Note: Descriptions are shown in the official language in which they were submitted.
CA 02528281 2005-12-06
WO 2005/001478 PCT/SE2004/001039
METHODS FOR IDENTIFYING AGENTS, WHICH REGULATE CYTOKINES.
TECHNICAL FIELD
The present invention relates to methods for identifying agents that modulate
the
effect of cytokine class I receptor binding compounds, said agents being
useful for
decreasing IGF-1 levels in a cell, and for the treatment of medical disorders
caused by
hormone dysregulation, such as growth hormone or prolactin dysregulation.
BACKGROUND
Growth hormone (GH) is secreted from the adenohypophysis (anterior pituitary
gland) and has a variety of target tissues. GH has a common range of actions
including
somatic growth, differentiation and intermediary metabolism, effects that are
mediated
by GH-induced insulin-like growth factor-1 (IGF-1) (Bichell et al., 1992). IGF-
1 is the
major regulator of post-natal growth, and has both endocrine and paracrine
action on
different tissues.
GH induces transcription of different genes by binding to a membrane-
associated receptor, the 'growth hormone receptor (GHR), which belongs to the
superfamily of cytokine receptors (Graichen et al., 2003). These receptors
lack intrinsic
catalytic activity but are associated to cytosolic proteins with tyrosine-
kinase activity.
The receptors possess a single membrane-spanning domain and they exist as
monomers
that dimerize and become activated upon ligand binding. Several intracellular
second
messengers have been implicated in the signal transduction of GH, including
calcium
ions, phospholipase C, phospholipase A2, G-proteins, protein kinase C (PKC),
Janus
kinase 2 (JAK2) and signal transducer and activator of transcription (STAT) 1,
3 and 5
(Wood, 1996).
The signal transduction of GH has been investigated in the serine protease
inhibitor (SPI) 2.1 gene, where activation is mediated through phosphorylation
of JAK2
and STATS (Wood, 1996). When GHR becomes activated upon ligand binding the
tyrosine kinase JAK2, which is associated to the GHR intracellular part,
becomes
phosphorylated and then phosphorylates the GHR itself. This leads to
phosphorylation
of STATS, which homodimerizes, translocates to the nucleus and binds a
specific
sequence in the SPI 2.1 promoter called the GH-response element (GHRE),
thereby
activating gene transcription.
CA 02528281 2005-12-06
WO 2005/001478 _ 2 _ PCT/SE2004/001039
To regulate the numbers of GHR on the cell surface, the GHR is internalized in
the cell by endocytosis and transported to lysosomal vesicles for destruction.
However,
the GHR has also been reported to get internalized and translocate to the
nucleus upon
GH-stimulation (Lobie, Wood 1994). It has been suggested that GHR itself might
be
involved in gene regulation. Interestingly, the nuclear translocation of both
GH and the
GHR is independent of JAK2 (Graichen et al., 2003), which suggests that this
nuclear
translocation might be an alternative signal transduction pathway independent
of the
JAK-STAT pathway.
Investigation of the two IGF-1 promoters reveals that no changes can be seen
in
DNA-protein interactions when rat hepatic IGF-1 is activated by GH (LeStunff
et al.,
1995, Thomas et al., 1994), and this together with the fact that GH induce a
rapid
activation of IGF-1 transcription (Bichell et al., 1992) suggests a GH-induced
modification of pre-existing transcription factors bound to the DNA. One of
the protein-
bound DNA-sites in promoter 2 has been found to be a possible binding site for
the
transcription factor AP2, and the transcription factor OCT1 has also been
suggested to
'bind to tlus promoter region (LeStunff et al., 1995). The transcription
factor AP2
belongs to a family with four members, which all have been implicated as
tissue-
specific effectors of proliferation and differentiation during embryogenesis
(Pfisterer et
al., 2002; Werling and Schorle 2002). OCT1 is a ubiquitous transcription
factor found
in most mammalian cell types, where it activates transcription of a variety of
genes.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1A depicts the results of electrophoresis mobility shift assays (EMSA) in
nuclear extracts from growth hormone receptor (GHR) transfected and non-
transfected
WRL-98 cells incubated with an anti-GHR antibody and OCTl DNA probes (SEQ ID
NOS:3 and 4).
Fig. 1B depicts the results of EMSA in nuclear extracts from GHR transfected
and non-transfected WRL-98 cells incubated with an anti-GHR antibody and AP2
DNA
probes (SEQ ID NOS:S and 6).
Fig. 1C depicts the results of EMSA in nuclear extracts from rat liver cells
incubated with an anti-GHR antibody and AP2 DNA probes (SEQ ID NOS:S and 6).
Fig. 1D depicts the results of EMSA in nuclear extracts from GHR transfected
and non-transfected WRL-98 cells incubated with an anti-GHR antibody and Pr2F
DNA
probes (SEQ ID NOS:9 and 10).
CA 02528281 2005-12-06
WO 2005/001478 - 3 _ PCT/SE2004/001039
Fig. 1E depicts the results of EMSA in nuclear extracts from prolactin
receptor
transfected WRL-98 cells incubated with Pr2F DNA probes (SEQ ID NOS: 9 and
10).
Fig. 1F depicts the results of EMSA in nuclear extracts from WRL-98 cells
incubated with an anti-GHR antibody and growth hormone response element DNA
probes (SEQ m NOS: 1 and 2). Free probe (P) was used as a negative control. To
confirm specific protein-DNA interaction, 400x excess of un-labeled specific
(+) or
non-specific (-) probe was added to the reaction. Investigation of possible
supershifts
was made by incubating the extracts with an anti-GHR antibody (MAb 263) prior
to the
addition of the labeled DNA-probe. Non-specific competition was not performed;
instead antibody only was incubated with the DNA-probe (Ab). S=BVTA (N-[5-
(aminosulfonyl)-2-methylphenyl]-5-bromo-2-furamide).
Fig. 1 G depicts the results of EMSA in nuclear extracts from GHR transfected
WRL-98 cells incubated with an anti-GHR antibody and growth hormone response
element DNA probes (SEQ ID NOS: 1 and 2). Free probe (P) was used as a
negative
control. To confirm specific protein-DNA interaction, 400x excess of un-
labeled
specific (+) or non-specific (-) probe was added to the reaction.
Investigation of
possible supershifts was made by incubating the extracts with an anti-GHR
antibody
(MAb 263) prior to the addition of the labeled DNA-probe. Non-specific
competition
was not performed; instead antibody only was incubated with the DNA-probe
(Ab).
S=BVTA.
