Note: Descriptions are shown in the official language in which they were submitted.
CA 02551057 2006-05-10
WO 2005!047239 PCT/EP2004/012393
1
Pentafluorosulfanyl benzoylguanidines, method for their production, their
use as medicaments or diagnostic agents and medicament containing the
same
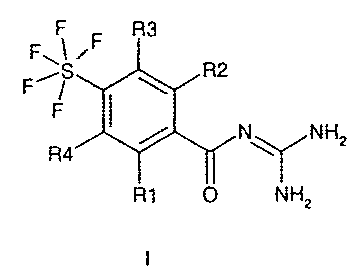
Pentafluorosulfanylbenzoylguanidines of the formula l
F R3
FBI iF
F~~ ~ R2
F
N~NH2
R4
R1 O NH2
in which R1 to R4 have the meanings indicated in the claims, and the
pharmaceutically acceptable salts thereof are substituted acylguanidines
+ +
and inhibit the cellular sodium-proton antiporker (Na /H exchanger, NHE).
Because of the NHE-inhibitory properties, the compounds of the formula I
and the pharmaceutically acceptable salts thereof are suitable for the
prevention and treatment of diseases caused by activation or activated
NHE, and of diseases caused secondarily by the NHE-related damage.
Compared with known compounds, the compounds of the invention are
distinguished by an extremely high activity in the inhibition of Na+/H+
exchange, and by improved ADMET properties. The xenobiotic structure (in
particular the introduction of the rather "unnatural/manmade" SF5
substituents) advantageously influences tissue distribution. This leads inter
alia to increased exposures in vivo. This involves no significant influence on
the absorption characteristics, and the high bioavailability of the
acylguanidines is retained.
In contrast to some acylguanidines described in the literature, the
compounds of the formula I described herein and their pharmaceutically
CA 02551057 2006-05-10
2
acceptable salts show no unwanted and disadvantageous saliduretic
properties.
The invention relates to pentafluorosulfanylbenzoylguanidines of the
formula I
F R3
F~I ~F
Fib ~ R2
F
R4 / ~NHz
R1 O NHZ
in which
R1 is hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms, alkoxy having 1,
2, 3 or 4 carbon atoms, F, CI, Br, I, -CN, NR5R6, -Op-(CH2)n-
(CF2)o-CF3 or -(SO,~,~,)q -(CH2)r(CF2)s-CF3~
R5 and R6
are, independently of one another, hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CF3;
I S m is zero, 1 or 2
n, o, p, q, r and s
are, independently of one another, zero or 1;
R2 is hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms, aikoxy having 1,
2, 3 or 4 carbon atoms, F, CI, Br, I, -CN, NR7R8,
-Ot-(CH2)u-(CF2)v-CF3 or -(SO~,v)x-(CH2)y-(CF2)z-CF3;
R7 and R8
are, independently of one another, hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CF3;
w is zero, 1 or 2
t, u, v,x,yandz
are, independently of one another, zero or 1;
CA 02551057 2006-05-10
3
R3 is CI, Br, I, -CN, -S02CH3, alkoxy having 1, 2, 3 or 4 carbon atoms,
NR9R10, -Oa-(CH2)b-(CF2)c-CF3, -(SOd)e-(CH2~-(CF2)g-CF3, alkyl
having 1, 2, 3, 4, 5 or 6 carbon atoms or cycloalkyl having 3, 4, 5, 6,
7 or 8 carbon atoms, in which 1, 2, 3 or 4 hydrogen atoms may be
replaced by fluorine atoms;
R9 and R10
are, independently of one another, hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CFg;
a, b and c
are, independently of one another, zero or 1;
d is zero, 1 or 2;
a is zero or 1;
f is zero, 1, 2, 3 or 4;
g is zero or 1;
or
R3 is -(CH2)h-phenyl or -O-phenyl,
in which the phenyl radicals are unsubstituted or substituted by 1, 2
or 3 radicals selected from the group consisting of F, CI, Br, I,
-Oj-(CHZ)k-CF3, alkoxy having 1, 2, 3 or 4 carbon atoms, alkyl
having 1, 2, 3 or 4 carbon atoms and -S02CH3;
j is zero or 1;
k is zero, 1, 2 or 3;
h is zero, 1, 2, 3 or 4;
or
R3 is -(CH2)aa-heteroaryl,
which is unsubstituted or substituted by 1, 2 or 3 radicals selected
from the group consisting of F, Cl, Br, !, -Obb-(CH2)cc-CF3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon
atoms and -S02CH3;
bb is zero or 1;
cc is zero, 1, 2 or 3;
CA 02551057 2006-05-10
as is zero, 1, 2, 3 or 4;
R4 is hydrogen, F, CI, Br, I, -CN, -S02CH3, alkoxy having 1, 2, 3 or 4
carbon atoms, NR11 R12, -Odd-(CH2)ee-(CF2~f-CF3; -(SOgg)hh-
(CH2)jj-(CF2)kk-CF3, alkyl having 1, 2, 3, 4, 5 or 6 carbon atoms or
cycloalkyl having 3, 4, 5, 6, 7 or 8 carbon atoms, in which 1, 2, 3 or 4
hydrogen atoms may be replaced by fluorine atoms;
R11 and R12
are, independently of one another, hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CF3;
dd, ee and ff
are, independently of one another, zero or 1;
gg is zero, 1 or 2;
hh is zero or 1;
jj is zero, 1, 2, 3 or 4;
kk is zero or 1;
or
R4 is -(CH2)II-Phenyl or -O-phenyl,
in which the phenyl radicals are unsubstituted or substituted by 1, 2
or 3 radicals selected from the group consisting of F, CI, Br, I,
-Omm-(CH2)nn-CF3. alkoxy having 1, 2, 3 or4 carbon atoms, alkyl
having 1, 2, 3 or 4 carbon atoms and -S02CH3;
mm is zero or 1;
nn is zero, 1, 2 or 3;
II is zero, 1, 2, 3 or 4;
or
R4 is -(CH2)oo-heteroaryl,
which is unsubstituted or substituted by 1, 2 or 3 radicals selected
from the group consisting of F, CI, Br, I, -Opp-(CH2)rrCF3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon
atoms and -S02CH3;
CA 02551057 2006-05-10
pp is zero or 1;
rr is zero, 1, 2 or 3;
0o is zero, 1, 2, 3 or 4;
and the pharmaceutically acceptable salts thereof.
5
Preference is given to compounds of the formula I, in which the meanings
are:
R1 hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms, alkoxy having 1,
2, 3 or 4 carbon atoms, F, CI, Br, I, -CN, NR5R6,
-Op-(CH2)n-(CF2)o-CF3 or -(SO,~n)q-(CH2)r(CF2)s-CF3>
R5 and R6
independently of one another hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CF3;
m zero, 1 or 2
n, o, p, q, r and s
independently of one another zero or 1;
R2 hydrogen or F;
R3 CI, Br, I, -CN, -S02CH3, alkoxy having 1, 2, 3 or 4 carbon atoms,
NR9R10, -Oa-(CH2)b-(CF2)c-CF3, -(SOd)e-(CH2)f-(CF2)g-CF3, alkyl
having 1, 2, 3, 4, 5 or 6 carbon atoms or cycloalkyl having 3, 4, 5, 6,
7 or 8 carbon atoms, in which 1, 2, 3 or 4 hydrogen atoms may be
replaced by fluorine atoms;
R9 and R10
independently of one another hydrogen, alkyl having 1,
2, 3 or 4 carbon atoms or -CH2-CF3;
a, b and c
independently of one another zero or 1;
d zero, 1 or 2;
a zero or 1;
f zero, 1, 2, 3 or 4;
g zero or 1;
or
CA 02551057 2006-05-10
6
R3 -(CH2)h-phenyl or -O-phenyl,
in which the phenyl radicals are unsubstituted or substituted by 1, 2
or 3 radicals selected from the group consisting of F, CI, Br, I,
-Oj-(CH2)k-CF3, alkoxy having 1, 2, 3 or 4 carbon atoms, alkyl
having 1, 2, 3 or 4 carbon atoms and -S02CH3;
j zero or 1;
k zero, 1, 2 or 3;
h zero, 1, 2, 3 or 4;
or
R3 -(CH2)aa-heteroaryl,
which is unsubstituted or substituted by 1, 2 or 3 radicals selected
from the group consisting of F, Cl, Br, I, -Obi-(CH2)cc-CF3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon
atoms and -S02CH3;
bb zero or 1;
cc zero, 1, 2 or 3;
as zero, 1, 2, 3 or 4;
R4 hydrogen or F;
and the pharmaceutically acceptable salts thereof.
Particular preference is given to compounds of the formula I, in which the
meanings ace:
R1 hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms, alkoxy having 1,
2, 3 or 4 carbon atoms, F, CI, Br, I, -CN, NR5R6, -O-CH2-CF3 or
-(SOm)q-(CH2)rCF3;
R5 and R6
independently of one another hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms or -CH2-CFg;
m zero, 1 or 2
q and r
independently of one another zero or 1;
CA 02551057 2006-05-10
7
R2 hydrogen or F;
R3 CI, Br, I, -CN, -S02CH3, alkoxy having 1, 2, 3 or 4 carbon atoms,
NR9R10, -O-CH2_CF3, -(SOd)e-CF3, alkyl having 1, 2, 3, 4, 5 or 6
carbon atoms or cycloalkyl having 3, 4, 5, 6, 7 or 8 carbon atoms, in
which 1, 2, 3 or 4 hydrogen atoms may be replaced by fluorine
atoms;
R9 and R10
independently of one another hydrogen, alkyl having 1,
2, 3 or 4 carbon atoms or -CH2-CF3;
d zero, 1 or 2;
a zero or 1;
or
R3 phenyl,
which is unsubstituted or substituted by 1, 2 or 3 radicals selected
from the group consisting of F, CI, Br, I, -Oj-(CH2)k-CF3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon
atoms and -S02CH3;
j zero or 1;
k zero, 1, 2 or 3;
or
R3 heteroaryl,
which is unsubstituted or substituted by 1, 2 or 3 radicals selected
from the group consisting of F, CI, Br, l, -Obb-(CH2)cc-CF3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon
atoms and -S02CH3;
bb zero or 1;
cc zero, 1, 2 or 3;
R4 hydrogen or F;
and the pharmaceutically acceptable salts thereof.
Very particular preference is given to compounds of the formula f in which
the meanings are.
