Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST ~.E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter 1e Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional vohxmes please contact the Canadian Patent Oi~ice.
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
COMPOSITIONS FOR TREATING PATHOLOGIES THAT NECESSITATE
SUPPRESSION OF GASTRIC ACID SECRETION
FIELD OF THE INVENTION
The present invention relates to novel oral compositions for inhibition of
gastric acid
secretion that possess fast onset, prolonged inhibition effect on gastric acid
secretion and are
meal-independent.
BACKGROUND OF THE INVENTION
A wide number of pathological conditions are characterized by the need to
suppress
gastric acid secretion. Such conditions include, but are not limited to
Zollinger/Ellison
syndrome (ZES), gastroesophageal reflux disease (GERD), peptic ulcer disease,
duodenal
ulcers, esophagitis, and the like. Conditions such as peptic ulcers can have
serious
complications and represent some of the most prevalent diseases in
industrialized nations.
Presently, the main therapies employed in the treatment of GERD and peptic
ulcer
diseases include agents for reducing the stomach acidity, for example by using
the histamine
HZ-receptor antagonists or proton pump inhibitors (PPI's). PPI's act by
inhibiting the parietal
cell H+/K+ ATPase proton pump responsible for acid secretion from these cells.
PPI's, such
as, omeprazole, and its pharmaceutically acceptable salts are disclosed for
example in EP
05129, EP 124495 and US Patent No. 4,255,431.
PPI agents are acid-labile pro-drugs that are usually administered in enteric-
coated
granules. Following their absorption in the small intestine PPIs, which are
weak bases,
preferentially accumulate within the acid milieu of parietal cells. The acid
environment
within the acid milieu of parietal cells causes the conversion of the pro-
drugs into the active
sulfenamids, which are the active agents that bind and inhibit the parietal
cell H+/K+ ATPase
pumps.
Despite their well-documented efficacy, PPIs have notable limitations. The
time of
dosing and ingestion of meals may influence the pharmacokinetics of these
agents as well as
their ability to suppress gastric acid secretion (Hatlebakk et al., Aliment
Pharmacol Ther.
2000;14(10):1267-72). Specifically, the PPI must be taken prior to ingestion
of food in order
to achieve optimal suppression of gastric acid secretion. Furthermore, PPIs
have a relatively
°slow onset of pharmacological action and may require several days to
achieve maximum acid
suppression and symptom relief, limiting their usefulness in on-demand GERD
therapy
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
(Sachs G, Eur J Gastroenterol Hepatol. 2001;13 Suppl 1:535-41). Moreover, PPIs
fail to
provide 24-h suppression of gastric acid and nocturnal acid breakthrough that
leads to
heartburn pain in GERD patients and occurs even with twice-daily dosing of
PPIs (Tytgat
GN, Eur J Gastroenterol Hepatol. 2001;13 Suppl 1:529-33). Finally, these drugs
exhibit
substantial inter-patient variability in pharmacokinetics and may have
significant interactions
with other drugs (Hatlebakk et al., Clin Pharmacokinet. 1996; 31(5):386-406).
Thus, an
improvement of PPI-mediated activity is a well-recognized challenge in
gastroenterology.
Pentagastrin (PG) ((3-alanyl-L-tryptophyl-L-methionyl-L- aspartyl-L-phenyl-
alanyl
amide; SEQ ID N0:2) is a pentapeptide containing the carboxyl terminal
tetrapeptide of
gastrin. This carboxyl terminal tetrapeptide is the active portion found in
essentially all
natural gastrins. In animals, PG acts to induce gastric acid secretion mainly
via induction of
histamine release from enterochromafin-like (ECL) cells residing in the
stomach. The release
of histamine and the consequent activation of histamine receptors residing on
the parietal
cells, leads to the activation of the parietal cells to actively secrete
proton ions to the gastric
lumen. It is also possible that PG acts directly on the parietal cells to
induce its activation.
PG is typically used in the art as a diagnostic agent for the evaluation of
gastric acid secretory
function.
The low solubility of PG in acidic environment and the fact that PG is prone
to pepsin
degradation in the stomach, rendered its use as an inducer of gastric acid
secretion following
oral administration clearly unexpected until Applicants discovery. Prior to
Applicants
discovery, PG was considered by anyone skilled in the art to only be
effectively active at
inducing acid secretion if administered via parenteral routes. Indeed, no
effect on acid
secretion was noted in four normal subjects subjected to oral administration
of PG, whereas
some effect was noted in three additional patients with gastrointestinal
abnormalities (Morrell
& Keynes Lancet. 1975; 2(7937):712). In fact, this study was cited in a
pharmacology
textbook as a proof of lack of PG activity when administered orally
(Martifzdale Thirty-
secorZd editiota, p1616, the Chapter: "Supplerneratary Drugs and Other
Substayzces").
W001/22985 to Pisegna et al. (the '985 publication) discloses the use of PG
administered systemically in conjunction with a proton pump inhibitor (PPI).
According to
the '985 publication, administration of PG in combination with a PPI increases
the efficacy of
the PPI in reducing/mitigating excess gastric acid secretion. The '985
publication discloses
and teaches that PG should preferably be administered by injection (e.g.,
subcutaneous
injection). The '985 publication does, however, disclose generically that PG
and PPI can be
2
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
administered by intravenous, parenteral, or oral means. The '985 publication
also generically
discloses that PPI and PG could be prepared in a tablet. The '985 publication,
however, does
not disclose any particular dosage or formulation that should be used and does
not provide
any working examples. Furthermore, the '985 publication does not provide any
teachings or
suggestion how one might avoid the prior art teachings that PG is not
effective if delivered
orally and provides no examples of suggested oral dosage amounts of PG that
would be
effective. In addition, the '985 publication also does not disclose that PG is
active locally in
the stomach, which the present inventors surprisingly discovered. The '985
publication also
does not teach the use of PG preservation agents to preserve the biological
activity of PG
activity in the stomach in order to achieve local effect in the gastric lumen.
In view of the
state of the art at the time of the present invention, the '985 publication's
generic disclosure
fails to motive one skilled in the art to prepare an oral composition
comprising PG for local
delivery.
De Graef et al., Gastroenterology, 91, 333-337 (1986) (De Graef publication)
discloses that omeprazole is more effective in inhibiting gastric acid
secretion when
administered to dogs pretreated intravenously with PG. There is no mention in
the De Graef
publication that oral administration of PG would be effective by acting
locally in the gastric
lumen to potentiate the effect of omeprazole.
US Patent Nos. 6,489,346; 6,645,988; and 6,699,885; to Phillips (jointly the
"Phillips
patents") disclose pharmaceutical compositions and methods of treating acid-
caused
gastrointestinal disorders using oral compositions consisting of a PPI, at
least one buffering
agent and specific parietal cell activators. The parietal cell activators
disclosed in the Phillips
patents include, for example, chocolate, sodium bicarbonate, calcium,
peppermint oil,
spearmint oil, coffee, tea and colas, caffeine, theophylline, theobromine and
amino acids
residues. As indicated in the Phillips patents, all these proposed parietal
cell activators
induce the release of endogenous gastrin that exerts both inhibitory and
stimulatory effects on
acid secretion. The Phillips patents, however, do not disclose or suggest the
use of PG,
which possesses a solely stimulatory activity, binding only to CCK-B
receptors, unlike the
parietal cell activators mentioned in the Phillips patents, which would
activate both CCK-A
and CCK-B receptors - including both inhibitory and stimulatory effects.
The development of an effective treatment for pathologies in which inhibition
of
gastric acid secretion is required would fulfill a long felt need. Despite the
wide-spread use
of PPI's, a need still exist for increasing the PPI efficacy, e.g., faster
effective onset,
3
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
prolonged effect including night time acid breakthrough, greater effect at
reduced dosage and
meal-independent administration.
SUMMARY OF THE INVENTION
It is the object of the present invention to provide oral compositions for
inhibition of
gastric acid secretion that are meal-independent and exhibit fast onset with
prolonged
inhibition effect on gastric acid secretion.
It is another object of the present invention to provide oral compositions for
inhibition
of gastric acid secretion comprising an irreversible gastric H-'-/K+-ATPase
proton pump
inhibitor (PPI) and a parietal cell activator, wherein the PPI anti-acid
activity is meal-
independent and exhibit fast onset and prolonged inhibitory effect on acid
secretion.
In one embodiment of the present invention the oral compositions comprises an
irreversible gastric H+/K+-ATPase proton pump inhibitor (PPI) as a gastric
acid secretion
inhibitor, pentagastrin (PG) andlor a PG analogue as an activator of parietal
cells and one or
more agents that preserve the availability of PG in the gastric fluids, so
that the biological
activity of PG is maintained thus enabling PG to act locally in the stomach.
Unexpectedly,
the compositions of the present invention possess anti-acid activity in the
stomach that is
meal-independent and exhibit fast onset and prolonged inhibition of acid
secretion. The
present compositions may be used for treating a subject suffering from chronic
or acute
disorders in which suppression of acid secretion in the stomach is required.
