Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST ~.E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional vohxmes please contact the Canadian Patent Oi~ice.
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Atty Docket No. 10271-119-228
EphA2 AGONISTIC MONOCLONAL ANTIBODIES AND
METHODS OF USE THEREOF
1. FIELD OF THE INVENTION
[0001] This application claims the benefit of U.S. Provisional Application No.
60/524,177, filed November 20, 2003, and is a continuation-in-part of U.S.
Nonprovisional
Application No. 10/436,783, filed May 12, 2003, which claims the benefit of
U.S.
Provisional Application Nos. 60/379,368, filed May 10, 2002, 60/418,204, filed
October 14,
2002, and 60/460,358, filed April 3, 2003, each of which is incorporated by
reference herein
in its entirety.
[0002] The present invention relates to methods and compositions designed for
the
treatment, management, or prevention of cancer. The methods of the invention
comprise the
administration of an effective amount of one or more antibodies specific for
EphA2,
preferably monoclonal antibodies, that are EphA2 agonists and/or
preferentially bind epitopes
on EphA2 that are selectively exposed or increased on cancer cells relative to
non-cancer
cells. The invention also provides pharmaceutical compositions comprising one
or more
monoclonal antibodies of the invention either alone or in combination with one
or more other
agents useful for cancer therapy. Diagnostic methods and methods for screening
for
therapeutically useful anti-EphA2 antibodies are also provided.
2. BACKGROUND OF THE INVENTION
Cancer
[0003] A neoplasm, or tumor, is a neoplastic mass resulting from abnormal
uncontrolled cell growth which can be benign or malignant. Benign tumors
generally remain
localized. Malignant tumors are collectively termed cancers. The term
"malignant"
generally means that the tumor can invade and destroy neighboring body
structures and
spread to distant sites to cause death (for review, see Robbins and Angell,
1976, Basic
Pathology, 2d Ed., W.B. Saunders Co., Philadelphia, pp. 68-122). Cancer can
arise in many
sites of the body and behave differently depending upon its origin. Cancerous
cells destroy
the part of the body in which they originate and then spread to other parts)
of the body where
they start new growth and cause more destruction.
[0004] More than 1.2 million Americans develop cancer each year. Cancer is the
second leading case of death in the United States and, if current trends
continue, cancer is
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
expected to be the leading cause of the death by the year 2010. Lung and
prostate cancer are
the top cancer killers for men in the United States. Lung and breast cancer
are the top cancer
killers for women in the United States. One in two men in the United States
will be
diagnosed with cancer at some time during his lifetime. One in three women in
the United
States will be diagnosed with cancer at some time during her lifetime.
[0005] A cure for cancer has yet to be found. Current treatment options, such
as
surgery, chemotherapy and radiation treatment, are often either ineffective or
present serious
side effects.
Metastasis
[0006] The most life-threatening forms of cancer often arise when a population
of
tumor cells gains the ability to colonize distant and foreign sites in the
body. These
metastatic cells survive by overriding restrictions that normally constrain
cell colonization
into dissimilar tissues. For example, typical mammary epithelial cells will
generally not
grow or survive if transplanted to the lung, yet lung metastases are a major
cause of breast
cancer morbidity and mortality. Recent evidence suggests that dissemination of
metastatic
cells through the body can occur long before clinical presentation of the
primary tumor.
These micrometastatic cells may remain dormant for many months or years
following the
detection and removal of the primary tumor. Thus, a better understanding of
the mechanisms
that allow for the growth and survival of metastatic cells in a foreign
microenvironment is
critical for the improvement of therapeutics designed to fight metastatic
cancer and
diagnostics for the early detection and localization of metastases.
Cancer Cell Signaling
[0007] Cancer is a disease of aberrant signal transduction. Aberrant cell
signaling
overrides anchorage-dependent constraints on cell growth and survival (Rhim,
et al., Critical
Reviews in Oncogenesis 8:305, 1997; Patarca, Critical Reviews in Oncogenesis
7:343, 1996;
Malik, et al., Biochimica et Biophysica Acta 1287:73, 1996; Cance, et al.,
Breast Cancer Res
Treat 35:105, 1995). Tyrosine kinase activity is induced by ECM anchorage and
indeed, the
expression or function of tyrosine kinases is usually increased in malignant
cells (Rhim, et
al., Critical Reviews in Oncogenesis 8:305,1997; Cance, et al., Breast Cancer
Res Treat
35:105, 1995; Hunter, Cell 88:333, 1997). Based on evidence that tyrosine
kinase activity is
necessary for malignant cell growth, tyrosine kinases have been targeted with
new
therapeutics (Levitzki, et al., Science 267:1782, 1995; Kondapaka, et al.,
Molecular &
Cellular Endocrinology 117:53, 1996; Fry, et al., Current Opinion in
BioTechnology 6: 662,
-2-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
1995). Unfortunately, obstacles associated with specific targeting to tumor
cells often limit
the application of these drugs. In particular, tyrosine kinase activity is
often vital for the
function and survival of benign tissues (Levitzki, et al., Science 267:1782,
1995). To
minimize collateral toxicity, it is critical to identify and then target
tyrosine kinases that are
selectively overexpressed in tumor cells.
EphA2
[0008] EphA2 is a 130 kDa receptor tyrosine kinase that is expressed in adult
epithelia, where it is found at low levels and is enriched within sites of
cell-cell adhesion
(Zantek, et al, Cell Growth & Differentiation 10:629, 1999; Lindberg, et al.,
Molecular &
Cellular Biology 10: 6316, 1990). This subcellular localization is important
because EphA2
binds ligands (known as EphrinsAl to AS) that are anchored to the cell
membrane (Eph
Nomenclature Committee, 1997, Cell 90:403; Gale, et al., 1997, Cell & Tissue
Research 290:
227). The primary consequence of ligand binding is EphA2 autophosphorylation
(Lindberg,
et al., 1990, supra). However, unlike other receptor tyrosine kinases, EphA2
retains
enzymatic activity in the absence of ligand binding or phosphotyrosine content
(Zantek, et al.,
1999, supra). EphA2 is upregulated on a large number of aggressive carcinoma
cells.
Cancer Therapy
[0009] One barrier to the development of anti-metastasis agents has been the
assay
systems that are used to design and evaluate these drugs. Most conventional
cancer therapies
target rapidly growing cells. However, cancer cells do not necessarily grow
more rapidly but
instead survive and grow under conditions that are non-permissive to normal
cells (Lawrence
and Steeg, 1996, World J. Urol. 14:124-130). These fundamental differences
between the
behaviors of normal and malignant cells provide opportunities for therapeutic
targeting. The
paradigm that micrometastatic tumors have already disseminated throughout the
body
emphasizes the need to evaluate potential chemotherapeutic drugs in the
context of a foreign
and three-dimensional microenvironment. Many standard cancer drug assays
measure tumor
cell growth or survival under typical cell culture conditions (i.e., monolayer
growth).
However, cell behavior in two-dimensional assays often does not reliably
predict tumor cell
behavior in vivo.
[0010] Currently, cancer therapy may involve surgery, chemotherapy, hormonal
therapy and/or radiation treatment to eradicate neoplastic cells in a patient
(see, for example,
Stockdale, 1998, "Principles of Cancer Patient Management," in Scientific
American:
Medicine, vol. 3, Rubenstein and Federman, eds., Chapter 12, Section IV).
Recently, cancer
-3-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
therapy may also involve biological therapy or immunotherapy. All of these
approaches can
pose significant drawbacks for the patient. Surgery, for example, may be
contraindicated due
to the health of the patient or may be unacceptable to the patient.
Additionally, surgery may
not completely remove the neoplastic tissue. Radiation therapy is only
effective when the
neoplastic tissue exhibits a higher sensitivity to radiation than normal
tissue, and radiation
therapy can also often elicit serious side effects. Hormonal therapy is rarely
given as a single
agent and, although it can be effective, is often used to prevent or delay
recurrence of cancer
after other treatments have removed the majority of the cancer cells.
Biological
therapies/immunotherapies are limited in number and each therapy is generally
effective for a
very specific type of cancer.
[0011] With respect to chemotherapy, there are a variety of chemotherapeutic
agents
available for treatment of cancer. A significant majority of cancer
chemotherapeutics act by
inhibiting DNA synthesis, either directly, or indirectly by inhibiting the
biosynthesis of the
deoxyribonucleotide triphosphate precursors, to prevent DNA replication and
concomitant
cell division (see, for example, Gilman et al., Goodman and Gilman's: The
Pharmacological
Basis of Therapeutics, Eighth Ed. (Pergamom Press, New York, 1990)). These
agents, which
include alkylating agents, such as nitrosourea, anti-metabolites, such as
methotrexate and
hydroxyurea, and other agents, such as etoposides, campathecins, bleomycin,
doxorubicin,
daunorubicin, etc., although not necessarily cell cycle specific, kill cells
during S phase
because of their effect on DNA replication. Other agents, specifically
colchicine and the
vinca alkaloids, such as vinblastine and vincristine, interfere with
microtubule assembly
resulting in mitotic arrest. Chemotherapy protocols generally involve
administration of a
combination of chemotherapeutic agents to increase the efficacy of treatment.
[0012] Despite the availability of a variety of chemotherapeutic agents,
chemotherapy
has many drawbacks (see, for example, Stockdale, 1998, "Principles Of Cancer
Patient
Management" in Scientific American Medicine, vol. 3, Rubenstein and Federman,
eds., ch.
12, sect. 10). Almost all chemotherapeutic agents are toxic, and chemotherapy
causes
significant, and often dangerous, side effects, including severe nausea, bone
marrow
depression, immunosuppression, etc. Additionally, even with administration of
combinations
of chemotherapeutic agents, many tumor cells are resistant or develop
resistance to the
chemotherapeutic agents. In fact, those cells resistant to the particular
chemotherapeutic
agents used in the treatment protocol often prove to be resistant to other
drugs, even those
agents that act by mechanisms different from the mechanisms of action of the
drugs used in
the specific treatment; this phenomenon is termed pleiotropic drug or
multidrug resistance.
-4-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Thus, because of drug resistance, many cancers prove refractory to standard
chemotherapeutic treatment protocols.
[0013] There is a significant need for alternative cancer treatments,
particularly for
treatment of cancer that has proved refractory to standard cancer treatments,
such as surgery,
radiation therapy, chemotherapy, and hormonal therapy. Further, it is uncommon
for cancer
to be treated by only one method. Thus, there is a need for development of new
therapeutic
agents for the treatment of cancer and new, more effective, therapy
combinations for the
treatment of cancer.
3. SUMMARY OF THE INVENTION
[0014] EphA2 is overexpressed and functionally altered in a large number of
malignant carcinomas. EphA2 is an oncoprotein and is sufficient to confer
metastatic
potential to cancer cells. EphA2 that is overexpressed on malignant cells
exhibits kinase
activity independent from ligand binding. The present inventors have found
that a decrease
in EphA2 levels decrease metastatic behavior of a cell. In particular, the
present invention
inventors' have discovered that, surprisingly, antibodies that agonize EphA2,
i.e., elicit
EphA2 signaling, actually decrease EphA2 expression and inhibit tumor cell
growth and/or
metastasis. Although not intending to be bound by any mechanism of action,
agonistic
antibodies may repress malignant cell behavior by inducing EphA2
autophosphorylation,
thereby causing subsequent EphA2 degradation to down-regulate expression. Thus
the
EphA2 antibodies of the invention agonize EphA2 signaling and increase
phosphorylation of
EphA2 ("EphA2 agonistic antibodies").
[0015) Differences in the subcellular localization, ligand binding properties
or protein
organization (e.g., structure, orientation in the cell membrane) can further
distinguish the
EphA2 that is present on cancer cells from EphA2 on non-cancer cells. In non-
cancer cells,
EphA2 is expressed at low levels and is localized to sites of cell-cell
contact, where it can
engage its membrane-anchored ligands. However, cancer cells generally display
decreased
cell-cell contacts and this can decrease EphA2-ligand binding. Furthermore,
the
overexpression of EphA2 can cause an excess of EphA2 relative to ligand that
increases the
amount of non-ligand bound EphA2. Consequently, changes in the subcellular
distribution or
membrane orientation of EphA2 can cause EphA2 to localize to sites in a cancer
cell where it
is inaccessible to ligand. Additionally, EphA2 may have altered ligand binding
properties
(e.g., due to an altered conformation) in cancer cells such that it is
incapable of stable
interactions with its ligand whether or not it is localized to the cell-cell
junction. In each
-5-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
case, these changes can expose certain epitopes on the EphA2 in cancer cells
that are not
exposed in non-cancer cells. Accordingly, the invention also provides
antibodies that
specifically bind EphA2 but preferably bind an EphA2 epitope exposed on cancer
cells but
not on non-cancer cells ("exposed EphA2 epitope antibodies"). Exposing cancer
cells to such
EphA2 antibodies that preferentially bind epitopes on EphA2 that are
selectively exposed or
increased on cancer cells but not non-cancer cells targets the
therapeutic/prophylactic
antibody to cancer cells and prevents or decreases the cells' ability to
proliferate while
sparing non-cancer cells.
[0016] The present invention provides for the screening and identification of
antibodies that bind to and agonize EphA2 and/or preferentially bind epitopes
on EphA2 that
are selectively exposed or increased on cancer cells but not non-cancer cells,
preferably
monoclonal antibodies. In particular, the antibodies of the invention bind to
the extracellular
domain of EphA2 and, preferably, elicit EphA2 signaling and EphA2
autophosphorylation.
In another particular embodiment, the antibodies of the invention bind to the
extracellular
domain of EphA2 and, preferably, bind an EphA2 epitope exposed on cancer cells
but not
non-cancer cells. In one embodiment, the antibodies of the invention are EA2,
EA3, EA4,
and EAS. In a preferred embodiment, the antibodies of the invention are human,
humanized
or chimeric.
[0017] In one embodiment, to identify antibodies that preferentially bind an
EphA2
epitope exposed on cancer cells but not non-cancer cells, antibodies may be
screened for the
ability to preferentially bind EphA2 not bound to ligand, e.g., Ephrin AI, and
that is not
localized to cell-cell contacts. Any method known in the art to determine
antibody
binding/localization on a cell can be used to screen candidate antibodies for
desirable binding
properties. In a specific embodiment, immunofluorescence microscopy or flow
cytometry is
used to determine the binding characteristics of an antibody. In this
embodiment, antibodies
that bind poorly to EphA2 when it is bound to its ligand and localized to cell-
cell contacts but
bind well to free EphA2 on a cell are encompassed by the invention. In another
specific
embodiment, EphA2 antibodies are selected for their ability to compete with
ligands (e.g.,
cell-anchored or purified ligands) for binding to EphA2 using cell-based or
ELISA assays.
[0018] In one embodiment, the antibodies of the invention are EA2, EA3, EA4,
or
EAS. In a more preferred embodiment, the antibodies of the invention are human
or
humanized. In a most preferred embodiment, the antibodies of the invention are
humanized
EA2, EA3, EA4, or EAS. In a specific embodiment, an antibody of the invention
is not EA2
or EAS.
-6-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0019] Accordingly, the present invention relates to pharmaceutical
compositions and
prophylactic and therapeutic regimens designed to prevent, treat, or manage
cancer,
particularly metastatic cancer, in a subject comprising administering one or
more antibodies
that specifically bind to and agonize EphA2 and/or preferentially bind
epitopes on EphA2
that are selectively exposed or increased on cancer cells but not non-cancer
cells. In one
embodiment, the cancer is of an epithelial cell origin. In another embodiment,
the cancer is a
cancer of the skin, lung, colon, breast, prostate, bladder, kidney, or
pancreas. In another
embodiment, the cancer cells in the cancer to be prevented, treated, or
managed overexpress
EphA2. In a preferred embodiment, some EphA2 is not bound to ligand, either as
a result of
decreased cell-cell contacts, altered subcellular localization, or increases
in amount of EphA2
relative to ligand. In a preferred embodiment, the methods of the invention
are used to
prevent, treat, or manage metastasis of tumors. The antibodies of the
invention can be
administered in combination with one or more other cancer therapies. In
particular, the
present invention provides methods of preventing, treating, or managing cancer
in a subject
comprising administering to said subject a therapeutically or prophylactically
effective
amount of one or more EphA2 antibodies of the invention in combination with
the
administration of a therapeutically or prophylactically effective amount of
one or more
chemotherapies, hormonal therapies, biological therapies/immunotherapies
and/or radiation
therapies other than the administration of an EphA2 antibody of the invention
or in
combination with surgery.
[0020] The methods and compositions of the invention are useful not only in
untreated patients but are also useful in the treatment of patients partially
or completely
refractory to current standard and experimental cancer therapies, including
but not limited to
chemotherapies, hormonal therapies, biological therapies, radiation therapies,
and/or surgery
as well as to improve the efficacy of such treatments. Accordingly, in a
preferred
embodiment, the invention provides therapeutic and prophylactic methods for
the treatment
or prevention of cancer that has been shown to be or may be refractory or non-
responsive to
therapies other than those comprising administration of EphA2 antibodies of
the invention.
In a specific embodiment, one or more EphA2 antibodies of the invention are
administered to
a patient refractory or non-responsive to a non-EphA2-based treatment to
render the patient
non-refractory or responsive. The treatment to which the patient had
previously been
refractory or non-responsive can then be administered with therapeutic effect.
[0021] In addition, the present invention provides methods of screening for
EphA2
antibodies of the invention. In particular, antibodies may be screened for
binding to EphA2,
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
particularly the extracellular domain of EphA2, using routine immunological
techniques. In
one embodiment, to identify agonistic EphA2 antibodies, EphA2 antibodies may
be screened
for the ability to elicit EphA2 signaling, e.g., increase EphA2
phosphorylation and/or to
degrade EphA2.
[0022] In another embodiment, to identify antibodies that preferentially bind
an
EphA2 epitope exposed on cancer cells but not non-cancer cells, antibodies may
be screened
for the ability to preferentially bind EphA2 that is not bound to ligand,
e.g., Ephrin A1, and
that is not localized to cell-cell contacts. Any method known in the art to
determine antibody
binding/localization on a cell can be used to screen candidate antibodies for
desirable binding
properties. In a specific embodiment, immunofluorescence microscopy or flow
cytometry is
used to determine the binding characteristics of an antibody. In this
embodiment, antibodies
that bind poorly to EphA2 when it is bound to its ligand and localized to cell-
cell contacts but
bind well to free EphA2 on a cell are encompassed by the invention. In another
specific
embodiment, EphA2 antibodies are selected for their ability to compete with
ligands (e.g.,
cell-anchored or purified ligands) for binding to EphA2 using cell-based or
ELISA assays.
[0023] The invention further provides diagnostic methods using the EphA2
antibodies
of the invention to evaluate the efficacy of cancer treatment, either EphA2-
based or not
EphA2-based. In general, increased EphA2 expression is associated with
increasingly
invasive and metastatic cancers. Accordingly, a reduction in EphA2 expression
with a
particular treatment indicates that the treatment is reducing the invasiveness
and/or metastatic
potential of cancer. In particular embodiments, the diagnostic methods of the
invention
provide methods of imaging and localizing metastases and methods of diagnosis
and
prognosis using tissues and fluids distal to the primary tumor site (as well
as methods using
tissues and fluids of the primary tumor), for example, whole blood, sputum,
urine, serum, fine
needle aspirates (i.e., biopsies). In other embodiments, the diagnostic
methods of the
invention provide methods of imaging and localizing metastases and methods of
diagnosis
and prognosis in vivo. In such embodiments, primary metastatic tumors are
detected using an
antibody of the invention, preferably an exposed EphA2 epitope antibody. The
antibodies of
the invention may also be used for immunohistochemical analyses of frozen or
fixed cells or
tissue assays.
[0024] In another embodiment, kits comprising the pharmaceutical compositions
or
diagnostic reagents of the invention are provided.
3.1 DEFINITIONS
_g_
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0025] As used herein, the term "agonist" refers to any compound, including a
protein, polypeptide, peptide, antibody, antibody fragment, large molecule, or
small molecule
(less than 10 kD), that increases the activity, activation or function of
another molecule.
EphA2 agonists cause increased phosphorylation and degradation of EphA2
protein. EphA2
antibodies that agonize EphA2 may or may not preferentially bind an EphA2
epitope that is
exposed in a cancer cell relative to a non-cancer cell.
[0026] The term "antibodies or fragments thereof that immunospecifically bind
to
EphA2" as used herein refers to antibodies or fragments thereof that
specifically bind to an
EphA2 polypeptide or a fragment of an EphA2 polypeptide and do not
specifically bind to
other non-EphA2 polypeptides. Preferably, antibodies or fragments that
immunospecifically
bind to an EphA2 polypeptide or fragment thereof do not non-specifically cross-
react with
other antigens (e.g., binding cannot be competed away with a non-EphA2
protein, e.g., BSA
in an appropriate immunoassay). Antibodies or fragments that
immunospecifically bind to an
EphA2 polypeptide can be identified, for example, by immunoassays or other
techniques
known to those of skill in the art. Antibodies of the invention include, but
are not limited to,
synthetic monoclonal antibodies, multispecific antibodies (including bi-
specific antibodies),
human antibodies, humanized antibodies, chimeric antibodies, synthetic
antibodies, single-
chain Fvs (scFv) (including bi-specific scFvs), single chain antibodies, Fab
fragments, F(ab')
fragments, disulfide-linked Fvs (sdFv), and anti-idiotypic (anti-Id)
antibodies, and epitope-
binding fragments of any of the above. In particular, antibodies of the
present invention
include immunoglobulin molecules and immunologically active portions of
immunoglobulin
molecules, i.e., molecules that contain an antigen binding site that
immunospecifically binds
to an EphA2 antigen (e.g., one or more complementarity determining regions
(CDRs) of an
anti-EphA2 antibody). Preferably agonistic antibodies or fragments that
immunospecifically
bind to an EphA2 polypeptide or fragment thereof only agonize EphA2 and do not
significantly agonize other activities.
[0027] As used herein, the term "cancer" refers to a disease involving cells
that have
the potential to metastasize to distal sites and exhibit phenotypic traits
that differ from those
of non-cancer cells, for example, formation of colonies in a three-dimensional
substrate such
as soft agar or the formation of tubular networks or weblike matrices in a
three-dimensional
basement membrane or extracellular matrix preparation, such as MATRIGELTM. Non-
cancer
cells do not form colonies in soft agar and form distinct sphere-like
structures in three-
dimensional basement membrane or extracellular matrix preparations. Cancer
cells acquire a
characteristic set of functional capabilities during their development, albeit
through various
-9-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
mechanisms. Such capabilities include evading apoptosis, self-sufficiency in
growth signals,
insensitivity to anti-growth signals, tissue invasion/metastasis, limitless
replicative potential,
and sustained angiogenesis. The term "cancer cell" is meant to encompass both
pre-
malignant and malignant cancer cells.
[0028] The term "derivative" as used herein refers to a polypeptide that
comprises an
amino acid sequence of an EphA2 polypeptide, a fragment of an EphA2
polypeptide, an
antibody that immunospecifically binds to an EphA2 polypeptide, or an antibody
fragment
that immunospecifically binds to an EphA2 polypeptide which has been altered
by the
introduction of amino acid residue substitutions, deletions or additions
(i.e., mutations). In
some embodiments, an antibody derivative or fragment thereof comprises amino
acid residue
substitutions, deletions or additions in one or more CDRs. The antibody
derivative may have
substantially the same binding, better binding, or worse binding when compared
to a non-
derivative antibody. In specific embodiments, one, two, three, four, or five
amino acid
residues of the CDR have been substituted, deleted or added (i. e., mutated).
The term
"derivative" as used herein also refers to an EphA2 polypeptide, a fragment of
an EphA2
polypeptide, an antibody that immunospecifically binds to an EphA2
polypeptide, or an
antibody fragment that immunospecifically binds to an EphA2 polypeptide which
has been
modified, i. e, by the covalent attachment of any type of molecule to the
polypeptide. For
example, but not by way of limitation, an EphA2 polypeptide, a fragment of an
EphA2
polypeptide, an antibody, or antibody fragment may be modified, e.g., by
glycosylation,
acetylation, pegylation, phosphorylation, amidation, derivatization by known
protecting/blocking groups, proteolytic cleavage, linkage to a cellular ligand
or other protein,
etc. A derivative of an EphA2 polypeptide, a fragment of an EphA2 polypeptide,
an
antibody, or antibody fragment may be modified by chemical modifications using
techniques
known to those of skill in the art, including, but not limited to specific
chemical cleavage,
acetylation, formylation, metabolic synthesis of tunicamycin, etc. Further, a
derivative of an
EphA2 polypeptide, a fragment of an EphA2 polypeptide, an antibody, or
antibody fragment
may contain one or more non-classical amino acids. In one embodiment, a
polypeptide
derivative possesses a similar or identical function as an EphA2 polypeptide,
a fragment of an
EphA2 polypeptide, an antibody, or antibody fragment described herein. In
another
embodiment, a derivative of EphA2 polypeptide, a fragment of an EphA2
polypeptide, an
antibody, or antibody fragment has an altered activity when compared to an
unaltered
polypeptide. For example, a derivative antibody or fragment thereof can bind
to its epitope
more tightly or be more resistant to proteolysis.
- 10-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0029] The term "epitopes" as used herein refers to a portion of an EphA2
polypeptide having antigenic or immunogenic activity in an animal, preferably
in a mammal,
and most preferably in a mouse or a human. An epitope having immunogenic
activity is a
portion of an EphA2 polypeptide that elicits an antibody response in an
animal. An epitope
having antigenic activity is a portion of an EphA2 polypeptide to which an
antibody
immunospecifically binds as determined by any method well known in the art,
for example,
by immunoassays. Antigenic epitopes need not necessarily be immunogenic.
[0030] The "fragments" described herein include a peptide or polypeptide
comprising
an amino acid sequence of at least 5 contiguous amino acid residues, at least
10 contiguous
amino acid residues, at least 15 contiguous amino acid residues, at least 20
contiguous amino
acid residues, at least 25 contiguous amino acid residues, at least 40
contiguous amino acid
residues, at least 50 contiguous amino acid residues, at least 60 contiguous
amino residues, at
least 70 contiguous amino acid residues, at least contiguous 80 amino acid
residues, at least
contiguous 90 amino acid residues, at least contiguous 100 amino acid
residues, at least
contiguous 125 amino acid residues, at least 150 contiguous amino acid
residues, at least
contiguous 175 amino acid residues, at least contiguous 200 amino acid
residues, or at least
contiguous 250 amino acid residues of the amino acid sequence of an EphA2
polypeptide or
an antibody that immunospecifically binds to an EphA2 polypeptide. Preferably,
antibody
fragments are epitope-binding fragments.
[0031] As used herein, the term "humanized antibody" refers to forms of non-
human
(e.g., murine) antibodies that are chimeric antibodies which contain minimal
sequence
derived from non-human immunoglobulin. For the most part, humanized antibodies
are
human immunoglobulins (recipient antibody) in which hypervariable region
residues of the
recipient are replaced by hypervariable region residues from a non-human
species (donor
antibody) such as mouse, rat, rabbit or non-human primate having the desired
specificity,
affinity, and capacity. In some instances, Framework Region (FR) residues of
the human
immunoglobulin are replaced by corresponding non-human residues. Furthermore,
humanized antibodies may comprise residues which are not found in the
recipient antibody or
in the donor antibody. These modifications are made to further refine antibody
performance.
In general, the humanized antibody will comprise substantially all of at least
one, and
typically two, variable domains, in which all or substantially all of the
hypervariable regions
correspond to those of a non-human immunoglobulin and all or substantially all
of the FRs
are those of a human immunoglobulin sequence. The humanized antibody
optionally also
will comprise at least a portion of an immunoglobulin constant region (Fc),
typically that of a
-11-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
human immunoglobulin that immunospecifically binds to an EphA2 polypeptide,
that has
been altered by the introduction of amino acid residue substitutions,
deletions or additions
(i.e., mutations). In some embodiments, a humanized antibody is a derivative.
Such a
humanized antibody comprises amino acid residue substitutions, deletions or
additions in one
or more non-human CDRs. The humanized antibody derivative may have
substantially the
same binding, better binding, or worse binding when compared to a non-
derivative
humanized antibody. In specific embodiments, one, two, three, four, or five
amino acid
residues of the CDR have been substituted, deleted or added (i.e., mutated).
For further
details in humanizing antibodies, see European Patent Nos. EP 239,400, EP
592,106, and EP
519,596; International Publication Nos. WO 91/09967 and WO 93/17105; U.S.
Patent Nos.
5,225,539, 5,530,101, 5,565,332, 5,585,089, 5,766,886, and 6,407,213; and
Padlan, 1991,
Molecular Immunology 28(4/5):489-498; Studnicka et al., 1994, Protein
Engineering
7(6):805-814; Roguska et al., 1994, PNAS 91:969-973; Tan et al., 2002, J.
Immunol.
169:1119-25; Caldas et al., 2000, Protein Eng. 13:353-60; Morea et al., 2000,
Methods
20:267-79; Baca et al., 1997, J. Biol. Chem. 272:10678-84; Roguska et al.,
1996, Protein
Eng. 9:895-904; Couto et al., 1995, Cancer Res. 55 (23 Supp):5973s-5977s;
Couto et al.,
1995, Cancer Res. 55:1717-22; Sandhu, 1994, Gene 150:409-10; Pedersen et al.,
1994, J.
