Note: Descriptions are shown in the official language in which they were submitted.
CA 02538009 2006-03-06
WO 2005/025674 PCT/EP2004/009837
Description
Combination of phenylcarboxamides with beta-adrenergic receptor
blockers and use thereof for the treatment of atrial arrhythmias
The invention relates to the combination of one or more (3-adrenoreceptor
blockers (abbreviation "beta-blockers"), such as, for example, atenolol,
carvedilol, nadolol, pindolol, acebutolol, metoprolol, oxprenolol,
propranolol,
alprenolol, pindolol, bisoprolol, esmolol, carteolol, bupranolol, mepindolol,
penbutolol, celiprolol or talinol, and one or more Kv1.5 blockers, in
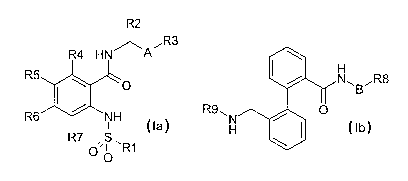
particular phenylcarboxamides of the formula la and/or Ib
R2
~ R3
R4 HN~A
R5 ~ N~B~R8
'O
Rfi ~ NH R9~
i la Ib
R7 O,S ~R~
O
and/or pharmaceutically tolerable salts thereof and the use of the
combination for the treatment of atrial arrhythmias.
Atrial fibrillation (AF) and atrial flutters are the most frequent, lasting
cardiac
arrhythmias. The occurrence increases with advancing age and frequently
leads to fatal, concomitant symptoms, such as, for example, cerebral
infarct. AF affects about 1 million Americans yearly and leads to more than
80,000 strokes each year in the USA. The antiarrhythmics of class I and III
customary at present reduce the reoccurrence rate of AF, but are only used
restrictively because of their potential proarrhythmic side effects. There is
therefore a great medical need for the development of better medicaments
for the treatment of atrial arrhythmias (S. Nattel, Am. Heart J. 130, 1995,
1094 - 1106; "Newer developments in the management of atrial
fibrillation")
It has been shown that most supraventricular arrhythmias are subject to
"reentry" excitation waves. Such reentries occur when the cardiac tissue
possesses a slow conductivity and at the same time very short refractory
periods. The increase in the myocardial refractory period due to
CA 02538009 2006-03-06
2
prolongation of the action potential is a recognized mechanism for ending
arrhythmias or preventing their formation (T. J. Colatsky et al., Drug Dev.
Res. 19, 1990, 129 - 140; "Potassium channels as targets for
antiarrhythmic drug action"). The length of the action potential is
essentially
determined by the extent of repolarizing K+ currents which flow out of the
cell via various K+ channels. Particularly great importance is ascribed here
to the "delayed rectifier" IK, which consists of 3 different components: IK~,
IKS and IK~~.
Most known class III antiarrhythmics (for example dofetilide, ibutilide,
almokalant,) mainly or exclusively block the rapidly activating potassium
channel IK~, which can be detected both in cells of the human ventricle and
in the atrium. It has been shown, however, that these compounds have an
increased proarrhythmic risk at low or normal heart rates, arrhythmias,
which are described as "torsades de pointes", in particular being observed
(D. M. Roden, Am. J. Cardiol. 72, 1993, 44B - 49B; "Current status of class
III antiarrhythmic drug therapy"). Beside this high and in some cases fatal
risk at a low rate, a decrease in the activity under the conditions of
tachycardia, in which the action is needed in particular, was found for the
IK~ blockers ("negative use dependence").
The "particularly rapidly" activating and very slowly inactivating component
of the delayed rectifier IK"~ (= ultra-rapidly activating delayed rectifier),
which corresponds to the Kv1.5 channel, plays a particularly large part for
the repolarization time in the human atrium. An inhibition of the IK~r
potassium outward current thus represents, in comparison to the inhibition
of IK~ or IKS, a particularly effective method for the prolongation of the
atrial
action potential and thus for the ending or prevention of atrial arrhythmias.
In contrast to IK~ and IKS, which also occur in the human ventricle, the IK~r
in fact plays an important part in the human atrium, but not in the ventricle.
For this reason, in the case of inhibition of the IK~~ current in contrast to
the
blockade of IK~ or IKS, the risk of a proarrhythmic action on the ventricle
should be excluded from the start. (Z. Wang et al, Circ. Res. 73, 1993,
1061 - 1076: "Sustained Depolarisation-Induced Outward Current in
Human Atrial Myocytes"; G.-R. Li et al., Circ. Res. 78, 1996, 689 - 696:
"Evidence for Two Components of Delayed Rectifier K+ Current in Human
Ventricular Myocytes"; G. J. Amos et al, J. Physiol. 491, 1996, 31 - 50:
CA 02538009 2006-03-06
3
"Differences between outward currents of human atrial and subepicardial
ventricular myocytes").
Antiarrhythmics which act via a selective blockade of the IK~~ current or
Kv1.5 channel have not been available hitherto on the market. A few patent
applications, however, describe compounds which on account of their
blocking action on the Kv1.5 channel act as atrial-selective antiarrhythmics.
For example, the patent application WO 0125189 describes, inter alia,
biphenylcarboxamides as Kv1.5 blockers. The applications WO 02088073
and WO 02100825 describe anthranilamides as Kv1.5 blockers for the
treatment of arrhythmias.
It has now surprisingly been found that the antiarrhythmic action on the
diseased atrium of the heart of Kv1.5 blockers such as, for example,
compounds of the formula la and Ib can be significantly enhanced by
simultaneous administration of a beta-blocker.
