Note: Descriptions are shown in the official language in which they were submitted.
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
DEHYDROPHENYLAHISTINS AND ANALOGS THEREOF AND THE SYNTHESIS
OF DEHYDROPHENYLAHISTINS AND ANALOGS THEREOF
Related Applications
[0001] This application is a continuation-in-part of U.S. Patent Application
No.
10/632,531 filed on August 1, 2003, and claims priority to U.S. Provisional
Application
No. 60/542,073 filed February 4, 2004 and to U.S. Provisional Application No.
60/624,262
filed November 1, 2004, all of which are entitled DEHYDROPHENYLAHISTINS AND
ANALOGS THEREOF AND THE SYNTHESIS OF DEHYDROPHENYLAHISTINS AND
ANALOGS THEREOF; and each of which is incorporated herein by reference in its
entirety.
Background of the Invention
Field of the Invention
[0002] The present invention relates to compounds and methods of synthetic
preparation in the fields of chemistry and medicine. More specifically, the
present invention
relates to compounds and procedures for making compounds useful in the
treatment of cancer
and the treatment of fungal infections.
Brief Description of the Related Art
[0003] It is thought that a single, universal cellular mechanism controls the
regulation of the eukaryotic cell cycle process. See, e.g., Harlwell, L.H. et
al., Science
(1989), 246: 629-34. It is also known that when an abnormality arises in the
control
mechanism of the cell cycle, cancer or an immune disorder may occur.
Accordingly, as is
also known, antiturnor agents and immune suppressors may be among the
substances that
regulate the cell cycle. Thus, new methods for producing eulearyotic cell
cycle inhibitors are
needed as antitumor and immune-enhancing compounds, and should be useful in
the
treatment of human cancer as chemotherapeutic, anti-tumor agents. See, e.g.,
Roberge, M. et
al., Cancer Res. (1994), 54, 6115-21.
[0004] Fungi, especially pathogenic fungi and related infections, represent an
increasing clinical challenge. Existing antifungal agents axe of limited
efficacy and toxicity,
-1-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
and the development and/or discovery of strains of pathogenic fungi that are
resistant to drugs
cuiTently available or under development. By way of example, fungi that are
pathogenic in
humans include among others Candida spp. including C. albicatzs, C.
t~~opicalis, C. kefyr, C.
krusei and C. galb~ata; Aspe~gillus spp. including A. fumigatus and A. flavus;
C~yptococcus
neoformans; Blastomyces spp. including Blastomyces dermatitidis; Pneumocystis
cap~inii;
Coccidioides immitis; Basidiobolus ranarum; Conidiobolus spp.; Histoplasnaa
capsulatuna;
Rhizopus spp. including R. oryzae and R. rnic~osporus; Cunninghamella spp.;
Rhizomucor
spp.; Paracoccidioides br~asiliensis; Pseudallesche~~ia boydii; Rhinosporidium
seebe~~i; and
Sporoth~~ix schenckii (Kwon-Chung, K.J. 8z Bennett, J.E. 1992 Medical
Mycology, Lea and
Febiger, Malvern, PA).
[0005] Recently, it has been reported that tryprostatins A and B (which are
diketopiperazines consisting of proline and isoprenylated tryptophan
residues), and five other
structurally-related diketopiperazines, inhibited cell cycle progression in
the M phase, see
Cui, C. et al., 1996 JAntibiotics 49:527-33; Cui, C. et al. 1996 JAntibiotics
49:534-40, and
that these compounds also affect the microtubule assembly, see Usui, T. et al.
1998 Biochem
J 333:543-48; Kondon, M. et al. 1998 J Antibiotics 51:801-04. Furthermore,
natural and
synthetic compounds have been reported to inhibit mitosis, thus inhibit the
eukaryotic cell
cycle, by binding to the colchicine binding-site (CLC-site) on tubulin, which
is a
macromolecule that consists of two 50 kDa subunits (a- and [3-tubulin) and is
the major
constituent of microtubules. See, e.g., Iwasaki, S., 1993 Med Res Rev 13:183-
198; Hamel, E.
1996 Med Res Rev 16:207-3I; Weisenberg, R.C. et al., 1969 Bioclaemist~y 7:4466-
79.
Microtubules are thought to be involved in several essential cell functions,
such as axonal
transport, cell motility and determination of cell morphology. Therefore,
inhibitors of
microtubule function may have broad biological activity, and be applicable to
medicinal and
agrochemical purposes. It is also possible that colchicine (CLC)-site ligands
such as CLC,
steganacin, see Kupchan, S.M. et al., 1973 JAm Chem Soc 95:1335-36,
podophyllotoxin, see
Sacleett, D.L., 1993 Phar~macol Tlae~~ 59:163-228, and combretastatins, see
Pettit, G.R. et al.,
1995 J Med Chena 38:166-67, may prove to be valuable as eukaryotic cell cycle
inhibitors
and, thus, may be useful as chemotherapeutic agents.
-2-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0006] Although diketopiperazine-type metabolites have been isolated from
various fungi as mycotoxins, see Horak R.M. et al., 1981 JCS Cherya Comm 1265-
67; Ali M.
et al., 1898 Toxicology Letters 48:235-41, or as secondary metabolites, see
Smedsgaard J. et
al., 1996 J Microbiol Meth 25:5-17, little is known about the specific
structure of the
diketopiperazine-type metabolites or their derivatives and their antitumor
activity,
particularly izz vivo. Not only have these compounds been isolated as
mycotoxins, the
chemical synthesis of one type of diketopiperazine-type metabolite,
phenylahistin, has been
described by Hayashi et al. in J. Or~g. Clzern. (2000) 65, page 8402. In the
art, one such
diketopiperazine-type metabolite derivative, dehydrophenylahistin, has been
prepared by
enzymatic dehydrogenation of its parent phenylahistin. With the incidences of
cancer on the
rise, there exists a particular need for chemically producing a class of
substantially purified
diketopiperazine-type metabolite-derivatives having animal cell-specific
proliferation-
inhibiting activity and high antitumor activity and selectivity. There is
therefore a particular
need for an efficient method of synthetically producing substantially
purified, and structurally
and biologically characterized, diketopiperazine-type metabolite-derivatives.
[0007] Also, PCT Publication WO/0153290 (July 26, 2001) describes a non-
synthetic method of producing dehydrophenylahistin by exposing phenylahistin
or a
particular phenylahistin analog to a dehydrogenase obtained from Str~eptomyces
albulus.
Summary of the Invention
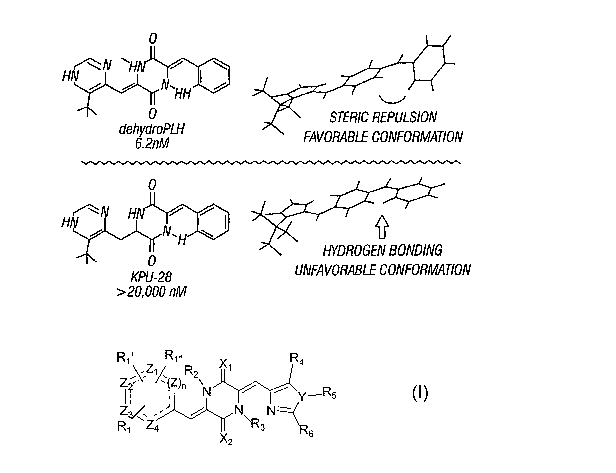
[0008] Compounds, and methods for the synthetic manufacture of compounds,
are disclosed for a class of compounds having the structure of Formula (I):
R~, R~,~ X R4
/ /
N ~ ~ ~Y~R5
N N-
R~ Z4r v ~3 6
[0009] The disclosed compounds have the structure of Formula (I) wherein:
-3-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0010] Rl, R4, and R6, are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated C~-Cz4
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, nitro, azido, substituted nitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R~, cyano, alkylthio, halogenated alkyl
including
polyhalogenated allcyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C1-C2~ alkyl, unsaturated
C~-Cz~
alkenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, nitro, azido, substituted
nitro, phenyl, and
substituted phenyl groups;
[0011] Rl' and Rl" are independently selected from the group consisting of a
hydrogen atom, a halogen atom, and saturated CI-C24 alkyl, unsaturated C1-Ca4
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, nitro, azido, substituted nitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, allcylthio, halogenated
alkyl including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C~-C24 alkyl, unsaturated
CI-C24
alkenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, nitro, azido, substituted
nitro, phenyl, and
substituted phenyl groups;
[0012] R2, R3, and RS are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated Ct-CIZ alkyl, unsaturated C1-CIZ
alkenyl, acyl,
cycloalkyl, alkoxy, cycloallcoxy, aryl, substituted aryl, heteroaryl,
substituted heteroaryl,
amino, substituted amino, nitro, and substituted nitro groups, sulfonyl and
substituted
sulfonyl groups;
[0013] Xl and X~ are separately selected from the group consisting of an
oxygen
atom, a nitrogen atom, and a sulfur atom, each either unsubstituted or
substituted with a RS
group, as defined above; a
-4-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0014] Y' is selected from the group consisting of a nitrogen atom, a
substituted
nitrogen atom with a RS group from above, an oxygen atom, a sulfur atom, a
oxidized sulfur
atom, a methylene group and a substituted methylene group;
[0015] n is an integer equal to zero, one or two;
[0016] Z, for each separate n, if non-zero, and Zl, Z2, Z3 and Z4 are each
separately selected from a carbon atom, a sulfur atom, a nitrogen atom or an
oxygen atom;
and
[0017] the dashed bonds may be either single or double bonds;
[0018] with the proviso that, in a particular compound, if Rl, Rl', R2, R3, R4
and
RS are each a hydrogen atom, then it is not true that Xl and X2 axe each an
oxygen atom and
Rg is either 3,3-dimethylbutyl-1-ene or a hydrogen atom.
(0019] The methods comprise the steps of:
[0020] reacting a diacyldiketopiperazine with a first aldehyde to produce an
intermediate compound; and
[0021] reacting said intermediate compound with a second aldehyde to produce
said class of compounds with said generic structure, wherein
[0022] said first aldehyde and said second aldehydes are selected from the
group
consisting of an oxazolecarboxaldeyhyde, imidazolecarboxaldehyde, a
benzaldehyde,
irnidazolecarboxaldehyde derivatives, and benzaldehyde derivatives, thereby
forming a
compound wherein
[0023] The disclosed compounds have the structure of Formula (I) wherein:
[0024] Rl, R4, and R6, are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated CI-C24 alkyl, unsaturated C1-C24
allcenyl,
cycloalkyl, cycloallcenyl, alleoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, nitro, azido, substituted nitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, allcylthio, halogenated
alkyl including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated
CI-C24
alkenyl, cyeloallcyl, cycloalkenyl, alkoxy, cycloallcoxy, aryl, substituted
aryl, heteroaryl,
-5-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
substituted heteroaryl, amino, substituted amino, vitro, azido, substituted
vitro, phenyl, and
substituted phenyl groups;
[0025] Rz' and Rl" are independently selected from the group consisting of a
hydrogen atom, a halogen atom, and saturated CI-C24 alkyl, unsaturated C1-C24
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, vitro, azido, substituted vitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, alkylthio, halogenated alkyl
including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated
C1-C24
allcenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, vitro, azido, substituted
vitro, phenyl, and
substituted phenyl groups;
[0026] R2, R3, and RS are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated C1-C12 alkyl, unsaturated CI-Cla
alkenyl, acyl,
cycloalkyl, alkoxy, cycloalkoxy, aryl, substituted aryl, heteroaryl,
substituted heteroaryl,
amino, substituted amino, vitro, and substituted vitro groups, sulfonyl and
substituted
sulfonyl groups;
[0027] Xl and X2 are separately selected from the group consisting of an
oxygen
atom, a nitrogen atom, and a sulfur atom, each either unsubstituted or
substituted with a RS
group, as defined above;
[0028] Y is selected from the group consisting of a nitrogen atom, a
substituted
nitrogen atom with a RS group from above, an oxygen atom, a sulfur atom, a
oxidized sulfur
atom, a methylene group and a substituted methylene group;
[0029] n is an integer equal to zero, one or two;
[0030] Z, for each separate n, if non-zero, and Z1, Z2, Z3 and Z4 are each
separately selected from a carbon atom, a sulfur atom, a nitrogen atom or an
oxygen atom;
and
[0031] the dashed bonds may be either single or double bonds.
[0032] In preferred embodiments of the compound and method, the
imidazolecarboxaldehyde is 5-(1,1-dimethyl-2-ethyl)imidazole-4-carboxaldehyde
and the
-6-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
benzaldehyde comprises a single methoxy group. Additional preferred
embodiments of the
compounds described herein include compounds having a t-butyl group, a
dimethoxy group,
a chloro- group, and a methylthiophen group, and methods of making such
compounds, as
well as the compounds described in Tables 2, 3 and 4, as well as methods of
making such
compounds.
[0033] Also disclosed are methods and materials for treating neoplastic tissue
or
preventing cancers or infection by a pathogenic fungus. These methods and
materials are
particularly well suited for treatment of mammalian subjects, more
particularly humans, and
involve administering to the subject a dehydrophenylahistin ox its analog. The
method
comprises administering to the subject a composition comprising an effective
antitumor or
antifungal amount of a dehydrophenylahistin or its analog.
[0034] Further embodiments relate to methods for treating a condition in an
animal, which :methods can include administering to the animal a compound as
described
herein in an amount that is effective to reduce vascular proliferation or in
an amount that is
effective to reduce vascular density. Exemplary conditions include neoplasms,
such as
cancers, as well as other conditions associated with or which rely upon
vascularization,
including for example, immune and non-immune inflammation, rheumatoid
arthritis, chronic
articular rheumatism, psoriasis, diabetic retinopathy, neovascular glaucoma,
retinopathy of
prematurity, macular degeneration, corneal graft rejection, retrolental
fibroplasia, rubeosis,
capillary proliferation in atherosclerotic plaques, osteoporosis, and the
like. In some
embodiments, the disease is not cancer.
[0035) Other embodiments relate to methods of inducing vascular collapse in an
animal. The methods can include treating said animal with a therapeutically
effective amount
of a compound of the Formula (n as described herein, for example. The
therapeutically
effective amount of said compound can cause tubulin depolyrnerization in the
vasculature.
[0036] Preferably the animal can be a human. Preferably the disease can be a
tumor, a diabetic retinopathy, an age-related macular degeneration, and the
like. W some
aspects the disease is not cancer or cancer can be specifically excluded from
the methods and
uses. Preferably, the compound is KPU-02.
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0037] Still further embodiments relate to pharmaceutical compositions for
treating or preventing vascular proliferation comprising a pharmaceutically
effective amount
of a compound disclosed herein together with a pharmaceutically acceptable
carrier therefor.
The vascular proliferation can be a symptom of a disease, for example, cancer,
age-related
macular degeneration and diabetic retinopathy. .
[0038] Some embodiments relate to methods of preferentially targeting tumor
vasculature over non-tumor tissue vasculature. The methods can include the
step of
administering to an animal, preferably a human, a compound having the
structure of Formula
(I) as described herein. The non-tumor tissue can be, for example, skin,
muscle, brain,
kidney, heart, spleen, gut, and the like. The tumor vasculature can be
preferentially targeted
over non-tumor tissue vasculature, for example, by about 10%, 20%, 30%, 40%,
50%, 60%,
70%, 80% and 90%.
[0039] Other embodiments relate to methods of preferentially targeting tumor
vasculature over non-tumor tissue vasculature, which methods can include
administering to
an animal an agent that preferentially targets tumor vasculature over non-
tumor tissue
vasculature.
(0040] Further embodiments relate to uses of a compound having the structure
of
Formula (I) in the preparation of a medicament for the treatment of a
condition associated
with increased vasculature or which relies upon vasculature. In some aspects
the condition
can be cancer, while in others, cancers particular types or all cancers are
specifically
excluded. The condition can be any other that is associated with
hypervascularization,
associated with vasculature or which relies upon vasculature. Examples include
immune and
non-immune inflammation, rheumatoid arthritis, chronic articular rheumatism,
psoriasis,
diabetic retinopathy, neovascular glaucoma, retinopathy of prematurity,
macular
degeneration, corneal graft rejection, retrolental fibroplasia, rubeosis,
capillary proliferation
in atherosclerotic plaques, osteoporosis, and the lilce.
Brief Description of the Drawings
[0041] The accompanying drawings, which are incorporated in and form part of
the specification, merely illustrate certain preferred embodiments of the
present invention.
Together with the remainder of the specification, they are meant to serve to
explain preferred
_g_
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
modes of making certain compounds of the invention to those of skilled in the
art. In the
drawings:
[0042] Figure 1 illustrates a reaction scheme for producing
dehydrophenylahistins
by reacting a diacyldiketopiperazine 1 with an imidazolecarboxaldeheyde 2 to
yield an
intermediate compound 3 which is reacted with a benzaldehyde 4 to produce a
dehydrophenylahistin.
[0043] Figure 2 depicts the HPLC profile of the synthetic crude
dehydrophenylahistin.
[0044] Figure 3 illustrates a reaction scheme for producing
dehydrophenylahistins
by reacting a diacyldiketopiperazine 1 with a benzaldehyde 4 to yield an
intermediate
compound 17 which is reacted with an imidazolecarboxaldeheyde 15 to produce a
dehydrophenylahistin.
[0045] Figure 4 depicts the HPLC profiles of the crude synthetic tBu-
dehyrophenylahistin produced from Route A and from Route B.
[0046] Figure 5 illustrates two modification strategies for dehydroPLH for
potent
cytotoxic activity.
[0047] Figure 6 depicts the putative active conformation of dehydroPLH at the
phenyl moiety.
[0048] Figure 7 depicts Cytochrome P450 metabolism of phenylahistin.
[0049] Figure 8 illustrates the Z-E migration of tBu-dehydroPLH.
[0050] Figure 9 depicts the synthesis and prodrug image of acyl-E-tBu-
dehydroPL,H.
[0051] Figure 10 depicts the temperature gradient of 3-Z-Benzylidene-6-[5"-
(l,l-
dimethylallyl)-1 H-imidazol-4"-Z-ylmethylene]-piperazine-2, 5-dione.
[0052] Figure 11 depicts the temperature gradient of 3-Z-benzylidene-6-(5"-
tert-
butyl-1H imidazol-4"-Z-ylmethylene)-piperazine-2,5-dione.
[0053] Figure 12 depicts the effect of I~PU-2, KPU-35 and t-butyl-
phenylahistin
in comparison to colchicine and taxol on HuVEC monolayer permeability to FITC-
Dextran.
[0054] Figure 13 depicts the effect of KPU-2 alone and in combination with CPT-
11 on estimated tumor growth in the HT-29 Human Colon Tumor Xenograft model.
-9-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0055] Figure I4 depicts the effect of KPU-2 alone and in combination with CPT-
11 on the weight of tumors excised at autopsy in individual mice in the HT-29
Human Colon
Tumor Xenograft model.
[0056] Figure 15 depicts the effect of KPU-2 alone and in combination with CPT-
11 on estimated tumor growth in the HT-29 Human Colon Tumor Xenograft model.
[0057] Figure 16 depicts the effect of KPU-2 alone and in combination with CPT-
11 on the weight of tumors excised at autopsy in individual mice in the HT-29
Human Colon
Tumor Xenograft model.
[0058] Figure 17 depicts the effects of: A. KPU-2, B. KPU-35 and C. t-butyl-
phenylahistin alone and in combination with CPT-11 on estimated tumor growth
in the HT-
29 human colon tumor xenograft model.
[0059] Figure 18 depicts the effects of A. KPU-2, B. KPU-35 and C. t-butyl-
phenylahistin alone and in combination with CPT-11 on the weight of tumors
excised at
autopsy in individual mice in the HT-29 Human Colon Tumor Xenograft model.
[0060] Figure 19 depicts the effects of KPU-2 alone and in combination with
CPT-I 1 on tumor growth in the HT-29 human colon tumor xenograft model:
comparison of
three studies.
[0061]. Figure 20 depicts the effects of KPU-2 alone and in combination with
CPT-11 on final tumor weights in the HT-29 human colon tumor xenograft model:
comparison of three studies.
[0062] Figure 2I depicts the effects of KPU-2 alone or in combination with
Taxotere on estimated tumor growth in the DU-145 Human Prostate Tumor
Xenograft
Model.
[0063] Figure 22 depicts the effects of A. KPU-2, B. KPU-35 and C. t-butyl-
phenylahistin alone and in combination with Taxotere on the estimated tumor
growth based
on observations made during the in-life portion of the DU-145 Human Prostate
Tumor
Xenograft Model.
[0064] Figure 23 depicts the effects of I~PU-2 alone and in combination with
Taxotere on the individual excised tumor weights at autopsy in the DU-145
Human Prostate
Tumor Xenograft Model.
-10-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0065] Figure 24 depicts the effects of KPU-35 alone and in combination with
Taxotere on the individual excised tumor weights at autopsy in the DU-145
Human Prostate
Tumor Xenograft Model.
[0066] Figure 25 depicts the effects of A. KPU-2, B. KPU-35 and C. t-butyl-
phenylahistin alone and in combination with Taxotere in MCF-7 Human Breast
Tumor
Xenograft model.
[0067] Figure 26 depicts the effects of KPU-2 alone and in combination with
Taxotere on estimated tumor growth in the A549 Human Lung Tumor Xenograft
model.
[0068] Figure 27 depicts the effects of KPU-2 alone and in combination with
Taxotere on the excised tumor weights at autopsy in the A549 Human Lung Tumor
Xenograft model.
[0069] Figure 28 depicts the effects of KPU-2 alone and in combination with
Paclitaxel on estimated tumor weight in the rnurine mammary fat pad implanted
MDA-231
Human Breast Tumor model.
[0070] Figure 29 depicts effects of A. KPU-2, B. KPU-35 and C. t-butyl-
phenylahistin alone and in combination with Paclitaxel in the Murine Melanoma
B16 F10
Metastatic Tumor Model.
[0071] Figure 30 depicts effects of I~PU-35 and KPU-02 on tumor vasculature in
the dorsal sl~infold chamber of Figure 30.
[0072] Figure 31 depicts effect of KPU-02 in combination with CPT-11 on the
estimated tumor weight in the HT-29 human colon tumor xenograft model.
[0073] Figure 32 depicts effect of Kl'U-02 in combination with CPT-11 on the
excised tumor weight in the HT-29 human colon tumor xenograft model.
[0074] Figure 33 depicts rapid tubulin depolymerization in HuVEC cells induced
by KPU-02 and KPU-35.
[0075] Figure 34 depicts effect of KPU-02 on monolayer permeability in HuVEC
cells.
[0076] Figure 35 depicts the effect of KPU-02 on tumor blood flow in the P22
rat
sarcoma model using the 125I-IAP technique.
-11-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0077] Figure 36 depicts the effect of KPU-02 IS mg/kg IP (expressed as
vehicle control) on blood flow in different tissues I and 24 hours post-dose.
[0078] Figure 37 depicts the tumor necrosis induced by KPU-02 7.5 and 15.0
mg/kg IP in the P22 rat sarcoma model
[0079] Figure 38 lists the activity of various tBu-dehydro-PLH derivatives at
HT-
29 cells.
[0080] Figure 39 depicts 3D QSAR (CoMFA) analysis of tBu-dehydro-PLH
derivatives.
[0081] Figure 40 depicts X-ray crystallographic analysis of tBu-dehydro-PLH
derivatives.
[0482] Figure 41 depicts the biologically activity of various phenylahistin
derivatives compared to colchicine.
[0083] Figure 42 depicts the effect on cell cycle progression of HeLa cells by
tBu-
dehydro-PLH (KPU-2) and KPU-3 5.
[0084] Figure 43 depicts the effect of dehydro-PLH and tBu-dehydro-PLH (KPU-
2) on drug-sensitive and drug-resistant tumor cell lines as compared to
paclitaxel.
[0085] Figure 44A depicts turbidity spectra of microtubule protein
polymerization
in the presence of DMSO drug vehicle (0), 1.25 ~.M (o), 2.5 p,M (-), and 5 pM
(o) KPU-02.
[0086] Figure 44B depicts turbidity spectra of microtubule protein
polymerization
in the presence of DMSO drug vehicle (0), 1.25 ~,M (o), 2.5 wM (-), and 5 ~.M
(o) CA4.
[0087] Figure 44C depicts turbidity spectra of microtubule protein
polymerization
in the presence of DMSO drug vehicle (0), I.25 ~.M (a), 2.5 ~M (-), and 5 ~.M
(o) CLC.
[0088] Figure 45 depicts inhibition of MT in the absence or presence of a
range of
KPU-02 (o),CA4 (o), and colchicine (0) concentrations.
[0089] Figure 46A depicts frequency histograms of mean microtubule lengths iya
vitro at steady state in the presence of KPU-02.
[0090] Figure 46B depicts , frequency histograms of mean microtubule lengths
in
vitro at steady state in the presence of CA4.
[0091] Figure 46C depicts frequency histograms of mean rnicrotubule lengths
ira
vitro at steady state in the presence of CLC.
-12-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
(0092] Figure 47A depicts electron micrographs of MAP-rich microtubules
formed in vitro at steady state in the presence of KPU-02.
[0093] Figure 47B depicts electron rnicrographs of MAP-rich microtubules
formed ih vitro at steady state in the presence of CA4.
(0094] Figure 47C depicts electron micrographs of MAP-rich microtubules
formed in vitro at steady state in the presence of CLC.
(0095] Figure 48 depicts a graphical summary of MT length decrease at steady
state in the presence of KPU-02, CA4, and colchicine.
[0096] Figure 49A depicts fluorescence emission spectra of tubulin in the
presence of increasing KPU-02.
(0097] Figure 49B depicts a fit to fluorescence emission maxima at 487 nm to
obtain the Kd of tubulin for KPU-02. The inset depicts residuals.
j0098J Figure 49C depicts double reciprocal transformation of the binding
data.
(0099] Figure 50 depicts the graphical results of a competitive inhibition
assay of
colchicine binding to tubulin with various concentrations of [3H]CLC in the
absence (0), or
presence of 10 ~M KPU-02 (o) or 10 ~M CA4 (o).
[0100] Figure 51 depicts log [compound] response curves for mitotic
progression
inhibition by KPU-02, CA4, and CLC in MCF7 cells cultured in the presence of
KPU-02 (o),
CA4 (a), and colchicine (0).
[0101] Figure 52 depicts immunofluorescence microscopy images of MCF7 cells.
a-d: Tubulin in control-(a) Tubulin in control, (b) KPU-02, (c) CA4, and' (d)
CLC treated
cells; e-h: DNA in control-(e) DNA in control, (f) KPU-02, (g) CA4, arid (h)
CLC treated
cells.
[0102] Figure 53A depicts immunofluorescence microscopy images of MCF7
cells treated with KPU-02
[0103] Figure 53B depicts immunofluorescence microscopy images of MCF7
cells treated with CA4.
[0104] Figure 53C depicts immunofluorescence microscopy images of MCF7
cells treated with CLC.
-13-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0105] Figure 54A depicts immunofluorescence microscopy images of MCF7
cells treated with KPU-02
[0106] Figure 54B depicts immunofluorescence microscopy images of MCF7
cells treated with CA4.
[0107] Figure 54C depicts irnmunofluorescence microscopy images of MCF7
cells treated with CLC.
[0108] In certain Figures, compounds are identified using an alternative
designation. A complete chart to convert these alternative designations is as
follows:
Alternative designationDesi nation used
herein
NPI-2350 ~- hen lahistin
NPI-2352 KPU-O 1
NPI-2353 ICPU-03
NPI-2354 KPU-04
NPI-2355 KPU-OS
NPI-2356 KPU-06
NPI-2357 KPU-07
NPI-2358 KPU-02
NPI-2359 I~PU-08
NPI-2360 I~3'U-09
NPI-2361 KPU-10
NPI-2362 KPU-11
NPI-2363 KPU-12
NPI-2364 I~1'U-13
NPI-2365 KPU-14
NPI-2366 KPU-15
NPI-2367 I~PU-16
NPI-2368 KPU-17
NPI-2369 KPU-I 8
NPI-2370 KPU-19
NPI-237.1 KPU-21
NPI-2372 KPU-22
NPI-23 73 KPU-23
NPI-2374 KPU-24
NPI-2375 KPU-25
NPI-2376 KPU-28
NPI-2377 KPU-26
NPI-2378 KPU-27
NPI-2379 KPU-29
NPI-23 80 I~PU-20
NPI-23 81 KPU-30
NPI-2382 KPU-31
NPI-23 83 KPU-32
NPI-2384 KPU-33
NPI-2385 KPU-34
-14-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Alternative desi Desi nation
nation used herein
NPI-2386 _
KPU_-35
NPI-2387 KPU-36
NPI-2388 KPU-37
NPI-2389 KPU-38
NPI-2390 KPU-39
NPI-2391 KPU-40
NPI-2392 I~PU-41
NPI-2393 KPU-42
NPI-2394 KPU-43
NPI-2395 KPU-44
NPI-2396 KPU-45
NPI-2397 KPU-46
NPI-2398 KPU-47
NPI-2399 KPU-48
NPI-2400 KPU-49
NPI-2401 KPU-50
NPI-2402 ICPU-51
NPI-2403 I~L'U-52
NPI-2404 KPU-53
NPI-2405 KPU-54
NPI-2406 I~PU-55
NPI-2407 I~I'U-56
NPI-2408 I~I'U-57
NPI-2409 KPU-58
NPI-2410 I~PU-59
NPI-2411 KPU-60
NPI-2412 KPU-61
NPI-2413 KPU-62
NPI-2414 KPU-63
NPI-2415 KPU-64
NPI-2416 KPU-65
NPI-2417 KPU-66
NPI-2418 KPU-67
NPI-2419 KPU-68
NPI-2420 KPU-69
NPI-2421 KPU-70
NPI-2422 KPU-71
NPI-2423 KPU-72
NPI-2424 KPU-73
NPI-2425 KPU-74
NPI-2426 K.PU-75
NPI-2427 KPU-76
NPI-2428 I~I'U-77
NPI-2429 ICPU-79
NPI-2430 KPU-80
NPI-2431 KPU-81
NPI-2432 KPU-82
-15-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Alternative deli Desi ~ nation used
nation herein
NPI-2433 ICPU-83
NPI-2434 ICPU-84
NPI-2435 KPU-86
NPI-2436 KPU-87
NPI-2437 KPU-88
NPI-2438 KPU-89
NPI-2439 KPU-90
NPI-2440 KPU-9I
NPI-2441 I~I'U-92
NPI-2442 KPU-80
NPI-2455 ICPU-94
NPI-2456 KPU-95
NPI-2457 KPU-96
NPI-2458 KPU-97
NPI-2459 KPU-98
NPI-2460 t-bu I hen Iahistin
Detailed Description of the Preferred Embodiment
[0109] Each reference cited herein, including the U.S. patents cited herein,
is to
be considered incorporated by reference in its entirety into this
specification, to the full extent
permissible by law. U.S. Patent Application No. 10/632,531, and PCT
Application No.
PCTUS03/24232, both filed on August 1, 2003, and both entitled,
"DEHYDROPHENYLAHIST1NS AND ANALOGS THEREOF AND THE SYNTHESIS
OF DEHYDROPHENYLAHISTINS AND ANALOGS THEREOF," are incorporated herein
by reference in their entireties.
[0110] The disclosure pxovides methods for the synthetic preparation of
compounds, including novel compounds, including dehydrophenylahistin and
dehydrophenylahistin analogs, and provides methods for producing
pharmaceutically
acceptable cell cycle inhibitors, antitumor agents and antifungal agents in
relatively high
yield, wherein said compounds and/or their derivatives are among the active
ingredients in
these cell cycle inhibitors, antitumor agents and antifungal agents. Other
objects include
providing novel compounds not obtainable by currently available, non-synthetic
methods. It
is also an object to provide a method of treating cancer, particularly human
cancer,
comprising the step of administering an effective tumor-growth inhibiting
amount of a
member of a class of new anti-tumor compounds. This invention also provides a
method for
preventing or treating a pathogenic fungus in a subject which involves
administering to the
-16-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
subject an effective anti-fungal amount of a member of a class of new anti-
fungal
compounds, e.g., administering a dehydrophenylahistin or its analog in an
amount and
manner which provides the intended antifungal effect. In the preferred
embodiment of the
compounds and methods of making and using such compounds disclosed herein, but
not
necessarily in all embodiments of the present invention, these objectives are
met.
[0111] Disclosed herein, also, are compounds, and methods of producing a class
of compounds, wherein the compounds are represented by Formula (I):
Z~~ ~1~'~~~ R2\ 1 / /
' N
/ N~~, N-
R1 ~4 X 'K3 6
2
(~
[0112] wherein:
[0113] Rl, R4, and R~, are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated C1-C24
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, vitro, azido, substituted vitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, alkylthio, halogenated alkyl
including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated
C1-C2a
allcenyl, cycloalkyl, cycloalkenyl, allcoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, vitro, azido, substituted
vitro, phenyl, and
substituted phenyl groups;
[0114] Rl' and Rl" are independently selected from the group consisting of a
hydrogen atom, a halogen atom, and saturated C1-CZ4 alkyl, unsaturated CI-Cz4
allcenyl,
cycloallcyl, cycloalkenyl, allcoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, vitro, azido, substituted vitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, alkylthio, halogenated alkyl
including
-17-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C2-C24 alkyl, unsaturated
C1-C24
alkenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, vitro, azido, substituted
vitro, phenyl, and
substituted phenyl groups;
[0115j R, Rl' and Rl" are either covalently bound to one another or are not
covalently bound to one another;
(0116] R2, R3, and RS are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated C~-C12 alkyl, unsaturated C1-C12
alkenyl, acyl,
cycloalkyl, alkoxy, cycloalkoxy, aryl, substituted aryl, heteroaryl,
substituted heteroaryl,
amino, substituted amino, vitro, and substituted vitro groups, sulfonyl and
substituted
sulfonyl groups;
[0117] Xl and X2 are separately selected from the group consisting of an
oxygen
atom, a nitrogen atom, and a sulfur atom, each either unsubstituted or
substituted with a RS
group, as defined above;
[011] Y is selected from the group consisting of a nitrogen atom, a
substituted
nitrogen atom with a RS group from above, an oxygen atom, a sulfur atom, a
oxidized sulfur
atom, a methylene group and a substituted methylene group;
[0119] n is an integer equal to zero, one or two;
[0120] Z, for each separate n, if non-zero, and Zl, Zz, Z3 and Z4 are each
separately selected from a carbon atom, a sulfur atom, a nitrogen atom or an
oxygen atom;
and
[0121] the dashed bonds may be either single or double bonds.
(0122] The method comprises a method of producing compounds of Formula (I)
by the steps of:
[0123] reacting a diacyldiketopiperazine with a first aldehyde to produce an
intermediate compound; and
[0124] reacting said intermediate compound with a second aldehyde to produce
said class of compounds with said generic structure, wherein
-18-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0125] said first aldehyde and said second aldehydes are selected from the
group
consisting of an oxazolecarboxaldeyhyde, imidazolecarboxaldehyde, a
benzaldehyde,
imidazolecarboxaldehyde derivatives, and benzaldehyde derivatives, thereby
forming a
compound of Formula (n wherein
[0126] Rl, R4, and R6, are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated CI-C24 alkyl, unsaturated Cl-C24
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloallcoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, nitro, azido, substituted nitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, alkylthio, halogenated
allcyl including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated C1-C24 alkyl, unsaturated
CI-C24
alkenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, nitro, azido, substituted
nitro, phenyl, and
substituted phenyl groups;
[0127] Rl' and Rl" are independently is selected from the group consisting of
a
hydrogen atom, a halogen atom, and saturated C~-Cz4 alkyl, unsaturated C~-C24
alkenyl,
cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted aryl,
heteroaryl, substituted
heteroaryl, amino, substituted amino, nitro, azido, substituted nitro, phenyl,
and substituted
phenyl groups, hydroxy, carboxy, -CO-O-R7, cyano, alkylthio, halogenated alkyl
including
polyhalogenated alkyl, halogenated carbonyl, and carbonyl -CCO-R7, wherein R7
is selected
from a hydrogen atom, a halogen atom, and saturated CI-Cz4 allcyl, unsaturated
CI-C24
alkenyl, cycloalkyl, cycloalkenyl, alkoxy, cycloalkoxy, aryl, substituted
aryl, heteroaryl,
substituted heteroaryl, amino, substituted amino, nitro, azido, substituted
nitro, phenyl, and
substituted phenyl groups;
[0128] RZ, R3, and RS are each separately selected from the group consisting
of a
hydrogen atom, a halogen atom, and saturated C1-C12 alkyl, unsaturated C1-C12
alkenyl, acyl,
cycloalkyl, alkoxy, cycloalkoxy, aryl, substituted aryl, heteroaryl,
substituted heteroaryl,
amino, substituted amino, nitro, and substituted nitro groups,' sulfonyl and
substituted
sulfonyl groups;
-19-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
(0129] X~ and X2 are separately selected from the group consisting of an
oxygen
atom, a nitrogen atom and a sulfur.atom, and
[0130] Y is selected from the group consisting of a nitrogen atom, a
substituted
nitrogen atom with a RS group from above, an oxygen atom, a sulfur atom, a
oxidized sulfur
atom, a methylene group and a substituted methylene group;
[0131] Z, for each separate n, if non-zero, and Zl, ZZ, Z3 and Z4 are each
separately selected from a carbon atom, a sulfur atom, a nitrogen atom or an
oxygen atom;
and
[0132] the dashed bonds may be either single or double bonds.
[0133] Also provided are pharmaceutically acceptable salts and pro-drug esters
of
the compound of Formulae (I) and (II) and provides methods of synthesizing
such
compounds by the methods disclosed herein.
(0134] The term "pro-drug ester," especially when referring to a pro-drug
ester of
the compound of Formula (I) synthesized by the methods disclosed herein,
refers to a
chemical derivative of the compound that is rapidly transformed ih vivo to
yield the
compound, for example, by~hydrolysis in blood or inside tissues. The term "pro-
drug ester"
refers to derivatives of the compounds disclosed herein formed by the addition
of any of
several ester-forming groups that are hydrolyzed under physiological
conditions. Examples
of pro-drug ester groups include pivoyloxymethyl, acetoxymethyl, phthalidyl,
indanyl and
methoxymethyl, as well as other such groups known in the art, including a (5-R-
2-oxo-1,3-
dioxolen-4-yl)methyl group. Other examples of pro-drug ester groups can be
found in, for
example, T. Higuchi and V. Stella, in "Pro-drugs as Novel Delivery Systems",
Vol. 14,
A.C.S. Symposium Series, American Chemical Society (1975); and "Bioreversible
Carriers
in Drug Design: Theory and Application", edited by E. B. Roche, Pergamon
Press: New
Yorlc, 14-21 (197) (providing examples of esters useful as prodrugs for
compounds
containing carboxyl groups).
