforrn-
CA 02525550 1997-04-09
- 13 -
° methanol (10:1). The main fraction was collected and
after evaporation of solvents gave 1.05g (55%) of yellow
( 4 ) , mp 2 8 9 - 2 9 3 ° C ( de c omp . ) . Ana 1 . ( C38H36N$Oz ~
Hz0 ) C , H ,
N.
1, 4-bis (3 - ( (6-oxo-6H-v- triazolo (4, 5, ] -de] -acridin-5-y1 )
(aminopropyl)piperazine (6)
A mixture of 5-chloro-6H-v-triazolo[4,5]-de]-
acridin-6-one (5) (5.12g, 0.02 mole), 60 mL of DMSO, 1,4-
bis(3-aminopropyl)piperazine (2) (2.003g, 0.01 mole), and
diisopropylethylamine (2.6g, 0.02 mole) was stirred at
100°C for 20 hours. To the reaction mixture 150 mL of 2%
aqueous sodium hydroxide solution was added, stirred
thoroughly for 10 minutes and left overnight in a
refrigerator. Precipitate was separated by filtration,
washed with water, transferred into 200 mL of 1% water
solution of methanesulfonic acid and stirred at room
temperature for 1 hour. Undissolved material was filtered
off. The filtrate was made alkaline with an aqueous
solution of sodium hydroxide. The precipitate of the free
base was separated by filtration, washed with water and
then crystallized twice from boiling DMA to give 3.848
( 6 0 % ) o f ye 11 ow ( 6 ) , mp 2 4 2 - 2 4 5 ° C . Ana 1. (
Cg6Hg4N10~2 ' Hz~ )
C, H, N. Structure of (6) was confirmed by a single
crystal X-ray structure analysis. For biological tests
the free bases of (4) and (6) were transformed water-
soluble salts such as methanesulfonate or hydrochloride.
Example 2
D3.hydrochloride of 6
6 (1.2g, 0.002 mole) was dissolved in boiling
mixture of 10% methanol in chloroform (400 mL). To the
hot solution, 10 mL of 0.4M hydrochloric acid in
methanol(generated by dissolving acetyl chloride in
methanol).was added and stirred for few minutes. The
solvents were concentrated by evaporation to about 100 mL.
CA 02525550 1997-04-09
- 14 -
° 100 mL of ether was added and yellow precipitate of the
salt was separated by filtration, washed with ether and
dried. Yield - 100%, mp>300°C (decomp. ) . Anal. (C36H34NIOG2
2HC1 ~ HZO) C, H, N.
The activity of compound (4) was tested against
several tumor xenografts in vitro. The experiments was
carried out on cell lines derived from these tumors,
utilizing a standard clonogenic assay protocol. The mean
IC~o (i.e. the concentration of the chemical which resulted
in 70% killing of the tumor cells) for all of the lines
tested was 2.3 ~.g/mL. The gastric carcinoma GFX (251/14)
was sensitive at the median dose, but the mammary tumor
MACL (MCF7X/13) required over twice the median dose. The
melanoma MEXF (514/12) was completely insensitive to the
drug, as was the renal carcinoma RXF (1220/5). The
prostate cancer cell lines PRXF (PX2M/27 and DU145X/2)
were very sensitive to the drug, especially PC3M which
required less than 1 nanogram/mL to achieve IC~0.. The
results are shown in figure 3.
Experiments were performed involving various
compounds. These compounds with their alternate
designations have the following formulas:
O H~,CHZCH~Z"yCHZCHrCH~~
CHI
/ ~ ~ I \
~i ~ WNC-26 ~i
H ~ N --~
:~ SC 682401
CA 02525550 1997-04-09
- 15 -
O H'\N ~H2y~~_ ~--CH2CF'.zCH~N~..~ 0
~i '
/ \ CHi /
f
/ \
W IiC-42
H v5C 682402
O ~ CH2C~HZ ~~H~CHzCN~N~ 0
/ i w CHi / ~ \
to f 1
\ / \
N~ ~4lfiC~43
sISC 682443
O ~,CH2CH2CH? ~ '-'~'~CH~CHZCH~ 0
1 w
\ /
WLiC-50 N N
H ~1SC 682444
O H~~CHzC~C~' ""~HZCHZCH~N~.,~ O
/ ( \ i ( \
\ i ~ /
wMC-~o N-..r!~
:~ SC 68245
Experiments involving WMC26 and WMC42 reveal
that these compounds inhibited both the integration and
disintegration steps in an in vitro experiment whioh
utilized the full length purified HIV-1 integrase. These
CA 02525550 1997-04-09
- 16 -
° compounds and several others were then examined in a
cytoprotection assay involving peripheral blood
lymphocytes which had been infected with HIV-1.
