Note: Descriptions are shown in the official language in which they were submitted.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
5-SUBSTITUTED 2-(PHENYLMETHYL)THIO-4-PHENYL-4H-1,2,4-TRIA20LE DERIVATIVES AND
RELATED COMPOUNDS AS GABA-AGONISTS FOR THE TREATMENT OF URINARY INCONTINENCE
AND
RELATED DISEASES
DETAILED DESCRIPTION OF INVENTION
TECH1~ICAL FIELD
The present invention relates to a phenyltriazole derivative which is useful
as an active ingredient
of pharmaceutical preparations. The phenyltriazole derivative of the present
invention has y-
aminobutyric acid receptor (GABAB receptor) agonistic activity, and can be
used for the
prophylaxis and treatment of diseases associated with GABAB receptor activity,
in particular for
the treatment of overactive bladder, urinary incontinence such as urge urinary
incontinence, benign
prostatic hyperplasia,spasticity and motor control, ,pain, epilepsy, cognitive
defects, psychiatric
.. disorders, alcohol dependence and withdrawal, feeding behaviour,
cardiovascular, respiratory
disorders and gastrointestinal disorders.
BACKGROUND ART
GABAB receptors are the first example of G protein-coupled receptors where
heteromerization of
t<vo receptor subt~~pes has been.demonstrated to be necessazy for normal-
function (Jones et a/.,
Nature, (1998) 396, 674- 679); Kaupmami et al., Nature, (1998) 396, 683-687;
Kuner et a/.,
Science, (1999) 283, Z4-77). Currently there are two GABAB receptor subtypes
lalown, GABABRl
and R2. In the brain there are two predominant N terminal splice variants
expressed from the
GABAB R1 gene, GABABRIa and Rlb, which heterodimerize with the R2 subunit.
Pharmacologically, the different splice forms of GABABR1 could not be
distinguished (Kaupmann
et al., Nature, (1997) 386;239-246.
GABAB receptors are located throughout the central and peripheral nervous
systems (see Ong and
Kerr, Life Sciences, (1990) 46,1489-1501; Bowery et al., Drug Res. (1992)
42(1), 2a, 215-223),
and are thus involved in the regulation of a wide variet~~ of neurally-
controlled physiological
responses, from memory and learning to muscle contraction. This makes the
GABAB receptor a
target for pharmaceutical agents intended to treat central and peripheral
neural disorders, and
indeed a variety of GABA$ agonists and antagonists are lrnown and have been
proposed for use in
therapy including pain, spasticity and motor conh~ol, epilepsy, cognitive
defects, psychiatric
disorders, alcohol dependence and withdrawal, feeding behaviour,
cardiovascular, respiratory
disorders and gastrointestinal disorders (Bittiger et al., in GABA: Receptors,
Transporters and
Metabolism, Tanalca, C., and Bowery, N.G. (Eds). Birkhauser Verlag
Basel/Switzerland (1996),
297-305; Bittiger et al., Trends Pharmacol. Sci., 14, 391-394,1993; Froestl et
al., J. Med. Chem.,
38, 3297-3312,1995; Froestl et al., Ibid., 3313-3331).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-2-
The GABA$ receptor agonist baclofen given intrathecally is used clinically for
reducing ~ii'ethral
resistance and detrusor overactivity associated with spasticity (Steers et
al.; J. Unol., 148, 1849-
1855,1992; Mertens et al., Acta Neuroclzir., 64, 17-25 1995). The main effect
of baclofen within
the central nervous system is to reduce transmitter release. In the spinal
cord it affects the activity
of motoneurons and interneurons that are important for micturition and
baclofen has previously
been reported to have an inhibitory action on rat micturition after
intrathecal administration (Igawa
et al. .I. Ur~ol., 150, 537-542, 1993; Pehrson et al. J. Urol., 168, 2700-2705
,2002).
Taken together, it is suggested that GABAB system is involved in the
micturition control, both in
animals and human. A potent and selective GABAR agonist can provide
therapeutic benefit in the
treatment of urinary bladder dysfunction as well as other indications
described above.
GABAB agonists are also known to have smooth muscle relaxation action, thus a
potent and
selective GABAB agonist can provide therapeutic benefit in the treatment of
BPH.
Urinary incontinence
UI is the involuntary loss of urine. UUI is one of the most common types of UI
together with stress .
urinary incontinence (SLTI~ which is usually caused by a defect in the
urethral closure mechanism.
UUI is often associated with neurological disorders or diseases causing
neuronal damages such as
dementia, Parkinson's disease, multiple sclerosis, stroke and diabetes,
although it also occurs in
individuals with no such disorders. One of the usual causes of UUI is
overactive bladder (OAB)
which is a medical condition referring to the symptoms of frequency and
urgency derived from
abnormal contractions and instability of the detrusor muscle.
There are several medications for urinary incontinence on the market today
mainly to help treating
UUI. Therapy for OAB is focused on drugs that affect peripheral neural control
mechanisms or
those that act directly on bladder detz-usor smooth muscle contraction, with a
major emphasis on
development of anticholinergic agents. These agents can inhibit the
parasympathetic nerves which
control bladder voiding or can exert a direct spasmolytic effect on the
detrusor muscle of the
bladder. This results in a decrease in intravesicular pressure, an increase in
capacity and a
reduction in the frequency of bladder contraction. Orally active
anticholinergic drugs are the most
commonly prescribed drugs. However, their most serious drawbacks are
unacceptable side effects .
such as dry mouth, abnormal visions, constipation, and central nervous system
disturbances. These
side effects lead to poor compliance. Dry mouth symptoms alone. are
responsible for a 70% non-
compliance rate with oxybutynin. The inadequacies , of present therapies
highlight the need for
novel, efficacious, safe, orally available drugs that have fewer side effects.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_3_
Benin prostatic hyperplasia BPH~
BPH is the benign nodular hyperplasia of the periurethral prostate gland
commonly seen in men
over the age of 50. The overgrowth occurs in the central area of the prostate
called the transition
zone, which wraps around the urethra. BPH causes variable degrees of bladder
outlet obstruction
resulting in progressive lower urinary tract syndromes (LUTS) characterized by
urinary frequency,
urgency, and nocturia due to incomplete emptying and rapid refilling of the
bladder. The actual
cause of BPH is unlrnown but may involve age-related alterations in balance of
steroidal sex
hormones.
The selective al-adrenoceptor antagonists, such as prazosin, indoramin and
tamsulosin are used as
an adjunct in the symptomatic treatment of urinary obstruction caused by BPH,
although they do
not affect on the underlying cause of BPH. In BPH, increased sympathetic tone
exacerbates the
degree of obstruction of the urethra through contraction of prostatic and
urethral smooth muscle.
These compounds inhibit sympathetic activity, thereby relaxing the smooth
muscle of the urinary
tract. Uroselective al-antagonists and al-antagonists with high tissue
selectivity for lower urinary
tract smooth muscle , that do not provoke hypotensive side-effects should be
developed for the
treatment.
Drugs bloclting dihydrotestosterone have been used to reduce the size of the.
prostate. Sa-reductase
inhibitors such as finasteride are prescribed for BPH. These agents
selectively inhibit Sa-reductase
which mediates conversion of testosterone to dihydrotestosterone, thereby
reducing plasma
dihydrotestosterone levels and thus prostate growth. The Sa-reductase
inhibitors do not bind to,
androgen receptors and do not affect testosterone levels nor do they possess
feminizing side-
effects.
Androgen receptor antagonists are used for the treatment of prostatic
hyperplasia due to excessive
action or production of testosterone. Various antiandrogens are under
investigation for BPH
including chlormadione derivatives with no estrogenic activity, orally-active
aromatase inhibitors,
luteinizing hormone-releasing hormone (LHRH) analogues.
WO01/87855 discloses phenyltriazole derivatives represented by the general
formula:
N-N
~N Ra
D
A
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-4-
wherein
A repz:esents optionally substituted aryl, etc;
B and D independently represent optionally substituted aryl, carbocyeles, or 5-
or 6-membered
heterocycles;
Ra .represents H, halgen-substituted alkyl, (un)substituted aryl,
(un)substituted heterocycles,
(un)substituted cycloalkyl, or-[Alkl]m :XP-[Alk2]n-YP-RlP;
wherein
RlP represents H, optionally substituted aryl, etc;
Xr represents direct bond, -O-, -S-, etc;
Y represents direct bond, m and n independently, represent an integer of 0
orl;
Alkl and Alk2 independently represent alkyl, etc,
as an inhibitor of glycine transporter.
Yamada, N. et al. discloses phenyltriazole derivatives represented by the
general formula:
Rb~
Rb3
N
Rb2
wherein
Rb 1 represents H, methyl, or ethyl;
Rb2 represents H, chloro, fluoro, dichloro, methyl, methoxy, or
trifluoromethyl;
Rb3 represents H, chloro, methyl, ethyl, methoxy, ethoxy, fluoro,
trifluoromethoxy, or dichloro,
as a bleaching herbicide. (Bioscience, Biotechnology, and Biochemistry (2002),
66(8), 1671-1676).
However, none of these references discloses phenyltriazole derivatives ha~~ing
GABAB receptor
agonistic activit~r.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-5-
The development of a compound which has effective GABAg agonistic activity and
can be used
for the prophylaxis and treatment of diseases associated with GABA$ receptor
activit~~, in
particular for the treatment of urinary incontinence, urge urinary
incontinence, overactive bladder
as well as pain, such as chronic pain, neuropathic pain, postoperative pain,
rheumatoid arthritic
pain, neuralgia, neuropathies, algesia or nerve injury induced pain,
spasticity and motor control,
epilepsy, cognitive defects, psychiatric disorders, alcohol dependence and
withdrawal, feeding
behaviour, cardiovascular, respiratory disorders and gastrointestinal
disorders has been desired.
SUMMARY OF THE INVENTION
This invention is to provide phenyltriazole derivatives of the formula (I),
their tautomeric and
stereoisomeric form, and salts thereof:
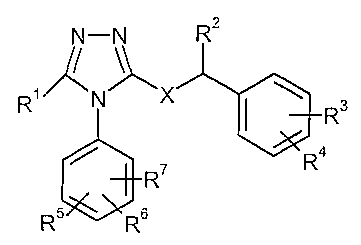
N-N Rz
s
R
R
R5 Rs
wherein
R' represents alkyl optionally substituted by one or tlvo substituents
selected from the group
consisting of alkoxy, amino, alkylamino, di(alkyl)amino, alkanoyloxy, hydroxy,
carboxy,
alkoxycarbonyl, cycloalkylphenyloxy, halogen, morpholino, carbamoyl,
alkylsulfonyl-
amimo, phenyloxy optionally substituted by cycloalkyl, and 3- R membered
saturated ring
optionally having one or two N atom which ring optionally substituted by
hydroxy or
alkanoyl,
or 3-~u membered saturated or unsaturated ring optionally having one or two
hetero atoms
selected from the group consisting of N and 0, and which ring is optionally
substituted by
one or two substituents selected from the group consisting of alkyl, halogen,
alkoxy, nitro,
amino, cyano, alkylamino, di(alkyl)amino, 4-7 membered saturated cyclic amine
optionally
substituted by hydroxy, and rizono-, di-, or tri- halogen substituted allyl;
R' represents -CORZ', -(CH~)~ R'' or tert-butyl,
wherein
Rz' is alkoxy, hydroxy, mono-, di-, or tri- halogen substituted alkyl,
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-6-
or 3-8 membered saturated or unsaturated ring optionally having one o'r two
heteroatoms selected from the group consisting of N, O, and S and which ring
is
optionally substituted by one or two substituents independently selected from
the
group consisting of alkanoyl, halogen, benzyl; alkoxycarbonyl, haloalkyloxy-
.carbonyl, cyano, hydroxy, amino, alkylamino, di(alkyl)amino, cycloalkylamino,
alkoxycarbonyl, sulfamoyl, alkylaminosulfonyl, di(alkyl)aminosulfonyl,
alkanoyl;
alkanoylamino, carbamoyl, alkylcarbamoyl, di-(allyl)carbamoyl, alkylsulfonyl,
alkyl optionally substituted by alkoxycarbonyl or mono-, di-, or tri-halogen,
alkoxy
optionally substituted by mono-, di-, or tri- halogen, and alkylthio
optionally
substituted by mono-, di-, or tri- halogen;
nis0orl;
R3 and R~ independently represent hydrogen, halogen, cyano, hydroxy, amino,
alkylamino,
di(alkyl)amino, cycloalkylamino, carboxy, alkoxycarbonyl, sulfamoyl,
alkylaminosulfonyl,
di(alkyl)aminosulfonyl; alkanoyl, alkanc~ylamino, carbamoyl, alkylcarbamoyl,
di-
(alkyl)carbamoyl, alkylsulfonyl, alkyl optionally substituted by hydroxy,
alkoxycarbonyl
or mono-, di-, or tri-halogen, alkoxy optionally substituted by mono-, di-, or
tri- halogen, or
alkylthio optionally substituted by mono-, di-, or tri- halogen;
RS represents hydxogen, hydroxy, vitro, cyano, halogen, sulfamoyl,
allrylsulfonyl, alkyl-
aminosulfonyl, di(allryl)aminosulfonyl, -(CHz)",-CO-Rs°, -(CHz)m Rs', -
NRszRs3, or -ORsa,
wherein
m is 0, 1, 2, or 3
Rs° is hydroxy, hydrogen, alkoxy, moipholino, di(phenyl)methyloxy,
di(halogen
substituted phenyl)methyloxy, -NRs°'Rsoz (,herein said Rs°' and
Rsoz
independently represent hydrogen, alkoxyalkyl, alkyl, hydroxyalkyl,
alkoxycarbonylalkyl, or carboxyalkyl or
Rs°' and Rs°z together form with the adjuscent N atom,
morpholino, piperazino optionally
substituted by oxo, or 4-7 membered saturated cyclic amino optionally
substituted
by one substituent selected from the group consisting of carboxy,
hydroxyalkyl,
hydroxy, and carbamoyl) or all'yl optionally substituted by halogen,
Rs' is hydrogen, hydroxy,. or -NRs"Rs'z (wherein said Rs" and Rs'z
independently
represent hydrogen, alkoxyalkyl, alkyl, hydroxyalkyl, alkox)~carbonylallyl, or
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ '7 _
carboxyalkyl, or RS" and RS'Z together forni with the adjuscent N atom, 4-7
membered ' saturated cyclic amino optionally substituted by one substituent
selected from the group consisting of carboxy, hydroxyalkyl, hydroxy, and
carbamoyl),
R52 and R53 independently represent hydrogen, alkyl, hydroxy,
cycloalkylcarbonyl,
hydroxyalkyl, alkylsulfonyl, hydroxyalkylcarbonyl, carboxyalkylcarbonyl,
alkanoyloxyalkylcarbonyl, or alkoxycarbonylalkylcarbonyl, or RSZ and R53
together
form with adjuscent N atom, morpholino, cyclic amino optionally substituted by
one
substituent selected from the group consisting of carboxy, hydroxyalkyl,
hydroxy,
and carbamoyl,
R54 represents alkyl optionally substituted by moipholino, amino,
di(alkyl)amino, carboxy,
alkoxycarbonyl, or mono-, di-, or tri- halogen, or piperazino substituted by
carboxy;
R6 and R' independently represents hydrogen, morpholino,
hydroxypyrrolidinylcarbonyl,
hydroxyalkylaminocarbonyl, cyano, hydroxy, hydroxyalkyl, hydroxyamino,
carboxy,
fluoro, chloro, bromo, nitro, amino, alkylamino, di(alkyl)amino,
cycloalkylanuno,
alkoxycarbonyyl, sulfamoyl, alkylaminosulfonyl, di(alkyl)aminosulfonyl,
alkanoyl,
alkanoylamino, carbamoyl, diphenylmethyloxycarbonyl, alkylcarbamoyl, di-
(alkyl)carb-
amoyl, alkylsulfonyl, alkyl optionally substituted by alkoxyalkyl(alkyl)amino,
di(alkyl)anuno, alkoxycarbonyl, carboxy, or mono-, di-, or tri-halogen, alkoxy
optionally
substituted by morpholino, di(alkyl)amino, or mono-, di-, or tri- halogen, or
Cl_6 alkylthio
optionally substituted by mono-, di-, or tri- halogen
or R6 and R' together form phenyl fused to adjacent phenyl; and
X represents GR'°R", NR'2, S, O, SO~, or SO
wherein~R'°, R", and R'' independently represent hydrogen or methyl.
. The phenyltriazole derivatives of formula (1), their tautomeric and
stereoisomeric form, and salts
thereof surprisingly show excellent GABA$ agonistic acti~nty. They are,
therefore, suitable
especially for the prophylaxis and treatment of diseases associated with GABAB
receptor activity,
in particular for the treatment of urinary incontinence, urge urinary
incontinence and/or overactive
bladder.