[0x35] The teen "pro-drug ester," as used herein, also refers 'to a chemical
derivative of the compound that is rapidly transformed i~z vivo to yield the
compound, for
example, by hydrolysis in blood. The term "pro-drug ester" refers to
derivatives of the
compounds disclosed herein formed by the addition of any of several ester-
forming groups
-20-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
that are hydrolyzed under physiological conditions. Examples of pro-drug ester
groups
include pivoyloxyrnethyl, acetoxymethyl, phthalidyl, indanyl and
methoxymethyl, as well as
other such groups known in the art, including a (5-R-2-oxo-1,3-dioxolen-4-
yl)methyl group.
Other examples of pro-drug ester groups can be found in, for example, T.
Higuchi and V.
Stella, in "Pro-drugs as Novel Delivery Systems", Vol. 14, A.C.S. Symposium
Series,
American Chemical Society (I975); and "Bioreversible Carriers in Drug Design:
Theory and
Application", edited by E. B. Roche, Pergamon Press: New York, 14-21 (1987)
(providing
examples of esters useful as prodrugs for compounds containing carboxyl
groups).
(0136] The term "pharmaceutically acceptable salt," especially when referring
to a
pharmaceutically acceptable salt of the compound of Formula (1) synthesized by
the methods
disclosed herein, refers to any pharmaceutically acceptable salts of a
compound, and
preferably refers to an acid addition salt of a compound. Preferred examples
of
pharmaceutically acceptable salt are the alkali metal salts (sodium or
potassium), the alkaline
earth metal salts (calcium or magnesium), or ammonium salts derived from
ammonia or from
pharmaceutically . acceptable organic amines, for example C1-C7 alkylamine,
cyclohexylamine, triethanolamine, ethylenediamine or tris-(hydroxyrnethyl)-
aminomethane.
With respect to compounds synthesized by the method that are basic amines, the
preferred
examples of pharmaceutically acceptable salts are acid addition salts of
pharmaceutically
acceptable inorganic or organic acids, for example, hydrohalic, sulfuric,
phosphoric acid or
aliphatic or aromatic carboxylic or sulfonic acid, for example acetic,
succinic, lactic, malic,
tartaric, citric, ascorbic, nicotinic, methanesulfonic, p-toluensulfonic or
naphthalenesulfonic
acid.
[0137] The term "pharmaceutically acceptable salt," as used herein, also
refers to
any pharmaceutically acceptable salts of a compound, and preferably refers to
an acid
addition salt of a compound. Preferred examples of pharmaceutically acceptable
salt are the
alkali metal salts (sodium or potassium), the alkaline earth metal salts
(calcium or
magnesium), or ammonium salts derived from ammonia or from pharmaceutically
acceptable
organic amines, for example C1-C7 allcylamine, cyclohexylamine,
triethanolamine,
ethylenediamine or tris-(hydroxymethyl)-aminomethane. With respect to
compounds that are
basic amines, the preferred examples of pharmaceutically acceptable salts are
acid addition
-21-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
salts of pharmaceutically acceptable inorganic or organic acids, for example,
hydrohalic,
sulfuric, phosphoric acid or aliphatic or aromatic carboxylic or sulfonic
acid, for example
acetic, succinic, lactic, malic, tartaric, citric, ascorbic, nicotinic,
methanesulfonic, p-
toluensulfonic or naphthalenesulfonic acid.
[0138] Preferred pharmaceutical compositions disclosed herein include
pharmaceutically acceptable salts and pro-drug esters of the compound of
Formula (I)
synthesized by the method disclosed herein. Accordingly, if the manufacture of
pharmaceutical formulations involves intimate mixing of the pharmaceutical
excipients and
the active ingredient in its salt form, then it is preferred to use
pharmaceutical excipients
which are non-basic, that is, either acidic or neutral excipients.
[0139] In preferred embodiments of the methods of the compounds disclosed
herein, a relatively rigid, planar pseudo three-ring structure may be formed.
To stabilize such
a relatively rigid, planar pseudo three-ring structure, R3 may preferably be
chosen to be
hydrogen.
[0140] In other preferable embodiments of the compounds and methods described
herein, n is equal to zero or one, more preferable one, and Z2, Z3, and Z4,
and each separately
selected from an oxygen atom, a nitrogen atom, and a carbon atom, more
preferable at one
least one of ZZ, Z3, and Z4 being a carbon atom, and most preferable at least
two of ZZ, Z3,
and Z4 being a carbon atom. All Z's may simultaneous be carbon atoms.
[0141] Still other preferred embodiments of the methods and compositions
disclosed herein involve compounds having the structures of Formulae (Ia) and
(Ib), below:
X~ R4
R~ Z; R2v
r ~ Z1 N ~ ~' y R
N N
i
R~ X2 ~3 6
(Ia)
R~ ~ R X~ R4
~Z= I:1~ 2 / i Y R
~N _ 5
Z2~ ,' / N N
R~ 'Z3 X ~3 6
2
(~)
-22-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[OI42] wherein the variable groups are as defined herein.
[0143] The term "halogen atom," as used herein, means any one of the radio-
stable atoms of column 7 of the Periodic Table of the Elements, i.e.,
fluorine, chlorine,
bromine, or iodine, with fluorine and chlorine being preferred.
[0144] The term "alkyl," as used herein, means any unbranched or branched,
substituted or unsubstituted, saturated hydrocarbon, with C1-C~ unbranched,
saturated,
unsubstituted hydrocarbons being preferred, with methyl, ethyl, iosbutyl, and
tert-butyl being
most preferred. Among the substituted, saturated hydrocarbons, C1-C6 mono- and
di- and
per-halogen substituted saturated hydrocarbons and amino-substituted
hydrocarbons are
preferred, with perfluromethyl, perchloromethyl, perfluoro-tert-butyl, and
perchloro-tert-butyl
being the most preferred. The term "substituted" has its ordinary meaning, as
found in
numerous contemporary patents from the related art. See, for example, U.S.
Patent Nos.
6,583,143, 6,509,331; 6,506,787; 6,500,825; 5,922,683; 5,886,210; 5,874,443;
and 6,350,759.
Specifically, the definition of substituted is as broad as that provided in
U.S. Patent No.
6,583,143, which defines the term substituted as any groups such as alkyl,
aryl, arylalkyl,
heteroaryl, heteroarylalkyl, hetexocycle and heterocyclealkyl, wherein at
least one hydrogen
atom is replaced with a substituent. The term "substituted" is also as broad
as the definition
provided in U.S. Patent No. 6,509,331, which defines the term "substituted
alkyl" such that it
refers to an alkyl group, preferably of from 1 to 10 carbon atoms, having from
1 to 5
substituents, and preferably 1 to 3 substituents, selected from the group
consisting of allcoxy,
substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl,
substituted cycloalkenyl,
acyl, acylamino, acyloxy, amino, substituted amino, arninoacyl, aminoacyloxy,
oxyacylamino, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, keto,
thioketo, thiol,
thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy,
heterocyclic,
heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted
alkyl, --SO-
aryl, --SO-heteroaryl, --S02-alkyl, --S02-substituted alkyl, --SO2-aryl and -
S02-heteroaryl.
The other above-listed patents also provide standard definitions for the term
"substituted" that
are well-understood by those of skill in the art. The term "cycloalkyl" refers
to any non-
aromatic hydrocarbon ring, preferably having five to twelve atoms comprising
the ring. The
-23-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
term "acyl" refers to alkyl or aryl groups derived from an oxoacid, with an
acetyl group being
preferred.
[0145] The term "alkenyl," as used herein, means any unbranched or branched,
substituted or unsubstituted, unsaturated hydrocarbon including
polyunsaturated
hydrocarbons, with C1-C6 unbranched, mono-unsaturated and di-unsaturated,
unsubstituted
hydrocarbons being preferred, and mono-unsaturated, di-halogen substituted
hydrocarbons
being most .preferred. In the RI and Rq positions, of the compound of
structure (~ a z-
isoprenyl moiety is particularly preferred. The term "cycloalkenyl" refers to
any non-
aromatic hydrocarbon ring, preferably having five to twelve atoms comprising
the ring.
[0146] The terms "aryl," "substituted aryl," "heteroaryl," and "substituted
heteroaryl," as used herein, refer to aromatic hydrocarbon rings, preferably
having five, six,
or seven atoms, and most preferably having six atoms comprising the ring.
"Heteroaryl" and
"substituted heteroaryl," refer to aromatic hydrocarbon rings in which at
least one
heteroatom, e.g., oxygen, sulfur, or nitrogen atom, is in the ring along with
at least one
carbon atom.
(0147] The term "alkoxy" refers to any unbranched, or branched, substituted or
unsubstituted, saturated or unsaturated ether, with CI-C6 unbranched,
saturated, unsubstituted
ethers being preferred, with methoxy being preferred, and also with dimethyl,
diethyl,
methyl-isobutyl, and methyl-tert-butyl ethers also being preferred. The term
"cycloalkoxy"
refers to any non-aromatic hydrocarbon ring, preferably having five to twelve
atoms
comprising the ring.
[0148] The terms "purified," "substantially purified," and "isolated" as used
herein refer to the compound being free of other, dissimilar compounds with
which the
compound is normally associated in its natural state, so that the compound of
the invention
comprises at least O.S%, 1%, 5%, 10%, or 20%, and most preferably at least 50%
or 75% of
the mass, by weight, of a given sample.
(0149] The compound of Formula (I] may be chemically synthesized or produced
from reagents known and available in the art. For example, modifications of
diacyldiketopiperazine (diacetyldiketopiperazine) have been described, for
example, by
Loughlin et al., 2000 Bioor~g Med Chen2 Lett 10:91 or by Brocchini et al. in
WO 95/21832.
-24-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
The diacyldiketopiperazine (diacetyldiketopiperazine) may be prepared, for
example, by
diacetylation of inexpensive 2,5-piperazinedione (TCI Cat. No. GO100, 25 g)
with sodium
acetate and sodium anhydride. The diacetyl structure of the activated
deketopiperazine can
be replaced with other acyl groups, to include carbamates such as Boc (t-
butoxycarbonyl), Z
(benzoyloxycarbonyl).
[0150] The imidazolecarboxaldehyde may be prepared, for example, according
the procedure disclosed in Hayashi et al., 2000 J Organic Chem 65: 8402 as
depicted below:
LL1A~, ,~~ Tas-~t ~~ C~BL
~'~"r GH~wrH~S~~~" ~~,..,r~
THF, ~H ~ pyridine,~'T~ ~ reflux~l~it)
10 G rt. ~)
68'~ 8B'~ 9B'~
~4~1 CC~CI~ Et~w3UCH~s3C5H
hJ~~H ~H ~ ~~t BuLi
EtOH,rt.~ ~H~CI~,rt~ THF,_70to-~ID~
98~ 6B~ 82~
~~~r SO~~Ig ~I H~~H~, ~,r. . DIBt~4LH
x~y~,,.QEt~ Et r, ~~~C~s3Et
I
I
O GHCh,reiuxC~ ~ retlu~940GH~~.,A-HH Toluene,-~~
~
77~ ~18'~ 59~
~~~~H MnQ2 _
~1.. HH acetone,rt ~ ~IH
''~r ~r~~
9.5~
t~i~Hii~,
OEt H~ ~I ~11~1e ~~ ~7~Me ~IBJ~LH
~HC~, retlux ~ ~ refluvy t ~~°G H".HH Toluene -5~°C
18 Z1 ~ (2 step) ~6~
Li~l H~
THF, rt
r41n02 E~~
~~~'~ (7 H -~ ~ C7
~I~NH acetan~ rt ~,.hJH
95°~ ~6
-25-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[OI52] The synthetic method disclosed herein may be preferably performed in
the
presence of cesium carbonate as a base in DMF and in a deoxygenated
atmosphere. The inert
atmosphere circumvents the probable oxidation of activated a-carbon atoms of
the
diketopiperazine ring during the treatment with cesium carbonate (see below)
as reported, for
example, by Watanabe et al., 18'h Intel°national Conga°ess of
Hete~ocyclic Chemistry i~a
Yokohama, Japan (30 July 2001), Abstract, page 225.
Air
Cs salt
~,.r > ' f.r
R H DMF R (OH ~
Air-oxidation of Activated Carbonyl Compounds with Cesium Salts
[0153] Other embodiments of the synthetic method involve modifications to the
compounds used in or otherwise involved in the synthesis of compounds
represented by
Formula (17. Such derivatives may include modifications to the phenyl ring,
introduction of
other aromatic ring systems, position of the aromatic ring, alterations to the
imidazole ring
system and/or further modifications to the 5-position on the imidazole ring.
Examples of
such modifications are discussed, for example, in Example 4. The result of
such
modifications includes increased nitrogen content of the phenyl ring and/or
the compound
which may increase compound solubility. Other modifications may incorporate
derivatives
of known tubulin inhibitors, thereby mimicking the activity of the tubulin
inhibitors. Other
modifications rnay simplify the synthesis of the (3-ketoester involved in the
production of the
irnidazolecarboxaldehyde used in the methods disclosed herein.
Pharmaceutical Compositions
[0154] The present invention also encompasses the compounds disclosed herein,
optionally and preferably produced by the methods disclosed herein, in
pharmaceutical
compositions comprising a pharmaceutically acceptable carrier prepared for
storage and
subsequent administration, which have a pharmaceutically effective amount of
the products
disclosed above in a pharmaceutically acceptable carrier or diluent.
Acceptable carriers or
diluents for therapeutic use are well known in the pharmaceutical art, and are
described, for
example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R.
Gennaro edit.
195). Preservatives, stabilizers, dyes and even flavoring agents may be
provided in the
-26-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
pharmaceutical composition. For example, sodium benzoate, ascorbic acid and
esters of p-
hydroxybenzoic acid may be added as preservatives. In addition, antioxidants
and
suspending agents may be used.
[0155) The dehydrophenylahistin or dehydrophenylahistin analog compositions
may be formulated and used as tablets, capsules, or elixirs for oral
administration;
suppositories for rectal administration; sterile solutions, suspensions for
injectable
administration; patches for transdermal administration, and sub-dermal
deposits and the like.
Injectables can be prepared in conventional forms, either as liquid solutions
or suspensions,
solid forms suitable for solution or suspension in liquid prior to injection
or infusion, or as
emulsions. Suitable excipients are, for example, water, saline, dextrose,
mannitol, lactose,
lecithin, albumin, sodium glutamate, cysteine hydrochloride, human serum
albumin and the
like. In addition, if desired, the injectable pharmaceutical compositions may
contain minor
amounts of nontoxic auxiliary substances, such as wetting agents, pH buffering
agents, and
the like. If desired, absorption enhancing preparations (for example,
liposomes), may be
utilized.
[0156] Pharmaceutical formulations for parentefal administration include
aqueous
solutions of the active compounds in water-soluble form. Additionally,
suspensions of the
active compounds may be prepared as appropriate oily injection suspensions.
Suitable
lipophilic solvents or vehicles include fatty oils such as sesame oil, or
other organic oils such
as soybean, grapefruit or almond oils, or synthetic fatty acid esters, such as
ethyl oleate or
triglycerides, or liposomes. Aqueous injection suspensions may contain
substances which
increase the viscosity of the suspension, such as sodium carboxymethyl
cellulose, sorbitol, or
dextran. Optionally, the suspension may also contain suitable stabilizers or
agents that
increase the solubility of the compounds to allow for the preparation of
highly concentrated
solutions.
j0157] Pharmaceutical preparations for oral use may be obtained by combining
the active compounds with solid excipient, optionally grinding a resulting
mixture, and
processing the mixture of granules, after adding suitable auxiliaries, if
desired, to obtain
tablets or dragee cores. Suitable excipients are, in particular, fillers such
as sugars, including
lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for
example, maize
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth,
methyl cellulose,
hydroxypropylmethyl-cellulose, sodium caxboxymethylcellulose, and/or
polyvinylpyrrolidone
(PVP). if desired, disintegrating agents may be added, such as the cross-
linked polyvinyl
pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
Dragee cores are
provided with suitable coatings. For this purpose, concentrated sugar
solutions may be used,
which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol
gel,
polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable
organic solvents
or solvent mixtures. Dyestuffs or pigments may be added to the tablets or
dragee coatings for
identification or to characterize different combinations of active compound
doses. For this
purpose, concentrated sugar solutions may be used, which may optionally
contain gum
arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or
titanium dioxide,
lacquer solutions, and suitable organic solvents or solvent mixtures.
Dyestuffs or pigments
may be added to the tablets or dragee coatings for identification or to
characterize different
combinations of active compound doses. Such formulations can be made using
methods
known in the art (see, for example, U.S. Patent Nos. 5,733,888 (injectable
compositions);
5,726,181 (pooxly water soluble compounds); 5,707,641 (therapeutically active
proteins or
peptides); 5,667,809 (lipophilic agents); 5,576,012 (solubilizing polymeric
agents); 5,707,615
(anti-viral formulations); 5,683,676 (particulate medicaments); 5,654,286
(topical
formulations); 5,688,529 (oral suspensions); 5,445,829 (extended release
formulations);
5653,987 (liquid formulations); 5,641,515 (controlled release formulations)
and 5,601,845
(spheroid formulations).
[0158] Further disclosed herein are various pharmaceutical compositions well
known in the pharmaceutical axt for uses that include intraocular, intranasal,
and
intraauricular delivery. Pharmaceutical formulations include aqueous
ophthalmic solutions
of the active compounds in water-soluble form, such as eyedrops, or in gellan
gum (Shedden
et al., 2001 Clin They 23(3):440-50) or hydrogels (Mayer et al., 1996
Ophthalmologica
210:101-3); ophthalmic ointments; ophthalmic suspensions, such as
microparticulates, drug-
containing small polymeric particles that axe suspended in a liquid carrier
medium (Joshi, A.,
1994 J Ocul Pharrraacol 10:29-45), lipid-soluble formulations (Alm et al.,
1989 Pr~og Clira
Biol Res 312:447-58), and microspheres (Mordenti, 1999 Toxicol Sci 52:101-6);
and ocular
-28-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
inserts. Such suitable pharmaceutical formulations are most often and
preferably forinulated
to be sterile, isotonic and buffered for stability and comfort. Pharmaceutical
compositions
may also include drops and sprays often prepared to simulate in many respects
nasal
secretions to ensure maintenance of normal ciliary action. As disclosed in
Remington's
Pharmaceutical Sciences (Mack. Publishing, 18t~' Edition), and well-known to
those skilled in
the art, suitable formulations are most often and preferably isotonic,
slightly buffered to
maintain a pH of S.5 to 6.5, and most often and preferably include
antimicrobial preservatives
and appropriate drug stabilizers. Pharmaceutical formulations for
intraauricular delivery
include suspensions and ointments for topical application in the ear. Common
solvents for
such aural formulations include glycerin and water.
(0159] When used as a cell cycle inhibitor, a tumor-growth-inhibiting, or a
fungus-growth-inhibiting compound, the compound of Formula (I) can be
administered by
either oral or a non-oral pathways. When administered orally, it can be
administered in
capsule, tablet, granule, spray, syrup, or other such form. When administered
non-orally, it
can be administered as an aqueous suspension, an oily preparation or the like
or as a drip,
suppository, salve, ointment or the like, when administered via injection or
infusion,
subcutaneously, intreperitoneally, intravenously, intramuscularly, or the
like. Similarly, it
may be administered topically, rectally, or vaginally, as deemed appropriate
by those of skill
in the art for bringing the compound into optimal contact with a tumor, thus
inhibiting the
growth of the tumor. Local administration at the site of the tumor is also
contemplated, either
before or after tumor resection, as are controlled release formulations, depot
formulations,
and infusion pump delivery.
Methods of Administration
[0160] The present invention also encompasses methods for making and for
administering the disclosed chemical compounds and the disclosed
pharmaceutical
compositions. Such disclosed methods include, among others, (a) administration
though oral
pathways, which administration includes administration in capsule, tablet,
granule, spray,
syrup, or other such forms; (b) administration through non-oral pathways,
which
administration includes administration as an aqueous suspension, an oily
preparation or the
like or as a drip, suppository, salve, ointment or the like; administration
via injection or
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
infusion, subcutaneously, intraperitoneally, intravenously, intramuscularly,
intradermally, or
the like; as well as (c) administration topically, (d) administration
rectally, or
(e) administration vaginally, as deemed appropriate by those of skill in the
art for bringing the
compound into contact with living tissue; arid (f) administration via
controlled released
formulations, depot formulations, and infusion pump delivery. As further
examples of such
modes of administration and as further disclosure of modes of administration,
disclosed
herein are various methods for administration of the disclosed chemical
compounds and
pharmaceutical compositions including modes of administration through
intraocular,
intranasal, and intraauricular pathways.
[OI61] The pharmaceutically effective amount of the dehydrophenylahistin or
dehydrophenylahistin analog composition required as a dose will depend on the
route of
administration, the type of animal, including human, being treated, and the
physical
characteristics of the specific animal under consideration. The dose can be
tailored to
achieve a desired effect, but will depend on such factors as weight, diet,
concurrent
medication and other factors which those skilled in the medical arts will
recognize.
[0162] In practicing the methods, the products or compositions can be used
alone
or in combination with one another, or in combination with other therapeutic
or diagnostic
agents. For example, as disclosed herein, the compounds disclosed herein are
effective in the
treatment of cancer when used in combination with other actives, specifically
other
chemotherapeutics, for example biologics and the specific chemotherapeutics
CPT-11,
Taxotene (docataxel) and paclitaxel. The compounds disclosed herein are also
effective in
the treatment of cancer when used in combination with other actives, including
anti-vascular
agents, anti-angiogenenic agents, such as Erbuitux (Imclone/bristol-Myers) and
Iressa
(AstraZeneca), other VEGF inhibitors and biologics, more specifically, at
least one anti-
VEGF antibodies, especially monoclonal antibodies to the VEGF receptor,
including DC101,
a rat monoclonal antibody, which blocks the mouse VEGF receptor 2 (flk-1).
Such
combinations may be utilized in vivo, ordinarily in a mammal, preferably in a
human, or in
vitro. T11 employing them in vivo, the disclosed compounds, alone or in
combination with
other chemotherapeutics or other biologic products, may be administered to the
mammal in a
variety of ways, including parenterally, intravenously, via infusion or
injection,
-30-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
subcutaneously, intramuscularly, colonically, rectally, vaginally, nasally or
intraperitoneally,
employing a variety of dosage forms. Such methods may also be applied to
testing chemical
activity ih wivo.
[0163] As will be readily apparent to one skilled in the art, the useful ire
vivo
dosage to be administered and the particular mode of administration will vary
depending
upon the age, weight and mammalian species treated, the particular compounds
employed,
and the specific use for which these compounds are employed. The determination
of
effective dosage levels, that is the dosage levels necessary to achieve the
desired result, can
be accomplished by one skilled in the art using routine pharmacological
methods. Typically,
human clinical applications of products are commenced at lower dosage levels,
with dosage
level being increased until the desired effect is achieved. Alternatively,
acceptable ih vita°o
studies can be used to establish useful doses and routes of administration of
the compositions
identified by the present methods using established pharmacological methods.
[0164] In non-human animal studies, applications of potential products are
commenced at higher dosage levels, with dosage being decreased until the
desired effect is no
longer achieved or adverse side effects disappear. The dosage may range
broadly, depending
upon the desired affects and the therapeutic indication. Typically, dosages
may be bettueen
about 10 microgramrkg and 100 mg/kg body weight, preferably between about 100
microgram/kg and 10 mg/kg body weight. Alternatively dosages may be based and
calculated upon the surface area of the patient, as understood by those of
skill in the art.
Administration may be oral on an every third day, every other day, daily,
twice daily, or thrice
daily basis.
[0165] The exact formulation, route of administration and dosage can be chosen
by the individual physician in view of the patient's condition. See for
example, Fingl et al., in
The Pharmacological Basis of Therapeutics, 1975. It should be noted that the
attending
physician would know how to and when to terminate, interrupt, or adjust
administration due
to toxicity, or to organ dysfunctions. Conversely, the attending physician
would also know to
adjust treatment to higher levels if the clinical response were not adequate
(precluding
toxicity). The magnitude of an administrated dose in the management of the
disorder of
interest will vary with the severity of the condition to be treated and to the
route of
-31-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
administration. The severity of the condition may, for example, be evaluated,
in part, by
standard prognostic evaluation methods. Further, the dose and perhaps dose
frequency, will
also vary according to the age, body weight, and response of the individual
patient. A
program comparable to that discussed above may be used in veterinary medicine.
[0166] Depending on the specific conditions being treated, such agents may be
formulated and administered systemically or locally. A variety of techniques
for formulation
and administration may be found in Remington's Pharmaceutical Sciences, 18th
Ed., Mack
Publishing Co., Easton, PA (1990). Suitable administration routes may include
oral, rectal,
transdermal, vaginal, transmucosal, or intestinal administration; parenteral
delivery, including
intramuscular, subcutaneous, intramedullary injections, as well as
intrathecal, direct
intraventricular, intravenous, via infusion, intraperitoneal, intranasal, or
intraocular
inj ections.
[0167] For injection or infusion, the agents may be formulated in aqueous
solutions, for example, in physiologically compatible buffers such as Hanks'
solution,
Ringer's solution, or physiological saline buffer. For such transmucosal
administration,
penetrants appropriate to the barrier to be permeated are used in the
formulation. Such
penetrants are generally known in the art. Use of pharmaceutically acceptable
carriers to
formulate the compounds herein disclosed for the practice of the invention
into dosages
suitable for systemic administration is within the scope of the invention.
With proper choice
of carrier and suitable manufacturing practice, the compositions disclosed
herein, in
particuXar, those formulated as solutions, may be administered parenterally,
such as by
intravenous injection or infusion. The compounds can be formulated readily
using
pharmaceutically acceptable carriers well known in the art into dosages
suitable for oral
administration. Such carriers enable the compounds to be formulated as
tablets, pills,
capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral
ingestion by a
patient to be treated.
[0168] Agents intended to be administered intracellularly may be administered
using techniques well known to those of ordinary slcill in the art. For
example, such agents
may be encapsulated into liposornes, then administered as described above. All
molecules
present in an aqueous solution at the time of liposome formation axe
incorporated into the
-32-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
aqueous interior. The Iiposornal contents are both protected from the external
micro-
environment and, because liposomes fuse with cell membranes, are efficiently
delivered into
the cell cytoplasm. Additionally, due to their hydrophobicity, small organic
molecules may
be directly administered intracellularly.
[0169] Determination of the effective amounts is well within the capability of
those skilled in the art, especially in light of the detailed disclosure
provided herein. In
addition to the active ingredients, these pharmaceutical compositions may
contain suitable
pharmaceutically acceptable carriers comprising excipients and auxiliaries
which facilitate
processing of the active compounds into preparations which can be used
pharmaceutically.
The preparations formulated for oral administration may be in the form of
tablets, dragees,
capsules, or solutions. The pharmaceutical compositions may be manufactured in
a manner
that is itself known, far example, by means of conventional mixing,
dissolving, granulating,
dragee-making, levitating, emulsifying, encapsulating, entrapping, or
lyophilizing processes.
[0170] Compounds disclosed herein can be evaluated for efficacy and toxicity
using known methods. For example, the toxicology of a particular compound, or
of a subset
of the compounds, sharing certain chemical moieties, may be established by
determining ih
vitro,toxicity towards a cell line, such as a mammalian, and preferably human,
cell line. The
results of such studies are often predictive of toxicity in animals, such as
mammals, or more
specifically, humans. Alternatively, the toxicity of particular compounds in
an animal model,
such as mice, rats, rabbits, or monkeys, may be determined using known
methods. The
efficacy of a particular compound may be established using several art
recognized methods,
such as ire vitro methods, animal models, ox human clinical trials. Art-
recognized irz vitro
models exist for nearly every class of condition, including the conditions
abated by the
compounds .disclosed herein, including cancer, cardiovascular disease and
various fungal
infections. Similarly, acceptable animal models may be used to establish
efficacy of
chemicals to treat such conditions. When selecting a model to determine
efficacy, the skilled
artisan can be guided by the state of the art to choose an appropriate model,
dose, and route of
administration, and regime. Of course, human clinical trials can also be used
to deterniine the
efficacy of a compound in humans.
-3 3-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[OI7Ij When used as an anti-cancer agent, or a tumor-growth-inhibiting
compound, the compounds disclosed herein may be administered by either oral or
a non-oral
pathways. When administered orally, it can be administered in capsule, tablet,
granule, spray,
syrup, or other such form. When administered non-orally, it can be
administered as an
aqueous suspension, an oily preparation or the like or as a drip, suppository,
salve, ointment
or the like, when administered via injection or infusion, subcutaneously,
intreperitoneally,
intravenously, intramuscularly, intradermally, or the like. Similarly, it may
be administered
topically, rectally, or vaginally, as deemed appropriate by those of skill in
the art for bringing
the compound into optimal contact with a tumor, thus inhibiting' the growth of
the tumor.
Local administration at the site of the tumor or other disease condition is
also contemplated,
either before or after tumor resection, or as part of an art-recognized
treatment of the disease
condition. Controlled release formulations, depot formulations, and infusion
pump delivery
are similarly contemplated.
(0172] When used as an anti-cancer agent or an anti-tumor agent, may be orally
or
non-orally administered to a human patient in the amount of about .0007 mg/day
to about
7,000 mg/day of the active ingredient, and more preferably about 0.07 mg/day
to about 70
mg/day of the active ingredient at, preferably, one time per day or, less
preferably, over two
to about ten times per day. Alternatively and also preferably, the compound
may preferably
be administered in the stated amounts continuously by, for example, an
intravenous drip.
Thus, for a patient weighing 70 kilograms, the preferred daily dose of the
active anti-tumor
ingredient would be about 0.0007 rng/lcg/day to about 35 mg/kg/day including
1.0 mg/kg/day
and 0.5 mg/kg/day, and more preferable, from 0.007 mg/kg/day to about 0.050
mg/kg/day,
including 0:035 mg/kg/day. Nonetheless, as will be understood by those of
skill in the art, in
certain situations it may be necessary to administer the anti-tumor compound
in amounts that
excess, or even far exceed, the above-stated, preferred dosage range to
effectively and
aggressively treat particularly advanced or lethal tumors.
[0173] When used as an antifungal agent the preferable amount of the
dehydxophenylahistin or its analog effective in the treatment or prevention of
a particular
fungal pathogen will depend in part on the characteristics of the fungus and
the extent of
infection, and can be determined by standard clinical techniques. hz vitro or
ih vivo assays
-34-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
may optionally be employed to help identify optimal dosage ranges. Effective
doses may be
extrapolated from dose-response curves derived from ih vitro analysis or
preferably from
animal models. The pxecise dosage level should be determined by the attending
physician or
other health care provider and will depend upon well known factors, including
route of
administration, and the age, body weight, sex and general health of the
individual; the nature,
severity and clinical stage of the infection; the use (or not) of concomitant
therapies.
[0174] The effective dose of the dehydrophenylahistin or its analog will
typically
be in the range of about 0.01 to about SO mg/kgs, preferably about 0.1 to
about 10 mg/kg of
mammalian body weight per day, administered in single or multiple doses.
Generally, the
compound may be administered to patients in need of such treatment in a daily
dose range of
about 1 to about 2000 mg per patient.
[0175] To formulate the dosage including the compounds disclosed herein as a
tumor-growth-inhibiting compound, known surface active agents, excipients,
smoothing
agents, suspension agents and pharmaceutically acceptable film-forming
substances and
coating assistants, and the like may be used. Preferably alcohols, esters,
sulfated aliphatic
alcohols, and the like may be used as surface active agents; sucrose, glucose,
lactose, starch,
crystallized cellulose, mannitol, light anhydrous silicate, magnesium
aluminate, magnesium
methasilicate aluminate, synthetic aluminum silicate, calcium carbonate,
sodium acid
carbonate, calcium hydrogen phosphate, calcium carboxymethyl cellulose, and
the like may
be used as excipients; magnesium stearate, talc, hardened oil and the like may
be used as
smoothing agents; coconut oil, olive oil, sesame oil, peanut oil, soya may be
used as
suspension agents or lubricants; cellulose acetate phthalate as a derivative
of a carbohydrate
such as cellulose or sugar, or methyiacetate-methacrylate copolymer as a
derivative of
polyvinyl 'may be used as suspension agents; and plasticizers such as ester
phthalates and the
like may be used as suspension agents. Tn addition to the foregoing preferred
ingredients,
sweeteners, fragrances, colorants, preservatives and the like may be added to
the administered
formulation of the compound, particularly when the compound is to be
administered orally.
[0176] The compositions disclosed herein in a pharmaceutical compositions may
also comprise a pharmaceutically acceptable carrier. Such compositions may be
prepared for
storage and for subsequent administration. Acceptable carriers or diluents for
therapeutic use
-35-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
are well known in the pharmaceutical art, and are described, for example, in
Remington's
Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro edit. 1985). For
example,
such compositions may be formulated and used as tablets, capsules or solutions
for oral
administration; suppositories for rectal or vaginal administration; sterile
solutions or
suspensions for injectable administration. Injectables can be prepared in
conventional forms,
either as liquid solutions or suspensions, solid forms suitable for solution
or suspension in
liquid prior to injection or infusion, or as emulsions. Suitable excipients
include, but are not
limited to, saline, dextrose, rnannitol, lactose, lecithin, albumin, sodium
glutamate, cysteine
hydrochloride, and the like. In addition, if desired, the injectable
pharmaceutical
compositions may contain minor amounts of nontoxic auxiliary substances, such
as wetting
agents, pH buffering agents, and the like. If desired, absorption enhancing
preparations (for
example, liposomes), may be utilized.
[0177] The pharmaceutically effective amount of the composition required as a
dose will depend on the route of administration, the type of animal being
treated, and the
physical characteristics of the specific animal under consideration. The dose
can be tailored
to achieve a desired effect, but will depend on such factors as weight, diet,
concurrent
medication and other factors which those skilled in the medical arts will
recognize.
[0178] The products or compositions, as described above, may be used alone or
in
combination with one another, or in combination with other therapeutic or
diagnostic agents.
Specifically, the disclosed compounds products may be used alone or in
combination with
other chemotherapeutics or biologics, including antibodies, for the treatment
of cancer, or in
combination with other anti-infective for the treatment of fungal infection.
These products or
compositions can be utilized ifa vivo or ih vitro. The useful dosages and the
most useful
modes of. administration will vary depending upon the age, weight and animal
treated, the
particular compounds employed, ~ and the specific use for which these
composition or
compositions are employed. The magnitude of a dose in the management or
treatment for a
particular disorder will vary with the severity of the condition to be treated
and to the route of
administration, and depending on the disease conditions and their severity,
the compositions
may be formulated and administered either systemically or locally. A variety
of techniques
-36-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
for formulation and administration may be found in Remington's Pharmaceutical
Sciences,
18th ed., Mack Publishing Co., Easton, PA (1990).
[0179] To formulate the compounds of Formula (17, preferably synthetically
produced according to the methods disclosed herein, as a cell cycle inhibitor,
a tumor-
growth-inhibiting, or an antifungal compound, known surface active agents,
excipients,
smoothing agents, suspension agents and pharmaceutically acceptable film-
forming
substances and coating assistants, and the like may be used. Preferably
alcohols, esters,
sulfated aliphatic alcohols, and the like may be used as surface active
agents; sucrose,
glucose, lactose, starch, crystallized cellulose, mannitol, light anhydrous
silicate, magnesium
aluminate, magnesium methasilicate aluminate, synthetic aluminum silicate,
calcium
carbonate, sodium acid carbonate, calcium hydrogen phosphate, calcium
carboxymethyl
cellulose, and the like may be used as excipients; magnesium stearate, talc,
hardened oil and
the like may be used as smoothing agents; coconut oil, olive oil, sesame oil,
peanut oil, soya
rnay.be used as suspension agents or lubricants; cellulose acetate phthalate
as a derivative of
a carbohydrate such as cellulose or sugar, or methyiacetate-methacrylate
copolymer as a
derivative of polyvinyl may be used as suspension agents; and plasticizers
such as ester
phthalates arid .the like may be used as suspension agents. In addition to the
foregoing
preferred ingredients, sweeteners, fragrances, colorants, preservatives and
the like may be
added to the administered formulation of the compound produced by the method,
particularly
when the compound is to be administered orally.
[0180] The cell cycle inhibitors, the antitumor agents, and the antifungal
agents
that may be produced by the method may be orally or non-orally administered to
a human
patient in the amount of about 0.001 mg/kg/day to about 10,000 mg/kg/day of
the active
ingredient, and more preferably about 0.1 mg/kg/day to about 100 mg/kg/day of
the active
ingredient at, preferably, once every three days on a cyclic basis, once every
other day, one
time per day, twice per day, or less preferably, over two to about ten times
per day.
Alternatively and also preferably, the compound produced by the method may
preferably be
administered in the stated amounts continuously by, for example, an
intravenous drip. Thus,
for the example of a patient weighing 70 kilograms, the preferred daily dose
of the active
anti-tumor ingredient would be about 0.07 mg/day to about 700 grams/day, and
more
-37-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
preferable; 7 mg/day to about 7 grams/day. Nonetheless, as will be understood
by those of
skill in the art, in certain situations it may be necessary to administer the
anti-tumor
compound produced by the method in amounts that excess, or even far exceed,
the above-
stated, preferred dosage range to effectively and aggressively treat
particularly advanced or
lethal tumors.
[0181] In the case of using the cell cycle inhibitor produced by methods as a
biochemical test reagent, the compound produced by methods of the invention
inhibits the
progression of the cell cycle when it is dissolved in an organic solvent or
hydrous organic
solvent and it is directly applied to any of various cultured cell systems.
Usable organic
solvents include, for example, methanol, methylsulfoxide, and the like. The
formulation can,
for example, be a powder, granular or other solid inhibitor, or a liquid
inhibitor prepared
using an organic solvent or a hydrous organic solvent. While a preferred
concentration of the
compound produced by the method of the invention for use as a cell cycle
inhibitor is
generally in the range of about 1 to about 100 ~g/ml, the most appropriate use
amount varies
depending on the type of cultured cell system and the purpose of use, as will
be appreciated
by persons of ordinary skill in the art. Also, in certain applications it may
be necessary or
preferred to persons of ordinary skill in the art to use an amount outside the
foregoing range.
[0182] From a pharmaceutical perspective, certain embodiments provide methods
for preventing or treating fungal infections and/or a pathogenic fungus in a
subject, involve
administering to the subject a composition including a.dehydrophenylahistin or
its analog, for
example, administering the dehydrophenylahistin or its analog in an amount and
manner
which provides the intended antifungal effect.
[0183] Other embodiments include the treatment or prevention of infection in a
patient by a pathogenic fungus such as those listed above or referred to
below.