As it was possible to show experimentally, the combined administration of a
Kv1.5 blocker with a beta-blocker such as, for example, atenolol leads to a
superadditive effect on the atrial refractory period (AERP). Since the
prolongation of the AERP is a recognized surrogate parameter for the
antiarrhythmic action of a substance, the superadditive action of the
combination as an antiarrhythmic is also confirmed hereby. Such an
enhanced action is surprising, because on the basis of previously published
data, which relate to the interaction of beta-adrenergic system and IK~R, no
action would have been expected (Li G.-R., Feng, J., Wang Z., Fermini B.,
Nattel S., "Adrenergic modulation of ultrarapid delayed rectifier K+ current
in human atrial myocytes", Circ-Res. 1996, 78: 903-915). Since beta-
adrenergic stimulation (sympathetic nerve activation or stimulation with
beta-adrenergic agonists such as isoprenaline) strongly activates the IK~R
current, it would have been expected that the action of a combination of a
beta-blocker with an IK~R blocker would not turn out to be stronger than
that of one substance on its own. If the action of the beta-blocker on the
refractory period was based on a beta-adrenergic stimulation of the IK~R,
this action would already be blocked by a Kv1.5 blocker and no additional
antiarrhythmic action of a beta-blocker would be expected. The cause of
the superadditive action of the combination of IK~R and beta-blocker on the
atrial refractory period remains to be investigated.
CA 02538009 2006-03-06
4
The advantage of the use of a beta-blocker lies in its good compatibility and
its recognized action on the total cardiovascular mortality. Since patients
with atrial fibrillation often simultaneously suffer from coronary heart
disease or coronary insufficiency, which simultaneously are a basis for a
great danger of ventricular arrhythmia with a usually fatal outcome
(ventricular fibrillation), a combination of a Kv1.5 blocker with another
active principle such as beta-blockade is particularly advantageous. Beta-
blockers show favourable actions on coronary heart disease in the sense of
an antianginal action, reduce the mortality in postinfarct patients and have
meanwhile become standard therapy for cardiac insufficiency.
Beta-blockers are already employed successfully in the prevention and
therapy of atrial fibrillation. In addition, they serve for the rate control
of the
ventricle, i.e. they protect the ventricle by means of its action on the AV
node against the high rates of the atrium if the sinus rhythm cannot be
restored or cannot be maintained. Beta-blockers are regarded as highly
tolerable and very efficacious cardiovascular medicaments: patients with
atrial fibrillation are often already suffering from other cardiovascular
diseases, for the therapy of which a beta-blocker is therapeutically useful. A
combination of a beta-blocker with an efficacious Kv1.5 blocker is therefore
particularly useful in relation to atrial fibrillation and to the basic
cardiac
disorder.
The combinations of Kv1.5 and beta-blockers described here can therefore
be used as highly efficacious antiarrhythmics having a particularly
advantageous safety profile. In particular, the compounds are suitable for
the treatment of supraventricular arrhythmias, for example atrial fibrillation
or atrial flutters. The combinations can be employed for the termination of
existing atrial fibrillation or flutters for regaining the sinus rhythm
(cardioversion). Owing to the markedly enhanced action of the
combination, patients having persistent fibrillation can also be cardioverted,
who were previously not accessible to medicinal treatment. Moreover, the
combinations reduce the susceptibility to the development of new fibrillation
events (retention of the sinus rhythm, prophylaxis).
The invention relates to the combination of one or more beta-blockers and
of one or more compounds of the formula la and/or Ib
CA 02538009 2006-03-06
R2
~ R3
R4 HN~A
R5 ~ N~B~R8
'O
R6 / NH R9~
i la Ib
R7 O,S ~R1
O
in which
R(1 ) is alkyl having 3, 4 or 5 carbon atoms or quinolinyl,
5 R(2) is alkyl having 1, 2, 3 or 4 carbon atoms or cyclopropyl;
R(3) is phenyl or pyridyl,
where phenyl and pyridyl are unsubstituted or substituted by 1 or 2
substituents selected from the group consisting of F, CI, CF3, OCF3,
alkyl having 1, 2 or 3 carbon atoms and alkoxy having 1, 2 or 3
carbon atoms;
A is -C~H2"-;
n is 0, 1 or 2;
R(4), R(5), R(6) and R(7)
independently of one another are hydrogen, F, CI, CF3, OCF3, CN,
alkyl having 1, 2 or 3 carbon atoms, alkoxy having 1, 2 or 3 carbon
atoms;
B iS -CmH2m-;
m is 1 or 2;
R(8) is alkyl having 2 or 3 carbon atoms, phenyl or pyridyl,
where phenyl and pyridyl are unsubstituted or substituted by 1 or 2
substituents selected from the group consisting F, CI, CF3, OCF3,
alkyl having 1, 2 or 3 carbon atoms and alkoxy having 1, 2 or 3
carbon atoms;
R(9) is C(O)OR(10) or COR(10);
R(10) is -CxH2x R(11 );
x is 0, 1 or 2;
R(11 ) is phenyl,
where phenyl is unsubstituted or substituted by 1 or 2 substituents
selected from the group consisting F, CI, CF3, OCF3, alkyl having 1,
2 or 3 carbon atoms and alkoxy having 1, 2 or 3 carbon atoms;
and/or their physiologically tolerable salts.
CA 02538009 2006-03-06
6
The combination of one or more beta-blockers and of one or more
compounds of the formula la and/or Ib and/or physiologically tolerable salts
thereof is preferred, the beta-blockers being selected from the group
consisting of atenolol, carvedilol, nadolol, pindolol, acebutolol, metoprolol,
oxprenolol, propranolol, alprenolol, pindolol, bisoprolol, esmolol, carteolol,
bupranolol, mepindolol, penbutolol, celiprolol, talinol.
The combination of one or more beta-blockers and of one or more
compounds of the formula la and/or Ib and/or physiologically tolerable salts
thereof is particularly preferred, the beta-blockers being selected from the
group consisting of atenolol, carvedilol, nadolol, pindolol, acebutolol,
metoprolol, oxprenolol, propranolol, alprenolol, pindolol, for example
atenolol.