CA 02551057 2006-05-10
8
R1 hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms, methoxy, ethoxy,
F, CI, NR5R6, -O-CH2-CF3 or -(SOm)q-(CH2),-CF3;
R5 and R6
independently of one another hydrogen, alkyl having 1,
2, 3 or 4 carbon atoms or -CH2-CFg;
m zero, 1 or 2
q and r
independently of one another zero or 1;
R2 hydrogen or F;
R3 CI, -CN, -S02CH3, methoxy, ethoxy, NR9R10, -O-CH2-CF3,
-(SOd)e-CF3, alkyl having 1, 2, 3, 4, 5 or 6 carbon atoms or
cycloalkyl having 3, 4, 5, 6 or 7 carbon atoms, in which 1, 2, 3 or 4
hydrogen atoms may be replaced by fluorine atoms;
R9 and R10
independently of one another hydrogen, methyl, ethyl
or -CHZ-CF3;
d zero, 1 or 2;
a zero or 1;
or
R3 phenyl,
which is unsubstituted or substituted by 1 or 2 radicals selected
from the group consisting of F, CI, -Oj-(CH2)k-CF3, methoxy,
ethoxy, alkyl having 1, 2, 3 or 4 carbon atoms and -S02CH3;
j and k
independently of one another zero or 1;
or
R3 heteroaryl,
which is unsubstituted or substituted by 1 or 2 radicals selected from
the group consisting of F, CI, -Obb-(CH2)cc-CF3, methoxy, ethoxy,
alkyl having 1, 2, 3 or 4 carbon atoms and -S02CH3;
bb and cc
CA 02551057 2006-05-10
9
independently of one another zero or 1;
R4 hydrogen or F;
and the pharmaceutically acceptable salts thereof.
In one embodiment, preference is given in this connection to the
compounds of the formula I in which R1 is described by hydrogen, alkyl
having 1, 2, 3 or 4 carbon atoms, alkoxy having 1, 2, 3 or 4 carbon atoms,
F, CI, Br, I, -CN, NR5R6, where R5 and R6 are, independently of one
another, hydrogen, alkyl having 1, 2, 3 or 4 carbon atoms or -CH2-CF3, or
-O-CH2-CF3 or -(SOm)q-(CH2)~CF3, where m is zero, 1 or 2, and q and r
are, independently of one another, are zero or 1; particular preference is
given to compounds in which R1 is described by hydrogen, alkyl having 1,
2, 3 or 4 carbon atoms, methoxy, ethoxy, F, CI, NR5R6, where R5 and R6,
are independently of one another, hydrogen, alkyl having 1, 2, 3, or 4
carbon atoms or -CH2-CF3, -O-CH2-CF3 or -(SO,n)q-(CH2)~CF3, where m
is zero, 1 or 2, and q and r are, independently of one another, zero or 1;
very particular preference is given to compounds in which R1 is described
by hydrogen, methyl, ethyl, CF3-CH2-O-, F, CI or CF3. In a further
embodiment, preference is given to compounds in which R1 is described
by hydrogen, methyl or ethyl, in particular methyl or ethyl.
In a further embodiment, preference is given to compounds of the formula I
in which R2 is described by hydrogen or F; particular preference is given to
compounds in which R2 is described by hydrogen.
In a further embodiment, preference is given to compounds of the formula I
in which R3 is described by CI, -CN, -S02CH3, methoxy, ethoxy, NR9R10,
where R9 and R10 are, independently of one another, hydrogen, methyl,
ethyl or -CH2-CF3, or -O-CH2-CF3, -(SOd)e-CF3, where d is zero, 1 or 2,
and a is zero or 1, alkyl having 1, 2, 3, 4, 5 or 6 carbon atoms or cycloalkyl
having 3, 4, 5, 6 or 7 carbon atoms, in which 1, 2, 3 or 4 hydrogen atoms
may be replaced by fluorine atoms, phenyl which is unsubstituted or
CA 02551057 2006-05-10
substituted by 1,2 or 3 radicals selected from the group consisting of F, CI,
Br, I, -Oj-(CH2)k-CF3, where j is zero or 1 and k is zero, 1, 2 or 3, alkoxy
having 1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon atoms
and -S02CH3, or heteroaryl which is unsubstituted or substituted by 1, 2 or
5 3 radicals selected from the group consisting of F, CI, Br, I, -Obb-
(CH2)~~-CFg, where bb is zero or 1 and cc is zero, 1, 2 or 3, alkoxy having
1, 2, 3 or 4 carbon atoms, alkyl having 1, 2, 3 or 4 carbon atoms and
-S02CH3; particular preference is given to compounds in which R3 is
described by Cl, -CN, -S02CH3, methoxy, ethoxy, NR9R10, where R9 and
10 R10 are, independently of one another, hydrogen, methyl, ethyl or
-CH2-CF3, or -O-CH2-CF3, -(SOd)e-CF3, where d is zero, 1 or 2, and a is
zero or 1, alkyl having 1, 2, 3, 4, 5 or 6 carbon atoms or cycloalkyl having
3,
4, 5, 6 or 7 carbon atoms, in which 1, 2, 3 or 4 hydrogen atoms may be
replaced by fluorine atoms, phenyl which is unsubstituted or substituted by
1-2 radicals selected from the group consisting of F, CI, -Oj-(CH2)k-CF3,
where j and k are, independently of one another, zero or 1, methoxy,
ethoxy, alkyl having 1, 2, 3 or 4 carbon atoms and -S02CH3, or heteroaryl
which is unsubstituted or substituted by 1-2 radicals selected from the
group consisting of F, CI, -Obb-(CH2)cc-CF3, where bb and cc are,
independently of one another, zero or 1, methoxy, ethoxy, alkyl having 1, 2,
3 or 4 carbon atoms and -S02CH3; very particular preference is given to
compounds in which R3 is described by CI, -CN or -SOZCH3.
In a further embodiment, preference is given to compounds of the
formulae I in which R4 is described by hydrogen and F, with particular
preference for compounds in which R4 is described by hydrogen.
In a further embodiment, preference is given to compounds of the formula I
in which p, t, a and dd are, independently of one another, 1.
CA 02551057 2006-05-10
11
If the substituents R1 to R4 contain one or more centers of asymmetry,
these may independently of one another have both the S and the R
configuration. The compounds may be in the form of optical isomers, of
diastereomers, of racemates or of mixtures thereof.
S
The present invention encompasses all tautomeric forms of the compounds
of the formula I.
Alkyl radicals may be straight-chain or branched. This also applies if they
carry substituents or occur as substituents of other radicals, for example in
fluoroalkyl radicals or alkoxy radicals. Examples of alkyl radicals are
methyl, ethyl, n-propyl, isopropyl (= 1-methylethyl), n-butyl, isobutyl (= 2-
methylpropyl), sec-butyl (= 1-methylpropyl), tert-butyl (= 1,1-dimethylethyl),
n-pentyl, isopentyl, tert-pentyl, neopentyl and hexyl. Preferred alkyl
radicals
1 S are methyl, ethyl, n-propyl and isopropyl. One or more, for example 1, 2,
3,
4 or 5, hydrogen atoms in alkyl radicals may be replaced by fluorine atoms.
Examples of such fluoroalkyl radicals are trifluoromethyl, 2,2,2-
trifluoroethyl
and pentafluoroethyl. Substituted alkyl radicals may be substituted in any
positions. Examples of cycloalkyl radicals are cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl. One or more, for example
1, 2, 3 or 4, hydrogen atoms in cycloalkyl radicals may be replaced by
fluorine atoms. Substituted cycloalkyl radicals may be substituted in any
positions.
2S Phenyl radicals may be unsubstituted or be substituted one or more times,
for example once, twice or three times, by identical or different radicals. If
a
phenyl radical is substituted, it preferably has one or two identical or
different substituents. This likewise applies to substituted phenyl radicals
in
groups such as, for example, phenylalkyl or phenyloxy. The substituent in
monosubstituted phenyl radicals may be in position 2, position 3 or position
4. Disubstituted phenyl may be substituted in the 2,3 position, 2,4 position,
2,5 position, 2,6 position, 3,4 position or 3,5 position. The substituents in
trisubstituted phenyl radicals may be in the 2,3,4 position, 2,3,5 position,
2,4,5 position, 2,4,6 position, 2,3,6 position or 3,4,5 position.
CA 02551057 2006-05-10
12
Heteroaryl radicals are aromatic ring compounds in which one or more ring
atoms are oxygen atoms, sulfur atoms or nitrogen atoms, e.g. 1, 2 or 3
nitrogen atoms, 1 or 2 oxygen atoms, 1 or 2 sulfur atoms or a combination
of various heteroatoms. The heteroaryl radicals may be attached by all
positions, for example by the 1 position, 2 position, 3 position, 4 position,
5
position, 6 position, 7 position or 8 position. Heteroaryl radicals may be
unsubstituted or be substituted one or more times, for example once, twice
or three times, by identical or different radicals. This applies likewise to
heteroaryl radicals such as, for example, in the radical heteroarylalkyl.
Examples of heteroaryl are furanyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl,
triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyridyl,
pyrazinyl, pyrimidinyl, pyridazinyl, indolyl, indazolyl, quinolyl,
isoquinolyl,
phthalazinyi, quinoxalinyl, quinazolinyl and cinnolinyl.
Heteroaryl radicals are, in particular, 2- or 3-thienyl, 2- or 3-furyl, 1-, 2-
or 3-
pyrrolyl, 1-, 2-, 4- or 5-imidazolyl, 1-, 3-, 4- or 5-pyrazolyl, 1,2,3-triazol-
1-, -
4- or -5-yl, 1,2,4-triazol-1-, -3- or -5-yl, 1- or 5-tetrazolyl, 2-, 4- or 5-
oxazolyl,
3-, 4- or 5-isoxazolyl, 1,2,3-oxadiazol-4- or -5-yl, 1,2,4-oxadiazol-3- or -5-
yl,
1,3,4-oxadiazol-2-yl or -5-yl, 2-, 4- or 5-thiazolyl, 3-, 4- or 5-
isothiazolyl,
1,3,4-thiadiazol-2- or -5-yl, 1,2,4-thiadiazol-3- or -5-yl, 1,2,3-thiadiazol-4-
or
-5-yl, 2-, 3- or 4-pyridyl, 2-, 4-, 5- or 6-pyrimidinyl, 3- or 4-pyridazinyl;
pyrazinyl, 1-, 2-, 3-, 4-, 5-, 6- or 7-indolyl, 1-, 2-, 4- or 5-
benzimidazolyl, 1-,
3-, 4-, 5-, 6- or 7-indazolyl, 2-, 3-, 4-, 5-, 6-, 7- or 8-quinolyl, 1-, 3-, 4-
, 5-, 6-,
7- or 8-isoquinolyl, 2-, 4-, 5-, 6-, 7- or 8-quinazolinyl, 3-, 4-, 5-, 6-, 7-
or 8-
cinnolinyl, 2-, 3-, 5-, 6-, 7- or 8-quinoxalinyl, 1-, 4-, 5-, 6-, 7- or 8-
phthalazinyl. Also encompassed are the corresponding N-oxides of these
compounds, i.e. for example 1-oxy-2-, 3- or 4-pyridyl.