[0184] Another embodiment relates to the treatment ~r prevention of infection
in
a patient by a pathogenic fungus which is resistant to one or more other
antifungal agents,
especially an agent other than dehydrophenylahistin or its analog, including
e.g. amphotericin
B or analogs or derivatives thereof (including 14(s)-hydroxyamphotericin B
methyl ester, the
hydrazide of amphotericin B with 1-amino-4-rnethylpiperazine, and other
derivatives) or
other polyene macrolide antibiotics, including, e.g., nystatin, candicidin,
pimaricin and
-3 8-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
natamycin; flucytosine; griseofulvin; echinocandins or aureobasidins,
including naturally
occurring and semi-synthetic analogs; dihydrobenzo[a~napthacenequinones;
nucleoside
peptide antifungals including the polyoxins and nikkornycins; allylarnines
such as naftifine
and other squalene epoxidase inhibitors; and azoles, imidazoles and triazoles
such as, e.g.,
clotrimazole, miconazole, ketoconazole, econazole, butoconazole, oxiconazole,
terconazole,
itraconazole or fluconazole and the like. For additional conventional
antifungal agents and
new agents under development, see e.g. Turner and Rodriguez, 1996 Current
Pharmaceutical
Design, 2:209-224. Another embodiment involves the treatment or prevention of
infection in
a patient by a pathogenic fungus in cases in which the patient is allergic to,
otherwise
intolerant of, or nonresponsive to one or more other antifungal agents or in
whom the use of
other antifungal agents is otherwise contra-indicated. Those other antifungal
agents include,
among others, those antifungal agents disclosed above and elsewhere herein.
[0185] In the foregoing methods for treatment or prevention, a
dehydrophenylahistin or its analog, is administered to the subject in an
effective antifungal
amount.
[0186] Other embodiments relate to the treatment or prevention of infection by
a
pathogenic fungus in a patient by administration of a dehydrophenylahistin or
its analog, in
conjunction with the administration of one or more other antifungal agents,
including for
example, any of the previously mentioned agents or types of agents (e.g. in
combination with
treatment with amphotericin B, preferably in a lipid or liposome formulation;
an azole or
triazole such as fluconazole, for example; an aureobasidin;
dihydrobenzo[alnapthacenequinone; or an echinocardin) as well as with a
different
dehydrophenylahistin or its analog.
(0187] The dehydrophenylahistin or its analog may be administered before,
after
or at the same time the other antifungal agent is administered. In certain
embodiments, the
combination therapy will permit the use of reduced amounts of one or both
antifungal
components, relative to the amount used if used alone.
[0188] Still other embodiments relate to administration of a
dehydrophenylahistin
or its analog to a subject for the treatment or prevention of infection by a
pathogenic fungus,
where the subject is immunosuppressed or immunocompromised, e.g, as the result
of genetic
-3 9-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
disorder, disease such as diabetes or HIV or other infection, chemotherapy or
radiation
treatment for cancer or other disease, or drug- or otherwise induced
immunosuppression in
coimection with tissue or organ transplantation or the treatment of an
autoimmune disorder.
Where the patient is being or will be treated with an immunosuppressive agent,
e.g., in
connection with a tissue or organ transplantation, a dehydrophenylahistin or
its analog may
be co-administered with the immunosuppressive agents) to treat or prevent a
pathogenic
fungal infection.
[0189] Another aspect of this invention is the treatment or prevention of
infection
by a pathogenic fungus in a patient infected, or suspected of being infected,
with HIV, by
administration of an antifungal dehydrophenylahistin or its analog, in
conjunction with the
administration of one or more anti-HIV therapeutics (including e.g. HIV
protease inhibitors,
reverse transcriptase inhibitors or anti-viral agents). The
dehydrophenylahistin or its analog
may be administered before, after or at the same time as administration of the
anti-HIV
agent(s).
[0190] Another aspect of this, invention is the. treatment or prevention of
infection
by a pathogenic fungus in a patient by administration of an antifungal
dehydrophenylahistin
or its analog, in conjunction with the administration of one or more other
antibiotic
compounds, especially one or more antibacterial agents, preferably in an
effective amount
and regiment to treat or prevent bacterial infection. Again, the
dehydrophenylahistin or its
analog may be administered before, after or at the same time as administration
of the other
agent(s).
[0191] Pathogenic fungal infections which may be treated or prevented by the
disclosed methods include, among others, Aspergillosis, including invasive
pulmonary
aspergillosis; Blastomycosis, including profound or rapidly progressive
infections and
blastornycosis in the central nervous system; Candidiasis, including
retrograde candidiasis of
the urinary tract, e.g. in patients with kidney stones, urinary tract
obstruction, renal
transplantation or poorly controlled diabetes mellitus; Coccidioidomycosis,
including chronic
disease which does not respond well to other chemotherapy; Cryptococcosis;
Histopolasmosis; Mucormycosis, including e.g. craniofacial mucormycosis and
pulmonary
mucormycosis; Paracoccidioidomycosis; and Sporotrichosis. It should be noted
that
-40-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
administration of a composition comprising an antifungal amount of one or more
dehydrophenylahistin or its analogs may be particularly useful for treating or
preventing a
pathogenic fungal infection in a mammalian subject where the fungus is
resistant to one or
more other antifungal therapies, or where the use of one or more other
antifungal therapies is
contraindicated, e.g., as mentioned above.
[0192] Antifungal pharmaceutical compositions containing at least one
antifungal
dehydrophenylahistin or its analog, are also provided for use in practicing
the disclosed
methods. Those pharmaceutical compositions may be packaged together with an
appropriate
package insert containing, iute~ alia, directions and information relating to
their antifungal
use. Pharmaceutical compositions are also provided which contain one or more
dehydrophenylahistin or its analog together with a second antifungal agent.
Methods of Treating Fungal Infections
[0193] Certain embodiments disclosed herein relate to methods for treating or
preventing a pathogenic fungal infection, including for example Aspergillosis,
including
invasive pulmonary aspergillosis; Blastomycosis, including profound or rapidly
progressive
infections. and blastomycosis in the central nervous system; Candidiasis,
including retrograde
candidiasis of the urinary tract, e.g. in patients with kidney stones, urinary
tract obstruction,
renal transplantaion or poorly controlled diabetes mellitus;
Coccidioidomycosis, including
chronic disease which does not respond well to other chemotherapy;
Cryptococcosis;
Histopolasmosis; Mucormycosis, including e.g. craniofacial mucormycosis and
pulmonary
mucormycosis; Paracoccidioidomycosis; and Sporotrichosis. The methods may
involve
administering at least one antifungal dehydrophenylahistin or its analog, as
described above,
to a human subject such that the fungal infection is treated or prevented. In
certain
embodiments the dehydrophenylahistin or its analog may be administered in
conjunction with
administration of one or more non-dehydrophenylahistin or its analog
antifungal agents such
as amphotericin B, or an imidazole or triazole agent such as those mentioned
previously.
[0194] The pathogenic fungal infection may be topical, e.g., caused by, among
other organisms, species of Candida, Trichophyton, Micf°osporum or
Epideriuophytoh or
mucosal, e.g., caused by Candida albicans (e.g. thrush and vaginal
candidiasis). The
infection may be systemic, e.g., caused by Candida albicaf2s, C~yptococcus
heoformaus,
-41-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Aspergillus fumigatus, Coccidiodes, Par°acocciciodes, Histoplasrrza or
Blastomyces spp. The
infection may also involve eumycotic mycetoma, chromoblastomycosis,
cryptococcal
meningitits or phycornycosis.
[0195] Further embodiments relate to methods for treating or preventing a
pathogenic fungal infection selected from the group consisting of Candida spp,
including C.
albicans, C. t>~opicalis, C. kefyr, C, kr°usei arid C. galbrata;
Aspergillus spp. including A.
fumigatus and A. flavus; Cryptococcus neoibrnzans; Blastomyces spp. including
Blastomyces
dermatitidis; Pneumocvstis car"inii; Coccidioides imrnitis; Basidiobolus
>~anar°unz;
Conidiobolus spp.; Histoplasma capsulatum; Rhizopus spp. including R. oryzae
and R.
microsporus; Cunninghamella spp.; Rhizoniucor spp.; Paracoccidioides
brasiliensis;
Pseudallescheria boydii; Rhinosporidium seeberi; and Sporothrix schenckii.
Again, the
method may involve administering a non-immunosuppressive antifungal
dehydrophenylahistin or its analog to a patient in need thereof such that the
fungal infection
is.treated or prevented without inducing an untoward immunosuppressive effect.
[0196] Further embodiments relate to methods for treating or preventing a
pathogenic fungal infection which is resistant to other antifungal therapy,
including
pathogenic fungal infections which are resistant to one or more antifungal
agents mentioned
elsewhere herein such as amphotericin B, flucytosine, one of the imidazoles or
triazoles
(including e.g. fluconazole, ketoconazole, itraconazole and the other
previously mentioned
examples). The methods may involve administering to the patient one or more
antifungal
dehydrophenylahistin or its analog, in an amount and dosing regimen such that
a fungal
infection resistant to another antifungal therapy in the subject is treated or
prevented.
[0197] Further embodiments relate to methods for treating or preventing a
pathogenic fungal infection in a patient who is allergic to, intolerant of or
not responsive to
another antifungal therapy or in whom the use of other antifungal agents is
otherwise contra-
indicated, including one or more other antifungal agents mentioned elsewhere
herein such as
amphotericin B, flucytosine, one of the imidazoles or triazoles (including
e.g. fluconazole,
ketoconazole, itraconazole and the other previously mentioned examples). The
methods may
involve administering to such patient one or more antifungal
dehydrophenylahistin or its
analog, in an amount such that a fungal infection is treated or prevented.
-42-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Packaged Dehydrobhenylahistin or its analogs
[0198] Certain embodiments relate to packaged dehydrophenylahistin or its
analogs, preferably packaged nonimmunosuppressive antifungal
dehydrophenylahistin or its
analogs, which term is intended to include at least one dehydrophenylahistin
or its analog, as
described above, packaged with instructions 'for administering the
dehydrophenylahistin or its
analogs) as an antifungal agent without causing a untoward immunosuppressive
effects
within a human subject. In some embodiments, the non-immunosuppressive
antifungal
dehydrophenylahistin or its analog is a member of one of the preferred subsets
of compounds
described above. The dehydrophenylahistin or its analog can be packaged alone
with the
instructions or can be packaged with another dehydrophenylahistin or its
analog, xaparnycin
or another ingredient or additive, e,g., one or more of the ingredients of the
pharmaceutical
compositions. The package can contain one or more containers filled with one
or more of the
ingredients of the phan-naceutical compositions. Optionally associated with
such
containers) can be a notice in the form prescribed by a governmental agency
regulating the
manufacture, use or sale of pharmaceutical or biological products, which
notice reflects
approval by the agency of manufacture, use or sale for human administration.
[0199] The following non-limiting examples are meant to describe the preferred
methods using certain preferred embodiments. Variations in the details of the
particular
methods employed and in the precise chemical compositions obtained will
undoubtedly be
appreciated by those of skill in the art.
EXAMPLE 1
A. Synthesis of Deh~dr~henylahistin
[0200] Dehydrophenylahistin was synthesized by condensation according to the
following basic reaction scheme, as shown in Figure 1:
N~NH
O / O
A~N~ 2 ~ A ~' / ,- ~ 4 ~ / HN / ~ NH'
~~Ac ~NH N~ H ~ \ I / NH N ~/
CsZC03/DMF [O Cs2C0~/DMF O
1 3 DehydroPLH
-43-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
N,N'-diacetyl-2,5-piperazinedione
[0201] 25.0 g of global 2,5-piperazinedione 1 [2,5-piperazinedione (Aldrich
6640-6), 25.0 g, 0.218 mol] in 100 xnL of acetic anhydride (Ac20) was mixed
with sodium
acetate (NaOAc) (17.96 g, .0218 mol). The mixture was heated at 110°C
for 8 h using a
double coiled condenser under an Ar atmosphere. After Ac20 was removed by
evaporation,
the residue was dissolved in AcOEt, washed with 10°/~ citric acid, 10%
NaHC03 and
saturated NaCI (three times each), dried over Na2S04, and concentrated in
vacuo. The residue
was ~ triturated with ether to form a solid. This solid was recrystallized
from EtOAc with
ether-hexane to afford 26.4 g (61°/~) of N,N'-diacetyl-2,5-
piperazinedione 1.
1-Acetyl-3-~,~~-1-f5-(1 1-dimethyl-2-propenyll-1H 4-imidazolyl]meth liy
dene~~2 S-
piperazinedione 2
[0202] To a solution of 5-(1,1-dimethyl-2-propenyl)imidazole-4-carboxaldehyde
(100 mg, 0.609 mmol) in DMF (2 mL) was added compound 1 (241 mg, 1.22 rnmol)
and the
solution was repeatedly evacuated in a short time to remove oxygen and flushed
with Ar,
followed by the addition of Cs2CO3 (198 mg, 0.609 mmol) and the evacuation-
flushing
process was repeated again. The resultant mixture was stirred for 5 h at room
temperature.
After the solvent was removed by evaporation, the residue was dissolved in the
mixture of
EtOAc and 10% Na2C03, and the organic phase was washed with 10% NaZC03 again
and
saturated NaCI for three times, dried over Na2S04 and concentrated in vacuo.
The residual
oil was purified by column chromatography on silica using CHCl3-MeOH (100:0 to
50:1) as
an eluant to give 60 mg (33 %) of a pale yellow solid 2.
Deh~phenylahistin
[0203] To a solution of 2 (30 mg, 0.099 mmol) in DMF (0.8 mL) was added
benzaldehyde (51 q,L, 0.496 mmol, 5 eq) arid the solution was repeatedly
evacuated in a short
time to remove oxygen and flushed with Ar, followed by the addition of CsZC03
(53 rng,
0.149 mmol, 1.5 eq) and the evacuation-flushing process was repeated again.
The resultant
mixture was heated for 2.5 h at 80°C. (The temperature must be
increased slowly. Rapid
heating increases the production of E-isomer at the benzylidene moiety.) After
the solvent
was removed by evaporation, the residue was dissolved in EtOAc, washed with
water for two
-44-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
times and saturated NaCI for three times, dried over NazS04 and concentrated
ih vacuo. On
TLC using CHCl3-MeOH (10:1), you can observe a spot with bright green-yellow
luminescence at 365 nm UV. The purity of this crude product was more than 75%
from
HPLC analysis. The resulting residue was dissolved in 90% MeOH aq and applied
to
reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 x 250 mm) and eluted using a
linear
gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min,
and the
desired fraction was collected and concentrated by evaporation to give a 19.7
mg (60%) of
yellow colored dehydrophenylahistin. The HPLC profile of the synthetic crude
dehydrophenylahistin is depicted in Figure 2.
[0204] In the purification of dehydrophenylahistin, as shown in Figure 4, a
major
peak was the desired Z- form compound of dehydrophenylahistin. The formation
of an E-
isomer was observed as a minor component (about 10%), which was eluted as a
more polar
peak than Z-isomer. As other minor peaks, the reduced Z- and E-compounds, in
which the
dimethylallyl part of dehydrophenylahistin was reduced, was also observed. The
formation
of these reduced compounds was due to the aldehyde 2 with a reduced impurity,
which was
generated during the reduction of with DIBAL-H and was not separated in the
subsequent
process.
[0205] These minor compounds could be removed by preparative HPLC
purification, afforded dehydrophenylahistin with the Z configuration at the
benzylidene part
in a 60% yield (20% yield in two steps) with more than 95% purity. The
compounds with E-
configuration at the imidazole side of the diketopiperazine ring were not
observed in this
HPLC chart, suggesting that the first reaction from compound 1 to 3 in Figure
1 is Z
selective.
B. Chemical Characteristics:
(0206] The above dehydrophenylahistin compound is a pale yellow solid. Its
structure is confirmed by standard NMR analyses.
-45-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
EXAMPLE 2
Synthesis and Physical Characterization of tBu-deh~drophenylahistin
Derivatives
(0207] Structural derivatives of dehydrophenylahistin were synthesized
according
to the following reaction schemes to produce tBu-dehydrophenylahistin.
Synthesis by Route
A (see Figure 1) is similar in certain respects to the synthesis of the
dehydrophenylahistin
synthesized as in Exarriple 1.
Route A
,O ~ HO
O ~NH I i
ANN 15 ~. ANN / -- 4 ~ / HN /
~N. NH N ~ NH ~ NH ~ NH
Ac Cs~C03/DMF ~ ~ Cs2CO3/DMF ~ / N~
O O O
1 33% (1st) 16 50% tBu-dehydroPLH
ROUte B
HO i~~0
O I ~ N~fJH
A
~N~ 4 ~ / HN 15 ~ / HN
NH
~N'Ac CsZC03/DMF ~ ( / 'Ac Cs2C031DMF ~ I / NH N~/
O O O
1 17 1-2 % tBu-dehydroPLH
-46-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Route A:
[0208] N,N'-diacethyl-2,S-piperazinedione 1 was prepared as in Example 1.
11 1-Acetyl-3-f(~-1-f5-tert-butyl-1H 4-imidazolvllmethvlidenell-2.5-
ninerazinedione (16
O
Ac'N /
~NH N.~NH
~O[
. [0209] ~ To a solution of S-tert-butylimidazole-4-carboxaldehyde 15 (3.02 g,
19.8.
mmol) in DMF (30 mL) was added compound 1 (5.89 g, 29.72 mmol) and the
solution was
repeatedly evacuated in a short time to remove oxygen and flushed with Ar,
followed by the
addition of CsaC03 (9.7 g, 29.72 mmol) and the evacuation-flushing process was
repeated
again. The resultant mixture was stirred for 5 h at room temperature. After
the solvent was
removed by evaporation, the residue was dissolved in the mixture of EtOAc and
10%
Na2C03, and the organic phase was washed with 10% Na2C03 again and saturated
NaCI for
three times, dried over Na2S04 and concentrated in vacuo. The residual oil was
purified by
column chromatography on silica using CHC13-MeOH (100:0 to 50:1) as an eluant
to give
1.90 g (33 %) of a pale yellow solid 16. 1H NMR (270 MHz, CDCl3) 8 12.14 (d,
br-s,
1H), 9.22 (br-s, 1H), 7.57 (s, 1H), 7.18, (s, 1H), 4.47 (s, 2H), 2.65 (s, 3H),
1.47 (s,
9H).
2) t-Bu-dehydrophenylahistin
O
/ HN /
/ NH N~NH
O
[0210] To a solution of 1-Acetyl-3-{(Z)-1-[S-tert-butyl-1H 4-
irnidazolyl]methylidene~]-2,S-piperazinedione (IG) (lI mg, 0.038 mmol) in DMF
(1.0 mL)
was added benzaldehyde (19 ~.L, 0.19 mmol, S eq) and the solution was
repeatedly evacuated
in a short time to remove oxygen and flushed with Ar, followed by the addition
of Cs2CO3
(43 mg, 0.132 mrnol, 3.S eq) and the evacuation-flushing process was repeated
again. The
resultant mixture was heated for 2.S h at 80°C. After the solvent was
removed by
-47-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
evaporation, the residue was dissolved in EtOAc, washed with water for two
times and
saturated NaCI foT three times, dried over Na2S04 and concentrated in vacuo.
The resulting
residue was dissolved in 90% MeOH aq and applied to reverse-phase HPLC column
(YMC-
Pack, ODS-AM, 20 x 250 .mm) and eluted using a linear gradient from 70 to 74%
MeOH in
water over 16 min at a flow rate of 12 mL/min, and the desired fraction was
collected and
concentrated by evaporation to give a 6.4 mg (50%) of yellow colored tert-
butyl-
dehydrophenylahistin. 1H NMR (270 MHz, CDCl3) S 12.34 br-s, 1H), 9.18 (br-s,
1H),
x.09 (s, 1H), 7.59 (s, 1H), 7.31 - 7.49 (m, SH), 7.01 s, 2H), 1.46 (s, 9H).
[0211] The dehydrophenylahistin reaction to produce tBu-dehydrophenylahistin
is
identical to Example 1.
[0212] The total yield of the tBu-dehydrophenylahistin recovered was 16.5%.
Route B:
[0213] N,N'-diacethyl-2,5-piperazinedione 1 was prepared as in Example 1.
1 1-Aeet~-3-[(~-benzylidenel]'-2,5-~nerazinedione (17)
O
ANN /
~NH
O
[0214] To a solution of benzaldehyde 4 (0.54 g, 5.05. mrnol) in DMF (5 mL) was
added compound 1 (2.0 g, 10.1 mmol) and the solution was repeatedly evacuated
in a short
time to remove oxygen and flushed with Ar, followed by the addition of Cs2C03
(1.65 g, S.OS
mrnol) and the evacuation-flushing process was repeated again. The resultant
mixture was
stirred for 3.5 h at room temperature. After the solvent was removed by
evaporation, the
residue was dissolved in the mixture of EtOAc and 10% NaZC03, and the organic
phase was
washed with 10% Na2C03 again and saturated NaCI for three times, dried over
Na2SO4 and
concentrated in vacuo. The residual solid was recrystalized from MeOH-ether to
obtain a
off white solid of 17; yield 1.95 g (79%).
-48-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
21 t-Bu-dehydrophenylahistin
O
/ I HN
/ NH N~NH
O
(0215] To a solution of 1-Acetyl-3-[(2)-benzylidenel~-2,5-piperazinedione (17)
(48 mg, 0.197 mmol) in DMF (1.0 rnL) was added 5-tert-butylimidazole-4-
carboxaldehyde
15 (30 mg, 0.197 mmol) and the solution was repeatedly evacuated in a short
time to remove
oxygen and flushed with Ar, followed by the addition of Cs2C03 (96 mg, 0.296
mmol) and
the evacuation-flushing process was repeated again. The resultant mixture was
heated for 14
h at 80°C. After the solvent was removed by evaporation, the residue
was dissolved in
EtOAc, washed with water for two times and saturated' NaCI for three times,
dried over
Na2S04 and concentrated in vacuo. The resulting residue was dissolved in 90%
MeOH aq
and applied to reverse-phase HPLC column (YMC-Pacl~, ODS-AM, 20 x 250 mm) and
eluted using a linear gradient from 70 to 74% MeOH in water over 16 min at a
flow rate of
12. mL/min, and the desired fraction was collected and concentrated by
evaporation to give a
0.8 mg (1.2%) of yellow colored tert-butyl-dehydrophenylahistin.
[0216] The total yield of the tBu-dehydrophenylahistin recovered was 0.9%.
[0217] The HPLC profile of the crude synthetic tBu-dehyrophenylahistin from
Route A and from Route B is depicted in 'Figure 4.
[0218] Two other tBu-dehydrophenylahistin derivatives were synthesized
according to the method of Route A. In the synthesis of the additional tBu-
dehydrophenylahistin derivatives, modifications to the benzaldehyde compound 4
were made.
[0219] Figure 4 illustrates the similarities of the HPLC profiles (Column: YMC-
Paclc ODS-AM (20 x 250mm); Gradient: 65% to 75% in a methanol-water system fox
20
min, then 10 min in a 100% methanol system; Flow rate: l2mL/min; O.D. 230 nm)
from the
synthesized dehydrophenylahistin of Example 1 (Fig 2) and the above
exemplified tBu-
dehydrophenylahistin compound produced by Route A.
[0220] The sequence of introduction of the aldehydes is a relevant to the
yield and
is therefore aspect of the synthesis. An analogue of dehydrophenylahistin was
synthesized, as
-49-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
a control or model, wherein the dimethylallyl group was changed to the tert-
butyl group with
a similar steric hindrance at the 5-position of the imidazole ring.
[0221] The synthesis of this "tert-butyl (tBu)-dehydrophenylahistin" using
"Route
A" was as shown above: Particularly, the sequence of introduction of the
aldehyde exactly
follows the dehydrophenylahistin synthesis, and exhibited a total yield of
16.5% tBu-
dehydrophenylahistin. This yield was similar to that of dehydrophenylahistin
(20%). Using
"Route B", where the sequence of introduction of the aldehydes is opposite
that of Route "A"
for the dehydrophenylahistin synthesis, only a trace amount of the desired tBu-
dehydroPLH
was obtained with a total yield of 0.9%, although in the introduction of first
benzaldehyde 4
gave a 76% yield of the intermediate compound 17. This .result indicated that
it may be
difficult to introduce the highly bulky imidazole-4-carboxaldehydes 15 with a
substituting
group having a quaternary-carbon on the adjacent 5-position at the imidazole
ring into the
intermediate compound 17, suggesting that the sequence for introduction of
aldehydes is an
important aspect for obtaining a high yield of dehydrophenylahistin or an
analog of
dehydrophenylahistin employing the synthesis disclosed herein:
[0222] From the HPLC analysis of the final crude products, as shown in Figure
4,
a very high content of tBu-dehydrophenylahistin and small amount of by-product
formations
were observed in the crude sample of Route A (left). However, a relatively
smaller amount
of the desired tBu-dehydrophenylahistin and several other by-products were
observed in the
sample obtained using Route B (right).
-50-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
EXAMPLE 3
Alternative, Larger-Scale Synthesis of Dehydrophenylahistin and Analogs
Syfathesis of 3-Z Benzylide~ae-6 (S"-(l,l-di~etlaylallyl)-IH iz~aidazol-4"-Z
yl~zethyleheJ-
pipe~azirae-2, 5-diohe ~Dehydt°ophenylahistinJ (1)
~O~ a ~~Ow
pO OH O 1Q
O= =O
n
[O~ pO __ II0 ''
n
OI O O~ OH
\ ~ .NH .--~--r. \ ~ NH
O O NJ N_/
i,
O O
O
\ NH N~ I ~NH N~l k
~ HN w ~ NH -~-.__. N w ~ NH ~ \
~~''\~NH
p / O O ~ N =~
1
-51-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0223] Reagents: a) LDA; CH3CH0; b) Tos-Cl, pyridine; c) DBU; d) NaOH; e)
CZC1202; f) KOOCCH2COOEt, BuLi; g) S02C12; h) HZNCHO, H20; i) LiAlH4; ,j)
MnOa; k)
1,4-diacetyl-piperazine-2,5-dione, Cs2C03;1) benzaldehyde, CsaC03
3-Hydroxy-2,2-dimethyl-butyric acid methyl ester
1 .
o. . o ._-~..
I.DA OH CO
O
[0224] A solution of LDA in heptane/THF/ethylbenzene (2 M, 196 ml, 0.39 rnol)
was added under argon to a solution of methyl isobutyrate (45 ml, 0.39 mol) in
THF (270 ml)
at -60° and the resultant mixture was stirred for 30 min. A solution of
acetaldehyde (27 ml,
0.48 mol) in THF (45 ml), precooled to -60°, was added slowly and 'the
resulting solution
stirred for a further 30 min. Saturated ammonium chloride (50 ml) was added
and the
solution was allowed'to warm to room temperature. The reaction mixture was
extracted with
ethyl acetate, and the extracts were washed with HCl (2 M), sodium
bicarbonate, then brine.
The organic layer was dried over magnesium sulfate, filtered, then evaporated
to give a clear
oil (52.6 g). Distillation 76-82°/30 mmHg gave pure 3-hydroxy-2,2-
dimethyl-butyric acid
methyl ester (42.3 g, 74%). (Burk et al., J. Am. Chem. Soc., 117:4423-4424
(1995)).
~oazs~ 1H NMR (400 MHz, CDC13) 8 1.15 (d, J= 6.2 Hz, 3H); 1.17 (s, 6H); 2.66
(d, J= 6.2 Hz, 1H, -OH); 3.71 (s, 3H, -OMe); 3.87 (app quintet, J= 6,4 Hz, 1H,
H3).
2,2-Dimethyl-3-(toluene-4-sulfonyloxy)-butyr~ie acid methyl ester
O~ TsCi Ov
-~...
OH O Pyridine O O
O=S=O
-52-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0226] To a cooled (0°) solution of 3-hydroxy-2,2-dimethyl-butyric acid
methyl
ester (52.0 g, 0.36 mol) in pyridine (100 ml) was added gradually, p-toluene
sulfonyl chloride
(69.0 g, 0.36 mol). The mixture was allowed to warm to room temperature and
was stirred
for 60 h. The reaction was again cooled in ice and was acidified by addition
of HCl (2 M).
The resultant solution was extracted with ethyl acetate, the extracts were
washed with HCl,
then brine, dried and evaporated to give an oil which formed a white
precipitate upon
standing. This mixture.was dissolved in the minimum amount of ethyl acetate
and then light
petroleum was added to afford a white precipitate which was collected and
washed with more
light petroleum. The filtrate was partially evaporated and a second crop of
crystals was
collected and added to the first to afford 2,2-difsaethyl-3-(toluene-4-
sulfouyloxy)-butyric acid
methyl ester (81.2 g, 76%).
~022~~ 1H NMR (400 NMz, CDCl3) ~ 1.12 (s, 3H); 1.13 (s, 3H); 1.24 (d, J= 6.4
Hz, 3H); 2.45 (s, 3H, -PhMe) 3.58 (s, 3H, -OMe); 4.94 (quartet, J= 6.4 Hz, 1H,
H3), 7.33 (d,
J= 8.0 Hz, 2H), 7.78 (d, J= 8.0 Hz, 2H).
[0228] Evaporation of the final filtrate afforded additional crude 2,2-
dimethyl-3-
(toluene-4-sulfonyloxy)-butyric acid methyl ester (19.0 g, 18%). ,
2,2-Dimethyl-but-3-enoic acid methyl ester
O ~O
D~U
---~~
., . . 0
[0229] A solution of 2,2-dirnethyl-3-(toluene-4-sulfonyloxy)-butyric acid
methyl
ester (18.06 g, 0.06 mol) in DBU (15 ml) was heated at 140-160° for 3.5
h. The mixture was
allowed to cool to room temperature and was then diluted with ether. The
mixture was
washed with HCl (1 M), sodium bicarbonate, then brine. The ethereal layer was
dried and
partially evaporated to give a concentrated solution of 2,2-dirnethyl-but-3-
enoic acid methyl
ester (10 g). (Save and Katzenellenbogen, J. Org. Claem, 46:239-250 (1981)).
Further
-53-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
evaporation was avoided due to the volatility of the product (bp 102°).
(Tsaconas et al., Aust.
J. Claem., 53:435-437 (2000)).
[0230] 1H NMR (400 NMz, CDCl3) 8 1.31 (s, 6H); 3.68 (s, 3H); 5.06 (d, J= 17.1
Hz, 1H, -CH=CH2); 5.11 (d, J= 10.7 Hz, 1H, -CH=CHZ); 6.03 (dd, J= 17.1, 10.7
Hz, 1H,
CH=CHI).
2,2-Dimetlayl-but-3-enoic acid
~~"~~0~ . ~ OH
4 ~ O
[0231] The above ethereal solution of 2,2-dimethyl-but-3-enoic acid methyl
ester
(10 g) was diluted with ethanol (25 rnl), sodium hydroxide (4 M, 22 ml) was
added and the
mixture was stirred overnight. The solution was partially evaporated to remove
the ethanol
and the resultant mixture was added to HCl (1M, 100 ml). The product was
extracted with
ethyl acetate and the extracts were dried and evaporated to give 2,2-dimethyl-
but-3-enoic acid
(6.01 g, 88% 2 steps). (Hayashi et al., J. Org. ClZem., 65:8402-8405 (2000).
~oasa~ 1H NMR (400 MHz, CDCl3) & 1.33 (s, 6H); 5.11 (d, J = 10.8 Hz, 1H, -
CH=CH2); 5.15 (d, J = 17.2 Hz, 1H, -CH=CHZ); 6.05 (dd, J = 17.2, 10.8 Hz, 1H, -
CH=CH2).
[0233] Monoethyl hydrogen malonate (Wierenga and Skulniclc, "Aliphatic and
Aromatic (3-keto Esters from Monoethyl Malonate: Ethyl 2-Butyrylacetate,"
Organic
S]~ntheses Collective Volume 7, 213).
O p O
y
K+ _O O HO O
[0234] Ethyl potassium malonate (25.0 g, 0.15 mol) was suspended in water
(15.6
ml) and cooled in an ice bath. Concentrated HCl (12.5 ml) was added dropwise
over 30 min,
-54-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
then the mixture was stirred for a further 10 min. The precipitate was
filtered, then washed
twice with ether. The filtrate was separated and the aqueous phase was
extracted with ether.
The combined ethereal solutions were dried (MgS04) and evaporated to afford,
as an oil,
monoethyl hydrogen malonate (I9.2 g, 99%) which was dried under vacuum
overnight (or
50°l1 mm for 1 h) prior to use.
4,4-Dimethyl-3-oxo-hex-S-enoic acid ethyl estef°
i) C~OaCt2 / DMF
--~. \ O~
O
O ii) ~O~ BuLi O O
NO
(0235] Oxalyl chloride (3.83 ml, 43.9 mmol) was added dxopwise to a cooled
(0°)
solution of 2,2-dirnethyl-but-3-enoic acid (5.0 g, 43.9 mmol) and DMF (1 drop)
in anhydrous
dichloromethane (25 mI). The mixture was stirred for 1 h at 0°, then
for 16 h at room
temperature. Fractional distillation (121°/760 mmHg) afforded 2,2-
dimethyl-but-3-enoyl
chloride (4.1 g, 71°Jo).
[0236] Monoethyl hydrogen malonate (7.2 g, 0.05 mol) and bipyridyl (few
milligrams) were dissolved in THF (90 rnl) and the system was flushed with
nitrogen. The
solution was cooled to -70°, then BuLi (2.5 M in hexanes, 37 ml, 0.09
mol) was added. After
the addition of only ~10 ml of BuLi the solution turned pink and additional
THF (15 rnl) was
required to enable magnetic stirring. The cooling bath was removed and the
remaining BuLi
was added, the temperature was allowed to reach -10°, upon which the
solution turned
colorless. The mixture was again cooled to -60° and a solution of 2,2-
dimethyl-but-3-enoyl
chloride (4.1 g, 0.03 mol) in THF (12 ml) was added dropwise. After addition
was complete
the mixture was allowed to warm to 0° and stir for 3 h, then it was
added to a 1:1 mixture of
ether/1M HCl (260 rnl) at 0° and stirred for a further 1.5 h. The
organic layer was removed,
washed with HCl (1 M), sodium bicarbonate solution, brine then dried and
evaporated to give
4,4-dimethyl-3-oxo-hex-5-enoic acid ethyl ester (5.6 g, 98%). (Hayashi et al.,
J. O~g. Chem.,
65:8402-8405 (2000). Distillation with a Kugelrohr oven (160°/1 mmHg)
afforded pure
material.
-55-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
~ozs~~ IH NMR (400 MHz, CDC13) 8 1.26 (s, 6H); 1.27 (t, J = 6.9 Hz, 3H, -
CH~CH3); 3.51 (s, 2H); 4.18 (q, J = 6.9 Hz, 2H, -CH2CH3); 5.20 (d, J = 17.7
Hz, 1H, -
CH=CH2); 5.21 (d, J= 9.6 Hz, 1H, -CH=CHZ); 5.89 (dd, J= 17.7, 9.6 Hz, 1H, -
CH=CH2).
~-Chloro-4,4-dinaethyl-3-oxo-hex-5-enoic acid ethyl ester
'''. O~ SOxCtz ~ ~i O
-- Y
O O O O
[0238] Sulfuryl chloride (0.84 ml, 10.4 mmol) was added to a cooled
(0°) solution
of 4,4-dirnethyl-3-oxo-hex-5-enoic acid ethyl ester (1.83 g, 9.93 mmol) in
chloroform (7 ml).
The resulting mixture was allowed to warm to room temperature and stir for 30
min, after
which it was heated under reflux for 2 h. After cooling to room temperature
the reaction
mixture was diluted with chloroform, then was washed with sodium bicarbonate,
water then
brine. The organic phase was dried and evaporated to afford, as a brown oil, 2-
chloro-4,4-
dimethyl-3-oxo-hex-5-enoic acid ethyl ester (2.01 g, 93°l0). (Hayashi
et al., J. O~g. Chem.,
65:8402-8405 (2000).
[0239] 1H NMR (400 MHz, CDCl3) 8 1.28 (t, J= 7.0 Hz, 3H, -CH2CH3); 1.33 (s,
3H); 1.34 (s, 3H); 4.24 (q, J= 7.0 Hz, 2H, -CHzCH3); 5.19 (s, 1H; 5.28 (d, J=
16.9 Hz, 1H, -
CH=CH2); 5.29 (d, J = 10.9 Hz, 1H, -CH=CH2); 5.96 (dd, J = 16.9, 10.9 Hz, 1H, -
CH=CH2).
j0240] LC/MS tR = 8.45 (219.3 jM(C137)+H]+ min.
[0241] This material was reacted without further purification.
S-(1,1-Dimethyl-allyl)-3H imidazole-4-carboxylic acid ethyl ester~
0 0.~. o off o o~
Cl NHZ
~ NH + \ ~ NH ~ ~ ~ N
0 o N=J N=J o-J
[0242] A suspension of 2-chloro-4,4-dimethyl-3-oxo-hex-5-enoic acid ethyl
ester
(19.4 g, 0.09 mol) and water (1.94 ml, 0.11 mol) in formamide (36.8 ml) was
shaken briefly,
then dispensed into 15 x 18 ml vials. The vials were sealed and heated at
150° for 5 h. After
-56-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
cooling to room temperature, the vials' contents were combined and extracted
exhaustively
with chloroform. The extracts were dried and evaporated to afford a
concentrated formamide
solution (14.7 g). This was added to a silica column (7 cm diameter, 11 cm
height).paclced in
I% MeOHII% Et3N in chloroform. Elution of the column with 2 L of this mixture
followed
by 2 L of 2% MeOH/1% Et3N in chloroform afforded, in the early fractions, a
compound
suspected ofbeing 5-(.l,l-dimethyl-allyl)-oxazole-4-cap°boxylic acid
ethyl ester (1.23 g. 7%).
[0243] HPLC (214nm) tR = 8.68 (50.4%) min.
[0244] 1H N~,IR (400 MHz, CDC13) 8 1.40 (t, J = 7.2 Hz, 3H, -CH2CH3); 1.54 (s,
6H); 4.38 (t, J = 7.2 Hz, 2H, -CH2CH3); 5.03 (d, J = 17.4 Hz, 1H, -CH=CHI);
5.02 (d, J =
10.4 Hz, 1H, -CH=CH2); 6.26 (dd, J= 17.4, 10.4 Hz, 1H, -CH=CH2); 7.83 (s, 1H).
[0245] LCMS tR = 8.00 (210.1 [M+H]+, 361.1 [2M+H]~ min.
[0246] Recovered from Iater fractions was the desired 5-(1,1-dirnethyl-allyl)-
3H-
imidazole-4-carboxylic acid ethyl ester (3.13 g, 17%). (Hayashi et al., J ..
Or g. Chem.,
65:8402-8405 (2000)).
[0247] HPLC (214nrn) tR = 5.52 (96.0%) rnin.