The compounds of the present invention are also effective for treating or
preventing a disease
selected from the group consisting of pain, such as chronic pain, neuropathic
pain, postoperative
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_8_
pain, rheumatoid artlwitic pain, neuralgia, neuropathies, algesia, or nerve
injury inducefi' pain,
spasticity and motor control, epilepsy, cognitive defects, psychiatric
disorders, alcohol dependence
and withdrawal, feeding behaviour, cardiovascular, respiratory disorders and
gastrointestinal
disorders since the diseases also relate to GA.BAB receptor activity.
In another embodiment, the phenyltriazole derivatives of formula (~ are those
wherein;
wherein
R' represents all'yl optionally substituted by one or two substituents
selected from the group
consisting of alkoxy, amino, alk~~lamino, di(alkyl)amino, alkanoyloxy,
hydroxy, carboxy,
alkoxycarbonyl, cycloalkylphenyloxy, halogen, morpholino, carbamoyl, phenyloxy
optionally substituted by cycloalkyl, and 3- 8 membered saturated ring
optionally having
one or two N atom which ring optionally substituted by hydroxy or alkanoyl,
or 3-8 membered saturated or unsaturated ring optionally having one or two
hetero atoms
selected from the group consisting of N and 0, and which ring is optionally
substituted by
one or tlvo substituents selected from the group consisting of alkyl, halogen,
alkoxy, nitro,
amino, cyano, alkylamino, di(allyl)amino, 4-7 membered saturated cyclic amine
optionally
substituted by hydroxy, and mono-, di-, or tri- halogen substituted allyl;
R'' represents -COR''' or -(CHz)"-R'',
wherein
R'' is alkoxy, hydroxy, mono-, di-, or tri- halogen substituted alkyl,
or 3-8 membered saturated or unsaturated ring optionally having one or hvo
heteroatoms selected from the group consisting of N, 0, and S and which ring
is
optionally substituted by one or t<vo substituents independently selected from
the
group consisting of alkanoyl, halogen, benzyl, alkoxycarbonyl, haloalkyloxy-
carbonyl, cyano, hydroxy, ari~ino, alkylamino, di(allyl)amino,
cycloalkylamino,
alkoxycarbonyl, sulfamoyl, alkylaminosulfon~rl, di(all'yl)aminosulfonyl,
alkanoyl,
alkanoylamino, carbamoyl, alkylcarbamoyl, di-(alkyl)carbamoyl, alkylsulfonyl,
alkyl optionally substituted by alkoxycarbonyl or mono-, di-, or tl~i-halogen,
alkoxy
optionally substituted by mono-, di-, or tri- halogen, and alkylthio
optionally
substituted by mono-, di-, or tri- halogen;
nis0orl;
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-9-
R' and R4 independently represent hydrogen, halogen, cyano, hydroxy, amino,
alkyrarnino,
di(allcyl)amino, cycloalkylamino, alkoxycarbonyl, sulfamoyl,
allcylaminosulfonyl,
di(alkyl)aminosulfonyl, alkanoyl, alkanoylamino, carbamoyl, alkylcarbamoyl, di-
(alkyl)carbamoyl, alkylsulfonyl, alkyl optionally substituted by
alkoxycarbonyl or mono-,
di-, or tri-halogen, alkoxy optionally substituted by mono-, di-, or tri-
halogen, or alkylthio
optionally substituted by mono-, di-, or tri- halogen;
Rs represents hydrogen, hydroxy, nitro, cyano, halogen, sulfamoyl,
alkylsulfonyl, alkyl-
aminosulforlyl, di(alkyl)aminosulfonyl, -(CH~)m CO-Rs°, -(CHz)m Rs', -
NR52R53~ or -ORs4,
wherein
mis0, 1,2,or3
Rs° is hydroxy, hydrogen, alkoxy, morpholino, di(phenyl)methyloxy,
di(halogen
substituted phenyl)methyloxy, -NRs°'Rs°z (wherein said
Rs°' and Rsoz
independently represent hydrogen, alkoxyalleyl, alkyl, hydroxyalkyl,
alkoxycarbonylalkyl, or carboxyalkyl or
Rs°' and Rsoz together form with the adjuscent N atom, rnorpholino, or
4-7 membered
saturated cyclic anuno optionally substituted by one substituent selected from
the
group consisting of carboxy, hydroxyalkyl, hydroxy, and carbamoyl) or alkyl
optionally substituted by halogen,
Rs' is hydrogen, hydroxy, or -NRs"Rs'z (wherein said Rs" and Rs'z
independently
represent hydrogen, alkoxyalkyl, alkyl, hydroxyalkyl, alkoxycarbonylalkyl, or
carboxyalkyl, ox Rs" and Rs'z together form with the adjuscent N atom, 4-7
membered saturated cyclic amino optionally substituted by one substituent
selected from the group consisting of carboxy, hydroxyalkyl, hydroxy, and
carbamoyl),
Rsz and Rs3 independently represent hydrogen, alkyl, hydroxy,
cycloalkylcarbonyl, or
hydroxyalkyl, or Rsz and Rs3 together form with adjuscent N atom, morpholino,
cyclic anuno optionally substiW ted by one substituent selected from the group
consisting of carboxy, hydroxyalkyl, hydroxy, and carbamoyl,
Rs~ represents alkyl optionally substituted by morpholino, amino, di(alkyl)
anuno, or
mono-, di-, or tri- halogen;
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-10-
R6 and R' independently represents hydrogen, morpholino,
hydroxypyrrolidinylcafhonyl,
hydroxyalkylaminoearbonyl, cyano, hydroxy, hydroxyalkyl, hydroxyamino,
carboxy,
fluoro, chloro, bromo, vitro, amino, alhylamino, di(alkyl)amino,
cycloalkylanuno,
alkoxycarbonyl, .sulfamoyl, alhylaminosulfonyl, di(alkyl)aminosulfonyl,
alkanoyl,
alkanoylamino, carbamoyl, diphenylmethyloxycarbonyl, alhylcarbamoyl, di-
(alkyl)carb-
amoyl, alkylsulfonyl, al)'yl optionally substituted by
alkoxyalkyl(alkyl)amino,
di(alkyl)amino, alkoxycarbonyl, carboxy, or mono-, di-, or tri-halogen, alkoxy
optionally
substituted by morpholino, di(alkyl)amino, or mono-, di-, or tri- halogen, or
C,_6 allc~~lthio
optionally substituted by mono-, di-, or tri- halogen
or R6 and R' together form phenyl fused to adj acent phenyl; and
X represents CR'°R", NR'2, S, O, SO~, or SO
wherein R'°, R", and R'z independently represent hydrogen or methyl.
Yet another embodiment of formula ()7 can be those wherein:
X represents CHI, NH, S, O, SOz, or SO;
R' represents C3 to C$ cycloalkyl,
C~-Cb alkyl optionally substituted by one or W o substituents selected from
the group
consisting of CI-C6 alkoxy, amino, C~-C6 alkylamino, di(C~-C6 alkyl)amino, C~-
C6
alkanoyloxy, hydroxy, C3-C8 cycloall'yl, carboxy, C~-C6 alkoxycarbonyl, C3-C$
cycloalkylphenyloxy, halogen, morpholino, and pyrrolidinyl,
pyridyl, pyrrolidinyl, piperidinyl optionally substituted by methyl, or
phenyl optionally substituted by one selected from the group consisting of
halogen, C~-C6
alkoxy, .vitro, amino, cyano, C~-Cbalkylanuno, di(C~-Cbalkyl)amino, and mono-,
di- or tri-
halogen substituted C,-Ctiall'yl,
R' represents -COR'' or -(CH~)"-R'', wherein RZ' represents mono-, di-, tri-
halogen sub-
stituted C,-C6 all..yl, morpholino, C,-C6 alkoxy, hydroxy, C3 to C8
cycloalkyl, pyridyl,
furanyl, thiophenyl, pyrrolidinyl, piperidinyl optionally substituted by one
substituent
selected from the group consisting of benzyl, C~-Cn alkoxycarbonyl, and halo
CI-C6
alkyloxycarbonyl, or phenyl optionally substituted by one substituent selected
fi~om the
group consisting of C~-Cb alkyl, halogen, C~-C6 alkoxy, and mono-, di-, or tri-
halogen
substituted C~-C6alkyl;
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-11-
nis0orl;
R3 and R'' independently represent hydrogen, halogen, cyano, hydroxy, amino,
C1_6 alkylamino,
di(C~_6 alkyl)amino, C3_$ cycloalkylamino, Cl_6 alkoxycarbonyl, sulfamoyl,
Cl_ti all'yl-
aminosulfonyl, di(C~_6 alkyl)aminosulfonyl, C~_6 alkanoyl, C1_b alkanoylamino,
carbamoyl,
C,_b alkylcarbamoyl, di-(C~_6 alkyl)carbamoyl, C~_6 alkylsulfonyl, C~_6 alkyl
optionally
substituted by C,_6 alkoxycarbonyl or mono-, di-, or tri-halogen, C~_6 alkoxy
optionally
substituted by mono-, di-, or tri- halogen, or C,_6 alkylthio optionally
substituted by mono-,
di-, or tri- halogen;
Rs ' represents hydrogen, nitro, cyano, hydroxy, halogen, sulfamoyl, C1-
C6alkylsulfonyl, CI
C6alhylaminosulfonyl, di(C~-C6a11'yl)aminosulfonyl, -(CHz)m-CO-Rs°, -
(CHz)m Rs',
-~szRsa or -ORsa,
wherein m is 0, 1, 2, or 3
Rs° is hydraxy, hydrogen, C~-Cbalkoxy, morpholino, diphenylmethyloxy, -
NRs°'Rs°z
(wherein said Rs°' and Rs°z independently represent hydrogen, CI-
C6alkoxyalkyl,
C~-C6alkyl, hydroxy C~-Cballyl, C~-C6alkoxycarbonyl C~-C6allyl, or carboxy C~-
Cbalkyl or Rs°' and Rs°'' together form with the adjacent N atom
morpholino, 4-6
membered saturated cyclic amino optionally substituted by one substituent
selected from the group consisting of carboxy, hydroxyalkyl, hydroxy, and
carbamoyl) or Cl-C6 alkyl optionally substituted by halogen,
Rs' is hydrogen, hydroxy, or -NRsi'Rsiz (,herein said Rs" and Rs'''
independently
represent hydrogen, C~-C6 alkoxyalk~~l, C~-C6 alkyl, hydroxyalkyl, C~-C6
alkoxy-
carbonylalkyl, or carboxyalkyl or Rs" and Rs'z together form with the adjacent
N
atom, 4-7 membered saturated cyclic anuno optionally substituted by one
substituent selected from the group consisting of carboxy, hydroxyalkyl,
hydroxy,
and carbamoyl)
Rsz and Rs3 independently represent hydrogen, C~-C6 allyl, hydroxy, C3-
CSCycloalkyl-
carbonyl, or hydroxy C,-C6 all.~~l or Rsz and Rs3 together forni with adjacent
N
atom, rnorpholino, 4-7 membered saturated cyclic amino optionally substituted
by
one substituent selected from the group consisting of carboxy, hydroxyalkyl,
hydroxy, and carbamoyl
Rs4 represents alkyl optionally substituted by morpholino, amino, or di(alkyl)
amino,
or mono-, di-, or tri- halogen; and
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-12-
R6 and R' independently represent hydrogen, morpholino, .
hydroxypyrrolidinylca~ljonyl,
hydroxyC~-C6alkylaminocarbonyl, cyano, hydroxy, hydroxyC,-G6alkyl,
hydroxyamino,
carboxy, fluoro, chloro, bromo, nitro, anuno, C,_~ alkylamino, di(C1_6
alkyl)anuno, C3_g
cycloalkylamino, C1_6 alkoxycarbonyl, sulfamoyl, C~_6 alkylaminosulfonyl,
di(C~_6
alkyl)anunosulfonyl, C,_6 alkanoyl, C~_a alkanoylamino, carbamoyl,
diphenylmeth
yloxycarbonyl, C,_6 alkylcarbamoyl, di-(C~_6 allcyl)carbamoyl, C~_a
alkylsulfonyl, C,_6 alkyl
optionally substituted by alkoxyall'yl(alkyl)amino, di(alkyl)amino, C1_E
alkoxycarbonyl,
carboxy, or mono-, di-, or tri-halogen, C1._6 alkoxy optionally substituted by
morpholino,
di(alkyl)amino, or mono-, di-, or tri- halogen, or C~_ti alkylthio optionally
substituted by
mono-, di-, or tri- halogen
or R6 and R' together form phenyl fused to adjacent phenyl.
Yet another embodiment of formula (1~ can be those wherein:
X represents CHz, NH, S, or SO;
R' represents cyclopropyl, pyridyl,
phenyl optionally substituted by halogen, C1-Cbalkoxy, nitro, amino, cyano, C~-
Ctiallylamino, di(Cl-C6alkyl)amino, or halogen substituted C1-Coalkyl,
C,-C6 alkyl optionally substituted by one or two substituents selected from
the group con
sisting of C~-G6alkoxy, amino, C~-C6 alhylamino, di(C~-C6 alkyl)amino, C~-Cb
alkanoyloxy,
hydroxy, C3-C$ cycloallyl, carboxy, C~-C6 alkoxycarbonyl, C3-Cs
cycloalhylphenyloxy,
halogen, morpholino, and pywolidinyl,
pyrrolidinyl, or piperidinyl optionally substituted by methyl;
R' represents -COR''' or -(CHI)"-Rz', wherein R'' represents mono-, di- or tri-
halogen substi-
tuted alkyl, morpholino, C,-Ctialkoxy, hydroxy, C3 to C$ cycloalkyl, pyridyl,
furanyl,
thiophenyl, pyrrolidinyl, piperidinyl optionally substituted by one selected
from the group
consisting from benzyl, C,-C6alkoxycarbonyl, and haloC,-Ctialkyloxycarbonyl,
or phenyl
optionally substituted by one selected from the group consisting of C,-C6
allyl, halogen,
C~-Cd alkoxy, and mono-, di- or tri- halogen substituted C~-C6alhyl;
nis0orl;
R3 and R4 independently represent hydrogen, halogen, methyl, or amino;
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-13-
RS represents hydrogen, morpholino, hydroxypyrrolidinylcarbonyl,
hydroxyalkylaminocaflionyl,
cyano, hydroxy, hydroxyall'yl, hydroxyamino, carboxy, fluoro, chloro, bromo,
vitro,
amino, C~_6 alkylamino, di(Cl_6 all'yl)amino, C3_$ cycloalkylamino, CI_6
alkoxycarbonyl,
sulfamoyl, .Cl_6 alkylaminosulfonyl, di(C~_~ alkyl)aminosulfonyl, C1_6
alkanoyl, Cl_6
alkanoylamino, carbamoyl, diphenylmethyloxycarbonyl, C~_6 alkylcarbamoyl, di-
(C,_6
all'yl)carbamoyl, C1_6 alkylsulfonyl, Gl_6 alkyl optionally substituted by
alkoxy-
alkyl(allyl)ami.no, di(allyl)amino, C~_6 alkoxycarbonyl, carboxy, or mono-, di-
, or tri-
halogen, C~_6 alkoxy optionally substituted by morpholino, di(alkyl)amino, or
substituted
by mono-, di-, or tri- halogen, or C~_6 alkylthio optionally substituted by
mono-, di-, or tri-
halogen; and
R6 and R' represent hydrogen,
or R° and R' together fornl phenyl fused to adj acent phenyl.
Yet another embodiment of fornmla (I) can be those wherein:
X represents CHz, NH, or S;
R' represents cyclopropyl, pyridyl, phenyl optionally substituted by halogen,
alkoxy, vitro,
amino, cyano, alkylamino, di(alkyl)amino, or halogen substituted allyl,
C,-C6 all'yl optionally substituted by one or t~~~o substituents selected from
the group
consisting of alkoxy, amino, C~-C6 alkylamino, di(C~-C6 alkyl)amino, C,-C6
alkanoyloxy,
hydroxy, C3-C8 cycloalkyl, carboxy, Cl-C6 alkoxycarbonyl, C~-CR
cycloalkylphenyloxy,
halogen, moipholino, and pyrrolidinyl,
pyrrolidiny, or piperidinyl optionally substituted by methyl.
Further, another embodiment of formula (~ can be those wherein:
X represents CHI, NH, or S;
RZ represents -COR''', -(CHZ)"R''', wherein RZ' is phenyl optionally
substituted by C~-Cb alkyl,
halogen, halogen substituted alkyl or alkoxy and n is 0 or 1.
Additional embodiment of formula (n can be those wherein:
X represents CH2, NH, or S;
R3 and R4 independently represent hydrogen, halogen, methyl, amino; and
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-14-
RS represents hydrogen, morpholino, hydroxypyrrolidinylcarbonyl,
hydroxyalkyfamino-
carbonyl, cyano, .hydroxy, hydroxyalkyl, hydroxyamino, carboxy, fluoro,
chloro, bromo,
vitro, amino, C,_6.alkylamino, di(C,_b alkyl)amino, C3_8 cycloalkylainino,
C,_ti alkoxy-
carbonyl, sulfamoyl, C,_6 alkylaminosulfonyl, di(Cl_6 alkyl)aminosulfonyl,
Cl_6 alkanoyl,
S C,_b alkanoylamino, carbamoyl, diphenylmethyloxycarbonyl, C~_ti
alkylcarbamoyl, di-(C~_6
alkyl)carbamoyl, C,_6 alkylsulfonyl, C1_d alkyl optionally substituted by
alkoxy-
alkyl(alkyl)amino, di(all'yl)amino, C~_6 alkoxycarbonyl, carboxy, or mono-, di-
, or tri-
halogen, C~_6 alkoxy optionally substituted by morpholino, di(alkyl)amino, or
substituted
by mono-, di-, or tri- halogen, or C~_6 alkylthio optionally substituted by
mono-, di-, or tri-
halogen; and
R° and R' represents hydrogen.
