Note: Descriptions are shown in the official language in which they were submitted.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
DERIVES DE N- " PHENYL(PIPERIDIN -2-YL)METHYL !BENZAMIDE, LEUR
PRÉPARATION ET LEUR APPLICATION EN THÉRAPEUTIQUE
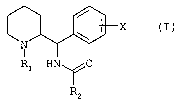
Les composés de l'ïnventïon répondent à la formule générale
(I)
X
N
1
R1 HN\ /'0
'R~~
dans laquelle
R1 représente soit un atome d'hydrogène, soit un groupe
(Ci-C7)alkyle linéaire ou ramifié éventuellement substitué
par un ou plusieurs atomes de flu or, soit un groupe
(C3-C?) cycloalkyle, soit un groupe (C3-C7) cycloalkyl-
(Ci-C3) alkyle, soit un groupe phényl (C1-C3) alkyle
éventuellement substituê par un o u deux groupes méthoxy,
soit un groupe (C2-CQ) alcényle, soit un groupe
(C2-C4) alcynyle,
X représente un atome d'hydrogëne ou un ou plusieurs
substituants choisis parmi les atomes d'halogènes et les
groupes trifluorométhyle, (C1-C4) alkyle et (C1-C4) alcoxy
linéaires ou ramifiés,
R~ représente un groupe choisi parmi les les groupes
naphtalényle, pyridinyle, pyrimid inyle, pyrazinyle,
pyridazïnyle, triâzinyle, indanyle, indényle, quinolëinyle,
isoquinoléinyle, quinazolinyle, quinoxalïnyle,
phtalazinyle, thïényle, furyle, p yrrolyle, imidazolyle,
pyrazolyle, oxazolyle, thiazolyle, isoxazolyle,
isothiazolyle, thïadïazolyle, oxa dïazolyle, triazolyle,
benzothiényle, benzofuryle, benzirnidazolyle,
benzothiazolyle, indolyle, isoïnd olyle, indazolyle,
benzoxazolyle, benzisoxazolyle, b enzotriazolyle,
benzisothïazolyle, dihydroindolyle, pyrrolopyridinyle,
furopyridinyle, thiénopyrïdinyle, imidazopyridinyle,
oxazolopyridinyle, thiazolopyridinyle, pyrazolopyridinyle,
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
2
isoxazolopyridinyle, isothiazolopy rïdinyle,
tétrahydroquinoléinyle, tétrahydro isoquinoléïnyle, et
éventuellement sunstitué par un ou plusieurs substituants
choisis parmi les atomes d'halogèn es et les groupes
(C1-C4) alkyles, (C1-C4) alcoxy, thio (C1-C4) alkyles ou phényles
éventuellement substitués par un o u plusieurs substituents
choisis parmi les atomes d'halogèn es et les groupes
trifluorométhyle, (C1-CQ) alkyles et (C1-CQ) alcoxy.
Les composés dé formule générale (I) peuvent exister sous
forme du racémate thréo (1R, 2R ; 1 S, 2S) ou sous forme
d'énantiomères (1R,2R) ou (1S,2S) ; ils peuvent exister à
l'état de bases libres ou de sels d'addition à des acides.
Des composés de structure analogue à celle des composés de
l'inventïon sont décrits dans le brevet US-5254569 comme
analgésiques, diurétiques, anticonvulsivants, anesthé-
siques, sédatifs, cérébroprotecteu rs, par un, mécanisme
d'action sur les récepteurs opiacé s. D'autres composés de
structure analogue sont décrits dans la demande de brevet
EP-0499995 comme antagonistes 5-HT3 utiles dans le
traitement des désordres psychotiques, des maladies
neurologiques, des symptômes gastriques, des nausées et des
vomissements.
Les composés préférés de l'invention sont dépourvus
d'activité sur les récepteurs opiacés ou 5-HT3 et
présentent une activité particulière comme inhibiteurs
spécifiques des transporteurs de 1 a glycine glytl et/ou
glyt2.
Les composés de formule générale (I) dans laquelle R1 est
différent d'un atome d'hydrogène peuvent être préparés par
un procédé illustré par le schéma 1 qui suit.
On effectue un couplage d'une diam ine de formule générale
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
3
(II), dans laquelle R1 et X sont tels que définis ci-dessus
(avec R1 différent d'un atome d'hydrogène) avec un acide
activé ou un chlorure d'acide de formule générale (III)
dans laquelle Y représente un groupe OH activé ou un
Schéma 1
Y \ /'0
(III)
R~
X
(I)
I
Rl NH2 ( I I )
atome de chlore et R2 est tel que défini ci-dessus, en
utilisant les méthodes connues de l'homme du métier.
La diamine de formule générale (II) peut être préparée par
un procédé illustré par le schéma 2 qui suit.