Particularly preferred heteroaromatic radicals are 2- or 3-thienyl, 2- or 3-
furyl, 1-, 2- or 3-pyrrolyl, 1-, 2-, 4- or 5-imidazolyl, 2-, 3-, 4-, 5-, 6-, 7-
or 8-
quinolyl, 1-, 3-, 4- or 5-pyrazolyl, 2-, 3- or 4-pyridyl, 2- or 3-pyrazinyl, 2-
, 4-,
5- or 6-pyrimidinyl and 3- or 4-pyridazinyl.
CA 02551057 2006-05-10
13
The invention further relates to a process for preparing the compounds of
the formula I which comprises reacting a compound of the formula II
F R3
FBI iF
F~~ ~ R2
F
/ L
R4
R1 O
in which R1 to R4 have the stated meaning, and L is a leaving group which
can undergo nucleophilic substitution, with guanidine.
The activated acid derivatives of the formula II in which L is an alkoxy,
preferably a methoxy, group, a phenoxy group, phenylthio, methylthio,
2-pyridylthio group, a nitrogen heterocycfe, preferably 1-imidazolyl, are
advantageously obtained in a manner known to those skilled in the art
from the underlying carbonyl chlorides (formula II; L = CI), which in turn can
themselves be prepared in a known manner from the underlying carboxylic
acids (formula II; L = OH), for example using thionyl chloride.
Besides the carbonyl chlorides of the formula II (L = CI) it is also possible
to
prepare other activated acid derivatives of the formula II in a known manner
directly from the underlying benzoic acids (formula II; L = OH), such as the
methyl esters of the formula II with L = OCH3 by treatment with gaseous
HCI in methanol, the imidazolides of the formula II by treatment with
carbonyldiimidazole, the mixed anhydrides of the formula II by treatment
with CI-COOC2H5 or tosyl chloride in the presence of triethylamine in an
inert solvent, and activations of benzoic acids with
dicyclohexylcarbodiimide (DCC) or with O-[(cyano(ethoxycarbonyl)-
methylene)amino]-1,1,3,3-tetramethyluronium tetrafluoroborate ("TOTU")
are also possible. A number of suitable methods for preparing activated
carboxylic acid derivatives of the formula II are indicated in J. March,
Advanced Organic Chemistry, third edition (John Wiley & Sons, 1985,
page 350), indicating source literature.
CA 02551057 2006-05-10
14
Reaction of an activated carboxylic acid derivative of the formula II with
guanidine preferably takes place in a known manner in a protic or aprotic
polar but inert organic solvent. Those which have proved suitable for the
reaction of the methyl benzoates (formula II; L = OCH3) with guanidine are
methanol, isopropanol or THF at temperatures from 20°C to the boiling
point of these solvents- Most reactions of compounds of the formula II with
salt-free guanidine are, for example, carried out in aprotic inert solvents
such as THF, dimethoxyethane, dioxane. However, it is also possible to
use water in the presence of a base such as, for example, NaOH as
solvent in the reaction of compounds of the formula 11 with guanidine.
If L is CI, it is advantageous to add an acid scavenger, for example in the
form of excess guanidine, to bind the hydrohalic acid.
The compounds of the formula Il can be prepared as follows, by
CA 02551057 2006-05-10
F F H F j,F H F F H
F~S~ R2 F~S ~ R2 F~S~ R2
F I \ F F I \
i
R4 / N~ ~ a R4 / NHZ b R4 i / NHZ
I _.
H O H X
III IV V
S~F ~ R2 F S~F ~ R2 F F'F H
\ R2
F
-- Y
R4 ~ / NHZ d R4 / Y a R4 i / ~ f
R1 R~ R1 \ N
VI VII
VIII
ow ,-O
F 1 ~F H F t ~F N F F ~F NHZ
F~S \ R2 ~ F~S ~ R2 F~S ~ R2
F, I --~ I
/F F
R4 / O 9 R4 / O h R4 i / O I
R1 OH R1 OH R1 OH
IX X XI
F R3
F F R3
F~S' \ R2 F~S~F \ R2
i
R4 i / O k F / O
R4
R1 OH R1 L
XII II
a) reducing a 4-nitrophenylsulfur pentafluoride derivative of the formula III
to the amine of the formula IV,
b) halogenating the compound of the formula IV in the ortho position to the
5 amino group with a halogenating agent to give the compound of the
formula V,
c) replacing the halogen substituent in the compound of the formula V with
a suitable nucleophile or an organoelement compound, for example an
alkylboron compound, where appropriate with catalysis, by a substituent R1
10 and
d) replacing the amino function in the compound of the formula VI by a
halogen substituent,
CA 02551057 2006-05-10
16
e) replacing the halogen substituent in the compound of the formula VII by
a nitrite function,
f) hydrolyzing the nitrite function of the compound of the formula VIII to the
carboxylic acid,
g) nitrating the compound of the formula lX in the ortho position to the
pentafluorosulfanyl group to give the compound of the formula X,
h) reducing the nitro compound of the formula X to the aniline,
i) replacing the amino function in the compound of the formula XI by R3
using a suitable nucleophile
and
k) converting the compound of the formula XII into the compound of the
formula II, where in the compounds of the formulae II, III, IV, V, VI, VII,
VIII,
IX, X, XI and XII
R1 to R4 are defined as in formula I
L is defined as in formula II and
X and Y are, independently of one another, F, CI, Br or I.
The procedure for preparing the compounds of the formula II is initially in
step a to convert the compounds of the formula III by methods known in
principle for the reduction of aromatic vitro compounds to aromatic amines
into compounds of the formula IV. Such methods are described, for
example, in: R.C. Larock, Comprehensive Organic Transformations: A
Guide to Functional Group Preparations, VCH Publishers, New York,
Weinheim, 1999, 821-828 and the literature cited therein.
Subsequently (step b), the compounds of the formula IV are dissolved in an
organic solvent A and reacted with a halogenating agent, for example a
brominating agent. The reaction temperature in this case is generally from
-30°C to +150°C, preferably 0°C to 40°C. The
reaction time is generally
from 10 min to 20 h, depending on the composition of the mixture and the
chosen temperature range. The resulting reaction mixture can be worked
up by subsequent filtration through a layer of silica gel, washing with
organic solvent A and, after removal of the solvent in vacuo, purifying the
product by conventional purification methods such as recrystallization,
distillation or chromatography.
From 0.1 to 10 mot of the compound of the formula IV for example are
CA 02551057 2006-05-10
17
dissolved in 1000 ml of organic solvent A. For example, from 0.8 to 1.2
equivalents of the halogenating agent are used for 1 mol of the compound
of the formula IV to be halogenated.
The term "halogenating agent" means for example elemental halogens,
halogen-amine complexes, cyclic and acyclic N-halogenated carboxamides
and -imides, and ureas, as described, for example, in R.C. Larock,
Comprehensive Organic Transformations: A Guide to Functional Group
Preparations, VCH Publishers, New York, Weinheim, 1999, 619-628, and
the literature cited therein or M.B. Smith and J. March, March's Advanced
Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New
York, 2001, 704-707, and the literature cited therein, such as, for example,
N-bromosuccinimide, N-chlorosuccinimide, HBr in H2S04 or 1,3-dibromo-
5,5-dimethylimidazolidine-2,4-dione. The term "brominating agent" means,
for example, elemental bromine, bromine-amine complexes, cyclic and
acyclic N-brominated carboxamides and -imides, and ureas, as described,
for example, in R.C. Larock, Comprehensive Organic Transformations: A
Guide to Functional Group Preparations, VCH Publishers, New York,
Weinheim, 1999, 622-624, and the literature cited therein or M.B. Smith
and J. March, March's Advanced Organic Chemistry: Reactions,
Mechanisms, and Structure, Wiley, New York, 2001, 704-707, and the
literature cited therein, for example N-bromosuccinimide, HBr in H2S04 or
1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione, the latter being able to
transfer 2 bromine atoms per molecule.
The term "organic solvent A" preferably means aprotic solvents such as, for
example, dichloromethane, chloroform, tetrachloromethane, pentane,
hexane, heptane, octane, benzene, toluene, xylene, chlorobenzene, 1,2-
dichloroethane, trichloroethylene or acetonitrile.
Any HX produced in the reaction can be trapped by organic or inorganic
bases.
In step c, the compounds of the formula V are subsequently dissolved in an
organic solvent B and reacted with a nucleophile R1 or an element
compound comprising the substituent R1 to give compounds of the formula
VI. It is possible in this case to add a base A and to add a catalyzing metal
salt A.
The reaction temperature in this case is generally between -20°C
and
+150°C, preferably between 30°C and 100°C. The reaction
time is
generally from 0.5 h to 20 h, depending on the composition of the mixture
and the chosen temperature range. The resulting reaction mixture can be
CA 02551057 2006-05-10
18
worked up by subsequent filtration through a layer of silica gel, washing
with an organic solvent B and, after removal of the solvent in vacuo,
purifying the product by conventional purification processes such as
recrystallization, chromatography, for example on silica gel, distillation or
steam distillation.
From 0.1 to 10 mol of the compound of the formula V for example are
dissolved in 1000 ml of organic solvent B. For example, from 0.8 to 3
equivalents of the nucleophile R1 or of the element compound comprising
the substituent R1 are used for 1 mol of the starting compound of the
formula V.
The term "nucleophile R1 " means compounds which result on
deprotonation of a compound R1-H with strong bases such as, for
example, alkyl- or aryllithium compounds, organomagnesium compounds,
alcoholates or lithium diisopropylamide.
"Organoelement compounds comprising the substituent R1" mean for
example organolithium compounds R1-Li, organomagnesium compounds
R1-Mg-Hal, with Hal = Cl, Br, I, organoboron compounds such as
R1-B(OH)2, R1-boronic esters such as, for example,
R1-~B~Q
t
O
R1-boronic anhydrides such as, for example,
R1.,B~O,~B~R1
f I
O,~B~O
1
R1
or organozinc compounds R1-Zn-Z, with Z = Cl, Br, I.
The term "base A" means bases like those used as auxiliary bases in
cross-coupling reactions and mentioned for example in A. Suzuki et al.,
Chem. Rev. 1995, 95, 2457-2483 or M. Lamaire et al., Chem. Rev. 2002,
702, 1359-1469 or S.P. Stanforth, Tetrahedron 1998, 54, 263-303 and the
literature cited therein in each case, for example Na2C03, Cs2C03, KOH,
NaOH, K3P04, N(ethyl)3.