~ozas~ 1H NMR (400 MHz, CDC13) b 1.38 (t, J= 7.0 Hz, 3H); 1.57 (s, 6H); 4.35
(q, J = 7.0 Hz, 2H); 5.04-S.I4 (m, 2H, -CH=CH2); 6.28 (dd, J = 18.0, 10.4 Hz,
1H, -
CH=CHz); 7.52 (s, 1H).
[0249] LC/MS tR = 5.30 (209.1 [M+H]+, 417.2 [2M+H]+) min.
[0250] Additional 5-(1,1-dimethyl-allyl)-3H-imidazole-4-carboxylic acid ethyl
ester was also recovered from the column (3.59 g, I9%) which was of lower
purity but still
sufficient for further reaction.
[0251] Another byproduct isolated from a similar reaction (smaller scale) by
further elution of the column with 5% MeOH/1% Et3N in chloroform was a
compound
suspected of being. 5-(1,1-dirnethyl-allyl)-3H imidazole-4-carboxylic acid
(0.27 g, 9%).
[0252] HPLC (245nm) tn = 5.14 (68.9%) min.
~ozss~ IH NMR (400 MHz, CD30D) & 1.45 (s, 6H); 4.97 (d, J= 10.6 Hz, 1H, -
CH=CHI); 5.01 (d, J = 17.7 Hz, 1H, -CH=CHZ); 6.28 (dd, J = 17.7, 10.6 Hz, 1H, -
CH=CHz); 7.68 (s, 1H).
[0254] LCMS tR = 4.72 (181.0 [M+H]+, 361.1 [2M+H]~ min.
-57-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
(S-(l,l-Dimethyl-allyl)-3H i»zidazol-4 ylJ-methanol
p~ p~ OH
LiP~iH4
''~ NH . .---~. ~ ~ H.
N=~ N
[0255] A solution of S-(1,1-dimethyl-allyl)-3H-irnidazole-4-carboxylic acid
ethyl
ester (3.13 g, I5.0 mmol) in THF (60 rnl) was added dropwise to a suspension
of lithium
aluminium hydride (95% suspension, 1.00 g, 25.0 mmol) in THF (40 ml) and the
mixture
was stirred at room temperature for 4 h. Water was added until the evolution
of gas ceased,
the mixture was stirred for 10 min, then was filtered through a sintered
funnel. The
precipitate was washed with THF, then with methanol, the filtrate and washings
were
combined, evaporated, then freeze-dried to afford [S-(1,1-dimethyl-allyl)-3H-
imidazol-4-y1]-
methanol (2.56 g, 102%). Residual water was removed by azeotroping with
chloroform prior
to further reaction. (See Hayashi et al., J. Org. Chem., 65:8402-8405 (2000)).
[0256] HPLC (240nm) tR = 3.94 (56.8%) min.
ioas7~ 1H NMR (400 MHz, CD30D) 8 1.43 (s, 6H); 4.57 (s, 2H); S.O1 (d, J =
10.5 Hz, 1H, -CH=CH2); 5.03 (d, .I= 17.7 Hz, 1H, -CH=CHZ); 6.10 (dd, J= 17.7,
10.5 Hz,
1H, -CH=CHa); 7.46 (s, 1H).
[0258] LC/MS tR = 3.77 (167.3 [M+H]+) min.
5-(1,1.-Dimethyl-allyl)-3H imidazole-4-carbaldehyde
OH .
Mn~2
~ NH .~Y ~ ~ NH
N_J N.~.l
[0259] Manganese dioxide (20 g, 0.23 mol) was added to a solution of [5-(1,1-
dimethyl-allyl)-3H-imidazol-4-yl]-methanol (2.56 g, 0.02 mol) in acetone (300
ml) and the
resulting mixtuxe was stirred at room temperature for 5 h: The mixture was
filtered through
filter paper and the residue was washed with acetone. The filtrate and
washings were
combined and evaporated to afford 5-(l,l-dimethyl-allyl)-3H-imidazole-4-
carbaldehyde (1.82
g, 51%). (Hayashi et al., J. Org. Chefsa., 65:8402-8405 (2000)).
[0260] HPLC (240nm) t~ = 4.08 (91.5%) min.
-5 8-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
wash 1H NMR (400 MHz, CDC13) S 1.56 (s, 6H); S.I6 (d, J = 10.6 Hz, 1H, -
CH=CH2); 5.19 (d, J = 17.3. Hz, 1H, CH=CH2); 6.22 (dd, J =17.3, 10.6 Hz, 1H, -
CH=CH2);
7.75 (s, 1H), 10.02 (s, 1H, HCO).
[0262] LC/MS tR = 3.75 (165.2 [M+H]~ min.
1-Acetyl-3-(5'-(1,1-dimethyl-allyl)-IH imidazol-4'-Z ylmethyleizeJ
pipes°azifae-2,5-dione
O O ~ O
~N ~NH N=~
N~ H + ~'N~ ~ ' N .y, NH
O 'O' ' O O
[0263] To a solution of 5-(1,1-dimethyl-allyl)-3H-imidazole-4-carbaldehyde
(1.78
g, 0.01 mol) in DMF (35 ml) was added 1,4-diacetyl-piperazine-2,5-dione (8.59
g, 0.04 mol)
and the mixture was evacuated, then flushed with argon. The evacuation-
flushing process
was repeated a further two times, then cesium carbonate (3.53 g, 0.01 mol) was
added. The
evacuation-flushing process was repeated a further three times, then the
resultant mixture was
heated at 45° for 5 h. The reaction mixture was partially evaporated
(heating under high
vacuum) until a small volume remained and the resultant solution was added
dropwise to ice-
water (50 ml). The yellow precipitate was collected, washed with water, then
freeze-dried to
afford 1-acetyl-3-[5'-(1,1-dimethyl-allyl)-1H-imidazol-4'-ylmethylene]-
piperazine-2,5-dione
(1.18 g, 36%). (Hayashi, Personal Communication (2001)).
(0264] HPLC (214nm) tR = 6.01 (72.6%) min.
~oass~ 1H NMR (400 MHz, CDCI~) ~ 1.53 (s, 6H); 2.64 (s, 3H); 4.47 (s, 2H);
5.19 (d, J = 17.3 Hz, 1H, -CH=CHZ); 5.23 (d, J = 10.7 Hz, 1H, -CH=CH2); 6.06
(dd, J =
17.3, 10.7 Hz, 1 H, -CH=CHZ); 7.16 (s, 1 H), 7.59 (s, 1 H), 9.47 (bs, 1 H);
12.11 (bs, 1 H)
[observed ~2% 1,4-diacetyl-piperazine-2,5-dione contamination 8 2.59 (s, 6H);
4.60 (s, 4H).]
[0266] LC/MS tR = 6.65 (303.3 [M+H]+, 605.5 [2M+H]+) min. (n.b. different
system used).
-59-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
3-Z-Benzylide>ze-G-~5 "-(1,1-dimethylallyl)-1 H-iryaidazol-4 "-Z ylrnethyleneJ
piperazi~ce-2, S-
diorze
O ' ~ ~0 O
~NH N~ ~ ~ ~ ~ ~ NH N NH
~N ~ ~ NH . -~- / HN
'~O(~ O ~ . O
[0267] To a solution of 1-acetyl-3-[5'-(l,l-dimethyl-allyl)-1H-imidazol-4'-
ylmethylene]-piperazine-2,5-dione (2.91 g, 9.62 mmol) in DMF (70 ml) was added
benzaldehyde (4.89 ml, 48.1 mmol) and the solution was evacuated, then flushed
with Argon.
The evacuation-flushing process was repeated a further two times, then cesium
carbonate
(4.70 g, 14.4 mmol) was added. The evacuation-flushing process was repeated a
further three
times, then the resultant mixture was heated undex the temperature gradient ad
shown below.
[0268] After a total time of 5 h the reaction was allowed to cool to room
temperature and the mixture was added to ice-cold water (500 ml). The
precipitate was
collected, washed with water (300 ml), then freeze-dried to afford a yellow
solid (2.80 g).
This material was dissolved in chloroform (250 ml) filtered thxough filter
paper and
evaporated to azeotrope remaining water. The residual yellow precipitate (2.70
g, HPLC
(2I4nm) tR = 7.26 (93.6%) min.) was partially dissolved in chloroform (20 ml),
the
suspension was sonicated for 5 min, then the solid was collected and air dried
to afford 3-Z-
benzylidene-6-[5"-(1,1-dimethylallyl)-1H imidazol-4"-Z ylmethylene]-piperazine-
2,5-dione
(1.82 g, 54%) (Hayashi, Personal Communication (2001)), m.p. 239-240°
(dec.).
[0269] HPLC (214nm) t~ = 6.80 (1.92) min, 7.33 (95.01%).
~oa~o~ 1H NMR (400 MHz, CDC13) b I.53 (s, 6H); 5.18 (d, J = 17.6 Hz, 1H, -
CH=CHZ); 5.21 (d, J = 11.0 Hz, 1H, -CH=CHa); 6.06 (dd, J = 17.6, 11.0 Hz, 1H, -
CH=CH2); 6.99 (s, 1H, -C-C=CH); 7.00 (s, 1H, -C-C=CH); 7.30-7.50 (m, S x ArH);
7.60 (s,
H2"); 8.07 (bs, NH); 9.31 (bs, NH); 12.30 (bs, NH).
[0271] LC/MS tR = 6.22 (349.3 [M+H]+, E isomer), 6.73 (349.5 [M+H]+, 697.4
[2M+H]+, Z isomer) min.
[0272] ESMS m/z 349.5 [M+H]+, 390.3 [M+CH~CN]t.
-60-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0273) Evaporation of the chloroform solution gave additional 3-Z benzylidene-
6-
[5"-(1,1-dimethylallyl)-lHimidazol-4"-Z-ylmethylene]-piperazine-2,5-dione
(0.76 g, 29%).
[0274) HPLC (214nm) tR = 7.29 (84.5%) min.
3-E-Benzylidene-6-[5"-(1,1-dimethylallyl)-1H-imidazol-4"-Z-ylmethylene]-
piperazine-2,5-
dione
O ~ ~ 0 ~~ O
r W
~NH N~'.1 NH N=~ H
'N ~ ~ NN ----~.,- HN '~- ~ N
~O O ~''~% O ~'~,%
[0275] Preparative HPLC purification of a crude sample of material synthesized
as above afforded the geometric isomer 3-E-Benzylidene-6-[5"-(l,l-
dirnethylallyl)-1H-
imidazol-4"-Z-ylrnethylene]-piperazine-2,5-dione (1.7 mg).
[0276] HPLC (214nm) tR = 6.75 (87.79) min.
~oa~7~ 1H N~ (400 MHz, CDC13) 8 1.52 (s, 6H); 5.19 (d, J = 20.8 Hz, 1H,
CH=CHZ); 5.22 (d, J= 14.0 Hz, 1H, CH=CH2); 6.05 (dd, J= 18.0, 10.4 Hz, 1H,
CH=CH2);
6.33 (s, 1H, C-C=CH); 6.90-7.65 (m, 7H).
[0278) ESMS m/z 349.5 [M+H]+, 390.4 [M+CH4CN]+.
-61-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Synthesis of 3-~ Be~tzylidehe-6-(S "-test-butyl-IH imidazol-4 "-Z ylmethyle~e)
pipe~azihe-
2,S-diohe (2)
o a
n
o O~"/. off
t i
_.- ~-~ / NH -" ~ NH
0 0 N=~
n
.a ° o
~'NH N~ .
NH N=~ I
/ HN w w 'NH , ~N ~~ . 'w. NH -' , _ ~°~H
a o.
0
z
[0279] Reagents: g) S02C12; h) HZNCHO, H20; 1'LiAlH4; j) Mn02; k) 1,4-
diacetyl-piperazine-2,S-dione, Cs2C03; 1) benzaldehyde, CsaC03
2-Chlo~°o-4,4-dimethyl-3-oxo pehtanoic acid ethyl ester
/ CI
C SfJ~Cl2 ~ ~\~~p~
0 0 0 0
[0280] Sulfuryl chloride (14.0 ml, 0.17 mol) was added to a cooled (0°)
solution
of ethyl pivaloylacetate (27.17 g, O.1G mol) in chloroform (100 ml). The
resulting mixture
was allowed to warm to room temperature and was stirred for 30 min, after
which it was
heated under reflux for 2.5 h. After cooling to room temperature, the reaction
mixture was
diluted with chloroform, then washed with sodium bicarbonate, water then
brine.
-62-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0281] The organic phase was dried and evaporated to afford, as a clear oil, 2-
chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester (33.1 g, 102%). (Durant
et al.,
"Aminoalkylimidazoles and Process for their Production." Patent No. GB 1341375
(Great
Britain, 1973)).
[0282] HPLC (214nm) tR = 8.80 (92.9%) min.
[0283] 1H NMR (400 MHz, CDCl3) 8 1.27 (s, 9H); 1.29 (t, J= 7.2 Hz, 3H); 4.27
(q, J= 7.2 Hz, 2H); 5.22 (s, 1H).
loasa~ i3C Nh,IR (100 MHz, CDCl3) 8 13.8, 26.3, 45.1, 54.5, 62.9, 165.1,
203.6.
5-te>~t-Butyl-3H irnidazole-4-carboxylic acid ethyl ester
O O~ O OH O O~
CI NHz
~~~~0~/. ~ J H * . /.=J H.
iO' IO' ' N N O
[0285] A solution of 2-chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester
(25.0
g, 0.12 mol) in formamide (47.5 ml) and water (2.5 ml) was shaken, then
dispensed into 15 x
8 ml vials. All vials were sealed and then heated at 150° for 3.5 h.
The vials were allowed to
cool to room temperature, then water (20 ml) was added and the mixture was
exhaustively
extracted with chloroform. The chloroform was removed to give a concentrated
formamide
solution (22.2 g) which was added to a flash silica column (6 cm diameter, 12
cm height)
packed in 1% MeOH/1% Et3N in chloroform. Elution of the column with 2.5 L of
this
mixture followed by 1 L of 2% MeOH/1% Et3N in chloroform gave, in the early
fractions, a
product suspected of being S-ter°t-butyl-oxazole-4-carboxylic acid
ethyl este>~ (6.3 g, 26%).
[0286] HPLC (214nm) tR = 8.77 min.
loas~~ 1H NMR (400 MHz, CDCl3) 8 1.41 (t, J = 7.2 Hz, 3H); 1.43 (s, 9H); 4.40
(q, J= 7.2 Hz, 2H); 7.81 (s, 1H).
lozss~ 13C NMR (100 MHz, CDCl3) 8 14.1, 28.8, 32.5, 61.3, 136.9, 149.9, 156.4,
158.3.
[0289] ESMS nzlz 198.3 [M+H]+, 239.3 [M+CH4CN]+.
[0290] LC/MS tR = 7.97 (198.1 [M+H]+) min.
-63-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[029I] Recovered from later fractions was 5-tert-butyl-3H-imidazole-4-
carboxylic
acid ethyl ester (6.20 g, 26%). (Durant et al., "Aminoalkylimidazoles and
Process for their
Production." Patent No. GB 1341375 (Great Britain, 1973)).
[0292] HPLC (2I4nm) tR = 5.4I (93.7%) min.
[0293] 1H NMR (400 MHz, CDC13) 8 1.38 (t, J= 7.0 Hz, 3H); 1.47 (s, 9H); 4.36
(q, J= 7.2 Hz, 2H); 7.S4 (s, 1H).
[0294] t3C ~R (100 MHz, CDCl3) S 13.7, 28.8, 32.0, 59.8, 124.2, 133.3, 149.2,
162.6.
[0295] ESMS m/z 197.3 [M+H]+, 238.3 [M+CH4CN]+.
[0296] Further elution of the column with 1L of 5% MeOh/1% Et3N gave a
compound suspected of being 5-test-butyl-3H imidazole-4-carboxylic acid (0.50
g, 2%).
[0297] HPLC (245nm) tR = 4.68 (83.1%) min.
~oa9s~ 1H NMR (400 MHz, CD30D) cS 1.36 (s, 9H); 7.69 (s, 1H).
(0299] 1H ~R (400 MHz, CDCl3) b 1.37 (s, 9H); 7.74 (s, 1H).
~osoo~ 1H ~R (400 MHz, CD3S0) ~ 1.28 (s, 9H); 7.68 (s, 1H).
[0301] ESMS m/z 169.2 [M+H]+, 210.4 [M+CH4CN]+.
(5=tent-Butyl-3H imidazol-4 yl)-methafaol
O ~~ OH
IiAIH,~
~~ti ---.-~. ~ H
N N
[0302] A solution of 5-tent-butyl-3-imidazole-4-carboxylic acid ethyl ester
(3.30
g, 16.8 mmol) in THF (60 ml) was added dropwise to a suspension of lithium
aluminium
hydride (95% suspension, 0.89 g, 22.2 mmol) in THF (40 ml) and the mixture was
stirred at
room temperature for 3 h. Water was added until the evolution of gas ceased,
the mixture
was stirred for 10 min, then was filtered through a sintered funnel. The
precipitate was
washed with THF, then with methanol, the filtrate and washings were combined
and
evaporated. The residue was freeze-dried overnight to afford, as a white solid
(5-tert-butyl-
-64-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
3H-irnidazol-4-yl)-methanol (2.71 g, IOS%). (Durant et al.,
"Arninoalkylimidazoles and
Process for their Production." Patent No. GB 1341375 (Great Britain, 1973)).
[0303] HPLC (240nm) tR = 3:70 (67.4%) min.
[03041 1H NMR (400 MHz, CD3OD) 8 1.36 (s, 9H); 4.62 (s, 2H); 7.43 (s, 1H).
(0305] 13C NMR (100 MHz, CD30D) ~ 31.1, 33.0, 57.9, 131.4, 133.9, 140.8.
[0306] LC/MS tR = 3.41 (1SS.2 [M+H]+) min.
[0307] This material was used without further purification.
5-tent Butyl-3H i~nida~ole-4-carbaldehyde
OH O
MnO2
JH ~JH
[0308] Manganese dioxide (30 g, 0.35 mol) was added to a heterogeneous
solution of (S-tent-butyl-3H-imidazol-4-yl)-methanol (4.97 g, 0.03 mol) in
acetone (700 ml)
and the resulting mixture was stirred at room temperature for 4 h. The mixture
was filtered
through a pad of Celite and the pad was washed with acetone. The, filtrate and
washings were
combined and evaporated. The residue was triturated with ether to afford, as a
colorless
solid, S-tert-butyl-3H-imidazole-4-carbaldehyde (2.50 g, S1%). (Hayashi,
Personal
Communication (2000)).
[0309] HPLC (240nm) tR = 3.71 (89.3%) min.
~o3io~ 1H j~R (400 MHz, CDC13) 8 1.48 (s, 9H); 7.67 (s, 1H); 10.06 (s, 1H).
[0311] LC/MS tR = 3.38 (153.2 [M+H]~ min.
[0312] Evaporation of the filtrate from the trituration gave additional S-tert-
butyl-
3H-imidazole-4-carbaldehyde (1.88 g, 38%).
-65-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
1-Acetyl-3-(5'-test-butyl-IH imdazol-4'-Z ylmethylehe) piperazihe-2,5-diofze
.O O O
O ~
~N~ ~NH
H .~ ~N~ --- ~N w. 'w NH
N O O O O
[0313) To a solution of 5-tent-butyl-3H imidazole-4-carbaldehyde (2.50 g,
164.4
mmol) in DMF (50 ml) was added 1,4-diacetyl-piperazine-2,5-dione (6.50 g, 32.8
mmol) and
the solution was evacuated; then flushed with argon. The evacuation-flushing
process was
repeated a further two times, then cesium carbonate (5.35 g, 16.4 mmol) was
added. The
evacuation-flushing process was repeated a further three times, then the
resultant mixture was
stirred at room temperature for 5 h. The reaction mixture was partially
evaporated (heat and
high vacuum) until a small volume remained and the resultant solution was
added dropwise
to water (100 ml). The yellow precipitate was collected, then freeze-dried to
afford 1-acetyl-
3-(5'-tert-butyl-1H-imidazol-4'-Z-ylmethylene)-piperazine-2,5-dione (2.24 g,
47%).
(Hayashi, Personal Communication (2000)).
[0314] HPLC (214nm) tR = 5.54 (94.4%) min.
[0315] 1H NMR (400 MHz, CDCl3) ~ 1.47 (s, 9H); 2.65 (s, 3H), 4.47 (s, 2H);
7.19 (s, 1H); 7.57 (s, 1H), 9.26 (s, 1H), 12.14 (s, 1H).
(0316] isC NMR (100 MHz, CDC13+CD30D) 8 27.3, 30.8, 32.1, 46.5, 110.0,
123.2, 131.4, 133.2, 141.7, 160.7, 162.8, 173.0
[0317] LC/MS tR = 5.16 (291.2 [M+H]+, 581.6 [2M+H]~ min.
3-Z-Benzylidene-6-(5 "-tef~t-butyl-1 H itnidazol-4 "-Z ylmethylene)
piper°azine-2, 5-dione
O ( ~ 'O O
~'NH N='y ~ ~ ~ NH N'-1
N ...~ ~ .NH ,. I / HN ~ ~.. ~ NH
a ~ o
[0318] To a solution of 1-acetyl-3-(5'-tert-butyl-1H-imidazol-4'-Z-
ylmethylene)-
piperazine-2,5-dione (2.43 g, 8.37 mmol) in DMF (55 ml) was added benzaldehyde
(4.26 ml,
41.9 mmol) and the solution was evacuated, then flushed with nitrogen. The
evacuation-
-66-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
flushing process was repeated a further two times, then cesium carbonate (4.09
g, 12.6 mmol)
was added. The evacuation-flushing process was repeated a further three times,
then the
resultant mixture was heated under the temperature gradient as shown below.
After a total
time of 5 h the reaction was allowed to cool to room temperature and the
mixture was added
to ice-cold water (400 ml). The precipitate was collected, washed with water,
then freeze-
dried to afford a yellow solid (2.57 g, HPLC (214nm) tR = 6.83 (83.1%) min.).
This material
was dissolved in chloroform (100 ml) and evaporated to azeotrope remaining
water, resulting
in a brown oil. This was dissolved in chloroform (20 ml) and cooled in ice.
After 90 min the
yellow precipitate was collected and air-dried to afford 3-Z-benzylidene-6-(5"-
tert-butyl-1H
imidazol-4"-Z-ylmethylene)-piperazine-2,5-dione (1:59 g, 56%). (Hayashi,
Personal
Communication (2000)).
[0319] HPLC (214nm) tR = 6.38 (2.1%), 6.80 (95.2) min.
j0320J 1H NMR (400 MHz, CDC13) 8 1.46 (s, 9H); 7.01 (s, 1H, -C-C=CI~; 7.03
(s, 1H, -C-C=CIA; 7.30-7.50 (m, SH, As'); 7.60 (s, 1H); 8.09 (bs, NH); 9.51
(bs, NH); 12.40
(bs, NH).
(0321] LC/MS tR = 5.84 (337.4 [M+H]+, E isomer), 6.25 (337.4 [M+H]+, 673.4
[2M+H]+, Z isomer) min.
(0322] ESMS m/z 337.3 [M+H]+, 378.1 [M+CH4CN]+.
[0323] Evaporation of the chloroform solution gave additional 3-Z benzylidene-
6-
(5"-tert-butyl-1H imidazol-4"-Z-ylmethylene)-piperazine-2,5-dione (0.82 g,
29%). HPLC
(214nrn) tR = 6.82 (70.6%) min.
General Experimental
[0324] Sodium bicarbonate refers to a 5% solution.
[0325] Organic solvents were dried over sodium sulfate unless otherwise
stated.
Analytical Conditions
NMR Conditions
[0326] 1H ~ (400 M$z) analysis was performed on a Varian Inova Unity 400
MHz NMR machine. Samples were run in deuterated chloroform containing 0.1% TMS
(unless otherwise specified). Chemical shifts (ppm) are referenced relative to
TMS (0.00
-67-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
pp~n) or
CH30H at
3.30 ppm
for samples
run CD30D.
Coupling
constants
are expressed
in
hertz (Hz).
Analytical Conditions
HPLC
[0327) System 6 conditions:
[0328] RP-HPLC was done on a Rainin Microsorb-MV CIS
(5 ~,m, 100A) 50 x
4.6 mm column.
[0329] Buffer A: 0.1 % aqueous TFA
[0330] Buffer B: 0.1% TFA in 90% aqueous MeCN
[0331] Gradient: 0 - 100% Buffer B over 11 min
[0332] Flow rate: I.5 mL/min
LCMS Conditions
[0333] LCMS were run on a Perkin-Elmer Sciex API-100
instrument.
[0334] LC conditions:
[0335] Reverse Phase HPLC analysis
(0336] Column: Monitor 5 ~.m C18 50x4.6 rnm
[0337] Solvent A: 0.1% TFA in water
(0338] Solvent B: 0.085% TFA in 90% aqueous MeCN
(0339] Gradient: 0-100% B over 11.0 min
[0340] Flow rate: I.5 mL/min
[0341] Wavelength: 214 nm
[0342] MS conditions:
(0343] Ion Source: Ionspray
(0344] Detection: Ion counting
(0345] Flow rate to the mass spectrometer: 300 ~.L/min
after split from
column (1.5
mL/min).
ESMS Conditions
[0346] ESMS was done on a Perkin Elmer/Sciex-API III
LC/MS/MS using an
electrospray
inlet.
[0347] Solvent: 0.1 % AcOH in 60% aqueous MeCN
[0348] Flow rate: 25 ~.L/min
-68-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0349] Ionspray: 5000 V
[0350] Orifice plate: SS V
[0351] Acquisition time: 2.30
min
[0352] Scan range: 100-1000
amu/z
[0353] Scan step size: 0.2
amu/z
Preparative
RP-HPLC
Purification
Conditions
[0354] Reverse phase HPLC purification was carried out using Nebula with the
Waters XterraMS column (19xS0 mm, S ~,m, C18) using the following conditions:
[0355] Solvent A: 0.1% aqueous TFA
[0356] Solvent B: 0.1% TFA in 90% aqueous MeCN
[0357] Gradient: S-9S% B over 4 min
[0358] Flow rate: 20 mL/min
[0359] Wavelength: 214 nm
[0360] Abbreviations are as follows: br s: broad singlet; BuLi : n-butyl
lithium; d:
doublet; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; ESMS: electrospray mass
spectrometry;
HCI: hydrochloric acid; HPLC: high performance liquid chromatography; LCMS:
liquid
chromatography mass spectrometry; LD: lithium diisopropylamide; M+: molecular
ion; m:
multiplet; MeCN: acetonitrile; M: mass spectrometry; MW: molecular weight;
NMR: nuclear
magnetic resonance; q: quartet; s: singlet; : triplet; tR: retention time;
TFA: trifluoroacetic
acid; THF: tetrahydrofuran
Detailed Procedure for the SytZthesis of Dehyd~ophenylahistih
0
0
o r~~ o ' I , o
Acs N~Nti Acv
N . N ' ._._.,:~, ~ HN ~ --"
H N~ NH ~ ~ / NH N~ H
Ac ~ ..!
o Cs2CO3 IDMF O ~ Cs2C03 7DMF o
2
-69-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
1-Acetyl-3- f (~-1-[5-(1,1-dimethyl-2-propenyl)-1H 4-imidazolyl]methylidene}]-
2,5-
piperazinedione (2)
A ',
i
~NH N-r/NH
0
[0361] To a solution of 5-(1,1-dimethyl-2-propenyl)imidazole-4-carboxaldehyde
(100 mg, 0.609 rnmol) in DMF (2 mL) was added compound 1 (241 mg, 1.22 mmol)
and the
solution was repeatedly evacuated in a short time to remove oxygen and flushed
with Ar,
followed by the addition of CsZC03 (198 mg. 0.609 rnrnol) and the evacuation-
flushing
process was repeated again. The removal of oxygen is preferred because such
removal is
believed to decrease oxidation of alpha-carbon at the position 6 of the
dilcetopiperazine ring.
The resultant mixture was stirred for 5 h at room temperature. After the
solvent was removed
by evaporation, the residue was dissolved in the mixture of EtOAc and 10%
Na2C03, and the
organic phase was washed with 10% Na2C03 again and saturated NaCl for three
times, dried
over Na2C03 and concentrated in vacuo. The residual oil was purified by column
chromatography on silica using CHC13-MeOH (100:0 to 50:1) as an eluant to give
60 mg
(33%) of a pale yellow solid 2.
Dehydrophenylahistin
[0362] To a solution of 2 (30 mg, 0.099 mmol) in DMF (0.8 mL) was added
benzaldehyde (51 ~,L, 0.496 mmol, 5 e~ and the solution was repeatedly
evacuated in a short
time to remove oxygen and flushed with Ar, followed by the addition of Cs2C03
(53 mg,
0.149 mmol, 1.5 ec~ and the evacuation-flushing process was repeated again.
The resultant
mixture was heated for 2.5 h at 80°C. (The temperature. must be
increased slowly. Rapid
heating increases the production of E-isomer at the benzylidene.moiety.) After
the solvent
was removed by evaporation, the residue was dissolved in EtOAc, washed with
water for two
times and saturated NaCI for three times, dried over NaZSOø and concentrated
in vacuo. On
TLC using CHCI3-MeOH (IO:I), you can observe a spot with bright green-yellow
luminescence at 365 nm UV. The purity of this crude product was more than 75%
from
-70-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
HPLC analysis. The resulting residue was dissolved in 90% MeOH aq and applied
to
reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 x 250 rnm) and eluted using a
linear
gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min,
and the
desired fraction was collected and concentrated by evaporation to give a I9.7
mg (60%),
although the yields are not optimized for each step,. of yellow colored
dehydrophenylahistin.
EXAMPLE 4
Bioloycal Characteristics of Deh dro hen,~lahistin and DehYd~henylahistin
Analogs
A. Biological Evaluation
[0363] The biological characteristics of synthesized tBu-dehydrophenylahistin
and dehydrophenylahistin were evaluated in both HT29 human colon cells, and PC-
3
prostatic adenocarcinoma cells.
[0364] HT-29 (ATCC HTB-38) a human colorectal adenocarcinoma was
maintained in McCoy's complete medium (McCoy's SA medium with L-glutamine and
25rnM HEPES supplemented with 10% FBS, 1mM Na pyruvate, 1X NEAR, 2mM L-
glutamine, and Pen/Strep at 100ILT/ml and 100p,g/ml, respectively). PC-3
.(ATCC CRL-
1435), a human prostate adenocarcinoma, was maintained in F12K complete medium
(F12K
medium supplemented with 10% FBS; 2mM Glutamine; 1% HEPES; and Pen/Strep at
100ICT/ml and 100~.g/ml, respectively). Cell lines were cultured at 37
°C, 5% C02 in a 95%
humidified incubator.
[0365] For tumor cytotoxicity assays HT-29 or PC-3 cells were seeded at 5,000
cells/well in 90 pl complete media into a Corning 3904 black-walled, clear-
bottom tissue
culture plate and the plate were incubated overnight to allow cells to
establish and enter log
phase growth. 20 mM stock solutions of dehydrophenylahistin and tBu-
dehydrophenylahistin
were prepared in 100% DMSO and stored at -20 °C. lOX concentrated
serial dilutions of the
two compounds were prepared in appropriate culture medium for final
concentrations ranging
from 2.0 x 10-S M to 2.0 x 10'1° M. Ten ~1 volumes of the l OX serial
dilutions were added to
the test wells in triplicate and the plates returned to the incubator for 48
hours. The final
concentration of DMSO was 0.25% in all samples.
[0366] Following 48 hours of drug exposure 10 ~.1 of 0.2 mg/ml resazurin
(obtained from Sigma-Aldrich Chemical Co.) in Mg2+, Ca2+ free PBS was added to
each well
-71-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
and the plates were returned to the incubator for 3-4 hours. The plates were
removed and
resazurin fluorescence was measured using 530 nm excitation and 590 nm
emission filters in
a Fusion fluorimeter (Packard Instruments). Resazurin dye without cells was
used to
determine the background, which was subtracted from the data for all
experimental wells.
The data were analyzed using Prism software (GraphPad Software). The data were
normalized to the average of the cells treated with media only (100% cell
growth) and ECso
values were determined using a standard sigmoidal dose response curve fitting
algorithm.
[0367] As indicated in Table 1 below, tBu-dehydrophenylahistin demonstrates
about a 4-times greater cytotoxic activity in comparison with
dehydrophenylahistin.
Table 1. Cytotoxic Effect of dehydrophenylahistin and derivative.
0 0
/ HN / i H / HN / ~ NH
NH N,/ w ~ i NH N,/
Dehydrophenylahistin tBu-dehydrophenylahistin
EC5o (nM)
cell ~PLH tBu-~PLH
HT29 43 13
PC-3 5.4 1.0
(0368] See also Figure 41 for additional data at HT-29, PC-3, and P-388 cells.
B. Structure and Activity Study of Deh~drophenylahistin Derivatives
[0369] The cytotoxic effects of phenylahistin, dehydrophenylahistin and
various
derivatives of dehydrophenylahistin were examined in P388 murine leukemia
cells, HT-29
human colon cells, and PC-3 prostatic adenocarcinoma cells.
[0370] As explained above, HT-29 a human colorectal adenocarcinoma was
maintained in McCoy's complete medium (McCoy's SA medium with L-glutamine and
25mM HEPES supplemented with 10% FBS, 1mM Na pyruvate, 1X NEAR, 2mM L-
glutamine, and Pen/Strep at 1001U/ml and 100~.g/ml, respectively). PC-3, a
human prostate
_72-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
adenocarcinoma, was maintained in F12K complete medium .(F12K medium
supplemented
with 10% FBS; 2mM Glutamine; 1% HEPES; and Pen/Strep at 100IU/ml and
100~,g/ml,
respectively). Cell lines were cultured at 37 °C, 5% C02 in a 95%
humidified incubator.
[0371] For tumor cytotoxicity assays HT-29 or PC-3 cells were seeded at 5,000
cells/well in 90 ~.l complete media into a Corning 3904 black-walled, clear-
bottom tissue
culture plates and the plates were incubated overnight to allow cells to
establish and enter log
phase growth. 20 mM stock solutions of dehydrophenylahistin and tBu-
dehydrophenylahistin
were prepared in 100% DMSO and stored at -20 °C. lOX concentrated
serial dilutions of the
two compounds were prepared in appropriate culture medium far final
concentrations ranging
from 2.0 x 10'5 M to 2.0 x 10'1° M. Ten wl volumes of the lOX serial
dilutions were added to
the test wells in triplicate and the plates returned to the incubator for 48
hours. The final
concentration of DMSO was 0.25% in all samples.
[0372] Following 48 hours of drug exposure 101 of 0.2 mg/ml resazurin
(obtained from Sigma-Aldrich Chemical Co.) in Mg2+, CaZ+ free PBS was added to
each well
and the plates were returned to the incubator for 3-4 hours. The plates were
removed and
resazurin fluorescence was measured using 530 nrn excitation and 590 nm
emission filters in
a Fusion fluorirneter (Packard Instruments). Resazurin dye without cells was
used to
determine the background, which was subtracted from the data for all
experimental wells.
The data were analyzed using Prism software (GraphPad Software). The data were
normalized to the average of the cells treated with media only (100% cell
growth) and ECso
values were determined using a standard sigmoidal dose response curve fitting
algorithm:
[0373] ECSO and ICso values of phenylahistin, dehydrophenylahistin and
dehydrophenylahistin derivatives are summarized in Table 2 below.
-73-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table Z. SAR. stud~of phen~lahistin or dehydrophenylahistin
arid of dehydronhenylahistin derivatiyes
ECsp n ICSO n
COMPOUNDS STRUCTURE
HT-29 PC-3 P-388
(-)-Phenylahistin O
NH~N NH~~ I ~ 1600 n.t. 833 ~ 153
i / NH~
\ _ . O (n=5)
KPU-1 O
OPLH ~=N NH ~/ ~ 4g 4.7 36 ~ 12.8
NH ~ / NH
., O (n-5)
KPU-2 O
tBu-~PLH /=N NH ~/ ~ 13 1 31.8 ~ 5.0
NH ~ .~ NH I i
(n=5)
O
KPU-6 O
tBu-OPLH-p-OMe /=N NH ~/ ~ >2000 n.t. 9333 ~
NH ~. / NH I /
OMe 5457
O
(n=3)
KpU_g O OMe
tBu-~PLH-o-OMe /=N NH ~/ ~ g9 315 +_ 137
NH ~ / NH
( (n=4)
O
KPU-9 O
tBu-~PLH-M-OMe /=N NH / ~ OMe
31 20.8 ~ 68
NH ~ / NH I /
(n=4)
O
Colchicine --- 208~68
(n=4)
-74-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0374] Modifications to the phenyl ring have a significant effect of the
cytotoxic
activities. In comparison with the activity of tBu-dehydrophenylahistin (#6),
the activity of
the methoxy group at the meta-position (I~.I'U-9) exhibited the highest
activity than the other
derivatives with an ICso of 20.83.3 nM. in P388 cells. The KPU-9 derivative
also exhibited
cytotoxicity in HT-29 cells (ECso 31 nM). Dehydrophenylahistin, tBu-
dehydrophenylahistin
(KPU-2) and the KPU-9 derivative all exhibited cytotoxicity in P388 cells.
C. Structure and Activity Study of Additional Dehydrophenylahistin Derivatives
[0375] The cytotoxic effects of phenylahistin, dehydrophenylahistin and
various
additional derivatives of dehydrophenylahistin were examined in HT-29 human
colon cells
and PC-3 prostatic adenocarcinoma cells using the methodology described above.
Table 3. SAR study of phen~ahistin, dehydrophenylahistin and of additional
dehydrophenylahistin derivatives
COMPOUNDS STRUCTURE F Mv'~' n
g~ ~ T ~9 pC-3
O
(-)-Phenylahistin/=N NH~' ( ~ - 350.41 1600 n.t.
~
NH
i ~ NH
O
KPU-1 /-=N NH ~~ - 348.40 48 4.7
~PLH ~~
NH ~ ~ NH
O
O
KPU-2 /~N NH ~~ - 336.39 13 1
tBu-~PLH ~
NH ~ ~' NH
O
O
KPU-6 /=N NH ~~ - 366.41 >2000 n.t.
tBu-OPLH-p-OMe~~
NH ~ ~ NH I ~ OMe
O
-75-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F ~ M:W -B-'
~ C5o
~
, HT-29 PC-3
O OMe
~'U-8 /=N NH ~ - 366.41 89
tBu-aPLH-o-OMe~
NH ~ ~ NH
O
O
.