The proton pump inhibitors (PPIs) according to the present invention are
compounds
that inhibit the activity of the H+lK+-adenosine triphosphatase (ATPase)
proton pump in the
gastric parietal cells. In its pro-drug form, PPI is non-ionized and therefore
is capable of
passing through the cellular membrane of the parietal cells. Once reaching the
parietal cells,
the non-ionized PPI moves into the acid-secreting portion of activated
parietal cells, the
secretory canaliculus. The PPI trapped in the canaliculus becomes protonated,
thus converted
to the active sulfenamide form that can form disulfide covalent bonds with
cysteine residues
in the alpha subunit of the proton pump, thereby irreversibly inhibiting the
proton pump.
As mentioned above, the present invention is based on the inventors surprising
discovery that PG is active locally when administered orally, preferably by
acting locally in
the gastric lumen to activate the parietal cells. Active parietal cells
possess acidic pH, which
is required for the conversion of the PPI to the active protonated sulfenamide
form.
4
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Therefore, the synchronized activation of the parietal cells by PG acting
directly in the gastric
lumen maximizes the inhibition of the pumps by the PPI.
The oral compositions of the present invention exhibit the following
advantages over
the known PPI-based compositions aimed to reduce gastric acid secretion. The
present
compositions permit activation of the parietal cells by PG without any side
effects associated
with systemic administration of PG due to the local effect of PG in the
gastric lumen. Pre-
activation of parietal cells by PG facilitates the conversion of the PPI to
the active
sulfenamide form leading to fast onset of the effect of PPI. Furthermore, the
present
compositions exhibit fast onset of anti-acid activity in the stomach in a meal-
independent
manner. Thus, the combined active agents in the oral compositions provide an
efficient
solution for acute conditions in which fast reduction of acid secretion is
required. Finally, the
present oral compositions provide prolonged suppression of gastric acid
secretion for at least
24 h using a single medication.
The oral compositions according to the present invention comprise PG or a PG
analogue as an local activator of parietal cells in the gastric lumen. In
addition to PG that
comprises the amino acid sequence (3Ala-Trp-Met-Asp-PheNH2 (SEQ ID N0:2), this
invention contemplates the use of gastrin or PG analogues or derivatives
thereof as parietal
cell activators. Such variants include, but are not limited to the 34-, 17-,
and 14-amino acid
species of gastrin, and other truncation variants comprising the active C-
terminal tetrapeptide
of gastrin Trp-Met-Asp-PheNH2 (SEQ ID N0:1), which is reported in the
literature to have
full pharmacological activity (see Tracey and Gregory (1964) Nature (London),
204: 935).
Also included are variants of gastrin and/or truncated gastrins where native
amino
acids are replaced with conservative substitutions. Various analogues of these
molecules are
also included, for example, but not limited to the N-protected derivative of
PG Boc-(3Ala-Trp-
Met-Asp-PheNH2 in which Boc is tent-butyloxycarbonyl group or F-Moc-(3Ala-Trp-
Met-Asp-
PheNHz in which Moc is methoxycarbonyl.
In a non-limiting embodiment, the oral compositions according to the present
invention further comprise one or more agents that preserve the availability
of PG in the
acidic gastric fluids. These agents preferably are in an amount sufficient to
preserve the
availability of PG in the gastric fluids by retaining the solubility of PG in
the gastric fluids
and preventing its degradation, so that the local biological activity of PG in
the stomach is
preserved. This enables PG to act locally in the stomach to activate the
parietal cells. Such
agents are preferably antacids or alkaline agents that when dissolved in the
gastric juice are
5
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
capable of temporally elevating the pH of the gastric fluids to a value in
which pepsin is
inhibited, thereby inhibiting the degradation of PG in the gastric fluids by
pepsin. Since PG
is soluble only in alkaline conditions, the temporal elevation of the pH in
the gastric fluids
ensures that at least significant proportion of PG remains soluble in the
gastric fluids.
It is noted that any weak or strong base (and mixtures thereof) can be
utilized as the
alkaline agent in the present oral compositions. The alkaline agent or the
antacid is present in
the composition in an amount sufficient to substantially preserve the
stability and the
solubility of PG in the acidic gastric fluids. Therefore, the alkaline agent
of the present
invention, when dissolved in the gastric juice, is capable of elevating the pH
of the stomach to
a value sufficient to achieve adequate availability of PG to effect
therapeutic action.
According to a preferred embodiment, the alkaline agent in the composition is
present
in an amount sufficient to elevate the pH of the gastric fluids to a value
above 4, and more
preferably above 5, for a time period sufficient for PG to reach and activate
the parietal cells
in the stomach. In more preferred embodiment, the alkaline agent is capable of
elevating the
pH of the gastric fluids to a value above 5 for a time period ranging from 5
to 60 minutes,
preferably for a time period ranging from 5 to 30 minutes. Thus, the alkaline
agent according
to the present invention preserves the solubility of PG in the gastric fluids
for a time period
sufficient for PG to activate the parietal cells. Furthermore, the temporal
alkali condition in
the gastric fluid prevents the degradation of PG by pepsin that is active only
in acidic pH.
According to various embodiments, the present compositions further comprise
other
agents that preserve the availability of PG in the acidic gastric fluids. Such
agents are for
example pepsin inhibitors (i.e., pepstain and its derivative bacitracin -
cyclic dodecapeptide)
that reduce the degradation of the peptide in the stomach or mucolytic agents
that reduce the
viscosity of the gastric mucosa, thereby accelerating the ability of PG to
reach the cells
responsible for acid secretion. Such mucolytic agents are for example reducing
agents such
as N-acetyl cysteine, dithiothreitol, citric acid or mannitol. The present
compositions may
further comprise an antibiotic effective against bacteria residing in the
stomach.
The active ingredients of the present invention may be formulated in a single
oral
dosage form, preferably a solid dosage form. Liquid dosage forms such as
suspensions may
be used as well. Thus, in one embodiment the PPI, PG and the agent that
preserves the
availability of PG in the gastric fluids may be formulated as multi-layered
tablets, suspension
tablets, effervescent tablets, powder, pellets, granules, hard gelatin
capsules comprising
multiple beads, or soft gelatin capsules containing a lipid-based vehicle.
6
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
According to one embodiment, the solid dosage form of the present invention is
a
capsule or a mufti-layered tablet containing PPI particles coated with either
enteric pH-
dependent release polymers or non-enteric time-dependent release polymers,
particles of PG
and particles of one or more alkaline agents. In order to ensure that the
activation of parietal
cells in the gastric lumen by PG is synchronized with the absorption of the
PPI in the
proximal part of the small intestine, the single oral dosage form may comprise
PG beads
coated with time-dependent release polymer that extends the PG releasing time
in the
stomach. Thus, the extension of PG release in the stomach permits the
synchronization
between the activity of PG and the activity of the PPI on the parietal cells.
The active ingredients of the present invention may also be formulated in
separate
dosage forms. For example, PG and the agent that preserves the availability of
PG in the
gastric fluids may be formulated in an oral suspension or a solid dosage form
such as
capsules, tablets, suspension tablets, or effervescent tablets and the PPI may
be formulated in
a separate solid dosage form, preferably capsules or tablets comprising beads
with enteric pH-
dependent release polymers or non-enteric time-dependent release polymers. The
separate
dosage forms may be provided as a kit containing PG and the agent that
preserves the
availability of PG in the gastric fluids in one dosage form and the PPI in a
separate dosage
form. In this case, the PG is administered in conjunction with the PPI so that
there is at least
some chronological overlap in their physiological activity. The PPI and PG can
be
administered simultaneously and/or sequentially.
The PPI particles used in the present invention may be coated with either
enteric pH-
dependent release polymer, non-enteric time-dependent release polymer or may
be without
coating layer. The stability of the non-coated PPI while passing the stomach
is preserved by
the one or more alkaline agents present in the composition. It was previously
demonstrated
that the absorption of buffered suspension of non-enteric-coated PPI in the
proximal part of
the small intestine is faster than the absorption of the enteric-coated PPI
granules (Pilbrant
and Cederberg, Scand. J. Gastroenterol 1985:20 (supp. 108): 113-120).
Therefore, it is not
necessary to delay the release of PG in the stomach if non-coated PPI
particles are used in the
composition. However, when coated PPI particles are used, it is required to
synchronize the
release of the PPI with the release of PG by delaying the release of PG in the
stomach for
example by using polymeric coated PG particles.
In another embodiment, the present invention is directed to a method of
treating a
subject suffering from a disorder in which suppression of gastric acid
secretion is required or
7
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
a disorder normally treated by suppression of gastric acid secretion. The
method comprising
administering to the subject a pharmaceutical composition comprising a PPI as
a gastric acid
secretion inhibitor, PG or a PG analogue as an activator of parietal cells in
the gastric lumen,
and at least one preservation agent in an mount sufficient to preserve the
availability of PG in
the gastric lumen.
The compositions of the present invention may be used for preventing or
treating
pathologies in a mammal in which inhibition of gastric acid secretion is
required. Preferably
the mammal is human. The compositions of the present invention are effective
both in
treating the pathologies and in minimizing the risk of development of such
pathologies before
onset of symptoms.
The pharmaceutical compositions of the present invention may be used in a wide
number of pathological conditions that are treated by suppression of gastric
acid secretion.
Such conditions include, but are not limited to Zollinger/Ellison syndrome
(ZES),
gastroesophageal reflux disease (GERD), esophagitis, peptic ulcer diseases,
duodenal ulcers,
gastritis and gastric erosions, dyspepsia, and the like.