Briefly, the infected cells in culture are
killed by the virus, and the cytotoxicity is detected by
the formation of color due to the oxidation of a formazan
dye (XTT) by enzymes released by the dying cells (Weislow
et al., J. Natl. Cancer Inst. 81:577-586, 1989). An
effective antiviral drug is able to protect the infected
cells from cytotoxicity by inhibiting some vital function
of the virus. Thus an in vitro demonstration of antiviral
activity depends on dose-dependent protection of the
cytopathic effects of the virus by the putative drugs.
Figures 4 and 5 show the cytoprotective activity of WMC26
and WMC42. Since many drugs can also be toxic to cells in
IS their own right, a parallel experiment is carried out
wherein uninfected cells are treated with the drugs until
a concentration is reached where the cells are killed by
the action of the drug. These data are indicated on
Figures 6 and 7. Clearly, both of these drugs were able
2p to protect the infected lymphocytes at concentrations far
lower than those which resulted in drug induced killing.
This means that the compounds had very good therapeutic
indices in this assay.
As a result of these preliminary data,
25 additional experiments were carried out to establish the
antiviral activity in other cell lines, using a variety of
protocols. The preliminary experiments in lymphocytes
were confirmed using infected macrophages. Much more
extensive experiments were carried out in HIV-1-infected
30 CEM SS leukemia cells. Unfortunately, compounds such as
WMC26 are very potently (nanomolar levels) cytotoxic to
leukemia (Cholody et al., J. Med. Chem. 55:2338-2245,
1995), and therefore could not be shown to be
cytoprotective in this assay. However, two derivatives
35 were found which had low antileukemic activity while
CA 02525550 1997-04-09
- 17 -
° maintaining high antiviral activity. Figures 8 and 9
demonstrate the cytoprotective activity of WMC50 and
WHC70, respectively, in this cell line. While both
compounds are clearly cytoprotective at very low
concentrations, it is clear that WMC-70 is likely to be a
better drug because of its superior therapeutic index.
Thus, while the concentration wherein WMC-70 inhibits the
cytotoxic effect of the virus to 50% (the EC50 value) is
less than 1 nanomolar, the drug kills 50% of the cells at
greater than micromolar concentrations. This gives a
therapeutic index of more than 3 orders of magnitude. The
corresponding therapeutic index for WMC50 is about 100-
200, which is quite acceptable. Additional experiments
were carried out on WMC-70. It was shown that the drug
did not inhibit either the HIV protease or the reverse
transcriptase, two other important enzymes involved in
viral replication. WMC-70 also did not damage the gag
protein zinc finger, another structural feature in the
virus which is considered to be crucial for viral
replication. Interestingly, treatment of the HIV-l virus
itself by the drug appeared to prevent its ability to
infect CEM cells. Further, WMC-70 appears to affect post-
transcription events in the virus. Treatment of cells in
which all of the virus is integrated by the drug, reveals
that apparently normal viral gag p55 polyprotein is
formed, but it is not processed further, i.e., no p24 can
be detected. While the inhibition of integration appears
to be the principal mechanism by which this compound
exerts its antiviral activity, there are perhaps other
mechanisms which are also involved in the therapeutic
action of this drug, and by extension, of other antiviral
drugs of this class.
Compounds WC-26, WMC-42, WMC-50 and WMC-70 are
potent, antiviral agents which have potential activity
against HIV infections and against AIDS. ("WMC-70" is
also referred to herein as "NSC 682405" or "temacrazine".)