The term "organic solvent B" means erotic or aprotic solvents such as
diethyl ether, dimethoxyethane, THF, alcohols, water or mixtures thereof. In
one embodiment, mixtures with water are preferred.
CA 02551057 2006-05-10
19
The term "catalyzing metal salt A" means inter alia Pd and Ni catalysts like
those used for Suzuki and Negishi reactions and described for example in
A. Suzuki et al., Chem. Rev. 1995, 95, 2457-2483 or M. Lamaire et al.,
Chem. Rev. 2002, 102, 1359-1469 or S.P. Stanforth, Tetrahedron 1998,
S 54, 263 or G.C. Fu et al., J. Am. Chem. Soc. 2001, 123, 10099 or G.C. Fu
et al., J. Am. Chem. Soc. 2002, 124, 13662 and the literature cited therein
in each case, including the added ligands such as Pd(OAc)2, PdCl2(dppf)
or Pd2(dba)3.
In step d, the compounds of the formula VI are subsequently converted into
the compounds of the formula VII by a diazotization-halogenation process
with a diazotizing-halogenating agent, for example with a diazotizing-
brominating agent, as described for other aromatic amines to replace the
amine function by a halogen function for example in M.B. Smith and
1 S J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms,
and Structure, Wiley, New York, 2001, 935-936 or R.C. Larock,
Comprehensive Organic Transformations: A Guide to Functional Group
Preparations, VCH Publishers, New York, Weinheim, 1999, 678-679 and
the literature cited therein, for example by the Sandmeyer or Gattermann
reaction. The process of M. Doyle et al., J. Org. Chem. 1977, 42, 2426 or of
S. Oae et al., Bull. Chem. Soc. Jpn. 1980, 53, 1065 is preferred.
In step e, the compounds of the formula VII are reacted in a solvent C with
a cyanidating agent, for example with addition of a catalyzing metal salt B.
2S The reaction temperature is generally from 20°C to 200°C,
preferably 80°C
to 150°C. The reaction time is generally from 1 h to 20 h, depending on
the
composition of the mixture and the chosen temperature range. The
resulting reaction mixtures can be filtered with suction through a layer of
silica gel or kieselguhr and the filtrate can be worked up by aqueous
extraction. After evaporation of the solvent in vacuo, the compound of the
formula VIII is purified by conventional purification processes such as
recrystallization, chromatography on silica gel, distillation or steam
distillation.
From 0.1 to 10 mol of the compound of the formula VII for example are
3S dissolved in 1000 ml of organic solvent C. For example, from 1 to 10
equivalents of the cyanidating agent are used for 1 mol of the compounds
having the formula VII to be reacted.
The term "cyanidating agent" means, for example, alkali metal cyanides or
Zn(CN)2 either alone or mixed with metallic zinc, preferably in the form of
CA 02551057 2006-05-10
zinc dust.
The term "organic solvent C" preferably means aprotic polar solvents such
as, for example, DMF, dimethylacetamide, NMP, DMSO.
The term "catalyzing metal salt B" means inter alia Pd and Ni catalysts like
5 those employed in Suzuki reactions and described for example in A. Suzuki
et al., Chem. Rev. 1995, 95, 2457-2483 or M. Lamaire et al., Chem. Rev.
2002, 102, 1359-1469 or S.P. Stanforth, Tetrahedron 1998, 54, 263 and
the literature cited therein, for example PdCl2(dppf), Pd(OAc)2, Pd2(dba)3.
10 The resulting compounds of the formula VIII are subsequently hydrolyzed
in step f to the carboxylic acids of the formula IX, for example in the
presence of a base. This can take place by processes known to the skilled
worker for hydrolyzing aromatic nitrites, as described, for example, in
R.C. Larock, Comprehensive Organic Transformations: A Guide to
15 Functional Group Preparations, VCH Publishers, New York, Weinheim,
1999, 1986-1987 or M.B. Smith and J. March, March's Advanced Organic
Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 2001,
1179-1180 and the literature cited therein.
20 In step g, compounds of the formula IX are nitrated with a nitrating agent
as
described, for example, in Houben-Weyl, Methoden der organischen
Chemie 4th edition, Organo-Stickstoff-Verbindungen IV, part 1, Georg
Thieme Verlag Stuttgart 1992, pages 262-341.
In step h, the vitro compounds of the formula X are converted into
compounds of the formula XI by methods known in principle for reducing
aromatic vitro compounds to aromatic amines. Such methods are
described for example in: R.C. Larock, Comprehensive Organic
Transformations: a Guide to Functional Group Preparations, VCH
Publishers, New York, Weinheim, 1999, 821-828 and the literature cited
therein.
In step i, the anilines of the formula XI are converted by the diazotization -
replacement route into the compounds of the formula XI1 with replacement
of the amine group by R3. Such methods are known to the skilled worker
and are described for example in Houben-Weyl, Methoden der organischen
Chemie 4th edition, Organo-Stickstoff-Verbindungen I, part 2, Georg
Thieme Verlag Stuttgart 1990, pages 1087-1136 and the references cited
therein.
CA 02551057 2006-05-10
21
For example, an aniline of the formula XI can be converted by the
diazotization-replacement route into a sulfochloride of the formula XII
(R3 = S02C1) as described, for example, in Houben-Weyl, Methoden der
organischen Chemie 4th edition, Organo-Schwefel-Verbindungen, part 2,
Georg Thieme Verlag Stuttgart 1985, pages 1069-1070.
In step k, the compounds of the formula XII are derivatized to the
compounds of the formula If by methods known to the skilled worker and as
described above.
It is possible in this step for example for the sulfochlorides of the formula
XII (R3 = S02C1) to be converted initially into the corresponding sulfinic
acids (as described for example in Houben-Weyl, Methoden der
organischen Chemie 4th edition, Organo-Schwefel-Verbindungen, part 1,
Georg Thieme Verlag Stuttgart 1985, pages 620-621 and Houben-Weyl,
Methoden der organischen Chemie, Schwefel-, Selen-, Tellur-
Verbindungen, Georg Thieme Verlag Stuttgart 1955, pages 304-309) and
subsequently alkylated to give the methyl sulfone as described for example
in Houben-Weyl, Methoden der organischen Chemie 4th edition, Organo-
Schwefel-Verbindungen, part 2, Georg Thieme Verlag Stuttgart 1985,
pages 1145-1149. Simultaneous esterification of the carboxylic acid to the
methyl ester takes place.
Compounds of the formula I in which R1 is hydrogen are prepared by
carrying out the synthesis without steps b and c.
Compounds of the formula I in which R3 is NR9R10 are prepared by
carrying out the synthesis without step i.
Functional groups in the starting compounds may also be present in
protected form or in the form of precursors, and then be converted into the
desired groups in the compounds of the formula II prepared by the process
described above. Corresponding protective group techniques are known to
the skilled worker.
It is likewise possible for appropriate functional groups to be derivatized by
methods known to the skilled worker. For example, compounds in which R3
is NH2 can be converted by reaction with appropriate alkyl halides or
2,2,2-trifluoroethyl halides, for example methyl iodide, ethyl iodide or
2,2,2-trifluoroethyl iodide, into compounds in which R3 is NR9R10, where
R9 and R10 are, independently of one another, hydrogen, alkyl having 1, 2,
3 or 4 carbon atoms or -CH2-CF3 and are not both simultaneously
hydrogen.
CA 02551057 2006-05-10
22
Pentafluorosulfanylbenzoylguanidines of the formula I are generally weak
bases and ace able to bind acids to form salts. Suitable acid addition salts
are salts of all pharmaceutically acceptable acids, for example halides, in
particular hydrochlorides, lactates, sulfates, citrates, tartrates, acetates,
phosphates, methylsulfonates, p-toluenesulfonates.
The compounds of the formula I are substituted acylguanidines and inhibit
the cellular sodium-proton antiporter (Na+/H+ exchanger, NHE), in
particular the subtype NHE-1.
Because of the NHE-inhibitory properties, the compounds of the formula 1
and/or the pharmaceutically acceptable salts thereof are suitable for the
prevention and treatment of diseases caused by activation of or by an
activated NHE, and of diseases caused secondarily by the NHE-related
damage.
The compounds of the formula I may also be used for treating and
preventing diseases by the NHE being only partially inhibited, for example
by use of a low dosage.
Since NHE inhibitors predominantly act via their effect on cellular pH
regulation, they can generally be combined beneficially with other
compounds which regulate the intracellular pH, with suitable combination
partners being inhibitors of the carbonic anhydrase enzyme group,
inhibitors of systems transporting bicarbonate ions, such as of the sodium
bicarbonate cotransporter (NBC) or of the sodium-dependent chloride-
bicarbonate exchanger (NCBE), and NHE inhibitors with inhibitory effect on
other NHE subtypes, because it is possible through them to enhance or
modulate the pharmacologically relevant pH-regulating effects of the NHE
inhibitors described herein.
The use of the compounds of the invention relates to the prevention and
treatment of acute and chronic diseases in veterinary and human medicine.
CA 02551057 2006-05-10
23
Thus, the NHE inhibitors of the invention are suitable for the treatment of
diseases caused by ischemia and by reperfusion.
The compounds described herein are suitable because of their
S pharmacological properties as antiarrhythmic medicaments.
Owing to their cardioprotective component, the NHE inhibitors are
outstandingly suitable for infarction prophylaxis and infarction treatment
and for the treatment of angina pectoris, in which cases they also
preventively inhibit or greatly reduce the pathophysiological processes
associated with the development of ischemia-induced damage, in particular
in the triggering of ischemia-induced cardiac arrhythmias. Because of their
protective effects against pathological hypoxic and ischemic situations, the
compounds of the formula I and/or the pharmaceutically acceptable salts
thereof used according to the invention can, because of inhibition of the
cellular Na+/H+ exchange mechanism, be used as medicaments for the
treatment of all acute or chronic ischemia-induced damage or diseases
induced primarily or secondarily thereby.
This also relates to their use as medicaments for surgical interventions.
Thus, the compounds can be used during organ transplantations, it being
possible to use the compounds both to protect the organs in the donor
before and during the removal, to protect removed organs for example
during treatment with or storage thereof in physiological bath liquids, and
during transference to the recipient organism.
The compounds of the invention are likewise valuable medicaments with a
protective effect when performing angioplastic surgical interventions, for
example on the heart as well as on peripheral organs and vessels.
The compounds of the invention may also be used when performing
bypass operations, for example bypass operations on coronary vessels and
in Coronary Artery Bypass Graft (CABG).
CA 02551057 2006-05-10
24
Depending on their activity with regard to ischemia-induced damage, the
compounds of the invention I may similarly be used in resuscitation after a
cardiac arest.