The present invention also includes an oral pharmaceutical kit. The kit
typically
comprises as active ingredients a pharmaceutically effective amount of: (i) a
peptide
comprising the amino acid sequence of SEQ )D NO:1; (ii) an irreversible
gastric I~/I~+-
ATPase proton pump inhibitor (PPI); and (iii) at least one agent that
preserves the availability
of the peptide in the gastric fluids. In one embodiment, the active
ingredients are formulated
in separate dosage unit forms. The kit may be used to treat or prevent a
disorder in a subject
in which suppression of gastric acid secretion is required by administering to
a subject the
active ingredients. The peptide is typically administered simultaneously,
prior to or following
the administration of the PPI.
These and further embodiments will be apparent from the detailed description
and
examples that follow.
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 demonstrates that NaHC03 preserves PG stability in artificial gastric
fluid;
Figure 2 demonstrates the percentage of non-degraded PG in various pH values;
Figure 3 is a schematic illustration of a double-layered tablet comprising PG,
non-
enteric-coated omeprazole and buffering agents;
Figure 4 is a schematic illustration of PG granules used in the multi
particulate
capsule formulation;
Figure 5 is a schematic illustration of a capsule comprising time release-
coated beads;
Figure 6 demonstrates that PG stimulates gastric acid secretion in rats in a
dose-
dependent manner ;
Figure 7 demonstrates that PG enhances PPI-mediated effect on gastric acid
secretion
in rats;
Figure 8 demonstrates that Lansoprazole inhibits gastric acid secretion in
conscious
animals in a dose-dependent manner;
Figure 9 demonstrates that PG increases the efficacy of Lansoprazole in the
blockade
of gastric acid secretion when Lansoprazole is administered prior to PG (A)
and not when
Lansoprazole is administered following PG (B);
Figure 10 demonstrates that administration of Lansoprazole in combination with
PG
during 3 consecutive days resulted in significantly higher intragastric pH (A)
and lower
gastric acid secretion (B) as compared to Lansoprazole alone.
DETAILED DESCRIPTION OF THE INVENTION
The term "alkaline agent" refers to any pharmaceutically appropriate weak base
or
strong base (and mixtures thereof) that, when formulated or delivered with
(e.g., before,
during and/or after) PG, functions to temporally elevate the pH in the gastric
lumen to a value
that substantially preserves the availability of PG in the stomach.
The term "an agent that preserves the availability of PG in the stomach"
refers to any
agent that is capable of maintaining the solubility and stability of PG in the
stomach.
Specifically, such an agent is capable of maintaining at least a substantial
amount of PG in a
soluble form and non-degraded in the gastric juice, so that the biological
activity of PG in the
stomach is maintained.
The term "biological activity of PG in the stomach" refers to its activation
of parietal
cells located in the gastric lumen.
9
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
The term "in conjunction with" means that when the PPI and the PG are
administered
in separate dosage forms, there is at least some chronological overlap in
their physiological
activity. Thus the PPI and PG can be administered simultaneously and/or
sequentially.
The present invention is based on the surprising discovery that PG is capable
of
remaining active following oral administration to activate the parietal cells,
preferably by
acting locally in the stomach. Importantly, parietal cell activation is
required for the
conversion of the PPI pro-drug to the active form that acts as an irreversible
inhibitor of the
gastric H+lI~+-ATPase proton pump. The oral compositions of the present
invention provide
a unique combination of active agents that increase the efficacy of the PPI in
inhibiting gastric
acid secretion.
The compositions of the present invention may be used for preventing or
treating
pathologies in a mammal in which inhibition of gastric acid secretion is
required. The
compositions of the present invention are effective both in treating the
pathologies and in
minimizing the risk of development of such pathologies before onset. Such
pathologies
include for example: reflux esophagitis, gastritis, duodenitis, gastric ulcer
and duodenal ulcer.
Furthermore, the compositions of the present invention may be used for
treatment or
prevention of other gastrointestinal disorders where gastric acid inhibitory
effect is desirable,
e.g. in patients on nonsteroidal anti-inflammatory drugs (NSAID) therapy
(including low
dose aspirin), in patients with Non Ulcer Dyspepsia, in patients with
symptomatic gastro-
esophageal reflux disease (GERD), and in patients with gastrinomas. They may
also be used
in patients in intensive care situations, in patients with acute upper
gastrointestinal bleeding,
in conditions of pre-and postoperatively to prevent aspiration of gastric acid
and to prevent
and treat stress ulceration. Further, they may be useful in the treatment of
Helicobacter
infections and diseases related to these. Other conditions well suited for
treatment include,
but are not limited to Zollinger-Ellison syndrome (ZES), Werner's syndrome,
and systemic
mastocytosis.
The parietal cell activator according to the present invention is preferably
PG having
the amino acid sequence denoted as SEQ ID N0:2. However, any PG analog that
comprises
the C-terminal tetrapeptide of gastrin Trp-Met-Asp-PheNH2 (denoted as SEQ ID
NO:1) may
be used as a parietal cell activator. Such analogues include, but are not
limited to the 34-,
17-, and 14-amino acid species of gastrin, and other truncation variants. Also
included are
variants of gastrin and/or truncated gastrins where native amino acids are
replaced with
conservative substitutions. Also included are various analogues of these
molecules, including
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
for example, but not limited to the N-protected derivatives of PG. Suitable
protecting groups
for PG include standard hydroxyl protecting groups known in the art, e.g.,
methoxymethyl
(MOM), (3-methoxyethoxymethyl (MEM), trialkylsilyl, triphenylmethyl (trityl),
tert-
butoxycarbonyl (t-BOC), ethoxyethyl (EE), f-MOC (methoxycarbonyl), TROC, etc.
The
protecting groups) may be removed by using standard procedures generally known
to those
skilled in the art to give the desired PG derivatives (T. W. Green, Protective
Groups in
Organic Synthesis, Chapter 2, pages 10-69 (1981)).
Gastrins, pentagastrins, or analogues thereof are commercially available. In
addition,
synthetic protocols are well known. Thus, for example, PG can be chemically
synthesized
using well-known peptide synthesis methodologies (see, e. g. Barany and
Merrifield Solid-
Phase Peptide Synthesis; pp. 3-284 in The Peptides: Analysis, Synthesis,
Biology. Vol. 2
Special methods in peptide synthesis, part a.; Merrifield et al. (1963) J. Am.
Chem. Soc.,
85: 2149-2156; and Stewart et al. (1984) Solid Phase Peptide Synthesis, 2nd
ed. Pierce
Chem. Co., Rockford, ILL.). Additionally, PG can be chemically synthesized,
for example,
by conjugation of a Boc-Ala residue to the tetrapeptide Trp-Met-Asp-PheNH2.
The compositions of the present invention comprise PG or an analog thereof in
an
effective amount to achieve a pharmacological effect on the parietal cells
without undue
adverse side effects. The standard approximate amount of PG present in the
compositions is
preferably in an amount of 1-100 mg, more preferably 2-60 mg, and most
preferably 4-40 mg
of PG (or an equivalent amount of a PG analogue).
The compositions of the present invention further comprise a PPI that acts as
an
irreversible inhibitor of the gastric Ii+/K+-ATPase proton pump. The PPI used
in the present
invention can be any substituted benzimidazole compound having Ii+, K+ -ATPase
inhibiting
activity. For the purposes of this invention, the term "PPI" shall mean any
substituted
benzimidazole possessing pharmacological activity as an inhibitor of H+,K+ -
ATPase,
including, but not limited to, omeprazole, lansoprazole, pantoprazole,
rabeprazole,
dontoprazole, perprazole (s-omeprazole magnesium), habeprazole, ransoprazole,
pariprazole,
and leminoprazole in neutral form or a salt form, a single enantiomer or
isomer or other
derivative or an alkaline salt of an enantiomer of the same.
Examples of gastric Ii+/K+-ATPase proton pump inhibitors that may be used in
the
present invention are disclosed for example in LTS Patent 6,093,738 that
describes novel
thiadiazole compounds that are effective as proton pumps inhibitors. European
Patent Nos.
322133 and 404322 disclose quinazoline derivatives, European Patent No. 259174
describes
11
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
quinoline derivatives, and WO 91/13337 and US Patent 5,750,531 disclose
pyrimidine
derivatives, as proton pump inhibitors. Suitable proton pump inhibitors are
also disclosed for
example in EP-A1-174726, EP-A1-166287, GB 2163 747 and W090/06925, W091/19711,
W091/19712, W094/27988 and W095/01977.
The PPI particles in the compositions according to the present invention may
be either
coated or non-coated. The preparation of enteric-coated particles comprising a
PPI such as
Omeprazole is disclosed for example in US Patents Nos. 4,786,505 and
4,853,230.
The compositions of the present invention comprise a PPI in an effective
amount to
achieve a pharmacological effect or therapeutic improvement without undue
adverse side
effects. A therapeutic improvement includes but is not limited to: raising of
gastric pH,
reduced gastrointestinal bleeding, or improvement or elimination of symptoms.
