Note: Descriptions are shown in the official language in which they were submitted.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-1-
TRIAZOLE DERIVATIVES WHICH INHIBIT VASOPRESSIN ANTAGONISTIC ACTIVITY
This invention relates to novel compounds useful in therapy and to processes
for the
preparation of such compounds. It also relates to compositions containing such
compounds, their use and intermediates used in their preparation.
WO 01/87855 discloses triazole derivatives as inhibitors of glycine
transporter activity.
WO 01/58880 and JP2000-63363 disclose triazole derivatives useful as arginine
Vasopressin VIA receptor antagonists. Kakefuda et al., Bioorg. Med. Chem. 10
(2002)
1905-1912 and Kakefuda et al., J.Med.Chem., 2002, 45, 2589-2598 discuss the
utility of
4,5-diphenyl-1,2,4-triazole derivatives as selective antagonists for the human
VIA receptor
and comment that the 4,5-diphenyl-1,2,4-triazole structure plays an essential
role in VIA
affi n ity.
The compounds of the present invention have been found to have useful
pharmaceutical
properties. They may be used to treat aggression, Alzheimer's disease,
anorexia
nervosa, anxiety, anxiety disorder, asthma, atherosclerosis, autism,
cardiovascular
disease (including angina, atherosclerosis, hypertension, heart failure,
edema,
hypernatremia), cataract, central nervous system disease, cerebrovascular
ischemia,
cirrhosis, cognitive disorder, Cushing's disease, depression, diabetes
mellitus,
dysmenorrhoea (primary and secondary), emesis (including motion sickness),
endometriosis, gastrointestinal disease, glaucoma, gynaecological disease,
heart
disease, intrauterine growth retardation, inflammation (including rheumatoid
arthritis),
ischemia, ischemic heart disease, lung tumor, micturition disorder,
mittlesmerchz,
neoplasm, nephrotoxicity, non-insulin dependent diabetes, obesity,
obsessive/compulsive
disorder, ocular hypertension, preclampsia, premature ejaculation, premature
(preterm)
labor, pulmonary disease, Raynaud's disease, renal disease, renal failure,
male or female
sexual dysfunction, septic shock, sleep disorder, spinal cord injury,
thrombosis, urogenital
tract infection or urolithiasis.
Particularly of interest are the following diseases or disorders:
anxiety, cardiovascular disease (including angina, atherosclerosis,
hypertension, heart
failure, edema, hypernatremia), dysmenorrhoea (primary and secondary),
endometriosis,
emesis (including motion sickness), intrauterine growth retardation,
inflammation
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-2-
(including rheumatoid arthritis), mittlesmerchz, preclampsia, premature
ejaculation,
premature (preterm) labor and Raynaud's disease.
In particular, the compounds of the present invention exhibit vasopressin
antagonistic
activity and can be used in the treatment of dysmenorrhoea (primary and
secondary).
There is a high unmet need in the area of menstrual disorders and it is
estimated that up
to 90% of all menstruating women are affected to some degree. Up to 42% of
women
miss work or other activities due to menstrual pain and it has been estimated
that around
600 million work hours a year are lost in the US as a result {Coco, A.S.
(1999). Primary
dysmenorrhoea. [Review] [30 refs]. American Family Physician, 60, 489-96.}.
Menstrual pain in the lower abdomen is caused by myometrial hyperactivity and
reduced
uterine blood flow. These pathophysiological changes result in abdominal pain
that
radiates out to the back and legs. This may result in women feeling nauseous,
having
headaches and suffering from insomnia. This condition is called dysmenorrhoea
and can
be classified as either primary or secondary dysmenorrhoea.
Primary dysmenorrhoea is diagnosed when no abnormality causing the condition
is
identified. This affects up to 50% of the female population {Coco, A.S.
(1999). Primary
dysmenorrhoea. [Review] [30 refs]. American Family Physician, 60, 489-96.;
Schroeder,
B. & Sanfilippo, J.S. (1999). Dysmenorrhoea and pelvic pain in adolescents.
[Review] [78
refs]. Pediatric Clinics of North America, 46, 555-71}. Where an underlying
gynaecological disorder is present, such as endometriosis, pelvic inflammatory
disease
(PID), fibroids or cancers, secondary dysmenorrhoea will be diagnosed.
Secondary
dysmenorrhoea is diagnosed in only approximately 25% of women suffering from
dysmenorrhoea. Dysmenorrhoea can occur in conjunction with menorrhagia, which
accounts for around 12% of referrals to gynaecology outpatients departments.
Currently, women suffering from primary dysmenorrhoea are treated with non-
steroidal
anti-inflammatory drugs (NSAID's) or the oral contraceptive pill. In cases of
secondary
dysmenorrhoea surgery may be undertaken to correct the underlying
gynaecological
disorder.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-3-
Women suffering from dysmenorrhoea have circulating vasopressin levels which
are
greater than those observed in healthy women at the same time of the menstrual
cycle.
Inhibition of the pharmacological actions of vasopressin, at the uterine
vasopressin
receptor, may prevent dysmenorrhoea.
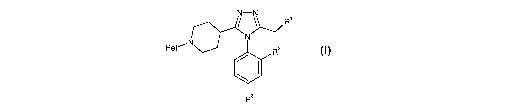
According to the present invention there is provided a compound of formula
(I),
Heto
or a pharmaceutically acceptable derivative thereof, wherein:
Het represents 2-pyridinyl or 2-pyrimidinyl;
R' represents H, C~~ alkyl or a nitrogen-containing heterocyclic ring;
R~ represents H, benzyl or C~_3 alkyl; and
R3 represents H, methyl, methoxy or chloro.
In the above definitions, alkyl groups containing three carbon atoms, except
where
indicated, can be unbranched or branched chain. Examples of alkyl include
methyl, ethyl,
n-propyl and i-propyl.
Unless otherwise stated, the term heterocyclic ring, heterocyclic or
heterocycle, means a
five- or six-membered saturated, unsaturated or aromatic ring containing one
or more
heteroatoms selected from N, S and O. Preferred heterocycles included within
the above
definition are triazolyl, piperidinyl and morpholinyl
Preferred aspects of the invention are as set out below:
(i) a compound according to formula (I) wherein Het represents 2-
pyridinyl;
(ii) a compound according to formula (I) or aspect 1 wherein R'
represents 1,2,3-triazolyl;
(iii) a compound according to formula (I) or either of aspects (i) or (ii)
wherein R2 represents H or methyl;
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-4-
l ..
(iv) a compound according to formula (1) or any of aspects (i) to (iii)
wherein R3 represents chloro;
(v) a compound according to formula (I) or aspect (iii) wherein at least one
of R', R~ and R3 represents a group other than H.
(vi) a compound according to formula (I) wherein R' represents 1,2,3-
triazolyl andlor R3 represents chloro.
Preferred compounds according to the present invention are:
2-{4-[4-(2-Ethyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-piperidin-1-yl}-
pyrimidine;
2-~4-[5-Methyl-4-(2-propyl-phenyl)-4H-[1,2,4]triazol-3-yl]-piperid in-1-yl}-
pyrimidine;
2-~4-[4-(2-Isopropyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-piperidin-1-yl}-
pyrimidine
4-(5-Morpholin-4-ylmethyl-4-phenyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-tetrahydro-
2H-
[1,2']bipyridinyl;
4-(5-Butyl-4-phenyl-4H-[1,2,4]trizaol-3-yl)-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl;
4-(4-Phenyl-5-piperidin-1-ylmethyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-tetrahydro-
2H-
[1,2']bipyridinyl;
4-(5-Methyl-4-phenyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl;
4-[4-(4-Methoxy-2-methyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-3,4,5,6-
tetrahydro-
2H-[1,2']bipyridinyl;
4-[4-(4~Chloro-2-methyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-3,4,5,6-
tetrahydro-
2H-[1,2']bipyridinyl;
4-(5-Methyl-4-o-tolyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl;
4-[4-(4-Chloro-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl;
4-[4-(4-Methoxy-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
3,4,5,6-
tetrahydro-2H-[1,2']bipyridinyl;
4-[4-(4-Methoxy-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl;
4-(4-o-Tolyl-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-
tatrahydro-2H-
[1,2']bipyridinyl;
4-[4-(4-Chloro-2-methyl-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-
3-yl]-
3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl;
4-(4-Phenyl-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-
tetrahydro-2H-
[1,2']bipyridinyl;
4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
3,4,5,6-
tetrahydro-2H-[1,2']bipyridinyl;
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-5-
4-(5-Methyl-4-p-tolyl-4H-[1,2,4]triazol-3-yl)-3,4,5,6-tetrahyd ro-2H-
[1,2']bipyridinyl;
4-[4-(2,4-Dimethyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-3,4,5,6-tetrahydro-
2H
[1,2']bipyridinyl;
4-[4-(4-Chloro-2-methyl-phenyl)-5-morpholin-4-ylmethyl-4H-[1,2,4]triazol-3-yl]-
3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl; and
pharmaceutically acceptable derivatives thereof.
Pharmaceutically acceptable derivatives of the compounds of formula (1)
according to the
invention include salts, solvates, complexes, polymorphs, prodrugs,
stereoisomers,
geometric isomers, tautomeric forms, and isotopic variations of compounds of
formula (I).
Preferably, pharmaceutically acceptable derivatives of compounds of formula
(I) comprise
salts, solvates, esters and amides of the compounds of formula (I). More
preferably,
pharmaceutically acceptable derivatives of compounds of formula (I) are salts
and
solvates.
The pharmaceutically acceptable salts of the compounds of formula (I) include
the acid
addition and base salts thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts.
Examples
include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate,
bisulphate,
borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate,
gluconate,
glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride,
hydrobromide/bromide, hydroiodide/iodide, isethionate, D- and L-lactate,
malate, maleate,
malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate,
nitrate, orotate,
oxalate, palmitate, palmoate, phosphate, hydrogen phosphate, dihydrogen
phosphate,
saccharate, stearate, succinate, sulphate, D- and L- tartrate, tosylate and
trifluoroacetate
salts. A particularly suitable salt is the besylate derivative of the
compounds of the
present invention.
Suitable base salts are formed from bases, which form non-toxic salts.
Examples include
the aluminium, arginine, benzathine, calcium, choline, diethylamine,
diolamine, glycine,
lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and
zinc
salts.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-6-
For a review on suitable salts see Stahl and Wermuth, Handbook of
Pharmaceutical
Salts: Properties, Selection and Use, Wiley-VCH, Weinheim, Germany (2002).
A pharmaceutically acceptable salt of a compound of formula (I) may be readily
prepared
by mixing together solutions of the compound of formula (I) and the desired
acid or base,
as appropriate. The salt may precipitate from solution and be collected by
filtration or
may be recovered by evaporation of the solvent. The degree of ionisation in
the salt may
vary from completely ionised to almost non-ionised.
The compounds of the invention may exist in both unsolvated and solvated
forms. The
term "solvate" is used herein to describe a molecular complex comprising the
compound
of the invention and one or more pharmaceutically acceptable solvent
molecules, for
example, ethanol. The term "hydrate" is employed when said solvent is water.
Included within the scope of the invention are complexes such as clathrates,
drug-host
inclusion complexes wherein, in contrast to the aforementioned solvates, the
drug and
host are present in stoichiometric or non-stoichiometric amounts. Also
included are
complexes of the drug containing two or more organic andlor inorganic
components what
may be in stoichiometric or non-stoichiometric amounts. The resulting
complexes may be
ionised, partially ionised, or non-ionised. For a review of such complexes,
see J Pharm
Sci, 64 (8), 1269-1288 by Haleblian (August 1975).
Hereinafter all references to compounds of formula (I) and pharmaceutically
acceptable
derivatives include references to salts, solvates and complexes thereof and to
solvates
and complexes of salts thereof.
The compounds of the invention include compounds of formula (I) as
hereinbefore
defined, polymorphs, prodrugs, and isomers thereof (including optical,
geometric and
tautomeric isomers) as hereinafter defined and isotopically-labelled compounds
of
formula (I).
As stated, the invention includes all polymorphs of the compounds of formula
(1) as
hereinbefore defined.