The combination of one or more beta-blockers and of one or more
compounds of the formula la and/or Ib and/or physiologically tolerable salts
thereof is very particularly preferred,
the beta-blockers being selected from the group consisting of atenolol,
carvedilol, nadolol, pindolol, acebutolol, metoprolol, oxprenolol,
propranolol,
alprenolol, pindolol, and the compounds of the formula la and/or Ib being
selected from the group consisting of
2'-~[2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
(2-pyridin-3-ylethyl)amide,
2'-(benzyloxycarbonylaminomethyl)biphenyl-2-carboxylic acid 2-(2-pyridyl)-
ethylamide,
2'-~(2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
2,4-difluorobenzylamide,
(S)-2'-(a-methylbenzyloxycarbonylaminomethyl)biphenyl-2-carboxylic acid
2-(2-pyridyl)ethylamide,
2-(butyl-1-sulfonylamino)-N-[1(R)-(6-methoxypyridin-3-yl)propyl]benzamide,
2-(butyl-1-sulfonylamino)-N-(cyclopropylpyridin-3-ylmethyl)-5-methylbenz-
amide,
(S)-5-fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenylpropyl)benzamide
and/or their physiologically tolerable salts.
The following combinations of beta-blockers and of compounds of the
formula la and/or Ib are especially preferred, it also being possible for the
components to be present in the form of their physiologically tolerable salts:
CA 02538009 2006-03-06
7
2'-{[2-(4-methoxyphenyl)acetylamino)methyl}biphenyl-2-carboxylic acid
(2-pyridin-3-ylethyl)amide and atenolol,
2-(butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl)benzamide
and atenolol,
2-(butyl-1-sulfonylamino)-N-(cyclopropylpyridin-3-ylmethyl)-5-methylbenz-
amide and atenolol,
(S)-5-fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenylpropyl)benzamide
and atenolol.
Furthermore, the invention relates to the use of one or more beta-blockers
together with one or more compounds of the formula la and/or Ib
R2
~ R3
R4 HN~A
R5 H
N~B~RB
R6 ~ NH R9~
la Ib
R7 O~S'R1
O
for the production of a medicament for the therapy or prophylaxis of atrial
fibrillation or atrial flutters,
in which
R(1 ) is alkyl having 3, 4 or 5 carbon atoms or quinolinyl,
R(2) is alkyl having 1, 2, 3 or 4 carbon atoms or cyclopropyl;
R(3) is phenyl or pyridyl,
where phenyl and pyridyl are unsubstituted or substituted by 1 or 2
substituents selected from the group consisting of F, CI, CF3, OCF3,
alkyl having 1, 2 or 3 carbon atoms and alkoxy having 1, 2 or 3
carbon atoms;
A is -C"H2~-;
n is 0, 1 or 2;
R(4), R(5), R(6) and R(7)
independently of one another are hydrogen, F, CI, CF3, OCF3, CN,
alkyl having 1, 2 or 3 carbon atoms, alkoxy having 1, 2 or 3 carbon
atoms;
B is -Cr,,H2m ;
m is 1 or 2;
CA 02538009 2006-03-06
8
R(8) is alkyl having 2 or 3 carbon atoms, phenyl or pydidyl,
where phenyl and pyridyl are unsubstituted or substituted by 1 or 2
substituents selected from the group consisting F, CI, CF3, OCF3,
alkyl having 1, 2 or 3 carbon atoms and alkoxy having 1, 2 or 3
carbon atoms;
R(9) is C(O)OR(10) or COR(10);
R(10) is -CXH2X R(11 );
x is 0, 1 or 2;
R(11 ) is phenyl,
where phenyl is unsubstituted or substituted by 1 or 2 substituents
selected from the group consisting of F, CI, CF3, OCF3, alkyl having
1, 2 or 3 carbon atoms and alkoxy having 1, 2 or 3 carbon atoms;
and/or their pharmaceutically acceptable salts.
The use of one or more beta-blockers together with one or more
compounds of the formula la and/or Ib and/or of a physiologically tolerable
salt thereof for the production of a medicament for the therapy or
prophylaxis of atrial fibrillation or atrial flutters is preferred, the beta-
blockers being selected from the group consisting of atenolol, carvedilol,
nadolol, pindolol, acebutolol, metoprolol, oxprenolol, propranolol,
alprenolol, pindolol, bisoprolol, esmolol, carteolol, bupranolol, mepindolol,
penbutolol, celiprolol, talinol.
The use of one or more beta-blockers together with one or more
compounds of the formula la and/or Ib and/or of a physiologically tolerable
salt thereof for the production of a medicament for the therapy or
prophylaxis of atrial fibrillation or atrial flutters is particularly
preferred, the
beta-blockers being selected from the group consisting of atenolol,
carvedilol, nadolol, pindolol, acebutolol, metoprolol, oxprenolol,
propranolol,
alprenolol, pindolol, for example atenolol.
The use of one or more beta-blockers together with one or more
compounds of the formula la and/or Ib and/or of a physiologically tolerable
salt thereof for the production of a medicament for the therapy or
prophylaxis of atrial fibrillation or atrial flutters is very particularly
preferred,
the beta-blockers being selected from the group consisting of atenolol,
carvedilol, nadolol, pindolol, acebutolol, metoprolol, oxprenolol,
propranolol,
alprenolol, pindolol,
CA 02538009 2006-03-06
9
and the compounds of the formula la andlor Ib being selected from the
group consisting of
2'-{[2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
(2-pyridin-3-ylethyl)amide,
2'-(benzyloxycarbonylaminomethyl)biphenyl-2-carboxylic acid 2-(2-pyridyl)-
ethylamide,
2'-{[2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
2,4-difluorobenzylamide,
(S)-2'-(a-methylbenzyloxycarbonylaminomethyl)biphenyl-2-carboxylic acid
2-(2-pyridyl)ethylamide,
2-(butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]benzamide,
2-(butyl-1-sulfonylamino)-N-(cyclopropylpyridin-3-ylmethyl)-5-methylbenz-
amide,
(S)-5-fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenylpropyl)benzamide
and/or their physiologically tolerable salts.
The use of the following combinations of beta-blockers together with
compounds of the formula la and/or Ib for the production of a medicament
for the therapy or prophylaxis of atrial fibrillation or atrial flutters is
especially preferred, it also being possible for the components to be
present in the form of their physiologically tolerable salts:
2'-{[2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
(2-pyridin-3-ylethyl)amide and atenolol,
2-(butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]benzamide
and atenolol,
2-(butyl-1-sulfonylamino)-N-(cyclopropylpyridin-3-ylmethyl)-5-methylbenz-
amide and atenolol,
(S)-5-fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenylpropyl)benzamide
and atenolol.