On fait réagir l'amide de Weinreb de formule (IV) avec le
dérivé de phényllithium de formule générale (V), dans
laquelle X est tel que défini ci-dessus, dans un solvant
éthéré tel que l'éther diéthylique, entre -30°C et la
température ambiante ; on obtient une cétone de formule
générale (VI) que l'on réduit en alcool de configuration
thréo de formule générale (VII) par un agent réducteur tel
que 1e K-Selectride0 ou le L-Selectride~ (tri-sec-butyl-
borohydrure de potassium ou de Lithium), dans un solvant
éthéré tel que le tétrahydrofurane, entre -78°C et la
température ambiante. Le carbamate de formule générale
(VTI) peut ensuite être réduit en N-mëthylaminoalcool thréo
de formule générale (VIII) par action d'un hydrure mixte
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
4
tel que l'hydrure double d'aluminium e t de lithium, dans un
Schéma 2
\ Z1
X
O~CH3 / (V) /
I X
N NwCH3 N \
Boc O (IV) Boc 0 (VI)
/ /
X X
( I I ) s N ~ ' -E-- N \
I
CH3 OH (VIII) Boc OH (VII)
/ /
X X
( I I ) -E \ .~-- \
N N
I H
Rl OH (X) OH ( IX)
solvant éthéré tel que le tétrahydrofurane, entre la
température ambiante et 1a température de reflux. On
transforme ensuite l'alcool thréo de formule générale
(VIII) en intermédiaire thréo de formu 1e générale (II) où
R1 représente un groupe méthyle en deux étapes . on
transforme d' abord- la fonction alcool en groupe nucléofuge,
par exemple un groupe méthanesulfonatee par action du
chlorure de méthylsulfonyle, dans un solvant chloré tel que
' le dichlorométhane, et en présence d'une base telle que la
triéthylamine, entre 0°C et la température ambiante, puis
on fait réagir le groupe nucléofuge ave c de l'ammoniac
liquéfié à -50°C, dans un alcool tel que l'éthanol, dans un
milieu clos tel qu'un autoclave, entre -50°C et la
température ambiante.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
On peut également déprotéger le carbamate de formule
générale (VII) au moyen d'une base forte telle que la
potasse aqueuse, dans un alcool tel que le méthanol pour
obtenir l'aminoalcool thréo de formule générale (IX),
5 procéder ensuite à une N-alkylation au moyen d'un dërivé
halogéné de formule R1Z, dans laque lle R1 est tel que défini
ci-dessus, mais différent d'un atome d'hydrogène, et Z
représente un atome d'halogène, en présence d'une base
telle que le carbonate de potassium, dans un solvant
polaire tel que le N,N-diméthylformamide, entre la
température ambiante et 100°C. On traite ensuite l'alcool
de formule générale (X) ainsi obtenu comme décrit à propos
de l'alcool de formule générale (VIIT).
ïlne autre variante de procédé, illustrée par le schéma 3
qui suit, peut être utilisée dans 1 e cas où R1 représente
un groupe méthyle et X représente un atome d'hydrogène.
On quaternarise la pyridineoxime de formule (XI), par
exemple par action du trifluorométhanesulfonate de méthyle,
dans un solvant éthéré tel que l'éther diéthylique à
température ambiante. On soumet ensuite le sel de
pyridinium ainsi obtenu, de formule (XII) à une
hydrogénation sous atmosphère d'hydrogène, en présence d'un
catalyseur tel que l'oxyde de platine, dans un mélange
d'alcool et d'acide aqueux tel que l'ëthanol et l'acide
chlorhydrique 1N. On obtient la diamine de formule générale
(II) où R1 reprësente un groupe méthyle et X représente un
atome d'hydrogène sous forme d'un mélange des deux
diastéréoisomères thréo/érythro 9/l. On peut la salifier,
par exemple avec de l'acide oxalique, puis la purifier par
recristallisation de l'oxalate formé dans un mélange
d'alcool et d'un solvant ëthéré tel que le méthanol et
l'éther diéthylique, pour obtenir 1e diastéréoisomère thréo
(1R, 2R ; 1S, 2S) pur.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
6
Schéma 3
I~ /I I~ 1
N -~ ...~ N --; ( II, Rl=CH3, X=H)
NEC ~ CH3 N~o
/ I /
(XI) (XII)
Les composés de formule générale (II) dans laquelle R1
représente un atome d'hydrogène peuvent é tre préparés
suivant le schéma 2, avec utilisation d'un composé de
formule générale (I) dans laquelle R1 représente un groupe
phénylméthyle éventuellement substitué, puis déprotection
de l'azote du cycle pipéridine, par exemp le par un agent
oxydant ou par un acide de Lewis, tel que le tribromure de
bore, ou par hydrogénolyse, soit un groupe alcényle, de
préférence allyle, et à déprotëger 1'azot e par un complexe
du Pd°, pour obtenir un composé de formul e générale (I)
dans laquelle R1 représente un atome d'hydrogène.