KPU-9 ~ OMe _ 366.41 31
H N NH /
tBu-OPLH-m-OMeI
N
~ ~ NH
O
O OMe
KPU-14 ~=-N NH / ~ OMe '1'FA 396.44 610 96%
tBu-OPLH-2,3- NH i / NH ~ S 10.46
diOMe 101 (+TFA)
O OMe
KPU-12 ~=N NH ~~ - 396.44 4980
tBu-OPLH-2,4- ~~
NH i ~ NH I ~
OMe
diOMe
O
O OMe
KPU-10 ~--N NH Y ~ - 396.44 1350
tBu-~PLH-2,S- NH .~ / NH I i
diOMe I I
O OMe
O OMe
KPU-1S /~N NH ~ TFA 396.44 4430 96%
fBu-OPLH-2,6- ~ S 10.46
NH .~ ~ NH ~ ,
v
diOMe ~ Me0 (+TFA)
O
KPU-13 ~N NH / ~ OMe _ 396.44 2130
tBu-~PLH-3,4- NH i / NH
~
diOMe OMe
O
-76-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
STRUCTURE F Mv'~' n
COMPOUNDS ~
g~ H 29 PC-3
O
KPU-16 /=N NH ~ ~ OMe - 396.44 42 82%
tBu-tIPLH-3,S-NH i / NH
diOMe
O OMe
O
KPU-1 I /=N NH , ~ OMe _ 426.47 > 20
tBu-~PLH-3,4,5-NH i / NH ~ !~M
triOMe OMe
p OMe
O OMe
KPU-17 /=N NH / ~ OMe TFA 426.47 4060 94%
tBu-~PLH-2,3,4-NH ~ ~ NH ~ 540.49
triOMe OMe (+TFA)
v ~
O CI
KPU-18 ~-N NH ~ ~ TFA 370.83 42 100
tBu-~PLH-o-CI NH i / NH ~ / 484.86
(+TFA)
O
O
KPU-19 /- N NH ~ ~ CI TFA 370.83 20 98%
tBu-OPLH-m-CI NH ~- / NH ~ 484.86
(+TFA)
O
O
KPU-20 /=N NH ~' ~ TFA 370.83 S4S
tBu-OPLH-p-Cl NH ~ r NH ~ / 484.86
CI (+TFA)
jj
O
O CI
KPU-21 /=N NH ~ ~ TFA 415.83 S 1 100
tBu-dPLH-2CI-S-NH i / NH ~ 529.85
NOa TI (+TFA)
O N02
_77_
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
STRUCTURE M
W
COMPOUNDS gOgM ' 2 PC
' 3
H - -
9
O
KPU-22 /=N NH ~ ~ ~ TFA 380.40 82 95%
tBu-dPLH-3,4- NH i / NH ~ 494.42
methylene-dioxyv ~ O (+TFA)
O
O OH
KPU-23 ~N NH ~ ~ OMe TFA 382.41 5870 86%
tBu-~PLH-2-OH-NH ~ 496.44
NH ~
3-OMe , (+TFA)
v ~
(o-vanillin) O
O
KPU-24 ~-N NH ~ ~ TFA 364.40 7040 100
tBu-~PLH- NH .~ / N~ 487.42
cyclized-3-Me0v O ~ (+TFA)
OMe
O
KPU-25 /~ (~ NH ~ ~ TFA 337.38 544 98%
tBu-~PLH-4- NH i i NH I ~ N 565.42
pyridyl ~ (+2TFA
O )
O
KPU-28 /-N NH ~ ~ TFA 337.38 > 20 99%
tBu-4PLH-2- NH i i NH N ~ 565.42 ~.M
py~dyl ~ o (+2TFA
)
O
KPU-26 O TFA 326.35 600 88%
/=N NH ~
tBu-OPLH-2-furyl~ 440.37
~
NH i i NH
(+TFA)
O
O
KPU-27 Me TFA 356.44 80 97%
S
/=N NH ~
tBu-dPLH-5-Me-/ 470.47
\
NH ~ i NH
2-thienyl v ~ (+TFA)
_78_
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE Fo~ M _ ECS
W n
- , AT-29 PC-3
,
O -"
KPU-29 H N NH ~ ~ S ~ TFA 356.44 44 81%
tBu-OPLH-3-Me-N 470.47
i
, NH >--~
2-thienyl _ (+TFA)
~ Me
[0376] Additional cytotoxicity assays were performed as described above under
this example, using Resazurin fluorescence as an indicator of cell viability.
The results are
shown below in Table 3.1.
Table 3.1. Study of phenylahistin, deh~phe~lahistin and of additional
dehydrophenylahistin derivatives
ECso % c
(M) totox
NPI Structure Chemical name Mean SD Mean SD n
0
NH (-)-(S)-Halimide
N (-)-
\ ,
/ HN
OH~ (S)-Phenylahistin,
H (-)-
2350 ~aO S -PLH 3.94E-079.49E-0887 2 5
_ Q- _
\ \ NH N=~
NH
/ HN \ \
CH
H ~ ' ' Dehydrophenylahistin,
~
2352 '~ delta-PLH, KPU-14.26E-081.08E-0886 2 5
OCH3
\
N N--~
/ N ~ \ \ NH
CH
OCH3HC KPU-4, D,L-bislactim-
2354 ~'3 PLH >2E-05 NA 4 NA 2
0
NH N
i~
H
/N~ \ \ KPU-5, D,L-
p 'CH2
CH~H
2355 ~ monolactim-PLH >2E-05 NA 13 NA 2
"~ .
0
\ \ NH N-~
~
NH
/ HN \ \
H3C0
CH
~ 2
~ H
2356 'H3~ KPU-6 6.57E-06NA 63 NA 2
H
NH N~,H
HNCH,
2357 "~~ KPU-7 8.25E-06NA 63 NA 2
0
\ \ NH N'~~
I o HN \ \ NH
o t-butyl-delta-PLH,
2358 "~ ~~H' KPU-2 1.49E-083.77E-0986 2 5
-79-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
oCH, o
I W W NH N=1
r HN ~ ~ tJH
2359 "3~"~"' KPU-8 7.55E-082.47E-0887 3 3
0
HaCO ~ ~ NH -1
~
r HN ~ ~
H
2360 ~ "a "~N' KPU-9 2:61 8.54E-0987 3 3
E-08
OCH3 O
NH N--~
r HN w WH
~
2361 oCH, o KPU-10 9.70E-07NA 86 NA 2
"a '~H'
0
H~CO ~ ~ NH
I r HN w WH
H~00
0~
ocH,
2362 ~"3 KPU-11 8.19E-06NA 78 NA 2
ocH3 0
~ w NH
I r HN ~ ~ NH
H,CO
2363 o H,C CHcH' KPU-12 5.08E-06NA 79 NA 2
a
H,CO ~ ~ NH N-
HN w W"
I
r
H3C0
~0
2364 '~H' KPU-13 2.02E-06NA 76 NA 2
O OMe
/~N HN / ~ OMe
HN ~ r NH ~ r
o KPU-14, tBu-delta-
2365 H'c ~H' PLH-2,3-diOMe 8.68E-07NA 84 NA 2
0
/~N HN r
HN ~
NH ~
/
r KpU-15, tBu-delta-
Me0
'2366 H'c '~~H' PLH-2,6-diOMe 5.17E-06NA 81 NA 2
o
/~N HN r ~ oMe
HN
~
~ r NH
r KPU-16, tBu-delta-
oMc
2367 H'c '~"' 3,5-diOMe 4.54E-OS1.21E-0887 2 3
0 oMe
~N HN r ~ oMe
HN ~ , NH I /'
OMe KPU-17, tBu-delta-
o
2368 H'c '~'H' PLH-2,3,4-triOMe4.80E-06NA 78 NA 2
0
/=N HN
HN ~ / NH ~ r
o KPU-18, tBu-delta-
2369 H'c '~'H' PLH-o-CI 4.63E-081.35E-0886 3 3
0
/~N HN r
HN ~ / NH ~ r
~~'' KPU-19, tBu-delta-
~
2370 H'c '~'H PLH-m-CI 2.45E-088.73E-0985 3 3
-~0-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
m
RN HN i
HN
~
~ ~ NH
i
.~ N KPU-21, tBu-delta-
2371 H PLH-2-CI-5-N025.34E-084.85E-0986 3 3
,
' '~"'
"N ;" ~ NN ~ ~ KPU-22
o tBu-delfa-
,
PLH-3,4-methylene-
2372 H' '~_"' diox 8.73E-081.10E-0884 4 3
O OH
HN ~N ~ NH I KPU-23
~e tBu-delta
s ,
PLH-2-OH-3-OMe
2373 "' '~_"' o-vanillin 5.36E-06NA 79 NA 2
~N HN ~
HN
I
~ / N
, KPU-24, tBu-delta-
~O OMe
2374 "' '~3"' PLH-c clized-3-Me06.92E-06NA 58 NA 2
0
~N HN i
HN
I
~ , NH
~N KPU-25, t8u-delta-
2375 H' "~~"' PLH-4- rid 5.48E-07NA 82 NA 2
I
/~N HN i
HN ~ o NH ~ i
KPU-28, tBu-delta-
2376 H' '~3"' PLH-2- rid >2E-05 NA 39 NA 2
!
0
HN~N HN ~ ~ ~ .
i NH
KPU-26, tBu-delta-
2377 "'c "~3"' PLH-2-fu I 7.12E-07NA 80 NA 2
/-N HN i S Me
HN ~ ~ NH
KPU-27, tBu-delta-
2378 "' " ~"' PLH-5-Me-2-thien8.52E-081.35E-0885 3 3
I
0
HN N HN ~
i NH
Me KPU-29, tBu-delta-
2379 H' '~3"' PLH-3-Me-2-thien4.70E-081.30E-0886 3 3
I
N HN
~NN
H~G KPU-20; t-butyl-delta-
C'
PLH-p-CI +
TFA salt
2380 .TFA MW:484 7.09E-07NA 81 NA 2
~ KPU-30, tBu-delta-
/~N HN i PLH-2
HN 3-
I
~ a N" ,
~ methylendioxy;
~ TFA
2381 "~ salt MW:494.425.20E-07NA 82 NA 2
~3 ~
/~N HN s
"" ~ , NH ~ N KPU-31, tBu-delta-
PLH-3-pyridyl;
2TFA
2382 H' '~~"' salt MW 565.421.03E-071.16E-0886 3 3
-81-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
O CHI
/=N HN s
HN , KPU-32, tl3u-delta-
NH I
~ PLH-o-Me; TFA
~ salt
~
2383 "3 MW: 464.44 4.58E-081.15E-0886 3 3
"~' '
_ KPU-33, tBu-delta-
"N~N sN NH N ~ PLH-3-Me-2-pyridyl;
2TFA salt (MW:
~ H
2384 3 579.45 >2E-05 NA 37 NA 2
H'c C'~H
HN ;N ,N H I ~ KPU-34, tBu-delta-
F PLH-4-F; TFA
~ salt
2385 H' '~a" MW:468.40 5.01 8.48E-0881 5 3
E-07
0
- F ~ I H N~ N~ KPU-35, tBu-delta-
NH
~o . PLH-m-F; TFA
salt
2386 3H' MW:468.40 1.31E-085.32E-0985 2 5
"~ KPU-36, tBu-delta-
r-N "N~ PLH-5-Me-4-im;
HN ~ , NH NJNH
O 2TFA salt
2387 H,C H MW:584.47 2.35E-06NA 84 NA 2
"~~ '
O F
HN ,N ~N N I ~ KPU-37, tBu-delta-
PLH-o-F, TFA
~ salt
2388 H' '~3" MW: 468.40 3.OOE-081.65E-0886 3 3
~
HN ;N ~ H I ~ H3 tBu-delta-
KPU-38,
PLH-m-Me; TFA
~ salt
23gg "' ~_" MW: 464.44 4.67E-089.64E-0986 3 3
HN ;N ~ H I ~ KPU-39, tBu-delta-
"' PLH-p-Me; TFA
~ Salt
2390 H,c c~" MW:464.44 4.83E-076.15E-0884 4 3
0
HN ,N /N H I ~ KPU-40, tBu-delta-
a' pLH-p-Br; TFA
Salt
2391 "' '~" MW: 529.31 7.14E-07NA 82 NA 2
0
HN,N ~N KPU-41, tBu-delta-
I ~ Br
N PLH-m-Br; TFA
Salt
~
2392 ' "' '~~" MW: 529.31 3.11 7.29E-0987 3 3
E-08
HN ~N / NH ~ S KPU-42, tBu-delta-
PLH-3-thienyl;
TFA
2393 H' '~~"' Salt MW: 456.445.58E-086.23E-0986 3 3
0
I tBu-delta-
HN ~N /N KPU-43
~ ,
NH PLH-p-CN; TFA
~~N Salt
2394 H' ~"' MW: 475.42 >2E-05 NA 14 NA 2
-82-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
0
/=N HN i w O~CH~
HN ~ , NH I i
~
o KPU-44, tBu-delta-
~
H,C C~'H pLH-m-EtOTFA
Salt
2395 MW:494.46 4.59E-081.02E-0885 4 3
0 0~'"' . tBu-delta-
KPU-45
!~N HN i ,
HN 6-TriOMe;
I PLH-2
4
, ~ N~ ,
~ o,~~ ,
TFA Salt (MW:
2396 H' '~~"' 540.49 >2E-05 NA 2 NA 2
O O,.N,A
AN HN i ~ KPU-46, tBu-defta-
I
~'UIfNH PLH-o-N02;
~ TFA
2397 H' C~"' ~ Salt MW: 495.415.03E-081.24E-0886 4 3
0 0
,~,
HN ,N tBu-delta-
I ~ N'o KPU-47
s H ,
PLH-m-N02;
TFA
2398 H'o '~3"' Salt MW: 495.414.47E-081.19E-0886 3 3
0
HN ,N ,N H I ~ KPU-48, tBu-delta-
~,
o PLH-p-NO2;
TFA
23gg H' o~H' Salt MW: 495.41>2E-05 NA 23 NA 2
~N
HN ;N ,N N; I ~ KPU-49, tBu-delta-
.~ o PLH-m-CN; TFA
~ Salt
2400 H'~ '~3H MW: 475.42 3.56E-081.40E-0885 3 3
0 6r
HN ~N , NH I i KPU-50, tBu-delta-
o PLH-o-Br; TFA
~ Salt
2401 H'o '~H MW: 529.31 4.05E-081.09E-0886 3 3
CH KPU-51, tl3u-delta-
HN ~N %N H I
~ PLH-m-OH; TFA
o Salt
~
2402 H'o o'~sH MW: 466.41 3.64E-071.86E-0786 2 4
O O'N p KPU-52, tBu-delta-
~N HN ~ ~ ~ PLH-2-N02-5-CI;
HN TFA salt (MW:
~NH ~
~
'
'
2403 O 529.85 9.97E-082.63E-0886 2 5
OI
HsO.O
~'Hs
o H
HN ~N / KPU-53, tBu-delta-
I s
N PLH-o-OH; TFA
~ Salt
2404 o MW: 466.41 6.10E-061.23E-0666 10 3
H'o '~~"
0 off KpU-54, tBu-delta-
HN ~N / NH I ~ PLH-2-OH-5-OMe;
o TFA Saft (MW:
2405 , 496.44 >2E-05 NA 25 12 4
H'o ~3"' "'
0
HN ,N jN N I o KPU-55, tBu-delta-
PLH-3-furanyl;
TFA
2406 H'c '~H' Sait MW: 440.373.59E-081.03E-0886 2 5
-83-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
o OH'
HN ~N ~N H I ~ KPU-56
tBu-delta-
,
~ Br PLH-2-OH-5-Br;
TFA
2407 H'o '~" Salt MW: 54 >2E-05 NA 13 6 3
5.31
- O i w OH _
KPU-57, tBu-delta-
~N HN PLH-3-OH-4-OMe;
HN ~ , NH ~ ~
~ ~H TFA Salt (MW;
2408 "~o 496.44 7.48E-068.62E-0766 4 3
~
'~3 '
0 0" KpU-58, tBu-delta-
HN ;N ~ H I ~ PLH-2-OH-4-OMe;
TFA Salt (MW:
~
2409 Ha0 c~a" 496.44 >2E-05 NA 8 10 4
oH3
O OH
HN ~N jN H I ~ KPU-59, tBu-delta-
PLH-2-OH-5-CI;
~ o~ TFA
2410 H'o '~" Salt MW: 500.86>2E-05 NA 37 17 3
O KPU-60, tBu-delta-
i O PLH-5-Me-2-furanyl;
~N HN
NH I , oH,
HN ,
~ TFA Salt (MW:
o
~
2411 "'o '~a" 454.40 5.72E-082.OOE-OS85 2 5
O KPU-61, tBu-delta-
i S PLH-5-CI-2-thlonyl;
/~N HN
HN ~ ~, N" I /
~
TFA Salt (MW:
~
2412 H'o "~~" 490.88 1.65E-073.32E-0885 3 3
0
i-N HN i
HN ~ / N" I m KPU-62, tBu-delta-
PLH-2-thionyl;
TFA
2413 "'o '~s"' Salt MW: 456.442.74E-086.29E-0988 2 3
o ~Ha KPU-63, tBu-deita-
/-N HN i N
HN i s NH I r PLH-N-Me-2-pyrrole;
o TFA Salt
2414 H'o '~'"' MW:453.42 1.17E-073.14E-0888 3 3
0
HN ;N rN tBu-deita-
" I ~ c~ KPU-64
N ,
PLH-3,5-diCl;
TFA
2415 ~ H'o '~'"' Salt MW 519.30 9.49E-085.05E-0987 3 3
_ F.
FF tBu-delta-
H~N , N I KPU-65
/ ,
o PLH-m-CF3; TFA
~ Salt
2416 H'o '~~" MW 518.41 4.40E-087.40E-0987 2 3
' I KPU-66, tBu-delta-
l-N HN i
HN PLH-1-napthalene;
I ~
~ ~ NH TFA Salt (MW
~ 500.47
2417 "'o ~" 1.49E-082.12E-0988 2 3
O KPU-67, fiBu-delta-
i w w PLH-2-napthalene;
/=N HN
HN , / NH I ~
~
TFA Salt (MW
2418 H'o '~'H' 500.47 1.73E-06NA 83 NA 2
-84-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
o ~
HN ~N / tBu-delta-
~ ~ ~ KPU-68
N ,
~ PLH-2,3-diCl;
TFA
2419 H3 Salt MW 519.302.99E-084.46E-0988 2 3
'~ '
RN HN i ~
HN
I
' ''Iz
~ ~ NH KPU-69, tBu-delta-
i
H' ~~H' PLH-m-Vinyl;
TFA
2420 Salt MW 476.451.70E-081.52E-0988 2 3
0
Q N ~N KpU-70
H t ~ tBu-delta-
N ,
PLH-oxazole;
TFA
2421 H' '~3H' Salt MW 451.402.22E-083.28E-0988 2 3
0
' ~ KPU-71, tBu-delta-
I
I
h", c ~ PLH-2-Hydroxybenzyl;
' N"
'
"3 TFA Salt (MW
2422 495.91 >2E-05 NA 14 NA 2
tBu-delta-
KPU-72
S ,
/-N HN i PLH-4.-Br-2-thionyl;
~ /
HN
~ > NH TFA Salt (MW
.~ e~
~
2423 "' '~aH 484.49 1.71 1.86E-0886 3 3
E-07
tBu-delta-
KPU-73
S ,
HN N "N ' \ / H, PLH-5-Et-2-thiOn
i NH I;
y
TFA Salt (MW
2424 H'c '~H' 535.34 1.32E-072.56E-0887 3 3
0
H iN HN ~ \/ B' KPU-74, tBu-delta-
NH
~ PLH-5-Br-2-furyi;
TFA
2425 H' '~H' Salt MW 519.273.05E-088.71 87 3 3
E-09
HN~N HN ~ \/ CH KPU-75, tBu-delta-
NH
PLH-5-Et-2-furyl;
TFA
2426 "' "~"' Salt MW 468.432.42E-07NA 86 NA 2
H ,N ~N N \/ ~ KPU-76, tBu-delta-
PLH-5-CI-2-furyl;
TFA
2427 H' '~sH' Salt MW 474.823.82E-088.88E-0987 2 3
O F
HN ~N /N N" ~ KPU-77, tBu-delta-
/ PLH-2-F-5-I;
TFA Salt
2428 H' ~a"3 MW 594.30 4.17E-087.78E-0987 2 3
S~"' tBu-delta-
KPU-79
RN HN i ,
"N PLH-2-(Methylthio);
,
~
_, _ TFA Salt (MW
N"
a
2429 H' ~~" 496.50 2.86E-083.59E-0987 2 3
0
/~N HN i w F F
HN ~
NH ~
i
i KPU-80, t8u-delta-
H3 H PLH-m-OCF3;
TFA
2430 Salt MW 1.43E-084.93E-0987 2 3
-85-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
o P
HN ~N jN H I tBu-delta-
KPU-81
a ,
o PLH-2-F5-OMe;
TFA
2431 , Salt MW 2.03E-082.12E-0988 2 3
H, ,~,H' H'
O CHI
/=N HN i
HN tBu-delta-
I KPU-82
~
~ / NH ,
~ PLH-4-F-3-OMe;
F TFA
2432 H' '~,H' Salt MW 4.93E-07NA 85 NA 2
o cH,
HN~N HN ~ I ~ CHHa
H
~i
O KPU-83, tBu-delta-
H~c O~H~ pLH-2-OH-5-tBU;
2433 TFA Salt MW 8.53E-06NA 80 NA 2
O
/-N HN i
HN ~ KPU-84, tBu-delta-
NH
J PLH-cyclohexane;
2434 H' '~,H' TFA Salt MW 7.89E-081.41 87 3 3
E-08
O CHI
/~N HN i ~ F
HN ~ - NH ~ KPU-86, tBu-delta-
~ PLH-2-Me-3-F;
~ TFA
2435 H,c c~H Salt MW 3.34E-086.66E-0986 3 3
O F
HN ~N ,N H I KPU-87, tBu-delta-
, PLH-2-F-5-Me;
~~~~'''' TFA
~ oH
2436 3 Salt MW 2.50E-083.52E-1086 3 3
H3c c~,H
O F
H ;N ~ N~ KPU-88, tBu-delta-
~ ~
~ PLH-2-CI-6-F;
~ TFA
2 Salt MW 2.49E-081.31E-0986 2 3
437 H' ~,H
_ O F
HN ;N ~ NH I KPU-89, tBu-delta-
~ PLH-2,5-di-F;
F TFA
2438 H' '~'H' Salt MW 3.07E-082.14E-0987 3 3
O CHI
HN ~N %N H I ~ KPU-90
cH, tBu-delta-
,
PLH-2,3-di-Me;
TFA
2439 H'o '~,H' Salt MW 2.96E-092.27E-1087 3 3
o cH,
/~N HN i
HN ~ , NH I / KPU-91, tBu-delta-
H, PLH-2,6-di-Me;
o TFA
2440 H' '~,H' Salt MW 4.30E-07NA 85 NA 2
O NOZ
~N HN i ~ O-cH,
HN ~ ~ NH ~ i
KPU-92, tBu-delta-
H' ~" PLH-2-N02-3-OMe;
2441 TFA Salt MW 6.63E-07NA 83 NA 2
O cH,
HN ~N , NH ~ i KPU-93, tBu-delta-
-~ PLH-2,5-diMe;
~ H TFA
2442 3 Salt MW 8.82E-081.40E-0887 3 3
H' '~,H
-86-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
O N"Z
RN HN i ~ 'OH,
HN
I
~ , NH
i KPU-94, tBu-delta-
"' p'~"~ PLH-2-NH2-3-OMe;
2455 TFA Salt 3.49E-07NA 87 NA 2
/~N HN i
HN ~ , N" ~ i tBu-delta-
KPU-95
~ ~" ,
PLH-3-OMe-2-furyl;
2456 "3p TFA Salt 1.25E-073.68E-0888 2 3
~
'~"'
O NHx
l~N HN i
HN ~
NH I
i
i KPU-96, tBu-delta-
2457 "' ~"~ ~ PLH-2-NH2; 4.32E-07NA 86 NA 2
TFA Salt
/~N HN i ~ NH,
HN
(
~ / NH KPU-97, tBu-delta-
~
2458 H~p PLH-m-NH2; 3.10E-081.71 87 3 3
'~' ~ TFA Salt E-09
NH ~S~ KPU-98, tBu-delta-
HN ,N
s PLH-3-B(OH)2-2-
~o Ho'g'OH
2459 "' "~"' thien I; TFA 2.53E-084.31 88 3 3
Salt E-09
NH N-~
WN
I
i HN W
"
2460 '"~ - -tBu-PLH 2.55E-071.24E-0787 2 5
~
EXAMPLE 5
Other Dehydrophenylahistin Analogs
A. Modifications for the s nn~is of dehydrophenylahistin derivatives
[0377] Other derivatives of dehydrophenylahistin are synthesized using the
foregoing techniques alone or in conjunction with other well known organic
synthesis
techniques.
[0378] Modifications to the diacyldiketopiperazine and the first and second
aldehydes involved in the synthesis method vary according to the desired
derivative to
produce. Derivatives are synthesized that:
A) modify the phenyl ring and/or introduce other aromatic ring systems,
B) alter the position of the aromatic ring,
C) alter the imidazole aromatic ring system, and/or
D) modify the 5-position on the imidazole ring.
(0379] The figure below depicts regions of the dehydrophenylahistin compound
modified to produce derivatives of dehydrophenylahistin. Non-limiting examples
of
_87_
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
modifications are disclosed, and based on this disclosure would be understood
by those of
skill in the art.
C
H~ HN i w
i NH
D A
A 1 ) Modification of the phenyl ring besed on the structure of known anti-
tubulin compounds
Alkyl, Halogen, Alkoxy, Acetyl, Sulfonamide, Amino, Hydroxyl, Nitro, etc.
H3C0 / O~--CH3 HaCO ~
....NH ~
H3C0 ~ \ H3C0 / ~ OH HZNOpS' v H
CH30 ~ CH30 ~ w
OCH3 Mol. Wt.: 385.85
'OCHO Combretastatin A-4 P7g~0 CI
Coichicine 0.15 uglmL
P-388 Phase II
2) Introduction of other aromatic ring systems
a ~ a ~ ~ /
etc
B Position of the aromatic ring
a
/ ~/ (/
C Change to the other ring systems
HN~N / NH /~ S/~N
' / ' ~ ' / I / /
etc
D Further modification of the 5-positin on the imidazole ring
HN~N H ~N HN~N
HN~N ' /
/ ~ etC
i
_g8_
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0297] Expanding on the above modifications to the dehydrophenylahistin
compound, derivatives of the compound may include the following substitutions
at the
phenyl ring (A): -CF3, -SOZNHz (-SOaNRIR2), -S03H, -CONH2 (-CONR1R2), -COON,
etc. Other ring systems (C) may also include the following:
~ ,r
~:-
N~. ~~ ~ N~ ~r
B. Examples of synthesized dehydrophenylahistin derivatives
[0380] Additional examples of synthesized dehydxophenylahistin derivatives are
disclosed in the Table 4.
Table 4. Additional synthesized derivatives of dehydrophenylahistin
COMPOUNDS STRUCTURE F ~ M.W.
O
KPU-20 /=N NH ~ ~ TFA 370.83
tBu-~PLH-p-Cl NH i / NH ~ i 484.86
C~ (+TFA)
- ~
O
O O---~
KPU-30 /-N NH -~ ~ O TFA 380.40
tBu-OPLH-2,3-methylendioxyNH ~ '~ NH ~ 494.42
(+TFA)
O
KPU-31 /=N NH ~ ~ 2TFA 337.38
~
NH
tBu-~PLH-3-pyridyl ~ ~ NH 565.42
N
(+2TFA)
O Me
KPU-32 ~'=N NH ~ ~ TFA 350.41
tBu-dPLH-o-Me NH ~ ~ NH I ~ 464.44
(+TFA)
-89-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE Fo~ M.W.
O
KPU-33 /=N NH ~y 2TFA 351.40
tBu-~PLH-3-Me-2-pyridyl~H ~ i NH N i 579.45
O Me (+2TFA)
O
KPU-34 ~=N NH ~~ TFA 354.38
tBu-OPLH-4-F ~ 468.40
NH ~ ~ NH I ~ F
(+TFA)
O
KPU-35 /=N NH -~ ~ F TFA 354.38
tBu-OPLH-rn-F NH ~ ~ NH ~ 468.40
(+TFA)
O Me
KPU-36 /=N NH ~ i 2TFA 356.42
NH ~ ~ NH N~NH . 584
47
tBu-~PLH-5-Me-4-im .
(+2TFA)
O F
KPU-37 ~=N NH ~ ~ TFA 354.38
tBu-~PLH-o-F NH ~ ~ NH I ~ 468.40
(+TFA)
O
KPU-38 /-N NH ~ ~ Me TFA 350.41
tBu-aPLH-m-Me NH ~ ~ NH ~ 464.44
(+TFA)
O
KPU-39 /=N NH ~' ~ TFA 350.41
tBu-dPLH-p-Me NH ~ ~ NH I ~ Me 464.44
(+TFA)
-90-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE FO~ M.W.
O
KPU-40 ~- N N H ~ ~ i~ TFA 415.28
tBu-OPLH-p-Br NH ~ ~ NH I ~ gr 529.31
(+TFA)
O
KPU-41 /'=N NH ~ w Br TFA 415.28
tBu-dPLH-m-Br NH ~ ~ NH ~ 529.31
(+TFA)
O
KPU-42 ~=N NH ~ ~ S TFA 342.42
NH ~.. , NH 456.44
tBu-~PLH-3-thienyl
(+TFA)
O
KPU-43 ~-N NH i ~ TFA 361.40
tBu-OPLH-p-CN NH ~ ~ NH ( ~ CN 475.42
(+TFAj
O
KPU-44 /=N NH ~' ~ ~E TFA 380.44
tBu-~PLH-m-Et0 NH ~ ~ NH ~ 494.46
(+TFA)
O OMe
KPU-4S /-N NH ~~~~ TFA 426.47
tBu-OPLH-2,4,6-TriOMe NH ~ ~ Me0 I ~ OMe 540.49
(+TFA)
~ NO2
KPU-46 /=N NH ~ ~ TFA 381.39
tBu-~PLH-o-NOa NH ~ ~ NH I ~ 495.41
(+TFA)
-91-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F~~ M.W.
O
KPU-47 /-N NH i ~ N02 TFA 381.39
tBu-OPLH-m-N02 NH ~ , NH ~ 495.41
p (+TFA)
O
KPU-48 ~=N NH ~ w TFA 381.39
tBu-~PLH-p-N02 NH ~ ~ NH I ~ N02 495.41
p (+TFA)
O
KPU-49 ~=-N NH ~ w CN TFA 361.40
tBu-~PLH-m-CN NH ~ ~ NH ~ 475.42
(+TFA)
O Br
LPU-50 ~N NH ~~ TFA 415.28
tBu-OPLH-o-Br ~ 529.31
NH ~ ~ NH I ~
p (+TFA)
O
KPU-51 ~=N NH i ~ OH TFA 352.39
tBu-~PLH-m-OH NH ~ ~ NH ~ 466.41
(+TFA)
O NOZ
KPU-52 /=N NH ~ ~ TFA 415.83
tBu-OPLH-2-NOZ-5-Cl NH ~ ~ NH I ~ 529.85
O CI (+TFA)
O OH
KPU-53 /-N NH ~~ TFA 352.39
tBu-OPLH-o-OH ~~ 4GG.41
NH ~ ~ NH I ~
p (+TFA)
-92-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE FO~ M.W.
O OH
KPU-54 /=N NH ~ ~ TFA 382.41
tBu-OPLH-2-OH-5-OMe NH ~ ~ NH ~ 496.44
O OMe (+TFA)
O
KPU-55 /=N NH ~ ~ TFA 326.35
tBu-OPLH-3-furanyl NH ~ ~ NH I O 440.37
(+TFA)
O OH
KPU-56 /-N NH ~ ~ TFA 431.28
tBu-~PLH-2-OH-5-Br NH ~ ~ NH ~ 545.31
O Br (+TFA)
O
KPU-57 /=N NH ~ ~ OH TFA 382.41
tBu-~PLH-2-OH-4-OMe NH ~ ~ NH ~OMe 496.44
(+TFA)
O -OH
KPU-58 /=N NH ~ ~ TFA 382.41
tBu-4PLH-2-OH-4-OMe NH ~ ~ NH I ~ OMe 496.44
(+TFA)
O OH
KPU-59 ~=N NH ~ ~ TFA 386.83
tBu-~PLH-2-OH-5-Cl NH ~ ~ NH ~ (+TFA)
O CI 500.86
O
KPU-60 ~-Nr NH ~ ~ ~ CH2 TFA 340.38
tBu-L1PLH-5-Me-2-furanyl NH ~ ~ NH 454.40
(+TFA)
-93-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F ~ M.W.
O
KPU-61 ~'-N NH ~ ~ ~ CI TFA 376.86
tBu-4PLH-5-Cl-2-thionyl NH ~ ~ NH 490.88
(+TFA)
O
KPU-62 NH N NH ~ ~ ~ TFA 342.42
tBu-~PLH-2-thionyl ~ ~ NH 456.44
0 (+TFA)
O CH3
KPU-63 ~=N NH ~ ~ ~ TFA 339.39
NH ~ ~ NH 453.42
tBu-~PLH-N-Me-2-pyrrole
O (+TFA)
O
KPU-64 ~N NH ~ ~ CI TFA 405.27
tBu-OPLH-3,5-diCl NH i ~ NH
O CI
O
KPU-65 ~N NH ~ ~ CF3 TFA 404.39
tBu-~PLH-m-CF3 NH -~ , NH
O
O
KPU-66 ~ TFA 386.44
/=N NH
tBu-~PLH-1-Naphthalene NH ~ , NH ~ i
O
O
I~PU-67 , ~ ~ TFA 386.44
/=N NH
tBu-~PLH-2-Naphthalene NH i ~ H
O
-94-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F~~ 1VLW,
O CI
KPU 68 CI
/=N NH i ~ TFA 405.27
TBu-OPLH-2,3-diCl NH i i NH
O
O
KPU-69
N NH ~- TFA 362.42
~ ~
TBu-OPLH-m-Vinyl NH
I
~ ~ NH
O
O F
~U ~~ /=N NH ~ ~ TFA 480.28
TBu-OPLH-2-F-5-I NH ~ i NH
O i
O S~
KPU-79
TFA 368
45
/-N NH ~ ~ .
tBu-~PLH-2-(Methylthio)NH ~
NH
,
O
O
KPU-80 ~N NH ~ ~ OCF3 TFA 420.38
TBu-OPLH-m-OCF3 NH ~
O
O F
KPU-81 , w TFA 3 84.3
/=N NH 8
TBu-OPLH-2-F-5-OMe NH i / H ~ ,/
O OMe
O
KPU-82 OMe
~N NH , ~ TFA 384.38
TBu-OPLH-4-F-3-OMe HN ~ / NH I /
F
O
-95-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F~~ M,W,
O
KPU-83
~N NH -~ ~ TFA 408.39
TBu-OPLH-2-OH-5-tBu HN ~ ~ NH
II OH
O
O
KPU-84
/=N NH ~~ TFA 341.39
TBu-OPLH-cyclohexane ~
HN ~' / NH
I I
O
O
KPU-86 F
~N NH i ~ TFA 368.39
TBu-OPLH-2-Me-3-F HN i ~ NH
O
KPU 87
/=N NH ~ ~ TFA 368.39
TBu-OPLH-2-F-5-Me HN i ~ NH '
I I
O
O F
KPU 88
/--N NH ~ ~ TFA 388.83
TBu-OPLH-2-Cl-6-F HN ~ ~ NH
II CI
O
O F
KPU-89
~N NH i ~ TFA 372.38
TBu-OPLH-2,5-di-F HN i ~ NH
O F
O
KPU-90
~N NH ~ ~ TFA 364.38
TBu-OPLH-2,3-di-Me HN i ~ NH
O
O
KPU-91
/=N NH Y~ ~ TFA 364.38
TBu-~PLH-2,6-di-Me HN ~ ~ NH
O
-96-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
COMPOUNDS STRUCTURE F~~ M.W.
O N02
KPU-92 ~N NH ~ ~ OMe TFA 411.39
tBu-~PLH-2-N02-3-OMe HN ~ ~ NH
O
O
KPU-93 ~N NH ~ ~ TFA 364.38
TBu-dPLH-2,5-diMe HN ~ ~ NH ~ i
O
-O. NH2
KPU-94 ~-N NH ~ ~ OMe TFA 381.399
tBu-OPLH-2-NH2-3-OMe HN ~ / NH
O
_ - O NH2
KPU-96 ~N NH ~ ~ TFA 351.41
TBu-~PLH-2-NH2 HN ~ , NH
I I
O
O
KPU-97 ~N NH i ~ NH2 TFA 351.41
TBu-~PLH-m-NH2 HN ~ , NH
O
C. Evaluation of dehydrophen~ahistin derivatives
[0381] Evaluation of derivatives described above is assessed according to the
methods described in Example 3. Additional evaluation of the derivatives are
extended to
specific activities such as determining the inhibiting effect on cell
proliferation, the effects on
a specific cellular mechanism (i.e. microtuble function), effects on cell
cycle progression,
evaluating i~z vitf~o anti-tumor activity against cancer cell lines, etc. Some
evaluation method
protocols are given below.
-97-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
1) Cell Proliferation Inhibiting Effect of Dehydrophenylahistin And Its
Analogs
[0382] Into each well of a 96-well microtiter plate, 100,1 of A-S49 cells
derived
from human lung cancer prepared to 105 cells/ml in a culture medium obtained
by adding
10% bovine fetus serum to EMEM culture medium (Nissui Seiyaku Co., Ltd.)
having
antitumor effect against A-S49 cells derived from human lung cancer are
placed. Methanol
solution of the derivative obtained by the above-listed examples are added to
the wells of the
uppermost row, specimens are diluted by the half log dilution method and
added, and the
plate is incubated in a carbon dioxide gas incubator at 37°C for 48
hours. The result is added
in Iots of IOpl with MTT reagent (3-(4,S-dimethyl-2-thiazole)-2,S-diphenyl-2H-
tetra
bromide)(lmg/ml ~ PBS), followed by incubation in a carbon dioxide gas
incubator at 37 °C
for 6 hours. The culture medium is discarded and the crystal of produced in
the cells are
dissolved in 100p.1/well of dimethylsulfoxide. Absorption of S9S nm light is
then measured
with a microplate reader. By comparing the light absorptions of the untreated
cells to that of
cells treated with a specimen of a known concentration, the specimen
concentration that
inhibited cell proliferation SO% (ICSO) is calculated.
2) CeII Cycle Inhibiting Activity of Dehydrophenylahistin And Its Analogs
[0383] Cell strain A431 is derived from human lung cancer. EMEM culture
medium containing 10% bovine fetal serum and 1% MEM nonessential amino acid
solution
(SIGMA M202S) is used to incubate A431 cells at 37 °C in an incubator
saturated with S%
carbon dioxide gas and water vapor. The refined specimen of
dehydrophenylahistin obtained
by the methods above is added to the cells in the log-growth phase and
progression of the cell
cycle is analyzed by flow cytometer and microscopic observation.