The compounds of the invention are of interest for medicaments for life-
threatening arrhythmias. Ventricular fibrillation is terminated and the
physiological sinus rhythm of the heart is restored.
Since NHE1 inhibitors of human tissue and organs, especially the heart,
protect effectively not only against damage caused by ischemia and
reperfusion but also against the cytotoxic effect of medicaments like those
used in particular in cancer therapy and the therapy of autoimmune
diseases, combined administration with compounds of the formula I and/or
the pharmaceutically acceptable salts thereof is suitable for inhibiting the
cytotoxic, especially cardiotoxic, side effects of said compounds. The
reduction in the cytotoxic effects, especially the cardiotoxicity, resulting
from comedication with NHE1 inhibitors makes it additionally possible to
increase the dose of the cytotoxic therapeutic agents and/or to prolong the
medication with such medicaments. The therapeutic benefits of such a
cytotoxic therapy can be considerably increased by combination with NHE
inhibitors.
In addition, the NHE1 inhibitors of the invention of the formula I and/or the
pharmaceutically acceptable salts thereof can be used when there is heart-
damaging overproduction of thyroid hormones, thyrotoxicosis, or on
external supply of thyroid hormones. The compounds of the formula I
and/or the pharmaceutically acceptable salts thereof are thus suitable for
improving therapy with cardiotoxic medicaments.
In accordance with their protective effect against ischemia-induced
damage, the compounds of the invention are also suitable as medicaments
for the treatment of ischemias of the nervous system, especially of the
central nervous system, being suitable for example for the treatment of
stroke or of cerebral edema.
CA 02551057 2006-05-10
The compounds of the formula I and/or the pharmaceutically acceptable
salts thereof are also suitable for the therapy and prophylaxis of diseases
and disorders induced by overexcitability of the central nervous system, in
particular for the treatment of epileptic disorders, centrally induced clonic
5 and tonic spasms, states of psychological depression, anxiety disorders
and psychoses. In these cases it is possible to use the NHE inhibitors
described herein alone or in combination with other substances with
antiepileptic activity or antipsychotic active ingredients, or carbonic
anhydrase inhibitors, for example with acetazolamide, and with other
10 inhibitors of NHE or of the sodium-dependent chloride-bicarbonate
exchanger (NCBE).
The compounds used according to the invention of the formula I and/or the
pharmaceutically acceptable salts thereof are additionally likewise suitable
1 S for the treatment of types of shock such as, for example, of allergic,
cardiogenic, hypovolemic and bacterial shock.
The compounds of the formula I and/or the pharmaceutically acceptable
salts thereof can likewise be used for the prevention and treatment of
20 thrombotic disorders because they, as NHE inhibitors, are able to inhibit
platelet aggregation themselves. They are additionally able to inhibit or
prevent the excessive release, occurring after ischemia and reperfusion, of
mediators of inflammation and coagulation, especially of von Willebrand
factor and of thrombogenic selectin proteins. It is thus possible to reduce
25 and eliminate the pathogenic effect of significant thrombogenic factors.
The
NHE inhibitors of the present invention can therefore be combined with
other anticoagulant andlor thrombolytic active ingredients such as, for
example, recombinant or natural tissue plasminogen activator,
streptokinase, urokinase, acetylsalicylic acid, thrombin antagonists, factor
Xa antagonists, medicinal substances with fibrinolytic activity, thromboxane
receptor antagonists, phosphodiesterase inhibitors, factor Vlla antagonists,
clopidogrel, ticlopidine etc. Combined use of the present NHE inhibitors
with NCBE inhibitors and/or with inhibitors of carbonic anhydrase such as,
for example, with acetazolamide, is particularly beneficial.
CA 02551057 2006-05-10
26
NHE1 inhibitors are additionally distinguished by a strong inhibitory effect
on the proliferation of cells, for example fibroblast proliferation and the
proliferation of smooth vascular muscle cells. The compounds of the
formula I and/or the pharmaceutically acceptable salts thereof are therefore
suitable as valuable therapeutic agents for diseases in which proliferation
represents a primary or secondary cause, and can therefore be used as
antiatherosclerotics, agents for chronic renal failure, cancers.
It was possible to show that cell migration is inhibited by NHE inhibitors.
The compounds of the formula I and/or the pharmaceutically acceptable
salts thereof are therefore suitable as valuable therapeutic agents for
dieases in which cell migration represents a primary or secondary cause,
such as, for example, cancers with a pronounced tendency to metastasis.
IS
NHE1 inhibitors are further distinguished by a retardation or prevention of
fibrotic disorders. Compounds of the formula I and/or the pharmaceutically
acceptable salts thereof are thus suitable as agents for the treatment of
cardiac fibroses, and of pulmonary fibrosis, hepatic fibrosis, renal fibrosis
and other fibrotic disorders. They can thus be used for the treatment of
organ hypertrophies and hyperplasias, for example of the heart and the
prostate. They are therefore suitable for the prevention and treatment of
heart failure (congestive heart failure = CHF) and for the treatment and
prevention of prostate hyperplasia or prostate hypertrophy.
Since there is significant elevation in NHE in essential hypertensives, the
compounds of the formula I and/or the pharmaceutically acceptable salts
thereof are suitable for the prevention and treatment of high blood pressure
and for the treatment of cardiovascular disorders. In these cases they can
be used alone or with a suitable combination and formulation partner for
the treatment of high blood pressure and for the treatment of
cardiovascular disorders. Thus, for example, one or more diuretics with a
thiazide-like action, loop diuretics, aldosterone and pseudoaldosterone
antagonists, such as hydrochlorothiazide, indapamide, polythiazide,
CA 02551057 2006-05-10
27
furosemide, piretanide, torasemide, bumetanide, amiloride, triamterene,
spironolactone or eplerone, can be combined. The NHE inhibitors of the
present invention can further be used in combination with calcium channel
blockers such as verapamil, diltiazem, amlodipine or nifedipine, and with
ACE inhibitors such as, for example, ramipril, enalapril, lisinopril,
fosinopril
or captopril. Further beneficial combination partners are also beta-blockers
such as metoprolol, albuterol etc., antagonists of the angiotensin receptor
and its receptor subtypes such as losartan, irbesartan, valsartan,
omapatrilat, gemopatrilat, endothelin antagonists, renin inhibitors,
adenosine receptor agonists, inhibitors and activators of potassium
channels such as glibenclamide, glimepiride, diazoxide, cromakalim,
minoxidil and derivatives thereof, activators of the mitochondrial ATP-
sensitive potassium channel (mitoK(ATP) channel), inhibitors of Kv1.5 etc.
It has emerged that NHE1 inhibitors have a significant antiinflammatory
effect and can thus be used as antiinflammatory drugs. Inhibition of the
release of mediators of inflammation is noteworthy in this connection. The
compounds can thus be used alone or in combination with an
antiinflammatory drug for the prevention or treatment of chronic and acute
inflammatory disorders. Combination partners advantageously used are
steroidal and non-steroidal antiinflammatory drugs. The compounds of the
invention can additionally be employed for the prevention or treatment of
disorders caused by protozoa, such as malaria and coccidiosis in poultry.
It has additionally been found that NHE1 inhibitors show a beneficial effect
on serum lipoproteins. It is generally acknowledged that blood fat levels
which are too high, called hyperlipoproteinemias, represent an essential
risk factor for the development of arteriosclerotic vascular lesions,
especially coronary heart disease. The reduction of elevated serum
lipoproteins therefore has exceptional importance for the prophylaxis and
regression of atherosclerotic lesions. Besides the reduction in total serum
cholesterol, it is particularly important to reduce the proportion of specific
atherogenic lipid fractions of this total cholesterol, in particular of the
low
density lipoproteins (LDL) and of the very low density lipoproteins (VLDL),
CA 02551057 2006-05-10
28
because these lipid fractions represent an atherogenic risk factor. By
contrast, a protective function against coronary heart disease is ascribed to
the high density lipoproteins. Accordingly, hypolipidemics should be able to
reduce not only total cholesterol but, in particular, the VLDL and LDL serum
cholesterol fractions. It has now been found that NHE1 inhibitors show
valuable therapeutically utilizable properties in relation to influencing the
serum lipid levels. Thus, they significantly reduce the elevated serum
concentrations of LDL and VLDL as are to be observed, for example, due
to increased dietary intake of a cholesterol- and lipid-rich diet or in cases
of
pathological metabolic alterations, for example genetically related
hyperlipidemias. They can therefore be used for the prophylaxis and
regression of atherosclerotic lesions by eliminating a causal risk factor.
Included herein are not only the primary hyperlipidemias but also certain
secondary hyperlipidemias occurring, for example, in association with
diabetes. In addition, the NHE1 inhibitors lead to a marked reduction in the
infarctions induced by metabolic abnormalities and, in particular, to a
significant reduction in the induced infarct size and the severity thereof.
The compounds of the formula I and/or the pharmaceutically acceptable
salts thereof are therefore advantageously used for producing a
medicament for the treatment of hypercholesterolemia; for producing a
medicament for the prevention of atherogenesis; for producing a
medicament for the prevention and treatment of atherosclerosis, for
producing a medicament for the prevention and treatment of diseases
induced by elevated cholesterol levels, for producing a medicament for the
prevention and treatment of diseases induced by endothelial dysfunction,
for producing a medicament for the prevention and treatment of
atherosclerosis-induced hypertension, for producing a medicament for the
prevention and treatment of atherosclerosis-induced thromboses, for
producing a medicament for the prevention and treatment of
hypercholesterolemia-induced and endothelial dysfunction-induced
ischemic damage and post-ischemic reperfusion damage, for producing a
medicament for the prevention and treatment of hypercholesterolemia-
induced and endothelial dysfunction-induced cardiac hypertrophies and
cardiomyopathies and of congestive heart failure (CHF), for producing a
CA 02551057 2006-05-10
29
medicament for the prevention and treatment of hypercholesterolemia-
induced and endothelial dysfunction-induced coronary vasospasms and
myocardial infarctions, for producing a medicament for the treatment of
said disorders in combinations with hypotensive substances, preferably
with angiotensin converting enzyme (ACE) inhibitors and angiotensin
receptor antagonists. A combination of an NHE inhibitor of the formula I
and/or the pharmaceutically acceptable salts thereof with an active
ingredient lowering the blood fat levels, preferably with an HMG-CoA
reductase inhibitor (for example lovastatin or pravastatin), the latter
bringing about a hypolipidemic effect and thus increasing the hypolipidemic
properties of the NHE inhibitor of the formula I and/or the pharmaceutically
acceptable salts thereof, proves to be a favorable combination with
enhanced effect and reduced use of active ingredients.