Mol. Biol. 235:959-73; Jones et al., 1986, Nature 321:522-525; Reichmann et
al:, 1988,
Nature 332:323-329; and Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
[0032] As used herein, the term "hypervariable region" refers to the amino
acid
residues of an antibody which are responsible for antigen binding. The
hypervariable region
comprises amino acid residues from a "Complementarity Determining Region" or
"CDR"
(i.e. residues 24-34 (L1), 50-56 (L2) and 89-97 (L3) in the light chain
variable domain and
31-35 (H1), 50-65 (H2) and 95-102 (H3) in the heavy chain variable domain;
Kabat et al.,
Sequences of Proteins of Immunological Interest, 5th Ed. Public Health
Service, National
Institutes of Health, Bethesda, MD. (1991)) and/or those residues from a
"hypervariable
loop" (i.e. residues 26-32 (L1), 50-52 (L2) and 91-96 (L3) in the light chain
variable domain
and 26-32 (H1), 53-55 (H2) and 96-101 (H3) in the heavy chain variable domain;
Chothia
and Lesk, 1987, J. Mol. Biol. 196:901-917). CDR residues for EA2 and EA5 are
listed in
Table 1. "Framework Region" or "FR" residues are those variable domain
residues other
than the hypervariable region residues as herein defined.
[0033] As used herein, the term "in combination" refers to the use of more
than one
prophylactic and/or therapeutic agents. The use of the term "in combination"
does not
restrict the order in which prophylactic and/or therapeutic agents are
administered to a subject
-12-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
with a hyperproliferative cell disorder, especially cancer. A first
prophylactic or therapeutic
agent can be administered prior to (e.g., 1 minute, 5 minutes, 15 minutes, 30
minutes, 45
minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72
hours, 96 hours, 1
week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks
before),
concomitantly with, or subsequent to (e.g., 1 minute, 5 minutes, 15 minutes,
30 minutes, 45
minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72
hours, 96 hours, 1
week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks after)
the
administration of a second prophylactic or therapeutic agent to a subject
which had, has, or is
susceptible to a hyperproliferative cell disorder, especially cancer. The
prophylactic or
therapeutic agents are administered to a subject in a sequence and within a
time interval such
that the agent of the invention can act together with the other agent to
provide an increased
benefit than if they were administered otherwise. Any additional prophylactic
or therapeutic
agent can be administered in any order with the other additional prophylactic
or therapeutic
agents.
[0034] As used herein, the phrase "low tolerance" refers to a state in which
the patient
suffers from side effects from treatment so that the patient does not benefit
from and/or will
not continue therapy because of the adverse effects and/or the harm from the
side effects
outweighs the benefit of the treatment.
[0035] As used herein, the terms "manage," "managing" and "management" refer
to
the beneficial effects that a subject derives from administration of a
prophylactic or
therapeutic agent, which does not result in a cure of the disease. In certain
embodiments, a
subject is administered one or more prophylactic or therapeutic agents to
"manage" a disease
so as to prevent the progression or worsening of the disease.
[0036] As used herein, the phrase "non-responsive/ refractory" is used to
describe
patients treated with one or more currently available therapies (e.g., cancer
therapies) such as
chemotherapy, radiation therapy, surgery, hormonal therapy and/or biological
therapy/immunotherapy, particularly a standard therapeutic regimen for the
particular cancer,
wherein the therapy is not clinically adequate to treat the patients such that
these patients
need additional effective therapy, e.g., remain unsusceptible to therapy. The
phrase can also
describe patients who respond to therapy yet suffer from side effects,
relapse, develop
resistance, etc. In various embodiments, "non-responsive/refractory" means
that at least
some significant portion of the cancer cells are not killed or their cell
division arrested. The
determination of whether the cancer cells are "non-responsive/refractory" can
be made either
in vivo or in vitro by any method known in the art for assaying the
effectiveness of treatment
-13-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
on cancer cells, using the art-accepted meanings of "refractory" in such a
context. In various
embodiments, a cancer is "non-responsive/refractory" where the number of
cancer cells has
not been significantly reduced, or has increased during the treatment.
[0037] As used herein, the term "potentiate" refers to an improvement in the
efficacy
of a therapeutic agent at its common or approved dose.
[0038] As used herein, the terms "prevent," " preventing" and "prevention"
refer to
the prevention of the recurrence or spread of a disease in a subject resulting
from the
administration of a prophylactic or therapeutic agent.
[0039] As used herein, the terms "prophylactic agent" and "prophylactic
agents" refer
to any agents) that can be used in the prevention of the onset, recurrence or
spread of a
disorder associated with EphA2 overexpression, particularly cancer. In certain
embodiments,
the term "prophylactic agent" refers to an EphA2 agonistic antibody or an
exposed EphA2
epitope antibody (e.g., EA2, EA3, EA4, or EAS). In certain other embodiments,
the terms
"prophylactic agent" and "prophylactic agents" refer to cancer
chemotherapeutics, radiation
therapy, hormonal therapy, biological therapy (e.g., immunotherapy), and/or
EphA2
antibodies of the invention. In other embodiments, more than one prophylactic
agent may be
administered in combination.
[0040] As used herein, a "prophylactically effective amount" refers to that
amount of
the prophylactic agent sufficient to result in the prevention of the
recurrence or spread of
cancer. A prophylactically effective amount may refer to the amount of
prophylactic agent
sufficient to prevent the recurrence or spread of cancer or the occurrence of
cancer in a
patient, including but not limited to those predisposed to cancer or
previously exposed to
carcinogens. A prophylactically effective amount may also refer to the amount
of the
prophylactic agent that provides a prophylactic benefit in the prevention of
cancer. Further, a
prophylactically effective amount with respect to a prophylactic agent of the
invention means
that amount of prophylactic agent alone, or in combination with other agents,
that provides a
prophylactic benefit in the prevention of cancer. Used in connection with an
amount of an
EphA2 antibody of the invention, the term can encompass an amount that
improves overall
prophylaxis or enhances the prophylactic efficacy of or synergies with another
prophylactic
agent.
[0041] A used herein, a "protocol" includes dosing schedules and dosing
regimens.
[0042] As used herein, the phrase "side effects" encompasses unwanted and
adverse
effects of a prophylactic or therapeutic agent. Adverse effects are always
unwanted, but
unwanted effects are not necessarily adverse. An adverse effect from a
prophylactic or
- 14-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
therapeutic agent might be harmful or uncomfortable or risky. Side effects
from
chemotherapy include, but are not limited to, gastrointestinal toxicity such
as, but not limited
to, early and late-forming diarrhea and flatulence, nausea, vomiting,
anorexia, leukopenia,
anemia, neutropenia, asthenia, abdominal cramping, fever, pain, loss of body
weight,
dehydration, alopecia, dyspnea, insomnia, dizziness, mucositis, xerostomia,
and kidney
failure, as well as constipation, nerve and muscle effects, temporary or
permanent damage to
kidneys and bladder, flu-like symptoms, fluid retention, and temporary or
permanent
infertility. Side effects from radiation therapy include but are not limited
to fatigue, dry
mouth, and loss of appetite. Side effects from biological
therapies/immunotherapies include
but are not limited to rashes or swellings at the site of administration, flu-
like symptoms such
as fever, chills and fatigue, digestive tract problems and allergic reactions.
Side effects from
hormonal therapies include but are not limited to nausea, fertility problems,
depression, loss
of appetite, eye problems, headache, and weight fluctuation. Additional
undesired effects
typically experienced by patients are numerous and known in the art. Many are
described in
the Physicians' Desk Reference (58~' ed., 2004).
[0043] As used herein, the terms "single-chain Fv" or "scFv" refer to antibody
fragments comprise the VH and VL domains of antibody, wherein these domains
are present
in a single polypeptide chain. Generally, the Fv polypeptide further comprises
a polypeptide
linker between the VH and VL domains which enables the scFv to form the
desired structure
for antigen binding. For a review of sFv see Pluckthun in The Pharmacology of
Monoclonal
Antibodies, vol. 113, Rosenburg and Moore eds. Springer-Verlag, New York, pp.
269-315
(1994). In specific embodiments, scFvs include bispecific scFvs and humanized
scFvs.
[0044] As used herein, the terms "subject" and "patient" are used
interchangeably.
As used herein, a subject is preferably a mammal such as a non-primate (e.g.,
cows, pigs,
horses, cats, dogs, rats etc.) and a primate (e.g., monkey and human), most
preferably a
human.
[0045] As used herein, the terms "treat," "treating" and "treatment" refer to
the
eradication, reduction or amelioration of symptoms of a disease or disorder,
particularly, the
eradication, removal, modification, or control of primary, regional, or
metastatic cancer tissue
that results from the administration of one or more therapeutic agents. In
certain
embodiments, such terms refer to the minimizing or delaying the spread of
cancer resulting
from the administration of one or more therapeutic agents to a subject with
such a disease.
[0046] As used herein, the terms "therapeutic agent" and "therapeutic agents"
refer to
any agents) that can be used in the prevention, treatment, or management of a
disorder
-15-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
associated with the overexpression of EphA2, particularly cancer. In certain
embodiments,
the term "therapeutic agent" refers to an EphA2 agonistic antibody and/ an
exposed EphA2
epitope antibody, e. g., EA2, EA3, EA4, or EAS. In certain other embodiments,
the terms
"therapeutic agent" and "therapeutic agents" refer to cancer
chemotherapeutics, radiation
therapy, hormonal therapy, biological therapy/immunotherapy, and/or EphA2
antibody of the
invention. In other embodiments, more than one therapeutic agent may be
administered in
combination.
[0047] As used herein, a "therapeutically effective amount" refers to that
amount of
the therapeutic agent sufficient to destroy, modify, control or remove
primary, regional or
metastatic cancer tissue. A therapeutically effective amount may refer to the
amount of
therapeutic agent sufficient to delay or minimize the spread of cancer. A
therapeutically
effective amount may also refer to the amount of the therapeutic agent that
provides a
therapeutic benefit in the treatment or management of cancer. Further, a
therapeutically
effective amount with respect to a therapeutic agent of the invention means
that amount of
therapeutic agent alone, or in combination with other therapies, that provides
a therapeutic
benefit in the treatment or management of cancer. Used in connection with an
amount of an
EphA2 antibody of the invention, the term can encompass an amount that
improves overall
therapy, reduces or avoids unwanted effects, or enhances the therapeutic
efficacy of or
synergies with another therapeutic agent.
4. DESCRIPTION OF THE FIGURES
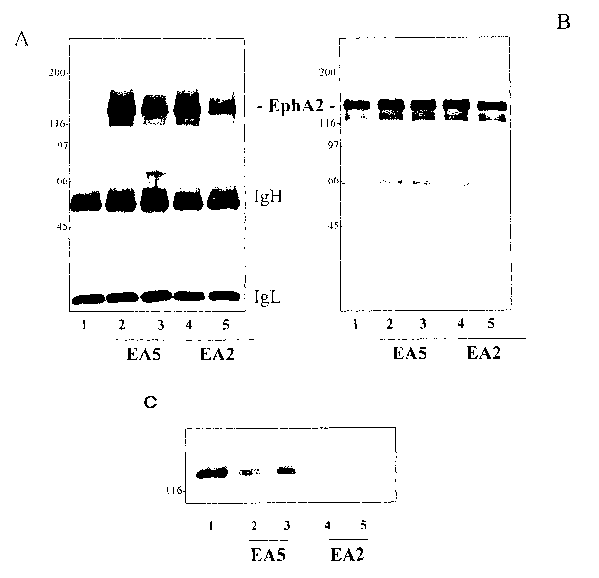
[0048] FIGS. lA-1C: EphA2 antibodies promote tyrosine phosphorylation and
degradation of EphA2 in MDA-MB-231 cells. (A, B) Monolayers of MDA-MB-231
cells
were incubated in the presence of EAS or EA2 or control for 8 minutes at
37°C. Cell lysates
were then immunoprecipitated with an EphA2-specific antibody, resolved by SDS-
PAGE and
subjected to western blot analysis with a phosphotyrosine-specific antibody
(A). The
membranes were stripped and re-probed with the EphA2-specific antibody used in
the
immunoprecipitation as a loading control (B). Levels of EphA2 phosphorylation
increase
with antibody incubation. (C) Monolayers of MDA-MB-231 cells were incubated in
the
presence of presence of 30 pg/ml EAS or EA2 or a control for 24 hours at
37°C. Cell lysates
were then resolved by SDS PAGE and subjected to western blot analysis with an
EphA2-
specific antibody. EphA2 protein levels decrease with antibody incubation. The
relative
mobility of molecular mass standards is shown on the left of each blot.
Antibody heavy
(IgH) and light (IgL) chains are indicated in (A).
- 16-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0049] FIGS. 2A-2D: EphA2 antibodies promote tyrosine phosphorylation and
degradation of EphA2 in A549 cells. Monolayers of A549 cells were incubated at
37°C in
the presence of EAS or EA2 or control (PBS) for either (A, B) 10 minutes or
(C, D) 5 hours.
Cell lysates were then immunoprecipitated with an EphA2-specific antibody D7,
resolved by
SDS-PAGE and subjected to western blot analysis with a phosphotyrosine-
specific antibody
(A, C). The membranes were stripped and re-probed with the EphA2-specific
antibody used
in the immunoprecipitation as a loading control (B, D).
[0050] FIGS. 3A-3B: EphA2 antibodies inhibit malignant tumor cell growth in
vitro.
Purified EphA2 antibodies were incubated with both malignant and benign tumor
cells for 7
days at 37°C in soft agar. (A) A549 malignant lung cancer cells were
incubated with either
pg/ml or 2.5 pg/ml of EAS or EA2 monoclonal antibodies or a control (PBS). All
amounts of antibodies used inhibited cell growth in soft agar. (B) Benign MCF-
7 breast
epithelial tumor cells were converted to malignant cells by the overexpression
of EphA2'
(MCF-7EPh~). Both tumor cell types were incubated with either EAS monoclonal
antibodies
or a control (PBS). EAS inhibits the ability of MCF-7Eph'~ cells to grow in
soft agar. Results
are reported as colonies per high-powered field (HPF).
[0051] FIGS. 4A-4D: The EphA2 antibody EAS inhibits tumor cell growth in vivo.
MDA-MB-231 breast cancer cells were implanted (A) orthotopically or (B)
subcutaneously
into athymic mice. (C) A549 lung cancer cells were implanted subcutaneously
into athymic
mice. After the tumors had grown to an average volume of 100mm3, mice were
administered
6 mg/kg of the indicated antibody or negative control (PBS or lA7 antibody)
intraperitoneally twice a week for 3 weeks. Tumor growth was assessed and
expressed as a
ratio of the tumor volume divided by initial tumor volume ( 100 mm3). (D) MDA-
MB-231
breast cancer cells were implanted subcutaneously into athymic mice. After the
tumors had
grown to an average volume of 100mm3, mice were administered 6 mg/kg of the
indicated
antibody or negative control intraperitoneally twice a week for 3 weeks. Total
tumor volume
was determined after sacrifice. Negative control is black and EAS is white.
[0052] FIGS. 5A-5B: EphA2 overexpression selectively increases malignant cell
growth. (A) 1x105 control (white bar) or MCF-7Eph~ cells (black bar) were
suspended in soft
agar in the presence of 1 mg/ml 17(3-estradiol for 14 days prior to
microscopic evaluation.
EphA2-transfected cells formed more colonies (47 colonies/high powered field
(HPF)) than
matched controls (1 colony/HPF; P<0.01). (B) Monolayer growth assays did not
distinguish
between the growth of control (white circles) and MCF-7EPnnz cells (black
squares).
-17-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0053] FIGS. 6A-6B: EphA2 overexpression increases tumorigenic potential. (A)
1x106 control (white circle) or MCF-7Ep"'~ cells (black square) were implanted
into the
mammary fatpad of athymic mice (n=20 mice per group) in the presence of
supplemental
estrogen (1 pM 17(3-estradiol). The tumors formed by MCF-7Ep"'~ cells were
significantly
larger than tumors formed by matched controls (P = 0.027). (B) Equal amounts
of protein
lysate, isolated from input cells or resected tumors (T) were evaluated by
western blot
analyses with an EphA2 antibody (D7). The membranes were stripped and re-
probed with a
(3-catenin-specific antibody as a loading control.
[0054] FIGS. 7A-7C: EphA2 overexpression decreases estrogen dependence. (A)
1 x 105 control (white bar) or MCF-7Ep"'~ cells (black bar) were suspended in
soft agar in the
absence of exogenous estrogen and colony formation was evaluated
microscopically after 14
days. The monolayer growth (B) and tumorigenic potential (C) Of MCF-7Ep"AZ
(black square)
cells were increased relative to matched controls (white circle) in the
absence of
supplemental estrogen (P<0.01 and P<0.004, respectively).
[0055] FIGS. 8A-8B: EphA2 overexpression decreases tamoxifen sensitivity. (A)
1x105 MCF-7 or MCF-7EP"''Z cells were suspended in soft agar in the presence
of 1~M
tamoxifen (TAM) and or 1~.M 17(3-estradiol and colony formation was evaluated
microscopically after 14 days. (B) MCF-7 (circles) or MCF-7EP"a2 cells
(squares) were
implanted into the mammary fatpad (n=15 mice per group) in the presence of
supplemental
estrogen. Tamoxifen treatment was initiated 17 days post-implantation. Tumor
volume of
tamoxifen treated (black circles and squares) and saline treated (white
circles and squares)
animals was measured at the indicated time. Note the lower inhibitory effects
of tamoxifen
on MCF-7EPnA? relative to control cells (P=0.01).
[0056] FIGS. 9A-9F: Estrogen receptor is expressed but functionally altered in
MCF-
7EphA2 Cells. (A) ERoc and (B) ER(3 levels were evaluated in MCF-7"e~ control
cells and
MCF-7Ep"~ cells by western blot analyses with an EphA2-specific antibody (D7).
(C, D)
The membranes were stripped and re-probed with a (3-catenin-specific antibody
as a loading
control. (E, F) Estrogen receptor activity was measured using a CAT reporter
system,
revealing comparable estrogen receptor activity in control and MCF-7EPn,a2
cells. The
average results from three experiments are graphed in (F). E2 indicates
estrogen treatment;
TAM indicates tamoxifen treatment; % conversion indicates the amount of
substrate
converted from non-acetylated substrate (non-AC) to acetylated substrate (AC)
by CAT
enzyme.
-18-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0057] FIGS. l0A-lOC: EphA2 agonistic antibody EAS decreases malignant
growth. MCF-7EPna,2 cells were incubated in the presence of 3 ~tg/ml of EAS
for the time
indicated prior to sample extraction and western blot analyses with an EphA2-
specific
antibody (D7). (B) The membrane was stripped and re-probed with a ~i-catenin-
specific
antibody as a loading control. (C) 1x105 control or MCF-7EPna2 cells were
suspended in soft
agar in the presence or absence of tamoxifen (TAM, 1N.M) and EphA2 agonistic
antibody
(EAS, 10~g/ml). Note that EAS increased the sensitivity OF MCF-7EPnA2 cells to
tamoxifen.
[0058] FIGS. 11A-11D: EAS and EA2 selectively bind to malignant cells. The
anti-
EphA2 monoclonal antibodies EAS (A, C) and EA2 (B, D) bind malignant MDA-MB-
231
breast epithelial tumor cells (A, B) more strongly than benign MCF-l0A breast
epithelial
tumor cells (C, D) as shown by immunofluorescent staining.
[0059] FIG. 12: EAS was immunoreactive against malignant prostate cells. The
anti-EphA2 monoclonal antibody EAS identified malignant prostate cancer cells
in formalin-
fixed, paraffin-embedded archival clinical specimens.
[0060] FIGS.13A-13D.: EphA2 EAS antibody preferentially binds cancer cells.
Non-transformed MCF-l0A (A, C) or transformed MDA-MB-231 (B, D) cells were
incubated with 10 pg/ml (A, B) Eph099B-233.152 or (C, D) EAS at 4°C
prior to fixation and
immunolabeling with fluorophore-conjugated anti-mouse IgG.
[0061] FIGS. 14A-14D: EphA2 EAS antibody preferentially binds EphA2 epitopes
exposed by decreasing cell-cell contacts. (A, B) Non-transformed MCF-l0A cells
were
labeled with EAS at 4°C either before (A) or after (B) treatment with
EGTA and prior to
fixation and immunolabeling with fluorophore-conjugated anti-mouse IgG. (C, D)
Non-
transformed MCF-l0A (C) or transformed MDA-MB-231 (D) cells were labeled with
EAS
either before (middle) or after (top) treatment with EGTA. Control cells were
incubated with
secondary antibody alone (bottom). The amount of EAS-EphA2 binding was
measured using
flow cytometry.
[0062] FIGS. 15A-15B: EphA2 EAS epitope is distinct from ligand binding site.
(A) EphA2- F~ was incubated with and bound to immobilized Ephrin A1-F~.
Labeled Ephrin
A1-F~ (black) or EAS (white) was incubated with the EphA2-Ephrin Al-F~ complex
and
amount of binding was measured. (B) EphA2-F~ was incubated with and bound to
immobilized Ephrin Al-F~. Labeled EAS was then incubated with the EphA2-Ephrin
A1
complex. Unlabeled competitor was incubated with EphA2-Ephrin A1-EAS complex
in the
indicated amount. Competitors were Ephrin A1-F~ (black) or EAS (white).
-19-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0063] FIG.16: Sequences of VL and VH of EA2. Amino acid and nucleic acid
sequences of EA2 (A) VL (SEQ m NOs:l and 9, respectively) and (B) VH (SEQ ID
NOs:S
and 13, respectively) are shown. Sequences of the CDRs are indicated in bold
and
underlined.
[0064] FIG.17: Sequences of VL and VH of EAS. Sequences of the CDRs are
indicated in bold and underlined. Shown in (A) are the amino acid and nucleic
acid
sequences of EAS VL (SEQ ID NOs:l7 and 25, respectively). Three clones
containing the
polynucleotide sequences of the EAS light chain variable region were
identified using
degenerate primers (i.e., oligonucleotides that wobble at the third position
base to allow for
identification of all codons that encode the corresponding amino acid)
designed from the
amino terminus of the light chain protein sequence. In the three clones
identified that
contained the variable light chain EAS polynucleotide sequence, the bases at
positions 6 and
9 were guanine (G) or tyrosine (T), designated by the letter "k" in bold; and
at position 15,
the base was G, T or cytosine (C), designated by the letter "b" in bold. Shown
in (B) are the
amino acid and nucleic acid sequences of EAS VH (SEQ m NOs:21 and 29,
respectively).
5. DETAILED DESCRIPTION OF THE INVENTION
[0065] The present invention is based, in part, on the inventors' discovery
that EphA2
monoclonal antibodies can inhibit cancer cell phenotypes. Decreased EphA2
activity
selectively inhibits malignant cancer cell growth. Decreased EphA2 activity
can be achieved
with EphA2 agonistic monoclonal antibodies. Although not intending to be bound
by any
mechanism of action, this inhibition of malignant cell growth is achieved by
stimulating (i.e.,
agonizing) EphA2 signaling thereby causing EphA2 phosphorylation which leads
to its
degradation. Malignant cell growth is decreased due to the decreased EphA2
levels and,
therefore, ligand-independent EphA2 signaling.
[0066] Accordingly, the present invention relates to methods and compositions
that
provide for the treatment, inhibition, and management of cancer, particularly
metastatic
cancer. A particular aspect of the invention relates to methods and
compositions containing
compounds that inhibit cancer cell proliferation and invasion, particularly
those cancer cells
that overexpress EphA2. The present invention further relates to methods and
compositions
for the treatment, inhibition, or management of metastases of cancers of
epithelial cell origin,
especially human cancers of the breast, lung, skin, and prostate, bladder,
kidney and
pancreas. Further compositions and methods of the invention include other
types of active
ingredients in combination with the EphA2 antibodies of the invention.
-20-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0067] The present invention also relates to methods for the treatment,
inhibition, and
management of cancer that has become partially or completely refractory to
current or
standard cancer treatment, such as chemotherapy, radiation therapy, hormonal
therapy, and
biological therapy.
[0068] The invention further provides diagnostic methods using the EphA2
antibodies
of the invention, particularly the exposed EphA2 epitope antibodies, to
evaluate the efficacy
of cancer treatment, either EphA2-based or not EphA2-based. The diagnostic
methods of the
invention can also be used to prognose or predict cancer progression. In
particular
embodiments, the diagnostic methods of the invention provide methods of
imaging and
localizing metastases and methods of diagnosis and prognosis using tissues and
fluids distal
to the primary tumor site (as well as methods using tissues and fluids of the
primary tumor).
In other embodiments, the diagnostic methods of the invention provide methods
of imaging
and localizing metastases and methods of diagnosis and prognosis in vivo.
5.1 Antibodies
[0069] As discussed above, the invention encompasses administration of
antibodies
(preferably monoclonal antibodies) or fragments thereof that
immunospecifically bind to and
agonize EphA2 signaling ("EphA2 agonistic antibodies") and/or preferentially
bind epitopes
on EphA2 that are selectively exposed or increased on cancer cells but not non-
cancer cells
("exposed EphA2 epitope antibodies"). In one embodiment, the antibody binds to
the
extracellular domain of EphA2 and, preferably, also agonizes EphA2, e.g.,
increases EphA2
phosphorylation. In another embodiment, the antibody binds to the
extracellular domain of
EphA2 and, preferably, also binds an epitope on EphA2 that is selectively
exposed or
increased on cancer cells but not non-cancer cells. In a more preferred
embodiment, the
antibody is EA2, EA3, EA4, or EAS. In another embodiment, the antibody binds
to an
epitope bound by EA2, EA3, EA4, or EAS and/or competes for EphA2 binding with
EA2,
EA3, EA4, or EAS, e.g. as assayed by ELISA. In other embodiments, the antibody
of the
invention immunospecifically binds to and agonizes EphA2 signaling and/or
preferentially
binds an epitope on EphA2 that is selectively exposed or increased on cancer
cells but not
non-cancer cells and may or may not compete for binding with an EphA2 ligand,
e.g., Ephrin
A1.
[0070] Hybridomas producing antibodies EA2 (strain EA2.31) and EA5 (strain
EA5.12) of the invention have been deposited with the American Type Culture
Collection
(ATCC, P.O. Box 1549, Manassas, VA 20108) on May 22, 2002 under the provisions
of the
-21-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Budapest Treaty on the International Recognition of the Deposit of
Microorganisms for the
Purposes of Patent Procedures, and assigned accession numbers PTA-4380 and PTA-
4381,
respectively and incorporated by reference. The amino acid sequence of VL and
VH of the
EA2 and EAS antibodies are shown in FIGS. 16A-16B and FIGS. 17A-17B,
respectively.
The sequences of the EA2 and EAS CDRs are indicated in Table 1. In a most
preferred
embodiment, the antibody is human or has been humanized.
[0071] Antibodies used in the methods of the invention include, but are not
limited to,
monoclonal antibodies, synthetic antibodies, multispecific antibodies
(including bi-specific
antibodies), human antibodies, humanized antibodies, chimeric antibodies,
single-chain Fvs
(scFv) (including bi-specific scFvs), single chain antibodies, Fab fragments,
F(ab')
fragments, disulfide-linked Fvs (sdFv), and epitope-binding fragments of any
of the above.
In particular, antibodies used in the methods of the present invention include
immunoglobulin
molecules and immunologically active portions of immunoglobulin molecules,
i.e., molecules
that contain an antigen binding site that immunospecifically binds to EphA2
and is an
agonist of EphA2 and/or preferentially binds an EphA2 epitope exposed on
cancer cells but
not non-cancer cells. The immunoglobulin molecules of the invention can be of
any type
(e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgGI, IgG~, IgG3, IgG4,
IgA~ and IgA2) or
subclass of immunoglobulin molecule.
[0072] The antibodies used in the methods of the invention may be from any
animal
origin including birds and mammals (e.g., human, murine, donkey, sheep,
rabbit, goat, guinea
pig, camel, horse, or chicken). Preferably, the antibodies are human or
humanized
monoclonal antibodies. As used herein, "human" antibodies include antibodies
having the
amino acid sequence of a human immunoglobulin and include antibodies isolated
from
human immunoglobulin libraries or from mice or other animal that express
antibodies from
human genes.
[0073] The antibodies used in the methods of the present invention may be
monospecific, bispecific, trispecific or of greater multispecificity.
Multispecific antibodies
may immunospecifically bind to different epitopes of an EphA2 polypeptide or
may
immunospecifically bind to both an EphA2 polypeptide as well a heterologous
epitope, such
as a heterologous polypeptide or solid support material. See, e.g.,
International Publication
Nos. WO 93/17715, WO 92/08802, WO 91/00360, and WO 92/05793; Tutt, et al.,
1991, J.
Immunol. 147:60-69; U.S. Patent Nos. 4,474,893, 4,714,681, 4,925,648,
5,573,920, and
5,601,819; and Kostelny et al., 1992, J. Immunol. 148:1547-1553.
-22-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0074] In a specific embodiment, an antibody used in the methods of the
present
invention is EA2, EA3, EA4, or EAS or an antigen-binding fragment thereof
(e.g., one or
more complementarity determining regions (CDRs) of the afore-mentioned
antibodies of the
invention, e.g., see Table 1). In another embodiment, an agonistic antibody
used in the
methods of the present invention binds to the same epitope as any of EA2, EA3,
EA4, or EAS
or competes with any of EA2, EA3, EA4, or EAS for binding to EphA2, e.g., in
an ELISA
assay.