Fig. 1H depicts the results of EMSA in nuclear extracts from HX rat liver
cells
incubated with an anti-GHR antibody and growth hormone response element DNA
probes (SEQ ID NOS: 1 and 2). Free probe (P) was used as a negative control.
To
confirm specific protein-DNA interaction, 400x excess of un-labeled specific
(+) or
non-specific (-) probe was added to the reaction. Investigation of possible
supershifts
was made by incubating the extracts with an anti-GHR antibody (MAb 263) prior
to the
addition of the labeled DNA-probe. Non-specific competition was not performed;
instead antibody only was incubated with the DNA-probe (Ab).
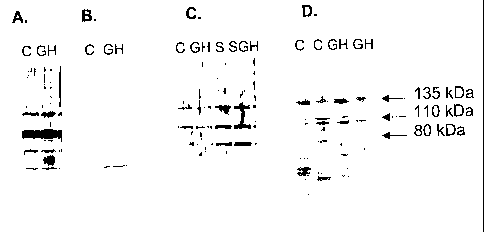
Fig. 2A depicts the total protein on filter as visualized by Ponceau staining.
Fig. 2B depicts a Western blot with an anti-GHR antibody.
Fig. 2C depicts an EMSA gel, showing that the GHR is present in the shifted
band.
CA 02528281 2005-12-06
WO 2005/001478 _ 4 _ PCT/SE2004/001039
Fig. 3A depicts a Western blot of nuclear extracts from GHR-transfected
WRL-68 cells, using a rabbit anti-GHR antibody as a primary antibody,
visualized by a
pig anti-rabbit secondary antibody coupled to HRP.
Fig. 3B depicts a control Western blot of nuclear extracts from GHR-
transfected
WRL-68 cells, using the pig anti-rabbit secondary antibody alone.
Fig. 3C depicts a Western blot of nuclear extracts from WRL-68 cells, using a
rabbit anti-GHR antibody as a primary antibody, visualized by a pig anti-
rabbit
secondary antibody coupled to HRP. Although GHR is present in nuclear extracts
from
both transfected and untransfected cells, regardless of treatment, the amount
of the
receptor is higher in transfected cells.
Fig. 3D depicts a Western blot of nuclear extracts from HX rat liver cells,
using
a rabbit anti-GHR antibody as a primary antibody, visualized by a pig anti-
rabbit
secondary antibody coupled to HRP. The amount of GHR in HX rat liver cells is
as
high as in transfected WRL-68 cells.
DISCLOSURE OF THE INVENTION
It has been found that the full-length growth hormone (GH) receptor is present
in isolated nuclei from rat hepatocytes and a cultured human liver cell-line
by
immunocytochemistry and Western blotting. Electrophoretic mobility shift
assays
indicate that the receptor interacts with other transcription factors, as
shown by an
increased amount of super shift observed in the presence of GH-receptor
firansfected
cells. This interaction is down regulated by treatment with BVTA (N-[5-
(aminosulfonyl)-2-methylphenyl]-5-bromo-2-furamide), a GH-receptor binding
compound, which causes a decrease of GH-inducible mRNA. Thus, it is proposed
that
the nuclear GH-receptor is functional and a part of the protein complexes
regulating the
level of transcription.
In a first aspect, this invention provides a method for identifying an agent
that
modulates an interaction between a cytokine class I receptor and a nuclear
factor, the
method comprising: (i) contacting a cell with a candidate agent; and (ii)
determining
whether the candidate agent modulates an interaction between a cytokine class
T
receptor and a nuclear factor that interacts with the receptor; with the
proviso that the
nuclear factor is other than STATS. An agent identified by such a method can
be used,
fox example, for the treatment or prevention of a medical disorder caused by
dysregulation of a cytokine class I receptor binding compound.
CA 02528281 2005-12-06
WO 2005/001478 - $ - PCT/SE2004/001039
The method can optionally include the following steps: (i) contacting the cell
with the candidate agent, wherein the candidate agent modulates the
interaction between
the cytokine class I receptor and the nuclear factor; (ii) measuring, in the
presence of the
candidate agent, a biological effect of a cytokine class I receptor binding
compound in
the cell; and (iii) determining whether the candidate agent modulates the
biological
effect of the cytokine class I receptor binding compound in the cell. In some
embodiments, the candidate agent inhibits the interaction between growth
hormone
receptor and the nuclear factor. In other embodiments, the candidate agent
stimulates
the interaction between growth hormone receptor and the nuclear factor.
The method can optionally include a step of determining the expression of a
reporter gene coupled to a promoter comprising a response element for a
nuclear factor
selected from the group consisting of AP2, OCT1, and Pr2F.
Candidate agents that can be used in the methods described herein include, for
example, polypeptides, peptides, antibodies or antibody fragments, non-peptide
compounds, carbohydrates, small molecules, lipids, single or double stranded
DNA,
single or double stranded RNA, antisense nucleic acid molecules, and
ribozymes.
The identification methods described herein can be carried out in vitro or i~
vivo. For ih vitYO methods, the identification can be made using a cell based
system or a
cell free system.
In another aspect, the invention features a method for identifying a nuclear
factor that interacts with a cytokine class I receptor, the method comprising:
(i)
transfecting a cell with a nucleic acid encoding a cytokine class I receptor;
(ii) preparing
a nuclear extract from the cell; (iii) incubating the nuclear extract with a
labeled
oligonucleotide probe that binds to a candidate nuclear factor; (iv)
separating the
reaction mixture in a polyacrylamide gel; and (v) detecting bands
corresponding to
protein-DNA complexes. The method can optionally include a step of, prior to
preparing the nuclear extract, stimulating the cell with a cytokine class I
receptor
binding compound.
Other methods for identifying nuclear factors include transfecting cells with
a
reporter construct wherein the nuclear factor oligonucleotide is part of the
promoter
regulating transcription of the reporter gene. The cells are stimulated with a
cytokine
class I receptor binding compound and the reporter gene activity is measured.
The nuclear factor is optionally a DNA binding protein, such as AP2, OCT1, or
Pr2F.
CA 02528281 2005-12-06
WO 2005/001478 _ ( _ PCT/SE2004/001039
A method according to the invention can comprise the determination whether
the candidate agent modulates the effect of growth hormone in the cell. In one
embodiment of the invention, such determination comprises determining the
expression
of a reporter gene coupled to a promoter, e.g., the SPI 2.1, AP2, OCT-1 or
Pr2F
promoter.