According to
a preferred embodiment, the typical daily dose of the PPI varies and will
depend on various
factory such as the individual requirements of the patients and the disease to
be treated. In
general, the daily dose of PPI will be in the range of 1-400 mg. A preferred
standard
approximate amount of a PPI present in the composition is typically about 20-
40 mg of
omeprazole, about 30 mg lansoprazole, about 40 mg pantoprazole, about 20 mg
rabeprazole,
and the pharmacologically equivalent doses of the following PPIs: habeprazole,
pariprazole,
dontoprazole, ransoprazole, perprazole (s-omeprazole magnesium), and
leminoprazole.
In a preferred embodiment, the compositions of the present invention further
comprise
one or more agents that preserve the availability of PG in the acidic gastric
fluids. More
specifically, the preservation agent maintains the stability or the solubility
of PG in the gastric
fluids. This enables PG to act locally in the stomach to activate the parietal
cells. Such
agents are preferably alkaline agents or antacids that when dissolved in the
gastric juice are
capable of elevating the pH of the gastric fluids to a pH in which the gastric-
residing
peptidases are inhibited and at least significant proportion of PG remains
soluble in the
gastric fluids.
Alkaline agents to be used in the present invention include for example:
sodium or
potassium bicarbonate, magnesium oxide, hydroxide or carbonate, magnesium
lactate,
magnesium glucomate, aluminum hydroxide, aluminium, calcium, sodium or
potassium
carbonate, phosphate or citrate, di-sodium carbonate, disodium hydrogen
phosphate, a
mixture of aluminum glycinate and a buffer, calcium hydroxide, calcium
lactate, calcium
carbonate, calcium bicarbonate, and other calcium salts. It is noted that
while sodium
bicarbonate dissolves easily in water, calcium carbonate is water-insoluble
and is slowly
12
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
soluble only in acidic environment. Therefore, calcium carbonate may be useful
when
sustained dissolution of the alkaline agent in the stomach is desired.
Examples of Antacids to be used in the present invention include one or more
of the
following: alumina, calcium carbonate, and sodium bicarbonate; alumina and
magnesia;
alumina, magnesia, calcium carbonate, and simethicone; alumina, magnesia, and
magnesium
carbonate; alumina, magnesia, magnesium carbonate, and simethicone; alumina,
magnesia,
and simethicone; alumina, magnesium alginate, and magnesium carbonate; alumina
and
magnesium carbonate; alumina, magnesium carbonate, and simethicone; alumina,
magnesium
carbonate, and sodium bicarbonate; alumina and magnesium trisilicate; alumina,
magnesium
trisilicate, and sodium bicarbonate; alumina and simethicone; alumina and
sodium
bicarbonate; aluminum carbonate, basic ; aluminum carbonate, basic, and
simethicone ;
aluminum hydroxide; calcium carbonate; calcium carbonate and magnesia; calcium
carbonate, magnesia, and simethicone; calcium carbonate and simethicone;
calcium and
magnesium carbonates; magaldrate; magaldrate and simethicone; magnesium
carbonate and
sodium bicarbonate; magnesium hydroxide; magnesium oxide.
Preferably, the compositions of the present invention comprise one or more
alkaline
agents or antacids in an effective amount to achieve a pharmacological effect.
Specifically,
the alkaline agents or antacids in the composition are present in an amount
sufficient to
elevate the pH of the gastric fluids to a pH above the pH optima for proteases
found in the
stomach for a time period sufficient for PG to activate the parietal cells in
the stomach. In a
preferred embodiment, the alkaline agents or antacids are present in an amount
sufficient to
elevate the pH of the gastric fluids to a pH above 5 for a time period ranging
from 5 to 60
minutes, preferably for a time period ranging from 5 to 30 minutes. The
quantity of alkaline
agents required in the compositions of the present invention will necessarily
vary with several
factors including the type of alkaline agent used and the equivalents of base
provided by a
given alkaline agent. In practice, the amount required to provide good
availability of PG in
the stomach is an amount which, when added to a solution of 200 milliliters of
artificial
gastric fluid (prepared according to the United States Pharmacopea (USP)
guideline), raises
the pH of that HCl solution to at least pH 5Ø Preferably, at least 100
milligrams, and more
preferably at least 300, and most preferably at least 500 milligrams of the
alkaline agents are
used in the pharmaceutical compositions of the invention.
In another embodiment, the compositions of the present invention further
comprise
other agents that preserve the availability of PG in the acidic gastric
fluids. For example, the
13
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
compositions may comprise pepsin inhibitors such as the activated pentapeptide
pepstatin and
its derivatives, either of natural or synthetic origin. These inhibitors might
decrease the
degradation of PG by pepsin. Furthermore, the compositions may comprise
mucolytic agents
that reduce the viscosity of the gastric mucosa, thereby accelerating the
ability of PG to reach
the parietal cells. Such mucolytic agents are for example reducing agents such
as N-acetyl
cysteine, dithiothreitol, citric acid or mannitol. The compositions
alternatively may also
comprise a polymeric coating for PG, such as, an enteric-coating of polymers
to protect the
PG from the acidic environment of the stomach.
The active ingredients of the present invention are preferably formulated in a
single
oral dosage form containing all active ingredients. The compositions of the
present invention
may be formulated in either solid or liquid form. It is noted that solid
formulations are
preferred in view of the improved stability of solid formulations as compared
to liquid
formulations.
In one embodiment, the PPI particles, PG and the one or more agents that
preserve the
availability of PG in the gastric fluids are formulated in a single solid
dosage form such as
mufti-layered tablets, suspension tablets, effervescent tablets, powder,
pellets, granules or
capsules comprising multiple beads. In another embodiment, the active agents
may be
formulated in a single liquid dosage form such as suspension containing all
active ingredients
or dry suspension to be reconstituted prior to use.
In the single dosage form, the PPI particles and the PG particles may be
coated with
either enteric pH-dependent release polymer or non-enteric, time-dependent
release polymer
in order to synchronize between the local biological activity of PG in the
stomach and the
systemic effect of the PPI on parietal cells. For example, if coated PPI
particles are used
resulting in delayed absorption in blood, it is desirable that the PG
particles be coated as well
to delay its release. In one specific embodiment, the PPI particles are coated
with a thick non-
enteric layer so as the release of the PPI is preferably delayed by between,
20-80 min, more
preferably 25-75 min, most preferably 30-60 min, and the PG particles are
coated with a thin
non-enteric polymer layer so as the release of PG is preferably delayed by 5-
60 min, more
preferably between 8-45 min, and most preferably 10-30 min. These conditions
permit pre-
activation of the parietal cells by PG prior to the achievement of a
pharmacological PPI
plasma concentration.
Non-limiting examples of suitable pH-dependent enteric polymers to be used in
the
present invention are: cellulose acetate phthalate,
hydroxypropylnethylcellulose phthalate,
14
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
polyvinylacetate phthalate, methacrylic acid copolymer, shellac,
hydroxypropylmethylcellulose succinate, cellulose acetate trimellitate, and
mixtures of any of
the foregoing. A suitable commercially available enteric material, for
example, is sold under
the trademark Eudragit L 100-55. This coating can be spray coated onto the
substrate.
Non-enteric time-dependent release polymers include, for example, one or more
polymers that swell in the stomach via the absorption of water from the
gastric fluid, thereby
increasing the size of the particles to create thick coating layer. The time-
dependent release
coating generally possesses erosion and/or diffusion properties that are
independent of the pH
of the external aqueous medium. Thus, the active ingredient is slowly released
from the
particles by diffusion or following slow erosion of the particles in the
stomach.
The erosion properties of the polymer in the stomach resulting from the
interaction of
fluid with the surface of the dosage form are determined mainly by the polymer
molecular
weight and the drug/polymer ratio. In order to ensure a delay of between about
10 min to
about 60 min in the release of PG and PPI, it is recommended that the
molecular weight of
the polymer be in the range from about 105 to about 10' gram/mol. Furthermore,
it is
recommended that the PG or PPI/polymer ratio be in the range of about 2:3 to
about 9:1,
preferably about 3:2 to 9:1, and most preferably about 4:1 to 9:1.
Suitable non-enteric time-dependent release coatings are for example: film-
forming
compounds such as cellulosic derivatives, such as methylcellulose,
hydroxypropyl
methylcellulose (HPMC), hydroxyethylcellulose, and/or acrylic polymers
including the non-
enteric forms of the Eudragit brand polymers. Other film-forming materials may
be used
alone or in combination with each other or with the ones listed above. These
other film
forming materials generally include poly(vinylpyrrolidone), Zein, polyethylene
glycol),
polyethylene oxide), polyvinyl alcohol), polyvinyl acetate), and ethyl
cellulose, as well as
other pharmaceutically acceptable hydrophilic and hydrophobic film-forming
materials.
These film-forming materials may be applied to the substrate cores using water
as the vehicle
or, alternatively, a solvent system. Hydro-alcoholic systems may also be
employed to serve
as a vehicle for film formation.