[0075] The present invention also encompasses antibodies or fragments thereof
that
immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially bind
an EphA2
epitope exposed in cancer cells, said antibodies comprising a VH CDR having an
amino acid
sequence of any one of the VH CDRs of EA2, EA3, EA4, or EAS. The present
invention
also encompasses the use of antibodies that immunospecifically bind to EphA2
and agonize
EphA2 and/or preferentially bind an EphA2 epitope exposed in cancer cells,
said antibodies
comprising a VL CDR having an amino acid sequence of any one of the VL CDRs of
EA2,
EA3, EA4, or EAS. The present invention also encompasses the use of antibodies
that
immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially bind
an EphA2
epitope exposed in cancer cells, said antibodies comprising one or more VH
CDRs and one or
more VL CDRs of EA2, EA3, EA4, or EAS. In particular, the invention
encompasses the use
of antibodies that immunospecifically bind to EphA2 and agonize EphA2 and/or
preferentially bind an EphA2 epitope exposed in cancer cells, said antibodies
comprising a
VH CDR1 and a VL CDR1; a VH CDR1 and a VL CDR2; a VH CDR1 and a VL CDR3; a
VH CDR2 and a VL CDR1; VH CDR2 and VL CDR2; a VH CDR2 and a VL CDR3; a VH
CDR3 and a VL CDR1; a VH CDR3 and a VL CDR2; a VH CDR3 and a VL CDR3; a VH1
CDRI, a VH CDR2 and a VL CDRI; a VH CDR1, a VH CDR2 and a VL CDR2; a VH
CDR1, a VH CDR2 and a VL CDR3; a VH CDR2, a VH CDR3 and a VL CDR1, a VH
CDR2, a VH CDR3 and a VL CDR2; a VH CDR2, a VH CDR3 and a VL CDR3; a VH1
CDRI, a VH CDR3 and a VL CDR1; a VH CDR1, a VH CDR3 and a VL CDR2; a VH
CDR1, a VH CDR3 and a VL CDR3; a VH CDR1, a VL CDR1 and a VL CDR2; a VH
CDR1, a VL CDRI and a VL CDR3; a VH CDRI, a VL CDR2 and a VL CDR3; a VH
CDR2, a VL CDR1 and a VL CDR2; a VH CDR2, a VL CDR1 and a VL CDR3; a VH
CDR2, a VL CDR2 and a VL CDR3; a VH CDR3, a VL CDRI and a VL CDR2; a VH
CDR3, a VL CDR1 and a VL CDR3; a VH CDR3, a VL CDR2 and a VL CDR3; a VH
CDRl, a VH CDR2, a VH CDR3 and a VL CDR1; a VH CDR1, a VH CDR2, a VH CDR3
and a VL CDR2; a VH CDR1, a VH CDR2, a VH CDR3 and a VL CDR3; a VH CDR1, a
-23-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
VL CDR1, a VL CDR2 and a VL CDR3; a VH CDR2, a VL CDR1, a VL CDR2 and a VL
CDR3; a VH CDR3, a VL CDR1, a VL CDR2 and a VL CDR3; a VH CDR1, a VH CDR2, a
VL CDR1 and a VL CDR2; a VH CDR1, a VH CDR2, a VL CDR1 and a VL CDR3; a VH
CDR1, a VH CDR2, a VL CDR2 and a VL CDR3; a VH CDR1, a VH CDR3, a VL CDR1
and a VL CDR2; a VH CDR1, a VH CDR3, a VL CDR1 and a VL CDR3; a VH CDR1, a
VH CDR3, a VL CDR2 and a VL CDR3; a VH CDR2, a VH CDR3, a VL CDR1 and a VL
CDR2; a VH CDR2, a VH CDR3, a VL CDR1 and a VL CDR3; a VH CDR2, a VH CDR3, a
VL CDR2 and a VL CDR3; a VH CDR1, a VH CDR2, a VH CDR3, a VL CDR1 and a VL
CDR2; a VH CDR1, a VH CDR2, a VH CDR3, a VL CDR1 and a VL CDR3; a VH CDR1, a
VH CDR2, a VH CDR3, a VL CDR2 and a VL CDR3; a VH CDR1, a VH CDR2, a VL
CDR1, a VL CDR2, and a VL CDR3; a VH CDR1, a VH CDR3, a VL CDR1, a VL CDR2,
and a VL CDR3; a VH CDR2, a VH CDR3, a VL CDR1, a VL CDR2, and a VL CDR3; a
VH CDR1, a VH CDR2, a VH CDR3, a VL CDRI, a VL CDR2, and a VL CDR3 or any
combination thereof of the VH CDRs and VL CDRs of EA2, EA3, EA4, or EAS. In
specific
embodiments, the VH CDR1 is SEQ >D N0:6; the VH CDR2 is SEQ >D N0:7; the VH
CDR3 is SEQ ID N0:8; the VL CDR1 is SEQ ID N0:2; the VL CDR2 is SEQ ID N0:3;
and
the VL CDR3 is SEQ ID N0:4. In other specific embodiments, the VH CDR1 is SEQ
ID
N0:22; the VH CDR2 is SEQ ID N0:23; the VH CDR3 is SEQ ID N0:24; the VL CDR1
is
SEQ 1D N0:18; the VL CDR2 is SEQ 1D N0:19; and the VL CDR3 is SEQ ID N0:20
(see,
e.g., Table 1). The invention also encompasses any of the foregoing with one,
two, three,
four, or five amino acid substitutions, additions, or deletions that bind
EphA2.
[0076] In one embodiment, an antibody that immunospecifically binds to EphA2
and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR1 having the amino acid sequence of SEQ ID N0:6 and a VL
CDR1
having the amino acid sequence of SEQ ID N0:2. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR1 having the amino
acid
sequence of SEQ ID N0:6 and a VL CDR2 having the amino acid sequence of SEQ ID
N0:3. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EpliA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR1 having the amino acid sequence of SEQ ID N0:6 and a VL
CDR3
having the amino acid sequence of SEQ ID N0:4.
[0077] In another embodiment, an antibody that immunospecifically binds to
EphA2
and agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in
cancer cells
-24-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
comprises a VH CDR1 having the amino acid sequence of SEQ ID N0:22 and a VL
CDR1
having the amino acid sequence of SEQ )D N0:18. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR1 having the amino
acid
sequence of SEQ ID N0:22 and a VL CDR2 having the amino acid sequence of SEQ
ID
N0:19. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR1 having the amino acid sequence of SEQ ID N0:22 and a VL
CDR3
having the amino acid sequence of SEQ ID N0:20.
[0078] In another embodiment, an antibody that immunospecifically binds to
EphA2
and agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in
cancer cells
comprises a VH CDR2 having the amino acid sequence of SEQ ID N0:7 and a VL
CDR1
having the amino acid sequence of SEQ ID N0:2. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR2 having the amino
acid
sequence of SEQ >D N0:7 and a VL CDR2 having the amino acid sequence of SEQ ID
N0:3. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR2 having the amino acid sequence of SEQ ID N0:7 and a VL
CDR3
having the amino acid sequence of SEQ ID N0:4.
[0079] In another embodiment, an antibody that immunospecifically binds to
EphA2
and agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in
cancer cells
comprises a VH CDR2 having the amino acid sequence of SEQ ID N0:23 and a VL
CDR1
having the amino acid sequence of SEQ ID N0:18. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR2 having the amino
acid
sequence of SEQ >D N0:23 and a VL CDR2 having the amino acid sequence of SEQ
ID
N0:19. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR2 having the amino acid sequence of SEQ 1Z7 N0:23 and a VL
CDR3
having the amino acid sequence of SEQ >D N0:20.
[0080] In another embodiment, an antibody that immunospecifically binds to
EphA2
and agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in
cancer cells
comprises a VH CDR3 having the amino acid sequence of SEQ ID N0:8 and a VL
CDR1
-25-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
having the amino acid sequence of SEQ 1D N0:2. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR3 having the amino
acid
sequence of SEQ ID N0:8 and a VL CDR2 having the amino acid sequence of SEQ ID
N0:3. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR3 having the amino acid sequence of SEQ ID N0:8 and a VL
CDR3
having the amino acid sequence of SEQ ID N0:4.
[0081] In another embodiment, an antibody that immunospecifically binds to
EphA2
and agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in
cancer cells
comprises a VH CDR3 having the amino acid sequence of SEQ ID N0:24 and a VL
CDR1
having the amino acid sequence of SEQ ID N0:18. In another embodiment, an
antibody that
immunospecifically binds to EphA2 and agonizes EphA2 and/or preferentially
binds an
EphA2 epitope exposed in cancer cells comprises a VH CDR3 having the amino
acid
sequence of SEQ ID N0:24 and a VL CDR2 having the amino acid sequence of SEQ
ID
N0:19. In another embodiment, an antibody that immunospecifically binds to
EphA2 and
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed in cancer
cells
comprises a VH CDR3 having the amino acid sequence of SEQ B7 N0:24 and a VL
CDR3
having the amino acid sequence of SEQ ID N0:20.
[0082] The antibodies used in the methods of the invention include derivatives
that
are modified, i.e, by the covalent attachment of any type of molecule to the
antibody. For
example, but not by way of limitation, the antibody derivatives include
antibodies that have
been modified, e.g., by glycosylation, acetylation, pegylation,
phosphorylation, amidation,
derivatization by known protecting/blocking groups, proteolytic cleavage,
linkage to a
cellular ligand or other protein, etc. Any of numerous chemical modifications
may be carried
out by known techniques, including, but not limited to, specific chemical
cleavage,
acetylation, formylation, metabolic synthesis of tunicamycin, etc.
Additionally, the
derivative may contain one or more non-classical amino acids.
[0083] The present invention also provides antibodies of the invention or
fragments
thereof that comprise a framework region known to those of skill in the art.
In a specific
embodiment, an antibody of the invention or a fragment thereof comprises a
human
framework region. Preferably, the antibody of the invention or fragment
thereof is human or
humanized. In a specific embodiment, the antibody of the invention or fragment
thereof
comprises one or more CDRs from any of EA2, EA3, EA4, or EAS (or any other
EphA2
-26-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
agonistic antibody or EphA2 antibody that preferentially preferentially binds
an EphA2
epitope exposed on cancer cells but not non-cancer cells), binds EphA2, and,
preferably,
agonizes EphA2 and/or preferentially binds an EphA2 epitope exposed on cancer
cells but
not non-cancer cells.
[0084] The present invention encompasses single domain antibodies, including
camelized single domain antibodies (see e.g., Muyldermans et al., 2001, Trends
Biochem. Sci.
26:230; Nuttall et al., 2000, Cur. Pharm. Biotech. 1:253; Reichmann and
Muyldermans,
1999, J. Immunol. Meth. 231:25; International Publication Nos. WO 94/04678 and
WO
94/25591; U.S. Patent No. 6,005,079; which are incorporated herein by
reference in their
entireties). In one embodiment, the present invention provides single domain
antibodies
comprising two VH domains having the amino acid sequence of any of the VH
domains of
EA2, EA3, EA4, or EAS (or any other EphA2 agonistic antibody or EphA2 antibody
that
preferentially binds an EphA2 epitope exposed on cancer cells but not non-
cancer cells) with
modifications such that single domain antibodies are formed. In another
embodiment, the
present invention also provides single domain antibodies comprising two VH
domains
comprising one or more of the VH CDRs of EA2, EA3, EA4, or EAS (or any other
EphA2
agonistic antibody or EphA2 antibody that preferentially binds an EphA2
epitope exposed on
cancer cells but not non-cancer cells).
[0085] The methods of the present invention also encompass the use of
antibodies or
fragments thereof that have half-lives (e.g., serum half-lives) in a mammal,
preferably a
human, of greater than 15 days, preferably greater than 20 days, greater than
25 days, greater
than 30 days, greater than 35 days, greater than 40 days, greater than 45
days, greater than 2
months, greater than 3 months, greater than 4 months, or greater than 5
months. The
increased half-lives of the antibodies of the present invention or fragments
thereof in a
mammal, preferably a human, result in a higher serum titer of said antibodies
or antibody
fragments in the mammal, and thus, reduce the frequency of the administration
of said
antibodies or antibody fragments and/or reduces the concentration of said
antibodies or
antibody fragments to be administered. Antibodies or fragments thereof having
increased in
vivo half-lives can be generated by techniques known to those of skill in the
art. For
example, antibodies or fragments thereof with increased in vivo half-lives can
be generated
by modifying (e.g., substituting, deleting or adding) amino acid residues
identified as
involved in the interaction between the Fc domain and the FcRn receptor (see,
e.g.,
International Publication Nos. WO 97/34631 and WO 02/060919, which are
incorporated
herein by reference in their entireties). Antibodies or fragments thereof with
increased in
-27-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
vivo half-lives can be generated by attaching to said antibodies or antibody
fragments
polymer molecules such as high molecular weight polyethyleneglycol (PEG). PEG
can be
attached to said antibodies or antibody fragments with or without a
multifunctional linker
either through site-specific conjugation of the PEG to the N- or C- terminus
of said antibodies
or antibody fragments or via epsilon-amino groups present on lysine residues.
Linear or
branched polymer derivatization that results in minimal loss of biological
activity will be
used. The degree of conjugation will be closely monitored by SDS-PAGE and mass
spectrometry to ensure proper conjugation of PEG molecules to the antibodies.
Unreacted
PEG can be separated from antibody-PEG conjugates by, e.g., size exclusion or
ion-exchange
chromatography.
[0086] The present invention also encompasses the use of antibodies or
antibody
fragments comprising the amino acid sequence of one or both variable domains
of EA2, EA3,
EA4, or EAS with mutations (e.g., one or more amino acid substitutions) in the
framework or
variable regions. Preferably, mutations in these antibodies maintain or
enhance the avidity
and/or affinity of the antibodies for the particular antigens) to which they
immunospecifically bind. Standard techniques known to those skilled in the art
(e.g.,
immunoassays) can be used to assay the affinity of an antibody for a
particular antigen.
[0087] Standard techniques known to those skilled in the art can be used to
introduce
mutations in the nucleotide sequence encoding an antibody, or fragment
thereof, including,
e.g., site-directed mutagenesis and PCR-mediated mutagenesis, which results in
amino acid
substitutions. Preferably, the derivatives include less than 15 amino acid
substitutions, less
than 10 amino acid substitutions, less than 5 amino acid substitutions, less
than 4 amino acid
substitutions, less than 3 amino acid substitutions, or less than 2 amino acid
substitutions
relative to the original antibody or fragment thereof. In a preferred
embodiment, the
derivatives have conservative amino acid substitutions made at one or more
predicted non-
essential amino acid residues.
[0088] The present invention also encompasses antibodies or fragments thereof
that
immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially bind
an EphA2
epitope exposed in cancer cells, said antibodies or antibody fragments
comprising an amino
acid sequence of a variable light chain and/or variable heavy chain that is at
least 45%, at
least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least
75%, at least 80%, at
least 85%, at least 90%, at least 95%, or at least 99% identical to the amino
acid sequence of
the variable light chain and/or heavy chain of EA2, EA3, EA4, or EAS. In some
embodiments, antibodies or antibody fragments of the invention
immunospecifically bind to
-28-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
EphA2 and comprise an amino acid sequence of a variable light chain that is at
least 45%, at
least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least
75%, at least 80%, at
least 85%, at least 90%, at least 95%, or at least 99% identical to SEQ ID
NO:1 or SEQ 117
N0:17. In other embodiments, antibodies or antibody fragments of the invention
immunospecifically bind to EphA2 and comprise an amino acid sequence of a
variable heavy
chain that is at least 45%, at least 50%, at least 55%, at least 60%, at least
65%, at least 70%,
at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at
least 99% identical
to SEQ >I7 N0:5 or SEQ ID N0:21. In other embodiments, antibodies or antibody
fragments
of the invention immunospecifically bind to EphA2 and comprise an amino acid
sequence of
a variable light chain that is at least 45%, at least 50%, at least 55%, at
least 60%, at least
65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at
least 95%, or at
least 99% identical to SEQ ID NO:1 or SEQ ID N0:17 and a variable heavy chain
that is at
least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least
70%, at least 75%, at
least 80%, at least 85%, at least 90%, at least 95%, or at least 99% identical
to SEQ 1D N0:5
or SEQ ID N0:21.
[0089] The present invention further encompasses antibodies or fragments
thereof
that immunospecifically bind to EphA2 and agonize EphA2 and/ preferentially
bind an
EphA2 epitope exposed in cancer cells, said antibodies or antibody fragments
comprising an
amino acid sequence of one or more CDRs that is at least 45%, at least 50%, at
least 55%, at
least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least
85%, at least 90%, at
least 95%, or at least 99% identical to the amino acid sequence of one or more
CDRs of EA2,
EA3, EA4, or EAS. In one embodiment, antibodies or antibody fragments of the
invention
immunospecifically bind to EphA2 and comprise an amino acid sequence of a CDR
that is at
least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least
70%, at least 75%, at
least 80%, at least 85%, at least 90%, at least 95%, or at least 99% identical
to SEQ >D N0:2,
3, 4, 18, 19, or 20. In another embodiment, antibodies or antibody fragments
of the invention
immunospecifically bind to EphA2 and comprise an amino acid sequence of a CDR
that is at
least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least
70%, at least 75%, at
least 80%, at least 85%, at least 90%, at least 95%, or at least 99% identical
to SEQ >D N0:6,
7, 8, 22, 23, or 24.
[0090] The determination of percent identity of two amino acid sequences can
be
determined by any method known to one skilled in the art, including BLAST
protein
searches.
-29-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0091] The present invention further encompasses antibodies or fragments
thereof
that immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially
bind an
EphA2 epitope exposed in cancer cells, said antibodies or antibody fragments
comprising an
amino acid sequence of one or more CDRs comprising amino acid residue
substitutions,
deletions or additions as compared to SEQ ID NO: 2, 3, 4, 6, 7, 8, 18, 19, 20,
22, 23, or 24.
The antibody comprising the one or more CDRs comprising amino acid residue
substitutions,
deletions or additions may have substantially the same binding, better
binding, or worse
binding when compared to an antibody comprising one or more CDRs without amino
acid
residue substitutions, deletions or additions. In specific embodiments, one,
two, three, four,
or five amino acid residues of the CDR have been substituted, deleted or added
(i.e.,
mutated).
[0092] The present invention also encompasses the use of antibodies or
antibody
fragments that immunospecifically bind to EphA2 and agonize EphA2 and/or
preferentially
bind epitopes on EphA2 that are selectively exposed or increased on cancer
cells but not non-
cancer cells, where said antibodies or antibody fragments are encoded by a
nucleotide
sequence that hybridizes to the nucleotide sequence of EA2, EA3, EA4, or EAS
under
stringent conditions. In one embodiment, the invention provides antibodies or
fragments
thereof that immunospecifically bind to EphA2 and agonize EphA2 and/or
preferentially bind
an epitope on EphA2 that is selectively exposed or increased on cancer cells
but not non-
cancer cells, said antibodies or antibody fragments comprising a variable
light chain encoded
by a nucleotide sequence that hybridizes under stringent conditions to the
nucleotide
sequence of the variable light chain of EA2, EA3, EA4, or EAS. In a preferred
embodiment,
the invention provides antibodies or fragments that immunospecifically bind to
EphA2 and
comprise a variable light chain encoded by a nucleotide sequence that
hybridizes under
stringent conditions to the nucleotide sequence of SEQ ID N0:9 or SEQ ID
N0:25. In
another embodiment, the invention provides antibodies or fragments thereof
that
immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially bind
an epitope
on EphA2 that is selectively exposed or increased on cancer cells but not non-
cancer cells,
said antibodies or antibody fragments comprising a variable heavy chain
encoded by a
nucleotide sequence that hybridizes under stringent conditions to the
nucleotide sequence of
the variable heavy chain of EA2, EA3, EA4, or EAS. In a preferred embodiment,
the
invention provides antibodies or fragments thereof that immunospecifically
bind to EphA2
and comprise a variable heavy chain encoded by a nucleotide sequence that
hybridizes under
stringent conditions to the nucleotide sequence of SEQ ID N0:13 or SEQ ID
N0:29. In
-30-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
other embodiments, antibodies or antibody fragments of the invention
immunospecifically
bind to EphA2 and comprise a variable light chain encoded by a nucleotide
sequence that
hybridizes under stringent conditions to the nucleotide sequence of SEQ m N0:9
or SEQ ~
N0:25 and a variable heavy chain encoded by a nucleotide sequence that
hybridizes under
stringent conditions to the nucleotide sequence of SEQ B7 N0:13 or SEQ ID
N0:29.
[0093] In another embodiment, the invention provides antibodies or fragments
thereof
that immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially
bind an
EphA2 epitope exposed on cancer cells but not non-cancer cells, said
antibodies or antibody
fragments comprising one or more CDRs encoded by a nucleotide sequence that
hybridizes
under stringent conditions to the nucleotide sequence of one or more CDRs of
EA2, EA3,
EA4, or EAS. In a preferred embodiment, the antibodies or fragments of the
invention
immunospecifically bind to EphA2 and comprise a CDR encoded by a nucleotide
sequence
that hybridizes under stringent conditions the nucleotide sequence of SEQ ID
NO:10, 11, 12,
26, 27, or 28. In another preferred embodiment, the antibodies or fragments of
the invention
immunospecifically bind to EphA2 and comprise a CDR encoded by a nucleotide
sequence
that hybridizes under stringent conditions the nucleotide sequence of SEQ ID
N0:14, 15, 16,
30, 31, or 32.
[0094] Stringent hybridization conditions include, but are not limited to,
hybridization
to filter-bound DNA in 6X sodium chloride/sodium citrate (SSC) at about
45°C followed by
one or more washes in 0.2X SSC/0.1% SDS at about 50-65°C, highly
stringent conditions
such as hybridization to filter-bound DNA in 6X SSC at about 45°C
followed by one or more
washes in O.1X SSC/0.2% SDS at about 60°C, or any other stringent
hybridization conditions
known to those skilled in the art (see, for example, Ausubel, F.M. et al.,
eds. 1989 Current
Protocols in Molecular Biology, vol. 1, Green Publishing Associates, Inc. and
John Wiley
and Sons, Inc., NY at pages 6.3.1 to 6.3.6 and 2.10.3).
[0095] The present invention further encompasses antibodies or fragments
thereof
that immunospecifically bind to EphA2 and agonize EphA2 and/or preferentially
bind an
EphA2 epitope exposed in cancer cells, said antibodies or antibody fragments
comprising one
or more CDRs encoded by a nucleotide sequence of one or more CDRs comprising
nucleic
acid residue substitutions, deletions or additions as compared to SEQ ll~
NO:10, 11, 12, 14,
15, 16, 26, 27, 28, 30, 31, or 32. The antibody comprising the one or more
CDRs comprising
nucleic acid residue substitutions, deletions or additions may have
substantially the same
binding, better binding, or worse binding when compared to an antibody
comprising one or
more CDRs without nucleic acid residue substitutions, deletions or additions.
In specific
-31-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
embodiments, one, two, three, four, five, six, seven, eight, nine, ten,
eleven, twelve, thirteen,
fourteen, or fifteen nucleic acid residues of the CDR have been substituted,
deleted or added
(i.e., mutated). The nucleic acid substitutions may or may not change the
amino acid
sequence of the mutated CDR.
TABLE 1
Antibody V chainCDR SEQ ID NO. SEQ ID NO. ATCC
(amino acids)(nucleic De osit
acids) No.
EA2 PTA-4380
VL 1 9
VL1 2 10
VL2 3 11
VL3 4 12
VH 5 13
VH1 6 14
VH2 7 15
VH3 8 16
EAS PTA-4381
VL 17 25
VLl 18 26
VL2 19 27
VL3 20 28
VH 21 29
VH1 22 30
VH2 23 31
VH3 24 32
5.1.1 Antibody coniu~ates
[0096] The present invention encompasses the use of antibodies or fragments
thereof
recombinantly fused or chemically conjugated (including both covalent and non-
covalent
conjugations) to a heterologous polypeptide (or portion thereof, preferably to
a polypeptide of
at least 10, at least 20, at least 30, at least 40, at least 50, at least 60,
at least 70, at least 80, at
least 90 or at least 100 amino acids) to generate fusion proteins. The fusion
does not
necessarily need to be direct, but may occur through linker sequences. For
example,
antibodies may be used to target heterologous polypeptides to particular cell
types, either in
vitro or in vivo, by fusing or conjugating the antibodies to antibodies
specific for particular
cell surface receptors. Antibodies fused or conjugated to heterologous
polypeptides may also
be used in in vitro immunoassays and purification methods using methods known
in the art.
See e.g., International Publication WO 93/21232; EP 439,095; Naramura et al.,
1994,
-32-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Immunol. Lett. 39:91-99; U.S. Patent 5,474,981; Gillies et al., 1992, PNAS
89:1428-1432;
and Fell et al., 1991, J. Immunol. 146:2446-2452, which are incorporated by
reference in their
entireties. In some embodiments, the disorder to be detected, treated,
managed, or monitored
is malignant cancer that overexpresses EphA2. In other embodiments, the
disorder to be
detected, treated, managed, or monitored is a pre-cancerous condition
associated with cells
that overexpress EphA2. In a specific embodiments, the pre-cancerous condition
is high-
grade prostatic intraepithelial neoplasia (PIN), fibroadenoma of the breast,
fibrocystic
disease, or compound nevi.
[0097] The present invention further includes compositions comprising
heterologous
polypeptides fused or conjugated to antibody fragments. For example, the
heterologous
polypeptides may be fused or conjugated to a Fab fragment, Fd fragment, Fv
fragment, F(ab)~
fragment, or portion thereof. Methods for fusing or conjugating polypeptides
to antibody
portions are known in the art. See, e.g., U.S. Patent Nos. 5,336,603,
5,622,929, 5,359,046,
5,349,053, 5,447,851, and 5,112,946; EP 307,434; EP 367,166; International
Publication
Nos. WO 96/04388 and WO 91/06570; Ashkenazi et al., 1991, PNAS 88: 10535-
10539;
Zheng et al., 1995, J. Immunol. 154:5590-5600; and Vil et al., 1992, PNAS
89:11337- 11341
(said references incorporated by reference in their entireties).
[0098] Additional fusion proteins, e.g., of any of EA2, EA3, EA4, or EAS
antibodies
(or any other EphA2 agonistic antibody or EphA2 antibody that preferentially
binds an
EphA2 epitope exposed on cancer cells but not non-cancer cells), may be
generated through
the techniques of gene-shuffling, motif-shuffling, exon-shuffling, and/or
codon-shuffling
(collectively referred to as "DNA shuffling"). DNA shuffling may be employed
to alter the
activities of antibodies of the invention or fragments thereof (e.g.,
antibodies or fragments
thereof with higher affinities and lower dissociation rates). See, generally,
U.S. Patent Nos.
5,605,793; 5,811,238; 5,830,721; 5,834,252; and 5,837,458, and Patten et al.,
1997, Curr.
Opinion Biotechnol. 8:724-33; Harayama, 1998, Trends Biotechnol. 16:76;
Hansson, et al.,
1999, J. Mol. Biol. 287:265; and Lorenzo and Blasco, 1998, BioTechniques
24:308 (each of
these patents and publications are hereby incorporated by reference in its
entirety).
Antibodies or fragments thereof, or the encoded antibodies or fragments
thereof, may be
altered by being subjected to random mutagenesis by error-prone PCR, random
nucleotide
insertion or other methods prior to recombination. One or more portions of a
polynucleotide
encoding an antibody or antibody fragment, which portions immunospecifically
bind to
EphA2 may be recombined with one or more components, motifs, sections, parts,
domains,
fragments, etc. of one or more heterologous molecules.
-33-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[0099] Moreover, the antibodies or fragments thereof can be fused to marker
sequences, such as a peptide to facilitate purification. In preferred
embodiments, the marker
amino acid sequence is a hexa-histidine peptide, such as the tag provided in a
pQE vector
(QIAGEN, Inc., 9259 Eton Avenue, Chatsworth, CA, 91311 ), among others, many
of which
are commercially available. As described in Gentz et al., 1989, PNAS 86:821,
for instance,
hexa-histidine provides for convenient purification of the fusion protein.
Other peptide tags
useful for purification include, but are not limited to, the hemagglutinin
"HA" tag, which
corresponds to an epitope derived from the influenza hemagglutinin protein
(Wilson et al.,
1984, Cell 37:767) and the "flag" tag.
[00100] In other embodiments, antibodies of the present invention or fragments
or
variants thereof are conjugated to a diagnostic or detectable agent. Such
antibodies can be
useful for monitoring or prognosing the development or progression of a cancer
as part of a
clinical testing procedure, such as determining the efficacy of a particular
therapy.
Additionally, such antibodies can be useful for monitoring or prognosing the
development or
progression of a pre-cancerous condition associated with cells that
overexpress EphA2 (e.g.,
high-grade prostatic intraepithelial neoplasia (PIN), fibroadenoma of the
breast, fibrocystic
disease ,or compound nevi). W one embodiment, an exposed EphA2 epitope
antibody is
conjugated to a diagnostic or detectable agent. In a more specific embodiment,
the antibody
is EA2. In another specific embodiment, the antibody is EAS.