Alkyl radicals and alkylene radicals can be straight-chained or branched.
This also applies for the alkylene radicals of the formulae C~H2", CmH2m
and CxH2X. Alkyl radicals and alkylene radicals can also be straight-chained
or branched if they are substituted or are contained in other radicals, for
example in an alkoxy radical. Examples of alkyl radicals are methyl, ethyl,
n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl or n-pentyl. The
divalent radicals derived from these radicals, for example methylene,
1,1-ethylene, 1,2-ethylene, 1,1-propylene, 1,2-propylene, etc are examples
of alkylene radicals.
CA 02538009 2006-03-06
Pyridyl stands both for 2-, 3- and 4-pyridyl.
Quinolinyl includes 2-, 3-, 4-, 5-, 6-, 7- or 8-quinolyl, the 8-quinolyl
radical
5 being preferred.
Monosubstituted phenyl radicals can be substituted in the 2-, the 3- or the
4-position, or disubstituted in the 2,3-, 2,4-, 2,5-, 2,6-, 3,4- or 3,5-
position.
The same analogously also applies for the pyridyl radicals.
In the case of disubstitution of a radical, the substituents can be identical
or
different.
If the compounds of the formula la or ib contain one or more acidic or basic
groups or one or more basic heterocycles, the invention also includes the
corresponding physiologically or toxicologically tolerable salts, in
particular
the pharmaceutically utilizable salts. Thus, the compounds of the formula la
can be deprotonated on the sulfonamide group and used, for example, as
alkali metal salts, preferably sodium or potassium salts, or as ammonium
salts, for example as salts with ammonia or organic amines or amino acids.
Compounds of the formula la or Ib which contain a pyridine or quinoline
substituent can also be used in the form of their physiologically tolerable
acid addition salts with inorganic or organic acids, for example as
hydrochlorides, phosphates, sulfates, methanesulfonates, acetates,
lactates, maleates, fumarates, malates, gluconates etc.
Correspondingly, the beta-blockers can be employed in the form of their
physiologically tolerable salts.
In the case of appropriate substitution, the compounds of the formula I can
be present in stereoisomeric forms. If the compounds of the formula la or Ib
contain one or more asymmetric centers, these can independently of one
another have the S configuration or the R configuration. The invention
includes all possible stereoisomers, for example enantiomers or
diastereomers, and mixtures of two or more stereoisomeric forms, for
example enantiomers and/or diastereomers, in any desired ratios.
Enantiomers, for example, are thus included in enantiomerically pure form,
both as levorotatory and as dextrorotatory antipodes, and also in the form
of mixtures of the two enantiomers in different ratios or in the form of
CA 02538009 2006-03-06
11
10
racemates, in the invention. The preparation of individual stereoisomers
can be carried out, if desired, by separation of a mixture according to
customary methods or, for example, by use of isomerically pure synthesis
units.
Suitable beta-blockers which can be used are, for example, the substances
shown in table 1.
Table 1: Names and structural formulae of exemplary beta-blockers
Name Structure
OH
O~N
O
atenolol I i
HzN ~ -.~
HO N~0
~H I
O O
carvedilol ~ /
l~
N ~
H
OH
OH \ OH
nadolol ~~o
H
N
I N
pindolol
O OH
O~~ O
HEN ~
acebutolol
O~H
OH
CA 02538009 2006-03-06
12
HN
metoprolol ~ ~ o
0
OH
OH
H
O N
oxprenolol
0
O~H
propranolol OH
/ ~ \
\ /
OH
atprenolol ~ O HN
i
H
N
N
pindolol
o ~oH
NH
bisoprolol
~OH
O
~O
Q
CA 02538009 2006-03-06
13
N O
carteolol
O
H
N
HO
OH
O~~N
bupranolol
-Cl
H
N
mepindolol
OH H
O N
HN
penbutoloi HO,,
O
i
O
HN~N~
celiprolol
OH
N~~O 0
CA 02538009 2006-03-06
14
N
talinol NH
O~ ~ \ O/ \OH
N
H
The compounds of the formulae la and Ib used according to the invention
and/or their physiologically tolerable salts can thus be used in an
advantageous manner as pharmaceuticals together with one or more beta-
s blockers in animals, preferably in mammals, and in particular in humans, in
particular for the treatment of atrial arrhythmias.
The combination of the two active compounds can be carried out in such a
way that active compounds of the formula la and/or Ib and one or more
beta-blockers are administered together in one medicament or that a
medicament which contains one or more active compounds of the formula
la and/or Ib and a separate medicament which contains one or more beta-
blockers are administered simultaneously or successively in any sequence.
An administration successively also includes a combination in which the
individual medicaments are administered at different times and in different
ways in order to achieve a better effect. However, it can also be expedient
first to administer a suitable dose of the one medicament and then to
administer the other medicament, for example by infusion, until the desired
combination effect, for example the cardioversion to the sinus rhythm, has
occurred. Depending on the conditions in the individual case, it can be
more favorable to administer the active compounds) of the formula la
and/or Ib and one or more beta-blockers in the form of a pharmaceutical
combination preparation in which the two active compounds are present in
a fixed quantitative ratio, or to administer them in the form of separate
pharmaceutical individual preparations. In the latter case, in which the
quantitative ratio of the two active compounds can be varied, the individual
preparations can be situated in suitable primary packaging and, if
appropriate, together with use instructions referring to the use according to
the invention in a common packaging, or the individual preparations can, if
appropriate, in each case be situated in separate packagings together with
use instructions referring to the use according to the invention. All products
and kinds of preparation of this type are included by the present invention.
The invention thus relates, for example, to a product comprising a
CA 02538009 2006-03-06
combination of one or more beta-blockers and of one or more compounds
of the formula la and/or Ib and/or physiologically tolerable salts thereof for
simultaneous, separate or sequential use for the therapy or prophylaxis of
atria) fibrillation or atria) flutters.