Other materials which are suitable for making the time-dependent release
coating of
the invention include, by way of example and without limitation, water soluble
polysaccharide gums such as carrageenan, fucoidan, gum ghatti, tragacanth,
arabinogalactan,
pectin, and xanthan; water-soluble salts of polysaccharide gums such as sodium
alginate,
sodium tragacanthin, and sodium gum ghattate; water-soluble
hydroxyalkylcellulose wherein
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
the alkyl member is straight or branched of 1 to 7 carbons such as
hydroxymethylcellulose,
hydroxyethylcellulose, and hydroxypropylcellulose; synthetic water-soluble
cellulose-based
lamina forrriers such as methyl cellulose and its hydroxyalkyl methylcellulose
cellulose
derivatives such as a member selected from the group consisting of
hydroxyethyl
methylcellulose, hydroxypropyl methylcellulose, and hydroxybutyl
methylcellulose; other
cellulose polymers such as sodium carboxymethylcellulose; and other materials
known to
those of ordinary skill in the art. Other lamina forming materials that can be
used for this
purpose include poly(vinylpyrrolidone), polyvinylalcohol, polyethylene oxide,
a blend of
gelatin and polyvinyl-pyrrolidone, gelatin, glucose, saccharides, povidone,
copovidone,
poly(vinylpyrrolidone)-polyvinyl acetate) copolymer.
Another approach for delaying the release of PG in the stomach is the use of
floating
particles having density lower than the gastric fluid, thereby delaying the
release of PG from
the particles. In one preferred embodiment, floating particles are obtained by
the release of
carbon dioxide within ethylcellulose-coated sodium bicarbonate beads upon
contacting with
the gastric juice. The release of carbon dioxide from the ethylcellulose-
coated sodium
bicarbonate core permits the buoyancy of the particles, thereby delaying the
release of PG
from the particles.
Other delayed gastric emptying approaches may be used in order to delay the
release
of PG in the stomach. These include the use of indigestible polymers or fatty
acid salts that
change the motility pattern of the stomach to a fed state, thereby decreasing
the gastric
emptying rate and permitting considerable prolongation of drug release
(disclosed for
example in Singh and Kim, J. of Controlled Release 63 (2000) 235-259).
In certain conditions, it is desirable to prolong the retention time of PG in
the stomach
by using dosage forms that unfold rapidly within the stomach to a size that
resists gastric
emptying. Such systems retain their integrity for an extended period and will
not empty from
the stomach at all until breakdown into small pieces occurs. Caldwell
(Caldwell, L. J.,
Gardener, C. R., Cargill, R. C. (1988), U.S. Pat. No. 4,767,627) describes a
cross shaped
device made of erodible polymer and loaded with drug which is folded and
inserted into a
hard gelatin capsule. Following oral administration the gelatin shell
disintegrates and the
folded device opens out. With a minimum size of 1.6 cm and a maximum size of 5
cm it will
not pass from the stomach through the pylorus until the polymer erodes to the
point where the
system is sufficiently small that it can be passed from the stomach.
16
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
An alternative approach to prolong the retention time of PG in the stomach is
to use a
hydrophilic erodible polymer system such as Polyethylene oxide) (Polyox) and
Hydroxypropyl-methylcellulose (HI'MC) that is of a convenient size for
administration to
humans. On imbibing fluid the system swells over a short period of time to a
size that will
encourage prolonged gastric retention, allowing sustained delivery of
contained drug to
absorption sites in the upper gastrointestinal tract. Because these systems
are made of an
erodible and hydrophilic polymer or polymer mixture, they readily erode over a
reasonable
time period to pass from the stomach. The time period of expansion is such
that this will not
occur in the esophagus and if the system passes into the intestine in a
partially swollen state,
the erodibility and elastic nature of the hydrated polymer will eliminate the
chance of
intestinal obstruction by the device.
In one specific example, the composition of the present invention is
formulated as a
single dosage form comprising multiple beads contained in hard or soft gelatin
capsules. The
capsules contain mixed population of beads selected from: beads comprising
enteric-coated
PPI or beads comprising PPI coated with time-dependent release polymer, beads
comprising
calcium carbonate and beads comprising ethylcellulose sodium bicarbonate beads
coated with
PG, calcium carbonate and hydroxypropyl methylcellulose. The cellulose-based
polymer in
the composition permits the floating of the PG beads, thus delaying the
release of PG from
the beads. The rate of PG release is determined by the thickness and the
erosion rate of the
hydroxypropyl methylcellulose.
In another specific example, the gelatin capsules contain mixed population of
beads
selected from: beads comprising enteric-coated PPI or beads comprising PPI
coated with
time-dependent release coating, beads comprising calcium carbonate and beads
comprising
alginate coated with PG, calcium carbonate and hydroxypropyl methylcellulose.
In yet another specific example, the gelatin capsules contain mixed population
of
beads selected from: beads comprising enteric-coated PPI, beads comprising PPI
coated with
time-dependent release polymer, beads comprising calcium carbonate and
particles in the
form of mini-tabs comprising PG, calcium carbonate and hydroxypropyl
methylcellulose.
In yet another example, the compositions of the present invention are
formulated as
press-coat or double-layered tablets comprising enteric-coated PPI in one
layer and PG,
calcium carbonate and hydroxypropyl methylcellulose in a second layer.
In yet another example, the compositions of the present invention may be
formulated
as two layer non-aqueous semi-solid fill into hard gelatin capsules in which
the PPI is
17
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
solubilized in a lipid base (non-aqueous, quick release) which is liquid above
room
temperature but forms a semi-solid on cooling and can therefore be filled into
hard gelatin
capsules. A lipid soluble alkaline agent such as an amine or a fine suspension
of sodium
bicarbonate may be included as well.
The single dosage form compositions of the present invention preferably
comprise
non-coated PPI instead of the enteric-coated PPI particles or the time-
dependent release
particles. The absorption of non-coated PPI in the upper portion of the small
intestine is
faster than the absorption of the coated PPI. Therefore, the use of non-coated
PPI in the
compositions permits more precise synchronization between the biological
activity of PG in
the stomach and the time period in which the PPI is active without the need
for delaying the
release of PG. Thus, according to various preferred embodiments, the
compositions
according to the present invention are formulated as double-layered tablets,
press-coat tablets,
effervescent tablets or suspension tablets comprising PG, non-coated PPI and
one or more
alkaline agents.
The active ingredients of the present invention may be formulated in a
multiple oral
dosage forms in which PG and the one or more agents that preserve the
availability of PG in
the gastric fluids are administered in a separate dosage form but in
conjugation with the PPI.
For example, PG and the one or more agents that preserve the availability of
PG in the gastric
fluids may be formulated in oral suspension or a solid dosage form such as
capsules, tablets,
suspension tablets, or effervescent tablets and the PPI may be formulated in a
separate solid
dosage form, preferably enteric-coated beads or time-dependent release beads
contained in
capsules or tablets.
When using multiple oral dosage forms, the PG and the one or more agents that
preserve the availability of PG in the gastric fluids can be administered
before,
simultaneously with, or after the PPI. In sequential administration, there may
be some
substantial delay (e. g., minutes or even few hours) between the
administration of PG and the
PPI as long as the PG has exerted some physiological effect when the PPI is
administered or
becomes active. In a preferred embodiment, the PPI administered is in the
enteric-coated or
the time-dependent release form. According to this embodiment, it is
preferable that the PPI
administration precedes the PG administration in order to ensure that the PPI
absorbed in the
proximal part of the small intestine will be available for inhibiting the
Ii+/K+-ATPase pumps
while PG is still active in the stomach.
18
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
The active ingredients of the present invention may be incorporated within
inert
pharmaceutically acceptable beads. In this case, the drugs) may be mixed with
further
ingredients prior to being coated onto the beads. Ingredients include, but are
not limited to,
binders, surfactants, fillers, disintegrating agents, alkaline additives or
other pharmaceutically
acceptable ingredients, alone or in mixtures. Binders include, for example,
celluloses such as
hydroxypropyl methylcellulose, hydroxypropyl cellulose and carboxymethyl-
cellulose
sodium, polyvinyl pyrrolidone, sugars, starches and other pharmaceutically
acceptable
substances with cohesive properties. Suitable surfactants include
pharmaceutically acceptable
non-ionic or ionic surfactants. An example of a suitable surfactant is sodium
lauryl sulfate.
The particles may be formed into a packed mass for ingestion by conventional
techniques. For instance, the particles may be encapsulated as a "hard-filled
capsule" using
known.encapsulating procedures and materials. The encapsulating material
should be highly
soluble in gastric fluid so that the particles are rapidly dispersed in the
stomach after the
capsule is ingested.
In another embodiment, the active ingredients of the present invention are
packaged in
compressed tablets. The term "compressed tablet" generally refers to a plain,
uncoated tablet
for oral ingestion, prepared by a single compression or by pre-compaction
tapping followed
by a final compression. Such solid forms can be manufactured as is well known
in the art.
Tablet forms can include, for example, one or more of lactose, mannitol, corn
starch, potato
starch, microcrystalline cellulose, acacia, gelatin, colloidal silicon
dioxide, croscarmellose
sodium, talc, magnesium stearate, stearic acid, and other excipients,
colorants, diluents,
buffering agents, moistening agents, preservatives, flavoring agents, and
pharmaceutically
compatible carriers. The manufacturing processes may employ one, or a
combination of, four
established methods: (1) dry mixing; (2) direct compression; (3) milling; and
(4) non-aqueous
granulation. Lachman et al., The Theory and Practice of Industrial Pharmacy
(1986). Such
tablets may also comprise film coatings, which preferably dissolve upon oral
ingestion or
upon contact with diluent.