Par ailleurs les composés chiraux de formule générale (I)
correspondant aux énantiomères (1R,2R) ou (1S,2S) du
diastéréoisomère thréo peuvent être également obtenus par
séparation des composés racémiques par chromatographie
liquide à haute performance (CLHP) sur colonne chirale, ou
par dédoublement de l'amine racémique de formule générale
(IT) par utilisation d'un acide chiral te 1 que l'acide
tartrique, l'açide camphorsulfonique, l'a vide
dibenzoyltartrique ou la N-acétylleucine, par la
recristallisation fractionnée et préfërentielle d'un sel
diastéréoisomérique, dans un solvant de type alcool, ou
encore par synthèse énantiosélective selo n le schéma 2 avec
utilisation d'un amide de Weinreb chiral de formule (IV).
L'amide de Weinreb de formule (IV) racémi que ou chiral,
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
7
ainsi que la cétone de formule générale (VI), peuvent être
préparés selon une méthode analogue à ce 11e décrite dans
Eur. J. Med. Chem., 35, (2000), 979-988 et J. Med. Chem.,
41, (1998), 591-601. Le composé phényllithié de formule
générale (V) où X représente un atome d'hydrogène est
disponible dans, le commerce. Ses dérivés substitués peuvent
être préparés selon une mëthode analogue à celle dëcrite
dans Tetrahedron Zett., 57, 33, (1996), 5905-5908. La
pyridineoxime de formule générale (XI) peut être préparée
selon une méthode analogue à celle décrit e dans la demande
de brevet EP-0366006. L'amine de formule générale (IX) dans
laquelle X représente un atome d'hydrogène peut être
préparée en série chirale selon une méthode décrite dans le
brevet US-2928835. Enfin, l'amine de formule générale
(XIII) peut être préparée selon une méthode analogue à
celle décrite dans Chem. Pharm. Bu11.,32, 12, (1984) ,
4893-4906 et Synthesis, (1976) , 593-595.
Les acides et chlorures d'acïdes de formule générale (III)
sont disponïbles dans le commerce, sauf l'acide
4-[2-chloro-3-(trifluorométhyl)phényl]-1H-imidazole-2-
carboxylique, qui peut être préparé dans des conditions
comparables à celles décrites dans la demande de brevet
EP-0365030 et dans le brevet US-3336300.
Les exemples qui vont suivre illustrent 1 a préparation de
quelques composés de l'invention. Les microanalyses élëmen-
taires, les spectres I.R. et R.M.N. et la CLHP sur colonne
chirale confirment les structures et les puretés
énantiomériques des composés obtenus.
Les numéros indiqués entre parenthèses dans les titres des
exemples correspondent à ceux de la lère colonne du tableau
donné plus loin.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
8
Exemple 1 (Composé N°4).
Chlorhydrate de thréo-2,5-dichloro-N-[(1 -méthyl-2-
pipéridinyl)(phényl)méthyl]-3-thiophènecarboxamide 1:1.
1.1. Trifluorométhanesulfonate de 2-(benzyloxyïmino-
phénylméthyl)-1-méthylpyridinium.
A une suspension de 35 g (120 mmoles) de phényl(pyridin-
2-yl)mëthanone O-benzyloxime dans 200 m1 d°éther
diéthylique on ajoute, goutte à goutte et à 0°C, 17,4 ml
(120 mmoles) de trifluorométhanesulfonat e de méthyle et on
agite le mélange â température ambiante pendant 3 h.
On recueille le précipité formé par filt ration et on le
sèche sous pression réduite.
On obtient 49 g de produit qu'on utilise tel quel dans
l'étape suivante.
1.2. Ethanedioate de thréo-(1-méthyle ipéridin-2-yl)-
phénylméthanamine 2:1.
Dans une fïole de Parr on place 14,8 g (31,89 mmoles) de
trifluorométhanesulfonate de 2-(benzyloxyiminophényl-
méthyl)-1-méthylpyridinium et 0,74 g d'oxyde de platine
dans 50 ml d'éthanol et 50 ml d'acide chlorhydrique 1N et
on effectue une hydrogénation pendant 5 h.
On évapore l'éthanol sous pressïon réduite, on extrait le
résidu avec du dichlorométhane, on sépare la phase aqueuse,
on y ajoute une solution d'ammoniaque et on l'extrait avec
du dichlorométhane. Après lavage des phases organiques
réunies, séchage sur sulfate de sodium, filtration et
évaporation du solvant sous pression réduïte, on obtient
6,7 g de produit huileux comprenant 10~ de diastérëoisomère
érythro.
On prépare l'éthanedioate en dissolvant ces 6,7 g de base
dans le méthanol, par action de deux équivalents d'acide
éthanedioïque dissous dans le minimum de méthanol.