More preferably, said phenyltriazole derivative of the formula (I) is selected
from the group
consisting of:
(4-{3-cyclopropyl-5-[(diphenyhnethyl)thio)-4H-1,2,4-triazol-4-yl}
phenyl)dimethylamine;
( .4-{3-[(diphenylmethyl)thioJ-5-ethyl-4H-1,2,4-triazol-4-
yl}phenyl)dimethylanune;
(4-{3-[(diphenylmethyl)thio]-5-propyl-4H-1,2,4-triazol-4-
yl}phenyl)dimethylamine;
[4-(3-cyclopropyl-5-{[(2-methylphenyl)(phenyl)methyl]thio} -4H-1,2,4-triazol-4-
yl)phenyl]dimeth-
ylamine;
[4-(3- { [bis(4-chlorophenyl)methyl]thin} -5-cyclopropyl-4H-1,2,4-triazol-4-
yl)phenyl]dimethyl-
30 amine;
[4-(3-cyclopropyl-5-{[(4-methylphenyl)(phenyl)methyl]thio}-4H-1,2,4-triazol-4-
yl)phenyl]dimeth-
ylamine;
[4-(3-{[bis(4-fluorophenyl)methyl]thio}-S-cyclopropyl-4H-1,2,4-triazol-4-
yl)phenyl]dimethyl-
amore;
[4-(3-{[(.4-chlorophenyl)(phenyl)methyl]thio~-5-cyclopropyl-4H-1,2,4-triazol-4-
yl)phenylJdimeth-
ylamine;
(4-{3-cyclobutyl-5-[(diphenylmethyl)thio]-4H-1,2,4-triazol-4-
yl}phenyl)dimethylamine;
(4-{3-buyl-5-[(diphenylmethyl)thin]-4H-1,2,4-ti-iazol-4-yl}
phenyl)dimethylamine;
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-15-
[4-(3- f [bis(4-methylphenyl)methyl]thio}-5-cyclopropyl-4H-1,2,4-triazol-4-
yl)phenyl]dimetH-
ylamine;
{4-[3-cyclopropyl-5-( {phenyl[4-(trifluoromethyl)phenyl]methyl} thio)-4H-1,2,4-
triazol-4-y1]phen-
yl} dimethylamine;
[4-(3-{[(4-chlorophenyl)(cycloheayl)methyl]thio}-5-cyclopropyl-4H-.1,2,4-
triazol-4-yl)phenyl]di-
methylamine;
3-[(diphenylmethyl)thio]-5-ethyl-4-(4-isopropylphenyl)-4H-1,2,4-triazole;
{4-[3-{[bis(4-chlorophenyl)methyl]thio}-5-(3-fluorophenyl)-4H-1,2,4-triazol-4-
yl]phenyl} dimeth-
ylamine;
[4-(3-{[bis(4-chlorophenyl)methyl]thio}-5-propyl-4H-1,2,4-triazol-4-
yl)phenyl]dimethylamine;
3-(3-{[bis(4-chlorophenyl)methyl]thio}-5-propyl-4H-1,2,4-triazol-4-yl)benzoic
acid;
3-{5-{[bis(4-chlorophenyl)methyl]thio}-4-[4-(dimethylamino)phenyl]-4H-1,2,4-
triazol-3-
yl}propan-1-ol;
3-[3-{[bis(4-chlorophenyl)methyl]thin}-5-(3-fluorophenyl)- 4H-1,2,4-triazol-4-
yl)benzoic acid;
3-[3-{[bis(4-chlorophenyl)methyl]thio}-5-(3-fluorophenyl)- 4H-1,2;4-triazol-4-
yl]phenol;
3-(3-{[bis(4-chlorophenyl)methyl]thio}-5-propyl-4H-1,2,3-triazol-4-yl)benzoic
acid;
3-(3-{[bis(4-chlorophenyl)methyl]thio}-5-cyclopropyl-4H-1,2,4-triazol-4-
yl)benzoic acid;
5-[3-{[bis(4-chlorophenyl)methyl]thio}-5-(3-fluorophenyl)-4H-1,2,4-triazol-4-
yl]-2-(dimethyl-
amino)benzoic acid;
1-[4-(3-{[bis(4-chlorophenyl)methyl]thio}-5-propyl-4H-1,2,4-triazol-4-
yl)phenyl]-piperidine-3-
carboxylic acid; and
1-{4-[3- { [bis(4-chlorophenyl)methyl]thio } -5-(3-fluorophenyl)-4H-1,2,4-
triazol-4-yl]-phenyl } -
piperidine-3-carboxylic acid
or the salt thereof.
Further, the present invention pro~~ides a medicament, tvhich includes one of
the compounds,
described above and optionally pharmaceutically acceptable e.xcipients.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-16-
Alkyl per se and "alk" and "allyl" in alkenyl, alkynyl, alkoxy, alkanoyl,
alhylamino, allylamino
carbonyl, alkylanunosulphonyl, alkylsulphonylamino, alkoxycarbonyl and
alkoxycarbonylamino
represent a linear, branched alkyl radical having generally 1 to 6, preferably
1 to 4 and particularly
preferably 1 to 3 carbon atoms, representing illustratively and preferably
methyl, ethyl, n-propyl,
S isopropyl, tert-butyl, n-pentyl and n-hexyl.
"Alk" in alkanoylamino represent a linear, branched and cyclo alkyl radical
having generally 1 to
6, preferably 1 to 4 and particularly preferably 1 to 3 carbon atoms,
representing illustratively and
preferably methyl, ethyl, n-propyl, isopropyl, text-butyl, n-pentyl, n-hexyl,
and cyclopropyl.
Alkoxy illustratively and preferably represents methoxy, ethoxy, n-propoxy,
isopropoxy, tert-
butoxy, n-pentoxy and n-hexoxy.
Alkylamino illustratively and preferably represents an alkylanuno radical
having one or two
(independently selected) alkyl substituents, illustratively and preferably
representing methylamino,
ethylamino, n-propylamino, isopropylamino, tort-butylannino, n-pentylamino, n-
hexyl-amino, N,N-
dimethylamino, N,N-diethylamino, N-ethyl-N-methylamino, N-methyl-N-n-
propylamino, N-
isopropyl-N-n-pxopylamino, N-t-buyl-N-methylamino, N-ethyl-N-n-pentylamino and
N-n-hexyl-
N-methylamino.
4-7 membered .saturated Cyclic amine, illustratively and preferably represent
pyrrolidine,
piperidine, azepane, and azetidine.
Heterocycle and/or heterocyclic as used herein, designate a closed ring
structure, in which one or
more of the atoms in the ring is a heteroatom such as sulfur, nitrogen,
oxygen, and the like.
Suitable examples include, without limitation, pyrrolidinyl, piperidino,
piperazinyl, homo-
piperidino, .morpholinyl, thiomorpholinyl, tetrahydrofuryl, furyl, thienyl,
pyrrolyl, imidazolyl,
pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, h-iazolyl,
oxadiazolyl, pyridyl, pyrazinyl,
pyrimidyl, pyridazinyl and the like.
EMBODIMENT OF THE INVENTION
The compound of the formula (I) .of the present invention can be, but not
limited to be, prepared by
combining various known methods. In some embodiments, one or more of the
substituents, such
as amino group, carboxyl group, and hydroxyl group of the compounds used as
starting materials
or intermediates are advantageously protected by a protecting group known to
those skilled in the
art. Examples of the protecting groups are described in "Protective Groups in
Organic Synthesis
(3rd Edition)" by Greene and Wuts, John Wiley and Sons, New'~'ork 1999.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-17-
The compound of the formula (I-a) of the present invention can be, but not
limited to be, prepared
by the Method [A] below.
[Method A]
N~N Rz
H R~ ~ ~X~ W Ra
R N h Rz N
~ R~ 1 \ R3 ~ / R7 Ra
L
-r Ra _I
R5 Rs Rs Rs
(I-a)
(II) (III)
S The compound of the formula (I-a) (wherein R', Rz, R3, Ra, RS, R6 and R' are
the same as defined
above and X' represents O, S or NR''') can be prepared by reacting the
compound of the formula
(II) (wherein R', R5, R6 and R' are the same as defined above) with the
compound of the formula
(~ (wherein Rz, R3 and Ra are the same as defined above and L, represents a
leaving group
including, for instance, halogen atom such as chlorine, bromine, or iodine
atom; Cti_lo
arylsulfonyloxy group such as benzenesulfonyloxy, or p-toluenesulfonyloxy; and
C~-a
alkylsulfonyloxy group such as methanesulfonyloxy, and the like.)
The reaction may be carried out in a solvent including, for instance,
halogenated hydrocarbons
such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as
diethyl ether, iso-
propyl ether, dioxane .and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aromatic hydrocarbons
such as benzene, toluene and xylene; nitriles such as acetonitrile; amides
such as N, N-dimethyl-
formamide .(DMF), N, N-dimethylacetamide (DMAC) and N-methylpyrrolidone (NMP);
urea such
as 1,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide
(DMSO); and
others. Optionally, two or more of the solvents selected from the listed above
can be mixed and
used.
The reaction temperature can be optionally set depending on the compounds to
be reacted. The
reaction temperature is usually, but not limited to, about 20°C to 50
°C. The reaction may be
conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
The reaction can be advantageously carried out in the presence of a base
including, for instance,
organic amines such as pyridine, triethylamine and N,N-diisopropylethylamine,
dimethylaniline,
diethylaniline, or 4-dimethylaminopyridine, and inorganic base such as sodium
hydride, potassium
hydride, sodium hydroxide, potassium hydroxide, potassium carbonate, sodium
carbonate, sodi~.un
bicarbonate, or potassium bicarbonate, and others.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-18-
R5, R6, and/or R' of compound of the formula (n can be further modified using
conventional
methods.
X' is further modified to be converted to SO or SO2.
Preparation of intermediate (lQ-a)
[Method (i)J
s
N-N
N-NHa NCS N-H~NH
R N SH
R~~p + R~ ~ R ~ ~ R~ Step i-2
5 Rs Step i-1 ~ R~
R s Rs
R
(VI) R5 Rs
(IV)
(V)
( I I-a )
The compound of the formula (II-a) (wherein R', R5, R6 and R' are the same as
defined above) can
be prepared by the following procedures in two steps.
In Step i-1, the compound of the formula (VI) (wherein R', R5, R6 and R' are
the same as defined
above) can be prepared by reacting the compound of the formula (IV) (wherein
R' is the same as
defined above) with the compound of the formula (V) (wherein R5, R6 and R' are
the same as
defined above).
The reaction may be carried out in a solvent including, for instance,
halogenated hydrocarbons
such as dichlorome.thane, chloroform and 1,2-dichloroethane; ethers such. as
diethyl ether, iso-
propyl ether, dioxane and tetrahydrofuran (TI-iF) and 1,2-dimethoxyethane;
aromatic hydrocarbons
such as benzene, toluene and xylene; nitriles such as acetonitrile; amides
such as N, N-
dimethylformamide (DMF), N, N-dimethylacetanude (DMAG) and N-methylpyrrolidone
(NI\~IP);
urea such as 1,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as
dimethylsulfoxide
(DMSO); and others. Optionally, two or more of the solvents selected from the
listed above can be
mixed and used.
The reaction temperature can be optionally set depending on the compounds to
be reacted. The
reaction temperature is usually, but not linuted to, about room temperature to
100°C. The reaction
may be conducted for, usually, 30 minutes to 4S hours and preferably 1 to 24
hours.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-19-
In Step i-2, the compound of the formula (lI-a) (wherein R', R5, R6 and R' are
the same as Refined
above) can be prepared by cyclization reaction of the compound of the formula
(VI) (wherein R',
R5, R° and R' are the same as defined above).
The reaction can be advantageously carried out in the presence of a base
including, for instance,
organic amines such as pyridine, triethylamine and N,N-diisopropylethylamine,
dimethylaniline,
die.thylaniline, or 4-dimethylaminopyridine, and inorganic base such as sodium
hydride, potassium
hydride, sodium hydroxide, potassium hydroxide, potassium carbonate, sodium
carbonate, sodium
bicarbonate, or potassium bicarbonate, and others.
The. reaction can be carried out in a solvent including, for instance,
alcohols such as methanol,
ethanol, 1-propanol, isopropanol and tert-butanol, water and others.
Optionally, two or more of the
solvents selected from the listed above can be mixed and used.
The reaction temperature is usually, but not limited to, about 0°C to
200°C and preferably about
20°C to 100°C. The reaction may be conducted for, usually, 30
minutes to 4S hours and preferably
2 hours to 24 hours.
The compound of the formula (V) is conunercially available or can be. prepared
by the use of
known techniques.
Preparation of intermediate (1~
[Method (ii)]
O
HZN-NHS ~ NHS
R'~N
Rv L2 ~ H
Step ii-1a (IV)
(VII)
Step ii-1b Step ii-2b
O
HzN~N~Ls R~~N~N~L
H a
(VIII) (IX)
In step ii-1a, the compound of the formula (IV) (wherein R' is the same as
defined above) can be
prepared by reacting the compound of the formula (VIl) (wherein R' is the same
as defined above
and L~ represents a leaving group including, for instance, halogen atom such
as chlorine, bromine,
or iodine atom, hydroxy and C~_6 alkoxy) with hycu~azine (free base, its salt
or its hydrate).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 20 -
The reaction can be carried out in a solvent including, for instance, alcohots
such as me'tnanol,
ethanol, 1-propanol, isopropanol and tert-butariol, water and others.
Optionally, two or more of the
solvents selected from the listed above can be mixed and used.
The reaction temperature is usually, but not limited to, about 0°C to
200°C and preferably about
0°C to 100°C. The reaction may be conducted for, usually, 30
minutes to 48 hours and prefei-ablyy
2 hours to 24 hours.
Alternatively, the compound of the fornmla (N) can be prepared by the
following procedures.
In Step ii-lb, the compound of the formula (IX) (wherein wherein R' is the
same as defined above
and L3 represents a protecting group including, for instance, tert-
butoxycarbonyl) can be prepared
by reacting the compound of the formula (\TIn (wherein R' and L~ are the same
as defined above)
with the compomd .of the fomnula (VJT~ (wherein L3 is the same as defined
above).
When L~ is hydroxy, the reaction can be done using a coupling agent including,
for instance,
carbodiimides .such as N, N-dicyclohexylcarbodiimide and 1-(3-
dimethylaminopropyl)-3-ethyl-
carbodiimide, benzophenyltriazole-1-yl-oxy-tris-pyrrolidino-phosphonium
hexafluorophosphate
(PyBOP), diphenylphosphoryl azide. N-hydroxysuccinimide, 1-hydroxybenzotiazole
monohydrate
(HOBt), and the like can be used as an accelerator of the reaction.
The reaction may be carried out in a solvent including, for instance,
hatogenate.d hydrocarbons
such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as
diethyl ether, iso-
propyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aromatic hydrocarbons
such as benzene, toluene and xylene; nitrites such as acetonitrile; amides
such as N, N-
dimethylformamide .(DMF), N, N-dimethylacetamide (DMAC) and N-
methylpyrrolidone (NMP);
urea such as 1,3-dimethyl-2-imidazolidinone (DMn; sulfoxides such as
dimethylsulfoxide
(DMSO); and others. Optionally, two or more of the solvents selected from the
listed above can be
mixed and used.
The reaction temperature is usually, but not limited to, about 0°C to
180°C and preferably about
20°C to 100°C. The reaction may be conducted for, usually, 30
minutes to 48 hours and preferably
2 hours to 12 hours.
In Step ii-2b, the compound of the fornmla (N) (wherein R' is the same as
defined above) can be
prepared by removing the protecting group L3 of the compound of the. formula
(LY) (wherein R'
and L3 are the same as defined above).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-21-
The removal of protective group L3 can be done by using a reagent including,
for instance, an acid
such as tritluoroacetic acid and hydrochlaric acid.
The reaction may be earned .out without solvent or in a .solvent including,
for instance, ethers such
as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-
dimethoxyethane;
aromatic hydrocarbons such as benzene, toluene and xylene; niti-iles such as
acetonitrile; amides
such as N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-
methylpyrrolidone (NMP); urea such as 1,3-dimethyl-2-in>idazolidinone (DMI);
and others.
Optionally, two or more of the solvents selected from the listed above can be
mixed and used.