[0384] The effect on cell cycle progression of HeLa cells is depicted in
Figure 42.
EXAMPLE 6
Structure-Activity Relationship Of Synthesized Dehydrophenylahistin
(DehydroPLH)
Deriyatives
1) Overview in derivative syntheses
[0385] Many, but not all, of the derivatives of dehydroPLH disclosed herein
include one, iwo, or three modifications at the phenyl ring (Figure S below).
The derivatives
were synthesized by the methods described above. As shown in Table S, certain
compounds
-98-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
showed more potent cytotoxic activity than dehydroPLH and tBu-dehydroPLH. The
most
potent compound exhibiting an EC50 value of 3 nM was KPU-90. This value was 16-
times
and 4-times higher than that of dehydroPLH and tBu-dehydroPLH, respectively.
These
derivatives have mono-substitution at the o- or era-position of the phenyl
ring with the halogen
atoms such as fluorine and chlorine atoms or the methyl, vinyl or rnethoxy
group.
Derivatives with substitutions to heteroaryl structures such as the
npahthalene, thiophene and
furan rings also elicited a potent activity.. KPU-35, 42, 69, 80 and 81 also
showed higher
activity than tBu-dehydroPLH.
Table 5. Synthetic potent dehydroPLH derivatives
Com ound Structure ECso n
O
KPU-9 ~--N NH ~ ~ OM 31
NH ~ / NH
O
O
KPU-35 ~=N NH ~ ~ F 10
NH ~ , NH
O
O CI
KPU-18 ~-N NH ~~ 42
~
NH ,. / NH
O
O
KPU-19 /-N NH ~ ~ ~i 20
NH ~ / NH
O
O
KPU-38 ~=N NH ~ ~ M~ 45
NH ~ / NH
O
-99-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure ECsa nM
O F
KPU-37 ~=N NH ~~ 21
NH ~ / NH I /
O
O
KPU-41 /=N NH / ~ Br 31
NH ~ / NH
O
O _
KPU-29 ~--N NH / ~ S ~ 44
NH ~ / NH
Me
O
O
I~'U-16 ~=N NH / ~ OM 42
NH i / NH
O OMe
O Me
KPU-32 /=N NH ~~~ 42
NH ~ / NH
O
O
I~PU-42 ~N NH / ~ S 54
NH ~ / NH
O
O N02
I~PU-46 /=N NH ~~~ 44
NH ~ / NH I /
0
0
KPU-44 /=N NH / ~ O~ 43
NH ~ / NH
O
-100-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure ECso n
O
tBu-~PLH /=N NH / ~ 13
(KPU-2) NH i / NH I /
O
O
KPU-69 /=N NH ~ ~~ ~ 16
NH / ~ NH I /
O
O
KPU-80 /=N NH i ~ OcF3 13
NH ~ , NH
O
O F
KPU,81 /=N NH / w 19
NH ~ / NH I /
O OMe
O
KPU-90 /= N N H ~/ ~ 3
HN ~ / NH I /
O
O
DehydroPL /-N NH /~~ 48
H (KPU-1) NH ~ / NH I /
O
2) Introduction of the methoxy groups to the phenyl ring
[0386] Colchicine recognizes the same binding site on [3-tubulin as PLH.
Colchicine has four characteristic methoxy groups on its A and B rings. A
series of
substitutions with the single or multiple methoxy groups was performed and the
results of
cytotoxic activity are shown in Table 6.
-101-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 6. Effect of the methox~~roup substitution on the proliferation of HT-29
cells
Com ound Structure EC50 (nlVn
O
DehydroPLH /=N NH ~ ~ 48
(KPU-1) NH ~ a NH I a
O
O
tBuOPLH /=N NH ~~ 13
(KPU-2) ~
NH ~ a NH
O
O OMe
KPU-8 /-N NH / w 89
NH ~ / NH
O
O
KPU-9 ~-N NH a ~ OMe 31
NH ~ a NH
O
O
KPU-6 ~=N NH ~~~ 6730
~
NH ~ / NH I /
OMe
0
O OMe
KPU-10 /-N NH / ~ 1350
NH ~ / NH
O OMe
O OMe
KPU-12 ~=N NH e~ 4980
~
NH ~ / NH I /
OMe
O
-102-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure EC50 (nlVn
O
KPU-13 /-N NH ~ ~ OMe 2130
NH ~ , NH ~
OMe
O
O OMe
KPU-14 /=.N NH ~ ~ OMe 610
NH ~. / NH
I I
O
O OMe
KPU-15 ~=N NH ~~ 4430
~~
NH ~ / NH
IOI Me0
O
KPU-16 /=N NH / ~ OMe 42
NH ~ / NH
O OMe
O
KPU-24 /=N 'NH ~ ~ 7040
NH ~ / N
O OMe
[0387] The result demonstrated that substitutions at the na- or o-position
increased
cytotoxic activity against HT-29 cells. KPU-9 and 16 showed high activity. The
methoxy-
derivatives with triple substitution (KPU-11, 17 and 45) also showed activity.
The structure
of KPU-24 was assigned by MASS analysis.
3) Modification with the electron-withdrawing groups
[0388] To study more expanded structure-activity relationship on the phenyl
ring,
a series of different functional groups were introduced, which include both
electron-
withdrawing and -donating groups. The result of cytotoxicity against HT-29
cells is shown in
Tables 7 and 8, respectively.
-103-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0389] Substitution at the o- or fn-position effectively increased activity.
These
results were well consistent with the case of the methoxy group.
Table 7. Effect of the electron-withdrawing ~rou~on proliferation of HT-29
cells
Com ound Structure
ECso (n~
O CI
KPU-18 /=N NH ~ ~ 42
~~
NH ~ / NH
O
O
KPU-19 /=N NH / ~ CI 20
NH ~ / NH
O
O
KPU-20 ~N NH Y ~ 545
~~
NH ~ / NH I ~
CI
O
O CI
KPU-21 ~N NH ~ ~ 51
NH ~ / NH
O N02
O N02
KPU-52 ~N NH ~ ~ 110
NH ,. / NH
O CI
O F
KPU-37 ~-N NH ~ ~ 21
~
NH ,. / NH
O
O
KPU-35 /=N NH / w F 10
NH ~ / NH
O
-104-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure
ECSO (nlV1)
O
KPU-34 ~=N NH ~ 466
~
NH ~ , NH
O
O Br
I~PU-50 /=N NH ~~' 38
~
NH ~ / NH
O
O
KPU-41 /=N NH ~ ~ Br 31
NH ~ , NH
O
O
KPU-40 ~=N NH ~ ~ 623
NH ~ ~ NH ~ ~
gr
O
O N02
KPU-46 ~=N NH ~ ~ 44
NH ~ / NH
O
O
KPU-47 ~=N NH ~ ~ N02 40
NH ~ / NH
O
O
KPU-48 /=N NH ~~~ > 20 ~.M
~
NH ~ ~ NH I ~
NO~
O
-105-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure
ECso ~n~
O
KPU-49 /-N NH / ~ CN 28
NH ~ , NH
O
O
KPU-43 '--N NH ~ ~ > 20 ~.M
~~
NH ~ / NH , ~
CN
O
Table 8. Effect of the electron-donatin~-Lgroup on proliferation of HT-29
cells
Com ound Structure
ECso ~n~
_
O OMe
KPU-8 ~=N NH ~~ 89
~
NH ~ / NH
O
O
KPU-9 /=N NH ~ ~ OMe 31
NH ~ / NH
O
O
KPU-6 /-N NH ~~ 6730
~
NH ~ ~ NH I ~
OMe
O
O
KPU-44 ~=N NH ~ ~ O~ 43
NH ~ , NH
O
-106-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
ECso (n~
Com ound Structure
O O--~
KPU-30 ~=N NH / ~ O 477
NH ~ , NH
O
O
KPU-22 /=N NH / w ~ 82
NH ~ / NH
O
O Me
KPU-32 /=N NH / ~ 42
NH ~ / NH I /
O
O
KPU-38 ~ Me 45
H N NH /
N
I
i / NH
O
O
KPU-39 /=-N NH ~~~ 460
~
NH ~ / NH
Me
O
O OH
KPU-53 /=N NH Y ~ > 20 ~M
~
NH ~ / NH I /
O
O
KPU-51 /=N NH / ~ OH 617
NH ~ / NH
O
-107-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure ECso (nlV~
O OH
KPU-23 /=N NH / ~ OMe 5870
NH ~ / NH
O
O OH
KPU-58 ~-N NH Y ~ >20 ~M
~
NH ~ , NH I ~
OMe
O
O OH
KPU-54 /-N NH ~ ~ > a0 I~
NH ~ , NH
O OMe
O
KPU-57 /=N NH ~ ~ OH > 20 ~M
NH ~ / NH ~
OMe
O
[0390) The present disclosure is not bound by or limited to any particular
scientific theory. Nonetheless, it is appreciated that persons of skill in the
art may interpret
the results presented herein to suggest that a relatively smaller functional
group, affecting less
steric hindrance, may be preferred to elicit more potent activity, and
slightly large groups
such as the ethoxy group (when compared to the methoxy group) or the Br atom
(when
compared to the Cl atom) may affect steric hindrance unfavorable to
interaction with, for
example, the tubulin binding site. Moreover, because the electrical property
of these
substituents did not affect the activity, it is suggested that these
relatively small substituents
do not directly interact with the binding site of (3-tubulin, but restrict the
conformation of
dehydroPLH suitable for the binding. Or, as another possible hypothesis, the
hydrophobic
property may be a more important factor at the binding site for o- or ~n-
position on (3-tubulin,
since introduction of the hydrophilic hydroxyl group, which can form the
hydrogen bonding
as a hydrogen-donor, drastically decreased the activity.
-108-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0391] As shown in Table 9, the effect of the substituents in the cytotoxic
activity
at the o-position may be ordered as in the case of m-position, as shown in
Table I0. The
compounds having effective functional groups, which showed higher activity
than tBu-
dehydroPLH, may also be further modified. And since the migration of the
stereochemistry
from Z to E under the visible light irradiation was observed, substituents
that decrease the
electron density in the conjugated double bonds may contribute to the
reduction of Z to E
migration by the light, results in more physicochemically stable structures.
Temperature can
also effect this migration.
[0392] Modification at two parts of the ring can be preferred for the
development
of potent but also biologically stable compounds. The phenyl ring of
phenylahistin is
oxidized by cytochrome P-450. Double modification that reduces the electron
density of the
phenyl ring may therefore be effective to avoid P-450 oxidation. Thus, the
combination of
the small electron withdrawing group such as the fluorine atom to the element
that can
increase the activity such as -OMe, -Me, -Cl, -F and Br, may result in more
potent and
biologically stable drug compounds.
Table 9. Summaryof modification at the o-position
ECso (n~
Com ound Structure
O
KPU-2 ~=N NH / w 4~
NH ~ / NH I /
O
O OMe
KPU-~ /=N NH / ~ 89
NH ~ / NH I /
O
O F
KPU-37 ~-N NH / ~ 21
NH ,. / NH i /
O
-109-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Com ound Structure
EC5o (nlVn
O CI
KPU-18 /-N NH ~~ 42
~
NH ~ / NH I /
O
O Br
KPU-50 ~=N NH ~~ 38
~~
NH ~ / NH
O
O NOa
KPU-46 ~=N NH ~~ 44
~~
NH ~ / NH I /
O
O Me
KPU-32 /=N NH Y ~ 42
~
NH ~ / NH I /
O
O OH
I~PU-53 ~-N NH / w > 20 ~.M
NH ~ / NH I /
O
Table 10. Summary of modification at the raa-position
Compound Structure
ECso (nlVn
O
KPU-2 ~--N NH ~ ~ 48
~
NH ~ / NH
O
O
KPU-9 /=N NH ~ ~ OM 31
NH ~ / NH
O
-110-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
ECso (nM)
Compound Structure
O
KPU-35 ~ F 10
H N NH /
N
(
i / NH
O
O
KPU-19 /=N NH / ~ O~ 20
NH ~ / NH
O
O
KPU-41 /=N NH / ~ Br 31
NH ~ / NH
O
O
KPU-47 /=N NH / ~ N02 40
NH ~ / NH
O
O
KPU-3~ /=N NH / ~ Me 45
NH ~ / NH
O
O
KPU-51 /=N NH / ~ OH 617
NH ~ / NH
O
O
KPU-49 /-N NH / ~ CN 28
NH ~ / NH
O
-111-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Compound Structure ECso (n~
O ,
KPU-44 ~=N NH ~ ~ O~ 43
NH ~ / NH
O
4) Substitution of the phenyl ring to aryl-heterocycles
[0393] The phenyl ring may also be replaced by heteroaryl groups. The result
of
such replacements in terms of the cytotoxic activity is shown in Table 11.
Since the arylic
nitrogen atoms can form a hydrogen bonding with a NH group of the
diketopiperazine ring
and restrict the conformation of the molecule between pyridine and
diketopiperazine rings to
an uniplanar structure, the active conformation of dehydroPLH would be
required a certain
level of dihedral angle formed by the steric repulsion between an amide
hydrogen atom of the
diketopiperazine ring and an o-hydrogen atom of the phenyl ring (Figure 6).
Table 11. Effect of the replacement with the heteroaryl ring on proliferation
of HT-29 cells
Compound Structure ~ ECso (nlV~
O
KPU-28 ~=N NH ~ >20 ~.M
~
I
NH ~ / NH N
I I
O
O
KPU-31 ~=N NH ~ ~ 96
NH ,- / NH I N
O
O
KPU-25 ~--N NH ~ ~ 544
NH ~. , NH I ~ N
O
-112-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
ECSO (nlVn
Compound Structure
O
KPU-33 /=N NH / ~ ~ >20 ~.M
NH ~ , NH N /
O Me
O
I~PU-26 ~=N NH / ~ O ~ 600
NH ~ / NH
O
O
KPU-60 /=N NH / ~ % CH2 71
NH ~ / NH
O
O
KPU-42 ~=N NH / ~ S 54
NH ~ / NH
O
O
KPU-27 /=N NH / ~ S ~ Me 80
NH ~ / NH
O
O
I~PU-29 /- N N H / ~ S ~ 44
NH ~ / NH >--J
~( Me
O
O
KPU-61 /-N NH ~ ~ ~ CI 184
NH ~ / NH
O
-113-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
ECso (nlVn
Compound Structure
O Me
KPU-36 ~-N NH ~ ~ 2790
NH ~ / NH N~/NH
O
O CH3
I~.PU-63 /=N NH ~ N 105
NH ~ / NH
O
[0394] Replacing the phenyl ring with a smaller furan or thiophene ring, for
example, KPU-29 or -42, exhibited activity. The phenyl ring can be changed to
other
aromatic structure while maintaining the potent activity.
5) Metabolism of phenylahistin
[0395] In the recent his study, (~)-phenylahistin was treated with rat hepatic
microsome or human hepatic P450s. In human case at least seven metabolites
were detected,
and two of them, i.e., P1 and P3, were major metabolites, represented more
than 60% of the
recovered metabolites.
[0396] Since there is no exo-olefin structure in tBu-dehydroPLH, present
synthesized derivatives have no oxidization like P 1 and P4. However,
oxidizations such as
P3 and PS are formed during the hepatic metabolism. Various derivatives, which
prevent
such metabolism, are effective to avoid P450 oxidization at the phenyl ring.
The imidazole
ring can also be modified to avoid the unfavorable oxidation.
6) Physicochemical stability of dehydroPLH
[0397] The physicochemical stability is one of the unfavorable problems of
dehydroPLH. In phenylahistin, since there is no additional olefin structure at
the benzyl part,
there is no such problem. However, in dehydroPLH, the benzylidene moiety can
be easily
activated, probably with the visible light, and the Z to E migration
frequently occurs due to
the existence of longer conjugation of the double bond. This migration
occurred even under
normal room light. In the cytotoxic assay, some of the compounds migrate to E-
form during
the incubation, although this migration probably equilibrates at the 1 : 1
ratio in the case of
-114-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
dehydroPLH, This migration can be controlled. The Z to E migration is also
known in
cornbretastatin A4, a same type of tubulin inhibitor, and a few studies for
improving this
problem were reported.
7) Prodrug Synthesis
(0398] The E-form may also be used as a prodrug of dehydroPLH or of one or
more of its analogs, including those analogs described herein. One of the
undesired
properties of anti-tubulin drugs involves its low selectivity between tumor
and intact tissues,
although these drugs belong to one of the molecular target therapies. This
causes undesired
side effects. However, if the compounds functions selectively only in tumor
tissues, negative
side effects of anti-microtubule drugs can be reduced. Since the dehydroPLH (Z
form) can
be produced from its E-isomer by visible light irradiation, the E-form is
administered and
photo irradiation is performed only at the tumor site, then only, the tumor is
damaged by
photo-produced Z-form and the adverse effect to the intact tissues is reduced.
[0399] The E-form can be protected chemically by the addition of a bulky but
biodegradable acyl group, which is introduced into the diketopiperazine ring
as a prodrug.
This acyl group can be cleaved by the protease in the body. Therefore, the
acylated-E-
compound is maintained before administration, then after administration it is
changed to the
real E-form, which can migrate to the bioactive Z-form by the local photo
irradiation.
[0400] The synthetic scheme of this acyl-E-form of tBu-dehydroPLH is
summarized in Figure 9.
EXAMPLE 7
Pharmaceutical Formulations of the Synthesized Dehydrophenylahistins
1) Formulations Administered Intravenous) ,~bx Drip, Injection, Infusion or
The Like
[0401] Vials containing 5 g of powdered glucose are each added aseptically
with
mg of a compound synthesized by the method and sealed. After being charged
with
nitrogen, helium or other inert gas, the vials are stored in a cool, dark
place. Before use, the
contents are dissolved in ethanol and added to 100 ml of a 0.85% physiological
salt water
solution. The resultant solution is administered as a method of inhibiting the
growth of a
cancerous tumor in a human diagnosed as having such a tumor at between
approximately 10
ml/day to approximately 1000 ml/day, intravenously, by drip, or via a
subcutaneous or
-115-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
intraperitoneal injection, as deemed appropriate by those of ordinary skill in
the art.
2) Formulation to be Administered Orally Or The Like
[0402] A mixture obtained by thoroughly blending 1 g of a compound synthesized
by the method, 98 g of lactose and 1 g of hydroxypropyl cellulose is formed
into granules by
any conventional method. The granules are thoroughly dried and sifted to
obtain a granule
preparation suitable for packaging in bottles or by heat sealing. The
resultant granule
preparations are orally administered at between approximately 100m1/day to
approximately
1000 ml/day, depending on the symptoms, as deemed appropriate by those of
ordinary skill in
the art of treating cancerous tumors in humans.
3) Formulation to be Administered TopicallX
[0403] Administration to an individual of an effective amount of the compound
can also be accomplished topically by administering the compounds) directly to
the affected
area of the skin of the individual. For this purpose, the compound
administered or applied is
in the form of a composition including a pharmacologically acceptable topical
carrier, such as
a gel, an ointment, a lotion, or a cream, which includes, without limitation,
such carriers as
water, glycerol, alcohol, propylene glycol, fatty alcohols, triglycerides,
fatty acid esters, or
mineral oils. Other topical carriers include liquid petroleum, isopropyl
palmitate,
polyethylene glycol, ethanol (95%), polyoxyethylene monolaurate (5%) in water,
or sodium
lauryl sulfate (5%) in water. Other materials such as anti-oxidants,
hurnectants, viscosity
stabilizers, and similar agents may be added as necessary. Percutaneous
penetration
enhancers such as Azone may also be included. In addition, in certain
instances, it is
expected that the compound may be disposed within devices placed' upon, in, or
under the
skin. Such devices include patches, implants, and injections which release the
compound
into the skin, by either passive or active release mechanisms.
EXAMPLE 8
In vitro Phannacoloay of KPU-2, KPU-35 and t-butyl phenylahistin
[0404] The irz vitro efficacy studies performed with I~PU-2, KPU-35 and t-
butyl
phenylahistin included: A) a panel of six tumor cell lines, B) studies in
multidrug-resistant
tumor cells, and C) studies to determine the mechanism of action.
-116-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
A). Study of KPU-2, KPU-35 and t-butyl phenylahistin in a panel of six tumor
cell lines
[0405] The following cell lines (source in parentheses) were used: HT29 (human
colon tumor; ATCC; HTB-38), PC3 (human prostate tumor; ATCC; CRL-1435), MDA-MB-
231 (human breast tumor; ATCC; HTB-26), NCI-H292 (human non-small cell lung
tumor;
ATCC; CRL-1848), OVCAR-3 (human ovarian tumor; ATCC; HTB-161), B16-F10 (murine
melanoma; ATCC; CRL-6475) and CCD-27sk (normal human fibroblast; ATCC; CRL-
1475). Cells were maintained at subconfluent densities in their respective
culture media.
[0406] Cytotoxicity assays were performed as described above in Example 4,
using Resazurin fluorescence as an indicator of cell viability.
[0407] The disclosed compounds are effective agents against a variety of
different
and distinct tumor cell lines. Specifically, for example, KPU-2 and I~PU-35
were most
effective on the HT-29 tumor cell line, both in terms of potency (active in
the low nanomolar
range) and efficacy (most responsive in terms of the maximum cytotoxic
effect); t-butyl-
phenylahistin exhibited its greatest potency against the PC-3 tumor cell line,
although the
greatest efficacy was displayed against the HT-29 cell line; KPU-2 and KPU-35
were
generally 10-40 fold more potent than t-butyl-phenylahistin whereas the
efficacy was similar
for all three compounds in the different tumor cell lines; the HT-29, PC-3,
MDA-MB-231
and NCI-H292 tumor cell lines all responded similarly to the NPI compounds,
whereas the
B16-F10 appeared to be somewhat. less sensitive. t-butyl-phenylahistin
displayed a marled
differential between normal fibroblasts and the tumor cell lines, with a ratio
ranging from
>20 - >100, except for the OVCAR-3 cell line.
-117-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 12 Activity of KPU-2, KPU-35 and t-but~phenXlahistin in the Tumor Panel
Screen
Cell Line KPU-2 KP U-35 t-bu hen
1- lahistin
Mean SD n Mean SD n Mean SD n
H'T-29 Colon
IC50 nM 9.8 2.4 4 8.2 2.0 4 420 473 3
Cytotoxicity 82.5 5.3 4 81.3 4.0 4 88 0.2 3
PC-3 Prostate
ICSO.nM 13.4 0.7 4 13.2 2.5 4 174 - 2
Cytotoxicity 60.3 2.1 4 56.8 1.0 4 59.5 - 2
MDA=MB-231
Breast 13.8 1.9 3 9.7 4.2 4 387 - 2
IC50 nM 56.7 7.2 3 59.3 5.6 4 65.5 - 2
! Cytotoxici
NCI-H292 Lung
IC50 nM 17.5 1.1 4 15.9 1.1 4 384 194 3
Cytotoxicity 70.5 2.9 4 68.5 2.9 4 65 5 3
OVCAR-3 Ovary
IC50 nM >20,00- 4 >20,000 4 >20,000- 2
Cytotoxicity 0 3.0 4 39 2.2 4 37 - 2
45.8
B 16-F 10 Melanoma
IC50 nM 37.1 26.34 32.3 19.94 736 "650 3
~
Cytotoxicity 71.8 2.5 4 72.0 2.2 4 74 2 3
CCD-27sk
Fibroblast 9.2 2.9 4 7.4 2.6 4 >20,000- 2
IC50 nM 64.3 2.4 4 60.8 1.9 4 45 - 2
% CytOtOXIClty
B). Studies in Dru~Resistant Cell Lines
[0408] One of the major challenges in the use of chemotherapeutic agents in
clinical oncology is the development of resistance to the drug effect by the
tumor cells. There
are several mechanisms for the development of resistance, each of which will
have
differential effects on chemotherapeutic drugs. These mechanisms include
increased
expression of ATP-dependent efflux pumps such as the P-glycoprotein encoded by
M.DRI or
the multidrug-resistance associated protein 1 encoded by MRPl. Reduced dmg
uptalce,
alteration of the drug's target, increasing repair of drug-induced DNA damage,
alteration of
the apoptotic pathway and the activation of cytochrome P450 enzymes are other
examples of
mechanisms by which cancer cells become resistant to anticancer drugs. The
selected
-118-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
compounds were studied in three different cell lines that exhibit two
different mechanisms of
resistance; the overexpression of the P-glycoprotein and altered topoisomerase
II activity.
1,) Human Uterine Sarcoma Tumor Cell Line Pair: MES-SA (Taxol Sensitive) and
MES-SA/Dx5 (Taxol Resistantl.
[0409] This cell line expresses elevated mdr-1 mRNA and P-glycoprotein (an
extrusion pump mechanism). Pretreatment with cyclosporin-A (CsA) blocks P-
glycoprotein
and reinstates activity in the resistant cell line for those compounds for
which the resistance is
due to elevated P-glycoprotein.
[0410] As can be seen from Table 13, KPU-2, and KPU-35 have the same
potency in the resistant cell line as in the sensitive line and the potency of
t-butyl-
phenylahistin was only slightly reduced. Cyclosporin A (CsA) pretreatment did
not alter the
potency of the selected compounds. In contrast, taxol was virtually inactive
in the MES-
SA/DXS resistant cell line, whereas this compound was very potent in the
sensitive cell line.
CsA treatment restored the sensitivity to taxol of the MES-SA/DXS cell Iine.
The MES-
SA/DXS cell line also showed reduced susceptibility to etoposide (60 fold),
doxorubicin (34
fold) and mitoxantrone (20 fold).
[0411] These data indicate that the effects of KPU-2, KPU-35 and t-butyl-
phenylahistin are not susceptible to the taxol-related resistance mechanism (p-
glycoprotein)
in this cell line, and that cross-resistance from taxol does not occur to
these selected
compounds in this model.
-119-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 13 Activity of KPU-2, KPU-35 t-butyl-phenylahistin and Taxol in MES-SA
Taxol
Sensitive and MES-SA/DX5 Taxol Resistant Human Uterine Sarcoma Tumor Cell
Lines
Compound MES-SA MES-SA/DX5
Sensitive Resistant
Study No CsA No CsA CsA
Pretreat Pretreat
CsA
IC50 ICSO Ratio IC50 Ratio ICSO Ratio
nM nM No CsA nM MES-SA nM No CsA
KPU-2
Study I 8.5 - - 10.5 1.2 - -
Study II 19.4 27.4 1.4 21.7 1.1 37.8 1.74
KPU-35
Study I 6.6 - 5.2 0.8 - -
Study III 3.9 2.0 0.5 2.5 0.6 6.7 2.7
t-butyl-
phenylahistin144 - - 825 5.7 - -
Study I 122 162 1.3 694 4.3 622 0.9
Study III
Taxol
Study I 4.4 - - >20,000>45S - -
Study II 13.3 7.6 0.6 100 8 40 0.25
Study III 7.3 2.8 0.4 > >24,000>3000 2.0 0.001
~
[0412] See also the additional data presented in Figure 43.
2) Human Acute Prom ey locytic Leukemia Cell Line Pair: HL-60 fMitoxantrone-
Sensitive)
and HL-601MX-2 (Mitoxantrone-Resistant)
[0413] This cell line is considered to have atypical drug resistance
properties with
altered topoisomerase II catalytic activity without overexpression of P-
glycoprotein.
[0414] As can be seen in Table 14, these results indicate that the potencies
of the
selected novel compounds are very similar in the sensitive and resistant HL-60
cell lines. In
contrast, Mitoxantrone loses efficacy by a factor of 24-fold in the resistant
HL-60/MX-2 cell
line.
[0415] Thus, KPU-2, KPU-35 and t-butyl-phenylahistin are not susceptible to
the
same resistance mechanisms as Mitoxantrone in this cell line, and there is no
cross-resistance
from Mitoxantrone to these selected novel compounds in this model.
-120-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 14. Activity of KPU-2, KPU-35, t-butyl~henylahistin and Mitoxantrone in
the HL-60
Human Acute Prom~elocytic Leukemia Tumor Sensitive and Resistant Cell Line
Pair
HL-60 SensitiveHL- 60 Resistant
Compound IC50 nM IC50 Ratio to Sensitive
nM
KPU-2 G.4 8.17 1.28
KPU-35 9.2 7.3 0.79
t-butyl- henylahistin255 175 0.69
Mitoxantrone 202 4870 24.1
3). Human Breast Carcinoma Cell Line Pair: MCF-7 (Taxol Sensitive) and MCF-
7/ADR
(Taxol Resistant)
[0416] This study involved KPU-2 in comparison to taxol. KPU-2 demonstrated
similar potencies in both the sensitive and resistant members of this cell
line pair. In contrast,
taxol was virtually inactive in the resistant cell line whereas there was low
nanomolar
potency in the sensitive cell line (Table 15).
[0417] These studies confirm in a different human tumor cell line that taxol
resistance does not transfer to KPU-2.
Table 15. Activity of KPU-2 and Taxol in the MCF-7 Human Breast Carcinoma
Sensitive
and Resistant Cell Line Pair
MCF-7 Sensitive CF-7/ADR
Resistant
M
Compound ___ Ratio
IC50 nM IC 50 nM
to Sensitive
KPU-2 39.6 27.4 0.69
Taxol 2.6 100 3 8
C) Studies of the Mechanism of Action
~. Action on Microtubule Function
[0418] Human umbilical vein endothelial cells (HuVEC from Cambrex) were
used in this study, for evaluating the effects of KPU-2 and t-butyl-
phenylahistin in
comparison to colchicine and taxol on tubulin by staining for a-tubulin.
[0419] Thirty minutes exposure to KPU-2, t-butyl-phenylahistin or colchicine
(all
at 2 ~.M) induced microtubule depolymerization as was indicated by the lack of
intact
microtubule structure in contrast to that observed in the DMSO Control and
cell membrane
blebbing (a clear indication of apoptosis) in the HuVEC cells, whereas taxol
did not induce
-121-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
microtubule depolymerization under these conditions. Colchicine is a known
microtubule
depolymerizing agent whereas taxol is a tubulin stabilizing agent. Similar
results were
obtained when CCD-27sk cells were exposed to KPU-2 or colchicine.
21. Induction of Apoptosis
[0420] Apoptosis and its dysregulation play an important role in oncology; the
selective induction of the programmed cell death cycle in tumor cells is the
goal of many
chemotherapeutic drug discovery programs. This induction of apoptosis can be
demonstrated
by different methods including the characteristic cell membrane blebbing, DNA
fragmentation, hyperphosphorylation of the antiapoptotic factor Bcl-2,
activation of the
caspase cascade and cleavage of poly (ADP ribose) polymerase (PARP).
[0421] The characteristic signs of apoptotic cell death include cell membrane
blebbing, disruption of nuclei, cell shrinkage and condensation and finally
cell death, very
distinctive from necrotic cell death. KPU-2 induced the typical morphological
changes
associated with early stages of apoptosis in human prostate tumor cells. A
similar finding
was also clear in the treatment of HuVEC cells with KPU-2.
3). DNA Fra~znentation
[0422] A late stage characteristic of apoptosis is internucleosomal DNA
cleavage
that results in a distinctive ladder pattern that can be visualized by gel
electrophoresis. This
approach was used to study the effect of KPU-2 on DNA laddering in Jurkat
cells (human T
cell leukemia line) in comparison to halimide and dehydrophenylahistin (KPU-
1). KPU-2
induced DNA laddering at the 1 nM concentration whereas halimide and KPU-1
were much
less potent.
4). Activation of the Cas~ase Cascade
[0423] Several enzymes in the caspase cascade are activated during apoptosis,
including Caspase-3, -8 and-9. The activity of Caspase-3 was monitored in
Jurkat cells
following treatment with KPU-2, KPU-35 and t-butyl-phenylahistin.
[0424] The results indicate that caspase-3 was activated in a dose-dependent
manner by treatment with all three compounds in a manner similar to halimide.
The caspase-
-122-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
3 activation occurred over a similar concentration range as for the ICSOs for
cytotoxicity in
the Jurkat cell line (Table 16).
Table 16 Cytotoxicity of KPU-2.~KPU-35 and t-butyl-phenylahistin in Jurkat
Cells
C totoxicit~.,
_
NPI Compound Potency Efficacy
IC50 nM % Cell Death
KPU-2 11 94
KPU-35 5 93
t-butyl-phenylahistin165 93
Mitoxantrone 41 99
5). Cleavage of Poly(ADP-ribose~Polymerase (PARP) in Jurkat Cells
[0425] In order to assess the ability of these compounds to induce apoptosis
in
Jurkat cells, cleavage of poly(ADP-ribose) polymerase (PARP) was monitored.
PARP is a
116 kDa nuclear protein that is one of the main intracellular targets of
Caspase-3. The
cleavage of PARP generates a stable 89 kDa product, and this process can be
easily
monitored by western blotting. Cleavage of PARP by caspases is one of the
hallmarks of
apoptosis, and as such serves as an excellent marker for this process. KPU-2
at 100 nM
induced cleavage of PARP in Jurkat cells 10 hours after exposure of the cells
to the
compound. KPU-2 appeared to be more active than either halimide or KPU-1.
6). Enhanced Vascular Permeability in HuVEC Cells
[0426] Compounds that depolymerize microtubules (e.g. combretastatin A-4-
phosphate, ZD6126) have been shown to induce vascular collapse in tumors i~.
vivo. This
vascular collapse is preceded by a rapid induction of vascular cell
permeability initially to
electrolytes arid soon after to large molecules. The enhanced permeability of
HuVEC cells to
a fluorescent-labeled dextran is used as a proxy assay for vascular collapse.
[0427] KPU-2, KPU-35 and t-butyl-phenylahistin all rapidly (within 1 hour)
induced significant HuVEC monolayer permeability, to an extent similar to
colchicine. The
microtubule stabilizing agent taxol was inactive in this assay (Figure 12).
7). Profile in A Broad Kinase Screen
[0428] KPU-2 was initially screened at a concentration of 10 pM in a panel of
60
different kinases; the ATP concentration was 10 ~,M. Four kinases were
inhibited by greater
-123-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
than SO% in the primary screen and the IC50's determined in secondary
screening are
presented in Table 17. All of the IC50 values are in the low micromolar range,
which
indicates that inhibition of these kinases is not related to the low nanomolar
activities
observed for tumor cell cytotoxicity.
Table 17. Activity of KPU-2 against Selected Kinases
Kinase IC50 (wM~
CDKIICyclin B (human -
10.1
c-R.AF (human) 8.9
JNK3 (rat) 6.8
Lyn (mouse) 11.1
EXAMPLE 9
In vivo Pharmacolo~y
[0429] Preliminary studies with KPU-2 were performed using the MX-1 (breast)
and HT-29 (colon) xenograft models and the P-388 murine leukemia tumor model,
in the
mouse. Other tumor models selected on the basis of activity in the i~ vitro
tumor panel were
the DU-145 (prostate), MCF-7 (breast), and the A549 (lung) cell lines. The
human
pancreatic tumor (MiaPaCa-2) was also included. The novel compounds were
studied as
monotherapy and in combination with a clinically used chemotherapeutic agent.
The doses
of the selected novel compounds were determined from the acute tolerability
testing
(Maximally Tolerated Dose, MTD) and were adjusted if necessary during each
study. The
doses of the clinically-used chemotherapeutic agents were selected on the
basis of historical
studies.
[0430] KPU-2 was the first compound to be studied in these five tumor models.
Following the initial results from this study, all three compounds were
compared in the HT-
29 human colon tumor, the DU-145 human prostate and the MCF-7 human breast
tumor
xenograft models.
[0431] The above models all use the subcutaneous xenograft implantation
technique and are potentially subject to selective effects of a compound on
the subcutaneous
vasculature producing a magnified (or apparent) antitumor activity. In order
to circumvent
this possibility, two other tumor models have been incorporated in the
research. One of these
is the observation of lung metastases following the intravenous injection of
B16-F10 mouse
-124-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
melanoma tumor cells. The other model is the implantation of MDA-231 human
breast
tumor cells in the mouse mammary fat pad. While this latter model is a
xenograft model, the
subcutaneous vasculature does not play a role.
Methods
1). Xenograft Models
[0432) Animals used were (exceptions are indicated for individual studies):
female nude mice (~ulrcu) between S and 6 weeks of age (~24g, Harlan); group
size was 9-10
mice per group unless otherwise indicated.
[0433] Cell lines used for tumor implantation were: HT-29 human colon tumor;
MCF-7 human breast tumor; A549 human non small cell lung tumor; MiaPaCa-2
human
pancreas tumor; DU-145 human prostate tumor.
[0434) Selected novel compounds were administered as monotherapy via the
intraperitoneal (i.p.) route at the doses indicated for the individual study;
for the combination
studies the selected reference chemotherapy agents were injected 15-30 min
prior to the
compound. .
[0435] Vehicles used in these studies were: 12.5% DMSO, 5% Cremaphor and
82.5% peanut oil for the selected novel compounds; (1:3) Polysorbate 80:13%
ethanol for
taxotere; (1:1) Cremaphor:ethanol for paclitaxel; for CPT-11 each mL of
solution contained
20 mg of irinotecan hydrochloride, 45 mg of sorbitol NF powder, and 0.9 mg of
lactic acid,
the pH being adjusted to 7.4 with NaOH or HCI. Saline dilutions are used to
achieve the
injection concentrations used for the reference compounds.
HT-29 Human Colon Tumor Model
[0436] Animals were implanted subcutaneously (s.c.) by trocar with fragments
of
HT-29 tumors harvested from s.c. growing tumors in nude mice hosts. When the
tumor size
reached 5 mm x 5 mm (about 10-17 days) the animals were matched into treatment
and
control groups. Mice were weighed twice weelcly and tumor measurements were
obtained
using calipers twice weekly, starting on Day 1. The tumor measurements were
converted to
estimated mg tumor weight using the formula (W2xL)/2. When the estimated tumor
weight
of the control group reached an average of 1000 mg the mice were weighed,
sacrificed and
the tumor removed. The tumors were weighed and the mean tumor weight per group
was
-125-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
calculated and the tumor growth inhibition (TGI) was determined for each group
(100%
minus the change in the mean treated tumor weight/the change in the mean
control tumor
weight x 100.
[0437] In this model unless otherwise noted for the individual study, the
selected
novel compounds were inj ected intraperitoneally every third day for 15 days [
1, 4, 8, 11 and
15 (q3dx5)]; CPT-11 was administered intraperitoneally on days 1, 8 and 15
(qwx3).
MCF-7 Human Breast Tumor Model
(0438] Female nude mice (~20 g) were implanted s.c. with 21-day release
estrogen (0.25 mg) pellets 24 hours prior to the s.c. implantation with MCF-7
tumor
fragments (harvested from s.c. tumors in nude mice hosts). The study then
proceeded as
described for the HT-29 model, using taxotere as the standard chemotherapy
agent.