On purifie le sel obtenu par recristallisation dans un
mélange de méthanol et d'éther diéthylique.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
9
On isole finalement 4,7 g d'éthanedioate du dia s téré-
oisomère thréo pur.
Point de fusion . 156-159°C.
1.3. Chlorhydrate de thréo-2,5-dichloro-N-[(1 -méthyl-2-
pipéridinyl)(phényl)méthyl]thiophène-3-carboxamide
1:1.
Dans un ballon de 100 ml on introduit 0,768 g (4 mmoles)
d'acide 2,5-dichlorothiophène-3-carboxylique en solution
dans 15 ml de dichlorométhane, puis on ajoute
successivement 0,651 ml (4,7 mmoles) de triéthyl amine et
0,447 ml (4,7 mmoles) de chloroformiate d'éthyle, et on
agite le mélange réactïonnel à température ambiante pendant
2 h.
On ajoute 0,80 g (3,9 mmoles) de thréo (1-méthylpipéridin-
2-yl)phénylméthanamine en solution dans 15 m1 de
dichlorométhane et on poursuit l'agitation à température
ambiante pendant 1ê h.
On traite le mélange avec de l'eau et on l'extrait
plusieurs fois avec du dichlorométhane. Après lavage des
phases organiques avec de l'eau puis avec une solution
aqueuse de soude 1N, séchage sur sulfate de sodium,
filtration et évaporation du solvant sous press i on rëduite
on purifie le rësidu par chromatographie sur col orme de gel
de silice en éluant avec un mélange 97/3 à 9515 de
dichlorométhane et de méthanol.
On obtient 0,6 g de produit huileux dont on prépare le
chlorhydrate par addition d'une solution 0,1N d'acide
chlorhydrique dans le propan-2-ol.
Après évaporation des solvants sous pression réduite on
recristallise 1e solide blanc obtenu dans un mél ange
d'éther isopropylique et de propan-2-ol.
On isole finalement 0,474 g de chlorhydrate sous forme de
solide blanc.
Point de fusion . 216-317°C.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
Exemple 2 (Composé N°5).
Chlorhydrate de 2,5-dichloro-N-[(S)-[(2S)-1-méthyl-2-
pipéridinyl](phényl)méthyl]thiophène-3-carboxamide 1:1.
5 2.1 (2S)-2-benzoylpipérïdine-1-carboxy late de
1,1-diméthyléthyle.
Dans un ballon.de 500 ml, sous atmosphère d'azote, on
introduit 11, 8 g (43, 3 mmoles) de (~S) -~- (N-méthoxy-
N-méthylcarbamoyl)pipéridine-1-carboxylate de
10 1,1-dimëthyléthyle dans 100 ml d'éther dïéthylïque anhydre,
on refroidït le milieu à -23°C, on ajoute, goutte à goutte,
21,6 ml (43,2 mmoles) d'une solution 1,8M de phényllïthium
dans un mélange 70J30 de cyclohexane et d'éther diéthylique
et on agïte le mélange à température ambiante pendant 3 h.
Après hydrolyse avec une solution aqueuse saturée de
chlorure de sodium on sépare la phase aqueuse et on
l'extrait avec de l'acétate d'éthyle. On rêche la phase
organique sur sulfate de sodium, on la filtre, on la
concentre sous pression réduite et on purifie le résidu par
chromatographie sur colonne de gel de sili ce en éluant avec
un mélange d'acétate d'éthyle et de cyclohexane.
On obtient 4,55 g de produit solide.
Point de fusion . 123-125°C.
[a] ~5p=-25, 4 ° (c=2, 22 ; CH2C12) ee=97, 2 ~ .
2 . 2 . ( 1S) -2- [ (2S) -hydroxy (phényl ) méthyl ] pipéridine-
1-carboxylate de 1,1-diméthyléthyle.
Dans un ballon de 500 m1, sous atmosphère d'azote, on
introduit 4,68 g (16,2 mmoles) de (2S)-2-benzoylpipérïdine-
1-carboxylate de 1,1-diméthylëthyle dans 170 ml de
tétrahydrofurane anhydre, on refroidit la solution à -78°C,
on ajoute, goutte à goutte , 48,5 ml (48,5 mmoles) d'une
solution 1M de L-Selectride~ (tri-sec-but ylborohydrure de
lithium) dans le tétrahydrofurane, et on agite le mélange à
température ambiante pandant 5 h.
On l'hydrolyse à froïd lentement avec 34 ml d'eau et 34 ml
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
11
d'une solution aqueuse à 35~ de peroxyde d'hydrogène, et on
laisse le mélange revenir à température ambiante en
l'agitant pendant 2 h.
On le dilue avec de l'eau et de l'acétate d'éthyle, , on
sépare la phase aqueuse, on l'extrait avec de l'acétate
d'éthyle. Après lavage des phases organiques réunies,
séchage, séchage sur sulfate de sodium, filtration et
évaporation on purifie le résidu par chromatographi e sur
colonne de gel de silice en éluant avec un mélange
d'acétate d'éthyle et de cyclohexane.