The reaction temperature can be optionally set depending on compoundss to be
reacted. The
reaction temperature is usually, but not limited to, about 20°C to
120°C. The reaction may be
conducted for, usually, 30 minutes to 60 hours and preferably 1 to 48 hours.
Hydrazine (free base, its salt or its hydrate), the compound of the formula
(V)~ and (VIII are
commercially available or can be prepared by the use of lrnown techniques.
Alternative procedures for the preparation of intermediate (VT)
[Method (iii)]
NCS ~ S S
3\ 3 _
L N-NHS ~ R~ H H~NH H~N-H~NH
H + ---t
Rs Rs Step iii-1 / R~ Step iii-2 / R~
(Vlll)
(V) R5 Rs Rs Rs
(X)
(XI)
S
l_" H
N-H~NH
R1.~ (vli) ,
O R O / R7
Step iii-3 Rs Rs
(VI)
The compound of the formula (VI) (wherein R', R5, R6 and R' are the same as
defined above) can
be alternatively prepared by the following procedures in three steps.
In Step iii-1, the compound of the formula (X) (vs~herein wherein L3, R5, R6
and R' are the same as
defined above) can be prepared by reacting the compound of the formula (VDT
(wherein L3 is the
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-~2-
same as defined above) with the compound of the formula (V) (wherein R5, R6
and R' are th'e same
as defined above) in a similar manner described in Step i-1 for the
preparation of compounds of the
formula (VI).
In Step iii-2, the compound of the formula (X17, (wherein R5, R6 and R' are
the same as defined
above) can be prepared by removing the protecting group L3 of the compound of
the formula (.k)
(wherein L3, R5, R6 and R' are are the same as defined above) in a similar
manner described in Step
ii-2b for the preparation of compounds of the formula (N).
In Step iii-3, the compound of the formula (VIA (wherein R', R5, R° and
R' are the same as defined
above) can be prepared by reacting the compound of the formula (.'~ (wherein
R5, R6 and R' are
the same as defined above) with the compound of the formula (Vff) (wherein R'
and L2 are the
same as defined above) in a similar manner described in Step ii-la for the
preparation of
compounds of the formula (N).
Preparation of intermediate (II-b)
[Method (iv)]
L2
(VII) R~ N R~N
HZN R ~ \ ~ R
\ R~ O ~ I R _ GI
O Step iv-2 s Rs
Rs Rs Step iv-1 Rs Rs R
(fill I) (XIV) ,
(XII)
N-N
HZN-NH2 R~ N Br~N R~ N NH
2
I R Step iv-4 ~ R~
Srep iv-3 HZN~N Rs Rs s s
R R
(I I-b)
1 s (xv)
The compound of the formula (II-b) (wherein R', R5, R6 and R' are the 'same as
defined above) can
be prepared by the following procedures.
In Step iv-1, the compound of the formula (XI~ (wherein R', R5, R° and
R' are the same as
defined above) can be prepared by reacting the compound of the formula (XII)
(wherein R5, R6 and
R' are the same as defined above) with the compound of the formula (VII)
(wherein R' and LZ are
the same as defined above).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 23 -
When LZ is hydroxy, the reaction can be done 'using a coupling agent
including, for ir(stance,
carbodiimides such as N, N-dicyclohexylcarbodiimide and 1-(3-
dimethylaminopropyl)-3-ethyl-
carbodiimide, benzophenyltriazole-1-yl-oxy-Iris-pyrrolidino-phosphonium
hexafluorophosphate
(PyBOP), diphenylphosphoryl azide. N-hydroxysuccininude, 1-hydroxybenzotiazole
monohydrate
(HOBt), and the like can be used as an accelerator of the reaction.
The reaction maybe carried out in a solvent including, for instance,
halogenated hydrocarbons
such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as
diethyl ether, iso-
propyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aromatic hydrocarbons
such as benzene, toluene and xylene; nitTiles such as acetonitrile; amides
such as N, N-dimethyl-
formamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpyrrolidone (NMl');
urea such
as 1,3-dimethyl-2-imidazolidinone (DMn; sulfoxides such as dimethylsulfoxide
(DMSO); and
others. Optionally, two or more of the solvents selected from the listed above
can be mixed and
used.
The reaction temperature is usually, but not limited to, about 0°C to
180°C and preferably 'about
20°C to 100°C. The reaction may be conducted fox, usually, 30
minutes to 48 hours and preferably
2 hours to 12 hours.
In Step iv-2, the compound of the formula (XIV) (where.in R', R5, Rb and R'
are the same as
defined above) can be prepared by reacting the compound of the .formula (XI>~
(wherein R', RS, R6
and R' are the same as defined above) with an appropriate halogenating reagent
including, for
instance, SOC12, POCK, and the like.
The reaction may be carried out without solvent or in a solvent including, for
instance, halogenated
hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane;
ethers such as dioxane
and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such
as benzene,
toluene and xylene, and others. Optionally, two or more of the solvents
selected. from the listed
above can be mixed and used.
The reaction temperature is usually, but not limited to, about 0°C to
200°C and preferably about
20°C to 100°C. The reaction rnay be conducted for, usually, 30
minutes to 48 hours and preferably
2 hours to 24 hours.
In Step iv-3, the compound of the formula (XV) (wherein R', R5, R6 and R' are
the same as defined
above) can be prepared by reacting the compound of the formula (XIV) (wherein
R', R5, R6 and R'
are the same as defined above) with hydrazine (free base, its salt or its
hydrate).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-24_
The reaction may be carried out in a solvent including, for instance,
halogenated hyydro6arbons
such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as
diethyl ether,
isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aromatic hydro-
carbons such as benzene, toluene and xylene; nitrile,s such as acetonitrile;
amides such as N, N-di-
S methylformamide (DMF), N, N-dimethylacetanude (DMAC) and N-methylpyrrolidone
(NMP);
urea such as 1,3-dimethyl-2-imidazolidinone (DMII; sulfoxides such as
dimethylsulfoxide
(DMSO); and others. Optionally, two or more of the solvents selected from the
listed above can be
mixed and used.
The reaction temperature is usually, but not limited to, about 0°C to
180°C and preferably about
20°C to 100°C. The reaction may be conducted fox, usually, 30
minutes to 48 hours and preferably
2 hours to 12 hours.
In Step iv-4, the compound of the formula (II-b) (wherein R', R5, R6 and R'
are the same as defined
above) can be prepared by reacting the compound of the formula (XV) (wherein
R', R$, R6 and R'
are the same as defined above) with cyanogen halides such as cyanogen bromide.
The reaction may be carried out in a solvent including, for instance, ethers
such as diethyl ether,
isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aromatic hyd~~o
carbons such as benzene, toluene and xylene; amides such as N, N-
dimethylformamide (DMF), N,
N-dimethylacetamide and N-methylpyrrolidone; alcohols such as methanol,
ethanol, 1-propanol,
isopropanol and tent-butanol; and others. Optionally, two or more of the
solvents selected from the
listed above can be mixed and used.
The reaction temperature is usually, but not limited to, about -10°C to
200°C. The reaction may be
conducted for, usually, 30 minutes to 48 hours and preferably 1 hour to 24
hours.
The compound of the formula (XI~ is commercially available or can be prepared
by the use of
laiown techniques.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ 25 _
Preparation of intermediate (~
[Method (v)]
H / Rs R? Lq
\ q \ (XVII)
R R' R
(XVI)
Step v-1 ~. R3 Step v-2 ~ Rs
HO 1 ~ L~
q R4
R
(XVIII) (III)
R \~R3 Step v'-1
\ 4
R
(XIX)
The compound of the fornmla ()T~ (wherein R2, R' and R4 are the same as
defined above) can be
prepared by the following procedures.
In Step v-1, the compound of the formula (XVII17 (wherein wherein RZ, R~ and
R4 are the same as
defined above) can be prepared by reacting the compound of the formula (XVfj
(wherein R3 and
R~ are the same as defined above) with the compound of the formula (.XVII)
(wherein RZ is the
same as defined above and L4 represents metal or metal complex including, for
instance, lithium,
magnesium chloride and magnesium bromide).
The reaction may be carried out in a solvent including, for instance, ethers
such as diethyl ether,
isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aliphatic
hydrocarbons such as n-hexane, cyclohexane; aromatic hydrocarbons such as
benzene, toluene and
xylene; and others. Optionally, two or more of the solvents selected from the
listed above can be
mixed and used.
The reaction temperature can be optionally set depending on the compounds to
be reacted. The
reaction temperature is usually, but not limited to, about -20°C to
50°C.
The reaction may be conducted for, usually, 30 minutes to 10 hours and
preferably 1 to 24 hours.
The compound of the formula (XVIB) (wherein wherein Rz, R3 and R4 are the same
as def'med
above) can be alternatively prepared by reacting the compound of the formula
(.~) (wherein R2,
R3 and R'' are the same as defined above) with a reducing agent including, for
instance, sodium
borohydride or lithium aluminum hydride as shown in Step v'-1.
The reaction may be carried out in a solvent including, for instance, ethers
such as diethyl ether,
isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane;
aliphatic hydro-
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-26-
carbons such as n-hexane, cyclohexane; aromatic hydrocarbons such as benzene,
toluene and
xylene; and others. Optionally, two or more of the solvents selected from the
listed above can be
mixed and used.
The reaction temperature can be optionally set depending on the compounds to
be reacted. The
reaction temperature is usually, but not limited to, about 20°C to
50°C.
The reaction may be conducted for, usually, 30 minutes to 24 hours and
preferably 1 to 10 hours.
In Step v-2, the compound of the fonnula (lII) (wherein L~, R'', R3 and R4 are
the same as defined
above) can be prepared by reacting the compound of the formula (XVIIl~
(wherein wherein Rz, R3
and R4 are the same as defined above) with an appropriate halogenating reagent
including, for
instance, POCl3, PC13, SOCK, and the like; or with the corresponding sulfonyl
chloride for instance
methanesulfonyl chloride.
The reaction may be carried out without solvent or in a solvent including, for
instance, halogenated
I~ydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane;
ethers such as dioxane
and tetrahydrofuran (THF)and 1,2-dimethoxyethane; aromatic hydrocarbons such
as benzene,
toluene and xylene, and others. Optionally, two or more of the solvents
selected from the listed
above can be mixed and used.
The reaction can be advantageously conducted in the presence of a base,
including, for instance,
pyridine, triethylamine and N,N-diisopropylethylamine, dimethylaniline,
diethylaniline, and
others.
The reaction temperature is usually, but not limited to, about 0°C to
200°C and preferably about
20°C to 100°C. The reaction may be conducted for, usually, 30
minutes to 48 hours and preferably
2 hours to 24 hours.
The compound of the formula (XVI), (XVIl~ and (XIX) are commercially available
or can be
prepared by the use of known techniques.
Preparation of compound of (I-b)
The compound of the formula (I-b) of the present invention can be, but not
limited to be, prepared
by the Method [B] below.
[Method B]
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 27 _
Rz '
R_
N-N
H w Rs ~ ~ w s
R~~N ~ . R7 ~a R~° Rya ~ Ra ~ Ro N ,° ~ R
Ray
H NON ~ s (~) / R Ra
z Rs R ~ R~
(XV) Rs Rs
The compound of the formula (I-b) (wherein R', R', R3, Ra, R5, R6, R',
R'° and R" are the same as
defined above) can be prepared by reacting the compound of the formula (XV)
(wherein R', R5, Rb
and R' are the same as defined above) with the compound of the formula (~'.~i)
(wherein Rz, R3,
Ra, R'° and R" are the same as defined above and La represents a
leaving group including, for
instance, halogen atom such as chlorine, bromine, or iodine atom).
The reaction can be carried out in a solvent including, for instance, alcohols
such as methanol,
ethanol, 1-propanol, isopropanol and tert-butanol, water and others.
Optionally, t<vo or more of the
solvents selected from the listed above can be mixed and used.
he reaction temperature can be optionally set depending on the compounds to be
reacted. The
reaction temperature is usually; but not limited to, about 20°C to 50
°C. The reaction may be
conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
The compound of the formula (X~.) is conunercially, available or can be
prepared by the use. of
lrno~m techniques..
When the compound shown by the formula (n or a salt thereof has an asymmetl-ic
carbon in the
structure, their optically active compounds and racenuc mixtures are also
included in the scope of
the present invention.
Typical salts of the compound shown by the formula (I~ include salts prepared
by reaction of the
compounds of the present invention with a mineral or organic acid, or an
organic or inorganic
hase. Such salts are la~own as acid addition and base addition salts,
respectively.
Acids to form acid addition .salts include inorganic acids such as, without
limitation, sulfuric acid,
phosphoric acid, hydrochloric acid, hydrobromic acid, hydriodic acid and the
like, and organic
acids, such as, without limitation, p-toluenesulfonic acid, methanesulfonic
acid, oxalic acid, p-
bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic
acid, acetic acid, and
the like.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_7g_
Base addition salts include those derived from inorganic bases, such as,
without limitation,
ammonium hydroxide, alkaline metal hydroxide, alkaline earth metal hydroxides,
carbonates,
bicarbonates, and the like, and organic bases, such as, without limitation,
ethanolamine,
triethylamine, tris(hydroxymethyl)aminomethane, and the like. Examples of
inorganic bases
include sodium hydroxide, potassium hydroxide, potassium carbonate, sodium
carbonate, sodium
4
bicarbonate, potassium bicarbonate, calcium hydroxide, calcium carbonate, and
the like.
The compound of the present invention or a salt thereof, depending on its
substituents, may be
modified to forni lower alkylesters or lmown other esters; and/or hydrates or
other solvates. Those
esters, hydrates, and solvates are included in the scope of the present
invention.
The compound of the present invention may be administered in oral forms, such
as, without
limitation normal and enteric coated tablets, capsules, pills, powders,
granules, elixirs, tinctures,
solution, suspensions, syrups, solid and liquid aerosols and emulsions. They
may also be
administered in parenteral forms, such as, without limitation, intravenous,
intrape.ritoneal,
subcutaneous, uitramuscular, and the like forms, well-known to those of
ordinary skill in the
pharmaceutical arts. The compounds of the present invention can be
administered in intranasal
form via topical use of suitable intranasal vehicles, or via transdermal
routes, using transdermal
delivery systems well-known to those of ordinary skilled in the art.
The dosage regimen with the use of the compounds of the present invention is
selected by one of
ordinary skill in the arts, in view of a variety of factors, including,
without limitation, age, weight,
sex, and medical condition of the recipient, the severity of the condition to
be treated, the route of
administration, the level of metabolic and excretory function of the
recipient, the dosage form
employed, the particular compound and salt thereof employed.
The compounds of the present invention are preferably formulated prior to
administration together
with one or more pharmaceutically-acceptable excipients. Excipients axe inert
substances such as,
without limitation carriers, diluents, flavoring agents, sweeteners,
lubricants, solubilizers,
suspending agents, binders, tablet disintegrating agents and encapsulating
material. ._
Yet another embodiment of the present invention is pharmaceutical formulation
comprising a
compound of the invention and one or more pharmaceutically-acceptable
excipients that are
compatible with the other ingredients of the formulation and not deleterious
to the recipient
thereof. Pharmaceutical formulations of the invention are prepared by
combining a therapeutically
effective amount of the compounds of the invention together with one or more
pharmaceutically-
acceptable excipients therefore. In malting the compositions of the present
invention, the active
ingredient may be mixed with a diluent, or enclosed within a carrier, which
may be in the form of a
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-29-
capsule, sachet, paper, or other container. The earner .may serve as a
diluent, which may be solid,
semi-solid, or liquid material which acts as a vehicle, or can be in the form
of tablets, pills
powders, lozenges, elixirs, suspensions, emulsions, solutions, syrups,
aerosols, ointments,
containing, for example, up to 10% by weight of the active compound, soft and
hard gelatin
capsules, suppositories, sterile injectable solutions and sterile packaged
powders.
For oral administration, the -active ingredient may be combined with an oral,
and non-toxic,
pharmaceutically-acceptable cawier, such as, without limitation, lactose,
starch, sucrose, glucose,
sodium carbonate, mannitol, sorbitol, calcium carbonate, calcium phosphate,
calcium sulfate,
methyl cellulose, and the like; together with, optionally, disintegrating
agents, such as, without
limitation, maize, starch, methyl cellulose, agar bentonite, xanthan gum,
alginic acid, and the like;
and optionally, binding agents, for example, without limitation, gelatin,
natural sugars, beta-
lactose, corn sweeteners, natural and synthetic gums, acacia, tragacanth,
sodium alginate,
carboxymethylcellulose, polyethylene glycol, waxes, and the like; and,
optionally, lubricating
agents, fox example, without limitation, magnesium stearate, sodium stearate,
stearic acid, sodium
oleate, .sodium benzoate, sodium acetate, sodium chloride, talc, and the like.
In powder forms, the carrier may be a finely divided solid which is in
admixture with the finely
divided active ingredient. The active ingredient may be mixed with a carrier
having binding
properties in suitable proportions and compacted in the shape and size desired
to produce tablets.