Thus, compounds of the formula I and/or the pharmaceutically acceptable
salts thereof lead to effective protection against endothelial damage of
various origins. This protection of the vessels against the syndrome of
endothelial dysfunction means that the compounds of the formula I and/or
the pharmaceutically acceptable salts thereof are valuable medicaments for
the prevention and treatment of coronary vasospasms, peripheral vascular
diseases, in particular intermittent claudication, atherogenesis and
atherosclerosis, left ventricular hypertrophy and dilated cardiomyopathy
and thrombotic disorders.
It has additionally been found that NHE1 inhibitors are suitable in the
treatment of non-insulin-dependent diabetes (NIDDM), with the insulin
resistance being restrained. It may in this connection be beneficial, to
enhance the antidiabetic activity and quality of the effect of the compounds
of the invention, to combine them with a biguanide such as mettormin, with
an antidiabetic sulfonylurea such as glyburide, glimepiride, tolbutamide
etc., with a glucosidase inhibitor, with a PPAR agonist such as
rosiglitazone, pioglitazone etc., with an insulin product of different
administration form, with a DB4 inhibitor, with an insulin sensitizor or with
meglitinide.
CA 02551057 2006-05-10
Besides the acute antidiabetic effects, the compounds of the formula I
andlor the pharmaceutically acceptable salts thereof counteract the
development of late complications of diabetes and can therefore be used
as medicaments for the prevention and treatment of late damage from
5 diabetes, such as diabetic nephropathy, diabetic retinopathy, diabetic
cardiomyopathy and other disorders occurring as a consequence of
diabetes. They can in this connection be advantageously combined with
the antidiabetic medicaments just described under NIDDM treatment. The
combination with a beneficial dosage form of insulin should be particularly
10 important in this connection.
NHE1 inhibitors show, besides the protective effects against acute
ischemic events and the subsequent equally acutely stressing reperfusion
events, also direct therapeutically utilizable effects against diseases and
15 disorders of the entire mammalian organism which are associated with the
manifestations of the chronically progressive aging process and which
occur independently of acute hypoperfusion states and under normal, non-
ischemic conditions. These pathological, age-related manifestations
induced over the long aging period, such as illness, invalidity and death,
20 which can now be made amenable to treatment with NHE inhibitors, are
diseases and disorders which are essentially caused by age-related
changes in vital organs and the function thereof and become increasingly
important in the aging organism.
Disorders connected with an age-related functional impairment or with age-
25 related manifestations of wear of organs are, for example, the inadequate
response and reactivity of the blood vessels to contraction and relaxation
reactions. This age-related decline in the reactivity of vessels to
constricting
and relaxing stimuli, which are an essential process of the cardiovascular
system and thus of life and health, can be significantly eliminated or
30 reduced by NHE inhibitors. One important function and a measure of the
maintenance of the reactivity of vessels is the blockade or retardation of the
age-related progression in endothelial dysfunction, which can be eliminated
highly significantly by NHE inhibitors. The compounds of the formula I
andlor the pharmaceutically acceptable salts thereof are thus outstandingly
CA 02551057 2006-05-10
31
suitable for the treatment and prevention of the age-related progression in
endothelial dysfunction, especially of intermittent claudication.
An example of another variable characterizing the aging process is the
decline in the contractability of the heart and the decline in the adaptation
of the heart to a required pumping output of the heart. This diminished
efficiency of the heart as a consequence of the aging process is in most
cases connected with a dysfunction of the heart which is caused inter alia
by deposition of connective tissue in the myocardial tissue. This deposition
of connective tissue is characterized by an increase in the weight of the
heart, by an enlargement of the heart and by restrictive cardiac function. ft
was surprising that it was possible almost completely to inhibit such aging
of the heart organ. The compounds of the formula I and/or the
pharmaceutically acceptable salts thereof are thus outstandingly suitable
for the treatment and prevention of heart failure, of congestive heart failure
(CHF).
Not only is it possible to cure a cancer which has already occurred through
inhibition of proliferation, but there is also reduction and highly
significant
retardation of the age-related incidence of cancer through NHE inhibitors. A
particularly noteworthy finding is that the disorders, occurring as a result
of
aging, of all organs and not only certain types of cancer are suppressed or
occur with a highly significant delay. The compounds of the formula I and/or
the pharmaceutically acceptable salts thereof are thus suitable for the
treatment and, in particular, the prevention of age-related types of cancer.
With NHE inhibitors, a delay, shifted highly significantly in time is found in
the occurrence of age-related disorders of all the organs investigated,
including the heart, vessels, liver etc., and a highly significant delay in
cancer of the elderly. On the contrary, there is also surprisingly a
prolongation of life to an extent which has to date been achievable by no
other group of medicaments or by any natural products. This unique effect
of NHE inhibitors also makes it possible, besides the use of the active
ingredients alone on humans and animals, to combine these NHE inhibitors
CA 02551057 2006-05-10
32
with other active principles, measures, substances and natural products
which are used in gerontology and which are based on a different
mechanism of action. Such classes of active ingredients used in
gerontological therapy are: in particular vitamins and substances with
antioxidant activity. Since there is a correlation between caloric load or
food
intake and the aging process, the combination with dietary measures can
take place for example with appetite suppressants. It is likewise possible to
consider a combination with hypotensive medicaments such as with ACE
inhiibitors, angiotensin receptor antagonists, diuretics, Ca+2 antagonists
etc.
or with metabolism-normalizing medicaments such as cholesterol-lowering
agents.
The compounds of the formula I and/or the pharmaceutically acceptable
salts thereof are thus outstandingly suitable for the prevention of age-
related tissue changes and for prolonging life while retaining a high quality
of life.
The compounds of the invention are effective inhibitors of the cellular
sodium-proton antiporter (Na/H exchanger) which in a large number of
disorders (essential hypertension, atherosclerosis, diabetes etc.) is also
increased in cells which are readily amenable to measurements, such as,
for example, in erythrocytes, platelets or leucocytes. The compounds used
according to the invention are therefore suitable as outstanding and simple
scientific tools, for example in their use as diagnostic agents for
determining and distinguishing different types of hypertension, but also of
atherosclerosis, diabetes and the late complications of diabetes,
proliferative disorders etc.
Also claimed is a medicine for human, veterinary or phytoprotective use,
comprising an effective amount of a compound of the formula i andlor the
pharmaceutically acceptable salts thereof, together with pharmaceutically
acceptable carriers and additives, alone or in combination with other active
pharmaceutical ingredients or medicaments.
Medicaments which comprise a compound of the formula I andlor the
pharmaceutically acceptable salts thereof can in this connection be
CA 02551057 2006-05-10
33
administered, for example, orally, parenterally, intravenously, rectally,
transdermally or by inhalation, the preferred administration being
dependent on the particular characteristics of the disorder. The compounds
of the formula I may moreover be used alone or together with
pharmaceutical excipients, both in veterinary medicine and in human
medicine. The medicaments generally comprise active ingredients of the
formula I and/or the pharmaceutically acceptable salts thereof in an amount
of from 0.01 mg to 1 g per dose unit.
The excipients suitable for the desired pharmaceutical formulation are
familiar to the skilled worker on the basis of his expert knowledge. Besides
solvents, gel formers, suppository bases, tablet excipients, and other active
ingredient carriers, it is possible to use, for example, antioxidants,
dispersants, emulsifiers, antifoams, flavorings, preservatives, solubilizers
or
colors.
For a form for oral administration, the active compounds are mixed with
additives suitable for this purpose, such as carriers, stabilizers or inert
diluents, and converted by conventional methods into suitable dosage
forms such as tablets, coated tablets, hard gelatin capsules, aqueous,
alcoholic or oily solutions. Examples of inert carriers which can be used are
gum arabic, magnesia, magnesium carbonate, potassium phosphate,
lactose, glucose or starch, especially corn starch. It is moreover possible
for the preparation to take place both as dry granules and as wet granules.
Examples of suitable oily carriers or solvents are vegetable or animal oils
such as sunflower oil or fish liver oil.
For subcutaneous, intramuscular or intravenous administration, the active
compounds used are converted, if desired with the substances customary
for this purpose, such as solubilizers, emulsifiers or other excipients, into
a
solution, suspension or emulsion. Examples of suitable solvents are. water,
physiological saline or alcohols, e.g. ethanol, propanol, glycerol, as well as
sugar solutions such as glucose or mannitol solutions, or else a mixture of
the various solvents mentioned.
CA 02551057 2006-05-10
34
Suitable as pharmaceutical formulation for administration in the form of
aerosols or sprays are, for example, solutions, suspensions or emulsions of
the active ingredient of the formula I and/or the pharmaceutically
acceptable salts thereof in a pharmaceutically acceptable solvent such as,
in particular, ethanol or water, or a mixture of such solvents. The
formulation may, if required, also contain other pharmaceutical excipients
such as surfactants, emulsifiers and stabilizers, and a propellant gas. Such
a preparation normally contains the active ingredient in a concentration of
about 0.1 to 10, in particular of about 0.3 to 3% by weight.
The dosage of the active ingredient of the formula I to be administered, and
the frequency of administration, depend on the potency and duration of
action of the compounds used; additionally also on the nature and severity
of the disorder to be treated and on the sex, age, weight and individual
responsiveness of the mammal to be treated.
On average, the daily dose of a compound of the formula I and/or the
pharmaceutically acceptable salts thereof for a patient weighing about
75 kg is at least 0.001 mg/kg, preferably 0.01 mg/kg, to a maximum of
10 mg/kg, preferably 1 mg/kg, of body weight. For acute episodes of the
disorder, for example immediately after suffering a myocardial infarction,
higher and, in particular, more frequent dosages may also be necessary,
e.g. up to 4 single doses a day. Up to 700 mg a day may be necessary, in
particular on i.v. administration, for example for a patient with infarction
in
the intensive care unit, and the compounds of the invention can be
administered by infusion.