CA 02525550 1997-04-09
- 18 -
° Example 3
Antiviral Properties of Test Compounds
The antiviral properties of the WMC series of
compounds were evaluated. First, all compounds were
tested for their ability to inhibit HIV-1 replication in
cell culture using the XTT cytoprotection assay. This
assay measures the concentration-dependent capacity of
compounds to protect CEM-SS cells from the cytopathic
effects of HIV-1~. The concentration of compound
providing 50% protection is the ECso antiviral value, while
the concentration of compound that results in 50°s cell
death is the ICso toxicity value. As shown in Table 1,
compounds NSC 682401-682403 were inactive, indicating that
no antiviral effect was observed in the absence of
toxicity. NSC 682404 exhibited an ECso = 0.41 nM and an
ICso = 158 nM, while temacrazine exhibited an ECso = 1.1 nM
and an ICso = 2.77 ~.M (see also Figures 8 and 9) . Thus,
temacrazine was the most efficacious compound in the XTT
cytoprotection assay. This conclusion was also reached
when the compounds were tested for anti-HIV-1 activity
against HIV-1~" in human monocyte/macrophage cultures
(Table 1).
Example 4
Mechanistic Properties of Test Comx~ounds
Data from mechanistic studies against the known
antiviral targets of HIV-1 are also shown in Table 1. The
compounds did not inhibit attachment of HIV-1 to host
cells, the enzymatic activities of HIV-1 reverse
transcriptase or protease, or the p7 nucleocapsid protein
zinc fingers. Based on the findings thus far our efforts
have focused on biological assays of the temacrazine
congener. In a time course assay the temacrazine did not
prevent the formation of proviral DNA that occurs~during
reverse transcription, as shown by the amplified LTR/gag
region of the proviral DNA (Figure 10). Temacrazine
CA 02525550 1997-04-09
- 19 -
inhibited 3'-processing and stand transfer activity in an
in vitro assay using purified oligomers and recombinant
HIV-1 integrase in a concentration dependent manner with
an ECSO between 10 and 100 nM (Table lA).
These findings are consistent with temacrazine
acting as an inhibitor of HIV-1 integrase, as the proviral
DNA formation is completed prior to the integration event
during the HIV-1 replication cycle. Experimental compounds
that inhibit virus attachment and fusion (e. g. dextran
sulfate) and compounds that inhibit reverse transcription
(DDC, AZT, Nevirapine, etc.) prevent formation of proviral
DNA in the time course assay.
Table 1
XTT Assay 'p7NC
1 S (~ Mo~~ "AttachmentRT Zinc
(nlVn Finger
Compound ECM ICS EC ICS IDS (N,1V1)IDS IDS
682401 Inactive 50 1000 NI,~~M NI,~,,MNI
682402 Inactive 160 10000 NI,~~,M NI,~~M NI
682403 Inactive ND NI~M NI,~~,MNI
682404 0.41 158 100 10000 N1,~~,M 35 NI
682405 1.1 2767 10 10000 NI,~~M NI,~,,MNI
a The XTT cytoprotection refers to data from the screening assay.
Mechanistic studies for virion attachment, RT and protease were performed as
previously described. The values represent IDsoS, which reflect the
concentration
that inhibited the indicated activity by 50% .
The p7NC ZF assay measured the percent reduction in relative fluorescence
units
(RFU) of the zinc finger after treatment of the p7NC protein with 25 ~.M of
each
compound for 10 min.
The "Inactive" indicates that the compound demonstrated no efficacy in the
screen.
' The "NI" reflects no inhibition of activity at the indicated high test
concentration.
CA 02525550 1997-04-09
- 20 -
Table lA Mechanism of action studies for Temacrazine
I~,(uM)'
Molecular target Temacrazine Control compound
Attachment NI4 1.1 uM (Farmatalia)
gp 120-CD4 NI 4.3 ~.M (Farmatalia)
RT Enzyme Activity
rAdT Template/primer NI 27 nM (A2TTP)
rCdG Template/primer NI 6 nM (UC38)
Protease activity
HPLC-based assay NI 3 nM (KNI-272)
Pr55B°g processing NI Z-10 nM (KNI-272)
Integrase 0.01 to 0.1 0.5 /cM (ISIS 5320)
NCp75
TSQ (Increase in NI 17 (NSC 624151)
RFU/90 min)
~U/ 10 min)
Trp3' (Decrease in NI 145 (NSC 624151)
' I~ are the drug concentrations inhibiting 50 % of the indicated activity.