5
The weight ratio of the active compounds of the formula la andlor Ib to the
beta-blocker(s) in the combinations according to the invention is
customarily in a range from 1000:1 to 1:10000, preferably between 50:1
and 1:250.
The present invention also relates to the use of compounds of the formulae
la and/or Ib and/or of a physiologically tolerable salt thereof and of one or
more beta-blockers for the production of pharmaceutical preparations
which contain one or more of the compounds la and/or Ib and one or more
of the beta-blockers as active components in addition to customary,
pharmaceutically innocuous vehicles, and their use as a medicament for
the treatment of, for example, atria) arrhythmias.
Furthermore, the present invention relates to pharmaceutical preparations
(combination preparation) which as active constituent contain an
efficacious dose of at least one compound of the formula 1a and/or Ib
and/or of a physiologically tolerable salt thereof and at least one beta-
blocker and/or of a physiologically tolerable salt thereof in addition to
customary, pharmaceutically innocuous vehicles and excipients and, if
appropriate, additionally one or more other pharmacological active
compounds. The pharmaceutical preparations normally contain 0.1 to 90
percent by weight of the compounds of the formula la andlor Ib and/or their
physiologically tolerable salts and of the beta-blockers and/or of their
physiologically tolerable salts.
The pharmaceutical preparations can be produced in a manner known per
se. For this, the active compounds and/or their physiologically tolerable
salts, together with one or more solid or liquid pharmaceutical vehicles
and/or excipients are brought into a suitable administration form or dose
form, which can then be used as a pharmaceutical in human medicine or
veterinary medicine. The same also applies for pharmaceutical
preparations which separately contain the two active compounds Kv1.5
blocker and beta-blocker and/or their pharmaceutically tolerable salts.
CA 02538009 2006-03-06
16
Pharmaceuticals which contain the combinations of compounds of the
formula la and/or Ib according to the invention and/or their physiologically
tolerable salts and of one or more beta-blockers and/or their physiologically
tolerable salts or the individual components employed in combination can
be administered, for example, orally, parenterally, intravenously, rectally,
by
inhalation or topically, the preferred administration being dependent on the
individual case.
In particular, combination preparations of compounds of the formula la
and/or Ib and/or their physiologically tolerable salts and one or more beta-
blockers and/or their physiologically tolerable salts are claimed for the
treatment of atrial arrhythmias such as atrial fibrillation and atrial
flutters.
The person skilled in the art is familiar on the basis of his/her expert
knowledge with excipients which are suitable for the desired
pharmaceutical formulation. In addition to solvents, gel-forming agents,
suppository bases, tablet excipients and other active compound carriers, it
is possible to use, for example, antioxidants, dispersants, emulsifiers,
antifoams, taste corrigents, preservatives, solubilizers, agents for achieving
a depot effect, buffer substances or colorants.
For an oral administration form, the active compounds are mixed with the
additives suitable therefor, such as vehicles, stabilizers or inert diluents,
and brought by means of the customary methods into the suitable
administration forms, such as tablets, coated tablets, hard gelatin capsules,
aqueous, alcoholic or oily solutions. The inert carriers which can be used
are, for example, gum arabic, magnesia, magnesium carbonate, potassium
phosphate, lactose, glucose or starch, in particular cornstarch. The
preparation can be carried out here both as dry and moist granules.
Suitable oily vehicles or solvents are, for example, vegetable or animal oils,
such as sunflower oil or cod-liver oil. Suitable solvents for aqueous or
alcoholic solutions are, for example, water, ethanol or sugar solutions or
mixtures thereof. Further excipients, also for other administration forms,
are, for example, polyethylene glycols and polypropylene glycols.
For subcutaneous, intramuscular or intravenous administration, the active
compounds, if desired with the substances customary therefor such as
solubilizers, emulsifiers or further excipients, are brought into solution,
suspension or emulsion. Suitable solvents are, for example, water,
CA 02538009 2006-03-06
17
physiological saline solution or alcohols, for example ethanol, propanol,
glycerol, in addition also sugar solutions such as glucose or mannitol
solutions, or alternatively mixtures of the various solvents mentioned.
Suitable pharmaceutical formulation for administration in the form of
aerosols or sprays are, for example, solutions, suspensions or emulsions of
the active compounds or their physiologically tolerable salts in a
pharmaceutically innocuous solvent, such as, in particular, ethanol or
water, or a mixture of such solvents. If required, the formulation can also
additionally contain other pharmaceutical excipients such as surfactants,
emulsifiers and stabilizers, and a propellant. Such a preparation
customarily contains the active compound in a concentration of
approximately 0.1 to 10, in particular of approximately 0.3 to 3, percent by
weight.
The dose of the active compounds of the active compounds to be
administered according to the invention or of the physiologically tolerable
salts thereof depends on the individual case and is to be adapted to the
conditions of the individual case as customary for an optimum action. Thus,
it depends, of course, on the frequency of administration and on the
potency and duration of action of the compounds in each case employed
for therapy or prophylaxis, but also on the nature and severity of the illness
to be treated and on the sex, age, weight and individual responsiveness of
the human or animal to be treated and on whether the therapy is to be
acute or chronic or prophylaxis is to be carried out. In particular in the
treatment of acute cases of cardiac arrhythmias, for example in an
intensive care unit, parenteral administration by injection or infusion, for
example by an intravenous continuous infusion, can also be advantageous.
If the inventions are used on animals, preferably on mammals, and in
particular on humans.
The dose of the Kv1.5 blocker of the formula la and/or Ib can customarily
vary in the range from 0.1 mg to 1 g per day and per person (in the case of
a body weight of approximately 75 kg), preferably from 0.5 to 750 mg per
day per person. In the case of the beta-blocker, the dose can customarily
vary between 5 and 300 mg per day per person, preferably between 25 and
100 mg per day per person. However, even higher doses may be
appropriate.
CA 02538009 2006-03-06
18
In the case of the combination treatment according to the invention, the
Kv1.5 blocker(s) and the beta-blocker(s) and/or their physiologically
tolerable salts can be administered in lower doses than in the case of
administration of only one of the two active compounds.
In the case of the combination treatment according to the invention, the
daily dose of the active compounds can be administered in one portion or it
can be divided into a number of, for example two, three or four,
administrations.