List of abbreviations:
ADMET absorption - distribution - metabolism - excretion
- toxicology
CDI diimidazol-1-ylmethanone
dba dibenzylideneacetone
DIP diisopropyl ether
DIPEA diisopropylethylamine
CA 02551057 2006-05-10
DME 1,2-dimethoxyethane
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
EA ethyl acetate
5 eq. equivalent
HOAc acetic acid
KOtBu potassium 2-methylpropan-2-olate
MeOH methanol
mp melting point
10 MTB tert-butyl methyl ether
NMP N-methyl-2-pyrrolidone
OAc acetate
dppf 1,1'-bis(diphenylphosphino)ferrocene
RT room temperature
15 THF tetrahydrofuran
TMEDA N,N,N',N'-tetramethylethane-1,2-diamine
Experimental part
Example 1: N-(5-Methanesulfonyl-2-methyl-4-pentafluorosulfanyl-
benzoyl)guanidine
F
F\~ ~F
F'S ~ \
OF
,,S / O
O' \
N \ /NH2
'N~hi2
a) 4-Aminophenylsulfur pentafluoride
FsS \
NH2
A solution of tin(II) chloride (1465 g, 7.73 mol) in concentrated (32 percent)
aqueous HCI solution was heated with stirring to 80°C and then, with
ice
CA 02551057 2006-05-10
36
cooling, 4-nitrophenylsulfur pentafluoride (584 g, 2.344 mol) was introduced
in 8 portions over the course of 1 h. The internal temperature was kept
below 100°C during this. Subsequently, the mixture was stirred at an
internal temperature of 85°C for 1.5 h and then cooled to 45°C
over the
course of a further hour. A mixture of ice (12 kg), NaOH (2 kg) and
dichloromethane (1.5 I) was prepared and the reaction mixture was added
with vigorous stirring. The phases were separated, the aqueous phase was
extracted 3 times with 1 I of dichloromethane each time, and the combined
organic phases were dried over Na2S04 and evaporated in vacuo. 510 g of
4-aminophenylsulfur pentafluoride were obtained as a pale yellow
crystalline powder, m.p. 63-65°C
b) 4-Amino-3-bromophenylsulfur pentafluoride
F5S .~ Br
NH2
4-Aminophenylsulfur pentafluoride (510 g, 2.327 mol) was dissolved in
dichloromethane (7 I), the solution was cooled to 5°C and, while
stirring,
1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione (326 g, 1.14 mol) was
introduced in several portions with ice cooling so that the internal
temperature was kept at 3-8°C (approx. 1 h). The mixture was then left
to
stir and warm to room temperature without external cooling for 1 h. The
mixture was filtered through a bed of silica gel (volume about 1 I) and
washed with dichloromethane (5.5 I), and the filtrate was evaporated in
vacuo. About 700 g of a red-brown crystalline mass was obtained and was
dissolved in n-heptane (600 ml) at 60°C and then crystallized in a
refrigerator at 4°C. Filtration with suction resulted in 590 g (85%) of
4-amino-3-bromophenylsulfur pentafluoride as brownish crystals,
m.p. 59-59.5°C.
c) 4-Amino-3-methylphenylsulfur pentafluoride
F5S ~ GH3
NH2
A mixture of Cs2COg (794 g, 2.7 mol), dimethoxyethane (2 I), water
(300 ml) and trimethylboroxine (50 percent solution in THF, 225 g, 0.9 mol)
CA 02551057 2006-05-10
37
was heated to 70°C, PdCl2 (dppf) X CH2C12 (37 g, 45 mmol) was added,
and a solution of 4-amino-3-bromophenylsulfur pentafluoride (270 g,
0.9 mol) in dimethoxyethane (400 ml) was added dropwise over the course
of 2 h while the reaction mixture was heated to reflux. It was subsequently
heated under reflux for a further 3 h and then cooled to room temperature,
diluted with MTB ether (500 ml), filtered through a silica gel column
(14 x 7 cm, 70-200 pm) and washed with MTB ether (2500 ml). The filtrate
was evaporated in vacuo. 490 g of a black, semicrystalline mass was
obtained and was subjected to a steam distillation. A total of 5.5 I of
condensate was collected, from which the crystals of the product separated
out. The condensate was extracted 3 X with MTB ether, and the combined
organic phases were dried over Na2S04 and evaporated in vacuo.
4-Amino-3-methylphenylsulfur pentafluoride (181 g, 76%) was obtained as
colorless crystals, m.p. 65-66°C,
d) 4-Bromo-3-methylphenylsulfur pentafluoride
F5S ~ CH3
Br
A mixture of tert-butyl nitrite (90 percent pure, 37 ml, 280 mmol) and CuBr2
(35.8 g, 160 mmol) in acetonitrile (260 ml) was cooled to 5°C and,
while
stirring and cooling with ice, a solution of 4-amino-3-methylphenylsulfur
pentafluoride (30.9 g, 132.5 mmol) in MTB ether (140 ml) was added
dropwise at 5-8°C over the course of 1 h. Evolution of nitrogen started
after
about 2 min. The mixture was then allowed to warm with stirring to room
temperature over the course of 1 h, a mixture of ice (250 g), 26 percent
aqueous NH3 solution (50 ml) and MTB ether (250 ml) was added, and the
mixture was stirred for 10 min. The phases were separated, the aqueous
was extracted 3 x with MTB ether (150 ml each time), and the combined
organic phases were shaken once with 400 ml of water. Drying with
Na2S04 and evaporation of the organic phase resulted in 39 g of 4-bromo-
3-methylphenylsulfur pentafluoride as a red-brown oil which was
contaminated with 8 mol% of 4,5-dibromo-3-methylphenylsulfur
pentafluoride, but was used further without further purification. Yield 89%
based on a purity of 90%.
CA 02551057 2006-05-10
38
e) 4-Cyano-3-methylphenylsulfur pentafluoride
F5S ~ CH3
CN
A mixture of 4-bromo-3-methylphenylsulfur pentafluoride (136.4 g, purity
80%, 0.367 mol), Zn(CN)2 (72.8 g, 0.62 mol) and Zn dust (7.2 g, 0.11 mol)
in dimethylacetamide (900 ml) and water (40 ml) was heated with stirring
and nitrogen blanketing to 125°C, and PdCl2(dppf) X CH2C12 (32.7 g,
40 mmol) was added. After stirring at 125°C for one hour,
PdCl2(dppf) X CH2C12 (16.3 g, 20 mmol) and Zn dust (3.6 g, 55 mmol)
were again added, and stirring was continued at 125°C for 2 h. The
mixture
was then cooled to room temperature, diluted with n-heptane (400 ml) and
stirred vigorously with addition of 5 N aqueous NH4C1 solution (250 ml) and
water (450 ml) for 15 min. The mixture was filtered with suction through a
layer of kieselguhr, the phases were separated, and the aqueous was
extracted 2 x with n-heptane (200 ml). The combined organic phases were
shaken with water (450 ml), dried over MgS04 and evaporated in vacuo.
The resulting black residue was dissolved in 200 ml of n-heptane, filtered
and again evaporated in vacuo. 78 g of a dark brown liquid were obtained
and were purified by chromatography on a silica gel column (7 X 55 cm,
60-200 Vim, n-heptane/dichloromethane 4:1 to 3:2). The first fraction
obtained was 6.5 g of 4-bromo-3-methylphenylsulfur pentafluoride
(precursor) as yellowish liquid, and then 71.1 g (80%) of 4-cyano-3-methyl-
phenylsulfur pentafluoride as pale yellow oil.
f) 2-Methyl-4-pentafluorosulfanylbenzoic acid
FMS ;,~ CH3
CO2H
A mixture of 4-cyano-3-methylphenylsulfur pentafluoride (41.2 g,
169.4 mmol), NaOH (20.4 g, 510 mmol) and water (60 ml) in ethylene
glycol (160 ml) was heated to 130°C and stirred at this temperature for
4 h.
It was then cooled to room temperature and diluted with MTB ether
(150 ml) and water (250 ml), and the mixture was filtered with suction. The
phases of the filtrate were separated, and the aqueous was acidified with
concentrated aqueous HCI solution, and the precipitated solid was filtered
off with suction. 41.1 g (93%) of 2-methyl-4-pentafluorosulfanylbenzoic acid
CA 02551057 2006-05-10
39
were obtained as colorless crystals, m.p. 138-139°C.
g) 2-Methyl-5-vitro-4-pentafluorosulfanylbenzoic acid
F
F\1 ~F
F~S ~ \
F
O
II
O OH
6.0 g of 2-methyl-4-pentafluorosulfanylbenzoic acid were dissolved in 60 ml
of a 90% aqueous HN03 solution and, at RT, 6 ml of a 96% H2S04 were
added dropwise. The mixture was left to stand at RT for 28 h and then
poured into 300 g of ice, 300 ml of water were added and, after stirring for
1 h, the product was filtered off. Drying in air resulted in 6.5 g of a pale
yellow solid, mp. 218-220°C.
Rf (DIP/2%HOAc) = 0.27 MS (ES ): 306
h) 5-Amino-2-methyl-4-pentafluorosulfanylbenzoic acid
F
F\I ~F
F~S \
F
/ O
H2N
OH
6.5 g of 2-methyl-5-vitro-4-pentafluorosulfanylbenzoic acid were dissolved
in 100 ml of MeOH and 20 ml of HOAc, and 500 mg of 10% Pd/C were
added. Hydrogenation was carried out under hydrogen at atmospheric
pressure and RT for 20 h. The reaction was incomplete and therefore
hydrogenation was continued under a pressure of 6 bar of hydrogen and at
RT for 48 h. The catalyst was then filtered off and the solvents were
removed in vacuo. 5.7 g of a pale grey solid were obtained, mp. 187-
189°C.
Rf (DIP/2%HOAc) = 0.23 MS (ES ): 276
CA 02551057 2006-05-10
i) 5-Chlorosulfonyl-2-methyl-4-pentafluorosulfanylbenzoic acid
F
F\I ,F
F~S \
F ~ / O
CI-S
yv
O O OH
1.0 g of 5-amino-2-methyl-4-pentafluorosulfanylbenzoic acid was dissolved
5 in 30 ml of HOAc, and 30 g of ice and 30 ml of a saturated aqueous HCI
solution were added. Then, at 0°C, a solution of 274 mg of NaN02 in 1
ml
of water was added dropwise over the course of one minute. The mixture
was stirred at 0°C for 15 minutes. The resulting suspension was then
added in portions to a suspension, cooled to 0°C, of 6.1 mg of CuCI and
10 61.5 mg of CuCl2 x 2H20 in 30 ml of a saturated solution of S02 in HOAc.
The mixture was stirred at 0°C for 1 h and then at RT for 1 h. The
reaction
mixture was subsequently extracted 3 times with 200 ml of diethyl ether
each time. MgS04 was used for drying, and the volatile constituents were
removed in vacuo. 1.3 g of the product were obtained and immediately
15 reacted further.
k) 2-Methyl-5-sulfino-4-pentafluorosulfanylbenzoic acid
F
F\~ ~F
F~S \
HO S I / O
I I
O OH
1.2 g of 5-chlorosulfonyl-2-methyl-4-pentafluorosulfanylbenzoic acid were
20 added in portions to a solution, heated to 70°C, of 4.2 g of Na2S03
in 50 ml
of water and, during this, the pH of the solution was kept between pH = 9
and pH = 11 with a 2N aqueous NaOH solution. The mixture was stirred at
70°C for 20 minutes, cooled to RT and adjusted to pH = 1-2 with an
aqueous HCI solution. The mixture was left to stand at RT for 16 h and then
25 the product was filtered off and dried in vacuo. 1.0 g of a white solid was
CA 02551057 2006-05-10
41
obtained, mp. 288-290°C (with decomposition).