[00101] Such diagnosis and detection can accomplished by coupling the antibody
to
detectable substances including, but not limited to various enzymes, such as
but not limited to
horseradish peroxidase, alkaline phosphatase, beta-galactosidase, or
acetylcholinesterase;
prosthetic groups, such as but not limited to streptavidin/biotin and
avidin/biotin; fluorescent
materials, such as but not limited to, umbelliferone, fluorescein, fluorescein
isothiocynate,
rhodamine, dichlorotriazinylamine fluorescein, dansyl chloride or
phycoerythrin; luminescent
materials, such as but not limited to, luminol; bioluminescent materials, such
as but not
limited to, luciferase, luciferin, and aequorin; radioactive materials, such
as but not limited to,
bismuth (z~3Bi), carbon (~4C), chromium (s~Cr), cobalt (s7Co), fluorine (~gF),
gadolinium
(issGd~ is9Gd), gallium (6gGa, 67Ga), germanium (6gGe), holmium (166Ho),
indium (IlsIn,
nsln, il2y, lilIn), iodine (13~h i2sh ~23I, ~2~I), lanthanium (~4°La),
lutetium (~~~Lu), manganese
(saMn), molybdenum (99Mo), palladium (lo3Pd), phosphorous (32P), praseodymium
(la2Pr),
promethium (~49Pm), rhenium (~86Re, ~BgRe), rhodium (~°sRh), ruthemium
(97Ru), samarium
(~s3Sm), scandium (47Sc), selenium (7sSe), strontium (gsSr), sulfur (3sS),
technetium (99Tc),
thallium (2o~Ti), tin (~ ~3Sn, Il7Sn), tritium (3H), xenon (~33Xe), ytterbium
(~69Yb, osYb),
-34-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
yttrium (9°Y), zinc (65Zn); positron emitting metals using various
positron emission
tomographies, and nonradioactive paramagnetic metal ions.
[00102] The present invention further encompasses uses of antibodies or
fragments
thereof conjugated to a therapeutic agent.
[00103] An antibody or fragment thereof may be conjugated to a therapeutic
moiety
such as a cytotoxin, e. g., a cytostatic or cytocidal agent, a therapeutic
agent or a radioactive
metal ion, e.g., alpha-emitters. A cytotoxin or cytotoxic agent includes any
agent that is
detrimental to cells. Examples include paclitaxel, cytochalasin B, gramicidin
D, ethidium
bromide, emetine, mitomycin, etoposide, tenoposide, vincristine, vinblastine,
colchicin,
doxorubicin, daunorubicin, dihydroxy anthracin dione, mitoxantrone,
mithramycin,
actinomycin D, 1-dehydrotestosterone, glucocorticoids, procaine, tetracaine,
lidocaine,
propranolol, puromycin, epirubicin, and cyclophosphamide and analogs or
homologs thereof.
Therapeutic agents include, but are not limited to, antimetabolites (e.g.,
methotrexate, 6-
mercaptopurine, 6-thioguanine, cytarabine, 5-fluorouracil decarbazine),
alkylating agents
(e.g., mechlorethamine, thioepa chlorambucil, melphalan, carmustine (BCNU) and
lomustine
(CCNU), cyclothosphamide, busulfan, dibromomannitol, streptozotocin, mitomycin
C, and
cisdichlorodiamine platinum (II) (DDP) cisplatin), anthracyclines (e.g.,
daunorubicin
(formerly daunomycin) and doxorubicin), antibiotics (e.g., dactinomycin
(formerly
actinomycin), bleomycin, mithramycin, and anthramycin (AMC)), and anti-mitotic
agents
(e.g., vincristine and vinblastine).
[00104] Further, an antibody or fragment thereof may be conjugated to a
therapeutic
agent or drug moiety that modifies a given biological response. Therapeutic
agents or drug
moieties are not to be construed as limited to classical chemical therapeutic
agents. For
example, the drug moiety may be a protein or polypeptide possessing a desired
biological
activity. Such proteins may include, for example, a toxin such as abrin, ricin
A,
pseudomonas exotoxin, cholera toxin, or diphtheria toxin; a protein such as
tumor necrosis
factor, a-interferon, (3-interferon, nerve growth factor, platelet derived
growth factor, tissue
plasminogen activator, an apoptotic agent, e.g., TNF-a, TNF-(3, AIM I (see,
International
Publication No. WO 97/33899), AIM II (see, International Publication No. WO
97/34911),
Fas Ligand (Takahashi et al., 1994, J. Iminunol., 6:1567), and VEGI (see,
International
Publication No. WO 99/23105), a thrombotic agent or an anti-angiogenic agent,
e.g.,
angiostatin or endostatin; or, a biological response modifier such as, for
example, a
lymphokine (e.g., interleukin-1 ("IL-1"), interleukin-2 ("IL-2"), interleukin-
6 ("IL-6"),
-35-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
granulocyte macrophage colony stimulating factor ("GM-CSF"), and granulocyte
colony
stimulating factor ("G-CSF")), or a growth factor (e. g., growth hormone
("GH")).
[00105] Moreover, an antibody can be conjugated to therapeutic moieties such
as a
radioactive materials or macrocyclic chelators useful for conjugating
radiometal ions (see
above for examples of radioactive materials). In certain embodiments, the
macrocyclic
chelator is 1,4,7,10-tetraazacyclododecane-N,N',N",N"-tetraacetic acid (DOTA)
which can
be attached to the antibody via a linker molecule. Such linker molecules are
commonly
known in the art and described in Denardo et al., 1998, Clin Cancer Res.
4:2483-90; Peterson
et al., 1999, Bioconjug. Chem. 10:553; and Zimmerman et al., 1999, Nucl. Med.
Biol. 26:943-
50 each incorporated by reference in their entireties.
[00106] In a specific embodiment, the conjugated antibody is an EphA2 antibody
that
preferably binds an EphA2 epitope exposed on cancer cells but not on non-
cancer cells (i.e.,
exposed EphA2 epitope antibody). In a more specific embodiment, the conjugated
antibody
is EA2. In another specific embodiment, the conjugated antibody is EAS.
[00107] Techniques for conjugating therapeutic moieties to antibodies are well
known.
Moieties can be conjugated to antibodies by any method known in the art,
including, but not
limited to aldehyde/Schiff linkage, sulphydryl linkage, acid-labile linkage,
cis-aconityl
linkage, hydrazone linkage, enzymatically degradable linkage (see generally
Garnett, 2002,
Adv. Drug Deliv. Rev. 53:171-216). Additional techniques for conjugating
therapeutic
moieties to antibodies are well known, see, e.g., Arnon et al., "Monoclonal
Antibodies For
Immunotargeting Of Drugs In Cancer Therapy," in Monoclonal Antibodies And
Cancer
Therapy, Reisfeld et al. (eds.), pp. 243-56 (Alan R. Liss, Inc. 1985);
Hellstrom et al.,
"Antibodies For Drug Delivery," in Controlled Drug Delivery (2nd Ed.),
Robinson et al.
(eds.), pp. 623-53 (Marcel Dekker, Inc. 1987); Thorpe, "Antibody Carriers Of
Cytotoxic
Agents In Cancer Therapy: A Review," in Monoclonal Antibodies '84: Biological
And
Clinical Applications, Pinchera et al. (eds.), pp. 475-506 (1985); "Analysis,
Results, And
Future Prospective Of The Therapeutic Use Of Radiolabeled Antibody In Cancer
Therapy,"
in Monoclonal Antibodies For Cancer Detection And Therapy, Baldwin et al.
(eds.), pp. 303-
16 (Academic Press 1985), and Thorpe et al., 1982, Immunol. Rev. 62:119-58.
Methods for
fusing or conjugating antibodies to polypeptide moieties are known in the art.
See, e.g., U.S.
Patent Nos. 5,336,603, 5,622,929, 5,359,046, 5,349,053, 5,447,851, and
5,112,946; EP
307,434; EP 367,166; International Publication Nos. WO 96/04388 and WO
91/06570;
Ashkenazi et al., 1991, PNAS 88: 10535-10539; Zheng et al., 1995, J. Immunol.
154:5590-
5600; and Vil et al., 1992, PNAS 89:11337- 11341. The fusion of an antibody to
a moiety
-36-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
does not necessarily need to be direct, but may occur through linker
sequences. Such linker
molecules are commonly known in the art and described in Denardo et al., 1998,
Clin Cancer
Res. 4:2483-90; Peterson et al., 1999, Bioconjug. Chem. 10:553; Zimmerman et
al., 1999,
Nucl. Med. Biol. 26:943-50; Garnett, 2002, Adv. Drug Deliv. Rev. 53:171-216,
each of which
is incorporated herein by reference in its entirety.
[00108] Alternatively, an antibody can be conjugated to a second antibody to
form an
antibody heteroconjugate as described by Segal in U.S. Patent No. 4,676,980,
which is
incorporated herein by reference in its entirety.
[00109] Antibodies may also be attached to solid supports, which are
particularly
useful for immunoassays or purification of the target antigen. Such solid
supports include, but
are not limited to, glass, cellulose, polyacrylamide, nylon, polystyrene,
polyvinyl chloride or
polypropylene.
5.1.2 Methods Of Producing Antibodies
[00110] The antibodies or fragments thereof can be produced by any method
known in
the art for the synthesis of antibodies, in particular, by chemical synthesis
or preferably, by
recombinant expression techniques.
[00111] Monoclonal antibodies can be prepared using a wide variety of
techniques
known in the art including the use of hybridoma, recombinant, and phage
display
technologies, or a combination thereof. For example, monoclonal antibodies can
be produced
using hybridoma techniques including those known in the art and taught, for
example, in
Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory
Press, 2nd
ed. 1988); Hammerling, et al., in: Monoclonal Antibodies and T Cell Hybridamas
563-681
(Elsevier, N.Y., 1981) (said references incorporated by reference in their
entireties). The
term "monoclonal antibody" as used herein is not limited to antibodies
produced through
hybridoma technology. The term "monoclonal antibody" refers to an antibody
that is derived
from a single clone, including any eukaryotic, prokaryotic, or phage clone,
and not the
method by which it is produced.
[00112] Methods for producing and screening for specific antibodies using
hybridoma
technology are routine and well known in the art. Briefly, mice can be
immunized with
EphA2 (either the full length protein or a domain thereof, e.g., the
extracellular or the ligand
binding domain) and once an immune response is detected, e.g., antibodies
specific for
EphA2 are detected in the mouse serum, the mouse spleen is harvested and
splenocytes
isolated. The splenocytes are then fused by well known techniques to any
suitable myeloma
-37-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
cells, for example cells from cell line SP20 available from the ATCC.
Hybridomas are
selected and cloned by limited dilution. Hybridoma clones are then assayed by
methods
known in the art for cells that secrete antibodies capable of binding a
polypeptide of the
invention. Ascites fluid, which generally contains high levels of antibodies,
can be generated
by immunizing mice with positive hybridoma clones.
[00113] Accordingly, monoclonal antibodies can be generated by culturing a
hybridoma cell secreting an antibody of the invention wherein, preferably, the
hybridoma is
generated by fusing splenocytes isolated from a mouse immunized with EphA2 or
fragment
thereof with myeloma cells and then screening the hybridomas resulting from
the fusion for
hybridoma clones that secrete an antibody able to bind EphA2.
[00114] Antibody fragments which recognize specific EphA2 epitopes may be
generated by any technique known to those of skill in the art. For example,
Fab and F(ab')2
fragments of the invention may be produced by proteolytic cleavage of
immunoglobulin
molecules, using enzymes such as papain (to produce Fab fragments) or pepsin
(to produce
F(ab')2 fragments). F(ab')2 fragments contain the variable region, the light
chain constant
region and the CH1 domain of the heavy chain. Further, the antibodies of the
present
invention can also be generated using various phage display methods known in
the art.
[00115] In phage display methods, functional antibody domains are displayed on
the
surface of phage particles which carry the polynucleotide sequences encoding
them. In
particular, DNA sequences encoding VH and VL domains are amplified from animal
cDNA
libraries (e.g., human or murine cDNA libraries of lymphoid tissues). The DNA
encoding
the VH and VL domains are recombined together with an scFv linker by PCR and
cloned into
a phagemid vector (e.g., p CANTAB 6 or pComb 3 HSS). The vector is
electroporated in E.
coli and the E. coli is infected with helper phage. Phage used in these
methods are typically
filamentous phage including fd and M13 and the VH and VL domains are usually
recombinantly fused to either the phage gene III or gene VIII. Phage
expressing an antigen
binding domain that binds to the EphA2 epitope of interest can be selected or
identified with
antigen, e.g., using labeled antigen or antigen bound or captured to a solid
surface or bead.
Examples of phage display methods that can be used to make the antibodies of
the present
invention include those disclosed in Brinkman et al., 1995, J. Immunol.
Methods 182:41-50;
Ames et al., 1995, J. Immunol. Methods 184:177; Kettleborough et al., 1994,
Eur. J.
Immunol. 24:952-958; Persic et al., 1997, Gene 187:9; Burton et al., 1994,
Advances in
Immunology 57:191-280; International Application No. PCT/GB91/01134;
International
Publication Nos. WO 90/02809, WO 91/10737, WO 92/01047, WO 92/18619, WO 93/1
-38-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
1236, WO 95/15982, WO 95/20401, and W097/13844; and U.S. Patent Nos.
5,698,426,
5,223,409, 5,403,484, 5,580,717, 5,427,908, 5,750,753, 5,821,047, 5,571,698,
5,427,908,
5,516,637, 5,780,225, 5,658,727, 5,733,743 and 5,969,108; each of which is
incorporated
herein by reference in its entirety.
[00116] Phage may be screened for EphA2 binding, particularly to the
extracellular
domain of EphA2. Agonizing EphA2 activity (e.g., increasing EphA2
phosphorylation,
reducing EphA2 levels) may also be screened.
[00117] As described in the above references, after phage selection, the
antibody
coding regions from the phage can be isolated and used to generate whole
antibodies,
including human antibodies, or any other desired antigen binding fragment, and
expressed in
any desired host, including mammalian cells, insect cells, plant cells, yeast,
and bacteria, e. g.,
as described below. Techniques to recombinantly produce Fab, Fab' and F(ab')2
fragments
can also be employed using methods known in the art such as those disclosed in
International
Publication No. WO 92/22324; Mullinax et al., 1992, BioTechniques 12:864;
Sawai et al.,
1995, AJRI 34:26; and Better et al., 1988, Science 240:1041 (said references
incorporated by
reference in their entireties).
[00118] To generate whole antibodies, PCR primers including VH or VL
nucleotide
sequences, a restriction site, and a flanking sequence to protect the
restriction site can be used
to amplify the VH or VL sequences in scFv clones. Utilizing cloning techniques
known to
those of skill in the art, the PCR amplified VH domains can be cloned into
vectors expressing
a VH constant region, e.g., the human gamma 4 constant region, and the PCR
amplified VL
domains can be cloned into vectors expressing a VL constant region, e.g.,
human kappa or
lambda constant regions. Preferably, the vectors for expressing the VH or VL
domains
comprise an EF-la promoter, a secretion signal, a cloning site for the
variable domain,
constant domains, and a selection marker such as neomycin. The VH and VL
domains may
also be cloned into one vector expressing the necessary constant regions. The
heavy chain
conversion vectors and light chain conversion vectors are then co-transfected
into cell lines to
generate stable or transient cell lines that express full-length antibodies,
e.g., IgG, using
techniques known to those of skill in the art.
[00119] For some uses, including in vivo use of antibodies in humans and in
vitro
detection assays, it may be preferable to use human or chimeric antibodies.
Completely
human antibodies are particularly desirable for therapeutic treatment of human
subjects.
Human antibodies can be made by a variety of methods known in the art
including phage
display methods described above using antibody libraries derived from human
-39-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
immunoglobulin sequences. See also U.S. Patent Nos. 4,444,887 and 4,716,111;
and
International Publication Nos. WO 98/46645, WO 98/50433, WO 98/24893, WO
98/16654,
WO 96/34096, WO 96/33735, and WO 91/10741; each of which is incorporated
herein by
reference in its entirety.
[00120] Human antibodies can also be produced using transgenic mice which are
incapable of expressing functional endogenous immunoglobulins, but which can
express
human immunoglobulin genes. For example, the human heavy and light chain
immunoglobulin gene complexes may be introduced randomly or by homologous
recombination into mouse embryonic stem cells. Alternatively, the human
variable region,
constant region, and diversity region may be introduced into mouse embryonic
stem cells in
addition to the human heavy and light chain genes. The mouse heavy and light
chain
immunoglobulin genes may be rendered non-functional separately or
simultaneously with the
introduction of human immunoglobulin loci by homologous recombination. In
particular,
homozygous deletion of the JH region prevents endogenous antibody production.
The
modified embryonic stem cells are expanded and microinjected into blastocysts
to produce
chimeric mice. The chimeric mice are then be bred to produce homozygous
offspring which
express human antibodies. The transgenic mice are immunized in the normal
fashion with a
selected antigen, e.g., all or a portion of a polypeptide of the invention.
Monoclonal
antibodies directed against the antigen can be obtained from the immunized,
transgenic mice
using conventional hybridoma technology. The human immunoglobulin transgenes
harbored
by the transgenic mice rearrange during B cell differentiation, and
subsequently undergo
class switching and somatic mutation. Thus, using such a technique, it is
possible to produce
therapeutically useful IgG, IgA, IgM and IgE antibodies. For an overview of
this technology
for producing human antibodies, see Lonberg and Huszar (1995, Int. Rev.
Immunol.
13:65-93). For a detailed discussion of this technology for producing human
antibodies and
human monoclonal antibodies and protocols for producing such antibodies, see,
e.g.,
International Publication Nos. WO 98/24893, WO 96/34096, and WO 96/33735; and
U.S.
Patent Nos. 5,413,923, 5,625,126, 5,633,425, 5,569,825, 5,661,016, 5,545,806,
5,814,318,
and 5,939,598, which are incorporated by reference herein in their entirety.
In addition,
companies such as Abgenix, Inc. (Fremont, CA) and Medarex (Princeton, NJ) can
be
engaged to provide human antibodies directed against a selected antigen using
technology
similar to that described above.
[00121] A chimeric antibody is a molecule in which different portions of the
antibody
are derived from different immunoglobulin molecules such as antibodies having
a variable
-40-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
region derived from a non-human antibody and a human immunoglobulin constant
region.
Methods for producing chimeric antibodies are known in the art. See, e.g.,
Morrison, 1985,
Science 229:1202; Oi et al., 1986, BioTechniques 4:214; Gillies et al., 1989,
J. Immunol.
Methods 125:191-202; and U.S. Patent Nos. 6,311,415, 5,807,715, 4,816,567, and
4,816,397,
which are incorporated herein by reference in their entirety. Chimeric
antibodies comprising
one or more CDRs from a non-human species and framework regions from a human
immunoglobulin molecule can be produced using a variety of techniques known in
the art
including, for example, CDR-grafting (EP 239,400; International Publication
No. WO
91/09967; and U.S. Patent Nos. 5,225,539, 5,530,101, and 5,585,089), veneering
or
resurfacing (EP 592,106; EP 519,596; Padlan, 1991, Molecular Immunology
28(4/5):489-
498; Studnicka et al., 1994, Protein Engineering 7:805; and Roguska et al.,
1994, PNAS
91:969), and chain shuffling (U.S. Patent No. 5,565,332). In one embodiment, a
chimeric
antibody of the invention immunospecifically binds EphA2 and comprises one,
two, or three
VL CDRs having an amino acid sequence of any of the V,, CDRs of EA2, EA3, EA4,
or EA5
within human framework regions. In a specific embodiment, a chimeric antibody
of the
invention immunospecifically binds EphA2 and comprises a VL CDR having the
amino acid
sequence of SEQ ID N0:2, 3, 4, 18, 19, or 20. In another embodiment, a
chimeric antibody
of the invention immunospecifically binds EphA2 and comprises one, two, or
three VH
CDRs having an amino acid sequence of any of the VH CDRs of EA2, EA3, EA4, or
EA5
within human framework regions. In a specific embodiment, a chimeric antibody
of the
invention immunospecifically binds EphA2 and comprises a VH CDR having the
amino acid
sequence of SEQ ID N0:6, 7, 8, 22, 23, or 24. In a preferred embodiment, a
chimeric
antibody of the invention immunospecifically binds EphA2 and comprises one,
two, or three
VL CDRs having an amino acid sequence of any of the VL CDRs of EA2, EA3, EA4,
or
EA5 and further comprises one, two, or three VH CDRs having an amino acid
sequence of
any of the VH CDRs of EA2, EA3, EA4, or EA5 within human framework regions. In
a
specific preferred embodiment, a chimeric antibody of the invention
immunospecifically
binds EphA2 and comprises a VL CDR having an amino acid sequence of SEQ ID NO:
2, 3,
4, 18, 19, or 20 and further comprises a VH CDR having an amino acid sequence
of SEQ ID
N0:6, 7, 8, 22, 23, or 24. In a more preferred embodiment, a chimeric antibody
of the
invention immunospecifically binds EphA2 and comprises three VL CDRs having an
amino
acid sequence of any of the VL CDRs of , EA2, EA3, EA4, or EA5 and three VH
CDRs
having an amino acid sequence of any of the VH CDRs of EA2, EA3, EA4, or EA5
within
human framework regions. In an even more preferred embodiment, a chimeric
antibody of
-41-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
the invention immunospecifically binds EphA2 and comprises VL CDRs having an
amino
acid sequence selected from the group consisting of SEQ ID NO: 2, 3, 4, 18,
19, or 20, and
further comprises VH CDRs having an amino acid sequence selected from the
group
consisting of SEQ ID N0:6, 7, 8, 22, 23, or 24.
[00122] Often, framework residues in the framework regions will be substituted
with
the corresponding residue from the CDR donor antibody to alter, preferably
improve, antigen
binding. These framework substitutions are identified by methods well known in
the art, e.g.,
by modeling of the interactions of the CDR and framework residues to identify
framework
residues important for antigen binding and sequence comparison to identify
unusual
framework residues at particular positions. (See, e.g., U.S. Patent No.
5,585,089; and
Riechmann et al., 1988, Nature 332:323, which are incorporated herein by
reference in their
entireties.)
[00123] A humanized antibody is an antibody or its variant or fragment thereof
which
is capable of binding to a predetermined antigen and which comprises a
framework region
having substantially the amino acid sequence of a human immunoglobulin and a
CDR having
substantially the amino acid sequence of a non-human immunoglobulin. A
humanized
antibody comprises substantially all of at least one, and typically two,
variable domains in
which all or substantially all of the CDR regions correspond to those of a non-
human
immunoglobulin (i.e., donor antibody) and all or substantially all of the
framework regions
are those of a human immunoglobulin consensus sequence. Preferably, a
humanized
antibody also comprises at least a portion of an immunoglobulin constant
region (Fc),
typically that of a human immunoglobulin. Ordinarily, the antibody will
contain both the
light chain as well as at least the variable domain of a heavy chain. The
antibody also may
include the CH1, hinge, CH2, CH3, and CH4 regions of the heavy chain. The
humanized
antibody can be selected from any class of immunoglobulins, including IgM,
IgG, IgD, IgA
and IgE, and any isotype, including IgGi, IgG2, IgG3 and IgG4. Usually the
constant domain
is a complement fixing constant domain where it is desired that the humanized
antibody
exhibit cytotoxic activity, and the class is typically IgG~. Where such
cytotoxic activity is not
desirable, the constant domain may be of the IgG2 class. The humanized
antibody may
comprise sequences from more than one class or isotype, and selecting
particular constant
domains to optimize desired effector functions is within the ordinary skill in
the art. The
framework and CDR regions of a humanized antibody need not correspond
precisely to the
parental sequences, e.g., the donor CDR or the consensus framework may be
mutagenized by
substitution, insertion or deletion of at least one residue so that the CDR or
framework
-42-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
residue at that site does not correspond to either the consensus or the import
antibody. Such
mutations, however, will not be extensive. Usually, at least 75% of the
humanized antibody
residues will correspond to those of the parental framework region (FR) and
CDR sequences,
more often 90%, and most preferably greater than 95%. Humanized antibodies can
be
produced using variety of techniques known in the art, including but not
limited to,
CDR-grafting (European Patent No. EP 239,400; International Publication No. WO
91/09967; and U.S. Patent Nos. 5,225,539, 5,530,101, and 5,585,089), veneering
or
resurfacing (European Patent Nos. EP 592,106 and EP 519,596; Padlan, 1991,
Molecular
Immunology 28(4/5):489-498; Studnicka et al., 1994, Protein Engineering
7(6):805-814; and
Roguska et al., 1994, PNAS 91:969-973), chain shuffling (U.S. Patent No.
5,565,332), and
techniques disclosed in, e.g., U.S. Patent Nos. 6,407,213, 5,766,886,
5,585,089, International
Publication No. WO 9317105, Tan et al., 2002, J. Immunol. 169:1119-25, Caldas
et al., 2000,
Protein Eng. 13:353-60, Morea et al., 2000, Methods 20:267-79, Baca et al.,
1997, J. Biol.
Chem. 272:10678-84, Roguska et al., 1996, Protein Eng. 9:895-904, Couto et
al., 1995,
Cancer Res. 55 (23 Supp):5973s-5977s, Couto et al., 1995, Cancer Res. 55:1717-
22, Sandhu,
1994, Gene 150:409-10, Pedersen et al., 1994, J. Mol. Biol. 235:959-73, Jones
et al., 1986,
Natecre 321:522-525, Riechmann et al., 1988, Nature 332:323, and Presta, 1992,
Curr. Op.
Struct. Biol. 2:593-596. Often, framework residues in the framework regions
will be
substituted with the corresponding residue from the CDR donor antibody to
alter, preferably
improve, antigen binding. These framework substitutions are identified by
methods well
known in the art, e.g., by modeling of the interactions of the CDR and
framework residues to
identify framework residues important for antigen binding and sequence
comparison to
identify unusual framework residues at particular positions. (See, e.g., Queen
et al., U.S.
Patent No. 5,585,089; and Riechmann et al., 1988, Nature 332:323, which are
incorporated
herein by reference in their entireties.)
[00124] Further, the antibodies of the invention can, in turn, be utilized to
generate
anti-idiotype antibodies using techniques well known to those skilled in the
art. (See, e.g.,
Greenspan & Bona, 1989, FASEB J. 7:437-444; and Nissinoff, 1991, J. Immunol.
147:2429-
2438). The invention provides methods employing the use of polynucleotides
comprising a
nucleotide sequence encoding an antibody of the invention or a fragment
thereof.
5.1.3 Polynucleotides Encoding An Antibody
[00125] The methods of the invention also encompass polynucleotides that
hybridize
under high stringency, intermediate or lower stringency hybridization
conditions, e.g., as
- 43 -
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
defined supra, to polynucleotides that encode an antibody of the invention. 1n
a specific
embodiment, the invention provides an isolated nucleic acid comprising a
nucleotide
sequence encoding a heavy chain variable domain or a light chain variable
domain of an
antibody of the invention (e.g., EA2, EA3, EA4 or EAS). In another specific
embodiment,
the invention provides an isolated nucleic acid comprising a nucleotide
sequence encoding a
heavy chain variable domain or a light chain variable domain of an antibody of
the invention
(e.g., EA2, EA3, EA4 or EAS) that has been humanized or chimerized.
[00126] The polynucleotides may be obtained, and the nucleotide sequence of
the
polynucleotides determined, by any method known in the art. Since the amino
acid
sequences of the antibodies are known, nucleotide sequences encoding these
antibodies can
be determined using methods well known in the art, i.e., nucleotide codons
known to encode
particular amino acids are assembled in such a way to generate a nucleic acid
that encodes the
antibody or fragment thereof of the invention. Such a polynucleotide encoding
the antibody
may be assembled from chemically synthesized oligonucleotides (e.g., as
described in
Kutmeier et al., 1994, BioTechniques 17:242), which, briefly, involves the
synthesis of
overlapping oligonucleotides containing portions of the sequence encoding the
antibody,
annealing and ligating of those oligonucleotides, and then amplification of
the ligated
oligonucleotides by PCR. For example, and not by way of limitation, three
clones containing
the polynucleotide sequences of the EAS light chain variable region were
identified using
degenerate primers (i.e., oligonucleotides that wobble at the third position
base to allow for
identification of all codons that encode the corresponding amino acid)
designed from the
amino terminus of the light chain protein sequence. In the three clones
identified that
contained the variable light chain EAS polynucleotide sequence, the bases at
positions 6 and
9 were guanine (G) or tyrosine (T); and at position 15, the base was G, T or
cytosine (C). All
three clones encoded the amino acid sequence of the EAS VL region having SEQ
1D N0:17.
[00127] Alternatively, a polynucleotide encoding an antibody may be generated
from
nucleic acid from a suitable source. If a clone containing a nucleic acid
encoding a particular
antibody is not available, but the sequence of the antibody molecule is known,
(see e.g., FIG.
16), a nucleic acid encoding the immunoglobulin may be chemically synthesized
or obtained
from a suitable source (e. g., an antibody cDNA library, or a cDNA library
generated from, or
nucleic acid, preferably poly A+ RNA, isolated from, any tissue or cells
expressing the
antibody, such as hybridoma cells selected to express an antibody of the
invention, e.g., clone
deposited in the ATCC as PTA-4380) by PCR amplification using synthetic
primers
hybridizable to the 3' and 5' ends of the sequence or by cloning using an
oligonucleotide
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
probe specific for the particular gene sequence to identify, e.g., a cDNA
clone from a cDNA
library that encodes the antibody. Amplified nucleic acids generated by PCR
may then be
cloned into replicable cloning vectors using any method well known in the art.