Also within the scope of the invention are so-called "prodrugs" of the
compounds of
formula (I). Thus certain derivatives of compounds of formula (I) which may
have little or
no pharmacological activity themselves can, when administered into or onto the
body, be
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-7-
converted into compounds of formula (I) having the desired activity, for
example,
hydrolytic cleavage. Such derivatives are referred to as "prodrugs". Further
information
on the use of prodrugs may be found in "Pro-drugs as Novel Delivery Systems,
Vol. 14,
ACS Symposium Series (T Higuchi and W Stella) and "Bioreversible Carriers in
Drug
Design", Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical
Association).
Prodrugs in accordance with the invention can, for example, be produced by
replacing
appropriate functionalities present in the compounds of formula (I) with
certain moieties
know to those skilled in the art as "pro-moieties" as described, for example,
in "Design of
Prodrugs" by H Bundgaard (Elsevier, 1985).
Some examples of prodrugs~in accordance with the invention include:
(i) where the compound of formula (I) contains a carboxylic acid functionality
(
COOH), an ester thereof, for example, replacement of the hydrogen with (C~
Cg)alkyl;
(ii) where the compound of formula (I) contains an alcohol functionality (-
OH), an
ether thereof, for example, replacement of the hydrogen with (C~-
C6)alkanoyloxymethyl; and
(iii) where the compound of formula (1) contains a primary or secondary amino
functionality (-NHS or -NHR where R ~ H), an amide thereof, for example,
replacement of one or both hydrogens with (C~-C~o)alkanoyl.
Further examples of replacement groups in accordance with the foregoing
examples and
examples of other prodrug types may be found in the aforementioned references.
Finally, certain compounds of formula (I) may themselves act as prodrugs of
other
compounds of formula (I).
Also within the scope of the invention are the metabolites of the compounds of
formula (I)
when formed in vivo following administration of a compound of formula (I).
Compounds of formula (I) containing one or more asymmetric carbon atoms can
exist as
two or more stereoisomers. Where a compound of formula (I) contains an alkenyl
or
alkenylene group, geometric cisltrans (or Z/E) isomers are possible, and where
the
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-g_
compound contains, for example, a keto ~or oxime group or an aromatic moiety,
tautomeric isomerism ('tautomerism') may occur. It follows that a single
compound may
exhibit more than one type of isomerism.
Included within the scope of the present invention are all stereoisomers,
geometric
isomers and tautomeric forms of the compounds of formula (I), including
compounds
exhibiting more than one type of isomerism, and mixtures of one or more
thereof. Also
included are acid addition or base salts wherein the counter ion is optically
active, for
example, D-lactate or L-lysine, or racemic, for example, DL-tartrate or DL-
arginine.
Cisltrans isomers may be separated by conventional techniques well known to
those
skilled in the art, for example, fractional crystallisation and
chromatography.
Conventional techniques for the preparationlisolation of individual
enantiomers include
chiral synthesis from a suitable optically pure precursor or resolution of the
racemate (or
the racemate of a salt or derivative) using, for example, chiral HPLC.
Alternatively, the racemate (or racemic precursor) may be reacted wifih a
suitable optically
active compound, for example, an alcohol, or, in the case where the compounds
of
formula (I) contains an acidic or basic moiety, an acid or base such as
tartaric acid or 1-
phenylethylamine. The resulting diastereomeric mixture may be separated by
chromatography and/or fractional crystallisation and one or both of the
diastereomers
converted to the corresponding pure enantiomer(s) by means well known to a
skilled
person.
Chiral compounds of the invention (and chiral precursors thereof) may be
obtained in
enantiomerically-enriched form using chromatography, typically HPLC, on an
asymmetric
resin with a mobile phase consisting of a hydrocarbon, typically heptane or
hexane,
containing from 0 to 50% isopropanol, typically from 2 to 20%, and from 0 to
5% of an
alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords
the enriched
mixture.
Stereoisomeric conglomerates may be separated by conventional techniques known
to
those skilled in the art - see, for example, "Stereochemistry of Organic
Compounds" by E
L Eliel (Wiley, New York, 1994).
The present invention also includes all pharmaceutically acceptable isotopic
variations of
a compound of the formula (1) one or more atoms is replaced by atoms having
the same
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
_g_
atomic number, but an atomic mass or mass number different from the atomic
mass or
mass number usually found in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention
include
isotopes of hydrogen such as 2H and 3H, carbon such as "C, '3C and '4C,
nitrogen such
as '3N and '5N, oxygen such as '5O, "O and '$O, phosphorus such as 32P,
sulphur such
as 35S, fluorine such as'$F, iodine such as '231 and '251, and chlorine such
as 36C1.
Certain isotopically-labelled compounds of formula (I), for example those
incorporating a
radioactive isotope, are useful in drug and/or substrate tissue distribution
studies. The
radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. '4C, are
particularly useful for this
purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford
certain
therapeutic advantages resulting from greater metabolic stability, for
example, increased
in vivo half-life or reduced dosage requirements, and hence may be preferred
in some
circumstances.
Substitution with positron emitting isotopes, such as "C,'$F,'50 and'3N, can
be useful in
Positron Emission Topography (PET) studies for examining substrate receptor
occupancy.
Isotopically-labelled compounds of formula (I) can generally be prepared by
conventional
techniques known to those skilled in the art or by processes analogous to
those described
in the accompanying Examples and Preparations using appropriate isotopically-
labelled
reagents in place of the non-labelled reagent previously employed.
Pharmaceutically acceptable solvates in accordance with the invention include
those
wherein the solvent of crystallisation may be isotopically substituted, e.g.
D20, ds-acetone
and d6-DMSO.
Unless otherwise provided herein:
Et3N means triethylamine;
AcOH means acetic acid;
MeOH means methanol, EtOH means ethanol, and EtOAc means ethyl acetate;
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-10-
THF means tetrahydrofuran, DCM means dichloromethane, DMF means N,N-
dimethylformamide and NMP means N-methyl-2-pyrrolidinone;
Boc means tert-butoxy carbonyl, CBz means benzyloxy carbonyl;
Me means methyl, Et means ethyl; Bu means butyl, CI means chloro; OH means
hydroxy; and LG means a suitable leaving group; ,
p-TSA means p-toluenesulphonic acid;
Pd2(Dba)3 means bis(dibenzylideneacetone)palladium;
NMM means N-Methylmorpholine;
WSCDI means 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride;
DCC means N,N'-dicyclohexylcarbodiimide;
HOAT means 1-hydroxy-7-azabenzotriazole;
HOBT means 1-hydroxybenzotriazole hydrate;
PyBOP~ means Benzotriazol-1-yloxytris(pyrrolidino)phosphoniumhexa
fluorophosphate;
PyBrOP~ means bromo-tris-pyrrolidino-phosphoniumhexafluoro phosphate;
Mukaiyama's reagent means 2-chloro-1-methylpyridinium iodide;
Hunig's base means N-ethyldiisopropylamine;
Prot means protecting group;
TFA means trifluoroacetic acid;
halo means halogen; and
triflic anhydride means Trifluoromethanesulfonic acid anhydride.
The following Schemes 1.0 to 6.2 illustrate the preparation of compounds of
the formula
(I), throughout which HET and R' to R3 are as hereinbefore defined:
I-~ NHz
a
~Ri + ~ Rz --~..
HET~N
R3
t°> ~m~
Scheme 1.0
Amines suitable for use as compound (III) are commercially available or are
known in the
literature.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-11-
NI-f2 ~ ~ ,
N-N
\ R ~ ~N R
~O R Prot~ -~/N
Prot~N~ / / R~
R3 \
(IV)
(III) (V)
R3
b
,
c
E
,HE1'
L~' (VII)
R' R~ _
(1) Nl)
Scheme 2.
Prot represents a suitable protecting group for nitrogen. Standard methodology
for
nitrogen protecting groups is used, such as that found in textbooks, (e.g.
"Protecting
Groups in Organic Synthesis" by T.W. Greene and P. Wutz).
LG represents a leaving group such as halogen, preferably Br.
Compounds suitable for use as compound (V!1) are commercially available or are
known
in the literature.
Step (a): Amine (11l) is reacted with oxadiazole (II & IV) to give a compound
of formula
(I). This reaction is carried out by heating the starting materials to
elevated temperatures
such as 100-200°C for 15 minutes to 18 hours and optionally at elevated
pressures, or
optionally under microwave radiation, with a suitable acidic catalyst such as
p-TSA or
trifluoroacetic acid, or Lewis acid catalyst such as magnesium chloride,
optionally using a
high boiling solvent such as xylene or toluene.
Preferred conditions are: 1.5 eq. of amine (III) with 0.1 eq. magnesium
chloride at 150°C
for 4 to 18 hours, optionally at elevated pressures; or
3 to 4 eq. amine (III) with 0.25 eq. pTSA, in xylene at 150°C for 18
hours; or
2 eq. amine (III), 1 eq. TFA in xylene or toluene, at between 110
to150°C.
Step (b): Deprotection of compound (V) is undertaken using standard
methodology, as
described in "Protecting Groups in Organic Synthesis" by T.W. Greene and P.
Wutz".
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-12-
When Prot is Boc the preferred method is hydrogen chloride in a suitable
solvent such as
1,4-dioxane at room temperature for 1 to 16 hours, or a solution of
trifluoroacetic acid in
dichloromethane for 1 to 2 hours.
When Prot is CBz the preferred method is hydrogenolysis using a suitable
palladium
catalyst in a solvent such as ethanol.
When Prot is an allyl carbamate, preferred conditions are thiobenzoic acid and
a suitable
palladium catalyst such as Pd2(Dba)3 with a suitable phosphine additive such
as 1,4-
bis(diphenylphosphino)butane in tetrahydrofuran for 20 minutes.
Step (c): Arylation of compound (VI) can be carried out by a palladium
catalysed cross-
coupling reaction with compound (VII) using a suitable base (t BuONa), a
catalytic
amount of suitable additive such as 2,2'-bis(diphenylphosphino)-1,1'-
binaphthyl and a
suitable palladium catalyst. The reaction is carried out in toluene at an
elevated
temperature for 1 to 24 hours, under an inert atmosphere, to give compound
(I).
Alternatively compound (I) can be prepared by reacting amine (VI) with
compound (VII) at
an elevated temperature, such as 50°C to 140°C, in a suitable
solvent, such as DMF,
NMP or 1,4-dioxan, for about 1 to 48 hours in the presence of a base such as
potassium
carbonate, sodium hydrogen carbonate or Hianig's base.
Preferred conditions are: 1.3 eq. halide (VII), 1 to 2 eq. potassium carbonate
in N,N-
dimethylformamide at 60 °C for 4 to 18 hours.
Compounds suitable for use as compounds (II) and (IV) are known in the
literature or can
be prepared as shown in Schemes 3.1 and 3.2.
c o
H
OH O d _ ~N R
N + HzN~ ~ ~ ~ H
HET~ ~ N R' HET~N O
(VIII) H
(IX) (x)
N-N
.N /O~R~
(II) HET
Scheme 3.1
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-13-
S
, o ,
0 0
SON H N ~ d ~N R'
z v
/N~ H R~ H
Prot + ~N~ O
Prot
(VIII') (IX) (x')
~ ~N~.N
(IV) Prot-N\'~/
~,J O Ri
Scheme 3.2
Compounds (VIII)/(VIII') and (IX) are either commercially available or are
known in
methodology such as the hydrolysis of the corresponding ester. (see
Preparation 2).
Step (d): The reaction of carboxylic acid (VIIIIVIII') with hydrazide (IX) can
be carried out
by standard methods.
Coupling may be undertaken by using either:
(i) an acyl chloride derivative of acid (VIIIIVIII') + hydrazide (IX), with an
excess of acid
acceptor in a suitable solvent; or
(ii) the acid (VI11/VIII') with a conventional coupling agent + hydrazide
(IX), optionally in the
presence of a catalyst, with an excess of acid acceptor in a suitable solvent.
Typically the conditions are as follows:
(i) acid chloride of acid (VIIIIVIII') (generated in-situ), an excess of
hydrazide,
optionally with an excess of 3° amine such as Et3N, Hunig's base or
NMM, in DCM or
THF, without heating for 1 to 24 hrs; or
(ii) acid (VIIIIVIII'), WSCDI /DCC and HOBT /HOAT, an excess of hydrazide,
with an
excess of NMM, Et3N, Hunig's base in THF, DCM or EtOAc, at room temperature
for 4 to
48 hours; or acid (VIII/VIII'), PYBOP~/PyBrOP~/Mukaiyama's reagent, an excess
of
hydrazide, with an excess of NMM, Et3N, Hunig's base in THF, DCM or EtOAc, at
room
temperature for 4 to 24 hours.