Experimental section
List of abbreviations
DMAP 4-dimethylaminopyridine
EDAC N-ethyl-N'-(3-dimethylaminopropyl)carbodiimide hydrochloride
HOBT 1-hydroxy-1 H-benzotriazole
RT room temperature
THF tetrahydrofuran
Example 1: 2'-{[2-(4-Methoxyphenyl)acetylamino]methyl}biphenyl-
2-carboxylic acid (2-pyridin-3-ylethyl)amide
H
,o ~ I o N l
N
15.5 g (0.115 mole) of HOBT and 21.9 g (0.115 mole) of EDAC were added
to a solution of 37.8 g (0.11 mole) of 2'-(tert-butoxycarbonylaminomethyl)-
biphenyl-2-carboxylic acid (Brandmeier, V.; Sauer, W.H.B.; Feigel, M.;
Helv. Chim. Acta 1994, 77(1), 70-85) in 550 ml of THF and the reaction
mixture was stirred at room temperature for 45 min. 14.0 g (0.115 mole) of
3-(2-aminoethyl)pyridine were then added and the mixture was stirred
overnight at RT. After addition of 400 ml of water and 500 ml of ethyl
acetate and intensive stirring, the phases were separated. The organic
phase was washed once with 400 ml of saturated sodium chloride solution
and twice with 400 ml each of saturated sodium hydrogencarbonate
CA 02538009 2006-03-06
19
solution. After drying over magnesium sulfate in the presence of activated
carbon, it was filtered and concentrated on a rotary evaporator. The
intermediate obtained (40.7 g) was dissolved in 600 ml of methylene
chloride and 100 ml of trifluoroacetic acid were then slowly added
dropwise. After stirring overnight, the reaction mixture was concentrated in
vacuo. The residue was treated with 250 ml of ethyl acetate and
concentrated again in order to distill out excess trifluoroacetic acid. 72.8
ml
(530 mmol) of triethylamine were added dropwise to the crude product
obtained dissolved in 170 ml of methylene chloride and 1 g of DMAP was
added. 18.7 g (100 mmol) of 4-methoxyphenylacetyl chloride were then
added dropwise at 5 - 10°C in the course of 30 min, and the batch was
stirred overnight at room temperature. After addition of 150 ml of water and
intensive stirring, the phases were separated and the organic phase was
washed once with 100 ml of sodium chloride solution, once with 25 ml of
1 M hydrochloric acid and twice with 100 ml each of saturated sodium
hydrogencarbonate solution. After drying over magnesium sulfate and
activated carbon, it was concentrated in vacuo. The oil obtained was
dissolved in hot acetonitrile and slowly allowed to crystallize out. 21.5 g of
2'-{[2-(4-methoxyphenyl)acetylamino]methyl}biphenyl-2-carboxylic acid
(2-pyridin-3-ylethyl)amide, melting point 116°C, were obtained.
Example 2:2'-(Benzyloxycarbonylaminomethyl)biphenyl-2-carboxylic acid
2-(2-pyridyl)ethylamide
H
N N\
~O
The compound was obtained according to the synthesis procedure
indicated in WO 0125189.
Example 3: 2'-{[2-(4-Methoxyphenyl)acetylamino]methyl}biphenyl-
2-carboxylic acid 2,4-difluorobenzylamide
CA 02538009 2006-03-06
F ~ F
/ O ~ ~ N ~ i
w H w ~ O
The compound was obtained according to the synthesis procedure
indicated in WO 0125189.
5
Example 4:(S)-2'-(a-Methylbenzyloxycarbonylaminomethyl)biphenyl-
2-carboxylic acid 2-(2-pyridyl)ethylamide
H
N N
~O
The compound was obtained according to the synthesis procedure
indicated in WO 0125189.
Example 5: 2-(Butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)-
propyl]benzamide
0 ~H
/ ,H ~ ~~
~NH N 0
0%O ~
a) 2-(Butyl-1-sulfonylamino)benzoic acid
20 g (188 mmol) of sodium carbonate were added to a suspension of 20 g
(146 mmol) of 2-aminobenzoic acid in 250 ml of water. 11.4 g (72.8 mmol)
of butylsulfonyl chloride were then added dropwise and the reaction mixture
was stirred at room temperature for 2 days. It was acidified with
concentrated hydrochloric acid, stirred at room temperature for 3 hours and
the deposited product was filtered off with suction. After drying in vacuo,
9.6 g of 2-(butyl-1-sulfonylamino)benzoic acid were obtained.
CA 02538009 2006-03-06
21
b) 1-(6-Methoxypyridin-3-yl)propylamine
3 ml (23.2 mmol) of 5-bromo-2-methoxypyridine were added at -70°C to a
solution of 10.2 ml of butyllithium (2.5 M solution in hexane; 25.5 mmol) in
50 ml of diethyl ether. After 10 min, 1.4 ml (19.5 mmol) of propionitrile were
added. After 2 hours at -70°C, the reaction mixture was slowly allowed
to
come to room temperature. 2.2 g of sodium sulfate decahydrate were then
added and allowed to stir for 1 hour. After subsequent addition of 5 g of
magnesium sulfate, the salts were filtered off after stirring briefly and the
filtrate was concentrated. The residue was dissolved in 70 ml of methanol
and 1.1 g (28 mmol) of sodium borohydride were added at 0°C. After
stirring overnight, the reaction mixture was adjusted to pH 2 using
concentrated hydrochloric acid and concentrated on a rotary evaporator.
The residue was treated with 10 ml of water, and extracted once with
diethyl ether. The aqueous phase was then saturated with sodium
hydrogencarbonate, concentrated in vacuo and the residue was extracted
with ethyl acetate. After drying and concentrating the ethyl acetate extracts,
1.4 g of racemic 1-(6-methoxypyridin-3-yl)propylamine were obtained.
c) 2-(Butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]-
benzamide
4.4 g (32.7 mmol) of 1-hydroxy-1 H-benzotriazole and 6.3 g (32.7 mmol) of
N-ethyl-N'-(3-dimethylaminopropyl)carbodiimide hydrochloride were added
to a solution of 8.0 g (31.1 mmol) of 2-(butyl-1-sulfonylamino)benzoic acid
in 250 ml of tetrahydrofuran and the reaction mixture was stirred for 90 min.