Positive controls for individual assays were; Attachment: Farmatalia
ICSO 1.1 ~cM (D.J. Clanton et al., J. Acquir. Immune Defic. Syndr.
5:771 ( 1992)), Reverse transcriptase inhibition: rAdT Template/primer-
AZTTP 27 nM, rCdG Template/primer-UC38 6 nM (J.P. Bader et al.,
Proc. Natl. Acad. Sci. USA, 88:6740 (199I)), protease inhibition
(HPLC Detection of cleavage of the Ala-Ser-Glu-Asn-Tyr-Pro-Ile-Val-
Glu-amide substrate): KNI-272 3 nM (S. Kageyama et al., Antimicrob.
. Agents Chemother, 37:810 (1993)) Integrase inhibitor, ISIS 5320 (R.W.
Buckheit, Jr., et al., AIDS Res. Hum. Retrovir. 10:1497 (1994)) and
NCp7 Zn2+ ejection NSC 625151, (Dithiane) (W.G. Rice, et al.,
Antimicrob. Agents and Chemother, 41 In Press (1997))
3 Attachment of HIV-1,~ to CEM-SS cells with attachment determined by
determination of the amount of cell-bound p24 by p24 ELISA assay of
cellular lysates.
° NI indicates no inhibition at the high test concentration.
Changes are expressed as relative fluorescence units per unit time.
35
CA 02525550 1997-04-09
- 21 -
° Example 5
Effects of temacrazine on the Late
Phase of the HIV-1 Replication Cycle
As a model of the late phase events during the
HIV-1 replication cycle, U1 cells were used that are
latently infected with two copies of HIV-1 proviral DNA.
The high level expression of virus production in these U1
cells can be stimulated by certain factors, such as TNF-a.
Ul cells were stimulated with TNF-a for 24 hours, after
which were added various concentrations of temacrazine,
and the cultures were allowed to incubate for 48
additional hours. At that time, the cells were evaluated
for viability and for their content of viral protein
synthesis and processing, while the cell-free supernatant
was evaluated for viral p24 content and for the infectious
titer of released virions. Figure 11 shows that the virus
production (p24) was inhibited at concentrations below 4
nM, and that the released viral particles were non-
infectious (expressed as infectious units per ml,
"IU/ml"). Cell viability was diminished above 30 nM with
g y, inspection of the viral
these U1 cells. Interestin 1
proteins by Western blotting with p7 and p24 antisera
(Figure 12) revealed that the Gag precursor polyprotein
(Pr55g~) was synthesized but was not processed by the
HIV-1 protease into mature viral proteins (p24 and p7NC).
In a similar study, cells were either treated
for 24h with TNFa followed by the addition of temacrazine,
or temacrazine and TNFa were added simultaneously, or
temacrazine was added 24 h post TNFa addition. After TNFa
induction and temacrazine treatment, cultures were
continued for 72 h after which virion-associated p24 and
cell viability (XTT dye reduction) were measured.
Temacrazine was an efficient inhibitor of HIV-1 virus
replication in both U1 (and also the latently infected
cell line, ACH-2) with an ECso in the 10 to 100 nM range.
In addition, temacrazine-mediated virus inhibition was not
significantly effected by the order of addition of
CA 02525550 1997-04-09
- 22 -
temacrazine and TNFa.
Also, it was found that a 30 min. pulse of
cultures with temacrazine presented subsequent production
of infectious virus following stimulation with TNFa.
Example 6
Direct Inactivation of HIV-1 by temacrazine
The mechanistic studies in Table 1 indicated
that the compound did not inhibit HIV-1 protease activity.