The preferred conditions are: acid chloride of acid (VIII/VIII') (generated in-
situ), 1.2 to 2
eq. Hydrazide (IX) in DCM at room temperature for 18 hours, in the presence of
1 to 1.2
eq. of N-methylmorpholine; or
the carboxylic acid (VIII/VIII'), 1eq. HOBT, 1 eq. WSCDI, 1.2 eq. hydrazide
(IX) in
dichloromethane at room temperature for 18 hours.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-14-
Step (e): Cyclisation of compound (XIX') is carried out under suitable
dehydrating
conditions, at elevated temperatures for up to 18 hours.
Typically, dehydrating agents such as polyphosphoric acid, phosphorous
oxychloride, or
triflic anhydride with pyridine, optionally in a suitable solvent such as
dichloromethane, are
used at temperatures from 50 to 120°C for 5 minutes to 12 hours.
Optionally the reaction
can be carried out under an inert atmosphere. Alternatively, the oxadiazole
(II & IV) may
be prepared according to the method of Rigo et. al. Synth. Commun. 16(13),
1665, 1986.
Preferred conditions are: Phosphorous oxychloride at 100 to 110°C for 1
to 2 hours.
Alternative routes to compound (X/X') are shown below in Schemes 4.1 and 4.2:
O H O
~N~ H
OH HzN Prot N~N~prot*
H
HET~N~ d HET~N
(VIII) (X111)
b
O ~ O
O
H
N~NH2 Ri~ N~N~R~
H J HH
HET~N d HET~N O
X is OH or CI
(X11)
Scheme 4.1
0
O HZN.N~Prot N~N~Prot*
,OH ---~. ~ H
N
N~ d Prot~
Prot~
(VIII') (X111')
b
O ~ O O
H
N~NHz R1~X NON R~
NJ H ~ H
N O
Prot d Prot
(xu') X is off or c1 (X')
Scheme 4.2
Carboxylic acid (VIIIIVIII') and a protected hydrazine, where Prot* is
typically Boc, may be
coupled to give compound (XIIIIXIII'), using the conditions described for the
preparation of
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-15-
(XIX') above. Prot* is then removed using standard methodology as described in
step
(b), to give (XIIIXII').
Compound (X/X') may then be obtained by the coupling of hydrazide (XIIIXII')
with a
carboxylic acid, or it's derivative [R'C(O)X, where X is OH or CI], under the
conditions
described previously for step,(d).
Alternative routes to compound (XII/XII') are shown below in Schemes 5.1 and
5.2:
0
0
~NHz
w0 HzN NiNHz
~N~ RS ~ H
HET HET~N
f
(XIV) (X11)
Scheme 5.1
0
0
~NHz
w0 HzN NiNHz
,N~ R5 ~ H
Prot /N
f Prot
(xlv'> (xu~)
Scheme 5.2
Step (t~: The ester (XIV/XIV') may be reacted with hydrazine in a suitable
solvent, such
as methanol at elevated temperature to provide the hydrazide (XIIIXII').
Preferred conditions: 3 eq, hydrazine, in methanol, at reflux for 1 ~ hours.
When R' represents a nitrogen containing heterocyclic ring (represented in the
following
N
schemes as ~ ~ ), compounds of the formula (11V') may be prepared according to
the
routes described in Schemes 6.1 and 6.2:
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-16-
0
O
N~NH2 x LG (X111) O
~N H ~ H~N~LG
HET ~ d ,N O
X is OH or chloro HET
(X11) LG is typically halo (XIV)
Preferably chloro or bromo
a
N ' ~,~
H-N~ (xvp N'N
O
iN g O' LG
HET
HET~N
(XVII) (XV)
a
Scheme 6.1
O v H
N~NHZ x (X111) N~N~LG
N H ~G /NJ H IIO
Prot~ ~ d Prot
X is OH or chloro (XIV')
(X11') LG is typically halo,
preferably chloro or bromo
a
N 'N ~,_
H N~ (~l) N'N
O N ~~----~~'
iN ~ g O i-G
Prot= ~ NJ
(XVII') Prot~
(~')
(v')
Scheme 6.2
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-17-
Compounds suitable for use as compound (X111) and (XVI) are commercially
available or
are known in the literature.
Step (d): Coupling of compound (X111) with hydrazide (XIIIXII') may be carried
out using
standard methodology as outlined above.
Step (e): Dehydration and cyclisation of compound (XIV/XIV') to give
oxadiazole (XVIXV')
is achieved by the methodology outlined above.
Step (g): Compound (XVIXV') is reacted with amine (XVI) to give compound
(XVII/IV') in
the presence of an excess of base, such as triethylamine, Hunig's base or
potassium
carbonate as proton acceptor. The reaction is conducted in a suitable high
boiling solvent,
such as toluene or DMF, at temperatures from 50°C to 100°C for 1
to 24 hours.
Alternatively a palladium catalysed cross-coupling reaction can be carried out
using a
suitable base (t BuONa), a catalytic amount of a suitable additive, such as
tri n-butyl
phosphine, and a suitable palladium catalyst in toluene at reflux from 12 to
24 hours
under an inert atmosphere.
Preferred conditions are: 1 eq. of amine, 2 eq. of potassium carbonate in DMF
at 60°C for
3 to 4 hours.
'
Step (a): Amination of compound (XVII/IV') to give compound (11V') is carried
out using
the methodology outlined above.
It will be appreciated by those skilled in the art that, when appropriate, the
order of steps
(a) and (g) may be reversed. Compound (V') may then be converted to compounds
of
formula (I), according to the reactions described in Scheme 2.
It will be apparent to those skilled in the art that sensitive functional
groups may need to
be protected and deprotected during synthesis of a compound of formula (11V').
This may
be achieved by conventional techniques, for example as described in
"Protective Groups
in Organic Synthesis" by T W Greene and P G M Wuts, John Wiley and Sons Inc,
1991.
The compounds of the present invention are useful because they possess
pharmacological
activity in animals. In particular they are useful in the treatment of a
number of conditions
including aggression, Alzheimer's disease, anorexia nervosa, anxiety, anxiety
disorder,
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-18-
asthma, atherosclerosis, autism, cardiovascular disease (including angina,
atherosclerosis, hypertension, heart failure, edema, hypernatremia), cataract,
central
nervous system disease, cerebrovascular ischemia, cirrhosis, cognitive
disorder,
Cushing's disease, depression, diabetes mellitus, dysmenorrhoea (primary and
secondary), emesis (including motion sickness), endometriosis,
gastrointestinal disease,
glaucoma, gynaecological disease, heart disease, intrauterine growth
retardation,
inflammation (including rheumatoid arthritis), ischemia, ischemic heart
disease, lung
tumor, micturition disorder, mittlesmerchz, neoplasm, nephrotoxicity, non-
insulin
dependent diabetes, obesity, obsessive/compulsive disorder, ocular
hypertension,
preclampsia, premature ejaculation, premature (preterm) labor, pulmonary
disease,
Raynaud's disease, renal disease, renal failure, male or female sexual
dysfunction, septic
shock, sleep disorder, spinal cord injury, thrombosis, urogenital tract
infection or
urolithiasis.sleep disorder, spinal cord injury, thrombosis, urogenital tract
infection,
urolithiasis. Particularly of interest is dysmenorrhoea (primary or
secondary), more
particularly, primary dysmenorrhoea.
Thus, according to another aspect of the invention, there is provided a method
of treatment
of dysmenorrhoea which comprises administering a therapeutically effective
amount of a
compound of the invention to a patient suffering from anxiety, cardiovascular
disease
(including angina, atherosclerosis, hypertension, heart failure, edema,
hypernatremia),
dysmenorrhoea (primary and secondary), endometriosis, emesis (including motion
sickness), intrauterine growth retardation, inflammation (including rheumatoid
arthritis),
mittlesmerchz, preclampsia, premature ejaculation, premature (preterm) labor
or
Raynaud's disease. The use of the compounds as a medicament and the use of the
compounds of the present invention in the manufacture of a medicament for the
treatment
of anxiety, cardiovascular disease (including angina, atherosclerosis,
hypertension, heart
failure, edema, hypernatremia), dysmenorrhoea (primary and secondary),
endometriosis,
emesis (including motion sickness), intrauterine growth retardation,
inflammation
(including rheumatoid arthritis), mittlesmerchz, preclampsia, premature
ejaculation,
premature (preterm) labor or Raynaud's disease, particularly dysmenorrhoea,
are also
provided.
Compounds of the invention intended for pharmaceutical use may be administered
as
crystalline or amorphous products. They may be obtained, for example, as solid
plugs,
powders, or films by methods such as precipitation, crystallisation, freeze
drying, spray
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-19-
drying, or evaporative drying. Microwave or radio frequency drying may be used
for this
purpose.
They may be administered alone or in combination with one or more other
compounds of
the invention or in combination with one or more other drugs (or as any
combination
thereof). The compounds of the present invention may be administered in
combination
with an oral contraceptive. Thus in a further aspect of the invention, there
is provided a
pharmaceutical product containing an V1 a antagonist and an oral contraceptive
as a
combined preparation for simultaneous, separate or sequential use in the
treatment of
dysmenorrhoea.
The compounds of the present invention may be administered in combination with
a
PDE5 inhibitor. Thus in a further aspect of the invention, there is provided a
pharmaceutical product containing a V1a antagonist according to the present
invention
and a PDEV inhibitor as a combined preparation for simultaneous, separate or
sequential
use in the treatment of dysmenorrhoea.
PDEV inhibitors useful for combining with V1a antagonists include, but are not
limited to:
( i ) The PDE5 inhibitors mentioned in International Patent Application
publication
nos. W003/000691; W002/64590; W002128865; W002/28859; W002/38563;
W002/36593; W002/28858; WO02/00657; W002/00656; WO02/10166;
W002/00658; W001/94347; WO01/94345; WO00/15639 and WO00/15228;
(ii) The PDE5 inhibitors mentioned in US Patents 6,143,746; 6,143,747 and
6,043,252;
(iii) the pyrazolo [4,3-d]pyrimidin-7-ones disclosed in EP-A-0463756; the
pyrazolo
[4,3-d]pyrimidin-7-ones disclosed in EP-A-0526004; the pyrazolo (4,3-
d]pyrimidin-7-ones disclosed in published international patent application WO
93/06104; the isomeric pyrazolo [3,4-d]pyrimidin-4-ones disclosed in published
international patent application WO 93/07149; the quinazolin-4-ones disclosed
in published international patent application WO 93/12095; the pyrido [3,2-
d]pyrimidin-4-ones disclosed in published international patent application WO
94/05661; the purin-6-ones disclosed in published international patent
application WO 94/00453; the pyrazolo (4,3-d]pyrimidin-7-ones disclosed in
published international patent application WO 98/49166; the pyrazolo [4,3-
d]pyrimidin-7-ones disclosed in published international patent application WO
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-20-
99/54333; the pyrazolo [4,3-d]pyrimidin-4-ones disclosed in EP-A-0995751; the
pyrazolo (4,3-d]pyrimidin-7-ones disclosed in published international patent
application WO 00/24745; the pyrazolo [4,3-d]pyrimidin-4-ones disclosed in
EP-A-0995750; the hexahydropyrazino [2',1':6,1]pyrido (3,4-b]indole-1,4-diones
disclosed in published international application WO95/19978; the pyrazolo [4,3-
d]pyrimidin-4-ones disclosed in WO00/27848; the imidazo[5,1-t][1,2,4]triazin-
ones disclosed in EP-A-1092719 and in published international application WO
99/24433 and the bicyclic compounds disclosed in published international
application WO 93/07124; the pyrazolo [4,3-d]pyrimidin-7-ones disclosed in
published international application WO 01/27112; the pyrazolo [4,3-d]pyrimidin-
7-ones disclosed in published international application WO 01/27113; the
compounds disclosed in EP-A-1092718 and the compounds disclosed in EP-A-
1092719; the tricyclic compounds disclosed in EP-A-1241170; the alkyl
sulphone compounds disclosed in published international application WO
02/074774; the compounds disclosed in published international application WO
02/072586; the compounds disclosed in published international application WO
02/079203 and the compounds disclosed in WO 02/074312.