Non-limiting examples of alkaline agents which could be utilized in such
tablets
include sodium bicarbonate, alkali earth metal salts such as calcium
carbonate, calcium
hydroxide, calcium lactate, calcium glycerophosphate, calcium acetate,
magnesium carbonate,
magnesium hydroxide, magnesium silicate, magnesium aluminate, aluminum
hydroxide or
aluminum magnesium hydroxide. A particular alkali earth metal salt useful for
making an
antacid tablet is calcium carbonate.
19
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
In another alternative, the compositions of the present invention are
formulated in
compressed forms, such as suspension tablets and effervescent tablets, such
that upon
reaction with water or other diluents, the aqueous form of the composition is
produced for
oral administration. These forms are particularly useful for medicating
children and the
elderly and others in a way that is much more acceptable than swallowing or
chewing a tablet.
The present pharmaceutical tablets or other solid dosage forms disintegrate
alkaline agent
with minimal shaking or agitation.
The term "suspension tablets" as used herein refers to compressed tablets
which
rapidly disintegrate after they are placed in water, and are readily
dispersible to form a
suspension containing a precise dosage of the PPI, the PG and the alkaline
agent. In one non-
limiting example, the suspension tablets may comprise 20-40 mg omeprazole, 4
mg PG and
about 1-4 grams of sodium or calcium bicarbonate as an alkaline agent. To
achieve rapid
disintegration of the tablet, a disintegrant such as Croscarmellose sodium may
be added to the
formulation. The disintegrant may be blended in compressed tablet formulations
either alone
or in combination with microcrystalline cellulose, which is well known for its
ability to
improve compressibility of difficult to compress tablet materials.
Microcrystalline cellulose,
alone or co-processed with other ingredients, is also a common additive for
compressed
tablets and is well known for its ability to improve compressibility of
difficult to compress
tablet materials. It is commercially available under the Avicel trademark.
The suspension tablet composition may, in addition to the ingredients
described
above, contain other ingredients often used in pharmaceutical tablets,
including flavoring
agents, sweetening agents, flow aids, lubricants or other common tablet
adjuvants, as will be
apparent to those skilled in the art. Other disintegrants, such as
crospividone and sodium
starch glycolate may be employed, although croscarmellose sodium is preferred.
In addition to the above ingredients, the oral dosage forms described above
may also
contain suitable quantities of other materials, e.g. diluents, lubricants,
binders, granulating
aids, colorants, flavorants and glidants that are conventional in the
pharmaceutical art. The
quantities of these additional materials will be sufficient to provide the
desired effect to the
desired formulation. Specific examples of pharmaceutically acceptable carriers
and
excipients that may be used to formulate oral dosage forms are described in
the Handbook of
Pharmaceutical Excipients, American Pharmaceutical Association (196),
incorporated by
reference herein.
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
The following examples are presented in order to more fully illustrate certain
embodiments of the invention. They should in no way, however, be construed as
limiting the
broad scope of the invention. One skilled in the art can readily devise many
variations and
modifications of the principles disclosed herein without departing from the
scope of the
invention.
EXAMPLES
Example l: NaHC03 preserves PG stability in artz'ficial gastric fluid
The stability of PG in acidic pH in the presence of NaHC03 was tested in vitro
using
artificial gastric fluid. Artificial gastric fluid was prepared in accordance
with U.S.
Pharmacopoeia (USP) 2000 Ed., P. 235. For preparing 200 ml of gastric fluid,
0.4 g of NaCI
and 0.64 g of Pepsin were dissolved in 16 ml 1M HCl and 184 ml of water. The
pH of the
gastric fluid was 1.2. Ten or twenty ml of 8.4% (1M) NaHC03 (final
concentration 3.72
mg/ml or 7.12 mg/ml, respectively) and 16 ml of 250 ppm PG solution (0.25
mg/ml) were
added to the solution. The concentration of PG in the final solution was 16
ppm. When
indicated, Omeprazole granules were added as well (solutions B and C). In
order to
determine the stability of PG in the final solution over time, HPLC analysis
was performed on
samples taken at the following time points post preparation: 0' (immediately
following
preparation), 5', 10', 20', 40', 60'. To stop the reaction, the pH was
adjusted to 7.5 - 8.5
using NH40H.
As demonstrated in Figure 1, fast degradation of PG was observed in solutions
A and
B that comprise PG in the presence of 3.72 mg/ml of NaHC03 (pH 1.2). However,
PG
remained stable for 1h in solution C that comprises 7.12 mg/ml of NaHC03 (pH
5.7). These
results indicate that the addition of an alkaline agent such as NaHC03 in a
concentration
sufficient to elevate the pH above 5.0 prevents the degradation of PG by
pepsin. Figure 2
further demonstrates that at least 80% of PG remains non-degraded for at least
15 min in pH
4.8.
A. For»zulatiou descri~ntiorz- Tablets corztainitz~ port-enteric-coated
Omenrazole:
Example 2: Press-coated or double-layered tablets cofnprisittg PG, rzon-
enteric-
coated omeprazole, sodium bicarbonate and calcium carbonate
Press-coated or double-layered tablets are formulated as a single dosage form
in which each tablet containing the following ingredients:
21
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Omeprazole (powder) 40 mg
PG 4 mg
NaHC03 500
mg
CaC03 500
mg
Croscarmellose sodium
hydroxypropyl methylcellulose
(HPMC)
Microcrystalline cellulose (Avicel)
Magnesium stearate
Starch
Press-coated or double-layered tablets are prepared in a two-step process. For
a single tablet, 4 mg PG, 250 mg calcium carbonate and microcrystalline
cellulose are
mixed and pre-compressed into the first layer of the tablet. The layer
containing the
PG is further coated with a thin layer of HPMC that permits a delay of 10-15
min in
the release of PG from the tablet. For the second layer, 40 mg of non-enteric-
coated
omeprazole powder together with 500 mg NaHC03, 250 mg CaC03 and the
appropriate binders are compressed onto the PG layer to form the second layer
of the
tablet. The second layer of the tablet disintegrates immediately after
digestion to
permit prompt release of omeprazole. A schematic illustration of a double-
layered
tablet comprising PG, non-enteric-coated omeprazole, sodium bicarbonate and
calcium carbonate is presented in Figure 3.
Example 3: Fast disintegrating tablets comprising PG, non-enteric-coated
onzeprazole, sodium bicarbonate and calcium carbonate
Fast disintegrating tablets are formulated as a single dosage containing the
following ingredients:
Omeprazole (powder) 40 mg
PG 4 mg
NaHC03 500 mg
CaC03 500 mg
Croscarmellose sodium
Microcrystalline cellulose
Magnesium stearate
22
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Starch
Non-enteric-coated omeprazole (40 mg), PG (4 mg), NaHC03, CaC03,
Croscarmellose sodium, Microcrystalline cellulose and Magnesium stearate are
mixed
and the resulting mixture is compressed into tablets using standard tablet
pressing to
yield a fast disintegrating tablet (intravescent).
Example 4: Effervescent sacs comprising PG, enteric-coated omeprazole, and
sodium bicarbonate
Effervescent tablets are formulated as a single dosage containing the
following
ingredients:
Omeprazole 40 mg
PG 4 mg
NaHC03 958 mg
Citric acid 832 mg
Potassium carbonate 312 mg
Magnesium stearate
Starch
Enteric-coated omeprazole (40 mg) and PG (4mg) are placed into a mortar and
triturated with a pestle to a fine powder. Sodium bicarbonate, citric acid,
potassium
carbonate and all other excipients are added to the mixture to form a
homogeneous mixture of
effervescent powder. The resulting powder is mixed with 40mg enteric-coated
omeprazole
and packed in packets of unit dose.
B. Formulation description- Multi particulate capsules coutaiuin~ coated
Omenrazole:
Example 5: Capsules comprising ethylcellulose-PG beads, enteric-coated
omeprazole
beads, and calcium carbonate.
This example illustrates the steps involved in manufacturing mufti particulate
hard
gelatin capsules. Hard gelatin capsules are formulated as a single dosage form
comprising
mixed population of particles. Each capsule contains the following
ingredients:
23
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
40 mg omeprazole as enteric-coated beads
4 mg PG loaded on ethylcelluiose-coated sodium bicarbonate beads
600 mg calcium carbonate (CaC03)
hydroxypropyl methylcellulose (HPMC)
PG solution is prepared by dissolving PG in ammonium carbonate buffer pH 8.
The
PG solution is sprayed on the ethylcellulose-coated sodium bicarbonate beads
in a fluidized
bed apparatus. After drying, the PG-sodium bicarbonate beads are further
coated with CaC03
and with hydroxypropyl methylcellulose (HPMC) to form the final PG particles.
The final
PG particles are packed together with enteric-coated omeprazole beads and
calcium carbonate
powder into size 0 hard gelatin capsules in an amount corresponding to 40 mg
omeprazole, 4
mg PG and 600 mg calcium carbonate per capsule.
Upon dissociation of the gelatin capsules in the gastric juice of the stomach,
the
HPMC layer of the PG-containing beads expands and the gastric acid reacts with
sodium
bicarbonate to form COZ inside the bead core. The release of carbon dioxide
from the
ethylcellulose-coated sodium bicarbonate core permits the buoyancy of the
particles, thereby
delaying the release of PG and calcium carbonate from the particles. The rate
of PG release is
determined by the thickness and the erosion rate of the HPMC layer of the PG
beads. CaC03
increases the gastric pH for a prolonged period of time, to protect PG upon
release. The
enteric-coated omeprazoie beads pass the stomach and omeprazole is absorbed in
the upper
part of the small intestine without any delay.