The powders and tablets preferably contain from about 1 to about 99 weight
percent of the active
ingredient which is the novel composition of the present invention. Suitable
solid carriers are
magnesium carboxymethyl cellulose, low melting waxes, and cocoa butter.
Sterile liquid formulations include suspensions, emulsions, syrups and
elixirs. The active
ingredient can be dissolved or suspended in a pharmaceutically acceptable
carriers, such as sterile
water, sterile organic solvent, or a mixture of both sterile water and sterile
organic solvent.
The active ingredient can also be dissolved in a suitable organic solvent, for
example, aqueous
propylene glycol. Other compositions can be. made by dispersing the finely
divided active ingre-
dient in aqueous starch or sodium carboxymethyl cellulose solution or in a
suitable oil.
The fornmlation may be in unit dosage form, which is a physically discrete
unit containing a unit
dose, suitable for administration in human or other mammals. A unit dosage
form can be a capsule
or tablets, or a number of capsules or tablets. A "unit dose" is a
predetermined quantity of the
active compound of the present invention, calculated to produce the desired
therapeutic effect, in
association with one or more excipients. The quantity of active ingredient in
a unit dose may be
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-30-
varied or adjusted from about 0.1 to about 1000 milligrams or more according
to the particular
treatment involved.
Typical oral dosages of the present invention, when used for the indicated
effects, will range from
about 0.01 mg/kg/day to about 100 mg/lcg/day, preferably from 0.1 mg/lcglday
to 30 mgll;g/day,
and most preferably from about 0.5 mgllcg/day to about 10 mg/kg/day. In the
case of parenteral
administration, it has generally proven advantageous to administer quantities
of about 0.001 to
100mg /lcg/day, preferably from 0.01 mg/kg/day to 1 mg/kg/day. The compounds
of the present
invention may be administered in a single daily dose, or the total daily dose
may be administered
in divided doses, two, three, or more times per day. Where delivery is via
transdermal forms, of
course, administration is continuous.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-31 -
EXA1~ZPLES
The present invention will be described as a form of examples, but they should
by no means be
construed as defining the metes and bounds of the present invention.
In the examples below, all quantitative data, if not stated otherwise, relate
to percentages by
weight.
Mass spectra were obtained using electrospray (ES) ionization techniques
(micromass Platform
LC). Melting points are uncorrected. TLC was performed on a precoated silica
gel plate (Merck
silica gel 60 F-254). Silica gel (WAKO-gel C-200 (75-150 pm)) was used for all
column
chromatography separations. All chemicals were reagent grade and were
purchased from Sigma-
Aldrich, Wako pure chemical industries, Ltd., Great Britain, Tokyo kasei kogyo
Co., Ltd., Nacalai
tesque, Ine., Watanabe Chemical lnd. Ltd., Maybridge plc, Lancaster Synthesis
Ltd., Merck KgaA,
Germany, Kanto Chemical Co., Ltd.
Analytical HPLC Retention times of intermediates and examples are measured as
follows:
Method A
Equipment: Waters 2690 separation module
Column temperature: 40 °C.
Mobile phase: water / acetonitrile (each of them containing lOmM ammonium
acetate)
Column: Chromolith Rash RP-18e, 25 * 4.6mm
Flow rate: 1.3 mL/min.
Injection volume: 5 ~L
Gradient (Time ) : (water / acetonitrile)
01\~Iinutes : 9 / 1
0.2 Minutes: 9 / 1
2.0 Minutes: 1 / 9
3.5 Minutes: 1 / 9
4.0 Minutes: 9 / 1
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
3~ _
Method B
Equipment: Hewlett Packard series 1100
Column temperature: 40 °C.
Mobile phase: water / acetonitrile (each of them containing l OmM ammonium
acetate)
Column: YMC PackPro C-18,35 * 4.6mm
Flow rate: 1.0 mL/min.
Injection volume: 5 microL
Gradient (Time ) : (water / acetonitrile)
1 Minutes : 9 / 1
0.1 Minutes: 9 / 1
1.5 Minutes: 1 / 9
3.5 Minutes: 1 / 9
4.5 Minutes: 9 / 1
Method C
Equipment: Hewlett Packard series 1100
Column temperature: 40 °C.
Mobile phase: water / acetonitrile (each of them containing lOmM ammonium
acetate)
Column: Phenomenex Luna 3u C18(2) 30 * 4.6mm
Flow rate: 1.0 mL/min.
~0 Injection volume: 10 microL
Gradient (Time ) : (water / acetonitrile)
2 Minutes : 9 / 1
0.5 Minutes: 9 / 1
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 33 -
4.5 Minutes: 1 / 9
6.5 Minutes: 1 / 9
8.5 Minutes: 9 / 1
HPLC-Methods:
Analytical HPLC as follows were determined on a HP 1100 with DAD-detection
(Hewlett
Packard) under the following conditions:
Method 2A
Column: Kromasil C1S 60*2 at 30 °C; injection volume: 1.00 ~1;
flowrate: 0.75 ml/min; eluent:
A= 0.01 M H3P0~ in HzO, B= CH3CN; gradient [t(min): A/B]: 0.0: 90/10; 0.5:
90/10; 4.5: 10/90;
8.0: 10/90; 8.5: 90/10 10.0: 90/10.
Method 2B
Column: kromasil C18 60*2 at 30 °C; injection volume: 0.20 - 0.30 ~,1;
flowrate: 0.75 ml/min;
eluent: A= 0.01 M H3P04 in HzO, B= CH3CN; gradient [t(min): AB]: 0.0: 90/10;
0.5: 90/10; 4.5:
10/90; 6.5: 10/90; 7.5: 90/10.
Method 2C
Column: Kromasil C18 60*2 at 30 °C; injection volume: 1.0 ~1; flowrate:
0.75 ml/nun; eluent: A=
5ml 70% HC104/1L H20, B= CH3CN; gradient [t(min): A/B]: 0.0: 98/2; 0.5: 98/2;
4.5: 10/90; 6.5:
10190; 6.7: 98/2; 7.5: 98/2.
LC/MS-Methods:
Retention times for peaks with the correct product mass were recorded as
follows:
Method 2D
Instrument MS: Micromass TOF (LCT); instrument HPLC: Waters2690; column: I'MC-
ODS-AQ,
50 mm x 2.0 mm, 3.0 Vim; eluent A: water + 0.1% formic acid, eluent B: CH3CN +
0.1% formic
acid; gradient: 0.0 min 100%A -~ 0.2 min 100%A ~ 2.9 min 30%A -~ 3.1 min 10%A -
~ 4.5 nun
10%A ~ 4.51 min 100%A-j 6.5 min 100%A; oven: 40°C; flow rate: 0.8
mllmin; UV-detection:
210 nm.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-34-
Method 2E
Instrument MS: Micromass ZQ; instrument HPLC: Waters Alliance 2790; column:
Uptisphere C
18, 50 mm x 2.0 mm, 3.0 Vim; eluent A: water + 0.05% formic acid, eluent B:
CH3CN + 0.05%
formic acid; gradient: 0.0 min 5%B -~ 2.0 min 40%B -~ 4.5 min 90%B~ 5.5 min
90%B; oven:
45°C; flow rate: 0.0 min 0.75 ml/min -~ 4.5 min 0.75 mllmin~ 5.5 min
1.25 ml/min; UV-
detection: 210 nm.
'H NMR spectra were recorded using either Bruker DRX-300 (300 1\~iz for 'H)
spectrometer or
Brucker 500 UltraShieled~ (S00 MHz for 1H) . Chemical shifts are reported in
parts per million
(ppm) with tetramethylsilane (TMS) as an internal standard at zero ppm.
Coupling constant (J) are
given in hertz and the abbreviations s, d, t, q, m, and br refer to singlet,
doublet, triplet, quartet;
multiplet; and broad, respectively. The mass determinations were carried out
by MAT95 (Finnigan
MAT).
All starting materials are commercially available or can be prepared using
methods cited in the
literature.
The effect of the present compounds was exanuned by 'the following assays and
pharmacological
tests.
[Measurement of change of intracellular cAMP accumulation by luciferase
detection in the human
GABAB receptor-transfected HEK293 cell line] (Assay 1)
(1) Cloning of GABAB receptors and generation of stable cell lines
The human GABAB~~a~, GABAB~~ba and GABAB~2~ receptor subunits were cloned into
pcDNA3 .(Invitrogen) as previously described (White J. H. et al., Nature 1998,
396(6712):679-S2). The cell culture and transfection of Human Embryonic Kidney
(HEK293) cells was done as follows. HEK293-Luc cells were grown in Dulbecco's
modified Eagle's medium (DMEM, Gibco BRL) supplemented with 5% modified bovine
serum (MBS, Gibco BRL) in fibronectin coated 96-well microtiter plates. For
transfe.ction (mammalian transfection kit; Stratagene) cells were grown at
20000 cells per
well at 35 °C with 3% CO~ for 24 h with 0.1 ml per well. DNA
suspension: 10 pg
expression plasmid DNA of each human GABAB~~aa and human GABAB~Z~ in pcDNA3
was dissolved in 450 p,l of water with 50 ml CaClz (2.5 M) + 500 pl 2x
phosphate
buffered saline (PBS, pH 6.95) and incubated for 10 to 20 nun at room
temperature. In
the meantime cell medium was aspirated and cells were washed twice with 200 pl
PBS
per well and then 200 ~.1 medium plus 5% MBS was added. For transfection 20
p.l of
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 35 -
suspended DNA was added and incubated for 3 h at 35 °C with 3% CO~,
cells were
washed with PBS and 200 ~1 of growth medium was added and cells were grown for
2
days. Cells were then trypsinized and diluted 1:10 in fibronectin coated.
wells and
incubated with growth medium supplemented with 1 mg/n~l G41 S (Gibco BRL) and
grown under selection pressure for 10 days with 2-3 medium changes. After 6418
selection cells were grown until colonies had formed.
(2) Folskolin-stimulated luciferase-reporter gene assay
GABAByniz>-HEK293/CRE-luc cells were seeded into poly-D-lysine-coated 3S4-well
white/opaque plates (BD BIOCOAT) at 4000 cells/well in 40 p.l DMEM/F12 medium
i0 supplemented with 2.5% FBS, and grown for 48 hours at 370 in a humidified
atmosphere
with 5% CO2. Test compounds dissolved in DMSO were diluted into DMEM/F12
medium
containing 0.1% BSA and transferred to the test cultures at 5 pllwell. 10
minutes after the
test compound addition, forskolin prepared in a manner similar to the test
compounds was
added at 5 pl/well (1.6 p.M of final concentration), and cells were then
incubated for.3
hours at 370 in 5% CO2. After the incubation, the medium was discarded,
followed by
addition of 20 ~l/well of 1:1 mixture of Steady-GIoTT' reagent (Promega) and
Phenol-red
free DMEM/F12 medium. The plates were incubated at least 5 minutes to ensure
complete
cell lysis and then luciferase activity was measured with ViewLux microplate
imager
(Perkin Elmer).
[Measurement of rhythmic bladder contraction in anesthetized rats] (Assay 2)
(1) Animals
Female Sprague-Dawley rats (200250 g / Charles River Japan) were used.
(2) Rhythmic bladder contraction in anesthetized rats
Rats were anesthetized by intraperitoneal administration of urethane (Sigma)
at 1.25 g/kg.
The trachea was cannulated with a polyethylene tube (HIBIKI, No.S) to
facilitate
respiration; and a cannula (BECTON DIChTNSON, PE-50) was placed in the left
femoral
vein for intravenous administration of testing compounds. The abdomen was
opened
through a midline incision, and after both ureters were cut, a water-filled
baloon (about 1
ml capacity) was inserted through the apex of the bladder dome. The baloon was
connected to a pressure transducer onto a polygraph. Rhythmic bladder
contraction was
elicited by raising up inti~avesical pressure to approximately 15 cm HzO.
After the
rhythmic bladder contraction was stable, a testing compound was administered
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-36-
intravenously. Activity was estimated by measuring disappearance time and
amplitude of
the rhythmic bladder contraction. The effect on amplitute of bladder
contractions was
expressed as a percent suppression of the amplitude of those after the
disappearance was
recovered. Experimental values were expressed as the mean~S.E.M. The testing
compounds-mediated inhibition of the rhythmic bladder contraction was
evaluated using
Student's t-test. A probability level less than 5% was 'accepted as
significant difference.
Results in folskolin-stimulated luciferase-reporter gene assay (Assay 1) are
shown in Examples
and tables of the Examples below. For practical reasons, the compounds are
grouped in four
classes based on activity as follows:
ICso=A(<or=)O.l~M<B(<or=)0.5 ~M<C(<or=) 1 pM<D
[Cystometry in anesthetized rats] (Assay 3)
Effect of a compound on cystometric parameters in rats were studied as
described previously
[Takeda H et al: J. Pharmacol. Exp. Ther. 126: 939- 945, 2000].
Female rats, weighing from 200 to 230 g, were anesthetized with wethane (1.2
g/kg i.p.). Through
a midline abdominal incision, the ureter on each side was ligated and cut
proximal to the ligature.
A polyethylene, catheter (PE-50) was inserted into the urinary bladder and
connected through a
three-way connector to: 1) a pressure transducer (Viggo-Spectramed Pte Ltd, DT-
~XA.D) for
measurement of bladder pressure, and 2) a syringe infusion pump (TERUMO) for
continuous
infusion of saline into the bladder. During cystometry, saline was infused at
a rate of 2.4 ml/h.
Bladder pressure was recorded continuously on a PowexLab systems (BioResearch
Center). The
following cystometric parameters were obtained: micturition interval and
micturition pressure
(maximum bladder pressure during micturition). Two reproducible micturition
cycles were
recorded before drug administration and used to provide a baseline value to be
compared with the
first two micturition cycles just after drug administration. Relative values
for the. various
cystometric parameters were calculated as follows: (mean value from two
micturition cycles just
after drug administration)/(mean value from two micturition cycles just before
drug
administration). A venous catheter was inserted into the left femoral vein for
drug injection.
Z used in Melting point in the following section indicates decomposition.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-37-
(Example 1-1]
Method A
3-(3-benzhydrylsulfanyl-S-cyclopropyl-[1,2,4]triazol-4-yl)-benzoic acid
Br
OH
A solution of 3-(3-cyclopropyl-5-thioxo-1,5-dihydro-4H-1,2,4-triazol-4-
yl)benzoic acid (152 mg,
0.58 mmol) iri N,N-dimethylformamide (1 mL) was added potassium carbonate (403
mg,
2.91 nunol) and bromodiphenylmethane (187 mg, 0.76 mmol), and the mixture was
stirred at 60 °C
for 16 hours. The inorganic salts were filtered, and the filtrate was diluted
with.aqueous sodium
bicarbonate solution. The mixture was washed with ethylacetate, and the
aqueous layer was
acidified to pH 2 with 1N aqueous HCl solution. The mixture was extracted with
ethylacetate, and
the organic layer was concentrated under reduced pressure. The obtained
residue was
recrystallized from the mixture of dichloromethane., diethylether, and hexane
to provide 3-{3-
cyclopropyl-5-[(diphenylmethyl)thio]-4H-1,2,4-triazol-4-yl~benzoic acid (65.9
mg).
'H NMR (DMSO-d6): 8 0.82-0.89 (m, 4H), 1.48-1.53 (m, ,1H), 5.88 (s, 1H), 7.21-
7.30 (m, lOH),
7.55 (d, J= 7.9 Hz, 1H), 7.70-7.74 (m, 2H), 8.12 (d, J= 7.9 Hz, 1H), 13.40
(s(br), 1H).
mp 193 °C;
Molecular weight : 427.53
MS (M+H): 428
Activity Class : B
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-38-
Preparation of intermediates
Method (i)
,,O
O O I/~(~/N
SCN .~ O~CH~ H
H-NH2
"-CHI
A mixture of cyclopropanecarbohydrazide (255 mg, 2.55 mmol) and methyl 3-
isothiocyanato-
benzoate (492 mg, 2.55 minol) in ethanol (3 mL) was stirred at refluxing
temperature for 16 hours.
The mixture was concentrated under reduced pressure, and to the obtained
residue was added a
solution mixture of diethylether and hexane. The precipitates were collected
and dried to afford
methyl 3-( {[2-(cyclopropylcarbonyl)hydrazine]carbonothioyl~ amino) benzoate
(399 mg).1H NMR
(DMSO-dti) 8 0.77-0.79 (m, 4H), 1.60-1064 (m, 1H), 3.32 (s, 3H), 7.47 (t, J=
7.9 Hz, 1H), 7.73 (d,
J= 7.3 Hz, 1H), 7.80 (d, J= 7.3 Hz, 1H), 8.08 (s(br), 1H), 9.86 (s(br), 1H),
10.10 (s(br), 1H); ; MS
n~/z 294 (1\~+1).