( iv) Preferably 5-[2-ethoxy-5-(4-methyl-1-piperazinylsulphonyl)phenyl]-1-
methyl-3
n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil, e.g. as
sold
as Viagra~) also known as 1-[(3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1 H
pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulphonyl]-4-methylpiperazine
(see EP-A-0463756);5-(2-ethoxy-5-morpholinoacetylphenyl)-1-methyl-3-n-
propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see EP-A-0526004);3-
ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxyphenyl]-2-(pyridin-2-
yl)methyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see W098/49166);3-
ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2-
(pyridin-2-yl)methyl-2,6-dihydro-7H-pyrazoto[4,3-d]pyrimidin-7-one(see
W099154333); (+)-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxy-
1 (R)-methylethoxy)pyridin-3-yl]-2-methyl-2,6-dihydro-7H-pyrazolo[4,3-
d]pyrimidin-7-one, also known as 3-ethyl-5-{5-[4-ethylpiperazin-1-ylsulphonyl]-
2-([(1 R)-2-methoxy-1-methylethyl]oxy)pyridin-3-yl}-2-methyl-2,6-dihydro-7H-
pyrazolo[4,3-d] pyrimidin-7-one (see W099/54333);5-[2-ethoxy-5-(4-
ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-
dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, also known as 1-{6-ethoxy-5-(3-
ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazoto[4,3-d]pyrimidin-5-yl]-3-
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-21-
'', pyridylsulphonyl}-4-ethylpiperazine'(see WO 01/27113, Example 8);5-[2-iso-
Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyrid in-3-yl]-3-ethyl-2-(1-
methylpiperidin-4-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one(see WO
01/27113, Example 15);5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-
yl]-3-ethyl-2-phenyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see WO
01/27113, Example 66);5-(5-Acetyl-2-propoxy-3-pyridinyl)-3-ethyl-2-(1-
isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see WO
01/27112, Example 124); 5-(5-Acetyl-2-butoxy-3-pyridinyl)-3-ethyl-2-(1-ethyl-3-
azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see WO 01/27112,
Example 132); (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-
methylenedioxyphenyl)pyrazino[2',1':6,1]pyrido[3,4-b]indole-1,4-dione
(tadalafil,
IC-351, Cialis~), i.e. the compound of examples 78 and 95 of published
international application W095/19978, as well as the compound of examples 1,
3, 7 and 8; 2-[2-ethoxy-5-(4-ethyl-piperazin-1-yl-1-sulphonyl)-phenyl]-5-
methyl-
7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one (vardenafil, LEVITRA ~) also
known as 1-[[3-(3,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1-f]-as-triazin-2-
yl)-4-ethoxyphenyl]sulphonyl]-4-ethylpiperazine, i.e. the compound of examples
20, 19, 337 and 336 of published international application W099/24433;the
compound of example 11 of published international application W093/07124
(EiSAI); compounds 3 and 14 from Rotella D P, J. Med. Chem., 2000, 43,
1257; 4-(4-chlorobenzyl)amino-6,7,8-trimethoxyquinazoline; N-[[3-(4,7-dihydro-
1-methyl-7-oxo-3-propyl-1 H-pyrazolo[4,3-d]-pyrimidin-5-yl)-4-
propxyphenyl]sulfonyl]-1-methyl2-pyrrolidinepropanamide ["DA-8159" (Example
68 of WO00/27848)]; and 7,8-dihydro-8-oxo-6-[2-propoxyphenyl]-1H-
imidazo[4,5-g]quinazoline and 1-[3-[1-[(4-fluorophenyl)methyl]-7,8-dihydro-8-
oxo-1 H-imidazo[4,5-g]quinazolin-6-yl]-4-propoxyphenyl]carboxamide.
(v) 4-bromo-5-(pyridylmethylamino)-6-[3-(4-chlorophenyl)-propoxy]-
3(2H)pyridazinone; 1-[4-[(1,3-benzodioxol-5-ylmethyl)amiono]-6-chloro-2-
quinozolinyl]-4-piperidine-carboxylic acid, monosodium salt; (+)-cis-
5,6a,7,9,9,9a-hexahydro-2-[4-(trifluoromethyl)-phenylmethyl-5-methyl-
cyclopent-4,5]imidazo[2,1-b]purin-4(3H)one; furazlocillin; cis-2-hexyl-5-
methyl-
3,4,5,6a,7,8,9,9a- octahydrocyclopent[4,5]-imidazo[2,1-b]purin-4-one; 3-acetyl-
1-(2-chlorobenzyl)-2-propylindole-6- carboxylate; 3-acetyl-1-(2-chlorobenzyl)-
2-
propylindole-6-carboxylate; 4-bromo-5-(3-pyridylmethylamino)-6-(3-(4-
chlorophenyl) propoxy)-3- (2H)pyridazinone; I-methyl-5(5-morpholinoacetyl-2-n-
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-22-
propoxyphenyl)-3-n-propyl-1,6-dihydro- 7H-pyrazolo(4,3-d)pyrimidin-7-one; 1-
[4-[(1,3-benzodioxol-5-ylmethyl)arnino]-6-chloro-2- quinazolinyl]-4-
piperidinecarboxylic acid, monosodium salt; Pharmaprojects No. 4516 (Glaxo
Wellcome); Pharmaprojects No. 5051 (Bayer); Pharmaprojects No. 5064
(ICyowa Hakko; see WO 96/26940); Pharmaprojects No. 5069 (Schering
Plough); GF-196960 (Glaxo Wellcome); E-8010 and E-4010 (Eisai); Bay-38-
3045 & 38-9456 (Bayer); FR229934 and FR226807 (Fujisawa); and Sch-
51866.
The contents of the published patent applications and journal articles and in
particular the
general formulae of the therapeutically active compounds of the claims and
exemplified
compounds therein are incorporated herein in their~entirety by reference
thereto.
Preferably the PDEV inhibitor is selected from sildenafil, tadalafil,
vardenafil, DA-8159 and
5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-
methoxyethyl]-2,6-
dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one.
Most preferably the PDE5 inhibitor is sildenafil and pharmaceutically
acceptable salts
thereof. Sildenafil citrate is a preferred salt.
The compounds of the present invention may be administered in combination with
an NO
donor. Thus in a further aspect of the invention, there is provided a
pharmaceutical
product containing a V1a antagonist according to the present invention and a
NO donor
as a combined preparation for simultaneous, separate or sequential use in the
treatment
of dysmenorrhoea.
The compounds of the present invention may be administered in combination with
L-
arginine, or as an arginate salt. Thus in a further aspect of the invention,
there is
provided a pharmaceutical product containing a V1a antagonist according to the
present
invention and L-arginine as a combined preparation for simultaneous, separate
or
sequential use in the treatment of dysmenorrhoea.
The compounds of the present invention may be administered in combination with
a COX
inhibitor. Thus in a further aspect of the invention, there is provided a
pharmaceutical
product containing a V1a antagonist according to the present invention and a
COX
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-23-
inhibitor as a combined preparation for simultaneous, separate or sequential
use in the
treatment of dysmenorrhoea.
COX inhibitors useful for combining with the compounds of the present
invention include,
but are not limited to:
(i) ibuprofen, naproxen, benoxaprofen, flurbiprofen, fenoprofen, fenbufen,
ketoprofen, indoprofen, pirprofen, carprofen, oxaprozin, prapoprofen,
miroprofen, tioxaprofen, suprofen, alminoprofen, tiaprofenic acid, fluprofen,
bucloxic acid, indomethacin, sulindac, tolmetin, zomepirac, diclofenac,
fenclofenec, alclofenac, ibufenac, isoxepac, furofenac, tiopinac, zidometacin,
acetyl salicylic acid, indometacin, piroxicam, tenoxicam, nabumetone,
ketorolac, azapropazone, mefenamic acid, tolfenamic acid, diflunisal,
podophyllotoxin derivatives, acemetacin, droxicam, floctafenine,
oxyphenbutazone, phenylbutazone, proglumetacin, acemetacin, fentiazac,
clidanac, oxipinac, mefenamic acid, meclofenamic acid, flufenamic acid,
niflumic acid, flufenisal, sudoxicam, etodolac, piprofen, salicylic acid,
choline
magnesium trisalicylate, salicylate, benorylate, fentiazac, clopinac,
feprazone, isoxicam and 2-fluoro-a-methyl[1,1'-biphenyl]-4-acetic acid, 4
(nitrooxy)butyl ester (See Wenk, et al., Europ. J. Pharmacol. 453:319-324
(2002));
(ii) meloxicam, (CAS registry number 71125-38-7; described in U.S. Patent No.
4,233,299), or a pharmaceutically acceptable salt or prodrug thereof;
(iii) Substituted benzopyran derivatives that are described in U.S. Patent No.
6,271,253. Also benzopyran derivatives described in U.S. Patent Nos.
25. 6,034,256 and 6,077,850 along with International Publication No's WO
98/47890 and WO 00/23433;
(iv) Chromene COX2 selective inhibitors described in U.S. Patent No. 6,077,850
and U.S. Patent No. 6,034,256;
(v) The compounds described in International Patent Application Publication
No's WO 95/30656, WO 95/30652, WO 96/38418 and WO 96/38442, and
the compounds described in European Patent Application Publication No.
799823, along with the pharmaceutically acceptable derivatives thereof;
(vi) celecoxib (US Patent No. 5,466,823), valdecoxib (US Patent No.
5,633,272),
deracoxib (US Patent No. 5,521,207), rofecoxib (US Patent No. 5,474,995),
etoricoxib (International Patent Application Publication No. WO 98/03484),
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-24-
JTE-522 (Japanese Patent Application Publication No. 9052882), or a
pharmaceutically acceptable salt or prodrug thereof;
(vii) Parecoxib (described in U.S. Patent No. 5,932,598), which is a
therapeutically effective prodrug of the tricyclic Cox-2 selective inhibitor
valdecoxib (described in U.S. Patent No. 5,633,272), in particular sodium
parecoxib;
(viii) ABT-963 (described in International Patent Application Publication No.
WO
00/24719)
(ix) Nimesulide (described in U.S. Patent No. 3,840,597), flosulide (discussed
in
J. Carter, Exp.Opin.Ther.Patents, 8(1 ), 21-29 (1997)), NS-398 (disclosed in
U.S. Patent No. 4,885,367), SD 8381 (described in U.S. Patent No.
6,034,256), BMS-347070 (described in U.S. Patent No. 6,180,651), S-2474
(described in European Patent Publication No. 595546) and MK-966
(described in U.S. Patent No. 5,968,974);
(x) The compounds and pharmaceutically acceptable derivatives described in
U.S. Patent No. 6,395,724, U.S. Patent No. 6,077,868, U.S. Patent No.
5,994,381, U.S. Patent No. 6,362,209, U.S. Patent No. 6,080,876, U.S.
Patent No 6,133,292, U.S. Patent No. 6, 369,275, U.S. Patent No.
6,127,545, U.S. Patent No. 6,130,334, U.S. Patent No. 6,204,387, U.S.
Patent No. 6,071,936, U.S. Patent No. 6,001,843, U.S. Patent No.
6,040,450, International Patent Application Publication No WO 96/03392,
International Patent Application Publication No WO 96/24585, U.S. Patent
No. 6,340,694, U.S. Patent No. 6,376,519, U.S. Patent No. 6,153,787, U.S.
Patent No. 6,046,217, U.S. Patent No. 6,329,421, U.S. Patent No.
6,239,137, U.S. Patent No. 6,136,831, U.S. Patent No. 6,297,282, U.S.
Patent No. 6,239,173, U.S. Patent No. 6,303,628, U.S. Patent No.
6,310,079, U.S. Patent No. 6,300,363, U.S. Patent No. 6,077,869, U.S.
Patent No. 6,140,515, U.S. Patent No. 5,994,379, U.S. Patent No.
6,028,202, U.S. Patent No. 6,040,320, U.S. Patent No. 6,083,969, U.S.
Patent No 6,306,890, U.S. Patent No. 6,307,047, U.S. Patent No. 6,004,948,
U.S. Patent No. 6,169,188, U.S. Patent No. 6,020,343, U.S. Patent No.
5,981,576, U.S. Patent No. 6,222,048, U.S. Patent No. 6,057,319, U.S.
Patent No. 6,046,236, U.S. Patent No. 6,002,014, U.S. Patent No.