[00128] Once the nucleotide sequence of the antibody is determined, the
nucleotide
sequence of the antibody may be manipulated using methods well known in the
art for the
manipulation of nucleotide sequences, e.g., recombinant DNA techniques, site
directed
mutagenesis, PCR, etc. (see, for example, the techniques described in Sambrook
et al., 1990,
Molecular Cloning, A Laboratory Manual, 2d Ed., Cold Spring Harbor Laboratory,
Cold
Spring Harbor, NY and Ausubel et al., eds., 1998, Current Protocols in
Molecular Biology,
John Wiley & Sons, NY, which are both incorporated by reference herein in
their entireties),
to generate antibodies having a different amino acid sequence, for example to
create amino
acid substitutions, deletions, and/or insertions.
[00129] In a specific embodiment, one or more of the CDRs is inserted within
framework regions using routine recombinant DNA techniques. The framework
regions may
be naturally occurring or consensus framework regions, and preferably human
framework
regions (see, e.g., Chothia et al., 1998, J. Mol. Biol. 278: 457-479 for a
listing of human
framework regions). Preferably, the polynucleotide generated by the
combination of the
framework regions and CDRs encodes an antibody that specifically binds to
EphA2.
Preferably, as discussed supra, one or more amino acid substitutions may be
made within the
framework regions, and, preferably, the amino acid substitutions improve
binding of the
antibody to its antigen. Additionally, such methods may be used to make amino
acid
substitutions or deletions of one or more variable region cysteine residues
participating in an
intrachain disulfide bond to generate antibody molecules lacking one or more
intrachain
disulfide bonds. Other alterations to the polynucleotide are encompassed by
the present
invention and within the skill of the art.
5.1.4 Recombinant Expression Of An Antibody
[00130] Recombinant expression of an antibody of the invention, derivative,
analog or
fragment thereof, (e.g., a heavy or light chain of an antibody of the
invention or a portion
thereof or a single chain antibody of the invention), requires construction of
an expression
vector containing a polynucleotide that encodes the antibody. Once a
polynucleotide
encoding an antibody molecule or a heavy or light chain of an antibody, or
portion thereof
(preferably, but not necessarily, containing the heavy or light chain variable
domain), of the
invention has been obtained, the vector for the production of the antibody
molecule may be
- 45 -
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
produced by recombinant DNA technology using techniques well known in the art.
Thus,
methods for preparing a protein by expressing a polynucleotide containing an
antibody
encoding nucleotide sequence are described herein. Methods which are well
known to those
skilled in the art can be used to construct expression vectors containing
antibody coding
sequences and appropriate transcriptional and translational control signals.
These methods
include, for example, in vitro recombinant DNA techniques, synthetic
techniques, and in vivo
genetic recombination. The invention, thus, provides replicable vectors
comprising a
nucleotide sequence encoding an antibody molecule of the invention, a heavy or
light chain
of an antibody, a heavy or light chain variable domain of an antibody or a
portion thereof, or
a heavy or light chain CDR, operably linked to a promoter. Such vectors may
include the
nucleotide sequence encoding the constant region of the antibody molecule
(see, e.g.,
International Publication Nos. WO 86/05807 and WO 89/01036; and U.S. Patent
No.
5,122,464) and the variable domain of the antibody may be cloned into such a
vector for
expression of the entire heavy, the entire light chain, or both the entire
heavy and light chains.
[00131] The expression vector is transferred to a host cell by conventional
techniques
and the transfected cells are then cultured by conventional techniques to
produce an antibody
of the invention. Thus, the invention includes host cells containing a
polynucleotide
encoding an antibody of the invention or fragments thereof, or a heavy or
light chain thereof,
or portion thereof, or a single chain antibody of the invention, operably
linked to a
heterologous promoter. In preferred embodiments for the expression of double-
chained
antibodies, vectors encoding both the heavy and light chains may be co-
expressed in the host
cell for expression of the entire immunoglobulin molecule, as detailed below.
[00132] A variety of host-expression vector systems may be utilized to express
the
antibody molecules of the invention (see, e.g., U.S. Patent No. 5,807,715).
Such host-
expression systems represent vehicles by which the coding sequences of
interest may be
produced and subsequently purified, but also represent cells which may, when
transformed or
transfected with the appropriate nucleotide coding sequences, express an
antibody molecule
of the invention in situ. These include but are not limited to microorganisms
such as bacteria
(e.g., E. coli and B. subtilis) transformed with recombinant bacteriophage
DNA, plasmid
DNA or cosmid DNA expression vectors containing antibody coding sequences;
yeast (e.g.,
Saccharomyces Pichia) transformed with recombinant yeast expression vectors
containing
antibody coding sequences; insect cell systems infected with recombinant virus
expression
vectors (e.g., baculovirus) containing antibody coding sequences; plant cell
systems infected
with recombinant virus expression vectors (e.g., cauliflower mosaic virus,
CaMV; tobacco
-46-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
mosaic virus, TMV) or transformed with recombinant plasmid expression vectors
(e.g., Ti
plasmid) containing antibody coding sequences; or mammalian cell systems
(e.g., COS,
CHO, BHK, 293, NSO, and 3T3 cells) harboring recombinant expression constructs
containing promoters derived from the genome of mammalian cells (e.g.,
metallothionein
promoter) or from mammalian viruses (e.g., the adenovirus late promoter; the
vaccinia virus
7.SK promoter). Preferably, bacterial cells such as Escherichia coli, and more
preferably,
eukaryotic cells, especially for the expression of whole recombinant antibody
molecule, are
used for the expression of a recombinant antibody molecule. For example,
mammalian cells
such as Chinese hamster ovary cells (CHO), in conjunction with a vector such
as the major
intermediate early gene promoter element from human cytomegalovirus is an
effective
expression system for antibodies (Foecking et al., 1986, Gene 45:101; and
Cockett et al.,
1990, BioTechnology 8:2). In a specific embodiment, the expression of
nucleotide sequences
encoding antibodies or fragments thereof which immunospecifically bind to and
agonize is
regulated by a constitutive promoter, inducible promoter or tissue specific
promoter.
[00133] In bacterial systems, a number of expression vectors may be
advantageously
selected depending upon the use intended for the antibody molecule being
expressed. For
example, when a large quantity of such a protein is to be produced, for the
generation of
pharmaceutical compositions of an antibody molecule, vectors which direct the
expression of
high levels of fusion protein products that are readily purified may be
desirable. Such vectors
include, but are not limited to, the E. coli expression vector pUR278 (Ruther
et al., 1983,
EMBO 12:1791), in which the antibody coding sequence may be ligated
individually into the
vector in frame with the lac Z coding region so that a fusion protein is
produced; pIN vectors
(Inouye & Inouye, 1985, Nucleic Acids Res. 13:3101-3109; Van Heeke & Schuster,
1989, J.
Biol. Chem. 24:5503-5509); and the like. pGEX vectors may also be used to
express foreign
polypeptides as fusion proteins with glutathione 5-transferase (GST). In
general, such fusion
proteins are soluble and can easily be purified from lysed cells by adsorption
and binding to
matrix glutathione-agarose beads followed by elution in the presence of free
glutathione. The
pGEX vectors are designed to include thrombin or factor Xa protease cleavage
sites so that
the cloned target gene product can be released from the GST moiety.
[00134] In an insect system, Autographa californica nuclear polyhedrosis virus
(AcNPV) is used as a vector to express foreign genes. The virus grows in
Spodoptera
frugiperda cells. The antibody coding sequence may be cloned individually into
non-
essential regions (for example the polyhedrin gene) of the virus and placed
under control of
an AcNPV promoter (for example the polyhedrin promoter).
-47-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00135] In mammalian host cells, a number of viral-based expression systems
may be
utilized. In cases where an adenovirus is used as an expression vector, the
antibody coding
sequence of interest may be ligated to an adenovirus transcription/translation
control
complex, e.g., the late promoter and tripartite leader sequence. This chimeric
gene may then
be inserted in the adenovirus genome by in vitro or in vivo recombination.
Insertion in a non-
essential region of the viral genome (e.g., region El or E3) will result in a
recombinant virus
that is viable and capable of expressing the antibody molecule in infected
hosts (e.g., see
Logan & Shenk, 1984, PNAS 8 1:355-359). Specific initiation signals may also
be required
for efficient translation of inserted antibody coding sequences. These signals
include the
ATG initiation codon and adjacent sequences. Furthermore, the initiation codon
must be in
phase with the reading frame of the desired coding sequence to ensure
translation of the
entire insert. These exogenous translational control signals and initiation
codons can be of a
variety of origins, both natural and synthetic. The efficiency of expression
may be enhanced
by the inclusion of appropriate transcription enhancer elements, transcription
terminators, etc.
(see, e.g., Bittner et al., 1987, Methods in Enzymol. 153:516-544).
[00136) In addition, a host cell strain may be chosen which modulates the
expression
of the inserted sequences, or modifies and processes the gene product in the
specific fashion
desired. Such modifications (e.g., glycosylation) and processing (e.g.,
cleavage) of protein
products may be important for the function of the protein. Different host
cells have
characteristic and specific mechanisms for the post-translational processing
and modification
of proteins and gene products. Appropriate cell lines or host systems can be
chosen to ensure
the correct modification and processing of the foreign protein expressed. To
this end,
eukaryotic host cells which possess the cellular machinery for proper
processing of the
primary transcript, glycosylation, and phosphorylation of the gene product may
be used.
Such mammalian host cells include but are not limited to CHO, VERO, BHK, HeLa,
COS,
MDCK, 293, 3T3, W138, BT483, Hs578T, HTB2, BT20, NS1, and T47D, NSO (a murine
myeloma cell line that does not endogenously produce any immunoglobulin
chains),
CRL7O30 and HsS78Bst cells.
[00137] For long-term, high-yield production of recombinant proteins, stable
expression is preferred. For example, cell lines which stably express the
antibody molecule
may be engineered. Rather than using expression vectors which contain viral
origins of
replication, host cells can be transformed with DNA controlled by appropriate
expression
control elements (e.g., promoter, enhancer, sequences, transcription
terminators,
polyadenylation sites, etc.), and a selectable marker. Following the
introduction of the
-48-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
foreign DNA, engineered cells may be allowed to grow for 1-2 days in an
enriched media,
and then are switched to a selective media. The selectable marker in the
recombinant plasmid
confers resistance to the selection and allows cells to stably integrate the
plasmid into their
chromosomes and grow to form foci which in turn can be cloned and expanded
into cell lines.
This method may advantageously be used to engineer cell lines which express
the antibody
molecule. Such engineered cell lines may be particularly useful in screening
and evaluation
of compositions that interact directly or indirectly with the antibody
molecule.
[00138] A number of selection systems may be used, including but not limited
to, the
herpes simplex virus thymidine kinase (Wigler et al., 1977, Cell 11:223),
glutamine synthase,
hypoxanthine guanine phosphoribosyltransferase (Szybalska & Szybalski, 1992,
Proc. Natl.
Acad. Sci. USA 48:202), and adenine phosphoribosyltransferase (Lowy et al.,
1980, Cell
22:8-17) genes can be employed in tk-, gs-, hgprt- or aprt- cells,
respectively. Also,
antimetabolite resistance can be used as the basis of selection for the
following genes: dhfr,
which confers resistance to methotrexate (Wigler et al., 1980, PNAS 77:357;
O'Hare et al.,
1981, PNAS 78:1527); gpt, which confers resistance to mycophenolic acid
(Mulligan & Berg,
1981, PNAS 78:2072); neo, which confers resistance to the aminoglycoside G-418
(Wu and
Wu, 1991, Biotherapy 3:87; Tolstoshev, 1993, Ann. Rev. Pharmacol. Toxicol.
32:573;
Mulligan, 1993, Science 260:926; and Morgan and Anderson, 1993, Ann. Rev.
Biochem. 62:
191; May, 1993, TIB TECH 11:155-); and hygro, which confers resistance to
hygromycin
(Santerre et al., 1984, Gene 30:147). Methods commonly known in the art of
recombinant
DNA technology may be routinely applied to select the desired recombinant
clone, and such
methods are described, for example, in Ausubel et al. (eds.), Current
Protocols in Molecular
Biology, John Wiley & Sons, NY (1993); Kriegler, Gene Transfer and Expression,
A
Laboratory Manual, Stockton Press, NY (1990); and in Chapters 12 and 13,
Dracopoli et al.
(eds), Current Protocols in Human Genetics, John Wiley & Sons, NY (1994);
Colberre-
Garapin et al., 1981, J. Mol. Biol. 150:1, which are incorporated by reference
herein in their
entireties.
[00139] The expression levels of an antibody molecule can be increased by
vector
amplification (for a review, see Bebbington and Hentschel, The use of vectors
based on gene
amplification for the expression of cloned genes in mammalian cells in DNA
cloning, Vol.3.
(Academic Press, New York, 1987)). When a marker in the vector system
expressing
antibody is amplifiable, increase in the level of inhibitor present in culture
of host cell will
increase the number of copies of the marker gene. Since the amplified region
is associated
-49-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
with the antibody gene, production of the antibody will also increase (Grouse
et al., 1983,
Mol. Cell. Biol. 3:257).
[00140] The host cell may be co-transfected with two expression vectors of the
invention, the first vector encoding a heavy chain derived polypeptide and the
second vector
encoding a light chain derived polypeptide. The two vectors may contain
identical selectable
markers which enable equal expression of heavy and light chain polypeptides.
Alternatively,
a single vector may be used which encodes, and is capable of expressing, both
heavy and
light chain polypeptides. In such situations, the light chain should be placed
before the heavy
chain to avoid an excess of toxic free heavy chain (Proudfoot, 1986, Nature
322:52; and
Kohler, 1980, PNAS 77:2197). The coding sequences for the heavy and light
chains may
comprise cDNA or genomic DNA.
[00141] Once an antibody molecule of the invention has been produced by
recombinant expression, it may be purified by any method known in the art for
purification of
an immunoglobulin molecule, for example, by chromatography (e.g., ion
exchange, affinity,
particularly by affinity for the specific antigen after Protein A, and sizing
column
chromatography), centrifugation, differential solubility, or by any other
standard technique
for the purification of proteins. Further, the antibodies of the present
invention or fragments
thereof may be fused to heterologous polypeptide sequences described herein or
otherwise
known in the art to facilitate purification.
5.2 Prophylactic/Therapeutic Methods
[00142] The present invention encompasses methods for treating, preventing, or
managing a disorder associated with overexpression of EphA2, preferably
cancer, in a subject
comprising administering one or more EphA2 agonistic antibodies and/or exposed
EphA2
epitope antibodies, preferably one or more monoclonal (or antibodies from some
other source
of a single antibody species) EphA2 agonistic antibodies and/or exposed EphA2
epitope
antibodies. In a specific embodiment, the disorder to be treated, prevented,
or managed is
malignant cancer. In another specific embodiment, the disorder to be treated,
prevented, or
managed is a pre-cancerous condition associated with cells that overexpress
EphA2. In more
specific embodiments, the pre-cancerous condition is high-grade prostatic
intraepithelial
neoplasia (PIN), fibroadenoma of the breast, fibrocystic disease, or compound
nevi.
[00143] In one embodiment, the antibodies of the invention can be administered
in
combination with one or more other therapeutic agents useful in the treatment,
prevention or
management of cancer. In certain embodiments, one or more EphA2 antibodies of
the
-50-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
invention are administered to a mammal, preferably a human, concurrently with
one or more
other therapeutic agents useful for the treatment of cancer. The term
"concurrently" is not
limited to the administration of prophylactic or therapeutic agents at exactly
the same time,
but rather it is meant that the EphA2 antibodies of the invention and the
other agent are
administered to a subject in a sequence and within a time interval such that
the antibodies of
the invention can act together with the other agent to provide an increased
benefit than if they
were administered otherwise. For example, each prophylactic or therapeutic
agent may be
administered at the same time or sequentially in any order at different points
in time;
however, if not administered at the same time, they should be administered
sufficiently close
in time so as to provide the desired therapeutic or prophylactic effect. Each
therapeutic agent
can be administered separately, in any appropriate form and by any suitable
route. In other
embodiments, the EphA2 antibodies of the invention are administered before,
concurrently or
after surgery. Preferably the surgery completely removes localized tumors or
reduces the size
of large tumors. Surgery can also be done as a preventive measure or to
relieve pain.
[00144] In preferred embodiments, the one or more EphA2 antibodies of the
invention
consist of EA2, EA3, EA4, or EAS. In a more preferred embodiment, the
antibodies consist
of EA2, EA3, EA4, or EAS that have been humanized. In other embodiments,
variants of
EA2, EA3, EA4, or EAS, e.g., with one or more amino acid substitutions,
particularly in the
variable domain, are provided that have increased activity, binding ability,
etc., as compared
to EA2, EA3, EA4, or EAS.
[00145] In various embodiments, the prophylactic or therapeutic agents are
administered less than 1 hour apart, at about 1 hour apart, at about 1 hour to
about 2 hours
apart, at about 2 hours to about 3 hours apart, at about 3 hours to about 4
hours apart, at about
4 hours to about 5 hours apart, at about 5 hours to about 6 hours apart, at
about 6 hours to
about 7 hours apart, at about 7 hours to about 8 hours apart, at about 8 hours
to about 9 hours
apart, at about 9 hours to about 10 hours apart, at about 10 hours to about 11
hours apart, at
about 11 hours to about 12 hours apart, no more than 24 hours apart or no more
than 48 hours
apart. In preferred embodiments, two or more components are administered
within the same
patient visit.
[00146] The dosage amounts and frequencies of administration provided herein
are
encompassed by the terms therapeutically effective and prophylactically
effective. The
dosage and frequency further will typically vary according to factors specific
for each patient
depending on the specific therapeutic or prophylactic agents administered, the
severity and
type of cancer, the route of administration, as well as age, body weight,
response, and the past
-51-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
medical history of the patient. Suitable regimens can be selected by one
skilled in the art by
considering such factors and by following, for example, dosages reported in
the literature and
recommended in the Physicians' Desk Reference (58'h ed., 2004).
5.2.1 Patient Population
[00147] The invention provides methods for treating, preventing, and managing
cancer
by administrating to a subject a therapeutically or prophylactically effective
amount of one or
more EphA2 antibodies of the invention. In another embodiment, the EphA2
antibodies of
the invention can be administered in combination with one or more other
therapeutic agents.
The subject is preferably a mammal such as non-primate (e.g., cows, pigs,
horses, cats, dogs,
rats, etc.) and a primate (e.g., monkey, such as a cynomolgous monkey and a
human). In a
preferred embodiment, the subject is a human.
[00148] Specific examples of cancers that can be treated by the methods
encompassed
by the invention include, but are not limited to, cancers that over express
EphA2. In a further
embodiment, the cancer is of an epithelial origin. Examples of such cancers
are cancer of the
lung, colon, prostate, breast, and skin. Additional cancers are listed by
example and not by
limitation in the following section 5.2.1.1. In particular embodiments,
methods of the
invention can be used to treat and/or prevent metastasis from primary tumors.
[00149] The methods and compositions of the invention comprise the
administration of
one or more EphA2 antibodies of the invention to subjects/patients suffering
from or
expected to suffer from cancer, e.g., have a genetic predisposition for a
particular type of
cancer, have been exposed to a carcinogen, or are in remission from a
particular cancer. As
used herein, "cancer" refers to primary or metastatic cancers. Such patients
may or may not
have been previously treated for cancer. The methods and compositions of the
invention may
be used as a first line or second line cancer treatment. Included in the
invention is also the
treatment of patients undergoing other cancer therapies and the methods and
compositions of
the invention can be used before any adverse effects or intolerance of these
other cancer
therapies occurs. The invention also encompasses methods for administering one
or more
EphA2 antibodies of the invention to treat or ameliorate symptoms in
refractory patients. In a
certain embodiment, that a cancer is refractory to a therapy means that at
least some
significant portion of the cancer cells are not killed or their cell division
arrested. The
determination of whether the cancer cells are refractory can be made either in
vivo or in vitro
by any method known in the art for assaying the effectiveness of treatment on
cancer cells,
using the art-accepted meanings of "refractory" in such a context. In various
embodiments, a
-52-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
cancer is refractory where the number of cancer cells has not been
significantly reduced, or
has increased. The invention also encompasses methods for administering one or
more
EphA2 agonistic antibodies to prevent the onset or recurrence of cancer in
patients
predisposed to having cancer. Preferably, the monoclonal antibody is EA2, EA3,
EA4, or
EAS.
[00150] In particular embodiments, the EphA2 antibodies of the invention, or
other
therapeutics that reduce EphA2 expression, are administered to reverse
resistance or reduced
sensitivity of cancer cells to certain hormonal, radiation and
chemotherapeutic agents thereby
resensitizing the cancer cells to one or more of these agents, which can then
be administered
(or continue to be administered) to treat or manage cancer, including to
prevent metastasis.
[00151] In alternate embodiments, the invention provides methods for treating
patients' cancer by administering one or more EphA2 antibodies of the
invention in
combination with any other treatment or to patients who have proven refractory
to other
treatments but are no longer on these treatments. Preferably, the EphA2
antibody is EA2,
EA3, EA4, or EAS. In certain embodiments, the patients being treated by the
methods of the
invention are patients already being treated with chemotherapy, radiation
therapy, hormonal
therapy, or biological therapy/immunotherapy. Among these patients are
refractory patients
and those with cancer despite treatment with existing cancer therapies. In
other
embodiments, the patients have been treated and have no disease activity and
one or more
agonistic antibodies of the invention are administered to prevent the
recurrence of cancer.
[00152] In preferred embodiments, the existing treatment is chemotherapy. In
particular embodiments, the existing treatment includes administration of
chemotherapies
including, but not limited to, methotrexate, taxol, mercaptopurine,
thioguanine, hydroxyurea,
cytarabine, cyclophosphamide, ifosfamide, nitrosoureas, cisplatin,
carboplatin, mitomycin,
dacarbazine, procarbizine, etoposides, campathecins, bleomycin, doxorubicin,
idarubicin,
daunorubicin, dactinomycin, plicamycin, mitoxantrone, asparaginase,
vinblastine, vincristine,
vinorelbine, paclitaxel, docetaxel, etc. Among these patients are patients
treated with
radiation therapy, hormonal therapy and/or biological therapy/immunotherapy.
Also among
these patients are those who have undergone surgery for the treatment of
cancer.
[00153] Alternatively, the invention also encompasses methods for treating
patients
undergoing or having undergone radiation therapy. Among these are patients
being treated or
previously treated with chemotherapy, hormonal therapy and/or biological
therapy/immunotherapy. Also among these patients are those who have undergone
surgery
for the treatment of cancer.
-53-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00154] In other embodiments, the invention encompasses methods for treating
patients undergoing or having undergone hormonal therapy and/or biological
therapy/immunotherapy. Among these are patients being treated or having been
treated with
chemotherapy and/or radiation therapy. Also among these patients are those who
have
undergone surgery for the treatment of cancer.
[00155] Additionally, the invention also provides methods of treatment of
cancer as an
alternative to chemotherapy, radiation therapy, hormonal therapy, and/or
biological
therapy/immunotherapy where the therapy has proven or may prove too toxic,
i.e., results in
unacceptable or unbearable side effects, for the subject being treated. The
subject being
treated with the methods of the invention may, optionally, be treated with
other cancer
treatments such as surgery, chemotherapy, radiation therapy, hormonal therapy
or biological
therapy, depending on which treatment was found to be unacceptable or
unbearable.
[00156] In other embodiments, the invention provides administration of one or
more
agonistic monoclonal antibodies of the invention without any other cancer
therapies for the
treatment of cancer, but who have proved refractory to such treatments. In
specific
embodiments, patients refractory to other cancer therapies are administered
one or more
agonistic monoclonal antibodies in the absence of cancer therapies.
[00157] In other embodiments, patients with a pre-cancerous condition
associated with
cells that overexpress EphA2 can be administered antibodies of the invention
to treat the
disorder and decrease the likelihood that it will progress to malignant
cancer. In specific
embodiments, the pre-cancerous condition is high-grade prostatic
intraepithelial neoplasia
(PIN), fibroadenoma of the breast, fibrocystic disease, or compound nevi.
5.2.1.1 Cancers
[00158] Cancers and related disorders that can be treated or prevented by
methods and
compositions of the present invention include but are not limited to cancers
of an epithelial
cell origin. Examples of such cancers include the following: leukemias, such
as but not
limited to, acute leukemia, acute lymphocytic leukemia, acute myelocytic
leukemias, such as,
myeloblastic, promyelocytic, myelomonocytic, monocytic, and erythroleukemia
leukemias
and myelodysplastic syndrome; chronic leukemias, such as but not limited to,
chronic
myelocytic (granulocytic) leukemia, chronic lymphocytic leukemia, hairy cell
leukemia;
polycythemia vera; lymphomas such as but not limited to Hodgkin's disease, non-
Hodgkin's
disease; multiple myelomas such as but not limited to smoldering multiple
myeloma,
nonsecretory myeloma, osteosclerotic myeloma, plasma cell leukemia, solitary
plasmacytoma
-54-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
and extramedullary plasmacytoma; Waldenstrom's macroglobulinemia; monoclonal
gammopathy of undetermined significance; benign monoclonal gammopathy; heavy
chain
disease; bone and connective tissue sarcomas such as but not limited to bone
sarcoma,
osteosarcoma, chondrosarcoma, Ewing's sarcoma, malignant giant cell tumor,
fibrosarcoma
of bone, chordoma, periosteal sarcoma, soft-tissue sarcomas, angiosarcoma
(hemangiosarcoma), fibrosarcoma, Kaposi's sarcoma, leiomyosarcoma,
liposarcoma,
lymphangiosarcoma, neurilemmoma, rhabdomyosarcoma, synovial sarcoma; brain
tumors
such as but not limited to, glioma, astrocytoma, brain stem glioma,
ependymoma,
oligodendroglioma, nonglial tumor, acoustic neurinoma, craniopharyngioma,
medulloblastoma, meningioma, pineocytoma, pineoblastoma, primary brain
lymphoma;
breast cancer including but not limited to adenocarcinoma, lobular (small
cell) carcinoma,
intraductal carcinoma, medullary breast cancer, mucinous breast cancer,
tubular breast
cancer, papillary breast cancer, Paget's disease, and inflammatory breast
cancer; adrenal
cancer such as but not limited to pheochromocytom and adrenocortical
carcinoma; thyroid
cancer such as but not limited to papillary or follicular thyroid cancer,
medullary thyroid
cancer and anaplastic thyroid cancer; pancreatic cancer such as but not
limited to, insulinoma,
gastrinoma, glucagonoma, vipoma, somatostatin-secreting tumor, and carcinoid
or islet cell
tumor; pituitary cancers such as but limited to Cushing's disease, prolactin-
secreting tumor,
acromegaly, and diabetes insipius; eye cancers such as but not limited to
ocular melanoma
such as iris melanoma, choroidal melanoma, and cilliary body melanoma, and
retinoblastoma; vaginal cancers such as squamous cell carcinoma,
adenocarcinoma, and
melanoma; vulvar cancer such as squamous cell carcinoma, melanoma,
adenocarcinoma,
basal cell carcinoma, sarcoma, and Paget's disease; cervical cancers such as
but not limited
to, squamous cell carcinoma, and adenocarcinoma; uterine cancers such as but
not limited to
endometrial carcinoma and uterine sarcoma; ovarian cancers such as but not
limited to,
ovarian epithelial carcinoma, borderline tumor, germ cell tumor, and stromal
tumor;
esophageal cancers such as but not limited to, squamous cancer,
adenocarcinoma, adenoid
cystic carcinoma, mucoepidermoid carcinoma, adenosquamous carcinoma, sarcoma,
melanoma, plasmacytoma, verrucous carcinoma, and oat cell (small cell)
carcinoma; stomach
cancers such as but not limited to, adenocarcinoma, fungating (polypoid),
ulcerating,
superficial spreading, diffusely spreading, malignant lymphoma, liposarcoma,
fibrosarcoma,
and carcinosarcoma; colon cancers; rectal cancers; liver cancers such as but
not limited to
hepatocellular carcinoma and hepatoblastoma; gallbladder cancers such as
adenocarcinoma;
cholangiocarcinomas such as but not limited to pappillary, nodular, and
diffuse; lung cancers
-55-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
such as non-small cell lung cancer, squamous cell carcinoma (epidermoid
carcinoma),
adenocarcinoma, large-cell carcinoma and small-cell lung cancer; testicular
cancers such as
but not limited to germinal tumor, seminoma, anaplastic, classic (typical),
spermatocytic,
nonseminoma, embryonal carcinoma, teratoma carcinoma, choriocarcinoma (yolk-
sac
tumor), prostate cancers such as but not limited to, adenocarcinoma,
leiomyosarcoma, and
rhabdomyosarcoma; penal cancers; oral cancers such as but not limited to
squamous cell
carcinoma; basal cancers; salivary gland cancers such as but not limited to
adenocarcinoma,
mucoepidermoid carcinoma, and adenoidcystic carcinoma; pharynx cancers such as
but not
limited to squamous cell cancer, and verrucous; skin cancers such as but not
limited to, basal
cell carcinoma, squamous cell carcinoma and melanoma, superficial spreading
melanoma,
nodular melanoma, lentigo malignant melanoma, acral lentiginous melanoma;
kidney cancers
such as but not limited to renal cell carcinoma, adenocarcinoma,
hypernephroma,
fibrosarcoma, transitional cell cancer (renal pelvis and/ or uterer); Wilms'
tumor; bladder
cancers such as but not limited to transitional cell carcinoma, squamous cell
cancer,
adenocarcinoma, carcinosarcoma. In addition, cancers include myxosarcoma,
osteogenic
sarcoma, endotheliosarcoma, lymphangioendotheliosarcoma, mesothelioma,
synovioma,
hemangioblastoma, epithelial carcinoma, cystadenocarcinoma, bronchogenic
carcinoma,
sweat gland carcinoma, sebaceous gland carcinoma, papillary carcinoma and
papillary
adenocarcinomas (for a review of such disorders, see Fishman et al., 1985,
Medicine, 2d Ed.,
J.B. Lippincott Co., Philadelphia and Murphy et al., 1997, Informed Decisions:
The Complete
Book of Cancer Diagnosis, Treatment, and Recovery, Viking Penguin, Penguin
Books
U.S.A., Inc., United States of America).