Next, a solution of methyl 3-({[2-
(cyclopropylcarbonyl)hydrazine]carbonothioyl; amino) benzoate
(399 mg, 1.36 mmol) in 4N aqueous solution of sodium hydroxide (7 mL) was
stirred at refluxing
temperature for 16 hours. After having cooled to ambient temperature, the
mixture. was acidified
to pH 2 with 1N aqueous solution of HCI. The mixture was extracted with
ethylacetate, dried
over NazS04, altered, and concentrated under reduced .pressure to obtain 3-(3-
cyelopropyl-5-
thioxo-1,5-dihydro-4H-1,2,4-triazol-4-yl)benzoic acid (355 mg).'H NMR (DMSO-
d6) S 0.77-0.92
(m, .4H), 1.47-1.52 (m, 1H), 7.70-7.77 (m, 2H), 8.01 (s, 1H), 5.09 (d, J= 6.3
Hz, 1H), 13.00 (s(br),
1H), 13.64 (s, 1H) ; MS m/z 262 (M++1).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-39-
Preparation of intermediates
Method (ii)
cyclopropanecarbohydrazide
O H , O
,N O CH3 N O CH
CI + HZN ~ ~ Ni ~ s
GH3 Hs H ~ ! HCH3
3
O
---~ N~NH
H
To a solution of tert-butyl hydrazinecarboxylate (6.38 g, 48.3 mmol) and
triethylamine (7.26 g,
71.5 nunol) in dichloromethane (10 mL) was added cyclopropanecarbonyl chloride
(5.00 g,
47.8 mrnol) at 0 °C. The mixture was stirred for 16 hours at ambient
temperature, and the resulting
suspension eras filtered and washed with dichloromethane. The filtrate was
concentrated under
reduced pressure to provide tert-butyl 2-
(cyclopropylcarbonyl)hydrazinecarboxylate (13.0 g). 1H
NMR (DMSO-d6) 8 0.67-0.72 (m, 4H), 1.39 (s, 1H), 1.52 -1.54 (m, 1H), 8.64 (s,
1H), 9.70 (s, 1H).
Next, to a stirred solution of tent-butyl 2-
(cyclopropylcarbonyl)hydrazinecarboxylate (3.00 g,
15.0 mrnol) in 1,4-dioxane (50 mL) was added 4N HCl in 1,4-dioxane (20 mL).
The mixture was
stirred at 80 °C for 1 hour, and after cooled to ambient temperature,
it was concentrated under
reduced pressure. To the obtained residue was added ethylacetate and
triethylamine (8.04 g,
79.4 mmol), and the organic layer was v,~ashed with saturated sodium
bicarbonate aqueous solution
and brine, dried over NazS04, filtered, and concentrated under reduced
pressure to obtain cyclo-
propanecarbohydrazide (0.89 g).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-40-
Preparation of intermediates
Method (iv)
5-(3-fluorophenyl)-4-phenyl-4H-1,2,4-triazol-3-amine
o / I o
H~N
CI I \ F --.~. \ y F
/ H ~ /
\ I / I F / I I NHz
\ F
N
N I / H I /
F N-N
/ N~NHz
i
/I
To a solution of aniline (1.00 g, 10.7 mmol) and pyridine (0.849 g, 10.7 mmol)
in dichloromethane
(20 mL) was added 3-fluorobenzoyl chloride, (1.70 g, 10.7 nunol) at 0
°C and stirred for 1 hour.
After water was added, the mixture was extracted with dichloromethane. The
organic layer was
washed with brine, dried over Na2S04, filtered, and concentrated i.mder
reduced pressure to obtain
3-fluoro-N-phenylbenzanude (2.45 g). 'H NMR (DMSO-db) 8: 7.09-7.15(m, 1H),
7.33-7.40 (m,
ZH), 7.41-7.49 (m, 1H), 7.55-7.64(m, 1H), 7.73-7.84 (m, 4H); m/z 216.15
(M++11.
Next, a nvxture of 3-fluoro-N-phenylbenzamide (1.OO g, 4.65 mmol) and thionyl
chloride (3.4 mL)
was heated at 80 °C for 16 hours. After cooled to ambient temperature,
excess of thionyl chloride
was removed under reduced pressure to obtain 3-fluoro-N-
phenylbenzenecarboximidoyl chloride
(1.00 g).
Next, to a solution of anhydrous hydrazine (2.72 g, 54.9 mmol) in benzene (15
mL) was added 3-
fluora-N-phenylbenzenecarboxinudoyl chloride (0.800 g, 3.39 mmol) at 0
°C. After ha~~ing stirred
at room temperature for 16 hours, water was added and the mixture was
extracted with
diethylether. The organic layer was washed with saturated aqueous sodium
bicarbonate solution
and brine, dried over MgS04, filtered, and concentrated under reduced pressure
to provide 3-
fluoro-N-phenylbenzenecarbohydrazonamide (0.822 g).
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-41-
Next, a mixture of 3-fluoro-N-phenylbenzenecarbohydrazonamide (200 mg, 0.65
mmdl) and
cyanogen bromide (69.3 mh, 0.65 mmol) in methanol (3 mL) was heated at 90
°C for 48 houxs.
After having cooled to ambient temperature, the mixture was concentrated under
reduced pressure,
and the obtained residue was purified by preparative TLC (eluent:
dichloromethane -/ methanol =
95 / 5) to provide 5-(3-fluorophenyl)-4-phenyl-4H-1,2,4-triazol-3-amine (lOS
mg).. 'H NMR
(DMSO-do) b: 5.82 (s, 2H), 6.90 (t, ,I--7.3Hz, 1H), 6.98-7.27 (W , 3H), 7.28-
7.42 (m, 3H), 7.52-7.54
(m, 2H); m/z 255.25 (M~+1).
Preparation of intermediates
Method (v)-1
Phenyl(pyridin-3-yl)methanol
O H
MgBr
NI \ ~H + ~ \ ~ i \ ~ \
/ / /
To a solution of 3-pyridinecarboxaldehyde (1.00 g, 9.34 mmol) in
tetrahydrofuran (50 mL) was
added 1.09 M phenyl magnesium bromide in tetrahydrofuran solution (10.3 mL,,
11.20 mmol).
After the mixture was stirred at room temperature for 2 hours, water was added
and extracted with
ethylacetate. The organic layer was washed with brine, dried over MgSO,,,
altered, and
concentrated under reduced pressure. The resulting residue was purified by
silica gel column
chromatography (eluent: dichloromethane / diethylether = 4 l 1) to provide
phenyl(pyridin-3-
yl)methanol (1.07 g).'H NMR (CDC13-d): c~ 3.85 (1H, s), 5.83 (1H, sz), 7.20-
7.37 (6H, m), 7.69
(1H, ddd, J = 2.5, 2.5, 7.9 Hz), 8.38 (1H, dd, J = 2.5, 4.8 Hz), 8.51 (1H, d,
J = 2.5 Hz); MS m/z
186 (M+1).
Method (v°)-1
cyclobutyl(phenyl)methanol
O OH
\ \
To a solution of cyclobutyl(phenyl)methanone (1.00 g, 6.24 mmol) in methanol
was added sodium
borohydride (0.315 g, 7.49 mmol) at 0 °C. After the mixture was stirred
for 1 hour at 0 °C, water
was added and extracted with ethylacetate. The organic Iayer was dried over
MgS04, filtered, and
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-42-
concentrated under reduced pressure to obtain cyclobutyl(phenyl)methanol (1.03
g). '~P NMR
(CDCl3-c~: 8 1.74-1.88 (4H, t, m), 1.91-2.15 (2H, m), 2.63 (1H, m), 4.57 (1H,
d, J= 7.9 Hz), 7.23-
7.35 (SH, m).
Method (v)-2
1,1'-(chloromethylene)bis(4-chlorobenzene)
OH Cl
\ \ \ \
CI ~ / ~ / . CI CI ~ / ~ / CI
To a solution of 4,4'-dichlorobenzhydrol (4.7S g, 18.9 mmol) in
dichloromethane (400 mL) was
added thionyl chloride (2.81 g, 23.6 mmol) and 1H-benzotriazole (2.81 g, 23.6
mmol) at room
temperature. After the mixture was stirred for 10 minutes, it was filtered,
and to the filtrate was
added water and extracted with dichloromethane. The organic layer was washed
with 3% aqueous
sodium hydroxide solution, dried over Na~SO~, filtered, and concentrated under
reduced pressure
to provide 1,1'-(chloromethylene)bis(4-chlorobenzene) (5.14 g). 'H NMR (CDCl3-
a~: 8 6.05 (1H,
s), 7.26-7.32 (8H, m).
In the similar manner as described in Example 1-1 and with the use of
intermediates
described above, compounds in Example 1-2 to 1-167 as shown in Table 1 were
synthesized.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 43 -
Table 1
Melting Point
Example No. Structure 11~'V MS (M+l) or PLC Activity
retention time class
'_J
O N
/-~. w
S
Example 1-2 ~N 1 , 463,603 464 189,1 D
i )
N
HaCi wCHs
Example 1-3 ~ ~ N~"S ~ ~ 531,721 (m tho'd2C) D
..
N-~ ~._%
m
r
Example 1-4 I ~ N ~ \ 420,538 (m thod 2B) B
NON
S
w N Rt=4.41
Example 1-5 ~ / 420,538 B
N \ (method 2B)
w
l,
N.. N
Rt=4.49
Example 1-6 F ~ ~ N ' ~ 481,549 B
(method 2A)
0 0
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-44-
Table 1
Melting Point
Example No. Structure MVV MS (M+1 or HPLC . Activity
retention time class
H3C ~ N~.CH3
\ ..
_ ~2
Example 1-7 N S ~ 426,SS5 427 (method A) '~
N-N
\
N~N
j S \
Rt=4.52
Example 1=8 ~ ~ N I ~ 481,549 (method 2A) A
1 0
OH
~.
s
Exam le 1-9 'N~N~ ~ '' 443 6185 444 Rt = 2,75
p ~ ~ ' (method A)
~N'
i
J _~s ! ~ 7
Example 1-10 / ~ \ ' 480,6118 481 (method A)
I
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 45 -
Table 1
Melting
Point
Example Structure MW MS (M+1or HPLC Activity
No. ,
retention class
time
Ht
\
,
~
CIW / -N
~ N ~ ~-
N S \ /
Example ~ ~ 516,5405444 207,8 B
1-11
~ N.~
CIH
~O~-~ w
S 85
~ Rt= 2
Example ~ N 458,55 459,2 . A
1-12 ~'
(method
w 1 B)
/N\
w
N-N
Example N S ~ ~ 414,57 415,2 82.1-86.5 A
1-13 I
/N~
H0~ ~ , w.
S
Example N 1 ~ 416,55 417,2 214.4-215.2B '
1-14
/N~
w
N S U
Example 428,6 429,3 81.2-84.6 A
1-15 i
w
,N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-46-
Table 1
Melting
Point
or HPLC Activity
Example Structure MW MS (M+1)
No.
retention class
time
/ \
F ~ ~ / N
l ~~~ S 437 25 156-157 A
1 54 438
1
6
E
- ~ \ , ,
, w
xamp
e
~' GH3
N- N
/ ~ w
~ S
'
Example '' 440,612441 48,7 A
1-17 " N
w
H3G.N.GH3
w
N-N
1 1 495 496 4 A
E '~s 476 57
l ~
xamp ,, ~~ , ,
e 1- N
8 ,
i ~
H3G.N.G N3
H3C
N-N
E w 440 441 60 A
1 S 1 612 8
l
19
- r , .
xamp N ,
e
w
H~C~N~CH3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ 47 _
Table 1
Melting
Point
or HPLC Activit
Example Structure MVO' MS (M+1 y
No.
, retention class
time
F
N-N
w
~
S ~
Example N 462,566463 41,3 A
1-20 / F
i I
H3G.N.CH
3
f
N- N
~
~ w
~
Example S 485,6998484 Rt = 2.58 B
1-21 ~~N
1
(method
A)
~N~
f
HO~~ .
w 54
S ~ ~t - 2
Example N 446,6192445 . A
1-22 f
(method
A)
~N~
~ /
N-N
w
S
Example ~~N ~ ' 558,6217484 203,8 B
1-23
CIH
H C
3
GIH
,N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 48 _
Table 1
Melting Point
Example No. Structure M'Vi' MS (M+~) or HPLC Activity
retention time class
N-N
/ ~ w
Example 1-24 ~N S 1 ~ ci 461,031 462 56,2 A
w
H C N CH
w
!,
N-N
Example 1-25 ~N S ' ~' 428,6 429,3 135.8-138.6 A
~ ~
i
~N~
N-N
Example 1-26 ~N S 1 ' 440,61 441,3 150.8-151.7 A
~. N ~
/ \
F I \ N_N~ .~
S
Exa le 1-27 ~ / \ 467 57 468 2 169-170 A
mp ~ -~ ' ' (method B)
0
CH3
s
Example 1-28 i ~' N / \ 467,5? 468,2 Rt =3.21 A
i I ~- (method B)
w
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-49-
Table 1
Melting Point
Example No. Structure MW MS (M+1) °r PLC Activity
retention time class
m
/ ~
v
Example 1-29 ~ ~ ~N ~ ~ 453,54 454,06 69-70 A
OH
F ~~- S \
Example 1-30 ~ ~' '~ ~ 519,65 ~.2 (-38[ >300 A
~ ~ 1 ~o
0
K
GOOEt
~Ph
\ N S
Example 1-31 ~ . / 463,53 464 method C) D
\
OMe
_ COOEt
~Ph
\ N S
r
Example 1-32 r I 447,54 448 141-142 D
Me
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-50-
Table 1
Melting Point
or HI'LC Activity
Eaaznple No. Structure MW MS (l~~I+~) retention time class
N-N COOH ,
\ ~ ~ ~Ph
~N
Example 1-33 / ' 435,48 436 110-111 D
OMe
_ COOH
/ ~ .,,~Ph
N S
Example 1-34 ~ I 419,48 420 88-89 D
Me
Ph
N
Example 1-35 ~ 397,55 398 Rt ~ 5.08
(method C)
Me
i
s
Example 1-36 I i 482,693 483 146,8 A
.
w
,N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-51-
Table 1
Melting Point
or HPLC Activity
Example No. Structure MW MS (M+1) retention time class
I w
0
HO'~~~
N S Rt = 2.2
Example 1-37 ~ I i 444,557 445 (method A) B
N
I
o i "
"o~_~
Example 1-3S ~ I i 472,61 473 (method A)
~I
iNy
Example 1-39 ~ ~N S / \. 4S2,54 483,07 79-80 B
NN~
Example 1-40 ~ S ~ ~ 480,57 481,12 81-82 A .
i
~ ~ o
N HZ
Example 1-41 N r '1 442,63 443,3 59.5-62.4 A
I
~N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 52 -
Table 1
Melting
Point
or HPLC Activity
Example Structure 11'IW MS (M+1
No.
retention class
tune
1
N-N
~S
~N, ~ ''
Example 442,63 443,3 194-198.8 B
1-42 I
w
~N~
N- N _
~S y
Example ~N 456,66 457,3 108.2-109.1D
1-43 /
~I
w
~N~
~S
N 1
'
Example 486,64 487,3 149.5-155.2A
1-44 '
I
w
/NW
w
/ N_~ w
N S ~
Example 456,66 457,3 126.4-127.2B
1-45 I
w
/N~
N-N
S
Example 432,633433 47,1 A
1-46 ~f
H3C~N~CH3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-53-
Table 1
Melting Point
Example No. Structure ~ MS ~+1 or HPLC Activity
retention time class
N-N
,
S _
Example 1-47 ~ 390,552 391 (method B) C
H3C~N~CH3
w
1
N-N
~ w
Example 1-48 " N \ ° 416,547 417 Rt = 3.03
~ (method B)
H3C~N~CH3
w
1
N-N
Example 1-49 ~ ~N 427,574 428 46,9 D
I
NaCiNwCHs
~°~,CH3
~N~S
Example 1-50 ° 422,55 423 50,9 D
w
H3C~N~°H3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-54-
Table 1
Melting Point
Example No. Structure ~ MS (~+1~ or HPLC Activity
retention time class
,OH
N S o 394,497 395 114,6 D
Example 1-51
I
N
H3G~ ~CH3
N-N
Rt = 3.48
Example 1-5? ~ N 404,579 405 (method B) A
N
H3C~ ~GH3
w
N-N
Example 1-53 ~ ~ 418,606 419 (method B) A
H3C~N~GH3
H
Example 1-54 ~N~S~ ~ \: cH3 454,639 455 46,6 A
I
H~C~N~GH~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 55 -
Table 1
Melting
Point
or PLC Actis~ity
Example Structure M'VV MS retention class
No. (M+1 time
R
1
N-N
\
\
Example N S 432,614433 45 A
1-55 S
I
H,C~N'CH3
\
I
N
566 17 -2
N S I 7 567
~
Example ~ , , method A)
1-56 ~
/
\ I o ~ ~%
/ \
N-N
S 452 453
N~ 56 08
~
Example / \ , , method A)
1-57 ~
I
NHZ
F \ % _~
S
/
/
Example ' I ~ 495,58 496,08method A)
1-58
O OH
i
,i
N-N
~, ~I. ~S. w F A
'
N 1
Example f 494,583495 54,2
1-59 F
"
/ F
I
H~C~N'CH3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ 56 _
Table 1
llZelting Point
Example No. Structure MVV MS (M+1 or HPLC Acti~~ity
retention time class
n1 .
i-~
Example 1-60 " N 440,612 441 54,8 B
I
w
H3C~N~CH~
N-N _
/ N~S, ~
Example 1-61 ~ N~' 427,574 42S 79,9 A
H3C~N~CH3
,\ /
Example 1-62 ~N s ~ ~ 493,589 494 201,5 B
w ~ o
OH
o_ ~ 1
.+
s
Example 1-63 ~ ~ 1 ~ 508,56 509 211,1 B
i
w ~ o
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-.57._
Table 1
Melting
Point
Example Structure NIW MS or HPLC Activity
No. (M+1 retention class
time
1
,,.