5,945,539, U.S. Patent No. 6,359,182, International Patent Application
Publication No. WO 97/13755, International Patent Application Publication
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-25-
1 No. WO 96/25928, International Patent Application Publication No. WO
96/374679, International Patent Application Publication No. WO 95/15316,
International Patent Application Publication No. WO 95/15315, International
Patent Application Publication No. WO 96/03385, International Patent
Application No. WO 95/00501, International Patent Application No. WO
94/15932, International Patent Application Publication No. WO 95/00501,
International Patent Application Publication No. WO 94/27980, International
Patent Application Publication No. WO 96/25405, International Patent
Application Publication No. WO 96/03388, International Patent Application
Publication No. WO 96/03387, U.S. Patent No. 5,344,991, International
Patent Application Publication No. WO 95/00501, International Patent
Application Publication No. WO 96/16934, International Patent Application
Publication No. WO 96/03392, International Patent Application Publication
No. WO 96/09304, International Patent Application Publication No. WO
98/47890, and International Patent Application Publication No. WO
00/24719.
The contents of any of the patent applications, and in particular the general
formulae of
the therapeutically active compounds of the claims and exemplified compounds
therein,
are incorporated herein in their entirety by reference thereto.
Generally, the compounds of the present invention will be administered as a
formulation
in association with one or more pharmaceutically acceptable excipients. The
term
"excipient" is used herein to describe any ingredient other than the
compounds) of the
invention. The choice of excipient will to a large extent depend on factors
such as the
particular mode of administration, the effect of the excipient on solubility
and stability, and
the nature of the dosage form.
Pharmaceutical compositions suitable for the delivery of compounds of the
present
invention and methods for their preparation will be readily apparent to those
skilled in the
art. Such compositions and methods for their preparation may be found, for
example, in
"Remington's Pharmaceutical Sciences", 19t" Edition (Mack Publishing Company,
1995).
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-26-
Thus, according to another aspect of the present invention, there is provided
a
pharmaceutical formulation comprising a compound of formula (1) in admixture
with a
pharmaceutically acceptable adjuvant, diluent or carrier.
The compounds of the invention may be administered orally. Oral administration
may
involve swallowing, so that the compound enters the gastrointestinal tract, or
buccal or
sublingual administration may be employed by which the compound enters the
blood
stream directly from the mouth.
Formulations suitable for oral administration include solid formulations such
as tablets,
capsules containing particulates, liquids or powders, lozenges (including
liquid-filled),
chews, multi- and nano-particulates, gels, solid solution, liposome, films
(including muco-
adhesive), ovules, sprays and liquid formulations.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such
formulations
may be employed as fillers in soft or hard capsules and typically comprise a
carrier, for
example water, ethanol, polyethylene glycol, propylene glycol,
methylcellulose, or a
suitable oil, and one or more emulsifying agents and/or suspending agents.
Liquid
formulations may also be prepared by the reconstitution of a solid, for
example from a
sachet. .
The compounds of the invention may also be used in fast-dissolving, fast
disintegrating
dosage forms such as those described in Expert (pinion in Therapeutic Patents,
11 (6),
981-986 by Liang and Chen (2001 ).
a
For tablet dosage forms, depending on dose, the drug may make up from 1 wt% to
80
wt% of the dosage form, more typically from 5 wt% to 60 wt% of the dosage
form. In
addition to the drug, tablets generally contain a disintegrant. Examples of
disintegrants
include sodium starch glycolate, sodium carboxymethyl cellulose, calcium
carboxymethyl
cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl
cellulose,
microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose,
starch,
pregelatinised starch and sodium alginate. Generally, the disintegrant will
comprise from
1 wt% to 25 wt%, preferably from 5 wt% to 20 wt%, of the dosage form.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-27-
Binders are generally used to impart cohesive qualities to a tablet
formulation. Suitable
binders include microcrystalline cellulose, gelatin, sugars, polyethylene
glycol, natural and
synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl
cellulose and
hydroxypropyl methylcellulose. Tablets may also contain diluents, such as
lactose
(monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol,
xylitol,
dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic
calcium
phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium
lauryl sulfate
and polysorbate 80, and glidants such as silicon dioxide and talc. When
present, surface
active agents may comprise from 0.2 wt% to 5 wt% of the tablet, and glidants
may
comprise from 0.2 wt% and 1 wt% of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium
stearate,
zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate
with sodium
lauryl sulphate. Lubricants generally comprise from 0.25 wt% to 10 wt%,
preferably from
0.5 wt% to 3 wt%, of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring
agents,
preservatives and taste-masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 wt% to about 90
wt%
binder, from about 0 wt% to about 85 wt% diluent, from about 2 wt% to about 10
wt%
disintegrant, and from about 0.25 wt% to about 10 wt% lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet
blends or
portions of blends may alternatively be wet-, dry-, or melt-granulated, melt
congealed, or
extruded before tabletting. The final formulation may comprise one or more
layers and
may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in "Pharmaceutical Dosage Forms:
Tablets, Vol.
1", by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-
8247-
6918-X).
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-28-
Solid formulations for oral administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted- and programmed release.
Suitable modified release formulations for the purposes of the invention are
described in
US Patent No. 6,106,864. Details of other suitable release technologies such
as high
energy dispersions and osmotic and coated particles are to be found in Verma
et al,
Pharmaceutical Technology On-line, 25(2), 1-14 (2001). The use of chewing gum
to
achieve controlled release is described in WO 00/35298.
The compounds of the invention may also be administered directly into the
blood stream,
into muscle, or into an internal organ. Suitable means for parenteral
administration
include intravenous, intraarterial, intreperitoneal, intrathecal,
intraventricular, intraurethral,
intrasternal, intracranial, intramuscular and subcutaneous. Suitable devices
for parenteral
administration include needle (including microneedle) injectors, needle-free
injectors and
infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain
excipients such
as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to
9), but, for
some applications, they may be more suitab4y formulated as a sterile non-
aqueous
solution or as a dried form to be used in conjunction with a suitable vehicle
such as
sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for
example, by
lyophilisation, may readily be accomplished using standard pharmaceutical
techniques
well known to those skilled in the art.
The solubility of compounds of formula (I) used in the preparation of
parenteral solutions
may be increased by suitable processing, for example, the use of high energy
spray-dried
dispersions (see WO 01/47495) andlor by the use of appropriate formulation
techniques,
such as the use of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate
andlor
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted- and programmed release. Thus, compounds of the
invention may
be formulated as a solid, semi-solid, or thixotropic liquid for administration
as an
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-29-
implanted depot providing modified release of the active compound. Examples of
such
formulations include drug-coated stents and PGLA microspheres.
The compounds of the invention may also be administered topically to the skin
or
mucosa, either dermally or transdermally. Typical formulations for this
purpose include
gels, hydrogels, lotions, solutions, creams, ointments, dusting powders,
dressings, foams,
films, skin patches, wafers, implants, sponges, fibres, bandages and
microemulsions.
Liposomes may also be used. Typical carriers include alcohol, water, mineral
oil, liquid
petrolatum, white petrolatum, glycerin, polyethylene glycol. and propylene
glycol.
Penetration enhancers may be incorporated - see, for example, J. Pharm. Sci.,
88 (10),
955-958 by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by iontophoresis,
electroporation,
phonophoresis, sonophoresis and microneedle or needle-free (e.g.
PowderjectT"",
BiojectT"", etc.) injection.
Formulations for topical administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
The compounds of the invention can also be administered intranasally or by
inhalation,
typically in the form of a dry powder (either alone, as a mixture, for
example, in a dry
blend with lactose, or as a mixed component particle, for example, mixed with
phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an
aerosol
spray from a pressurised container, pump, spray, atomiser (preferably an
atomiser using
electrohydrodynamics to produce a fine mist), or nebuliser, with or without
the use of a
suitable propellant, such as 1,1,1,2-tetrafluoroethane or 1,1,1,2,3,3,3-
heptafluoropropane.
For intranasal use, the powder may comprise a bioadhesive agent, for example,
chitosan
or cyclodextrin.
The pressurised container, pump, spray, atomizer or nebuliser contains a
solution or
suspension of the compounds) of the invention comprising, for example,
ethanol,
aqueous ethanol, or a suitable alternative agent for dispersing, solubilising
or extending
release of the active, the propellants) as solvent and an optional surfactant,
such as
sorbitan trioleate, oleic acid, or an oligolactic acid.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-30-
Prior to use in a dry powder of suspension formulation, the drug product is
micronised to
a size suitable for delivery by inhalation (typically less than 5 microns).
This may be
achieved by any appropriate comminuting method, such a spiral jet milling,
fluid bed jet
milling, supercritical fluid processing to form nanoparticles, high pressure
homogenisation
or spray drying.
Capsules (made, for example, from gelatin or HPMC), blisters and cartridges
for use in an
inhaler or insufflator may be formulated to contain a powder mix of the
compound of the
invention, a suitable powder base such as lactose or starch and a performance
modifier
such as l-leucine, mannitol, or magnesium stearate. The lactose may be
anhydrous or in
the form of the monohydrate, preferably the latter. other suitable excipients
include
dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomiser using
electrohydrodynamics to
produce a fine mist may contain from 1 pg to 20mg of the compound of the
invention per
actuation and the actuation volume may vary from 1 p1 to 100 p1. A typical
formulation
may comprise a compound of formula (I), propylene glycol, sterile water,
ethanol and
sodium chloride. Alternative solvents which may be used instead of propylene
glycol
include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as
saccharin or
saccharin sodium, may be added to those formulations of the invention intended
for
inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be
immediate
and/or modified release using, for example, poly-DL-lactic-coglycolic acid
(PGLA).
Modified release formulations include delayed-, sustained-, pulsed-,
controlled-, targeted
and programmed release.
The compounds of the invention may be administered rectally or vaginally, for
example, in
the form of a suppository, pessary or enema. Cocoa butter is a traditional
suppository
base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be
immediate and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-31-
The compounds of the invention may also be administered directly to the eye or
ear,
typically in the form of drops of a micronised suspension or solution in
isotonic, pH-
adjusted, sterile saline. Other formulations suitable for ocular and aural
administration
include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and
non-
biodegradable (e.g. silicone) implants, wafers, lenses and particulate or
vesicular
systems, such as niosomes or liposomes. A polymer such as crossed-linked
polyacrylic
acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example,
hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a
heteropolysaccharide polymer, for example, gelan gum, may be incorporated
together
with a preservative, such as benzalkonium chloride. Such formulations may also
be
delivered by iontophoresis.,
Formulations for ocular/aural administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted, or programmed release.
The compounds of the invention may be combined with soluble macromolecular
entities
such as cyclodextrin or polyethylene glycol-containing polymers to improve
their solubility,
dissolution rate, taste-masking, bioavailability and/or stability for use in
any of the
aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for
most
dosage forms and administration routes. Both inclusion and non-inclusion
complexes
may be used. As an alternative to direct complexation with the drug, the
cyclodextrin may
~ be used as an auxiliary additive, i.e. as a carrier, diluent or solubiliser.
Most commonly
used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of
which
a
may be found in International Patent Applications Nos. WO 91/11172, WO
94/02518 and
W O 98/55148.
Inasmuch as it may be desirable to administer a combination of active
compounds, for
example, for the purpose of treating a particular disease or condition, it is
within the scope
of the present invention that two or more pharmaceutical compositions, at
least one of
which contains a compound in accordance with the invention, may conveniently
be
combined in the form of a kit suitable for coadministration of the
compositions.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-32-
Thus the kit of the invention comprises two or more separate pharmaceutical
compositions, at least one of which contains a compounds of formula (I) in
accordance
with the invention, and means for separately retaining said, compositions,
such as a
container, divided bottle, or divided foil packet. An example of such a kit is
the familiar
blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different
dosage forms, for
example, oral and parenteral, for administering the separate compositions at
different
dosage intervals, or for titrating the separate compositions against one
another. To assist
compliance, the kit typically comprises directions for administration and may
be provided
with a so-called memory aid.
For administration to human patients, the total daily dose of the- compounds
of the
invention will typically be in the range of from about 0.01 to about 15 mg/kg
of body
weight, depending on the mode of administration. The total daily dose may be
administered in a single dose or divided doses throughout the day. These
dosages are
based on an average human subject having a weight of about 65kg to 70kg. The
physician will readily be able to determine doses for subjects whose weight
falls outside
this range, such as infants and the elderly.