Rf (EA/MeOH 1:1 ) = 0.52
I) Methyl 5-methanesulfonyl-2-methyl-4-pentafluorosulfanylbenzoate
F
F~1 ~F
F' S ~ \
F
\S ~ O
O O
1.0 g of 2-methyl-5-sulfino-4-pentafluorosulfanylbenzoic acid was
suspended in 10 ml of water, and 3.1 ml of an aqueous 2N NaOH solution
were added (phenolphthalein: basic). The water was removed in vacuo and
then coevaporated twice with 20 ml of toluene each time. The disodium salt
was then dissolved in 40 ml of anhydrous DMF and, after addition of
0.69 ml of methyl iodide, stirred initially at 60°C for 4 h and then at
RT for
h. The reaction mixture was poured into 100 ml of water and a first
portion of the product (500 mg) was filtered off with suction. The filtrate
was
adjusted to pH = 2 with aqueous HCI solution and extracted 3 times with
15 30 ml of EA each time. MgS04 was used for drying, and the solvent was
removed in vacuo. Chromatography on silica gel with DIP afforded a further
460 mg of white crystals, mp 127°C.
Rf(DIP)=0.36
m) N-(5-Methanesulfonyl-2-methyl-4-pentafluorosulfanylbenzoyl)guanidine
0.70 g of guanidine chloride and 0.68 g of KOtBu were stirred in 20 ml of
anhydrous DMF at RT for 30 minutes. This suspension was then added to
0.43 g of methyl 5-methanesulfonyl-2-methyl-4-pentafluorosulfanyl-
benzoate and stirred at RT for 16 h. The reaction mixture was then poured
into 200 ml of water, adjusted to pH = 8 with aqueous HCI solution and
extracted 3 times with 100 ml of EA each time. MgS04 was used for drying,
and the solvent was removed in vacuo. The residue was suspended in 5 ml
of CH2C12 and the product was filtered off. 190 mg of colorless crystals
CA 02551057 2006-05-10
42
were obtained, mp. 254-256°C.
Rf (EA) = 0.22 MS (ES+): 382
Example 2: N-(5-Methanesulfonyl-2-methyl-4-pentafluorosulfanylbenzoyl)-
guanidine methanesulfonic acid salt
F
F~~ ~F y
F~S \ O-S-
OF I / O HO
,.
O ~S\
N \ /NH2
~NH2
9.3 g of the title compound of example 1 were suspended in 100 ml of
water and a solution of 2.3 g of methanesulfonic acid in 10 ml of water was
added. The mixture was subsequently stirred at RT for 30 minutes and then
the water was removed under reduced pressure to obtain 11.7 g of the
methanesulfonic acid salt, which was subsequently recrystallized from
110 ml of water to obtain 10.0 g of N-(5-methanesulfonyl-2-methyl-
4-pentafluorosulfanylbenzoyl)guanidine methanesulfonic acid salt as white
crystals, m.p. 230°C.
Example 3: N-(5-Methanesulfonyl-2-methyl-4-pentafluorosulfanylbenzoyl)-
guanidine hydrochloride
F
F\~ ~F
F~s I \
OF
OiS\ / O HCI
N \ /NH2
NHZ
300 mg of the title compound of example 2 were suspended in 50 ml of a
saturated aqueous Na2C03 solution and extracted twice with 40 ml of EA
each time. The EA phase was subsequently dried over MgS04 and the
CA 02551057 2006-05-10
43
solvent was removed under reduced pressure. The residue was dissolved
in 10 ml of MeOH and admixed with 2 ml of a 10% aqueous HCI solution.
The volatiles were removed under reduced pressure to leave 230 mg of
white crystals, m.p. 276-278°C.
Determination of the NHE inhibition
The inhibitory concentration ICSO for NHE-1 inhibition was determined as
follows:
ICSp for NHE-1 inhibition was determined in an FLIPR assay by
measurement of the pHi recovery in transfected cell lines which express
human NHE-1.
The assay was carried out in an FLIPR (fluorometric imaging plate reader)
with black-walled 96-well microtiter plates with clear bases. The transfected
cell lines expressing the various NHE subtypes (the parental cell line LAP-1
shows no endogenous NHE activity as a result of mutagenesis and
subsequent selection) were seeded the preceding day at a density of
~25 000 cells/well.
The growth medium for the transfected cells (Iscove +10% fetal calf serum)
additionally contained 6418 as selection antibiotic in order to ensure the
presence of the transfected sequences.
The actual assay started with the removal of the growth medium and
addition of 100 ~I of loading buffer per well (5 NM BCECF-AM [2',7'-
bis(carboxyethyl)-5- (and -6-)carboxyfluorescein, acetoxymethyl ester] in
20 mM NH4C1, 115 mM choline chloride, 1 mM MgCl2, 1 mM CaCl2, 5 mM
KCI, 20 mM HEPES, 5 mM glucose; pH 7.4 [adjusted with KOH]). The cells
were then incubated at 37°C for 20 minutes. This incubation led to
loading
of the cells with the fluorescent dye whose fluorescence intensity depends
on pHi, and with NH4CI which made the cells slightly alkaline.
The nonfluorescent dye precursor BCECF-AM is, as ester, membrane-
CA 02551057 2006-05-10
44
permeable. The actual dye BCECF is not membrane-permeable but is
liberated inside cells by esterases.
After this incubation for 20 minutes, the loading buffer which contained
NH4CI and free BCECF-AM was removed by washing three times in a cell
washer (Tecan Columbus) with in each case 400 NI of washing buffer
(133.8 mM choline chloride, 4.7 mM KCI, 1.25 mM MgCl2, 1.25 mM CaCl2,
0.97 mM K2HP04, 0.23 mM KH2P04, 5 mM HEPES, 5 mM glucose; pH
7.4 [adjusted with KOH}). The residual volume which remained in the wells
was 90 NI (50-125 NI possible). This washing step removed the free
BCECF-AM and results, as a consequence of the removal of the external
NH4+ ions, in intracellular acidification (- pHi 6.3 - 6.4).
Since the equilibrium of intracellular NH4+ with NH3 and H+ was disturbed
by the removal of the extracellular NH4+ and by the subsequent
instantaneous passage of the NH3 through the cell membrane, the washing
process resulted in H+ remaining inside the cells, which was the cause of
the intracellular acidification. This may eventually lead to cell death if it
persists long enough.
It was important at this point that the washing buffer was sodium-free
(<1 mM) because extracellular sodium ions would lead to an instantaneous
recovery of the pHi through the activity of the cloned NHE isoforms.
It was likewise important for all the buffers used (loading buffer, washing
buffer, recovery buffer) not to contain any HC03 ions, because the
presence of bicarbonate would lead to activation of interfering bicarbonate-
dependent pHi regulatory systems present in the parental LAP-1 cell line.
The microtiter plates with the acidified cells were then (up to 20 minutes
after the acidification) transferred to the FLIPR. In the FLIPR, the
intracellular fluorescent dye was excited by light with a wavelength of
488 nm generated by an argon laser, and the measured parameters (laser
power, illumination time and aperture of the CCD camera incorporated in
CA 02551057 2006-05-10
the FLIPR) were chosen so that the average fluorescence signal per well is
between 30 000 and 35 000 relative fluorescence units.
The actual measurement in the FLIPR started with a photograph being
5 taken by the CCD camera every two seconds under software control. After
ten seconds, the recovery of the intracellular pH was initiated by adding
90 NI of recovery buffer (133.8 mM NaCI, 4.7 mM KCI, 1.25 mM MgCl2,
1.25 mM CaCl2, 0.97 mM K2HP04, 0.23 mM KH2P04, 10 mM HEPES,
5 mM glucose; pH 7.4 [adjusted with NaOH]) by means of the 96-well
10 pipettor incorporated in the FLIPR.
Positive control wells (100% NHE activity) were those to which pure
recovery buffer is added, while negative controls (0% NHE activity)
received washing buffer. Recovery buffer with twice the concentration of
test substance was added to all the other wells. Measurement in the FLIPR
15 terminated after 60 measurements (two minutes).
The raw data are exported into the ActivityBase program. This program
firstly calculates the NHE activities for each tested substance concentration
and, from these, the ICSp values for the substances. Since the progress of
20 pHi recovery was not linear throughout the experiment, but fell at the end
owing to decreasing NHE activity at higher pH; values, it was important to
select for evaluation of the measurement the part in which the increase in
fluorescence of the positive controls was linear.
Example NHE1 inhibition ICSp [nM]
-
1 49
In vivo pharmacokinetics - profiling with the "n in one method"
The exposure data and the volumes of distribution were determined as
characteristic pharmacokinetic data as follows:
CA 02551057 2006-05-10
46
The NHE-1 inhibitor of example 1 of the invention and, as reference
substance, the known NHE-1 inhibitor cariporide with the formula
O
i1
O=S-
O I \ OH
/ O
O~ \
N \ /NH2
~NH2
were dissolved in an aqueous, slightly acidic medium (water, pH 4,
adjusted with 1 M hydrochloric acid). The concentration of the aqueous
formulation prepared in this way was about 1.5 mg of each substance per
1 g of solution. 10 ml of this formulation were administered as a single
bolus by catheter into the jugular vein of a fasting male beagle dog (dose
about 1 mg of each substance administered per kg of the dog's body
weight). Blood samples were taken by means of a second catheter after
5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h, and heparinized plasma
was prepared by centrifugation at 1000 G in appropriate plasma tubes.
The plasma samples were worked up and, after an HPLC separation,
quantified by MS/MS. The high specificity of this method permitted
simultaneous determination of a plurality of substances. The exposures
could be calculated using the WinNonlin computer program from the
concentration-time plots (see figure 1 ) and compared with the exposure of
the known NHE-1 reference substance. Since the various substances were
measured in the same animal at the same time, the result was an accurate
comparison of the compounds, and a ranking of the volumes of distribution
was possible.
Compound Volume of distribution
[I/kg of body weight]
Example 1 1.67
Reference substance 2.94
cariporide
CA 02551057 2006-05-10
47
It is clearly evident from the concentration-time plots in figure 1 that the
compound of the invention is retained in the blood also over a longer period
and thus the exposure is about 2-3 times higher than with the reference
substance cariporide. Cariporide is no longer detectable in the plasma after
24 hours.
The captions and signs in the figure were as follows:
Fig.1: concentration-time plots in the blood plasma of dogs after
administration of in each case approx. 1 mg/kg of the compound of
example 1 and of cariporide.
y axis: concentration of the measured compound in the pg/ml in plasma
x axis: time in h