(0439] In this model unless otherwise noted for the individual study, the
novel
compounds were injected via the intraperitoneal route daily on Days 1-5,
inclusive (qdx5);
taxotere was administered intravenously on Days l, 3 and 5 (qodx3).
A549 Human Luna Tumor Model
(0440] Animals were implanted s.c. by trocar with fragments of A549 tumors
harvested from s.c. growing tumors in nude mice hosts. When the tumor size
reached 5 mm
x 5 mm (about 10-17 days) the animals were matched into treatment and control
groups. The
rest of the study proceeded as described for the HT-29 model, using taxotere
and CPT-11 as
the standard chemotherapy agents.
[0441] In this model unless otherwise noted for the individual study, the
tested
compounds were administered via the intraperitoneal route on a q3dx5 dose
schedule for the
CPT-l I combination or on a qdx5 dose regimen for the combination with
taxotere; CPT-11
was administered via the intraperitoneal route on a qwx3 schedule; taxotere
was administered
intravenously on a qodx3 dose regimen.
MiaPaCa-2 Human Pancreas Tumor Model
[0442] Animals were implanted s.c. by trocar with fragments of MiaPaCa-2
tumors harvested from s.c. growing tumors in nude mice hosts. When the tumor
size reached
mrn x 5 mrn (about 10 -17 days) the animals were matched into treatment and
control
-126-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
groups. The rest of the study proceeded as described for the HT-29 model,
using gemcitabine
as the standard chemotherapy agent.
[0443] In this model unless otherwise noted for the individual study, test
compounds were administered every third day via the intraperitoneal route on
Days l, 4, 7,
and 15 (q3dx5); gemcitabine was administered via the intraperitoneal route on
Days 1, 4,
7 and 10 (q3dx4).
DU-145 Human Prostate Tumor Model
[0444] Male mice were implanted s.c. by trocar with fragments of DU-145 tumors
harvested from s.c. growing tumors in nude male mice hosts. When the tumors
reached ~ S
mm x S mm ( at about 13 -17 days) the animals were matched into treatment and
control
groups. The remainder of the study proceeded as for the HT-29 model, using
taxotere as the
standard chemotherapy agent.
[0445] In this model unless otherwise noted for the individual study, test
compounds were administered via the intraperitoneal route on Days 1, 3, 5, 8
and 1 I (q3dx5);
taxotere was administered intravenously on Days l, 3 and 5 (q2dx3).
2). Non Subcutaneous Implantation Tumor Models
[0446] The animals used were: female nude mice (nulhu) (MDA-231 study) or
B6D2F1 (B16-FIO studies) mice between 5 and 6 weeks of age (~20g, Harlan);
group size
was 10 mice per group unless otherwise indicated.
(0447] The cell lines used were: MDA-MB-231 human breast tumor and B16-F10
murine melanoma cells.
[0448] NPI compounds were administered as monotherapy via the intraperitoneal
route at the doses indicated for the individual study; for the combination
studies the selected
reference chemotherapy agents were injected 15-30 min prior to the NPI
compound.
MDA-23I Human Breast Tumor
[0449] Female nude mice were injected in the mammary fat pad with 2x106
MDA-231 cells harvested from i~c vitro cell culture. When the tumor size
reached 5 mm x 5
mm (about 14-28 days) the animals were matched into treatment and control
groups. The
study then proceeded as described for the HT-29 model, using paclitaxel as the
standard
chemotherapy agent.
-127-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
(0450] In this model unless otherwise noted for the individual study, the test
compounds were administered via the intraperitoneal route on Days 1, 4, 8, 11
and 15
(q3dx5); paclitaxel was administered via the intraperitoneal route on Days 1-5
(qdx5).
B16-F10 Metastatic Murine Melanoma Model
[0451] Mice received B16-F10 cells (prepared from an ih vitro cell culture of
B 16-F10 cells) by the iv route on Day 0. On Day 1 mice were randomized into
treatment and
control groups and treatment commenced. Mice were weighed twice weekly,
starting on Day
1. All mice are sacrificed on Day 16, the lungs removed, weighed and the
surface colonies
counted. Results are expressed as mean colonies of treated mice/mean colonies
of control
mice (T/C) x 100%). The metastasis growth inhibition (MGI) is this number
subtracted from
100%. Paclitaxel was the standard chemotherapy agent used in this study.
[0452] In this model unless otherwise noted for the individual study, the test
compounds were administered via the intraperitoneal route on Days 1-5 (qdx5);
paclitaxel
was administered intravenously on Days 1-5(qdx5).
[0453] When appropriate (n >_ 3), results are presented as means ~ SEM.
Statistical analysis of studies with several groups was performed using ANOVA
with
Neuman-Keuls post test, unless otherwise indicated. A one-tailed t-test was
also used based
on the hypothesis that the compound or drug, or the combination, would reduce
tumor
growth.
Results
Studies in the HT-29 Human Colon Tumor Xenoaraft Model
1. In Vivo Evaluation of KPU-2 +/- CPT-11 in the HT-29 Human Colon Tumor
Xeno~raft
Model
[0454] This study assessed changes in dosage strength and dosing regimen for
KPU-2 alone and in combination with a relevant chemotherapeutic CPT-11 in the
HT-29
model.
[0455] KPU-2 was administered at doses of 7.5 mg/kg ip daily for five days
(qdx5), 3.75 rng/kg ip bid for five days, 7.5 mg/kg ip every second day for 10
days (qodx5)
and 7.5 mg/lcg ip every third day for 15 days (q3dx5). The combination of CTP-
11 with NPI-
2358 at a dose of 7.5 mg/kg ip q3dx5 resulted in a significantly greater
effect than for either
-128-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
compound alone, which lasted for the duration of the study (Figure 13). These
observations
during the in-life portion of the study were confirmed,by the mean group final
tumor weights
at autopsy for which only the combination group exhibited a statistically
significant lower
tumor weight than controls. Tn addition the difference between the mean tumor
weights of
the combination therapy and CPT-11 monotherapy groups was statistically
significant (Figure
14). When the individual final tumor weights at autopsy are examined the
greater effect of
cotherapy is clear (Figure 14). The TGI of cotherapy was 78% as compared to
38.9% for
CPT-11 alone, The TGI for the combined therapy group exceeds the NCI criterion
of 58%
for a positive result.
2. Study of KPU-2 +/- Standard Chemotherapy vs. Five Human Tumor XenoQxaft
Models
[0456] This study consists of five different arms, each with its own protocol,
timing, dosing regimen and reference compound. Each arm will be considered
within the
presentation of the particular tumor model.
[0457] The aim of the HT-29 arm of the study was to investigate a slightly
higher
dose of KPU-2 (10 mg/kg ip q3dx5) in the HT-29 human colon tumor xenograft
model as
compared to those used in the study described above, in which a marked synergy
was
observed between KPU-2 (7.5 mg/kg ip q3dx5) and CPT-11 (100 mg/kg ip qwx3).
[0458] As can be observed in Figure 15, the combination of I~I'U-2 and CPT-11
in this model resulted in a marked synergy in the inhibition of tumor growth,
with the tumor
growth being almost completely inhibited up to Treatment Day 29 in the
combination therapy
group. The combined therapy maintained efficacy and the estimated tumor growth
for this
group was significantly lower than for either rnonotherapy group. Accordingly,
administration of KPU-2 and CPT-I1 inhibited tumor growth and is an effective
anti-tumor
treatment.
[0459] The observations of the in-life portion of the study (estimated tumor
growth, Figure 15) are supported by measurement of the weights of the tumors
excised at
autopsy (Figure 16). The tumor weights for the combination group was
significantly less
than the Controls (p<0.01), as were the tumor weights for CPT-11 alone
(p<0.05).
[0460] When the individual final tumor weights are considered (Fig 16), the
tumor size for the combination group was generally smaller than for the other
treated or
-129-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
control groups. The TGI of the combination group was 65.8%, indicating a
positive effect by
the NCI criterion, while monotherapy did not reach the NCI criterion of TGI >
58%.
3. Study of activity of KPU-2, KPU-35 and t-butyl-phenylahistin in the HT-29
Human Colon
Tumor Xeno~raft Study
[0461] The results of this study are presented in Figure 17 and Table 18. The
combination therapy groups all indicated a marked synergy between the novel
compounds
and CPT-11. The individual tumor weights demonstrate the effectiveness of the
cotherapy
treatment (Figure 18). In each case the TGI for the combination group
surpasses the NCI
criterion for a positive effect, whereas the TGI for CPT-11 monotherapy did
not reach this
level.
-130-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
I~ M l~
>C
N
N N N N ~ N
~ A
V
H
V ~
o
C'
pp
O O O O O
.
HI
~'''
a t~ ~' ~
o a~ ~ a
'
~ U
~
. : ;. ;.n
a W E o 0 0 0 0
U U U U U
0
V
m o
o
o
~,
U a~
~,7
H ~ 00 t~ M ~ l~
~
ooa~ ~n~ oa~ M~ t~~ U
N ~ ~ o
H
V
.~."~ M M M M M
1~
3?
C7
t","F A7
. . O
~ Qr Q.' Q" Q. 'S~
~" ~ ~ ~ M ~ . ~" M
M . ~ . , M ~ .
M
~+..~U Z wo a.ro .wo wo a. o
~ ~ ~ ~
U U U U U
a~
U
0
~o ~ U' W W W W W W
o ~~' zz z z z z
O
V ~; .
~4
'' .
n
nn ~n
,.
a~ ~ ~ N v" N N ~n t~ ~, ~
~ ~n ~n
~n
r-~ ~ r~ r-~ ~ G~ p
~
O ~ ~ ~ O ~ ~O
M M M M
r'' 3E W
~ ,S".,
'' O a~
H H H H H
a~
l0 00 M_ M_ M
N N N N N
- 131 -
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
4. Summary of the Effects of KPU-2,KPU-35 and t-butyl_phenylahistin in
Combination with
CPT-11 in the HT-29 Human Colon Tumor XenoQraft Model
[0462] When combined with CPT-11, KPU-2 enhanced the effect of CPT-11, the
standard chemotherapeutic agent, to a level well in excess of the NCI
criterion of a TGI >
5S% for a positive effect. The results generated in the three studies are very
comparable for
both the in-life observations (Figure 19) and for the weights of the tumors
excised at autopsy
(Figure 20).
Studies in the DU-145 Human Prostate Tumor Xeno~raft Model
[0463] Two studies have been completed with this model: the first study
involved
KPU-2 alone and in combination with taxotere; the second study compared KPU-2,
KPU-35
and t-butyl-phenylahistin alone and in combination with taxotere.
1. Effect of KPU-2 in Combination with Taxotere in the DU-145 Human Prostate
Tumor
Xeno~raft Model
[0464] As can be seen from the data obtained during the in-Iife portion of
this
study (Fig 21), the most effective treatment of the DU-145 human prostate
tumor was the
combined therapy of KPU-2 plus taxotere. The treatment effect was most
pronounced at the
beginning of the study and appeared to be reduced as the study progressed.
From treatment
Days 20 -27, the combination therapy did provide an apparent TGI that exceeded
the NCI
criterion (TGI > 58%), and the estimated tumor weight of the combined therapy
was
significantly less than for either monotherapy.
2. Activity of KPU-2, KPU-35 and t-butyl-phenylahistin Alone or in Combination
with
Taxotere in the DU-145 Human Prostate Xeno -~r~aft Model
[0465] Based on the data obtained with KPU-2 in combination with taxotere in
the Study described above a second study comparing KPU-2 to KPU-35 and t-butyl-
phenylahistin alone and in combination with taxotere was initiated.
[0466] The observations made during the in-life portion of this study indicate
that
the combination of either KPU-2 or KPU-35 with taxotere has a greater
reduction on tumor
growth than for taxotere alone (Figure 22). The tumor growth was almost
completely
blocked by KPU-35 in combination with taxotere.
-132-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0467] The excised tumor weights at autopsy confirmed the observations made
during the in-life segment of the study. The combination of either KPU-2
(Figure 23) or
KPU-35 (Figure 24) with taxotere was significantly more effective than
taxotere alone in
blocking tumor growth. In the case of KPU-35, three of ten mice showed
evidence for tumor
shrinkage. The tumor growth inhibition indices indicated a marked inhibition
of tumor
growth for KPU-2 (group mean = 74.1 %) and an almost total block for KPU-35
(group mean
= 92.5%). Taxotere alone did not reach the NCI established criterion for a
positive effect
(TGA >_ 58%).
5. Studies in the MCF-7 Human Breast Turnor Xeno~raft Model
[0468] This study compared the effects of KPU-2, KPU-35 and t-butyl-
phenylahistin in the MCF-7 human breast tumor xenograft model. The doses of
the
compounds were administered on Days 1, 2, 3, 4, and 7; Taxotere was
administered on Days
1, 3 and 7.
[0469] The selected novel compounds have early onset, statistically
significant
effects when used in combination with taxotere in this model, apparently
almost completely
blocking estimated tumor growth (Figure 25). Of the three compounds, KPU-2
appeared to
be the most effective, with t-butyl-phenylahistin also exhibiting a
significant potentiation of
taxotere.
6. Studies in the A549 Human Non Small Cell Luna Tumor Xeno~raft Model
[0470] The in-life observations during this study (Figure 26) indicated that
the
combination of KPU-2 (7.5 mg/kg ip, qdx5) with taxotere resulted in a marked
inhibition of
tumor growth as compared to the Control or either monotherapy group. This was
confirmed
by the autopsy tumor weights, as the mean of the cotherapy group was
significantly less than
that of taxotere alone or the Control group (Figure 27). The cotherapy group
tumor weights
form a cluster of low tumor weights, indicating the consistency of the effect.
[0471] When the tumor growth index is calculated, the cotherapy group had a
TGI
of 74.4% as compared to the control group well in excess of the NCI criterion
for a positive
effect (TGI > 58%). Taxotere alone had a TGI of 26.1 %.
-133-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
7. Studies in the MDA-231 Human Breast Tumor Orthoto~ic Xenoaraft Model
[0472] This model involves the placement of the human tumor tissue into the
mouse mammary fat pad, a surrogate of the natural environment. In this manner
the
possibility of a positive effect due to a specific action on the subcutaneous
vascular bed is
avoided. This study compared the effect of KPU-2 (7.5 mg/kg ip, q3dx5) alone
and in
combination with paclitaxel (16 mg/kg ip, qdx5).
[0473] Three weeks into the study there was a significant inhibition of tumor
growth in the combination therapy group, a highly significant effect. This
effect appeared to
be more marked than for taxotere alone (Figure 28).
8. Studies in the Murine Melanoma B16 F10 Metastatic Tumor Model
[0474] This study examined the effect of I~PU-2, KPU-3S and t-butyl-
phenylahistin alone and in combination with paclitaxel on the number of
metastases
appearing on the surface of the lung 16 days after the intravenous inj ection
of B 16 F 10
melanoma cells to the mouse. This model is not a xenograft model; however, it
does not
involve a high degree of vascularization into the tumor mass.
[0475] In this model the most effective treatment was KPU-2 alone (Figure 29),
having a mean metastases count about 10% less than that for paclitaxel (MGIs
of 41.6% and
35.0 %, respectively). While this study does not itself establish that
combination therapy is
more effective than monotherapy, it does indicate that. KPU-2, KPU-35 and t-
butyl-
phenylahistin are most effective in highly vascularized tumors.
EXAMPLE 10
Assays For Activity Against Pathogenic Fungi
[0476] Comparative activity of a dehydrophenylahistin or its analog against a
pathogenic fungus, relative to known antifungal compounds recited above, for
use in
determining the dehydrophenylahistin or its analog's AF/IS value is measured
directly against
the fungal organism, e.g. by microtiter plate adaptation of the NCCLS broth
macrodilution
method described in Diagn Mice°o afad bifect Diseases 21:129-133
(1995). Antifungal
activity can also be determined in whole-animal models of fungal infection.
For instance,
one may employ the steroid-treated mouse model of pulmonary mucormycosis
(Goldaill, L.Z.
& Sugar, A.M. 1994 J Afatimic~°ob Cher~aother 33:369-372). By way of
illustration, in such
-134-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
studies, a number of animals are given no dehydrophenylahistin or its analog,
various doses
of dehydrophenylahistin or its analog (and/or combinations with one or more
other antifungal
agents), or a positive control (e.g. Amphotericin B), respectively, beginning
before, at the
time of, or subsequent to infection with the fungus. Animals may be treated
once every 24
hours with the selected dose of dehydrophenylahistin or its analog, positive
control, or
vehicle only. Treatment is continued for a predetermined number of days, e.g.
up to ten days.
Animals are observed for some time after the treatment period, e.g. for a
total of three weeks,
with mortality being assessed daily. Models can involve systemic, pulmonary,
vaginal and
other models of infection with or without other treatments (e.g. treatment
with steroids)
designed to mimic a human subject susceptible to infection.
[0477] To further illustrate, one method for determining the is vivo
therapeutic
efficacies (EDso, e.g. expressed in mg dehydrophenylahistin or its analog/kg
subject), is a
rodent model system. For example, a mouse is infected with the fungal pathogen
such as by
intravenous infection with approximately 10 times the 50% lethal dose of the
pathogen (10~
C. albicans cells /mouse). Immediately after the fungal infection,
dehydrophenylahistin
compounds are given to the mouse at a predetermined dosed volume. The EDSO is
calculated
by the method of Van der Waerden (A~ch Exp Patlaol Pharmakol 195:389-412,
1940) from
the survival rate recorded on 20th day post-infection. Generally, untreated
control animals
die 7 to 1 3 days post-infection.
[0478] In another illustrative embodiment, C. albicahs Wisconsin (C43) and C.
tropicalis (C112), grown on Sabouraud dextrose agar (SDA) slants for 48 h at
28°C, are
suspended in saline and adjusted to 46% transmission at 550 nm on a
spectrophotometer.
The inoculum is further adjusted by hemacytometer and confirnied by plate
counts to be
approximately 1 or 5 x 107 CFU/ml. CF-1 mice are infected by injection 1 or 5
x 10~ CFU
into the tail vein. Antifungal agents are administered intravenously or
subcutaneously in
ethanol:water (10:90), 4 h post infection and once daily thereafter for 3 or 4
more days.
Survival is monitored daily. The EDso can be defined as that dose which allows
for 50%
survival of mice.
-135-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
EXAMPLE 11
Evaluating Antimicotic Activity
[0479] Benzimidazoles and griseofulvin are anti-tubulin agents capable of
binding
to fungal microtubules. Once bound, these compounds interfere with cell
division and
intracellular transport in sensitive organisms, resulting in cell death.
Commercially,
benzimidazoles are used as fungicidal agents in veterinary medicine and plant
disease
control. A wide variety of fungal species, including Botsytis cine~°ea,
Beauve~°ia bassiana,
Helminthosporium solani, S'acchaf°omyces cep°evisiae and
Aspergillus are susceptible to these
molecules. Toxicity concerns and increasing drug resistance, however, have
negatively
impacted their usage. Griseofulvin is used clinically to treat ringworm
infections of the skin,
hair and nails, caused by Trichophyton sp., Nlicrosporum sp., and
Epidermophyton
fZoccosuyn. Its antifungal spectrum, however, is restricted to this class of
fungal organisms.
Genotoxicity is also a significant side effect. Terbinafine, while an
alternative first-line
treatment, is more costly. Further, clinical resistance recently has been
observed in
Trichophyton rubs°um (the major causative agent for all dermatophyte
infections).
(0480] In Candida albicans, microtubule/microfilament formation is affected
where cells are exposed to the microtubule inhibitors nocodazole and
chloropropham. These
results further validate the exploration of cytoskeleton inhibitors as
effective antimycotic
agents. Accordingly, several of the compounds disclosed herein were evaluated
for
antimycotic activity.
[0481] Specifically, disclosed compounds were evaluated alongside commercially
available microtubulin inhibitors as well as recognized antifungal agents. The
test
compounds and controls used in this study: (-)-Phenylahistin, KPU-1, KPU-2,
KPU-11 and
KPU-17, KPU-35, t-butyl phenylahistin, Colchicine (commercial microtubulin
inhibitor
tested versus 3 Candida isolates), Benomyl (commercial microtubulin inhibitor
tested versus
3 Candida isolates), Griseofulvin (commercial microtubulin inhibitor and
antibiotic control
for testing versus 6 dermatophyte isolates), Amphotericin B (antibiotic
control for testing
versus 3 Candida isolates), Itraconazole (antibiotic control for testing
versus 2 Aspergillus
isolates).
-136-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0482] Microorganisms against which these compounds were tested included:
Candida albicans, Candida glabrata, Aspefgillus fumigatus, Ti~ichophyton
~ubrum,
Trichophyton mentagrophytes, Epide~°mophyton floccosurn. With the
exception of Candida
glabr~ata (one isolate), two isolates of each species were tested.
[0483] Antifungal susceptibility testing was accomplished according to the
methods outlined in the National Committee for Clinical Laboratory Standards,
M38-A
"Reference Method for Broth Dilution Antifungal Susceptibility Testing of
Conidium-
Forming Filamentous Fungi; Approved Standard." This includes testing in RPMI-
1640 with
glutamine and without bicarbonate, an inoculum size of 0.4 - 5 X 104, and
incubation at 30 or
35°C for 48 hours. The minimum inhibitory concentration (MIC) was
defined as .the lowest
concentration that resulted in an 80% , reduction in turbidity as compared to
a drug-free
control tube. Drug concentrations were 0.03-16 wglml for the investigational
compounds,
0.015-8 ~.glrnl for itraconazole and griseofulvin.
[0484] The minimum inhibitory concentration (MIC) at which a compound
prevented the growth of the target microorganism was assessed according to the
modified
version of the NCCLS protocol. Minimum inhibitory concentrations (MIC) were
determined
at the first 24-hour interval where growth could be determined in the drug-
free control tube.
The defined MIC was the lowest concentration that exhibited an 80% reduction
in turbidity
as compared to the growth control. The minimum lethal concentration (MLC) was
determined by plating 0.1 ~,1 from the MIC concentration and each
concentration above the
MIC. The MLC was called at the first concentration that exhibited five or
fewer colonies of
fungal growth representing a 99.95% kill. When a MIC was obtained, a minimum
fungicidal
concentration (MFC) was determined to assess the fungistatic/fungicidal nature
of the
compound. This procedure entails diluting drug-treated cell samples (removed
from test
wells containing compound at and above the MIC) to compound concentrations
significantly
below the inhibitory concentration and depositing them on agar plates. The
compound is
scored as fungistatic if the cells are able to resume growth and fungicidal if
no regrowth is
possible because the compound had killed the organisms. .
-137-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0485] Compounds disclosed herein were shown to be effective against two
Ti~ichoplaytoh species. T. ~ubrum is the principal causative agent for human
dermatophytic
infections, and would be the key organism to target in the development of a
clinical agent.
[0486] Compounds KPU-2, KPU-11 and CPU-17, KPU-35 & t-
butylphenylahistin were equivalent in potency or in some cases more potent
than
griseofulvin, a current, standard pharmaceutical agent used for treating
dermatophytic
infections.
[0487] Compounds (-)-Phenylahistin and KPU-1 were significantly less potent
than the other compounds when tested versus T. ~ubf°um and weaker but
more comparable to
the others versus the sensitive T. mentagr~ophytes isolate.
[0488] In those instances when an MFC could be determined, the results
indicate
that these compounds are fungistatic in nature (see Tables 19 and 20).
-138-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
0 0
.~o
'~ U ~ ~ ~ o c~ cn
~ ~ ~ o
~C~~
n n n o o ~
.~;o
p ~ U w o vo ~
-~'
~ n n n n n n
H
0
d
U
a
V v o ~ m cNnm cNn~ N d-
n n n n n n '
~
00 N N N N ~ N d.
n '
V ~ n n n n n o ~
U
U ~ ~ ~ o v~
o n
,
.~ ~
v
U ~ n n n n o ~ ~ ~'
~
U n n
H
,
..
cr3..,
'
O ; i~ ~ '' N gyp~ ~f';
~ ~
r ~ ~ .,.p " 's.O
~1 N cd ~,, ~ OV ~~ N
p
U ~ ~ ~,p,,.~ ;~ o ,.fl
-139-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Z ~ ~ ~ ~ ~ z~ ~
Z Z z z
'" n n n n
W n n
o
-. U vo m o ~ ~ ~ N
~
~
"'
W ~ n n n n n n
'~~
N
o
~
~.~ v ~
~ '~'z
y ~ ~ ~ n n n n n n
U
Q.
~ ~
v o n n n n n n
ao
~
0
O ~
W d' N ,--~,~ d-
.,.,
U
V ~ ~ N c o 0
n ~ o
o V v
wa
0 0 0
0 ~ ~ o
c
V w V V V
N
~ ~ ~ ~
~
H ~k ~ /~ 0 0
~D
s
cd
~ \0 ~ ~0 0 0
v v
O O ~~ ~ ~ U
R.i ~ M M M M <t'+O~'O ~ ~ O
~ ~ ~ ~ ~
o , r ,~ cd . N
O
V
-140-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
EXAMPLE 12
Evaluating vascular tar~etin activity
[0489] Tumors and neoplastic conditions can be treated using the compounds
disclosed herein. The occlusion of the blood supply in tumors with vascular
targeting agents
(VTAs) induces regression of the tumors. The compounds disclosed herein,
including NPU-
02 and KPU-35, for example, can be as VTAs. Many VTAs exhibit their vascular
effects by
interacting at the colchicine-binding site on microtubules. This interaction
induces a
characteristic, rapid collapse and occlusion of established vasculature in the
tumor and
therefore compromises the integrity of existing vessels leading to necrosis.
[0490] Vascular collapse can occur, for example, within 30-60 minutes of
exposure to the VTA and involves changing the shape of the immature and
proliferating, but
riot the quiescent and mature, endothelial cells in the central portion of the
tumor. This
differential effect on vascular cells provides a rationale for the selective
effects on the tumor
due to the higher percentage of proliferating immature endothelial cells in
the tumor blood
vessels versus normal blood vessels. VTAs can be classified into three
overlapping spectra
of activity: (1) potent vascular and cytotoxic effects, (2) potent vascular
with weak cytotoxic
effects, and (3) potent cytotoxic with weak vascular effects.
Ih Yivo Vascular Targeting Activity of I~PU-02 and KPU-35
[0491] Animal models are essential to investigate new therapies that inhibit
tumor-induced angiogenesis, target the established tumor vasculature, and
inhibit tumor
growth.
[0492] A murine syngeneic "pseudo-orthotopic" breast cancer model was used to
address these issues. Tomes Filho et al., Microvascular Research (1995)49, 212-
226, which
is incorporated herein by reference in its entirety. To create the "pseudo-
orthotopic milieu,"
the coverslip of a dorsal skinflap chamber was removed and small pieces of
mammary fatpad
from donor mice were implanted into the chamber. On top of the fatpad graft,
tumor
spheroids containing N202 mammary tumor cells transduced with Histone (H2B)-
green
fluorescent protein (GFP) were applied. The use of H2B-GFP transduced cells
allows for
visualizing tumor growth and monitoring mitosis and apoptosis.
-141-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
(0493] Fluorescence video microscopy allows for the relatively non-invasive
study of tumor microcirculation in conscious mice. This model can provide data
regarding
the effects of compounds on tumor vasculature, tumor growth, mitosis and
apoptosis, and is
useful to examine the activity of compounds either alone or in combination
with other
therapeutics. Utilizing this model, KPU-02 and KPU-35 were shown to induce a
rapid
vascular collapse leading to central necrosis, and the regression of
established tumors after a
single i.v. administration.
[0494] On day 12 of tumor growth, mice were treated i.v. with a 2-minute
infusion of 5 mg/kg KPU-35, a 5 minute i.v. infusion of 10 mg/kg KPU-02, or
bolus of
vehicle (10% solutol (w/w) + 2% DMSO in water). On day 13, 5-minute infusions
of 10
rng/kg KPU-02, KPU-35 or vehicle were administered. Treatments with KPU-02 or
KPU-35
were well tolerated. Mice were observed for two additional days. Tumor area,
blood flow
rate, and vascular density within and surrounding the umor were visualized.
Real-time
observations were recorded at various time-points using still photos and video
microscopy.
(0495] This study demonstrates the rapid collapse of the central vasculature
after
the single i.v. treatment with either KPU-02 or I~PU-35. The changes in
vascular functions
resulted in a significant central tumor necrosis, without an observed effect
on the vasculature
in the surrounding fat pad or skin (Figure 30). These observations support the
selectivity and
specificity of KPU-02 and KPU-35, which both individually can disrupt
established tumor
vasculature.
Ih Yivo Activity of KPU 02 in Human Tumo~° Xenogs~afts
(0496] When KPU-02 was administered with CPT-11 (Irinotecan), Taxotere or
Paclitaxel, marked antitumor activity was seen in the human colon (HT-29),
breast (MCF-7;
MDA-MB231) and lung (A549) tumor xenograft models (Table 21). The effect of
KPU-02
in the HT-29 model was robust, reproducible in three studies, and showed a
dose-dependent
effect i.e., 7.5 mg/kg was statistically greater than 2.5 mg/lcg (Figures 32,
33).
Ih vitro Activity of KPU-02 and KPU-35 in HuVEC cells
(0497] The above-described i~a vivo effects of I~.PU-02 and KPU-35 on tumor
vasculature were supported by the ifa vitro effects of the same compounds in
HuVEC cells.
Human umbilical vein endothelial cells are considered a good ifa vitf°o
model of tumor
-142-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
endothelium, which is considered "immature". Tumor endothelium lacks
supporting vascular
mural cells and is increasingly reliant on microtubule network for integrity
of the tumor
vasculature. Therefore, disruption of the microtubule network tumor causes
vascular
collapse.
KPU OZ induces rapid tubulin depolymeri~ation in HuVEC cells.
[0498] Human umbilical vein endothelial cells (HuVECs; Carnbrex CC2519A)
were maintained at subconfluent densities in EGM-2 (Cambrex) media. The cells
were
cultured in an incubator at 37°C in 5% CO2 and 95% humidified air. For
tubulin staining
assays, HuVEC cells were seeded at a density of 3x104 cells/rnl in EGM-2 on
tissue culture
compatible coverslips (Fisher). The plates were returned to the incubator for
2 days.
[0499] Stock (20 mM) solutions of the test compounds were prepared in 100%
DMSO. 400X concentrated dilutions of the compounds were prepared in 100% DMSO.
5 ~.1
volumes of the dilutions were added to individual wells resulting in a final
concentration of
200 nM. The final concentration of DMSO was 0.25% in all samples. The plates
were
returned to the incubator for 30 minutes. HuVEC cells were treated for 30 min
with 200 nM
KPU-02 or I~PU-35.
[0500] The cells were rinsed in dPBS before fixation in 10%(v/v) neutral
buffered
formalin for 10 minutes at room temperature. Following fixation, a-tubulin was
visualized
by indirect immunofluorescence. Specifically, the cells were permeabilized in
0.2%(v/v)
triton X-100/dPBS for 10 minutes. The cells were washed prior to transferring
the coverslips
to a humidified chamber, the coverslips were blocked for two hours in antibody
buffer
(2%(w/v) BSA/ 0.1%(v/v) Tween 20/ dPBS). The coverslips were incubated with SO
~,l of
0.1 ~.g/ml mouse a-tubulin (Molecular Probes) in antibody buffer for 1 hour
before washing
and incubation with 50 ~.1 of 1 ~g/ml goat anti-mouse FITC (Jackson
ImmunoResearch
Laboratories) for one hour in the dark. Finally, the cells were washed and
treated with 2
~g/ml DAPI (Molecular Probes) for 10 minutes before rinsing in HZO and
mounting with
Vectashield (Vector Labs) mounting media. The cells were imaged using a 60x
oil
immersion objective on an upright microscope (Olympus BX51). The images were
digitally
captured using a CCD camera and Magnafire 2.0 software (Olympus). Post image
processing
was performed in Photoshop Elements 2.0 (Adobe) and in Microsoft Powerpoint.
-143-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0501] Figure 33 shows that KPU-02 and KPU-35 rapidly induce tubulin
depolymerization in HuVEC cells.
KPU 02 induces dose dependent monolayet° permeability ire HuVEC
cells.
[0502] Human umbilical vein endothelial cells (HuVECs; Cambrex CC2519A)
were maintained at subconfluent densities in EGM-2 (Carnbrex) media. The cells
were
cultured in an incubator at 37°C in 5% CO2 and 95% humidified air. For
monolayer
permeability assays, HuVEC cells were seeded at 1x105 cells/ ml in EGM-2 media
on
Fibronectin-coated 3.0 ~m Fluoroblolc inserts (Becton Dickinson) in 24-well
plates. The
plates were returned to the incubator for 4 days to allow the cells to reach
confluency.
[0503] Stock solutions (20 mM) of the test compounds were prepared in 100%
DMSO. l OX concentrated serial dilutioris of the compounds were prepared in
EGM-2. 10 ~.1
volumes of the serial dilutions were added to the test inserts in duplicate
resulting in final
concentrations ranging from 2 ~.M to 2 nM. The final concentration of DMSO was
0.25% in
all samples. The cells were treated with 2 nM-2 ~,M IfPU 02 for 15 minutes.
[0504] FITC-Dextran (50 mg/ml) in dPBS (38.2 kDa; Sigma) was diluted 2.5 fold
in EGM-2, 10 ~1 of FITC-Dextran was added to each insert. The final
concentration of
FTTC-Dextran was 1 mg/ml. The plates were returned to the incubator and 30
minutes later
the fluorescence of the lower chambers of the 24 well plates was read using a
Fusion
fluorimeter (Packard Bioscience) with ~X = 485 nm and ?~e", = 530 nm filters.
[0505] Figure 34 shows that KPU 02 is able to induce monolayer permeability in
a dose dependent manner. The results shown in Figure 34 represent the mean ~
S.D. of three
independent experiments.
Blood Flom in the P22 Rat Sarcoma Model with I Z5I IAP
[0506] Tumor blood flow was assessed in a model using a quantitative Iasl_
iodoantipyrine (IAP) technique in rats bearing a P22 rat sarcoma. KPU-02 (15
mg/kg, IP)
markedly and selectively reduced tumor blood flow to 23% of vehicle at 1 hour
after
administration; blood flow remained markedly reduced 24 hours later (59%
vehicle). In
contrast, blood flow in non-tumor tissues was affected to a much lesser extent
at 1 hour (see
Figure 35).
-144-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0507] The reduction in blood flow at 24 hours post-dose was more variable
between tissues for KPU-02 compared to vehicle, as shown in Figure 36. The
blood flow to
the tumor was the most affected. other tissues exhibited a small.reduction in
blood flow.
skeletal muscle blood flow appeared to be increased at 24 hours post-dose.
[050~J The effects of KPU-02 observed at 1 hour appear to be longer lasting
and
more selective for tumor blood flow than that previously reported for CA4P
using the same
technique.
[0509) In an experiment with the P22 rat sarcoma model, it was demonstrated
that
KPU-02 7.5 andl5 mg/kg IP (n=2 per dose) produced a dose-dependent tumor
necrosis by 24
hours post-dose, with the highest dose resulting in an almost total necrosis'
of the tumor as
shown in Figure 37. All tumors in the I~PU-02-treated rats showed evidence of
necrosis,
whereas tumors in vehicle-treated rats did not. The VTAs that have entered
into the clinic
(e.g., CA4P, ZD6126, AVE8062) show similar qualitative effects on tumor blood
using the
IAP methodology (or similar technology) to demonstrate reduced blood flow in
the P22 rat
sarcoma tumor and in humans using the dce-MRI technique. See Stevenson JP,
Rosen M,
Sun W, Gallagher M, Haller DG, Vaughn D, et al.,. "Phase I trial of the
antivascular agent
combretastatin A4 phosphate on a 5-day schedule to patients with cancer:
magnetic resonance
imaging evidence for altered tumor blood flow," J Clih Ofzcol 2003;21(23):4428-
38;
Evelhoch JL, LoRusso PM, He Z, DelProposto Z, Polin L, Corbett TH, et al.,
"Magnetic
resonance imaging. measurements of the response of murine and human tumors to
the
vascular-targeting agent ZD6126," Clifa Cancer Res 2004;10(11):3650-7; and
Gadgeel SM,
LoRusso PM, Wozniak AJ, Wheeler C. "A dose-escalation study of the novel
vascular-
targeting agent, ZD6126, in patients with solid tumors," Proc Am Soc Clin
OrZCOI
2002;21:abstract 438; each of which is hereby incorporated by reference in its
entirety.
Combination Therapy with Microtubule Tar~eting~A ,ents
[0510] The findings that VTAs selectively damage the vasculature in the
central
part of the tumor versus the periphery, which recovers functionality, support
using these
agents in combination with chemotherapeutics (Taxol, Vinblastine and
Cisplatin), radiation
and angiogenesis inhibitors directed against VEGF and EGF. The new VTAs will
-145-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
supplement rather than supplant these therapies and should provide for greater
antitumor
activities.
Treatment of other conditions
[0511] In addition to cancer, other diseases may be treated using the VTAs
disclosed herein. Conditions include other neoplasms, retinopathies, and any
other condition
or disease that relies upon blood supply, preferably blood supply from new
vasculature in
order to remain viable and/or proliferate.
(0512] Many conditions are associated with excessive or inappropriate
vasculature. Examples of conditions associated with excessive vasculature
include
inflammatory disorders such as immune and non-immune inflammation; rheumatoid
arthritis,
chronic articular rheumatism and psoriasis; disorders associated with
inappropriate or
inopportune invasion of vessels such as diabetic retinopathy, neovascular
glaucoma,
retinopathy of prematurity, macular degeneration, corneal graft rejection,
retrolental
fibroplasia, rubeosis, capillary proliferation in atherosclerotic plaques and
osteoporosis; and
cancer associated disorders, including for example, solid tumors, tumor
metastases, blood
born tumors such as leukemias, angiofibromas, Kaposi sarcoma, benign tumors
such as
hemangiomas, acoustic neurornas, neurofibrornas, trachomas, and pyogenic
granulomas, as
well as other cancers which require vascularization to support tumor growth.
Additional
examples of vasculature-dependent diseases include, for example, Osler-Webber
Syndrome;
myocardial angiogenesis; plaque neovascularization; telangiectasia;
hemophiliac joints and
wound granulation. Furthermore, excessive vasculature is also associated with
clinical
problems as part of biological and mechanical implants (tissue/organ implants,
stems, etc.).
The instant compounds and compositions can be used to target vasculature, in
preferably to
preferentially target disease vasculature over non disease tissue vasculature,
and thus the
compounds and compositions can be used in the treatment of such conditions.
Other diseases
in which vascularization plays a role, and to which the instant compounds and
compositions
can be used, are known by those of skill in the art.