Reporter genes such as, for example, luciferase, (3-galactosidase, alkaline
phosphatase, chloramphenicol acetyl transferase (CAT), Green Fluorescent
Protein and
other members of the Reef Coral Fluorescent Protein (RCFP) family, can be used
to
determine transcriptional activity in screening assays according to the
invention (see,
for example, Goeddel (ed.), Methods Enzymol., Vol. 185, San Diego: Academic
Press,
Inc. (1990)).
Those agents identified according to the methods described herein that
modulate
the effect of growth hormone in a cell can be used, for example, for the
treatment or
prevention of a medical disorder caused by growth hormone dysregulation. Such
disorders include, e.g., acromegaly, growth hormone deficiency, growth
retardation
associated with the Prader-Willi syndrome and Turner's syndrorile, growth
hormone
insensitivity, wasting disorders associated with Acquired Immunodeficiency
Syndrome
(AmS), and osteoporosis. Further, the agents can be used for decreasing or
inhibiting
the IGF-1 levels or IGF-1 production in a cell.
For the treatment of acromegaly, it is expected that the identified agent will
inhibit or decrease the interaction between GHR and the nuclear factor. For
the
treatment of disorders related to growth hormone deficiency, such as Prader-
Willi
syndrome, Turner's syndrome, growth hormone insensitivity, and osteoporosis,
it is
expected that the identified agent will stimulate or increase the interaction
between
GHR and the nuclear factor.
Those agents identified according to the methods described herein that
modulate
the effect of prolactin in a cell can be used, for example, fox the treatment
or prevention
of a medical disorder caused by prolactin dysregulation. Hyperprolactinemia is
a
disease caused by excess production and secretion of prolactin, and results in
clinical
symptoms such as suppression of reproductive function and galactorrhea. As a
cause of
hyperprolactinemia, prolactin-secreting pituitary tumor (prolactinoma) is
frequently
observed. Further, it is known in the art that metabolic disorders such as
obesity,
hyperglycemia, hyperinsulinemia, hypercholesterolemia, hyperlipidemia and Type
II
diabetes are associated with aberrant patterns in the daily levels (and
fluctuations) of
CA 02528281 2005-12-06
WO 2005/001478 - ~ - PCT/SE2004/001039
prolactin. A subject in need of prolactin is, e.g., a person in need of
stimulation of
lactation, e.g., a mother; a person in need of stimulation of the immune
system, e.g., a
person at risk for an immune disorder, e.g., a person at risk of AIDS, or a
person
infected with a human immunodeficiency virus (HIV), or a person having a
nutritional
deficiency (see, e.g., US Patent No. 6,545,198).
For the treatment of hyperprolactinemia or prolactinoma, it is expected that
the
identified agent will inhibit or decrease the interaction between the
prolactin receptor
and the nuclear factor. For the treatment of disorders related to prolactin
deficiency it is
expected that the identified agent will stimulate or increase the interaction
between the
prolactin receptor and the nuclear factor.
In another aspect, the invention features a method for treating or preventing
a
medical disorder caused by dysregulation of a cytokine class I receptor
binding
compound, the method comprising administering to a subject in need thereof an
effective amount of an agent that modulates an interaction between a cytokine
class I
receptor and a nuclear factor. In some embodiments, the nuclear factor is
other than
STATE.
The method can optionally include a step of identifying a subject as having or
being at risk of having a medical disorder described herein prior to the
administration of
the agent. In addition, or alternatively; the method can include a step of,
following the
administration of the agent, evaluating the subject for the presence of
severity of one or
more symptoms of the medical disorder. The amount of the agent achninistered
to the
subject can optionally be selected based upon the results of such an
evaluation.
In some embodiments, the agent inhibits the interaction between the cytokine
class I receptor and the nuclear factor. In other embodiments, the agent
stimulates the
interaction between the cytokine class I receptor and the nuclear factor.
In another aspect, the invention features a method for modulating IGF-1
transcription in a cell, the method comprising contacting a cell with an
effective amount
of an agent that modulates an interaction between growth hormone receptor and
a
nuclear factor, thereby modulating IGF-1 transcription in the cell. In some
embodiments, the agent inhibits the interaction between growth hormone
receptor and
the nuclear factor, thereby decreasing IGF-1 transcription in the cell. Tn
other
embodiments, the agent stimulates the interaction between growth hormone
receptor
and the nuclear factor, thereby increasing IGF-1 transcription in the cell.
CA 02528281 2005-12-06
WO 2005/001478 - $ - PCT/SE2004/001039
In another aspect, the invention features a method for modulating
transcription
in a cell, the method comprising contacting a cell with an effective amount of
an agent
that modulates an interaction between a cytokine class I receptor and a
nuclear factor,
thereby modulating transcription induced by the cytokine class I receptor in
the cell,
with the proviso that the nuclear factor is other than STATS. The agent can
be, for
example, a compound that binds to the cytokine class I receptor and prevents
or reduces
the ability of the cytokine class I receptor to bind to the nuclear factor. In
some
embodiments, the agent inhibits the interaction between the cytokine class I
receptor
and the nuclear factor, thereby decreasing transcription induced by the
cytokine class I
receptor in the cell. In other embodiments, the agent stimulates the
interaction between
the cytokine class I receptor and the nuclear factor, thereby increasing
transcription
induced by the cytokine class I receptor in the cell.
In the methods and compositions described herein, the cytokine class I
receptor
can be, for example growth hormone receptor or prolactin receptor. In some
embodiments, the cytokine class I receptor binding compound is growth hormone
and
the cytokine class I receptor is growth hormone receptor. In other
embodiments, the
cytokine class I receptor binding compound is prolactin and the cytokine class
I receptor
is prolactin receptor.
The nuclear factor used in the methods and compositions described herein can
be, for example, AP2, OCT1, or Pr2F.
The agent used in the methods described herein can be, for example, a
polypeptide, peptide, antibodiy or antibody fragment, non-peptide compound,
carbohydrate, small molecule, lipid, single or double stranded DNA, single or
double
stranded RNA, antisense nucleic acid molecule, or ribozyme.
Unless otherwise defined, all technical and scientific terms used herein have
the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Suitable methods and materials are described below,
although
methods and materials similar or equivalent to those described herein can also
be used
in the practice or testing of the present invention. All publications, patent
applications,
patents, and other references mentioned herein are incorporated by reference
in their
entirety. In case of conflict, the present specification, including
definitions, will control.
In addition, the materials, methods, and examples are illustrative only and
not intended
to be limiting.
CA 02528281 2005-12-06
WO 2005/001478 _ 9 - PCT/SE2004/001039
The invention will now be further illustrated through the description of
examples of its practice. The examples are not intended as limiting in any way
of the
scope of the invention.