A solution of 5.4 g (32.7 mmol) of racemic 1-(6-methoxypyridin-3-yl)-
propylamine in 20 ml of tetrahydrofuran was then added dropwise and the
mixture was stirred overnight. The reaction mixture was treated with 250 ml
of water and extracted with 300 ml of ethyl acetate. The organic phase was
extracted 5 times with 100 ml each of saturated sodium hydrogencarbonate
solution and then dried over magnesium sulfate. 9.0 g of 2-(butyl-
1-sulfonylamino)-N-[1-(6-methoxypyridin-3-yl)propyl]benzamide were
obtained. The enantiomers were separated by preparative HPLC on a
Chiralpak ADH column (250 x 4.6 mm); eluent: heptane/ethanol/methanol
10:1:1; temperature: 30°C; flow rate: 1 ml/min. First, 4.0 g of 2-
(butyl-
1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]benzamide were
eluted at a retention time of 5.9 min. After a mixed fraction, 3.0 g of
2-(butyl-1-sulfonylamino)-N-[1 (S)-(6-methoxypyridin-3-yl)propyl]benzamide
were obtained at a retention time of 7.2 min.
CA 02538009 2006-03-06
22
2 g of the 2-(butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]-
benzamide were dissolved in 9 ml of isopropanol in the presence of heat,
then 8 ml of warm water were added and the reaction mixture was slowly
allowed to cool overnight. After filtering off with suction at 0°C, 1.5
g of
2-(butyl-1-sulfonylamino)-N-[1 (R)-(6-methoxypyridin-3-yl)propyl]benzamide
were obtained as colorless needle-shaped crystals; melting point 97°C.
Example 6: 2-(Butyl-1-sulfonylamino)-N-(cyclopropylpyridin-3-ylmethyl)-
5-methylbenzamide
O
_N ~ i
H
NH N
i
O,O w/~/
The compound was obtained according to the synthesis procedure
indicated in WO 0288073.
Example 7: (S)-5-Fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenyl-
propyf)benzamide
O
F
~NH
I
O=S=O
~ N~
/ /
a) 5-Fluoro-2-(quinoline-8-sulfonylamino)benzoic acid
A reaction mixture of 10.0 g (64 mmol) of 5-fluoro-2-aminobenzoic acid,
16.3 g (193 mmol) of sodium hydrogencarbonate and 16.3 g of 8-quinoline
sulfonyl chloride in 325 ml of water and 325 ml of ethyl acetate was stirred
overnight at RT. The aqueous phase was separated off and extracted once
CA 02538009 2006-03-06
23
with 50 ml of ethyl acetate. The aqueous phase was then rendered acidic
using conc. hydrochloric acid and stirred for 2 h. The precipitate deposited
was filtered off with suction, dried in vacuo and 19.5 g of 5-fluoro-
2-(quinoline-8-sulfonylamino)benzoic acid were obtained.
b) 5-Fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenylpropyl)benzamide
From 5.5 g (15.9 mmol) of 5-fluoro-2-(quinoline-8-sulfonylamino)benzoic
acid and 2.3 g (16.7 mmol) of (S)-phenylpropylamine, 5.7 g of the title
compound were obtained according to the procedure in WO 02100825.
M. p.: 163°C
Example 8: (S)-5-Fluoro-2-(quinoline-8-sulfonylamino)-N-(1-phenyl-
propyl)benzamide sodium salt
2 ml of a 30 percent sodium methoxide solution were added to a solution of
5 g of the compound of example 7 in 120 ml of ethyl acetate. The sodium
salt deposited was filtered off with suction and recrystallized from 25 ml of
ethanol and 3.3 g of the title compound were obtained.
Pharmacological investigations
Determination of the activity on the Kv1.5 channel
Kv1.5 channels from humans were expressed in Xenopus oocytes. For
this, oocytes were first isolated from Xenopus laevis and defolliculated.
RNA encoding Kv1.5 synthesized in vitro was then injected into these
oocytes. After Kv1.5 protein expression for 1 - 7 days, Kv1.5 currents were
measured using the two microelectrode voltage clamp technique. The
Kv1.5 channels were in this case as a rule activated using voltage jumps to
0 mV and 40 mV lasting 500 ms. The bath was rinsed using a solution of
the following composition: NaCI 96 mM, KCI 2 mM, CaCl2 1.8 mM, MgCl2
1 mM, HEPES 5 mM (titrated to pH 7.4 using NaOH). These experiments
were carried out at room temperature. The following were employed for
data acquisition and analysis: Geneclamp amplifier (Axon Instruments,
Foster City, USA) and MacLab D/A converter and software
(ADlnstruments, Castle Hill, Australia). The substances according to the
invention were tested by adding them to the bath solution in different
concentrations. The effects of the substances were calculated as
percentage inhibition of the Kv1.5 control current which was obtained when
CA 02538009 2006-03-06
24
no substance was added to the solution. The data were then extrapolated
using the Hill equation in order to determine the inhibitory concentrations
ICSO for the respective substances.
In this manner, the following ICSO values were determined for the
compounds listed below:
Example No. IC50 [NM]
1 4.7
2 0.7
3 1.4
4 0.2
5 10
6 1.0
7 1.1
Investigation of the refractory period in anaesthetized pigs
The investigations were carried out in the anaesthetised pig as described
by Knobloch et al. (Knobloch K., Brendel J., Peukert S., Rosenstein B.,
Busch A.-E., Wirth K.-J., "Electrophysiological and antiarrhythmic effects of
the novel IK~R channel blockers, S9947 and S20951, on the left vs. right
pig atrium in vivo in comparison with the IKr blockers dofetilide, azimilide,
d,
I-sotalol and ibutilide", Naunyn-Schmiedeberg's Arch Pharmacol 2002, 366:
482-487). The action of beta-blockers and IK~R blockers on the refractory
period of the left atrium was investigated. First, the action of the
individual
substances was tested, then the action of the combination of both
individual substances.