On obtient 4,49 g d'une huile jaune pâle.
[cc] 25D=+63, 75 ° (~c=0, 8 ; CH2C12) ee=97, 8 ~ .
2.3. (1S)-[(2S)-(1-méthylpipéridin-2-yl)]phénylmëthanol.
Dans un bicot de 200 ml, sous atmosphère d'azote, on
introduit 2,96 g (78,1 mmoles) d'hydrure double d'aluminium
et de lithium dans 50 ml de tétrahydrofurane anhydr e, on
chauffe le mëlange au reflux, on ajoute 4,49 g (15, 4
mmoles ) d' une solution de (1S) -2- [ (2S) -hydroxy (phényl) -
méthyl]pipéridine-1-carboxylate de 1,1-diméthyléthyle dans
35 m1 de tétrahydrofurane et on maintient le mélange au
reflux pendant 3,5 h.
On le refroidït, on l'hydrolyse lentement avec une solution
0,1M de tartrate double de potassium et de sodium et on
laisse le mélange sous agitation pendant une nuit.
On le filtre et on rince le précipité avec du
tétrahydrofurane, puïs on concentre le filtrat sous
pression réduite.
On obtient 2,95 g d'un produit huileux incolore.
2.4. (1S)-[(2S)-(1-méthylpipéridin-2-yl)]phényl-
méthanamine.
Dans un ballon de 250 ml, sous atmosphère d'azote, on
introduit 2, 95 g ( 14, 4 mmoles ) de ( 1S) - [ ( 2S) - ( 1-méthyl-
pipéridin-2-yl)]phénylméthanol et 2 ml (14,4 mmoles) de
triéthylamine dans 70 ml de dichlorométhane anhydre, on
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
12
refroidit le milieu à 0°C, on ajoute 1,l ml (14,4 mmoles)
de chlorure de méthanesulfonyle, on laisse le mélange
revenir lentement à température ambiante pendant 2 h et on
le concentre sous pression réduite.
Dans un autoclave muni d'une agitation magnétique et
refroidi à -50°C on introduit de l'ammoniac liquéfié, on
ajoute une solution du méthanesulfonate précédemment
préparé dans 30 ml d'éthanol absolu, on ferme l'autoclave
et on maintient l'agitation pendant 48 h.
On transvase le mélange dans un ballon, on évapore les
solvants sous pression réduite et on isole l'amine sous
forme de produit huileux qu'on utilise tel quel dans
l'étape suivante.
2.5. Chlorhydrate de 2,5-dichloro-N-[(1S)-[(2 S)-1-
méthyl-2-pipéridinyl](phényl)méthyl]thiophène-3-
carboxamide 1:1.
Dans un ballon de 250 ml on introduit 0,37 g (1,88 mmole)
d'acide 2,5-dichlorothiophène-3-carboxylique dans 15 ml de
dichlorométhane, on ajoute successivement 0,31 ml (2,25
mmoles) de triéthylamine et 0,21 ml (2,25 mmoles) de
chloroformiate d'éthyle et on agite le mëlange à
tempërature ambiante pendant 1 h.
On ajoute 0, 38 g (1, 88 mmole) de (1S) - [ (2S) -(1-méthylpipé-
ridin-2-yl)]phénylméthanamine en solution dans 10 ml de
dichlorométhane et on poursuit l'agitation à température
ambiante pendant 12 h.
On traite le mélange avec de l'eau, on l'extrait plusieurs
fois avec du dichlorométhane, on réunit les phases
organiques, on les lave avec une solution aqueuse 1N de
soude, on les sèche sur sulfate de sodium, on (filtre et on
concentre le filtrat sous pression réduite.
On purifie le résïdu brut par chromatographie sur colonne
de gel de silice en éluant avec un mélange 98/2 de
dichloromëthane et de méthanol contenant 0,1~ d'ammoniaque.
On obtient 0,368 g de produit huileux dont on p répare le
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
13
chlorhydrate par addïtïon d'une solution 0,1N d'acide
chlorhydrique dans le propan-2-ol.
Après évaporatïon du solvant sous pression réduit e on
recristallise le solide dans un mélange de propan-2-ol et
d'éther isopropylique.
On isole finalement 0,36 g de chlorhydrate sous forme de
solide jaune pâle.
Point de fusion . 134-136°C [a]25D=+45,6 (c=0,99) ;CH30H.
Exemple 3 (Composé N°18).
Chlorhydrate de thrëo-4-[2-chloro-3-(trifluorométhyl)-
phényl ] -N- [ ( 1-méthyl-2-pipérïdinyl ) (phényl ) méthyl ] -1H-
imidazole-~-carboxamide 1:1.