[00159] Accordingly, the methods and compositions of the invention are
alsowseful in
the treatment or prevention of a variety of cancers or other abnormal
proliferative diseases,
including (but not limited to) the following: carcinoma, including that of the
bladder, breast,
colon, kidney, liver, lung, ovary, pancreas, stomach, cervix, thyroid and
skin; including
squamous cell carcinoma; hematopoietic tumors of lymphoid lineage, including
leukemia,
acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T-
cell
lymphoma, Burkitt's lymphoma; hematopoietic tumors of myeloid lineage,
including acute
and chronic myelogenous leukemias and promyelocytic leukemia; tumors of
mesenchymal
origin, including fibrosarcoma and rhabdomyoscarcoma; other tumors, including
melanoma,
seminoma, tetratocarcinoma, neuroblastoma and glioma; tumors of the central
and peripheral
nervous system, including astrocytoma, neuroblastoma, glioma, and schwannomas;
tumors of
mesenchymal origin, including fibrosarcoma, rhabdomyoscarama, and
osteosarcoma; and
-56-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
other tumors; including melanoma, xeroderma pigmentosum, keratoactanthoma,
seminoma,
thyroid follicular cancer and teratocarcinoma. It is also contemplated that
cancers caused by
aberrations in apoptosis would also be treated by the methods and compositions
of the
invention. Such cancers may include but not be limited to follicular
lymphomas, carcinomas
with p53 mutations, hormone dependent tumors of the breast, prostate and
ovary, and
precancerous lesions such as familial adenomatous polyposis, and
myelodysplastic
syndromes. In specific embodiments, malignancy or dysproliferative changes
(such as
metaplasias and dysplasias), or hyperproliferative disorders, are treated or
prevented in the
skin, lung, colon, breast, prostate, bladder, kidney, pancreas, ovary, or
uterus. In other
specific embodiments, sarcoma, melanoma, or leukemia is treated or prevented.
[00160] In some embodiments, the cancer is malignant and overexpresses EphA2.
In
other embodiments, the disorder to be treated is a pre-cancerous condition
associated with
cells that overexpress EphA2. In a specific embodiments, the pre-cancerous
condition is
high-grade prostatic intraepithelial neoplasia (PIN), fibroadenoma of the
breast, fibrocystic
disease, or compound nevi.
[00161] In preferred embodiments, the methods and compositions of the
invention are
used for the treatment and/or prevention of breast, colon, ovarian, lung, and
prostate cancers
and melanoma and are provided below by example rather than by limitation.
5.2.1.2 Treatment of Breast Cancer
[00162] In specific embodiments, patients with breast cancer are administered
an
effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for breast cancer therapy
including but
not limited to: doxorubicin, epirubicin, the combination of doxorubicin and
cyclophosphamide (AC), the combination of cyclophosphamide, doxorubicin and 5-
fluorouracil (CAF), the combination of cyclophosphamide, epirubicin and 5-
fluorouracil
(CEF), herceptin, tamoxifen, the combination of tamoxifen and cytotoxic
chemotherapy,
taxanes (such as docetaxel and paclitaxel). In a further embodiment,
antibodies of the
invention can be administered with taxanes plus standard doxorubicin and
cyclophosphamide
for adjuvant treatment of node-positive, localized breast cancer.
[00163] In a specific embodiment, patients with pre-cancerous fibroadenoma of
the
breast or fibrocystic disease are administered an EphA2 antibody of the
invention to treat the
disorder and decrease the likelihood that it will progress to malignant breast
cancer.
-57-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
5.2.1.3 Treatment of Colon Cancer
[00164] In specific embodiments, patients with colon cancer are administered
an
effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for colon cancer therapy
including but
not limited to: the combination of 5-FU and leucovorin, the combination of 5-
FU and
levamisole, irinotecan (CPT-11) or the combination of irinotecan, 5-FU and
leucovorin (IFL).
5.2.1.4 Treatment of Prostate Cancer
[00165] In specific embodiments, patients with prostate cancer are
administered an
effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for prostate cancer
therapy including but
not limited to: external-beam radiation therapy, interstitial implantation of
radioisotopes (i. e.,
I~25, palladium, iridium), leuprolide or other LHRH agonists, non-steroidal
antiandrogens
(flutamide, nilutamide, bicalutamide), steroidal antiandrogens (cyproterone
acetate), the
combination of leuprolide and flutamide, estrogens such as DES,
chlorotrianisene, ethinyl
estradiol, conjugated eslrogens U.S.P., DES-diphosphate, radioisotopes, such
as strontium-
89, the combination of external-beam radiation therapy and strontium-89,
second-line
hormonal therapies such as aminoglutethimide, hydrocortisone, flutamide
withdrawal,
progesterone, and ketoconazole, low-dose prednisone, or other chemotherapy
regimens
reported to produce subjective improvement in symptoms and reduction in PSA
level
including docetaxel, paclitaxel, estramustine/docetaxel,
estramustine/etoposide,
estramustine/vinblastine, and estramustine/paclitaxel.
[00166] In a specific embodiment, patients with pre-cancerous high-grade
prostatic
intraepithelial neoplasia (PIN) are administered an EphA2 antibody of the
invention to treat
the disorder and decrease the likelihood that it will progress to malignant
prostate cancer.
5.2.1.5 Treatment of Melanoma
[00167] In specific embodiments, patients with melanoma are administered an
effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for melanoma cancer
therapy including
but not limited to: dacarbazine (DTIC), nitrosoureas such as carmustine (BCNU)
and
lomustine (CCNU), agents with modest single agent activity including vinca
alkaloids,
-58-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
platinum compounds, and taxanes, the Dartmouth regimen (cisplatin, BCNU, and
DTIC),
interferon alpha (IFN-A), and interleukin-2 (IL-2). In a specific embodiment,
an effective
amount of one or more agonistic monoclonal antibodies of the invention can be
administered
in combination with isolated hyperthermic limb perfusion (ILP) with melphalan
(L-PAM),
with or without tumor necrosis factor-alpha (TNF-alpha) to patients with
multiple brain
metastases, bone metastases, and spinal cord compression to achieve symptom
relief and
some shrinkage of the tumor with radiation therapy.
[00168] In a specific embodiment, patients with pre-cancerous compound nevi
are
administered an EphA2 antibody of the invention to treat the disorder and
decrease the
likelihood that it will progress to malignant melanoma.
5.2.1.6 Treatment of Ovarian Cancer
[00169] In specific embodiments, patients with ovarian cancer are administered
an
effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for ovarian cancer therapy
including but
not limited to: intraperitoneal radiation therapy, such as P3z therapy, total
abdominal and
pelvic radiation therapy, cisplatin, the combination of paclitaxel (Taxol) or
docetaxel
(Taxotere) and cisplatin or carboplatin, the combination of cyclophosphamide
and cisplatin,
the combination of cyclophosphamide and carboplatin, the combination of 5-FU
and
leucovorin, etoposide, liposomal doxorubicin, gemcitabine or topotecan. It is
contemplated
that an effective amount of one or more agonistic monoclonal antibodies of the
invention is
administered in combination with the administration Taxol for patients with
platinum-
refractory disease. Included is the treatment of patients with refractory
ovarian cancer
including administration of: ifosfamide in patients with disease that is
platinum-refractory,
hexamethylmelamine (HMM) as salvage chemotherapy after failure of cisplatin-
based
combination regimens, and tamoxifen in patients with detectable levels of
cytoplasmic
estrogen receptor on their tumors.
5.2.1.7 Treatment of Lune Cancers
[00170] In specific embodiments, patients with small lung cell cancer are
administered
an effective amount of one or more monoclonal antibodies of the invention. In
another
embodiment, the antibodies of the invention can be administered in combination
with an
effective amount of one or more other agents useful for lung cancer therapy
including but not
limited to: thoracic radiation therapy, cisplatin, vincristine, doxorubicin,
and etoposide, alone
-59-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
or in combination, the combination of cyclophosphamide, doxorubicin,
vincristine/etoposide,
and cisplatin (CAV/EP), local palliation with endobronchial laser therapy,
endobronchial
stems, and/or brachytherapy.
[00171] In other specific embodiments, patients with non-small lung cell
cancer are
administered an effective amount of one or more monoclonal antibodies of the
invention in
combination with an effective amount of one or more other agents useful for
lung cancer
therapy including but not limited to: palliative radiation therapy, the
combination of cisplatin,
vinblastine and mitomycin, the combination of cisplatin and vinorelbine,
paclitaxel, docetaxel
or gemcitabine, the combination of carboplatin and paclitaxel, interstitial
radiation therapy
for endobronchial lesions or stereotactic radiosurgery.
5.2.2 Other Pronhylactic/Therapeutic Agents
[00172] In some embodiments, therapy by administration of one or more
monoclonal
antibodies is combined with the administration of one or more therapies such
as, but not
limited to, chemotherapies, radiation therapies, hormonal therapies, andlor
biological
therapies/immunotherapies. Prophylactic/therapeutic agents include, but are
not limited to,
proteinaceous molecules, including, but not limited to, peptides,
polypeptides, proteins,
including post-translationally modified proteins, antibodies etc.; or small
molecules (less than
1000 daltons), inorganic or organic compounds; or nucleic acid molecules
including, but not
limited to, double-stranded or single-stranded DNA, or double-stranded or
single-stranded
RNA, as well as triple helix nucleic acid molecules. Prophylactic/therapeutic
agents can be
derived from any known organism (including, but not limited to, animals,
plants, bacteria,
fungi, and protista, or viruses) or from a library of synthetic molecules.
[00173] In a specific embodiment, the methods of the invention encompass
administration of an antibody of the invention in combination with the
administration of one
or more prophylactic/therapeutic agents that are inhibitors of kinases such
as, but not limited
to, ABL, ACK, AFK, AKT (e.g., AKT-1, AKT-2, and AKT-3), ALK, AMP-PK, ATM,
Auroral, Aurora2, bARKl, bArk2, BLK, BMX, BTK, CAK, CaM kinase, CDC2, CDK, CK,
COT, CTD, DNA-PK, EGF-R, ErbB-1, ErbB-2, ErbB-3, ErbB-4, ERK (e.g., ERK1,
ERK2,
ERK3, ERK4, ERKS, ERK6, ERK7), ERT-PK, FAK, FGR (e.g., FGF1R, FGF2R), FLT
(e.g., FLT-1, FLT-2, FLT-3, FLT-4), FRK, FYN, GSK (e.g., GSK1, GSK2, GSK3-
alpha,
GSK3-beta, GSK4, GSKS), G-protein coupled receptor kinases (GRKs), HCK, HER2,
HKII,
JAK (e.g., JAK1, JAK2, JAK3, JAK4), JNK (e.g., JNK1, JNK2, JNK3), KDR, KIT,
IGF-1
receptor, IKK-1, IKK-2, INSR (insulin receptor), IRAK1, IRAK2, IRK, ITK, LCK,
LOK,
-60-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
LYN, MAPK, MAPKAPK-1, MAPKAPK-2, MEK, MET, MFPK, MHCK, MLCK, MLK3,
NEU, NIK, PDGF receptor alpha, PDGF receptor beta, PHK, PI-3 kinase, PKA, PKB,
PKC,
PKG, PRK1, PYK2, p38 kinases, p135tyk2, p34cdc2, p42cdc2, p42mapk, p44mpk,
RAF,
RET, RIP, RIP-2, RK, RON, RS kinase, SRC, SYK, S6K, TAKI, TEC, TIE1, TIE2,
TRKA,
TXK, TYK2, UL13, VEGFR1, VEGFR2, YES, YRK, ZAP-70, and all subtypes of these
kinases (see e.g., Hardie and Hanks (1995) The Protein Kinase Facts Book, I
and II,
Academic Press, San Diego, Calif.). In preferred embodiments, an antibody of
the invention
id administered in combination with the administration of one or more
prophylactic/therapeutic agents that are inhibitors of Eph receptor kinases
(e.g., EphA2,
EphA4). In a most preferred embodiment, an antibody of the invention is
administered in
combination with the administration of one or more prophylactic/therapeutic
agents that are
inhibitors of EphA2.
[00174] In another specific embodiment, the methods of the invention encompass
administration of an antibody of the invention in combination with the
administration of one
or more prophylactic/therapeutic agents that are angiogenesis inhibitors such
as, but not
limited to: Angiostatin (plasminogen fragment); antiangiogenic antithrombin
III; Angiozyme;
ABT-627; Bay 12-9566; Benefin; Bevacizumab; BMS-275291; cartilage-derived
inhibitor
(CDI); CAI; CD59 complement fragment; CEP-7055; Col 3; Combretastatin A-4;
Endostatin
(collagen XVIII fragment); fibronectin fragment; Gro-beta; Halofuginone;
Heparinases;
Heparin hexasaccharide fragment; HMV833; Human chorionic gonadotropin (hCG);
IM-862;
Interferon alpha/betalgamma; Interferon inducible protein (IP-10); Interleukin-
12; Kringle 5
(plasminogen fragment); Marimastat; Metalloproteinase inhibitors (TIMPs); 2-
Methoxyestradiol; MMI 270 (CGS 27023A); MoAb IMC-1C11; Neovastat; NM-3;
Panzem;
PI-88; Placental ribonuclease inhibitor; Plasminogen activator inhibitor;
Platelet factor-4
(PF4); Prinomastat; Prolactin l6kD fragment; Proliferin-related protein (PRP);
PTK 787/ZK
222594; Retinoids; Solimastat; Squalamine; SS 3304; SU 5416; SU6668; SU11248;
Tetrahydrocortisol-S; tetrathiomolybdate; thalidomide; Thrombospondin-1 (TSP-
1); TNP-
470; Transforming growth factor-beta (TGF-(3); Vasculostatin; Vasostatin
(calreticulin
fragment); ZD6126; ZD6474; farnesyl transferase inhibitors (FTI); and
bisphosphonates.
[00175] In another specific embodiment, the methods of the invention encompass
administration of an antibody of the invention in combination with the
administration of one
or more prophylactic/therapeutic agents that are anti-cancer agents such as,
but not limited to:
acivicin, aclarubicin, acodazole hydrochloride, acronine, adozelesin,
aldesleukin, altretamine,
ambomycin, ametantrone acetate, aminoglutethimide, amsacrine, anastrozole,
anthramycin,
-61-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
asparaginase, asperlin, azacitidine, azetepa, azotomycin, batimastat,
benzodepa, bicalutamide,
bisantrene hydrochloride, bisnafide dimesylate, bizelesin, bleomycin sulfate,
brequinar
sodium, bropirimine, busulfan, cactinomycin, calusterone, caracemide,
carbetimer,
carboplatin, carmustine, carubicin hydrochloride, carzelesin, cedefingol,
chlorambucil,
cirolemycin, cisplatin, cladribine, crisnatol mesylate, cyclophosphamide,
cytarabine,
dacarbazine, dactinomycin, daunorubicin hydrochloride, decarbazine,
decitabine,
dexormaplatin, dezaguanine, dezaguanine mesylate, diaziquone, docetaxel,
doxorubicin,
doxorubicin hydrochloride, droloxifene, droloxifene citrate, dromostanolone
propionate,
duazomycin, edatrexate, eflornithine hydrochloride, elsamitrucin, enloplatin,
enpromate,
epipropidine, epirubicin hydrochloride, erbulozole, esorubicin hydrochloride,
estramustine,
estramustine phosphate sodium, etanidazole, etoposide, etoposide phosphate,
etoprine,
fadrozole hydrochloride, fazarabine, fenretinide, floxuridine, fludarabine
phosphate,
fluorouracil, flurocitabine, fosquidone, fostriecin sodium, gemcitabine,
gemcitabine
hydrochloride, hydroxyurea, idarubicin hydrochloride, ifosfamide, ilmofosine,
interleukin 2
(including recombinant interleukin 2, or rIL2), interferon alpha-2a,
interferon alpha-2b,
interferon alpha-nl, interferon alpha-n3, interferon beta-I a, interferon
gamma-I b, iproplatin,
irinotecan hydrochloride, lameotide acetate, letrozole, leuprolide acetate,
liarozole
hydrochloride, lometrexol sodium, lomustine, losoxantrone hydrochloride,
masoprocol,
maytansine, mechlorethamine hydrochloride, megestrol acetate, melengestrol
acetate,
melphalan, menogaril, mercaptopurine, methotrexate, methotrexate sodium,
metoprine,
meturedepa, mitindomide, mitocarcin, mitocromin, mitogillin, mitomalcin,
mitomycin,
mitosper, mitotane, mitoxantrone hydrochloride, mycophenolic acid,
nitrosoureas,
nocodazole, nogalamycin, ormaplatin, oxisuran, paclitaxel, pegaspargase,
peliomycin,
pentamustine, peplomycin sulfate, perfosfamide, pipobroman, piposulfan,
piroxantrone
hydrochloride, plicamycin, plomestane, porfimer sodium, porfiromycin,
prednimustine,
procarbazine hydrochloride, puromycin, puromycin hydrochloride, pyrazofurin,
riboprine,
rogletimide, safingol, safingol hydrochloride, semustine, simtrazene,
sparfosate sodium,
sparsomycin, spirogermanium hydrochloride, spiromustine, spiroplatin,
streptonigrin,
streptozocin, sulofenur, talisomycin, tecogalan sodium, tegafur, teloxantrone
hydrochloride,
temoporfin, teniposide, teroxirone, testolactone, thiamiprine, thioguanine,
thiotepa,
tiazofurin, tirapazamine, toremifene citrate, trestolone acetate, triciribine
phosphate,
trimetrexate, trimetrexate glucuronate, triptorelin, tubulozole hydrochloride,
uracil mustard,
uredepa, vapreotide, verteporfin, vinblastine sulfate, vincristine sulfate,
vindesine, vindesine
sulfate, vinepidine sulfate, vinglycinate sulfate, vinleurosine sulfate,
vinorelbine tartrate,
-62-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
vinrosidine sulfate, vinzolidine sulfate, vorozole, zeniplatin, zinostatin,
zorubicin
hydrochloride. Other anti-cancer drugs include, but are not limited to: 20-epi-
1,25
dihydroxyvitamin D3, 5-ethynyluracil, abiraterone, aclarubicin, acylfulvene,
adecypenol,
adozelesin, aldesleukin, ALL-TK antagonists, altretamine, ambamustine, amidox,
amifostine,
aminolevulinic acid, amrubicin, amsacrine, anagrelide, anastrozole,
andrographolide,
angiogenesis inhibitors, antagonist D, antagonist G, antarelix, anti-
dorsalizing morphogenetic
protein-1, antiandrogens, antiestrogens, antineoplaston, aphidicolin
glycinate, apoptosis gene
modulators, apoptosis regulators, apurinic acid, ara-CDP-DL-PTBA, arginine
deaminase,
asulacrine, atamestane, atrimustine, axinastatin 1, axinastatin 2, axinastatin
3, azasetron,
azatoxin, azatyrosine, baccatin III derivatives, balanol, batimastat, BCR/ABL
antagonists,
benzochlorins, benzoylstaurosporine, beta lactam derivatives, beta-alethine,
betaclamycin B,
betulinic acid, bFGF inhibitor, bicalutamide, bisantrene,
bisaziridinylspermine, bisnafide,
bistratene A, bizelesin, breflate, bropirimine, budotitane, buthionine
sulfoximine, calcipotriol,
calphostin C, camptothecin derivatives, canarypox IL-2, capecitabine,
carboxamide-amino-
triazole, carboxyamidotriazole, CaRest M3, CARN 700, cartilage derived
inhibitor,
carzelesin, casein kinase inhibitors (ICOS), castanospermine, cecropin B,
cetrorelix,
ehloroquinoxaline sulfonamide, cicaprost, cis-porphyrin, cladribine, clomifene
analogues,
clotrimazole, collismycin A, collismycin B, combretastatin A4, combretastatin
analogue,
conagenin, crambescidin 816, crisnatol, cryptophycin 8, cryptophycin A
derivatives, curacin
A, cyclopentanthraquinones, cycloplatam, cypemycin, cytarabine ocfosfate,
cytolytic factor,
cytostatin, dacliximab, decitabine, dehydrodidemnin B, deslorelin,
dexamethasone,
dexifosfamide, dexrazoxane, dexverapamil, diaziquone, didemnin B, didox,
diethylnorspermine, dihydro-5-azacytidine, dihydrotaxol, dioxamycin, diphenyl
spiromustine,
docetaxel, docosanol, dolasetron, doxifluridine, droloxifene, dronabinol,
duocarmycin SA,
ebselen, ecomustine, edelfosine, edrecolomab, eflornithine, elemene, emitefur,
epirubicin,
epristeride, estramustine analogue, estrogen agonists, estrogen antagonists,
etanidazole,
etoposide phosphate, exemestane, fadrozole, fazarabine, fenretinide,
filgrastim, finasteride,
flavopiridol, flezelastine, fluasterone, fludarabine, fluorodaunorunicin
hydrochloride,
forfenimex, formestane, fostriecin, fotemustine, gadolinium texaphyrin,
gallium nitrate,
galocitabine, ganirelix, gelatinase inhibitors, gemcitabine, glutathione
inhibitors, hepsulfam,
heregulin, hexamethylene bisacetamide, hypericin, ibandronic acid, idarubicin,
idoxifene,
idramantone, ilmofosine, ilomastat, imidazoacridones, imiquimod,
immunostimulant
peptides, insulin-like growth factor-1 receptor inhibitor, interferon
agonists, interferons,
interleukins, iobenguane, iododoxorubicin, ipomeanol, iroplact, irsogladine,
isobengazole,
-63-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
isohomohalicondrin B, itasetron, jasplakinolide, kahalalide F, lamellarin-N
triacetate,
lanreotide, leinamycin, lenograstim, lentinan sulfate, leptolstatin,
letrozole, leukemia
inhibiting factor, leukocyte alpha interferon,
leuprolide+estrogen+progesterone, leuprorelin,
levamisole, liarozole, linear polyamine analogue, lipophilic disaccharide
peptide, lipophilic
platinum compounds, lissoclinamide 7, lobaplatin, lombricine, lometrexol,
lonidamine,
losoxantrone, lovastatin, loxoribine, lurtotecan, lutetium texaphyrin,
lysofylline, lytic
peptides, maitansine, mannostatin A, marimastat, masoprocol, maspin,
matrilysin inhibitors,
matrix metalloproteinase inhibitors, menogaril, merbarone, meterelin,
methioninase,
metoclopramide, MIF inhibitor, mifepristone, miltefosine, mirimostim,
mismatched double
stranded RNA, mitoguazone, mitolactol, mitomycin analogues, mitonafide,
mitotoxin
fibroblast growth factor-saporin, mitoxantrone, mofarotene, molgramostim,
monoclonal
antibody, human chorionic gonadotrophin, monophosphoryl lipid A+myobacterium
cell wall
sk, mopidamol, multiple drug resistance gene inhibitor, multiple tumor
suppressor 1-based
therapy, mustard anticancer agent, mycaperoxide B, mycobacterial cell wall
extract,
myriaporone, N-acetyldinaline, N-substituted benzamides, nafarelin, nagrestip,
naloxone+pentazocine, napavin, naphterpin, nartograstim, nedaplatin,
nemorubicin,
neridronic acid, neutral endopeptidase, nilutamide, nisamycin, nitric oxide
modulators,
nitroxide antioxidant, nitnillyn, 06-benzylguanine, octreotide, okicenone,
oligonucleotides,
onapristone, ondansetron, ondansetron, oracin, oral cytokine inducer,
ormaplatin, osaterone,
oxaliplatin, oxaunomycin, paclitaxel, paclitaxel analogues, paclitaxel
derivatives,
palauamine, palmitoylrhizoxin, pamidronic acid, panaxytriol, panomifene,
parabactin,
pazelliptine, pegaspargase, peldesine, pentosan polysulfate sodium,
pentostatin, pentrozole,
perflubron, perfosfamide, perillyl alcohol, phenazinomycin, phenylacetate,
phosphatase
inhibitors, picibanil, pilocarpine hydrochloride, pirarubicin, piritrexim,
placetin A, placetin B,
plasminogen activator inhibitor, platinum complex, platinum compounds,
platinum-triamine
complex, porfimer sodium, porfiromycin, prednisone, propyl bis-acridone,
prostaglandin J2,
proteasome inhibitors, protein A-based immune modulator, protein kinase C
inhibitor, protein
kinase C inhibitors, microalgal, protein tyrosine phosphatase inhibitors,
purine nucleoside
phosphorylase inhibitors, purpurins, pyrazoloacridine, pyridoxylated
hemoglobin
polyoxyethylene conjugate, raf antagonists, raltitrexed, ramosetron, ras
farnesyl protein
transferase inhibitors, ras inhibitors, ras-GAP inhibitor, retelliptine
demethylated, rhenium Re
186 etidronate, rhizoxin, ribozymes, RII retinamide, rogletimide, rohitukine,
romurtide,
roquinimex, rubiginone B 1, ruboxyl, safingol, saintopin, SarCNU, sarcophytol
A,
sargramostim, Sdi 1 mimetics, semustine, senescence derived inhibitor 1, sense
-64-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
oligonucleotides, signal transduction inhibitors, signal transduction
modulators, single chain
antigen binding protein, sizofiran, sobuzoxane, sodium borocaptate, sodium
phenylacetate,
solverol, somatomedin binding protein, sonermin, sparfosic acid, spicamycin D,
spiromustine, splenopentin, spongistatin 1, squalamine, stem cell inhibitor,
stem-cell division
inhibitors, stipiamide, stromelysin inhibitors, sulfinosine, superactive
vasoactive intestinal
peptide antagonist, suradista, suramin, swainsonine, synthetic
glycosaminoglycans,
tallimustine, tamoxifen methiodide, tauromustine, taxol, tazarotene, tecogalan
sodium,
tegafur, tellurapyrylium, telomerase inhibitors, temoporfin, temozolomide,
teniposide,
tetrachlorodecaoxide, tetrazomine, thaliblastine, thalidomide, thiocoraline,
thioguanine,
thrombopoietin, thrombopoietin mimetic, thymalfasin, thymopoietin receptor
agonist,
thymotrinan, thyroid stimulating hormone, tin ethyl etiopurpurin,
tirapazamine, titanocene
bichloride, topsentin, toremifene, totipotent stem cell factor, translation
inhibitors, tretinoin,
triacetyluridine, triciribine, trimetrexate, triptorelin, tropisetron,
turosteride, tyrosine kinase
inhibitors, tyrphostins, UBC inhibitors, ubenimex, urogenital sinus-derived
growth inhibitory
factor, urokinase receptor antagonists, vapreotide, variolin B, vector system,
erythrocyte gene
therapy, velaresol, veramine, verdins, verteporfin, vinorelbine, vinxaltine,
vitaxin, vorozole,
zanoterone, zeniplatin, zilascorb, and zinostatin stimalamer. Preferred
additional anti-cancer
drugs are 5-fluorouracil and leucovorin.
[00176] In more particular embodiments, the present invention also comprises
the
administration of one or more monoclonal antibodies of the invention in
combination with
the administration of one or more therapies such as, but not limited to anti-
cancer agents such
as those disclosed in Table 2, preferably for the treatment of breast, ovary,
melanoma,
prostate, colon and lung cancers as described above.
TABLE 2
Therapeutic AdministrationDose Intervals
Agent
doxorubicin Intravenous 60-75 mg/m' on 21 day intervals
Day 1
hydrochloride
(Adriamycin
RDF~
and Adriamycin
PFS~)
epirubicin Intravenous 100-120 mg/m' on 3-4 week cycles
Day 1 of
hydrochloride each cycle or divided
equally
(EllenceTM) and given on Days
1-8 of the
cycle
fluorousacil Intravenous How supplied:
5 ml and 10 ml
vials
(containing 250
and S00 mg
flourouracil respectively)
docetaxel Intravenous 60- 100 mg/m' overOnce every 3 weeks
1 hour
-65-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Therapeutic AdministrationDose Intervals
Agent
(TaxotereO)
paclitaxel Intravenous 175 mg/m' over Every 3 weeks for
3 hours 4 courses
(Taxol~) (administered sequentially
to
doxorubicin-containing
combination chemotherapy)
tamoxifen citrateOral 20-40 mg Daily
(Nolvadex~) (tablet) Dosages greater
than 20 mg
should be given
in divided
doses (morning
and evening)
leucovorin Intravenous How supplied: Dosage is unclear
calcium or from text.
for injection intramuscular350 mg vial PDR 3610
injection
luprolide acetateSingle I mg (0.2 ml or Once a day
20 unit mark)
(Lupron~) subcutaneous
injection
flutamide Oral (capsule)250 mg 3 times a day at
8 hour
(Eulexin~) (capsules contain intervals (total
125 mg daily dosage
flutamide each) 750 mg)
nilutamide Oral 300 mg or 150 mg 300 mg once a day
for 30
(Nilandron~) (tablet) (tablets contain days followed by
50 or l50 mg 150 mg
nilutamide each) once a day
bicalutamide Oral 50 mg Once a day
(Casodex~) (tablet) (tablets contain
50 mg
bicalutamide each)
progesterone Injection USP in sesame oil
50 mg/ml
ketoconazole Cream 2% cream applied
once or
(Nizoral~) twice daily depending
on
symptoms
prednisone Oral Initial dosage
may vary from
(tablet) 5 mg to 60 mg per
day
depending on the
specific
disease entity
being treated.
estramustine Oral 14 mg/ kg of body Daily given in
weight 3 or 4 divided
phosphate sodium(capsule) (i.e. one 140 mg doses
capsule for
(Emcyt~) each 10 kg or 22
lb of body
weight)
etoposide or Intravenous 5 ml of 20 mg/
VP-16 ml solution
( 100 mg)
dacarbazine Intravenous 2-4.5 mg/kg Once a day for
10 days.