N-N
Example. N* ~ ~ N , %' 508,56 509 98,7 A
1-64
0
w I o
OH
,i
-N ~ w
Example ,~c, ~ ~ ~ NHS' \ ~ 506,6313507,15229.8-232.0B
1-65 N i
hf~C
OH
0
f
N-N ~w
Example c~N~sr 1 ,1 401,4908402,09158.0-160.2D
1-66 H
,
oN
0
1
,,
r~
1 ~
N 427,574428 45,8 A
Example 7
1-6 I
w
H~C~N~CH3
i
N
=7
~ N
Example F 418,485419 (Rethod B
1-68 I B)
F~aC~NvCHa
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
__Sg _
Table 1
Melting
Point
Example Structure MW MS (1~~+1)or HPLC Activity
No. retention class
time
ci
1
N-N
~ i 46S 52 A
~ 7
S
69 i 467,078 ,
Example -
1 N
I
w
H3e'N~cH,
Ph
S- 'Ph
Et N
413,59 414 Rt = 5.64 A
Example (method
1-70 C)
H3C CH3
f
F \ / _~ w
S ~ .2
f
/
Example ~ ~ 468,56 469,09 (method A
1-71 I A)
NH
i
OH
I
F \ N
N I
~
Example ~ 462,55 463,09 62-63 A
1-72 ~
I
fl
N
/ ~
i
~ ~ N~S 538 27 l B
69 539
Example ~ ~ , , (method
1-73 B)
CH3 i
I
\ N\/~O~CHa
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
Table 1
Melting
Point
or HPLC Acti~~ity
Example Structure MW MS (11Z+1retention class
No. time
\ ,
F
w
I -~-
-
\
~N S \
~
Example ~ ~ 494,64 495,11 method B
1-74 I A)
HaCiNwCHa
CI
N-N
H3
Example N S ~ 546,519547 126-127 B
1-75
cl
w
OBn
CI~
N S ~~ 126 B
5
127
3
Example 434,99 435,2 .
1-76 -
.
/N~
~N
~
N S
~
Example 485,65 486,3 54.0-57.9 B
1-77
~N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-6~-
Table 1
Melting Point -
Example No. Structure ~ MS ~+1 or HPLC Activity
retention time class
t
1
N-N
~~ ~N~S~ ; ~ Rt = 3.17 B
Example 1-78 ( 469,65 470,3
(method B)
I
,N~
N_\ w
Example 1-79 l ~ N~S~ '~ 464,547 465,07 196-198 D
i
w I off
0
° N N
Example 1-80 ~ \ / N~S/ ~ 492,65 493 Rt = 2.96 A
(method A)
/N~
r
-N~~ _
\ / ~S.
Example 1-81 °~~N* ~ -~ N ' ~~ 507,62 508 179,1 A
/N~
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-61-
Table 1
Melting
Point
Example Structure MW MS (M+1or HPLC Activity
No. retention class
time
I
/
~N ~ ~~~
. w
\ ~S~ 506 507 177 C
~N. I ~., 63 3
Example ~ , ,
1-S? i
0
0
N-N _
1 \ / ~ , f 477 47S 1 A
83 N S ~ :' 59
l ~
E
- ~ , (meth d
e i A)
xamp
0
0
CI
N-N
HO~
~
Example S ~ ' CI 514,431515 114,4 D
1-S4 N
~
OH
O
1
F N _
Example / ~ ~ N~S ~ f c~ 549,499550 69,5 A
1-SS
I
H3C~N~CH3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
~- 62-
Table I
Melting
Point
Example Structure MW MS (M-~-1r PLC Activity
No. retention class
time
cl
1,
Example N-N 497,491498 53,7 A
1-86 H'~~'N~S ~~c~
I
H,C~N~CH~
CI
~ N
Example H~~~ ~S' ~ 498,432499 201,6 A
1-87 N ~ /
CI
I
OH
\
O
CI
/,
N-N
Example ~ / N~S ~,~ 496,416497 139,4 A
1-8S
el
I
off
0
F \ N-~ w
S \
f
/
Example ' I ~ 520,63 521,2 95-96 A
1-S9
'NH
O
90 N S ~ \ 44 05 61-62 A
l ~ 516 518
1
E
e , ,
-
xamp
~
/
Br
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
~- 63 -
.Ta- ble 1
lVlelting
Point
or HPLC Acti~~ity
Example Structure ~ MS retention class
No. ~+1 time
cl
f
H0,
N S ~ ~ 49 514 4 A
513 59
Example cl , ,
1-91 I
H C'N~CH3
a
CI
i
,
S ~~ 530 58 A
~ 1
92 cl 529,92 ,
Example N .
1-
cl
H3C'N~CH3
CI
N-N
le 1-93 iw 529,92 530 67,2 A
Exam ~N S 1 ~
p cl
cl
H3C'N~CH~
CI
-N
Example F / ~ / N~S~ 1 \; cl 550,439551 235,4 A
1-94
I
.
OH
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
._ 64 _
Table 1
Melting Point
Example No. Structure ~~ MS ~+1 or HPLC Activity
retention time class
C4
;'
N-N
Exam le 1-95 ~ ~ ~ ' ' ~~ 522,429 523 212 A
p ~ N S 1'~ci
OH
, w
f
HO %~
Example 1-96 0~ ~N '~ 458,58 459,2 98.7-103.7 B
w
/N~
CI
HC ;
S
Example 1-97 N 1 ' e' 536,522 499 95,7 A
I
o-
0
K
CI
i
N- N
Example 1-98 ~N~S r / CI 534,506 467 127,2 A
i
w I O_
O
K
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
._ 65
Table 1
Melting
Point
r PLC Acti~~ity
Example Structure MW MS (l~Z+1retentionclass
No. time
CI
1 fJ
F N'N ,
99 ~ ~ ~ NO'S ICI 635,588636 51,2 A
Example -
1
i ~O
-~NJ
0
N-N
Example ~ ~ v / N~S~ ~~ 593,551594 48,5 A
1-100 ci
/ I CH3
N
O~ ~CH3
CI
w
Example 564,466567 90,4 A
1-101 N S l~ci
I
0 off
cl'
1
HO,
~~
Example N S 485,093486 53,4 A
1-102 I
H3C~N~CH3
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 66--
Table 1
Melting Point
Example No. Structure ~ MS ~+1) or HPLC Activity
retention time class
cl
1
,,
N-N
HC
Example 1-103 N 470,034 471 89,3 A
OH
O
CI
1 /
HO~ //
Example 1-104 ~N>'Sf 1 ; ~ cl 527,474 528 109,5 D
I -
w
N
H~C~ ~CH3
CI
/~
"a°~ ~ -' ' ~~ Rt = 6.13
Example 1-105 N S ~~cl 482,48 482 (method C) A
I
H,c cH,
Cl
1 ;
F N- N
Example 1-106 / ~ ~N~S ~ / CI 588,529 551 164,5 A
O K
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
.~ 67;
Table 1
Melting
Point
or HPLC Activity
Example Structure NIW MS (1~~I+1retention class
No. time
ci
N-N ! _ '
Example ~s \ 470,42 470,06
1-107 "3~
, c~ (method
N A)
I
~oH
ci
N-N
"3C
5
N
/
Example ~ 539,53 539,11 60-61 B
1-108
c~
I
Cy
,,
S
~
Example '~ I 591,54 591,11 84-85 B
1-109 ci
CI
F NN
Example / ~ ~N~S ~ ! CI 549,455550 204 A
1-110
NHZ
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
:- 6s_
Table 1
Melting Point -
Example No. Structure MW MS (M+1 or HPLC Acti~~ity
retention time class
cr
Example 1-111 N S N 558,19 559 57,6 A
'f 1
H3C N CH3
Cf
N-N
Example 1-112 ~N S ,NH 468,066 469 146,5 D
~I
w
N
H3C \CH3
Cr
/.
Example 1-113 ~N~S ~ ~~ 526,102 527 59,4 A
O.~CH
3
N
HyC~ ~CH3
Cf
N-N
Example 1-114 H'e~N~'S l ~ef 567,535 568 60,5 A
i ('~
N' j-OH
'~'0
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
6~
Table 1
Melting
Point
Example Structure MW MS (M+1 or HPLC Activity
No. l
retention c
time ass
cl
HOC' '~
Example N S 1 ~cl 541,5 54 70,3 A
1-115 i
H
N~OH
0
CI
I i
HOC '~
l S l~ 448 49S 90 A
116 497 3
1
e N , ,
Examp cl
- i
N Hz
0
CI
H3C~ r
N-N
N S \
117 ~ ~ ~ ' 539,53539,09 84-85 A
Example .
1-
cl
OH
CI
/ \
p / \ N~
S \
Example ' ~ 591,54591,06 108-109 A
1-118 ~
I
cl
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_'70:_
Table 1
Melting Point
Example No. Structure MW MS (M+1) or HFLC Activity
retention time class
cl
1 ,
N~ ,N-N _
~N~S, I~cl 526,49 526(Ivl~, 204 4 A
Example 1-119 0 528(M+2) '
I
/N~
CI
1 /
Example 1-120 ~N~s '~ c 510,103 511 74,6 B
/ I CHa
N
H3~s \CH3
CI
F
Example 1-121 ~ ~ NHS 1 / CI 563,482 564 75,5 A
I
O N H.,
a
1
,.;
%\
Example 1-122 ~'°~N'~s~ 1 , cl 512,459 513 75,5 A
I
O OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
71-
Tabled
Melting
Point
Example Structure MW MS (M+1~or PLC Activity
No. retention class
time
a
,r~
~/
N-N
H
C ~
Example a 511,475512 61,4 A
1-123 N S ' r~c,
I
0 NH,
ci
"'~N~S/ 1~
~~
Example ~ ~ 581,565582 82,3 A
1-124
O N
[
/
~
OH
CI
~r
N-N
H / ~
Example.l-125N , ' ci 555,527556 53 A
I
0 N'~/OH
H
ci
1
'
-N
H,c~N~~s
~
oi 581,565582 56,4 A
Example ,
1-126 ~I
w
o ~
o
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
Table 1
Melting
Point
or HPLC Activity
Example Structure ~ MS ~+1 retention class
No. time
cl
\
i
N-N 36
HOC Rt=2
S
E:~ample ~ \ 541,5 541,1 . A
1-127 N (method
A)
I /, C CI
H3C~N~CH~H
CI
\
/
N-N 77
c'~ Rt =2
~ i~
H
Example 3 527,52 527,11 . A
1-128 - (method
s A)
N / \
I ~ a
H3C~N~OH
CI
/
N-N
H C'
Example 3 N S / \ 527,47 527,04 Rt =2.42 A
1-129 w
(method
A)
O GI
,NH OH
H3C
Cl
i
F 51 08 Rt =2.49 A
/ \ N~S 3 593
5
Example \ , , )
1-130 ~ / 9 (
w ~ method
A
I , o a
H3C~N~CH~H
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ 73 ._
_Ta- ble 1
Melting Point
Example No. Structure MW MS (M+1 or HPLC Activity
retention time class
a
/ \
Exam le 1-131 ~ I~S 579,52 579,09 Rt =2.82 A
p / \ (method A)
w
I ~ cl
H3C~N~OH
CI
/ \
Example 1-132 / ~ N~S / \ 579,48 579,09 (method A) '~
I ~ o cl
,NH OH
H3C
a
~ /N/-N\\
~C~N~S
/ \
Example 1-133 ~ ~ o ~ ~Im 776,613 777,01 Rt =3.46
(method A)
H~C~N1CH~
CI
C~
/~
Example 1-134 / N S ~ 550,409 551 247-249 A
ci
COOH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
~- 74-
Table 1
Melting
Point
Example Structure MVV MS (l~Z+1)or HPLC Activity
No.
retention class
time
cl
1,
N-N _
H C
/ '
N
Example S 1 / CI 599,58 600 44,8 B
1-135 ~ I
~oH
0
OH
CI
w
1 /
-N
C
H
O
N S 1 ~ CI
Example ~ ~ 622,618623 100,4 B
1-136
O N
~~-~I
/NH;
~
0
CI Chiral
w
1 f
HOC
N 1 ,
CI
Example i I 595,592596 58,9 A
1-137
0
OH
CI
N-N
HO~
~
Example N S ~ 485,44 485,1 205.2 L10
1-138 cl
,Ny
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- .75.~
Table 1
llZelting
Point
Example Structure 11~' MS (M+1or HPLC Activity
No.
retention class
time
t
ci
i \
N-N
H3C~
~
N
Example S 567,54 568 122 A
I-139 ., / ~
ci
0
N
'~OH
Cf
~N" i
~~S
Example -' ~ \ 619,54 620 87 A
1-140
cl
0
,N
~OH
CI
H
N ~ \
Example I ~ ~ cl 581,57 582 86 A
1-141
N
~~O
''''
O~rH
CI
/ \
S
Example I ~ " 'ol 633,57 A
1-142
0
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-'76 _
Table 1
Melting Point
Example No. Structure MW MS (M+1 or HPLC Activity
retention time class
Ht
cl
,
N-N
H,c~ ~~s ~~cl 569 5144 570 33.6-86.1 A
Example 1=143 N
0
\ ~ N~o~c~
0
cl
. -,
_ / -,
Example 1-144 H3°~N' S l~cl 583,5415 584 61.1-62.9 A
\ ~ N~°'cH
II 3
O O
CI
,
N-N
H G~ ~S~
Example.l-145 3 N ~ ~ cl 555,4873 556 110-113 B
0
\ ~ N~OH
0
CI
,
,.
Example 1-146 H3c~N~s~ ~~cl 569,5144 570 95-98 A
N~OH
1~ ~f\
0 O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-.77_
Table ~1.:
Melting Point
Example No. Structure MW MS (M+11 or HPLC Activity
retention time class
ci
I
~N~ ~ , w.
Example 1-147 '~~N S \ .~' c~ 554,54 555 66.7-72-3 A
i
/N~
l
N,N
Example 1-148 / ~ N'~'~N ~ 420,49 421 177.5-178.4
H3C~N~CH3
Example 1-149 N ~ Rt=4.44
N S (method 2C)
N-N
f
N-N
~>'-S
N Rt=5.01
Example 1-150 ~ , / 1 / \ (method 2A)
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
~g._,
Table l
Melting Point
Example No. Structure MW MS (M+1 or HPLC Activity
retention time class
f
~N
I ~,~--s
Example 1-151 ~ N Rt=4.52
/ ~ (method 2B)
N
C H3
o \ \
I
S N F Rt=4.48
Example 1-152 ~ 11 , ~ ~ F (method ZD)
\ I ~ N F
O
CH3 O-CH3
O
\ /
Example 1-153 ~ S Rt=4.25 B
~N ~ ~ (method 2D)
N~ ~ O
H~O~O \ I O N CHI
CH3 GI
O _
GI
Exam le 1-154 ~ S N Rt=4.64 B
p ~ 1j . ~/ (method 2D)
H3C~ / O N~N~~~CI
O ~~\
~O
0 ~N~ CH3
WS I \ NwCHs
Rt=3.61 D
Example 1-155 ~ ~,~ N,~N (method 2D)
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-~79~--
Table 1
Melting.Point
Example No. Structure 1\'IW MS (11~I+1) or HPLC Activity
retention time class
H3C
CH3 N-CH3
O
Example 1-156 ~ \ s \ ~ Rt=4.56
A
~N ~ \ F (method 2D)
H30,0 \ ' O N.N~F
CH3
O
~-CH3
Example 1-157 s ,~ ~ Rt=4.08 A
HaCO - ~-N (method 2D)
O N~N
CH3
O HaC
/ \ N-CH3
' " Rt=4.19
Exam le 1-158
p H3~ - S\- (method 2D)
o \ / N
o N~ ~~
N
CHI H3C~N_CH~
O _
\ /
Exam le 1-159 ~ s Rt=4.35 A
p rN (method 2D)
HOC, ~ / O N~N; ~ O
0 \ CH
CH3 O-CH3
O
Exam le 1-160 ~ ~ ~ ~ Rt=4.47 A
p s~N ~ \ (method 2D)
H C ~ ' O N,N~ F F
~0
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ g0.~
Table 1
Melting Point
Example No. Structure MW MS (M+1) or HPLC Activity
retention time class
O H' O_CH3
Example 1-16I ~- S ~ ~ Rt=4.43
N (method 2D)
H3o. ~ ~ O Nr r ~ ~ ci
O \
CH3
O
!\
Rt=4.35
Example 1-162 H~o S ~ (method 2D)
O ~ ~ O N// N,
N
C Ha
C(
I
Example 1-163 ~ ~ c~ Rt=4.37 E
(method 2D)
H3C, r ~N
O ~ I O N~i
CH3
O
Exam le 1-164 ~ ~ ~ Rt=4.06
(method 2D)
/ o
c~
G~ .