Dans un ballon de 50 ml on ïntroduit 0,1 g (0,344 mmole)
d'acide 4-(2-chloro-3-(trifluorométhyl)phényl]-1H-
imïdazole-2-carboxylique, 0,066 g (0,344 mmole) de
chlorhydrate de 1-[3-(dïméthylamino)propyl]-3-
éthylcarbodiimide, 0,047 g (0,344 mmole) de
1-hydroxybenzotriazole en solution dans 10 ml de
dichlorométhane, et on agite le mélange â température
ambiante pendant 5 min.
On ajoute 0,072 g (0,344 mmole) de thréo-(1-méthy 1-
pïpéridin-2-yl)phénylméthanamine (préparée selon l'exemple
1,2) dans quelques ml de dichlorométhane et on poursuit
l'agitation pendant 6 h.
On traite le mélange avec de l'eau, on l'extrait plusieurs
fois avec du dichlorométhane, on lave les phases organiques
avec de l'eau puis avec une solution aqueuse 1N de soude,
puis avec une solution aqueuse saturée de chlorure de
sodium, on les sèche sur sulfate de sodium et on évapore le
solvant sous pression réduite. On purifie le rési du par
chromatographie sur colonne de gel de silïce en é luant avec
un mélange de dichlorométhane et de méthanol.
On obtient 91 mg de produit dont on prépare le chlorhydrate
par addition d'une solution 0,1N d'acide chlorhydrique dans
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
14
le propan-2-ol. On évapore partiellement le solvant sous
pression réduite pour obtenir, après cristallisatïon,
104 mg de composé solide blanc.
Point de fusion . 188-195°C.
Le tableau de la page suivante illustre les structures
chimiques et les propriétés physiques de quelques composés
de l'invention.
Dans la cotonné "Sel", "-" désigne un composé à l'état de
base et "HCl" désigne un chlorhydrate.
Le pouvoir rotatoire du composé N°5 est [a]25D=+45,6°
(c=0, 99) ;CH30H.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
N t.t~ I~- lfl
y -I f-I N c-I
M N c-1 N c-i
o
y -I N lO
-I- rl N c-I (Y7
N c-i N r-I
I I
M x ~ x
U U U
pn y
w w
U ~
O
~~ x x x x x x
z
x
H
v
x
U U U U U
N N N N
' '
O
r-I r1 ~--I ~-I N
~. ~.
U N N N N v-1
O ' ' '
~N ~ t~ 0.i ~' O
~-I c-I r1 v--1 '-I ~O
,O
cn O O O O
,O ,O
~-I ~-I ~-I S-t
.I-~ ~ +~ .1~
o c-I N c~ V'
z
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
16
[) M ~ ~ N ~' ~ r1 l9
TI c-I TI N ~ ~ N
I I I I I I
cet' cet' N 61 lfl
w m u».n ~ ~ ~ ,.-~-I f
~--i c-f c-I N N ~,'
'-1 .-I ~i r-I
O I I I U 1 I U
x x
M
z \ z \ ü z \ w / \ xz ~ xz ~ xz'~ z
m / \ / \
U U / \
x x x x x x x x
m m m m m m m c.~
U ~ ~ ~ ~ ~ ~ ~
N N N N N N N CV
O
r-I ~I r1 r~ ~-W -I r1 r-I
.r-I .. .. _.
~,
U N N N N N N N CV
O
~, ~ ~i ~ ~i
S-1 c--I r-I r-1 r-I r-I r-I c--I c-1
'O ~ ~ ~ ~ ~. ~ .r
+~
M O O O O O O O
'O 'N 'N '~ 'O 'N 'N
~-I ~-I ~I f-I S-1 ~-I ~-I ~I
,.s~ ..~ ..s~
I~ 00 Ol O - N c~
r-I r-1 r1 r--i
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
17
~r
M
° ~ N N N ~-I
I I ( 1 I
O C~ l0 00 CO
I~ M lfl OD
N N N ~-I
I ~ I t U
x
~ w
U
xz z'z / \ z \ z \
i
/ \ ~ / \ / \ / \
z~
~z
x
U U
x x x x x x
M M M M f~'1
~ ~ ~ ~ ~
N N N N N
O
r-1 v--I r-1 ~-I i--I
.r1 ~. .. .. .. ..
~ N N N N N
O
,O .r
O O O O O
'N
~ ~ +~
° ~' ~ lfl I~ N
-~-n r~ t-I v-I ri r-1
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
18
Les composés de l'invention ont été soumis à une série
d'essais pharmacologiques qui ont mis en évidence leur
intérêt comme substances à activités thérapeutiques.
Etude du transport de la glycine dans les cellules SK-N-MC
exprimant le transporteur humaïn glyt 1 natif.