. EXAMPLES
EXPERIMENTAL METHODS
Cell cultuYe: WRL-68 cells, a human fetal hepatic cell-Line, were cultured in
EMEM medium with NaHC03 (Statens Veterinarmedicinska Anstalt), supplemented
with 10% fetal bovine serum (FBS), 2% L-Glutamine, 1 % Pyruvate and 1 % Non
essential amino acids (NEA), all from GIBCO. The cells were sub-cultivated
twice
weekly by trypsination to maintain a cell density of approximately 80%.
Ti~ansfection: Cells were transfected with human full-length GH-receptor
(pMB1288, 2 ~.g/p.l) by using DOTAP Liposomal Transfection Reagent (Roche),
according to manufacturers manual. With this reagent a cationic liposome-
mediated
transfection method (lipofection) is used. T75 flasks with WRL-68 cells were
transfected with 10 pg DNA for each flask. 5 p,1 (lOpg) DNA was diluted with
250 p1
OPTIMEM1 medium (GIBCO) and mixed gently with 75 ~.l DOTAP reagent diluted in
175 ~.1 OPT1MEM1 medium. The mixture was incubated for 10 minutes and then
mixed
gently with 10 ml OPTIMEM1 medium. Cells were washed once and then incubated
for
4 hours with the DNA/DOTAP transfection mix, which was then changed to fresh
culture medium. Calls were re-seeded into new culture dishes the following
day.
Stimulation: Cells were stimulated two days after transfection at a confluence
of
approximately 80%. Starvation in serum free medium for one hour were followed
by
stimulation with 10 nM hGH (Genotropin, Pharmacia) for 60 minutes, 1 p,M BVTA
(N-
[5-(aminosulfonyl)-2-methylphenyl]-5-bromo-2-furamide) for 60 minutes or with
1 p,M
BVTA for 60 minutes followed by 10 nM hGH for 60 minutes. Starvation in serum
free
medium for three hours (GHR-transfected cells) or sixteen hours (non-
transfected
WRL-68 cells) were followed by stimulation with 100 nM hGH for 20 minutes, 1
p,M
CA 02528281 2005-12-06
WO 2005/001478 _ IQ _ PCT/SE2004/001039
BVTA for 20 minutes or 1 ~M BVTA for 20 minutes followed by 100 nM hGH for 20
minutes.
Prepaf~ation of hucleaf- aszd cytosolic fractioyas: Nuclear extracts were
prepared
according to the methods described in Dignam et al. (1983) and Ausubel et al.
(1993),
with the following modifications:
WRL-68 cells were scraped into ice cold PBS, pooled in 50 ml Falcon tubes and
centrifuged 3000 rpm for 5 minutes. Cell pellets were re-suspended in
hypotonic buffer
with protease and phosphatase inhibitors (10 mM HEPES pH 7.9, 0.2mM
phenylmethylsulfonyl-fluoride (PMSF), both from Sigma, 1.5 mM MgCl2, 10 mM
KCI,
0.1 % Nonidet P-40 (Amersham), 0.2 mM sodium ortho-vanadate, 2 nM okadaic acid
(Calbiochem), lxcornplete protease inhibitor (Roche)), centrifuged 3000 rpm
for 5
minutes and re-suspended again in hypotonic buffer. The cells were allowed to
swell on
ice for ten minutes and then homogenized in a bounce homogenizes with a Teflon
pestle type C. Cell lysis was confirmed in a microscope by adding trypan blue
(0.4%,
Sigma) to an aliquot of cells, since nuclei will stain blue in broken cells.
Approximately
40 strokes were necessary to get 70-80% clean nuclear fractions. The nuclei
were
collected by centrifugation at 4000 rpm for I S minutes and immediately frozen
in
-70°C. The supernatant was also saved in -70°C as the cytosolic
fraction.
Nuclear and cytosolic fractions were prepared from hypophysectomized
Sprague Dawley rat livers (ethical license N176/02). The rats had minipumps
implanted at 5 weeks of age for administration of hGH. Control animals were
not
stimulated and thus completely GH-deficient while GH-animals were stimulated
continuously with hGH 0.12 mg/kg/day for five days. Animals were anaesthetized
and
livers were cut out and placed in ice-cold PBS. Livers were then cut into
smaller pieces
and transferred to 8 ml ice-cold hypotonic buffer with protease and
phosphatase
inhibitors and were allowed to swell on ice for 10 minutes. The small pieces
of liver
were homoge~uzed in a bounce homogenizes with Teflon pestle type C and the
suspension was filtered through a sterile compress to get a cell-suspension.
The cell-
suspension was further homogenized in a bounce homogenizes with a glass
pestle. Cell
lysis was confirmed in a microscope by adding trypan blue (0.4%, Sigma) to an
aliquot
of cells, since nuclei will stain blue in broken cells. Approximately 10-20
strokes were
necessary to achieve 70-80% clean nuclear fractions. The nuclei were collected
by
centrifugation at 4000 rpm and 4°C for IS minutes. The supernatant was
removed and
CA 02528281 2005-12-06
WO 2005/001478 _ 11 _ PCT/SE2004/001039
saved as the cytosolic fraction. Pelleted nuclei and cytosol were immediately
frozen in -
70°C.
Extraction of nuclear proteins: To extract the proteins from the nuclei, the
nuclear pellets were re-suspended in 2xlysis buffer with protease and
phosphatase
inhibitors (100 mM HEPES, pH 7.6, 300 mM NaCI, 10 mM EDTA (GIBCO), 2%
Triton X-100 (Sigma), 0.2 mM PMSF, 0.2 mM sodium orto-vanadate, 2 nM okadaic
acid, lxcomplete), or EMSA buffer with inhibitors (20 mM Tris pH 8.0, I.SmM
MgCl2,
0.2 mM EDTA, 25% glycerol, 0.5 mM.PMSF, 0.2 mM sodium ortho-vanadate, 2 nM
okadaic acid, lxcomplete protease inhibitor), depending on whether the samples
should
also be used in electrophoresis mobility shift assays (EMSA) or not. For an
optimal
protein extraction, three different methods were evaluated. Samples were left
on ice for
30 minutes, treated with 1 p.I DNAse at 37°C for 15 minutes or treated
with an injection
needle to shear the DNA. Optimal extraction was evaluated by Western blot and
protein
concentration determination as described below. The samples were centrifuged
at 14000
rpm and 4°C for 10 minutes and supernatants with nuclear proteins were
divided in
aliquots and stored in -70°C or used in Western Blot or EMSA assays.