As used herein, the terms "treating" and "to treat", mean to alleviate
symptoms, eliminate
the causation either on a temporary or permanent basis, or to prevent or slow
the
appearance of symptoms. The term "treatment" includes alleviation, elimination
of
causation (either on a temporary or permanent basis) of, or prevention of
symptoms and
disorders associated with primary and/or secondary dysmenorrhoea. The
treatment may
be a pre-treatment as well as a treatment at the on-set of symptoms.
The compounds of the present invention may be tested in the screens set out
below:
1.0 VIA Filter Binding Assay
1.1 Membrane Preparation
Receptor binding assays were performed on cellular membranes prepared from CHO
cells stably expressing the human VIA receptor, (CHO-hV~A). The CHO-hV~A cell
line was
kindly provided under a licensing agreement by Marc Thibonnier, Dept. of
Medicine, Case
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-33-
Western,, Reserve University School of Medicine, Cleveland, Ohio. CHO-hV~A
cells were
routinely maintained at 37°C in humidified atmosphere with 5% C02 in
DMEM/Hams F12
nutrient mix supplemented with 10 % fetal bovine serum, 2 mM L-glutamine, 15
mM
HEPES and 400 pg/ml 6418. For bulk production of cell pellets, adherent CHO-
hV~A cells
were grown to confluency of 90-100% in 850 cm2 roller bottles containing a
medium of
DMEM/Hams F12 Nutrient Mix supplemented with 10 % fetal bovine serum, 2 mM L-
glutamine and 15 mM HEPES. Confluent CHO-hV~A cells were washed with phosphate-
buffered saline (PBS), harvested into ice cold PBS and centrifuged at 1,000
rpm. Cell
pellets were stored at -80°C until use. Cell pellets were thawed on ice
and homogenised
in membrane preparation buffer consisting of 50 mM Tris-HCI, pH 7.4, 5 mM
MgCl2 and
supplemented with a protease inhibitor cocktail, (Roche). The cell homogenate
was
centrifuged at 1000 rpm, 10 min, 4°C and the supernatant was removed
and stored on
ice. The remaining pellet was homogenised and centrifuged as before. The
supernatants
were pooled and centrifuged at 25,000 x g for 30 min at 4°C. The pellet
was resuspended
in freezing buffer consisting of 50 mM Tris-HCI, pH 7.4, 5 mM MgClz and 20 %
glycerol
and stored in small aliquots at -80°C until use. Protein concentration
was determined
using Bradford reagent and BSA as a standard.
1.2 V,a Filter binding
Protein linearity followed by saturation binding studies were performed on
each new batch
of membrane. Membrane concentration was chosen that gave specific binding on
the
linear portion of the curve. Saturation binding studies were then performed
using various
concentrations of [3H]-arginine vasopressin, [3H]-AVP (0.05 nM - 100 nM) and
the Kd and
Bmax determined.
Compounds were tested for their effects on [3H]-AVP binding to CHO-hV~A
membranes,
(3H-AVP; specific activity 65.5 Ci / mmol; NEN Life Sciences). Compounds were
solubilised in dimethylsulfoxide (DMSO) and diluted to working concentration
of 10%
DMSO with assay buffer containing 50 mM Tris-HCL pH 7.4, 5 mM MgCh and 0.05%
BSA. 25 p1 compound and 25 p1 [3H]-AVP, (final concentration at or below Kd
determined
for membrane batch, typically 0.5 nM - 0.6 nM) were added to a 96-well round
bottom
polypropylene plate. The binding reaction was initiated by the addition of 200
p1
membrane and the plates were gently shaken for 60 min at room temperature. The
reaction was terminated by rapid filtration using a Filtermate Cell Harvester
(Packard
Instruments) through a 96-well GF/B UniFilter Plate which had been presoaked
in 0.5%
polyethyleneimine to prevent peptide sticking. The filters were washed three
times with 1
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-34-
ml ice cold wash buffer containing 50 mM Tris-HCL pH 7.4 and 5 mM MgCh. The
plates
were dried and 50 p1 Microscint-0 (Packard instruments) was added to each
well. The
plates were sealed and counted on a TopCount Microplate Scintillation Counter
(Packard
Instruments). Non-specific binding (NSB) was determined using 1 pM unlabelled
d(CH2)STyr(Me)AVP (((3-mercapto-[3,(3-cyclopentamethylenepropionyl,0-Me-
Tyrz,Arg$]-
vasopressin ) ((3MCPVP), (Sigma). The radioligand binding data was analysed
using a
four parameter logistic equation with the min forced to 0%. The slope was free
fitted and
fell between -0.75 and -1.25 for valid curves. Specific binding was calculated
by
subtracting the mean NSB cpm from the mean Total cpm. For test compounds the
amount of ligand bound to the receptor was expressed as % bound = (sample cpm -
mean NSB cpm)lspecific binding cpm x100. The % bound was plotted against the
concentration of test compound and a sigmoidal curve was fitted. The
inhibitory
dissociation constanfi (K) was calculated using the Cheng-Prusoff equation:
f~C=IC50/(1+[L]/Kd) where [L] is the concentration of ligand present in the
well and Kd is the
dissociation constant of the radioligand obtained from Scatchard plot
analysis.
2.0 VIA Functional Assay; Inhibition of AVP I V,A-R mediated Ca2+ mobilization
by
FLIPR (Fluorescent Imaaina Plate Reader) (Molecular Devices)
Intracellular calcium release was measured in CHO-hV~A cells using FLIPR,
which allows
the rapid detection of calcium following receptor activation. The CHO-hV~A
cell line was
kindly provided under a licensing agreement by Marc Thibonnier, Dept. of
Medicine, Case
Western Reserve University School of Medicine, Cleveland, Ohio. CHO-VIA cells
were
routinely maintained at 37°C in humidified atmosphere with 5% COZ in
DMEM/Hams F12
nutrient mix supplemented with 10 % fetal bovine serum, 2 mM L-glutamine, 15
mM
HEPES and 400 pg/ml 6418. On the afternoon before the assay cells were plated
at a
density of 20,000 cells per well into black sterile 96-well plates with clear
bottoms to allow
cell inspection and fluorescence measurements from the bottom of each well.
Wash
buffer containing Dulbecco's phosphate buffered saline (DPBS) and 2.5 mM
probenecid
and loading dye consisting of cell culture medium containing 4 NM Fluo-3-AM
(dissolved
in DMSO and pluronic acid),(Molecular Probes) and 2.5 mM probenecid was
prepared
fresh on the day of assay. Compounds were solubilised in DMSO and diluted in
assay
buffer consisting of DPBS containing 1 % DMSO, 0.1 % BSA and 2.5 mM
probenecid. The
cells were incubated with 100 p1 loading dye per well for 1 hour at
37°C in humidified
atmosphere with 5% CO2. After dye loading the cells were washed three times in
100 p1
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-35-
wash buffer using a Denley plate washer. 100 p1 wash buffer was left in each
well.
Intracellular fluorescence was measured using FLIPR. Fluorescence readings
were
obtained at 2s intervals with 50 NI of the test compound added after 30s. An
additional
155 measurements at 2s intervals were then taken to detect any compound
agonistic
activity. 50 p1 of arginine vasopressin (AVP) was then added so that the final
assay
volume was 200 p1. Further fluorescence readings were collected at 1 s
intervals for 120s.
Responses were measured as peak fluorescence intensity (Fl). For
pharmacological
characterization a basal FI was subtracted from each fluorescence response.
For AVP
dose response curves, each response was expressed as a % of the response to
the
highest concentration of AVP in that row. For ICSO determinations , each
response was
expressed as a % of the response to AVP. 1C50 values were converted to a
modified Kb
value using the Cheng-Prusoff equation which takes into account the agonist
concentration, [A], the agonist EC5o and the slope: Kb=ICSO/(2+[A]/A5o]")'~"-1
where [A] is
the concentration of AVP, A5~ is the ECSO of AVP from the dose response curve
and
n=slope of the AVP dose response curve.
The compounds of the invention may have the advantage that they are more
potent, have
a longer duration of action, have a broader range of activity, are more
stable, have fewer
side effects or are more selective, or have other more useful properties than
the
compounds of the prior art.
The invention is illustrated by the following preparations and examples:
Preparation 1: 2-[4-(5-Methyl-[1,3,4]oxadiazol-2-yl)-piperidin-1-yl]-
pyrimidine
N N~N
C'>-N;
N o cH3
4-(5-Methyl-[1,3,4]oxadiazol-2-yl)-piperidine hydrochloride (203 mg, 1 mmol)
(see
reference WO 0039125, preparation 43) was added to a solution of 2-
bromopyrimidine
(207 mg, 1.3 mmol) and potassium carbonate (207 mg, 1.5 mmol) in N,N-
dimethylformamide (0.5 mL). The solution was heated to 60°C for 4
hours, before cooling
and stirring at room temperature for 18 hours. The solvent was evaporated
under
reduced pressure, and the residue partitioned between water and
dichloromethane. The
aqueous layer was extracted with dichloromethane, and the combined organic
layers
were washed with brine, before being dried over magnesium sulphate and
evaporated
under reduced pressure. The residue was purified by chromatography on silica
gel (25 g)
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-36-
eluting with dichloromethane and methanol (97.5:2.5) to give the title
compound as a
crystalline solid (75 mg).
'H NMR (400MHz, CDCI3): 8 1.76-1.92 (m, 2H), 2.02-2.17 (m, 2H), 2.45 (s, 3H),
3.05-3.19
(m, 3H), 4.63-4.76 (m, 2H), 6.44 (t, 1 H), 8.26 (d, 2H); LRMS: mlz ES+ 246
[M+H]+
Preparation 2: 3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid
0
N
~~---~OH
N
Sodium hydroxide solution (5M, 24.8 ml, 0.12 mol) was added drop wise to a
solution ofi 3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid ethyl
ester (5.8
g, 24 mmol)(see reference Farmaco, 1993, 48(10), 1439) in 1,4-dioxane (100
mL).
The mixture was stirred at room temperature for 72 hours and then evaporated ,
under reduced pressure. The residue was purified by ion exchange
chromatography on Dowex~ 50 WX8 resin using methano1:0.88 ammonia:water as
eluant (gradient from 0:0:100 to 0:5:95 to 5:5:90). The material obtained was
triturated with diethyl ether to give the title compound (4.42 g); LCMS: m/z
ES*
288 [M+H]+
Preparation 3: 3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid N'-(2-
morpholin-4
yl-acetyl)-hydrazide
O H
I
~NiN
\ N\~ I ~N
H O
i N
The acid of Preparation 2 (0.5 g, 2.4 mmol) was suspended in dichloromethane
(20 mL)
containing N,N-dimethylformamide (2 drops) and oxalyl chloride (1.27 mL, 14
mmol) in
dichloromethane (5 mL) was added dropwise. The mixture was stirred for 2 hours
at
room temperature and then was evaporated under reduced pressure. The yellow
solid
was suspended in dichloromethane (5 mL) and N-methylmorpholine (0.32 mL, 2.9
mmol)
added cautiously. Morpholin-4-yl-acetic acid hydrazide (462 mg, 2.9 mmol) (see
reference Bull. Soc. Chim. Fr. 1962, 250) was added, and the mixture was then
stirred at
room temperature for 18 hours. The reaction was diluted into dichloromethane
(100 mL)
and washed with aqueous sodium hydrogen carbonate. The aqueous layer was
extracted
with dichloromethane and the combined organic layers were dried over magnesium
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-37-
sulphate, filtered and evaporated under reduced pressure. The residue was
triturated
with ethyl acetate, to give the title compound as a solid (306 mg)
'H NMR (400MHz, CDCI3): 8 1.94 (m, 4H), 2.60 (s, 5H), 2.79 (s, 2H), 2.99 (m,
2H), 3.72
(m, 4H), 4.33 (d, 2H), 6.63 (m, 1 H), 6.73 (d, 1 H), 7.51 (m, 1 H), 8.14 (m, 1
H), 8.82 (s, 1 H),
9.22 (broad s, 1 H); LRMS m/z ES+ 348 [M+H]+
Preparation 4: 4-(5-Morpholin-4-ylmethyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-
tetrahydro-2H
[1,2']bipyridinyl
I
-o
N N
I ,1' ~o
Phosphorus oxychloride (1.0 mL, 10.73 mmol) was added to the hydrazide of
Preparation
3 (200 mg, 0.58 mmol) and the suspension was heated to 110°C for 90
minutes. After
cooling, acetonitrile was added to give a brown solution to which water was
added, and
then basified using aqueous sodium carbonate. The mixture was then extracted
with
ethyl acetate (2 x 100 mL), and the combined extracts dried over magnesium
sulphate
and evaporated under reduced pressure. The residue was crystallised from ethyl
acetate
and pentane to give the title compound as a sandy coloured solid.