Exanzple 6: Capsrcles cofnprising alginate-PG beads, entez-ic-coated
ofneprazole
beads, arid calcium carbonate
Hard gelatin capsules are formulated as a single dosage form comprising mixed
population of particles. Each capsule contains the following ingredients:
40 mg omeprazole as enteric-coated beads
4 mg PG loaded on alginate particles
600 mg calcium carbonate (CaC03)
hydroxypropyl methylcellulose (HPMC)
24
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Alginate particles are made by dropping an alginate solution into calcium
chloride
solution following by freeze-drying to yield alginate particles. The PG
solution prepared as in
Example 5 is sprayed on the alginate particles in a fluidized bed apparatus.
After drying, the
PG-alginate beads are further coated with CaC03 and with hydroxypropyl
methylcellulose
(HPMC) to form the final PG particles. The final PG particles together with
the enteric-
coated omeprazole beads and calcium carbonate powder are packed into size 0
hard gelatin
capsules in an amount corresponding to 40 mg omeprazole, 4 mg PG and 600 mg
calcium
carbonate per capsule.
Upon dissociation of the gelatin capsules in the stomach, the PG beads are
expanded
IO due to the contact of the HPMC layer with the gastric juice. The freeze-
dried alginate
particles permit the buoyancy of the particles due to their low density
thereby delaying the
release of PG from the particles. The rate of PG release is determined by the
thickness and
the erosion rate of the HPMC layer of the PG beads. The enteric-coated
omeprazole beads
pass the stomach and omeprazole is absorbed in the upper part of the small
intestine without
any delay.
Example 7: Capsules co»tprisiug sucrose-PG beads, enteric-coated o»teprazole
beads, and calciu»z carbo»ate
Hard gelatin capsules are formulated as a single dosage form comprising mixed
population of particles. Each capsule contain the following ingredients:
40 mg omeprazole as enteric-coated beads
4 mg PG loaded on inert sugar beads
600 mg calcium carbonate (CaC03)
hydroxypropyl methylcellulose (HPMC)
The PG solution is sprayed on inert sugar pellets (Nu-Pareils, 25/30) in a
fluidized bed
apparatus. After drying, the PG-sugar beads are further coated with CaC03 and
with
hydroxypropyl methylcellulose (HPMC) to form the final PG particles. A
schematic
illustration of the PG granules is presented in Figure 4. The final PG
particles together with
the enteric-coated omeprazole beads and calcium carbonate powder are packed
into size 0
hard gelatin capsules in an amount corresponding to 40 mg omeprazole, 4 mg PG
and 600 mg
calcium carbonate per capsule.
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Upon dissociation of the gelatin capsules in the stomach, the PG beads are
expanded
due to the contact of the HPMC layer of the PG-containing beads with the
gastric juice,
thereby delaying the release of PG from the particles. The rate of PG release
is determined by
the thickness and the erosion rate of the HPMC layer of the PG beads. The
enteric-coated
omeprazole beads pass the stomach and omeprazole is absorbed in the upper part
of the small
intestine without any delay.
Exa»anle 8: Hard gelatin capsules comprising HPMC-PG »ainitabs, enteric-coated
omeprazole beads, and calcium carbonate
Hard gelatin capsules are formulated as a single dosage form comprising mixed
population of particles. Each capsule contains the following ingredients:
40 mg omeprazole as enteric-coated omeprazole beads
4 mg PG loaded on inert sugar beads
600 mg calcium carbonate (CaC03)
hydroxypropyl methylcellulose (HPMC)
PG is granulated in combination with HPMC and CaC03 and compressed into mini-
tabs. The mini-tabs possess the ability of fast swelling upon contact with the
gastric juice of
the stomach, thereby enabling gastric retention. The release of PG into the
stomach is
controlled by the erosion rate of the polymeric matrix of the swelled mini-
tabs. The PG mini-
tabs together with the enteric-coated omeprazole beads are packed into size 0
hard gelatin
capsules in an amount corresponding to 40 mg omeprazole, 4 mg PG and 600 mg
calcium
carbonate per capsule.
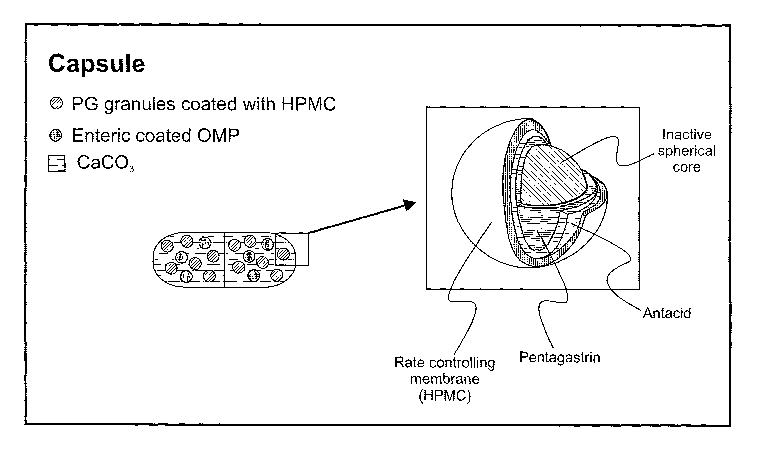
Example 9: Multi particulate capsules containing Omeprazole and PG beads
coated
with non-enteric tifne-dependent release coating:
This example illustrates the steps involved in manufacturing multi particulate
hard
gelatin capsules. Capsules are formulated as a single dosage form comprising
mixed
population of particles: PG beads coated with time-dependent release coating,
omeprazole
beads coated with time-dependent release coating, and calcium carbonate. A
schematic
illustration of the capsule is present in Figure 5. Each capsule contains the
following
ingredients:
~ 40 mg omeprazole beads coated with thick HPMC layer
26
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
~ 4 mg PG loaded on sugar spheres and coated with thin HPMC layer
~ 600 mg calcium carbonate (CaC03)
The composition of the coating is designed such that the core is rapidly
disintegrated
into an aqueous environment when the media come into contact with the core.
For this
purpose Sugar sphere will be coated with an antacid (NaHC03 or CaC03) layer.
PG solution
is prepared by dissolving PG in ammonium carbonate buffer pH ~. The PG
solution is
sprayed on to the above antacid-coated beads in a fluidized bed apparatus.
After drying, the
beads are further coated with a thin layer of HPMC to create PG particles with
approx. 10
min delayed release. Omeprazole is layered over the antacid-coated Sugar
spheres and is
covered with a thick time-release HPMC coating. A disintegrant also may be
added to the
core of the particle to facilitate the prompt release of omeprazole after the
HPMC is
dissolved. The coated Omeprazole beads are aimed to pass the stomach and are
absorbed at
the upper parts of the small intestine after the HPMC is dissolved and the
Omeprazole is
released at once. The final PG particles are packed together with the
omeprazole beads and
calcium carbonate powder into size 0 hard gelatin capsules in an amount
corresponding to 40
mg omeprazole, 4 mg PG and 600 mg calcium carbonate per capsule. The rate of
PG and
OMP release is determined by the thickness and the erosion rate of the HPMC
layer of the
beads. CaCO3 increases the gastric pH for a prolonged period of time, to
preserve PG upon
release.
C For»tulatio~t descrintiou- Tablets cofttainin~ enteric-coated Omenra ole:
Exa»tnle 10: Press-coated tablets comprising PG, enteric-coated o»teprazole
beads,
and calciu»a carbonate
Press-coated tablets are formulated as a single dosage form containing the
following
ingredients:
40 mg omeprazole as enteric-coated omeprazole beads
4 mg PG granules
Calcium carbonate
hydroxypropyl methylcellulose (HPMC)
Press-coated tablets are prepared in a two-step process. For a single tablet,
4 mg PG,
900 mg calcium carbonate and HPMC are mixed and pre-compressed into the
central core of
27
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
the tablet. 40 ing of enteric-coated omeprazole beads are press-coated onto
the PG core to
form the external layer of the tablet. The final tablet is composed of
controlled-release PG
core layer and immediate release outer layer of omeprazole enteric-coated
beads. In another
example, the active ingredients are compressed into double-layered tablet
wherein the first
layer comprises 4 mg PG, 900 mg calcium carbonate and HPMC and the second
layer
comprises 40 mg of enteric-coated omeprazole beads.
The compressed tablet may include one or more of the following excipients:
lactose,
mannitol, corn starch, potato starch, microcrystalline cellulose, acacia,
gelatin, colloidal
silicon dioxide; croscarmellose sodium, talc, magnesium stearate, stearic
acid, and other
excipients, colorants, diluents, buffering agents, moistening agents,
preservatives, flavoring
agents, and pharmaceutically compatible carriers.
Exaznnle 1l: Fast disintegrating tablets comprising PG, enteric-coated
onzeprazole
beads atzd calciunz carbonate
Fast disintegrating suspension tablets are formulated as a single dosage
containing the
following ingredients:
40 mg omeprazole as enteric-coated omeprazole beads
4 mg PG granules
900 mg calcium carbonate
Croscarmellose sodium
Microcrystalline cellulose
Magnesium stearate
hydroxypropyl methylcellulose (HPMC).