Protein concentrations were determined by using a BCA Protein Assay Reagent
Kit (Pierce), which uses the Biuret reaction (reduction of Cu2+ to Cul+ by
proteins in an
alkaline medium) and a reagent containing bicinchoninic acid (BCA). Two
molecules of
BCA form a complex with one Cul~ ion, which gives a purple color and has a
strong
absorbance at 562 nm. Bovine serum albumin (BSA) was used as standard.
Inzmuraoprecipitation: Protein G Sepharose (Amersham) was used to
immunoprecipitate GHR and phospho-STATSb from the total nuclear extracts.
Protein
G is immobilized on sepharose beads and binds to the F~ region of IgG, leaving
the Fab
region available for binding the antigen. For each sample, 60 ~,l protein G
Sepharose
slurry was used (approximately 30 ~.l clean sepharose). The sepharose was
pooled in
one 2 ml Eppendorf tube and washed in 3x 1 ml of phosphate buffer saline (PBS)
by
centrifuging 3000 rpm for 2 minutes. Then the antibody was added, 5 ~,1/sample
of
either anti-GHR (MAb 263, AGEN) or anti phosphoSTATSa/b, Y694/Y699 (Upstate
Biotechnology). The sepharose with antibody was diluted with PBS and mixed end
over
end at room temperature for 2 hours. After that the sepharose slurry was
divided in
CA 02528281 2005-12-06
WO 2005/001478 _ 12 _ PCT/SE2004/001039
clean 2 ml Eppendorf tubes and washed in 1x1 ml PBS. Nuclear lysates were then
thawed and added to the sepharose, I00-200 ~,l lysate from each sample.
Dilution was
made with S00 ~Cl 2xlysis buffer with protease and phosphatase inhibitors as
above,
followed by mixing end over end at 4°C over night. Next day each sample
was washed
S two times with lxlysis buffer and once with lxlysis buffer and 12S mM Tris
pH 6.~ in a
mix 1:1. The proteins were then dissociated from the sepharose by adding
4xNuPAGE
sample buffer (sodium dodecyl sulphate (SDS), bromphenol blue (BFB), glycerol)
and
SOmM of the reducing agent dithiothreitol (DTT) (Sigma) to the samples and
thereafter
heating them at 70°C for 10 minutes. The irrnnunoprecipitated proteins
in the
supernatants could then be separated with gel electrophoresis.
Gel Elect~opho~esis: Extracted nuclear proteins were treated with 4xNuPAGE
sample buffer azid were then heated at 70°C for 10 minutes. Samples
analyzed in
reduced form were also treated with SO mM DTT in order to reduce disulfide
bonds and
1 S break protein-protein interactions. Proteins were then separated on NuPAGE
4-12%
Bis-Tris gel (Invitrogen). The gels were run at 200V and room temperature for
1' hour,
running buffer Mops (SO mM MOPS, SO mIVI Tris, 3.S mM SDS, 1mM EDTA). As a
molecular weight marker SeeBlue Standard (Invitrogen) was used.
Weste~fa blot: Proteins were transferred to Hybond ECL nitrocellulose
membranes (Atnersham) at 4°C and 100V for 1 hour with lxNuPAGE transfer
buffer
(2SmM Bis-Tris, 2SmM Bicine, 1mM EDTA, 10% EtOH). Membranes were then
blocked with blocking buffer consisting of 1 % milk in Tris Buffered Saline-
Tween
(TBST) (130 mM NaCI, 10 mM Tris-HCl pH 7.5, O.OS % Tween 20 (Amersham)) over
2S night at 4°C to prevent unspecific binding of antibodies to the
membrane. Membranes
were washed in TBST 2xS minutes and then incubated with primary antibody for 1
hour
at room temperature. After that, membranes were washed again in TBST 3x10
minutes,
incubated with secondary antibody for 1 hour at room temperature and washed in
TBST
4x10 minutes. The Horse Radish Peroxidase (HRP) coupled secondary antibodies
were
detected with ECL+ Plus and exposed on ECL Hyperfilm, both from Amersham.
St~ippi~rg ahd y~e-blottirag.~ Membranes were stripped from antibodies by
incubation in stripping buffer (62.5 mM Tris pH 6.5, 2% SDS, 100 mM (3-
CA 02528281 2005-12-06
WO 2005/001478 _ 13 _ PCT/SE2004/001039
Mercaptoethanol) at 50°C for 30 minutes. They were washed for 2x10
minutes in large
volumes of TBST and then blocked in blocking buffer (1% milk in TBST) at
4°C over
night. Re-blotting could then be performed with another antibody as described
above.
Primary antibodies used: Mouse monoclonal anti-GHR MAb 263 #174A-021 1:1000
(AGED, rabbit polyclonal antisera anti-GHR directed to the intracellular part
of GHR
1:5000 (Zhang et aL, 2001), rabbit polyclonal antisera anti-GHR directed to
the
extracellular part of GHR (Biovitrum), rabbit polyclonal IgG anti-STATSb (C-
17) Lot
#252 1:X000 (Santa Cruz Biotechnology), rabbit polyclonal IgG anti-
phosphoSTATSa/b
(Y694/Y699) 1:1000 (Upstate Biotechnology). Secondary antibodies used: Goat
anti-
mouse IgG-HRP 1:2000 (Dako A/S), Sheep anti-mouse Ig-HRP 1:2000 (Amersham),
Swine anti-rabbit IgG-HRP 1:3000 (Dako A/S).
Electrophoretic Mobility Shift Assay: Electrophoretic Mobility Shift Assay
(EMSA) is a method used to investigate protein-DNA interactions. A DNA probe
with a
known sequence is end-labeled with 33P-ATP and then incubated with nuclear
extracts.
Proteins that bind specifically to the DNA-probe will reduce the mobility of
the
complex when separated on a non-denaturing poly acryl-amid gel and a shifted
band
can be seen. To identify proteins bound to the DNA-probe, an antibody can be
added to
the nuclear extract before incubation with the DNA-probe. If the antibody
recognizes a
protein that binds to the DNA, the mobility of the complex will be reduced
even further
and cause a supershift.