Description of method:
Male pigs of native German breed of age 2-3 months and weight 23-30 kg
were used.
The pigs were premedicated with 3 ml of Rompun~ 2% (xylocaine 23.3
mg/ml - 3 mg/kg; injected intramuscularly) and 6 ml of Hostaket~
(ketamine 115 mg/ml = 20 mg/kg; injected intramuscularly). Anesthesia
was induced using an intravenous bolus injection of 5 ml of Narcoren~
(pentobarbital 160 mg/ml = 25-30 mg/kg; i.v.) and continuously supplied
intravenously by means of a pentobarbital perfusor at 12-17 mg/kg/h. After
CA 02538009 2006-03-06
tracheotomy and intubation, the animals were ventilated with oxygen by
means ofi a respirator. A left-lateral thoracotomy was carried out in the
fifth
intercostal space. The lungs were retracted using sutures, the pericardium
was opened and held using sutures so that the heart rocked in this. The tip
5 of a MAP PacingT"" electrophysiology catheter (EP Technologies, Model
1675, Boston Scientific Corporation, 92257 La Garenne-Colombes Cedex,
France) were then placed on the free wall of the left atrium in a right-angled
position and fixed to the left atrium under constant pressure in a stand. The
electrical stimulation was carried out using an external heart stimulator from
10 Biotronik (UHS 20, Universal heart stimulator, Biotronik GmbH, 12359
Berlin, Germany).
Measurement of the atrial effective refractory period (AERP): The electrical
atrial response to the external pacemaker stimulation was visualized by
means of a mono phasic action potential (MAP), which was derived from
15 the left atrium by means of the electrophysiology catheter. A conditioning
stimulation cycle of 10 base intervals (S1) in a double stimulation amplitude
followed a diastolic, prematurely coupled extrastimulus (S2, 1 ms pulse
duration, 200 ms refractory period), which originated during a 5 ms
decrement of a coupling interval, which was 30 ms above the expected
20 effective refractory period (AERP). The 5 ms decrement in the coupling
interval of the extrastimulus ran until the atrium no longer produced any
response in the form of an action potential. The longest coupling interval
which was no longer able to induce any atrial action potential was used as
the effective refractory period. The refractory period was in each case
25 investigated at three basal cycle length, 240, 300 and 400 ms).
Experimental groups: The sole action of atenolol (bolus administration of
1 mg/kg) on the refractory period was investigated in a separate group of
pigs (n = 5). In two other groups (both n = 6), the action of the combination
of Example 1 or 5 and the beta-blocker atenolol was investigated: After a
basal period, vehicle was administered and the refractory periods were
determined. After this, the compounds of Example 1 or 5 was introduced in
an infusion of 3 mg/kg/h in order to achieve a stable plateau of action. This
investigation allowed the assessment of the action of the sole
administration of Example 1 or 5. On the stable plateau of action of
Example 1 or 5, it was possible to assess the action of atenolol (bolus
administration of 1 mg/kg). As a rule, after 1 h infusion with the compounds
of Example 1 or 5, the refractory periods were indeed increased to a stable
level.
CA 02538009 2006-03-06
26
10
Results: Both beta-blockers (table 2) and IK~R blockers (tables 3 and 4)
showed a prolongation of the refractory period. The combination of both
active principles leads to a marked prolongation of action, which was
superadditive at the basal cycle length of 400 ms (tables 3 and 4).
Basal 240 300 400
C cle len ms ms ms
th
Mean SEM Mean SEM Mean SEM
value value value
Control 101 3 108 6 118 4
Atenolol 136 6 129 8 129 7
Increase in 25* 5 ~ 21 * 2 ~ 11 * 4
ms ~ ~ ~ ~
Table 2: Refractory periods in milliseconds after administration of atenolol
(1 mg/kg as bolus; i.v.) to the left atrium of the pig (n = 5) at three basal
cycle lengths (240, 300 and 400 ms). *p < 0.05 vs. control.
Basal 240 300 400
ms ms ms
c cle len
th
Mean SEM Mean SEM Mean SEM
value value value
Control 101 5.8 108.8 7.4 113.8 7.2
Exam le 1 136 4.3 149.5 3.8 166.2 4.2
Example 1 159.5 2.5 176.2 4.2 183.7 3.3
lus atenolol
Increase after35# 5.8 40.7# 7.9 52.3# 9.8
Exam le 1
in ms
Additional 23.5* 4.3 26.7* 10.2 17.5* 8.9
increase after
atenolol in
ms
Table 3: Refractory periods in milliseconds after combined administration of
the IK~R blocker compounds of Example 1 and atenolol beta-blocker to the
left atrium of the pig (n = 6) at three basal cycle lengths (240, 300 and 400
ms). The compound of example 1 was infused in a dose of 3 mg/kg/h. After
infusion for 1h, on the plateau of action of the compound of Example 1,
1 mg/kg of atenolol was administered as a bolus. # p < 0.01 vs. control, *p <
0.01 atenolol vs. compound of Example 1.
CA 02538009 2006-03-06
27
Basal 240 300 400
ms ms ms
c cle len th
Mean SEM Mean SEM Mean SEM
value value value
Control 99 7.7 112 6.8 135 3.3
Exam le 5 133 6 147 4.3 162 3.7
Example 5 156 8 169 5.4 179 6.5
lus atenolol
Increase after34# 4 35# 3.9 27# 2.5
Exam le 5 in
ms
Additional 23* 4.2 22* 2.8 17* 4.0
increase after
atenolol in
ms
Table 4: Refractory periods in milliseconds after combined administration of
the IKur blocker compound of Example 5 and atenolol beta-blocker to the
left atrium of the pig (n=6) at three basal cycle lengths (240, 300 and 400
ms). The compound of Example 5 was infused in a dose of 3 mg/kg/h. After
infusion for 1 h, on the plateau of action of the compound of Example 5,
1 mg/kg of atenolol was administered as a bolus. #p<0.05 vs. control,
*p<0.05 atenolol vs. compound of Example 5.