PG granules are coated with CaC03 and with hydroxypropyl methylcellulose
(HPMC)
to form the final PG particles. The final PG particles are mixed with enteric-
coated
omeprazole beads and the excipients listed above and the resulting mixture is
compressed
into tablets using standard tablet pressing. The resulting tablets possess
rapid disintegration
time and may be swallowed with water for fast disintegration in the stomach.
Upon disintegration of the suspension tablet, the PG particles are expanded
due to the
contact of the HPMC layer of the PG-containing beads with aqueous environment,
thereby
delaying the release of PG from the particles. The rate of PG release is
determined by the
thickness and the erosion rate of the HPMC layer of the PG beads. The enteric-
coated
2~
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
omeprazole beads pass the stomach and omeprazole is absorbed in the upper part
of the small
intestine without any delay.
D In vivo experiments
Example 12: Stimulation of Gastric Acid Secretion Following Oral
Ad»zifzistration
of PG i» Rats
Inhibition of gastric acid secretion by a combination of PG and PPI is based
on the
ability of orally administered PG to trigger acid secretion locally within the
stomach. To
address this issue anesthetized rats were administered (per os) with
increasing amounts of PG
and gastric acid secretion was monitored in a pylorus-ligated stomachs.
Increasing amounts
(10, 30, and 90p.g/kg) of PG were administered by oral gavage to pylorus-
ligated rats.
Following 30 min treatment, gastric juice was collected from the gastric
lumen, and acid
concentration was determined by titration with NaOH and total acid output
expressed in ~.Eq
HCl was calculated by multiplying the sample volume by the acid concentration.
Results are
expressed as means ~ SEM of 7-~ animals from each experimental group. As
demonstrated
in Figure 6, orally administered PG significantly enhanced gastric acid
secretion in a dose-
dependent manner, suggesting that orally administered PG successfully induces
gastric acid
secretion in a local manner.
Example 13: The effect of PG administered with o»zeprazole o» intragastric pH
To test the effect of the PG-PPI combination on suppression of gastric acid
secretion,
anesthetized rats were subjected to intragastric injection of either
omeprazole (10 mg/kg)
alone or in combination with PG (350 ~,g/kg). Rats treated with the
combination received PG
15 minutes before omeprazole. The gastric juice was collected by suction at
30, 45, and 60
minutes after the treatment and an effect of drugs on gastric acid secretion
was detected by
monitoring pH. The data demonstrated that the intragastric pH value at all
time points was
markedly higher in rats treated with combination of PG and omeprazole than
with omeprazole
alone (Figure 7). These results indicate that PG enhances the anti-secretory
activity of PPI in
rats.
Example 14: Larzsoprazole inhibits gastric acid secretion in conscious
ani»zals in a
dose-dependent manner.
29
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
In this experiment, a different model of pylorus-ligated rats that permits the
analysis
of the effect of drugs on gastric acid secretion in conscious animals was
used. This model
eliminates the effect of anesthesia on gastric acid secretion. The study drugs
alone or in
combination were administered per os. One or two hours later the animals were
anesthetized
using anesthetic gas machine for a short period (5 minutes) that is sufficient
to perform the
pylorus ligation procedure and to close the abdomen. The animals were then
placed back into
its cage for recovery. Several hours later the animals were sacrificed, the
ligature was placed
around the esophagus, the stomach removed and gastric content was collected.
Following
centrifugation the gastric juice samples were automatically titrated with 0.01
N NaOH to
endpoint pH 7 and titratable acid output was calculated.
Lansoprazole was administered by oral gavage as a simplified suspension (SLS).
SLS
was prepared as follows: the content of one 30 mg capsule (Zoton) was
suspended in 8.4%
sodium bicarbonate. Rats were treated with three doses of Lansoprazole (20, 5
and 1.25
mg/kg) 2 hours before pylorus ligation. 8.4% NaHC03 was administered into the
control
group as a placebo. Figure 8 demonstrates that Lansoprazole inhibited the
gastric acid
secretion in a dose-dependent manner.
Example I5: The effect of Lansoprazole administered in combination with PG on
gastric acid secretion in conscious pylorus-ligated rats.
In this experiment, rats were treated with SLS at a dose 5 mg/kg either 15
minutes
before (A) or after (B) PG (300 ~g/kg). The control rats were injected with
combination of
8.4% NaHC03 and PG-vehicle as a placebo. All drugs were administered by oral
gavage 2
hours before pylorus ligation. The gastric juice was collected during 3 hours.
Data is
presented as mean~SEM. Number of animals is 8-9 in each experimental group.
As can be seen in Figure 9A the administration of SLS 15 minutes before PG led
to a
greater extent of acid inhibition as compared to Lansoprazole alone, whereas
acid output in
rats pretreated with PG and then treated with SLS did not differ from that of
Lansoprazole
alone-treated rats (Figure 9B). These results indicate that PG increases the
efficacy of
Lansoprazole in the blockade of gastric acid secretion. Moreover, the timing
between the two
compounds is important in order to get increased effectiveness of
PG/Lansoprazole combined
treatment.
In another experiment, rats were treated once daily during 3 consecutive days
with
either SLS at a dose 2.5 mg/kg and vehicle or SLS and PG (300 ~,g/kg). SLS was
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
administered 15 minutes before PG or vehicle. The control rats were injected
with
combination of 8.4% NaHC03 and PG-vehicle as a placebo. All drugs were
administered by
oral gavage. The pylorus ligation was performed on third day 2 hours following
treatment.
The gastric juice was collected during 3 hours. Data is presented as mean~SEM.
Number of
animals is 8 in each experimental group. As demonstrated in Figures 10A and
10B,
administration of SLS in combination with PG during 3 consecutive days
resulted in
significantly higher intragastric pH as compared to SLS alone. Similarly, the
gastric acid
secretion in rats treated with SLS/PG combination for three consecutive days
was lower than
that following administration of SLS alone.
Example 16: The effect of a CCK B Ahtagohist oh PG-Mediated gastric acid
secretioya ih
pats
As PG is a gastrin hormone homologue, its local effect is thought to be
mediated via
gastrin pathway, i.e. an activation of gastrin receptor (CCKB). To test this
hypothesis the
effect of the specific CCKB antagonist (Itriglumide) on PG-mediated acid
secretion in rats
was examined.
In this study, rats were anesthetized with I~etamine and Domitor mixture and
provided with 20 mg/kg of Itriglumide that was administered intraduodenally
(i.d.).
Following 15 min, gastric pylorus was ligated and 300 ~ug/kg PG was
administered into the
stomach (i.g.). After 30 min, gastric juice was obtained, centrifuged and the
volume and pH
of the supernatants were measured. The acid concentration (titratable acidity)
was analyzed
by titration the gastric juice samples with NaOH and total acid output
expressed in E,~Eq HCl
was calculated by multiplying the sample volume by the acid concentration. As
revealed from
the results presented in Table 1 below, intraduodenal injection of CCKB
antagonist (ant.)
inhibits the local effect of PG on gastric acid secretion in rats.
Table 1:
31
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Group Acid Output
MEAN SEM
PG (i.g.), 300 uglkg60.056 10.43
CCI~B ant. (i.d.) 15.24 2.82
20 mglkg
Placebo of PG (i.g.)-19.25 3.03
NH4HC03
Placebo of CCKB ant.12.93 1.55
-
saline (i.d.)
PG (i.g.), 300 ug/kg22.884 2.70
and
CCKB ant.(i.d.) 20
mg/ml
PG (i.g.), 300 ug/kg51.74 9.35
and
Placebo of CCKB ant.
-
saline (i.d.)
Student t-test
PG vs.ant.+PG P=0.0023
P=0.0042
P=0.0016
Example 17: The effect of intraduodehal iujectio~a of PG oh acid secretion iu
anesthetized pylorus-ligated rats
The effect of intraduodenal injection of PG on acid secretion in anesthetized
pylorus-
ligated rats was examined. In this study, 300 pg/kg PG was administered
intraduodenaly in
anesthetized pylorus-ligated rats and the level of gastric acid secretion was
determined 30
minutes later. Gastric juice was obtained, centrifuged and the volume and pH
of the
supernatants were measured. The acid concentration (titratable acidity) was
analyzed by
titration gastric juice samples with NaOH and total acid output expressed in
~.Eq HCl was
calculated by multiplying the sample volume by the acid concentration. As a
control the equal
amount of PG was injected intragastrically and the effect of PG on gastric
secretion was
determined. As demonstrated in Table 2, both intragastric and intraduodenal
injection of PG
induce gastric acid secretion in anesthetized pylorus-ligated rats.
32
CA 02536906 2006-02-24
WO 2005/020879 PCT/IB2004/002745
Tahle 2:
Grou Acid Out ut
MEAN
SEM
PG (i. .), 300 a 45.89 6.37
/k
Placebo (i. .) 12.46 2.65
PG (i.d.), 300 a 42.26 6.95
/k
Placebo (i.d.) 11.65 1.44
Student t-test
(i.g.) vs. Placebo P= 0.000125
P= 0.000243
P= 1.981x105
It will be appreciated by a person skilled in the art that the present
invention is not
limited by what has been particularly shown and described hereinabove. Rather,
the scope of
the invention is defined by the claims that follow.
33
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST L,E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter 1e Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional valumes please contact the Canadian Patent Office.