Labeling of oligonucleotides with 33P: Oligonucleotides Were end-labeled with
[Y 33P~ ATP (2500 Gi/mmol, Amersham or 3000 Ci/mmol, Perkin Elmer) by mixing
2~,1
consensus oligonucleotide (1.75 pmol/~,1, Promega or SGS DNA), 1 p,1 T4
Polynucleotide Kinase l Ox Buffer (Promega), 1 ~1 T4 Polynucleotide Kinase
(Promega), 1 ~1 [y-33P] ATP (Amersham or Perkin Elmer) and 5 ~.1 nuclease free
water
(DEPC medium). The mixture was incubated at 37°C for 10 minutes and the
reaction
was stopped by adding 1 ~,1 O.S M EDTA. The volume was adjusted to 100 ~cl by
adding
89 X10.05 M EDTA. To remove unincorporated [y-33P] ATP, the 100 ~,1 aliquot
was
loaded on a NICI~TM Column (Amersham Pharmacia Biotech AB) and labeled DNA
was eluted with 2x400 x,10.05 M EDTA. Specific activity was measured by mixing
2 ~.1
CA 02528281 2005-12-06
WO 2005/001478 _ 14 _ PCT/SE2004/001039
33P-labeled sample with 200 p,1 scintillation fluid and counting was performed
in a Beta
counter (Trilux1450).
Oligonucleotide sequences
NO:l)
NO: 2)
GHRE:
5'-TAC GCT TCT ACT AAT CCA TGT TCT GAG AAA TCA T-3' (SEQ m
3'-ATG CGA AGA TGA TTA GGT ACA AGA CTC TTT AGT A-5' (SEQ ID
OCT 1:
5'-TGT CGA ATG CAA ATC ACT AGA A-3' (SEQ ID NO: 3)
3'-ACA GCT TAC GTT TAG TGA TCT T-5' (SEQ ID NO: 4)
AP2: .
5'-GAT CGA ACT GAC CGC.CCG CGG CCC GT-3' (SEQ ID NO: 5)
3'-CTA GCT TGA CTG GCG GGC GGC GGG CA-5' (SEQ ID NO: 6)
AP1:
5'-CGC TTG ATG AGT CAG CCG GAA-3' (SEQ ID NO: 7)
3'-GCG AAC TAC TCA GTC GGC CTT-5' (SEQ ID NO: 8)
Sequences of oligonucleotides that were annealed and used in EMSA or cloned
to reporter vectors
Pr2F linker with BgIIIlHindIII overhangs, used in EMSA:
5'-GATCTAGATGCTTTCACAAACCCCACCCACAAA-3' (SEQ ID NO: 9)
S'-AGCTTTTGTGGGTGGGGTTTGTGAAAGCATCTA-3' (SEQ ID NO: 10)
Linker containing two potential AP2 sites, with KpnIlXhoI overhangs for
cloning to Luc- and SEAP-reporter vectors:
5'-CTAGATGCTTTCACAAACCCCACCCACAA.AATAGATGCTTTCACA
AACCCCACCCACAA.AAC-3' (SEQ ID NO: 11)
CA 02528281 2005-12-06
WO 2005/001478 _ 15 _ PCT/SE2004/001039
5'-TCGAGTTTTGTGGGTGGGGTTTGTGAAAGCATCTATTTTGTGGGTG
GGGTTTGTGAAAGCATCTAGGTAC-3' (SEQ ID NO: 12)
Linker containing two "Promega" AP2 sites, with KprlIlXhoI overhangs for
cloning to Luc- and SEAP-reporter vectors.
S'-CGATCGAACTGACCGCCCGCGGCCCGTGATCGAACTGACCGCCCG
CGGCCCGTC-3' (SEQ ID NO: 13)
S'-TCGAGACGGGCCGCGGGCGGTCAGTTCGATCACGGGCCGCGGGCG
GTCAGTTCGATCGGTAC-3' (SEQ ID NO: 14)
P~eparatiofz of DNA binding ~eactiorxs: For each reaction, 3-6 ~.g of nuclear
protein extracted in EMSA buffer was mixed with 2 ~.l Gel Shift 5x Binding
Buffer
(Promega) and nuclease free water (DEPC medium) to 9 ~1 and then incubated for
10
minutes at room temperature. For supershift analysis, nuclear extracts were
pre-
IS incubated with 1 ~,l of anti-GHR antibody or antisera for 1 hour at room
temperature.
To control the specificity of the protein-DNA binding, 400x excess of specific
or
unspecific un-labeled oligonucleotide was'added to control reactions. The
reactions
were then incubated with 1 ~.1 of 33P-end labeled GHRE (SGS DNA), AP 1, AP2 or
OCT1 consensus oligonucleotides (Promega) for 20 minutes at room temperature.
Gel electrophoresis of Proteiya-DNA complexes: 2 ~1 of 6x loading buffer (3x
TBE buffer, 32% glycerol, 0.06% BFB) was added to each sample, and the samples
were then analyzed on a Novex 6% DNA retardation gel (Invitrogen). As running
buffer
O.SxTBE (50 mM Tris pH 8.4, 45 mM Boric Acid, O.S mM EDTA (GIBCO)) was used
and gels were run at 250 volt and room temperature for 19 minutes. Gels were
fixed in a
fix solution (30% ethanol, 10% acetic acid) and dried in a gel dryer. They
were then
analyzed with phosphor imager instrumentation (STORM 860 (Molecular Dynamics)
and Image Quant S.0).
EMSA - YYester~rz Blot: EMSA was performed as described above, but instead of
drying the geI and expose it to a phosphor imager screen, the proteins in the
gel were
transferred to a nitrocellulose membrane and blotted with an anti-GHR antibody
as in a
regular Western Blot, as described. To confirm that the proteins were
transferred to the
CA 02528281 2005-12-06
WO 2005/001478 _ 16 _ PCT/SE2004/001039
membrane, the membrane was immersed in Ponceau S Solution (Sigma), which
stains
all proteins red.
EXAMPLE l: GHR and STATE are~resent in nuclear extracts
Cultured cell lines usually exhibit low amounts of endogenously produced GHR
(Fig. 3C), whereas liver tissue contains high amounts of receptors (Fig. 3D).
To detect
the GHR in the nucleus, nuclear proteins were separated with gel
electrophoresis and
visualized in Western Blot. When blotted with a polyclonal antibody against
the
intracellular part of the receptor, the GHR could be detected as three
distinct bands in
all fractions of the examined cell types (WRL-68, GHR-transfected WRL-68 and
HX
rat livers. None of the bands appeared as a result of unspecific binding of
the secondary
antibody (pig anti-rabbit, Fig. 3B).
The Western Blot membrane with non-transfected WRL-68 total nuclear
extracts was re-blotted with a polyclonal antibody against STATE, and two
bands of 60
and 100 kDa, respectively, could be seen. The 100-kDa band probably
represented
intact STATE monomers, while the 60-kDa band could be a cleaved form of STATE
(Data not shown).