'H NMR (400MHz, CD30D): b 1.91 (m, 2H), 2.16 (m, 2H), 2.57 (m, 4H), 3.11 (m,
2H),
3.30 (m, 1 H), 3.70 (m, 4H), 3.80 (s, 2H), 2.24 (m, 2H), 6.64 (m, 1 H), 6.86
(m, 1 H), 7.56
(m, 1 H), 8.05 (m, 1 H); LRMS m/z ES+ 330 [M+H]+
Preparation 5: 3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid N'-
acetyl
hydrazide
0
H
I ~ N~H~N CHa
~N
O
Oxalyl chloride (17 mL, 0.18 mol) was added drop wise to a suspension of the
acid from
Preparation 2 (8.0 g, 38.79 mmol) in dichloromethane (150 mL) at 0°C.
The mixture was
stirred at room temperature for 2.5 hours. Oxalyl chloride and dichloromethane
were
evaporated under reduced pressure. The residue was dissolved in
dichloromethane (200
mL) and acetic hydrazide (3.45 g, 45.57 mmol) was added in small portions,
followed by
N-methylmorpholine (4.26 mL, 38.75 mmol). The mixture was then stirred at room
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-38-
temperatGre fior 18 hours. The mixture was evaporated under reduced pressure,
and the
residue partitioned between ethyl acetate and aqueous hydrochloric acid (3M).
The
aqueous layer was washed with ethyl acetate, then basified to pH10 with sodium
hydroxide pellets before extracting with ethyl acetate. The combined organic
layers were
evaporated under reduced pressure, and the resulting white solid was
triturated with
diethyl ether to give the title compound (4.6 g).
'H NMR (400MHz, CD30D): 8 1.70-1.82 (m, 2H), 1.90 (m, 2H), 1.98 (s, 3H), 2.53
(m, 1H),
2.90 (t, 2H), 4.29 (d, 2H), 6.63 (m, 1 H), 6.83 (d, 1 H), 7.53 (m, 1 H), 8.05
(m, 1 H); LRMS:
m/z APCI'~ 263 [M+H]+
Preparation 6: 4-(5-Methyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl
N._N
/ ~ N
O CHs
N
The title compound was obtained as a yellow solid (1,38 g, 82% yield) from the
hydrazide
of Preparation 5 (1.81 g, 6.9 mmol) and phosphorus oxychloride (12 mL, 128
mmol)
following the procedure described in Preparation 4. The residue obtained was
purifiied by
flash chromatography using a 20 g Isolute° cartridge, eluting with a
solvent gradient of
dichloromethane:methanol (100:0 to 95:5 by volume).
'H NMR (4001VIHz, CD30D): 8 1.88 (m, 2H), 2.15 (m, 2H), 2.52 (s, 3H), 3.09 (t,
2H), 3.23
(m, 1 H), 4.27 (d, 2H), 6.66 (m, 1 H), 6.86 (d, 1 H), 7.55 (m, 1 H), 8.08 (m,
1 H); LRMS: m/z
APCI+ 245 [M+H]+
Preparation 7: 3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid N'-
pentanoyl
hydrazide
0
-NH
N~ HN
~~~CH3
IIIf IT '' _O
The carboxylic acid from Preparation 2 (1.5 g, 7.3 mmol) was suspended in
dichloromethane (40 mL) containing N,N-dimethylformamide (2 drops) and oxalyl
chloride
(1.27 mL, 14 mmol) in dichloromethane (5 mL) was added dropwise. The mixture
was
stirred for 5 hours at room temperature and then evaporated under reduced
pressure.
The residue was suspended in hexane and evaporated (3x20 mL). The residue was
dissolved in dichloromethane and cooled to 0°C and pentanoic acid
hydrazide (1.7 g, 14.6
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-39-
mmol) was added. 1-Methyl-pyrrolidin-2-one (1.6 mL, 14.6 mmol) in
dichloromethane (10
mL) was added drop wise and the mixture was stirred at room temperature for 16
hours.
The solvent was evaporated under reduced pressure and the residue was
triturated with
diethyl ether. The material obtained was dissolved in water and acidified to
pH 2 by
addition of 2N hydrochloric acid. The acidic solution was filtered and the
filtrate was
washed with ethyl acetate (3x20 mL) then basified with sodium carbonate. The
solid
formed was triturated with diethyl ether and isolated by filtration to give
the title compound
as a white solid (0.68 g).
LCMS: m/z ES- 303 [M-H]-
Preparation 8: 4-(5-Butyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-tetrahydro-2H-
[1,2']bipyridinyl
/ N-.N
/ ~ ~
N~~~C~%~CHs
The hydrazide from Preparafiion 7 (2.5 g, 8.71 mmol) was combined with
phosphorus
oxychloride (25 mL) and heated to 100°C for 2 hours, after which time
the mixture was
poured onto iced water with care. The mixture was then extracted into ethyl
acetate, and
then dried over magnesium sulphate. The residue was evaporated under reduced
pressure to give a beige solid which was triturated with ethyl acetate, and
then purified by
chromatography on silica gel, eluting with a gradient~of ethyl acetate:pentane
(20:80 to
25:75). The title compound was obtained as an off-white solid (500 mg).
LRMS: m/z ES+ 287 [M+H]+
Preparation 9: 3,4,5,6-Tetrahydro-2H [1,2']bipyridinyl-4-carboxylic acid
hydrazide
0
N~\__~~
N H-NHx
3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid ethyl ester (35 g,
150 mmol)(see
reference Farmaco, 1993, 48(10), 1439) was dissolved in methanol (700 mL)
containing
hydrazine hydrate (34 mL, 150 mmol) and was heated under reflux for 18 hours.
After this
time, more hydrazine hydrate (22.7 mL, 100 mmol) was added and the mixture
stirred for
a further 18 hours. The mixture was cooled to room temperature and evaporated
under
reduced pressure. The solid formed was triturated with ethyl acetate to give
the title
compound as a white solid.
'H NMR (400MHz, CD30D): 8 1.99 (m, 4H), 2.20 (m, 1H), 2.85 (t, 2H), 4.50 (d,
2H), 6.30
(m, 1 H), 7.30 (d, 1 H), 7.70 (t, 1 H), 8.40 (d, 1 H).
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-40-
Preparation 10: 3,4,5,6-Tetrahydro-2H-[1,2']bipyridinyl-4-carboxylic acid N'-
(2-chloro
acetyl)-hydrazide
0
N r ~N.r-N
N ~CI
The hydrazide of Preparation 9 (23.6 g, 0.11 mol) was suspended in
dichloromethane
(500 mL) and 4-methylmorpholine (17.7 mL, 0.16 mol) was added. The mixture was
cooled using an ice bath and chloroacetyl chloride (12.8 mL, 0.16 mol) was
added
dropwise. The reaction was warmed to room temperature and was stirred for 3
hours.
The solid formed was isolated by filtration, washed with dichloromethane and
diethyl
ether, and dried under vacuum to give the title compound (20.4 g).
LCMS: m/z ES+ 297 [M+H]+
Preparation 11: 4-(5-Chloromethyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-tetrahydro-2H
[1,2']bipyridinyl
N~N
N~~~p~Cl
N
The hydrazide of Preparation 10 (20.4 g, 69 mmol) was suspended in phosphorus
oxychloride (150 mL) at 100°C for 4 hours. The mixture was cooled and
the solvent was
evaporated under reduced pressure. The residue was dissolved in ethyl acetate
and was
added to water. The aqueous layer was basified by addition of solid sodium
hydrogen
carbonate and the phases were separated. The aqueous phase was extracted with
ethyl
acetate (x2) and the combined organic layers were dried over magnesium
sulphate and
evaporated under reduced pressure. The material isolated was triturated with
diethyl
ether to give the title compound as a beige solid (15 g).
'H NMR (400MHz, CD30D): 8 1.91 (m, 2H), 2.19 (m, 2H), 3.14 (m, 2H), 3.30 (m,
1H),
4.29 (m, 2H), 4.86 (s, 2H), 6.69 (m, 1 H), 6.89 (d, 1 H), 7.58 (m, 1 H), 8.08
(d, 1 H)
Preparation 12: 4-(5-Piperidin-1-ylmethyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-
tetrahydro-2H
[1,2']bipyridinyl
/ N~~O~N
N
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-41-
The chloromethyl compound of Preparation 11 (0.5 g, 1.8 mmol) was added to
piperidine
(0.18 mL, 1.8 mmol) and potassium carbonate (0.5 g, 3.6 mmol) in N,N-
dimethylformamide (8 mL) and the mixture was heated at 60°C for 3
hours. The reaction
mixture was evaporated under reduced pressure and the residue was partitioned
between
water and ethyl acetate. The organic solution was washed with water and then
with 2N
hydrochloric acid and the combined aqueous solutions were basified with solid
sodium
hydrogen carbonate. The aqueous mixture was extracted with ethyl acetate (x3)
and the
combined organic layers were dried over magnesium sulphate and evaporated
under
reduced pressure. The residue was purified by chromatography on silica gel
using
methano1:0.88 ammonia:dichloromethane as eluant (2:0.25:98) to give the title
compound as a pale pink solid (0.48 g).
LCMS: m/z ES* 328 [M+H]* : --
Preparation 13: 4-(5-[1,2,3]Triazol-2-ylmethyl-[1,3,4]oxadiazol-2-yl)-3,4,5,6-
tetrahydro-
2H-[1,2']bipyridinyl
NON
N~WO~Nv
N N
The chloromethyl compound of Preparation 11 (5 g, 17.9 mmol) was combined with
potassium carbonate (4.97 g, 36.0 mmol), 1,2,3-triazole (1.04 mL, 17.9 mmol)
and N,N-
dimethylformamide (70 mL), and then heated to 60°C with stirring for 4
hours. The
solvent was evaporated under reduced pressure and the residue was partitioned
between
dichloromethane and water. The aqueous layer was washed with dichloromethane
and
the combined organic layers were dried over magnesium sulphate and then
evaporated
under reduced pressure to give an orange oil. This oil was purified by flash
chromatography on silica gel eluting with a solvent gradient of
dichloromethane:methanol:
0.88 ammonia (98:2:0.25 to 97:3:0.25 by volume). The title compound was
obtained after
trituration from diethyl ether as a white solid (2.0g).
'H NMR (400MHz, CD30D): 8 1.82 (m, 2H), 2.11 (m, 2H), 3.07 (t, 2H), 3.23 (m,
1H), 4.22
(d, 2H), 4.80 (s, 2H), 6.64 (d, 1 H), 6.84 (d, 1 H), 7.54 (t, 1 H), 7.76 (s,
2H), 8.07 (d, 1 H).
LRMS: m/z APCI* 312 [M+H]+
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-42-
t t
Example 1: 2-{4-[4-(2-Ethyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-piperidin-
1-yl}-
pyrimidine
N~N
~--CH3
'N
N / CH3
~N w
The oxadiazole of Preparation 1 (100 mg, 0.41 mmol) was combined with 2-
ethylphenylamine (0.075 mL, 0.61 mmol), and magnesium (II) chloride (10 mg,
0.10
mmol) in a sealed vessel and heated to 150°C for 18 hours. The reaction
mixture was
dissolved in dichloromethane and washed with citric acid (10% aqueous), and
sodium
hydrogen carbonate (saturated aqueous solution). The organic layer was
evaporated
under reduced pressure, and the residue was purified on silica gel using Isco
Systems
Combiflash~ Sg100c on an Isco Systems Redisep 10g cartridge, eluting with
dichloromethane and methanol (gradient from 100:0 to 95:5). This afforded the
title
compound (43.6 mg).
'H NMR (400MHz, CDCI3): s 1.15 (t, 3H), 1.76-1.89 (m, 3H), 2.00-2.12 (m, 1 H),
2.16 (s,
3H), 2.28 (q, 2H), 2.56 (m, 1 H), 2.85 (q, 2H), 4.71 (dd, 2H), 6.42 (t, 1 H),
7.09 (d, 1 H),
7.37 (t, 1 H), 7.49 (m, 2H), 8.24 (d, 2H); LRMS: m/z ES+ 349 [M+H]+
Examples 2 to 7:
The compounds of the following tabulated examples of the general formula:
were prepared by a similar method to that of Example 1 using the appropriate
oxadiazole
starting material, and the corresponding anilines.