However, to insure that the HPLC-based assay (used in
Table 1) was providing accurate findings, the action of
temacrazine in the Gag processing assay was evaluated. In
this assay, purified recombinant HIV-1 protease enzyme was
pretreated with the test reagent for one hour at 37°C, and
then exposed the recombinant Pr55g~ precursor polyprotein
to the treated enzyme. As shown in Figure 13, the
protease alone was able to efficiently process the
precursor. In addition, pretreatment of the enzyme with
the compound had no inhibitory effect. Together, these
findings demonstrated that temacrazine does not inhibit
the HIV-1 protease enzyme.
Significance of Findings
Temacrazine demonstrated potent anti-HIV-1
activity in models of acute infection against
l~phocytotropic HIV-1 strains in proliferating cells and
against monocytotropic HIV-1 strains in non-proliferating
normal cells. Moreover, the compound exerted an antiviral
effect on previously infected cells. The exact nature of
the late phase effect is under investigation.
Nevertheless, temacrazine potently inhibited the
production of new infectious virus from previously
infected cells. This is a very important action of the
compound given that any means for reducing the viral
burden in HIV-1 infected persons would result in decreases
in subsequent rounds of infection in vivo. In addition,
the ability of the compound to directly inactivate virions
CA 02525550 1997-04-09
- 23 -
would be of great significance, since this would
effectively reduce the infectious titer in plasma and
prevent later rounds of replication and seeding of the
virus.
S Example 7
Initiation and regulation of HIV-1 transcription
requires a complex series of interactions with cellular
transcription factors, such as NFkB, SP-1 and AP-l, and
the participation of cellular factors in the
transcriptional complex. In addition, the post-
integrative model used here requires TNFa stimulated U1
and ACH-2 cells, this model requires not only the
participation of the NFkB transcriptional complex, and
also components of the TNFa signaling pathway. In order
1S to rule out that temacrazine was non-specifically
effecting transcription or down-regulating replication by
interfering with the TNFa signal pathway, LTR-directed
chloramphenicol acetyltransferase (CAT) activity was
measured in BF-24 cells. BF-24 cells are derived from the
THP-1 monocytic cell line and carry a stably integrated
CAT gene under the transcriptional control of the HIV-1
LTR (S. Schwartz, et al., Proc. Natl. Acad. Sci. USA
86:7200 (1989); B.K. Felber, et al., Science 239:184
(1988)). The LTR in this monocytic cell line is inducible
2S by IL-6, PMA and TNFa, and produces enzymatically active
chloramphenicol transferase ("CAT"). Induction of CAT
enzyme activity by TNFa occurs in the absence of any viral
components, and as a result will identify mechanisms of
action that are virus regulatory protein independent. BF-
24 cells were stimulated with TNFa and treated with
temacrazine for 24 h, and cellular proteins were then
collected for determination of CAT enzyme activity (Figure
14) .
CAT activity was determined using a fluorescent
3S Bodipy conjugated chloramphenicol after 18 h of reaction
with 50 ~cg of protein extract containing Acetyl CoA.
CA 02525550 1997-04-09
- 24 -
Figure 14 shows that BF-24 cells alone have a low
intrinsic CAT activity that is inducible by at least 10
fold by TNFa. Treatment of BF-24 cells with up to 5 ~M
temacrazine did not significantly alter CAT induction.
Similarly, AZT which does not effect temacrazine's
activity because reverse transcription is not required for
CAT expression, was inactive. Even though, these data
argue against a non-specific transcriptional/translational
effect, specific experiments were conducted to verify this
hypothesis. The incorporation of 3H Leucine (to measure
protein metabolism), 3H Thymidine (to measure DNA
replication) and 3H Uridine (to measure RNA production) in
CEM-SS cells was determined and it was found that
temacrazine had no effect on these cellular parameters.
These data strongly suggest that the antiviral activity of
temacrazine is not due to a non-specific downregulation of
transcription, and requires the participation of viral
factor(s).
Example 8
Temacrazine completely inhibited the expression
of unspliced or singly spliced mRNA at 500 and 50 nM when
given either prior to or 24 h after the TNFa induction.
Similar results were seen when temacrazine and TNFa were
added simultaneously. In contrast, temacrazine did not
alter the expression of HIV-1 multispliced transcripts
when using RT-PCR primers designed to detect all multi-
spliced rev independent mRNAs (Figure 15).