N
Rt=4.40
Example 1-165 o S N /
11 ~ F (method 2D)
N,N
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
_ g~ _
Table l
_,_ _ Melting Point
Example No. Structure MW MS (M+1 °r PLC Activity
retention time class
~~CHa
COQ
N F Rt=4.07
Example 1-166 o s~N ~ \ F (method 2D)
N_N F
O
O~F
F
O
Example 1-167 S \ '~ Rt~4.y7
_ ~ N (method 2Dj
N~N
'' CI
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
8~_
[Example' 2-1]
Method B
3-[3-(2,2-diphenylethyl)-5-(3-fluorophenyl)-4H-1,2,4-triazol-4-yl]benzoic acid
o / o
HZN \ O~CH3 CI \ F HsC~O \ I N \ F
/ I / O H I /
I CI F
H3C~0 \ \ F
N
H
o I/
O
F
To solution of ethyl m-anunobenzoate (5.36 g, 32.4 mmol) in tetrahydrofuran
(100 mL,) was added
1-hydroxybenzotriazole (7.67 g, 56.8 mmol), triethylamine (3.61 g, 35.7 mmol),
1-ethyl-3-(3-
dimethylaminopropyl) carbodiimide (10.9 g, 56.8 mmol), and m-fluorobenzoic
acid (5.00 g, 35.7
rnmol) at room temperature and stirred for 4 hours. After water was added, the
mixture was ex-
tracted with ethylacetate. The organic layer was washed with brine, dried over
Na~SO~, filtered,
and concentrated under reduced pressure to obtain ethyl 3-[(3-
fluorobenzoyl)amino]benzoate.
Next, a mixture of ethyl 3-[(3-fluorobenzoyl)amino]benzoate (1.11 g, 3.86
mmol) and thionyl
chloride was heated at 80 °C for 16 hours. After cooled to room
temperature, the excess of thionyl
chloride was removed underreduced pressure and obtained ethyl 3- f [(IE,Z)-
chloro(3-
fluorophenyl)methyl ene] amino ] benzoate
(1.11 g).
Next, to a solution of ethyl 3-{[(IE,Z)-chloro(3-
fluorophenyl)methylene]amino]benzoate (320 mg,
1.04 xnmol) in acetonitrile (5 mL) Was added 3,3-diphenylpropanohydrazide (300
mg, 1.25 mmol)
and the mixture was heated to 90 °C for 16 hours. After having cooled
to ambient temperature, the
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 83._ '
iiiixture was concentrated under reduced pressure. The obtained residue was
purified t~~ice by
preparative TLC (eluent: dichloromethane / methanol = 95 /.5 and then with
ethylacetate / hexane
1 / T) toprovide ethyl 3-[3-(2,?-diphenylethyl)-5-(3-fluorophenyl)-4H-1,2,4-
triazol-4-yl]benzoate
(78.0 mg). 'H NMR (DMSO-d6) 8: 1.31 (t, J--7.3Hz, 3H), 3.36 (d, J--7.6Hz, 2H),
4.32 (q, J--7.3Hz,
2H), 4.45 (t, J--8.2Hz, 1H), 7.04 (d, J--7.9Hz, 1H), 7.08-7.24 (m, 12H), 7.33-
7.35 (m, 1H), 7.57-
7.59 (m, 1H), 7.68 (t, J--l.9Hz, 1H), 7.72 (t, J--7.9Hz, 1H), 8.15 (d, J--
7.9Hz, 1H); mlz 492.2
(M"+1).
To a solution of ethyl 3-[3-(2,2-diphenylethyl)-5-(3-fluorophenyl)-4H-1,2,4-
triazol-4-yl]benzoate
(72.0 mg, 0.15 mmol) in ethanol (2 mL) was added 1N aqueous sodium hydroxide
solution at room
temperature and stirred for 16 hours. The mixture was concentrated under
reduced pressure,
neutralized with 1N HCl aqueous solution, and extracted with ethylacetate. The
organic layer was
dried over Na~S04, filtered, and concentrated under reduced pressure. The
obtained residue was
purified by preparative TLC (eluent: dichloromethane / methanol = 95 / 5) to
provide 3-[3-(2,2-di-
phenylethyl)-5-(3-fluorophenyl)-4H-1,2,4-triazol-4-yl]benzoic acid (37.0 mg).
'H NMR (DMSO-d6) 8: 3.36 (d, J--7.8Hz, 2H), 4.44 (t, J--7.9Hz, 1H), 7.03-7.22
(m, 13H), 7.35 (q,
J--7.9Hz, 1H), 7.52 (d, .l--7.9Hz, 1H), 7.67-7.72 (m, 2H), S.l 1 (d, J--7.9Hz,
1H).
mp 233-234 °C;
Molecular weight : 463.51
MS (M+H): 464
Activity Class : D
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
s4..
In the siiriilar manner as described in Example 2-1, compounds in Example 2-2
to 2-3 as
shown in Table 2 were synthesized.
Table 2
Example No. Structure MW MS (M+1) Melting Point Activity
or HPLC class
retention time
Example 2-2. I \ 419,5 420,3 128-129 D
i
F I ~ I,N~ \
Example 2-3. w 491,56 492,2 Rt =2.92 D
1 (method B)
v r y,,
I N 1
~I
0
CH3
Example 3-1
(4-{3-cyclopropyl-5-[(diphenylmethyl)sulfinyl]-4H-1,2,4-triazol-4-
yhphenyl)dimethylamine
1 ~1 1
N-N N-N
~ w / ~ ,
V N/ 'S I N S I
O
,N
H3C~N~CH3 H3C ~CH3
To a stirred suspension of (4-{3-cyclopropyl-5-[(diphenylmethyl)thio]-4H-1,2,4-
triazol-4-yl}phen-
yl)dimethylamine (370 mg, 0.87 mmol) in dichloromethane was added m-
chloroperoxybenzoic
acid (374 mg, 2.17 nunol) at room temperature. After the mixture was stirred
for 16 hours, it was
filtered, and the filtrate was washed with sodium bicarbonate solution, water,
and then with brine.
The organic layer was dried over NaZS04, filtered, and concentrated under
reduced pressure to
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
85 ~-
obtain (4-{3-cyclopropyl-5-[(diphenylmethyl)sulfinyl]-4H-1,3,4-triazol-4-
yl}phenyl)dimethyl-
amine (326 rng),
1H NMR (DMSO- .d6) 8 0.83-0.90 (m, 4H), 1.49-1.54 (m, 1H), 3.54(s, 6H), 5.87
(s, 1H), 7.?2-
7.31(m, lOH), 7.44 (d, J= 8.6 Hz, 2H), 8.30 (d, J= 5.6 Hz, ?H).
Molecular weight : 44?.58
MS (M+H): 443
Activity Class : A
In the similar manner as described above and with the use of intermediates
described above,
compounds in Example 4-1 to 4-73 as shown in Table 3 were synthesized.
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
:_ 86-
Table 3
Example Structure MolecularActivity
No.
Wei ht Class
cl
p !,
4_1 H~~N~S~ ~~cl 542,48 A
O. GH
3
O
GI
4-2 ~s~ ~1~GI 528,46 A
H3c'~-~
N
i
OH
~
'IO
O
CI
CHI _
O=S.N~ 5
4-3 " N I ~ 576,57 A
GI
I
HaG.N.GH3
CI
\
/
N-N
H3C~\~, ~.S
4_4 N i \ 541, 50 A
cl
0
HaC'N~OH
CI
\
/
N-N
H~C''~C~
~'S
4-5 N 583,54 A
i ~
~-
~
o cl
OH
OH
CI
\
/
N-N
H3C~~
~-.5
4-6 N 572,46 A
~ / \
GI
OH
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
87-_
Table 3
Example Structure MolecularActivity
No.
Wei ht Class
Cf
HzN~_ ~~~,
I~
4-7
cl 484,45 A
H3C~N'CH3
C1
1
H C'~-L ~ i ~~,
3 N S l~
4_8 cl 527,47 A
~
I
NH
OOH
GI
w
1 f
HC~
3
S I~
4-9 N 569,51 A
GI
~~
I
v 'NH
O~O~CH3
I I
O
N-N
4-10 "~~N~'S ~ ~ G, 597,56 A
0
\ ~ ~~O~CHa
II
Fi
O
CI
~
1
.
N-N _
4-11 H3G~N~5 ~ ~~ 51 A
569
c, ,
0
w I N~oH
H ~~O
CI
i
1
f
N-N
HC~
S 1
3
4-12 ~ 585,55 A
~ GI
I
o
\.
,
N
HOI 'OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
gg._
Table 3
MolecularActivity
Example Structure Wei Ht Class
No.
cl
1
l
N-
S ~
~
4-13 ~ cl 496,46 A
N
I
w
H
C.N.CHa
3
cl
1
4-14 "~~ 581 A
~S~ ~~ci 57
N ,
~,.OH
~~'(N
O
CI
4-15 "~c~ 54 A
~'S~ ~ ~ 580
c, ,
N
~N~H
w N~O
O
_ CI
.~ ~NJ-N'\
H
C~
~
3
4-16 N 541,50 A
S
~ \
ci
HN~OH
II
O
-- CI
N-N
H
C~
~
4-17 S j \ 513,49 A
'
N
cl
HN,fOH
CI
\
/
N-N
C'~C ~
H
~
4-18 S 571,53 A
N / \
r
c ci
"O~.N.CHOH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-, 89--
'Table 3
MolecularActivity
Example Structure Wei ht Class
No.
cl
4-19 "3c'~ 51 A
~'s \~- 556
N ,
cl
OH
fIO
O
C~
i
H~C~
~S
N
4-20 I 533,95 A
~ ~ cl
~
w
N.CH~
CHy
CIN
CI
\
/
N-N
H3C'''~-~
S
4-21 N / \ 555,53 A
i
I cl
~
NH O
C' v -OH
H
3
CI
\
/
N-N
H
C
3
4-22 N S 526,49 A
/ ~
CI
OH
O
OI
\
/
N-N
H
4-23 N / \ 499,42 A
~ I cl
NOZ
CI
H
C
i
N-N
4-24 3 579,59 A
s
N / ~
o ci
H30.N,OHO N'
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
90-
'Table 3
Example No. Structure Molecular Activity
Wei ht Class
cl
~r
N-N
4-25 ~N S N~.," cl 496,46 A
I
w
H3G,N.CH3
CI
/ \
N-N
H3G'~.rC ~S
4-2g N / \ 469,44 A
cl
NHZ
CI
/ \
N-N
H3C'~-C,N~S
4-27 / \ 528,46 A
o cl
H C~O OH
3
GI
~ rNr-N\\
HsC~ ~S
4-28 N / ~ 528,46 A
cl
0
O OHCH3
CI
l \
N-N '
H~C'~,~N~S
4-29 / \ 484,45 A
~ I cl
O.CH3
CI
i \
N-N
H~C''~ N ~ S
4-30 / \ 569,59 A
~ I cl
0~~
HN~O~CH3
IIO
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
:_:91-
'Table 3
Example No. Structure Molecular Activity
Wei ht Class
cl
/ \
~ ~N~-N
H3C
4-31 N / \ 514,43 A
c cl
OH OH
CI
/ \
~ /N/-N\\
HaC~ ~S
4-32 N ~ \ 514,43 A
cl
OH
O DH
CI
/ \
~ /N/-N\\
H3G~ ~S
4-33 N / \ 470,42 A
I
cl
OH
C!
N-N _
H3C'\~N~g'
4-34 °' 563,94 A
~ NH
~OH GIH
[~O
GI
- ~-N _
4-35 H3G~N~~ ~ ,. cl 581,57 A
0
~N OH
CI
/ \
.~ jN~-N '
H3c~N~'S
4-36 ~ I ~ 527,47 A
cl
HN~OH
IIO
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
._ 92-
'Table 3
MolecularActivity
Example Structure Wei ht Class
No.
cl
~ !Nj-N
H
C~
~
3
N
S
4-37 ~ ~ 542,48 A
\ I
GI
~- o
o~o
CH3
CL
\
N-N
H~C~
~S
4-38 N 537,51 A
~
~ ~
I
cl
w
HN
flO
CI
\
i
N-N
H3C~N'"S
4-39 ~ \ 547,53 A
cl
HN. ,O
O CHa
CI
\
/
~ /N/-N
C~
~
H
a
4-40 N 625,62 A
S
~ / ~
cl
O~. ,N. .,O
s S'
CH
H C'
3 O O '
GI
~ /N/-N
H
C~
~
3
4-41 N 528,46 A
S
/ \
I
GI
O
O~ OH
r F
N
-N
~ /
/
H
C~ ~
3
4-42 S 491, 67 B
N
r
_
I
O
w
K
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
..:93-
'Table 3
MolecularActivity
Example Structure Wei ht Class
No.
\ F
I,
N-N
H3C'~L -
4-43 N 491,67 A
_
o
Ky
O
F
r
N-N
4-44 H'~~N~S 491,67 A
p _ K.
O
I\
/ F
N-N
C ~''~, ~.
H
3
4-45 S 467,61 B
N
i
\ I O.CH
a
O
F
N-N S I
H3C'~C~ Y
4-46 N 467,61 A
/
I
\ .CH
O
F
N_N S I /
H C~~ y
4-47 467,61 A
I
\
.CHa
O
\ F
/
N-N
H
C'
4-48 3 453,58 A
N
i
\ I OH
O
-- F
N-N\\
H
C'~ ~
a
4-4g S 453,58 A
N
/
\ I OH
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
:_.94-
'Table 3
Example No. Structure Molecular Activity
Wei ht Class
i
F
~ jN~-N
H3C~ ~.S
4-50 N 453,58 B
i
w I off
0
H'C 1
O O
N-N i
4-51 N S 504,70 A
HyC~N~CH3
HO O
I w
N-N
N S
4-52 ~ 513,10 B
H3C~N~CH3
CIH
CI
~ ~N~-N~~
HsC~ ~S
4-53 N / ~ 555,53 A
o cl
H3C.N.C O.CH3
C1
H3C~\~CN~S~
4-54 °' 513,49 A
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
- 95=,
Table 3
MolecularActivity
Example Structure Weic Class
No. ht
ci
~ f
N-N
H
C~
~
~.
~
a
N
S 1
f , c~
4-55 ~- ~ 625,62 A
1
N
off
0
ci
s
.
N-N
H3C~
~S
l-
4-56 N 625,62 A
'
~
l
w
0
~'~' N~ off
h
c
i
I
N-N
4-57 H3c'~N' I ~ 480,40 B
ci
~ j off
0
F
~r
H'~ 8 A
~'S ~
4-58 N 480,5
' F
i I
W
HN~OH
H C
~
S 1~
4-59 ~. 506,62 A
F
l
w
OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
'Table 3
Example No. Structure Molecular Activity
Wei ht Glass
cl
~.
N-N _
H3C~N~~S~ ~ f
CI
4-60 ~ ~ 581,57 A
N
O OH
F
N-N g
H3C~NY
4-61 , H~c c,~Ha 427,54 A
~ ~ off
0
F
N-N S
H3C~Ny
4-62 ~ H3c cH3H3 441,57 A
I °~cH
3
HO
N_N
N S
4-63 462,66 A
w
H3C'N~CH3
F
1 /
N-N
H Cue,
4-64 3 N S ~ ' F 493,58 A
~I
OH
O
F
N-N
H C''
4-65 ' N S ~ / F 494,61 A
~i
H3C'N~OH
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
~- 97--
Table 3
MolecularActivity
Example Structure Wei ht Class
No.
F
N-N
H C ~~
~-
N
S
f' F
4-66 ~ ~ 548,66 A
N
~OH
[~O
F
N-N
HC~
4-67 3 N S , / F 538,62 A
~
w
OH
HO~N'CH~
F
N-N
HC~
4-68 3 N S ~ / F 522,62 A
~~
I
~NH O
H~C~OH
F
N-N
H
e~
~.'
' 508,59 A
69 N
4 S ~ f F
-
~
w
OH
H3C.N.CHO
F
r
N-N
HC~
4-70 ' N S ~ ' F 522,62 A
I
~'
O'CH
3
H
C.N.CHO
3
CH3
N-N
c'~! ~-. ,,
H
3 A
4-71 S 449,6
N
w ~ OH
O
CA 02542682 2006-04-13
WO 2005/039569 PCT/EP2004/011101
-98
'Table 3
Example Structure MolecularActivity
No. Wei ht Cfass
cl
1
HC~
4-72 3 N S ~ '' ' 581,57 A
~
I
w ru
OH
O
CI
1 /
N-N
H3c~
~s ~ '
4-73
N 563,98 A
' cl
H CIH
N~O
CH3