(DTIC-Dome)
May be repeated
at 4 week
intervals
polifeprosan wafer placed8 wafers, each
20 with in containing 7.7
carmustine resection mg of carmustine,
implant cavity for a total
(BCNU) (nitrosourea) of 61.6 mg, if
size and shape
(Gliadel~) of resection cavity
allows
cisplatin Injection [n/a in PDR 86l]
How supplied:
solution of 1 mg/ml
in multi-
dose vials of 50mL
and
-66-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Therapeutic AdministrationDose Intervals
Agent
100mL
mitomycin Injection supplied in 5 mg
and 20 mg
vials (containing
5 mg and 20
mg mitomycin)
gemcitabine Intravenous For NSCLC- 2 schedules4 week schedule-
HCl
(Gemzar~) have been investigatedDays 1,8 and 15
and of each 28-
the optimum scheduleday cycle. Cisplatin
has not
been determined intravenously at
100 mg/m2
4 week schedule- on day 1 after
the infusion of
administration Gemzar.
intravenously
at 1000 mg/m2 over3 week schedule-
30
minutes on 3 week Days 1 and 8 of
schedule- each 21 day
Gemzar administeredcycle. Cisplatin
at dosage of
intravenously at 100 mg/m2 administered
1250 mg/m2
over 30 minutes intravenously after
administration
of Gemzar on
day I.
carboplatin Intravenous Single agent therapy:Every 4 weeks
(Paraplatin~) 360 mg/m2 LV. on
day 1
(infusion lasting
IS minutes
or longer)
Other dosage calculations:
Combination therapy
with
cyclophosphamide,
Dose
adjustment recommendations,
Formula dosing,
etc.
ifosamide Intravenous 1.2 g/mZ daily 5 consecutive days
(Ifex~) Repeat every 3
weeks or
after recovery
from
hematologic toxicity
topotecan Intravenous 1.5 mg/m' by intravenous5 consecutive days,
starting
hydrochloride infusion over 30 on day 1 of 21
minutes day course
(Hycamtin~) daily
~VVl / / J l ne invention also encompasses administration of the EphA2
antibodies of the
invention in combination with radiation therapy comprising the use of x-rays,
gamma rays
and other sources of radiation to destroy the cancer cells. In preferred
embodiments, the
radiation treatment is administered as external beam radiation or teletherapy
wherein the
radiation is directed from a remote source. In other preferred embodiments,
the radiation
treatment is administered as internal therapy or brachytherapy wherein a
radioactive source is
placed inside the body close to cancer cells or a tumor mass.
[00178] Cancer therapies and their dosages, routes of administration and
recommended
usage are known in the art and have been described in such literature as the
Physicians' Desk
Reference (58~' ed., 2004).
5.3 Identification of Antibodies of the Invention
-67-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
5.3.1 Agonistic Antibodies
[00179] Antibodies of the invention may preferably agonize (i.e., elicit EphA2
phosphorylation) as well as immunospecifically bind to the EphA2 receptor.
When agonized,
EphA2 becomes phosphorylated and then subsequently degraded. Any method known
in the
art to assay either the level of EphA2 phosphorylation, activity, or
expression can be used to
assay candidate EphA2 antibodies to determine their agonistic activity (see,
e. g., Section
6.2.1 infra).
[00180] Thus, the invention provides methods of assaying and screening for
EphA2
antibodies of the invention by incubating antibodies that specifically bind
EphA2, particularly
that bind the extracellular domain of EphA2, with cells that express EphA2,
particularly
cancer cells, preferably metastatic cancer cells, that overexpress EphA2
(relative to non-
cancer cells of the same cell type) and then assaying for an increase in EphA2
phosphorylation and/or EphA2 degradation, thereby identifying an EphA2
antibody of the
invention.
5.3.2 Antibodies That Preferentially Bind EphA2 Epitopes Exposed on Cancer
Cells
[00181] Antibodies of the invention may preferably bind to EphA2 epitopes
exposed
on cancer cells (e.g., cells overexpressing EphA2 and/or cells with
substantial EphA2 that is
not bound to ligand) but not non-cancer cells or cell where EphA2 is bound to
ligand. In this
embodiment, antibodies of the invention are antibodies directed to an EphA2
epitope not
exposed on non-cancer cells but exposed on cancer cells (see, e.g., Section
6.6 infra).
Differences in EphA2 membrane distribution between non-cancer cells and cancer
cells
expose certain epitopes on cancer cells that are not exposed on non-cancer
cells. For
example, normally EphA2 is bound to its ligand, EphrinAl, and localizes at
areas of cell-cell
contacts. However, cancer cells generally display decreased cell-cell contacts
as well as
overexpress EphA2 in excess of its ligand. Thus, in cancer cells, there is an
increased
amount of unbound EphA2 that is not localized to cell-cell contacts. As such,
in one
embodiment, an antibody that preferentially binds unbound, unlocalized EphA2
is an
antibody of the invention.
[00182] Any method known in the art to determine candidate EphA2 antibody
binding/localization on a cell can be used to screen candidate antibodies for
desirable binding
properties. In a one embodiment, immunofluorescence microscopy is used to
determine the
binding characteristics of an antibody. Standard techniques can be used to
compare the
-68-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
binding of an antibody binding to cells grown in vitro. In a specific
embodiment, antibody
binding to cancer cells is compared to antibody binding to non-cancer cells.
An exposed
EphA2 epitope antibody binds poorly to non-cancer cells but binds well to
cancer cells. In
another specific embodiment, antibody binding to non-cancer dissociated cells
(e.g., treated
with a calcium chelator such as EGTA) is compared to antibody binding to non-
cancer cells
that have not been dissociated. An exposed EphA2 epitope antibody binds poorly
non-cancer
cells that have not been dissociated but binds well to dissociated non-cancer
cells.
[00183] In another embodiment, flow cytometry is used to determine the binding
characteristics of an antibody. In this embodiment, EphA2 may or may not be
crosslinked to
its ligand, Ephrin AI. An exposed EphA2 epitope antibody binds poorly
crosslinked EphA2
but binds well to uncrosslinked EphA2.
[00184] In another embodiment, cell-based or immunoassays are used to
determine the
binding characteristics of an antibody. In this embodiment, antibodies that
can compete with
an EphA2 ligand (e.g., Ephrin A1) for binding to EphA2 displace Ephrin A1 from
EphA2.
The EphA2 ligand used in this assay can be soluble protein (e.g.,
recombinantly expressed) or
expressed on a cell so that it is anchored to the cell.
5.4 Characterization And Demonstration Of Therapeutic Or Prophylactic
Utility
[00185] Toxicity and efficacy of the prophylactic and/or therapeutic protocols
of the
instant invention can be determined by standard pharmaceutical procedures in
cell cultures or
experimental animals, e.g., for determining the LDSO (the dose lethal to
50°l0 of the
population) and the EDSO (the dose therapeutically effective in 50% of the
population). The
dose ratio between toxic and therapeutic effects is the therapeutic index and
it can be
expressed as the ratio LDSO/EDSO. Prophylactic and/or therapeutic agents that
exhibit large
therapeutic indices are preferred. While prophylactic and/or therapeutic
agents that exhibit
toxic side effects may be used, care should be taken to design a delivery
system that targets
such agents to the site of affected tissue in order to minimize potential
damage to uninfected
cells and, thereby, reduce side effects.
[00186] The data obtained from the cell culture assays and animal studies can
be used
in formulating a range of dosage of the prophylactic and/or therapeutic agents
for use in
humans. The dosage of such agents lies preferably within a range of
circulating
concentrations that include the EDSO with little or no toxicity. The dosage
may vary within
this range depending upon the dosage form employed and the route of
administration utilized.'
-69-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
For any agent used in the method of the invention, the therapeutically
effective dose can be
estimated initially from cell culture assays. A dose may be formulated in
animal models to
achieve a circulating plasma concentration range that includes the ICSO (i.e.,
the concentration
of the test compound that achieves a half-maximal inhibition of symptoms) as
determined in
cell culture. Such information can be used to more accurately determine useful
doses in
humans. Levels in plasma may be measured, for example, by high performance
liquid
chromatography.
[00187] The anti-cancer activity of the therapies used in accordance with the
present
invention also can be determined by using various experimental animal models
for the study
of cancer such as the SCID mouse model or transgenic mice where a mouse EphA2
is
replaced with the human EphA2, nude mice with human xenografts, animal models
described
in Section 6 infra, or any animal model (including hamsters, rabbits, etc.)
known in the art
and described in Relevance of Tumor Models for Anticancer Drug Development
(1999, eds.
Fiebig and Burger); Contributions to Oncology (1999, Karger); The Nude Mouse
in Oncology
Research (1991, eds. Boven and Winograd); and Anticancer Drug Development
Guide (1997
ed. Teicher), herein incorporated by reference in their entireties.
5.4.1 Demonstration of Therapeutic Utility
[00188] The protocols and compositions of the invention are preferably tested
in vitro,
and then in vivo, for the desired therapeutic or prophylactic activity, prior
to use in humans.
For example, in vitro assays which can be used to determine whether
administration of a
specific therapeutic protocol is indicated, include in vitro cell culture
assays in which a
patient tissue sample is grown in culture, and exposed to or otherwise
administered a
protocol, and the effect of such protocol upon the tissue sample is observed,
e.g., increased
phosphorylation/degradation of EphA2. A lower level of proliferation or
survival of the
contacted cells indicates that the therapeutic agent is effective to treat the
condition in the
patient. Alternatively, instead of culturing cells from a patient, therapeutic
agents and
methods may be screened using cells of a tumor or malignant cell line. Many
assays standard
in the art can be used to assess such survival and/or growth; for example,
cell proliferation
can be assayed by measuring 3H-thymidine incorporation, by direct cell count,
by detecting
changes in transcriptional activity of known genes such as proto-oncogenes
(e.g., fos, myc) or
cell cycle markers; cell viability can be assessed by trypan blue staining,
differentiation can
be assessed visually based on changes in morphology, increased
phosphorylation/degradation
of EphA2, etc.
-70-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00189] Compounds for use in therapy can be tested in suitable animal model
systems
prior to testing in humans, including but not limited to in rats, mice,
chicken, cows, monkeys,
rabbits, hamsters, etc., for example, the animal models described above. The
compounds can
then be used in the appropriate clinical trials.
[00190] Further, any assays known to those skilled in the art can be used to
evaluate
the prophylactic and/or therapeutic utility of the combinatorial therapies
disclosed herein for
treatment or prevention of cancer.
5.5 Pharmaceutical Compositions
[00191] The compositions of the invention include bulk drug compositions
useful in
the manufacture of pharmaceutical compositions (e. g., impure or non-sterile
compositions)
and pharmaceutical compositions (i.e., compositions that are suitable for
administration to a
subject or patient) which can be used in the preparation of unit dosage forms.
Such
compositions comprise a prophylactically or therapeutically effective amount
of a
prophylactic and/or therapeutic agent disclosed herein or a combination of
those agents and a
pharmaceutically acceptable carrier. Preferably, compositions of the invention
comprise a
prophylactically or therapeutically effective amount of one or more EphA2
antibodies of the
invention and a pharmaceutically acceptable carrier. In a further embodiment,
the
composition of the invention further comprises an additional anti-cancer
agent. In aspecific
embodiment, additional anti-cancer agent include, but are not limited to,
chemotherapeutic
agents, radiation therapeutic agents, hormonal therapeutic agents, biological
therapeutics and
and immunotherapeutic agents.
[00192] In a specific embodiment, the term "pharmaceutically acceptable" means
approved by a regulatory agency of the Federal or a state government or listed
in the U.S.
Pharmacopeia or other generally recognized pharmacopeia for use in animals,
and more
particularly in humans. The term "carrier" refers to a diluent, adjuvant
(e.g., Freund's
adjuvant (complete and incomplete) or, more preferably, MF59C.1 adjuvant
available from
Chiron, Emeryville, CA), excipient, or vehicle with which the therapeutic is
administered.
Such pharmaceutical carriers can be sterile liquids, such as water and oils,
including those of
petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean
oil, mineral oil,
sesame oil and the like. Water is a preferred carrier when the pharmaceutical
composition is
administered intravenously. Saline solutions and aqueous dextrose and glycerol
solutions can
also be employed as liquid carriers, particularly for injectable solutions.
Suitable
pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin,
malt, rice, flour,
-71-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium
chloride, dried skim
milk, glycerol, propylene, glycol, water, ethanol and the like. The
composition, if desired,
can also contain minor amounts of wetting or emulsifying agents, or pH
buffering agents.
These compositions can take the form of solutions, suspensions, emulsion,
tablets, pills,
capsules, powders, sustained-release formulations and the like.
[00193] Generally, the ingredients of compositions of the invention are
supplied either
separately or mixed together in unit dosage form, for example, as a dry
lyophilized powder or
water free concentrate in a hermetically sealed container such as an ampoule
or sachette
indicating the quantity of active agent. Where the composition is to be
administered by
infusion, it can be dispensed with an infusion bottle containing sterile
pharmaceutical grade
water or saline. Where the composition is administered by injection, an
ampoule of sterile
water for injection or saline can be provided so that the ingredients may be
mixed prior to
administration.
[00194] The compositions of the invention can be formulated as neutral or salt
forms.
Pharmaceutically acceptable salts include those formed with anions such as
those derived
from hydrochloric, phosphoric, acetic, oxalic, tartaric acids, etc., and those
formed with
canons such as those derived from sodium, potassium, ammonium, calcium, ferric
hydroxides, isopropylamine, triethylamine, 2-ethylamino ethanol, histidine,
procaine, etc.
[00195] Various delivery systems are known and can be used to administer an
agonistic monoclonal antibody of the invention or the combination of an
agonistic
monoclonal antibody of the invention and a prophylactic agent or therapeutic
agent useful for
preventing or treating cancer, e.g., encapsulation in liposomes,
microparticles, microcapsules,
recombinant cells capable of expressing the antibody or antibody fragment,
receptor-
mediated endocytosis (see, e.g., Wu and Wu, 1987, J. Biol. Chem. 262:4429-
4432),
construction of a nucleic acid as part of a retroviral or other vector, etc.
Methods of
administering a prophylactic or therapeutic agent of the invention include,
but are not limited
to, parenteral administration (e.g., intradermal, intramuscular,
intraperitoneal, intravenous
and subcutaneous), epidural, and mucosal (e.g., intranasal, inhaled, and oral
routes). In a
specific embodiment, prophylactic or therapeutic agents of the invention are
administered
intramuscularly, intravenously, or subcutaneously. The prophylactic or
therapeutic agents
may be administered by any convenient route, for example by infusion or bolus
injection, by
absorption through epithelial or mucocutaneous linings (e.g., oral mucosa,
rectal and
intestinal mucosa, etc.) and may be administered together with other
biologically active
agents. Administration can be systemic or local.
-72-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00196] In a specific embodiment, it may be desirable to administer the
prophylactic or
therapeutic agents of the invention locally to the area in need of treatment;
this may be
achieved by, for example, and not by way of limitation, local infusion, by
injection, or by
means of an implant, said implant being of a porous, non-porous, or gelatinous
material,
including membranes, such as sialastic membranes, or fibers.
[00197] In yet another embodiment, the prophylactic or therapeutic agent can
be
delivered in a controlled release or sustained release system. In one
embodiment, a pump
may be used to achieve controlled or sustained release (see Langer, supra;
Sefton, 1987, CRC
Crit. Ref. Biomed. Eng. 14:20; Buchwald et al., 1980, Surgery 88:507; Saudek
et al., 1989, N.
Engl. J. Med. 321:574). In another embodiment, polymeric materials can be used
to achieve
controlled or sustained release of the antibodies of the invention or
fragments thereof (see
e.g., Medical Applications of Controlled Release, Langer and Wise (eds.), CRC
Pres., Boca
Raton, Florida (1974); Controlled Drug Bioavailability, Drug Product Design
and
Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and
Peppas, 1983, J.
Macromol. Sci. Rev. Macromol. Chem. 23:61; see also Levy et al., 1985, Science
228:190;
During et al., 1989, Ann. Neurol. 25:351; Howard et al., 1989, J. Neurosurg. 7
1:105); U.S.
Patent Nos. 5,679,377; 5,916,597; 5,912,015; 5,989,463; 5,128,326;
International
Publication Nos. WO 99/15154 and WO 99/20253. Examples of polymers used in
sustained
release formulations include, but are not limited to, poly(2-hydroxy ethyl
methacrylate),
poly(methyl methacrylate), poly(acrylic acid), polyethylene-co-vinyl acetate),
poly(methacrylic acid), polyglycolides~(PLG), polyanhydrides, poly(N-vinyl
pyrrolidone),
polyvinyl alcohol), polyacrylamide, polyethylene glycol), polylactides (PLA),
poly(lactide-
co-glycolides) (PLGA), and polyorthoesters. In a preferred embodiment, the
polymer used in
a sustained release formulation is inert, free of leachable impurities, stable
on storage, sterile,
and biodegradable. In yet another embodiment, a controlled or sustained
release system can
be placed in proximity of the prophylactic or therapeutic target, thus
requiring only a fraction
of the systemic dose (see, e.g., Goodson, in Medical Applications of
Controlled Release,
supra, vol. 2, pp. 115-138 (1984)).
[00198] Controlled release systems are discussed in the review by Langer
(1990,
Science 249:1527-1533). Any technique known to one of skill in the art can be
used to
produce sustained release formulations comprising one or more therapeutic
agents of the
invention. See, e.g., U.S. Patent No. 4,526,938; International Publication
Nos. WO 91/05548
and WO 96/20698; Ning et al., 1996, Radiotherapy & Oncology 39:179-189; Song
et al.,
1995, PDA Journal of Pharmaceutical Science & Technology 50:372-397; Cleek et
al., 1997,
-73-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
Pro. Int'l. Symp. Control. Rel. Bioact. Mater. 24:853-854; and Lam et al.,
1997, Proc. Int'l.
Symp. Control Rel. Bioact. Mater. 24:759-760, each of which is incorporated
herein by
reference in its entirety.
5.5.1 Formulations
[00199] Pharmaceutical compositions for use in accordance with the present
invention
may be formulated in conventional manner using one or more physiologically
acceptable
carriers or excipients.
[00200] Thus, the EphA2 antibodies of the invention and their physiologically
acceptable salts and solvates may be formulated for administration by
inhalation or
insufflation (either through the mouth or the nose) or oral, parenteral or
mucosal (such as
buccal, vaginal, rectal, sublingual) administration. In a preferred
embodiment, local or
systemic parenteral administration is used.
[00201] For oral administration, the pharmaceutical compositions may take the
form
of, for example, tablets or capsules prepared by conventional means with
pharmaceutically
acceptable excipients such as binding agents (e.g., pregelatinised maize
starch,
polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.,
lactose,
microcrystalline cellulose or calcium hydrogen phosphate); lubricants (e.g.,
magnesium
stearate, talc or silica); disintegrants (e. g., potato starch or sodium
starch glycolate); or
wetting agents (e.g., sodium lauryl sulphate). The tablets may be coated by
methods well
known in the art. Liquid preparations for oral administration may take the
form of, for
example, solutions, syrups or suspensions, or they may be presented as a dry
product for
constitution with water or other suitable vehicle before use. Such liquid
preparations may be
prepared by conventional means with pharmaceutically acceptable additives such
as
suspending agents (e.g., sorbitol syrup, cellulose derivatives or hydrogenated
edible fats);
emulsifying agents (e.g., lecithin or acacia); non-aqueous vehicles (e.g.,
almond oil, oily
esters, ethyl alcohol or fractionated vegetable oils); and preservatives
(e.g., methyl or propyl-
p-hydroxybenzoates or sorbic acid). The preparations may also contain buffer
salts,
flavoring, coloring and sweetening agents as appropriate.
[00202] Preparations for oral administration may be suitably formulated to
give
controlled release of the active compound.
[00203] For buccal administration the compositions may take the form of
tablets or
lozenges formulated in conventional manner.
-74-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00204] For administration by inhalation, the prophylactic or therapeutic
agents for use
according to the present invention are conveniently delivered in the form of
an aerosol spray
presentation from pressurized packs or a nebulizer, with the use of a suitable
propellant, e.g.,
dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane,
carbon dioxide
or other suitable gas. In the case of a pressurized aerosol the dosage unit
may be determined
by providing a valve to deliver a metered amount. Capsules and cartridges of
e.g., gelatin for
use in an inhaler or insufflator may be formulated containing a powder mix of
the compound
and a suitable powder base such as lactose or starch.
[00205] The prophylactic or therapeutic agents may be formulated for
parenteral
administration by injection, e.g., by bolus injection or continuous infusion.
Formulations for
injection may be presented in unit dosage form, e.g., in ampoules or in multi-
dose containers,
with an added preservative. The compositions may take such forms as
suspensions, solutions
or emulsions in oily or aqueous vehicles, and may contain formulatory agents
such as
suspending, stabilizing and/or dispersing agents. Alternatively, the active
ingredient may be
in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-
free water,
before use.
[00206] The prophylactic or therapeutic agents may also be formulated in
rectal
compositions such as suppositories or retention enemas, e.g., containing
conventional
suppository bases such as cocoa butter or other glycerides.
[00207] In addition to the formulations described previously, the prophylactic
or
therapeutic agents may also be formulated as a depot preparation. Such long
acting
formulations may be administered by implantation (for example subcutaneously
or
intramuscularly) or by intramuscular injection. Thus, for example, the
prophylactic or
therapeutic agents may be formulated with suitable polymeric or hydrophobic
materials (for
example as an emulsion in an acceptable oil) or ion exchange resins, or as
sparingly soluble
derivatives, for example, as a sparingly soluble salt.
[00208] The invention also provides that a prophylactic or therapeutic agent
is
packaged in a hermetically sealed container such as an ampoule or sachette
indicating the
quantity. In one embodiment, the prophylactic or therapeutic agent is supplied
as a dry
sterilized lyophilized powder or water free concentrate in a hermetically
sealed container and
can be reconstituted, e.g., with water or saline to the appropriate
concentration for
administration to a subject.
[00209] In a preferred embodiment of the invention, the formulation and
administration of various chemotherapeutic, biological/immunotherapeutic and
hormonal
-75-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
therapeutic agents are known in the art and often described in the Physicians'
Desk
Reference, (58'~ ed., 2004). For instance, in certain specific embodiments of
the invention,
the therapeutic agents of the invention can be formulated and supplied as
provided in Table 2.
[00210] In other embodiments of the invention, radiation therapy agents such
as
radioactive isotopes can be given orally as liquids in capsules or as a drink.
Radioactive
isotopes can also be formulated for intravenous injections. The skilled
oncologist can
determine the preferred formulation and route of administration.
[00211] In certain embodiments the agonistic monoclonal antibodies of the
invention,
are formulated at 1 mg/ml, 5 mg/ml, 10 mg/ml, and 25 mg/ml for intravenous
injections and
at 5 mg/ml, 10 mg/ml, and 80 mg/ml for repeated subcutaneous administration
and
intramuscular injection.
[00212] The compositions may, if desired, be presented in a pack or dispenser
device
that may contain one or more unit dosage forms containing the active
ingredient. The pack
may for example comprise metal or plastic foil, such as a blister pack. The
pack or dispenser
device may be accompanied by instructions for administration.
5.5.2 Dosages
[00213] The amount of the composition of the invention which will be effective
in the
treatment, prevention or management of cancer can be determined by standard
research
techniques. For example, the dosage of the composition which will be effective
in the
treatment, prevention or management of cancer can be determined by
administering the
composition to an animal model such as, e.g., the animal models disclosed
herein or known
to those skilled in the art. In addition, in vitro assays may optionally be
employed to help
identify optimal dosage ranges.
[00214] Selection of the preferred effective dose can be determined (e.g., via
clinical
trials) by a skilled artisan based upon the consideration of several factors
which will be
known to one of ordinary skill in the art. Such factors include the disease to
be treated or
prevented, the symptoms involved, the patient's body mass, the patient's
immune status and
other factors known by the skilled artisan to reflect the accuracy of
administered
pharmaceutical compositions.
[00215] The precise dose to be employed in the formulation will also depend on
the
route of administration, and the seriousness of the cancer, and should be
decided according to
the judgment of the practitioner and each patient's circumstances. Effective
doses may be
extrapolated from dose-response curves derived from in vitro or animal model
test systems.
-76-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00216] For antibodies, the dosage administered to a patient is typically 0.1
mg/kg to
100 mg/kg of the patient's body weight. Preferably, the dosage administered to
a patient is
between 0.1 mg/kg and 20 mg/kg of the patient's body weight, more preferably 1
mg/kg to 10
mg/kg of the patient's body weight. Generally, human and humanized antibodies
have a
longer half-life within the human body than antibodies from other species due
to the immune
response to the foreign polypeptides. Thus, lower dosages of human antibodies
and less
frequent administration is often possible.
[00217] For other cancer therapeutic agents administered to a patient, the
typical doses
of various cancer therapeutics known in the art are provided in Table 2. Given
the invention,
certain preferred embodiments will encompass the administration of lower
dosages in
combination treatment regimens than dosages recommended for the administration
of single
agents.
[00218] The invention provides for any method of administrating lower doses of
known prophylactic or therapeutic agents than previously thought to be
effective for the
prevention, treatment, management or amelioration of cancer. Preferably, lower
doses of
known anti-cancer therapies are administered in combination with lower doses
of agonistic
monoclonal antibodies of the invention.
5.6 Kits
[00219] The invention provides a pharmaceutical pack or kit comprising one or
more
containers filled with an EphA2 antibody of the invention. Additionally, one
or more other
prophylactic or therapeutic agents useful for the treatment of a cancer can
also be included in
the pharmaceutical pack or kit. The invention also provides a pharmaceutical
pack or kit
comprising one or more containers filled with one or more of the ingredients
of the
pharmaceutical compositions of the invention. Optionally associated with such
containers)
can be a notice in the form prescribed by a governmental agency regulating the
manufacture,
use or sale of pharmaceuticals or biological products, which notice reflects
approval by the
agency of manufacture, use or sale for human administration.
[00220] The present invention provides kits that can be used in the above
methods. In
one embodiment, a kit comprises one or more EphA2 antibodies of the invention.
In another
embodiment, a kit further comprises one or more other prophylactic or
therapeutic agents
useful for the treatment of cancer, in one or more containers. Preferably the
EphA2 antibody
of the invention is EA2, EA3, EA4, or EAS. In certain embodiments, the other
prophylactic
_77_
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
or therapeutic agent is a chemotherapeutic. In other embodiments, the
prophylactic or
therapeutic agent is a biological or hormonal therapeutic.
6. EXAMPLES
6.1 Preparation of Monoclonal Antibodies
Anti eng~Pr_eparation
[00221] Ras-transformed MCF-l0A cells were extracted in RIPA buffer. Tyrosine
phosphorylated proteins were partially purified using immobilized PY20
antibodies (Kanner
et al., 1989, J. Immunol. Meth. 120:115-124). Bound proteins were
competitively eluted with
25 mM phenylphosphate. Fractions containing PY20-reactive proteins were
confirmed by
western blot analysis using phosphotyrosine-specific antibodies.
Antibody Screening
[00222] As a preliminary screen for EphA2-immunoreactivity, supernatants from
bulk
culture hybridomas were screened for immunoreactivity against EphA2.
Immunization
strategy was designed to identify extracellular EphA2 epitopes on viable tumor
cells. Thus, a
fluorescence-based ELISA protocol (FluorELISA), which selects for antibody
reactivity
against live cells, was utilized. This screening approach was preferable to
western blot
analyses, which might have biased against antibodies that recognize
conformation-restricted
epitopes.
[00223] Cell surface binding by anti-EphA2 antibodies to the EphA2 receptor
was
monitored using modifications to a reported assay (Kilpatrick et al., 1998,
Hybridoma
17:576). 96 well, flat-bottom tissue culture treated plates (Costar,
Cambridge, MA) were
treated with 100p1 of poly-L-lysine hydrobromide (Sigma, St. Louis, MO)
diluted to 10
p,g/ml in O.1M sodium phosphate (pH 8.0) for 1 hour. Poly-L-lysine was removed
from the
wells before the addition of 100p1 of a cell suspension of MDA-MB-23 1
(positive for
EphA2) or BT474 cells (negative controls) at a concentration of 3 x 104 cells
per well. After
incubation overnight at 37°C, 5% C02, the culture media was gently
removed, and 100p1 of
supernatants from hybridomas were incubated on cells at room temperature for 1
hour. The
samples were washed three times with 1X Dulbecco's phosphate buffered saline
(pH 7.1)
(GIBCO, Grand Island, NY). Goat anti-mouse Alexa Fluor 488 antibody (100p1;
Molecular
Probes, Eugene, OR), diluted to 2pg/ml in PBS, was added for one hour at room
temperature.