Eg. R R X Analytical Data
2 CH3 ~cH N 'H NMR (400MHz, CDCI3): s 0.89
3 (t, 3H),
1.54 (m, 2H), 1.75-1.87 (m, 4H),
2.17 (s, 3H),
2.23 (t, 2H), 2.55 (m, 1 H), 2.79-2.90
(m, 2H),
4.65-4.79 (dd, 2H), 6.42 (t, 1
H), 7.09 (d, 1 H),
7.37 (t, 1 H), 7.47 (m, 2H), 8.25
(d, 2H).
LRMS m/z ES+ 363 [M+H]+
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-43-
3' CH3 ~CH3 N 'H NMR (400MHz, CDCI3): 8 1.17
Y (dd, 6H),
I 1.72-1.88 (m, 4H), 2.18 (s, 3H),
2.43 (m, 1 H),
CH3
2.52 (m, 1 H), 2.77-2.90 (m, 2H),
4.67 (d, 1 H),
4.80 (d, 1 H), 6.43 (t, 1 H),
7.06 (d, 1 H), 7.36
(t, 1 H), 7.53 (m, 2H), 8.26 (d,
2H).
LRMS m/z ES+ 363 [M+H]+
4' ~C H C 'H NMR (400MHz, CD30D): 8 1.84
(m, 4H),
2.31 (m, 4H), 2.74 (m, 2H), 2.83
(m, 1 H),
3.50 (s, 6H), 4.24 (d, 2H), 6.62
(m, 1 H), 6.81
(d, 1 H), 7.49 (m, 3H), 7.61 (m,
3H), 8.02 (m,
1 H).
LRMS m/z ES+ 405 [M+H]+
~ H C 'H NMR (400MHz, CD30D): 8 0.84
(t, 3H),
cH 1.28 (m, 2H), 1.54 (m, 2H), 1.89
3 (m, 4H),
2.59 (t, 2H), 2.79 (m, 3H), 4.27
(m, 2H), 6.63
(t, 1 H), 6.80 (d, 1 H), 7.45
(m, 2H), 7.51 (t,
1 H), 7.64 (m, 3H), 8.02 (d, 1
H).
LRMS m/z ES+ 384 [M+Na]+
H C 'HNMR (400MHz, CD30D): S 1.33-1.44
(m,
6H), 1.90 (m, 4H), 2.27 (m, 4H),
2.76 (m, 2H),
2.86 (m, 1 H), 3.44 (s, 2H), 4.27
(m, 2H), 6.62
(t, 1 H), 6.81 (d, 1 H), 7.47-7.54
(m, 3H), 7.61
(m, 3H), 8.02 (d, 1 H).
LRMS m/z ES+403 [M+H]+
T' CH3 H C 'HNMR (400MHz, CD30D): 8 1.85
(m, 4H),
2.25 (s, 3H), 2.70-2.85 (m, 3H),
4.25 (d, 2H),
6.62 (m, 1 H), 6.80 (d, 1 H),
7.44-7.55 (m, 3H),
7.62-7.69 (m, 3H), 8.03 (d, 1
H).
LRMS m/z ES+ 320 [M+H]+
ivon sewed vessel
Z 3 equivalents of aniline. Non-sealed vessel, 4 hours heating. Product was
triturated
from ethyl acetate.
3 3 equivalents of aniline (1.26 mmol), 15 mg of MgCl2 used. Non-sealed
vessel. After
5 chromatography product was triturated from diethyl ether.
4 equivalents of aniline used (1.24 mmol). Non-sealed vessel. After
chromatography
product was crystallised from ethyl acetate:pentane.
2 equivalents of aniline used (2.26 mmol)
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-44-
Example 8: 4-[4-(4-Methoxy-2-methyl-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-
3,4,5,6
tetrahydro-2H-[1,2']bipyridinyl
The oxadiazole of Preparation 6 (250 mg, 1.025 mmol) was combined with 4-
methoxy-2-
methylaniline (0.39 mL, 3.07 mmol), p-toluenesulphonic acid monohydrate (50
mg, 0.26
mmol), and xylene (3 mL). The mixture was heated to 150°C with stirring
for 18 hours.
The solvent was then evaporated under reduced pressure, and then purified by
flash
chromatography on silica gel, eluting with ethyl acetate:methanol (90:10). The
title
compound was obtained as a colourless gum (257 mg) after trituration with
diethyl ether.
'H NMR (400MHz, CD3OD): b 1.81 (m, 2H), 1.90 (m, 2H), 1.97 (s, 3H), 2.12 (s,
3H), 2.63
(m, 1 H), 2.79 (m, 2H), 3.85 (s, 3H), 4.24 (m, 2H), 6.61 (m, 1 H), 6.79 (d, 1
H), 6.98 (m,
1 H), 7.04 (s, 1 H), 7.33 (d, 1 H), 7.51 (m, 1 H), 8.03 (m, 1 H); LRMS: m/z
ES+ 364 [M+H]+
Examples 9 to 20:
The compounds of the following tabulated examples of the general formula:
were prepared by a similar method to that of Example 8, as described in the
footnotes to
the table below, using the appropriate oxadiazole starting material and the
corresponding
anilines.
Eg. R R2 R Analytical Data
9b CH3 CH3 CI 'HNMR (400MHz, CD30D): S 1.82 (m,
2H), 1.96
(m, 2H), 2.01 (s, 3H), 2.14 (s, 3H),
2.63 (m, 1 H),
2.78 (m, 2H), 4.25 (br m, 2H), 6.61
(m, 1 H), 6.90
(d, 1 H), 7.35 (d, 1 H), 7.45 (m,
1 H), 7.50 (m, 1 H),
7.56 (m, 1 H), 8.03 (m, 1 H).
LRMS m/z 368 [M+H]+
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-45-
10' CH3 CH3 H 'HNMR (400MHz, CD3OD): 8 1.76-2.00
(m, 4H),
2.03 (s, 3H), 2.16 (s, 3H), 2.64 (m,
1 H), 2.74 (m,
2H), 4.25 (m, 2H), 6.62 (t, 1 H),
6.79 (d, 1 H), 7.34
(d, 1 H), 7.44-7.55 (m, 4H), 8.04
(d, 1 H).
LRMS m/z 334 [M+H]+
11' CH3 H CI 'HNMR (400MHz, CD30D): 8 1.87 (m,
4H), 2.23 (s,
3H), 2.75-2.83 (m, 3H), 4.26 (m, 2H),
6.62 (t, 1 H),
6.80 (d, 1 H), 7.47 (d, 2H), 7.52
(t, 1 H), 7.66 (d,
2H), 8.04 (d, 1 H).
LRMS m/z 354 [M+H]+
12' N~ H OCH3 'HNMR (400MHz, CD3OD): s 1.87 (m,
, 4H), 2.76
~N~N (m, 3H), 3.85 (s, 3H), 4.25 (m, 2H),
5.65 (s, 2H),
6.62 (t, 1 H), 6.79 (d, 1 H), 7.02
(d, 2H), 7.15 (d,
2H), 7.51 (t, 1 H), 7.59 (s, 2H),
8.03 (d, 1 H).
LRMS m/z 312 [M+H]+
13 CH3 H OCH3 'HNMR (400MHz, CD~OD): s 1.87 (m,
4H), 2.21 (s,
3H), 2.73-2.84 (m, 3H), 3.89 (s, 3H),
4.26 (m, 2H),
6.62 (t, 1 H), 6.80 (d, 1 H), 7.15
(d, 2H), 7.34 (d,
2H), 7.51 (t, 1 H), 8.03 (d, 1 H).
LRMS m/z 350 [M+H]+
14 N~ CH3 H 'HNMR (400MHz, CD30D): s 1.72-1.85
(m, 3H),
~N~N 1.88 (s, 3H), 2.04 (m, 1 H), 2.61
(m, 1 H), 2.68-2.81
(m, 2H), 4.20-4.30 (m, 2H), 5.55-5.68
(m, 2H), 6.62
(t, 1 H), 6.79 (d, 1 H), 7.15 (d,
1 H), 7.34 (t, 1 H), 7.42
(m, 1 H), 7.46-7.53 (m, 2H), 7.56
(s, 2H), 8.04 (d,
1 H).
LRMS m/z 401 [M+H]+
15 N~ CH3 CI 'HNMR (400MHz, CD30D): 8 1.75-1.88
> (m, 6H),
~N~N 2.03 (m, 1 H), 2.62 (m, 1 H), 2.71-2.85
(m, 2H),
4.21-4.31 (m, 2H), 5.59-5.70 (m, 2H),
6.63 (t, 1 H),
6.80 (d, 1 H), 7.17 (d, 1 H), 7.35
(d, 1 H), 7.45 (s,
1 H), 7.52 (t, 1 H), 7.58 (s, 2H),
8.03 (d, 1 H).
LRMS m/z 435 [M+H]+
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-46-
16 N~ H H 'HNMR (400MHz, CDsOD): 8 1.88 (m,
4H), 2.71-
~N~N> 2.83 (m, 3H), 4.25 (m, 2H), 5.67 (s,
2H), 6.62 (t,
1 H), 6.79 (d, 1 H), 7.25 (d, 2H),
7.49-7.56 (m, 6H),
8.03 (d, 1 H).
LRMS m/z 387 [M+H]+
17 N~ H CI 'HNMR (400MHz, CDsOD): 8 1.87 (m,
, 4H), 2.74-
~N~N 2.83 (m, 3H), 4.25 (m, 2H), 5.69 (s,
2H), 6.62 (t,
1 H), 6.80 (d, 1 H), 7.26 (d, 2H),
7.51 (m, 3H),), 7.58
(s, 2H), 8.03 (d, 1 H).
LRMS m/z 421 [M+H]+
18~ CHs H CHs Retention time 0.30 minutes
19' CHs CHs CHs LRMS m/z ES+ 373.99 [M+Na]+
Retention time 0.30 minutes'o
20" ~o CHs CI 'HNMR (400MHz, DMSO-d6): 8 1.45-1.90
(m, 4H),
2.13 (m, 2H), 2.21 (m, 2H), 2.55 (m,
1 H), 2.70 -
2.87 (m, 2H), 3.20-3.39 (m, 3H), 3.28
(s, 3H), 4.21
(m, 5H), 6.56 (m, 1 H), 6.79 (m, 1
H), 7.44 (m, 3H),
_ 7.58 (s, 1 H), 8.06 (m, 1 H).
LRMS m/z 453 [M+H]+
a m i . of xyene used and heated for 18 hours, then volume reduced to 3 mL and
heated for a
further 12 hours.
2 mL of xylene used, 20 mg of p-toluene sulphonic acid, and 4 equivalents of
aniline. After
heating reaction mixture partitioned against dichloromethane and sodium
hydrogen carbonate
(aqueous saturated) before chromatography.
$ 3 mL of xylene used, 20 mg of p-toluene sulphonic acid, and 4 equivalents of
aniline.
After heating reaction mixture partitioned against dichloromethane and sodium
hydrogen
carbonate (aqueous saturated) before chromatography.
9 4.9 mg (20 ~mol) Oxadiazole starting material, 0.5 mg p-toluene sulphonic
acid, 3 equivalents of
aniline, and 0.2 mL of xylene heated to 120°C in a sealed vessel for 24
hours. Purred by prep
HPLC
1° Purified using a Phenomenex C18 column eluting with aqueous (A):
[trifluoroacetic acid in
water (0.1% v/v):acetonitrile (95/5 v/v)]; organic(B): acetonitrile. Gradient
0-0.5 minutes 5% B;
0.5-0.6 minutes 5-10% B; 0.6-7.0 minutes 10-95 %; 7.0-9.8 minutes 95%; 9.8-9.9
minutes 95-5%;
9.9-11 minutes 5%. Wavelength 225 nM; flow rate 8 mL/minute, ambient
temperature
11 4 equivalents of aniline used and no xylene.
All of the compounds exemplified above showed a Ki value of less than 400 nM
when
tested in screen 1.0 (VIA filter binding assay) as described above. Examples
of specific
compounds are illustrated in Table 2 below.
CA 02554090 2006-07-20
WO 2005/079808 PCT/IB2005/000079
-47-
Table 2.
Example No. Ki (nM)
3.32
11 5.15
15 0.86
17 0.47