La capture de [19C]glycine est étudiée dans les cellules
SK-N-MC (cellules neuro-épithéliales humaines) exprïmant le
transporteur humain glytl natif par la~mesure de la
radioactivité incorporée en présence ou en absence du
composé à tester. Les cellules sont cultivées en monocouche
pendant 48 h dans des plaques prétraitées à la fibronectine
à 0,02. Le jour de l'expérience, le milieu de culture est
éliminé et les cellules sont lavées par un tampon Krebs-
HEPES (acide [4-(2-hydroxyéthyl)pipérazine-1-éthanesul-
fonique) à pH 7,4. Après 10 min de préincubation à 37°C en
présence soit de tampon (lot témoin), soit de composé à
tester à différentes concentrations ou de 10 mM de glycine
(détermination de la capture non spécifique), 10 ~,M de
[14C]glycine (activité spécifique 112 mCi/mmole) sont
ensuite ajoutés. L'ïncubation se poursuit pendant 10 min à
37°C, et la réaction est arrêtée par 2 lavages avec un
tampon Krebs-HEPES à pH 7,4. La radio activité incorporée
par les cellules est alors estimée après ajout de 100 ~,l de
scintillant liquide et agitation pendant 1 h. Le comptage
est réalisé sur compteur Microbeta Tri-luxTM. L'efficacité
du composé est déterminée par 1a CISO, concentration du
composé qui diminue de 50% la capture spécifique de
glycine, définie par la différence de radioactivité
incorporée par le lot témoin et le lot qui a reçu la
glycine à 10 mM.
Les composés de l'invention, dans ce test, ont une CI5o de
l'ordre de 0, 01 à 10 ~~I.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
19
Étude du transport de la glycine dans l'homogénat de moelle
épinière de souris.
La capture de [1qC]glycine par le transporteur glyt2 est
étudiée dans l'homogénat de moelle épinière de souris par la
mesure de radioactivité incorporée en p rësence ou en
absence de composé à étudier.
Après euthanasie des animaux (souris mât es OFl Iffa Crédo
pesant 20 à 25 g le jour de l'expérience), la moelle
épinière de chaque animal est rapidement prélevée, pesée et
conservée sur glace. Les échantillons sont homogénéisés
dans un tampon Krebs-HEPES (acide [4-(2- hydroxyéthyl)pipé-
razine-1-éthanesulfonique), pH 7,4, à raison de 25 ml/g de
tissu.
50 ~.l d'homogénat sont pré-incubés pendant 10 min à 25°C en
présence de tampon Krebs-HEPES , pH 7,4 et de composé à
étudier à différentes concentrations, ou de 10 mM de
glycine pour déterminer la capture non spécifique. La
[14C] glycine (activité spécifiqué = 112mCi/mmole) est
ensuite ajoutée pendant 10 min à 25°C à 1a concentration
finale de 10 ~M. La réaction est arrêtée par filtration
sous vide et la radioactivité est estimé e par scintillation
solide par comptage sur un compteur Microbeta Tri-luxTM.
L'efficacité du composé est déterminée pa r la concentration
CISO capable de diminuer de 50~ la capture spécifique de
glycine, définie par la différence de radioactivité
incorporée par le lot témoin et le lot qui a reçu la
glycine 10 mM.
Les composés de l'invention, dans ce tes t, ont une CISO de
l'ordre de 0,01 à 10 ~M.
Ces résultats suggèrent que les composés de l'invention
peuvent être utilisés pour le traitement des troubles
comportementaux associés à la démence, des psychoses, en
particulier de 1a schizophrénie (forme déficitaire et forme
productive) et des symptômes extrapyrami daux aigus ou
chroniques induits par les neuroleptique s, pour le
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
traitement des diverses formes d'anxiété, des attaques de
panique, des phobies, des troubles obsessïonnels
compulsifs, pour le traitement des différentes formes de
dépression, y compris la dépression psychotique, pour le
5 traitement des troubles dus à l'abus ou au sevrage
d'alcool, des troubles du comportement sexuel, des troubles
de la prise de nourriture, et pour le traïtement de la
migraine.
10 De plus, les composés de l'invention peuvent être utilisés
pour le traitement des sont ractures musculaires
douloureuses en rhumatologie et en pathologie rachidienne
aiguë, pour le traitement des contractures spastiques
d'origine mëdullaïre ou cérébrale, pour le traitement
15 symptomatique des douleurs aiguës et subaiguës d'intensité
légère à modérée, pour le traitement des douleurs intenses
et/ou chroniques, des douleurs neurogènes et algies
rebelles, pour le traitement de la maladie de Parkinson et
des symptômes parkinsoniens d'origine neurodégénérative ou
20 induits par des neuroleptiques, pour le traïtement des
épilepsies généralisées primaires et secondaires,
partielles à symptomatologie simple ou complexe, des formes
mixtes et autres syndromes épileptiques en complément d'un
autre traitement antïépileptique, ou en monothérapie, pour
le traitement des apnées du sommeil, et pour la
neuroprotection.