[0513] Examples of retinopathies include age-related macular degeneration
(ARMD), diabetic retinopathy, and the like. Pathological angiogenesis is a
major
contributing factor to a number of retinopathies that collectively are major
cause of blindness
-146-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
in the developed world. Kahn and Hiller Am J Ophthalmol (1974) 78, 58-67,
which is
incorporated herein by reference in its entirety. For example, retinal and
disk
neovascularization occurs in 30-50% of patients with diabetic retinopathy for
more than 20
years. Yanko et al Retina (2003) 23, 518-522, which is incorporated herein by
reference in
its entirety. Furthermore, subretinal neovascularization is a serious
complication in ~10% of
patients with macular degeneration. Ferris et al Arch Ophthalmol (1984), 102,
1640-1642,
which is incorporated herein by reference in its entirety.
[0514] Vascular targeting agents such as Combretastatin A-4 (CA-4) have been
shown to cause the disruption. of neoeessels in non-neoplastic tissue. Griggs
et al Br J
Cancer (2001) 84, 832-835, which is incorporated herein by reference in its
entirety.
Additionally, CA-4P was shown to inhibit the retinal neovascularization that
occurs during
proliferative retinopathy. Griggs et al Am J Path (2002) 160, 1097-1103, which
is
incorporated herein by reference in its entirety. Finally, CA-4P Phosphate was
demonstrated
to suppress the development of VEGF induced retinal neovascularization and
inhibit the
development and/ or cause partial regression of choroidal neovascularization.
Nambu et al
Invest Ophthalmolbgy & Visual Sci (2003) 44, 3650-3655, which is incorporated
herein by
reference in its entirety. The compounds disclosed herein can be used to treat
retinopathies.
For example, the methodologies of Griggs (2001 and 2002) and Nambu are used to
treat
retinopathies. Furthermore, the compounds and compositions disclosed herein
can be used to
treat such retinopathies by applying the compounds and/or compositions to the
target area in
an effective amount for reducing vascular density and/or vascular
proliferation.
-147-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
a
0 0 0 0
O ~ M N ~ y
d' M O O
f~
O
~--
...
N
'TJ~
O
~ O
y ~ O O O O_
O y
L1 p p .~-~ O
r
O ,",
y
a + V
r~O o ~ .+. ~ ,-.. d'
y ~
.~,~ W N
t~
N ~
O
N p V
.~ +I
,..pi G) by l~ N ~ N
p M
O
G
O
p V Cdr .--~--~ O v, ~n N ~n N m ~n
o~ ~ ~C ~
U o cd cri.~~ o cd cri
U
0
U
O
.,.,
N ~ p x "'~ oo V, ~, ~n~ oo 'n O,
~ ~ ~ ~ l~ l~~ h
H
M
H ~ p O ~' M ~ ~ NI ~ N ~ O
~ v ~ 'r as ~ " ra~ 'r a
-148-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
EXAMPLE 13
Structure-Activit~Relationship
[0515] The effect of activity from various modifications on the phenyl ring of
tBu-dehydroPLH is illustrated by the data in Figure 38. It is apparent that
substitution with
relatively hydrophobic and smaller functional groups at the m- or o- position
increased or
maintained the cytotoxic activity at HT-29 cells while substitutions at the p-
position
decreased activity. While not being bound to any particular theory, this data
suggests a
rigorous recognition of the phenyl ring by tubulin.
[0516] 3D-QSAR (CoMFA) analysis (see Figure 39) also supports the existence
of sterically favorable fields at m- and o- positions and sterically
unfavorable fields exist at
the p- position. X-ray crystal analysis (see Figure 40) indicates that the
conformation of
potent derivatives require a certain amount of dihedral angle between the
phenyl ring and the
pseudo-tricyclic cor template formed by DKP and imidazole rings. Thus,
modification with
the appropriate conformational restriction of the phenyl ring may elicit
potent activity. While
not being bound to any particular theory, it may be that the binding mode of
PLH derivatives
at the chochicine binding site of tubulin is different from that of
colchicines and its known
homologues.
EXAMPLE 14
Ire Tjit~o Action on Microtubules
Purification of Microtubule Protein and Tubulin
[0517] Microtubule protein (MTP) was prepared as previously described (Farrell
KW and Wilson L. (1987) Tubulin-colchicine complexes differentially poison
opposite
microtubule ends. Biochemistry 23(16):3741-8, which is incorporated herein by
reference in
its entirety). MTP preparations consisting of 70% tubulin and 30% microtubule-
associated
proteins (MAPS) were isolated from bovine brain by three cycles of wamn
polymerization and
cold depolymerization in PEM100 (100 mM 1-4 piperazinediethansulfonic acid
(Pipes), 1
mM MgS04, 1 mM EGTA, pH 6.8) and 1 mM GTP. MTP was drop-frozen in liquid
nitrogen and stored at -70°C until use. Tubulin was purified from
microtubule protein by
phosphocellulose chromatography (PC-tubulin) and stored in PEM50 (50 mM Pipes,
1 mM
-149-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
MgS04, 1 mM EGTA, pH 6.8). Protein concentration was determined by a Bradford
assay
(Sigma Chemicals, St. Louis, MO) using bovine serum albumin as the standard
(Bradford,
1976).
Test Agents .
[0518] Stock solutions of KPU-02 were prepared at a concentration of 20 mM in
DMSO. Stock solutions of Combretastatin A4 (National Cancer Institute,
Bethesda, MD)
(CA4) was prepared at a concentration of 5 mM in DMSO. Colchicine (Sigma
Chemicals,
St. Louis, MO) (CLC) was prepared at a concentration of 3 mM in water. All
agents were
shielded from ambient light with amber Eppendorf tubes. Serial dilutions were
made in
DMSO andlor PEM50 to the desired concentrations.
Determination of Steady-State Microtubule Polymer Mass
[0519] MTP (2 rng/ml) was polymerized into microtubules in the presence of a
range of drug concentrations in PEM100 containing 1 mM GTP and a final DMSO
concentration of 0.5%. Samples were monitored by light scattering at 350 nm at
37°C for 75
minutes.
[0520] Polymerization reactions were centrifuged and the microtubule protein
concentrations in the supernatant, a measure of the soluble tubulin at steady
state, and the
pellet, a measure of the microtubule polymer, were used to calculate the
inhibition of
polymerization. After incubation, polymerized microtubules were separated and
sedimented
from unpolymerized MTP by centrifugation (150,000 x g, 45 minutes,
37°C). The
supernatant was removed, and the microtubule pellets were depolymerized in
deionized Ha0
(24 hours, 0°C) before protein determination by the Bradford assay.
[0521] The percent inhibition was calculated in two ways and the values
obtained
from the two ways were compared. In one way, a ratio of the microtubule
protein in the
pellet, drug to no drug, was calculated. Another ratio of microtubule protein
in the pellet to
the supernatant, drug to no drug, was also calculated. The numbers were in
close agreement
and the former values were used because they were subject to less variance and
experimental
perturbation.
-150-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Microtubule Mean Length Distributions
[0522] Transmission electron microscopy was used to determine the mean length
distribution of microtubules in the absence or presence of tested agent. At 75
minutes and
prior to sedimentation, 10 ~,l aliquots from the polymer mass experiments were
fixed by
dilution into 290 pl PEM100-buffered 0.2% glutaraldehyde. Thirty microliters
of fixed
sample was settled onto formvar-coated 150 ICG mesh electron microscope grids
for 90
seconds. Excess sample was wicked off with Whatman Blter paper. Thirty
microliters of
cytochrome C (1 mg/ml) was applied for 30 seconds to enhance protofilament
resolution and
facilitate negative staining. Uranyl acetate (1.5%) was applied for 20 seconds
and the excess
was wicked off. Grids were viewed in a Jeol electron microscope-1200 EX11 at
2000X and
30,OOOX magnification: The Zeiss MOPIII was used to determine microtubule
length
distributions and mean lengths for at least 100 rnicrotubules per sample.
CLC Competition Assays
[0523] PC-tubulin (0.2 mg/ml) was incubated in PEM50 with 1 mM GTP, 1%
DMSO, 10 ~M of tested agent and 7-25 wM [3H] CLC for 120 minutes at
37°C.
Measurement of [3H] CLC binding was followed by DEAF-cellulose filter-binding
assay as
described previously (Wilson, 1970). This method depends on the adsorption of
tubulin to
filter paper impregnated with DEAF-cellulose. Whatman DE81 filter paper was
pre-wet with
PEM50 prior to sample application. The total 100 ~1 reaction volume was
applied to 2.5 cm
disks of filter paper, over parafilm, on ice. The paper disks were washed by
immersion in
five successive 50 ml changes of PEM50, 5 min/wash, 4°C, to remove all
unbound
colchicine. The paper disks with adhering tubulin-bound colchicine were then
counted
directly in a scintillation vial containing 2 ml of Beckman Coulter Ready
Protein solution
(Fullerton, CA). All of the disks were washed together. Negligible binding of
unbound CLC
to the paper disks occurred in controls, either in the presence of absence of
tubulin.
[0524] The K; values were calculated by linear regression of a double
reciprocal
plot of the experimental data in Microsoft Excel. The Kr" value of tubulin for
CLC under the
experimental conditions was first determined, with x intercept equal to -1/Km.
Km app, Km in
-151-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
the presence of drug, was determined experimentally. The K; was determined
using the
relationship Km app = a, Km, and for competitive inhibition a = K ",(1 +
[I]/K;).
Fluorescence Spectroscopy
[0525] Fluorescence measurements were performed using a Perkin-Elrner LSSOB
spectrofluorirneter. PC-tubulin (0.2 mg/ml) was incubated in PEM50, 2 mM GTP,
3%
DMSO, with 0 - 30 wM KPU-02. The interaction of KPU-02 with tubulin was
reported by
4,4'-dianilino- l,l'-binaphthyl- S,5'-disulfonic acid, dipotassium salt (bis-
ANS; Molecular
Probes, Eugene, OR) fluorescence, with an excitation wavelength of 395 nm and
an emission
wavelength maximum of 4~7 nrn. Excitation and emission band passes were 10 nm.
This
experiment was performed twice.
[0526] The bis-ANS fluorophore probes the hydrophobic surface of proteins and
a
change in intensity of the bis-ANS fluorescence signal is a result of a change
in the solvent
accessible surface area of a protein. If there is some conformational change
that changes the
tubulin-bis-ANS interaction upon ligand binding, then bis-ANS can be used to
report
binding.
[0527] PC-tubulin (0.2 rng/ml) was incubated with 0 - 30 ~.M KPU-02 at
25°C
for 20 minutes. Bis-ANS (25 ~M) was then added and relative fluorescence
intensities of
samples were measured at 25°C within 15 minutes. Buffer blank spectra
were collected and
showed that KPU-02 plus bis-ANS produced negligible fluorescence in the
experimental
wavelength range.
[0528] The Ka was determined by fitting experimental data in Sigmaplot and
Microsoft Excel using the equation F = ((-FmaX x L)/(Ka + L)) + Fo where F is
the
fluorescence intensity of bis-ANS-tubulin in the presence of total ligand
concentration L, Fmax
is the bis-ANS fluorescence intensity of fully liganded tubulin, and Fo is bis-
ANS
fluorescence in the absence of drug. Fmax was determined by plotting 1/(Fo-F)
versus 1/L and
extrapolating to 1/L = 0. The fraction of binding sites B occupied by KPU-02
was
determined using the following relationship: B = (Fo-F)/(Fo-Fmax). The
concentration of free
ligand was determined with Lfree = L - B[C] in which [C] is the molar
concentration of
ligand-binding sites, assuming a single binding site per tubulin dirner.
-152-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Inhibition of microtubule polymerization by KPU-02
[0529] KPU-02, CA4, and CLC were assayed for their ability to alter the
polymerization of MAP-rich tubulin (MTP) (2 mg/ml) in a cell-free system ih
vitro. Initially,
inhibition of polymerization was assayed using phosphocellulose-purified,
microtubule-
associated protein-free tubulin (data not shown). KPU-02 was a more potent
inhibitor
towards MTs assembled with glycerol and DMSO seeds as compared to MTs
assembled in
the presence of MAPS that copurify with tubulin. Although microtubule polymer
in the
absence of stabilizing MAPS did not reach.steady state over a 2-hour period,
these assays
demonstrated that KPU-02 interacts directly with purified tubulin and that it
does not exert its
primary effect through a MAP.
[0530] KPU-02 and CA4 inhibited MT polymerization more powerfully than
CLC as measured by light scattering (Figure 44) and sedimentation analysis
(Figure 45).
MTP (2 mg/ml) was polymerized into rnicrotubules in the presence of a range of
drug
concentrations and allowed to reach steady state as monitored by light
scattering at 350 nm.
Figure 41 depicts turbidity spectra of microtubule protein polymerization in
the presence of
DMSO drug vehicle (0), 1.25 ~M (o), 2.5 ~M (-), and 5 ~M (o) NPI-2358 (a), CA4
(b) and
CLC (c).KPU-02 and CA4 inhibited MT polymerization with comparable potencies.
Figure
45 depicts inhibition of microtubule polymerization in the absence or presence
of a range of
KPU-02 (o),CA4 (o), and colchicine (0) concentrations. The total polymer mass
after 75
minutes of assembly was determined by sedimentation. Error bars are standard
deviation
values from three experiments. The concentration at which polymerization was
inhibited 50%
(ICSO), is 2.4 ~ 0.4 ~,M for KPU-02, 2.2 ~ 0.3 ~M for CA4, and 7.6 ~ 2.4 ~.M
for CLC (Table
22). (Variances obtained by statistical analysis are reported as standard
deviation values
unless stated otherwise). At concentrations over the ICso for in vitro
polymerization of MAP-
rich tubulin, MTP displays aggregation kinetics, suggesting that KPU-02 and
CA4 sequester
protein to prevent microtubule assembly.
-153-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 22. Microtubule polymerization inhibition concentrations.
Compound Polymer n
Mass
Ave. IC50
sd
(~M)
KPU-02 2.4 0.4 4
CA4 2.2 0.3 3
CLC 7.6 2.4 3
[0531] As shown in Figure 44, all three of the tested agents produced a
concentration dependent inhibition of the extent of microtubule polymerization
from 1.25 - 5
~M. There are two important differences to note among the spectra. First, the
initial rate of
increase in absorbance over time decreases with increasing drug concentration
(Figure 44A
and 44B). The spectra indicate that there is a lag period for MT formation in
the presence of
KPU-02 and CA4. Drugs that significantly and rapidly reduce the soluble,
assembly-
competent pool of tubulin would decrease the initial rate of polymerization.
In contrast, the
initial rate of polymerization is unchanged at all concentrations of CLC
(Figure 44C).
Second, MTP in the presence of I~PU-02 or CA4 does not reach steady state at
high drug
concentrations (above 5 ~,M), as shown by the absorbance values that increase
linearly with
time (Figure 44A and 44B). In contrast, MTP in the presence of CLC reaches
steady state at
high drug concentrations (Figure 44C).
[0532] The amount of drug required to inhibit polymerization by 50% (ICSO) was
deterinined from the analysis of the linear relationship between the decrease
in microtubule
polymer sedimented by centrifugation with the increase in drug concentration
(Figure 45).
The error bars in Figure 45 represent standard deviation values from at least
three
independent experiments.
Decrease in mean microtubule length measured by transmission electron
microscopy
[0533] Transmission electron microscopy was performed on agent-microtubule
polymerization reactions to describe the polymer formed at steady state and to
evaluate
conclusions drawn from the light scattering spectra. KPU-02, CA4, and GLC all
decreased
-154-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
the lengths of the microtubules formed at steady state. MTs were progressively
shorter with
increasing drug concentration (Figures 46, 47 and 48). Figure 46 depicts
frequency
histograms of mean microtubule lengths ih vitf°o at steady state in the
presence of (A) KPU-
02, (B) CA4, and (C) CLC. The Zeiss M~PIII was used to 'determine microtubule
length
distributions and mean lengths. At least 100 microtubules per drug
concentration were
counted. Figure 47 depicts electron microscopy used to record microtubules in
the absence
or presence of tested compounds. At 75 minutes, samples from polymer mass
experiments
were fixed and stained and viewed in a Jeol electron microscope-1200 EXll at
2000x
magnification. Representative electron micrographs of MAP-rich microtubules
formed in
vitro at steady state in the presence of (A) KPU-02, (b) CA4, and (C) CLC.
Scale bar, 10
wM. Figure 48 depicts a graphical summary. of MT length decrease at steady
state in the
presence of I~PU-02, CA4, and colchicine. Black bars, 1.25 ~,M, and shaded
bars, 2.5 wM
drug. In the presence of KPU-02, and CA4, MTs are progressively shorter with
increasing
drug concentration, until the drug concentration at which MTP displays
aggregation kinetics
as detected by turbidity, and no MTs are observed. Error bars are standard
deviation values
from the measurement of at least 100 rnicrotubules.
[0534] KPU-02, CA4 and CLC did not affect MT nucleation. The numerous,
short microtubules formed in the polymerization reactions evidence that the
presence of
KPU-02, CA4, or CLC does not affect nucleation. If nucleation were affected,
then fewer,
longer microtubules, as opposed to numerous, shorter microtubules would have
been
observed in drug-treated versus control samples.
[0535] KPU-02 and CA4 were comparably potent in decreasing the average MT
length. At 1.25 ~,M, the lowest drug concentration analyzed by electron
microscopy, KPU-02
and CA4 decreased mean MT length by approximately 70%, and CLC by 40% (Figure
48).
[0536] At drug concentrations over the ICSO for ih vitro microtubule
polymerization, rnicrotubules are not observed by electron microscopy for KPU-
02 and CA4.
W contrast, microtubules were observed by electron microscopy for all
concentrations of
CLC assayed. At concentrations over the ICso, rnicrotubule protein in the
presence of KPU-
02 and CA4 displays aggregation kinetics, characterized by a linear increase
in light
absorbance over time (Figure 44A and 44B), whereas in the presence of CLC,
light scattering
-155-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
polymer reaches steady state (Figure 44C). Despite the observation that MTP
with KPU-02
or CA4 increases absorbance at 350nm over time, drug-specific protein
aggregates were not
observed.
Fluorescence Spectroscopy
[0537] Tubulin (0.2 mg/ml) was incubated with a range of KPU-02
concentrations for 20 minutes at 25°C in PEM50 and 2 mM GTP. KPU-02
quenched bis-
ANS fluorescence in a concentration-dependent manner (Figure 49A). For KPU-02
and
tubulin as measured by non-linear regression analysis of bis-ANS fluorescence
intensity at
the emission maximum, Ka = 10 ~ 1.6 p,M (standard error) (Figure 49B). The
double
reciprocal plot of the binding data, assuming a single binding site for KPU-02
per tubulin
dimer, yielded a dissociation constant of 6.4 ~M (Figure 49C). The two
different Kd values
obtained by nonlinear and linear regression analysis methods were sufficiently
close and the
values were considered approximately equivalent. Figure 49A depicts
fluorescence emission
spectra of tubulin in the presence of increasing KPU-02. Drug binding results
in quenching
of bis-ANS fluorescence. Figure 49B depicts fluorescence emission rnaxirna at
487 nrn fit to
obtain the Kd of tubulin for KPU-02, 10 ~.M, standard deviation 1.6 p,M.
Inset, residuals.
Figure 49C depicts the double reciprocal transformation of the binding data
assuming a one
mole drug/rnole tubulin. dirner.
Competitive Inhibition of CLC Binding
[0538] KPU-02 and CA4 competitively inhibited CLC binding to tubulin (Figure
50). Figure 50 depicts the results of an inhibition assay where
phosphocellulose-purified
tubulin (0.2 mg/ml) was incubated with various concentrations of [3H]CLC in
the absence
(0), or presence of 10 ~,M KPU-02 (o) or 10 ~M CA4 (o). Tubulin-CLC Km was 11
~ 4.4
wM and inhibition constants for KPU-02 and CA4 were 3.2 ~ 1.7 p,M and 2.4 ~
0.3 uM,
respectively. Constants were calculated from three independent experiments.
The
colchicine-tubulin binding reaction is time and temperature dependent and the
binding
dissociation constant is Kd = 0.1-1 wM, depending on the conditions of the
assay (Wilson L
and Meza I. (1973) The mechanism of action of colchicine. Colchicine binding
properties of
-156-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
sea urchin sperm tail outer doublet tubulin. Jou~hal of Cell Biology 58(3):709-
19, which is
incorporated herein by reference in its entirety). Under the test conditions,
the Km of tubulin
for CLC is 11 ~ 4.4 ~.M. The Km may be considered the overall Ka of tubulin
for CLC,
however, due to the time-dependence of CLC binding, the Km is greater than the
reported
values for the Kd. The K; for KPU-02 and CA4 was 3.2 ~ 1.7 ~,M and 2.4 ~ 0.3
~M,
respectively. The K; is defined as the amount of drug required to inhibit CLC
binding by
50% and it is based on the amount of radioactive CLC bound to tubulin. The K;
is a measure
of the drugs' ability to compete with CLC; it is not a direct measurement of
drug-tubulin
binding dissociation because of the method in which binding affinity is
reported.
Results
At all concentrations of CLC assayed, MAP-rich tubulin reached steady state.
In
contrast, at higher KPU-02 or CA4 drug concentrations, MAP-rich tubulin did
not
polymerize to steady state and microtubules were not observed by electron
microscopy.
KPU-02 and CA4 effectively decreased the concentration of available tubulin.
This decrease
in the pool of soluble tubulin increased the MT critical concentration and
prevented
polymerization. The stoichiometric amounts of KPU-02 and CA4 required to
decrease ih
vitro polymer mass coupled with the data that microtubule protein did not
reach steady state
above those concentrations, suggesting that KPU-02 and CA4 act by a
sequestering
mechanism in which soluble tubulin is bound and prevented from polymerization.
Observations by electron microscopy on the steady state, MAP-rich microtubules
formed in the presence of the tested agents were consistent with the proposed
mechanism that
KPU-02 and CA4 sequester tubulin. There was a concentration-dependent decrease
in the
average microtubule length in the presence of KPU-02, CA4, and CLG. In the
presence of
KPU-02 and CA4, there was a drug concentration dependent decrease in the
initial rate of
polymerization, indicating that these drugs reduce the tubulin available for
elongation. This
decrease in the initial polymerization rate was not seen with CLC due to its
slow association
with tubulin. Furthermore, microtubules were formed at CLC concentrations over
its ICso for
polymerization, but microtubules were not formed at KPU-02 or CA4
concentrations over
their ICso for polymerization. While not being bound by any particular theory,
the
-157-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
concentration of soluble tubulin bound by KPU-02 or CA4 must be under the
critical
concentration required for tubulin polymerization to proceed.
[0539] Binding studies indicated that tubulin has a lower affinity for KPU-02
than
it has for CLC. Tnhibition of CLC binding to tubulin by KPU-02 and CA4
occurred within a
20-minute incubation period, indicating that KPU-02 and CA4 association with
tubulin
approaches equilibrium relatively faster than for CLC (data not shown). KPU-02
competitively inhibited CLC binding to tubulin at a site overlapping with the
CLC-binding
site, consistent with studies characterizing phenylahistin (halimide) (Kanoh
K, Kohno S,
Kataka J, Takahashi J and Uno I. (1999) (-)-Phenylahistin arrests cells in
mitosis by
inhibiting tubulin polymerization. The Journal of Antibiotics 52(2):134-141,
which is
incorporated herein by reference in its entirety). CA4, a structural analog of
CLC, also
competitively inhibited CLC binding. Without being bound to any particular
henry, it
appears that despite sharing a tubulin binding region with CLC, KPU-02 and CA4
interact
with tubulin and inhibit microtubules by a mechanism distinct from that of
CLC.
EXAMPLE 15.
In Vivo Action on Microtubules
Cell culture studies
[0540] MCF7 human breast, carcinoma cells (American Type Culture Collection,
Manassas, VA) stably transfected with GFP-alpha-tubulin (Clontech, Palo Alto,
CA) were
cultured in Dulbecco's modified Eagle's medium supplemented with 5% fetal
bovine serum,
0.1% penicillin/streptorilycin and nonessential amino acids (Sigma) in 250-ml
tissue culture
flasks or 35-mm six-well plates (doubling time, 29 hours) at 37°C in 5%
COa. Cells were
incubated with KPU-02, CA4, or CLC, prepared as described in Example 14, by
replacing the
original medium with an equal volume of medium containing the required
concentration of
tested agent or DMSO vehicle, and incubation was continued at 37°C for
20 hours.
Mitotic Progression
[0541] The fraction of cells in mitosis at a given drug concentration (mitotic
index) was determined in the breast cancer cell line MCF7. Cells were plated
at a density of
-158-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
3 x 104 cells/ml in six-well plates. After 24 hours, cells were incubated in
the absence or
presence of drug over a range of concentrations (1 nM to 1 wM) for 20 hours.
Media were
collected and cells were rinsed with versene (137 mM NaCI, 2.7 mM KCI, 1.5 mM
KH2P04,
8.1 mM Na2P04, and 0.5 mM EDTA), detached with trypsin, and added back to the
media to
ensure that floating and poorly attached. mitotic cells were included in the
analysis. Cells
were fixed with 10% formalin in PBS overnight at 37°C, permeabilized in
methanol for 10
minutes, and stained with 4,6-diamidino-phenylindole (DAPI) to visualize
nuclei. Stained
cells were spread on coverslips in Vectashield mounting media (Burlingame, CA)
and sealed
onto slides with nail polish. Fluorescence microscopy was used to determine
mitotic indices.
Results were the mean and standard deviation of 4-7 experiments in which 300
cells were
counted for each concentration. The ICso was the drug concentration that
experimentally
induced 50% of the maximal mitotic accumulation at 20 hours.
Immunofluorescence microscopy
[0542] Cells were prepared as for mitotic progression, except that cells were
seeded onto poly-L-lysine (50 ~.g/ml, Sigma) treated coverslips. On the day of
staining, cells
were rinsed in PBS and fixed in 10% formalin overnight at 37°C. Cells
were rinsed in PBS,
permeabilized in methanol at -20°C, and hydrated with PBS. Coverslips
were treated with
20% normal goat serum in 1'BS/ BSA (1%) for 1 h at room temperature. Cells
were
incubated in a mouse monoclonal cocktail of anti-alpha- and beta-tubulin, DM1A
lDMlB
diluted in PBS/BSA for 1 hour at room temperature, then stained with FITC-
conjugated
secondary antibody and DAPI. Coverslips were mounted using Prolong antifade
media
(Molecular Probes, Eugene OR).
Preparation of cells for analysis of microtubule dynamics
[0543] Cells were prepared as for mitotic progression, except that to promote
cell
spreading, cells were seeded onto glass coverslips that had been pretreated
with poly-L-lysine
(50 ~.g/ml, Sigma) for 2 hours, followed by laminin and fibrouectin (10
p.g/ml, Sigma) for 1
hour at 37°C. Cells were incubated with drug or DMSO for 20 hours and
serum-starved.
Before analysis, coverslips were transferred to recording media (culture media
lacking phenol
-159-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
red and sodium bicarbonate buffered with 25 mM HEPES and supplemented with 3.5
g/L
sucrose). To prevent photobleaching, Oxyrase (30 ~,1/ml, Oxyrase Inc.,
Mansfield, OH) was
added to the recording media immediately before sealing cells in a double
coverslip-enclosed
chamber.
Time-lapse microscopy and image acquisition
[0544] Microtubules were observed using a Nikon Eclipse E800 fluorescence
microscope with a plan apochromat 1:4 N:A. x 100 objective lens. The stage was
enclosed in
a Pyrex box and maintained at 36 ~ 1°C by a forced air heating system.
Thirty images of
each cell were acquired at 4-s intervals using a Photometrics CooISNAP HQ
digital camera
(Tucson, AZ) driven by Metamorph software (Universal Imaging, Media, PA) at 10
MHz,
with a 300 ms exposure time, a gain of 2, and 2 x 2 binning to enhance
brightness.
Analysis of microtubule dynamics
[0545] The positions of the plus ends of microtubules over time were tracked
using the Metamorph Track Points application exported to Microsoft Excel and
analyzed
using Real Time Measurement software. The lengths of individual microtubules
were
graphed as a function of time. Individual growth and shortening rates were
determined by
linear regression. Changes of > 0.5 ~m between two points were considered to
be growth or
shortening events, and changes of < 0.5 ~.m between two points were considered
to be
periods of attenuated dynamics or pause. At least 25 microtubules were
analyzed for each
condition. Results are the mean and standard deviation of at least three
independent
experiments.
[0546] The time-based catastrophe frequency for each microtubule was
calculated
by dividing the number of catastrophes per microtubule by the time spent in
growth or
attenuation. The time-based rescue frequency per microtubule was calculated by
dividing the
total number of rescues per microtubule by the time spent shortening. The
distance-based
catastrophe and rescue frequencies were calculated similarly by dividing the
number of
transitions by the length grown or shortened, respectively. Microtubules that
were visible for
< 2 min were included in the frequency analysis. Dynamicity per microtubule
was calculated
-160-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
as the length grown and shortened divided by the total life span of the
microtubule.
Microtubules that.were visible for>_ 0.3 min were included in the dynamicity
analysis.
Cell cycle progression blocked at prometaphase
[0547] The concentration range for KPU-02, CA4 and CLC over which cells
accumulate in mitosis were determined. After 20 hours, 60-70% of cells were
inhibited at
prornetaphase, compared to 30-40% of cells at metaphase in studies on MT
depolymerizers
such as the vinca alkaloids and 2-methoxyestradiol, and MT stabilizers such as
taxol,
epothilone B, and discodermolide (Jordan MA (2002) Mechanism of action of
antitumor
drugs that interact with microtubules and tubulin. Current Medicinal Chemistry
- Ahti-
Car~cer Agents 2: 1-17, which is incorporated herein by reference in its
entirety). The drug
concentration necessary for 50% maximal mitotic block (ICSO) was evaluated
between 1 nM
and 1 wM drug (Figure 51). Figure S1 depicts log [Drug] response curves for
mitotic
progression inhibition by KPU-02, CA4, and.CLC. MCF7 cells were cultured in
the presence
of NPI-2358 (o), CA4 (o), and colchicine (0). To evaluate mitotic indices,
MCF7 cells were
plated at a density of 3 x 104 cellslml in six-well plates. After 24 hours,
cells were incubated
in the absence or presence of drug over a range of concentrations (1 nM to 1
p.M) for 20
hours. Cells were fixed and stained with DAPI to visualize nuclei.
Fluorescence microscopy
was used to determine mitotic indices. Results are the mean and standard
deviation of three
or four experiments in which 300 cells were counted for each drug
concentration. The
mitotic block ICSO for KPU-02 was 17.4 ~ 1.2 nM, CA4 was 5.4 ~ 0.7 nM, and CLC
was 23.8
~ 3.1 nM (Table 23).
Table 23. Inhibition of mitotic progression.
Compound Mitotic blockn
Ave.
IC50 sd (nM)
KPU-02 17.4 1.2 4
CA4 5.4 0.7 3
CLC 23.8 3.1 4
-161-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0548] Most MT-targeting agents block mitosis at the metaphase to anaphase
transition. Mitotic block at the metaphase to anaphase transition is
associated with
suppression of MT dynamics. Without being bound to any particular theory, the
earlier
prometaphase block, together with the depletion of MT polymer, suggests a
distinct
mechanism of action for KPU-02 as compared with other MT depolymerizing drugs,
e.g.,
vinblastine, which at low concentrations stabilize MT dynamics.
Depolymerization of the mitotic spindle and the interphase array MTs
[0549] KPU-02, CA4, and CLC were observed to be potent rnicrotubule
depolymerizers in MCF7 cells. Although mitotic spindle microtubules are more
susceptible
to depolymerization and/or inhibition of polymerization than interphase array
microtubules,
both microtubule populations were affected (Figure 52). Figure 52 depicts
imrnunofluorescence 'microscopy images of MCF7 cells. Interphase arrays are
relatively
more stable to depolymerization by KPU-02, CA4 and CLC than mitotic spindles,.
Cells
were prepared and seeded as for mitotic progression and treated with the
mitotic block ICso
for each drug for 20 hours. Cells were incubated in a mouse monoclonal
cocktail of anti-
alpha- and beta-tubulin, DMlA/DM1B then stained with FITC-conjugated secondary
antibody and DAPI. a-d, Tubulin in control (a), KPU-02 (b), CA4 (c), and CLC
(d) treated
cells, and e-h, DNA in control (e), KPU-02 (f), CA4 (g), and CLC (h) treated
cells. Narrow
arrows indicate mitotic spindle polymer and mitotic chromosomes and thicker
arrows
indicate interphase arrays and nuclei.
[0550] At the ICas for mitotic block, KPU-02 dramatically altered spindle
morphology. Figure 53A-C depicts immunofluorescence microscopy images of MCF7
cells
treated .with KPU-02 (A), CA4 (B), and CLC (C) for 20 hours. Mitotic spindle
destruction
with increasing drug concentration. 1-4, Alpha and beta tubulin in control
(1), a
concentration of IC2s for mitotic block (2), the ICso for mitotic blocl~ (3),
and twice the ICso
for mitotic block (4); 5-8, corresponding images of DNA for the adjoining
panels. There
were no normal, bipolar spindles at the ICZS for KPU-02 or CA4 (Figures 53A
and B).
Compound-treated cells had monopolar or bipolar spindles with uncongressed
chromosomes.
In contrast, normal bipolar spindles persist at the ICas for CLC (Figure 53C).
At the ICso for
-162-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
KPU-02, 75% of the mitotic cells contained asters or foci of tubulin, and the
remaining cells
had no detectable mitotic polymer. In the presence of CLC, half of the cells
were bipolar
with uncongressed chromosomes and the remaining half were monopolar. At
concentrations
of twice the ICSO for mitotic block, there was little detectable MT polymer in
mitotic cells
treated with KPU-02, CA4, or CLC.
[0551] Microtubule interphase arrays were more resistant to depolymerization
than mitotic spindles for all of the compounds examined (Figure 52). However,
a qualitative
decrease in polymer was observed in a dose-dependent manner for all three
compounds
(Figure 54 A-C). Figure 54A-C depicts immunofluorescence microscopy images of
MCF7
cells treated with KPU-02 (a), CA4 (b), and CLC (c) for 20 hours. Interphase
MT
depolymerization with increasing drug concentration. 1-4, Alpha and beta
tubulin in control
(1), a concentration of IC25 for mitotic block (2), the ICSO for mitotic block
(3), and 2XICSo
for mitotic block (4); 5-8, corresponding images of DNA for the adjoining
panels.
Presumably, tubulin is sequestered in these interphase cells, despite the
presence of
intracellular stabilizing MAPS, just as MAP-rich tubulin is sequestered in ire
vitro polymer
mass assays.
Lack of suppression or modulation of MT dynamic instability in living MCF7
cells
[0552] I~PU-02, as well as CA4, did not have a measurable effect on MT dynamic
instability at concentrations effecting 25% (Table 24) or 50% (Table 25) of
the maximal
mitotic block in MCF7 cells. Without being bound by any particular theory,
these data
suggest that the antiproliferative mechanism of action of KPU-02 (and CA4) is
primarily due
to inhibition of MT polymerization, rather than suppression of rnicrotubule
dynamics.
-163-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
Table 24. MT dynamic instability at the mitotic block.
P'.nn+rnl er+ AIPI_'JRGR cri C:Ad c(1 ~'rL.C Sd
Mean rates
(Nm/min) 9.03 4.6611.89 6.2310.59 4.7511.31 8.46
Growth
Shortenin 32.85 18.0631.87 15.1240.21 18.9029.17 17.13
Mean duration
(min) 0.41 0.270.31 0.150.27 0.120.26 0.14
Growth
Shortenin 0.20 0.070.20 0.070.21 0.090.23 0.29
Attenuation0.58 0.380.73 0.360.45 0.320.56 0.43
time spent
Growth 41.83 29.66 35.31 33.07
Shortenin 18.57 20.53 25.19 25.96
Attenuation39.60 49.81 39.50 40.97
Freq. of
(min-1 1.15 1.27 1.52 1.57
)
Catastro
he
Rescue 3.05 2.79 _1.90 2.95
D n. m/min 9.87 10.07 13.87 11.31
MTs/cells 16/30 ~ 11133 ~ 13/39 8/26
Minutes 40.66 43.69 39.62 35.29
Table 25. MT dynamic instability at the mitotic block.
Control sd NPI-2358sd CA4 sd
Mean rates (pm/min)
Growth 9.03 4.66 10.63 6.78 11.21 5.64
Shortening 32.85 18.0634.06 12.8429.88 18.87
Mean duration
(min) ~ 0.41 0.27 0.30 0.18 0.33 0.18
Growth
Shortenin 0.20 0.07 0.22 0.08 0.22 0.08
Attenuation 0.58 0.38 0.61 0.41 0.55 0.44
time spent
Growth 41.83 33.02 43.64
Shortenin 18.57 20.92 20.13
Attenuation 39.60 46.06 36.23
Freq. of (min-1
) 1.15 1.22 1.09
Catastro he
Rescue 3.05 2.60 2.79
D namicit mlmin9.87 10.64 10.91
MTs/cel Is 30/16 25/11 29/9
Minutes 40.66 33.13 39.14
[0553] The examples described above are set forth solely to assist in the
understanding of the invention. Thus, those slcilled in the art will
appreciate that the
disclosed methods and compounds encompass and may otherwise provide further
derivatives
of dehydrophenylahistins.
-164-
CA 02553630 2006-07-18
WO 2005/077940 PCT/US2005/003636
[0554] One skilled in the art would readily appreciate that the present
invention is
weh adapted to obtain, for example, the ends and advantages mentioned, as well
as others
inherent. The methods and procedures described herein are presently
representative of
preferred embodiments and are exemplary and are not intended as limitations on
the scope of
the invention. Changes therein and other uses will occur to those skilled in
the art which are
encompassed within the spirit of the inventioxi.
[0555] It will be readily apparent to one skilled in the art that varying
substitutions and modifications may be made to the invention disclosed herein
without
departing from the scope and spiritof the invention.
[0556] As noted above, all patents and publications mentioned in the
specification
are indicative of the levels of those skilled in the art to which the
invention pertains. All
patents and publications are hereby incorporated by reference herein to the
extent allowable
by law, such that each individual patent and publication may be treated as
specifically and
individually indicated to be incorporated by reference.
[0557] The invention illustratively described herein suitably may be practiced
in
the absence of any element or elements, limitation or limitations which is not
specifically
disclosed herein. The terms and expressions which have been employed are used
as terms of
description and not of limitation, and there is rlo intention that in the use
of such terms and
expressions indicates the exclusion of equivalents of the features shown and
described or
portions thereof. It is recognized that various modifications are possible
within the scope of
the invention. Thus, it should be understood that although the present
invention has been
specifically disclosed by preferred embodiments and optional features,
modification and
variation of the concepts herein disclosed may be resorted to by those skilled
in the art, and
that such modifications and variations are considered to be falling within the
scope of the
invention.
-165-