As a control the house keeping gene
porphobilinogen deaminase (Hydroxymethylbilane synthase,
"PBGD") was amplified to determine uniformity of loading.
Since the drug did not decrease PBGD expression, this
demonstrates that temacrazine did not non-specifically
downregulate transcription.
Example 9
To assess the possibility that temacrazine was
CA 02525550 1997-04-09
- 25 -
0
regulating HIV-1 mRNA expression, the expression of total
HIV-1 mRNA by Northern blotting was examined (Figure 16).
Figure 16 shows that upon stimulation with TNFa,
HIV-1 specific mRNA expression in U-1 cells increases
dramatically. Treatment of TNFa stimulated U-1 cells with
and 100 nM temacrazine suppressed mRNA production
globally, generating a phenotype that is very close in
appearance to that of the unstimulated U1 cells ("rev"
independent phenotype). However, treatment with 1 nM
temacrazine resulted in a decrease in single spliced and
10 unspliced mRNA with no apparent decrease in the multi-
spliced mRNA. These patterns of RNA inhibition suggest
that temacrazine is inhibiting or altering the expression
of rev-responsive element (RRE) containing mRNAs.
Example 10
IN VIVD .DATA
The novel mechanism of action of temacrazine and
its broad range of activity against HIV-1 and its high
therapeutic indexes led us to assess its in vivo and anti-
viral properties in a nude mouse hollow fiber model of
HIV-1 replication. Briefly, CEM-SS cells are infected
with HIV-1~ and placed into hollow fibers and implanted
intraperitoneal or subcutaneously into nude mice.
Temacrazine in a DMSO vehicle was intraperitoneally
administered either 3X, 2X or 1X per day with total dosage
of 25 mg/kg per day for 6 days. Similarly, mice are being
treated by oral and intravenous administration of
temacrazine. Peritoneal washes and serum samples were
collected and p24 expression determined by antigen
capture. The results are shown in Table 2.
Intraperitoneal administration of temacrazine reduced the
amount of detectable p24 in peritoneal washes by 5 to 10
fold independently of the dosage schedule. Temacrazine
also reduced the amount of p24 in the serum. Thus;
temacrazine reduces HIV-1 replication in vivo.
Experiments presented herein indicate that
CA 02525550 1997-04-09
- 26 -
temacrazine functions by interacting with a post-
integrative event in the HIV life cycle in infected cells.
This interaction leads to the loss of viral transcripts
with a selective depletion of unspliced and singly spliced
transcripts. This drug functions by a new mechanism of
antiviral action. Nevertheless, the utility of
temacrazine and its congeners as potential antivirals is
further strengthened by the ability of temacrazine to
inhibit HIV-1 replication in an in vi.vo model.
Table 2. In Vivo activity of temacrazine in the hollow fiber mouse model
Peritoneal Wash Serum (pg p24)
(pg p24)
1 S Animals mean SD pl Mean SD P
Treatment2
uninfected 3 0 0 - 0 0 -
Saline q8h3 S 2185 67S - 2694 717 -
DMSO q8h S 24S 868 - 3716 1287 -
1
ddC 40 mg/Kg q8h 3 7 12 0.0024 87 21 0.0009
Temacrazine
8.Smg/kg q8h 2 460 396 0.026 1790 198 0.1
12.5 mg/kg 3 190 97 0.004 1863 601 0.06
ql2h
2S ~c ."~i~,rt ~~dt, 2 coy ~cn n nn~ ~nzn nnz n "~
3S
CA 02525550 1997-04-09
- 27 -
Nude mice were implanted with hollow fibers containing HIV-1RF infected
peripheral blood mononuclear cells and dosed with temacrazine (total dose 25
mg/kg).
1 Students t-test.
2 AlI compounds were given intraperitoneally.
3 q is frequency of dosage.
4 Groups significantly different from animals injected with DMSO alone.
While the foregoing invention has been described
in some detail for purposes of clarity and understanding,
it will be appreciated by one skilled in the art from a
reading of the disclosure that various changes in form and
detail can be made without departing from the true scope
of the invention.