EXAMPLE 2: Transfection of WRL-68 cells with full-length GH-receptor increase
protein interactions with GHRE, AP2, OCT1 and Pr2F DNA probes
GH-receptor transfected and non-transfected WRL-68 nuclear extracts were
incubated with anti-GHR antibody and 33P-GHRE (SEQ ID NOS: 1 and 2), 33P-AP2
(SEQ ID NOS: 5 and 6) or 33P-OCT1 (SEQ 1D NOS: 3 and 4) or 33P-Pr2F (SEQ ID
NOS: 9 and 10) consensus oligo-nucleotides and analyzed in EMSA to elucidate
any
possible interactions of the GHR with proteins binding to these DNA probes.
For
control, nuclear extracts not incubated with anti-GHR were also analyzed EMSA
with
the GHRE probe showed several shifted bands in both GHR-transfected and non-
transfected cells. The specificity of the bands was shown by incubating with
400 times
excess of unlabelled specific or unspecific probe (see Fig. 1 and Fig. 2). No
effect of
hGH stimulation could be identified. Interestingly, transfection with full-
length GHR
seemed to increase the intensity of the top shifted band. The protein-DNA
interaction in
this band was disrupted by BVTA (N-[5-(aminosulfonyl)-2-methylphenyl]-S-bromo-
2-
furamide), since the intensity of the band was decreased in extracts from BVTA
stimulated cells, and a lower band appeared representing a smaller complex
with higher
CA 02528281 2005-12-06
WO 2005/001478 - 1'7 - PCT/SE2004/001039
mobility. No difference in binding could be seen when incubating these
extracts with
anti-GHR antibody. In non-transfected cells, incubation of nuclear extracts
with anti-
GHR seemed to increase protein binding to the GHRE probe. The increased
intensity of
these bands could also be due to a supershift of the weak, lower bands. There
was also a
vague indication of a supershift of the GHRE top weak band in non-transfected
cells,
but in this case no effect from BVTA could be seen. Incubation with antibody
only and
labeled GHRE probe showed that the antibody did not interact with DNA itself.
EMSA with AP2 DNA probe and non-transfected WRL-68 cells showed one
shifted band that could not be competed out (Fig. 1B). In EMSA with APZ and
GHR
transfected WRL-68, an additional band could be seen, which was specific since
it was
competed by 400x excess of unlabeled AP2 probe (Fig. 1B, arrow). When
incubating
these nuclear extracts with an anti-GHR antibody, this specific band increased
in
intensity, suggesting an enhanced interaction between the protein complex and
the AP2
DNA probe. As with GHRE, BVTA broke the protein-DNA interaction represented by
the top specific band, since this band disappeared in extracts from BVTA
stimulated
cells. As before, no effects could be seen from hGH stimulation compared to
control
extracts. The antibody itself showed no interaction with the DNA probe.
EMSA with OCT1 DNA probe and the two WRL-68 cell types showed two
shifted bands (Fig. 1A). The top shifted band represented specific binding,
since it was
competed with 400 times excess of un-labeled OCT1 probe. As with GHRE and AP2,
the intensity of the top band was increased in extracts from GHR-transfected
WRL-68
cells (Fig. 1A, arrow), but in contrast to AP2, incubation of extracts with an
anti-GHR
antibody did not affect the binding of nuclear proteins to the OCT1 DNA probe.
No
effects could be seen in nuclear extracts from hGH stimulated cells compared
to un-
stimulated control cells. Also in this case BVTA broke some protein-DNA
interactions;
the intensity of the top specific band was decreased in extracts from BVTA
treated cells.
As with GHRE and AP2, the antibody itself showed no interaction with the DNA
probe.
Non-transfected WRL-68 nuclear extracts were analyzed with APl DNA probe
(SEQ ll~ NOS: 7 and 8) as a control since AP1 is activated via GH-activated
MAP
kinase. One shifted band could be seen in all fractions showing an activated
AP1
transcription factor, and no effects could be seen from either hGH or BVTA.
Incubation
with a GHR-antibody resulted in somewhat fainter bands.
EMSA was performed with AP2 and OCT1 probes together with nuclear extract
from GHR-transfected WRL-68 cells. The separated complexes were transferred to
a
CA 02528281 2005-12-06
WO 2005/001478 - 1$ ~ PCT/SE2004/001039
nitrocellulose membrane. Blotting the membranes with an anti-GHR antibody
indicated
the presence of GHR in a blurred band that, when compared to the EMSA gels,
seemed
to correspond to the specific shifted bands of OCT1 and AP2 oligonucleotide.
The
correlation of the GHR bands and the OCTl- and AP2-shifted bands indicated
that
GHR is present in the OCT1 shifted complex, and possibly also in the AP2
shifted
complex.
EXAMPLE 3 : Incubation of rat liver nuclear extracts with anti-GHR antibodies
enhances protein binding to an AP2 DNA~robe
HX rat liver nuclear extracts were incubated with anti-GHR and 33P-GHRE, 33P-
AP2 or 33P-OCT1 consensus oligonucleotides and analyzed in EMSA. For control,
nuclear extracts not incubated with anti-GHR were also analyzed. Extracts from
two
control animals and two hGH stimulated animals were used in the assays.
EMSA with GHRE and OCT1 DNA probes did not show any differences in
protein-DNA binding either with hGH stimulation or with pre-incubation of
nuclear
extracts with an anti-GHR antibody. In contrast, EMSA with an AP2 DNA probe
showed an enhanced protein-DNA interaction when nuclear extracts were
incubated
with an anti-GHR antibody (Fig. 1 C). As mentioned abave,. this effect was
also seen
with GHR-transfected WRL-68 cells and AP2 probe (Fig. 1B). No difference in
protein-
DNA binding could be seen in extracts from hGH stimulated animals compared to
control animals though. To confirm that the increased protein-DNA interactions
seen in
anti-GHR incubated HX rat liver nuclear extracts were specific for anti-GHR
antibodies, other antibodies against the GHR were tested. The experiment was
repeated
with two different rabbit polyclonal antibodies, one directed against the
extracellular
part and one against the intracellular part of the GHR. Both of these
antibodies showed
the same enhancing effect of protein-DNA interaction as the first antibody
used which
was a mouse monoclonal antibody against the extracellular part of the GHR.
None of
the antibodies interacted with the DNA probe themselves. To confirm that the
enhanced
protein-DNA interaction seen was specific for anti-GHR antibodies and not
caused by
any antibody, the same experiment was performed with an anti-PKC antibody.