After washing cells with PBS, 50 pl of PBS containing 2% FCS was added to each
well
_78_
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
before observation using an inverted fluorescence microscope (Model DM-IRB,
Leica,
Deerfield, IL).
[00224] FluorELISA identified 44 bulk hybridoma populations that stained EphA2-
overexpressing tumor cells (MDA-MB-231), but not EphA2-deficient cells (BT474)
(data not
shown). Immunoreactivity was confirmed using fluorescence microscopy, which
revealed a
pattern of diffuse membrane staining that was consistent with our previous
studies (e.g.,
Zelinski et al., 2001, Cancer Res. 61:2301 and Zantek, et al., 1999, Cell
Growth Diff. 10:629)
of EphA2 subcellular localization. Hybridoma bulk cultures were initially
selected for
subcloning by flow cytometry based on strong immunostaining of target-
positive, but not
target deficient, cells. Bulk culture populations of hybridomas were then
subcloned by flow
cytometry and FluorELISA was repeated with supernatants from the subcloned
hybridomas.
6.2 EphA2 Monoclonal Antibodies Decrease Metastatic Properties of Tumor
Cells
6.2.1 EphA2 Phosphorylation and Degradation
[00225] EphA2 antibodies promoted tyrosine phosphorylation and degradation of
EphA2 in MDA-MB-231 (FIGS. lA-1C) cells. Monolayers of cells were incubated in
the
presence of EAS (FIGS. lA-1B, lanes 2, 3) or EA2 (FIGS. lA-1B, lanes 4, 5) or
control
(FIGS. lA-1B, lane 1) for 8 minutes at 37°C. Cell lysates were then
immunoprecipitated
with an EphA2-specific antibody (D7, purchased from Upstate Biologicals, Inc.,
Lake Placid,
NY and deposited with the American Type Tissue Collection on December 8, 2000,
and
assigned ATCC number PTA 2755), resolved by SDS-PAGE and subjected to western
blot
analysis with a phosphotyrosine-specific antibody (4610, purchased from
Upstate
Biologicals, Inc., Lake Placid, NY) (FIG. lA). The membranes were stripped and
re-probed
with the EphA2-specific antibody used in the immunoprecipitation (D7) as a
loading control.
[00226] Western blot analyses and immunoprecipitations were performed as
described
previously (Zantek et al., 1999, Cell Growth Diff. 10:629-38). Briefly,
detergent extracts of
cell monolayers were extracted in Tris-buffered saline containing 1% Triton X-
100 (Sigma,
St. Louis, MO). After measuring protein concentrations (BioRad, Hercules, CA),
1.5 mg of
cell lysate was immunoprecipitated, resolved by SDS-PAGE and transferred to
nitrocellulose
(Protran, Schleicher and Schuell, Keene, NH). Antibody binding was detected by
enhanced
chemiluminescence (Pierce, Rockford, IL) and autoradiography (Kodak X-OMAT;
Rochester, NY).
-79-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00227] Levels of EphA2 phosphorylation were found to increase with EphA2
agonistic EAS and EA2 antibody incubation (FIG. 1B). Monolayers of MDA-MB-231
cells
were incubated in the presence of 30 pg/ml EAS (FIG. 1C, lanes 2, 3) or EA2
(FIG. 1C, lanes
4, 5) or a control (FIG. 1C, lane 1) for 24 hours at 37°C. Cell lysates
were then resolved by
SDS-PAGE and subjected to western blot analysis with an EphA2-specific
antibody (D7).
EphA2 protein levels decrease with antibody incubation.
[00228] Similar experiments were conducted with A549 cells. Monolayers of A549
cells were incubated at 37°C in the presence of EAS or EA2 or control
(PBS) for either 10
minutes (FIGS. 2A-2B) or 5 hours (FIGS. 2C-2D). Cell lysates were then
immunoprecipitated with an EphA2-specific antibody (D7), resolved by SDS-PAGE
and
subjected to western blot analysis with a phosphotyrosine-specific antibody
(4610, purchased
from Upstate Biologicals, Inc., Lake Placid, NY) (FIGS. 2A, 2C). The membranes
were
stripped and re-probed with the EphA2-specific antibody used in the
immunoprecipitation
(D7) as a loading control (FIGS. 2B, 2D). At 10 minutes, antibody incubation
caused an
increase in phosphorylation (FIG. 2A). With continued incubation of 5 hours,
these
antibodies caused degradation of the EphA2 protein. (FIG. 2D).
6.2.2 Growth in Soft Agar
[00229] Tumor cells were suspended in soft agar. Colony formation in soft agar
was
assayed as described in Zelinski et al. (2001, Cancer Res. 61:2301-6).
Antibodies or a
control solution (PBS) was included in bottom and top agar solutions. Cells
were suspended
in soft agar for 7 days at 37°C in the presence of purified antibody or
control solution (PBS),
administered at the time of suspension. Colony formation was scored
microscopically using
an Olympus CK-3 inverted phase-contrast microscope outfitted with a 40x
objective.
Clusters containing at least three cells were scored as a positive. The
average number of
colonies per high-powered field is shown. Ten separate high-power microscopic
fields were
averaged in each experiment, and the results shown are representative of at
least three
separate experiments.
[00230] A549 malignant lung cancer cells were incubated with either 10 pg/ml
or 2.5
pg/ml of EAS or EA2 monoclonal antibodies or a control (PBS). All amounts of
antibodies
used inhibited cell growth in soft agar (FIG. 3A). Benign MCF-7 breast
epithelial tumor
cells were converted to malignant cells by the overexpression of EphA2 (MCF-
7Ep~). Both
tumor cell types were incubated with either EAS monoclonal antibodies or a
control (PBS).
EAS inhibits the ability of MCF-7EPh'~ cells to grow in soft agar. Benign MCF-
7 did not
-80-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
form colonies in soft agar either with or without antibody incubation (FIG.
3B). Results are
reported as colonies per high-powered field (HPF). Control experiments
confirmed that
neither isotype-matched (IgG,) controls (e.g., anti-paxillin) nor antibodies
against
intracellular epitopes on EphA2 (e.g., D7) decreased soft agar colonization
(data not shown).
6.2.3 Tubular Network Formation in MATRIGELTM
[00231] Tumor cell behavior within a three-dimensional microenvironment, such
as
MATRIGELTM, can reliably predict the differentiation state and aggressiveness
of breast
epithelial cells. Monolayer cultures of benign (MCF-l0A) or malignant (MDA-MB-
231)
breast epithelial cells are incubated on MATRIGELTM in the presence of EphA2
antibodies
(10 pg/ml) or control solution (PBS). The behavior of cells on MATRIGELTM is
analyzed as
described in Zelinski et al. (2001, Cancer Res. 61:2301-6). Briefly, tissue
culture dishes are
coated with MATRIGELTM (Collaborative Biomedical Products, Bedford, MA) at
37°C
before adding 1 x 105 MDA-MB-231 or MCF-l0A cells that had been incubated on
ice for 1
hour with an EphA2 agonistic antibody or control solution (PBS). Cells are
incubated on
MATRIGELTM for 24 hours at 37°C, and cell behavior is assessed using an
Olympus IX-70
inverted light microscope. All images are recorded onto 35 mm film (T-Max-400.
Kodak.
Rochester, NY).
[00232] Within 24 hours, non-transformed MCF-l0A epithelial cells organize
into
acinus-like spheres on MATRIGELTM while MDA-MB-231 cells quickly assemble into
tubular networks. These networks progressively invade all throughout the
MATRIGELTM.
With the addition of EphA2 agonistic antibodies, the formation of tubular
networks is
prevented.
6.2.4 Growth in vivo
[00233] EAS can inhibit tumor cell growth in vivo. Sx 106 MDA-MB-231 breast
cancer
cells were implanted orthotopically or subcutaneously and Sx 106 A549 lung
cancer cells were
implanted subcutaneously into athymic mice. After the tumors had grown to an
average
volume of 100mm3, mice were administered 6mg/kg of an EAS or negative control
(PBS or
lA7 antibody) intraperitoneally twice a week for 3 weeks. Animals were
generally sacrificed
at least two weeks after the last treatment or when tumors exceeded 2000 mm~.
Tumor
growth was assessed and expressed either as a ratio of the tumor volume
divided by initial
tumor volume (100 mm3) or as total tumor volume. EAS inhibited growth of MDA-
MB-231
-81-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
cells implanted orthotopically (FIG. 4A) or subcutaneously (FIG. 4B, D).
Growth of A549
cells implanted subcutaneously was also inhibited by EAS (FIG. 4C).
6.3 Estrogen Dependence in Breast Cancer Cells
[00234] Estrogen-sensitive breast cancer cells, MCF-7 cells, were transfected
with and
stably overexpressed human EphA2 (MCF-7EPhaz) (pNeoMSV-EphA2 provided by Dr.
T.
Hunter, Scripps Institute). Western blot analyses confirmed the ectopic
overexpression of
EphA2 in transfected cells relative to matched controls (data not shown).
[00235] EphA2 overexpression increased malignant growth (FIGS. SA-SB). Growth
assays were conducted as follows. MCF-7"~° (control cells) or MCF7EP"'~
cells were seeded
in 96-well plates. Cell growth was measured with Alamar blue (Biosource
International,
Camarillo, CA) following the manufacture's suggestion. Colony formation in
soft agar was
performed as previously described (Zelinski et al., 2001, Cancer Res. 61:2301-
6) and scored
microscopically, defining clusters of at least three cells as a positive. The
data represent the
average of ten separate high-power microscopic fields from each sample and
representative
of at least three separate experiments. Error bars represent the standard
error of the mean of
at least three different experiments as determined using Microsoft Excel
software.
[00236] Although MCF-7 control cells were largely unable to colonize soft agar
(an
average of 0.1 colony/field), MCF-7EPnA2 cells formed larger and more numerous
colonies
(4.7 colonies/field; P < 0.01) that persisted for at least three weeks (FIG.
SA and data not
shown). Despite increased colonization of soft agar, the growth of MCF-7Eph'~
cells in
monolayer culture did not differ from matched controls (FIG. SB), thus
indicating that the
growth promoting activities of EphA2 were most apparent using experimental
conditions that
model anchorage-independent (malignant) cell growth.
[00237] Consistent with increased soft agar colonization, orthotopically
implanted
MCF-7Eph'~ cells formed larger, more rapidly growing tumors in vivo. Six to
eight week-old
athymic (nulnu) mice were purchased from Harlan Sprague Dawley (Indianapolis,
IN).
When indicated, a controlled release estradiol pellet (0.72 mg 17(3-estradiol,
60-day
formulation) was injected subcutaneously via a sterile 14-gauge trocar 24
hours prior to
tumor implantation and pellets were replaced every 60 days for those
experiments spanning >
60 days in duration. 1x106 MCF-7"~° or MCF7EPt'~'2 cells were injected
into the mammary fat
pad under direct visualization. When indicated, tamoxifen ( 1 mg) was
administered by oral
gavage 6 days per week.
-82-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00238] In the presence of supplemental estrogen (173-estradiol purchased from
Sigma), the MCF-7EPh'~ cells demonstrated a two-fold increase in tumor volume
relative to
matched controls (FIG. 6A). EphA2-overexpressing tumors differed
phenotypically from
control tumors in that they were more vascular and locally invasive at the
time of resection
(data not shown). To confirm that these tumors expressed EphA2, whole cell
lysates of
resected tumors were subjected to western blot analyses with EphA2-specific
antibodies
(FIG. 6B). The membranes were then stripped and reprobed with (3-catenin
antibodies to
verify equal sample loading. The relative amount of EphA2 was higher in tumor
samples
than in the input cells (prior to implantation), suggesting that tumors arose
from cells with
' high levels of EphA2. Comparable findings with in vitro and in vivo models
indicate that
EphA2 overexpression results in a more aggressive phenotype.
[00239] Parallel studies were performed in the absence of exogenous estrogen.
Experimental deprivation of estrogen amplified differences between the
cellular behaviors of
control and MCF-7EpnA2 cells. While MCF-7EPna2 cells continued to colonize
soft agar more
efficiently than matched controls (FIG. 7A), these cells did grow in the
absence of exogenous
estrogen (FIG. 7B). In contrast, supplemental estrogen was required for
monolayer growth of
control cells (FIG. 7B). Additionally, MCF-7EPnnz cells retained tumorigenic
potential in the
absence of supplemental estrogen. While control MCF-7 cells rarely formed
palpable
tumors, the MCF-7EPna,z cells formed tumors that persisted for over 12 weeks
(FIG. 7C and
data not shown). Thus, both in vitro and in vivo assay systems confirm that
EphA2
overexpression decreases the need for exogenous estrogen.
[00240] Sensitivity of MCF-7EP"'~ cells to tamoxifen was measured. Tamoxifen
(4-
hydroxy tamoxifen purchased from Sigma) reduced soft agar colonization of
control MCF-7
cells by at least 60%. The inhibitory actions of tamoxifen on MCF-7EPnA2 cells
were less
pronounced (25% inhibition, FIG. 8A). Notably, excess estradiol overcame the
inhibitory
effects of tamoxifen, which provided additional evidence for the specificity
of this finding
(FIG. 8A). Similarly, the tumorigenic potential of MCF-7EPh'~ cells was less
sensitive to
tamoxifen as compared with control (MCF-7"e~) cells (FIG. 8B).
[00241] Since tamoxifen sensitivity often relates to estrogen receptor
expression,
estrogen receptor expression and activity was assayed in MCF-7EPh~. Western
blot analyses
revealed comparable levels of ERa and ER~i in control and MCF-7EPh'~ cells
(FIGS. 9A-9B)
(ERa and ER(3 antibodies were purchased from Chemicon, Temecula, CA).
Moreover,
comparable levels of estrogen receptor activity were detected in control and
MCF-7EPna,2 cells
-83-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
and this enzymatic activity remained sensitive to tamoxifen (FIGS. 9E-9F).
Estrogen
receptor activity was measured using ERE-TK-CAT vector (which encodes a single
ERE; a
generous gift from Dr. Nakshatri, Indiana University School of Medicine) in
the unstimulated
state, after estradiol (10-8 M) stimulation and tamoxifen (10-6 M) inhibition.
Cells were plated
in phenol red free, charcoal stripped sera for 2 days and transfected with ERE-
TK-CAT (5
fig) using calcium phosphate method. The (3-galactosidase expression vector
RSV/~3-galactosidase (2 fig, Dr. Nakshatri's gift) was cotransfected as a
control. Fresh
media including the appropriate selection drugs were added 24 hours after
transfection. Cells
were harvested after 24 hours and CAT activity was evaluated as described
(Nakshatri et al.,
1997, Mol. Cell. Biol. 17:3629-39). These results indicate that the estrogen
receptor in MCF-
7E~'~'p'2 cells is expressed and remains sensitive to tamoxifen, thus
suggesting that the defect
which renders MCF-7EP~ less dependent on estrogen lies downstream of estrogen
signaling.
[00242] Growth MCF-7EP~ cells which had decreased EphA2 expression levels was
assayed in soft agar. The EphA2 monoclonal antibody EAS induced EphA2
activation and
subsequent degradation. Decreased levels of EphA2 expression were observed
within two
hours of EAS treatment and EphA2 remained undetectable for at least the
following 24 hours
(FIG. l0A). The soft agar colonization of control MCF-7 cells was sensitive to
tamoxifen
(FIG. lOC) and EAS did not further alter this response (since these cells lack
of endogenous
EphA2). The MCF-7Epna,2 cells were less sensitive to tamoxifen (25% inhibition
by
tamoxifen) as compared to the matched controls (75% inhibition by tamoxifen).
Whereas
EAS decreased soft agar colonization (by 19%), the combination of EAS and
tamoxifen
caused a much more dramatic (>80%) decrease in soft agar colonization. Thus,
EAS
treatment restored a phenotype that was comparable to control MCF-7 cells.
These findings
suggest that antibody targeting of EphA2 can serve to re-sensitize the breast
tumor cells to
tamoxifen.
[00243] All statistical analyses were performed using Student's t-test using
Microsoft
Excel (Seattle, WA), defining P<0.05 as significant. In vivo tumor growth
analyses were
performed using GraphPad Software (San Diego, CA).
6.4 Expression of EphA2 in Prostatic Intraepithelial Neoplasia
[00244] EphA2 immunoreactivity distinguished neoplastic prostatic epithelial
cells
from their non-neoplastic counterparts. Ninety-three cases of radical
retropubic
prostatectomy were obtained from the surgical pathology files of Indiana
University Medical
Center. Patients ranged in age from 44 to 77 years (mean = 63 years). Grading
of the
-84-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
primary tumor from radical prostatectomy specimens was performed according to
the
Gleason system (Bostwick "Neoplasms of the prostate" in Bostwick and Eble,
eds., 1997,
Urologic Surgical Pathology St. Louis:Mosby page 343-422; Gleson and
Mellinger, 1974, J.
Urol. 111:58-64). The Gleason grade ranged from 4 to 10. Pathological stage
was evaluated
according to the 1997 TNM (tumor, lymph nodes, and metastasis) standard
(Fleming et al.,
1997, AJCC Cancer Staging Manual. Philadelphia:Raven and Lippincott).
Pathological
stages were T2a (n= 9 patients), T2b (n= 43), T3a (n= 27), T3b (n=14).
Thirteen patients had
lymph node metastasis at the time of surgery.
[00245] Serial 5 pm-thick sections of formalin-fixed slices of radical
prostatectomy
specimens were used for immunofluorescent staining. Tissue blocks that
contained the
maximum amount of tumor and highest Gleason grade were selected. One
representative
slide from each case was analyzed. Slides were deparaffinized in xylene twice
for 5 minutes
and rehydrated through graded ethanols to distilled water. Antigen retrieval
was carried out
by heating sections in EDTA (pH 8.0) for 30 minutes. Endogenous peroxidase
activity was
inactivated by incubation in 3% H202 for 15 minutes. Non-specific binding
sites were
blocked using Protein Block (DAKO) for 20 minutes. Tissue sections were then
incubated
with a mouse monoclonal antibody against human EphA2 (IgGI, 1:100 dilution)
overnight at
room temperature, followed by biotinylated secondary antibody (DAKO
corporation,
Carpintera, CA) and peroxidase-labeled streptavidin, and 3,3-diaminobenzidine
was used as
the chromogen in the presence of hydrogen peroxide. Positive and negative
controls were
run in parallel with each batch.
[00246] The extent and intensity of staining were evaluated in benign
epithelium, high-
grade prostatic intraepithelial neoplasia (PIN) and adenocarcinoma from the
same slide for
each case. Microscopic fields with highest degree of immunoreactivity were
chosen for
analysis. At least 1000 cells were analyzed in each case. The percentage of
cells exhibiting
staining in each case was evaluated semiquantitatively on a 5% incremental
scale ranging
from 0 to 95%. A numeric intensity score is set from 0 to 3 (0, no staining; 1
weak staining;
2 moderate staining; and 3, strong staining) (Jiang et al., 2002, Am. J.
Pathol. 160:667-71;
Cheng et al., 1996, Am J. Pathol. 148:1375-80).
[00247] The mean percentage of immunoreactive cells in benign epithelium, high-
grade PIN and adenocarcinoma were compared using the Wilcoxon paired signed
rank test.
The intensity of staining for EphA2 in benign epithelium, high-grade PIN, and
adenocarcinoma was compared using Cochran-Mantel-Haenszel tests for correlated
ordered
-85-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
categorical data. Pairwise comparisons were made if the ANOVA revealed
significant
differences. A p-value<0.05 was considered significant, and all p-values were
two-sided.
[00248] EphA2 immunoreactivity was observed in all cases of high-grade
prostatic
intraepithelial neoplasia (PIN) and cancers but not in benign epithelial
cells. For example,
EphA2 expression (both the mean percentage of immunoreactive cells and
staining intensity)
was increased in both high-grade PIN and cancers relative to benign epithelial
cells (Tables 3
and 4). Similarly, EphA2 immunoreactivity (both the mean percentage of
immunoreactive
cells and staining intensity) was increased in prostatic carcinomas compared
with high-grade
PIN (Tables 3 and 4). This immunoreactivity was evident at the membrane and
cytoplasm of
the neoplastic epithelial cells (data not shown). In contrast, no EphA2
immunoreactivity was
observed in tumor-proximal stromal cells. In the high-grade PIN group, 22%
showed grade 1
staining intensity, 73% showed grade 2 staining intensity, and 5% showed grade
3 staining
intensity (Table 3). In the adenocarcinoma group, 13% of cases showed grade 1
staining
intensity, 50% showed grade 2 staining intensity, and 37% showed grade 3
staining intensity.
In contrast, the normal epithelium group showed grade 1 stain in 66% of the
cases, the
remaining cases showed no immunoreactivity for EphA2 protein (grade 0 staining
intensity)
(Table3). The mean percentage of EphA2 immunoreactive cells was 12% in the
normal
epithelial cells, 67% in the high-grade PIN, and 85% in the prostatic
adenocarcinoma (Table
4).
[00249] Although high levels of EphA2 could distinguish neoplastic from benign
prostatic epithelial cells, EphA2 did not correlate with other histologic and
pathologic
parameters of disease severity. For example, high levels of EphA2 were
observed in most
prostatic carcinomas and did not relate to Gleason grade, pathologic stage,
lymph node
metastasis, extraprostatic extension, surgical margins, vascular invasion,
perineural invasion,
or the presence of other areas of the prostate with high-grade PIN (Table 5).
TABLE 3
Staining
Intensity
Grade
0 1 2 3
Cell T a
Benign epithelium31(33%) 61(66%) 1 (1%) 0 (0%)
High-grade PINa 0 (0%) 20 (22%) 68 (73%) 5 (S%)
Adenocarcinoma~'b0 (0%) 12 (13%) 47 (50%). 34 (37%)
-86-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
a Indicates percentage of staining intensity was statistically lower compared
to that of
the normal cells with a P-value = 0.0001 using a Wilcoxon paired signed rank
test.
b The staining intensity was significantly higher compared to high-grade PIN
(P<0.01,
Cochran-Mantel-Henszel test).
TABLE 4
Mean % of Cells
Cell Type Range (%)
Staining SD
Normal Cells
12 + 17 0 - 90
High-grade PIN 67 + 18a 5 - 95
Adenocarcinoma g5 + 12a'b 30 - 95
a Indicates percentage of staining statistically lower compared to that of the
normal cells with a P-value = 0.0001 using a Wilcoxon paired signed rank test.
b The percentage of staining was statistically higher compared to high-grade
PIN
(P<0.01, ANOVA).
TABLE 5
Patient Characteristic % of Total Mean % of Cells Mean EphA2
Patients Staining w/EphA2 Antibody Staining
(n=93) Antibody (~SD) Intensity (~SD)
Primary Gleason Grade
2 12 832 2.00.6
3 43 8610 2.30.7
4 23 8416 2.30.7
15 8611 2.30.6
Secondary Gleason
Grade
2 15 8216 2.30.5
3 29 8515 2.10.6
4 35 859 2.30.7
5 14 888 2.40.8
Gleason Sum <7 28 8312 2.20.6
7 35 8514 2.20.7
>7 30 8710 2.40.7
T Classification T2a 9 896 2.30.5
T2b 43 8412 2.20.7
_87_
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
T3a 27 8415 2.20.7
T3b 14 6310 2.40.6
Lymph Node Metastasis
Positive 13 889 2.30.6
Negative 80 8413 2.20.7
Extraprostatic Extension
Positive 53 8611 2.30.7
Negative 40 8414 2.20.7
Surgical Margin Positive50 8611 2.10.6
Negative 43 8413 2.40.7
Vascular Invasion Positive30 8511 2.10.8
Negative 63 8613 2.30.6
PerineuralInvasion 82 8215 2.40.5
Positive
Negative 11 8512 2.20.7
High-grade PIN Positive89 8512 2.30.7
Negative 4 859 2.00.8
6.5 Treatment Of Patients With Metastatic Cancer
[00250] A study is designed to assess pharmacokinetics and safety of agonistic
monoclonal antibodies of the invention in patients with metastatic breast
cancer. Cancer
patients currently receive Taxol or Taxotere. Patients currently receiving
treatment are
permitted to continue these medications.
[00251] Patients are administered a single IV dose of a monoclonal antibody of
the
invention and then, beginning 4 weeks later, are analyzed following
administration of
repeated weekly IV doses at the same dose over a period of 12 weeks. The
safety of
treatment with the agonistic monoclonal antibody of the invention is assessed
as well as
potential changes in disease activity over 26 weeks of IV dosing. Different
groups of patients
are treated and evaluated similarly but receive doses of 1 mg/kg, 2 mg/kg, 4
mg/kg, or 8
mg/kg.
[00252] Antibodies of the invention are formulated at 5 mg/ml and 10 mg/ml for
IV
injection. A formulation of 80 mg/ml is required for repeated subcutaneous
administration.
The antibodies of the invention are also formulated at 100 mg/ml for
administration for the
purposes of the study.
[00253] Changes are measured or determined by the progression of tumor growth.
6.6 E~itope Analysis of EnhA2 Antibodies
_88_
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
[00254] The epitope of the EphA2 antibodies were characterized. EAS and EA2
selectively bind to malignant cells. The anti-EphA2 monoclonal antibodies EAS
and EA2
bind malignant MDA-MB-231 breast epithelial tumor cells (FIGS. 11A-11B) more
strongly
than benign MCF-l0A breast epithelial tumor cells (FIGS. 11C-11D) as shown by
immunofluorescent staining. Furthermore, EAS was immunoreactive against
malignant
prostate cells. The anti-EphA2 monoclonal antibody EAS identified malignant
prostate
cancer cells in formalin-fixed, paraffin-embedded archival clinical specimens
(FIG. 12).
[00255] EAS preferentially binds an EphA2 epitope exposed on cancer cells but
not
non-cancer cells. Non-transformed MCF-l0A cells or transformed MDA-MB-231
cells were
incubated with 10 pg/ml EA2 at 4°C for 30 min. prior to fixation in a
3% formalin solution
and immunolabeling with fluorophore-conjugated anti-mouse IgG. EAS
preferentially binds
EphA2 on transformed cells (FIG. 13D). In contrast, another EphA2 antibody
Eph099B-
233.152 (ATCC deposit no. PTA-5194; see co-pending US Patent Application
Serial No.
10/436,782, entitled "EphA2 Monoclonal Antibodies and Methods of Use Thereof,"
filed
May 12, 2003, binds EphA2 expressed on both transformed and non-transformed
cells (FIGS.
13A-13B). Treatment of non-transformed MCF-l0A cells with 4mM EGTA for 20 min.
dissociated the cells. EAS bound EphA2 on the EGTA dissociated cells but not
the untreated
cells (FIGS. 14A-14B).
[00256] An equivalent experiment was performed using MCF-l0A or MDA-MB-231
cells. The amount of EAS binding to EphA2 was measured using flow cytometry
(FIGS.
14C-l4Dl. Cells were either treated by incubation in 4 mM EGTA for 10-15
minutes on ice
(top panel) or were not treated with EGTA (middle panel) before incubation
with 10 pg/ml
EAS. Cells were then fixed with 3% formalin and labeled with fluorophore-
labeled donkey
anti-mouse IgG. Control cells were incubated only with secondary antibody
(fluorophore-
labeled donkey anti-mouse IgG) in the absence of primary antibody (EAS)
(bottom panel).
The samples were then evaluated using flow cytometry (Becton Dickinson FACStar
Plus).
EGTA treatment did not affect EAS binding to transformed cells (FIG. 14D, top
and middle
panels). In contrast, EAS binding to non-transformed cells was increased by
incubation in
EGTA (FIG. 14C, top and middle panels).
[00257] EAS does not bind the same epitope as the EphA2 ligand Ephrin A1. A
microtiter plate was coated with 10 mg/ml Ephrin A1-F~ overnight at
4°C. A fusion protein
consisting of the extracellular domain of EphA2 linked to human IgG, constant
region
(EphA2-F~) was incubated with and bound to the immobilized Ephrin A1-F~.
Biotinylated
Ephrin A1-F~ or EAS was incubated with the EphA2-Ephrin A1-F~ complex and
amount of
-89-
CA 02546763 2006-05-19
WO 2005/051307 PCT/US2004/039112
binding was measured. Very little additional Ephrin A1-F~ bound the EphA2-
Ephrin A1-F
complex while, in contrast, considerable levels of EAS bound the EphA2-phrinAl-
F~
complex (FIG. 15A).
[00258] The EphA2-Ephrin A1-F~ complex was prepared as described above.
Biotinylated EAS (10 g,g/ml) was then incubated with the complex for 30 min.
Unlabeled
competitor was incubated with EphA2-Ephrin Al-F~-EA5 complex in the indicated
amount.
Unlabeled EAS could displace the labeled EAS at concentrations of 100 ng/ml or
greater.
Unlabeled Ephrin A1-F~ did not significantly displace labeled EAS (FIG 15B).
7. EQUIVALENTS
[00259] Those skilled in the art will recognize, or be able to ascertain using
no more
than routine experimentation, many equivalents to the specific embodiments of
the invention
described herein. Such equivalents are intended to be encompassed by the
following claims.
All publications, patents and patent applications mentioned in this
specification are herein
incorporated by reference into the specification to the same extent as if each
individual
publication, patent or patent application was specifically and individually
indicated to be
incorporated herein by reference.
-90-
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST L,E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional valumes please contact the Canadian Patent Office.