C'est pourquoi la présente invention a également pour objet
des compositions pharmaceutiques contenant une dose
efficace d'au moins un composé selon l'invention, à l'état
de base ou de sel ou de solvat pharmaceutiquement
acceptable, et en mélange, le cas échéant, avec des
excipients convenables.
Lesdits excipients sont choisis selon la forme
pharmaceutique et le mode d'administration souhaité.
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
21
Les compositions pharmaceutiques selon l'invention peuvent
ainsi être destinées à l'administratïon orale, sublinguale,
sous-cutanée, intramusculaire, intraveineuse, topique,
intratrachéale,~ intranasa 1 e, transdermique, rectale,
intraocculaire.
Les formes unitaires d'administration peuvent être, par
exemple, des comprimés, de s gélules, des granules, des
poudres, des solutions ou suspensions orales ou
injectables, des timbres t ransdermiques ("patch"), des
suppositoires. Pour l'administration topique on peut
envisager des pommades, lotions et collyres.
Lesdites formes unitaires sont dosées pour permettre une
administration journalière de 0,01 à 20 mg de principe
actif par kg de poids corporel, selon la forme galénique.
Pour préparer des comprima s on ajoute au principe actif,
micronisé ou nôn, un véhicule pharmaceutique qui peut être
composé de diluants, comme par exemple le lactose, 1a
cellulose microcristalline, l'amidon, et des adjuvants de
formulation comme des liants, (polyvinylpyrrolidone,
hydroxypropylméth~lcellulo se, etc), des agents d'écoulement
comme la silice, des lubr.ifïants comme le stéarate de
magnésium, l'acide stéarique, le tribehenate de glycerol,
le stéarylfumarate de sodium. Des agents mouïllants ou
tensioactifs tels que le 1 aurylsulfate de sodium peuvent
aussi être ajoutés.
Les techniques de réalisai ion peuvent être la compression
directe, la granulation sèche, la granulation humide ou la
fusion à chaud.
Les comprimés peuvent être nus, dragéifiés, par exemple par
du saccharose, ou enrobés avec divers polymères ou autres
matières appropriées. Il peuvent être conçus pour permettre
une libération rapide, retardée ou prolongée du principe
actif grâce à des matrice s polymères ou à des polymères
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
22
spécifiques utilisés dans l'enrobage.
Pour préparer des gélules on mélange le principe actif avec
des véhicules pharmaceutiques secs (simple mélange,
granulation sèche ou humide, ou fusion à chaud), liquides
ou semi-solide s.
Les gélules peuvent être dures ou molles, pelliculées ou
non, de manïèr e à avoir une activité rapide, prolongée ou
retardée (par exemple pour une forme entérique).
Une compositio n sous forme de sirop ou d'élixir ou pour
l'administrati on sous forme de gouttes peut contenir le
principe actif conjointement à un édulcorant, de préférence
acalorique, du méthylparaben ou du propylparaben comme
antiseptique, un agent de sapidité et un colorant.
Les poudres et granules dispersibles dans de l'eau peuvent
contenir le principe actif en mélange avec des agents de
dispersion ou des agents mouillants, ou des agents
dispersants comme la polyvinylpyrrolidone, de même qu'avec
des édulcorant s et des agents correcteurs de goût.
Pour l'adminïs tration rectale, on recourt à des
suppositoires prëparës avec des liants fondant à la
température rectale, par exemple du beurre de cacao ou des
polyéthylènegl ycols.
Pour une administration parentérale, on utilise des
suspensions aqueuses, des solutions salines isotoniques ou
des solutions stériles injectables contenant des agents de
dispersion et/ ou des mouillants pharmacologiquement
compatibles, par exemple le propylèneglycol ou le butylène-
glycol.
Le principe actif peut être formulé également sous forme de
microcapsules, éventuellement avec un ou plusieurs supports
ou additifs, o u bien avec une matrice polymère ou avec une
CA 02542373 2006-04-11
WO 2005/037792 PCT/FR2004/002640
23
cyclodextrine (timbres transdermiques, formes à libération
prolongée ) .
Les compositions topiques selon l'invention comprennent un
milïeu compatible avec la peau. Elles peuvent se présenter
notamment sous forme de solutions aqueuses, alcooliques ou
hydroalcooliques, de gels, d'émulsions eau-dans-huile ou
huile-dans-eau ayant l'aspect d'une crème ou d'un gel, de
microémulsions, d'aérosols, ou encore sous forme de
dispersïons vésiculaires contenant des lipides ioniques
et/ou non ioniques. Ces formes galéniques sont préparées
selon les méthodes usuelles des domaines considérés.
Enfin, les compositions pharmaceutiques selon l'invention
peuvent contenir, à côtë d'un composë de formule générale
(I), d'autres principes actifs qui peuvent être utiles dans
le traitement des troubles et maladies ïndiqués ci-dessus.