Anti-
PKC did not enhance the protein-DNA interaction.
CA 02528281 2005-12-06
WO 2005/001478 - 19 .- PCT/SE2004/001039
REFERENCES
Ausubel, F.M., et al. (1993). In: Current Protocols in Molecular Biology,
Vol.2, John
Wiley and Sons, New York.
Bichell, D.P., Kikuchi, I~., Rotwein, P. (1992). Growth Hormone Rapidly
Activates
Insulin-Like Growth Factor I Gene Transcription irz Tlivo. Mol. Endocrin.
6:1899-1908
Clevenger, C.V., Furth, P., Hankinson, S.E., Shuler, L. (2003). The role
ofprolactin in
mammary carcinoma. Endocrine Rev. 24:1-27
Dignam, J.D., Lebowitz, R. M., Roeder, R. (1983). Accurate transcription
initiation by
RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucl.
Acids
Res. 11:1475-1489.
,. . Graichen, R., Sandstedt, J., Goh, E. L. K., Isaksson, O. G. P.,
Tornell,.J., Lobie, P. E.
(2003). the Growth Hormone-binding Protein Is a Location-dependent Cytokine
Receptor Transcriptional Enhancer. J Biol. Chern. 278:6346-6354
Helander, H., Gustafsson, J-~., Mode, A. (2002). Possible involvement of
truncated
signal transducer and activator of transcription-5 in the GH pattern-dependent
regulation of CYP2C12 gene expression in rat liver. Mol. Eradocrinology
16:1598-1611
Jiang, L.-W., Schindler, M. (1990). Nucloecytoplasmic transport is enhanced
concomitant with nuclear accumulation of epidermal growth factor (EGF) binding
activity in both 3T3-1 and EGF receptor reconstituted NR-6 fibroblasts. J.
Cell Biol.
110:559-568
Le Stunff, C., Thomas, M. J., Rotwein, P. (1995). Rapid Activation of Rat
Insulin-Like
Growth Factor-I Gene Transcription by Growth Hormone Reveals No Changes in
Deoxyribonucleic Acid-Protein Interactions within the Second Promoter.
Endocrinology 136:2230-2237
CA 02528281 2005-12-06
WO 2005/001478 _ 2~ _ PCT/SE2004/001039
Lobie, P. E., Wood, T. J. J., Chen, C. M., Waters, M. J., Norstedt, G. (1994).
Nuclear
Translocation and Anchorage of the Growth Hormone Receptor. J. Biol. Chem.
269:31735-31746
Nolten, L.A., Steenbergh, P.H., Sussenbach, J.S. (1995). Hepatocyte nuclear
factor la
activates promoter 1 of the human insulin-like growth factor 1 gene via two
distinct
binding sites. Mol. Endocrinology 9: 1488-1499
Pfisterer, P., Ehlennann, J., Hegen, M., Schorle, H. (2002). A Subtractive
Gene
Expression Screen Suggests a Role of Transcription Factor AP-2cc in Control of
Proliferation and Differentiation. J. Biol. Cheni. 277:6637-6644
Podlecki, D.A, Smith, R.M., Kao, M., Tsa, M., Huecksteadt, T., Brandenburg,
D.,
Lasher, R.S., Jarret, L., Olefsky, J.M. (1987). Nuclear translocation of the
insulin
receptor. J. Bi~l. Chem. 262:3362-3368
Thomas, M. J., Kikuchi, K., Bichell, D. P., Rotwein, P. (1994) Rapid
activation ~f rat
insulin-like growth factor-1 gene transcription by growth hormone reveals no
alterations
in deoxyribonucleic acid-protein interactions within the major promoter.
Endocrinology
135:1584-1592
Werling, U., Schorle, H. (2002). Transcription Factor gene AP-2yEssential for
Early
Murine Development. Mol. Cell. Biol. 22:3149-3156
Wood, T. (1996). Growth hormone, JAKs and STATs: a model cytokine signal
transduction system. Thesis.
Zhang, Y., Guan, R., Jiang, J., Kopchick, J.J., Black, R.A., Baumann, G.,
Frank, S.
(2001). Growth hormone (GH)-induced dimerization inhibits phorbol ester-
stimulated
GH receptor proteolysis. J. Biol. Chem. 276: 24565-24573
CA 02528281 2005-12-06
WO 2005/001478 1 PCT/SE2004/001039
SEQUENCE LISTING
<110> Biovitrum AB
<120> Methods for identifying active compounds
<130> BV-1045
<160> 14
<170> Patentln version 3.1
<210> 1
<211> 34
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 1
tacgcttcta ctaatccatg ttctgagaaa tcat 34
<210> 2
<211> 34
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 2
atgcgaagat gattaggtac aagactcttt agta 34
<210> 3
<211> 22
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 3
tgtcgaatgc aaatcactag as 22
<210> 4
<211> 22
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 4
acagcttacg tttagtgatc tt 22
CA 02528281 2005-12-06
WO 2005/001478 ~ PCT/SE2004/001039
<210> 5
<211> 24
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 5
24
gatcgaactg accgcccgcg gccc
<210> 6
<211> 26
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 6
26
ctagcttgac tggcgggcgc cgggca
<210> 7
<211> 21
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 7
21
cgcttgatga gtcagccgga a
<210> 8
<211> 21
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 8
gcgaactact cagtcggcct t 21
<210> 9
<211> 33
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 9
gatctagatg ctttcacaaa ccccacccac aaa 33
CA 02528281 2005-12-06
WO 2005/001478 3 PCT/SE2004/001039
<210> 10
<211> 33
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 10
agcttttgtg ggtggggttt gtgaaagcat cta 33
<210> 11
<211> 62
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 11
ctagatgctt tcacaaaccc cacccacaaa atagatgctt tcacaaaccc cacccacaaa 60
ac
<210> 12
<211> 70
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
62
<400> 12
tcgagttttg tgggtggggt ttgtgaaagc atctattttg tgggtggggt ttgtgaaagc 60
atctaggtac
<210> 13
<211> 54
<212> DNA
<213> unknown
<220>
<223> PCR oligonucleotide
<400> 13
cgatcgaact gaCCgCCCgC ggcccgtgat cgaactgacc gcccgcggcc cgtc 54
<210> 14
<211> 62
<212> DNA
<213> unknown
CA 02528281 2005-12-06
WO 2005/001478 4 PCT/SE2004/001039
<220>
<223> PCR oligonucleotide
<400> 14
tcgagacggg ccgcgggcgg tcagttcgat cacgggccgc gggcggtcag ttcgatcggt 60
62
ac